Способ получения кислотных солей гемифлоксацина
Номер патента: 7222
Опубликовано: 25.08.2006
Авторы: Чои Санг-Чул, Чои Хоон, Чои Бо-Сеунг, Нам До Хиун







Формула / Реферат
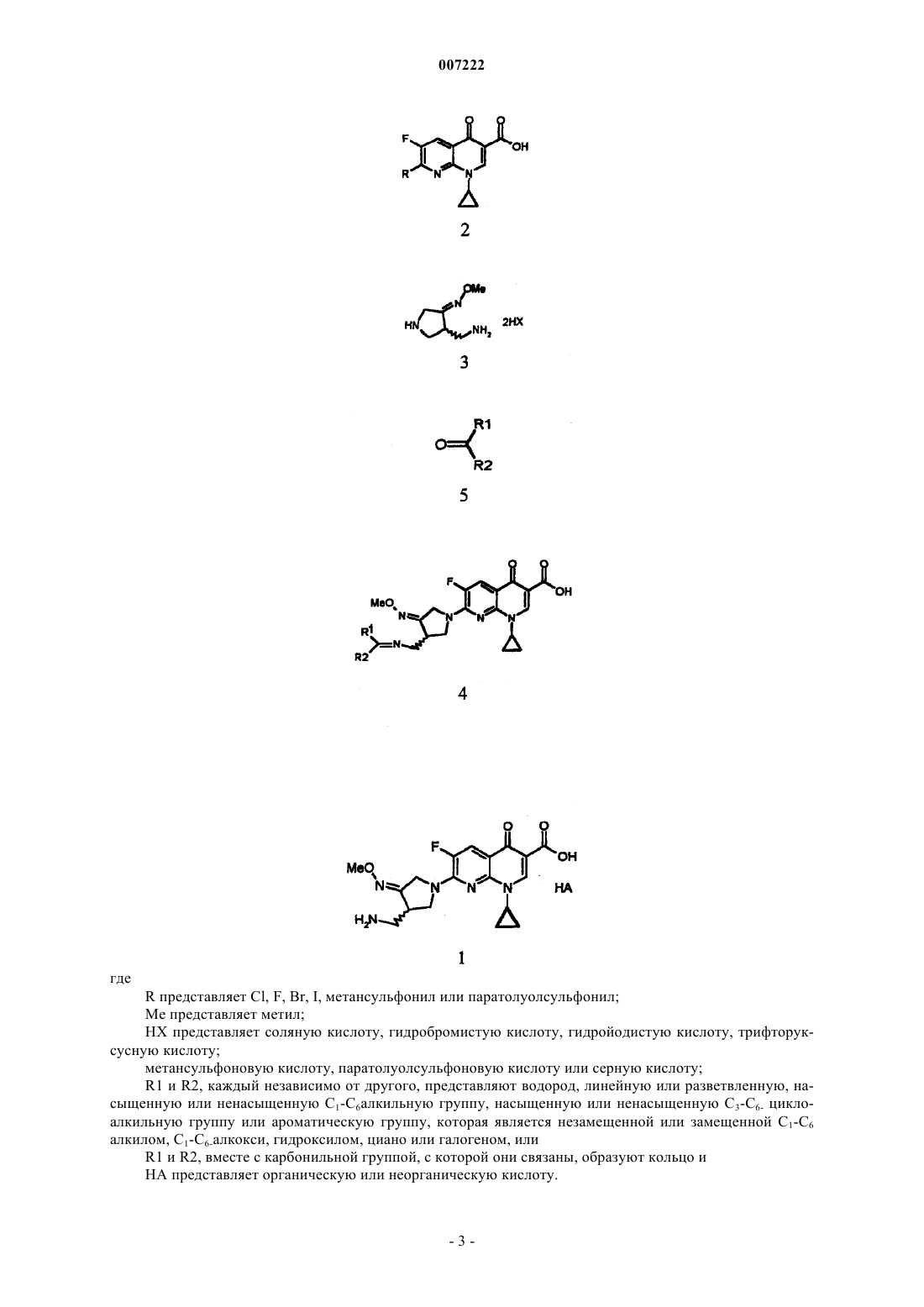
1. Способ получения солей гемифлоксацина с кислотами, представляемых формулой 1, который включает стадии:
a) осуществление реакции сочетания путем добавления соединения формулы 5 к нафтиридинкарбоновой кислоте формулы 2 и 3-аминометил-4-метоксииминопирролидиновой соли формулы 3 в воде, органическом растворителе или их смеси в присутствии органического основания и
b) одновременное удаление защиты и образование соли посредством добавления кислоты формулы НА к полученному соединению формулы 4 в воде, органическом растворителе или их смеси


где R представляет Cl, F, Br, I, метансульфонил или паратолуолсульфонил;
Me представляет метил;
НХ представляет соляную кислоту, гидробромистую кислоту, гидройодистую кислоту, трифторуксусную кислоту, метансульфоновую кислоту, паратолуолсульфоновую кислоту или серную кислоту;
R1 и R2, каждый независимо, представляет водород, линейную или разветвленную, насыщенную или ненасыщенную С1-С6алкильную группу, насыщенную или ненасыщенную С3-С6циклоалкильную группу или ароматическую группу, которая является незамещенной или замещенной C1-C6алкилом, C1-C6алкокси, гидроксилом, циано или галогеном, или
R1 и R2 вместе с карбонильной группой, с которой они связаны, образуют кольцо и
НА представляет органическую кислоту или неорганическую кислоту.
