Фторзамещенные циклоалканоиндолы, композиции, содержащие такие соединения, и способы лечения
Номер патента: 6134
Опубликовано: 27.10.2005
Авторы: Ванг Заойин, Ли Лианхай, Ляшанс Николя, Бертелетт Карл, Стурино Клаудио

















Формула / Реферат
1. Соединение формулы I

и его фармацевтически приемлемые соли, где n равно 0 или 1; m равно 1, 2 или 3; R1 представляет собой H, C1-C3алкил, галогенированный C1-C3алкил или циклопропил; R2 представляет собой 4-хлорфенил или 2,4,6-трихлорфенил.
2. Соединение по п.1, имеющее стереоконфигурацию, показанную ниже

3. Соединение по п.2, где m равно 1.
4. Соединение (-)-[4-(4-хлорбензил)-7-фтор-5-(метансульфонил)-1,2,3,4-тетрагидроциклопента[b]индол-3-ил]уксусной кислоты и его фармацевтически приемлемые соли.
5. Фармацевтическая композиция, содержащая соединение по любому из пп.1-4 и фармацевтически приемлемый носитель.
6. Композиция по п. 5, которая, кроме того, содержит второй активный ингредиент, выбранный из антигистамина, антагониста лейкотриена и ингибитора биосинтеза лейкотриена.
7. Применение соединения или его соли по любому из пп.1-4 для изготовления лекарственного средства для лечения заболевания, опосредуемого простагландином D2.
8. Применение по п.7, где указанным заболеванием является застойная гиперемия носа, ринит или астма.
Текст
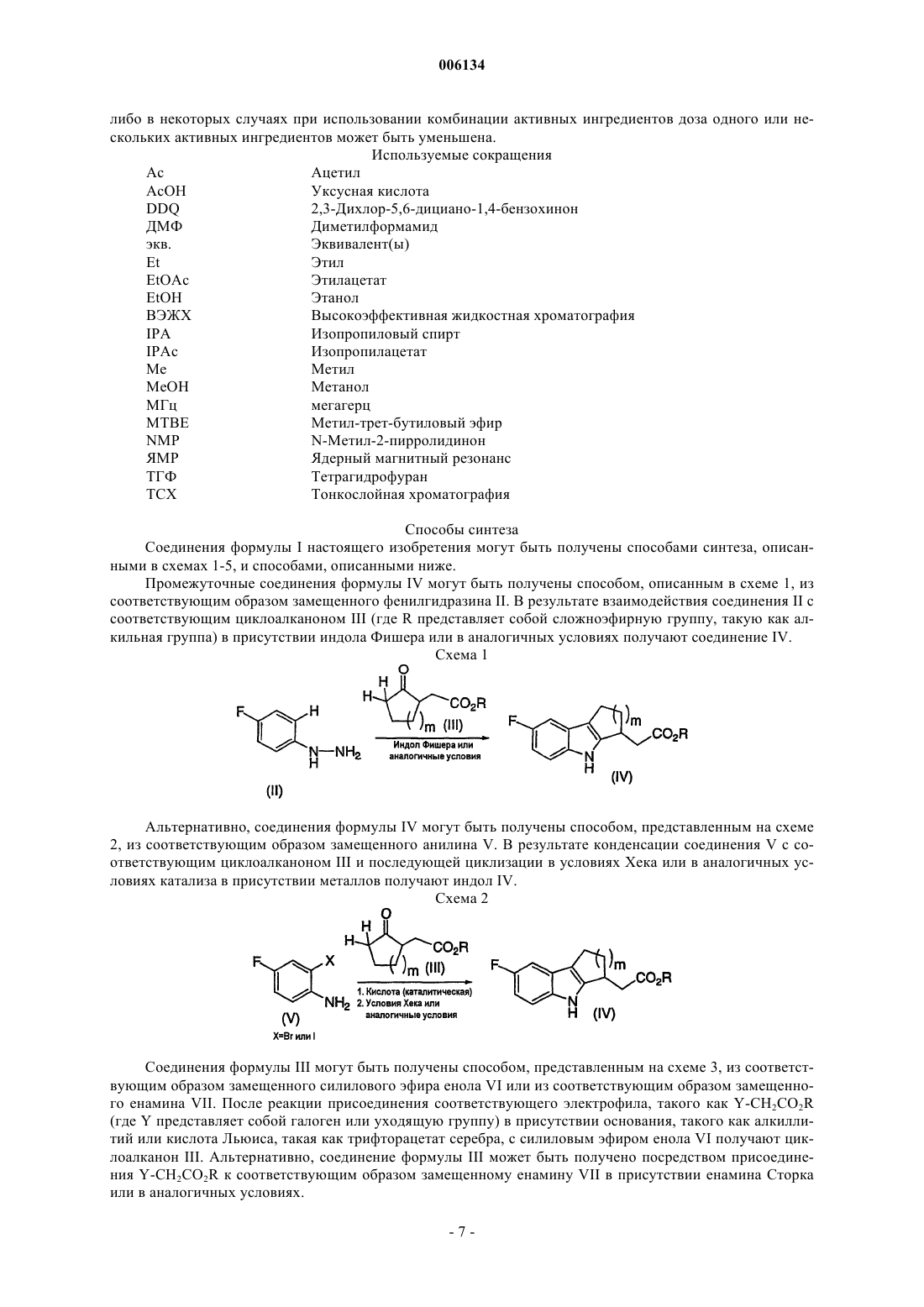
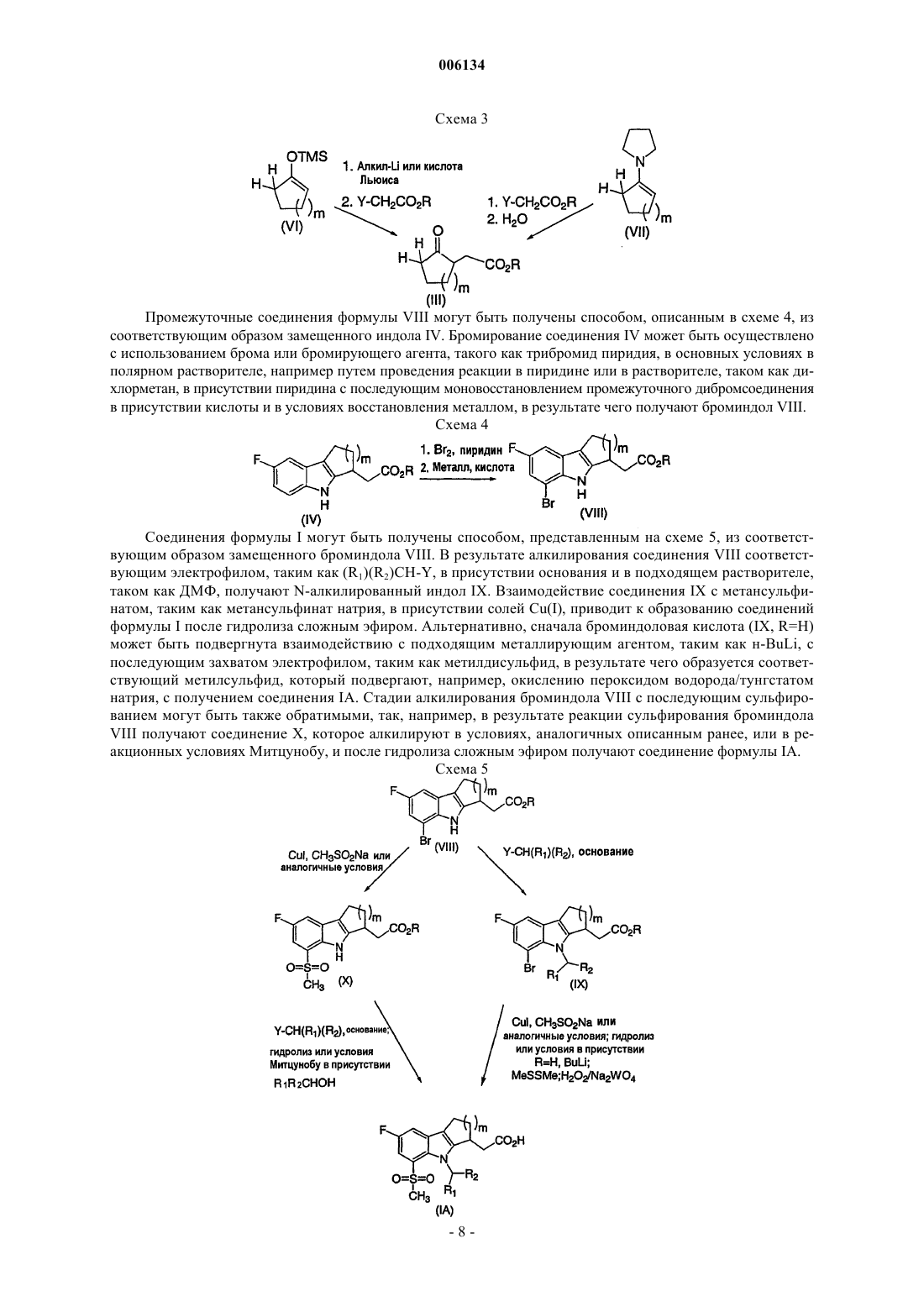
006134 Настоящее изобретение относится к соединениям и к способам лечения опосредованных простагландином заболеваний, а также к некоторым фармацевтическим композициям, содержащим эти соединения. Более конкретно, соединения настоящего изобретения структурно отличаются от стероидов, антигистаминов или адренергических агонистов и представляют собой антагонисты, подавляющие носовые и легочные застойные эффекты, индуцируемые простагландином D-типа. В двух обзорных статьях описана характеристика и терапевтическая релевантность простаноидных рецепторов, а также наиболее широко используемые селективные агонисты и антагонисты: Eicosanoids:T. Tsuri et al., опубликованной в 1997 в Journal of Medicinal Chemistry, vol.40, pp.3504-3507, указывается,что "PGD2, очевидно, является важным медиатором в различных аллергических заболеваниях, таких как аллергический ринит, атопическая астма, аллергический конъюнктивит и атопический дерматит". Совсем недавно в статье Matsuoka et al. Science (2000), 287:2013-7, сообщалось, что PGD2 является ключевым медиатором при аллергической астме. Кроме того, в патентах, таких как патент США 4808608,описаны антагонисты простагландина, которые могут быть использованы для лечения аллергических заболеваний, а особенно аллергической астмы. Антагонисты PGD2 описаны, например, в европейской патентной заявке 837052 и в заявке РСТ WO 98/25919, а также в WO 99/62555. В патенте США 4808608 описаны производные тетрагидрокарбазол-1-алкановой кислоты, используемые в качестве антагонистов простагландина. В заявке РСТ WO 0179169 описаны антагонисты PGD2, имеющие формулу В европейской патентной заявке 468785 описано соединение, 4-[(4-хлорфенил)метил]-1,2,3,4-тетрагидро-7-(2-хинолинилметокси)циклопент[b]индол-3-уксусная кислота, которое является соединением,относящимся к типу соединений-ингибиторов биосинтеза лейкотриена. В патенте США 3535326 описаны противовоспалительные соединения формулы Настоящее изобретение относится к новым соединениям, которые представляют собой антагонисты рецептора простагландина; более конкретно, они представляют собой антагонисты рецептора простагландина D2 (рецептора DP). Соединения настоящего изобретения могут быть использованы для лечения различных опосредованных простагландином заболеваний и расстройств; в соответствии с этим настоящее изобретение относится к способу лечения опосредованных простагландином заболеваний с использованием описанных здесь новых соединений, а также к их фармацевтически приемлемым композициям,содержащим такие соединения. Настоящее изобретение относится к соединениям формулы I и к их фармацевтически приемлемым солям, где n равно 0 или 1; m равно 1, 2 или 3; R1 представляет собой Н, C1-С 3 алкил, галогенированный C1-С 3 алкил или циклопропил; R2 представляет собой 4-хлорфенил или 2,4,6-трихлорфенил. В одном из вариантов настоящее изобретение относится к соединениям формулы I, где n равно 0. В другом варианте настоящее изобретение относится к соединениям формулы I, где n равно 1. В другом варианте настоящее изобретение относится к соединениям формулы I, где m равно 1.-1 006134 В другом варианте настоящее изобретение относится к соединениям формулы I, где m равно 2. В другом варианте настоящее изобретение относится к соединениям формулы I, где R1 представляет собой Н. В другом варианте настоящее изобретение относится к соединениям формулы I, где R1 представляет собой СН 3. В другом варианте настоящее изобретение относится к соединениям формулы I, где R2 представляет собой 4-хлорфенил. В другом варианте настоящее изобретение относится к соединениям формулы I, где R2 представляет собой 2,4,6-трихлорфенил. В другом варианте настоящее изобретение относится к соединениям формулы I, имеющим показанную ниже стереоконфигурацию (т.е., где хиральный центр имеет R-конфигурацию) В другом аспекте настоящее изобретение относится к фармацевтическим композициям, содержащим соединение формулы I и фармацевтически приемлемый носитель. В одном из вариантов осуществления изобретения фармацевтические композиции, кроме того, содержат второй активный ингредиент, выбранный из антигистамина, антагониста лейкотриена, ингибитора биосинтеза лейкотриена, антагонистов рецептора простагландина или ингибиторов биосинтеза простагландина, кортикостероидов, модуляторов цитокинов, антител против IgE, антител против холинергических рецепторов или НСПВС. В другом аспекте настоящее изобретение относится к способу лечения или предупреждения заболеваний, опосредуемых простагландином D2, где указанный способ предусматривает введение пациенту,нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы I. В одном из вариантов настоящее изобретение относится к способу лечения или предупреждения заболевания, опосредуемого простагландином D2, где указанный способ предусматривает введение пациенту млекопитающему, нуждающемуся в таком лечении, соединения формулы I в количестве, эффективном для лечения или предупреждения заболевания, опосредуемого простагландином D2, и где опосредуемыми простагландином заболеваниями являются застойная гиперемия носа, ринит, включая сезонный аллергический ринит и хронический аллергический ринит, и астма, включая аллергическую астму. В другом варианте настоящее изобретение относится к способу лечения застойной гиперемии носа у пациента, нуждающегося в таком лечении, где указанный способ предусматривает введение указанному пациенту терапевтически эффективного количества соединения формулы I. Еще в одном варианте настоящее изобретение относится к способу лечения астмы, включая аллергическую астму, у пациента, нуждающегося в таком лечении, где указанный способ предусматривает введение указанному пациенту терапевтически эффективного количества соединения формулы I. Еще в одном варианте настоящее изобретение относится к способу лечения аллергического ринита(сезонного и хронического) у пациента, нуждающегося в таком лечении, где указанный способ предусматривает введение указанному пациенту терапевтически эффективного количества соединения формулы I. Нумерация внутренней трициклической системы, где m равно 1, осуществляется, как показано ниже Нумерация внутренней трициклической системы, где m равно 2, осуществляется, как показано ниже Оптические изомеры - Диастереомеры - Таутомеры Соединения формулы I содержат один или несколько асимметрических центров, и, таким образом,они могут присутствовать в виде рацематов и рацемических смесей, отдельных энантиомеров, диастереомерных смесей и отдельных диастереомеров. Настоящее изобретение относится ко всем указанным изомерным формам соединений формулы I. Некоторые из описанных здесь соединений могут иметь различные положения присоединения водорода, и такие соединения называются таутомерами. В качестве примера может служить кетон или его-2 006134 енольная форма, известные как кетоноенольные таутомеры. Соединения формулы I охватывают отдельные таутомеры, а также их смеси. Соединения формулы I могут быть разделены на диастереомерные пары энантиомеров, например,посредством фракционной кристаллизации в подходящем растворителе, например в метаноле или в этилацетате или в их смеси. Полученная таким образом пара энантиомеров может быть разделена на отдельные стереоизомеры стандартными методами, например с использованием оптически активной кислоты или оптически активного основания в качестве разделяющего агента, либо методом хирального разделения, такого как разделение с помощью ВЭЖХ на хиральной колонке. Альтернативно, любой энантиомер соединения общей формулы I или Iа может быть получен путем стереоспецифического синтеза с использованием оптически чистых исходных материалов или реагентов с известной конфигурацией. Соли Термин "фармацевтически приемлемые соли" означает соли, полученные из фармацевтически приемлемых нетоксичных оснований, включая неорганические основания и органические основания. Солями, полученными из неорганических оснований, являются соли алюминия, аммония, кальция, меди, железа (III), железа (II), лития, магния, марганца, марганцевая соль (III), калия, натрия, цинка и т.п. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Солями, полученными из фармацевтически приемлемых органических нетоксичных оснований, являются соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин,N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин,лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, тиобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Если соединение настоящего изобретения является основным, то его соли могут быть получены из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такими кислотами являются уксусная, бензолсульфоновая, бензойная, камфорсульфоновая, лимонная,этансульфоновая, фумаровая, глюконовая, глутаминовая, бромисто-водородная, хлористо-водородная,изетионовая, молочная, малеиновая, яблочная, миндальная, метансульфоновая, слизевая, азотная, памовая, пантотеновая, фосфорная, янтарная, серная, винная, п-толуолсульфоновая кислоты и т.п. Особенно предпочтительными являются лимонная, бромисто-водородная, хлористо-водородная, малеиновая, фосфорная, серная и винная кислоты. При этом, следует отметить, что если это не оговорено особо, то при упоминании соединения формулы I подразумевается также и его фармацевтически приемлемая соль. Применение Соединения формулы I являются антагонистами простагландина D2. Способность соединений формулы I взаимодействовать с рецептором простагландина D2 позволяет использовать эти соединения для предупреждения или устранения нежелательных симптомов, вызываемых простагландинами у млекопитающего, в частности у человека. Антагонизм против действия простагландина D2 позволяет использовать данные соединения и их фармацевтические композиции для лечения, предупреждения или ослабления симптомов заболеваний у млекопитающих, а в частности у человека, таких как респираторные заболевания, аллергические состояния, боли, воспалительные состояния, нарушения выделения секрета слизистой, заболевания костей, расстройства сна, нарушения оплодотворяющей способности, нарушения свертывания крови, нарушение зрения, а также иммунные и аутоиммунные заболевания. Кроме того,такое соединение может ингибировать трансформацию клеточных опухолей и метастатический рост опухоли, а поэтому оно может быть использовано для лечения рака. Соединения формулы I могут быть также использованы для лечения и/или предупреждения опосредованных простагландином D2 пролиферативных расстройств, таких, которые могут возникать при диабетической ретинопатии и ангиогенезе опухоли. Соединения формулы I могут также ингибировать индуцированное простаноидом сокращение гладкой мышцы посредством подавления сократительных простаноидов или имитации релаксирующих простаноидов, а поэтому они могут быть использованы для лечения дисменореи и предотвращения преждевременных родов и расстройств, ассоциированных с эозинофилами. В соответствии с этим в другом аспекте настоящее изобретение относится к способу лечения или предупреждения заболевания, опосредуемого простагландином D2, где указанный способ предусматривает введение пациенту-млекопитающему, нуждающемуся в таком лечении, соединения формулы I в количестве, эффективном для лечения или предупреждения указанного заболевания, опосредуемого простагландином D2. Заболеваниями, опосредуемыми простагландином D2, являются, но не ограничиваются ими, аллергический ринит, застойная гиперемия, ринорея, хронический ринит, воспаление носовой полости, астма, включая аллергическую астму, хроническая обструктивная болезнь легких и другие формы воспаления легких; легочная гипотензия; расстройства сна и нарушения цикла "сонпробуждение"; индуцированное простаноидами сокращение гладкой мышцы, ассоциированное с дисменореей и преждевременными родами; расстройство, ассоциированное с эозинофилами; тромбоз; глауко-3 006134 ма и нарушения зрения; окклюзионные сосудистые заболевания, такие как, например, атеросклероз; застойная сердечная недостаточность; заболевания или состояния, требующие лечения антикоагулянтами,такие как посттравматические состояния или состояния после хирургического вмешательства; ревматоидный артрит и другие воспалительные заболевания; гангрена; болезнь Рейно; нарушения выделения секрета слизистой, включая нарушение цитопротективных функций; боли и мигрень; заболевания, требующие регуляции остеогенеза и резорбции кости, такие как, например, остеопороз; инсульт; нарушение терморегуляции, включая повышение температуры; реакция отторжения при трансплантации органов и при операции по шунтированию и иммунные расстройства или состояния, при которых желательна иммунорегуляция. Более конкретным заболеванием, требующим лечения, является одно из заболеваний,опосредуемых простагландином D2, таких как застойная гиперемия носа, аллергический ринит, застой легких и астма, включая аллергическую астму. Интервалы доз Очевидно, что величина профилактической или терапевтической дозы соединения формулы I варьируется в зависимости от природы и тяжести состояния, подвергаемого лечению, от конкретно используемого соединения формулы I и способа его введения. Эта доза также варьируется в зависимости от ряда факторов, включая возраст, массу, общее состояние здоровья, пол, режим питания, время введения,скорость экскреции, комбинацию лекарственных средств и ответ данного конкретного пациента. В основном, суточная доза составляет примерно от 0,001 до 100 мг на кг, а предпочтительно примерно от 0,01 до 10 мг на кг массы тела млекопитающего. С другой стороны, в некоторых случаях, если это необходимо, то могут быть использованы дозы, выходящие за вышеуказанные пределы. Количество активного ингредиента, которое может быть объединено с материалами-носителями для приготовления разовой лекарственной формы, будет варьироваться в зависимости от конкретного хозяина, подвергаемого лечению, и от конкретного способа введения. Так, например, препарат, предназначенный для перорального введения человеку, может содержать от 0,05 мг до 5 г активного агента,взятого вместе с соответствующим и стандартным количеством материала-носителя, которое может варьироваться примерно от 5 до 99,95% от всей композиции. Разовые лекарственные формы, в основном,содержат примерно от 0,1 мг до 0,4 г активного ингредиента, обычно 0,5, 1, 2, 5, 10, 25, 50, 100, 200 или 400 мг. Фармацевтические композиции В другом аспекте настоящее изобретение относится к фармацевтическим композициям, содержащим соединение формулы I и фармацевтически приемлемый носитель. Термин "композиция", как и фармацевтическая композиция, означает продукт, содержащий активный(е) ингредиент(ы) и инертный(е) ингредиент(ы) (фармацевтически приемлемые наполнители), которые входят в состав носителя, а также любой продукт, который образуется, прямо или опосредованно, в результате комбинации, комплексообразования или агрегации любых двух или более ингредиентов, либо в результате диссоциации одного или нескольких ингредиентов, либо в результате осуществления других типов реакций или взаимодействий одного или нескольких ингредиентов. В соответствии с этим фармацевтическими композициями настоящего изобретения могут быть любые композиции, полученные путем смешивания соединения формулыI, дополнительного(ых) активного(ых) ингредиента(ов) и фармацевтически приемлемых наполнителей. Для лечения любых опосредованных простаноидами заболеваний соединения формулы I могут быть введены перорально, путем ингаляции, местно, парентерально или ректально в виде разовых лекарственных форм, содержащих стандартные нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители. Используемый здесь термин "парентерально" означает введение, включая подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или вливания. Помимо лечения теплокровных животных, таких как мыши, крысы, лошади, крупный рогатый скот, овцы, собаки,кошки и т.п., соединения настоящего изобретения эффективны и для лечения человека. Фармацевтические композиции, содержащие активный ингредиент, могут быть получены в форме,подходящей для перорального использования, например в виде таблеток, пастилок, лепешек, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, жестких или мягких капсул либо сиропов или эликсиров. Композиции, предназначенные для перорального введения, могут быть приготовлены любым методом, известным специалистам в области фармацевтики, и такие композиции могут содержать один или несколько агентов, выбранных из группы, состоящей из подслащивающих агентов,ароматизаторов, красителей и консервантов, для придания им фармацевтически эстетических и приятных вкусовых качеств. