Ингибиторы протеинкиназы с.
Номер патента: 598
Опубликовано: 29.12.1999
Авторы: Джироусек Майкл Р., Макдональд Джон Х., Хит Уильям Ф., Рито Кристофер Дж.



















Формула / Реферат
1. Соединение формулы

где
W является О, -S- или NH,
R1 представляет собой независимо водород, галоген, С1-С4-алкил, гидрокси, С1-С4-алкокси, галогеналкил, нитро, -NH(С1-С4-алкил), N(С1-С4-алкил)2 или -NНСО(С1-С4-алкил),
R2 является водородом, СН3СО-, NН2 или гидроксигруппой,
Z является -(СН2)р- или (СН2)р-O-(СН2)p-,
R6 является -NH(CF3) или -N(CF3)(СН3),
m независимо является 0, 1, 2 или 3,
р независимо является 0, 1 или 2
или его фармацевтически приемлемая соль или сольват.
2. Соединение по п.1, в котором R2 является водородом.
3. Соединение по п.2, в котором W является -О-.
4. Соединение по п.3, которое имеет формулу

где Z является -СН2-, R6 является -NН(СF3) или N(СF3)(СН3).
5. Способ лечения сахарного диабета и его осложнений, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4.
6. Способ ингибирования протеинкиназы С, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, фармацевтически эффективного количества соединения любого из пп.1-4.
7. Способ лечения центрального ишемического повреждения мозга, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4.
8. Способ лечения сердечно-сосудистой ишемии, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4.
9. Способ лечения воспалениия, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4.
10. Способ лечения рестеноза, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4.
11. Способ лечения атеросклероза, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4.
12. Способ лечения псориаза, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4.
13. Способ лечения болезни Альцгеймера, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4.
14. Способ лечения рака, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4.
15. Фармацевтические композиции, отличающиеся тем, что содержат соединение по любому из пп.1-4 вместе с одним или несколькими фармацевтически приемлемыми наполнителями, носителями или разбавителями.
16. Способ получения соединения любого из пп.1-4, отличающийся тем, что предусматривает взаимодействие соединения формулы

где
V является -О- или N-СН3;
W является O,-S- или -NH;
R1 представляет собой независимо водород, галоген, С1-С4-алкил, гидрокси, С1-С4-алкокси, галогеналкил, нитро, -NH(С1-С4-алкил), -N(С1-С4-алкил)2 или NHCO(С1-С4-алкил),
Z является -(СН2)p или -(СН2)p-O-(СН2)p,
R6 является -NН(СF3) или -N(СF3)(СН3);
m независимо является 0, 1, 2 или 3,
р независимо является 0, 1 или 2,
с избытком смеси реагента и С1-С4-спирта в полярном апротонном растворителе, причем указанный реагент выбирают из гексаметилдисилазана или соли аммония.
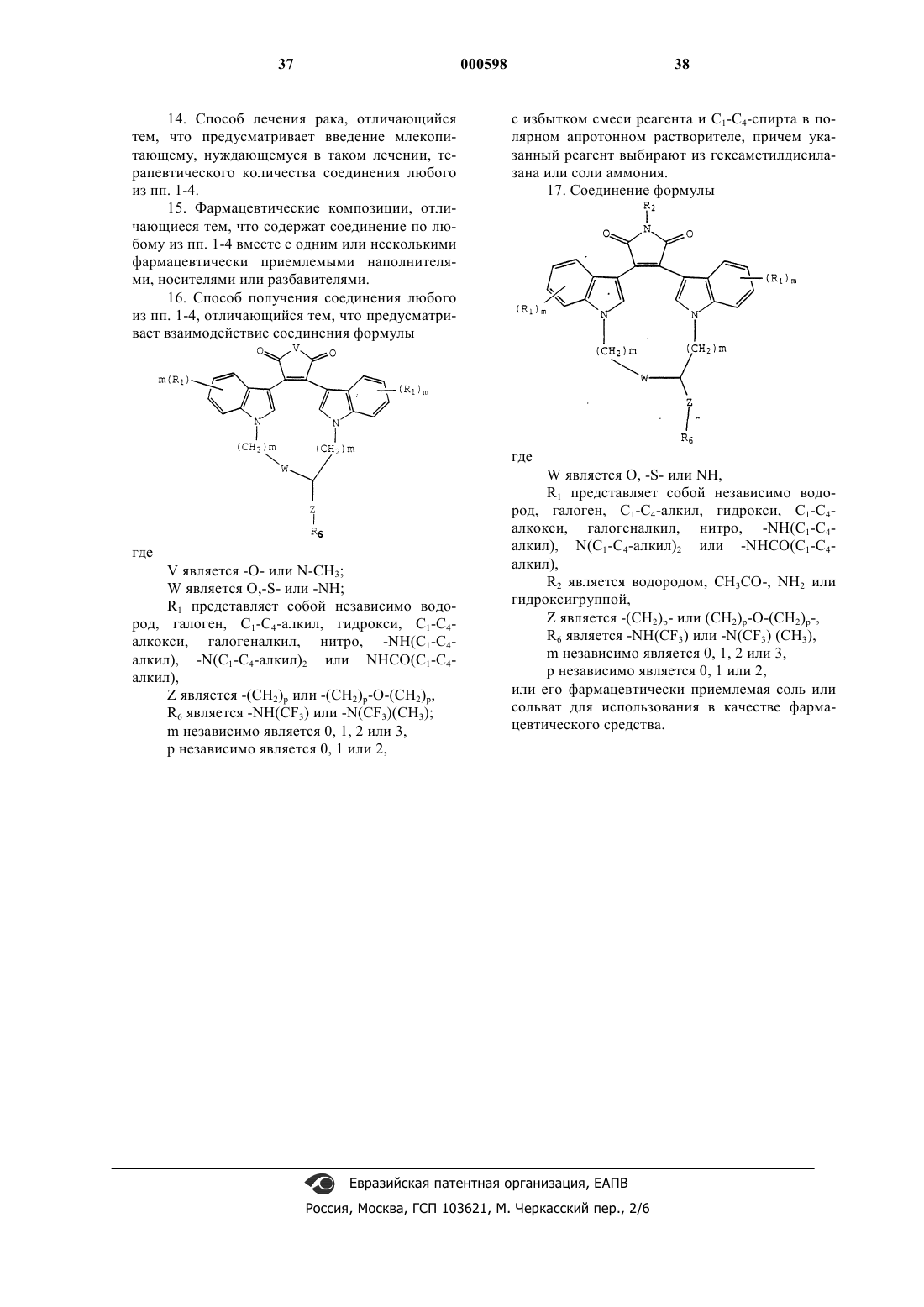
17. Соединение формулы

где
W является О, -S- или NH,
R1 представляет собой независимо водород, галоген, С1-С4-алкил, гидрокси, С1-С4-алкокси, галогеналкил, нитро, -NH(С1-С4-алкил), N(С1-С4-алкил)2 или -NHCO(C1-C4-алкил),
R2 является водородом, СН3СО-, NH2 или гидроксигруппой,
Z является -(СН2)р- или (СН2)p-O-(СН2)p-,
R6 является -NH(CF3) или -N(CF3) (СН3),
m независимо является 0,1,2 или 3,
р независимо является 0, 1 или 2,
или его фармацевтически приемлемая соль или сольват для использования в качестве фармацевтического средства.
Текст
1 Протеинкиназа С (ПКС) состоит из семейства близкородственных ферментов, которые функционируют как сериновые/треониновые киназы. Протеинкиназа С играет важную роль в передаче сигнала от клетки к клетке, экспрессии генов и регуляции дифференцировки и роста клеток. В настоящее время известны, по меньшей мере, десять изозимов (изоферментов) ПКС, которые отличаются по их распределению в ткани, ферментативной специфичности и регуляции. Nishizuka Y. Annu. Rev. Biochem. 58:31-44 (1989); Nishizuka Y. Science 258:607614 (1992). Изозимы протеинкиназы С представляют собой одиночные полипептидные цепи с длиной в пределах от 592 до 737 аминокислот. Изозимы содержат регуляторный домен и каталитический домен, соединенные линкерным пептидом. Регуляторные и каталитические домены могут быть подразделены далее на константные и вариабельные области. Каталитический домен протеинкиназы С очень сходен с каталитическими доменами других протеинкиназ, тогда как регуляторный домен является уникальным для изозимов ПКС. Изозимы ПКС обнаруживают 40-80% гомологию на уровне аминокислот среди этой группы. Однако гомология отдельного изозима между различными видами обычно более 97%. Протеинкиназа С является мембраносвязанным ферментом, который регулируется аллостерически рядом факторов, включающих мембранные фосфолипиды, кальций и некоторые мембранные липиды, такие как диацилглицерины, которые высвобождаются в ответ на активность фосфолипаз. Bell, R. M. and Burns,D. J. J. Biol. Chem. 266:4661-4664 (1991); Nishizuka Y. Science 258:607-614 (1992). Изозимы протеинкиназы С, альфа, бета-1, бета-2 и гамма,требуют для полной активации мембранного фосфолипида кальция и диацилглицерина/форболовых эфиров. Дельта-, эпсилон-, этаи тета-формы ПКС являются кальцийнезависимыми в способе их активации. Дзета- и лямбдаформы ПКС независимы как от кальция, так и от диацилглицерина, и считается, что они требуют только мембранного фосфолипида для их активации. Только один или два из изозимов протеинкиназы С могут участвовать в конкретном патологическом состоянии. Например, повышенные уровни глюкозы в крови, обнаруженные при диабете, приводят к изозим-специфическому повышению уровня бета-2-изозима в васкулярных тканях. Inoguchi et al., Proc. Natl. Acad. Sci.USA 89: 11059-11065 (1992). Связанное с диабетом повышение бета-изозимов в тромбоцитах человека коррелировано с их измененным ответом на агонисты. Bastyr III, E. J. and Lu, J. Diabetes 42: (Suppl. 1) 97A (1993). Было показано,что рецептор витамина D человека избирательно фосфорилируется протеинкиназой С-бета. 2 Это фосфорилирование было связано с изменениями в функционировании этого рецептора.Hsieh et al., Proc. Natl. Acad. Sci. USA 88:93159319 (1991); Hsieh et al., J. Biol. Chem. 268: 15118-15126 (1993). Кроме того, недавнее исследование показало, что бета-2 изозим ответственен за пролиферацию клеток эритролейкоза,тогда как альфа-изозим участвует в дифференцировке мегакариоцитов в тех же клетках.(1993). Повсеместное распространение изозимов протеинкиназы С и их важная роль в физиологии является поводом к получению высокоизбирательных ингибиторов ПКС. При условии наличия доказательства, демонстрирующего связь определенных изозимов с патологическими состояниями, резонно предположить, что ингибиторные соединения, избирательные в отношении одного или двух изозимов протеинкиназы С и других протеинкиназ, являются превосходными терапевтическими агентами. Такие соединения должны демонстрировать более высокую эффективность и меньшую токсичность, благодаря их специфичности. Микробный индолкарбазол, стауроспорин,является сильным ингибитором протеинкиназы С, который взаимодействует с каталитическим доменом этого фермента. Tamaoki et al., Biochem. Biophys. Res. Commun. 135: 397-402(1986); Gross et al., Biochem. Pharmacol. 40: 343350 (1990). Однако терапевтическая полезность этой молекулы и близкородственных соединений ограничена вследствие отсутствия специфичности в отношении протеинкиназы С по сравнению с другими протеинкиназами. Ruegg,U.T. and Burgess, G.M., Trends Pharmacol. Sci. 10: 218-220 (1989). Отсутствие избирательности приводит к неприемлемой токсичности в этом классе молекул. Дополнительный класс соединений, родственных стауроспорину, бис-индолмалеимиды,был в центре внимания недавней работы. Davisal., Biochem. Biophys. Res. Commun. 171: 10871092 (1990); Toullec et al., J. Biol. Chem: 266: 15771-15781 (1991); Davis et al., J. Med. Chem. 35: 994-1001 (1992); Bit et al., J. Med. Chem. 36: 21-29 (1993). Некоторые из этих соединений обнаружили избирательность в отношении протеинкиназы С по сравнению с другими протеинкиназами. Хотя были обнаружены соединения, которые демонстрируют специфичность относительно протеинкиназы С, очень мало известно в отношении избирательности относительно изозимов. Например, анализ изозимной избирательности стауроспорина показывает низкую избирательность в отношении изозимов, за исключением слабого ингибирования дзетаизозима по сравнению с другими изозимами.Pharmacol, 41: 387-392 (1992). Исследования ПКС-избирательного соединения,3-[1-(3 диметиламинопропил)-индол-3-ил]-4-(1Hиндол-3-ил)-1H-пиррол-2,5-диона, предполагает слабую избирательность в отношении кальцийзависимых изозимов. Toullec et al., J. Biol. Chem. 266: 15771-15781 (1991). Последующие исследования этого соединения не обнаружили различия или, возможно, показали лишь слабую избирательность для альфа-изозимов по сравнению с бета-1- и бета-2-изозимами. Martiny-Baronet al., J. Biol. Chem. 268: 9194-9197 (1993); Wilkinson, et al., Bichem. J. 294: 335 -336 (1993). Таким образом, несмотря на годы исследований и идентификацию классов соединений, которые ингибируют протеинкиназу С сильнее, чем другие протеинкиназы, остается потребность в терапевтически эффективных изозим-избирательных ингибиторах. Данное изобретение обеспечивает новые сильные ингибиторы протеинкиназы С. Соединения данного изобретения являются избирательными в отношении протеинкиназы С по сравнению с другими протеинкиназами и являются, совершенно неожиданно, высокоизбирательными в отношении изозимов. В качестве избирательных ингибиторов эти соединения применимы в лечении состояний, связанных с сахарным диабетом и его осложнениями, ишемии, воспаления, расстройств центральной нервной системы, сердечно-сосудистого заболевания, дерматологического заболевания и рака. Сущность изобретения Данное изобретение обеспечивают соединения формулы Ip независимо является 0, 1 или 2. Также обеспечены новые промежуточные продукты указанных выше соединений. Эти 4 промежуточные продукты представляют собой соединения формулы II.R1 представляет собой независимо водород, галоген, С 1-С 4-алкил, гидрокси, С 1-С 4 алкокси, галогеналкил, нитро-NH(С 1-С 4-алкил),-N(С 1-С 4-алкил)2 или -NHCO(С 1-С 4-алкил);m, независимо, является 0, 1, 2 или 3; р, независимо, является 0, 1 или 2. Одним из дальнейших аспектов данного изобретения является способ ингибирования протеинкиназы С, предусматривающий введение млекопитающему, нуждающемуся в таком лечении, фармацевтически эффективного количества соединения формулы I. Также обеспечен способ избирательного ингибирования бета-1- и бета-2-изозимов протеинкиназы С, предусматривающий введение млекопитающему, нуждающемуся в таком лечении, фармацевтически эффективного количества соединения формулыI. Далее изобретение обеспечивает способы лечения состояний, для которых была показана роль протеинкиназы С в патологии, таких как ишемия, воспаление, расстройства центральной нервной системы, сердечно-сосудистое заболевание, дерматологическое заболевание, предусматривающие введение млекопитающему, нуждающемуся в таком лечении, фармацевтически эффективного количества соединения формулыI. В частности, данное изобретение применимо в лечении осложнений диабета. Поэтому данное изобретение обеспечивает также способ лечения сахарного диабета, предусматривающий введение млекопитающему, нуждающемуся в таком лечении, фармацевтически эффективного количества соединения формулы I. Последним аспектом данного изобретения являются фармацевтические готовые формы,содержащие соединение формулы I вместе с одним или несколькими фармацевтически приемлемыми наполнителями, носителями или разбавителями. 