Способ получения замещенных производных бензола






Формула / Реферат
1. Способ получения соединения, представленного формулой (2)

где R1 и R2 каждый независимо представляет атом водорода, атом галогена, гидроксильную группу или C1-6 алкильную группу, Х представляет аминогруппу, имеющую защитную группу, и n представляет целое число от 0 до 4, отличающийся тем, что он включает гидрирование в присутствии палладиевого катализатора соединения, представленного формулой (1)

где X, R1, R2 и n имеют такие же значения, как определено выше.
2. Способ по п.1, отличающийся тем, что R1 представляет метильную группу, R2 представляет атом фтора, Х представляет ацетиламиногруппу и n = 2.
Текст
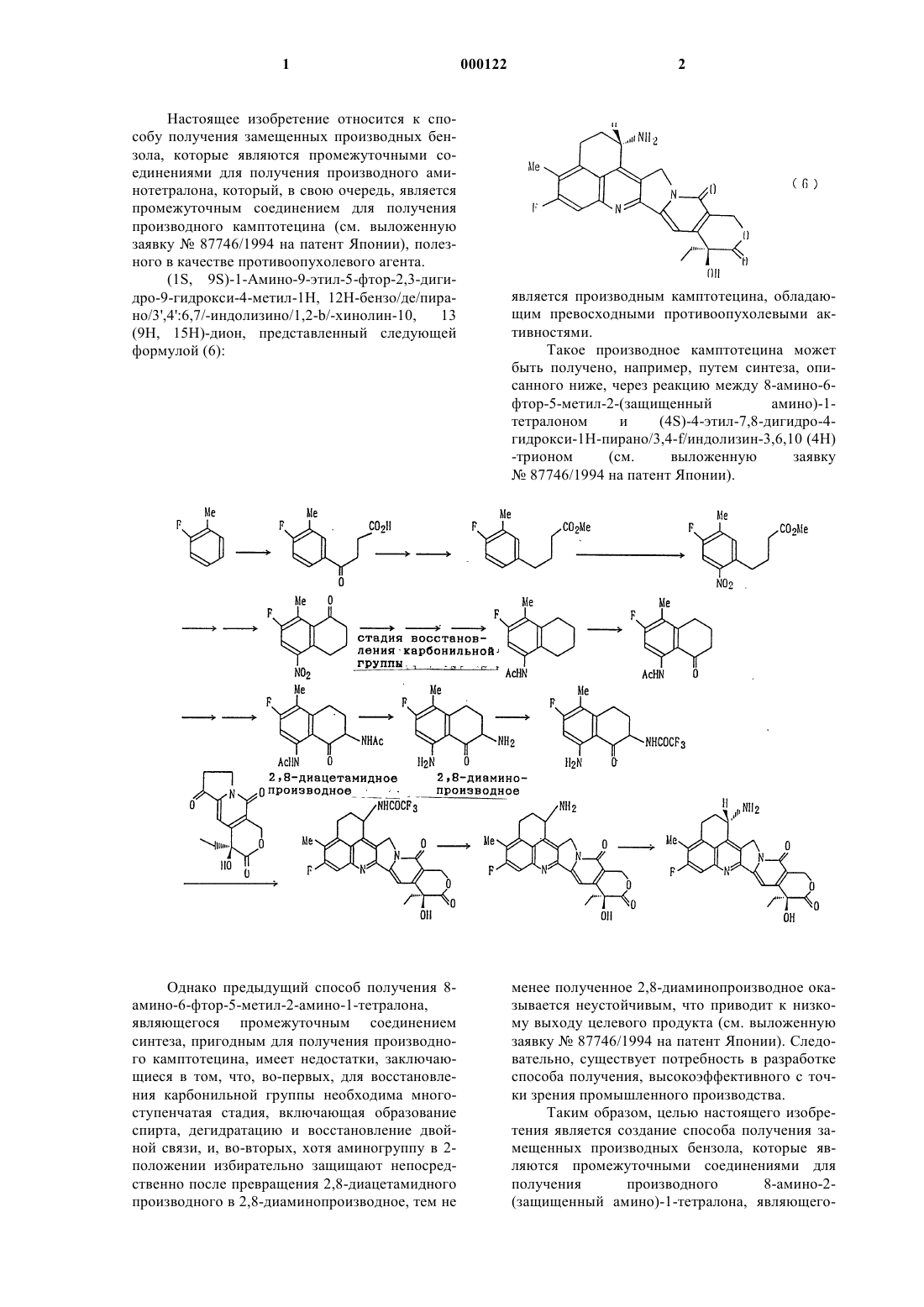
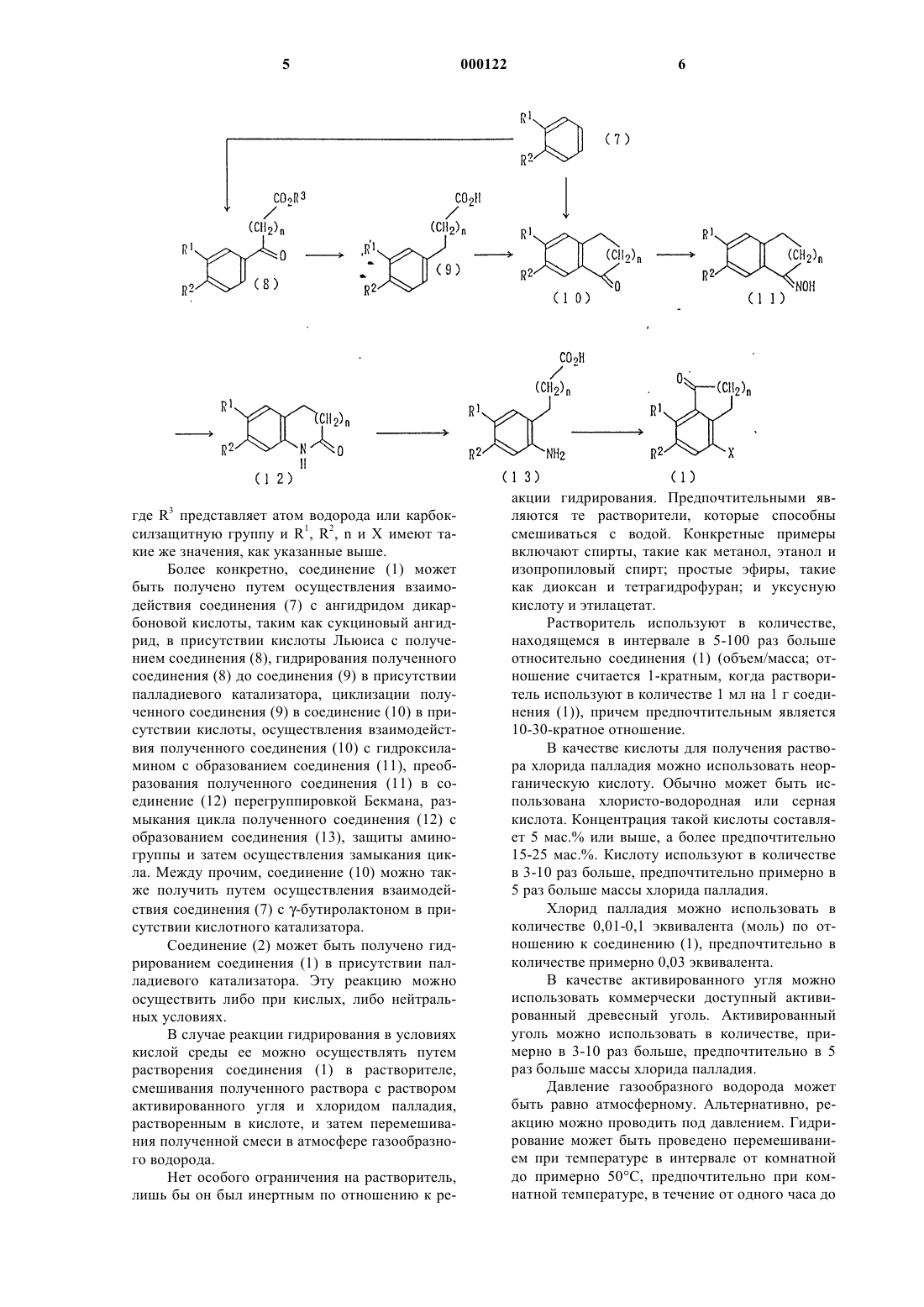
1 Настоящее изобретение относится к способу получения замещенных производных бензола, которые являются промежуточными соединениями для получения производного аминотетралона, который, в свою очередь, является промежуточным соединением для получения производного камптотецина (см. выложенную заявку 87746/1994 на патент Японии), полезного в качестве противоопухолевого агента.(9 Н, 15 Н)-дион, представленный следующей формулой (6): Однако предыдущий способ получения 8 амино-6-фтор-5-метил-2-амино-1-тетралона,являющегося промежуточным соединением синтеза, пригодным для получения производного камптотецина, имеет недостатки, заключающиеся в том, что, во-первых, для восстановления карбонильной группы необходима многоступенчатая стадия, включающая образование спирта, дегидратацию и восстановление двойной связи, и, во-вторых, хотя аминогруппу в 2 положении избирательно защищают непосредственно после превращения 2,8-диацетамидного производного в 2,8-диаминопроизводное, тем не является производным камптотецина, обладающим превосходными противоопухолевыми активностями. Такое производное камптотецина может быть получено, например, путем синтеза, описанного ниже, через реакцию между 8-амино-6 фтор-5-метил-2-(защищенный амино)-1 тетралоном и менее полученное 2,8-диаминопроизводное оказывается неустойчивым, что приводит к низкому выходу целевого продукта (см. выложенную заявку 87746/1994 на патент Японии). Следовательно, существует потребность в разработке способа получения, высокоэффективного с точки зрения промышленного производства. Таким образом, целью настоящего изобретения является создание способа получения замещенных производных бензола, которые являются промежуточными соединениями для получения производного 8-амино-2(защищенный амино)-1-тетралона, являющего 3 ся, в свою очередь, промежуточным соединением синтеза, пригодным для промышленного получения производного камптотецина, подходящим образом и с высоким выходом. Раскрытие сущности изобретения Для решения указанной задачи было проведено обширное исследование. В результате было обнаружено, что восстановление карбонильной группы можно проводить эффективно,используя палладиевый катализатор, и можно получить производное 8-амино-2-(защищенный амино)-1-тетралона с использованием короткой стадии и с высоким выходом без выделения неустойчивого 2,8-диаминопроизводного, подвергая 2,8-ди(защищенный амино)-производное обработке кислотой для избирательного отщепления защитной группы от 8-(защищенной амино) группы, что и привело к созданию настоящего изобретения. Способ получения производного 8-амино 2-(защищенный амино)-1-тетралона (5) по настоящему изобретению можно представить следующей схемой реакций: где R1 и R2 каждый независимо представляет атом водорода, атом галогена, гидроксильную группу или C1-6 алкильную группу, Х и У каждый независимо представляет аминогруппу,имеющую защитную группу, и n представляет целое число от 0 до 4. 