Макроциклическое хиноксалиновое соединение в качестве ингибитора протеазы вгс ns3
Номер патента: 19327
Опубликовано: 28.02.2014
Авторы: Харпер Стивен, Макколи Джон А., Сумма Винченцо, Ливертон Найджел Дж.


















Формула / Реферат
1. Соединение формулы (I) или его фармацевтически приемлемая соль
2. Фармацевтическая композиция для лечения пациента, инфицированного ВГС, содержащая эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
3. Фармацевтическая композиция по п.2, дополнительно содержащая второй терапевтический агент, выбранный из группы, состоящей из ингибитора протеазы ВГС и ингибиторов полимеразы ВГС NS5B.
4. Применение соединения по п.1 для профилактики или лечения инфекции ВГС.
5. Применение соединения по п.1 для получения лекарственного средства для ингибирования активности протеазы ВГС NS3 у нуждающегося в этом субъекта.
6. Применение композиции по любому из пп.2, 3 для получения лекарственного средства для ингибирования активности протеазы ВГС NS3 у нуждающегося в этом субъекта.
7. Применение соединения по п.1 для получения лекарственного средства для профилактики или лечения инфекции ВГС у нуждающегося в этом субъекта.
8. Применение композиции по любому из пп.2, 3 для получения лекарственного средства для профилактики или лечения инфекции ВГС у нуждающегося в этом субъекта.
9. Способ лечения пациента, инфицированного ВГС, включающий введение указанному пациенту эффективного количества соединения по п.1.
10. Способ лечения пациента, инфицированного ВГС, включающий введение указанному пациенту эффективного количества фармацевтической композиции, содержащей соединение по п.1.
11. Соединение по п.1, имеющее структуру
12. Фармацевтическая композиция для лечения пациента, инфицированного ВГС, содержащая эффективное количество соединения по п.11 и фармацевтически приемлемый носитель.
13. Фармацевтическая композиция по п.12, дополнительно содержащая второй терапевтический агент, выбранный из группы, состоящей из ингибитора протеазы ВГС и ингибиторов полимеразы ВГС NS5B.
14. Способ лечения пациента, инфицированного ВГС, включающий введение указанному пациенту эффективного количества соединения по п.11.
15. Способ лечения пациента, инфицированного ВГС, включающий введение указанному пациенту эффективного количества композиции по п.12.
16. Способ лечения пациента, инфицированного ВГС, включающий введение указанному пациенту эффективного количества композиции по п.13.
Текст
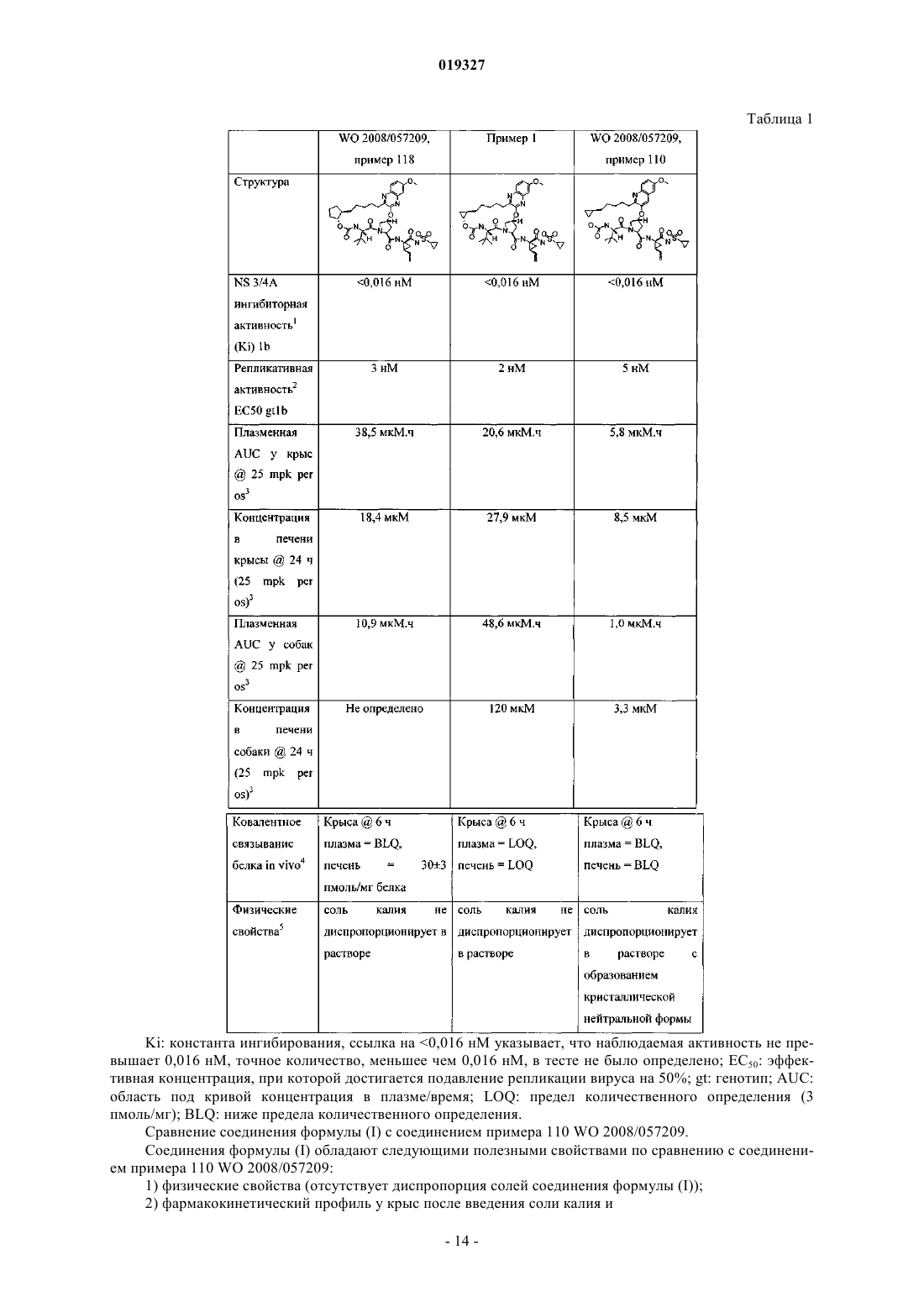
МАКРОЦИКЛИЧЕСКОЕ ХИНОКСАЛИНОВОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕАЗЫ ВГС NS3 Настоящее изобретение относится к макроциклическому соединению формулы (I) и его применению в качестве ингибиторов протеазы вируса гепатита С (ВГС) NS3, а также для лечения или профилактики инфекции ВГС.(71)(73) Заявитель и патентовладелец: МЕРК ШАРП ЭНД ДОМЭ КОРП.(US); ИСТИТУТО ДИ РИЧЕРКЕ ДИ БИОЛОДЖИА МОЛЕКОЛАРЕ П. АНДЖЕЛЕТТИ С.П.А. (IT) Область техники, к которой относится изобретение Настоящее изобретение относится к макроциклическим соединениям, которые можно использовать в качестве ингибиторов NS3 протеазы вируса гепатита С (ВГС), синтезу таких соединений и их применению для лечения инфекции ВГС и/или уменьшения вероятности возникновения или тяжести инфекции ВГС. Уровень техники Инфекция, вызываемая вирусом гепатита С (ВГС), является основной проблемой здоровья, поскольку в результате этой инфекции значительное число зараженных людей страдает хроническим заболеванием печени, таким как цирроз печени и гепатоцеллюлярная карцинома. Существующая профилактика инфекции ВГС включает иммунотерапию только рекомбинантным интерфероном- или в комбинации с нуклеозидным аналогом рибавирином. Несколько кодируемых вирусом ферментов являются предполагаемыми мишенями для терапевтического вмешательства, включающими металлопротеазу (NS2-3), серинпротеазу (NS3), геликазу (NS3) и РНК-зависимую РНК-полимеразу (NS5B). Протеаза NS3 находится в N-терминальной области белкаNS3. NS4A обеспечивает кофактор для активности NS3. Потенциальные способы лечения инфекции ВГС обсуждаются в различных источниках, включаяResearch 58:1-16, 2003. Сущность изобретения Настоящее изобретение относится к макроциклическому соединению формулы (I) и его фармацевтически приемлемым солям. Данное соединение и его соли представляют собой ингибиторы протеазы ВГС NS3. Это соединение и его соли применяются как в терапии, так и в научных исследованиях. Таким образом, первый аспект настоящего изобретения описывает соединение формулы (I) или его фармацевтически приемлемую соль Настоящее изобретение также включает фармацевтические композиции, содержащие соединение по настоящему изобретению, и способы получения таких фармацевтических композиций. Настоящее изобретение также включает способы лечения или уменьшения вероятности возникновения или тяжести инфекции ВГС. Другие варианты осуществления, аспекты и признаки настоящего изобретения либо описаны в настоящем описании ниже, либо являются очевидными из приведенного ниже описания, примеров и прилагаемой формулы изобретения. Подробное описание изобретения Настоящее изобретение включает соединение формулы (I) и его фармацевтически приемлемые соли. Данное соединение и его фармацевтически приемлемые соли могут использоваться для ингибирования протеазы ВГС NS3, лечения инфекции ВГС и/или уменьшения вероятности возникновения или тяжести инфекции ВГС. Профилактическое применение включает, например, лечение после появления подозрения на наличие ВГС в результате, например, переливания крови, замены жидкости тела, укуса,случайного укола иглой или заражения пациента через кровь во время хирургического вмешательства. В качестве компонентов фармацевтической композиции данные соединения и соли могут представлять собой основной активный терапевтический агент. Данное соединение также может быть объединено с другими терапевтическими агентами, включая, без ограничений, другие анти-ВГС агенты, противоинфекционные агенты, иммуномодуляторы, антибиотики или вакцины. Ингибиторы NS3 также пригодны для подготовки и осуществления скрининговых тестов антивирусных соединений. Например, такие соединения могут использоваться для выделения ферментов-мутантов,которые являются отличными инструментами для скрининга более сильных антивирусных соединений. Кроме того, эти соединения можно использовать для обнаружения или определения сайта связывания другого антивирусного агента с протеазой ВГС, например, путем конкурентого ингибирования. Как описано ниже в примере 2, при проведении сравнения соединения формулы (I) с соединением,описанным в примерах 110 и 118 WO 2008/057209, показано, что заявленное соединение обладает несколькими преимуществами. WO 2008/057209 не может рассматриваться в качестве ближайшего аналогаI. Композиции и способы. Различные варианты осуществления включают следующее:(a) фармацевтическую композицию, содержащую эффективное количество соединения формулы (I) и фармацевтически приемлемый носитель;(b) фармацевтическую композицию (а), дополнительно содержащую второй терапевтический агент,выбранный из группы, состоящей из анти-ВГС агентов, иммуномодуляторов и противоинфекционных агентов;(c) фармацевтическую композицию (b), в которой анти-ВГС агент представляет собой антивирусный агент, выбранный из группы, состоящей из ингибиторов протеазы ВГС и ингибиторов полимеразы ВГС NS5B;(d) фармацевтическую комбинацию из (i) соединения формулы (I) и (ii) второго терапевтического агента, выбранного из группы, состоящей из анти-ВГС агентов, иммуномодуляторов и противоинфекционных агентов; причем и соединение формулы (I), и второй терапевтический агент используется в количестве, которое делает комбинацию эффективной для ингибирования протеазы ВГС NS3 или для лечения инфекции ВГС и/или уменьшения вероятности возникновения или тяжести инфекции ВГС;(e) комбинацию (d), в которой анти-ВГС агент представляет собой антивирусный агент, выбранный из группы, состоящей из ингибиторов протеазы ВГС и ингибиторов полимеразы ВГС NS5B;(f) способ ингибирования протеазы ВГС NS3 у нуждающегося в этом субъекта, который включает введение субъекту эффективного количества соединения формулы (I);(g) способ лечения инфекции