Способ получения транс-1-((1r,3s)-6-хлор-3-фенилиндан-1-ил)-3,3-диметилпиперазина
Номер патента: 18059
Опубликовано: 30.05.2013
Авторы: Сюте Кристина, Робен Давид, Вехльк Нильсен Христина, Даль Аллан Карстен, Бресен Петер















Формула / Реферат
1. Способ получения соединения формулы I (соединение I) или его соли, включающий
получение соединения IVa разделением рацемического соединения IV с использованием хиральной хроматографии

превращение соединения формулы IVa в соответствующий спирт Va с цис-конфигурацией

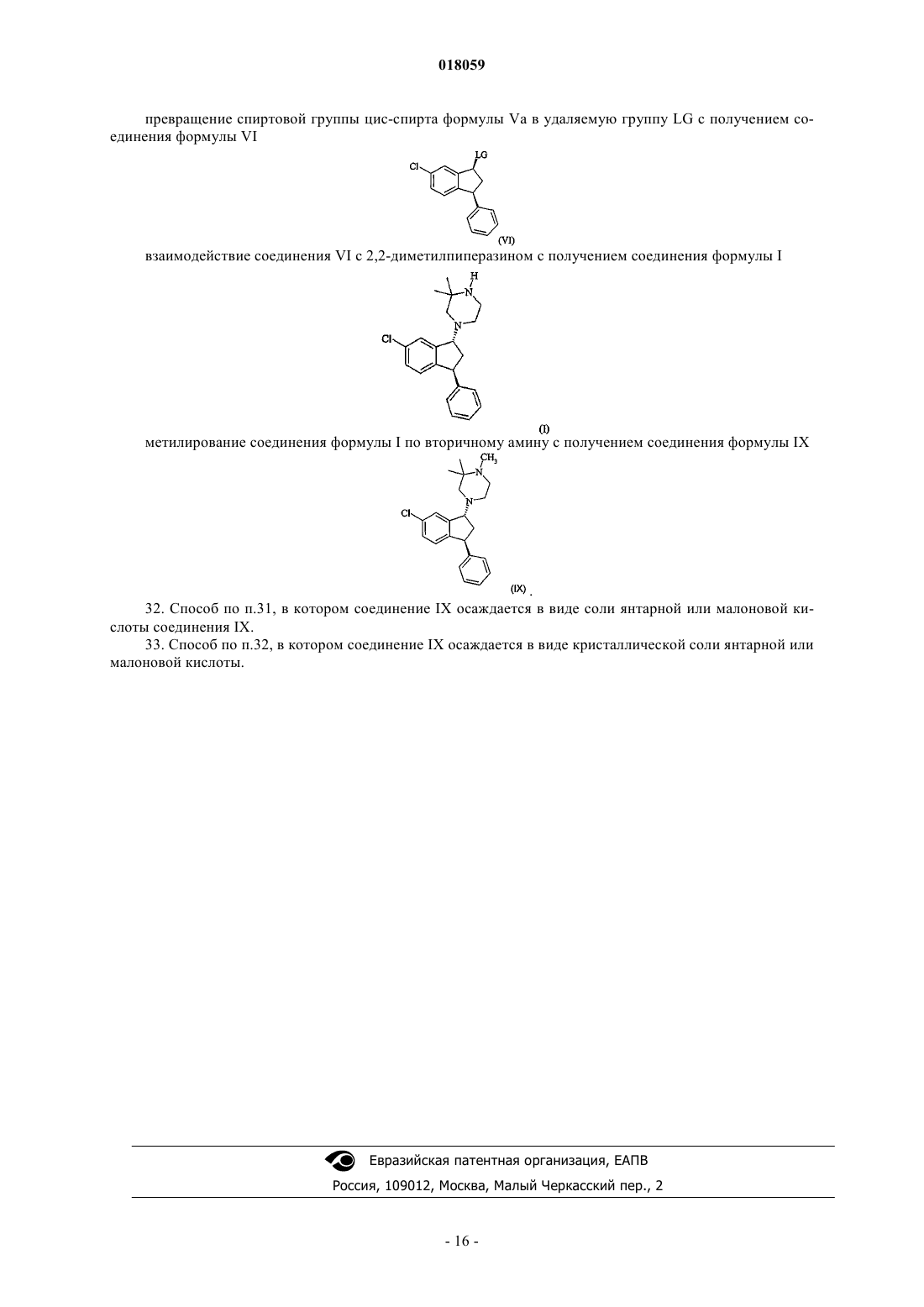
превращение спиртовой группы цис-спирта формулы Va в удаляемую группу LG с получением соединения формулы VI

взаимодействие соединения VI с 2,2-диметилпиперазином с получением соединения формулы I

2. Способ по п.1, в котором LG является галогеном или сульфонатом.
3. Способ по п.2, в котором LG является Cl, Br, тозилатом или мезилатом.
4. Способ по п.1, в котором соединение VI осаждают из подходящего растворителя.
5. Способ по п.4, в котором LG является галогеном и растворитель представляет собой алкан.
6. Способ по п.5, в котором соединение I осаждают в виде соли.
7. Способ по п.6, в котором образовавшаяся соль является солью фумаровой кислоты, солью малеиновой кислоты или солью соляной кислоты и соединения I.
8. Способ по п.5, в котором соединение I выделяют в виде свободного основания.
9. Способ по любому одному из пп.1-8, включающий
взаимодействие соединения VI с 1-защищенным 2,2-диметилпиперазином VII, в котором PG является защитной группой, с получением соединения формулы VIII; и
снятие защитной группы соединения VIII для получения соединения I,
где соединения VII и VIII являются следующими:

10. Способ по любому одному из пп.1-9, в котором хиральную хроматографию осуществляют с использованием хиральной жидкостной хроматографии.
11. Способ по любому одному из пп.1-10, в котором хиральную хроматографию осуществляют на хиральной неподвижной фазе.
12. Способ по п.11, в котором хиральную хроматографию осуществляют на колонке с силикагелем, покрытым оболочкой модифицированной амилозы.
13. Способ по п.12, в котором хиральную хроматографию осуществляют на колонке с силикагелем, покрытым оболочкой трис-[(S)-α-метилбензилкарбамат]амилозы.
14. Способ по п.13, в котором для хиральной хроматографии применяют растворитель, представляющий собой смесь н-гептана и этанола, и, необязательно, N,N-диэтиламин.
15. Способ по любому одному из пп.1-9, в котором хиральную хроматографию осуществляют с использованием хроматографии с суб- или суперкритической подвижной фазой.
16. Способ по п.15, в котором хиральную хроматографию осуществляют с использованием хроматографии с суперкритической подвижной фазой.
17. Способ по п.15 или 16, в котором хиральную хроматографию осуществляют на хиральной неподвижной фазе.
18. Способ по п.17, в котором хиральную хроматографию осуществляют на колонке с силикагелем, покрытым оболочкой, состоящей из хирального полимера, или на колонке с силикагелем с иммобилизованным хиральным полимером, или на колонке с силикагелем с ковалентно связанным хиральным мономером.
19. Способ по п.18, в котором хиральную хроматографию осуществляют на колонке с силикагелем, покрытым оболочкой трис-(3,5-диметилфенилкарбамат)амилозы или трис-[(S)-α-метилбензилкарбамат]амилозы, или на колонке с силикагелем, с иммобилизованной трис-(3,5-диметилфенилкарбамат)амилозой, или на колонке с силикагелем, покрытым оболочкой трис-(3,5-диметилфенилкарбамат)целлюлозы или трис-(4-метилбензоат) целлюлозы, или на колонке с силикагелем с ковалентно связанным амином 3,5-динитробензоилтетрагидрофенантрена.
20. Способ по любому одному из пп.18 или 19, в котором хиральную хроматографию осуществляют на колонке с силикагелем, покрытым оболочкой трис-(3,5-диметилфенилкарбамат)амилозы.
21. Способ по любому одному из пп.18-20, в котором хиральную хроматографию осуществляют с модификатором, выбранным из группы: метанол, этанол, 2-пропанол или ацетонитрил, необязательно содержащей диэтиламин.
22. Способ по п.21, в котором модификатор содержит 0,1% диэтиламина.
23. Способ по любому одному из пп.1-22, включающий рециклирование соединения IVb превращением энантиомерно обогащенного соединения IVb в рацемическое соединение IV, где IVb и IV являются:

24. Способ по п.23, в котором рацемизацию осуществляют с использованием основания или смеси двух или более оснований.
25. Способ по п.23 или 24, в котором рацемизацию осуществляют с использованием одного или более эквивалента ненуклеофильного основания ("первого основания"), за которым следует добавление каталитического количества одного или больше эквивалента того же самого или другого основания ("второго основания").
26. Способ превращения энантиомерно обогащенного соединения IVb в рацемическое соединение IV, где IVb и IV являются:

и в котором рацемизацию осуществляют с использованием одного или более эквивалента ненуклеофильного основания, за которым следует добавление каталитического количества одного или более эквивалента, того же самого или другого основания.
27. Способ по п.25 или 26, в котором рацемизацию осуществляют с использованием одного или более эквивалента ненуклеофильного основания ("первое основание"), выбираемого из группы, состоящей из диалкиламида металла, такого как диэтиламид лития, диизопропиламид лития (LDA) и тетраметилпиперидид лития;
бис-силиламида металла, такого как бис-(триметилсилил)амид лития (LiHMDS); и
алкоксида металла, такого как трет-бутоксид лития, за которым следует добавление каталитического количества одного или более эквивалента того же самого или другого ("второго основания"), выбираемого из группы, состоящей из
диалкиламида металла, такого как диэтиламид лития, диизопропиламид лития (LDA) и тетраметилпиперидид лития;
бис-силиламида металла, такого как бис-(триметилсилил)амид щелочного металла;
алкоксида металла, такого как трет-бутоксид калия, и
алкила металла, такого как бутиллитий или трет-бутиллитий.
28. Способ по пп.25-27, в котором "первое основание" и "второе основание" присутствуют сначала.
29. Способ по любому одному из пп.23-28, в котором рацемическое соединение IV перекристаллизовывается из подходящего растворителя.
30. Способ по п.29, в котором растворитель выбирают из группы, состоящей из C1-6спиртов, включая этанол или 2-пропанол или их смеси.
31. Способ получения соединение IX, включающий
получение соединения IVa разделением рацемического соединения IV, используя хиральную хроматографию

