Способ получения 2,4-дигидроксипиридина и 2,4-дигидрокси-3-нитропиридина
Номер патента: 884
Опубликовано: 26.06.2000
Авторы: Трусдейл Ларри К., Шербайн Джеймс П., Ванасс Бенуа Дж.




























Формула / Реферат
1. Способ получения 2,4-дигидроксипиридина, включающий нагревание соединения формулы А

где R является Н, алкилом или аралкилом, и фосфорной кислоты, когда массовое соотношение фосфорной кислоты к воде составляет не менее чем примерно 27:1.
2. Способ по п.1, отличающийся тем, что используют разбавленную фосфорную кислоту и массовое соотношение фосфорной кислоты к воде, равное 27:1, достигается посредством удаления воды из реакционной смеси.
3. Способ по п.2, отличающийся тем, что удаление производят посредством перегонки.
4. Способ по п.1, отличающийся тем, что фосфорную кислоту и соединение формулы А нагревают примерно до 210шС.
5. Способ получения 2,4-дигидрокси-3-нитропиридина, включающий реакцию 2,4-дигидроксипиридина с азотной кислотой.
6. Способ получения 2,4-дигидрокси-3-нитропиридина, включающий реакцию продукта по п.1 без его выделения с азотной кислотой.
7. Способ по п.5, отличающийся тем, что до реакции с азотной кислотой добавляют органическую кислоту.
8. Способ по п.7, отличающийся тем, что органическая кислота является уксусной кислотой.
9. Способ по п.6, отличающийся тем, что до реакции с азотной кислотой добавляют органическую кислоту.
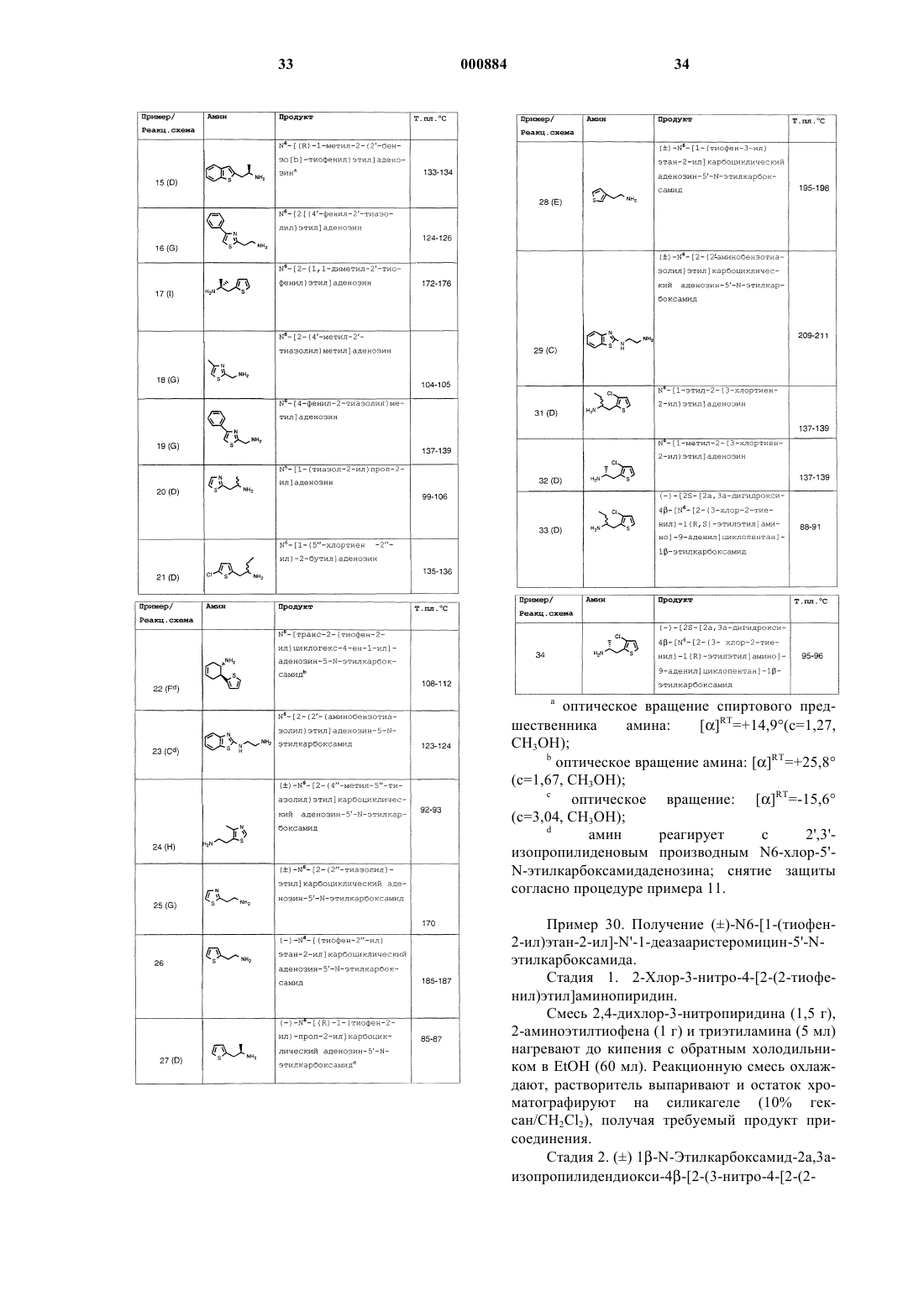
10. Способ по п.9, отличающийся тем, что органическая кислота является уксусной кислотой.
Текст
1 Предпосылки изобретения 1. Область изобретения Данное изобретение касается способов получения 2,4-дигидроксипиридина и 2,4-дигидрокси-3-нитропиридина, которые являются промежуточными продуктами, полезными в получении соединений аденозина и их аналогов,полезных при лечении гипертензии или ишемии миокарда, в качестве кардиозащитных агентов,уменьшающих ишемическое поражение или инфаркт миокарда вследствие ишемии миокарда, и в качестве антилиполитических агентов,которые снижают содержание липидов в плазме, содержание триглицеридов в сыворотке и содержание холестерина в плазме. Гипертензия Гипертензия, состояние повышенного кровяного давления, затрагивает существенное количество населения. Последствия устойчивой гипертензии включают сосудистые заболевания глазной, почечной, сердечной и церебральной систем, и риск этих осложнений повышается с повышением кровяного давления. Основными факторами регулирования кровяного давления являются минутный сердечный выброс и периферическое сопротивление сосудов, при том,что последнее является общим доминирующим механизмом, регулируемым различными влияниями. Симпатическая нервная система регулирует периферическое сопротивление сосудов посредством прямого действия на альфа- и бетаадренергические рецепторы, а также посредством непрямых воздействий на выделение ренина. При лекарственной терапии имеют в виду специфические компоненты этих регуляторных систем кровяного давления с различными механизмами действия, определяя несколько классов лекарств, включая диуретики, агонисты бетаадренергических рецепторов (бета-блокаторы),ингибиторы ангиотензин-превращающего фермента (АСЕ) и антагонисты кальциевого канала. Диуретики тиазидного типа применяют при гипертензии для снижения периферического сопротивления сосудов, благодаря их действию на выделение натрия и воды. Этот класс лекарственных средств включает гидрохлортиазид, хлортиазид, метиклотиазид и циклотиазид,а также аналогичные средства индапамид, метолазон и хлорталидон. Хотя прежде полагали,что механизмом действия бета-блокатора является блокада подтипа бета 1-адренергических рецепторов в сердце для снижения частоты сердечных сокращений и минутного сердечного выброса, более новые бета-блокаторы с характеристической симпатомиметической активностью (ISA), включая пиндолол, ацебутолол,пенбутолол и картеолол, являются эффективными не-ISA бета-блокаторами, вызывая меньшее снижение частоты сердечных сокращений и минутного сердечного выброса. Другие постулированные механизмы действия этих лекарств включают ингибирование выделения ренина, 000884 2 центральное действие и действие на пресинаптические бета-адренергические рецепторы, дающее в результате ингибирование выделения норэпинефрина. Кардиоселективные бетаблокаторы метопролол (Lopressor-Geigy), ацебутолол (Sectral-Wyeth) и атенолол (Tenormin-ICI) при низких дозах оказывают большее действие на бета 1-адренергические рецепторы, чем на подтип бета 2-адренергических рецепторов, расположенных в бронхах и кровяных сосудах. Неселективные бета-блокаторы действуют на оба подтипа адренергических рецепторов и включают пропранолол (Inderal-Ayerst), тимолол (Blocadren-Merck), надолол (CorgardSquibb), пиндолол (Visken-Sandoz), пенбутолол(Levatol-Hoechst-Roussel) и картеолол (CartrolAbbott). Побочные эффекты бета-блокаторов включают асимптоматическую брадикардию,обострение застойной сердечной недостаточности, желудочно-кишечные расстройства, повышенное сопротивление дыхательных путей,скрытые симптомы гипогликемии и депрессию. Они могут вызывать повышение содержания триглицеридов в сыворотке и могут снижать холестерин в липопротеинах высокой плотности. АСЕ ингибиторы предотвращают образование ангиотензина II и ингибируют разрушение брадикинина. Ангиотензин II является сильнодействующим сосудосуживающим агентом, а также стимулирует секрецию альдостерона. При блокаде ренин-ангиотензин-альдостеронной системы эти агенты понижают периферическое сопротивление сосудов, а также удерживание натрия и воды. Кроме того, АСЕ ингибиторы увеличивают уровень брадикинина и простагландинов, эндогенных сосудорасширяющих агентов. Ведущими АСЕ ингибиторами являются каптоприл (Capoten-Squibb) и эналаприл (Vasotec-Merck). Побочные эффекты АСЕ ингибиторов включают сыпь, нарушение вкусовых ощущений, протеинурию и нейтропению. Антагонисты кальциевого канала снижают приток кальция в клетки гладких мышц сосудов и вызывают общее расширение сосудов, результатом которого является гипотензивное действие. Другие эффекты антагонистов кальциевого канала, которые можно добавить к их гипотензивному действию, включают подавление действия ангиотензина II и блокады альфа 2 адренергических рецепторов. Антагонисты кальциевого канала не оказывают побочного метаболического и фармакологического действия триазидов или бета-блокаторов и, следовательно, полезны для пациентов с диабетом, заболеванием периферических сосудов или хроническим закупоривающим легочным заболеванием. Два антагониста кальциевого канала верапамил и дилтиазем оказывают серьезные побочные сердечно-сосудистые эффекты на атривентрикулярную сердечную проводимость у пациентов с ранее существовавшими наруше 3 ниями проводимости, и они могут ухудшать брадикардию, блокировку сердца и застойную сердечную недостаточность. Другие отрицательные побочные эффекты антагонистов кальциевого канала включают периферический отек,головокружение, состояние умственного расстройства, головную боль, тошноту и прилив крови, в особенности, в случае нифедипина и никардипина. Многие другие агенты способны лечить эссенциальную гипертензию. Такие агенты включают празозин и теразоцин, антагонисты альфа 1-адренергических рецепторов, гипотензивное действие которых обусловлено расширением артериальных сосудов; клонидин, агонист альфа 2-адренергических рецепторов, который действует центрально и периферически по ингибирующим альфа 2-адренергическим рецепторам, снижая симпатическую реакцию. Другие агенты центрального действия включают метилдопа, гуанбенз и гуанфацин; резерпин, который действует при истощении запасов катехоламинов; гуанадрел, периферический, адренергический антагонист, аналогичный гуанетидину с меньшим периодом действия; и сосудорасширяющие агенты прямого действия, такие как гидралазин и миноксидил. Эти агенты, хотя и являются эффективными, производят заметные побочные действия, включая рефлекторную симпатическую стимуляцию и задержку жидкости, ортостатическую гипотензию и импотенцию. Многие гипотензивные агенты активируют компенсаторные сосудосуживающие механизмы, такие как повышенное выделение ренина,усиленная секреция альдостерона и повышенный симпатический сосудосуживающий тонус,которые предназначены для возвращения артериального давления к уровню, предшествующему лечению, и которые могут вести к удерживанию соли и воды, отеку и, в конечном счете, к устойчивости действия гипотензивных агентов. Кроме того, вследствие широкого разнообразия побочных эффектов, испытываемых в случае данного комплекта гипотензивных лекарственных препаратов, и возникающих к тому же проблем при наблюдении особых категорий пациентов с повышенным артериальным давлением (включая пожилых, чернокожих и пациентов с хроническим закупоривающим легочным заболеванием, диабетом или болезнями периферических сосудов) существует потребность в дополнительных классах лекарственных препаратов для лечения гипертензии. Ишемия Ишемия миокарда является результатом дисбаланса снабжения миокарда кислородом и потребности в кислороде и включает физическую нагрузку и вазоспастическую дисфункцию миокарда. Ишемию, связанную с физической нагрузкой, обычно приписывают наличию кри 000884 4 тического атеросклеротического стеноза, включающего большие коронарные артерии, в результате которого происходит снижение субэндокардиального тока. Вазоспастическую ишемию связывают со спазмом, приступ которого не связан с физической нагрузкой или стрессом. Спазм лучше определяется как увеличение прерывания в сосудистом тонусе. Механизм вазоспастической ишемии включает: (I) усиленный сосудистый тонус на месте стеноза вследствие увеличенного выделения катехоламина; (II) временная внутрипросветная закупорка и (III) выделение вазоактивных веществ, образованных тромбоцитами на месте эндотелиальных повреждений. Коронарное