4 Более конкретно, соединение (5) получают путем гидрирования соединения (1) до соединения (2) в присутствии палладиевого катализатора, окисления полученного соединения (2) до соединения (3), защиты аминогруппы полученного соединения (3) с образованием соединения(4) и осуществления взаимодействия полученного соединения (4) с кислотой для отщепления только защитной группы аминогруппы в 8 положении. В числе вышеописанных стадий в выложенной заявке 87764/1994 на патент Японии описаны стадии получения соединения(4) из соединения (2). Таким образом, настоящее изобретение обеспечивает стадию получения соединения (2) из соединения (1). Наилучшие способы осуществления изобретения В вышеприведенной схеме реакций предпочтительные примеры R1 и R2 включают метильную группу, этильную группу, нпропильную группу, изопропильную группу,атом фтора, атом хлора и атом брома. Особенно предпочтительным является случай, когда R1 иR2 представляют метильную группу и атом фтора соответственно. Особенно предпочтительным является n = 2. Примеры защитной группы для защищенной аминогруппы, представленной Х или У,включают алкоксикарбонильные группы, такие как третичный бутоксикарбонил и 2,2,2 трихлорэтоксикарбонил; аралкилоксикарбонильные группы, такие как бензилоксикарбонил, пара-метоксибензилоксикарбонил и паранитробензилоксикарбонил; ацильные группы,такие как ацетил, метоксиацетил, трифторацетил, хлорацетил, пивалоил, формил и бензоил; алкильные или аралкильные группы, такие как третичный бутил, бензил, пара-нитробензил,пара-метоксибензил и трифенилметил; алкилсульфонильные или галогеналкилсульфонильные группы, такие как метансульфонил и трифторметансульфонил; и арилсульфонильные группы, такие как бензолсульфонил и толуолсульфонил. Из них предпочтительной является ацильная группа, особенно алканоильная или бензоильная группа, которая может быть замещена атомом галогена. Далее дается конкретное описание со ссылкой на приведенную выше схему реакций. Исходное соединение (1) можно получить,например, по следующей схеме: где R3 представляет атом водорода или карбоксилзащитную группу и R1, R2, n и Х имеют такие же значения, как указанные выше. Более конкретно, соединение (1) может быть получено путем осуществления взаимодействия соединения (7) с ангидридом дикарбоновой кислоты, таким как сукциновый ангидрид, в присутствии кислоты Льюиса с получением соединения (8), гидрирования полученного соединения (8) до соединения (9) в присутствии палладиевого катализатора, циклизации полученного соединения (9) в соединение (10) в присутствии кислоты, осуществления взаимодействия полученного соединения (10) с гидроксиламином с образованием соединения (11), преобразования полученного соединения (11) в соединение (12) перегруппировкой Бекмана, размыкания цикла полученного соединения (12) с образованием соединения (13), защиты аминогруппы и затем осуществления замыкания цикла. Между прочим, соединение (10) можно также получить путем осуществления взаимодействия соединения (7) с -бутиролактоном в присутствии кислотного катализатора. Соединение (2) может быть получено гидрированием соединения (1) в присутствии палладиевого катализатора. Эту реакцию можно осуществить либо при кислых, либо нейтральных условиях. В случае реакции гидрирования в условиях кислой среды ее можно осуществлять путем растворения соединения (1) в растворителе,смешивания полученного раствора с раствором активированного угля и хлоридом палладия,растворенным в кислоте, и затем перемешивания полученной смеси в атмосфере газообразного водорода. Нет особого ограничения на растворитель,лишь бы он был инертным по отношению к ре 000122 акции