2. Способ по п.1, в котором стадию а), стадию b) или как стадию а), так и стадию b) осуществляют в смешанном растворителе, представляющем смесь органического растворителя с водой.
3. Способ по п.1, в котором соединение формулы 5 выбрано из группы, состоящей из бензальдегида, 2-хлорбензальдегида, 2-гидроксибензальдегида, 4-метоксибензальдегида и 2-метилбензальдегида.
4. Способ по п.2, в котором органический растворитель на стадии а) является ацетонитрилом, а органический растворитель на стадии b) является изопропанолом или тетрагидрофураном (ТГФ).
5. Способ по п.1, в котором органическое основание выбрано из группы, состоящей из триэтиламина, триметиламина, диизопропилэтиламина, 1,8-диазабицикло[5,4,0]ундец-7-ена и 1,5-диазабицикло[4,3,0]нон-5-она.
6. Способ по п.1, в котором соединение формулы 5 используют в 1-3-кратном количестве по отношению к количеству соединения формулы 2.
7. Способ по п.1, в котором органическое основание на стадии а) используют в 3-4-кратном количестве по отношению к количеству соединения формулы 2 и реакцию осуществляют при температуре реакции от 0 до 30шС.
8. Способ по п.7, в котором органическим основанием является триэтиламин.
9. Способ по п.1, в котором кислоту формулы НА используют в количестве от 80 до 120 моль.% по отношению к количеству соединения формулы 4, причем температура реакционной смеси при добавлении кислоты находится в интервале 40-50шС, а температура после добавления кислоты находится в интервале 0-20шС.
10. Способ по любому одному из пп.1-9, в котором кислота формулы НА является метансульфоновой кислотой.
11. Промежуточное соединение, представляемое следующей ниже формулой 4, для получения солей гемифлоксацина с кислотой по п.1

где Me, R1 и R2 имеют определения, представленные в п.1.
