Способ ферментативного синтеза (7s)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновой кислоты или ее сложных эфиров и применение для синтеза ивабрадина и его соли









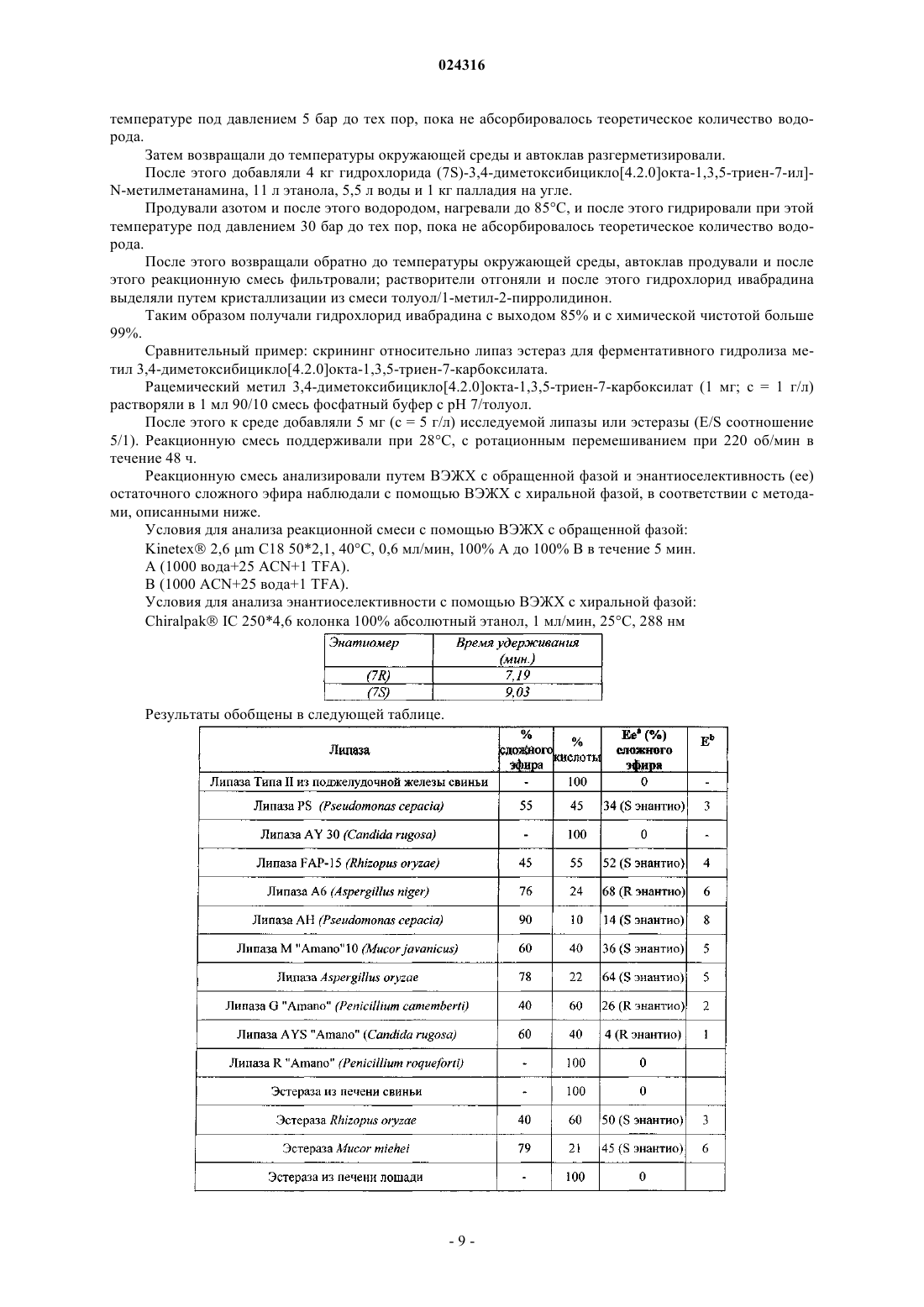




Формула / Реферат
1. Способ синтеза оптически чистого соединения формулы (Ia)

путем энантиоселективной ферментативной эстерификации рацемической или другой, оптически нечистой, кислоты формулы (X)

используя липазу Candida antarctica или Pseudomonas fluorescens, в смеси спирта ROH, где R представляет линейную или разветвленную C1-С6-алкильную группу, и органического сорастворителя, при концентрации от 5 до 500 г/л соединения формулы (X) на 1 л смеси растворителей, при E/S соотношении от 10/1 до 1/100, при температуре от 25 до 40°С.
2. Способ синтеза в соответствии с п.1, где E/S соотношение составляет от 1/5 до 1/10.
3. Способ синтеза в соответствии с любым из пп.1 или 2, где спирт ROH представляет собой метанол и со-растворитель представляет собой ацетонитрил.
4. Способ синтеза в соответствии с п.3, где соотношение ацетонитрил/метанол составляет от 8/2 до 9/1.
5. Способ синтеза в соответствии с любым из пп.1-3, где сложный эфир конфигурации (R), вторичный продукт реакции

подвергают гидролизу путем воздействия основания с образованием рацемической кислоты формулы (X) для повторного использования в процессе ферментативной эстерификации.
6. Способ синтеза в соответствии с п.5, где основание представляет собой KOH.
7. Способ синтеза в соответствии с п.5, где стадию гидролиз/рацемизация осуществляют in situ.
8. Способ синтеза в соответствии с любым из пп.1-3, где кислоту формулы (Ia) выделяют после одного или нескольких циклов ферментативной эстерификации.
9. Способ синтеза оптически чистого соединения формулы (Ib)

где R представляет линейную или разветвленную C1-C6-алкильную группу,
путем энантиоселективного ферментативного гидролиза рацемического или другого, оптически нечистого, сложного эфира формулы (XI)

где R представляет линейную или разветвленную C1-C6-алкильную группу,
используя липазу Candida antarctica или Pseudomonas fluorescens, в воде в буферном растворе с рН 7 или в смеси органического растворителя и воде или буфера с рН 7, при концентрации от 1 до 200 г/л соединения формулы (XI) на 1 л растворителя или смеси растворителей, при E/S соотношении от 10/1 до 1/100, при температуре от 25 до 40°С, с последующим выделением сложного эфира формулы (Ib).
10. Способ синтеза в соответствии с п.9, где E/S соотношение составляет от 1/5 до 1/10.
11. Способ синтеза в соответствии с любым из пп.9 или 10, где R представляет собой метильную группу.
12. Способ синтеза в соответствии с любым из пп.9 или 10, где реакцию осуществляют в смеси ацетонитрила и буфера с рН 7.
13. Способ синтеза в соответствии с п.12, где соотношение ацетонитрил/буфер с рН 7 составляет от 8/2 до 9/1.
14. Способ синтеза в соответствии с любым из пп.9 или 10, где кислоту конфигурации (R)

вторичный продукт реакции, рацемизируют путем воздействия основания и после этого рацемическую кислоту, полученную таким образом, алкилируют с образованием рацемического сложного эфира формулы (XI) для повторного использования в процессе ферментативного гидролиза.
15. Способ синтеза в соответствии с п.14, где кислоту конфигурации (R) рацемизируют путем воздействия KOH в горячем состоянии.
16. Способ синтеза в соответствии с любым из пп.9 или 10, где кислоту конфигурации (R)