гидрирования. Предпочтительными являются те растворители, которые способны смешиваться с водой. Конкретные примеры включают спирты, такие как метанол, этанол и изопропиловый спирт; простые эфиры, такие как диоксан и тетрагидрофуран; и уксусную кислоту и этилацетат. Растворитель используют в количестве,находящемся в интервале в 5-100 раз больше относительно соединения (1) (объем/масса; отношение считается 1-кратным, когда растворитель используют в количестве 1 мл на 1 г соединения (1, причем предпочтительным является 10-30-кратное отношение. В качестве кислоты для получения раствора хлорида палладия можно использовать неорганическую кислоту. Обычно может быть использована хлористо-водородная или серная кислота. Концентрация такой кислоты составляет 5 мас.% или выше, а более предпочтительно 15-25 мас.%. Кислоту используют в количестве в 3-10 раз больше, предпочтительно примерно в 5 раз больше массы хлорида палладия. Хлорид палладия можно использовать в количестве 0,01-0,1 эквивалента (моль) по отношению к соединению (1), предпочтительно в количестве примерно 0,03 эквивалента. В качестве активированного угля можно использовать коммерчески доступный активированный древесный уголь. Активированный уголь можно использовать в количестве, примерно в 3-10 раз больше, предпочтительно в 5 раз больше массы хлорида палладия. Давление газообразного водорода может быть равно атмосферному. Альтернативно, реакцию можно проводить под давлением. Гидрирование может быть проведено перемешиванием при температуре в интервале от комнатной до примерно 50 С, предпочтительно при комнатной температуре, в течение от одного часа до 7 нескольких дней, предпочтительно в течение примерно 5 ч. В случае проведения реакции гидрирования в нейтральных условиях способ с использованием катализатора палладий на углероде(PL/C) может быть дан в качестве примера. Более конкретно, гидрирование может быть осуществлено путем растворения соединения (1) в растворителе и затем перемешивания смеси полученного раствора и катализатора палладий на углероде в атмосфере газообразного водорода под давлением. Нет особого ограничения на растворитель,лишь бы он был инертным по отношению к реакции гидрирования. Конкретные примеры включают спирты, такие как метанол, этанол и изопропиловый спирт; простые эфиры, такие,как диоксан и тетрагидрофуран; и уксусную кислоту и эфиры уксусной кислоты, такие как этилацетат. Растворитель можно использовать в таком же количестве, как в вышеописанном способе с использованием хлорида палладия. В качестве катализатора палладий на углероде можно использовать палладий, нанесенный на углерод. Содержание палладия предпочтительно составляет 5-10% и предпочтительно примерно 0,2 эквивалента (молярное) относительно соединения (1). Реакцию гидрирования можно проводить в герметически закрытом реакторе, таком как автоклав. Является предпочтительным осуществлять реакцию в атмосфере газообразного водорода под давлением 10-100, особенно примерно 40 атмосфер при температуре в интервале от комнатной до 100 С, особенно примерно 50 С, в течение от одного часа до нескольких дней. Далее настоящее изобретение будет описано более подробно примерами. Следует, однако, иметь в виду, что примеры являются лишь иллюстративными и не ограничивают настоящее изобретение. Ссылочный пример 1. Способ получения 4-(4-фтор-3-метилфенил)-4-оксобутановой кислоты. К смешанному раствору 2,0 г сукцинового ангидрида и 50 мл 1,2-дихлорэтана добавляют 6,7 г хлорида алюминия, после чего смесь перемешивают при комнатной температуре в течение 40 мин. К полученной смеси добавляют по каплям при комнатной температуре 20 мл 2 фтортолуола и смесь перемешивают в течение 20 мин, после