5 Подробное описание и предпочтительные варианты Для целей данного изобретения, описанного и обсуждаемого здесь, определены следующие термины и аббревиатуры. Термин "галоген" обозначает фтор, хлор,бром или иод. Термин "С 1-С 4-алкил" обозначает алкильную группу с циклической, прямой или разветвленной цепью, такую как метил, этил, нпропил, изопропил, циклопропил, н-бутил, изобутил, втор-бутил, трет-бутил и т.п. Галогеналкил является одним из таких алкилов, замещенных одним или несколькими атомами галогена. Примером галогеналкила является трифторметил. С 1-С 4-алкокси является С 1-С 4-алкильной группой, ковалентно связанной с исходной частью молекулы связью -O-. Термин "уходящая группа" в применении к данной заявке понятен специалистам в данной области. Как правило, уходящей группой является любая группа или атом, которые увеличивают электрофильность атома, к которому они присоединены, для вытеснения. Предпочтительными уходящими группами являются трифлат, мезилат, тозилат, имидат, хлорид, бромид и йодид. Если алкилирующий агент содержит аминокислотный остаток (т.е. X, W и Y объединяются с образованием -(СН 2)n-АА-), то уходящая группа, соединенная с карбокси, предпочтительно представляет собой пентафторфениловый эфир или паранитрофениловый эфир. Термин "карбоксизащитная группа" в применении к данной заявке относится к одному из эфирных производных группы карбоновой кислоты, обычно применяемому для блокирования или защиты группы карбоновой кислоты при проведении реакций на других функциональных группах этого соединения. Виды карбоксизащитных групп не являются критическими, пока производное карбоновой кислоты является стабильным к условиям последующей (последующих) реакций и могут удаляться в нужный момент без разрушения остальной части молекулы. T.W. Greene and P. Wuts, Protective Groups inYork, N.Y., 1991, Chapter 5 дают список обычно применяемых защитных групп. См. также Е.Haslam, Protective Croups in Organic Chemistry,J.G.W. McOmie, Ed., Plenum Press, New York,N.Y., 1973. Родственный термин "защищенная карбокси" обозначает защищенную карбоксигруппу. Термин "гидроксизащитная группа" в применении к данной заявке относится к одному из эфирных или сложноэфирных производных гидроксигруппы, обычно применяемых для блокирования или защиты гидроксигруппы при проведении реакций на других функциональных группах данного соединения. Виды гидроксизащитных групп не являются критическими,пока производное гидроксигруппы стабильно к 6 условиям последующей реакции (реакций), и могут удаляться в нужный момент без разрушения остальной части молекулы. T.W. Greene andP. Wuts, Protective Groups in Organic Synthesis,John Wiley and Sons, New York, N.Y., 1991 обеспечивают список обычно используемых защитных групп. Предпочтительными гидроксизащитными группами являются трет-бутилдифенилсилилокси (TBDPS), трет-бутилдиметилсилилокси (TBDMS), трифенилметил (тритил),метокситритил или алкиловый или ариловый эфир. Родственным термином является "защищенная гидрокси", который относится к гидроксизащитной группе. Термин "аминозащитная группа" в применении к данной заявке относится к заместителям аминогруппы, обычно применяемым для блокирования или защиты аминогруппы при реакции других групп на этом соединении. Виды аминозащитных групп не являются критическими, пока производное аминогруппы является стабильным к условиям последующей реакции(реакций), и могут удаляться в нужный момент без разрушения остальной части молекулы.Organic Synthesis, Chapter 7 дают список обычно используемых защитных групп. См. также J. W.Barton, Protective Groups in Organic Chemistry,Chapter 2. Предпочтительными аминозащитными группами являются трет-бутоксикарбонил,фталимид, циклический алкил и бензилоксикарбонил. Родственный термин "защищенная амино" определяет аминогруппу, замещенную описанной аминозащитной группой. Термин "-NH-защитные группы" в применении к данной заявке относится к подклассу аминозащитных групп, обычно применяемых для блокирования или защиты -NH-групп при реакции других функциональных групп данного соединения. Виды используемой защитной группы не являются критическими, пока производное аминогруппы стабильно к условиям последующей реакции (реакций), и могут удаляться в нужный момент без разрушения остальной части молекулы. T.W. Greene and P. Wuts, Protective Groups in Organic Synthesis, Chapter 7, p. 362-385, дают список обычно используемых защитных групп. Предпочтительными -NHзащитными группами являются карбамат, амид,алкил- или арилсульфонамид. Родственный термин "защищенная -NH" определяет группу,замещенную описанной -NH-защитной группой. Термин "фармацевтически эффективное количество" в применении к данной заявке обозначает количество соединения изобретения,которое способно ингибировать активность ПКС у млекопитающих. Конкретная доза соединения, вводимого согласно данному изобретению, должна, конечно, определяться конкретными обстоятельствами случая заболевания, в том числе вводимым соединением, способом введения, конкретным подлежащим лечению 7 состоянием и подобными рассмотрениями. Соединения можно вводить различными способами, в том числе пероральным, ректальным,трансдермальным, подкожным, местным, внутривенным, внутримышечным или интраназальным способами. Для всех показаний типичная суточная доза содержит от 0,01 мг/кг до 20 мг/кг активного соединения данного изобретения. Предпочтительные суточные дозы составляют 0,05 - 10 мг/кг, идеально 0,1 - 5 мг/кг. Однако для местного введения обычная доза равна 1 - 500 мкг соединения на см 2 пораженной ткани. Предпочтительное наносимое количество соединения находится в диапазоне от 30 до 300 мг/см 2, более предпочтительно от 50 до 200 мкг/см 2 и наиболее предпочтительно от 60 до 100 мкг/см 2. Термин "лечение" здесь описывает помощь больному и лечение больного как борьбу с заболеванием, состоянием или расстройством и включает в себя введение соединения данного изобретения для предотвращения появления симптомов или осложнений, ослабления симптомов или осложнений или устранения заболевания, состояния или расстройства. Термин "изозим-селективный" означает предпочтительное ингибирование бета-1- или бета-2-изозима протеинкиназы С по сравнению с альфа-, гамма-, дельта-, эпсилон, дзета и этаизозимами протеинкиназы С. В общем, соединения демонстрируют минимально восьмикратное различие (предпочтительно десятикратное различие) в дозе, требующейся для ингибирования бета-1- или бета-2-изозима ПКС, и в дозе,требующейся для равного ингибирования альфа-изозима протеинкиназы С, как измерено в тесте ПКС. Соединения демонстрируют такое различие во всем диапазоне ингибирования и в качестве примера IC50, т.е. 50% ингибировании. Таким образом, изозим-селективные соединения ингибируют бета-1- и бета-2-изозимы протеинкиназы С при гораздо более низких концентрациях с меньшей токсичностью благодаря их минимальному ингибированию других изозимов ПКС. Как указано выше, изобретение обеспечивает соединения формулы I, которые избирательно ингибируют протеинкиназу С. Предпочтительными соединениями данного изобретения являются соединения формулыNН(СF3) или N(СН 3)(СF3), или m, независимо является 0, 1, 2 или 3 и р является 1. Благодаря кислотным частям их молекул соединения формулы I включают их фармацевтически приемлемые основно-аддитивные соли. Такие соли включают соли, образованные из неорганических оснований, таких как аммоний,гидроксиды щелочных и щелочно-земельных металлов, карбонатов, бикарбонатов и т.п., а также соли, образованные из основных органических аминов, таких как алифатические и аро 000598 8 матические амины, алифатические диамины,гидроксиалкамины и т.п. Такие основания, применимые в получении солей данного изобретения, включают, следовательно, гидроксид аммония, карбонат калия, бикарбонат натрия, гидроксид кальция, метиламин, диэтиламин, этилендиамин, циклогексиламин, этаноламин и т.п. Благодаря основной части молекулы соединения формулы I могут также существовать в виде фармацевтически приемлемых кислотноаддитивных солей. Кислоты, обычно используемые для образования таких солей, включают неорганические кислоты, такие как соляная,бромистоводородная, иодистоводородная, серная и фосфорная кислоты, а также органические кислоты, такие как пара-толуолсульфокислота,метансульфоновая, щавелевая, пара-бромфенилсульфоновая, карбоновая, янтарная, лимонная,бензойная, уксусная кислоты, и родственные им неорганические и органические кислоты. Так,такие фармацевтически приемлемые соли включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, фосфат, первичный кислый фосфат, вторичный кислый фосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, ацетат, пропионат, деканоат, каприлат, акрилат,формиат, изобутират, гептаноат, пропиолат,оксалат, малонат, сукцинат, субецат, фумарат,малеат, 2-бутин-1,4-диоат, 3-гексин-2,5-диоат,бензоат, суберат, хлорбензоат, гидроксибензоат,метоксибензоат, фталат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутират,цитрат, лактат, гиппурат, -гидроксибутират,гликоллат, малеат, тартрат, метансульфонат,пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделат, и т.п. соли. Кроме фармацевтически приемлемых солей в данное изобретение включены другие соли. Они могут служить в качестве промежуточных продуктов в очистке соединений, в получении других солей или в идентификации и характеристике соединений или промежуточных соединений. Фармацевтически приемлемые соли соединений формулы I существуют также в виде различных сольватов, например, с водой, метанолом, этанолом, диметилформамидом, этилацетатом и т.