превращение соединения формулы IVa в соответствующий спирт Va с цис-конфигурацией

превращение спиртовой группы цис-спирта формулы Va в удаляемую группу LG с получением соединения формулы VI

взаимодействие соединения VI с 2,2-диметилпиперазином с получением соединения формулы I

метилирование соединения формулы I по вторичному амину с получением соединения формулы IX

.
32. Способ по п.31, в котором соединение IX осаждается в виде соли янтарной или малоновой кислоты соединения IX.
33. Способ по п.32, в котором соединение IX осаждается в виде кристаллической соли янтарной или малоновой кислоты.
Текст
Описан способ получения соединений формулы (I) и (IX) Даль Аллан Карстен, Вехльк Нильсен Христина (DK), Сюте Кристина, Робен Давид (FR), Бресен Петер (DK) Медведев В.Н. (RU) включающий разделение исходных веществ с использованием хиральной хроматографии, замену спировой группы на удаляемую группу, взаимодействие с 2,2-диметилпиперазином с получением соединения формулы I и дальнейшее метилирование соединения формулы I по вторичному амину с получением соединения формулы IX соответственно. Изобретение относится к способу получения транс-1-1R,3S)-6-хлор-3-фенилиндан-1-ил)-3,3 диметилпиперазина (соединение I) и его солей. Уровень техники изобретения Соединение, которое является предметом настоящего изобретения (соединение I, транс-1-1R,3S)6-хлор-3-фенилиндан-1-ил)-3,3-диметилпиперазин) имеет общую формулу (I) Соединение I и его соли, включая фумараты и малеаты, и его применения в медицине, например,при шизофрении или других заболеваниях, вызывающих психотические симптомы, описаны в PCT/DK 04/000546 (WO 05/016901). Как описано в PCT/DK 04/000546, изобретатели установили, что соединение I проявляет высокое сродство к рецепторам дофамина (DA), D1 рецепторам, DA D2 рецепторам и к альфа 1 адреноцепторам. Кроме того, установлено, что соединение I является антагонистом рецепторов дофамина D1 и D2 и рецепторов серотонина 5-НТ 2 а. Как далее описано в PCT/DK 04/000546, соединение I является относительно слабым ингибитором CYP2D6 (т.е снижало возможность взаимодействия лекарств между собой) и обладает относительно слабым воздействием на QT интервал на модели кроликов (т.е. снижало возможность установления пролонгирования QT интервала, вызванного лекарством, и возникновения летальной аритмии сердца, двунаправленной веретенообразной желудочковой тахикардии (TdP) у людей). Кроме того, активность соединения I как 5-НТ 2 антагониста свидетельствует о том, что соединение I может иметь относительно небольшой риск экстрапирамидального побочного эффекта. Вышеотмеченные свойства, например, тесты связывания (включающие альфа-1, DA D1 или D2, или рецепторы серотонина 5-НТгд), ингибирование CYP2D6 и QT-интервал могут быть определены, как описано в PCT/DK04/000546, проведите сравнение именно раздела "Пример" стр. 19-24 текста настоящей заявки в качестве области применения PCT/DK 04/000546. Кроме того, изобретатели установили, что соединение I не вызывает дистонию при тестировании на свиньях, сенсибилизированных галоперидолом, что указывает на то, что соединение I не обладает EPSPCT/DK04/00054 6 описывает следующие медицинские применения соединения I: заболевание центральной нервной системы, включающее психоз, а именно, шизофрения (например, позитивный, негативный и/или депрессивные симптомы) или другие заболевания, включающие психотические симптомы,такие, как, например, шизофрения, шизофреноморфное расстройство, шизоаффективное расстройство,галлюцинаторное расстройство, кратковременное психотическое расстройство, общее психотическое расстройство, кроме того другие психотические расстройства или заболевания, которые вызывают психотические симптомы, например расстройство как маниакальный биполярный синдром. Также описано применение соединения I для лечения тревожных расстройств, аффективных расстройств, включающих депрессию, нарушения сна, мигрень, нейролептически-индуцированный паркинсонизм, или зависимости от кокаина, от никотина, от алкоголя и других расстройств, обусловленных зависимостью. Как указано в PCT/DK04/000546, группа соединений, структурно родственных соединению I, т.е. транс-изомеры 3-арил-1-(1-пиперазинил)инданов, замещенные во 2- и/или 3-положении пиперазинового кольца, описаны в патенте EP 638 073; Bogeso и др. в J. Med. Chem., 1995, 38, 4380-4392 и Klaus P.Bogeso в "Drug Hunting, the Medical Chemistry of 1-Piperazino-3-phenilindans and Related Compounds",1998, ISBN 87-88085-10-41. Например, энантиомерно чистое соединение, соответствующее формуле (I),но отличающееся тем, что оно имеет N-метильную группу вместо IV-водородной группы в пиперазине,раскрыто в Bogeso и др. в J. Med. Chem., 1995, 38, 4380-4392, см. табл. 5, соединение (-)-38. Ни одна из отдельно взятых ссылок из PCT/DK 04/000546 не раскрывает специфическую энантиомерную форму вышеупомянутого соединения (соединение I) или его медицинское применение. Только транс-изомер в виде рацемата соединения I неявно раскрыт как промежуточное соединение в синтезе соединения 38 в Bogeso и др. в J. Med. Chem., 1995, 38, 4380-4392, в то время, как не описано медицинское применение соединения I или соответствующего ему рацемата. Соединение I как промежуточное соединение раскрыто в PCT/DK 04/000545 (WO 05/016900). Подробное описание изобретения Настоящее изобретение в одном аспекте относится к новому способу получения соединения I, в котором хиральность вводится на более ранней стадии получения, по сравнению со способом, описанным вPCT/DK 04/000546. Введение хиральности на одну стадию ранее является преимуществом, так как в этом случае следующая стадия проходит эффективно, т.е. по величине выхода и по расходованию реагентов и растворителей, и по снижению количества побочных продуктов. В PCT/DK 04/000546 хиральность вво-1 018059 дится разделением рацемического нижеприведенного промежуточного соединения V либо ферментативно, либо хиральной ВЭЖХ. Настоящие изобретатели разработали способ синтеза, в котором энантиомер формулы (I) получают последовательными превращениями из исходного чистого энантиомера IV, т.е. соединения IVa S)-6-хлор-3-фенилиндан-1-она, см. ниже). Таким образом, в этом способе промежуточное соединение формулы IV разделяется, т.е. подвергается хиральной хроматографии, с получением энантиомера формулы IVa. Кроме того, настоящие изобретатели разработали способ рацемизации нежелательного энантиомера(соединение IVb, см. ниже), который затем может быть повторно использован при разделении. Это оказывает огромное влияние на эффективность всего синтеза, поскольку эффективность стадий до разделения повышается так же, как и последующих стадий. Таким образом, энантиомер формулы (I) может быть получен в способе, включающем следующие стадии: Цианистый бензил реагирует с 2,5-дихлорбензонитрилом в присутствии основания; подходящим является трет-бутоксид калия (t-BuOK), в подходящем растворителе, таком как 1,2-диметоксиэтан(DME), дальнейшая реакция с метилхлорацетатом (МСА) приводит к спонтанному закрытию цикла и одностадийному образованию соединения формулы (II). Соединение формулы (II) затем подвергается кислотному гидролизу с образованием соединения формулы (III), подходящими для этого условиями являются нагревание в смеси уксусной кислоты, серной кислоты и воды, с последующим декарбоксилированием, т.е. нагреванием соединения формулы (III) в подходящем растворителе, таком, как толуол с триэтиламином или N-метилпирролидин-2-оном (NMP),с образованием соединения формулы (IV) Соединение формулы (IV) разделяется для получения требуемого энантиомера (формула IVa) для дальнейшего синтеза соединения I, и нежелательного энантиомера (формула IVb), который может быть подвергнут рацемизации и рециклированию Разделение IV может быть осуществлено, например, с использованием хиральной хроматографии,предпочтительна жидкостная хроматография, или хроматография с суб- или суперкритической подвижной фазой. Хиральная жидкостная хроматография может осуществляться, например, на хиральной неподвижной фазе, целесообразно на колонке с силикагелем, на котором иммобилизован хиральный полимер, или предпочтительно на колонке с силикагелем, покрытым оболочкой, состоящей из хирального полимера,например, модифицированной целлюлозы или модифицированной амилозы, такой, как трис-[(S)-метилбензилкарбамат]амилозы, предпочтительно, на колонке с силикагелем, покрытым оболочкой трис[(S)метилбензилкарбамат]амилозы. Подходящим растворителем для применения в хиральной жидкостной хроматографии является такой, как, например, спирт (предпочтительно, C1-4-спирт), нитрил, простой эфир или алкан (предпочтительно, C5-10-алкан), или их смеси, целесообразно использовать этанол, метанол, изопропанол, ацетонитрил, метил-трет-бутиловый эфир или н-гептан, или смеси их. Кислотные или основные модификаторы могут добавляться в растворитель, например, муравьиная кислота, уксусная кислота, трифторуксусная кислота, триэтиламин или N,N-диэтиламин. Хроматография с суб- или суперкритической подвижной фазой может осуществляться, например,на хиральной неподвижной фазе, целесообразно на колонке с силикагелем, на котором иммобилизован хиральный полимер, или на колонке с силикагелем, покрытым оболочкой, состоящей из хирального полимера, например модифицированной амилозы, такой как трис-[(S)метилбензилкарбамат]амилозы,или предпочтительно трис-(3,5-диметилфенилкарбамат)амилозы, наиболее предпочтительно трис-(3,5 диметилфенилкарбамат)амилозы, или модифицированной целлюлозы, такой как трис-(4-метилбензоат) целлюлозы или предпочтительно трис-(3,5-диметилфенилкарбамат) целлюлозы, наиболее предпочтительно трис-(3,5-диметилфенилкарбамат) целлюлозы, оболочкой, покрывающей силикагель. Могут при-2 018059 меняться другие типы хиральных неподвижных фаз, например, колонки типа Pirkle, целесообразна колонка с силикагелем, который ковалентно связан с амин 3,5-динитробензоилтетрагидрофенантреном. В качестве элюента для хроматографии с суб- или суперкритической подвижной фазой может применяться суб- или суперкритический подвижный диоксид углерода, целесообразен суперкритический диоксид углерода, содержащий модификатор. Модификатор выбирается из числа низших спиртов, таких,как метанол, этанол, пропанол и изопропанол, или, например, может применяться ацетонитрил. Могут быть добавлены амин, такой, как диэтиламин, при желании, 0,1% диэтиламин, триэтиламин, пропиламин и диметилизопропиламин, и при желании, кислота, такая, как муравьиная кислота, уксусная кислота и трифторуксусная кислота. В дополнительном примере осуществления изобретения модификатор выбирается из числа низших спиртов, таких, как метанол, этанол, пропанол и изопропанол, или, например, может применяться ацетонитрил, при условии, что модификатор совместим с колонкой. Хиральная хроматография может быть увеличена в масштабе при использовании подходящих технологий, например, технологии псевдодвижущегося слоя (SMB), или технологии суб- или суперкритической подвижной фазы (сравн. G. В. Сох (ed.) Preparative Enantioselective Chromatography, Blackwell Publishing Ltd., Oxford, UK, 2005). Соединение формулы (IVa) затем восстанавливается, например, с помощью комплексного металлогидрида, такого как борогидрид, целесообразно с помощью борогидрида натрия (NaBH4), или такого как литийалюмогидрид, в растворителе, таком как спирт (например, C1-5-спирт), например этанол или изопропанол, и предпочтительно при температуре в интервале от около -30 до +30C, например при ниже 30C, ниже 20C, ниже 10C, или предпочтительно ниже 5C, с образованием соединения формулы (Va) с цис-конфигурацией Спиртовая группа цис-спирта формулы (Va) превращается в подходящую удаляемую группу, такую как, например, галоген, например, Cl или Br, предпочтительно Cl, или сульфонат, например мезилат (метансульфонилат) или тозилат (4-толуолсульфонилат), по реакции с таким реагентом, как тионилхлорид,мезил(метансульфонил)хлорид или тозил(4-толуолсульфонил)хлорид в инертном растворителе, например, в простом эфире, целесообразно, в тетрагидрофуране. Получающееся в результате соединение имеет формулу (VI), где LG является удаляемой группой В предпочтительном примере осуществления изобретения LG является Cl, т.е. дис-хлорид формулы Соединение VI, например, с LG являющейся Cl, далее реагирует с 2,2-диметилпиперазином в подходящем растворителе, например, кетоне, таком как метилизобутилкетон или метилэтилкетон, предпочтительно метилизобутилкетон в присутствии основания, такого как, например, карбонат калия, с получением соединения I. Альтернативно пиперазиновая часть молекулы может быть введена реакцией соединения VI с соединением нижеприведенной формулы (VII), где PG является защитной группой, такой как, но не ограничивается только ими, например, фенилметоксикарбонил (часто обозначаемая как Cbz или Z), третбутилоксикарбонил (часто обозначаемая как ВОС), этоксикарбонил или бензил, таким образом получается соединение нижеприведенной формулы (VIII). Последующее снятие защитных групп соединенияVIII позволяет получить соединение I Дополнительный пример осуществления изобретения связан со способом получения соединения[соединение IX: 4-1R,3S)-6-хлор-3-фенилиндан-1-ил)1,2,2-триметилпиперазин], имеющего нижеследующую формулу (IX), или его солей(ii) превращение соединения I в соединение IX, предпочтительно метилированием функциональной группы вторичного амина, целесообразно восстановительным алкилированием, используя подходящие агенты, такие как, например, формальдегид, параформальдегид, триоксан или диэтоксиметан (DEM). Термин восстановительное алкилирование относится к вышеупомянутым реагентам в сочетании с таким восстанавливающим агентом, как муравьиная кислота. Таким образом, дополнительный пример осуществления изобретения связан со способом, как описано в этом документе, получения соединения I, в котором соединение I "заменяется" соединением IX. Соединение IX в общем описано в патенте EP 638 073, тогда как энантиомер формулы (IX) описанBges и др. в J. Med. Chem., 1995, 38, 4380-4392, в виде соли фумаровой кислоты, см. табл. 5, соединение (-)-38. Соединение IX и способ получения соединения IX из соединения I, и соли соединения IX (в частности, кристаллическая соль янтарной кислоты и кристаллическая соль малоновой кислоты) кроме того описаны в PCT/DK2004/00545. Как указано выше, изобретение также связано со способом получения соединения I или соединенияIX, как описано в этом документе, в котором соединение IVb рециклируется, таким образом оно может использоваться для синтеза соединения I или соединения IX, соответственно, смотрите также нижеприведенные иллюстрации: Как ни удивительно, рацемизация соединения IVb может быть достигнута с использованием оснований различных типов, например, амида, предпочтительно, диалкиламида, например, но не ограничиваясь только ими, диэтиламида лития, диизопропиламида лития, тетраметилпиперидида лития, целесообразно диизопропиламида лития (LDA), или бис-силиламида металла, например, бис-(триметилсилил) амида щелочного металла, или алкоксида металла, например, но не ограничиваясь ими, метоксида металла, этоксида металла, трет-бутоксида металла, целесообразно, алкоксида щелочного металла, предпочтительно, алкоксида калия, наиболее предпочтительно трет-бутоксида калия или металлалкила, целесообразно алкиллития, предпочтительно бутиллития или трет-бутиллития. После охлаждения реакционной смеси может быть выделен рацемический кетон IV. Рацемизация также может быть достигнута использованием двух различных оснований; к тому же,различные типы ненуклеофильных оснований могут