ВГС и/или уменьшения вероятности возникновения или тяжести инфекции ВГС у нуждающегося в этом субъекта, который включает введение субъекту эффективного количества соединения формулы (I);(h) способ (g), в котором соединение формулы (I) вводят в комбинации с эффективным количеством по меньшей мере одного второго терапевтического агента, выбранного из группы, состоящей из антиВГС агентов, иммуномодуляторов и противоинфекционных агентов;(i) способ (h), в котором анти-ВГС агент представляет собой антивирусный агент, выбранный из группы, состоящей из ингибиторов протеазы ВГС и ингибиторов полимеразы ВГС NS5B;(j) способ ингибирования протеазы ВГС NS3 у нуждающегося в этом субъекта, который включает введение субъекту фармацевтической композиции (а), (b) или (с) или комбинации (d) или (е);(k) способ лечения инфекции ВГС и/или уменьшения вероятности возникновения или тяжести инфекции ВГС у нуждающегося в этом субъекта, который включает введение субъекту фармацевтической композиции (а) (b) или (с) или комбинации (d) или (е);(l) соединение формулы (I) для применения в медицине, для профилактики или лечения инфекции ВГС или для применения (i), (ii) в качестве лекарственного средства или (iii) для приготовления лекарственного средства для (а) ингибирования протеазы ВГС NS3 или (b) лечения инфекции ВГС и/или уменьшения вероятности возникновения или тяжести инфекции ВГС. В таких применениях соединения по настоящему изобретению необязательно используются в комбинации с одним или несколькими вторыми терапевтическими агентами, выбранными из анти-ВГС агентов, противоинфекционных агентов и иммуномодуляторов. Во всех этих вариантах осуществления соединение необязательно используется в виде фармацевтически приемлемой соли. Термин "или", как используется здесь, обозначает альтернативные варианты, которые, при необходимости, могут быть объединены. Таким образом, термин "или" включает каждую перечисленную альтернативу отдельно, а также их комбинацию, если такая комбинация не является взаимоисключающей. Ссылка на соединение также включает стабильные комплексы этого соединения, такие как стабильный гидрат. "Стабильное" соединение представляет собой соединение, которое может быть получено и выделено, и структура, и свойства которого остаются или могут оставаться без существенных изменений в течение периода времени, достаточного для возможности использовать это соединение с описанной в данной заявке целью (например, терапевтическое или профилактическое применение субъектом).II. Применение и композиции. Термин "применение" и его варианты (например, "введение" соединения) означает предоставление соединения или пролекарства данного соединения нуждающемуся в лечении индивидууму. Если соединение по изобретению или его пролекарство предоставляется в комбинации с одним или несколькими другими активными агентами (например, антивирусными агентами, пригодными для лечения инфекции ВГС), "применение" и его варианты следует интерпретировать как включающие параллельное и последовательное предоставление этого соединения или его соли и других агентов. Соединения по настоящему изобретению можно вводить в виде фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль" относится к соли родительского соединения, которое обладает активностью и которое не является нежелательным, ни биологически, ни каким-либо иным образом (например, не является ни токсическим, ни каким-либо иным образом вредным для того, кто его принимает). Подходящие соли включают кислотно-аддитивные соли, которые, например, могут быть образованы путем смешения раствора соединения с раствором фармацевтически приемлемой кислоты,такой как соляная кислота, серная кислота, уксусная кислота, трифторуксусная кислота или бензойная кислота. Соединения, несущие кислотный фрагмент, могут быть смешаны с подходящими фармацевтически приемлемыми солями для получения, например, солей щелочного металла (например, солей натрия или калия), солей щелочно-земельного металла (например, солей кальция или магния), и солей, образованных подходящими органическими лигандами, таких как соли четвертичного аммония. Кроме того, при наличии кислотной (-СООН) группы или группы спирта для изменения растворимости или характеристик гидролиза данного соединения можно использовать фармацевтически приемлемые эфиры. Термин "пролекарство", как используется здесь, охватывает неактивную форму лекарственного вещества или соединения, которая превращается в активную форму лекарственного вещества или соединения под воздействием ферментов, химикатов или метаболических процессов в теле человека, которому его вводят. Термин "композиция", как используется здесь, охватывает продукт, содержащий указанные компоненты, а также любой продукт, который получается, непосредственно или опосредованно, из объединения указанных компонентов."Фармацевтически приемлемый" означает, что компоненты фармацевтической композиции должны быть совместимыми друг с другом и безвредными для реципиента. Термин "субъект" (альтернативно упоминаемый здесь как "пациент"), используемый в настоящей заявке, относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, который был объектом лечения, наблюдения или эксперимента. Термин "эффективное количество" указывает на количество, достаточное для проявления терапевтического или профилактического эффекта. Для пациента, инфицированного ВГС, эффективным количеством является количество, достаточное для достижения одного или нескольких из следующих эффектов: уменьшения способности ВГС к репликации, уменьшения нагрузки ВГС и усиления элиминации вируса. Для пациента, не инфицированного ВГС, эффективным количеством является количество, достаточное для достижения одного или нескольких из следующих эффектов: сниженной восприимчивости к инфекции ВГС и ослабленной способности инфицирующего вируса вызывать персистентную инфекцию в случае хронического заболевания. С целью ингибирования протеазы ВГС NS3 и лечения инфекции ВГС и/или уменьшения вероятности возникновения или тяжести симптомов инфекции ВГС соединения по настоящему изобретению, необязательно в виде соли, могут вводиться таким образом, чтобы возникал контакт активного агента с участком его воздействия. Соединения могут вводиться с помощью обычных средств, доступных для применения совместно с фармацевтическими препаратами, или в виде отдельных терапевтических агентов или в комбинации с терапевтическими агентами. Они могут вводиться отдельно, но обычно вводятся с фармацевтическим носителем, выбираемым в зависимости от выбранного режима введения и стандартной фармацевтической практики. Соединения могут вводиться, например, одним или несколькими из следующих способов: перорально, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, интрастернальные инъекции или технику инфузии), с помощью ингаляции (такой как в форме спрея) или ректально в виде единичных доз фармацевтической композиции, содержащей эффективное количество соединения и обычные нетоксичные фармацевтически приемлемые носители, адъюванты и разбавители. Жидкие препараты, подходящие для перорального введения (например, суспензии, сиропы, эликсиры и т.п.), могут быть приготовлены согласно методам, известным в данной области, при этом можно использовать любую из обычных сред, таких как вода, гликоли, масла, спирты и т.п. Твердые препараты, подходящие для перорального введения (например, порошки, пилюли, капсулы и таблетки), могут быть приготовлены согласно методам, известным в данной области, при этом можно использовать такие твердые наполнители, как крахмалы, сахара, каолин, лубриканты, связующие агенты, дезинтегрирующие агенты и т.