п. Могут быть также получены смеси таких сольватов. Источник такого сольвата может исходить из растворителя кристаллизации, быть свойственным растворителю получения или кристаллизации или побочным для такого растворителя. Такие сольваты также находятся в сфере действия данного изобретения. Понятно, что могут существовать различные стереоизомерные формы соединений формулы I, например, W может содержать хиральный атом углерода в замещенной алкиленовой части молекулы. Эти соединения обычно получают в виде рацематов и их можно удобно использовать в виде таковых, но можно выделить или синтезировать отдельные энантиомеры при 9 помощи стандартных способов, если это желательно. Такие рацематы и отдельные энантиомеры и их смеси составляют часть данного изобретения. Данное изобретение охватывает также фармацевтически приемлемые пролекарства соединений формулы I. Пролекарство представляет собой лекарственное средство, которое было химически модифицировано и может быть биологически неактивным в месте его действия,но которое может быть деградировано или модифицировано посредством одного или нескольких ферментативных или иных процессовin vivo до исходной биоактивной формы. Это пролекарство должно иметь фармакокинетический профиль, отличающийся от такового исходного соединения, делающий возможным более легкое всасывание через эпителий слизистой оболочки, лучшее образование солей или лучшую растворимость и/или улучшенную системную стабильность (например, увеличение периода полувыведения из плазмы). В типичном случае такие химические модификации включают следующее: 1) эфирные или амидные производные, которые могут расщепляться эстеразами или липазами,2) пептиды, которые могут узнаваться специфическими или неспецифическими протеазами, или 3) производные, которые накапливаются в месте действия через мембранный отбор формы пролекарства или модифицированной формы пролекарства, или комбинации 1-3, supra. Общепринятые способы для выбора и получения подходящих пролекарств описаны, например, в Н. Bundgaard, Design of Prodrugs, (1985). Синтез некоторых производных бисиндол-N-малеимида описан в Davis et al., U.S.Parent 5057614, включенном здесь в качестве ссылки. В общем виде, соединения данного изобретения могут быть получены следующим образом: Схема 1R1, m и галоген являются такими же, как описано ранее. Галоген предпочтительно обозначает хлор, бром или йод. Соединение III является предпочтительно 2,3-дихлор-N-метилмалеимидом. Реакция между соединением III и индолом,соединением IV, известна, как правило, как реакция Гриньяра. Реакцию проводят в инертном органическом растворителе, таком как толуол,при температуре от комнатной до температуры перегонки реакционной смеси. Наиболее важно,что реакция, изображенная в схеме 1, зависит от растворителей. При проведении в системе рас 000598 10 творителей толуол : ТГФ : эфир реакция дает соединение V с выходом более 80% и с чистотой более 95%. Продукт осаждают из реакционной смеси хлоридом аммония, NH4Cl. Полученное промежуточное соединение, соединение V,можно выделить стандартными способами. Бис-3, 4-(3'-индолил)-1N-метилпиррол-2,5 дион, соединение V, может быть затем превращено щелочным гидролизом в соответствующий ангидрид формулы VI способами, известными в данной области и описанными Brenner etal., Tetrahedron 44: 2887-2892 (1988). Предпочтительно, соединение V реагирует с 5 и КОН в этаноле при температуре в пределах от 25 С до температуры дефлегмации. Соединения формулы V обычно более стабильны, чем соединения формулы VI. Поэтому предпочтительно, чтобы соединения V взаимодействовали согласно схеме 2 с образованием соединений формулы I. Однако специалисту в данной области будет понятно, что соединения формулы VI могут также реагировать согласно схеме 2. Схема 2Z, R6 и m имеют приведенные ранее значения, L является хорошей уходящей группой,такой как хлор, бром, йод, мезил, тозил и т.п., L может также быть гидрокси или другим предшественником, который может быть легко превращен в хорошую уходящую группу способами, известными выданной области. Например,гидрокси может легко превращаться в эфир сульфокислоты, такой как мезиловый эфир, реакцией гидрокси с метансульфонилхлоридом с образованием мезилатной уходящей группы. Реакцию, представленную схемой 2, выполняют любым из известных способов получения N-замещенных индолов. Эта реакция обычно включает приблизительно эквимолярные количества двух реагентов, хотя возможны также другие соотношения, в частности, такие, при которых алкилирующий реагент находится в избытке. Лучше всего эту реакцию проводить в полярном апротонном растворителе с использованием соли щелочного металла или других подобных условий алкилирования, как это понятно специалистам в данной области. Когда уходящей группой является бром или хлор, может быть добавлено каталитическое количество 11 такой соли, как йодид, например йодид калия,для ускорения реакции. Условия реакции включают следующее: гексаметилдисилазид калия в диметилформамиде или тетрагидрофуране, гидрид натрия в диметилформамиде. Предпочтительно, реакцию проводят при медленном реверсивном добавлении карбоната цезия в ацетонитриле, диметилформамиде(ДМФ) или тетрагидрофуране (ТГФ). Температура реакции предпочтительно находится в диапазоне от приблизительно температуры окружающей среды до приблизительно температуры дефлегмации реакционной смеси. Специалисту в данной области понятно,что реакцию, описанную в схеме 2, можно использовать с соединениями формулы VIIа:LХ',VIIa где X' и Y' представляют собой защищенную карбокси-, защищенную гидроксигруппу или защищенный амин. После алкилирования по схеме 2 Х' и Y' могут быть превращены в части молекулы, способные связываться с образованием заявленных соединений. Этот способ является предпочтительным способом получения соединений формулы I, где W является -S-, -Оили NH. Связывание X' и Y' с образованием различных производных простых эфиров, тиоэфирных или аминоэфирных производных известно в данной области и описано, например, вEvans, et al., J. Org. Chem. 50:1830 (1985). Специалисту в данной области понятно,что реакцию схемы 2 можно проводить также с использованием подходящих защитных групп в виде двухстадийного синтеза. То есть, при помощи подходящих защитных групп алкилирование каждого азота индолила соединения формулы V и VI может протекать, как описано здесь, в виде двух стадий (алкилирования одного индолила при медленном реверсивном добавлении и последующего закрывания макроцикла алкилированием второго индолила). Наиболее неожиданно, соединения данного изобретения можно получать со значительно более высоким выходом при проведении алкилирования при медленном реверсивном добавлении к СS2 СО 3 в полярном апротонном растворителе. Медленное реверсивное добавление включает соединение смеси соединения и алкилирующего агента с основанием при скорости от приблизительно 0,1 мл/ч до приблизительно 2,0 мл/ч. Концентрация каждого реагента в смеси равна приблизительно 1,5 молярной 0,001 молярной. Специалисту в данной области понятно, что при более высокой скорости добавления в реакции могла бы использоваться более низкая концентрация реагентов. Подобно 12 этому при более медленной скорости реакции более высокая концентрация реагентов могла бы использоваться в реакции. Предпочтительно,соединение добавляют со скоростью приблизительно 0,14 мл/ч с соединением и алкилирующим агентом при 0,37 молярной концентрации. Предпочтительно, чтобы CS2 СО 3 добавлялся в избытке, наиболее предпочтительно отношениеCS2 СО 3 к алкилирующему агенту, равное 4:1. Предпочтительными полярными апротонными растворителями являются ацетонитрил, диметилформамид (ДМФ), ацетон, диметилсульфоксид (ДМСО), диоксан, метиловый эфир диэтиленгликоля (диглим), тетрагидрофуран (ТГФ) или другие полярные апротонные растворители,в которых эти реагенты растворимы. Реакцию проводят при температурах в пределах от приблизительно 0 С до температуры дефлегмации. Специалисту в данной области понятно, что соотношение смеси соединения и алкилирующего агента не является критическим. Однако,предпочтительно, смешивать эти реагенты в эквивалентном отношении друг к другу, равном 0,5:3. Наиболее предпочтительно соотношение,равное 1:1. Когда V является N-СН 3, соединение II превращается в соответствующий ангидрид (V является О) щелочным гидролизом. Щелочной гидролиз включает реакцию соединения с основанием, таким как гидроксид натрия или гидроксид калия, в С 1-С 4-спирте (предпочтительно этаноле), смеси ДМСО/вода, диоксан/вода или ацетонитрил/вода при температуре от приблизительно 25 С до приблизительно температуры дефлегмации. Концентрация этих реагентов не является критической. Ангидрид (V является О) превращают в малеимид формулы I аммонолизом. Аммонолиз включает реакцию ангидрида с избытком гексаметилдисилазана или соли аммония (ацетата,бромида или хлорида аммония) и С 1-С 4-спирта(предпочтительно метанола) в полярном апротонном растворителе, таком как ДМФ, при комнатной температуре. Предпочтительно, гексаметилдисилазан или соль аммония реагирует при соотношении, большем чем 5:1 экв. ангидрида. Еще один способ получения соединений формулы I представлен в схеме 3. Этот способ применим, в частности, когда W является -NH.Ac представляет собой ацетил, R6, Z и m имеют приведенные ранее значения. Алкилирование соединения VI и VIII имеет место при условиях, описанных ранее и известных в данной области. Алкилирование соединения IX галогенкетоном, соединением X, происходит при обсужденных выше условиях. Превращение ангидрида в малеимид соединения ХI происходит, как описано ранее. Например, ангидрид может превращаться в бис-индолмалеимид реакцией ангидрида с гексаметилдисилазаном и метанолом в инертном органическом растворителе, таком как ДМФ, при комнатной температуре. Защищенная гидроксигруппа, представленная ОАс, легко гидролизуется с образованием спирта (например, К 2 СО 3 в водном метаноле и ТГФ). Полученный спирт превращается в уходящую группу способами, известными в данной области, такими как реакция спирта с мезилхлоридом в триэтиламине при 0 С. Уходящая группа заменяется азидом, таким как NaN3, в ДМФ при 50 С. Полученный азид восстанавливается с образованием амина с использованием катализатора Линдлера в присутствии Н 2. Макроциклу позволяют замкнуться через внутримолекулярное основание Шиффа. Основание Шиффа восстанавливается при стандартных условиях в присутствии, например, NaCNBH3 или других восстанавливающих агентов с образованием макроциклов формулы I. Промежуточное соединение данного изобретения получают согласно схеме 4. Эта схема применима, в частности, для получения соединений, в которых W является -О-.R8 является N3, NH-защитной группой,аминозащитной группой или гидроксизащитной группой, m является независимо 0, 1, 2 или 3 и L является хорошей уходящей группой, такой как хлор, бром, йод, мезил, тозил и т.