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, которые являются подходящими для производства таблеток. Такими наполнителями могут быть, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие агенты, например кукурузный крахмал или альгиновая кислота; связующие агенты, например крахмал, желатин или аравийская камедь, и замасливающие агенты, например стеарат магния, стеариновая кислота или тальк. Эти таблетки могут быть изготовлены без покрытия, либо, для замедления дезинтеграции и абсорбции в желудочно-кишечном тракте и, таким образом, для обеспечения их пролонгированного действия в течение длительного периода времени, они могут иметь покрытие, наносимое известными методами. Напри-4 006134 мер, для этих целей может быть использован материал пролонгированного действия, такой как глицерилмоностеарат или глицерилдистеарат. Для получения осмотических терапевтических таблеток для регулируемого высвобождения на них может быть нанесено покрытие способом, описанным в патентах США 4256108, 4166452 и 4265874. Препараты для перорального введения могут быть также изготовлены в виде жестких желатиновых капсул, где активный ингредиент смешивают с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, либо в виде мягких желатиновых капсул, где активный ингредиент смешивают с растворителями, смешивающимися с водой, такими как пропиленгликоль, ПЭГ и этанол, или с масляной средой, например с арахисовым маслом, вазелиновым маслом или с оливковым маслом. Водные суспензии содержат активный материал в смеси с наполнителями, подходящими для получения водных суспензий. Такими наполнителями являются суспендирующие агенты, например натрийсодержащая карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь; диспергирующие или смачивающие агенты, которые могут представлять собой природный фосфатид, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, происходящими от жирных кислот и гексита, такие как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с неполными сложными эфирами, происходящими от жирных кислот и ангидридов гексита, например полиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один или несколько консервантов, например этил- или н-пропил, п-гидроксибензоат, один или несколько красителей, один или несколько ароматизаторов и один или несколько подсластителей, таких как сахароза, сахарин или аспартам. Масляные суспензии могут быть получены путем суспендирования активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле, кокосовом масле, или в минеральном масле, таком как вазелиновое масло. Масляные суспензии могут содержать загуститель,например пчелиный воск, твердый парафин или цетиловый спирт. Для придания пероральному препарату приятных вкусовых качеств в данную композицию могут быть добавлены подсластители, такие как подсластители, указанные выше, и ароматизаторы. Для сохранения этих композиций в них могут быть добавлены антиоксиданты, такие как аскорбиновая кислота. Диспергируемые порошки и гранулы, подходящие для получения водной суспензии путем добавления воды, содержат активный ингредиент в смеси с диспергирующим или смачивающим агентом, суспендирующим агентом и с одним или несколькими консервантами. Примеры подходящих или смачивающих агентов и суспендирующих агентов были указаны выше. Могут также присутствовать и другие наполнители, например подсластители, ароматизаторы и красители. Фармацевтические композиции настоящего изобретения могут быть также получены в виде эмульсии типа "масло-в-воде". Масляная фаза может представлять собой растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например вазелиновое масло, или их смеси. Подходящими эмульгаторами могут быть природные фосфатиды, например соевые бобы, лецитин и сложные эфиры или неполные сложные эфиры, происходящие от жирных кислот и ангидридов гексита,например сорбитанмоноолеат, и продукты конденсации указанных неполных эфиров с этиленоксидом,например полиоксиэтиленсорбитанмоноолеат. Указанные эмульсии могут также содержать подсластители и ароматизаторы. В сиропы и эликсиры могут быть добавлены подсластители, например глицерин, пропиленгликоль,сорбит или сахароза. Такие препараты могут также содержать деэмульгатор, консервант, отдушки и красители. Фармацевтические композиции могут быть приготовлены в форме стерильных водных или масляных суспензий для инъекций. Такая суспензия может быть получена известными методами с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов, упомянутых выше. Стерильный препарат для инъекций может также представлять собой стерильный раствор или суспензию для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе,например раствор в 1,3-бутандиоле. Наряду с приемлемыми наполнителями и растворителями могут быть использованы вода, раствор Рингера и изотонический раствор хлорида натрия. Могут быть также использованы сорастворители, такие как этанол, пропиленгликоль или полиэтиленгликоли. Кроме того, в качестве растворителя или суспендирующей среды обычно используются стерильные малоподвижные масла. Для этих целей может быть использована любая смесь жирных масел, включая синтетические моно- или диглицериды. Кроме того, в препаратах для инъекций могут быть также использованы жирные кислоты, такие как олеиновая кислота. Соединения формулы I могут быть также введены в форме суппозиториев для ректального введения лекарственного средства. Эти композиции могут быть получены путем смешивания лекарственного средства с подходящим нераздражающим наполнителем, который является твердым при комнатной тем-5 006134 пературе и жидким при температуре прямой кишки, где он расплавляется с высвобождением лекарственного средства. Такими материалами являются масло какао и полиэтиленгликоли. Для местного применения используются кремы, мази, гели, растворы или суспензии, содержащие,например, соединение формулы I. (Для такого местного применения могут быть использованы жидкости для полоскания рта и для полоскания горла.) Композиции для местного применения могут, в основном,состоять из фармацевтически приемлемого носителя, сорастворителя, эмульгатора, агента, улучшающего смачивание, системы консервантов и умягчителя. Комбинации с другими лекарственными средствами Для лечения и предупреждения опосредованных простагландином заболеваний соединение формулы I может быть введено вместе с другими терапевтическими агентами. Таким образом, в другом аспекте настоящее изобретение относится к фармацевтическим композициям для лечения опосредованных простагландином D2 заболеваний, содержащим терапевтически эффективное количество соединения формулы I и одного или нескольких других терапевтических агентов. Подходящими терапевтическими агентами для комбинированной терапии с использованием соединения формулы I являются (1) антагонист рецептора простагландина; (2) кортикостероид, такой как ацетонид триамцинолона; (3) -агонист, такой как салметерол, формотерол, тербуталин, метапротеренол, альбутерол и т.п.; (4) модификатор лейкотриенов,такой как антагонист лейкотриенов или ингибитор липооксигеназы, такой как монтелукаст, зафирлукаст,пранлукаст или зилейтон; (5) антигистамин (антагонист гистамина H1), такой как бромфенирамин, хлорфенирамин, дексхлорфенирамин, трипролидин, клемастин, дифенгидрамин, дифенилпиралин, трипеленамин,гидроксизин, метдилазин, прометазин, тримепразин, азатадин, ципрогептадин, антазолин, фенирамин,пириламин, астемизол, норастемизол, терфенадин, лоратадин, цетиризин, левоцетиризин, фексофенадин,деслоратадин, и т.п.; (6) противозастойное лекарственное средство, включая фенилэфрин, фенилпропаноламин, псевдофедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин, пропилгекседрин или леводезоксиэфедрин; (7) средство против кашля, включая кодеин, гидрокодон, карамифен, карбетапентан или декстраметорфан; (8) другой лиганд простагландина, включая агонист простагландина F, такой как латанопрост; мизопростол, энпростил, риопростил, орнопростол или розапростол; (9) диуретик; (10) нестероидные противовоспалительные средства (НСПВС), такие как производные пропионовой кислоты(индометацин, ацеметацин, алклофенак, клиданак, диклофенак, фенклофенак, фенклозиновая кислота,фентиазак, фурофенак, ибуфенак, изоксепак, окспинак, сулиндак, тиопинак, толметин, зидометацин и зомепирак), производные фенаминовой кислоты (флуфенаминовая кислота, меклофенаминовая кислота,мефенаминовая кислота, нифлуминовая кислота и толфенаминовая кислота), производные бифенилкарбоновой кислоты (дифлунизал и флуфенизал), оксикамы (изоксикам, пироксикам, судоксикам и теноксикан), салицилаты (ацетилсалициловая кислота, сульфасалазин) и пиразолоны (апазон, безпиперилон,фепразон, мофебутазон, оксифенбутазон, фенилбутазон); (11) ингибиторы циклооксигеназы-2 (СОХ-2),такие как целекоксиб, рофекоксиб, эторикоксиб и вальдекоксиб; (12) ингибиторы фосфодиэстеразы типаCCR-1, CCR-2 и CCR-3; (14) агенты, снижающие уровень холестерина, такие как ингибиторы HMG-CoAредуктазы (ловастатин, симвастатин и правастатин, флувастатин, аторвастатин и другие статины), секвестранты (холестирамин и колестипол), никотиновая кислота, производные фенофибриновой кислоты(гемфиброзил, клофибрат, фенофибрат и бензафибрат) и пробукол; (15) противодиабетические средства,такие как инсулин, сульфонилмочевины, бигуаниды (метформин), ингибиторы -глюкозидазы (акарбоза) и глитазоны (троглитазон, пиоглитазон, энглитазон, розиглитазон и т.