вторичный продукт реакции, сначала алкилируют и после этого сложный эфир конфигурации (R), полученный таким образом, рацемизируют путем воздействия основания для повторного использования в процессе ферментативного гидролиза.
17. Способ синтеза в соответствии с п.16, где сложный эфир конфигурации (R) рацемизируют путем воздействия DBU в горячем состоянии или KOH при температуре окружающей среды.
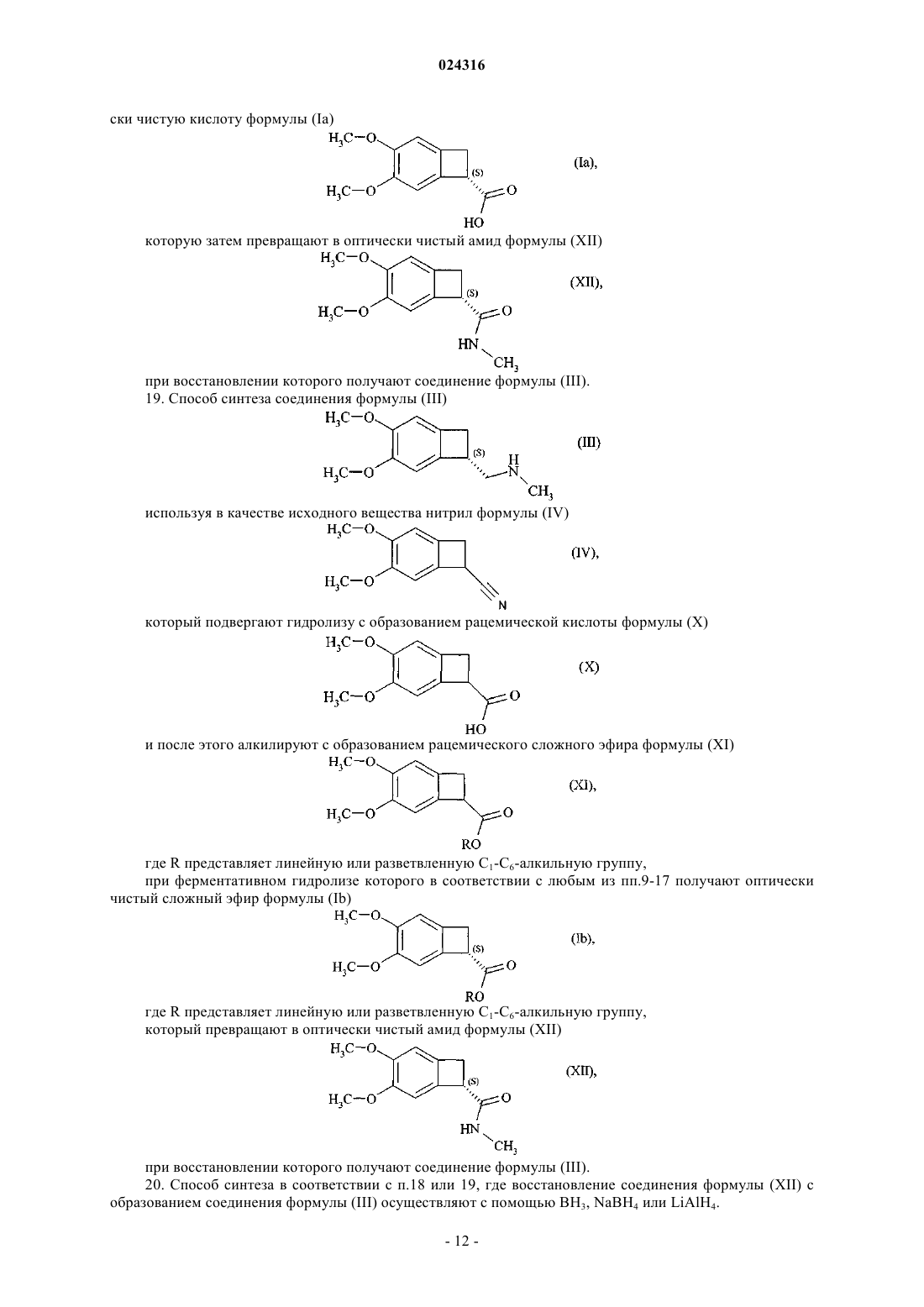
18. Способ синтеза соединения формулы (III)

используя в качестве исходного вещества нитрил формулы (IV)

который подвергают гидролизу с образованием рацемической кислоты формулы (X)

при ферментативной эстерификации которой в соответствии с любым из пп.1-8 получают оптически чистую кислоту формулы (Ia)

которую затем превращают в оптически чистый амид формулы (XII)

при восстановлении которого получают соединение формулы (III).
19. Способ синтеза соединения формулы (III)

используя в качестве исходного вещества нитрил формулы (IV)

который подвергают гидролизу с образованием рацемической кислоты формулы (X)

и после этого алкилируют с образованием рацемического сложного эфира формулы (XI)

где R представляет линейную или разветвленную C1-C6-алкильную группу,
при ферментативном гидролизе которого в соответствии с любым из пп.9-17 получают оптически чистый сложный эфир формулы (Ib)

где R представляет линейную или разветвленную C1-C6-алкильную группу,
который превращают в оптически чистый амид формулы (XII)

при восстановлении которого получают соединение формулы (III).
20. Способ синтеза в соответствии с п.18 или 19, где восстановление соединения формулы (XII) с образованием соединения формулы (III) осуществляют с помощью BH3, NaBH4 или LiAlH4.
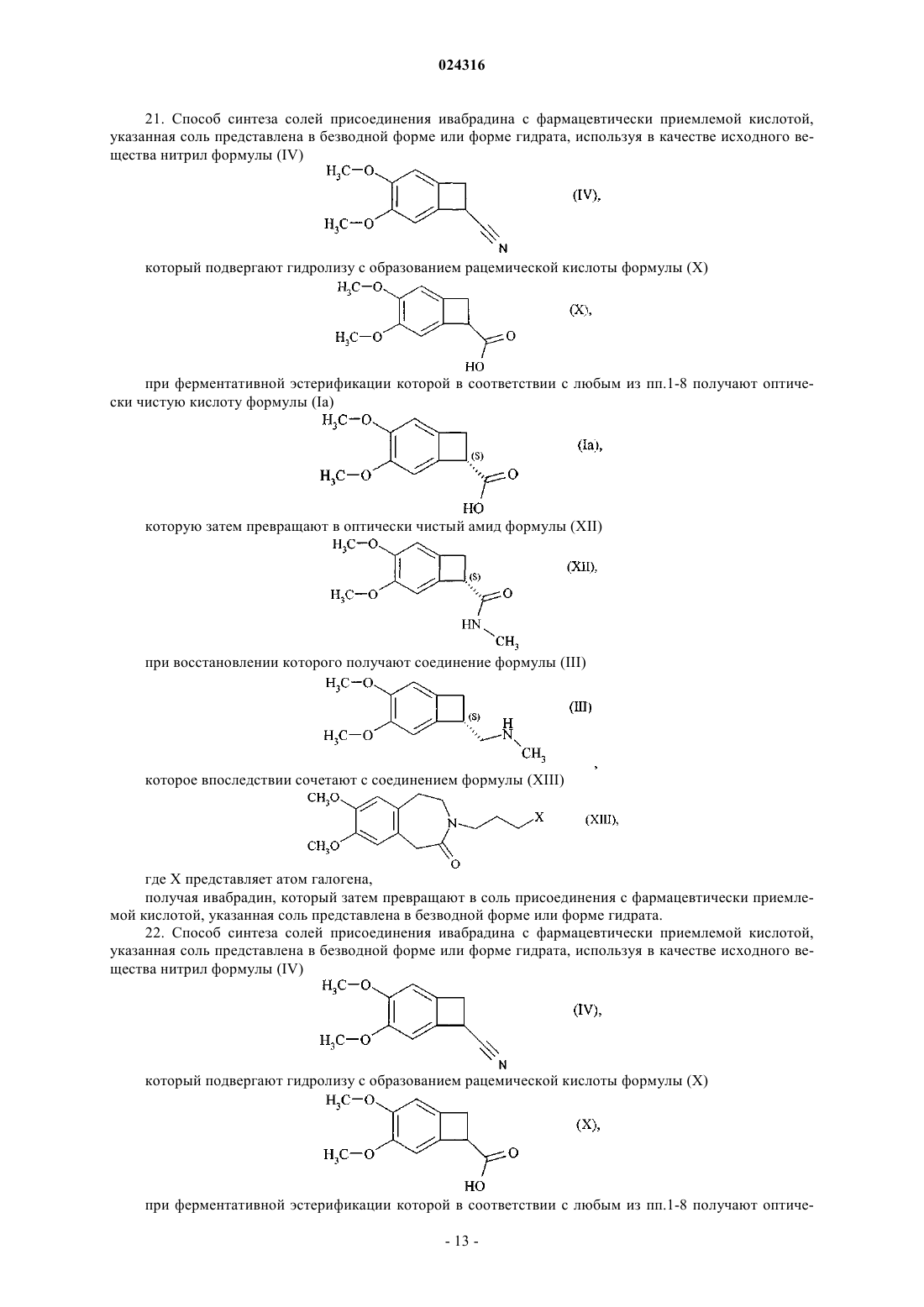
21. Способ синтеза солей присоединения ивабрадина с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата, используя в качестве исходного вещества нитрил формулы (IV)

который подвергают гидролизу с образованием рацемической кислоты формулы (X)

при ферментативной эстерификации которой в соответствии с любым из пп.1-8 получают оптически чистую кислоту формулы (Ia)

которую затем превращают в оптически чистый амид формулы (XII)

при восстановлении которого получают соединение формулы (III)

которое впоследствии сочетают с соединением формулы (XIII)

где X представляет атом галогена,
получая ивабрадин, который затем превращают в соль присоединения с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата.
22. Способ синтеза солей присоединения ивабрадина с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата, используя в качестве исходного вещества нитрил формулы (IV)

который подвергают гидролизу с образованием рацемической кислоты формулы (X)

при ферментативной эстерификации которой в соответствии с любым из пп.1-8 получают оптически чистую кислоту формулы (Ia)

которую затем превращают в оптически чистый амид формулы (XII)

при восстановлении которого получают соединение формулы (III)