п. L предпочтительно является мезилом. R8 является предпочтительно защищенной гидроксигруппой наиболее предпочтительно -О-тритилом. Схема 4 представляет стереоселективный синтез линкерной части макромолекулы. S-энантиомер представлен выше, однако, специалисту в данной области будет понятно, что соответствующий энантиомер или смесь энантиомеров можно было бы получать аналогичным образом. Кроме того, специалисту в данной области будет понятно, что аналогичная реакция с метилзамещенным эпоксидом или реактивом Гриньяра могла бы быть использована для получения различных линкеров, содержащих метилзамещенный алкилен. В приведенной выше реакции эпоксид, соединение (ХIV), открывают при помощи реактива Гриньяра. Реакцию проводят в присутствии содержащего медь комплексообразующего агента, однако можно использовать другие алкилирующие условия. Реакцию проводят в инертном растворителе при температуре между-30 С и температурой дефлегмации реакционной смеси. Реакция дает соединение (ХV), которое может реагировать далее без очистки. Соединение (ХV) аллилируют при обычных условиях, известных в данной области для получения простых эфиров. Реакция, иллюстрированная в схеме 4, является синтезом Уильямсона. Образование алкоксида натрия с использованием NaN, NaOH или КОН с последующим аллилированием аллилбромидом дает диен, соединение (ХVI). Соединение (ХVI) превращают в спирт, соединение (ХVII), стандартными способами. Например, соединение (ХVI) можно превратить в озонид обработкой озоном при низкой температуре. Затем озонид восстанавливаютNaBH4, LiAlH4, BH3 или каталитическим гидрированием с избытком Н 2 с получением спирта,соединения (ХVII). Гидроксигруппы соединения(ХVII) превращают в уходящую группу L стандартными способами, такими как реакция спирта с мезилхлоридом в триэтиламине. 15 Во всех приведенных выше схемах предпочтительно проведение реакций с подходящими защитными группами. В частности, предпочтительно, чтобы R1 был защищен во время алкилирования и/или ацилирования, после чего защитная группа должна быть удалена. Подобным образом, если R6 должен быть -NH(CF3),реакции лучше всего проводить с аминозащитной группой. Однако специалисту в данной области понятно, что многие из этих реакций можно проводить без защитных групп, если использовать подходящие условия реакции,блокирующие реагенты или т.п. Предпочтительно, что, когда макроцикл содержит гидроксиостаток, он защищен группами третбутилдифенилсилилокси (TBDPS) или трифенилметил (тритил) во время алкилирования или ацилирования индола. Полученные соединения формулы I могут быть выделены и очищены стандартными способами. Соединения схем 1-4 и другие реагенты,требующиеся для приведенных выше реакций,либо являются коммерчески доступными, известными в данной области, либо могут быть получены известными способами. Например,соединение III может быть получено способами,описанными Edge et al., Chem. and Ind. 130(1991), соединение IV предпочтительно получают in situ реакцией подходящим образом замещенного индола с алкилмагнийгалогенидом,таким как этилмагнийбромид, известным образом. Следующие примеры и получения приведены лишь для дальнейшей иллюстрации данного изобретения. Объем данного изобретения не ограничивается только следующими далее примерами. Для помощи специалисту обеспечена следующая структура для иллюстрации с номенклатурой характерного соединения, принятой здесь: В следующих далее примерах и получениях точка плавления, спектры ядерно-магнитного резонанса, масс-спектры, жидкостная хроматография высокого давления на силикагеле, N,Nдиметилформамид, палладий на древесном угле,тетрагидрофуран и этилацетат обозначают в виде аббревиатур: Т.пл., ЯМР, МС, ВЭЖХ,ДМФ, Рd/С, ТГФ и EtOAc, соответственно. Термины "ЯМР" и "МС" показывают, что спектр соответствует желаемой структуре. 16 Получение 1. 2,3-бис-(3'-индолил)фуран 1,4-дион. Этоксид натрия (3,56 г, 50 ммоль) добавляли к раствору, содержащему 2,3-дихлормалеиновый ангидрид (5,56 г, 33,3 ммоль) и гидрохлорид метиламина (3,50 г, 55,0 ммоль) в 40 мл уксусной кислоты. Смесь перемешивали под осушительной трубкой с CaCl2 при 25 С в течение 16 ч и затем нагревали с обратным холодильником в течение 4 ч. Охлажденную смесь выливали в воду (350 мл) и экстрагировалиEtOAc (3 x 75 мл). Объединенные органические экстракты промывали порциями по 100 мл насыщенного водного NaHCO3, воды и рассола и сушили (MgSO4). Растворитель выпаривали при пониженном давлении. Остаток перекристаллизовывали из этанола с получением 3,82 г (64%) 2,3-дихлор-N-метилмалеимида в виде белых кристаллов. Концентрирование маточного раствора и хроматография остатка радиальной препаративной хроматографией(Chromatron,Harrison Research) дали дополнительно 0,81 г 2,3-дихлор-N-метилмалеимида, что повысило выход до 77%. Раствор индола (10,5 г, 90 ммоль) в 175 мл сухого толуола обрабатывали по каплям на протяжении 1 ч в атмосфере N2 раствором этилмагнийбромида (1,0 М в ТГФ, 90 мл, 90 ммоль). После завершения добавления светло-зеленый раствор нагревали при 40 С в течение 30 мин и затем охлаждали до 25 С. Раствор 2,3-дихлорN-метилмалеимида (3,8 г, 21 ммоль) в 50 мл толуола добавляли на протяжении 30-минутного периода. Реакционную смесь нагревали при 100 С в течение 3 ч, затем охлаждали до 25 С и гасили 100 мл 20%-ного водного раствора лимонной кислоты. Слои разделяли. Водную фазу экстрагировали EtOAc (50 мл). Объединенные органические слои сушили над безводнымMgSO4. Растворитель выпаривали при пониженном давлении. Остаток поглощали 30 мл ацетона и отстаивали при 5C в течение 40 ч. Твердую массу собирали и промывали смесью льда с эфиром с получением 5,25 г (73%) 3,4 бис-(3'-индолил)-1-метилпиррол-2,5-диона в виде красного твердого вещества, т.пл. 276-278 С. К раствору 3,4-бис-(3'-индолил)-1-метилпиррол-2,5-диона в 150 мл этанола добавляли 5 н КОН (50 мл). Смесь перемешивали в течение 4 ч при 25 С и разбавляли 150 мл воды. Большую часть этанола выпаривали при пониженном давлении. Затем смесь подкисляли до рН 1. Осажденный продукт отфильтровывали и промывали водой. Неочищенный продукт растворяли в минимальном объеме СН 2 Сl2 и медленно фильтровали через двухдюймовую колонку силикагеля, элюируя 50% EtOAc в гексане, с получением целевого соединения (3,10 г, 79%) в виде красного твердого вещества. Т.пл. 225228 С. 17 Получение 2. 1-(трет-бутилдиметилсилилокси)-4-(трет-бутилдифенилсилилоксибутан-3 ол. К безводному раствору в CH2Cl2 (110 мл) 3-бутен-1-ола (15 г, 0,21 моль) добавляли имидазол (28,6 г, 0,42 моль, 2 экв.) и затем третбутилдиметилсилилхлорид (32 г, 0,22 моль). После 90 мин реакция завершалась, как показала ТСХ (10% EtOAc/гексан). Раствор в CH2Cl2 переносили в делительную воронку, разбавляли СН 2 Сl2 (110 мл), промывали водой (200 мл) и рассолом (200 мл). Органический слой собирали, сушили над MgSO4, фильтровали и растворитель удаляли в вакууме с получением масла[1-(O-TBDMS)-3-бутена], которое использовали в следующей реакции. Полученное выше масло растворяли в смеси ацетона (400 мл) и воды (50 мл). Затем добавляли N-метилморфолин-N-оксид (85,2 г, 0,63 моль, 3 экв.). Полученную суспензию охлаждали до 0 С и через 10 мин добавляли каталитическое количество OsO4 (0,3 г). Полученную суспензию перемешивали в течение ночи, постепенно подогревая до комнатной температуры. ТСХ (25% EtOAc/гексан) показала завершение реакции. Реакционную смесь гасили бисульфитом натрия, разбавляли эфиром (1 л), промывали водой (400 мл) и рассолом (400 мл). Органический слой собирали. Водный слой экстрагировали эфиром (2 х 500 мл). Объединенные органические слои сушили, фильтровали и концентрировали с получением 4-(O-TBDMS)-1,2 бутандиола в виде масла, которое использовали для следующей реакции. Масло растворяли в безводном CH2Cl2 (250 мл). К раствору добавляли имидазол (30 г, 0,44 моль, 2,5 экв.) в виде твердого вещества. Полученный раствор охлаждали до 0 С. После охлаждения в течение 15 мин раствор в CH2Cl2 (50 мл) трет-бутилдифенилсилилхлорида (50 г, 0,18 моль, 1 экв.) добавляли по каплям в течение 45 мин. После завершения добавления перемешивание продолжали при 0 С в течение 2,5 ч. Раствор переносили в делительную воронку, разбавляли CH2Cl2 (250 мл), промывали водой, рассолом, сушили над MgSO4 и фильтровали. Растворитель удаляли при пониженном давлении с получением неочищенного продукта в виде масла. Неочищенный продукт очищали, элюируя его (10% Et ОАс/гексан) через короткую колонку силикагеля. Элюирующий растворитель удаляли в вакууме с получением в виде вязкого масла целевого промежуточного продукта (78,1 г, общий выход 93%). МС. Получение 3. 1-(трет-бутилдиметилсилилокси)-3-(3-иодпропилокси)-4-(трет-бутилдифенилсилилокси)бутан. К раствору спирта из получения 2 в смеси метиленхлорид (20 мл)/циклогексан (100 мл) в атмосфере N2 из баллона добавляли аллилтрихлорацетимидат (17,82 г, 88 ммоль, 2,2 экв.),после чего добавляли трифторметансульфоно 000598 18 вую кислоту (50 мкл/г исходного материала,0,92 мл). После 20 ч раствор фильтровали и фильтрат промывали насыщенным водным раствором NaHCO3, водой и затем рассолом. Органический слой собирали и сушили над MgSO4. Растворитель удаляли с получением масла, которое очищали флэш-хроматографией на силикагеле, элюируя гексаном и увеличивая полярность мобильной фазы до 5% этилацетата в гексане в объеме более нескольких литров, с получением 19,27 г аллилового эфира 1-(третбутилдиметилсилилокси)-3-(пропенокси)-4(трет-бутилдифенилсилилокси)бутана в виде светло-коричневого масла (выход 97%). К раствору аллилового эфира (14,16 г,28,38 ммоль, 1 экв.) в ТГФ (60 мл) добавляли 9BBN (9-борабицикло [3,3,1]-нонан, 0,5 М раствор в ТГФ, 60 мл, 30 ммоль, 1,1 экв.) по каплям в атмосфере азота. После 3 ч реакции ТХС (10%EtOAc в гексане) показала, что исходный материал был потреблен. К этому раствору добавляли 3 М водный раствор NаОН (10,41 мл, 31,22 ммоль, 1,1 экв.), а затем медленно по каплям(1,5 ч) добавляли 30% раствор пероксида водорода (10,3 мл, 90,82 ммоль, 3,2 экв.). Температуру реакции во время гашения пероксидом поддерживали ниже 50 С (баня со льдом). После 30 мин добавляли хлорид натрия до насыщения раствора. Органический слой удаляли, водный слой экстрагировали эфиром, объединенные органические слои сушили и фильтровали, фильтрат концентрировали с получением масла. Неочищенное масло очищали флэшхроматографией на силикагеле, элюируя смесью 10% EtOAc/гексан и увеличивая полярность до 20% EtOAc/гексан, добавляя приблизительно 1,5 л растворителя, с получением 9,53 г светложелтого масла (выход 65%). МС. К раствору укзанного выше спирта в безводном эфире (150 мл) при 0 С добавляли триэтиламин (2,93 г, 28,91 ммоль, 1,5 экв.), а затем при интенсивном перемешивании добавляли по каплям мезилхлорид (3,31 г, 28,91 ммоль, 1 экв.). После 3 ч пермешивания при 0 С ТСХ(10% EtOAc в гексане) показала, что исходный материал был потреблен. Реакционную смесь разбавляли эфиром, промывали водой, рассолом, сушили над MgSO4 и растворитель удаляли. Полученное масло пропускали через подушку диоксида кремния, элюируя смесью 25%EtOAc/гексан и элюат концентрировали. К раствору в ацетоне (200 мл) полученного масла добавляли NaHCO3 (0,17 г, 1,93 ммоль, 0,1 экв.) и NaI (28,88 г, 192,7 ммоль, 10 экв.). После перемешивания в течение 30 мин при комнатной температуре в атмосфере азота реакционную смесь нагревали до 50 С на водяной бане. После 2,5 ч ТСХ (10% EtOAc в гексане) показала, что мезилат был потреблен. Реакционную смесь разбавляли эфиром (500 мл), промывали