чего ее перемешивают еще в течение 20 мин при внешней температуре 50 С. Реакционную смесь охлаждают и затем выливают в ледяную воду, в которую была добавлена 5%-ная хлористо-водородная кислота. Полученную смесь экстрагируют хлороформом. Хлороформный слой промывают водой и сушат над безводным сульфатом магния. Затем выпаривают растворитель. Полученный при этом остаток перекристаллизовывают из хлороформа, в ре 000122 8 зультате чего получают 3,2 г названного соединения. Ссылочный пример 2. Способ получения 4-(4-фтор-3-метилфенил) бутановой кислоты. В метаноле растворяют 10,0 г 4-(4-фтор-3 метилфенил)-4-оксобутановой кислоты с последующим добавлением 1,5 г активированного древесного угля (Norit EXW) и 12,4 мл раствора хлорида палладия (раствор, полученный растворением 2,2 мл концентрированной хлористоводородной кислоты и 2,5 мл воды на 1,0 г хлорида палладия при нагревании, чтобы получить общий объем 50 мл). Полученную смесь подвергают каталитическому восстановлению при комнатной температуре и атмосферном давлении в течение 6 ч. По окончании реакции отфильтровывают катализатор с помощью фильтровальной бумаги из стекловолокна с последующей промывкой 12 мл метанола. К фильтрату добавляют 100 мл 5%-ного водного раствора гидроксида натрия и смесь перемешивают в течение 1 ч. По окончании реакции метанол выпаривают и остаток подкисляют 12 мл концентрированной хлористо-водородной кислоты при охлаждении льдом. Кристаллы, осажденные при этом, собирают путем фильтрования, получая в результате 8,8 г указанного соединения . Ссылочный пример 3. Способ получения 4-(4-фтор-3-метилфенил) бутановой кислоты. В автоклав емкостью 50 мл загружают 1,0 г 4-(4-фтор-3-метилфенил)-4-оксобутановой кислоты и 2,5 мл метанола с последующим добавлением 0,1 г 10%-ного палладия на углероде. После повышения давления водорода до 30 кг/см 2 при комнатной температуре полученную смесь перемешивают при 50 С в течение 3,5 ч. По окончании реакции реакционную смесь фильтруют с последующей промывкой метанолом. После этого фильтрат концентрируют при пониженном давлении. К полученному при этом остатку добавляют воду. Осажденные таким образом кристаллы фильтруют, промывают водой и высушивают при пониженном давлении с получением 0,82 г указанного соединения. Ссылочный пример 4. Способ получения 7-фтор-6-метил-1-тетралона (способ 1). К смешанному раствору 1,5 мл 2 фтортолуола и 0,5 мл -бутиролактона добавляют при комнатной температуре 1,3 г хлорида алюминия с последующим перемешиванием при той же самой температуре в течение 20 ч. По окончании реакции реакционную смесь выливают в 5%-ный водный раствор хлористоводородной кислоты. Полученный раствор экстрагируют хлороформом. Хлороформный слой промывают водой и сушат над безводным сульфатом магния. Удаляют при пониженном давлении растворитель. К остатку добавляют при охлаждении льдом 5 мл концентрированной серной кислоты с последующим перемешиванием в течение 1 ч. По окончании реакции реакционную смесь выливают в воду и затем экстраги 9 руют хлороформом. Хлороформный слой промывают водой и сушат над безводным сульфатом магния. Удаляют при пониженном давлении растворитель с получением 1 г остатка. Как было установлено по 1 Н-ЯМР-спектру, полученный остаток представлял собой смесь (1:1) указанного соединения и его изомера.