использоваться в качестве "первого основания" (основание 1), например амид, предпочтительно диалкиламид, например, но не ограничиваясь ими, диэтиламид лития, диизопропиламид лития, тетраметилпиперидид лития, целесообразен диизопропиламид лития (LDA), или бис-силиламид металла, например, бис-силиламид лития, целесообразно, бис(триметилсилил)амид лития или алкоксид металла, например, но не ограничиваясь ими, метоксид металла, этоксид металла, трет-бутоксид металла, целесообразно, алкоксид щелочного металла, предпочтительно, алкоксид лития, наиболее предпочтительно трет-бутоксид лития. После того, как первое основание (основание 1) смешали с кетоном, добавляли "второе основание" (основание 2). Как и в случае с первым основанием, различные типы оснований могут использоваться; например, амид, предпочтительно,диалкиламид, например, но не ограничиваясь ими, диэтиламид лития, диизопропиламид лития, тетраметилпиперидид лития, целесообразен диизопропиламид лития (LDA), или бис-силиламид металла, например, бис-(триметилсилил)амид щелочного металла, или алкоксид металла, например, но не ограничиваясь ими, метоксид металла, этоксид металла, трет-бутоксид металла, целесообразно, алкоксид щелочного металла, предпочтительно алкоксид калия, наиболее предпочтительно трет-бутоксид калия или металлалкил, целесообразен алкиллитий, предпочтительно бутиллитий или трет-бутиллитий. Кроме того, рацемизация может быть достигнута использованием двух или более различных оснований добавлением их всех именно вначале, предпочтительно добавлением двух различных оснований именно вначале. В дополнительном примере осуществления изобретения рацемизация может быть достигнута использованием нуклеофильного основания. Альтернативно, рацемический спирт V может быть разделен хиральной хроматографией, как это описано в PCT/DK04/000546, с получением Va для синтеза соединения I и Vb, которое может подвергаться рацемизации и повторно использоваться для разделения, как указано на нижеприведенной фигуре. Рацемизация Vb достигается окислением Vb до IVb, например, с использованием хлорхромата пиридина(РСС), рацемизацией IVb до IV, как это описано выше, и затем восстановлением IV до V обычным способом, как это описано выше. При изучении рацемизации на 10 г загрузки вещества можно определить примесь как побочный продукт с помощью LC-MS (жидкостной хроматографии - масс-спектроскопии); аналитические данные свидетельствуют о том, что примесь является димером IV и/или энантиомерами IVa и IVb. Аналитические данные, кроме того, указывают, что для димера возможно элиминирование воды, в зависимости от метода исследования. Кропотливая работа показала, что образование димера можно в значительной мере подавлять тщательным подбором условий рацемизации, и содержание димера в продукте может быть,кроме того, снижено перекристаллизацией продукта из подходящего растворителя, например, спирта,предпочтительно, этанола или 2-пропанола. В способе синтеза соединения I может образоваться некоторое количество диастериомера соединения I (т.е. 1-1S,3S)-6-хлор-3-фенилиндан-1-ил)-3,3-диметилпиперазина) в качестве примеси к конечному продукту. Эта примесь в основном обусловлена образованием некоторого количества транс формы(VI) (т.е. (1S,3R)-3,5-дихлор-1-фенилиндана, когда LG является Cl) на стадии, когда образуется соединение VI. Поэтому примесь можно свести к минимуму кристаллизацией требуемой цис-формы соединенияVI из смеси транс и цис (VI); в случае, когда LG является Cl в соединении VI, это может быть сделано перемешиванием смеси с подходящим растворителем, например, алканом (C5-10-элканом), таким как геп-5 018059 тан, в результате чего требуемая цис-форма VI выпадает в осадок и нежелательная транс форма соединения VI переходит в раствор. Требуемая цис-форма соединения VI (т.е., когда LG является Cl) выделяется,например, фильтрованием и, предпочтительно, промывается обсуждаемым растворителем и высушивается. Цис-форма соединения I также может быть отделена выделением соединения I в виде свободного основания из подходящего растворителя. Изобретение в дополнительных аспектах также связано с промежуточными соединениями, как описано в этом документе для синтеза соединения I, в частности, с промежуточными соединениями IVa иIVb. В этом контексте понятно при установлении стереоизомерных форм, что стереоизомер является основным компонентом. В частности, при установлении энантиомерной формы, когда соединение имеет энантиомерный избыток, обсуждаемого энантиомера. Таким образом, один из примеров осуществления изобретения связан с соединением формулы(IVa), предпочтительно имеющим энантиомерный избыток по крайней мере 60% (60% энантиомерный избыток означает, что отношение соединения IV к его энантиомеру составляет 80:20 в обсуждаемой смеси), по крайней мере 70%, по крайней мере 80%, по крайней мере 85%, по крайней мере 90%, по крайней мере 96%, предпочтительно по крайней мере 98%. Один из примеров осуществления изобретения связан,в основном, с чистым соединением IVa. Дополнительный пример осуществления изобретения связан с соединением формулы (IVb), предпочтительно имеющим энантиомерный избыток по крайней мере 60%. Изобретение в дополнительном аспекте связано с соединением I или его солью (т.е. его солью с кислотами HCl, фумаровой, малеиновой), полученными способом изобретения и с медицинским применением их, в частности, по медицинским показаниям, как это раскрыто в данном документе, например, в качестве антипсихотического средства, как например при шизофрении. Также в изобретении присутствует фармацевтическая композиция соединения I или его соли, полученная способом изобретения. В настоящем контексте, в частности для фармацевтического применения соединения I, понятно, что когда определена энантиомерная форма как соответствующая формуле (I), тогда соединение является предпочтительно, в общем и целом, стехиометрически чистым, предпочтительно энантиомерный избыток составляет по крайней мере 60%, по крайней мере 70% и более предпочтительно по крайней мере 80% (80% энантиомерный избыток означает, что отношение I к его энантиомеру составляет 90:10 в обсуждаемой смеси), по крайней мере 90%, по крайней мере 96%, предпочтительно по крайней мере 98%. В предпочтительном примере осуществления изобретения диастереомерный избыток соединения I составляет по крайней мере 90% (90% диастереомерная чистота обозначает отношение соединения I к цис 1-1S,3S)-6-хлор-3-фенилиндан-1-ил)-3,3-диметилпиперазину, равное 95:5), по крайней мере 95%, по крайней мере 97% или по крайней мере 98%. Таким образом, способ по изобретению может включать стадию, в результате которой соединение I или его соль составляют фармацевтическую композицию. Соединение, соль или композиция соединенияI могут вводиться каким-либо удобным способом, например, перорально, трансбуккально, сублингвально, парентерально, и соединение или соль могут быть представлены в любой удобной форме для такого введения, например, в форме таблеток, капсул, порошков, сиропов или растворов, или дисперсий для инъекций. В одном из примеров осуществления изобретения, соединение или соль изобретения вводится в виде твердой фармацевтической формы, целесообразно, как таблетка или капсула. Способы приготовления твердых фармацевтических препаратов хорошо известны из уровня техники. Так, таблетки могут быть приготовлены смешиванием активного ингредиента с традиционными вспомогательными лекарственными веществами, наполнителями и разбавителями и с последующим прессованием смеси в подходящей машине для таблетирования. Примеры вспомогательных лекарственных средств, наполнителей и разбавителей включают кукурузный крахмал, лактозу, тальк, стеарат магния, желатин, лактозу, камеди и им подобные. Какие-либо другие вспомогательные лекарственные вещества или добавки, такие как красящие вещества, ароматизирующие вещества, консервирующие вещества и т.д. также могут использоваться, при условии, что они совместимы с активными ингредиентами. Растворы для инъекций могут быть приготовлены растворением соли изобретения и, по возможности, добавок в образце растворителя для инъекции, предпочтительно, стерильной воде, доведение раствора до желаемого объема, стерилизации раствора и заполнение им подходящих ампул или флаконов. Любые подходящие добавки, стандартно применяемые в соответствии с уровнем техники, могут быть добавлены, такие как тонизирующие агенты, консервирующие вещества, антиоксиданты, солюбилизирующие агенты и т.д. Ежедневная доза соединения вышеприведенной формулы (I), рассчитанная для свободного основания, целесообразна в пределах от 1,0 до 160 мг/день, более целесообразна в пределах от 1 до 100 мг, например в пределах от 2 до 55 мг. Термин "лечение", как он используется в этом документе в связи с заболеванием или расстройствами, включает также профилактику как возможный случай. Изобретение будет проиллюстрировано нижеследующими, не ограничивающими его объема, примерами. Примеры Аналитические методы. Энантиомерный избыток соединений (IV), (IVa) и (IVb) определяли хроматографией с суперкритической подвижной фазой, используя хроматограф Gilson SF3 Supercritical Fluid Chromatography System,детекцию осуществляли использованием детектора Gilson UV/VIS-831 при 254 нм. Также либо использовали хроматографическую колонку CHIRALPAKAD-H, 0,46 см ID25 см L при комнатной температуре при следующих условиях: Элюент:Этанол с 0,1% диэтиламином в качестве модификатора (30%),скорость потока 3 мл/мин и давление 200 бар. Время удерживания двух энантиомеров составляет 2,36 мин (IVa) и 2,99 мин (IVb). Либо использовали хроматографическую колонку CHIRALCELOD-H, 0,46 см ID25 см L при комнатной температуре при следующих условиях: Элюент:Этанол(30%) в качестве модификатора, скорость потока 4 мл/мин и давление 200 бар. Энантиомерный избыток соединения (Va) в примере 8 определяли хроматографией с суперкритической подвижной фазой, используя хроматограф Gilson SF3 Supercritical Fluid Chromatography System с хроматографической колонкой CHIRALPAKAD-H, 0,46 см ID25 см L при комнатной температуре. Элюент:Этанол с 0,1% диэтиламином в качестве модификатора (30%), скорость потока 3 мл/мин и давление 200 бар. Детекцию осуществляли использованием детектора Gilson UV/VIS-831 при 254 нм. Время удерживания двух энантиомеров составляет 2,41 мин (Va) и 3,06 мин (Vb). Энантиомерный избыток соединения (Va) в примере 1 а определяли хиральной ВЭЖХ, используя хроматографическую колонкуCHIRALCELOD, 0,46 см ID25 см L, 10 мкм при 40C. В качестве подвижной фазы применяли нгексан/этанол 95:5 (об./об.) при скорости потока 1,0 мл/мин, детекцию осуществляли с использованием УФ-детектора при 220 нм. Энантиомерный избыток соединения (I) определяли с помощью капиллярного электрофореза (СЕ) на плавленом кварце, используя следующие условия: Капилляр: 50 мкм ID48,5 смL, прогоняемый буфер: 1,25 мМ -циклодекстрин в 25 мМ дигидрофосфате натрия, pH 1,5, напряжение 16 кВ, температура: 22C, ввод пробы: 40 мбар за 4 с, детекция: колоночная диодная матричная детекция при 195 нм, концентрация образца: 500 мкг/мл. В этой системе соединение I имеет время удерживания приблизительно 10 мин, и другой энантиомер имеет время удерживания приблизительно 11 мин. Энантиомерный избыток соединения (IX) определяли с помощью капиллярного электрофореза (СЕ) на плавленом кварце, используя следующие условия: Капилляр: 50 мкм ID64,5 см L, прогоняемый буфер: 3,0 мМ -циклодекстрин и 10 мМ гидроксипропилциклодекстрин в 50 мМ дигидрофосфате натрия, pH 1,5, напряжение 15 кВ, температура: 22C, ввод пробы: 40 мбар за 4 с, детекция: колоночная диодная матричная детекция при 192 нм, концентрация образца: 100 мкг/мл. В этой системе соединениеIX имеет время удерживания приблизительно 47 мин, и энантиомер имеет время удерживания приблизительно 46 мин. Другие два диастереомера 4-1R,3R)-6-хлор-3-фенилиндан-1-ил)-1,2,2-триметилпиперазин и 4-1S,3S)-6-хлор-3-фенилиндан-1-ил)-1,2,2-триметилпиперазин имеют времена удерживания приблизительно 49 мин и 52 мин, соответственно. 1 Н-ЯМР спектры снимали при 500,13 МГц на приборе Bruker Avance AV-500, или на прибореBruker Avance DRX500, или при 250,13 МГц на приборе Bruker Avance DPX-250, или на приборе BrukerAC 250. Хлороформ (99,8%D) или диметилсульфоксид (99,8%D) применяли в качестве растворителей, и тетраметилсилан(TMS) использовали в качестве внутреннего стандарта. Отношение цис/транс соединения (I) и соединения (IX) определяли, используя 1 Н-ЯМР, как это описано в Bogeso и др. в J. Med. Chem., 1995, 38, 4380-4392 (стр.4388, правая колонка). Отношение цис/транс соединения (VIa) определяли с помощью 1 Н-ЯМР в DMSO-d6, используя интегралы сигнала при 5,6 м.д. для цис-изомера и сигнала при 5,75 м.д. для транс-изомера, или с помощью 1 Н-ЯМР в хлороформе, используя интегралы сигнала при 5,3 м.д. для цис-изомера и сигнала при 5,5 м.д. для трансизомера. Обычно, содержание нежелательного изомера, приблизительно составляющее 1%, может быть определено с помощью ЯМР. Температуры плавления измеряли с использованием Дифференциального Сканирующего Калориметра (DSC). Прибор представлял собой TA-Instruments DSC-Q1000 или TA-Instruments DSC-2920, откалиброванный при 5/мин, чтобы показывать температуру плавления как начальное значение температуры процесса плавления. Около 2 мг образца нагревали со скоростью 5/мин в неплотно закрытом резервуаре в токе азота. Элементный анализ осуществляли, используя Vario El анализатор, настроенный на измерение содержания элементов С, Н и N. Полученная величина представляла собой среднее из двух определений, в каждом из которых использовалось приблизительно 4 мг вещества. Оптическое вращение измеряли, используя поляриметр Perkin Elmer модель 241, устанавливалась концентрация вещества 1% в метаноле, за исключением иных случаев. Жидкостная хроматография - масс-спектрометрия (LC-MS) проводилась с использованием колонкиWaters Symmetry C-18, 0,46 см ID3 см L, 3,5 мкм, при 60C. Элюентом является градиент (А) вода с 0,05% трифторуксусной кислотой и (В) ацетонитрил с 5% водой и 0,035% трифторуксусной кислотой,начинающийся с 90% Аи 10% В и заканчивающийся 100% В за 2 мин, при скорости элюирования 3 мл/мин. Детекцию осуществляли с использованием детектора Shimadzu при 254 нм. Масс-спектр регист-7 018059 рировали на масс-спектрометре Sciex API300. Синтез Синтез ключевых исходных веществ Соединение V синтезировали из IV восстановлением борогидридом натрия (NaBH4), применение метода описано в Bogeso J. Med. Chem., 1983, 26, 935, с использованием этанола в качестве растворителя и проведением реакции приблизительно при 0C. Оба соединения описаны в Bogeso и др., J. Med. Chem.,1995, 38, 4380-4392. Соединение IV синтезировали из II, применяя общую методику, описанную в Sommer и др., J. Org. Chem. 1990, 55, 4822, который также описывает II и его синтез. Пример 0 а. Синтез (S)-6-хлор-3-фенилиндан-1-она (IVa) и (R)-6-хлор-3-фенилиндан-1-она (IVb) с применением хиральной хроматографии. Рацемический 6-хлор-3-фенилиндан-1-он (IV) разделяли препаративной хроматографией, используя колонку CHIRALPAK AS-V. Смесь н-гептана, этанола и N,N-диэтиламина применяли в качестве подвижной фазы, детекцию осуществляли с использованием УФ-детектора при 220 нм. Рацемический кетон(IV) вводился в виде раствора в элюенте; приемлемые объемы раствора вводили через целесообразные интервалы. Все фракции, которые содержат соединение (IVa) с более чем 98% энантиомерным избытком, объединялии упаривали досуха, используя роторный испаритель. Все фракции, которые содержат соединение (IVb) или смесь соединений (IVa) и (IVb) объединяли и упаривали досуха, используя роторный испаритель. Пример 0b. Синтез энантиомерно чистого (R)-6-хлор-3-фенилиндан-1-она (IVb) окислением(1R,3R)-6-хлор-3-фенилиндан-1-ол (Vb), выделенный как в примере 1 а, (20 г) растворяли в дихлорметане (400 мл) и добавляли пиридинхлорхромат (РСС) (26,5 г). Смесь перемешивали 11/2 ч при комнатной температуре. Смесь фильтровали, маслообразный остаток в реакционном сосуде промывали дихлорметаном. Объединенные органические фракции упаривали досуха на роторном испарителе, получали масло черного цвета (25 г). Добавляли этилацетат (200 мл) и гидроксид натрия (2 М в воде, 200 мл). Фазы разделяли, водную фазу экстрагировали дважды этилацетатом (200 мл). Объединенные органические фазы промывали трижды гидроксидом натрия (2 М в воде, 100 мл), дважды - водой (100 мл) и один раз солевым раствором (100 мл), и в заключение сушили сульфатом натрия. За упариванием досуха следовала сушка в вакуумном термостате при 40C, в результате получали 15 г кристаллического вещества.