п. Парентеральные композиции можно готовить согласно методам, известным в данной области, при этом в качестве носителя обычно используется стерильная вода и необязательно другие компоненты, такие как способствующие растворимости вещества. Инъецируемые растворы могут быть приготовлены согласно способам, известным в данной области, где носитель содержит физиологический раствор, раствор глюкозы или раствор, содержащий смесь физиологического раствора и глюкозы. Дополнительные инструкции относительно способов, подходящих для использования при приготовлении фармацевтических композиций по настоящему изобретению, и компонентов, подходящих для использования в указанных композициях, можно найти в Remington's Pharmaceutical Sciences, 20th edition (ed. A.R. Gennaro, Mack Publishing Co., 2000). Соединения по настоящему изобретению можно вводить перорально дозой от 0,001 до 1000 мг/кг массы тела млекопитающего (например, человека) в сутки в виде одной дозы или в виде небольших доз. Один из диапазонов доз составляет 0,01-500 мг/кг массы тела в сутки перорально в виде одной дозы или в виде небольших доз. Другой диапазон доз составляет 0,1-100 мг/кг массы тела в сутки перорально в виде одной дозы или в виде небольших доз. Для перорального введения композиции могут находиться в виде таблеток или капсул, содержащих 1,0-500 мг активного компонента, в частности 1, 5, 10, 15, 20, 25,50, 75, 100, 150, 200, 250, 300, 400, 500 и 750 мг активного компонента для подбора дозировки в зависимости от симптомов для пациента, находящегося на лечении. Определенный уровень дозы и частота приема доз для любого конкретного пациента могут быть различными и будут зависеть от множества факторов, включая активность конкретного используемого соединения, метаболическую стабильность и длительность воздействия этого соединения, возраст, массу тела, общее состояние здоровья, пол, диету,способ и время введения, скорость выведения, комбинацию лекарственных веществ, тяжесть конкретного состояния и основную проводимую терапию.III. Комбинированная терапия. Макроциклические хиноксалиновые соединения, описанные здесь, могут использоваться в комбинированной терапии, включающей один или несколько дополнительных терапевтических агентов. Дополнительные терапевтические агенты также включают агенты, направленные на ВГС, направленные на агенты, вызывающие другие заболевания, или включают агенты, усиливающие иммунную систему. Агенты, усиливающие иммунную систему, включают агенты, в основном усиливающие функцию иммунной системы, и агенты, вызывающие определенный иммунный ответ против ВГС. Дополнительные терапевтические агенты, направленные на ВГС, включают агенты, направленные на NS3, и агенты, направленные на другие активности ВГС, такие как NS5A и NS5B, и агенты, направленные на те виды активности клетки-хозяина, которые участвуют в репликации ВГС. Различные ингибиторы ВГС описаны в различных публикациях. Макроциклические соединения,используемые в качестве ингибиторов протеазы ВГС, описаны в WO 06/119061, WO 7/015785, WO 7/016441, WO 07/148135, WO 08/051475, WO 08/051477, WO 08/051514, WO 08/057209. Дополнительные ингибиторы протеазы ВГС NS3 раскрыты в публикациях международных заявок на патент WO 98/22496,WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888, WO 99/50230, WO 99/64442, WO 00/09543, WO 00/59929, WO 02/48116, WO 02/48172, британский патент GB 2337262 и патент США 6323180. Дополнительные примеры терапевтических агентов, которые могут присутствовать в комбинации,включают рибавирин, левовирин, вирамидин, тимозин -1, интерферон-, интерферон-, пегилированный интерферон- (пегинтерферон-), комбинацию интерферона- и рибавирина, комбинацию пегинтерферона- и рибавирина, комбинацию интерферона- и левовирина и комбинацию пегинтерферонаи левовирина. Интерферон- включает рекомбинантный интерферон-2 а (такой как интерферон РОФЕРОН, выпускаемый компанией Hoffmann-LaRoche, Nutley, Нью-Джерси), пегилированный интерферон 2 а (PEGASYS), интерферон-2b (такой как интерферон ИНТРОН-А, выпускаемый компанией Schering,Kenilworth, Нью-Джерси), пегилированный интерферон-2b (ПЕГИНТРОН), рекомбинантный консенсусный интерферон (такой как интерферон альфакон-1) и очищенный продукт интерферона-. Рекомбинантный консенсусный интерферон компании Amgen имеет торговое название ИНФЕРГЕН. Левовирин представляет собой L-энантиомер рибавирина, который проявляет иммуномодуляторную активность,аналогичную рибавирину. Вирамидин представляет собой аналог рибавирина, раскрытый в WO 01/60379. Отдельные компоненты комбинации можно вводить раздельно в разное время в ходе лечения или одновременно в виде раздельных форм или в виде одной комбинации. Рибавирин, левовирин и вирамидин могут проявлять анти-ВГС эффекты путем модуляции внутриклеточных пулов гуаниновых нуклеотидов через ингибирование внутриклеточного фермента инозинмонофосфат-дегидрогеназы (IMPDH). IMPDH представляет собой фермент, ограничивающий скорость биосинтеза de novo гуаниновых нуклеотидов в биосинтетическом пути. Рибавирин осуществляет быстрое внутриклеточное фосфорилирование, а производное монофосфата является ингибитором IMPDH. Таким образом, ингибирование IMPDH является другой полезной мишенью для обнаружения ингибиторов репликации ВГС. Таким образом, соединения по настоящему изобретению также можно вводить в комбинации с ингибитором IMPDH, таким как VX-497, который раскрыт в публикациях международных заявок на патент WO 97/41211 и WO 01/00622; другим ингибитором IMPDH, как описано в WO 00/25780; или микофенолат мофетилом. См. А.C. Allison и Е.М. Eugui, 44 (Suppl). Agents Action 165 (1993). Для лечения инфекции ВГС соединения по настоящему изобретению также можно вводить в комбинации с противовирусным средством амантадином (1-аминоадамантан). Для получения всестороннего описания этого агента см. J. Kirschbaum, 12 Anal. Profiles Drug Subs. 1-36 (1983). Для лечения инфекции ВГС соединения по настоящему изобретению также можно вводить в комбинации с противовирусным средством ингибитором полимеразы R7128 (Roche). Для лечения инфекции ВГС соединения по настоящему изобретению также можно комбинировать с противовирусными 2'-С-разветвленными рибонуклеозидами, раскрытыми у R.E. Harry-O'Kuru et al., 62 J.Org. Chem. 1754-59 (1997); M.S. Wolfe et al., 36 Tet. Lett. 7611-14 (1995); патенте США 3480613 и публикациях международных заявок на патент WO 01/90121, WO 01/92282, WO 02/32920, WO 04/002999, WO 04/003000 и WO 04/002422; содержание каждого из которых включено в настоящее описание во всей полноте в качестве ссылки. Такие 2'-С-разветвленные рибонуклеозиды включают, без ограничений, 2'-Сметилцитидин, 2'-С-метилуридин, 2'-С-метиладенозин, 2'-С-метилгуанозин и 9-(2-С-метилD-4 019327 рибофуранозил)-2,6-диаминопурин и соответствующий сложный эфир аминокислоты и С-2'-, С-3'- и С 5'-гидроксилов рибозы и соответствующие сложные эфиры необязательно замещенного циклического 1,3-пропандиола с производными 5'-фосфата. Для лечения инфекции ВГС соединения по настоящему изобретению также можно комбинировать с другими нуклеозидами, имеющими анти-ВГС свойства, такие как раскрыто в публикациях международных заявок на патент WO 02/51425, WO 01/79246, WO 02/32920, WO 02/48165 и WO 2005/003147 (включая R1656, (2'R)-2'-дезокси-2'-фтор-2'-С-метилцитидин, показанный в виде соединений 3-6 на стр.77);WO 01/68663; WO 99/43691; WO 02/18404 и WO 2006/021341 и заявке на патент US 2005/0038240, включая 4'-азидонуклеозиды, такие как R1626, 4'-азидоцитидин; публикациях заявок на патент US 2002/0019363, US 2003/0236216, US 2004/0006007 и US 2004/0063658 и публикациях международных заявок на патент WO 02/100415, WO 03/026589, WO 03/026675, WO 03/093290, WO 04/011478, WO 04/013300 и WO 04/028481; содержание каждой из которых включено в данное описание во всей полноте в качестве ссылки. Для лечения инфекции ВГС соединения по настоящему изобретению также можно объединять с агентом, который является ингибитором полимеразы ВГС NS5B. Такие ингибиторы полимеразы ВГСNS5B, которые могут использоваться в виде комбинированной терапии, включают, без ограничения, ингибиторы, описанные в публикациях международных заявок на патент WO 02/057287, WO 02/057425,WO 03/068244, WO 2004/000858, WO 04/003138 и WO 2004/007512; патенте США 6777392 и публикации заявки на патент US 2004/0067901; содержание каждого из которых включено в настоящее описание во всей полноте в качестве ссылки. Другие ингибиторы полимеразы ВГС включают, без ограничения, валопицитабин (НМ 283; Idenix), и 2'-F-2'метилцитидин (см. также WO 2005/003147). В одном из вариантов осуществления нуклеозидные ингибиторы полимеразы ВГС NS5B, которые используются в комбинации с ингибиторами протеазы ВГС NS3 по настоящему изобретению, выбирают из следующих соединений: 