кровообращение уникально,так как оно перфузирует орган, который генерирует давление перфузии для всего кровообращения. Таким образом, вмешательства, которые изменяют состояние периферического кровообращения и сократительную способность, будут оказывать глубокое действие на коронарное кровообращение. Регулирующим компонентом коронарной сосудистой сети являются небольшие коронарные артериолы, которые могут значительно менять свой внутренний диаметр. Изменение внутреннего радиуса является результатом присущего им сокращения сосудистой гладкой мышцы (авторегуляция) или экстраваскулярного сжатия вследствие внесосудистого сокращения. Полезное влияние терапии на ишемические проблемы включает комплексное взаимодействие противоположных факторов,которые определяют снабжение кислородом и потребность в нем. Защита сердца и предотвращение ишемического поражения Разработка новых терапевтических средств, способных ограничить степень ишемического поражения, а именно, степень инфаркта миокарда вслед за острой ишемией миокарда,является основной темой современной кардиологии. Появление в последние десять лет тромболитической (растворяющей сгустки) терапии демонстрирует, что раннее вмешательство во время сердечного приступа может привести к существенному снижению поражения ткани миокарда. Широкие клинические испытания подтверждают документально, что тромболитическая терапия снижает риск развития нарушений в сердечном сокращении, а также сохраняет способность сердца действовать в качестве насоса. Показано, что это сохранение нормальной сердечной функции снижает отдаленную смертность после инфаркта. Существует также интерес в развитии способов терапии, способных обеспечить дополнительную защиту сердца, которые можно назначать в сочетании с тромболитической терапией или сами по себе, так как ретроспективные эпидемиологические исследования показали, что, 5 по-видимому, смертность в течение первых нескольких лет после инфаркта связана с первоначальной величиной инфаркта. В преклинических исследованиях инфаркта, проведенных на разнообразных моделях животных, показано, что многие типы фармакологических агентов, такие как блокаторы кальциевого канала, аналоги простациклина и агенты,способные ингибировать определенные метаболические пути, могут снижать степень ишемического поражения у некоторых видов животных. Последние исследования показали, что подвергание миокарда кратким периодам ишемии (прерывание кровотока в сердце) с последующей повторной перфузией (возобновление кровотока) способно защитить сердце от последующих ишемических поражений, что иначе случилось бы вследствие воздействия более длинных периодов ишемии. Это явление называют предварительной закалкой миокарда и считают, что оно частично свойственно для выделения аденозина во время периода предварительной закалки. Другие исследования показали, что аденозин и агонисты аденозина снижают степень поражения ткани, которое наблюдают после прерывания кровотока к сердцу на различных моделях ишемического поражения у некоторых видов (смотри, например, Toombs, С. и др.(1992. По способам настоящего изобретения получают промежуточные соединения, полезные в получении соединений, которые симулируют предварительную закалку миокарда, уменьшая при этом ишемическое поражение или давая снижение величины инфаркта миокарда, являющегося результатом ишемии миокарда, и полезные в качестве кардиозащитных агентов. Антилиполиз Известно, что гиперлипидемия и гиперхолестеринемия являются двумя из основных факторов риска для атеросклероза и коронарной болезни сердца, главной причиной смерти и потери трудоспособности в западных странах. Хотя этиология атеросклероза многофакторна,развитие атеросклероза и состояний, включающих болезнь коронарной артерии, заболевание периферических сосудов и церебрососудистые заболевания, являющихся результатом ограничения кровотока, связано с нарушениями содержания холестерина и липидов в сыворотке. Этиология гиперхолестеринемии и гиперлипи 000884 6 демии, главным образом, генетическая, хотя свой вклад могут вносить такие факторы как употребление с пищей насыщенных жиров и холестерина. Антилиполитическая активность аденозина и его аналогов является результатом активации рецепторов подтипа А 1 (Lohse, M.J., и др.,Recent Advances in Receptor Chemistry, Melchiorre, C. and Gianella, Eds, Elsevier Science Publishers B.V., Amsterdam, 1988, 107-121). Стимуляция рецепторов этого подтипа снижает концентрацию внутриклеточного циклического АМФ (аденозинмонофосфата) в адипоцитах. Циклический АМФ является необходимым кофактором для фермента липопротеинлипазы,которая гидролитически расщепляет триглицериды в адипоцитах до свободных жирных кислот и глицерина (Egan, J.J. и др., Proc. Natl.Acad. Sci. 1992 (89), 8357-8541). Соответственно, уменьшение концентрации внутриклеточного циклического АМФ в адипоцитах снижает активность липопротеинлипазы и, следовательно, гидролиз триглицеридов. Повышенное кровяное давление и содержание липидов в плазме, включая триглицериды, обычно являются двумя распространенными факторами риска, связанными со смертностью в результате сердечно-сосудистых заболеваний. Для пациентов с диабетом, когда вероятность смертности от сердечно-сосудистого заболевания существенно выше, повышается риск, связанный с этими факторами (Bierman,E.L., Arteriosclerosis and Thrombosis, 1992 (12),647-656). Кроме того, данные говорят, что чрезмерный липолиз является характеристикой инсулиннезависимого диабета и, возможно, вносит вклад в устойчивость к инсулину и гипергликемию (Swislocki, A.L., Horm. Metab. Res. 1993(25), 90-95). По способам настоящего изобретения получают промежуточные продукты, полезные в получении соединений, которые являются гипотензивными и антилиполитическими агентами и полезны при лечении и ослаблении сосудистых и метаболических факторов риска. Соединения аденозина и их активность Аденозин обладает широким разнообразием физиологического и фармакологического действия, включая существенное изменение сердечно-сосудистой и почечной функции. У животных и человека внутривенная инъекция нуклеотида аденозина вызывает гипотензию. Физиологическое и фармакологическое действие аденозина опосредовано специфическими рецепторами, расположенными на поверхностях клеток. Идентифицируют два подтипа рецепторов аденозина, обозначенные какA1 рецепторы ингибируют образование цАМФ путем подавления активности аденилатциклазы, в то время как А 2 рецепторы повышают активность аденилатциклазы и внутрикле 7 точного цАМФ. По-видимому, каждый рецептор опосредует специфические действия аденозина в различных тканях: например, сосудистое действие аденозина, по-видимому, опосредовано через стимуляцию A2 рецепторов, подкрепленную позитивной корреляцией между генерированием цАМФ и расширением сосудов в обработанной аденозином выделенной гладкой мышце сосуда; при этом стимуляция A1 рецепторов сердца снижает в сердце генерацию цАМФ, которая вносит вклад в негативное дромотропное, инотропное и хронотропное действие сердца. Следовательно, в отличие от большинства сосудорасширяющих факторов, введение аденозина не продуцирует рефлекторную тахикардию. Аденозин также оказывает заметное влияние на почечную функцию. Внутрипочечное вливание аденозина вызывает временное ослабление почечного кровотока и повышение сопротивления сосудов почек. При длительном вливании аденозина почечный кровоток восстанавливается до контрольного уровня, и снижается сопротивление сосудов почек. Начальные почечные сосудосуживающие реакции на аденозин происходят не в результате прямого сосудосуживающего действия нуклеотида, а включают взаимодействие между аденозином и системой ренин-ангиотензин. Аденозин считают основным физиологическим медиатором реактивной гиперемии и авторегуляции коронарной поддерживающей ткани в ответ на ишемию миокарда. Сообщается, что коронарный эндотелий обладает А 2 рецепторами аденозина, связанными с аденилатциклазой, которые активируются параллельно с повышением коронарного тока, и что рецепторы кардиомиоцита преимущественно являются A1 подтипом рецепторов аденозина и связаны с брадикардией. Соответственно, аденозин предлагает уникальный механизм терапии ишемии. Сердечно-сосудистые реакции на аденозин кратковременны вследствие быстрого усвоения и метаболизма эндогенного нуклеотида. Аналоги аденозина, наоборот, более устойчивы к метаболическому разрушению, и сообщается, что они вызывают длительные изменения артериального давления и частоты сердечных сокращений. Синтезировано несколько сильнодействующих метаболически стабильных аналогов аденозина, которые демонстрируют разную степень селективности относительно двух подтипов рецепторов. Агонисты аденозина обычно показывают большую селективность относительно A1 рецепторов по сравнению с А 2 рецепторами. Циклопентиладенозин (СРА) и Rфенилизопропиладенозин (R-PIA) являются стандартными агонистами аденозина, которые показывают заметную селективность относительно A1 рецепторов (соотношение А 2/А 1=780 и 106, соответственно). N-5'-этилкарбоксамидо 000884 8 аденозин (NECA), напротив, является сильнодействующим агонистом A2 рецепторов (Ki-12 нМ), но имеет равное сродство относительно A1 рецепторовA2/A1=1,87). До настоящего времени CV-1808 был самым селективным доступным агонистом А 2 (A2/A1=0,19), даже, несмотря на то, что это соединение в 10 раз менее действенно, чемNECA, в своем сродстве к A2 рецепторам. В современных разработках раскрыты более новые соединения, которые являются очень сильнодействующими и селективными агонистами A2(Ki=3-8 нМ для A1; соотношение A2/A1=0,0270,042). В литературе сообщается о различных N6 арил- и N6-гетероарилалкилзамещенных аденозинах и замещенных-(2-амино и 2-гидрокси) аденозинах как обладающих различной фармакологической активностью, включая кардиотоническую и циркуляторную активность. Смотри, например, описание Патента Великобритании 1,123,245,German 67/7630, патент США 4,501,735, публикация Европейского патента 0139358 (раскрытыN6-[геминально диарилзамещенный алкил] аденозины), публикация Европейской Патентной заявки сер. 88106818.3 (раскрыто, что N6 гетероциклически замещенные производные аденозина демонстрируют кардиотоническую сосудорасширяющую активность), German Offen. 