Текст
007222 Данное изобретение относится к новому способу получения солей хинолонкарбоновой кислоты, то есть 7-(3-аминометил-4-метоксииминопирролидин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8 нафтиридин-3-карбоновой кислоты (далее называемой гемифлоксацином), обладающих высокой антимикробной активностью и представляемых следующей формулой 1Me представляет метил; НА является органической кислотой или неорганической кислотой. Указанный гемифлоксацин и его соли являются соединениями, описанными в корейском патенте 131999, принадлежащем авторам данного изобретения (корейская патентная заявка 94-13604, иностранных патентах, соответствующих этому патенту: ЕР 688722 А 1, JP патенте 41050/1996, российском патенте 2120940, канадском патенте 2151890, китайском патенте 1114959 и патентах США 5962468, 5869670, 5840916, 5776944, 5698570 и 5633262). Эти соединения обладают высокой антимикробной активностью и, более того, могут эффективно использоваться в качестве средств для лечения людей и животных, инфицированных бактериями. Указанные кислотные соли гемифлоксацина получены трехстадийным реакционным процессом, то есть посредством реакции сочетания, образования соли и перекристаллизацией, как представлено следующей схемой реакций 1: Схема реакций 1R представляет Cl, F, Br, I, метансульфонил или паратолуолсульфонил; НХ представляет соляную кислоту, гидробромистую кислоту, гидройодистую кислоту, трифторуксусную кислоту; метансульфоновую кислоту, паратолуолсульфоновую кислоту или серную кислоту; НА представляет органическую кислоту или неорганическую кислоту. Как показано на представленной выше схеме реакций 1, соединение (1) получают трехстадийным реакционным процессом, то есть реакцией сочетания, образования соли и перекристаллизации. Причиной осуществления трехстадийного реакционного процесса является то, что в реакции, в результате протекания побочной реакции, образуется в качестве примеси соединение (8) в количестве примерно 6-12%,и соединение (8) остается в соединении (6) в количестве от примерно 0,3 до 1,0%. Для удаления получающейся при реакции сочетания примеси в пределах 0,1% или менее, нужно осуществлять вторую стадию, то есть процесс образования соли. И, наконец, органический растворитель, использованный в процессе образования соли, нужно удалить со стадии перекристаллизации. По трехстадийному способу получали кислотную соль гемифлоксацина (1) в виде сырого лекарственного средства, имеющего высокую чистоту с общим выходом примерно 65%. Так как получающуюся в результате реакции сочетания представленного выше способа примесь (8) трудно удалить, необходимо осуществлять стадии образования соли и перекристаллизации для удаления примеси. Были проведены интенсивные исследования с целью сокращения стадии вышеуказанного трехстадийного способа до двухстадийного способа. В результате, было обнаружено, что двухстадийный способ получения, представленный ниже, имеет ряд преимуществ, таких как простота процесса получения,улучшение производительности и повышение выхода. Поэтому осуществлено данное изобретение. Данное изобретение представляет способ получения солей гемифлоксацина с кислотами, представляемых формулой 1, который включает следующие стадии:a) добавление соединения формулы 5 к нафтиридинкарбоновой кислоте формулы 2 и 3 аминометил-4-метоксииминопирролидиновой соли формулы 3 в воде, органическом растворителе или смешанном растворителе в присутствии органического основания для осуществления реакции сочетания,иb) добавление кислоты формулы НА к полученному соединению формулы 4 в воде, органическом растворителе или смешанном растворителе для осуществления реакций удаления защиты и образования соли в одно и то же времяMe представляет метил; НХ представляет соляную кислоту, гидробромистую кислоту, гидройодистую кислоту, трифторуксусную кислоту; метансульфоновую кислоту, паратолуолсульфоновую кислоту или серную кислоту;R1 и R2, каждый независимо от другого, представляют водород, линейную или разветвленную, насыщенную или ненасыщенную C1-C6 алкильную группу, насыщенную или ненасыщенную С 3-С 6- циклоалкильную группу или ароматическую группу, которая является незамещенной или замещенной C1-C6 алкилом, C1-C6-алкокси, гидроксилом, циано или галогеном, илиR1 и R2, вместе с карбонильной группой, с которой они связаны, образуют кольцо и НА представляет органическую или неорганическую кислоту.