4-амино-7-(2-С-метилD-арабинофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-метиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-диметиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-циклопропиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-7-(2-С-винилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-7-(2-С-гидроксиметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-7-(2-С-фторметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-5-метил-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-7-(2-С-метилD-рибофуранозил)-7H-пирроло[2,3-d]пиримидин-5-карбоксильная кислота; 4-амино-5-бром-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-5-хлор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-5-фтор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 2,4-диамино-7-(2-С-метилD-рибофуранозил)-7H-пирроло[2,3-d]пиримидин; 2-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 2-амино-4-циклопропиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 2-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4(3 Н)-он; 4-амино-7-(2-С-этилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-7-(2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 7-(2-С-метилD-рибофуранозил)-7H-пирроло[2,3-d]пиримидин-4(3 Н)-он; 2-амино-5-метил-7-(2-C,2-О-диметилD-рибофуранозил)-7H-пирроло[2,3-d]пиримидин-4(3 Н)-он; 4-амино-7-(3-дезокси-2-С-метилD-рибофуранозил)-7H-пирроло[2,3-d]пиримидин; 4-амино-7-(3-дезокси-2-С-метил-(-D-арабинофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-2-фтор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-7-(3-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-7-(3-С-метилD-ксилофуранозил)-7 Н-пирроло[2,3-d]пиримидин; 4-амино-7-(2,4-ди-С-метилD-рибофуранозил)-7H-пирроло[2,3-d]пиримидин; 4-амино-7-(3-дезокси-3-фтор-2-С-метилD-ксилофуранозил)-7 Н-пирроло[2,3-d]пиримидин и соответствующие 5'-трифосфаты; или их фармацевтически приемлемая соль. Для лечения инфекции ВГС соединения по настоящему изобретению также можно комбинировать с ненуклеозидными ингибиторами полимеразы ВГС такими, как раскрыто в публикациях международных заявок на патент WO 01/77091; WO 01/47883; WO 02/04425; WO 02/06246; WO 02/20497; WO 2005/016927 (в частности, JTK003); содержание каждой из которых включено в настоящее описание во всей полноте в качестве ссылки; и HCV-796 (Viropharma Inc). В одном из вариантов осуществления ненуклеозидные ингибиторы полимеразы ВГС NS5B, кото-5 019327 рые используются в комбинации с ингибиторами протеазы ВГС NS3, выбирают из следующих соединений: 14-циклогексил-6-[2-(диметиламино)этил]-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксильная кислота; 14-циклогексил-6-(2-морфолин-4-илэтил)-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11 карбоксильная кислота; 14-циклогексил-6-[2-(диметиламино)этил]-3-метокси-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксильная кислота; 14-циклогексил-3-метокси-6-метил-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11 карбоксильная кислота; метил([(14-циклогексил-3-метокси-6-метил-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11 ил)карбонил]аминосульфонил)ацетат;[2,5]бензодиазоцин-11-карбоксильная кислота; 14-циклогексил-6-[3-(диметиламино)пропил]-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксильная кислота; 14-циклогексил-7-оксо-6-(2-пиперидин-1-илэтил)-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксильная кислота; 14-циклогексил-6-(2-морфолин-4-илэтил)-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксильная кислота; 14-циклогексил-6-[2-(диэтиламино)этил]-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксильная кислота; 14-циклогексил-6-(1-метилпиперидин-4-ил)-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксильная кислота; 14-циклогексил-N-[(диметиламино)сульфонил]-7-оксо-6-(2-пиперидин-1-илэтил)-5,6,7,8 тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксамид; 14-циклогексил-6-[2-(диметиламино)этил]-N-[(диметиламино)сульфонил]-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксамид; 14-циклопентил-6-[2-(диметиламино)этил]-7-оксо-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксильная кислота; 14-циклогексил-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11-карбоксильная кислота; 6-аллил-14-циклогексил-3-метокси-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11 карбоксильная кислота; 14-циклопентил-6-[2-(диметиламино)этил]-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11 карбоксильная кислота; 14-циклогексил-6-[2-(диметиламино)этил]-5,6,7,8-тетрагидроиндоло[2,1-а][2,5]бензодиазоцин-11 карбоксильная кислота; 13-циклогексил-5-метил-4,5,6,7-тетрагидроиндоло[3',2':6,7][1,4]диазоцино[1,8-а]индол-10 карбоксильная кислота; 15-циклогексил-6-[2-(диметиламино)этил]-7-оксо-6,7,8,9-тетрагидро-5 Н-индоло[2,1-а][2,6]бензодиазонин-12-карбоксильная кислота; 15-циклогексил-8-оксо-6,7,8,9-тетрагидро-5 Н-индоло[2,1-а][2,5]бензодиазонин-12-карбоксильная кислота; 13-циклогексил-6-оксо-6,7-дигидро-5 Н-индоло[2,1-d][1,4]бензодиазепин-10-карбоксильная кислота; и их фармацевтически приемлемые соли.IV. Оценка соединений. Описанные здесь соединения могут быть оценены в отношении различных видов активности, таких как способность ингибировать активность ВГС NS3, активность репликона ВГС и активность репликации ВГС, используя методы, известные в данной области. (См., например, Carroll et al., J. Biol. Chem. 278:11979-11984, 2003). Одним из таких видов анализа является флуоресцентный анализ протеазы ВГС NS3 с временным разрешением (TRF), как описано ниже, а также у Мао et al., Anal. Biochem. 373:1-8, 2008 и в публикации международной заявки на патент WO 2006/102087. Анализ протеазы NS3 может быть выполнен, например, в буфере для анализа с конечным объемом 100 мкл, содержащем 50 мМ HEPES (рН 7,5), 150 мМNaCl, 15% глицерина, 0,15% тритона X-100, 10 мМ DTT и 0,1% ПЭГ 8000. Протеазу NS3 и NS4A предварительно инкубируют с различными концентрациями ингибиторов в ДМСО в течение 30 мин. Реакцию инициируют добавлением TRF пептидного субстрата (конечная концентрация 100 нм). NS3 опосредованный гидролиз субстрата завершают через 1 ч при комнатной температуре, используя 100 мкл 500 мМ MES (рН 5,5). Флюоресценцию продукта детектируют с помощью флуорофотометра либо VICTOR V2, либо FUSION (Perkin Elmer Life и Analytical Sciences) с возбуждением на 340 нм и излучением на 615 нм с 400 мкс задержкой. Тестируемые концентрации различных форм фермента выбирают таким образом, чтобы в результате получилось отношение сигнала к фону (S/B), находящееся в пределах 10-30. Значения IC50 получают, используя стандартную четырехпараметрическую подгонку данных. ЗначенияKi выводят из значений IC50 с помощью следующей формулы: где [S] - концентрация пептидного субстрата в реакции, а КМ - константа Михаэлиса. См. P. Gallinari et al., 38 BIOCHEM. 5620-32 (1999); P. Gallinari et al., 72 J. VIROL. 6758-69 (1998);V. Общая схема получения соединений. Настоящее изобретение также включает способы получения соединений формулы (I). Соединения по настоящему изобретению можно легко получить согласно следующим схемам реакций и примерам или путем их модификации, используя легко доступные исходные вещества, реактивы и обычные способы синтеза. В этих реакциях также можно использовать варианты, которые хорошо известны специалистам в данной области. Другие способы получения соединений по изобретению являются очевидными для специалиста из нижеприведенных схем реакции и примеров. Если не указано иное, все переменные следует понимать согласно приведенным выше определениям. Следующие схемы реакции и примеры служат только в качестве иллюстрации изобретения и его практического применения. Катализаторы метатезиса олефинов включают следующие разновидности на основе рутения: F.Scholl et al., 1 Org. Lett. 953 (1999); публикация заявки на патент США US 2002/0107138; K. Furstner et al.,64 J. Org. Chem. 8275 (1999). Польза этих катализаторов в замкнутом цикле метатезиса известна из литературы (например, Trnka and Grubbs, 34 Acc. Chem. Res. 18 (2001). Следующие примеры служат только в качестве иллюстрации изобретения и его практического применения. Примеры не следует рассматривать как ограничивающие объем или сущность изобретения. Синтез промежуточных соединений Промежуточные соединения А.Cu(I)BrSMe2 (0,05 экв.) и HMPA (2,4 экв.). Смесь перемешивали в течение 10 мин, затем раствор (1 М) акролеина (1 экв.) и TMSCl (2 экв.) в THF добавляли в течение более 1 ч таким образом, чтобы внутренняя температура оставалась ниже -68 С. Полученную смесь перемешивали при -78 С в течение 2 ч, затем обрабатывали избытком Et3N и разбавляли гексаном. После того как температура смеси достигла ком-8 019327 натной, смесь обрабатывали небольшим количеством Н 2 О и фильтровали через целит. Фильтрат промывали 10 раз, используя Н 2 О, а затем рассол. Органический слой сушили и летучие компоненты удаляли, а полученный остаток дистиллировали при пониженном давлении (20 мбар). При 80-86 С собирали фракцию, в состав которой входило названное соединение (58%) в виде бесцветной жидкости. 