2,131,938 (раскрывает арил- и гетероарилалкилгидразинильные производные аденозина),German Offen. 2,151,013 (раскрывает N6-арил- и гетероарил-замещенные аденозины), GermanOffen. 2,205,002 (раскрывает аденозины с N6 заместителями, включая структуры с мостиковым кольцом, связывающие N6-азот с заместителями, включая тиенил) и южно-африканский патент 68/5477 (раскрывает N6-индолилзамещенные-2-гидроксиаденозины). Патент США 4,954,504 и публикация Европейского Патента 0267878 раскрывают в общем виде, что соединения карбоциклической рибозы, аналоги аденозина, и их фармацевтически приемлемые сложные эфиры, замещенные по 2 и/или N6-положению арил(низший алкил)группами, включая тиенил, тетрагидропиранил,тетрагидротиопиранил, и бициклические бензоконденсированные 5- или 6-членные насыщенные гетероциклические производные низших алкилов, демонстрируют свойства агонистов рецепторов аденозина. Аналоги аденозина с заместителями тиенильного типа описаны в публикации европейского патента 0277917WO 85/04882 (раскрывает, что N6-гетероцикли 9 ческий алкилзамещенные производные аденозина, включая N6-[2-(2-тиенил)этил]амино-9(D-рибофуранозил)-9 Н-пурин, демонстрируют сердечно-сосудистую сосудорасширяющую активность, и что N6-хиральные заместители проявляют повышенную активность), опубликованной европейской патентной заявке 0232813 (раскрывает, что N6(1-замещенный тиенил)циклопропилметилзамещенные аденозины демонстрируют сердечно-сосудистую активность), патенте США 4,683,223 (раскрывает, что N6-бензотиопиранилзамещенные аденозины демонстрируют гипотензивные свойства), РСТ WO 88/03147 и WO 88/03148 (раскрывают, что N6-[2-арил-2-(тиен-2-ил)]этилзамещенные аденозины демонстрируют гипотензивные свойства), патентах США 4,636,493 и 4,600,707 (раскрывают, что N6-бензотиенилэтилзамещенные аденозины демонстрируют гипотензивные свойства). В патенте США 3,914,415 раскрыты амиды аденозин-5'-карбоновой кислоты как соединения, полезные в качестве гипотензивных агентов и агентов против стенокардии, а патент США 4,738,954 раскрывает, что N6-замещенный арил- и арилалкиладенозин-5'-этилкарбоксамиды демонстрируют разные кардиотонические и гипотензивные свойства.N6-алкил-2'-0-алкиладенозины раскрыты в публикации европейского патента 0,378,518 и патентной заявке Великобритании 2,226,027 как соединения, обладающие гипотензивной активностью. Патент США 4,843,066 сообщает также,что N6-aлкил-2',3'-ди-0-алкиладенозины применимы в качестве гипотензивных агентов. Сообщают, что аденозин-5'(N-замещенные)карбоксамиды, и их эфиры карбоксилаты иN1-оксиды являются коронарными сосудорасширяющими средствами.Stein и др.,J.Med.Chem. 1980, 23, 313-319 и J.Med.Chem. 19(10), 1180 (1976). В Патенте США 4,167,565 также сообщают, что аденозин-5'-карбоксамиды и их N1-оксиды являются ядами для мелких животных. Антилиполитическая активность аденозина описана в работе Dole, V.P., J.Biol.Chem. 236and Metabolic Functions; Jeanrenaud, В and Hepp,D.Eds., George Thieme, Stuttgart, 47-54 (1970). Патенты США 3,787,391, 3,817,981,3,838,147, 3,840,521, 3,835,035, 3,851,056,3,880,829, 3,929,763, 3,929,764, 3,988,317 и 5,032,583 раскрывают N6-моно-и дизамещенные аналоги аденозина как обладающие антилиполитической, антигиперхолестеринемической и антигиперлипемической активностью. Считают, что указанная токсичность, свойства по отношению к ЦНС и повышение частоты сердечных сокращений связаны с трудностями, привносимыми аналогами аденозина, 000884 10 препятствуя развитию коммерческого аналога аденозина,гипотензивного/противоишемического агента. Патентные заявки США с серийными 08,484,811 и 08,316/761, которые заявляют пользу опубликованной РСТ заявки PCT/US 91/06990, раскрывают класс метаболически стабильных агонистов аденозина и их производных, неожиданно обладающих желательными фармакологическими свойствами, а именно,гипотензивные, кардиозащитные, противоишемические и антилиполитические агенты, имеющие уникальный терапевтический профиль. 2. Представленные разработки Краткое описание изобретения Настоящее изобретение касается способа получения 2,4-дигидроксипиридина, включающего нагревание соединения формулы А где R является Н, алкилом или аралкилом, и фосфорной кислоты при соотношении фосфорной кислоты к воде не менее чем примерно 27-1 весовой %. Данное изобретение касается также способа получения 2,4-дигидрокси-3-нитропиридина, включающего реакцию 2,4-дигидроксипиридина с азотной кислотой. Способы настоящего изобретения позволяют получать промежуточные соединения, полезные в получении соединений, которые полезны для лечения сердечно-сосудистых заболеваний, охарактеризованных как гипертензия или ишемия миокарда, для уменьшения ишемического поражения или величины инфаркта миокарда, или полезны при лечении гиперлипидемии или гиперхолестеринемии. Подробное описание изобретения Использованные выше и на протяжении всего описания данного изобретения термины,если не указано по-другому, имеют следующие значения:"Ацил" обозначает линейную или разветвленную алкил-С(=0)-группу. Предпочтительными ацильными группами являются низшие алканоилы, имеющие от 1 до 6 атомов углерода в алкильной группе."Алкил" обозначает насыщенную алифатическую углеводородную группу, которая может быть линейной или разветвленной и имеет примерно от 1 до 20 атомов углерода в цепи."Разветвленная" означает, что низшая алкильная группа, такая как метил, этил или пропил, присоединена к линейной алкильной цепи."Алкилен" обозначает линейную или разветвленную бивалентную углеводородную цепь,имеющую от 1 до примерно 20 атомов углерода. Предпочтительными алкиленовыми группами 11 являются низшие алкиленовые группы, имеющие от 1 до примерно 6 атомов углерода. Наиболее предпочтительными алкиленовыми группами являются метилен, этилен, этилэтилен,метилэтилен и диметилэтилен."Циклоалкилен" обозначает 1,2- или 1,3 бивалентную карбоциклическую группу,имеющую примерно от 4 до 8 атомов углерода. Предпочтительные циклоалкиленовые группы включают 4,5-цис- или транс-циклогексенилен,1,2-циклогексанилен и 1,2-циклопентилен."Произвольно замещенный" обозначает,что данный заместитель или заместители могут присутствовать, а могут отсутствовать."Алкиламино" обозначает аминогруппу,замещенную одним или двумя алкильными группами. Предпочтительными группами являются (низший алкил)аминогруппы."Алкилкарбамоил" обозначает карбамоильную группу, замещенную одной или двумя алкильными группами. Предпочтительными являются (низший алкил)карбамоильные группы."Алкокси" обозначает алкилоксигруппу, в которой "алкил" является таким, как описано ранее. Предпочтительны низшие алкоксигруппы. Примеры таких групп включают метокси-, этокси-, н-пропокси-, изопропокси- и нбутоксигруппы."Алкоксиалкил" обозначает алкильную группу (как описано ранее), замещенную алкоксигруппой (как описано ранее)."арил" обозначает фенил или фенил, замещенный одним заместителем или более, которые могут быть алкилом, алкоксигруппой, аминогруппой, нитрогруппой, карбоксилом, карбоалкоксигруппой, цианогруппой, алкиламиногруппой, галогеном, гидроксилом, гидроксиалкилом, меркаптилом, алкилмеркаптилом, карбалкилом или карбамоилом."Карбалкокси" обозначает карбоксильный заместитель, этерифицированный спиртом формулы CnH2n+1OH, где n равно примерно от 1 до 6."Гетероциклический" обозначает 4-10 членную циклическую структуру, в которой один атом в кольце или более являются отличными от углерода элементами, например, N,0 или S."Формула I" обозначает соединение, описанное следующей формулой и определением: или R3 О-СН 2; Х является линейной или разветвленной алкиленовой, циклоалкиленовой или циклоалкениленовой группой, каждая из которых произвольно замещена, по крайней мере, одним СН 3, СН 3 СН 2, Cl, F, СF3 или СН 3 О; где Rc является водородом или алкилом,где Rd и Re независимо являются водородом, алкилом или вместе с атомом углерода, к которому они присоединены, могут образовывать 1,1-циклоалкильную группу; при условии,что когда Х является линейным алкиленом, a Q является кислородом, то Z представляет гетероциклил, включающий, по крайней мере, два гетероатома; или его фармацевтически приемлемую соль. Примеры гетероциклических фрагментов,включающих N6 заместитель, в соединениях формулы I включают следующие: Предпочтительные гетероциклические группы включают незамещенный и замещенный тиенил, тиазолил и бензотиазолил, с одним заместителем или более из группы, включающей алкоксигруппу, алкиламиногруппу, арил, карбалкоксигруппу, карбамоил, цианогруппу, галоген, гидроксил, меркаптил, алкилмеркаптил или нитрогруппу."Гидроксиалкил" обозначает алкильную группу, замещенную гидроксилом. Предпочтительными являются гидрокси(низший алкил)группы. Примеры предпочтительных групп включают гидроксиметил, 2-гидроксиэтил, 2 гидроксипропил и 3-гидроксипропил."Пролекарство" обозначает соединение,которое само может или не может быть биологически активным, но которое метаболическим,сольволитическим или другим физиологическим способом превращается в биологически активное химическое соединение."Кардиозащитный" относится к эффекту,при котором миокард становится менее восприимчивым к ишемическим поражениям и инфаркту миокарда вследствие ишемии миокарда."Уменьшение ишемического поражения" означает предупреждение или уменьшение ишемического поражения миокарда вследствие ишемии миокарда."