-3 007222 Наилучший способ осуществления изобретения Способ получения по данному изобретению подробно описан следующей схемой реакций 2: Схема реакций 2Me, R, R1, R2, НХ и НА, имеют значения, представленные выше. Как показано на представленной выше схеме реакций 2, чтобы сократить стадии образования соли и перекристаллизации до одной стадии путем предотвращения образования примеси (8), соединение (5),имеющее карбонильную группу, может быть добавлено к соединению (3) в реакции сочетания, для защиты первичной аминогруппы в соединении (3) с помощью соединения (5). Благодаря этой защите может быть в значительной степени предотвращена продукция побочного продукта (8) в количестве 0,1% или менее. Полученное в результате соединение (4), которое получают в соответствии с вышеуказанным способом с выходом примерно 90% или более, обрабатывают кислотой формулы НА для удаления защиты и образования соли в одну стадию. Посредством представленной выше методики, исключая стадию реакции перекристаллизации, получают соль гемифлоксацина с кислотой - соединение (1) с выходом 90% или более, при этом стадии процесса получения упрощаются. За счет упрощения способа получения могут быть получены следующие преимущества: снижение времени производства, улучшение производительности и повышение выхода. Способ по данному изобретению объяснен подробно в виде примера ниже. Во-первых, способ синтеза соединения (4) (стадия а) состоит из растворения соединения (3), соединения (5) и органического основания, например, триэтиламина в реакционном растворителе, например,смешанном растворителе ацетонитрил с водой, и добавления туда соединения (2), а затем взаимодействия полученной смеси. Условия данной реакции являются следующими. 1) В качестве реакционного растворителя можно использовать воду, органический растворитель,такой как ацетонитрил, тетрагидрофуран (ТГФ), метанол и этанол, или смешанный растворитель из органического растворителя с водой. Для увеличения чистоты и выхода, предпочтительно использовать смешанный растворитель из ацетонитрила с водой. 2) В качестве соединения (5) можно использовать кетоновое или альдегидное соединение, такое как формальдегид, ацетон, бензальдегид, 1-бутилальдегид и тому подобное. Для увеличения чистоты и выхода, предпочтительно использовать бензальдегидное производное, такое как бензальдегид, 2 гидроксибензальдегид, 2-хлорбензальдегид и тому подобное. В частности, более предпочтителен бензальдегид, позволяющий снизить стоимость процесса и стабильность. Предпочтительно, когда бензальдегид используют в молярном количестве, равном молярному количеству соединения (2) или большем. 3) Реакционная температура при этом может находиться в широком интервале от 0 до 80 С. Однако наиболее предпочтительная температура находится в интервале 20-30 С с точки зрения скорости реакции, выхода и чистоты. 4) В качестве органического основания можно использовать триэтиламин, триметиламин, диизопропилэтиламин, ДБУ (1,8-диазабицикло[5,4,0]ундец-7-ен), ДБН (1,5-диазабицикло[4,3,0]-нон-5-он), а также ряд других оснований. С точки зрения стоимости и выхода наиболее подходящим основанием является триэтиламин. Используемое количество его составляет 3 молярных количества или более по отношению к соединению (2). Вышеприведенным реакционным процессом с высоким выходом (более 90%), может быть получено соединение (4), имеющее высокую чистоту. Во-вторых, способ (стадия b) получения соединения (1) из соединения (4) включает растворение соединения (4) в реакционном растворителе, например смешанном растворителе, таком как изопропанол с водой, нагревание полученной смеси, добавление кислоты формулы НА, например метансульфоновой кислоты, для одновременного снятия защиты и образования соли, и последующее охлаждение реакционного продукта с получением соединения (1), минуя стадию перекристаллизации. Данную реакцию осуществляют в следующих условиях:-4 007222 1) В качестве растворителя в реакции можно использовать воду, спирт, такой как изопропанол,ТГФ, метанол, этанол, бутанол и тому подобное, или смешанный растворитель - смесь спирта с водой. Для увеличения чистоты и выхода целевого продукта предпочтительно использовать смешанный растворитель, представляющий смесь изопропанола с водой. 