п.); (16) препараты интерферонабета (интерферон бета-1a, интерферон бета-1b); (17) антихолинергические агенты, такие как мускариновые антагонисты (бромид ипратропия и бромид тиотропия), а также селективные мускариновые антагонисты М 3; (18) стероиды, такие как беклометазон, метилпреднизолон, бетаметазон, преднизон, дексаметазон и гидрокортизон; (19) триптаны, обычно используемые для лечения мигрени, такие как сумитриптан и ризатриптан; (20) алендронат и другие средства для лечения остеопороза; (21) другие соединения, такие как 5-аминосалициловая кислота и их пролекарства, антиметаболиты, такие как азатиоприн и 6-меркаптопурин, цитотоксические химиотерапевтические агенты для лечения злокачественной опухоли, антагонисты брадикинина (ВK2 или ВK1), антагонисты рецептора ТР, такие как сератродаст, антагонисты нейрокинина (NK1/NK2), антагонисты VLA-4, описанные, например, в патенте США 5510332, в WO 97/03094,WO 97/02289, WO 96/40781, WO 96/22966, WO 96/20216, WO 96/01644, WO 96/06108, WO 95/15973 иWO 96/31206. Кроме того, настоящее изобретение относится к способу лечения заболеваний, опосредованных простагландином D2, где указанный способ предусматривает введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы I, вводимого вместе с одним или несколькими вышеперечисленными ингредиентами. Активные ингредиенты могут быть введены в количествах, которые обычно используются для каждого активного ингредиента, вводимого отдельно,-6 006134 либо в некоторых случаях при использовании комбинации активных ингредиентов доза одного или нескольких активных ингредиентов может быть уменьшена. Используемые сокращения Ас Ацетил АсОН Уксусная кислотаEtOH Этанол ВЭЖХ Высокоэффективная жидкостная хроматографияN-Метил-2-пирролидинон ЯМР Ядерный магнитный резонанс ТГФ Тетрагидрофуран ТСХ Тонкослойная хроматография Способы синтеза Соединения формулы I настоящего изобретения могут быть получены способами синтеза, описанными в схемах 1-5, и способами, описанными ниже. Промежуточные соединения формулы IV могут быть получены способом, описанным в схеме 1, из соответствующим образом замещенного фенилгидразина II. В результате взаимодействия соединения II с соответствующим циклоалканоном III (где R представляет собой сложноэфирную группу, такую как алкильная группа) в присутствии индола Фишера или в аналогичных условиях получают соединение IV. Схема 1 Альтернативно, соединения формулы IV могут быть получены способом, представленным на схеме 2, из соответствующим образом замещенного анилина V. В результате конденсации соединения V с соответствующим циклоалканоном III и последующей циклизации в условиях Хека или в аналогичных условиях катализа в присутствии металлов получают индол IV. Схема 2 Соединения формулы III могут быть получены способом, представленным на схеме 3, из соответствующим образом замещенного силилового эфира енола VI или из соответствующим образом замещенного енамина VII. После реакции присоединения соответствующего электрофила, такого как Y-CH2CO2R(где Y представляет собой галоген или уходящую группу) в присутствии основания, такого как алкиллитий или кислота Льюиса, такая как трифторацетат серебра, с силиловым эфиром енола VI получают циклоалканон III. Альтернативно, соединение формулы III может быть получено посредством присоединения Y-CH2CO2R к соответствующим образом замещенному енамину VII в присутствии енамина Сторка или в аналогичных условиях. Промежуточные соединения формулы VIII могут быть получены способом, описанным в схеме 4, из соответствующим образом замещенного индола IV. Бромирование соединения IV может быть осуществлено с использованием брома или бромирующего агента, такого как трибромид пиридия, в основных условиях в полярном растворителе, например путем проведения реакции в пиридине или в растворителе, таком как дихлорметан, в присутствии пиридина с последующим моновосстановлением промежуточного дибромсоединения в присутствии кислоты и в условиях восстановления металлом, в результате чего получают броминдол VIII. Схема 4 Соединения формулы I могут быть получены способом, представленным на схеме 5, из соответствующим образом замещенного броминдола VIII. В результате алкилирования соединения VIII соответствующим электрофилом, таким как (R1)(R2)CH-Y, в присутствии основания и в подходящем растворителе,таком как ДМФ, получают N-алкилированный индол IX. Взаимодействие соединения IX с метансульфинатом, таким как метансульфинат натрия, в присутствии солей Cu(I), приводит к образованию соединений формулы I после гидролиза сложным эфиром. Альтернативно, сначала броминдоловая кислота (IX, R=H) может быть подвергнута взаимодействию с подходящим металлирующим агентом, таким как н-BuLi, с последующим захватом электрофилом, таким как метилдисульфид, в результате чего образуется соответствующий метилсульфид, который подвергают, например, окислению пероксидом водорода/тунгстатом натрия, с получением соединения IA. Стадии алкилирования броминдола VIII с последующим сульфированием могут быть также обратимыми, так, например, в результате реакции сульфирования броминдолаVIII получают соединение X, которое алкилируют в условиях, аналогичных описанным ранее, или в реакционных условиях Митцунобу, и после гидролиза сложным эфиром получают соединение формулы IA. Схема 5-8 006134 Соединение IB может быть получено из защищенного соединения IA, например сложного эфира IA,путем окисления подходящим окислителем с последующим гидролизом, как проиллюстрировано на схеме 6. Схема 6 Альтернативно, соединение IB может быть получено, как проиллюстрировано на схеме 7, путем окисления соединения IX подходящим окислителем, таким как DDQ, с последующим метилсульфированием, как показано на схеме 5, и с последующим гидролизом. Схема 7 Анализы для определения биологической активности Соединения формулы I могут быть проанализированы с помощью нижеследующих анализов для определения их активности в качестве антагонистов или агонистов простаноидов in vitro и in vivo и их селективности. Продемонстрированными активностями рецепторов простагландинов являются DP, EP1,ЕP2, ЕР 3, ЕP4, FP, IP и ТР. Стабильная экспрессия простаноидных рецепторов в эмбриональной клеточной линии почки человека (НЕK) 293(ebna) кДНK простаноидного рецептора, соответствующие полноразмерным кодирующим последовательностям, субклонировали в соответствующие сайты экспрессирующих векторов млекопитающих и трансфецировали в клетки НЕK 293(ebna). Клетки НЕK 293(ebna), экспрессирующие отдельные кДНК, культивировали в селективных условиях и после 2-3-недельного культивирования отдельные колонии выделяли методом с использованием клонзапирающего кольца, а затем размножали с получением клональных клеточных линий. Анализы на связывание с простаноидным рецептором Клетки НЕK 293(ebna) поддерживали в культуре, собирали и путем дифференциального центрифугирования получали мембраны, после чего клетки лизировали в присутствии ингибиторов протеазы для использования в анализах на связывание с рецептором. Анализы на связывание с простаноидным рецептором осуществляли в 10 мМ MES/KOH (рН 6,0) (ЕР, FP и ТР) или 10 мМ HEPES/KOH (рН 7,4) (DP иIP), содержащем 1 мМ EDTA, 10 мМ двухвалентный катион и соответствующий радиоактивный лиганд. Реакцию инициировали добавлением мембранного белка. Лиганды добавляли в диметилсульфоксиде,который поддерживали постоянным при 1% (об./об.) в течение всего периода инкубирования. Уровень неспецифического связывания определяли в присутствии 1 мкМ соответствующего нерадиоактивного простаноида. Инкубирование проводили в течение 60 мин при комнатной температуре или при 30 С и прекращали путем быстрой фильтрации. Специфическое связывание вычисляли путем вычитания неспецифического связывания из общего связывания. Затем для построения сигмоидальной кривой "концентрация-ответ" в целях определения аффинности лиганда вычисляли остаточное специфическое связывание для каждой концентрации лиганда, которое выражали как функцию концентрации лиганда. Анализ на агонисты и антагонисты простаноидного рецептора Анализ на вторичный мессенджер в целых клетках, где оценивали стимуляцию (ЕР 2, ЕP4, DP и IP в клетках НЕK 293(ebna или ингибирование (ЕР 3 в клетках человеческого эритролейкоза (HEL накопления внутриклеточного сАМР или мобилизации внутриклеточного кальция (EP1, FP и ТР в клетках НЕK 293(ebna), стабильно трансфецированных апоэкворином), осуществляли для того, чтобы определить, являются ли лиганды рецептора агонистами или антагонистами. Для анализов на сАМР клетки собирали и ресуспендировали в буфере HBSS, содержащем 25 мМ HEPES, рН 7,4. Смесь для инкубирования содержала 100 мкМ RO-20174 (ингибитор фосфодиэстеразы типа IV, поставляемый фирмой Biomol), а в случае анализа только на ингибирование ЕР 3 она содержала 15 мкМ форсколина для стимуляции продуцирования сАМР. Образцы инкубировали при 37 С в течение 10 мин, реакцию прекращали, а затем измеряли уровни сАМР. Для анализов на мобилизацию кальция клетки нагружали кофакторами, восстанов-9 006134 ленным глутатионом и коэлентерацином, собирали и ресуспендировали в среде Хэмса F12. Мобилизацию кальция измеряли путем мониторинга люминесценции, индуцированной связыванием кальция с внутриклеточным фотопротеином экворином. Лиганды добавляли в диметилсульфоксиде, который поддерживали постоянным при 1% (об./об.) в течение всего периода инкубирования. Для агонистов ответ в форме вторичного мессенджера выражали как функцию концентрации лиганд, и вычисляли величинуEC50 и величину максимального ответа по сравнению со стандартным простаноидом. Для антагонистов способность лиганда ингибировать ответ агониста оценивали с помощью анализа Шилда и вычисляли величину KB и угол наклона кривой. Предупреждение застойной гиперемии носа, индуцируемой PGD2 или аллергеном, у овец