Уменьшение размера инфаркта миокарда" означает снижение размера инфаркта или его профилактику вследствие ишемии миокарда. Соединения формулы I преимущественно включают хиральный (асимметрический) центр. Например, предпочтительные соединения с таким асимметрическим центром включают соединения, в которых Х является изопропиленом,и имеют либо R, либо S конфигурацию, причем более предпочтительна R конфигурация. Соединения формулы I включают индивидуальные стереоизомеры и их смеси. Индивидуальные изомеры получают или выделяют известными в практике способами или способами, описанными здесь. Соединения, полученные здесь из промежуточных продуктов, приготовленных согласно данному изобретению, можно использовать в виде свободного основания, в виде соли присоединения кислоты или в виде гидратов. Все эти формы включены в область соединений формулы I. Соли присоединения кислот являются более удобной формой для применения. На практике количество применяемой солевой формы соответствует количеству основной формы. Кислоты, которые можно использовать для получения солей присоединения кислот, предпочтительно включают кислоты, которые при объединении со свободным основанием дают фармацевтически приемлемые соли, то есть соли, 000884 14 анионы которых нетоксичны для пациента при фармацевтических дозах этих солей, так что полезные гипотензивные, кардиозащитные, противоишемические и антилиполитические эффекты, производимые свободным основанием,не портятся побочными эффектами, приписываемыми этим анионам. Хотя здесь предпочтительными являются фармацевтически приемлемые соли соединений, все соли присоединения кислот полезны в качестве источников форм свободных оснований, даже если конкретная соль сама по себе требуется только как промежуточный продукт, например, когда соль получают только в целях очистки и идентификации,или когда ее используют как промежуточный продукт в получении фармацевтически приемлемой соли путем ионообменных процедур. Фармацевтически приемлемые соли, попадающие в область данного изобретения, являются солями, производными следующих кислот: минеральные кислоты, такие как соляная кислота,серная кислота, фосфорная кислота, и сульфаминовая кислота; и органические кислоты, такие как уксусная кислота, лимонная кислота,молочная кислота, винная кислота, малоновая кислота, метансульфоновая кислота, фумаровая кислота, этансульфоновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота, циклогексилсульфаминовая кислота,хинная кислота и подобные. Соответствующими солями присоединения кислот являются следующие: гидрохлорид, сульфат, фосфат, сульфамат, ацетат, цитрат, лактат, тартрат, метансульфонат, фумарат, этансульфонат, бензолсульфонат, паратолуолсульфонат, циклогексилсульфамат и хинат, соответственно. Удобно получать соли присоединения кислот соединений формулы I путем растворения свободного основания в водном или водноспиртовом растворе или других подходящих растворителях, содержащих подходящую кислоту, и выделения соли при выпаривании раствора, или путем реакции свободного основания и кислоты в органическом растворителе, в этом случае соль выделяется прямо или может быть получена посредством концентрирования данного раствора. Область соединений формулы I включает классы соединений, которые обычно можно охарактеризовать как N6-гетероциклическизамещенные аденозины; N6-гетероциклическизамещенные карбоциклические аденозины (или,по-другому,дигидрокси[4,5-b]имидазопиридил]циклопентаны. Область соединений формулы I включает также 5'-алкилкарбоксамидные производные аденозина, карбоциклические аденозины и 1-деазааристеромицины, производные соедине 15 ний указанных выше классов, в которых одна или обе из 2- или 3-гидроксильных групп циклопентанового кольца (или в случаях классов соединений, содержащих рибозную часть, 2'или 3'-гидроксильные группы рибозного кольца) замещены. Такие производные сами заключают в себе биологически активную сущность, полезную при лечении гипертезии и ишемии миокарда, и в качестве кардиозащитных и антилиполитических средств, или могут действовать как пролекарства таких биологически активных соединений, которые получаются из них в физиологических условиях. Примеры продуктов, полученных из соединений данного изобретения, включают:N6-[транс-5-(2 тиенил)циклогекс-1-ен-4-ил]-2'-0-метиладенозин. Предпочтителен класс соединений формулы I, где R' и R" являются Н. Другим предпочтительным классом соединений формулы I являются 5'-N-алкилкарбоксамидные производные N6-гетероциклическизамещенных карбоциклических аденозинов,другими словами, соединений формулы I, в которых К является N, Q является CH2, и Т является R1R2N-C=0, или их фармацевтически приемлемые соли,Еще одним предпочтительным классом соединений формулы I являются 5'-N-алкилкарбоксамидные производныеN6-гетероциклическизамещенных-N'-1-деазааристеромицинов, то есть 4-[7-[гетероциклиламино]-3 Нимидазо[4,5-b]пиридин-3-ил]алкил-2,3-дигидроксициклопентанкарбоксамиды, другими словами, соединения формулы I, в которых К является СН, Q является CH2, и Т является R1R2N-C=0,или их фармацевтически приемлемые соли. 17 Наиболее предпочтительный класс соединений формулы I отличается присутствием хирального центра в альфа-положении к N6 атому пуринового или 1-деазапуринового кольца, тогда как специальная группа соединений этого класса включает соединения, отличающиеся наличием хиральной этильной группы, присоединенной к атому углерода в альфа-положении к N6 атому азота. Особо предпочтительный класс соединений отличается наличием в качестве заместителя группы N6-[1-(низший алкил)2-(3-галогентиен -2-ил)этил]. Наиболее предпочтительная группа соединений формулы I включает соединения: (-)-[2S[2 а,3 а-дигидрокси-4-[N6-[2-(5-хлор-2-тиенил)1-(R)-1-метилэтил]амино]-9-аденил]циклопентан]-1-этилкарбоксамид; (-)-[2S-[2a,3 а-дигидрокси-4-[N6-[1-(R)-этил-(3-хлор-2-тиенил) этил]амино]-9-аденил]циклопентан]-1-Nэтилкарбоксамид; [1S-[1a,2b,3b,4a(S)-4-[7-2(5-[хлор-2-тиенил)-1-метилэтил]амино]-3 Нимидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид; [1S-[1a,2b,3b,4a(S)-4-[7-2-(3-хлор-2-тиенил)-1 этилэтил]амино]-3 Н-имидазо[4,5-b]пиридин-2(3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид; и их фармацевтически приемлемые соли. Соединения формулы I можно получить известными способами или в соответствии с реакционными последовательностями, описанными ниже. Используемые в получении соединений формулы I материалы известны, или доступны коммерчески, или их можно получить известными способами или по специальным описанным здесь реакционным схемам, которые включают способы данного изобретения. Способы получения 2,4-дигидроксипиридина и 2,4-дигидрокси-3-нитропиридина согласно данному изобретению показаны на схеме А 1. Реакционная схема А 1R1 является алкилом или аралкилом; предпочтительно низшим алкилом и более предпочтительно метилом. Схема А 1 показывает первоначальное образование (алкил или аралкил) 4,6-дигидроксиникотината путем взаимодействия ди(алкил или аралкил)ацетондикарбоксилата с триметилортоформиатом и уксусным ангидридом в инертной атмосфере, такой как азот; дистилляции уксусной кислоты/(алкил или аралкил) ацетата (предпочтительно при пониженном давлении, примерно 20 мм рт.ст.) и последующего взаимодействия полученной смеси с гидроксидом аммония и соляной кислотой. Согласно одному аспекту данного изобретения вслед за получением (алкил или аралкил)4,6-дигидроксиникотината по схеме А 1 этот продукт превращают в 2,4-дигидроксипиридин путем нагревания с фосфорной кислотой при отношении фосфорной кислоты к воде не менее,чем примерно 27 к 1% по весу (Н 3 РO4: Н 2 О=27:1 вес.%). Такое соотношение можно получить посредством нагревания, после чего из реакционной смеси удаляют достаточное количество воды. После удаления достаточного количества воды температура реакционной смеси достигает примерно 210 С (5 С). Затем эту реакционную смесь выдерживают в течение примерно 4-5 ч при температуре, близкой к этой, до исчезновения (алкил или аралкил) 4,6 дигидроксиникотината или промежуточной 4,6 гидроксиникотиновой кислоты. Согласно этому аспекту изобретения реакция включает декарбоксилирование от пиридильного остатка,катализируемое фосфорной кислотой по существу в безводных условиях. Согласно другому аспекту данного изобретения по схеме А 1 можно дополнительно получить 2,4-дигидроксипиридин путем превращения (алкил или аралкил) 4,6-дигидроксиникотината в 4,6-дигидроксиникотиновую кислоту посредством гидролиза с сильным основанием, таким как NaOH или КОН, и последующей обработки 4,6-дигидроксиникотиновой кислоты таким же образом как (алкил или аралкил)4,6-дигидроксиникотината. Согласно еще одному аспекту данного изобретения на схеме А 1 также показано, что(алкил или аралкил) 4,6-дигидроксиникотинат или 4,6-дигидроксиникотиновую кислоту можно превратить в 2,4-дигидрокси-3-нитропиридин, не выделяя 2,4-дигидроксипиридин. Эта реакция включает проведение декарбоксилирования, как описано выше, и последующую обработку реакционной смеси азотной кислотой. Предпочтительнее обработки азотной кислотой является добавление органической кислоты в качестве растворителя. Нитрование предпочтительно проводят при нагревании, например, при температуре примерно от 80 до 100 С, более предпочтительно при 90 С до тех пор, пока уве 19 личивается количество воды, и прекращают нагревание. Согласно еще одному аспекту данного изобретения на схеме А 1 показано, что 2,4 дигидроксипиридин можно превратить в 2,4 дигидрокси-3-нитропиридин, используя предшествующие способы нитрования. На схеме А 1 показано также превращение 2,4-дигидрокси-3-нитропиридина в 2,4-дихлор 3-нитропиридин по реакции оксихлорида фосфора и 2,4-дигидрокси-3-нитропиридина в присутствии диизопропилэтиламина (DIPEA). Реакция происходит примерно при 100 С. Как здесь показано, 2,4-дихлор-3-нитропиридин можно использовать вместо других дигалогеннитрогетероарилов для образования промежуточных продуктов, как на схеме К. Наконец, на схеме А 1 показано превращение 2,4-дихлор-3-нитропиридина в 3-амино-2,4 дихлорпиридин в восстановительных условиях,таких как Zn/HCl, или в условиях гидрирования. Как здесь показано, 3-амино-2,4-дихлорпиридин можно использовать вместо других аминодигалогенгетероарилов, как на схеме В. Соединения формулы I, где К является N,Q является О, и Т является R3O-CH2, можно получить по реакции коммерчески доступного 6 хлорпуринрибозида с различными гетероциклическими аминами, как показано ниже. Соединения формулы I, где К является N,Q является О, и Т-R1R2N-C=0, получают аналогичным образом, исходя из продукта реакционной схемы А. В этой реакции 6-хлорпуринрибозид с защищенными 2'- и 3'-гидроксильными группами рибозного кольца обрабатывают окислителем, например, таким как реагент Джонса (Jones) и полученную кислоту обрабатывают дициклогексилкарбодиимидом (DCC) или ВОР-С 1 (хлорид бис(2-оксо-3-оксазоладинил)фосфиновой кислоты) в присутствии выбранного амина, получая 5'-алкилкарбоксамидное производное. 