2) В качестве кислоты можно использовать ряд кислот, представляющих НА, такие как соляная кислота, метансульфоновая кислота, серная кислота, фосфорная кислота, уксусная кислота, лимонная кислота, виннокаменная кислота, пергидрохлорная кислота, пикриновая кислота, (+)-камфорсульфоновая кислота и тому подобное. В частности, наиболее подходящей является метансульфоновая кислота. Для увеличения чистоты и выхода целевого продукта предпочтительно использовать молярное количество кислоты, равное молярному количеству соединения (4), до примерно + 20% или около этого от равного молярного количества. 3) Температура реакции может использоваться в широком интервале от 0 до 100 С. Однако для увеличения скорости реакции по конечному соединению (1), выхода и чистоты целевого продукта предпочтительной температурой являются температура реакции 40-50 С при добавлении кислоты и температура реакции 020 С после добавления кислоты. Посредством осуществления вышеприведенного реакционного процесса может быть получено соединение (4), имеющее высокую чистоту, с высоким выходом (более 90%). Как описано выше, новый двухстадийный способ синтеза позволяет достигнуть улучшенный эффект, а именно упростить способ получения, повысить выход (с примерно 65% до по меньшей мере 80%), улучшить производительность процесса, снизить стоимость производства и тому подобное за счет сокращения стадий общепринятого способа синтеза (с трех до двух стадий). В частности, преимуществом данного способа является то, что способ может быть применен к антибиотикам хинолоновой группы, имеющим строение, сходное со строением гемифлоксацина. Поэтому данное изобретение является усовершенствованным способом получения. Данное изобретение подробно раскрыто с помощью следующих примеров, которые не предназначены для ограничения объема данного изобретения. Примеры Пример 1. Получение 7-(3-бензилидинаминометил-4-метоксиимино-1-пирролидинил)-1 циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты. Ацетонитрил (1900 мл), 3-аминометил-4-метоксиимино-пирролидина диметансульфонат (248,0 г) и воду (100 мл) последовательно вводили в реакционный сосуд и охлаждали до 0-5 С. К реакционной смеси последовательно добавляли бензальдегид (97,6 г) и триэтиламин (229,1 г). После перемешивания смеси в течение 0,5 ч в нее вводили 7-хлор-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3 карбоновую кислоту (200,0 г). Полученную реакционную смесь медленно нагревали до комнатной температуры, перемешивая ее. Затем реакционную смесь перемешивали в течение примерно 3 ч при комнатной температуре. Продукт реакции, который образовался в виде дисперсного раствора при получении соединения, указанного в заголовке, отфильтровывали, промывали водой и ацетонитрилом, а затем сушили с получением 320,3 г соединения, названного в заголовке (Выход: 94,8%). 1 Н ЯМР (, CDCl3): 8,66 (с, 1 Н), 8,32 (с, 1 Н), 7,98 (д, J=12,4 Гц, 1 Н), 7,60 (д, J=7,0 Гц, 2 Н), 7,37 (t,J=7,4 Гц, 1 Н), 7,31 (t, J=7,4 Гц, 2 Н), 4,58 (с, 2 Н), 4,214,15 (м, 2 Н), 4,00 (м, 1 Н), 3,93 (с, 3 Н), 3,83 (м, 1 Н),3,56 (м, 1 Н), 3,40 (м, 1 Н), 1,21 (м, 2 Н), 1,00 (м, 2 Н) Масс (FAB): 478 (М+Н) Пример 2. Получение 7-[3-(2-хлорбензилидин)аминометил-4-метоксиимино-1-пирролидинил]-1 циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты.-5 007222 Ацетонитрил (100 мл), диметансульфонат 3-аминометил-4-метоксииминопирролидина (12,5) г, 2 хлорбензальдегид (10,0 г) и триэтиламин (12,2 г) последовательно вводили в реакционный сосуд при комнатной температуре. После перемешивания смеси в течение примерно 0,5 ч туда же вводили 7-хлор 1-циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновую кислоту (10,0 г). Полученную реакционную смесь перемешивали в течение примерно 15 ч при комнатной температуре, охлаждали до 05 С и вновь перемешивали в течение примерно 3 ч. Соединение, указанное в заголовке, в виде твердого вещества отфильтровывали, промывали ацетонитрилом и сушили с получением 16,3 г соединения,указанного в заголовке (выход: 90,0%). 