1 Н ЯМР (400 МГц, CDCl3)6,19 (д, J=11,6 Гц, 1 Н), 5,85-5,75 (м, 1 Н), 5,02-4,92 (м, 3 Н), 2,08-2,02 (м,2 Н), 1,94-1,88 (м, 2 Н), 1,46-1,38 (м, 2 Н), 0,18 (с, 9H). Стадия 2. транс-2-Пент-4-ен-1-илциклопропанол Раствор (0,45 М) предыдущего соединения в гексане обрабатывали раствором (15%) Et2Zn (1,2 экв.) в толуоле и полученный раствор охлаждали в ледяной бане. Дийодметан (1,2 экв.) добавляли по каплям,затем перед тем, как нагреть до 20 С, раствор перемешивали в течение 1 ч. Добавляли пиридин (6 экв.) и кашицу перемешивали в течение 15 мин, затем выливали в петролейный эфир. Смесь фильтровали через целит несколько раз до тех пор, пока не получили прозрачный раствор. Эту смесь концентрировали при 100 мбар и оставшийся раствор(содержащий триметил[(транс-2-пент-4-ен-1-илциклопропил]оксисилан, толуол и пиридин) дополнительно разбавляли THF. Смесь охлаждали до 0 С, затем по каплям добавляли раствор (1M) TBAF (1,2 экв.) в THF. Через 10 мин оставляли смесь с тем, чтобы она нагрелась до 20 С, а затем еще через 1 ч выливали в Н 2 О. Водную фазу экстрагировали с помощьюEtOAc и объединенные органические экстракты промывали рассолом, а затем сушили. После удаления летучих компонентов получали остаток, который очищали с помощью флеш-хроматографии (элюент 066% Et2O/петролейный эфир), получая названное соединение (71%) в виде бесцветной жидкости. 1 Раствор (0,39 М) метил 3-метил-L-валината в смеси насыщенного водного раствора NaHCO3 иCH2Cl2 (2:1) охлаждали в ледяной бане и быстро перемешивали. Смесь обрабатывали трифосгеном (0,45 экв.) в виде одной порции и полученную смесь перемешивали в течение 0,5 ч. Реакционную смесь разбавляли CH2Cl2 и слои отделяли. Водную фазу экстрагировали, используя CH2Cl2, затем объединенные органические слои промывали рассолом и сушили. После удаления растворителя получили названное соединение в виде прозрачного масла, которое выдерживали в течение 12 ч под вакуумом (0,1 мбар), затем сразу использовали на следующей стадии. 1 Раствор (0,45 М) транс-2-пент-4-ен-1-илциклопропанола в толуоле обрабатывали метил 3-метил-N(оксометилен)-L-валинатом (1,1 экв.), а затем DMAP (1 экв.). Полученную смесь нагревали с обратным холодильником в течение 12 ч, затем охлаждали до 20 С. Добавляли Н 2 О и EtOAc и органический слой отделяли и промывали 1N HCl, рассолом и сушили. После удаления летучих компонентов получали остаток, который дважды очищали с помощью флеш-хроматографии (элюент 0-30% Et2O/петролейный эфир). Первые фракции содержали метил 3-метил-N-([(1R,2R)-2-пент-4-ен-1-илциклопропил]оксикарбонил)-L-валинат (38%) в виде масла. MS (ES+) m/z 298 (М+Н)+. Более поздние фракции содержали метил 3-метил-N-([(1S,2S)-2-пент-4-ен-1-илциклопропил]окси карбонил)-L-валинат (28%) в виде масла. MS (ES+) m/z 298 (М+Н)+. Стадия 5. 3-Метил-N-([(1R,2R)-2-пент-4-ен-1-илциклопропил]оксикарбонил)-L-валин Раствор (0,1 М) метил 3-метил-N-([(1R,2R)-2-пент-4-ен-1-илциклопропил]оксикарбонил)-Lвалината в смеси МеОН/Н 2 О (2:1) обрабатывали LiOHH2O (4 экв.) и затем нагревали до 60 С в течение 4 ч. Смесь охлаждали и концентрировали до половины объема, затем разбавляли, используя EtOAc, и подкисляли водным раствором HCl (1N). Органический слой отделяли и промывали рассолом, затем суши-9 019327 ли. После удаления летучих компонентов получили названное соединение (98%) в виде масла. MS (ES+)Et3N (2 экв.) и затем нагревали до 150 С в течение 2 ч. Смесь охлаждали и фильтровали, затем собранное твердое вещество промывали, используя Н 2 О и EtOH. Полученный остаток сушили и получали названное соединение (69%). MS (ES+) m/z 193 (М+Н)+. Стадия 2. 3-Хлор-6-метоксихиноксалин-2-ол Раствор (1,53 М) 6-метоксихиноксалин-2,3-диола в DMF обрабатывали SOCl2 (1 экв.) и нагревали до 110 С. После 1,5 ч реакционную смесь охлаждали и наливали в водный раствор HCl (1N). Полученный осадок фильтровали и промывали, используя Н 2 О и Et2O. Высушенное твердое вещество состояло в основном из названного соединения в виде смеси с 6-метоксихиноксалин-2,3-диолом и 2,3-дихлор-6 метоксихиноксалином. Это вещество сразу использовали на следующей стадии. MS (ES+) m/z 211 Раствор (0,35 М) 3-хлор-6-метоксихиноксалин-2-ола в NMP обрабатывали Cs2CO3 (1,5 экв.) и 1-третбутил 2-метил-(2S,4S)-4-[(4-бромфенил)сульфонил]оксипирролидин-1,2-дикарбоксилатом (1,1 экв.). Полученную смесь перемешивали при 50 С в течение 18 ч, затем добавляли еще одну часть (0,1 экв.) 1 трет-бутил 2-метил-(2S,4S)-4-[(4-бромфенил)сульфонил]оксипирролидин-1,2-дикарбоксилата. После перемешивания в течение 2 ч смесь охлаждали и разбавляли, используя Н 2 О и EtOAc. Органические фазы промывали водным раствором HCl (1N), насыщенным водным раствором NaHCO3 и рассолом. Высушенную органическую фазу концентрировали до получения остатка, который очищали с помощью флеш-хроматографии (0-60% EtOAc/петролейный эфир) и получали названное соединение (35% за два прохода) в виде твердого вещества. MS (ES+) m/z 438 (М+Н)+. Стадия 4. Метил-(4R)-4-[(3-хлор-7-метоксихиноксалин-2-ил)окси]-L-пролината гидрохлорид Раствор (0,62M) 1-трет-бутилметил-(2S,4R)-4-[(3-хлор-7-метоксихиноксалин-2-ил)окси]пирролидин-1,2-дикарбоксилата в CH2Cl2 обрабатывали раствором (4M) HCl в диоксане (5 экв.). Смесь перемешивали при 20 С в течение 2 ч, затем обрабатывали раствором (4 М) HCl в диоксане (2 экв.). Через 5 ч после начала реакции было решено, что реакция завершилась, после чего смесь концентрировали при пониженном давлении. Полученный остаток растирали в порошок с Et2O для получения названного соединения (95%) в виде твердого вещества. MS (ES+) m/z 338 (М+Н)+. Пример 1. КалийDMF обрабатывали 3-метил-N-([(1R,2R)-2-пент-4-ен-1-илциклопропил]оксикарбонил)-L-валином (1,1 экв.), DIEA (5 экв.) и HATU (1,2 экв.). Полученную смесь перемешивали при 20 С в течение 5 ч, затем разбавляли EtOAc. Органический слой отделяли и промывали водным раствором HCl (1N), насыщенным водным раствором NaHCO3 и рассолом. Высушенную органическую фазу концентрировали при пониженном давлении для получения остатка, который очищали флэш-хроматографией (элюент 10-30%EtOAc/петролейный эфир), получая названное соединение (96%) в виде масла. MS (ES+) m/z 604 (M+Н)+. Стадия 2. Метил 3-метил-N-([(1R,2R)-2-пент-4-ен-1-илциклопропил]оксикарбонил)-L-валил-(4R)4-[(7-метокси-3-винилхиноксалин-2-ил)окси]-L-пролинат Раствор (0,1 М) метил 3-метил-N-([(1R,2R)-2-пент-4-ен-1-илциклопропил]оксикарбонил)-L-валил(4R)-4-[(3-хлор-7-метоксихиноксалин-2-ил)окси]-L-пролинат в EtOH обрабатывали калий трифтор(винил)боратом (1,5 экв.) и триэтиламином (1,5 экв.). Полученную смесь дегазировали, затем добавляли аддукт PdCl2(dppf)-CH2Cl2 (0,1 экв.). Смесь нагревали с обратным холодильником в течение 1 ч,- 11019327 затем охлаждали до комнатной температуры и разбавляли, используя Н 2 О и EtOAc. Органическую фазу отделяли, промывали Н 2 О и рассолом, затем сушили. После удаления летучих компонентов получали остаток, который очищали флэш-хроматографией (20-30% EtOAc/петролейный эфир), получая названное соединение в виде желтой пены, которую сразу использовали на следующей стадии. MS (ES+) m/z 595 Раствор (0,02 М) метил 3-метил-N-([(1R,2R)-2-пент-4-ен-1-илциклопропил]оксикарбонил)-Lвалил-(4R)-4-[(7-метокси-3-винилхиноксалин-2-ил)окси]-L-пролината в DCE нагревали до 80 С, затем обрабатывали катализатором Zhan 1 (0,15 экв.). Полученную смесь перемешивали при 80 С в течение 1 ч, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали флэш-хроматографией (20-50% EtOAc/петролейный эфир) и получали названное соединение (25% за 2 прохода) в виде пены. MS (ES+) m/z 567 (M+Н)+. Стадия 4. Метил (1aR,5S,8S,10R,22aR)-5-трет-бутил-14-метокси-3,6-диоксо-1,1 а,3,4,5,6,9,10,18,19,20,21,22,22 а-тетрадекагидро-8 Н-7,10-метанциклопропа[18,19][1,10,3,6]диоксадиазациклононадецино[11,12-b]хиноксалин-8-карбоксилата в смеси МеОН/диоксан (1:1) обрабатывали Pd/C (8 вес.