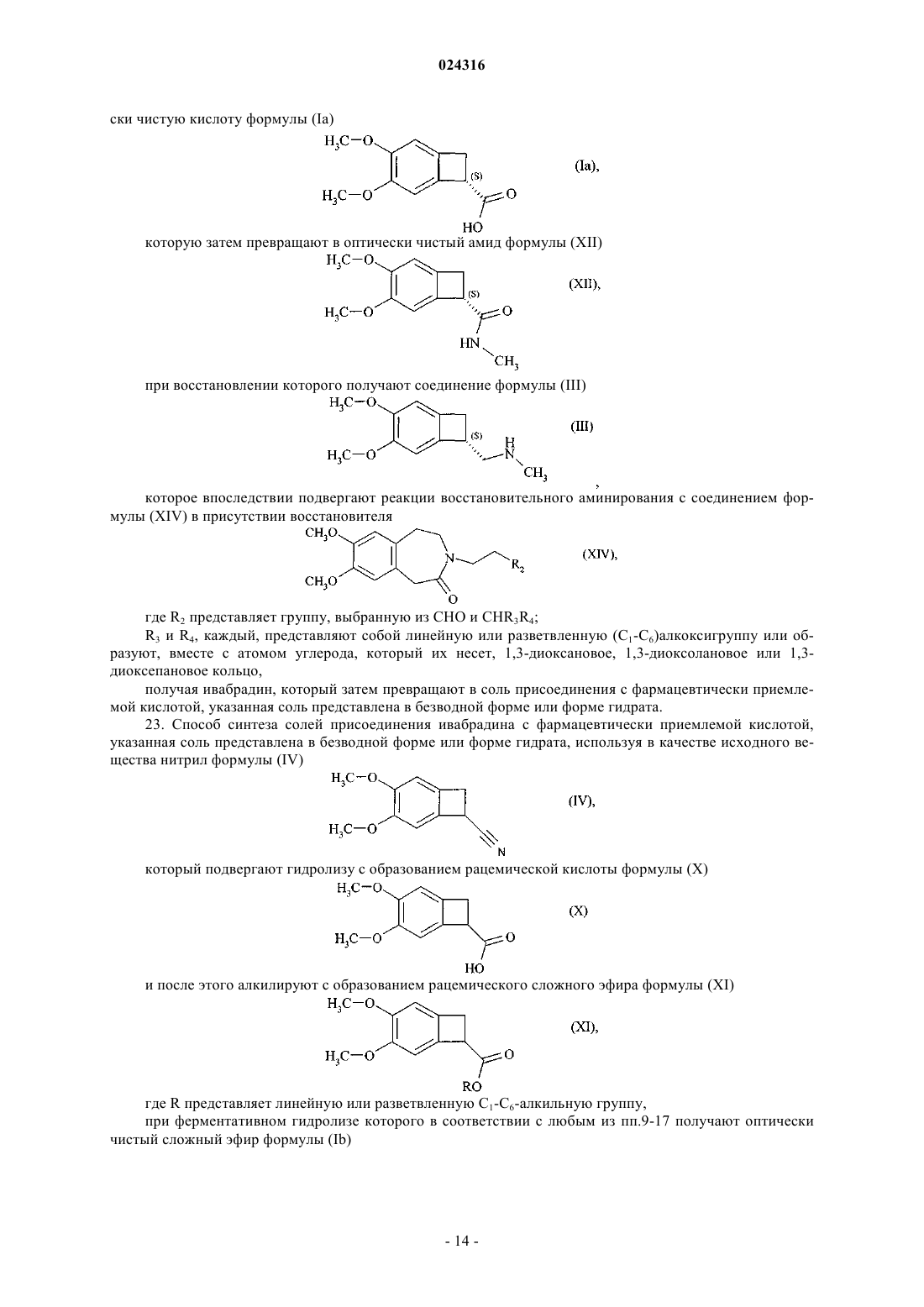
которое впоследствии подвергают реакции восстановительного аминирования с соединением формулы (XIV) в присутствии восстановителя

где R2 представляет группу, выбранную из СНО и CHR3R4;
R3 и R4, каждый, представляют собой линейную или разветвленную (C1-С6)алкоксигруппу или образуют, вместе с атомом углерода, который их несет, 1,3-диоксановое, 1,3-диоксолановое или 1,3-диоксепановое кольцо,
получая ивабрадин, который затем превращают в соль присоединения с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата.
23. Способ синтеза солей присоединения ивабрадина с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата, используя в качестве исходного вещества нитрил формулы (IV)

который подвергают гидролизу с образованием рацемической кислоты формулы (X)

и после этого алкилируют с образованием рацемического сложного эфира формулы (XI)

где R представляет линейную или разветвленную C1-C6-алкильную группу,
при ферментативном гидролизе которого в соответствии с любым из пп.9-17 получают оптически чистый сложный эфир формулы (Ib)

где R представляет линейную или разветвленную C1-C6-алкильную группу,
который превращают в оптически чистый амид формулы (XII)

при восстановлении которого получают соединение формулы (III)

которое впоследствии сочетают с соединением формулы (XIII)

где X представляет атом галогена,
получая ивабрадин, который затем превращают в соль присоединения с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата.
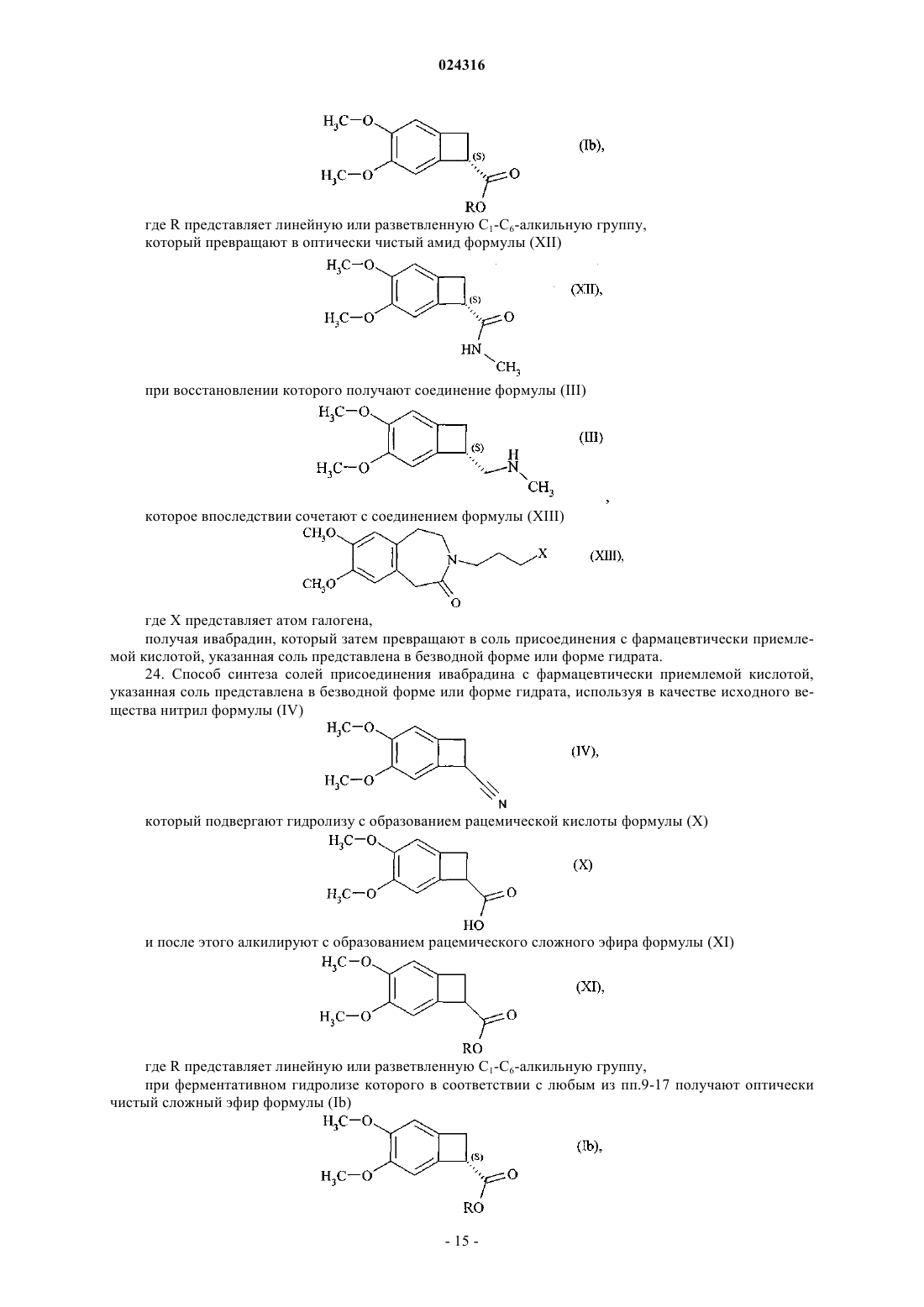
24. Способ синтеза солей присоединения ивабрадина с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата, используя в качестве исходного вещества нитрил формулы (IV)

который подвергают гидролизу с образованием рацемической кислоты формулы (X)

и после этого алкилируют с образованием рацемического сложного эфира формулы (XI)

где R представляет линейную или разветвленную C1-C6-алкильную группу,
при ферментативном гидролизе которого в соответствии с любым из пп.9-17 получают оптически чистый сложный эфир формулы (Ib)

где R представляет линейную или разветвленную C1-C6-алкильную группу,
который превращают в оптически чистый амид формулы (XII)

при восстановлении которого получают соединение формулы (III)

которое впоследствии подвергают реакции восстановительного аминирования с соединением формулы (XIV) в присутствии восстановителя