[]D20-61 (с=1,0, метанол). 90% энантиомерный избыток, в соответствии с данными хирального анализа. Пример 0 с. Рацемизация (R) -6-хлор-3-фенилиндан-1-она (IVb). Диизопропиламин (5,1 мл) растворяли в сухом тетрагидрофуране (THF) (50 мл) и раствор перемешивали под азотом при охлаждении в бане ацетон/сухой лед. Медленно добавляли бутил-литий (1,6 М в гексане, 22,6 мл), причем после охлаждения баню заменяли на лед/вода. После перемешивания в течение 11/2 ч добавляли в течение 30 мин синтезированный в примере 0b (R)-6-хлор-3-фенилиндан-1-он (IVb)(7,05 г, 90% энантиомерный избыток), растворенный в сухом THF (60 мл), и перемешивание на охлаждающей бане продолжали 17 мин. Затем добавляли трет-бутоксид калия (1,0 М в THF, 28,8 мл) за 17 мин,и затем перемешивание продолжалось в течение еще двух часов на бане лед/вода. Реакционную смесь нейтрализовали соляной кислотой (4 М, 50 мл) и затем THF удаляли из смеси на роторном испарителе. Добавляли воду (200 мл) и диэтиловый эфир (350 мл), и фазы разделяли. Водную фазу дважды экстрагировали диэтиловым эфиром (200 мл, затем 100 мл). Объединенные органические фазы промывали дважды водой (100 мл), один раз -солевым раствором (100 мл) и сушили сульфатом натрия. За упариванием досуха на роторном испарителе следовало высушивание в вакууме при 40C, в результате получали 6,70 г твердого вещества красного цвета. []D20-2,34 (c=1,0, метанол). Продукт имел энантиомерный избыток 2% в соответствии с хиральным анализом, и содержал 6% побочного продукта (см. основной текст) в соответствии с данными ВЭЖХ. Неочищенный продукт (4,99 г) перекристаллизовывали из абсолютного этанола (40 мл), в результате получали 3,71 г твердого вещества красного цвета. []D20-0,84 (c=1,0, метанол). Содержание побочного продукта составляло 2,6% (см. основной текст) в соответствии с данными ВЭЖХ. Пример 1a. Синтез (1S,3S)-6-хлор-3-фенилиндан-1-ола (Va) и (1R,3R)-6-хлор-3-фенилиндан-1-ола(Vb) с использованием хиральной хроматографии. Рацемический цис-6-хлор-3-фенилиндан-1-ол (Vb) (492 г) разделяли препаративной хроматографией, используя колонку CHIRALPAK AD, 10 см ID50 см L, 10 мкм при 40C. Метанол использовали в качестве мобильной фазы при скорости потока 190 мл/мин, детекцию осуществляли с использованием УФ-детектора при 287 нм. Рацемический спирт (V) вводился в виде 50000 м.д. раствора в метаноле; 90 мл вводились с интервалами в 28 мин. Все фракции, которые содержали соединение (Va) с энантиомерным избытком более чем 98%, объединяли и упаривали досуха, используя роторный испаритель, затем следовало высушивание в вакууме при 40C. Выход твердого вещества составлял 220 г. Данные элементного анализа и ЯМР-спектра соответствовали структуре, энантиомерный избыток превышал 98% в соответствии с данными хиральной ВЭЖХ, []D20+44,5 (c=1,0, метанол). Аналогичным образом, фракции, которые содержат соединение (Vb), объединяли и упаривали досуха, получая 214 г (Vb). Пример 1b. Синтез (15,3S)-6-хлор-3-фенилиндан-1-ола (Va) путем восстановления энантиомерно чистого (IVa).(5)-6-Хлор-3-фенилиндан-1-он (IVa) путем восстановления борогидридом натрия может быть превращен в соединение (Va), применяя способ, описанный в Bogeso J. Med. Chem., 1983, 26, 935, используя этанол в качестве растворителя и осуществляя реакцию приблизительно при 0C. Пример 2. Синтез (1S,3S)-3,5-дихлор-1-фенилиндана (VI, LG=Cl). Цис-(1S,3S)-6-хлор-3-фенилиндан-1-ол (Va) (204 г), полученный, как описано в примере 1 а, растворяли в THF (1500 мл) и охлаждали до -5C. Тионилхлорид (119 г) добавляли по каплям в виде раствора вTHF (500 мл) в течение 1 ч. Смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли лед (100 г). Когда лед растаял, разделяли водную фазу (А) и органическую фазу(В), и органическую фазу В дважды промывали насыщенным раствором бикарбоната натрия (200 мл). Фазы с бикарбонатом натрия объединяли с водной фазой А, доводили до pH 9 гидроксидом натрия(28%), и использовали еще раз для однократной промывки органической фазы В. Полученную водную фазу (С) и органическую фазу В разделяли, и водную фазу С экстрагировали этилацетатом. Этилацетатную фазу объединяли с органической фазой В, сушили сульфатом магния и упаривали досуха, используя роторный испаритель, получали упомянутое в заглавии соединение в виде масла. Выход 240 г, которые полностью использовали в примере 5. Цис/транс отношение 77:23, в соответствии с данными ЯМР. Пример 3. Синтез 3,3-диметилпиперазин-2-она. Карбонат калия (390 г) и этилендиамин (1001 г) перемешивали с толуолом (1,50 л). Раствор этил-2 бромизобутирата (500 г) в толуоле (750 мл) добавляли. Суспензию нагревали с обратным холодильником в течение ночи и фильтровали. Осадок на фильтре промывали толуолом (500 мл). Объединенные фильтраты (объем 4,0 л) нагревали на водяной бане и перегоняли при 0,3 атм, используя насадку Кляйзена; первые 1200 мл дистиллята собирали при 35C (температура в смеси 75C). Дополнительно добавляли толуол (600 мл) и другие 1200 мл дистиллята собирали при 76C (температура в смеси 80C). Опять добавляли толуол (750 мл) и 1100 мл дистиллята собирали при 66C (температура в смеси 71C). Смесь перемешивали на ледяной бане и вводили кристалл-затравку, в результате чего продукт выпадал в осадок. Продукт отделяли фильтрованием, промывали толуолом, сушили в течение ночи в вакуумном термостате при 50C. Выход 3,3-диметилпиперазин-2-она 171 г (52%). ЯМР-спектр соответствует структуре. Пример 4. Синтез 2,2-диметилпиперазина. Смесь 3,3-диметилпиперазин-2-она (8,28 кг, 64,6 моль) и тетрагидрофурана (THF) (60 кг) нагревали до 50-60C, получали слегка мутный раствор. THF (50 кг) перемешивали под азотом и LiAlH4 (250 г в растворимой пластиковой упаковке) добавляли, что приводило к слабому выделению газа. После прекращения выделения газа добавляли еще LiAlH4 (общее использованное количество 3,0 кг, 79,1 моль), и вследствие экзотермической реакции температура возрастала от 22C до 50C. Раствор 3,3-диметилпиперазин-2-она медленно добавляли в течение 2 ч при 41-59C. Суспензию перемешивали еще в течение часа при 59C (температура нагревательной оболочки 60C). Смесь охлаждали и добавляли воду (3 л) в течение 2 ч, поддерживая температуру ниже 25C (необходимо охлаждать внешней охлаждающей оболочкой с температурой 0C). Затем добавляли гидроксид натрия (15%, 3,50 кг) в течение 20 мин при 23C, охлаждение необходимо. Еще дополнительно добавляли воду (9 л) в течение получаса (охладжение необходимо), и смесь перемешивали в течение ночи под азотом. Добавляли фильтрационное веществоCelit (4 кг) и смесь фильтровали. Осадок на фильтре промывали THF (40 кг). Объединенные фильтраты концентрировали в реакторе, пока температура в реакторе не достигала 70C (температура дислилляции 66C) при 800 мбар. Остаток (12,8 кг) далее концентрировали на роторном испарителе приблизительно до 10 л. В заключение, смесь фракционировали дистилляцией при атмосферном давлении и продукт собирали при 163-4C. Выход 5,3 кг (72%). ЯМР-спектр соответствует структуре. Пример 5. Синтез транс-1-1R,3S)-6-хлор-3-фенилиндан-1-ил)-3,3-диметилпиперазина (соединениеI) кислой соли малеиновой кислоты. Цис-(1S,3S)-3,5-дихлор-1-фенилиндана (VI, LG=Cl) (240 г) растворяли в бутан-2-оне (1800 мл). Карбонат калия (272 г) и 2,2-диметилпиперазин (полученный в примере 4) (113 г) добавляли и смесь нагревали с обратным холодильником до температуры кипения в течение 40 ч. К реакционной смеси добавляли диэтиловый эфир (2 л) и соляную кислоту (1 М, 6 л). Фазы разделяли и pH водной фазы понижали от 8 до 1 с помощью концентрированной соляной кислоты. Водная фаза использовалась для еще одной промывки органической фазы, чтобы удостовериться, что весь продукт в водной фазе. Гидроксид натрия (28%) добавляли к водной фазе до тех пор, пока pH не станет равным 10, и водную фазу дважды экстрагировали диэтиловым эфиром (2 л). Экстракты с диэтиловым эфиром объединяли, сушили сульфатом натрия и упаривали досуха, используя роторный испаритель. Выход упомянутого в заглавии соединения составил 251 г в виде масла. Цис/транс отношение составляет 18:82, в соответствии с данными ЯМР-спектра. Неочищенное масло (прибл. 20 г) дополнительно очищали флэш-хроматографией на силикагеле (элюент:этилацетат/этанол/триэтиламин 90:5:5), за которой следовало упаривание досуха на роторном испарителе. Выход 12 г озаглавленного соединения в виде масла [цис/транс отношение составляет 10:90, в соответствии с данными ЯМР-спектра). Масло растворяли в этаноле (100 мл), и к раствору добавляли раствор малеиновой кислоты в этаноле до достижения pH 3. Полученную смесь перемешивали при комнатной температуре в течение 16 ч, и образовавшийся осадок отделяли фильтрованием. Объем этанола уменьшали и другую порцию осадка отделяли. Выход твердого вещества (цис-изомер не обнаруживался, в соответствии с данными ЯМР-спектра) упомянутого в заглавии соединения составил 3,5 г. Энантиомерный избыток в соответствии с данными СЕ составляет 99%. Температура плавления 175178C. ЯМР-спектр соответствует структуре. Пример 6. Скрининг условий разделения 6-хлор-3-фенилиндан-1-она (IV) хроматографией с суперкритической подвижной фазой. Проводили скрининг серии колонок на возможность разделения (IV), используя хроматограф GilsonSF3 Supercritical Fluid Chromatography System. Элюент: Различные растворители с 0,1% диэтиламином в качестве модификатора (30%), скорость потока 4 мл/мин и давление 200 бар, и колонка работала при комнатной температуре. Детекцию осуществляли с использованием детектора Gilson UV/VIS-831 при 254 нм. Время удерживания двух энантиомеров (RT1 и RT2) и ширину каждого из двух пиков на середине высоты (1 и 2) рассчитывали с использованием программного обеспечения Gilson Unipoint, версия 3,2. Нижеприведенная таблица содержит рассчитанные величины разделения (Rs) для отдельных колонок, как серийных, так и модифицированных; Rs рассчитывали по формуле Rs=2(RT2-RT1)/(1 + 2).CHIRALPAKAD-H, 20 мм ID 250 мм L, 5 мкм. Элюент: Этанол использовали в качестве модификатора (20%), скорость потока 50 мл/мин и давление 100 бар, и колонка работала при 35C. Детекцию осуществляли с использованием УФ-детектора при 230 нм. Применяли Berger устройство для сбора фракций и для декомпрессии. Работа аппаратуры контролировалась программным обеспечением SFC Pronto. Два энантиомера имели время удерживания 3,9 мин (IVa) и 4,8 мин (IVb). Рацемический кетон (IV) вводился в виде раствора в ацетонитриле (55 г (IV) в 800 мл ацетонитрила) ; 500 мл этого раствора вводились с интервалами 132 с. Все фракции, содержащие соединение (IVa), объединяли и декомпрессия позволяла получить раствор (IVa) в этаноле, и все фракции, содержащие соединение (IVb), объединяли и также подвергали декомпрессии. Соединение (IVa) выделяли упариванием раствора на роторном испарителе и высушиванием остатка в вакуумном термостате при 40C. Выход 25,6 г (47%) твердого вещества. Температура плавления 110,8C, ЯМР-спектр соответствует структуре, [] D20+72,65 (с=1,0, метанол). Содержание С, Н, рассчитано для C15H11OCl: С 74,23, Н 4,57; найдено: С 74,09, Н 4,70. Энантиомерный избыток 99% в соответствии с данными хирального анализа. Соединение (IVb) выделяли таким же образом, получили 23,9 г (43%) твердого вещества. Температура плавления 110,6C. ЯМР-спектр соответствует структуре, [] D20-70,33 (с=1,0, метанол). Содержание С, Н, рассчитано для C15H11OCl: С 74,23, Н 4,57; найдено: С 73,79, Н 4,70. Энантиомерный избыток 99% в соответствии с данными хирального анализа. Пример 8. Синтез (1S,3S)-6-хлор-3-фенилиндан-1-ола (Va) восстановлением энантиомерно чистого(S)-6-хлор-3-фенилиндан-1-он (IVa) (выделенный как в примере 7(23 г добавляли маленькими порциями к суспензии борогидрида натрия (1,6 г) в этаноле (160 мл) при 3-5C. После того как добавление было завершено, смесь оставляли до достижения комнатной температуры. Реакционную смесь перемешивали 2,7 5 ч, после чего упаривали досуха. Остаток растворяли в смеси воды (150 мл) и этилацетата(200 мл), фазы разделяли и водную фазу экстрагировали этилацетатом (100 мл). Органические фазы объединяли, промывали водой (100 мл), сушили сульфатом магния, фильтровали и упаривали досуха. Остаток перекристаллизовывали из гептана (250 мл), в результате получали 20,9 г (90%) упомянутого в заглавии продукта в виде твердого вещества. Температура плавления 108,9C, ЯМР-спектр соответствует- 10018059 структуре, []D20+48,30 (с=1,0, метанол). Содержание С, Н, рассчитано для C15H13OCl: С 73,62, Н 5,35; найдено: С 73,55, Н 5,29. Энантиомерный избыток 99% в соответствии с данными хирального анализа. Пример 9. Синтез (1S,3S)-3,5-дихлор-1-фенилиндана (VI, LG=Cl). Раствор (1S,3S)-6-хлор-3 фенилиндан-1-ола (Va) (17 г) (синтезированный, как в примере 8) в тетрагидрофуране (130 мл) охлаждали в ледяной бане. Тионилхлорид (9,9 г) в тетрагидрофуране (50 мл) добавляли по каплям при 4-5C, и затем смесь перемешивали в течение ночи при температуре окружающей среды. Добавляли смесь воды со льдом (примерно 25 мл) и перемешивание продолжали до тех пор, пока растаял весь лед. Фазы разделяли, органическую фазу дважды промывали бикарбонатом натрия (5% в воде, 25 мл). Водные фазы затем объединяли, экстрагировали органической фазой и затем экстрагировали этилацетатом (50 мл). Органические фазы затем объединяли, сушили сульфатом магния и упаривали досуха, используя роторный испаритель, получали озаглавленное соединение в виде масла. Выход 18,7 г (102%) озаглавленного соединения в виде масла, которое частично сделалось твердым. Содержание (1S,3S)-3,5-дихлор-1 фенилиндана составляет 18% в соответствии с данными ЯМР-спектра. Пример 10. Синтез транс-1-1R,3S)-6-хлор-3-фенилиндан-1-ил)-3,3-диметилпиперазина (соединение I). Смесь (1S,3S)-3,5-дихлор-1-фенилиндана (VI, LG=Cl) (18 г) (синтезированного как в примере 9),карбоната калия (20,8 г), 2,2-диметилпиперазина и метилэтилкетона (135 мл) нагревали до кипения с обратным холодильником в течение ночи. После охлаждения до комнатной температуры добавляли диэтиловый эфир (150 мл) и соляную кислоту (1 М, 450 мл), и смесь перемешивали несколько минут. Фазы разделяли и pH водной фазы доводили от 1 до 12, используя гидроксид натрия (28%). Водную фазу экстрагировали диэтиловым эфиром (два раза 17 0 мл). Все органические фазы объединяли, сушили сульфатом магния, фильтровали и упаривали, используя роторный испаритель. Выход 20,7 г (89%) озаглавленного соединения в виде масла. Содержание цис-изомера составляет 19% в соответствии с данными ЯМРспектра. Пример 11. Синтез транс-4-1R,3S)-6-хлор-3-фенилиндан-1-ил)-1,2,2-триметилпиперазина (IX) кислой соли фумаровой кислоты. Транс-1-1R,3S)-6-хлор-3-фенилиндан-1-ил)-3,3-диметилпиперазина (I, синтезированный как в примере 10) перемешивали с муравьиной кислотой (15,2 мл) и формальдегидом (37% в воде, 12,5 мл) и нагревали на масляной бане (температура 110C) в течение 11/2 ч. Воду добавляли к реакционной смеси после охлаждения до комнатной температуры, pH доводили примерно до 14 гидроксидом натрия (28%). Продукт экстрагировали диэтиловым эфиром и затем этилацетатом, добавляя гидроксид натрия (28%) между экстракциями, если pH становился ниже 12. Органические фазы объединяли, сушили сульфатом натрия, фильтровали и упаривали досуха, используя роторный испаритель. Выход 10,9 г (100%) (IX) в виде масла, содержащего 20% цис-формы, в соответствии с данными ЯМР-спектра. Маслообразное вещество (10 г) нагревали с 1-пропанолом (150 мл), получая раствор. Фумаровую кислоту (3,3 г) добавляли и нагревание продолжали до полного растворения. Смесь охлаждали до комнатной температуры и вносили кристалл-затравку, в результате чего продукт выпадает в осадок. Твердое вещество отделяли фильтрованием, промывали небольшим количеством 1-пропанола и сушили в вакуумном термостате при 40C. Выход 6,85 г (52%). Температура плавления 193,3C, ЯМР-спектр соответствует структуре, []D20-15,2 (с=1,0, метанол). Содержит 4% цис-формы в соответствии с CE, два другие диастериомера не детектировались (т.