(Способ 2) К 150 мл концентрированной серной кислоты добавляют порциями в течение 40 мин при охлаждении льдом 20,0 г 4-(4-фтор-3 метилфенил) бутановой кислоты и смесь перемешивают один час при тех же самых условиях. По окончании реакции реакционную смесь выливают в ледяную воду. Осажденные таким образом кристаллы собирают путем фильтрования с последующей достаточной промывкой. Полученный продукт передают для использования на следующей стадии во влажном виде. 1 Н-ЯМР (CDCl3): 2,07-2,15(2 Н, м), 2,30(2 Н, т, J=6,l Гц) 7,08 (1 Н, д, J=7,6 Гц), 7,63(1 Н,д, J=7,9 Гц). Ссылочный пример 5. Способ получения 7-фторметил-1-тетралоноксима. К раствору 10,6 г хлорида гидроксиаммония и 12,6 г ацетата натрия в воде (50 мл) добавляют все количество 7-фтор-6-метил-1 тетралона, полученного в способе 2 примера 4, к которому добавляют 300 мл этанола. Полученную смесь перемешивают 3 ч при внешней температуре 70-75 С. По окончании реакции удаляют при пониженном давлении растворитель. К остатку добавляют воду и осажденные таким образом кристаллы собирают путем фильтрования. После промывки водой кристаллы высушивают при пониженном давлении, получая в результате 15,3 г указанного соединения. 1 Н-ЯМР (CDCl3): 1,80-1,90 (2 Н, м), 2,25(3 Н, д, J=1,7 Гц), 2,69(2 Н, т, J=6,1 Гц), 2,78(2 Н,т, J=6,6 Гц) 6,96 (1 Н, д, J= 7,6 Гц), 7,50 (1 Н, д,J=10,9 Гц). Ссылочный пример 6. Способ получения 3,4-дигидро-8-фтор-7-метил-2-оксо-1-бензазепина. К 70 мл 85%-ной фосфорной кислоты добавляют порциями 100 г фосфорного ангидрида. После полного растворения фосфорного ангидрида к полученному раствору добавляют в течение 20 мин при наружной температуре 90 С 10,0 г 7-фтор-6-метил-1-тетралоноксима с последующим перемешиванием в течение 4 ч при тех же самых условиях. По окончании реакции реакционную смесь выливают в ледяную воду. Кристаллы, осажденные таким образом, собирают путем фильтрования. Полученные кристаллы перекристаллизовывают из смеси хлороформ-диэтиловый эфир с получением 8,3 г указанного соединения. 1(4-(2-ацетиламино-4-фтор-5-метилфенил) бутановой кислоты. К 1,0 г 3,4-дигидро-8-фтор-7-метил-2-оксо 1-бензазепина добавляют 15 мл метанола и 0,7 мл концентрированной хлористо-водородной кислоты и смесь нагревают с обратным холодильником в течение 3 ч. По окончании реакции реакционной смеси дают остыть и удаляют при пониженном давлении растворитель. К полученному при этом белому остатку добавляют 20 мл метиленхлорида с последующим добавлением 1,8 мл триэтиламина и затем 0,5 мл уксусного ангидрида при охлаждении льдом. Смесь перемешивают при комнатной температуре 2,5 ч. По окончании реакции к реакционной смеси добавляют воду и 5%-ный водный раствор хлористо-водородной кислоты, после чего смесь экстрагируют хлороформом. Хлороформный слой промывают насыщенным водным раствором бикарбоната натрия и затем сушат над безводным сульфатом магния. Затем выпаривают растворитель. К полученному при этом остатку добавляют 8 мл метанола и 5 мл 5%-ного водного раствора гидроксида натрия и смесь перемешивают при комнатной температуре в течение 30 мин. Затем отгоняют при пониженном давлении растворитель. К полученному остатку добавляют 5%-ный водный раствор хлористоводородной кислоты, после чего смесь экстрагируют этилацетатом. Этилацетатный слой промывают насыщенным рассолом и затем сушат над безводным сульфатом магния. Затем выпаривают растворитель. Полученный остаток перекристаллизовывают из смеси этилацетатхлороформ с получением 0,8 г указанного соединения. 1 Н-ЯМР (СDСl3): 1,71-1,85(2 Н, м), 2,20(3 Н, д, J=1,5 Гц), 2,20(3 Н, с), 2,47-2,59 (4 Н, м),6,91 (1 Н, д, J=8,6 Гц), 7,92 (1 Н, д, J=12,2 Гц),8,43 (1 Н, ушир.