где R2 представляет группу, выбранную из СНО и CHR3R4;
R3 и R4, каждый, представляют собой линейную или разветвленную (C1-C6)алкоксигруппу или образуют, вместе с атомом углерода, который их несет, 1,3-диоксановое, 1,3-диоксолановое или 1,3-диоксепановое кольцо,
получая ивабрадин, который затем превращают в соль присоединения с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата.
25. Способ синтеза в соответствии с п.21 или 23, где X представляет собой атом йод.
26. Способ синтеза в соответствии с п.22 или 24, где соединение формулы (III) используют в реакции восстановительного аминирования в форме его гидрохлорида, получая ивабрадин в форме гидрохлорида.
27. Способ синтеза в соответствии с пп.22, 24 или 26, где реакцию восстановительного аминирования с соединением формулы (XIV) осуществляют в присутствии диводорода, катализируемого палладием-на-угле.
Текст
СПОСОБ ФЕРМЕНТАТИВНОГО СИНТЕЗА (7S)-3,4 ДИМЕТОКСИБИЦИКЛО[4.2.0]ОКТА-1,3,5-ТРИЕН-7-КАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ СЛОЖНЫХ ЭФИРОВ И ПРИМЕНЕНИЕ ДЛЯ СИНТЕЗА ИВАБРАДИНА И ЕГО СОЛИ Способ ферментативного синтеза соединения формулы (I) где R1 представляет собой атом водорода или алкильную группу. Применение для синтеза ивабрадина и его солей присоединения с фармацевтически приемлемой кислотой.(2), 106-109. Найдено из базы данных Cas on STN(71)(73) Заявитель и патентовладелец: ЛЕ ЛАБОРАТУАР СЕРВЬЕ (FR) Настоящее изобретение относится к способу ферментативного синтеза соединения формулы (I) где R1 представляет собой атом водорода или C1-C6-алкильную группу, предпочтительно метил,а также его применения для синтеза ивабрадина формулы (II) или 3-3-(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил 7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-она, его солей присоединения с фармацевтически приемлемой кислотой и их гидратов. Ивабрадин и его соли присоединения с фармацевтически приемлемой кислотой, и более специфически его гидрохлорид, обладают чрезвычайно ценными фармакологическими и терапевтическими свойствами, в особенности брадикардическими свойствами, которые делают эти соединения пригодными для лечения или предотвращения различных клинических состояний ишемии миокарда, таких как стенокардия, инфаркт миокарда и ассоциированные нарушения ритма, а также различных патологий, связанных с нарушением ритма, в особенности суправентрикулярных нарушений ритма, и сердечной недостаточности. Приготовление и терапевтическое применение ивабрадина и его солей присоединения с фармацевтически приемлемой кислотой, и более специфически его гидрохлорида, было описано в описании к Европейскому патенту ЕР 0534859. В этом патенте описан синтез гидрохлорид ивабрадина, используя в качестве исходного вещества соединение формулы (III), (7S)-1-(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил) N-метил метанамин Соединение формулы (III) является ключевым промежуточным соединением для синтеза ивабрадина и его фармацевтически приемлемых солей. В уровне техники описаны несколько методов получения соединения формулы (III). В описании к патенту ЕР 0534859 описан синтез соединения формулы (III) путем восстановления рацемического нитрила формулы (IV) с помощью ВН 3 в тетрагидрофуране, с последующим добавлением соляной кислоты, получая гидрохлорид рацемического амина формулы (V) который подвергают реакции с этил хлорформиатом, получая карбамат формулы (VI) при восстановлении которого с помощью LiAlH4 получают рацемический метилированный амин формулы (VII)(III). Недостатком этого метода является получение соединения формулы (III) только в очень низком выходе от 2 до 3%, используя в качестве исходного вещества рацемический нитрил формулы (IV). Этот чрезвычайно низкий выход обусловлен низким выходом (4-5%) стадии разделения вторичного амина формулы (VII). В описании к патенту ЕР 1598333 описано получение соединения формулы (III) путем разделения рацемического первичного амина формулы (V) на оптически активный амин формулы (VIII) используя N-ацетил-L-глутаминовую кислоту, с последующим метилированием, используя аналогичную последовательность реакции, что и описана выше (превращение в карбамат, с последующим восстановлением). Выход стадии разделения составляет 39%. В описании к патенту ЕР 2166004 описано получение соединения формулы (III) путем оптического разделения рацемического нитрила формулы (IV) путем хиральной хроматографии, получая оптически чистый нитрил формулы (IX) который восстанавливают с помощью NaBH4, получая первичный амин формулы (VIII), который затем метилируют, используя аналогичную последовательность реакции, что и описана выше (превращение в карбамат, с последующим восстановлением). Выход стадии разделения составляет 45%. Задачей настоящего изобретения является получение соединения формулы (III), используя эффективный процесс, в особенности имеющий хороший выход, более специфически для стадии разделения. Применение биокатализа для предоставления возможности получения хиральных молекул является,по-видимому, чрезвычайно ценным в качестве альтернативы традиционному органическому синтезу. В действительности, применение ферментов, которые имеют собственные природные свойства, такие как хемо-, регио- и стереоселективность, предоставляет возможность использовать ферменты в качестве реагентов для экологически безопасной химии, которая является щадящей для окружающей среды. В случае, описанном в настоящем изобретении, применение гидролитических ферментов (гидролаз), которые функционируют без кофакторов, таких как липазы (ЕС 3.1.1.3 в международной классификации ферментов) или эстеразы (ЕС 3.1.1.1), предоставляет возможность получить хиральные соединения - ключевые промежуточные соединения для синтеза фармацевтически активных ингредиентов - с высоким энантиомерным избытком и хорошим выходом. Более специфически, настоящее изобретение относится к способу синтеза оптически чистого соединения формулы (Ia) путем энантиоселективной ферментативной эстерификации рацемической, или другой оптически нечистой, кислоты формулы (X) используя липазу или эстеразу, в смеси спирта ROH, где R представляет линейную или разветвленную C1-C6-алкильную группу, и органического со-растворителя,-2 024316 при концентрации от 5 до 500 г/л, предпочтительно от 100 г до 200 г соединения формулы (X) на 1 л смеси растворителей, при E/S соотношении от 10/1 до 1/100, предпочтительно от 1/5 до 1/10, при температуре от 25 до 40 С. В качестве липаз и эстераз, которые можно использовать в процессе ферментативной эстерификации в соответствии с настоящим изобретением, могут быть упомянуты, без какого-либо ограничения,липазы Candida antarctica, Pseudomonas fluorescens, Pseudomonas cepacia, Rhizopus oryzae, Aspergillus niger, Mucor javanicus, Aspergillus oryzae и Penicillium camemberti и эстеразы Rhizopus oryzae, Mucor miehei и Rhizopus niveus. Липазы, которые являются предпочтительными в соответствии с изобретением, представляют собой липазы Candida antarctica и Pseudomonas fluorescens. В качестве липаз Candida antarctica могут быть упомянуты, в качестве примера, липазы, иммобилизированные на полимерном матриксе, в особенности на акриловой смоле, такие как Novozym 435 от компании Novozymes или SPRIN absorbed CALB от компании Sprin Technologies, или на полистирольной смоле, такие как SPRIN actiplus CALB, SPRIN acti CALB или SPRIN lipo CALB от компанииSprin Technologies, или на акриловой эпокси смоле, такие как SPRIN epobond CALB от компании SprinTechnologies. Если спирт ROH представляет собой метанол, то со-растворитель представляет собой предпочтительно ацетонитрил. Предпочтительное соотношение ацетонитрил/метанол составляет 8/2 до 9/1. Схема ферментативной эстерификации в соответствии с изобретением является следующей: Благоприятно, сложный эфир конфигурации (R), вторичный продукт реакции, может быть подвержен гидролизу путем воздействия основания, предпочтительно KOH, с образованием рацемической кислоты формулы (X) для повторного использования в процессе ферментативной эстерификации. Если стадию гидролиз/рацемизация осуществляют in situ, то в соответствии с изобретением представляет собой процесс динамического кинетического разрешения (DKR), который предоставляет возможность получить S кислоту формулы (Ia) в ее 98% и с выходом 65%. Кислоту формулы (Ia) предпочтительно выделяют из реакционной смеси после одного или нескольких циклов ферментативной эстерификации. Другой аспект изобретения относится к способу синтеза оптически чистого соединения формулы где R представляет линейную или разветвленную C1-C6-алкильную группу, предпочтительно метил,путем энантиоселективного ферментативного гидролиза рацемического, или другого оптически нечистого, сложного эфира формулы (XI) где R представляет линейную или разветвленную C1-C6-алкильную группу,используя липазу или эстеразу, в буферном растворе с рН 7 или в смеси органического растворителя и буфера с рН 7, при концентрации от 1 до 200 г/л, предпочтительно приблизительно 100 г соединения формулы (XI) на 1 л растворителя или смеси растворителей, при E/S соотношении от 10/1 до 1/100,предпочтительно от 1/5 до 1/10, при температуре от 25 до 40 С, с последующим выделением сложного эфира формулы (Ib). В качестве липаз и эстераз, которые можно использовать в процессе ферментативного гидролиза в соответствии с настоящим изобретением, могут быть упомянуты, без какого-либо ограничения, липазыniveus. Липазы, которые являются предпочтительными в соответствии с этим аспектом изобретения, представляют собой липазы Candida antarctica и Pseudomonas fluorescens. В качестве липаз Candida antarctica могут быть упомянуты, в качестве примера, липазы, иммобилизированные на полимерном матриксе, в особенности на акриловой смоле, такие как Novozym 435 от компании Novozymes или SPRIN adsorbed CALB от компании Sprin Technologies, или на полистирольной смоле, такие как SPRIN actiplus CALB, SPRIN acti CALB или SPRIN lipo CALB от компанииSprin Technologies, или на акриловой эпокси смоле, такие как SPRIN epobond CALB от компании SprinTechnologies. Если реакцию осуществляют в присутствии органического растворителя, то он предпочтительно представляет собой ацетонитрил. Предпочтительное соотношение ацетонитрил/буфер с рН 7 находится в интервале от 8/2 до 9/1. Схема ферментативный гидролиз в соответствии с изобретением является следующей: Благоприятно, кислота конфигурации (R), вторичный продукт реакции, может быть рацемизирована путем воздействия основания, предпочтительно путем воздействия KOH в горячем состоянии, и после этого рацемическая кислота, полученная таким образом, может быть алкилирована с образованием рацемического сложного эфира формулы (XI) для повторного использования в процессе ферментативного гидролиза. Альтернативно, кислота конфигурации (R), вторичный продукт реакции, сначала может быть алкилирована и после этого сложный эфир конфигурации (R), полученный таким образом, может быть рацемизирован путем воздействия основания, предпочтительно путем воздействия DBU в горячем состоянии или KOH при температуре окружающей среды, для повторного использования в процессе ферментативного гидролиза. Если рацемизацию осуществляют в горячем состоянии, то температура предпочтительно составляет от 50 до 80 С. Определения. Под оптически чистым соединением понимают соединение, которое имеет энантиомерный избыток, больше или равный 90%. Под кислотой или сложным эфиром, которая(ый) не является оптически чистой(ым), понимают кислоту или сложный эфир, которая(ый) имеет энантиомерный избыток меньше 90%. Под рацемической кислотой или сложным эфиром понимают кислоту или сложный эфир в форме смеси двух энатиомеров в соотношении от 55:45 до 45:55. Под энантиоселективной эстерификацией рацемеческой, или другой оптически нечистой, кислоты понимают избирательную эстерификацию одного из энатиомеров смеси. Под энантиоселективным гидролизом рацемического, или другого оптически нечистого, сложного эфира понимают избирательный гидролиз одного из энатиомеров смеси. Другой аспект изобретения относится к способу синтеза соединения формулы (III), используя в качестве исходного вещества нитрил формулы (IV), который подвергают гидролизу с образованием рацемической кислоты формулы (X), при ферментативной эстерификации которой в соответствии с изобретением получают оптически чистую кислоту формулы (Ia), которую затем превращают в оптически чистый амид формулы (XII) при разделении которого, предпочтительно с помощью ВН 3, NaBH4 или LiAlH4, получают соединение формулы (III). Другой аспект изобретения относится к способу синтеза соединения формулы (III), используя в качестве исходного вещества нитрил формулы (IV), который подвергают гидролизу с образованием рацемической кислоты формулы (X), и после этого алкилируют с образованием рацемического сложного эфира формулы (XI), при ферментативном гидролизе которого в соответствии с изобретением получают оптически чистый сложный эфир формулы (Ib), который превращают в оптически чистый амид формулы при разделении которого, предпочтительно с помощью BH3, NaBH4 или LiAlH4, получают соединение формулы (III). Соединение формулы (III) впоследствии либо сочетают с соединением формулы (XIII) где