20 3-амино-2,4-дихлорпиримидином. Затем продукт этой начальной реакции нагревают с альдегидиламидинацетатом, например, формамидинацетатом, в диоксане и метоксиэтаноле в течение времени, достаточного для осуществления замыкания кольца (примерно от 30 мин до 4 ч),получая при этом продукт, который может удобно реагировать с различными гетероциклическими аминами описанным ниже образом,порядок проведения реакции не является критичным. Например, полученный по реакционной схеме В промежуточный продукт может реагировать с гетероциклическим амином с последующим замыканием цикла, давая требуемый конечный продукт. Реакционная схема В Различные гетероциклические амины, полезные при получении соединений данного изобретения, можно получить посредством одной или более реакций, показанных здесь ниже на реакционных схемах C-J и в примерах получения B-G и 50-74 (Неt=гетероциклическая группа; На 1 о=галоген; R=H или низший алкил; Ra и(Р-защитная группа) Подходящие исходные материалы для соединений формулы I, где К является N, Q является CH2, и Т является R1R2N-С=0, можно получить, как описано в работе Chen и др., Tetrahedron Letters 30: 5543-46 (1989). По-другому, для получения таких исходных материалов можно использовать реакционную схему В. При осуществлении реакционной схемы В 4-этилкарбоксамидное производное 2,3-дигидроксициклопентиламина,полученное, как описано Chen и др., реагирует с Последовательность реакций приведенной выше схемы I описана в Патенте США 4,321,398, относящаяся к делу информация включена здесь в качестве ссылки. Пример В. Получение 1-(тиофен-3-ил)этиламина. 3-тиофенкарбоксальдегид (1 ммоль), нитрометан (1,5 ммоля) и бета-аланин (0,1 ммоля) перемешивают в бутаноле в течение 6 ч, получая 3-нитровинилентиофен, который восстанавливают литийалюминийгидридом (2,5 ммоля),получая требуемый продукт амин. 3-замещенные тиенилалкиламины получают путем замены 3-замещенных тиофенов,таких как 3-хлортиофен, на тиофеновые исходные материалы в приведенном выше примере В. Пример С. Получение транс-2-(тиофен-2 ил)циклогекс-4-ениламина. Смесь 1,3-бутадиена (5 мл) и 2-нитровинилентиофена (7 г) в толуоле нагревают в закупоренной пробирке при 140 С в течение ночи. Полученный нитроциклогексен гидрируют (35 фунт/дюйм 2=2,461 кг/см 2 Н 2) (5% Pd/C MeOH) и обрабатывают литийалюминийгидридом (2,5 г). Посредством стандартной переработки получают рацемический транс-2-(тиофен-2-ил)циклогексиламин. Пример D. Основной способ получения 2 замещенных тиазоламинов. Бензоилхлорид и аминоэтилцианид реагируют, давая N-бензоиламиноэтилцианин, который реагирует с сероводородом в аммиаке, давая тиоамид, который реагирует с подходящим-галогенкетоном, давая требуемый тиазол. В результате обработки 5N соляной кислотой уда 000884 22 ляют защитную бензоильную группу, получая требуемый продукт - амин. Пример Е. Общий способ получения 4 замещенных тиазолиламинов. Предпочтительным синтезом для 2-(2'метил-4'-тиазолил)этиламина является реакция тиоацетамида с этилмонобромацетоацетатом с получением эфира тиазола, который восстанавливают предпочтительно боргидридом натрия,получая спирт, который переводят в амин. Для амина предпочтительный способ обработки включает обработку (1) диэтилазодикарбоксилатом (DEAD), трифенилфосфином и фталимидом и (2) гидразингидратом. Получение 4-замещенных тиазоламинов можно также проводить, используя предшествующую реакционную схему, посредством реакции замещенного тиоамида и этилмонобромацетоацетата. Превращение полученного эфира тиазолила в амид выполняют при помощи водного аммония, а амин получают посредством восстановления бораном. Пример получения 2(1,1-диметил-1'-тиофенил)этиламина описан в Патенте США 4,321,398. Диастереомерные смеси соединений или промежуточных продуктов, полученных по приведенным выше реакционным схемам A-I,можно разделить на отдельные рацемические или оптически активные энантиомеры способами, известными в практике; например, способом хроматографии, фракционной перегонки или фракционной перекристаллизации d- или 1 солей (тартратов, дибензоилтартратов, манделатов или камфорсульфонатов). Пример F. Получение (+) и (-)транс-2(тиофен-2-ил)циклогекс-4-ениламина.(S)-(+)-миндальную кислоту (0,55 экв) добавляют к изопропанольному раствору рацемического амина (3,4 г), полученного в примере С. Осадок перекристаллизовывают из изопропанола, получая 1,78 г соли ([]DRT=+4,13 (с=1,3,МеОН. Амины выделяют посредством экстракции нейтрализованных солей (насыщ. NaHCO3) CH2Cl2, сушки (Na2SO4) и концентрирования, получая частично разделенные свободные амины. Приблизительно 1 г левовращающего амина ([]DRT=-25,8 (с=1,54, МеОН обрабатывают 2 г 1-(-)-дибензоилвинной кислоты в метаноле и полученную соль обрабатывают, получая 0,64 г левовращающего амина ([]DRТ=-28,8 (с=1,65,МеОН. Исследование методом ЯМР в высоком поле МРТА-амида левовращающего амина показывает 96% энантиомерный избыток. Приблизительно 1,6 г обогащенной правовращающей смеси аминов обрабатывают 3,2 гd(+)-дибензоилвинной кислоты в метаноле. После обработки получают 0,87 г правовращающего амина ([]DRT=+25,8 (с=1,67, МеОН. 23 изобретения можно получить по реакции 6 хлорпуринрибозида или продуктов реакционной схемы А или В с различными гетероциклическими аминами в соответствии со способом синтеза, показанным ниже в реакционной схемеN'алкилдеазааристеромицины данного изобретения можно получить, как показано в реакционной схеме К. Реакционная схема К Соединения формулы I, которые могут действовать в качестве пролекарств, включают те соединения, в которых гидроксильные группы на рибозном или циклопентановом кольце замещены группами R' и R", как определено выше для формулы I. Их можно получить известными способами, и они представлены примерами способов получения, показанными ниже в реакционной схеме L. Реакционная схема L 24 Обработка дигидроксисоединений эфиром хлорформиата в присутствии органического основания, например, триэтиламина дает соответствующий бискарбонат. Алкоксиметиленацеталь можно получить посредством обработки соответствующим ортофиэром в присутствии каталитического количества пара-толуолсульфокислоты. Карбонат можно получить посредством обработки 1,1'-карбонилдиимидазолом, а тиокарбонат посредством обработки тиокарбонилдиимидазолом. Алкильные и диалкилкарбамоильные производные можно получить посредством обработки соответствующим алкилизоцианатом или диалкилкарбамоилхлоридом,соответственно, в присутствии органического основания. Соединения формулы I, в которых К является NО, то есть N-оксидами, можно получить путем окисления соответствующего аденозина или карбоциклического аденозина известными способами, например, посредством обработки перекисью водорода в уксусной кислоте. 2'-0-алкильные производные можно получить известными способами, например, по реакции подходящего гетероциклического амина с 6-хлор-9-(2'-0-метил-b-D-рибофуранозил)-9 Нпурином. Функциональные группы исходных соединений и промежуточных продуктов, которые используются в получении соединений формулы I, можно защитить обычными защитными группами, известными в практике. Общеизвестные защитные группы для функциональных амино- и гидроксильных групп описаны, например, в работе T.W.Green "Protective Groups inOrganic Synthesis", Wiley, New York (1984). Гидроксильные группы можно защитить в виде сложных эфиров, таких как ацильные производные, или в виде простых эфиров. Гидроксильные группы на соседних атомах углерода можно защитить преимущественно в виде кеталей или ацеталей. На практике соседние 2' и 3' гидроксильные группы исходных соединений в реакционных схемах А и B обычно защищают посредством образования 2',3'-изопропилиденовых производных. Свободные гидроксилы можно восстановить, например, по реакции кислотного гидролиза, или соль-волиза, или гидрогенолиза, обычно применяемой в органической химии. После синтеза соединения формулы I обычно чистят способом жидкостной хроматографии при среднем давлении (MPLC), на хроматотроне, способом тонкослойной хроматографии с радиальным ускорением, флэшхроматографии или колоночной хроматографии на силикагеле или матрице из Florisil с последующей кристаллизацией. Для соединений формулы I, где К является N, Q является 0, и Т является R3 О-СН 2, типичные системы растворителей включают хлороформ:метанол, этилацетат:гексан и метиленхлорид:метанол. Элюаты 25 можно кристаллизовать из метанола, этанола,этилацетата гексана или хлороформа. Для соединений формулы 1, где К являетсяN,Q является 0, и Т является R1R2N-C=0, типичные системы растворителей включают хлороформ:метанол. Элюаты можно кристаллизовать из 50-100% этанола (водного). Для соединения формулы I, где Q является СН 2, К является N или СН, и Т является R1R2NC=0, типичные системы растворителей включают метиленхлорид:метанол. Элюаты можно кристаллизовать из этилацетата в присутствии или без метанола, этанола или гексана. Соединения, требующие нейтрализации,можно нейтрализовать слабым основанием, таким как бикарбонат натрия, с последующим промыванием метиленхлоридом и насыщенным солевым раствором. Продукты, которые в очищенном виде представляют собой масла, перед окончательной кристаллизацией иногда растирают с гексаном/этанолом. Здесь описан также усовершенствованный способ получения по существу оптически чистых производных 2-замещенного-2-амино-1(гетероар-2-или 3-ил)этана. 