с.). Ссылочный пример 8. Способ получения 5-ацетиламино-7-фтор-8-метил-1-тетралона. В 50 мл метиленхлорида суспендируют 5,0 г 4-(2-ацетиламино-4-фтор-5-метилфенил) бутановой кислоты. К суспензии добавляют по каплям 4,3 мл тионилхлорида в течение 2 мин при внутренней температуре 3-4 С с последующим перемешиванием при той же самой температуре в течение 15 мин и затем при комнатной температуре в течение 45 мин. К полученной смеси добавляют 6,6 г хлорида алюминия в течение 5 мин при внутренней температуре 4-6 С с последующим перемешиванием при той же самой температуре в течение 1 ч и затем при перемешивании при комнатной температуре в течение 24 ч. По окончании реакции к реакционной смеси постепенно добавляют 5%-ный водный раствор хлористо-водородной кислоты и лед и 11 смесь перемешивают в течение некоторого времени. Экстрагируют реакционную смесь хлороформом. Хлороформный слой промывают в достаточной степени водой и насыщенным водным раствором бикарбоната натрия и затем сушат над карбонатом калия. Удаляют при пониженном давлении растворитель. К полученному при этом остатку добавляют изопропиловый эфир. Осажденные при этом кристаллы собирают путем фильтрования с получением 3,5 г указанного соединения. 1 Н-ЯМР (CDC13): 2,05-2,14 (2 Н, м), 2,22(3 Н, с), 2,50(3 Н, д, J=2,3 Гц), 2,64(2 Н, т, J=6,6 Гц), 2,77 (2 Н, т, J=6,4 Гц), 7,07 (1 Н, ушир. с),7,64 (1 Н, д, J=10,8 Гц). Пример 1. Способ получения 5 ацетиламино-7-фтор-8-метил-1,2,3,4-тетрагидронафталина. К 20,0 г 5-ацетиламино-7-фтор-8-метил-1 тетралона добавляют 1000 мл метанола, 7,2 г активированного древесного угля (Norit EXW) и 60 мл раствора хлорида палладия (100 мл раствора, полученного путем добавления 4,5 мл концентрированной хлористо-водородной кислоты и 4,5 мл воды к 2,0 г хлорида палладия и растворения их с нагреванием). Полученную смесь подвергают каталитическому восстановлению при комнатной температуре и атмосферном давлении в течение 8 ч. По окончании реакции отфильтровывают катализатор с помощью фильтровальной бумаги из стекловолокна с последующей тщательной промывкой смесью хлороформ-метанол. Фильтрат затем концентрируют досуха при пониженном давлении. Полученный при этом остаток перекристаллизовывают из смеси хлороформ-метанол с получением 14,4 г указанного соединения. Пример 2. Способ получения 5 ацетиламино-7-фтор-8-метил-1,2,3,4-тетрагидронафталина. В автоклав емкостью 50 мл загружают 1,0 г 5-ацетиламино-7-фтор-8-метил-1-тетралона и 10 мл метанола. К полученной смеси добавляют 0,5 г 10%-ного палладия на углероде. Давление водорода повышают до 40 кг/см 2 при комнатной температуре, при котором полученную смесь перемешивают при 50 С в течение 18 ч. По окончании реакции реакционную смесь фильтруют. Полученный при этом катализатор промывают хлороформом и затем фильтрат концентрируют при пониженном давлении. К остатку добавляют изопропиловый эфир. Кристаллы,осажденные при этом, собирают путем фильтрования и затем высушивают при пониженном давлении, получая в результате 0,76 г указанного соединения. Ссылочный пример 9. Способ получения 8-ацетиламино-6-фтор-5-метил-1-тетралона. В ацетоне суспендируют 5,0 г 5 ацетиламино-7-фтор-8-метил-1,2,3,4-тетрагидронафталина с последующим добавлением 2,3 12 г бикарбоната натрия. К полученной смеси добавляют порциями 13,9 г перманганата калия в течение 3,5 ч при внутренней температуре 1015 С и смесь перемешивают при комнатной температуре в течение 1 ч. К полученной смеси добавляют еще 0,8 г перманганата калия с последующим перемешиванием в течение 1,5 ч. Добавив небольшое количество 5%-ного водного раствора бисульфита натрия и убедившись в полном исчезновении перманганата калия, отфильтровывают осажденный таким образом диоксид марганца. Диоксид марганца тщательно промывают хлороформом и фильтрат удаляют при пониженном давлении. Полученный при этом остаток растворяют в хлороформе. Полученный раствор промывают три раза насыщенным водным раствором бикарбоната натрия и сушат над карбонатом калия. Выпаривают при пониженном давлении растворитель. Полученный при этом остаток перекристаллизовывают из смеси хлороформ-диэтиловый