X представляет атом галогена, предпочтительно атом йода,или подвергают реакции восстановительного аминирования с соединением формулы (XIV) в присутствии восстановителя где R2 представляет группу, выбранную из СНО и CHR3R4; где R3 и R4, каждый, представляют собой линейную или разветвленную (C1-C6)алкоксигруппу или образуют, вместе с атомом углерода, который их несет, 1,3-диоксановое, 1,3-диоксолановое или 1,3 диоксепановое кольцо,получая ивабрадин, который затем превращают в соль присоединения с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата. Соединение формулы (III) также можно использовать в реакции восстановительного аминирования в форме его соли присоединения с фармацевтически приемлемой кислотой, предпочтительно его гидрохлорида. В этом случае, ивабрадин получают непосредственно в форме гидрохлорида. В качестве фармацевтически приемлемых кислот могут быть упомянуты, без какого-либо ограничения, соляная, бромисто-водородная, серная, фосфорная, уксусная, трифторуксусная, молочная, пировиноградная, малоновая, янтарная, глутаровая, фумаровая, винная, малеиновая, лимонная, аскорбиновая,щавелевая, метансульфоновая, бензолсульфоновая и камфорная кислоты. В качестве восстановителей, которые можно использовать в реакции восстановительного аминирования между соединением формулы (III) и соединением формулы (XIV), могут быть упомянуты, без какого-либо ограничения, соединения доноры гидрада, такие триацетоксиборогидрид натрия или цианоборогидрид натрия, и диводород в присутствии катализатора, такого как палладий, платина, никель, рутений, родий или их соединения, в особенности на подложке или в форме оксидов. Предпочтительным восстановителем для реакции восстановительного аминирования между соединением формулы (III) и соединением формулы (XIV) является диводород, катализируемый палладий на угле. Примеры, представленные ниже, иллюстрируют изобретение. Сокращения:op - оптическая или энантиомерная кислота;E/S соотношение - соотношение фермент/субстрат соотношение (г/г); ЯМР - ядерный магнитный резонанс (спектроскопия); МС - масс-спектрометрия; ТГФ - тетрагидрофуран; ТМС - тетраметилсилан. Пример 1: 3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновая кислота. Суспендировали 3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрил (11 г, 58,1 ммоль) в 1 н. растворе гидроксида натрия (70 мл) и реакционную смесь нагревали в колбе с обратным холодильником (110 С) в течение 2 ч. Позволяли возвратиться до температуры окружающей среды и после этого смесь подкисляли, используя концентрированную соляную кислоту. Наблюдали осаждение. Растворяли продукт в 200 мл дихлорметана и после этого водную фазу экстрагировали. Высушивали над MgSO4 и упаривали, получая указанный в заглавии продукт (11,6 г) с выходом 95,9%. Пример 2: (7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновая кислота. 0,5 г (с = 200 г/л) рацемической кислоты, полученной в примере 1, растворяли в 2,5 мл 8/2 смеси ацетонитрил/метанол. После этого к смеси добавляли 0,1 г (с = 40 г/л) липазы Candida antarctica NOVOZYM 435 (Novozymes Denmark) (E/S соотношение 1/5). Реакционную смесь поддерживали при 30 С, с ротационным перемешиванием при 220 об/мин, в течение 48 ч. За осуществлением реакции наблюдали с помощью ВЭЖХ с хиральной фазой в условиях, предоставляющих возможность определения энантиомерных избытков как сложного эфира, так и кислоты: Хроматограммы ВЭЖХ с хиральной фазой рацемических соединений и продукта через 48 ч представлены на фиг. 1 и 2. Через 48 ч наблюдали присутствие оптически чистых сложного эфира и кислоты в оптимальном соотношении кислота/сложный эфир, близком к 50/50. Реакционную смесь фильтровали, фермент промывали с помощью 5 мл метанола и после этого фильтрат упаривали в вакууме. Оптически чистые S кислоту и R сложный эфир разделяли путем хроматографии на колонка диоксида кремния (элюант: дихлорметан/метанол 98/1).(ее 96%) в изопропаноле (2,5 мл) и добавляли диазабициклоундецен (58 мкл - 1,5 экв.). Реакционную смесь нагревали при 65 С в течение 2 ч. Наблюдали полную рацемизацию по истечении 2 ч реакции сложного эфира. Условия анализа:(ее 96%) в метаноле (1 мл) и добавляли гидроксид калия (56,1) (25 мг - 2 экв.). Реакционную смесь нагревали при 65 С в течение 6 ч. Наблюдали гидролиз сложного эфира до рацемической кислоты. Условия анализа:Chiralpak IC 2504,6 колонка; 30% абсолютный этанол + 0,1% TFA + 70% гептан + 0,1% TFA; 1 мл/мин, 25 С, 288 нм. Пример 5: (7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновая кислота. 2 г (с = 200 г/л) рацемической 3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновой кислоты растворяли в 20 мл смеси ацетонитрил/метанол (9/1). После этого к смеси добавляли 0,4 г (с = 20 г/л) липазы Candida antarctica SPRIN actiplus CALB (Sprin Technologies). Реакционную смесь поддерживали при 30 С, с ротационным перемешиванием при 220 об/мин в течение 24 ч. Фермент отфильтровали и после этого промывали метанолом, после этого к смеси добавляли 0,5 г KOH (2 экв.) (фильтрат) и продолжали перемешивали при 6 ч при 30 С. После этого смесь упаривали в вакууме. Это предоставляет возможность завершить рацемизацию и гидролиз R сложного эфира без рацемизации S кислоты. Остаток ресуспендировали в этилацетате и после этого промывали с помощью 10% раствора лимонной кислоты. Экстракция с помощью этилацетата не является достаточной, растворимую в воде кислоту повторно экстрагировали с помощью раствора бутан-1-ола. Экстракты высушивали над MgSO4, получая, после упаривания, 1,9 г кислоты, имеющей соотношение 75:25 (S:R). Эту энантиомерно обогащенную кислоту использовали во второй ферментативной реакции в присутствии 0,2 г липаза. Через 24 ч при 30 С фермент отфильтровали и после этого промывали метанолом. После упаривания остаток хроматографировали на колонке диоксида кремния (элюант CH2Cl2/МеОН от 99/1 до 99/2), получая следующие продукты:(R) сложный эфир: 0,42 г; ор 96%; выход сложного эфира (теоретически 25%): 21%; Суммарный выход реакции составляет 88%. ЯМР и МС характеристика кислоты. 1 Н ЯМР (ДМСО-d6, част. на млн/ТМС): 3,17 (dd; 1H; 13,6 Гц; 2,4 Гц); 3,27 (dd; 1H; 13,6 Гц; 5,3 Гц); 4,13 (dd; 1H); 3,71 (s;3H); 6,78 (s; 1 Н); 6,80 (s; 1H); 12,40 (s; 1H).MC (EI+) Молекулярный ион М+ при m/z 208. ЯМР и МС характеристика сложного эфира. 1 Н ЯМР (ДМСО-d6, част. на млн/ТМС) = 3,19 (dd; 1H; 13,6 Гц; 2,4 Гц); 3,33 (dd; 1H; 13,6 Гц; 5,5 Гц); 3,65 (s; 3H); 3,71 (s; 6H); 4,23 (dd; 1H); 6,79 (s; 1H); 6,82 (s;1H). МС (EI+) Молекулярный ион М+ при m/z 222. За последовательностью реакций наблюдали с помощью ВЭЖХ с хиральной фазой в условиях, предоставляющих возможность определения энантиомерных избытков как сложного эфира, так и кислоты:Chiralpak IC 2504,6 колонка; 30% абсолютный этанол + 0,1% TFA + 70% гептан + 0,1% TFA; 1 мл/мин, 25 С, 288 нм. Пример 6: метил 3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоксилат. Растворяли 3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновую кислоту (3 г -14,4 ммоль) в метаноле и добавляли ацетил хлорид (1,65 г - 21,1 ммоль). Нагревали в колбе с обратным холодильником реакционную смесь в течение 2 ч. Анализ с помощью ТСХ (элюант: дихлорметан) показал отсутствие исходного материала - рацемической кислоты. Упаривали реакционную смесь, ресуспендировали остаток в этилацетате и органическую фазу высушивали с помощью NaHCO3. Упаривали насухо и высушивали, получая указанный в заглавии продукт с выходом 97%. Пример 7: метил (7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоксилат. 2 г (с = 100 г/л) рацемического метил 3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоксилата растворяли в 20 мл 0/20 смеси ацетонитрил/буфер с рН 7. После этого к смеси добавляли 0,4 г (с = 20 г/л) липазы Candida antarctica NOVOZYM435 (Novozymes Denmark) (E/S соотношение 1/5). Реакционную смесь поддерживали при 30 С, с ротационным перемешиванием при 230 об/мин. После осуществления реакции в течение 4 ч (рН 5,8) значение рН доводили до 7,2. Через 24 ч фермент отфильтровали и промывали путем перемешивания в метаноле. Все фильтраты собирали, упаривали и лиофилизировали. Лиофилизат ресуспендировали в этилацетате, перемешивали в течение ночи и после этого реакционную смесь фильтровали и фильтрат упаривали. Остаток очищали на колонке с диоксидом кремния (элюант дихлорметан/метанол), получая 0,81 г сложного эфира указанного в заглавии (7S), т.е. с выходом 41%.[]20D при 589 нм: -58,8 (5 мг/мл в МеОН). Пример 8: метил (7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоксилат. Рацемический метил 3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоксилат (1 мг; с = 1 г/л) растворяли в 1 мл 90/10 смеси фосфатный буфер с рН 7/толуол. После этого к смеси добавляли 5 мг (с = 5 г/л) липазы Pseudomonas fluorescens (E/S соотношение 5/1). Реакционную смесь поддерживали при 28 С, с ротационным перемешиванием при 220 об/мин, в течение 48 ч. Реакционную смесь анализировали путем ВЭЖХ с обращенной фазой и за энантиоселективностью(ее) остаточного сложного эфира наблюдали с помощью ВЭЖХ с хиральной фазой, в соответствии с методами, описанными ниже. Условия для анализа реакционной смеси с помощью ВЭЖХ с обращенной фазой:A (1000 вода+25 ACN+1 TFA). В (1000 ACN+25 вода+1 TFA). Условия для анализа энантиоселективности с помощью ВЭЖХ с хиральной фазой: Анализ реакционной смеси показал хорошую гидролитическую активность (процент остаточного сложного эфира: 25%). Анализ энантиоселективности показал ее 90% для сложного эфира (7S). Пример 9: 3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновая кислота. Суспендировали (7R)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновую кислоту (50 мг ее 95%) в метаноле (1 мл) и добавляли гидроксид калия (20 мг). Реакционную смесь нагревали при 65 С в течение 24 ч. Наблюдали полноценную рацемизацию кислоты. Условия анализа:Chiralpak IC 2504,6 колонка; 30% абсолютный этанол + 0,1% TFA + 70% гептан + 0,1% TFA; 1 мл/мин, 25 С, 288 нм. Пример 10: (7S)-3,4-диметокси-N-метилбицикло[4.2.0]окта-1,3,5-триен-7-карбоксамид. Суспендировали (7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновую кислоту, полученную в примере 5 (300 мг), в ТГФ (3 мл) при температуре окружающей среды и после этого добавляли триэтиламин (200 мкл). К смеси медленно добавляли этил хлорформиат (150 мкл). Реакционная смесь осаждалась (смесь I). В другой колбе метиламин, в виде 2 М раствора в ТГФ (2,25 мл), перемешивали с водой (1 мл) и триэтиламином (300 мкл). Продолжали перемешивать 20 мин и после этого полученную смесь добавляли к смеси I и перемешивали при температуре окружающей среды в течение ночи. После этого реакционную смесь упаривали и очищали путем препаративной ВЭЖХ. Получали (7S)3,4-диметокси-N-метилбицикло[4.2.0]окта-1,3,5-триен-7-карбоксамид с выходом 60%. 1 Н ЯМР (ДМСО-d6, част. на млн/ТМС) = 2,61 (m; 3H); 3,16 (m; 2 Н); 3,71 (s; 6H); 4,05 (m; 1 Н); 6,78(s; 1 Н); 6,81 (s; 1 Н); 7,78 (s; 1H). Пример 11: (7S)-3,4-диметокси-N-метилбицикло[4.2.0]окта-1,3,5-триен-7-карбоксамид. Суспендировали метил (7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоксилат (500 мг) в воде и затем медленно добавляли, при температуре окружающей среды, 20 мл 33% раствора метиламина в абсолютном этаноле. После перемешивания в течение 3 ч реакционную смесь упаривали. Полученный остаток очищали путем препаративной ВЭЖХ (элюант: вода/ацетонитрил/трифторуксусная кислота от 98/2/0,2 до 20/80/0,2) в течение 30 мин, получая указанный в заглавии продукт с выходом 70%. Пример 12: (7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]-N-метилметанамин. Суспендировали (7S)-3,4-диметокси-N-метилбицикло[4.2.0]окта-1,3,5-триен-7-карбоксамид (450 мг) в тетрагидрофуране (20 мл) и затем к реакционной смеси медленно добавляли 1,6 мл 2 М раствораLiAlH4 в тетрагидрофуране при температуре окружающей среды. Наблюдали заметное выделение газа и реакционная смесь становилась прозрачной. Реакционную смесь нагревали в колбе с обратным холодильником в течение 30 мин. После возвращения до температуры окружающей среды, гидролизировали и после этого экстрагировали с помощью этилацетата. Высушивали над MgSO4 и после этого упаривали. Полученный остаток очищали путем препаративной ВЭЖХ (элюант: вода/ацетонитрил/трифторуксусная кислота от 98/2/0,2 до 20/80/0,2) в течение 30 мин получая указанный в заглавии продукт с выходом 46%. 1 Н ЯМР (ДМСО-d6, част. на млн/ТМС) = 2,60 (m; 3H); 2,85 (m; 1 Н); 3,15 (m; 1H); 3,25 (dd; 1 Н); 3,30(m; 1 Н); 3,62 (m; 1 Н); 3,70 (s; 6H); 6,82 (s; 1H); 6,89 (s; 1H); 8,48 (s; 1H). Пример 13: (7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]-N-метилметанамин гидрохлорид. 