с аллергией Подготовка животных: использовали взрослых здоровых овец (18-50 кг). Этих животных отбирали,исходя из природой положительной кожной реакции на внутрикожную инъекцию экстракта Ascarissuum. Определение степени застойной гиперемии носа: этот эксперимент осуществляли на неанестезированных животных. Этих животных фиксировали на столике в положении лежа на животе с неподвижно закрепленной головой. Резистентность носовых дыхательных путей (NAR) измеряли методом ринометрии с использованием модифицированной маски. Для введения назотрахеальной трубки носовые ходы подвергали местной анестезии (2% лидокаином). Максимальный конец этой трубки подсоединяли к пневмотахографу и сигнал потока и давления регистрировали на осциллоскопе, подсоединенном к компьютеру для вычисления NAR в режиме реального времени. Назальную провокацию осуществляли путем введения аэрозольного раствора (10 впрысков на ноздрю). Изменения в застойной гиперемии носовых ходов регистрировали до введения провокационной дозы и в течение 60-120 мин после провокации. Предупреждение обструкции носа, индуцируемой PGD2 или аллергеном, у собакоподобных обезьян Подготовка животных: использовали взрослых здоровых самцов собакоподобных обезьян (4-10 кг). Этих животных отбирали, исходя из их природной положительной кожной реакции на внутрикожную инъекцию экстракта Ascaris suum. Перед началом каждого эксперимента обезьян, отобранных для исследования, держали в условиях голодания в течение ночи со свободным доступом к воде. На следующее утро животным вводили кетамин (10-15 мг/кг, i.m.), а затем их забирали из клеток, в которых они содержались. Обезьян помещали на нагретый столик (36 С) и инъецировали ударную дозу (5-12 мг/кг, i.v.) пропофола. Животным интубировали эндотрахеальную трубку с надувной манжетой (внутренний диаметр 4-6 мм) и анестезию поддерживали путем непрерывного внутривенного вливания пропофола (25-30 мг/кг/ч). В течение всего эксперимента проводили мониторинг параметров жизненных функций (частоты сердечных сокращений, кровяного давления, частоты дыхания, температуры тела). Определение степени застойной гиперемии носа: измерение сопротивления дыхательных путей осуществляли на пневмотахографе, подсоединенном к эндотрахеальной трубке для гарантии ее нормального функционирования. Для оценки застойной гиперемии носа использовали акустический ринометрEcovision. Эта техника позволяет получать неинвазивную 2-размерную эхограмму внутренней полости носа. Носовой объем и площадь поперечного сечения по всей длине носовой полости вычисляли через 10 с на лэптоп-компьютере (типа ноутбука), снабженном специально разработанным пакетом программ(Hood Laboratories, Mass, U.S.A.). Введение назальной провокационной дозы осуществляли непосредственно в носовую полость животного (объем 50 мкл). Изменение в застойной гиперемии носовых ходов регистрировали до введения провокационной дозы и в течение 60-120 мин после введения провокационной дозы. Если имеет место застойная гиперемия носа, то на это будет указывать снижение носового объема. Биомеханика легких у "обученных" беличьих обезьян Процедура этого теста заключалась в том, что "обученных" беличьих обезьян помещали на специальное кресло в аэрозольные камеры. Для контроля пульмонально-механические измерения параметров дыхательных путей проводили в течение примерно 30 мин в целях установления нормальных контрольных значений для каждой обезьяны на этот день. Для перорального введения соединения растворяли или суспендировали в растворе 1% метоцела (метилцеллюлозы, 65HG, 400 сП) и вводили в объеме 1 мл/кг массы тела. Для аэрозольного введения соединений использовали ультразвуковой аэрозольный ингалятор DeVilbiss. Периоды предварительной обработки варьировались от 5 мин до 4 ч, после которой обезьянам вводили провокационные аэрозольные дозы либо PGD2, либо антигена Ascaris suum при разведении 1:25. После введения провокационной дозы через каждую минуту на компьютере вычисляли данные, выраженные в процентах от контрольного значения для каждого параметра дыхательных путей, включая сопротивление дыхательных путей (RL) и динамическую податливость (Сdyn). После этого для каждого тестируемого соединения получали результаты за период минимум 60 мин после введения провокационной дозы, которые затем сравнивали с предварительно измеренными исходными базальными контрольными значениями для данной обезьяны. Кроме того, все величины, полученные в течение 60 мин после введения провокационной дозы, для каждой обезьяны (исходные базальные значения и тест-величины) отдельно усредняли и использовали для вычисления общего процента ингибирования ответа медиатора или антигена Ascaris тестируемым соединением. Для статистического анализа использовали парный т- 10006134 критерий. (Работы: McFarlane, C.S. et al., Prostaglandins, 28, 173-182 (1984) и McFarlane C.S. et al., AgentsActions, 22, 63-68 (1987. Предупреждение индуцированного бронхостеноза у аллергических овец Подготовка животных: использовали взрослых здоровых овец со средней массой 35 кг (в интервале от 18 до 50 кг). Все используемые животные удовлетворяли двум критериям: а) они давали природную кожную реакцию на разведения 1:1000 или 1:10000 экстракта Ascaris suum (Greer, Diagnostics, Lenois,NC); и b) они ранее обнаруживали реакцию на провокационную ингаляцию экстракта Ascaris suum, выражающуюся острым бронхостенозом и поздней обструкцией бронхов (W.M. Abraham et al., Am. Rev.Resp. Dis., 128, 839-44 (1983. Измерение механических параметров: неанестезированную овцу фиксировали на столике в положении лежа на животе с неподвижно закрепленной головой. После местной анестезии носовых ходов раствором 2% лидокаина через одну ноздрю вставляли баллонный катетер в нижний отдел пищевода. Затем через другую ноздрю животным интубировали эндотрахеальную трубку с надувной манжетой с использованием гибкого фибробронхоскопа в качестве волновода. Плевральное давление оценивали с помощью эзофагального баллонного катетера (наполненного 1 мл воздуха), который был установлен так,чтобы вдох давал сдвиг отрицательного давления с явно различимыми кардиогенными осцилляциями. Латеральное давление в трахее измеряли с помощью катетера с боковым отверстием (внутренний диаметр 2,5 мм), проходящего через назотрахеальную трубку и расположенного на определенном расстоянии от конца этой назотрахеальной трубки. Транспульмонарное давление, которое определяется как разность между трахеальным давлением и плевральным давлением, измеряли с помощью датчика дифференциального давления (DP45; Validyne Corp., Northridge, CA). Для измерения резистентности легких (RL) максимальный конец назотрахеальной трубки подсоединяли к пневмотахографу (Fleish, Dyna Science, BlueBell, PA). Сигналы потока и транспульмонарного давления регистрировали на осциллоскопе (Model DR-12;Electronics for Medicine, White Plains, NY), который был подсоединен к цифровому компьютеру PDP-11(Digital Equipment Corp., Maynard, MA) для вычисления RL из транспульмонарного давления, дыхательного объема, полученного путем интегрирования, и потока в режиме реального времени. Для определения RL использовали анализ 10-15 вдохов. Для получения удельного сопротивления легких (SRL=RLVtg) измеряли объем газа в грудной клетке (Vtg) на плетизмографе. Для иллюстрации настоящего изобретения ниже приводятся примеры, которые не должны рассматриваться как ограничение объема настоящего изобретения. Если это не оговорено особо, то в этих примерах все конечные продукты соединения формулы I были проанализированы с помощью ЯМР, ТСХ и элементного анализа или масс-спектроскопии; промежуточные соединения анализировали с помощью ЯМР и ТСХ; большинство соединений очищали флэш-хроматографией на силикагеле, перекристаллизацией и/или заменой растворителя (суспензия в растворителе с последующей фильтрацией твердых веществ); за ходом реакции следили с помощью тонкослойной хроматографии (ТСХ), а время реакции приводится лишь для иллюстрации; энантиомерный избыток измеряли на ВЭЖХ с нормальной фазой и с использованием хиральной колонки: ChiralPak AD; 250 х 4,6 мм. Нижеследующие промежуточные соединения получали в соответствии с процедурами, описанными в литературе, или закупали у нижеследующих поставщиков: этил-2-(2-оксоциклопентил)ацетат: Acros/Fisher Scientific; 4-фтор-2-иоданилин: Beugelmans, R.; Chbani, M. Bull. Soc. Chim. Fr. 1995, 132, 306-313. Пример 1. (-)-[4-(4-Xлорбензил)-7-фтор-5-(метансульфонил)-1,2,3,4-тетрагидроциклопента[b]индол 3-ил]уксусная кислота. Раствор 10,00 г 4-фтор-2-иоданилина, 6,57 г этил 2-(2-оксоциклопентил)ацетата и 121 мг птолуолсульфоновой кислоты в 100 мл бензола кипятили с обратным холодильником с использованием ловушки Дина-Старка в атмосфере N2 в течение 24 ч. По истечении этого времени бензол удаляли путем перегонки. Затем добавляли 60 мл ДМФ и раствор дегазировали, после чего последовательно добавляли 19 мл основания Хьюнига и 405 мг Pd(OAc)2. Раствор нагревали до 115 С в течение 3 ч, а затем охлаждали до комнатной температуры. Реакцию гасили добавлением 300 мл 1 н НСl и 200 мл этилацетата и смесь- 11006134 фильтровали через целит. Фазы разделяли и кислотную фазу 2 раза экстрагировали 200 мл этилацетата. Органические слои объединяли, промывали насыщенным раствором соли, сушили над безводнымNa2SO4, фильтровали через целит и концентрировали. Затем неочищенный продукт очищали флэшхроматографией, элюируя 100% толуолом, и получали 5,36 г указанного в заголовке соединения в виде желтого твердого вещества. 