%). Полученную смесь перемешивали в атмосфере водорода в течение 4 ч. Катализатор отфильтровывали и фильтрат концентрировали при пониженном давлении, получая названное соединение (98%) в виде твердого вещества. MS (ES+) m/z 569 (M+Н)+. Стадия 5. (1aR,5S,8S,10R,22aR)-5-трет-бутил-14-метокси-3,6-диоксо-1,1a,3,4,5,6,9,10,18,19,20,21,22,22 а-тетрадекагидро-8 Н-7,10-метанциклопропа[18,19][1,10,3,6]диоксадиазациклононадецино[11,12b]хиноксалин-8-карбоксильная кислота Раствор (0,1 М) метил (1aR,5S,8S,10R,22aR)-5-трет-бутил-14-метилокси-3,6-диоксо-1,1 а,3,4,5,6,9,10,18,19,20,21,22,22 а-тетрадекогидро-8 Н-7,10-метанциклопропа[18,19][1,10,3,6]диоксадиазациклононадецино[11,12-b]хиноксалин-8-карбоксилата в смеси 1:1 H2O/THF обрабатывали LiOHН 2 О (3 экв.). Полученную смесь перемешивали при 20 С в течение 18 ч, подкисляли водным раствором HCl (0,2M) и разбавляли EtOAc. Органическую фазу отделяли, промывали водным раствором HCl (0,2M) и рассолом,затем сушили. После удаления летучих компонентов получили названное соединение (98%) в виде твердого вещества. MS (ES+) m/z 555 (M+Н)+.[11,12-b]хиноксалин-8-карбоксильной кислоты в CH2Cl2 обрабатывали (1R,2S)-1-[(циклопропилсульфонил)амино]карбонил-2-винилциклопропанаминий хлоридом (1,3 экв.), DIEA (3 экв.), DMAP (1,5 экв.) и TBTU (1,45 экв.). Полученную смесь перемешивали при 20 С в течение 18 ч, а затем разбавлялиEtOAc. Раствор промывали водным раствором HCl (0,2 М), насыщенным водным раствором NaHCO3 и рассолом. Органические фазы сушили и концентрировали до получения остатка, который очищали флэш-хроматографией (элюент 2,5% MeOH/CH2Cl2), получая названное соединение (89%) в виде твердого вещества. 13 С ЯМР (100 МГц, DMSO-d6)172,32, 170,63, 169,04, 159,86, 156,95, 154,74, 148,10, 140,41, 133,55,(2 сигнала), 128,94, 118,21, 117,58, 105,89, 74,88, 59,75, 58,71, 55,68, 54,13, 54,01, 40,13, 34,49, 34,04,33,76, 32,68, 30,71, 30,43, 28,55, 27,69, 27,28, 26,38, 21,98, 18,49, 10,67, 5,69, 5,46; MS (ES+) m/z 767 Предыдущее вещество использовали в EtOH и полученный раствор (0,025 М) охлаждали до 0 С. Добавляли раствор (0,02 М) трет-BuOK (1,5 экв.) в EtOH, что приводило к образованию осадка. Смесь перемешивали при 20 С в течение 18 ч, затем фильтрованием собирали твердое вещество. Это вещество промывали EtOH и сушили, получая названное соединение (93%) в виде белого кристаллического твердого вещества. MS (ES+) m/z 767 (M+Н)+. Пример 2. Сравнение различных соединений. Соединение, описанное в примере 1, сравнивали с соединением, раскрытым в примерах 110 и 118WO 2008/057209. Результаты показаны ниже в табл. 1 и 2. Как можно видеть из таблиц и обсуждения результатов, оказалось, что соединение формулы (I) обладает несколькими полезными свойствами по сравнению как с соединением примера 118 документа WO 2008/057209, так и соединением примера 110 документа WO 2008/057209.Ki: константа ингибирования, ссылка на 0,016 нМ указывает, что наблюдаемая активность не превышает 0,016 нМ, точное количество, меньшее чем 0,016 нМ, в тесте не было определено; ЕС 50: эффективная концентрация, при которой достигается подавление репликации вируса на 50%; gt: генотип; AUC: область под кривой концентрация в плазме/время; LOQ: предел количественного определения (3 пмоль/мг); BLQ: ниже предела количественного определения. Сравнение соединения формулы (I) с соединением примера 110 WO 2008/057209. Соединения формулы (I) обладают следующими полезными свойствами по сравнению с соединением примера 110 WO 2008/057209: 1) физические свойства (отсутствует диспропорция солей соединения формулы (I; 2) фармакокинетический профиль у крыс после введения соли калия и 3) воздействие на печень (целевой орган). Различия в свойствах, выявленные при сравнении с соединением примера 110 WO 2008/057209,особенно предпочтительны в случае применения соединения формулы (I) в лекарственных составах и применения этого соединения. Отсутствие диспропорции солей соединения формулы (I) дает возможность использовать разбавление в воде 1,8 мг/мл соли K+ соединения примера 1. Хотя соль K+ соединения примера 110 WO 2008/057209 демонстрирует лучшую растворимость в воде (9,7 мг/мл), это растворенное таким образом соединение диспропорционирует с образованием кристаллической цвиттерионной формы, имеющей низкую растворимость в воде (0,009 мг/мл). Отсутствие такого поведения у соединения примера 1 дает неожиданное преимущество для применения в лекарственном составе для фармацевтического введения и в результате обнаруживает улучшенные фармакокинетические свойства,как показано в табл. 1 (плазменная AUC и воздействие на печень крыс и собак). Высокий уровень воздействия в плазме и на печень в доклинических испытаниях является преимуществом с точки зрения отбора безопасных и эффективных доз для применения при лечении пациентов. Сравнение соединения формулы (I) с соединением примера 118 WO 2008/057209. Наблюдаемым преимуществом соединения формулы (I) по сравнению с соединением примера 118WO 2008/057209 является его профиль резистентности к различным мутантным ферментам. Согласно данным клинических исследований, проводимых с использованием противовирусных агентов родственных классов (например, ингибиторы протеазы ВИЧ), а также исследований, проводимых с использованием ингибиторов протеазы ВГС NS3 (например, VX-950, телапревир), предполагается, что резистентность к вирусам может развиться в ответ на лечение соединениями по настоящему изобретению. Соединение примера 1 показало улучшенную ферментативную аффинность (Ki) к различным мутантным ферментам, которые, как известно, обеспечивают резистентность к ингибиторам протеазы ВГС NS3. В табл. 2 суммированы данные по активности в отношении различных мутантных ферментов. Таким образом,преимуществом соединения 1 может быть более высокий барьер к развитию устойчивого вируса при введении пациентам. Оно также дает потенциальное преимущество в лечении пациентов, которым не помогли другие методы лечения из-за развития резистентности, поскольку соединение 1 может ингибировать такой устойчивый вирус. Таблица 2 Значения Ki1 к 1b мутантному ферменту (нМ) Сравнительные данные собраны в одной серии анализа ферментов По сравнению с соединением примера 110 WO 2008/057209 соединения формулы (I) имеют следующие дополнительные ожидаемые полезные свойства: 1) низкое ковалентное связывание in vivo и 2) высокий уровень воздействия в плазме и на печень. Было обнаружено, что соединения формулы (I) имеют очень хорошие характеристики ковалентного связывания in vivo и фармакокинетические свойства. Исходя из наблюдаемых фармакокинетических свойств и ковалентного связывания in vivo соединения примера 1 и соединения примера 118 WO 2008/057209, соединение формулы (I) имеет значительно более лучшие характеристики ковалентного связывания in vivo и фармакокинетические свойства. Соединения, образующие ковалентные связи с белками или образующие метаболиты, которые затем образуют ковалентные связи с белками, потенциально способствуют увеличению нежелательных явлений у пациентов, таких как иммунологическая токсичность, опосредованная антительными ответами на конъюгат белок-лекарственное вещество, и идиосинкратическая токсичность (см. Chem. Res. Toxicol. 