20 мл молярного раствора BH3 в тетрагидрофуране добавляли, при температуре окружающей среды, к смеси 2,2 г (10 ммоль) (7S)-3,4-диметокси-N-метилбицикло[4.2.0]окта-1,3,5-триен-7-карбоксамида в 45 мл тетрагидрофурана. После перемешивания в течение 1 ч добавляли 10 мл раствора BH3 в тетрагидрофуране. После перемешивания в течение ночи при температуре окружающей среды, по каплям добавляли 20 мл этанола и смесь перемешивали до тех пор, пока не прекращалось выделение газа (около 1 ч). После этого по каплям добавляли 20 мл раствора соляной кислоты в этаноле. После перемешивания в течение 4 ч полученный осадок (1,2 г указанного в заглавии продукта) отфильтровали. Фильтрат концентрировали и дополнительно 0,65 г указанного в заглавии продукта получали путем перевода его в твердое вещество в смеси 80/20 этилацетат/этанол. Два осадка объединяли, получая 1,85 г указанного в заглавии продукта (выход: 77%). Пример 14: гидрохлорид ивабрадина. Загружали 5,5 кг 3-[2-(1,3-диоксолан-2-ил)этил]-7,8-диметокси-1,3-дигидро-2H-3-бензазепин-2-она,27,5 л этанола и 550 г палладия на угле в автоклав. Продували азотом и после этого водородом, нагревали до 55 С, и после этого гидрировали при этой температуре под давлением 5 бар до тех пор, пока не абсорбировалось теоретическое количество водорода. Затем возвращали до температуры окружающей среды и автоклав разгерметизировали. После этого добавляли 4 кг гидрохлорида (7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]N-метилметанамина, 11 л этанола, 5,5 л воды и 1 кг палладия на угле. Продували азотом и после этого водородом, нагревали до 85 С, и после этого гидрировали при этой температуре под давлением 30 бар до тех пор, пока не абсорбировалось теоретическое количество водорода. После этого возвращали обратно до температуры окружающей среды, автоклав продували и после этого реакционную смесь фильтровали; растворители отгоняли и после этого гидрохлорид ивабрадина выделяли путем кристаллизации из смеси толуол/1-метил-2-пирролидинон. Таким образом получали гидрохлорид ивабрадина с выходом 85% и с химической чистотой больше 99%. Сравнительный пример: скрининг относительно липаз эстераз для ферментативного гидролиза метил 3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоксилата. Рацемический метил 3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоксилат (1 мг; с = 1 г/л) растворяли в 1 мл 90/10 смесь фосфатный буфер с рН 7/толуол. После этого к среде добавляли 5 мг (с = 5 г/л) исследуемой липазы или эстеразы (E/S соотношение 5/1). Реакционную смесь поддерживали при 28 С, с ротационным перемешиванием при 220 об/мин в течение 48 ч. Реакционную смесь анализировали путем ВЭЖХ с обращенной фазой и энантиоселективность (ее) остаточного сложного эфира наблюдали с помощью ВЭЖХ с хиральной фазой, в соответствии с методами, описанными ниже. Условия для анализа реакционной смеси с помощью ВЭЖХ с обращенной фазой:A (1000 вода+25 ACN+1 TFA). В (1000 ACN+25 вода+1 TFA). Условия для анализа энантиоселективности с помощью ВЭЖХ с хиральной фазой: Энантиомерный избыток ее (en%) = % энантиоЕ 2 - % энантиоЕ 1 / % энантио Е 2 + % энантио Е 1 (энантио Е 2 представляет собой преобладающий энатиомер).ee(S)]; с = уровень превращения = ее(сложный эфир) / ее(сложный эфир) + ее(кислота). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ синтеза оптически чистого соединения формулы (Ia) путем энантиоселективной ферментативной эстерификации рацемической или другой, оптически нечистой, кислоты формулы (X) используя липазу Candida antarctica или Pseudomonas fluorescens, в смеси спирта ROH, где R представляет линейную или разветвленную C1-С 6-алкильную группу, и органического сорастворителя, при концентрации от 5 до 500 г/л соединения формулы (X) на 1 л смеси растворителей, при E/S соотношении от 10/1 до 1/100, при температуре от 25 до 40 С. 2. Способ синтеза в соответствии с п.1, где E/S соотношение составляет от 1/5 до 1/10. 3. Способ синтеза в соответствии с любым из пп.1 или 2, где спирт ROH представляет собой метанол и со-растворитель представляет собой ацетонитрил. 4. Способ синтеза в соответствии с п.3, где соотношение ацетонитрил/метанол составляет от 8/2 до 9/1. 5. Способ синтеза в соответствии с любым из пп.1-3, где сложный эфир конфигурации (R), вторичный продукт реакции подвергают гидролизу путем воздействия основания с образованием рацемической кислоты формулы (X) для повторного использования в процессе ферментативной эстерификации. 6. Способ синтеза в соответствии с п.5, где основание представляет собой KOH. 7. Способ синтеза в соответствии с п.5, где стадию гидролиз/рацемизация осуществляют in situ. 8. Способ синтеза в соответствии с любым из пп.1-3, где кислоту формулы (Ia) выделяют после одного или нескольких циклов ферментативной эстерификации. 9. Способ синтеза оптически чистого соединения формулы (Ib) где R представляет линейную или разветвленную C1-C6-алкильную группу,путем энантиоселективного ферментативного гидролиза рацемического или другого, оптически нечистого, сложного эфира формулы (XI) где R представляет линейную или разветвленную C1-C6-алкильную группу,используя липазу Candida antarctica или Pseudomonas fluorescens, в воде в буферном растворе с рН 7 или в смеси органического растворителя и воде или буфера с рН 7, при концентрации от 1 до 200 г/л соединения формулы (XI) на 1 л растворителя или смеси растворителей, при E/S соотношении от 10/1 до 1/100, при температуре от 25 до 40 С, с последующим выделением сложного эфира формулы (Ib). 10. Способ синтеза в соответствии с п.9, где E/S соотношение составляет от 1/5 до 1/10. 11. Способ синтеза в соответствии с любым из пп.9 или 10, где R представляет собой метильную группу. 12. Способ синтеза в соответствии с любым из пп.9 или 10, где реакцию осуществляют в смеси ацетонитрила и буфера с рН 7. 13. Способ синтеза в соответствии с п.12, где соотношение ацетонитрил/буфер с рН 7 составляет от 8/2 до 9/1. 14. Способ синтеза в соответствии с любым из пп.9 или 10, где кислоту конфигурации (R) вторичный продукт реакции, рацемизируют путем воздействия основания и после этого рацемическую кислоту, полученную таким образом, алкилируют с образованием рацемического сложного эфира формулы (XI) для повторного использования в процессе ферментативного гидролиза. 15. Способ синтеза в соответствии с п.14, где кислоту конфигурации (R) рацемизируют путем воздействия KOH в горячем состоянии. 16. Способ синтеза в соответствии с любым из пп.9 или 10, где кислоту конфигурации (R) вторичный продукт реакции, сначала алкилируют и после этого сложный эфир конфигурации (R),полученный таким образом, рацемизируют путем воздействия основания для повторного использования в процессе ферментативного гидролиза. 17. Способ синтеза в соответствии с п.16, где сложный эфир конфигурации (R) рацемизируют путем воздействия DBU в горячем состоянии или KOH при температуре окружающей среды. 18. Способ синтеза соединения формулы (III) используя в качестве исходного вещества нитрил формулы (IV) который подвергают гидролизу с образованием рацемической кислоты формулы (X) при ферментативной эстерификации которой в соответствии с любым из пп.1-8 получают оптиче- 11024316 ски чистую кислоту формулы (Ia) которую затем превращают в оптически чистый амид формулы (XII) при восстановлении которого получают соединение формулы (III). 19. Способ синтеза соединения формулы (III) используя в качестве исходного вещества нитрил формулы (IV) который подвергают гидролизу с образованием рацемической кислоты формулы (X) и после этого алкилируют с образованием рацемического сложного эфира формулы (XI) где R представляет линейную или разветвленную C1-C6-алкильную группу,при ферментативном гидролизе которого в соответствии с любым из пп.9-17 получают оптически чистый сложный эфир формулы (Ib) где R представляет линейную или разветвленную C1-C6-алкильную группу,который превращают в оптически чистый амид формулы (XII) при восстановлении которого получают соединение формулы (III). 20. Способ синтеза в соответствии с п.18 или 19, где восстановление соединения формулы (XII) с образованием соединения формулы (III) осуществляют с помощью BH3, NaBH4 или LiAlH4. 21. Способ синтеза солей присоединения ивабрадина с фармацевтически приемлемой кислотой,указанная соль представлена в безводной форме или форме гидрата, используя в качестве исходного вещества нитрил формулы (IV) который подвергают гидролизу с образованием рацемической кислоты формулы (X) при ферментативной эстерификации которой в соответствии с любым из пп.1-8 получают оптически чистую кислоту формулы (Ia) которую затем превращают в оптически чистый амид формулы (XII) при восстановлении которого получают соединение формулы (III) которое впоследствии сочетают с соединением формулы (XIII) где X представляет атом галогена,получая ивабрадин, который затем превращают в соль присоединения с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата. 22. Способ синтеза солей присоединения ивабрадина с фармацевтически приемлемой кислотой,указанная соль представлена в безводной форме или форме гидрата, используя в качестве исходного вещества нитрил формулы (IV) который подвергают гидролизу с образованием рацемической кислоты формулы (X) при ферментативной эстерификации которой в соответствии с любым из пп.1-8 получают оптиче- 13024316 ски чистую кислоту формулы (Ia) которую затем превращают в оптически чистый амид формулы (XII) при восстановлении которого получают соединение формулы (III) которое впоследствии подвергают реакции восстановительного аминирования с соединением формулы (XIV) в присутствии восстановителя где R2 представляет группу, выбранную из СНО и CHR3R4;R3 и R4, каждый, представляют собой линейную или разветвленную (C1-С 6)алкоксигруппу или образуют, вместе с атомом углерода, который их несет, 1,3-диоксановое, 1,3-диоксолановое или 1,3 диоксепановое кольцо,получая ивабрадин, который затем превращают в соль присоединения с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата. 23. Способ синтеза солей присоединения ивабрадина с фармацевтически приемлемой кислотой,указанная соль представлена в безводной форме или форме гидрата, используя в качестве исходного вещества нитрил формулы (IV) который подвергают гидролизу с образованием рацемической кислоты формулы (X) и после этого алкилируют с образованием рацемического сложного эфира формулы (XI) где R представляет линейную или разветвленную C1-C6-алкильную группу,при ферментативном гидролизе которого в соответствии с любым из пп.9-17 получают оптически чистый сложный эфир формулы (Ib) где R представляет линейную или разветвленную C1-C6-алкильную группу,который превращают в оптически чистый амид формулы (XII) при восстановлении которого получают соединение формулы (III) которое впоследствии сочетают с соединением формулы (XIII) где X представляет атом галогена,получая ивабрадин, который затем превращают в соль присоединения с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата. 24. Способ синтеза солей присоединения ивабрадина с фармацевтически приемлемой кислотой,указанная соль представлена в безводной форме или форме гидрата, используя в качестве исходного вещества нитрил формулы (IV) который подвергают гидролизу с образованием рацемической кислоты формулы (X) и после этого алкилируют с образованием рацемического сложного эфира формулы (XI) где R представляет линейную или разветвленную C1-C6-алкильную группу,при ферментативном гидролизе которого в соответствии с любым из пп.9-17 получают оптически чистый сложный эфир формулы (Ib) где R представляет линейную или разветвленную C1-C6-алкильную группу,который превращают в оптически чистый амид формулы (XII) при восстановлении которого получают соединение формулы (III) которое впоследствии подвергают реакции восстановительного аминирования с соединением формулы (XIV) в присутствии восстановителя где R2 представляет группу, выбранную из СНО и CHR3R4;R3 и R4, каждый, представляют собой линейную или разветвленную (C1-C6)алкоксигруппу или образуют, вместе с атомом углерода, который их несет, 1,3-диоксановое, 1,3-диоксолановое или 1,3 диоксепановое кольцо,получая ивабрадин, который затем превращают в соль присоединения с фармацевтически приемлемой кислотой, указанная соль представлена в безводной форме или форме гидрата. 25. Способ синтеза в соответствии с п.21 или 23, где X представляет собой атом йод. 26. Способ синтеза в соответствии с п.22 или 24, где соединение формулы (III) используют в реакции восстановительного аминирования в форме его гидрохлорида, получая ивабрадин в форме гидрохлорида. 27. Способ синтеза в соответствии с пп.22, 24 или 26, где реакцию восстановительного аминирования с соединением формулы (XIV) осуществляют в присутствии диводорода, катализируемого палладием-на-угле. Фиг. 1 Ферментативная эстерификация через 48 ч
МПК / Метки
МПК: C07C 51/353, C12P 7/40, C07C 62/34, C07C 69/757, C07C 67/333, C07C 213/02, C12P 7/62, C07C 51/42, C12P 7/42, C07B 57/00, C07C 217/74
Метки: применение, соли, 7s)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновой, эфиров, сложных, ивабрадина, синтеза, способ, кислоты, ферментативного
Код ссылки
<a href="https://eas.patents.su/18-24316-sposob-fermentativnogo-sinteza-7s-34-dimetoksibiciklo420okta-135-trien-7-karbonovojj-kisloty-ili-ee-slozhnyh-efirov-i-primenenie-dlya-sinteza-ivabradina-i-ego-soli.html" rel="bookmark" title="База патентов Евразийского Союза">Способ ферментативного синтеза (7s)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновой кислоты или ее сложных эфиров и применение для синтеза ивабрадина и его соли</a>
Способ ферментативного синтеза (7s)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновой кислоты и применение в синтезе ивабрадина и его солей