холодным насыщенным водным раствором Na2CO3, 19 водой, рассолом, сушили (MgSO4) и растворитель удаляли. Полученное масло пропускали через подушку диоксида кремния, элюируя смесью 5% ЕtОАс в гексане с получением 10,3 г очищенного целевого соединения в виде бесцветного масла (выход 85%). Получение 4. (+)3,4-[(N,N'-1,1'-(3-3-третбутилдифенилсилилоксиметилен)гексан)-бис(3,3'-индолил)]-1-(метил)пиррол-2,5-дион. Раствор в ДМФ (50 мл) бис-(3,3'-индолил)1-(метил)-пиррол-2,5-диона (3,41 г, 10,0 ммоль),содержащий дибромид-3-трет-бутилдифенилсилилоксиметилен-1,6-дибромгексан (5,64 г, 11 ммоль, полученный аналогично получению бензоилпроизводного в получении 2), добавляли при помощи насоса со шприцем в течение 15 ч к суспензии Сs2 СО 3 (11,2 г, 34,3 ммоль) при 60 С. Через 4 ч после завершения добавления реакционную смесь охлаждали до комнатной температуры, выливали в воду (1,5 л) и экстрагировали СН 2 Сl2 (3 х 300 мл). Органическую фазу промывали водой, сушили, фильтровали и концентрировали. Концентрат очищали флэш-хроматографией, элюируя смесью 10-25% этилацетат/гексан, с получением 2,95 г (выход 43%) макроцикла 3,4-[(N,N'-1,1'-(3-3-трет-бутилдифенилсилилоксиметилен)гексан)-бис-(3,3'-индолил)]-1-(метил)-пиррол-2,5-диона в виде красного масла. MC. Получение 5. (S)-метил-4-трет-бутилдифенилсилилокси-3-(аллилокси)бутират. К раствору в циклогексане (400 мл) (S)метил 4-трет-бутилдифенилсилилокси-3-(гидрокси)бутирата (20,0 г, 53,7 ммоль) добавляли аллилтрихлорацетимидат (21,74 г, 107,4 ммоль), а затем трифторметансульфоновую кислоту (1 мл,50 мл/г спирта) в виде 5 порций на протяжении 30 мин с перемешиванием в атмосфере азота. После 70 ч образовавшуюся твердую массу отфильтровывали, фильтрат промывали циклогексаном и летучие вещества удаляли в вакууме. Полученное масло помещали на пробку из диоксида кремния и промывали гексаном, продукт элюировали смесью 10% этилацетат/гексан. ЯМР показал присутствие остаточного имидата(10%), однако эту массу использовали без дополнительной очистки. Остаток дал выход 24,76 г вещества, 22,2 г которого было целевым продуктом (100%). МС. Получение 6. (S)-4-трет-бутилдифенилсилилокси-3-(2-иодэтокси)-1-йодбутан.DIBAL-H (231 мл, 1,0 М в толуоле, 231 ммоль) добавляли по каплям на протяжении 40 мин к раствору (S)-метил 4-трет-бутилдифенилсилилокси-3-(аллилокси)бутирата (23,8 г, 57 ммоль) в безводном ТГФ (1,0 л) при -75 С в атмосфере N2. После перемешивания в течение 1,5 ч смеси давали нагреться до -10 С и гасили 5% водным раствором метанола и большим количеством цеолита. Погашенную реакционную смесь фильтровали через подушку цеолита,фильтрат концентрировали и распределяли ме 000598 20 жду эфиром и 20% раствором лимонной кислоты. Эфирный слой сушили и концентрировали в вакууме. Оставшееся масло пропускали через подушку диоксида кремния, элюируя хлороформом с получением 20,6 г (93%) (S)-4-третбутилдифенилсилилокси-3-аллилоксибутан-1 ола. К раствору в метаноле (500 мл) (S)-4-третбутилдифенил-силилокси-3-аллилоксибутан-1 ола (20,6 г, 53,6 ммоль) добавляли озон при-78 С в течение приблизительно 12 мин. Реакционную смесь, приобретшую слабоголубую окраску, добавляли в реакционный сосуд. Реакционной смеси давали нагреться до комнатной температуры. Летучие вещества удаляли в вакууме. Остаток пропускали через пробку диоксида кремния, элюируя этилацетатом, с получением 16,4 г (79%) (S)-4-третбутилдифенилсилилокси-3-(2-гидроксиэтокси) бутан-1-ола в виде бесцветного масла. К раствору в эфире (600 мл) (S)-4-третбутилдифенилсилилокси-3-(2-гидроксиэтокси) бутан-1-ола (15,7 г, 40,4 ммоль) при 0 С в атмосфере азота добавляли триэтиламин (16,8 мл,121 ммоль), а затем мезилхлорид (9,38 г, 121 ммоль). После 3 ч раствор фильтровали, фильтрат промывали водой (2 х) , рассолом (2 х), сушили над Na2SO4 и концентрировали в вакууме. Остаток дал 21,9 г (99%) бисмезилата в виде желтого масла, которое непосредственно использовали далее. Бисмезилат растворяли в ацетоне (1,4 л), который был подвергнут перегонке над карбонатом калия. К этому раствору добавляли Nal (90,4 г, 603 ммоль) и 0,05 экв. NaHCO3(170 мг, 2 ммоль). Реакционную смесь выдерживали при 56 С в течение 24 ч и фильтровали,фильтрат концентрировали в вакууме. Остаток распределяли между эфиром и 10% NaSO4,эфирный слой промывали рассолом, сушили над(73,2%) (S)-4-трет-бутилдифенилсилилокси-3(2-йодэтокси)-1-йодбутана в виде бесцветного масла. Общий выход был 54%. МС: MW = 608,39,наблюдали: 559 (М-трет-бутил, FD, CHCl3) . Получение 7. (S)-3-(трет-бутилдифенилсилилоксиметилен)-1,6-дибромгексан. После процедуры, описанной для получения рацемического дибромида 3-(трет-бутилдифенилсилилоксиметил)-1,6-дибромгексана, (S)(-)-3-(третбутилдифенилсилилоксиметил)-1,6 гександиол (4,85 г, 12,53 ммоль) взаимодействовал с N-бромсукцинимидом (5,35 г, 30,1 ммоль) и трифенилфосфином (7,87 г, 30,1 ммоль) в СН 2 Сl2 (150 мл) при 0 С с получением 4,81 г(75%) (S)-(-)-3-(трет-бутилдифенилсилилоксиметил)-1,6-дибромгексана в виде прозрачного бесцветного масла, которое было гомогенным согласно ТСХ (Rf= 0,8, 10% EtOAc в гексане). Как ТСХ-свойства, так и 1 Н-ЯМР-спектры этого соединения были идентичными во всех отноше 21 ниях со свойствами рацемического изомера. МС. 1H-ЯМР (300 Мгц, СDСl3) 1,06 (с,9 Н), 1,352,10 (м,7 Н), 3,55 (м,4 Н), 3,56 (приб. д., 2 Н, J = 4 Гц), 7,40 и 7,64 (м, 10 Н). Получение 8. 1-(трет-бутилдиметилсилилокси)-3-(2-йодэтокси)-4-(трет-бутилдифенил) бутан. Аллиловый эфир 1-(трет-бутилдиметилсилилокси)-3-(аллилокси)-4-(трет-бутилдифенил) бутана (21,6 г, 43,4 ммоль) растворяли в метаноле (500 мл) и охлаждали до -78 С в атмосфере азота. Озон барботировали в реакционную смесь, после 11 мин реакция завершалась согласно ТСХ (гексан/этилацетат, 9:1). Добавляли боргидрид натрия (9,9 г, 6 экв.) и после 5 мин реакционной смеси давали нагреться до комнатной температуры. Метанол удаляли в вакууме. Остаток суспендировали в эфире (800 мл). Эфир промывали водой, а водный раствор вновь промывали эфиром. Объединенные органические слои промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением масла. Материал пропускали через подушку диоксида кремния со смесью 5% этилацетат/гексан с последующим элюированием продукта смесью 25% этилацетат/гексан с получением 11,0 г (выход 50%) 1-трет-бутилдиметил-силилокси)-3-(2 гидрокси)этокси)-4-(трет-бутилдифенил)бутана в виде светло-желтого масла. МС. ЯМР. К раствору в безводном эфире (200 мл) 1(трет-бутилдиметилсилилокси)-3-(2-гидрокси) этокси)-4-(трет-бутилдифенил)бутана, (11,0 г,21,9 ммоль) в атмосфере азота при 5 С добавляли триэтиламин (4,6 мл, 1,5 экв.) и метансульфонилхлорид (2,5 мл, 1,5 экв.). После 1,5 ч реакция завершалась согласно ТСХ (5% этилацетат/дихлорметан). Реакционную смесь разбавляли эфиром (250 мл), промывали водой (2 х),рассолом (2 х), сушили ( Na2SO4) фильтровали и концентрировали в вакууме с получением масла. Этот материал пропускали через подушку диоксида кремния, элюируя смесью 5% этилацетат/гексан, а затем смесью 25% этилацетат/гексан с получением 11,6 г (выход 91%) мезилата 1-(трет-бутилдиметилсилилокси)-3-(2(метансульфонилокси)этокси)-4-(трет-бутилдифенил)-бутана в виде масла. МС. ЯМР. К раствору в ацетоне (300 мл) мезилата 1(трет-бутилдиметилсилилокси)-3-(2-(метансульфонилокси)этокси)-4-(трет-бутилдифенил)бутана (11,6 г, 20 ммоль) в атмосфере азота добавляли йодид натрия (44 г, 15 экв.) и бикарбонат натрия (170 мг, 0,1 экв.). Смесь нагревали с обратным холодильником в течение 18 ч с последующим удалением ацетона в вакууме. Полученный остаток суспендировали в эфире, промывали водой (2 х) и водный раствор вновь промывали эфиром. Объединенные эфирные слои промывали 10% раствором сульфита натрия, рассолом (2 х), сушили (МgSО 4), фильтро 000598 22 вали и концентрировали в вакууме с получением 10,7 г (выход 87%) целевого йодида в виде масла, которое использовали без дополнительной очистки. МС. ЯМР. Получение 9. 3,4-[(N,N'-1,1'-2-этокси)3-(О)-4"'-(гидрокси)бутан)-бис-(3,3'-индолил)]1(Н)-пиррол-2,5-дион. К раствору в диметилформамиде (250 мл) бис-(3,3'-индолил)-1-(метил)пиррол-2,5-диона(17,9 г, 52,5 ммоль, 3 экв.) в атмосфере азота добавляли карбонат цезия (68,4 г, 4 экв.). К полученной суспензии добавляли 1-(третбутилдиметилсилилокси)-3-(2-йодэтокси)-4(трет-бутилдифенил-силилокси)бутан (10,7 г,17,5 ммоль). Реакционную смесь перемешивали в течение 18 ч при комнатной температуре. ТСХ(5% этилацетат/гексан) показала исчезновение йодида. Реакционную смесь выливали в этилацетат (1200 мл) и промывали 1 н. НСl (400 мл) с последующей дополнительной промывкой этилацетатом (2 х). Объединенные порции этилацетата промывали насыщенным раствором бикарбоната натрия, (2 х), сушили (MgSO4), фильтровали и концентрировали в вакууме. Диметилформиат удаляли азеотропной перегонкой с ксилолом. Полученную красную смолу суспендировали в дихлорметане и ацетонитриле с получением суспензии твердого вещества. Ее концентрировали, добавляли еще раз дихлорметан,охлаждали и фильтровали с получением красного твердого вещества. Часть целевого продукта экстрагировали из этого твердого вещества растиранием в дихлорметане, а затем в этилацетате. Фильтраты концентрировали в вакууме, полученный осадок абсорбировали на диоксиде кремния и наносили на большую флэш-колонку. Диалкилированный побочный продукт удаляли,элюируя смесью гексан/этилацетат (5:1), и затем элюируя продукт смесью гексан/этилацетат(3:1) получали 8,2 г (57%) моноалкилированного продукта, 3-[(N-1-(2-этокси-(3-(О)-4-(третбутилдифенилсилилокси)-1-(трет-бутилдиметилсилилокси)бутаниндол-3-ил]-4-[индол-3 ил]-1N-(метил)-пиррол-2,5-дион. МС. ЯМР. К раствору в метаноле (450 мл) третбутилдиметилсилилового эфира, 3-[(N-1-(2 этокси-(3-(О)-4-(трет-бутилдифенилсилилокси)-1-(трет-бутилдиметилсилилокси)бутаниндол-3-ил]-4-[индол-3-ил]-1N-(метил)пиррол 2,5-диона (8,2 г, 9,9 ммоль) в атмосфере азота при 5C добавляли моногидрат п-толуолсульфокислоты (0,16 г, 0,085 экв.). Спустя 2 ч ТСХ(50% этилацетат/гексан) показала, что реакция близка к завершению. Реакцию гасили твердым бикарбонатом натрия (0,14 г). Метанол удаляли в вакууме. Полученный остаток растворяли в этилацетате, промывали 0,1 н. гидроксидом натрия, рассолом (2 х) , сушили (MgSO4), фильтровали и концентрировали в вакууме с получением красной пены. Это вещество абсорбировали на диоксиде кремния и помещали на подушку диоксида кремния. Элюируя смесью гек 23 сан/этилацетат (2:1) удаляли остаточный исходный материал и затем, элюируя смесью гексан/этилацетат (1:1) и смесью гексан/этилацетат(1:2), получили 6,4 г (91%) 3-[N-1-(2-этокси-(3(0)-4-трет-бутилдифенилсилилокси)-1-(гидрокси)бутаниндол-3-ил]-4-[индол-3-ил]-1N-(метил)-пиррол-2,5-дион. МС. ЯМР. К раствору в безводном эфире (500 мл) этого 3-[(N-1-(2-этокси-(3-(O)-4-(трет-бутилдифенилсилилокси)-1-(гидрокси)бутаниндол 3-ил]-4-[индол-3-ил]-1N-(метил)пиррол-2,5 диона (6,36 г, 8,9 ммоль) в атмосфере