1 К раствору 1,24 г сложного эфира, полученного на стадии 1, в 14 мл тетрагидрофурана (ТГФ) при комнатной температуре добавляли 7 мл МеОН, а затем 7 мл 2 н NaOH. Через 2,5 ч реакционную смесь выливали в делительную воронку, содержащую этилацетат (EtOAc)/1 н НСl. Фазы разделяли и кислотную фазу 2 раза экстрагировали EtOAc. Органические слои объединяли, промывали насыщенным раствором соли, сушили над безводным Na2SO4 и упаривали досуха с получением 1,08 г неочищенного и нестабильного восковидного коричневого масла, которое использовали в следующей стадии без очистки К раствору 2,20 г кислоты, полученной на стадии 2 (чистота 90%), в 30 мл пиридина добавляли 6,85 г трибромида пиридиния (чистота 90%) при -40 С. Суспензию перемешивали в течение 10 мин при 0 С и нагревали до комнатной температуры в течение 30 мин. Затем растворитель удаляли без нагревания в высоком вакууме. Неочищенный продукт растворяли в 40 мл АсОН и к холодному раствору при 0 С порциями добавляли 2,88 г цинковой пыли. Суспензию перемешивали в течение 15 мин при 15 С и нагревали до комнатной температуры еще 15 мин. По истечении этого времени реакционную смесь гасили добавлением 1 н НСl и смесь выливали в делительную воронку, содержащую насыщенный раствор соли/EtOAc. Слои разделяли и органический слой промывали водой, насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали. Этот продукт использовали в следующей стадии без дополнительной очистки. 1 К раствору 2,13 г кислоты, полученной на стадии 3, в 10 мл ТГФ добавляли избыток раствора диазометана в простом эфире до тех пор, пока ТСХ не указывала на полное израсходование кислоты. Затем растворители удаляли в вакууме. К полученному таким образом раствору неочищенного метилового эфира в 20 мл ДМФ при -78 С добавляли 539 мг суспензии NaH (60% в масле). Суспензию перемешивали в течение 10 мин при 0 С, снова охлаждали до -78 С и обрабатывали 1,70 г 4-хлорбензилбромида. Через 5 мин температуру повышали до 0 С и смесь перемешивали в течение 20 мин. По истечении этого времени реакцию гасили добавлением 2 мл АсОН и эту смесь выливали в делительную воронку, содержащую 1 н HCl/EtOAc. Слои разделяли и органический слой промывали насыщенным раствором соли,сушили над безводным Na2SO4 и концентрировали. Алкилированный продукт гидролизовали в соответствии с процедурой, описанной на стадии 2. Затем неочищенный продукт очищали путем растирания со смесью EtOAc/гексаны, в результате чего получали 2,35 г указанного в заголовке соединения в виде светло-коричневого твердого вещества. 1 К раствору 2,35 г кислоты, полученной на стадии 4, в 130 мл EtOH при 80 С добавляли 780 мкл (S)(-)-1-(1-нафтил)этиламина. Раствор охлаждали до комнатной температуры и перемешивали в течение ночи. Выделенную соль (1,7 г) снова перекристаллизовывали из 200 мл EtOH. После фильтрации полученную соль в виде белого твердого вещества нейтрализовали 1 н НСl и продукт экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали. Полученный продукт фильтровали через слой SiO2, элюируя EtOAc, в результате чего получали 500 мг указанного в заголовке энантиомера в виде белого твердого вещества. Время удерживания двух энантиомеров составляло, соответственно, 7,5 мин и 9,4 мин [колонка ChiralPak AD, смесь гексан/2 пропанол/уксусная кислота (95:5:0,1)]. Более полярный энантиомер имел энантиомерный избыток (э.и.) 98%. Э.и.=98%; время удерживания=9,4 мин [колонка ChiralPak AD, 250 х 4,6 мм, смесь гексаны/2-пропанол/уксусная кислота (75:25:0,1)]; []D21=+39,2 (с 1,0, МеОН). Стадия 6. (-)-[4-(4-Xлорбензил)-7-фтор-5-(метансульфонил)-1,2,3,4-тетрагидроциклопента[b]индол 3-ил]уксусная кислота и ее натриевая соль. Кислоту, полученную на стадии 5 (15,4 г), сначала эстерифицировали диазометаном. Сульфирование осуществляли путем смешивания полученного таким образом сложного эфира с 16,3 г натриевой соли метансульфиновой кислоты и 30,2 г CuI(I) в N-метилпирролидиноне. Суспензию дегазировали в потоке N2, нагревали до 150 С и перемешивали в течение 3 ч, после чего ее охлаждали до комнатной температуры. Реакцию гасили добавлением 500 мл этилацетата и 500 мл гексанов, и смесь фильтровали через слой SiO2, элюируя EtOAc. Органические фазы концентрировали. Неочищенное масло растворяли в EtOAc, 3 раза промывали водой и 1 раз насыщенным раствором соли, сушили над безводным Na2SO4,фильтровали и концентрировали. Затем неочищенный продукт очищали флэш-хроматографией, элюируя градиентом 100% толуол 50% толуол в EtOAc, и получали 14 г сульфированного сложного эфира, который гидролизовали в соответствии в процедурой, описанной со стадии 2. После двух последовательных перекристаллизаций: из смеси изопропилацетат/гептан, а затем из смесей СH2 Сl2/гексаны - получали указанное в заголовке соединение (9,8 г) в виде белого твердого вещества. 1MS (-APCI) m/z 436,1, 434,1 (М-Н)-. Э.и.=97%; время удерживания=15,3 мин [колонка ChiralCel OD: 250 х 4,6 мм, смесь гексаны/2-пропанол/этанол/уксусная кислота (90:5:5:0,2)]; []D21=-29,3 (с 1,0, МеОН). Т.пл. 175,0 С. Натриевую соль получали путем обработки 6,45 г (14,80 ммоль) вышеуказанного кислотного соединения в EtOH (100 мл) 14,80 мл водного 1 н раствора NaOH. Органический растворитель удаляли в вакууме и неочищенное твердое вещество растворяли в 1,2 л изопропилового спирта при кипячении с обратным холодильником. Конечный объем снижали до 500 мл путем перегонки растворителя. Натриевую соль кристаллизовали путем охлаждения до комнатной температуры. Кристаллическую натриевую соль суспендировали в Н 2 О, замораживали в бане с сухим льдом и лиофилизовали в высоком вакууме с получением 6,00 г указанного в заголовке соединения в виде натриевой соли. 1H ЯМР (500 МГц, ДМСО-d6):7,63 (дд, 1 Н, J1=8,5 Гц, J2=2,6 Гц), 7,47 (дд, 1 Н, J1=9,7 Гц, J2=2,6 Гц),7,33 (д, 2 Н, J=8,4 Гц), 6,70 (д, 2 Н, J=8,4 Гц), 6,06 (д, 1 Н, JAB=17,9 Гц), 5,76 (д, 1 Н, JAB=17,9 Гц), 3,29 (м,1 Н), 3,08 (с, 3 Н), 2,80 (м, 1 Н), 2,69 (м, 1 Н), 2,55 (м, 1 Н), 2,18 (м, 2 Н), 1,93 (дд, 1 Н, J1=14,4 Гц, J1=9,7 Гц). Пример 1 А. Альтернативная процедура получения -[5-бром-4-(4-хлорбензил)-7-фтор-1,2,3,4 тетрагидроциклопента[b]индол-3-ил]уксусной кислоты (пример 1, стадия 4). Стадия 1. Дициклогексиламиновая (DCHA) соль -(7-фтор-1,2,3,4-тетрагидроциклопента[b]индол 3-ил)уксусной кислоты. 0,526 М раствор 2-бром-4-фторанилина в ксилоле вместе с этил-(2-оксоциклопентил)ацетатом (1,5 экв.) и серной кислотой (0,02 экв.) нагревали до температуры дефлегмации в течение 20 ч. Затем осуществляли азеотропное удаление воды на аппарате Дина-Старка. За ходом реакции следили с помощью ЯМР, и через 20 ч обычно наблюдалось 80-85% превращение в нужное иминовое промежуточное соединение. Реакционную смесь промывали 1 М бикарбонатом натрия (0,2 объема) в течение 15 мин и органическую- 13006134 фракцию выпаривали. Остаточный сироп подвергали вакуумной перегонке (0,5 мм рт.ст.). Остаточный ксилол подвергали перегонке при 30 С, затем избыточный кетон и непрореагировавший анилин выделяли при температуре в пределах 50-110 С; при этом имин выделяли в 110-180 С-фракции в виде светлокоричневой прозрачной жидкости с чистотой 83%. Затем иминовое промежуточное соединение добавляли к дегазированной смеси ацетата калия (3 экв.),моногидрата хлорида тетра-н-бутиламмония (1 экв.), ацетата палладия (0,03 экв.) и N,N-диметилацетамида (конечная концентрация имина=0,365 М). Реакционную смесь нагревали до 115 С в течение 5 ч и оставляли для охлаждения до комнатной температуры. После этого добавляли 3 н KОН (3 экв.) и смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли водой (1,0 объема) и промывали толуолом (3 х 0,75 объема). Водную фазу подкисляли 3 н НСl до рН 1 и экстрагировали трет-бутилметиловым эфиром (2 х 0,75 объема). Объединенные органические фракции промывали водой(0,75 объема). К прозрачному светло-коричневому раствору добавляли дициклогексиламин (1 экв.) и раствор перемешивали при комнатной температуре в течение 16 ч. Соль фильтровали, промывали этилацетатом, трет-бутилметиловым эфиром и оставляли для сушки, в результате чего получали указанное в заголовке соединение в виде коричневато-желтого твердого вещества. Анализ: 94 А%. 