2-(Гетероарил) этиламины и их алкильные и фенильные производные получают различными способами, включая восстановление 2 нитровинилгетероарильных соединений, полученных из гетероарилформальдегидов (смотри, например, W. Foye и S.(1970; восстановление цианометилгетероарильных соединений (смотри, например, B.(1969; реакцию разложения по Хоффману(Hoffman) 2-(2-тиенил)пропиламида (смотри,например, G. Barger и A. Easson, J. Chem. Soc. 1938, 2100); и аминирование 2-(2-тиенил) этилпаратолуолсульфонатов. Патент США 4,128,561. Настоящий способ включает реакцию хирального производного 2-замещенного этиленоксида с 2- или 3-ил-анионом гетероарильного соединения и превращение гидроксильной группы, образовавшейся в данной реакции, стереоспецифическими способами в аминогруппу. Этот способ показан ниже на реакционной схеме M. Реакционная схема М 26 Преимуществом этого способа над способами получения производных 2-замещенного-2 амино-1-(гетероар-2- или 3-ил)этана, известными в практике, является то, что непосредственно получают по существу оптически чистое производное, в отличие от рацемической смеси, которую нужно затем разделять другими способами для получения оптически чистых изомеров. Предпочтительным классом этого способа является тот, в котором гетероар-2- или 3 ильная группа является замещенным или незамещенным тиен-2- или 3-илом или замещенным или незамещенным бензотиофен-2- или 3-илом. Более предпочтительным классом этого способа является тот, в котором указанный анион образуется по реакции замещенного или незамещенного тиофена или бензотиофена,имеющего водородный заместитель во 2- или 3 положении, с металлорганическим основанием в апротонном органическом растворителе. Другим более предпочтительным классом этого способа является тот, в котором указанный хиральный 2-замещенный этиленоксид замещен по 2-положению заместителем, выбранным из группы, состоящей из алкила, арила,тригалогенметила и бензилоксигруппы. Наиболее предпочтительным классом этого способа является тот, в котором указанное металлорганическое основание является алкиллитий- или литийдиизопропиламидом, указанный апротонный органический растворитель является тетрагидрофураном, эфиром, гексаном или смесью этих растворителей, а указанный хиральный 2-замещенный этиленоксид является 2-алкилэтиленоксидным производным. Способы стереоспецифического превращения гидроксильной группы в аминогруппу хорошо известны в практике (смотри, например,Mitsunobu, Synthesis 1981 (1), 1). Должно быть ясно, что производные (R)или(S)-2-замещенного-2-гидрокси-1-гетероарилэтана можно получить непосредственно, как описано выше, при использовании соответствующего производного (S)- или (R)-2 замещенного этиленоксида в качестве исходного материала или, если требуется или необходимо, полученный (R)- или (S)-2-замещенный-2 гидрокси-1-гетероарилэтан можно превратить в соответствующее производное (S)- или (R)-2 замещенного-2-гидрокси-1-гетероарилэтана,соответственно, хорошо известными в практике способами инвертирования конфигурации при гидроксильной группе (смотри, например, Mitsunobu, Synthesis 1981 (1), 1). Специфическим вариантом данного способа является такой, в котором: (а) замещенный или незамещенный тиофен или бензотиофен с водородным заместителем во 2- или 3 положении обрабатывают бутиллитием в смеси тетрагидрофурана и гексана при пониженной температуре, например, около -30 С в течение времени, достаточного для образования аниона 27 указанного тиофена или бензотиофена; (b) после этого добавляют(R)-2 алкилэтиленоксид и смесь держат при более высокой температуре, например, около 0 С в течение времени, достаточного для образования соответствующего (R) или (S)-2-алкил-2 гидрокси-1-тиенильного или бензотиофенильного производного этана; и (с) затем стереоспецифическими способами превращают гидроксильную группу указанного производного этана в аминогруппу. Этот способ дополнительно иллюстрируют и объясняют приведенные здесь ниже примеры с 50 по 74. Приведенные ниже примеры 1-3 описывают получение соединений-предшественников,использованных в способах получения соединений формулы I. Пример 1. Получение 6-хлор-2',3'-диметилметилендиокси-N-5'-этилкарбоксамидоаденозина. Стадия 1. 2',3'-Диметилметиленовое производное 6-хлорпуринрибозида. 6-Хлорпуринрибозид (31,5 г), триэтилортоформиат (73 мл) и ТsОН (19,8 г) перемешивают в 600 мл ацетона в течение 2 ч при комнатной температуре. Реакционную смесь концентрируют в вакууме, объединяют с этилацетатом и промывают насыщенным растворомNаНСО 3 и соляным раствором, сушат (Na2SO4) и концентрируют, получая 2',3'-диметилметиленовое производное 6-хлорпуринрибозида в виде белого твердого вещества. Стадия 2. 6-Хлор-2',3'-диметилметилендиоксиаденозин-5'-карбоновая кислота. Продукт стадии 1 (10 г) подвергают окислению по Джонсу (Jones), кислоту экстрагируют из этилацетата 2,5% раствором NaOH и водную часть промывают этилацетатом, подкисляют концентрированной НСl и экстрагируют этилацетатом. Органический слой промывают H2O и насыщенным солевым раствором, сушат(Nа 2SO4), фильтруют и концентрируют в вакууме до сухости, получая требуемую 5'карбоновую кислоту. Стадия 3. 6-Хлор-2',3-диметилметилендиокси-N-5'-этилкарбоксамидоаденозин. Продукт из стадии 2 (5,7 г) перемешивают с ВОР-Сl (хлорид бис(2-оксо-3-оксазоладинил) фосфиновой кислоты) (4,26 г) и триэтиламином(2,33 мл) в 100 мл метиленхлорида в течение 20 мин при комнатной температуре. Этиламин(3,46 г) подмешивают в раствор, который перемешивают в течение 2 ч при комнатной температуре. Органическую часть промывают разбавленным раствором НСl, разбавленным NaOH,H2O, насыщенным солевым раствором и сушат(Na2SO4), получая конечный продукт в виде пены. 28 Пример 2. Получение (+)-2S-[2 а,3 адиметилметилендиокси]-4-[6-хлор-9-аденил] циклопентан-1-N-этилкарбоксамида. Стадия 1. 5,6-Диметилендиокси-2-азабицикло[2.2.1]гептан-3-он. 5,6-дигидрокси-2-азабицикло[2.2.1]гептан 3-он (23,5 г) от Aldrich или полученного согласно процедуре, описанной в Cermak и Vince, Nucleic Acid Chemistry, Improved and New SyntheticProcedures, Methods and Techniques, Part Three,стр.26 (J.Wiley 1986), растворяют в ацетоне (150 мл), содержащем 2,2-диметоксипропан (185 мл) и пара-толуолсульфоновую кислоту (5,25 г),смесь кипятят с обратным холодильником в течение 10 мин, охлаждают, обрабатывают NaHCO3 (9,3 г) и концентрируют в вакууме. Остаток растворяют в CH2Cl2, промывают насыщенным солевым раствором, сушат над МgSO4 и выпаривают растворитель, получая масло. Это масло хроматографируют на SiO2 (4:1, этилацетат/гексан), получая 17,0 г (63%) белого твердого вещества (т.пл. 153-154 С). Стадия 2. (+)-4-амино-2 а,3 а-диметилендиоксициклопентан-1-N-этилкарбоксамид.[2.2.1]гептан-3-он (5 г), полученный на стадии 1,обрабатывают этиламином (15 мл) при 140 С в течение примерно 7 ч. Полученный продукт чистят способом флэш-хроматографии(B) Обработка рацемического амина (13,1 г), полученного, как описано в части А,Dдибензоилвинной кислотой (21,6 г) дает 15,1 г энантиомерно чистой соли. []DRT=+70,1(c=1,77, СН 3 ОН). Соль растворяют в 10% водном NaOH и водную фазу экстрагируют этилацетатом. Объединенную органическую фазу промывают насыщенным солевым раствором,сушат над MgSO4 и удаляют растворитель с получением оптически чистого соединения(2,10 г), полученного на стадии 2 (часть В), с 3 амино-2,4-дихлорпиридином (1,5 г) в нбутаноле (70 мл), содержащем триэтиламин (3 мл), в течение примерно 14 ч при кипячении с обратным холодильником и последующее удаление растворителя в вакууме дают масло, которое растворяют в этилацетате и промывают водным NаНСО 3. Органический экстракт сушат над Na2SO4 и концентрируют в вакууме, получая оптически чистое соединение. []DRТ=+15,8(c=41,48, CH3OH). Стадия 4. (+)-4-(3-Амино-4-хлор-2-пиримидиниламино)-2 а,3 а-диметилендиоксицикло 29 пентан (2,10 г), формамидинацетат (1,85 г) в метоксиэтаноле (2 мл) и диоксане (80 мл) перемешивают при 70 С в течение примерно 3 ч. Смесь охлаждают до комнатной температуры и удаляют растворитель в вакууме. Остаток растворяют в этилацетате, который промывают водным NаНСО 3 и насыщенным солевым раствором, органический экстракт сушат надNa2SO4, концентрируют в вакууме и чистят способом флэш-хроматографии (метиленхлорид/ метанол=95:5), получая чистый (+)-[2 а,3 адиметилметилендиокси]-4-[6-хлор-9-аденил]циклопентан-1-N-этилкарбоксамид (1,45 г). По-другому, оптически чистые производные 2 а,3 а-дизащищенного диокси-4-6-замещенного-9-аденил-циклопентан-1-N-этилкарбоксамида можно получить по реакционной схеме, представленной в примере 3. Пример 3. Получение 2S-[2 а,3 а-циклогексилидендиокси]-4-[N6-(2-тиенэтан-2-ил)-9 аденил]циклопентан-1-N-этилкарбоксамида. Стадия 1. 4-этилен-2 а,3 а-[циклогексилидендиокси]циклопентанон.Org.Chem. 1987, 52, 5457, добавляют в виде раствора в ТГФ (5 мл) к смеси винилмагнийбромида (15,2 ммоля) и Cul (15,2 ммоля) в ТГФ (100 мл). Эту смесь оставляют при -78 С в инертной атмосфере в течение примерно 2 ч, нагревают до 0 С и гасят, смешивая с насыщенным водным NH4Cl. Органическую фазу промывают насыщенным солевым раствором, сушат надMgSO4 и концентрируют в вакууме, получая желтое масло, которое чистят способом флэшхроматографии (метиленхлорид, 100%), получая 2,9 г требуемого соединения в виде масла. Стадия 2: 4-этилен-1-гидрокси-2 а,3 а[циклогексилидендиокси]циклопентан. 