азота при 5C добавляли триэтиламин (1,9 мл, 1,5 экв.) и метансульфонилхлорид (1,0 мл, 1,5 экв.). После 3 ч добавляли дополнительно триэтиламин (1,25 мл, 1,0 экв.) и метансульфонилхлорид (0,7 мл,1,0 экв.). Спустя 1 ч ТСХ (50% этилацетат/гексан) показала завершение реакции. Реакционную смесь разбавляли эфиром(250 мл), промывали водой, 0,1 н. НСl и рассолом (2 х). Эфир сушили (МgSO4), фильтровали и концентрировали в вакууме с получением 7,0 г мезилата, 3-[N-1-(2-этокси-(3-(О)-4-(трет-бутилдифенилсилилокси)-1-(метансульфонилокси)-бутаниндол-3-ил]-4-[индол-3-ил]-1N-(метил)пиррол-2,5-диона. МС. К раствору в ацетоне (200 мл) мезилата, 3[(N-1-(2-этокси-(3-(O)-4-(трет-бутилдифенилсилилокси)-1-(метансульфонилокси)бутаниндол-3-ил]-4-[индол-3-ил]-1N-(метил) пиррол-2,5-диона, (7,0 г, 8,9 ммоль) в атмосфере азота добавляли йодид натрия (13,3 г, 10 экв.) и бикарбонат натрия (75 мг, 0,1 экв.). Смесь перемешивали при 50 С в течение 13 ч. Реакционную смесь концентрировали в вакууме, остаток растворяли в эфире и промывали 10% раствором сульфита натрия. Слои разделяли и эфирную часть промывали 10% раствором сульфита натрия, водой, рассолом (2 х), сушили и концентрировали в вакууме. Остаток пропускали через подушку диоксида кремния, элюируя смесью гексан/этилацетат (1:1) и смесью гексан/этилацетат (1:2) с получением 7,6 г йодида, 3-[(N-1(2-этокси-(3-(О)-4-(трет-бутилдифенилсилилокси)-1-(йод)бутаниндол-3-ил]-4-[индол-3 ил]-1N-(метил)пиррол-2,5-диона, в виде красного твердого вещества (количественный выход для этих двух стадий). МС. ЯМР. К суспензии в диметилформамиде (1 л) карбоната цезия (12,0 г, 4 экв.) в атмосфере азота добавляли этот йодид, 3-[(N-1-(2-этокси-(3(О)-4-(трет-бутилдифенилсилилокси)-1-(йод) бутаниндол-3-ил]-4-[индол-3-ил]-1N-(метил) пиррол-2,5-дион (7,6 г, 9,2 ммоль), растворенный в диметилформамиде (25 мл), через насос со шприцем на протяжении 65 ч. Через три часа после завершения добавления реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате (700 мл),промывали водой (2 х 300 мл) и водный слой вновь промывали этилацетатом (2 х 200 мл). Объединенные этилацетатные порции промыва 000598 24 ли рассолом (2 х 200 мл), сушили (MgSO4),фильтровали и концентрировали в вакууме с получением пурпурного остатка. Этот материал абсорбировали на диоксиде кремния и наносили на флэш-колонку, элюировали смесью гексан/этилацетат (3:1) и затем гексан/этилацетат(300 мл) перемешивали при комнатной температуре в течение 65 ч, а затем в течение 1 ч при 60 С. Реакционную смесь концентрировали (150 мл) в вакууме, остаток суспендировали в воде,охлаждали до 5 С и подкисляли (рН 3) концентрированной соляной кислотой. Красную водную суспензию экстрагировали этилацетатом(4 х 200 мл), сушили и концентрировали в вакууме с получением 3,3 г неочищенного гидроксиангидрида,2,3-[(N,N'-1,1'-2-этокси)-3-(O)4-(гидрокси)бутан)-бис-(3,3'-индолил)]фуран 1,4-диона, в виде пурпурного твердого вещества. МС. К раствору в диметилформамиде (250 мл) этого ангидрида, 2,3-[(N,N'-1,1'-2-этокси)3(О)-4-(гидрокси)-бутан)-бис-(3,3'-индолил)]фуран-1,4-диона, (3,3 г, 7,5 ммоль) в атмосфере азота добавляли 1,1,1,3,3,3-гексаметилдисилазан(32 мл, 2 экв.) и метанол (3 мл, 10 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, а затем нагревали при 60 С в течение 2 ч. Диметилформамид удаляли в вакууме и полученный остаток растворяли в ацетонитриле (250 мл). Добавляли 1 н. НСl(50 мл). Реакцию перемешивали в течение 15 мин. Реакционную смесь концентрировали, распределяли между этилацетатом (1 л) и водой(250 мл). Продукт в виде твердого вещества осаждался. Получено 0,92 г (28%) гидроксималеимида,3,4-[(N,N'-1,1'-2-этокси)-3-(O)-4-(гидрокси)бутан)-бис-(3,3'-индолил)]-1(H)-пиррол 2,5-диона. Небольшое количество (50 мг) абсорбировали на диоксиде кремния и наносили на флэш-колонку. Элюировали дихлорметаном,5% ацетонитрилом и затем смесью 10% ацетонитрил/дихлорметан с получением 38 мг аналитически чистого материала. Этилацетат концентрировали и хроматографировали с получением дополнительно 8% неочищенного продукта. МС. 1 Н-ЯМР (D6-ДMCO):1,96 (1 Н,м), 2,09(140 мл) спирта, 3,4-[(N,N'-1,1'-2'-этокси)-3(О)-4-(гидрокси)бутан)-бис-(3,3'-индолил)]1(Н)-пиррол-2,5-диона, (472 мг, 1,07 ммоль) в атмосфере азота добавляли пиридин (260 мкл, 3 экв.) и метансульфоновый ангидрид (242 мг, 1,3 экв.). После 4 ч реакцию разбавляли дихлорметаном, промывали 0,1 н. НСl (2 х), фильтровали для удаления исходного материала (54 мг). Дихлорметановые порции промывали рассолом(2 х), сушили и концентрировали с получением неочищенного мезилата в виде пурпурного твердого вещества. Этот материал абсорбировали на диоксиде кремния и наносили на флэшколонку, которую последовательно элюировали дихлорметаном, смесью 5% ацетонитрил/ дихлорметан и смесью 10% ацетонитрил/дихлорметан с получением 288 мг (52%) мезилата, 3,4-[(N,N'-1,1'-2-этокси)-3-(О)-4(метансульфонилокси)бутан)-бис-(3,3'-индолил)]-1(Н)-пиррол-2,5-диона. МС. ЯМР. К раствору в тетрагидрофуране (20 мл) мезилата, 3,4-[(N,N'-1,1'-2-этокси)-3-(О)-4-(метансульфонилокси)-бутан)-бис-(3,3'-индолил)]1(Н)-пиррол-2,5-диона, (304 мг, 0,59 ммоль) добавляли 8,9 М раствор диметиламина в тетрагидрофуране (7 мл, 100 экв.). После нагревания(65 С) в течение 24 ч в запаянной пробирке реакцию разбавляли этилацетатом (200 мл), промывали рассолом (2 х), сушили и концентрировали с получением неочищенного производного диметиламина в виде твердого вещества. Этот материал абсорбировали на диоксиде кремния и наносили на флэш-колонку, которую последовательно элюировали смесью этилацетат/гексан(3:1), этилацетатом и смесью 2% изопропиламин/этилацетат с получением 193 мг (выход 70%) производного диметиламина, которое имело 90% чистоту согласно ВЭЖХ. Производное диметиламина, 3,4-[(N,N'-1,1'-2-этокси)3-(О)-4-(N,N-диметиламино)бутан)-бис-(3,3'индолил)]-1(Н)-пиррол-2,5-дион, oчищали до чистоты более 95% в виде соли трифторуксусной кислоты гель-фильтрационной ВЭЖХ с обращенной фазой, элюируя смесью ацетонитрил/(0,01% ТФА/вода) (85:15). Соль трифторуксусной кислоты 3,4-[(N,N'1,1'-2-этокси)-3-(О)-4-(N,N'-диметиламино) бутан)-бис-(3,3'-индолил)]-1(Н)-пиррол-2,5-диона превращали в соль НСl суспендированием 26 этой соли в этилацетате и осторожным промыванием 0,1 н. NaOH (5 х 50 мл). Этилацетатную часть промывали рассолом (2 х), сушили и концентрировали с получением свободного основания, 3,4-[(N,N'-1,1'-2-этокси)-3-(О)-4-(N,N'диметиламино)бутан)-бис-(3,3'-индолил)]-1(Н)пиррол-2,5-диона. К суспензии в безводном метаноле (50 мл) свободного основания, 3,4[(N,N'-1,1'-2-этокси)-3-(O)-4-(N,N-диметиламино)бутан)-бис-(3,3'-индолил)]-1(Н)-пиррол 2,5-диона, добавляли 1 н НСl в безводном эфире(13 мл, 50 экв.). Эфир выпаривали и остаток сушили в вакууме с получением 143 мг (выход 52%) гидрохлорида 3,4-[(N,N'-1,1'-2-этокси)3-(O)-4-(N,N-диметиламино)бутан)-бис-(3,3'индолил)]-1(Н)-пиррол-2,5-диона в виде красного твердого вещества. МС. 1 Н-ЯМР ( D6 -ДМСО):2,03 (1 Н,м), 2,26(2 Н,м), 10,33 (1H, шир.с.), 10,92 (1 Н,с), Пример 1. 3,4-[(N,N'-1,1'-(2-этокси]-(3(O)-4-[N-трифторметиламино)бутан)-бис-(3,3'индолил)]-1(Н)-пиррол-2,5-дион. 3,4-[N,N'-1,1'-2"-этокси)-3-(O)-4'"-(Nметиламин)бутан-бис(3,3'-индолил)]-1-(метил)пиррол-2,5-дион, полученный аналогично описанному получению 10, (20 мг, 0,04 ммоль) растворяли в ТГФ (10 мл), содержащем триэтиламин (6,1 мкл, 0,044 ммоль) в атмосфере азота. К этому раствору добавляли сероуглерод (3 мкл,0,05 ммоль) и после 15 мин добавляли метилйодид. Реакция завершалась после 12 ч согласно ТСХ (10% МеОН в CH2Cl2). Реакционную смесь разбавляли этилацетатом, промывали водой,рассолом сушили и концентрировали с получением дитиокарбамата (23 мг, ожидаемая масса)IS/MS 559 (M1) ожидаемая мол. масса 558. К раствору в дихлорметане этого дитиокарбамата добавляют тетрабутиламмонийдигидрогентрифторид и N-бромсукцинимид. Обработка и очистка при помощи хроматографии дает 3,4-[N,N'-1,1'-2-этокси)-3-(O)-4-(N,N+(трифторметил)метиламино)бутан-бис-(3,3'-индолил)]-1-(метил)-пиррол-2,5-дион,который превращают в NН-малеимид. Производное трифторметиламина можно также получить следующим образом. К раствору в ДМСО монометиламина добавляют дибромдифторметан и тетракис(диметиламин)этилен. Стандартная обработка дает целевое производное трифторметиламина. Это производное превращают в NH-малеимид,как описано выше. Как отмечалось ранее, соединения данного изобретения являются сильными ингибиторами протеинкиназы С. Эти соединения являются избирательными в отношении протеинкиназы С по сравнению с другими киназами. Способность соединений данного изобретения селективно ингибировать протеинкиназу С определяли в тесте на кальций-кальмодулинзависимую протеинкиназу, в тесте на казеиновую протеинкиназу 11, в тесте на каталитическую субъединицу цАМФ-зависимой протеинкиназы и в тесте на протеин-тирозин-киназу. Тест на кальций-кальмодулинзависимую протеинкиназу (СаМ) Тест на кальций-кальмодулинзависимую протеинкиназу описан в Journal of Neuroscience,3: 818-831 (1983). Компоненты теста находятся в общем объеме 250 мкл: 55 мМ HEPES (4-(2 гидроксиэтил)-1-пиперазинэтансульфоновая кислота), рН 7,5, 2,75 мМ дитиотреитол, 2,2 мМ ЭГТК (этиленбис (окси-этиленнитрило)тетрауксусная кислота, используемая в "слепом" буфере), 1,1 ммоль кальция (Sigma, St. Louis, Missouri) (используемый в контрольном буфере), 10 мМ хлорида магния (Sigma, St. Louis, Missouri),200 мкг/мл гистона типа HL (Worthington), 10 мкл ДМСО или ДМСО/ингибитор и 30 мкМ(гамма-32 Р)-АТФ (DuPont). Реакцию инициируют добавлением кальций-кальмодулинзависимой протеинкиназы (выделенной из гомогената мозга крысы), инкубируют при комнатной температуре в течение 10 мин и останавливают добавлением 0,5 мл охлажденной на льду трихлоруксусной кислоты (Amresco) с последующим добавлением 100 мкл 1 мг/мл бычьего сывороточного альбумина (Sigma, St. Louis, Missouri). Осадок собирают вакуумфильтрованием на фильтрах из стекловолокна и количественные измерения выполняют на бета-сцинтилляционном счетчике. Компоненты буфера: Контрольный буфер 200 MM HEPES рН 7,5 3125 мкл 50 мМ ДТТ 625 мкл Гистон 1250 мкл 100 мМ кальций 125 мкл. 100 мМ ЭГТК Деиониз.вода 2375 мкл Компоненты теста: 165 мкл буфера; 25 мкл кальмодулина (250 мкг/мл); 10 мкл ДМСО или ДМСО/ингибитора; 25 мкл киназы;25 мкл 32 Р-АТ. 28 Тест на казеиновую протеинкиназу 11(СК-11) Тест на казеиновую протеинкиназу 11 описан в Neurochem. Res. 13: 829-836 (1988). Компоненты теста находятся в общем объеме 250 мкл: 20 мМ трис-HCl, рН 7,5, 5 мМ фторид натрия, 50 мг/мл казеина (Sigma, At. Louis, Missouri), 10 мМ хлорида магния (Sigma, St. Louis,Missouri), 10 мкл ДМСО или ДМСО/ингибитора и 30 мкм гамма-32 Р-АТ (DuPont). Реакцию инициируют добавлением казеиновой протеинкиназы 11, выделенной из гомогената мозга крысы),инкубируют при комнатной температуре в течение 10 мин и останавливают добавлением 0,5 мл ледяной трихлоруксусной кислоты (Amresco) с последующим добавлением 100 мкл 1 мг/мл бычьего сывороточного альбумина (Sigma, St.Louis, Missouri). Осадок собирают вакуумфильтрованием на фильтрах из стекловолокна и количественные измерения выполняют на бетасцинтилляционном счетчике. Компоненты теста в порядке добавления: 175 мкл буфера; 10 мкл ДМСО или ДМСО/ингибитора; 25 мкл 32 Р-АТ в 300 мкМ хлориде магния; 40 мкл фермента (неразбавленного). Буфер, приготовлен следующим образом:(Конечный объем = 3,5 мл: количество 20 тестов). 