Номер патента: 23883
Опубликовано: 29.07.2016
Авторы: Лефулон Франсуа, Педрагоза-Моро Сандрин
МПК: C07B 57/00, C07C 213/02, C07C 217/56...
Метки: применение, синтеза, кислоты, ферментативного, ивабрадина, 7s)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновой, способ, солей, синтезе
Формула / Реферат:
1. Способ синтеза оптически чистого соединения формулы (I)путем энантиоселективного ферментативного гидролиза рацемического, или не оптически чистого, нитрила формулы (IV)с применением нитрилазы Rhodococcus rhodochrous NCIMB 11216, которая сверхэкспрессируется в другом организме, имеющем компетентную биологическую систему, в смеси органического растворителя и водного раствора, имеющего рН от 5 до 10, при концентрации от 1 до 500 г нитрила...
Новый способ синтеза (2s, 3аs, 7аs )-пергидроиндол-2-карбоновой кислоты и её сложных эфиров, а также их применение для синтеза периндоприла

Номер патента: 7948
Опубликовано: 27.02.2007
Авторы: Дюбюффе Тьерри, Ланглуа Паскаль
МПК: C07D 209/42
Метки: периндоприла, применение, новый, кислоты, эфиров, сложных, также, 3аs, способ, пергидроиндол-2-карбоновой, синтеза
Формула / Реферат:
1. Способ синтеза соединений формулы (I) в которой R представляет собой атом водорода или защитную группу для кислотной группы, характеризующийся тем, что 1-(1-циклогексен-1-ил)пирролидин формулы (III) подвергают реакции с соединением формулы (IV) в которой R является таким, как определено для формулы (I), a R' представляет собой защитную группу для аминовой группы, которая является отличной от R, с получением соединения формулы (V) в...
Способ разделения энантиомеров (3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)нитрила и применение при синтезе ивабрадина