3,95 мл 1 М раствора диизобутилалюминийгидрида в тетрагидрофуране (ТГФ) добавляют к раствору ТГФ (75 мл) и кетона, полученного на стадии 1 (0,73 г), который охлаждают до-78 С. Смесь нагревают до -40 С в течение приблизительно 2,5 ч, обрабатывают 2N NaOH (5 мл), нагревают до комнатной температуры и перемешивают в течение приблизительно 1,5 ч. Водную фазу экстрагируют диэтиловым эфиром и объединенные органические фазы промывают насыщенным солевым раствором, сушат надMgSO4 и концентрируют в вакууме до желтого масла, которое чистят способом колоночной флэш-хроматографии 30 метиленхлориде (5 мл) и пиридине (0,24 мл) добавляют к перемешиваемому раствору трифторметилсульфонилангидрида (0,49 мл) в метиленхлориде (25 мл) при 0 С под аргоном. Примерно через 20 мин к реакционной смеси добавляют насыщенный солевой раствор, органическую фазу сушат над Nа 2SO4 и удаляют растворитель в вакууме, получая требуемый продукт в виде оранжевого масла, которое используют без дополнительной очистки. Стадия 4. 1-этилен-[2 а,3 а-циклогексилидендиокси]-4-[N6-(2-тиенилэтан-2-ил)-9 аденил]циклопентан. Раствор N6-тиофенилэтилпурина (2,13 г),NaH (50% дисперсия в масле, 0,35 г) и 18-краун 6 (0,15 г) в ДМФ (60 мл) добавляют к раствору 4-этилен-1-трифторметилсульфонил-2 а,3 а[циклогексилидендиокси]циклопентана, полученного на стадии 4, в ДМФ (2 мл) при 0 С. Смесь перемешивают при 0 С в течение примерно 8 ч, гасят, смешивая с насыщеннымNH4Cl, растворитель удаляют в вакууме и остаток объединяют с этилацетатом (100 мл) и насыщенным солевым раствором. Органический слой сушат над МgSO4 и концентрируют в вакууме, сырой продукт чистят способом флэшхроматографии (метиленхлорид/метанол=99:1),получая 0,85 г чистого продукта. Стадия 5. 2S-[2 а,3 а-Циклогексилидендиокси]-4-[N6-(2-тиенилэтан-2-ил)-9-аденил] циклопентан-1-N-этилкарбоксамид. Раствор 1-этилен-[2 а,3 а-циклогексилидендиокси]-4-[N6-(2-тиенилэтан-2-ил)-9-аденил]циклопентана (0,32 г) в 2 мл бензола добавляют к бензольному раствору перманганата калия (0,29 г) и 18-краун-6 (0,016 г) при 0 С. Реакционную смесь оставляют при комнатной температуре примерно в течение 6 ч, добавляют 5% водный NaOH (15 мл) и водную фазу фильтруют через Celite, подкисляют 1N HCl до рН 5 и экстрагируют этилацетатом. Органические экстракты сушат над MgSO4 и концентрируют в вакууме, получая 0,1 г [2 а,3 а-циклогексилидендиокси]-4-[N6-(2-тиенилэтан-2-ил)-9-аденил]-циклопентан-1-карбоксилата в виде желтого масла, которое растворяют в метиленхлориде (4 мл), содержащем дициклогексилкарбодиимид (DCC) (0,044 г). Этиламин (0,4 мл) добавляют к этой смеси, которую перемешивают при комнатной температуре в течение примерно 18 ч, растворитель удаляют в вакууме и сырой продукт чистят способом флэш-хроматографии(метиленхлорил/метанол=98:2), получая 0,077 г чистого продукта. Пример 4. Получение N6-[транс-2-(тиофен-2-ил)циклогекс-4-ен-ил]аденозина. Транс-2-(2'-тиофенил)-циклогекс-4-ениламин (0,3 г), полученный согласно способу, описанному выше в примере С, 6-хлорпуринрибозид (0,28 г) и триэтиламин (0,27 мл) в 20 мл этанола нагревают до кипения с обратным хо 31 лодильником в течение ночи под аргоном. Реакционную смесь охлаждают до комнатной температуры и удаляют растворитель, а остаток чистят способом жидкостной хроматографии со средним давлением (MPLC) (хлороформ:метанол=95:5) с последующей сушкой в вакууме приблизительно при 80 С, получая конечный продукт в виде твердого вещества, т.пл. 105110 С; элементный анализ С 20 Н 23N5O4S. Пример 5. Получение N6-[транс-2-(тиофен-2-ил)циклогекс-4-ен-1-ил]аденозин-5'-Nэтилкарбоксамида. Стадия 1. (+)Транс-2-(тиофен-2-ил)циклогекс-4-ениламин и 2',3'-диметилметилендиоксипроизводное соединения 6-Cl-NECA реагируют в условиях, описанных в примере 4, давая 2',3'диметилметилендиокси-производное конечного продукта. Стадия 2. 2',3'-Диметилметилендиоксипроизводное требуемого продукта перемешивают с трифторуксусной кислотой/водой (90/10) в течение 30 мин при комнатной температуре,нейтрализуют, медленно выливая смесь в насыщенный раствор бикарбоната натрия, и экстрагируют метиленхлоридом. Водный слой экстрагируют метиленхлоридом и объединяют органические слои, промывают их насыщенным солевым раствором, сушат над сульфатом магния и фильтруют, профильтрованный прозрачный раствор выпаривают. Остаток чистят способом флэш-хроматографии (метиленхлорид:метанол= 9:1), получая после сушки в вакууме конечный продукт в виде белой стекловидной пены, т.пл 112-117 С; С 22 Н 26N6O4S. Пример 6. Получение (-)-[2S-[2 а,3 адигидрокси-4-[N6-[2-(5-[хлор-2-тиенил)-(1R)1-метилэтил]амино]-9-аденил]циклопентан]-1N-этилкарбоксамида. Стадия 1. Оптически чистый (+)-[2S[2 а,3 а-диметилметилендиокси]-4-(6-хлор-9 аденил)]циклопентан]-1-N-этилкарбоксамид,полученный, как описано в примере 2, и 2'R-(5 хлортиен-2-ил)-2-пропиламин,[]DRТ=-15,6(с=3,7, CH3OH), полученный, как описано в примере 4, объединяют, как описано в примере 4,получая 2,3-диметилметилендиоксипроизводное конечного продукта. Стадия 2. Диметилметилендиоксипроизводное стадии 1 нагревают в 5 мл 50% водной муравьиной кислоты с обратным холодильником в течение 3 ч. Охлажденную реакционную смесь выпаривают, к твердому остатку добавляют толуол и выпаривают растворитель. Остаток растворяют в этилацетате, промывают раствором бикарбоната натрия и насыщенным солевым раствором, сушат, фильтруют и выпаривают, получая после сушки в печи в течение ночи белый твердый продукт (0,240 г), т.пл. 1884 С; C20H25N6SO3Cl. []DRТ=-86,49 (с=5,5, МеОН). 32 Примеры 7-29, 31-34. Согласно основным процедурам приведенных выше примеров с 1 по 6 получают соединения, приведенные далее в таблице I. В примерах с 7 по 21, 31 и 32 гетероциклический амин реагирует с коммерчески доступным 6 хлорпуринрибозидом; в примерах 22 и 23 гетероциклический амин реагирует с N6-хлор-5'-Nэтилкарбоксамидоаденозином; и в примерах с 24 по 31, 33 и 34 гетероциклический амин реагирует с эфиромили (+)-[2S-[2 а,3 а-диметилметилен-диокси-4-(6-хлор-9-аденил)циклопентан]-1-N-этилкарбоксамидом. Таблица I оптическое вращение спиртового предшественника амина:d амин реагирует с 2',3'изопропилиденовым производным N6-хлор-5'N-этилкарбоксамидаденозина; снятие защиты согласно процедуре примера 11. Пример 30. Получение -N6-[1-(тиофен 2-ил)этан-2-ил]-N'-1-деазааристеромицин-5'-Nэтилкарбоксамида. Стадия 1. 2-Хлор-3-нитро-4-[2-(2-тиофенил)этил]аминопиридин. Смесь 2,4-дихлор-3-нитропиридина (1,5 г),2-аминоэтилтиофена (1 г) и триэтиламина (5 мл) нагревают до кипения с обратным холодильником в EtOH (60 мл). Реакционную смесь охлаждают, растворитель выпаривают и остаток хроматографируют на силикагеле (10% гексан/СН 2 Сl2), получая требуемый продукт присоединения. Стадия 2.1-N-Этилкарбоксамид-2 а,3 аизопропилидендиокси-4-[2-(3-нитро-4-[2-(2 35 тиофенил)этил]аминопиридил)амино]циклопентан. Смесь тиофениламинопиридина стадии (1)(0,3 г) и триэтиламина (0,3 мл) нагревают с обратным холодильником в нитрометане (15 мл) в течение примерно 5 ч. Удаляют растворитель и остаток помещают в метиленхлорид, хроматографируют на силикагеле (2% метанол/хлороформ), получая твердый продукт, который используют, как он есть, в следующей стадии. Стадия 3:1-N-этилкарбоксамид-2 а,3 аизопропилидендиокси-4-[2-(3-амино-4-[2-(2 тиофенил)этил]аминопиридил)амино]-циклопентан. Смесь нитросоединения стадии (2) (0,39 г),Pd/C (0,01 г) в этаноле (7 мл) перемешивают в атмосфере водорода в течение примерно 5 ч. Катализатор отфильтровывают, а фильтрат выпаривают, получая масло, которое чистят на флорисиле = florisil (10% метанол/ метиленхлорид), получая требуемый продукт в виде твердого вещества. Стадия 4:-N6- [1-тиофен-2-ил) этан-2 ил]-N'-1-деазаарис-теромицин-5'-N-этилкарбоксамид. Смесь аминосоединения стадии (3) (0,31 г) и формамидинацетата (0,72 г) в метоксиэтаноле(30 мл) нагревают при кипячении с обратным холодильником примерно в течение 3 ч. Реакционную смесь охлаждают, растворитель выпаривают и к остатку добавляют воду (5 мл) и муравьиную кислоту (5 мл). Кислую смесь нагревают до 50 С в течение примерно 5 ч, после чего удаляют растворитель и хроматографируют остаток на силикагеле (10% метанол/метиленхлорид), получая требуемый продукт в виде кристаллического твердого вещества, т.пл.=155156 С. Оптически чистое соединение получают,используя на стадии (2) + или - энантиомер циклопентанамина. Пример 35. Получение (2S)-2 а,3 а-дигидрокси-4-[N6[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-9 аденил]циклопентан-1-N-этилкарбоксамид-N1 оксида. Раствор (2S)-2 а,3 а-дигидрокси-4-[N6-[2(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-9 аденил]циклопентан-1-N-этилкарбоксамида(0,25 г) и ледяной уксусной кислоты (20 мл) в 30% перекиси водорода (1 л) перемешивают в течение 4 дней при комнатной температуре и смесь концентрируют в вакууме. Остаток чистят способом флэш-хроматографии, элюируя 20% метанолом в этилацетате, затем перемешивают с теплым метанолом и фильтруют, получая требуемый продукт, т.пл.240 С. Пример 36. Получение [1S-[1a,2b,3b,4a 36 амино]-3 Н-имидазо[4,5-b]пиридин-3-ил]-Nэтил-2,3-дигидроксициклопентанкарбоксамида. Стадия 1. Получение 2-хлор-4-[2-(5-хлор 2-тиенил)-(1R)-1-метилэтил]амино-3-нитропиридина. Применяя по существу процедуру примера 30, стадии 1, и чистя сырой продукт способом флэш-хроматографии с использованием градиентного элюирования от 10 до 30% этилацетата в гексане, получают требуемый продукт из 2-(5 хлор-2-тиенил)-(1R)-1-метил-этиламина. Стадия 2: получение (-)-1-N-этил-2 а,3 аизопропилидендиокси-4-[4-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-3-нитро-2-пиридил]аминоциклопентанкарбоксамида. 