е. их содержание ниже 1%). Содержание С, Н, N рассчитано дляC26H31N2O4Cl: С 66,30, Н 6,63, N 5,95; найдено: С 65,96, Н 6,61, N 5,57. Энантиомерный избыток 98% в соответствии с данными СЕ. Пример 12. Синтез энантиомерно чистого (R)-6-хлор-3-фенилиндан-1-она (IVb) окислением(1R,3R)-6-хлор-3-фенилиндан-1-ола (Vb). Хлорхромат пиридина (66,1 г) добавляли к раствору (1R,3R)-6-хлор-3-фенилиндан-1-ола (Vb) (выделенному, как в примере 1 а) (50,0 г) в дихлорметане (840 мл), и смесь перемешивали при температуре окружающей среды 2 ч. Смесь фильтровали и остаток в реакционном сосуде промывали дважды дихлорметаном (200 мл), который также применяли для промывки осадка на фильтре. Объединенные фильтраты упаривали досуха, используя роторный испаритель. Остаток перемешивали с гидроксидом натрия (2 М, 1 л) и этилацетатом (750 мл) в течение 1/2 ч. Фазы разделяли и водную фазу экстрагировали этилацетатом (500 мл). Объединенные органические фазы промывали дважды гидроксидом натрия (2 М,250 мл) и 25% хлоридом натрия (250 мл). Затем органическую фазу перемешивали с сульфатом магния(60 г), активированным углем (1,4 г) и силикагелем 60 (0,06-0,2 мм, 5 г), фильтровали и упаривали досуха, используя роторный испаритель. Остаток (31,5 г) перекристаллизовывали из 2-пропанола (125 мл); продукт выделяли фильтрованием и промывали 2-пропанолом (40 мл). Высушивание в вакуумном термостате при 50C приводило к получению 26,0 г (53%) продукта в виде твердого вещества. Температура плавления 110,8C, ЯМР-спектр соответствует структуре, []D20-75, 6 (с=1,0, метанол). Содержание С, Н, N, рассчитано для C15H11OCl: С 74,23, Н 4,57, N 0,00; найдено: С 73,89, Н 4,71, N 0,05. Энантиомерный избыток 99,2% в соответствии с данными хирального анализа. Реакцию повторяли дважды, получая 48 г продукта с энантиомерным избытком 99,6% и 48 г про- 11018059 дукта с энантиомерным избытком 98,9%, соответственно. Пример 13. Скрининг оснований для рацемизации (R)-6-хлор-3-фенилиндан-1-она (IVb). Использовали основания фирмы Aldrich: бутил-литий (BuLi) номер по каталогу 18,617-1, третбутил-литий (tBuLi) номер по каталогу 18,619-8, трет-бутоксид калия (KOtBu) номер по каталогу 32,8 650, трет-бутоксид лития (LiOtBu) номер по каталогу 398195, трет-бутоксид натрия (NaOtBu) номер по каталогу 35,927-0, бис-(триметилсилил)амид лития (LiHMDS) номер по каталогу 225770, бис(триметилсилил)амид натрия (NaHMDS) номер по каталогу 24,558-5, и бис-(триметилсилил)амид калия(KHMDS) номер по каталогу 324671. Диизопропиламид лития получали из диизопропиламида (SigmaAldrich номер по каталогу 386464) и BuLi непосредственно перед применением в каждом эксперименте. Нижеследующая методика является типичной для экспериментов и иллюстрирует применение LDA и применение двух различных оснований: Смесь диизопропиламида (437 мкл) и тетрагидрофурана (THF) (4 мл) перемешивали в токе азота и охлаждали на бане с сухим льдом и ацетоном. Бутил-литий (BuLi) (1,6 М в гексане, 1,60 мл) добавляли в течение 5 мин. Перемешивание продолжали 10 мин и затем заменяли охлаждающую баню на водноледяную баню. После перемешивания еще в течение 10 мин добавляли раствор (R)-6-хлор-3 фенилиндан-1-она (IVb) (синтезированного как в примере 12) (0,50 г) в THF (4 мл) по каплям в течение 5 мин к раствору LDA ("первое основание", основание 1), и перемешивание на водно-ледяной бане продолжали 1/2 ч. Затем добавляли BuLi (1,6 М в гексане) (1,61 мл) ("второе основание", основание 2) по каплям в течение 5 мин, а после перемешивания на водно-ледяной бане, продолжавшегося 21/2 ч, затем добавляли соляную кислоту (4 М, 4 мл). После перемешивания в течение 10 мин фазы разделяли и водную фазу экстрагировали этилацетатом (два раза по 10 мл). Объединенные органические фазы промывали хлоридом натрия (25%, 10 мл), сушили сульфатом магния, фильтровали и упаривали досуха, используя роторный испаритель. Выход 0,47 г (94%) маслообразного вещества, химическая чистота 83%, в соответствии с данными LC-MS, и энантиомерный избыток составляет 1% в соответствии с данными хирального анализа. Нижеприведенная таблица суммирует полученные результаты. Указано время перемешивания при 0C после смешения всех компонентов. Энантиомерный избыток; отрицательный знак указывает, что IV в избытке, примеси в образце могут мешать анализу. 3)LDA получали, используя 1,25 эквивалентов диизопропиламина и 1,50 эквивалентов BuLi. 6) В этом эксперименте весь BuLi, указанный также как количество основания 2, добавляли в начале эксперимента, до добавления соединения IVb. Пример 14. Увеличение рацемизации (R)-6-хлор-3-фенилиндан-1-она (IVb). Использованные основания такие же, как в примере 13. Типичная методика является следующей. Диизопропиламин (6,25 г) растворяли в тетрагидрофуране (THF) (160 мл) и смесь охлаждали на бане сухой лед/ацетон во время перемешивания под азотом. Бутил-литий (1,6 М в гексане, 33 мл) медленно добавляли, поддерживая температуру ниже -60C. Перемешивание продолжали 5 мин на бане сухой лед/ацетон, которую затем заменяли баней лед/вода. Смесь перемешивали 10 мин при температуре от -10 2) до 0C, после чего медленно добавляли раствор (R)-6-хлор-3-фенилиндан-1-она (IVb) (синтезированный как в примере 12) (10,0 г) в THF (80 мл), поддерживая температуру ниже 5C. После перемешивания приблизительно в течение 1/2 ч медленно добавляли бутил-литий (1,6 М в гексане, 33 мл), поддерживая температуру ниже 5C. После перемешивания при 0-5C в течение 21/2 ч медленно добавляли соляную кислоту (4 М, 100 мл). Фазы разделяли и водную фазу экстрагировали два раза этилацетатом (100 мл). Объединенные органические фазы промывали 25% хлоридом натрия (100 мл) и перемешивали 10 мин с сульфатом магния (26 г), активированным углем (1 г) и силикагелем (2,6 г). После фильтрования органическую фазу упаривали досуха, используя роторный испаритель. Остаток перекристаллизовывали из 2 пропанола (40 мл). Продукт отделяли фильтрованием, промывали ледяным 2-пропанолом (20 мл) и сушили в вакуумном термостате при 50C в течение ночи. Выход 5,85 г (60%). Температура плавления 95,2C, ЯМР-спектр соответствует структуре, []D20-1,1 (с=1,0, метанол). Содержание С, Н, рассчитано для C15H11OCl: С 74,23, Н 4,57; найдено: С 74,29, Н 4,62. Энантиомерный избыток -1,0% в соответствии с данными хирального анализа. Чистота 97% в соответствии с LC-MS. Нижеприведенная таблица суммирует полученные результаты. Указано время перемешивания при 0C после смешения всех компонентов. Энантиомерный избыток; отрицательный знак указывает, что IV в избытке. 3)LDA получали, используя 1,25 эквивалентов диизопропиламина и 1,50 эквивалентов BuLi. 6) В этом эксперименте весь BuLi, указанный также как количество основания 2, добавляли в начале эксперимента, до добавления соединения IVb, т.е. 1,25 эквивалентов диизопропиламина и всего 2,50 эквивалентов BuLi применяли. 2) ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы I (соединение I) или его соли, включающий получение соединения IVa разделением рацемического соединения IV с использованием хиральной хроматографии превращение соединения формулы IVa в соответствующий спирт Va с цис-конфигурацией превращение спиртовой группы цис-спирта формулы Va в удаляемую группу LG с получением соединения формулы VI взаимодействие соединения VI с 2,2-диметилпиперазином с получением соединения формулы I 2. Способ по п.1, в котором LG является галогеном или сульфонатом. 3. Способ по п.2, в котором LG является Cl, Br, тозилатом или мезилатом. 4. Способ по п.1, в котором соединение VI осаждают из подходящего растворителя. 5. Способ по п.4, в котором LG является галогеном и растворитель представляет собой алкан. 6. Способ по п.5, в котором соединение I осаждают в виде соли. 7. Способ по п.6, в котором образовавшаяся соль является солью фумаровой кислоты, солью малеиновой кислоты или солью соляной кислоты и соединения I. 8. Способ по п.5, в котором соединение I выделяют в виде свободного основания. 9. Способ по любому одному из пп.1-8, включающий взаимодействие соединения VI с 1-защищенным 2,2-диметилпиперазином VII, в котором PG является защитной группой, с получением соединения формулы VIII; и снятие защитной группы соединения VIII для получения соединения I,где соединения VII и VIII являются следующими: 10. Способ по любому одному из пп.1-9, в котором хиральную хроматографию осуществляют с использованием хиральной жидкостной хроматографии. 11. Способ по любому одному из пп.1-10, в котором хиральную хроматографию осуществляют на хиральной неподвижной фазе. 12. Способ по п.11, в котором хиральную хроматографию осуществляют на колонке с силикагелем,покрытым оболочкой модифицированной амилозы. 13. Способ по п.12, в котором хиральную хроматографию осуществляют на колонке с силикагелем,покрытым оболочкой трис-[(S)метилбензилкарбамат]амилозы. 14. Способ по п.13, в котором для хиральной хроматографии применяют растворитель, представляющий собой смесь н-гептана и этанола, и, необязательно, N,N-диэтиламин. 15. Способ по любому одному из пп.1-9, в котором хиральную хроматографию осуществляют с использованием хроматографии с суб- или суперкритической подвижной фазой. 16. Способ по п.15, в котором хиральную хроматографию осуществляют с использованием хроматографии с суперкритической подвижной фазой. 17. Способ по п.15 или 16, в котором хиральную хроматографию осуществляют на хиральной неподвижной фазе. 18. Способ по п.17, в котором хиральную хроматографию осуществляют на колонке с силикагелем,покрытым оболочкой, состоящей из хирального полимера, или на колонке с силикагелем с иммобилизованным хиральным полимером, или на колонке с силикагелем с ковалентно связанным хиральным мономером. 19. Способ по п.18, в котором хиральную хроматографию осуществляют на колонке с силикагелем,покрытым оболочкой трис-(3,5-диметилфенилкарбамат)амилозы или трис-[(S)-метилбензилкарбамат]амилозы, или на колонке с силикагелем, с иммобилизованной трис-(3,5 диметилфенилкарбамат)амилозой, или на колонке с силикагелем, покрытым оболочкой трис-(3,5 диметилфенилкарбамат)целлюлозы или трис-(4-метилбензоат) целлюлозы, или на колонке с силикагелем с ковалентно связанным амином 3,5-динитробензоилтетрагидрофенантрена. 20. Способ по любому одному из пп.18 или 19, в котором хиральную хроматографию осуществляют на колонке с силикагелем, покрытым оболочкой трис-(3,5-диметилфенилкарбамат)амилозы. 21. Способ по любому одному из пп.18-20, в котором хиральную хроматографию осуществляют с модификатором, выбранным из группы: метанол, этанол, 2-пропанол или ацетонитрил, необязательно содержащей диэтиламин. 22. Способ по п.21, в котором модификатор содержит 0,1% диэтиламина. 23. Способ по любому одному из пп.1-22, включающий рециклирование соединения IVb превращением энантиомерно обогащенного соединения IVb в рацемическое соединение IV, где IVb и IV являются: 24. Способ по п.23, в котором рацемизацию осуществляют с использованием основания или смеси двух или более оснований. 25. Способ по п.23 или 24, в котором рацемизацию осуществляют с использованием одного или более эквивалента ненуклеофильного основания ("первого основания"), за которым следует добавление каталитического количества одного или больше эквивалента того же самого или другого основания("второго основания"). 26. Способ превращения энантиомерно обогащенного соединения IVb в рацемическое соединение и в котором рацемизацию осуществляют с использованием одного или более эквивалента ненуклеофильного основания, за которым следует добавление каталитического количества одного или более эквивалента того же самого или другого основания. 27. Способ по п.25 или 26, в котором рацемизацию осуществляют с использованием одного или более эквивалента ненуклеофильного основания ("первое основание"), выбираемого из группы, состоящей из диалкиламида металла, такого как диэтиламид лития, диизопропиламид лития (LDA) и тетраметилпиперидид лития; бис-силиламида металла, такого как бис-(триметилсилил)амид лития (LiHMDS); и алкоксида металла, такого как трет-бутоксид лития, за которым следует добавление каталитического количества одного или более эквивалента того же самого или другого основания ("второго основания"), выбираемого из группы, состоящей из диалкиламида металла, такого как диэтиламид лития, диизопропиламид лития (LDA) и тетраметилпиперидид лития; бис-силиламида металла, такого как бис-(триметилсилил)амид щелочного металла; алкоксида металла, такого как трет-бутоксид калия, и алкила металла, такого как бутиллитий или трет-бутиллитий. 28. Способ по пп.25-27, в котором "первое основание" и "второе основание" присутствуют сначала. 29. Способ по любому одному из пп.23-28, в котором рацемическое соединение IV перекристаллизовывается из подходящего растворителя. 30. Способ по п.29, в котором растворитель выбирают из группы, состоящей из C1-6 спиртов, включая этанол или 2-пропанол или их смеси. 31. Способ получения соединение IX, включающий получение соединения IVa разделением рацемического соединения IV, используя хиральную хроматографию превращение соединения формулы IVa в соответствующий спирт Va с цис-конфигурацией превращение спиртовой группы цис-спирта формулы Va в удаляемую группу LG с получением соединения формулы VI взаимодействие соединения VI с 2,2-диметилпиперазином с получением соединения формулы I метилирование соединения формулы I по вторичному амину с получением соединения формулы IX. 32. Способ по п.31, в котором соединение IX осаждается в виде соли янтарной или малоновой кислоты соединения IX. 33. Способ по п.32, в котором соединение IX осаждается в виде кристаллической соли янтарной или малоновой кислоты.
МПК / Метки
МПК: C07C 49/697, C07D 295/073
Метки: получения, способ, транс-1-((1r,3s)-6-хлор-3-фенилиндан-1-ил)-3,3-диметилпиперазина
Код ссылки
<a href="https://eas.patents.su/17-18059-sposob-polucheniya-trans-1-1r3s-6-hlor-3-fenilindan-1-il-33-dimetilpiperazina.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения транс-1-((1r,3s)-6-хлор-3-фенилиндан-1-ил)-3,3-диметилпиперазина</a>
Сукцинатная и малонатная соли транс-4-((1r,3s )-6 хлор-3-фенилиндан -1-ил)-1,2,2-триметилпиперазина и их применение в качестве лекарственного средства