эфир с получением 2,6 г указанного соединения. Ссылочный пример 10. Способ получения 2,8-диацетиламино-6-фтор-5-метил-1-тетралона. В 550 мл тетрагидрофурана суспендируют 19,1 г третичного бутоксида калия. К полученной суспензии добавляют по каплям в течение 35 мин раствор, который был получен путем растворения 20,0 г 8-ацетиламино-6-фтор-5 метил-1-тетралона в 250 мл тетрагидрофурана при внутренней температуре 4-5 С в потоке газообразного азота, и смесь перемешивают при той же температуре в течение 10 мин. К полученной смеси добавляют 20 мл н-бутилнитрида при внутренней температуре 5-7 С в течение 15 мин с последующим перемешиванием при той же температуре в течение 1 ч. По окончании реакции к реакционной смеси добавляют воду. Полученный водный раствор доводят до рН 3-4 с помощью 5%-ного водного раствора хлористоводородной кислоты и затем фильтруют через фильтровальную бумагу из стекловолокна. Фильтрат выпаривают при пониженном давлении и осажденные при этом кристаллы собирают путем фильтрования. Собранные кристаллы промывают водой, высушивают и затем растворяют в смешанном растворе 200 мл уксусного ангидрида и 200 мл уксусной кислоты. При охлаждении льдом к полученному раствору при внутренней температуре 6-15 С добавляют в течение 1,5 ч 25 г цинкового порошка. Полученную смесь перемешивают при той же температуре в течение 30 мин, после чего отфильтровывают твердые вещества. Полученные твердые вещества промывают последовательно хлороформом и этилацетатом. Фильтрат затем концентрируют при пониженном давлении. К полученному при этом остатку добавляют воду и затем по существу нейтрализуют 5%-ным водным раствором гидроксида натрия. Кристаллы,осажденные при этом, некоторое время перемешивают в состоянии суспензии, после чего собирают путем фильтрования. Собранные кристаллы промывают водой и высушивают при пониженном давлении, получая в результате 16,5 г указанного соединения. Пример 3. Способ получения 2 ацетиламино-8-амино-6-фтор-5-метил-1-тетралона. К 8,0 г 2,8-диацетиламино-6-фтор-5-метил 1-тетралона добавляют 120 мл 20%-ного водного раствора хлористо-водородной кислоты и смесь перемешивают при наружной температуре 60 С в течение 2 ч. По окончании реакции реакционную смесь охлаждают и добавляют к ней 100 мл воды. Полученную смесь фильтруют через воронку Кириямы. К фильтрату добавляют еще 100 мл воды, после чего смесь экстрагируют хлороформом. К полученному при этом хлороформному слою добавляют карбонат калия и Флоризил и некоторое время их перемешивают. Полученную смесь фильтруют и затем удаляют при пониженном давлении растворитель. Полученный при этом остаток перекристаллизовывают из смеси хлороформ/этиловый эфир с получением 3,4 г указанного соединения. Температура плавления: 212-214C. 1 Н-ЯМР (CDCl3) : 1,66-1,85 (1 Н, м), 2,05 где R1 и R2 каждый независимо представляет атом водорода, атом галогена, гидроксильную группу или C1-6 алкильную группу, Х представляет аминогруппу, имеющую защитную группу,и n представляет целое число от 0 до 4, отличающийся тем, что он включает гидрирование в присутствии палладиевого катализатора соединения, представленного формулой (1) где X, R1, R2 и n имеют такие же значения, как определено выше. 2. Способ по п.1, отличающийся тем, что
МПК / Метки
МПК: B01J 23/44, C07C 233/15
Метки: бензола, замещенных, получения, производных, способ
Код ссылки
<a href="https://eas.patents.su/8-122-sposob-polucheniya-zameshhennyh-proizvodnyh-benzola.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения замещенных производных бензола</a>
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Ансель Жан-Эрик, Мейллян Пьер, Бьенейм Юг
МПК: C07C 39/19, A61K 31/355, B01J 31/24...