Номер патента: 15777
Опубликовано: 30.12.2011
Авторы: Фан Маризе, Гожон Эрик, Лерестиф Жан-Мишель, Дрон Даньель, Лекув Жан-Пьерр
МПК: C07B 57/00, C07D 223/16, C07C 255/47...
Метки: синтезе, 3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)нитрила, применение, энантиомеров, разделения, ивабрадина, способ
Формула / Реферат:
1. Способ разделения оптических изомеров (3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)нитрила формулы (I), где рацемически или энантиомерно обогащенную смесь (3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)нитрила разделяют на два его энантиомера (S)-(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)нитрил формулы (Ia) и (R)-(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)нитрил формулы (Ib) путем хиральной хроматографии.2. Способ по...
Соли карбоновой и серной кислот сложных эфиров 3-(2,2,2-триметилгидразиниум) пропионата и их использование для синтеза дигидрата 3-(2,2,2-триметилгидразиниум) пропионата

Номер патента: 16937
Опубликовано: 30.08.2012
Авторы: Лебедев Антон, Чернобровый Александр, Левина Агния, Пугович Освальд, Калвиньш Иварс
МПК: C07C 243/40, C07C 241/02
Метки: соли, дигидрата, кислот, использование, карбоновой, пропионата, эфиров, серной, синтеза, сложных, 3-(2,2,2-триметилгидразиниум
Формула / Реферат:
1. Соединение формулы (I)где n является целым числом 1 или 2;R представляет собой СН3, С2Н5, С3Н7, i-C3H7, C4H9, i-C4H9 и С6Н5СН2; аX представляет собой HCO3-, CO32-, HSO42- или SO42- или его моногидрат.2. Соединение по п.1, которое представляет собой гидрокарбонат метил-3-(2,2,2-триметилгидразиниум) пропионата.3. Соединение по п.1, которое представляет собой моногидрат гидрокарбоната метил-3-(2,2,2-триметилгидразиниум) пропионата.4. Соединение...
Новый способ синтеза сложных эфиров n-[(s)-1-карбоксибутил]-(s)-аланина и их применение для синтеза периндоприла

Номер патента: 9980
Опубликовано: 28.04.2008
Авторы: Бреар Фабьенн, Фуже Клод
МПК: C07C 229/12, C07C 227/32, C07C 229/16...
Метки: периндоприла, сложных, способ, новый, эфиров, применение, n-[(s)-1-карбоксибутил]-(s)-аланина, синтеза
Формула / Реферат:
1. Способ синтеза соединений формулы (I) в которой R представляет собой линейную или разветвленную С1-С6-алкильную группу, который характеризуется тем, что морфолинон формулы (III) в которой P представляет собой защитную группу для функциональной аминогруппы, подвергают реакции с аллилбромидом или аллилтрифлатом в присутствии основания, получая соединение формулы (IV), имеющее (3S,5S)-конфигурацию в которой P имеет значения, указанные выше,...
Предыдущий патент: Светодиодное осветительное устройство и устройство уличного освещения, содержащее светодиодное осветительное устройство
Следующий патент: Способ и система для динамического перераспределения спектральных ресурсов в беспроводной системе связи
Случайный патент: Способ извлечения углеводородов из резервуаров для хранения