1 Н ЯМР (, CDCl3) : 8,74 (с, 1 Н), 8,66 (с, 1 Н), 7,96 (д, J=12,4 Гц,1 Н), 7,84 (д, J=7,3 Гц, 1 Н), 7,29 (м,2 Н), 7,16 (м, 1 Н), 4,59 (шир.с, 2 Н), 4,18 (м, 2 Н), 4,02 (м, 1 Н), 3,94 (с, 3H), 3,93 (м, 1 Н), 3,59 (м, 1 Н), 3,42 Ацетонитрил (100 мл), диметансульфонат 3-аминометил-4-метоксииминопирролидина (12,5) г, 2 гидроксибензальдегид (8,6 г) и триэтиламин (12,2 г) последовательно вводили в реакционный сосуд при комнатной температуре. После перемешивания смеси в течение примерно 0,5 ч туда же вводили 7-хлор 1-циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновую кислоту (10,0 г). Полученную реакционную смесь перемешивали в течение примерно 15 ч при комнатной температуре, охлаждали до 0-5 С и вновь перемешивали в течение примерно 3 ч. Соединение, указанное в заголовке, в виде твердого вещества отфильтровывали, промывали ацетонитрилом и сушили с получением 16,0 г соединения,указанного в заголовке (выход: 91,8%). 1 Н ЯМР (, CDCl3): 8,68 (с, 1 Н), 8,42 (с, 1 Н), 8,01 (д, J=12,4 Гц, 1 Н), 7,30-7,20 (м, 3H), 6,90-6,82 (м,2 Н), 4,68-4,53 (м, 2 Н), 4,32-4,24 (м, 1 Н), 4,06 (дд, J1=11,9 Гц, J2=5,5 Гц, 1 Н), 4,02-3,85 (м, 3H), 3,95 (с, 3H),3,60 (м, 1 Н), 3,40 (м, 1 Н), 1,29-1,21 (м, 2 Н), 1,07-1,0 (м, 2 Н). Масс (FAB): 494 (М+Н) Пример 4. Получение 7-[3-(4-цианобензилидин)аминометил-4-метоксиимино-1-пирролидинил]-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты. Ацетонитрил (100 мл), диметансульфонат 3-аминометил-4-метоксииминопирролидина (12,5 г) и 1 нафтальдегид (11,1 г) последовательно вводили в реакционный сосуд при комнатной температуре и охлаждали до 0-5 С. К реакционной смеси по каплям добавляли триэтиламин (12,2 г). После перемешивания смеси в течение примерно 0,5 ч реакционную смесь разбавляли добавлением этанола (30 мл). В реакционную смесь вводили 7-хлор-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3 карбоновую кислоту (10,0 г). После медленного повышения температуры реакционной смеси до комнатной температуры реакционную смесь перемешивали в течение примерно 15 ч. Соединение, указанное в заголовке, в виде твердого вещества отфильтровывали, промывали водой и этанолом и сушили с получением 15,7 г соединения, указанного в заголовке (Выход: 84,4%). 1 Н ЯМР (, CDCl3.): 8,86 (м, 2 Н), 8,55 (с, 1 Н), 7,82 (м, 3H), 7,73 (м, 1 Н), 7,40 (м, 3H), 4,60 (м, 2 Н),4,24 (м, 2 Н), 4,08 (м, 1 Н), 3,99 (м, 1 Н), 3,95 (с, 3H), 3,45 (м, 2 Н), 1,13 (м, 2 Н), 0,89 (м, 2 Н). Масс (FAB): 528 (М+Н) Пример 7. Получение соли метансульфоновой кислоты с 7-(аминометил-4-метоксиимино-1 пирролидинил)-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислотой. Воду (60 мл), соединение (30,0 г), синтезированное в примере 1, и изопропанол (210 мл) последовательно вводили в реакционный сосуд и нагревали до 40-45 С. В реакционную смесь по каплям добавляли метансульфоновую кислоту (6,22 г). После перемешивания реакционной смеси в течение примерно 0,5 ч при температуре 40-45 С ее охлаждали до 27-35 С. К реакционной смеси добавляли соединение (1) (0,03 г). Реакционную смесь медленно охлаждали до комнатной температуры и перемешивали в течение примерно 17 ч с выпадением в осадок указанного в заголовке соединения в виде твердого вещества. Реакционную смесь в виде дисперсного раствора охлаждали до -1-1 С, перемешивали в течение 3 ч, фильтровали, промывали изопропанолом, сушили и адсорбировали с получением 29,0 соединения, указанного в заголовке (выход: 95,1%). 1 Н ЯМР (, CDCl3): 8,59 (с, 1 Н), 8,06 (д, J=12,4 Гц, 1 Н), 4,58 (шир.с, 2 Н), 4,37 (м, 1 Н), 3,90 (с, 3H),3,83 (шир.с, 1 Н), 3,71 (м, 1 Н), 3,40 (м, 1 Н), 3,243,10 (м, 2 Н), 2,32 (с, 3H), 1,201,05 (м, 2 Н), 1,031,02 (м,2 Н). Масс (FAB): 486 (М+Н) Пример 8. Получение соли метансульфоновой кислоты с 7-(аминометил-4-метоксиимино-1 пирролидинил)-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислотой.-7 007222 Получали соединение, указанное в заголовке, с выходом, равным 91,7% в соответствии с методикой, которая описана в примере 7, за исключением того, что использовали ТГФ (240 мл) вместо изопропанола (210 мл), а метансульфоновую кислоту использовали в количестве 6,04 г вместо 6,22 г. Новый двухстадийный способ получения в соответствии с данным изобретением позволяет упростить способ получения, повысить выход, улучшить производительность процесса, снизить стоимость производства и тому подобное, за счет сокращения общепринятого трехстадийного способа синтеза до двухстадийного способа. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения солей гемифлоксацина с кислотами, представляемых формулой 1, который включает стадии:a) осуществление реакции сочетания путем добавления соединения формулы 5 к нафтиридинкарбоновой кислоте формулы 2 и 3-аминометил-4-метоксииминопирролидиновой соли формулы 3 в воде, органическом растворителе или их смеси в присутствии органического основания иb) одновременное удаление защиты и образование соли посредством добавления кислоты формулы НА к полученному соединению формулы 4 в воде, органическом растворителе или их смеси-8 007222 НХ представляет соляную кислоту, гидробромистую кислоту, гидройодистую кислоту, трифторуксусную кислоту, метансульфоновую кислоту, паратолуолсульфоновую кислоту или серную кислоту;R1 и R2, каждый независимо, представляет водород, линейную или разветвленную, насыщенную или ненасыщенную С 1-С 6 алкильную группу, насыщенную или ненасыщенную С 3-С 6 циклоалкильную группу или ароматическую группу, которая является незамещенной или замещенной C1-C6 алкилом, C1C6 алкокси, гидроксилом, циано или галогеном, илиR1 и R2 вместе с карбонильной группой, с которой они связаны, образуют кольцо и НА представляет органическую кислоту или неорганическую кислоту. 2. Способ по п.1, в котором стадию а), стадию b) или как стадию а), так и стадию b) осуществляют в смешанном растворителе, представляющем смесь органического растворителя с водой. 3. Способ по п.1, в котором соединение формулы 5 выбрано из группы, состоящей из бензальдегида, 2-хлорбензальдегида, 2-гидроксибензальдегида, 4-метоксибензальдегида и 2-метилбензальдегида. 4. Способ по п.2, в котором органический растворитель на стадии а) является ацетонитрилом, а органический растворитель на стадии b) является изопропанолом или тетрагидрофураном (ТГФ). 5. Способ по п.1, в котором органическое основание выбрано из группы, состоящей из триэтиламина,триметиламина,диизопропилэтиламина,1,8-диазабицикло[5,4,0]ундец-7-ена и 1,5 диазабицикло[4,3,0]нон-5-она. 6. Способ по п.1, в котором соединение формулы 5 используют в 1-3-кратном количестве по отношению к количеству соединения формулы 2. 7. Способ по п.1, в котором органическое основание на стадии а) используют в 3-4-кратном количестве по отношению к количеству соединения формулы 2 и реакцию осуществляют при температуре реакции от 0 до 30 С. 8. Способ по п.7, в котором органическим основанием является триэтиламин. 9. Способ по п.1, в котором кислоту формулы НА используют в количестве от 80 до 120 моль.% по отношению к количеству соединения формулы 4, причем температура реакционной смеси при добавлении кислоты находится в интервале 40-50 С, а температура после добавления кислоты находится в интервале 0-20 С. 10. Способ по любому одному из пп.1-9, в котором кислота формулы НА является метансульфоновой кислотой. 11. Промежуточное соединение, представляемое следующей ниже формулой 4, для получения солей гемифлоксацина с кислотой по п.1
МПК / Метки
МПК: C07D 471/04
Метки: солей, получения, кислотных, способ, гемифлоксацина
Код ссылки
<a href="https://eas.patents.su/10-7222-sposob-polucheniya-kislotnyh-solejj-gemifloksacina.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения кислотных солей гемифлоксацина</a>
Способ получения кислотных солей тровафлоксацина

Номер патента: 2340
Опубликовано: 25.04.2002
Авторы: Роуз Питер Роберт, Норрис Тимоти, Ди Вриз Кит Майкл
МПК: C07D 471/04
Метки: тровафлоксацина, солей, кислотных, получения, способ
Формула / Реферат:
1. Способ получения кислотной соли тровафлоксацина, имеющей формулу (IV) где ZH представляет собой минеральную кислоту, включающий в себя стадию, на которой соединение формулы (I) где R представляет собой C1-С6алкильную группу и где бензилиденовое кольцо соединения формулы (I) возможно замещено одной или более чем одной фторо, хлоро, бромо, иодо, C1-С6алкильной или C1-С6алкоксигруппой, приводят в контакт с композицией, содержащей минеральную...