Номер патента: 14641
Опубликовано: 30.12.2010
Авторы: Сване Хенрик, Нильсен Оле, Хауэллз Марк, Даль Аллан Карстен, Лопес Де Диего Хейди, Бан-Андерсен Бенни, Ринггар Лоне Мюнк, Люнгсе Ларс Оле
МПК: C07C 25/22, A61K 31/495, A61P 25/18...
Метки: малонатная, 1-ил)-1,2,2-триметилпиперазина, средства, соли, хлор-3-фенилиндан, качестве, лекарственного, сукцинатная, транс-4-((1r,3s, применение
Формула / Реферат:
1. Сукцинатная соль или малонатная соль транс-4-((1R,3S)-6-хлор-3-фенилиндан-1-ил)-1,2,2-триметилпиперазина формулы (I)2. Сукцинатная соль по п.1, которая представляет собой гидросукцинатную соль соединения формулы (I).3. Кристаллическая гидросукцинатная соль соединения I, определенного в п.1.4. Соль по п.3, которая представляет собой кристаллическую альфа-форму.5. Соль по п.3 или 4, кристаллическую форму которой характеризуют порошковой...
Кристаллическое основание транс-1-((1r,3s)-3-фенил-6-хлороиндан-1-ил)-3,3-диметилпиперазина