2004, 17, 3-16). Соединение примера 1 показало недетектируемое связывание с белками плазмы после перорального введения крысам единичной дозы 20 мг/кг (см. табл. 1). При аналогичных условиях соединение примера 118 WO 2008/057209 продемонстрировало детектируемое связывание с белками печени крысы (см. табл. 1) и поэтому его можно считать менее полезным соединением для введения человеку по сравнению с соединением формулы (I). В табл. 3 приведены некоторые дополнительные данные по ковалентному связыванию in vivo, полученные для родственных соединений из WO 2008/057209, которые содержат фрагмент (R,R)-транс-2- 15019327 Наличие высокого уровня воздействия в плазме и на печень в доклинических испытаниях является преимуществом для эффективной демонстрации того, что потенциальное лекарственное веществокандидат не проявляет нежелательную токсичность. Также более вероятно, что соединение с высоким уровнем воздействия в плазме и на печень животных имеет такое же поведение в организме человека в сравнении с соединением, у которого такое воздействие отсутствует. Необходимое эффективное воздействие такого соединения у человека может быть достигнуто при более низкой дозе, что является преимуществом как с точки зрения стоимости, так и легкости изготовления лекарственного средства, а также с точки зрения потенциального уменьшения вероятности возникновения побочных эффектов. Воздействие на целевой орган у многих видов, участвовавших в доклинических испытаниях, подтверждает, что для этого соединения можно достичь высокого уровня воздействия на целевой орган у пациентов, а высокий уровень воздействия на печень как у крыс, так и у собак дает возможность достоверно оценить доклиническую токсичность. Высокий уровень воздействия на печень является особенно полезным для ВГС, поскольку она является целевым органом для данного лекарственного средства. Соединение примера 1 обладает очень хорошим воздействием в плазме и на печень крыс. Наблюдаемое воздействие на печень крыс является более сильным по сравнению с соединением примера 118 и соединением 110 WO 2008/057209 (см. табл. 1). На основании этих результатов и результатов тестирования нескольких различных соединений WO 2008/057209 путем их перорального введения как крысам (25mpk), так и собакам (5 mpk), также ожидалось, что соединение формулы (I) будет сильнее воздействовать на печень собак по сравнению с соединением примера 118 и соединением примера 110 WO 2008/057209. Методы. Ингибиторная активность 1 (Ki) NS3/4A. Ингибиторную активность (Ki) для NS3/4A определяли, как описано в секции IV. Оценка соединений supra и Мао et al., Anal. Biochem 373:1-8, 2008. Активность репликона 2 EC50. Активность репликона определяли, используя процедуры, описанные у Carroll et al., J. Biol. Chem. 278:11979-11984, 2003 и Olsen et al., Anti Microb. Agents 45:3944-3953, 2004. Плазменная AUC у крыс 25 mpk per os3. Тестируемые соединения растворяли в подходящем растворителе для iv введения (например, 20:60:20% DMSO:PEG400:вода), или per os введения (например,10% полисорбат 80:90% вода или 100% PEG400), которые вводили животным (n=3), используя перекрестное исследование, разработанное для негрызунов. Образцы плазмы отбирали во временных точках между 2 мин и 24 ч и определяли уровни соединений методом ВЭЖХ с обращенной фазой. Образцы печени крыс брали после смерти животного, а образцы печени собак брали с помощью анестезии (за 0,5 ч до биопсии). Образцы печени взвешивали, гомогенизировали и разбавляли, используя методы, хорошо известные специалистам в данной области, а уровни соединений определяли методом ВЭЖХ с обращенной фазой. Фармакокинетические параметры вычисляли методом некомпартментного анализа (например, Watson, WinNolin). Предозовые концентрации, которые были ниже предела количественного определения (BLQ), устанавливали в значение 0. Для оценки пероральной AUC первые значения BLQ в терминальной фазе задавали в виде значения, равного 1/2 самого низкого предела количественного определения, в то время как последующие значения в терминальной фазе задавали равными 0. Вычисляли стандартные фармакокинетические параметры CLp, Vdss, период полувыведения (только для IV), %F, Cmax,Tmax, AUC0-last, AUC0-infinity. Значения AUC вычисляли, используя линейный метод трапеций для увеличения концентраций и логарифмический метод трапеций для уменьшения концентраций. Ковалентное связывание 4 in vivo. Тестируемые соединения соответствующим образом метили радиоактивными метками (3 Н) и готовили 20 мг/кг дозу, содержащую радиоактивность 25-75 мКю/крыса(чистота 98,5%), путем комбинации холодного соединения и полученного выпариванием исходного раствора с радиоактивной меткой. Эту смесь растворяли в растворителе, подходящем для введения per os(см. выше), затем вводили перорально крысе (n=3 на 1 временную точку, 2 ч, 6 ч, 24 ч). Отбирали плазму и печень и до проведения анализа быстро замораживали и хранили при -80 С. Измерение радиоактивности образцов плазмы: 200 мкл аликвоты помещали в 20 мл сцинтилляционный флакон. Добавляли 500 мкл Solvable и инкубировали, встряхивая, в течение 1 ч при 55 С. Вынимали, давали остыть перед добавлением 15 мл сцинтилляционного коктейля и измеряли радиоактивность. Затем образцы плазмы (200 мкл аликвоты) обрабатывали, как описано ниже для белков печени. Гомогенизация ткани: взвешенные образцы печени разбавляли 2 об. 100 мМ фосфатного буфера(рН 7,4) и гомогенизировали на льду. Измерение радиоактивности гомогената печени: аликвоты помещали в 20 мл сцинтилляционный флакон, разбавляли, используя 1 мл Solvable, и инкубировали, встряхивая, в течение 1 ч при 55 С. Затем вынимали из инкубатора и после охлаждения добавляли 15 мл сцинтилляционного коктейля и 30% Н 2 О 2 и измеряли радиоактивность. Преципитация белка: брали 500 мкл аликвоты, добавляли гомогенат:ацетонитрил (1:8) (если имеется подозрение, что соединение имеет низкую растворимость в ацетонитриле, может быть выбран другой растворитель), встряхивали и центрифугировали (3500 об./мин в течение 20 мин). Супернатант удаляли. Ресуспендирование осажденного белка: обрабатывали ультразвуком (минимальная интенсивность,5 с) и встряхивали в смеси 80% МеОН:20% воды до разрушения осадка. Промывка белкового осадка: 2-5 мл МеОН:вода (80:20). При необходимости, следует удалить 1,0 мл супернатанта, добавляли 15 мл сцинтилляционного коктейля и измеряли радиоактивность. Белковый осадок продолжали промывать до тех пор, пока радиоактивность в супернатанте не стала 200 DPM, или до тех пор, пока значение DPM не перестало снижаться в результате последовательных промываний,превышая при этом 200. Растворение конечного осадка: 1 мл 1N NaOH или Solvable, инкубировали при 50 С в течение ночи или до полного растворения. Измерение радиоактивности конечного осадка: 1 мл растворенного осадка, 15 мл сцинтилляционного коктейля (если используется коктейль, отличный от ULTIMA GOLD, может возникнуть необходимость в нейтрализации осадка с помощью 1N HCl), и выполняли измерение радиоактивности. Концентрация белка в конечном осадке: в качестве стандарта BSA использовали набор ВСА илиBIO-Rad. Измерение радиоактивности пустых образцов: 15 мл сцинтилляционного коктейля, два образца. Измерение радиоактивности растворителя: использовали известный объем растворителя, три образца. Анализ данных: усредняли результаты измерения радиоактивности (DPM) растворителя и вычисляли определенную активность растворителя, выраженную в мкКю/моль. Усредняли результаты измерения радиоактивности пустых образцов. Вычитали результат измерения радиоактивности усредненного пустого образца из результатов измерения радиоактивности для каждого осадка, полученного из образцов печени и плазмы. Вычисляли величину радиоактивности (мкКю) на единичный объем (L) для каждого осадка, полученного из образцов печени и плазмы. Вычисляли концентрацию радиоактивности в каждом осадке, полученном из образцов плазмы и печени, путем деления полученного выше значения (мкКю/L) на удельную активность (мкКю/моль). Вычисляли величину радиоактивности ковалентных связей с белком в пмоль/мг белка. Измерение радиоактивности пустых образцов: 15 мл сцинтилляционного коктейля, два образца. Измерение радиоактивности растворителя: использовали известный объем растворителя, три образца. Физические свойства 5: кристаллические тестируемые соединения (соль калия, приблизительно 5 мг) взвешивали в стеклянном флаконе и добавляли воду или водный буфер (100 мкл). Полученную кашицу перемешивали в течение 24 ч при комнатной температуре. После центрифунгирования супернатант анализировали методом ВЭЖХ с обращенной фазой и определяли равновесную растворимость