500 мкл каждого: 200 мМ трис-НСl рН 7,5 50 мМ фторид натрия 50 мг/мл казеина+ 2 мл деион. воды Общий объем: 3,5 мл Тест на каталитическую субъединицу цАМФзависимой протеинкиназы (РКА) Компоненты теста находятся в общем объеме 250 мкл: 20 мМ HEPES (Sigma, St. Louis,Missouri) буфера рН 7,5, 200 мкг/мл гистона типа HL (Worthington), 10 мМ хлорида, магния(Sigma, St. Louis, Missouri), 10 мкл ДМСО или ДМСО/ингибитора и 30 мкМ (гамма-32 Р) -АТФ(DuPont). Реакцию инициируют добавлением каталитической единицы цАМФ-зависимой киназы бычьего сердца (Sigma, St. Louis, Missouri),инкубируют до 30 С в течение 10 мин и останавливают добавлением ледяной трихлоруксусной кислоты (Amresco) с последующим добавлением 100 мкл 1 мг/мл бычьего сывороточного альбумина (Sigma). Осадок собирают вакуумфильтрованием на фильтрах из стекловолокна с применением ТОМТЕС и количественно измеряют подсчетом в бета-сцинтилляционном счетчике. Этот тест проводят идентично тесту на фермент протеинкиназу С (ПКС), за исключением того, что в этом тесте не используют фосфолипиды или диацилглицерин, и субстрат гистон является специфическим для фермента с цАМФ-зависимой каталитической субъединицей. 29 Тест на протеинтирозинкиназу (src) Компоненты теста являются следующими: 10 мкл Raytide; 10 мкл Киназы; 4 мкл ДМСО или ДМСО/ингибитора; 6 мкл 200 мМ HEPES рН 7,5; 10 мкл 32 Р-АТ. Этот тест описан в Onogene Science. Inc.Cat. PK02 and РК 03 (1990). Неожиданно, соединения данного изобретения являются также изозим-селективными ингибиторами, то есть, эти соединения селективно ингибируют бета-1- и бета-2-изозимы протеинкиназы С. Эта селективность относительно изозимов была определена в тесте на ПКС-ферменты. Тест на ПКС-ферменты ПКС-ферменты = альфа-, бета-1-, бета-11-,гамма-, дельта-, эпсилон-, эта- и дзетапротеинкиназы С. Компоненты теста находятся в общем объеме 250 мкл. Везикулы, состоящие из 120 мкг/мл фосфатидилсерина (Avanti Polar Lipids) и достаточного количества диацилглицерина (Avanti PolarLipids) для активации фермента до максимальной активности, в 20 мМ HEPES-буфере (Sigma,St. Louis, Missouri), pH 7,5, 940 мкМ хлорида кальция (Sigma, St. Louis, Missouri) только для определения альфа-, бета-1, бета-2- и гаммафермента, 1 мМ ЭГТК для всех ферментов, 10 мМ хлорида магния (Sigma, St. Louis, Missouri) и 30 мкМ (гамма-32 Р)-АТФ (DuPont). Для всех ферментов используют в качестве субстрата либо гистон типа HL (Worthington), либо основной белок миелин. Тест инициируют добавлением фермента протеинкиназы С, инкубируют при 30 С- в течение 10 мин и останавливают добавлением 0,5 мл холодной трихлоруксусной кислоты (Amresco) с последующим добавлением 100 мкл 1 мг/мл бычьего сывороточного альбумина (Sigma, St. Louis, Missouri). Осадок собирают вакуум-фильтрованием на фильтрах из стекловолокна с применением системы для фильтрования ТОМТЕС и количественные измерения выполняют подсчетом в бетасцинтилляционном счетчике. Соединения данного изобретения ингибируют протеинкиназу С с величиной IC50 ниже 100 мкм. Кроме того, соединения данного изобретения селективно ингибируют бета-1- и бета 2-изозимы протеинкиназы С и имеют величинуIC50 относительно этих изозимов ниже 10 мкм. В качестве ингибитора протеинкиназы С эти соединения применимы в лечении состояний, в которых протеинкиназа С играет роль в патологии. Состояниями, известными в данной области, являются: сахарный диабет и его осложнения, ишемия, воспаление, расстройства центральной нервной системы, сердечнососудистое заболевание, болезнь Альцгеймера,дерматологическое заболевание и рак. 30 Было показано, что ингибиторы протеинкиназы С блокируют воспалительные реакции,такие как окислительный выброс нейтрофилов,отрицательная регуляция CD3 в Т-лимфоцитах и индуцируемый форболом отек лапок. Twoemy,В, et al., Bichem. Biophys. Res. Commun. 171: 1087-1092 (1990); Mulqueen, M.J. et al., AgentsActions 37: 85-89 (1992). Таким образом, в качестве ингибиторов ПКС соединения данного изобретения применимы в лечении воспаления. Активность протеинкиназы С играет центральную роль в функционировании центральной нервной системы. Huang, К.Р. Trends Neurosci. 12: 425-432 (1989). Кроме того, было показано, что ингибиторы протеинкиназы С предотвращают повреждение, наблюдаемое в фокальном и центральном ишемическом повреждении мозга и при отеке мозга. Нага, Н. et al., J. Cereb.al., Neurology 43: 1407-1413 (1993). Поэтому,соединения данного изобретения применимы в лечении болезни Альцгеймера и ишемического повреждения мозга. Активность протеинкиназы С давно связывали с ростом клеток, ускорением роста опухолей и раком. Rotenberg, S.A. and Weinstein, I.В.(1991). Ahmad et al., Molecular Pharmacology: 43: 858-862 (1993). Известно, что ингибиторы протеинкиназы С эффективны в предотвращении роста опухолей у животных. Меуег, Т. et al Int.J. Cancer 43: 851-856 (1989); Akinagaka, S. et al.,Cancer Res. 51: 4888-4892 (1991). Соединения данного изобретения действуют также как агенты реверсии множества лекарственных средств(MDR), превращающие их в эффективные соединения при введении вместе с другими химиотерапевтическими агентами. Активность протеинкиназы С играет также важную роль в сердечно-сосудистом заболевании. Было показано, что повышенная активность протеинкиназы С в сосудистой сети вызывает увеличенное сужение сосудов и гипертензию. Bilder, G.E. et al., J. Pharmacol. Exp.Ther. 252: 526-530 (1990). Поскольку ингибиторы протеинкиназы С демонстрируют ингибирование окислительного выброса нейтрофилов,они также применимы в лечении сердечнососудистой ишемии и в улучшении сердечной функции после ишемии. Muid, R.E. et al., FEBSLett. 293: 169-172 (1990); Sonoki, H. et al., Kokyu-To Junkan 37: 669-674 (1989). Роль протеинкиназы С в функции тромбоцитов также исследовалась, и показано, что повышенные уровни протеинкины С коррелируют с увеличенным ответом на агонисты. Bastyr III,E.J. and Lu, J. Diaberes 42: (Suppl. 1) 97A (1993). ПКС была причастна к биохимическому пути в модулировании проницаемости капилляров 31 фактором тромбоцитарной активности. Kobayashi et al., Amer. Phys. Soc. H1214-H1220 (1994). Было показано, что сильные ингибиторы протеинкиназы С влияют на индуцируемую агонистами агрегацию в тромбоцитах. Toulec, D. etal., J. Biol.Chem. 266: 15771-15781 (1991). Ингибиторы протеинкиназы С блокируют также индуцируемую агонистами пролиферацию клеток гладких мышц. Matsumoto, H. and Sasaki, Y.(1989) . Таким образом, соединения данного изобретения применимы в лечении сердечнососудистого заболевания, атеросклероза и рестеноза. Было показано, что аномальная активность протеинкиназы С связана также с дерматологическими расстройствами, такими как псориаз.Dermatol, 124: 542-546 (1991). Псориаз характеризуется аномальной пролиферацией кератиноцитов. Было показано, что известные ингибиторы протеинкиназы С ингибируют пролиферацию кератиноцитов таким образом, что степень ингибирования пролиферации параллельна их силе в качестве ингибиторов ПКС. Hegemann, L.Bollag, W.B. et al., J. Invest. Dermatol. 100: 240246 (1993). Таким образом, эти соединения в качестве ингибиторов ПКС применимы в лечении псориаза. Протеинкиназу С связывали с несколькими различными аспектами диабета. Избыточную активность протеинкиназы С связывали с дефектами передачи сигналов инсулина и, следовательно, с инсулинорезистентностью, наблюдаемой в случае диабета типа II. Karasik, A. etChem. 268: 6338-6347 (1993). Кроме того, исследования показали заметное увеличение активности протеинкиназы С в тканях, которые, как известно, являются чувствительными к диабетическим осложнениям при гипергликемических состояниях. Lee, T.-S. et al., J. Clin. Invest. 83: 90-94 (1989); Lее, Т.-S. еt al. Ргос. Natl. Acad.(1991); Tesfamariam, В. et al. J. Clin.Invest. 87: 1643-1648 (1991). Соединения данного изобретения являются также изозимелективными. Эти соединения предпочтительно ингибируют бета 1- и бета-2-изозимы протеинкиназы С в сравнении с ингибированием других изозимов протеинкиназы С, т.е. альфа-гамма-, дельта-, эпсилон-, дзета- и эта-изозимов. В общем, эти соединения демонстрируют минимально 10 кратное различие в дозе, требующейся для ингибирования бета-1- и бета-2-изозимов протеинкиназы С, и дозе, требующейся для равного 32 ингибирования альфа-изозима протеинкиназы С в тесте на ПКС. Поэтому соединения данного изобретения ингибирутют бета-1- и бета-2 изозимы протеинкиназы С при гораздо более низких концентрациях с минимальным ингибированием других изозимов ПКС. Вследствие этой избирательности эти соединения применимы, в частности, в лечении таких патологических состояний, в которых участвуют бета-1- и бета-2-изозимы протеинкиназы С. Например, повышенные уровни глюкозы в крови, обнаруживаемые при диабете, приводят к изозим-специфическому повышению бета-2-изозима в сосудистых тканях. Inoguchi et(1992). Связанное с диабетом увеличение бетаизозима в тромбоцитах человека коррелировало с их измененной реакцией на агонисты. Bastyr(1993). Было показано, что рецептор витамина D человека избирательно фосфорилируется протеинкиназой С бета. Это фосфорилирование было связано с изменениями в функционировании рецептора. Hsieh et al., Proc. Natl. Acad. Sci. USA 88: 9315-9319 (1991); Hsieh et al., J. Biol. Chem. 268: 15118-15126 (1993). Кроме того, недавно показано, что бета-2-изозим ответственен за пролиферацию клеток эритролейкоза, тогда как альфа-изозим участвует в дифференцировке мегакариоцитов в тех же самых клетках. Murrayet al., J. Biol. Chem. 268: 15847-15853 (1993). Соединения формулы I предпочтительно готовят перед введением. Следовательно, еще одним вариантом данного изобретения является фармацевтическая готовая форма, содержащая соединение формулы I и один или несколько фармацевтически приемлемых носителей, разбавителей или наполнителей. Данные фармацевтические готовые формы готовят известными способами с использованием хорошо известных и легко доступных ингредиентов. При приготовлении композиций данного изобретения активный ингредиент обычно смешивают с носителем или разбавляют носителем или заключают в носитель, который может быть в форме капсулы, мешочка, бумажного или иного контейнера. Когда носитель служит в качестве разбавителя, он может быть твердым, полутвердым или жидким материалом,который действует как носитель, наполнитель или среда для активного ингредиента. Таким образом, композиции могут