Номер патента: 17631
Опубликовано: 28.02.2013
Авторы: Лопес Де Диего Хейди, Банг-Андерсен Бенни
МПК: A61K 31/495, C07D 295/073, A61P 25/18...
Метки: транс-1-((1r,3s)-3-фенил-6-хлороиндан-1-ил)-3,3-диметилпиперазина, кристаллическое, основание
Формула / Реферат:
1. Кристаллическое основание соединения формулы (I) транс-1-((1R,3S)-3-фенил-6-хлороиндан-1-ил)-3,3-диметилпиперазинахарактеризующееся порошковой рентгеновской дифрактограммой, полученной с использованием CuKα1 облучения (l=1,5406 Å), показывающей сигналы при следующих 2θ углах: 6,1, 11,1, 12,1, 16,2, 16,8, 18,3, 18,6, 20,0.2. Кристаллическое основание по п.1, характеризующееся наличием ДСК термограммы в соответствии с фиг....
Способ получения хирально чистого n-(транс-4- изопропилциклогексилкарбонил)-d-фенилаланина и его кристаллических модификаций

Номер патента: 8883
Опубликовано: 31.08.2007
Авторы: Хегедуш Бела, Газдаг Мария, Бабьяк Моника, Тарканьи Габор, Тёрли Йожеф, Семзё Аттила, Гизур Тибор
МПК: C07C 231/24, C07C 231/12, C07C 233/63...
Метки: способ, модификаций, получения, изопропилциклогексилкарбонил)-d-фенилаланина, чистого, n-(транс-4, хирально, кристаллических
Формула / Реферат:
1. Способ получения кристаллической модификации "G" N-(транс-4-изопропилциклогексилкарбонил)-D-фенилаланина (натеглинида) формулы (I) путем обработки соединения общей формулы (II) где R означает низшую алкильную группу (C1-С4) или водород, основанием с образованием соли щелочного металла и высвобождения продукта из данной соли кислотой, отличающийся тем, что кислотное высвобождение продукта проводится при температуре ниже 20шС, предпочтительно...
Способ простого получения (3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6-нитрохиназолин-4 -ил] амина или (3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6 -аминохиназолин-4-ил] амина