1H ЯМР (500 МГц, СDСl3):9,24 (с, 1 Н), 7,16-7,08 (м, 2 Н), 6,82 (т, 1 Н), 6,2 (ушир., 2 Н), 3,6-3,5 (м,1 Н), 3,04-2,97 (м, 2 Н), 2,88-2,70 (м, 3 Н), 2,66 (дд, 1 Н), 2,45-2,37 (м, 1 Н), 2,13-2,05 (м, 2,05), 1,83 (д, 4 Н),1,67 (д, 2 Н), 1,55-1,43 (м, 4 Н), 1,33-1,11 (м, 6 Н). Стадия 2. -(5-Бром-7-фтор-1,2,3,4-тетрагидроциклопента[b]индол-3-ил)уксусная кислота. Суспензию DCHA-соли, полученной в вышеописанной стадии 1, в дихлорметане (0,241 М раствора) охлаждали до температуры от -20 до -15 С. Затем одной порцией добавляли пиридин (2 экв.) и к этой суспензии по каплям добавляли бром (2,5 экв.) в течение 30-45 мин, поддерживая температуру от -20 до-15 С. (При добавлении примерно 1/3 брома реакционная смесь становится густой и нуждается в эффективном размешивании. Затем, при добавлении 1/2 брома, смесь снова становится "рыхлой"). После завершения добавления реакционную смесь оставляли еще на 1 ч при -15 С. Затем в течение 5 мин добавляли уксусную кислоту (3,04 экв.) и порциями добавляли цинковую пыль (3,04 экв.). (Порцию цинка добавляли при -15 С и смесь оставляли примерно на 5 мин для гарантии прохождения экзотермической реакции (примерно от -15 С до -10 С).) Эту процедуру повторяли с добавлением 5 порций цинка в течение примерно 30 мин. Если экзотермическая реакция больше не наблюдалась, то быстро добавляли оставшийся цинк. Вся процедура занимала примерно 30-45 мин. После завершения добавления эту партию нагревали до комнатной температуры, выдерживали в течение 1 ч и концентрировали. Реакционную смесь переводили в метил-трет-бутиловый эфир (МТВЕ,0,8 объема) и добавляли 10% водный раствор уксусной кислоты (0,8 объема). Смесь (после кристаллизации солей, например пиридия) оставляли на 1 ч при комнатной температуре и фильтровали через solkafloc. Слой solka-floc промывали МТВЕ (приблизительно 0,2 объема) и фильтрат (двухфазный,МТВЕ/вода) переносили в экстракционный аппарат. Органическую фазу промывали водой (0,8 объема). МТВЕ-экстракт концентрировали и переводили в изопропиловый спирт (IPA, 0,25 объема) для кристаллизации соединения. После этого добавляли воду (0,25 объема) и партию оставляли на 1 ч. Затем в течение 1 ч снова добавляли воду (0,33 объема). После завершения добавления воды партию оставляли еще на 1 ч, фильтровали и промывали смесью IPA/вода, 30/70 (0,15 объема). Кристаллизованную бромкислоту сушили в печи при +45 С. Стадия 3. -[5-Бром-4-(4-хлорбензил)-7-фтор-1,2,3,4-тетрагидроциклопента[b]индол-3-ил]уксусная кислота. Бромкислоту, полученную на стадии 2, растворяли в диметилацетамиде (0,416 М раствор) и одной порцией добавляли карбонат цезия (2,5 экв.). К взвеси одной порцией добавляли 4-хлорбензилхлорид(2,5 экв.) и партию нагревали до 50 С в течение 20 ч. Эту партию охлаждали до комнатной температуры и в течение 5 мин добавляли 5 н гидроксид натрия (4,00 экв.) (температура повышалась до +40 С). Реакционную смесь выдерживали при 50 С в течение примерно 3 ч, охлаждали до комнатной температуры и переносили в экстракционный аппарат емкостью 1 л. Раствор разбавляли изопропилацетатом (IPAc, 2 объема) и охлаждали до +15 С. Раствор подкисляли 5 н НСl до рН 2. Слои разделяли и органический слой промывали водой (2 х 2 объема). Раствор IPAc концентрировали и переводили в IPA (0,8 объема) для кристаллизации продукта. После этого в течение 2 ч добавляли воду (8 л) и эту партию фильтровали с получением указанного в заголовке соединения с выходом 88%. Полученная партия может быть подвергнута сушке в печи при +40 С в течение 24 ч. Пример 2. -4-[1-(4-Xлорфенил)этил]-7-фтор-5-метансульфонил-1,2,3,4-тетрагидроциклопента[b] индол-3-илуксусная кислота.- 14006134 К раствору 1,5 г метилового эфира кислоты примера 1 (стадия 3) (который был получен путем эстерификации соответствующей кислоты с использованием диазометана в тетрагидрофуране) добавляли 2,03 г 1-(1-бромэтил)-4-хлорбензола в 50 мл ацетонитрила и 6,01 г карбоната цезия. Полученную смесь кипятили с обратным холодильником в течение 3 ч при тщательном перемешивании. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли 50 мл этилацетата, фильтровали и растворитель выпаривали. Остаток очищали флэш-хроматографией (силикагель, 4% EtOAc/гексан) с получением 1,41 г нужного продукта N-бензилирования в виде смеси диастереомеров приблизительно 1:1, на что указывал 1H ЯМР-анализ. К полученному, как описано выше, сложному эфиру (1,2 г), растворенному в 80 мл NMP, последовательно добавляли 2,63 г натриевой соли метансульфиновой кислоты и 3,7 г Cu(I)Br. Полученную суспензию дегазировали в потоке N2, нагревали до 140 С и тщательно перемешивали в течение 8 ч. Затем реакционную смесь охлаждали до комнатной температуры и разбавляли 500 мл этилацетата и 500 мл гексана. Полученную смесь фильтровали через слой силикагеля, а затем элюировали EtOAc. Фильтрат концентрировали до объема примерно 300 мл и промывали водой и насыщенным раствором соли. Органическую фазу отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле, элюируя 30% EtOAc/гексан, в результате чего получали 1,0 г сульфированного продукта. Этот продукт гидролизовали при комнатной температуре в течение 3 ч до соответствующей кислоты с использованием 10 мл 2 н NaOH в смеси растворителей, состоящей из 10 мл ТГФ и 10 мл МеОН. Реакционную смесь нейтрализовали водным раствором 1 М НСl и экстрагировали EtOAc. Отделенную органическую фазу сушили над безводным сульфатом натрия,фильтровали и упаривали с получением неочищенной кислоты. Два диастереомера разделяли с использованием препаративной ВЭЖХ (Zobax, 30% EtOAc/гексан с 0,2% АсОН) и получали 300 мг диастереомера А (более короткое время удерживания) и 210 мг диастереомера В (более длительное время удерживания). Диастереомер В: 1 Н ЯМР (ацетон-d6):10,70 (ушир.с, 1 Н), 7,66 (дд, 1 Н), 7,56 (дд, 1 Н), 7,32 (д, 2 Н),6,95 (д, 2 Н), 6,91 (кв, 1 Н), 3,39 (с, 3 Н), 3,05-3,00 (м, 1 Н), 2,90-2,75 (м, 2 Н), 2,70 (дд, 1H), 2,44 (дд, 1H),2,43-2,34 (м, 1 Н), 2,21 (дд, 1 Н), 2,11 (д, 3 Н).MS (-APCI) m/z 448,0 (M-H)-. Пример 2 А. Альтернативный синтез -4-[1-(4-хлорфенил)этил]-7-фтор-5-метансульфонил 1,2,3,4-тетрагидроциклопента[b]индол-3-илуксусной кислоты. К раствору 6,52 г метилового эфира кислоты примера 1 (стадия 3) (который был получен путем эстерификации соответствующей кислоты с использованием диазометана в тетрагидрофуране) в 160 млNMP последовательно добавляли 10,2 г натриевой соли метансульфиновой кислоты и 19 г CuI. Полученную суспензию дегазировали в потоке N2, нагревали до 150 С и тщательно перемешивали в течение 4 ч. Затем реакционную смесь охлаждали до комнатной температуры и разбавляли 500 мл этилацетата и 500 мл гексана. Полученную смесь фильтровали через слой силикагеля, а затем элюировали EtOAc. Фильтрат концентрировали до объема примерно 300 мл и промывали водой и насыщенным раствором соли. Органическую фазу отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле, элюируя 30% EtOAc/гексан, в результате чего получали 4,7 г сульфированного продукта, который растворяли в 200 мл дихлорметана. К полученному раствору добавляли 3,39 г 4-хлорфенилметилкарбинола и 5,68 г трифенилфосфина, а затем порциями добавляли 4,99 г ди-трет-бутил азодикарбоксилата. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, а затем концентрировали. Остаток загружали на колонку с силикагелем и элюировали смесью 5% EtOAc/гексан с получением 5,1 г метилового эфира указанного в заголовке соединения в виде смеси диастереомеров приблизительно 1:1, на что указывал 1H ЯМР-анализ. После проведения стадий гидролиза и очистки, описанных в примере 2, получали указанную в заголовке кислоту. Пример 3. -[9-(4-Xлорбензил)-6-фтор-8-метансульфонил-2,3,4,9-тетрагидро-1 Н-карбазол-1-ил] уксусная кислота. К суспензии 7,24 г гидрохлоридной соли (2-бром-4-фторфенил)гидразина в 100 мл уксусной кислоты добавляли 5,5 г этил 2-(2-оксоциклогексил)ацетата. Полученную смесь нагревали до температуры- 15006134 дефлегмации в течение 1 ч. Затем добавляли 10 мл этанола и реакционную смесь кипятили с обратным холодильником в течение ночи. Растворитель выпаривали, остаток разбавляли EtOAc, а затем последовательно промывали насыщенным водным раствором NaHCO3, водой и насыщенным раствором соли. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали. Остаток очищали флэш-хроматографией на силикагеле (5% EtOAc/гексан) и получали 3,12 г нужного соединения. 1 К раствору 3,12 г сложного эфира, полученного на стадии 1, и 3,62 г 1-бромметил-4-хлорбензола в 30 мл ацетонитрила добавляли 5,74 г карбоната цезия. Полученную смесь тщательно перемешивали при кипячении с обратным холодильником в течение 3 ч. Затем ее охлаждали до комнатной температуры,разбавляли минимальным количеством EtOAc, фильтровали и выпаривали. Остаток очищали флэш-хроматографией на силикагеле (50% толуол/гексан) с получением 4,1 г указанного в заголовке соединения. 1H ЯМР (ацетон-d6):7,32 (д, 2 Н), 7,24 (дд, 1 Н), 7,13 (дд, 1 Н), 6,86 (д, 2 Н), 6,00 и 5,65 (АВ кв, 2 Н),4,15-4,05 (м, 2 Н), 3,44-3,35 (м, 1 Н), 2,88-2,76 (м, 1 Н), 2,65-2,52 (м, 3 Н), 2,00-1,80 (м, 4 Н), 1,22 (т, 3 Н). Стадия 3. -[9-(4-Xлорбензил)-6-фтор-8-метансульфонил-2,3,4,9-тетрагидро-1 Н-карбазол-1-ил]уксусная кислота. К раствору 478 мг сложного эфира, полученного на стадии 2 в 8 мл NMP, последовательно добавляли 510 мг натриевой соли метансульфиновой кислоты и 950 мг CuI(I). Полученную смесь дегазировали в потоке N2, затем нагревали при 140 С в течение 8 ч при тщательном перемешивании. Реакционную смесь охлаждали до комнатной температуры и разбавляли минимальным количеством смесиEtOAc/гексан, 1:1. Полученную смесь фильтровали через слой силикагеля, а затем элюировали EtOAc. Фильтрат концентрировали до объема примерно 50 мл и промывали водой и насыщенным раствором соли. Органическую фазу собирали, сушили над безводным сульфатом натрия, фильтровали и выпаривали. Остаток очищали флэш-хроматографией на силикагеле (30% EtOAc/гексан) с получением 320 мг нужного сульфированного продукта, который растворяли в 5 мл ТГФ + 5 мл метанола. К полученному раствору добавляли 5 мл 2 н NaOH и полученную смесь перемешивали при комнатной температуре в течение 6 ч. Реакционную смесь нейтрализовали 1 М водным раствором НСl и экстрагировали EtOAc. Отделенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и выпаривали. Остаток кипятили с обратным холодильником в гексане при тщательном перемешивании в течение 0,5 ч. Полученную смесь охлаждали до комнатной температуры при тщательном перемешивании и фильтровали с получением 278 мг нужной кислоты. 