Метки: замещенных, способ, cпособ, использованием, витамина, фенолов, получения
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Трициклические соединения, способ их получения, способы получения оптически активных или рацемических производных колхицина и тиохолкицина с использованием трициклических соединений и промежуточныепродукты синтеза

Номер патента: 93
Опубликовано: 25.06.1998
Авторы: Диолез Кристиан, Брион Франсис, Мазюри Алан, Пронин Дидье, Мари Кристиан, Тороманофф Эдмон, Шаппер Бернадетт, Миддендорп Мишель
МПК: C07D 317/44, C07C 43/21
Метки: колхицина, активных, способ, производных, тиохолкицина, рацемических, способы, трициклические, промежуточныепродукты, использованием, получения, оптически, синтеза, соединений, трициклических, соединения
Формула / Реферат:
1. Трициклические соединения общей формулы I в которой либо а) оба R1 и R2 представляют собой алкильную группу, a R3 представляет собой атом водорода или группу A-SO2-, либо б) оба R2 и R3 представляют собой атом водорода или алкил, a R1 представляет собой группу A-SO2-, либо в) все три: R1, R2 и R3 представляют собой атом водорода или все три представляют собой алкил, либо г) R1 представляет собой группу А-SO2- или атом водорода, a...
Производные полипирролкарбоксамидонафталина, способ их получения и их применение

Номер патента: 6
Опубликовано: 30.12.1997
Авторы: Анджелуччи Францеско, Пезенци Энрико, Монджелли Никола, Бьясоли Джиованни, Ломбарди Борджиа Андреа, Чомеи Марина
МПК: C07D 207/34, A61K 31/40, C07H 15/252...
Метки: производные, получения, полипирролкарбоксамидонафталина, способ, применение
Формула / Реферат:
1. Соединение формулы (II): где R является кислотной группой; m - целое число от 1 до 3; n - ноль или целое число от 1 до 3; А представляет собой ферментативно гидролизуемый спейсер; Х является биологически активным соединением; или его фармацевтически приемлемые соли. 2. Соединение формулы (II) по п.1, где R является кислотной группой, выбранной из сульфоновой, карбоксильной и фосфоновой кислотных групп. 3. Соединение формулы (II)...
Безводный хлоргидрат цилпатерола в кристаллизованной форме, способ его получения (его варианты), моногидрат и тригидрат хлоргидрата цилпатерола в качестве промежуточных продуктов

Номер патента: 11
Опубликовано: 30.12.1997
Авторы: Шевремон Ив, Годар Жан-Ив
МПК: C07D 487/04, A23K 1/16
Метки: способ, качестве, варианты, получения, безводный, хлоргидрат, кристаллизованной, продуктов, цилпатерола, промежуточных, тригидрат, его, моногидрат, хлоргидрата, форме
Формула / Реферат:
1. Безводный хлоргидрат цилпатерола в кристаллизованной форме с кристаллами размером менее 250 мкм, содержащий менее 5% кристаллов размером менее 15 мкм. 2. Способ получения продукта по п.1, характеризующийся тем, что получают пересыщенный раствор хлоргидрата цилпатерола в воде или 50%-ном водном растворе этанола при температуре 50-70°С, охлаждают полученный раствор до 45°С и выдерживают при этой температуре до окончания образования моногидрата...
Способ получения 5-[2-этокси-5-(4-метилпиперазин-1-илсульфонил)-фенил]-1-метил-3-н-пропил,- 1,6-дигидро-7н-пиразоло[4,3-d]пиримидин-7-она

Номер патента: 102
Опубликовано: 27.08.1998
Авторы: Данн Питер Джеймз, Вуд Элберт Шо
МПК: C07D 487/04
Метки: 5-[2-этокси-5-(4-метилпиперазин-1-илсульфонил)-фенил]-1-метил-3-н-пропил, способ, 1,6-дигидро-7н-пиразоло[4,3-d]пиримидин-7-она, получения
Формула / Реферат:
1. Способ получения 5-[2-этокси-5-(4-метилпиперазин-1-ил-сульфонил)-фенил]-1-метил-3-н-пропил-1,6-дигидро-7Н-пиразоло-[4,3-d] пиримидин-7-она формулы (I) отличающийся тем, что соединение формулы (II) подвергают реакции циклизации в щелочной, нейтральной или кислой среде. 2. Способ по п.1, отличающийся тем, что циклизацию осуществляют в присутствии основания, предпочтительно в растворителе, возможно в присутствии перекиси водорода или...
Предыдущий патент: Новые производные стафилокиназы
Следующий патент: Многодисковое соединительное устройство, автоматическая трансмиссия, снабженная этим устройством, и способ изготовления этого устройства.
Случайный патент: Способы лечения аутоиммунных заболеваний при использовании антагониста нейтрокина-альфа