Способ получения (1r, 2s, 4r)-(-)-2-[n, n-(диметиламиноэтокси)]-2-фенил-1,7,7 -триметилбицикло[2,2,1] гептана и его фармацевтически приемлемых кислотных аддитивных солей

Номер патента: 2163
Опубликовано: 24.12.2001
Авторы: Суладьи Янош, Будаи Золтан, Порч-Маккаи Марта, Немет Норберт, Лукач Дьюла, Сабо Тибор, Шимиг Дьюла, Краснаи Дьёрдь, Мезеи Тибор, Надь Калман
МПК: C07C 217/12
Метки: солей, гептана, приемлемых, аддитивных, кислотных, n-(диметиламиноэтокси)]-2-фенил-1,7,7, триметилбицикло[2,2,1, 4r)-(-)-2-[n, способ, фармацевтически, получения
Формула / Реферат:
1. Способ получения (1R,2S,4R)-(-)-2-[N,N-(диметиламиноэтокси)]-2-фенил-1,7,7-триметилбицикло-[2,2,1]гептана высокой степени чистоты по формуле и его фармацевтически приемлемых кислотных аддитивных солей, который включает превращение (+)-1,7,7-триметилбицикло[2,2,1]гептан-2-она {(+)-камфоры} по формуле в (1R, 2S, 4R)-(-)-2-фенил-1,7,7-триметилбицикло[2,2,1]гептан-2-ол по формуле путем реакции соединения по формуле II с...
Способ получения солей цианобензиламинов

Номер патента: 3286
Опубликовано: 24.04.2003
Авторы: Мураками Масатоси, Ямагами Исао, Ясуда Хироси, Йосида Тору
МПК: C07C 253/30
Метки: цианобензиламинов, получения, солей, способ
Формула / Реферат:
1. Способ получения солей цианобензиламина, включающий взаимодействие цианобензиламина, который представляет собой соединение следующей формулы в которой X1, X2, X3 и X4, каждый независимо, представляет атом водорода, алкильную группу, содержащую от 1 до 3 атомов углерода, или атом галогена, и -CH2NH2 группа может находиться в любом из орто-, мета- или пара-положений относительно -CN группы с кислотой в присутствии воды в качестве растворителя....
Способ получения солей гидроксиламмония.

Номер патента: 1393
Опубликовано: 26.02.2001
Авторы: Леффертс Леонардус, Ван Лисхаут Ламбертус Хюбертус Вильхельмус Мария, Схевелир Петер Арнольд Сесилиан
МПК: C01B 21/14, B01J 23/42
Метки: гидроксиламмония, получения, солей, способ
Формула / Реферат:
1. Способ получения соли гидроксиламмония каталитическим восстановлением нитрат ионов водородом в кислой среде в присутствии палладиевого и/или платинового катализатора на носителе, отличающийся тем, что катализатор содержит ионы галогена в количестве, по меньшей мере, от 0,00025 до не более 0,004 ммоль ионов галогена на 1 м2 площади поверхности палладия и/или платины. 2. Способ по п.1, отличающийся тем, что ионы галогена содержатся в...
Способ получения аммониевых солей 3-изопропил-2,1,3-бензотиадиазин-4-он-2,2-диоксида

Номер патента: 2236
Опубликовано: 28.02.2002
Авторы: Хансен Ханспетер, Дурайн Альфонс, Меркле Ханс Руперт, Егер Карл-Фридрих
МПК: C07D 285/16
Метки: способ, солей, 3-изопропил-2,1,3-бензотиадиазин-4-он-2,2-диоксида, получения, аммониевых
Формула / Реферат:
1. Способ получения аммониевых солей 3-изопропил-2,1,3-бензотиадиазин-4-он-2,2-диоксида (бентазона) общей формулы I в которой R1, R2, R3 и R4 независимо друг от друга представляют собой водород, C1-C8 алкил или гидрокси C1-C8 алкил, отличающийся тем, что бентазон формулы IIа подвергают взаимодействию с амином общей формулы IIIa или его солью формулы IIIb где Х представляет собой анион кислоты с рКs больше 4 или означает гидроксильный ион, а...
Следующий патент: Стабилизированные фармацевтические композиции на основе полиоксиэтилированного касторового масла и способ их получения
Случайный патент: Расширитель полости тела