1- 16006134 Метиловый эфир соединения примера 1, стадии 5 (1,00 г, полученного путем обработки соответствующей кислоты избытком диазометана) в 10 мл раствора ТГФ/Н 2 О, 9:1, обрабатывали 2,52 г DDQ. Реакционную смесь оставляли на ночь с перемешиванием при комнатной температуре. По истечении этого времени реакционную смесь выливали в делительную воронку, содержащую EtOAc и насыщенный раствор соли. Объединенные органические слои промывали водой, насыщенным раствором соли, сушили над безводным MgSO4 и концентрировали. Затем полученный продукт очищали флэш-хроматографией,элюируя смесью 30% EtOAc/гексан. Эту процедуру хроматографии повторяли еще 2 раза. Таким образом, было получено 350 мг вышеуказанного кетона в виде серого твердого вещества. Стадия 2. [4-(4-Xлорбензил)-7-фтор-5-метансульфонил-1-оксо-1,2,3,4-тетрагидроциклопента[b]индол-3-ил]уксусная кислота. Бромид, полученный, как описано на стадии 1 (200 мг), в 4 мл NMP, обрабатывали 320 мг CuI и 175 мгCH3SO2Na. Через реакционную смесь в течение примерно 1 мин барботировали азот, а затем смесь нагревали в течение 6 ч при 130 С. По истечении этого времени реакционную смесь охлаждали до комнатной температуры, разбавляли EtOAc и фильтровали через слой силикагеля, после чего остаток снова промывали EtOAc. Органические слои промывали водой, насыщенным раствором соли, сушили над безводным MgSO4 и концентрировали. Полученное масло очищали флэш-хроматографией, элюируя смесью 30% EtOAc/гексан, и получали 54 мг соответствующего метилсульфона в виде не совсем белого твердого вещества. Метиловый эфир, полученный как описано выше, в 5 мл раствора ТГФ/Н 2 О (1:1) и 5 мл МеОН обрабатывали 1 мл 1 н раствора НСl. Эту смесь перемешивали при комнатной температуре в течение 2 ч. По истечении этого времени реакционную смесь подкисляли 1 н раствором НСl и выливали в делительную воронку, содержащую воду и EtOAc. Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические слои промывали водой, насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали. Затем полученный продукт очищали флэш-хроматографией, элюируя 100% EtOAc,содержащим 1% АсОН, в результате чего получали 26 мг указанной в заголовке кислоты в виде не совсем белого твердого вещества. 1 и его фармацевтически приемлемые соли, где n равно 0 или 1; m равно 1, 2 или 3; R1 представляет собой Н, C1-С 3 алкил, галогенированный C1-С 3 алкил или циклопропил; R2 представляет собой 4-хлорфенил или 2,4,6-трихлорфенил. 2. Соединение по п.1, имеющее стереоконфигурацию, показанную ниже 3. Соединение по п.2, где m равно 1. 4. Соединение (-)-[4-(4-хлорбензил)-7-фтор-5-(метансульфонил)-1,2,3,4-тетрагидроциклопента[b] индол-3-ил]уксусной кислоты и его фармацевтически приемлемые соли. 5. Фармацевтическая композиция, содержащая соединение по любому из пп.1-4 и фармацевтически приемлемый носитель. 6. Композиция по п. 5, которая, кроме того, содержит второй активный ингредиент, выбранный из антигистамина, антагониста лейкотриена и ингибитора биосинтеза лейкотриена.- 17006134 7. Применение соединения или его соли по любому из пп.1-4 для изготовления лекарственного средства для лечения заболевания, опосредуемого простагландином D2. 8. Применение по п.7, где указанным заболеванием является застойная гиперемия носа, ринит или астма.
МПК / Метки
МПК: A61K 31/403, C07D 209/80, A61P 43/00
Метки: циклоалканоиндолы, фторзамещенные, композиции, способы, содержащие, лечения, соединения, такие
Код ссылки
<a href="https://eas.patents.su/19-6134-ftorzameshhennye-cikloalkanoindoly-kompozicii-soderzhashhie-takie-soedineniya-i-sposoby-lecheniya.html" rel="bookmark" title="База патентов Евразийского Союза">Фторзамещенные циклоалканоиндолы, композиции, содержащие такие соединения, и способы лечения</a>
Антибактериальные соединения карбапенема, содержащие их фармацевтические композиции и способы лечения

Номер патента: 1296
Опубликовано: 25.12.2000
Авторы: Рэтклифф Рональд В., Близзард Тимоти А., Вилкенинг Роберт Р.
МПК: A61K 31/428, C07D 477/14
Метки: карбапенема, содержащие, соединения, способы, композиции, фармацевтические, антибактериальные, лечения
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемые соли, где R1 обозначает Н или метил; СО2М обозначает карбоновую кислоту, карбоксилатный анион, фармацевтически приемлемую сложноэфирную группу или карбоновую кислоту, защищенную защитной группой; Р обозначает водород, гидроксил, F или гидроксил, защищенный гидроксилзащитной группой; каждый R независимо выбирают из -R*; -Q; водорода; галогена; -CN; -NO2; -NRaRb; -ORc; -SRc; -C(O)NRaRb;...
Четырехциклические конденсированные соединения с гетероатомами, замещенные арилом, промежуточные соединения, способы получения, композиции и методы лечения

Номер патента: 1649
Опубликовано: 25.06.2001
Автор: Гриз Тимоти А.
МПК: A61P 5/30, C07D 471/00, A61K 31/35...
Метки: способы, гетероатомами, четырехциклические, промежуточные, композиции, методы, получения, соединения, конденсированные, лечения, арилом, замещенные
Формула / Реферат:
1. Соединение формулы I или II где Y обозначает -О-, -S-, -СН2-, -СН2СН2-, -СН=СН- или -NR4-; В обозначает -СН2- или -СО-; R1 и R2 обозначают каждый независимо -Н, -ОН, -O(C1-C4алкил), -ОСОС6Н5, -ОСО(С1-С6алкил), -ОSО2(С4-С6алкил), OSO2СF3, Сl или F; n равно 1 или 2; W обозначает -СН2- или >С=O; R3 обозначает 1-пиперидинил, 2-оксо-1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 2-оксо-1-пирролидинил,...
Циклоалкильные соединения, лактамы, лактоны и родственные соединения, содержащие их фармацевтические композиции и способы ингибирования высвобождения и/или синтеза β-амилоидного пептида с помощьюуказанных соединений

Номер патента: 2100
Опубликовано: 24.12.2001
Авторы: Портер Варрен Дж., Мабри Томас Э., Торсетт Юджин Д., Джон Варгес, Скотт Уильям Леонард, Фридман Стефен, Дрессман Брюс А., Стаки Расселл Д., Бриттон Томас К., Дрост Джеймс Дж., Ниссен Джеффри С., Кви Синтия Л., Танг Джей С., Латимер Ли Х., Рил Джон К., Макданиел Стейси Л., Одия Джеймс Е., Ву Джинг, Нейц Джеффри, Плейсс Майкл А., Генри Стивен С.
МПК: A61K 31/55, A61P 25/28, C07D 243/10...
Метки: фармацевтические, композиции, ингибирования, родственные, способы, лактамы, соединений, beta;-амилоидного, высвобождения, помощьюуказанных, циклоалкильные, синтеза, лактоны, пептида, соединения, содержащие
Формула / Реферат:
1. Способ ингибирования высвобождения и/или синтеза b -амилоидного пептида в клетке, который заключается в том, что в такую клетку вводят соединение или смесь соединений в количестве, эффективном для ингибирования высвобождения и/или синтеза b -амилоидного пептида в клетке, и указанные соединения имеют формулу I в которой R1 выбирают из группы, включающей C1-С10алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из...
Мутантный il-6 человека и его внутренний фрагмент, кодирующие их последовательности днк, способы их получения, содержащие их фармацевтические композиции, содержащие их векторы, линии клеток- хозяев испособ лечения il-6 опосредованных заболеваний

Номер патента: 852
Опубликовано: 26.06.2000
Авторы: Элерс Марк, Гротзингер Йоахим, Розе-Йон Штефан
МПК: A61P 19/10, A61K 38/20, A61P 35/00...
Метки: векторы, хозяев, кодирующие, фрагмент, заболеваний, содержащие, внутренний, опосредованных, клеток, получения, человека, последовательности, испособ, линии, композиции, способы, лечения, фармацевтические, днк, мутантный
Формула / Реферат:
1. Мутантный интерлейкин-6 (IL-6) человека, имеющий аминокислотную последовательность содержащую следующие точечные мутации по сравнению с природным IL-6 человека: Pro 54, Glu 159, Pro 162, Leu 170 и Аrg 176. 2. Внутренний фрагмент мутантного IL-6 человека по п.1 формулы, обладающий аналогичной биологической активностью. 3. Последовательность ДНК, кодирующая мутантный IL-6 человека по п.1 формулы. 4. Последовательность ДНК, кодирующая...
Производные пиразолотриазинов, фармацевтические композиции, содержащие их, способы лечения

Номер патента: 4403
Опубликовано: 29.04.2004
Авторы: Хорват Роберт Джон, Арванитис Арджириос Георгиос
МПК: A61P 1/04, A61K 31/4162, A61K 31/53...
Метки: производные, способы, фармацевтические, пиразолотриазинов, композиции, лечения, содержащие, их
Формула / Реферат:
1. Соединение формулы (1) и его изомеры, его стереоизомерные формы или смеси его стереоизомерных форм и его фармацевтически приемлемые соли или пролекарства, где A обозначает N; Z обозначает CR2; Ar выбран из группы фенил, пиридил, где каждый Ar необязательно замещен 1-5 группами R4; R1 независимо выбран в каждом случае из группы H, C1-C4-алкил; R2 независимо выбран из группы H, C1-C4-алкил; R3 выбран из группы -H, OR7, NR6aR7a, или фенил,...
Предыдущий патент: 7 – аминотриазолопиримидины, способ и промежуточные продукты для их получения, содержащие их средства, а также их применение для борьбы с фитопатогенными грибами
Следующий патент: Замещенные тиоацетамиды
Случайный патент: Сульфамоильные соединения и фунгициды для сельского хозяйства и садоводства