путем сравнения с калибровочной кривой. Твердое вещество частично переносили на пластину XRPD, сушили, а затем анализировали методом рентгеновской порошковой дифракции. Образец XRPD сравнивали с положительным контролем для кристаллической K+ соли, кристаллических цвиттер-ионных (или подкисленных) и аморфных форм тестируемого соединения. Дальнейшее определение формы соли получали из второй части твердого вещества, которое анализировали, используя 400 МГц ЯМР (Bruker) после растворения в DMSO-d6. Спектры 1H-ЯМР сравнивали с положительными контролями, описанными выше. Ни один из документов, представленных в настоящей заявке в виде ссылок, не рассматривается в качестве предшествующего уровня техники для заявленного изобретения. Другие варианты осуществления раскрыты в приведенной ниже формуле изобретения. Хотя показано и описано несколько вариантов осуществления, могут быть сделаны различные модификации без отступления от сущности и объема настоящего изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) или его фармацевтически приемлемая соль 2. Фармацевтическая композиция для лечения пациента, инфицированного ВГС, содержащая эффективное количество соединения по п.1 и фармацевтически приемлемый носитель. 3. Фармацевтическая композиция по п.2, дополнительно содержащая второй терапевтический агент,выбранный из группы, состоящей из ингибитора протеазы ВГС и ингибиторов полимеразы ВГС NS5B. 4. Применение соединения по п.1 для профилактики или лечения инфекции ВГС. 5. Применение соединения по п.1 для получения лекарственного средства для ингибирования активности протеазы ВГС NS3 у нуждающегося в этом субъекта. 6. Применение композиции по любому из пп.2, 3 для получения лекарственного средства для ингибирования активности протеазы ВГС NS3 у нуждающегося в этом субъекта. 7. Применение соединения по п.1 для получения лекарственного средства для профилактики или лечения инфекции ВГС у нуждающегося в этом субъекта. 8. Применение композиции по любому из пп.2, 3 для получения лекарственного средства для профилактики или лечения инфекции ВГС у нуждающегося в этом субъекта. 9. Способ лечения пациента, инфицированного ВГС, включающий введение указанному пациенту эффективного количества соединения по п.1. 10. Способ лечения пациента, инфицированного ВГС, включающий введение указанному пациенту эффективного количества фармацевтической композиции, содержащей соединение по п.1. 11. Соединение по п.1, имеющее структуру 12. Фармацевтическая композиция для лечения пациента, инфицированного ВГС, содержащая эффективное количество соединения по п.11 и фармацевтически приемлемый носитель. 13. Фармацевтическая композиция по п.12, дополнительно содержащая второй терапевтический агент, выбранный из группы, состоящей из ингибитора протеазы ВГС и ингибиторов полимеразы ВГСNS5B. 14. Способ лечения пациента, инфицированного ВГС, включающий введение указанному пациенту эффективного количества соединения по п.11. 15. Способ лечения пациента, инфицированного ВГС, включающий введение указанному пациенту эффективного количества композиции по п.12. 16. Способ лечения пациента, инфицированного ВГС, включающий введение указанному пациенту эффективного количества композиции по п.13.
МПК / Метки
МПК: C07K 5/12, A61K 38/08, A61K 38/06, A61K 38/07, C07K 5/10, C07K 5/08, A61P 31/14
Метки: вгс, соединение, хиноксалиновое, качестве, протеазы, ингибитора, макроциклическое
Код ссылки
<a href="https://eas.patents.su/19-19327-makrociklicheskoe-hinoksalinovoe-soedinenie-v-kachestve-ingibitora-proteazy-vgs-ns3.html" rel="bookmark" title="База патентов Евразийского Союза">Макроциклическое хиноксалиновое соединение в качестве ингибитора протеазы вгс ns3</a>
(4r,5s,6s,7r)-гексагидро-1-[5-(3-аминоиндазол)метил]-3-бутил-5,6-дигидрокси-4,7-бис[фенилметил]-2н-1,3-диазепин-2-он и его применение в качестве ингибитора вич-протеазы.

Номер патента: 1154
Опубликовано: 30.10.2000
Авторы: Роджерс Джеймс Дэвид, Лэм Патрик Юк-Сун
МПК: A61K 31/5513, C07D 403/06
Метки: применение, ингибитора, качестве, 4r,5s,6s,7r)-гексагидро-1-[5-(3-аминоиндазол)метил]-3-бутил-5,6-дигидрокси-4,7-бис[фенилметил]-2н-1,3-диазепин-2-он, вич-протеазы
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль, или его пролекарственная форма, где пролекарство формулы I является соединением, в котором две гидроксигруппы соединены с образованием эпоксида, -OCH2SCH2O-; -ОС(=O)O-; -OCH2O-; -OC(=S)O-; -ОС(=O)С(=O)О-; -ОС(СН3)2O-; -ОС((СН2)3NН2)(СН3)О-; -ОС(ОСН3) (СН2СН2СН3)О-; или -OS(=O)О- группы. 2. Соединение по п.1, где соединение является соединением формулы I. 3. Фармацевтическая...
Кремнийсодержащее соединение в качестве ингибитора коррозии в полиолефиновых композициях

Номер патента: 15093
Опубликовано: 30.06.2011
Авторы: Султан Бернт-Оке, Воигт Бьёрн, Фагрелль Ола, Даммерт Рут, Карлссон Рогер
МПК: C08K 3/24, C08K 5/00, C08K 5/42...
Метки: кремнийсодержащее, коррозии, полиолефиновых, композициях, ингибитора, качестве, соединение
Формула / Реферат:
1. Применение кремнийсодержащего соединения в качестве ингибитора коррозии в полиолефиновой композиции, включающей кислоту Брэнстеда, где кремнийсодержащее соединение имеет структуру формулы(R1)x[Si(R2)y(R3)z]m (I)где R1может быть одинаковым или различным, если присутствует больше одной группы (x больше 1), и представляет собой алкильную, арилалкильную, алкиларильную или арильную группу, содержащую от 1 до 40 атомов углерода, при условии, что,...
Соединение альфа-(n-сульфонамидо) ацетамида в качестве ингибитора продуцирования бета-амилоидного пептида

Номер патента: 16447
Опубликовано: 30.05.2012
Авторы: Джиллман Кевин В., Джон Е., Олсон Ричард Е.
МПК: A61P 25/28, C07C 235/30, A61K 31/4245...
Метки: альфа-(n-сульфонамидо, beta;-амилоидного, ингибитора, продуцирования, качестве, пептида, ацетамида, соединение
Формула / Реферат:
1. Соединение (2R)-2-[[(4-хлорфенил)сульфонил][[2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил] амино]-5,5,5-трифторпентанамид.2. Фармацевтическая композиция, включающая терапевтически эффективное количество (2R)-2-[[(4хлорфенил)сульфонил][[2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида совместно с фармацевтически приемлемым адъювантом, носителем или разбавителем.3. Способ лечения или отсрочки начала болезни...
Жидкая композиция, содержащая соединение кальция и подкислитель, для применения в качестве ингибитора эрозии зубов, способ ее приготовления и способы уменьшения эрозии зубов

Номер патента: 2852
Опубликовано: 31.10.2002
Автор: Паркер Дэвид Мьятт
МПК: A61K 33/06, A23L 2/52
Метки: качестве, способ, уменьшения, соединение, подкислитель, способы, композиция, ингибитора, применения, эрозии, приготовления, содержащая, зубов, кальция, жидкая
Формула / Реферат:
1. Применение жидкой композиции, содержащей соединение кальция и подкислитель в качестве ингибитора эрозии зубов, характеризующейся тем, что кальций присутствует в композиции в интервале от 0,3 до 0,8 моль на моль кислоты и рН композиции составляет от 3,5 до 4,5; исключая содержащие фрукты жидкие пищевые продукты с содержанием фруктов 25-100 мас.%, содержанием кислоты, рассчитанным на винную кислоту, по меньшей мере, 5 г/л, и содержащие, по...
Способ получения ингибитора протеазы вич из(2-s)-4-пиколил-2-пиперазин-трет-бутилкарбоксамида

Номер патента: 178
Опубликовано: 24.12.1998
Авторы: Аскин Дэвид, Енг Кан К., Ридер Пол, Воланте Ральф П.
МПК: A61K 31/495, C07D 401/06
Метки: способ, из(2-s)-4-пиколил-2-пиперазин-трет-бутилкарбоксамида, вич, протеазы, получения, ингибитора
Формула / Реферат:
1. Способ получения ингибитора протеазы ВИЧ структурной формулы включающий следующие стадии: (а) нагревание, по крайней мере, в течение одного часа одного эквивалента (S)-4-(3-пиколил)-2-трет-бутилкарбоксамидпиперазина (1) с приблизительно одним эквивалентом соединения в интервале температур между около 25 и около 150°С, при этом указанная смесь необязательно содержит приемлемый растворитель; (б) снятие защиты у полученного...
Предыдущий патент: Корпус банки и способ его изготовления
Следующий патент: Способ получения волокон на основе целлюлозы и волокна, полученные этим способом
Случайный патент: Регулирующий клапан для газовых сред