быть в форме таблеток, пилюль, порошков, лепешек, мешочков,крахмальных капсул, эликсиров, суспензий,эмульсий, растворов, сиропов, аэрозолей (как в твердой, так и в жидкой среде), мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильных упакованных порошков. Некоторые примеры удобных носителей,наполнителей и разбавителей включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахма 33 лы, аравийскую камедь, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция,микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, водный сироп, метилцеллюлозу метил- и пропилгидроксибензоаты,тальк, стеарат магния и минеральное масло. Готовые формы могут дополнительно включать смазывающие агенты, увлажняющие агенты,эмульгирующие и суспендирующие агенты,консерванты, подслащивающие агенты или корригирующие вкус и запах агенты. Композции данного изобретения могут быть приготовлены таким образом, чтобы обеспечивать быстрое,замедленное или пролонгированное высвобождение активного ингредиента после введения пациенту. Композции предпочтительно готовят в форме стандартной дозы, причем каждая доза содержит от 1 до 500 мкг, чаще 5 - 300 мг активного ингредиента. Однако должно быть понятно, что вводимая терапевтическая доза будет определяться врачом в связи с соответствующими обстоятельствами, в том числе в зависимости от состояния, которое должно быть подвергнуто лечению, выбора вводимого соединения и избранного способа введения и, следовательно, указанные выше диапазоны доз не предназначены для какого-либо ограничения объема данного изобретения. Термин "форма стандартной дозы" относится к физически дискретным единицам, пригодным в качестве стандартных доз для одного приема для человека и других млекопитающих, причем каждая единица содержит заранее определенное количество активного материала, рассчитанное для получения желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим носителем. Кроме указанных выше готовых форм соединения данного изобретения можно вводить местно. Готовые формы для местного введения представляют собой мази, кремы и гели. Мази, как правило, готовят с использованием либо (1) маслянистой основы, то есть, основы, состоящей из фиксированных масел или углеводородов, таких как белый вазелин или минеральное масло, либо (2) абсорбирующей основы, то есть, основы, состоящей из безводного вещества или веществ, которые могут поглощать воду, например, безводного ланолина. Обычно, после образования основы, маслянистой или абсорбентной, добавляют активный ингредиент (активное соединение) до количества, дающего желаемую концентрацию. Кремы представляют собой эмульсии масло/вода. Они состоят из масляной фазы и т.п.,такой как воски, вазелин, минеральное масло и т.п., и водной фазы (непрерывной фазы), содержащей воду и любые водорастворимые вещества, такие как добавленные соли. Эти две фазы стабилизируются эмульгирующим агентом, например, поверхностно-активным веществом,таким как лаурилсульфат натрия, гидрофиль 000598 34 ными коллоидами, такими как коллоидные глины на основе аравийской камеди, veegum и т.п. При образовании эмульсии активный ингредиент (соединение) обычно добавляют до количества, достаточного для достижения желаемой концентрации. Гели содержат основу, выбранную из маслянистой основы, воды или состоящей из эмульсии-суспензии основы. К этой основе добавляют гелирующий агент, который образует матрикс в основе, увеличивая ее вязкость. Примерами гелирующих агентов являются гидроксипропилцеллюлоза, полимеры акриловой кислоты и т.п. Обычно, активный ингредиент (соединения) добавляют к готовой форме при желаемой концентрации в момент перед добавлением гелирующего агента. Количество соединения, включенного в готовую форму для местного применения, не является критическим, концентрация лишь должна быть диапазоном, достаточным для легкого нанесения препарата на пораженную зону ткани в количестве, которое будет поставлять желаемое количество соединения. Обычное количество препарата для местного нанесения, которое должно быть нанесено на пораженную ткань, будет зависеть от размера пораженной ткани и концентрации соединения в препарате. Как правило, готовую форму наносят на пораженную ткань в количестве, доставляющем от 1 до 500 мкг соединения на см 2 пораженной ткани. Предпочтительно, наносимое количество соединения должно находиться в диапазоне от 30 до 300 мкг/см 2, более предпочтительно от 50 до 200 мкг/см 2 и, наиболее предпочтительно, от 60 до 100 мкг/см 2. Следующие далее примеры готовых форм являются только иллюстративными и не предназначены для какого-либо ограничения объема данного изобретения. Готовая форма 1. Твердые желатиновые капсулы получают с использованием следующих ингредиентов: Количество(мг/капсулу) Активный агент 250 Крахмал, высушенный 200 Стеарат магния 10 Всего 460 Указанные выше ингредиенты смешивают и наполняют ими твердые желатиновые капсулы в количестве 460 мг. Готовая форма 2. Таблетку готовят с использованием указанных ниже ингредиентов: Активный агент Целлюлоза, микрокристаллическая Коллоидальный диоксид кремния Стеариновая кислота 10 5 Всего 665 Компоненты смешивают и прессуют с получением таблеток весом 665 мг каждая. Готовая форма 3. Аэрозольный раствор готовят из следующих компонентов: Количество(хлордифторметан) 70,00 Всего 100,00 Активное соединение смешивают с этанолом. Смесь добавляют к части пропеллента 22,охлаждают до -30 С и переносят в заполняющее устройство. Затем необходимое количество подают в контейнер из нержавеющей стали и разбавляют остальной частью пропеллента. Затем к контейнеру присоединяют клапанные элементы. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулыW является О, -S- или NH,R1 представляет собой независимо водород, галоген, С 1-С 4-алкил, гидрокси, С 1-С 4 алкокси, галогеналкил, нитро, -NH (С 1-С 4 алкил), N (С 1-С 4-алкил)2 или -NНСО (С 1-С 4 алкил),R2 является водородом, СН 3 СО-, NН 2 или гидроксигруппой,Z является -(СН 2)р- или (СН 2)р-O-(СН 2)p-,R6 является -NH(CF3) или -N(CF3)(СН 3),m независимо является 0, 1, 2 или 3, р независимо является 0, 1 или 2, или его фармацевтически приемлемая соль или сольват. 2. Соединение по п.1, в котором R2 является водородом.N(СF3)(СН 3). 5. Способ лечения сахарного диабета и его осложнений, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4. 6. Способ ингибирования протеинкиназы С, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, фармацевтически эффективного количества соединения любого из пп.1-4. 7. Способ лечения центрального ишемического повреждения мозга, отличающийся тем,что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.14. 8. Способ лечения сердечно-сосудистой ишемии, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4. 9. Способ лечения воспалениия, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении,терапевтического количества соединения любого из пп.1-4. 10. Способ лечения рестеноза, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении,терапевтического количества соединения любого из пп.1-4. 11. Способ лечения атеросклероза, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп.1-4. 12. Способ лечения псориаза, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении,терапевтического количества соединения любого из пп.1-4. 13. Способ лечения болезни Альцгеймера,отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп. 1-4. 14. Способ лечения рака, отличающийся тем, что предусматривает введение млекопитающему, нуждающемуся в таком лечении, терапевтического количества соединения любого из пп. 1-4. 15. Фармацевтические композиции, отличающиеся тем, что содержат соединение по любому из пп. 1-4 вместе с одним или несколькими фармацевтически приемлемыми наполнителями, носителями или разбавителями. 16. Способ получения соединения любого из пп. 1-4, отличающийся тем, что предусматривает взаимодействие соединения формулы 38 с избытком смеси реагента и С 1-С 4-спирта в полярном апротонном растворителе, причем указанный реагент выбирают из гексаметилдисилазана или соли аммония. 17. Соединение формулыR1 представляет собой независимо водород, галоген, С 1-С 4-алкил, гидрокси, С 1-С 4 алкокси, галогеналкил, нитро, -NH(С 1-С 4 алкил), -N(С 1-С 4-алкил)2 или NHCO(С 1-С 4 алкил),Z является -(СН 2)p или -(СН 2)p-O-(СН 2)p,R6 является -NН(СF3) или -N(СF3)(СН 3);W является О, -S- или NH,R1 представляет собой независимо водород, галоген, С 1-С 4-алкил, гидрокси, С 1-С 4 алкокси, галогеналкил, нитро, -NH(С 1-С 4 алкил), N(С 1-С 4-алкил)2 или -NHCO(C1-C4 алкил),R2 является водородом, СН 3 СО-, NH2 или гидроксигруппой,Z является -(СН 2)р- или (СН 2)p-O-(СН 2)p-,R6 является -NH(CF3) или -N(CF3) (СН 3),m независимо является 0, 1, 2 или 3,р независимо является 0, 1 или 2,или его фармацевтически приемлемая соль или сольват для использования в качестве фармацевтического средства.
МПК / Метки
МПК: C07D 471/22, A61K 47/22
Метки: протеинкиназы, ингибиторы
Код ссылки
<a href="https://eas.patents.su/20-598-ingibitory-proteinkinazy-s.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы протеинкиназы с.</a>
Ингибиторы тромбина, основанные на аминокислотной последовательности гирудина

Номер патента: 88
Опубликовано: 25.06.1998
Автор: Димэйо Джон
МПК: A61K 38/58, C07K 14/815
Метки: тромбина, основанные, аминокислотной, гирудина, ингибиторы, последовательности
Формула / Реферат:
1. Производное пептида (I): (D)-Фен-Про-Арг-(СН2)4(СО)-[NН(СН2)4СО]2-Асп-Фен-Глу-Про-Иле-Про-Лей и его фармацевтически приемлемые соли. 2. Состав для лечения тромботических заболеваний, содержащий эффективное количество производного пептида (I) (D)-Фен-Про-Арг-(СН2)4(СО)-[NН(СН2)4СО]2-Асп-Фен-Глу-Про-Иле-Про-Лей и его фармацевтически приемлемые соли. 3. Способ лечения или профилактики сосудистых заболеваний, относящихся к тромбозу, который...
Фармацевтическая композиция, содержащая ингибиторы протонного насоса

Номер патента: 87
Опубликовано: 25.06.1998
Авторы: Дэйв Кошик Дж., Уилльямс Джеймс Б.
МПК: A61K 31/44
Метки: ингибиторы, содержащая, насоса, фармацевтическая, протонного, композиция
Формула / Реферат:
1. Фармацевтическая композиция для перорального введения, которая включает ингибитор протонного насоса, загуститель, подщелачивающий агент и гидрофобный масляный жидкий носитель. 2. Композиция по п.1, в которой указанный ингибитор протонного насоса представляет собой омепразол. 3. Композиция по п.1, в которой указанный загуститель представляет собой гидрогенизированное касторовое масло. 4. Композиция по п.1, в которой указанный гидрофобный...
Предыдущий патент: Способ стерилизации поверхности предмета, устройство для его осуществления и установка для стерилизации бутылок
Следующий патент: Парентеральная фармацевтическая композиция, содержащая соль 2-арилпропиновой кислоты с основанием, выбранным из альфа-аминокислоты, дропропизина и трометамина
Случайный патент: Способ и установка для подготовки линий сгиба