Номер патента: 5213
Опубликовано: 30.12.2004
Авторы: Шнайдер Зимон, Барт Хуберт, Штайнер Клаус
МПК: C07D 239/94
Метки: 3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6, аминохиназолин-4-ил, способ, ил, 3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6-нитрохиназолин-4, простого, амина, получения
Формула / Реферат:
1. Способ получения (3-хлор-4-фторфенил)-[7-(3-морфолин-4-илпропокси)-6-нитрохиназолин-4-ил]амина (I) или (3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6-аминохиназолин-4-ил]амина (VII) отличающийся тем, что в реакции, осуществляемой в одном реакционном сосуде в 3, соответственно 4 стадии, получают вначале 7-фтор-6-нитрохиназолин-4(3H)-он (III) с помощью тионилхлорида превращают в 4-хлор-7-фтор-6-нитрохиназолин (IV) который с помощью...
Способ получения [is-[1a,2b,3b,4a(s*)]]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3н-имидазо[4,5-b]пиридин-3-ил]-n-этил-2,3-дигидроксициклопентанкарбоксамида.

Номер патента: 1989
Опубликовано: 22.10.2001
Авторы: Цуей Чинг Т., Шах Харшавадан К., О'брайен Майкл К, Рейлли Лоренс В., Леон Патрик, Паунер Тори Х., Томпсон Майкл Д., Гарсиа Эрве, Вальтер Фрэнсис Л., Ванасс Бенуа Дж.
МПК: C07D 409/12
Метки: способ, is-[1a,2b,3b,4a(s*)]]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3н-имидазо[4,5-b]пиридин-3-ил]-n-этил-2,3-дигидроксициклопентанкарбоксамида, получения
Формула / Реферат:
1. Способ получения [1S-[1a,2b,3b,4a(S*)]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3Н-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамида (соединение (I)), включающий взаимодействие [1S-[1a,2b,3b,4a(S*)]]-4-[[3-амино-4-[[1-[(3-хлор-2-тиенил)метил]пропил]амино]-2-пиридинил]амино]-N-этил-2,3-дигидроксициклопентанкарбоксамида (соединение (IX)) со сложным эфиром ортоформиата, ацетатом формамидина или диметилацеталем...
Предыдущий патент: Устройство для контроля подлинности банкнот
Следующий патент: Усовершенствованная система катализаторов
Случайный патент: Фильтры табачного дыма для курительных устройств с пористыми массами, имеющими наполнение частицами углерода и перепад давления в капсуле