2-хлор-4-[2-(5-хлор-2-тиенил)-(1R)-1 метилэтил]амино-3-нитропиридин (0,68 г), (-)1-N-этил-2 а,3 а-изопропилидендиокси-4 аминоциклопентанкарбоксамид (0,381 г) и триэтиламин (0,85 мл) объединяют в этаноле (50 мл) и смесь нагревают при кипячении с обратным холодильником в течение примерно 18 ч. Смесь концентрируют в вакууме и сырой продукт чистят способом флэш-хроматографии,элюируя 0,5% метанолом в метиленхлориде,получая требуемый продукт. Стадия 3. Получение (-)-1-N-этил-2 а,3 аизопропилидендиокси-4-[3-амино-4-[2-(5 хлор-2-тиенил)-(1R)-1-метилэтил]-2-пиридил]аминоциклопентанкарбоксамида.(II) (2,1 г) объединяют в этаноле (20 мл) и смесь нагревают при 70 С в течение примерно 30 мин. Смесь выливают на лед, слегка подщелачивают при помощи водного бикарбоната натрия и водную фазу экстрагируют этилацетатом. Раствор этилацетата сушат над сульфатом магния,фильтруют и концентрируют в вакууме, получая требуемый продукт, который используют в следующей стадии без дополнительной очистки. Стадия 4. Получение [1S-[1a,2b,3b,4a(S)-4-2-(5-хлор-2-тиенил)-1-метилэтил] амино]-3 Н-имидазо[4,5-b]пиридин-3-ил]-Nэтил-2,3-дигидроксициклопентанкарбоксамида. Применяя в основном процедуру примера 30, стадии 4, получают требуемый продукт с т.пл 164-165 С из (-)-1-N-этил-2 а,3 а-изопропилидендиокси-4-[3-амино-4-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-2-пиридил]аминоциклопентанкарбоксамида. Применяя в основном процедуру примера 30, получают соединения примеров из подходящих исходных материалов. Пример 37. [1S-[1a,2b,3b,4a-4-[7-2-(3 хлор-2-тиенил)-1-этилэтил]амино]-3 Н-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид, т.пл 75-85 С. Пример 39. [1S-[1a,2b,3b,4a(S)-4-[7-2(3-[хлор-2-тиенил)-1-этилэтил]амино]-3 Нимидазо[4,5-b]пиридин-3-ил]-N-этил-2,3 дигидроксициклопентанкарбоксамид, т.пл 7578 С. Пример 40. [1S-[1a,2b,3b,4a(S)-4-[7-2(2-тиенил)-1-метилэтил]амино]-3 Н-имидазо[4,5b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид, т.пл. 155-160 С. Пример 41. Получение [1S-[1a,2b,3b,4a-4[7-2-(5-хлор-2-тиенил)-1-этилэтил]амино]-3 Нимидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамид. Применяя в основном процедуру примера 36, получают требуемый продукт, т.пл.77-85 С,из 2-(5-хлор-2-тиенил)-(1R)-1-этилэтиламина. Пример 42. Получение (2S)-2 а,3 а-бисметоксикарбонилокси-4-[N6-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-9-аденил]-циклопентан-1-N-этилкарбоксамида. К раствору (2S)-2 а,3 а-дигидрокси-4-[N6[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-9 аденил]циклопентан-1-N-этилкарбоксамида(0,56 г), триэтиламина (0,5 мл) и 4 диметиламинопиридина (1 мг) в тетрагидрофуране (25 мл) добавляют метилхлорформиат (0,21 мл) и раствор перемешивают при комнатной температуре в течение 1 ч. Смесь разбавляют этилацетатом, промывают насыщенным солевым раствором и органический раствор сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Сырой продукт перекристаллизовывают из гексана/этилацетата, получая требуемый продукт, т.пл. 74-76 С. Пример 43. Получение(0,14 г), триэтилортоформиата (3 мл) и паратолуолсульфокислоты (1 мг) нагревают при кипячении с обратным холодильником примерно в течение 1 ч и затем удаляют растворитель в вакууме. Остаток растворяют в этилацетате и раствор промывают насыщенным солевым раствором, сушат над сульфатом натрия, фильтруют,концентрируют в вакууме. Сырой продукт чистят способом флэш-хроматографии, элюируя 5% метанолом в метиленхлориде, затем перекристаллизовывают из гексана/этилацетата, получая требуемый продукт, т.пл.67-70 С. Пример 44. Получение(0,17 г) и 1,1'-карбонилдиимидазола (0,071 г) в бензоле (5 мл) кипятят с обратным холодильником в течение 5 ч, затем перемешивают при 60 С в течение примерно 18 ч. Раствор промывают насыщенным солевым раствором, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток чистят способом флэш-хроматографии, элюируя 5% метанолом в метиленхлориде, затем кристаллизуют из гексана/этилацетата, получая требуемый продукт,т.пл.87-89 С. Пример 45. Получение (2S)-2 а,3 а-бисметилкарбамоилокси-4-[N6-[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-9-аденил]циклопентан-1-N-этил-карбоксамида. К раствору (2S)-2 а,3 а-дигидрокси-4-[N6[2-(5-хлор-2-тиенил)-(1R)-1-метилэтил]амино-9 аденил]циклопентан-1-N-этилкарбоксамида[5.4.0]ундец-7-ена (1 каплю). Раствор перемешивают при 50 С в течение примерно 2,5 ч, охлаждают до комнатной температуры, разбавляют этилацетатом и промывают насыщенным солевым раствором. Органический раствор промывают насыщенным солевым раствором, сушат над сульфатом магния и концентрируют в вакууме. Остаток чистят способом флэшхроматографии, элюируя 5% метанолом в метиленхлориде, затем кристаллизуют из гексана/этилацетата, получая требуемый продукт,т.пл. 97-99 С. Пример 46. Получение(0,35 г) и тиокарбонилдиимидазола (0,134 г) в бензоле (10 мл) нагревают при 45 С в течение примерно 2 ч. Раствор промывают насыщенным солевым раствором, сушат над сульфатом магния и концентрируют в вакууме. Остаток чистят способом флэш-хроматографии, элюируя 5% метанолом в гексане, затем кристаллизуют из гексана, получая требуемый продукт, т.пл. 115117 С. Пример 47. Получение N6-[2-(3-хлор-2 тиенил)-(1R)-1-метилэтил]-2'-0-метиладенозина. Раствор 6-хлор-9-(2'-0-метилD-рибофуранозил)-9 Н-пурина (полученного так, как в публикации Европейского Патента 0378518) 39 флэш-хроматографии, элюируя 10% метанолом в метиленхлориде, затем кристаллизуют из гексана/этилацетата, получая требуемый продукт,т.пл. 75-76 С. Пример 48. Получение N6-[2-(5-хлор-2 тиенил)-(1R)-1-метилэтил]-2-'0-метиладенозина. Применяя в основном процедуру примера 47, получают требуемый продукт с т.пл. 84-85 С из 2-(5-хлор-2-тиенил)-(1R)-1-метилэтиламина. Пример 49. Получение N6-[транс-5-(2 тиенил)циклогекс-1-ен-4-ил]-2'-0-метиладенозина. Применяя в основном процедуру примера 47, получают требуемый продукт с т.пл. 86-89 С из транс-2-(2-тиенил)циклогекс-4-ениламина. Пример 50. Получение 1(R)-2-(5-хлор-2 тиенил)-1-метилэтиламина. Стадия 1. Получение (1S)-2-(5-хлор-2 тиенил)-1-гидрокси-1-метилэтана. Раствор 2-хлортиофена (8,17 г) в тетрагидрофуране (80 мл) охлаждают до -30 С и добавляют по капле 1,6 М н-бутиллитий в гексане(43,0 мл). Смесь перемешивают при -30 С примерно 1 ч, добавляют (S)-пропиленоксид (4 г) и смесь нагревают до 0 С и перемешивают при этой температуре около 3 ч. Реакцию гасят,смешивая с насыщенным водным раствором хлорида аммония, разбавляют эфиром и этот слой отделяют. Органический слой промывают насыщенным солевым раствором, сушат над сульфатом магния и концентрируют в вакууме,получая требуемый продукт. Стадия 2. Получение 1(R)-2-(5-хлор-2 тиенил)-1-метил-1-фталимидоэтана. К раствору 1(S)-2-(5-хлор-2-тиенил)-1 гидрокси-1-метилэтана (8,8 г), трифенилфосфина (13,1 г) и фталимида (7,35 г) в тетрагидрофуране (80 мл) добавляют по капле диэтилазодикарбоксилат (7,9 мл). Раствор перемешивают примерно 18 ч и удаляют растворитель в вакууме. Остаток чистят способом флэшхроматографии, элюируя 20% гексаном в метиленхлориде и получая требуемый продукт. Стадия 3. Получение 1(R)-2-(5-хлор-2 тиенил)-1-метилэтиламина. 1(R)-2-(5-хлор-2-тиенил)-1-метил-1 фталимидоэтан (13 г) растворяют в этаноле (75 мл) и добавляют гидразингидрат (2,5 мл) и смесь перемешивают при кипячении с обратным холодильником примерно 1 ч. Смесь охлаждают до комнатной температуры, удаляют фильтрацией твердые вещества и фильтрат концентрируют в вакууме. Остаток растворяют в этилацетате и этот раствор перемешивают с 5N водной соляной кислотой. Слои отделяют и доводят рН водного слоя до рН 10 при помощи 10% раствора гидроксида натрия, затем экстрагируют этилацетатом. Органический раствор промывают насыщенным солевым раствором, сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая требуемый продукт. []D=22,96 (с=11,5, метанол). Пример 51. Получение 1(R)-2-(2-тиенил)1-метилэтиламина. Стадия 1. Получение 1(S)-2-(2-тиенил)-1 гидрокси-1-метилэтана. Применяя в основном процедуру примера 50, стадии 1, получают требуемый продукт из тиофена. Стадия 2. Получение 1(R)-2-(2-тиенил)-1 метил-1-фталимидоэтана. Применяя в основном процедуру примера 50, стадии 2, получают требуемый продукт из 1(S)-2-(2-тиенил)-1-гидрокси-1-метилэтана. Стадия 3. Получение 1(R)-2-(2-тиенил)-1 метилэтиламина. Применяя в основном процедуру примера 50, стадии 3, получают требуемый продукт(2,49 г). Добавляют по капле диэтилазодикарбоксилат (3,22 мл) и смесь перемешивают при комнатной температуре в течение примерно 18 ч. Растворитель удаляют в вакууме. Остаток чистят способом флэш-хроматографии, элюируя 30% гексаном в метиленхлориде и получая (R)3-(5-хлор-2-тиенил)-2-пропилбензоат. Этот эфир (3,91 г) растворяют в диоксане (50 мл) и добавляют 20% водный гидроксид натрия (15 мл). Смесь нагревают при 55 С в течение 3 ч и концентрируют в вакууме. Остаток помещают в этилацетат (200 мл) и органический слой промывают насыщенным солевым раствором, сушат над сульфатом магния, фильтруют, концентрируют в вакууме, получая требуемый продукт. Стадия 2. Получение 1(S)-2-(5-хлор-2 тиенил)-1-метилэтиламина. Применяя в основном процедуры примера 50, стадий 2 и 3, получают требуемый продукт([]D=+21,71 (с=1,1, метанол из 1(S)-2-(5 хлор-2-тиенил)-1-гидрокси-1-метилэтана. Применяя в основном процедуры примеров 50, 51 и 52, получают из подходящих исходных материалов следующие соединения. Пример 53. 1(R)-2-(бензотиофен-2-ил)-1 метилэтиламин. Пример 54. 1(S)-2-(2-тиенил)-1-метилэтиламин, []D=15,5 (с=1, метанол). Пример 55. 1(R)-2-(3-бром-2-тиенил)-1 метилэтиламин. Пример 56. 1(R)-2-[5-(2-пиридил)-2-тиенил)]-1-метилэтиламин. 41 Пример 57. 1(R)-2-[5-(2-тиенил)-2-тиенил)]-1-метилэтиламин. Пример 58. 1(R)-2-(5-фенил-2-тиенил)-1 метилэтиламин Пример 59. 1(R)-2-(5-метокси-2-тиенил)-1 метилэтиламин. Пример 60. 1(R)-2-(5-метил-2-тиенил)-1 метилэтиламин. Пример 61. 1 (R)-2-(5-бром-2-тиенил)-1 метилэтиламин. Пример 62. 1(R)-2-(5-иод-2-тиенил)-1 метилэтиламин. Пример 63. 1(R)-2-(5-метилтио-2-тиенил)1-метилэтиламин. Пример 64. 1(R)-2-(5-метилсульфонил-2 тиенил)-1-метилэтиламин. Пример 65. 1(R)-2-(5-этил-2-тиенил)-1 метилэтиламин. Пример 66. 1(R)-2-(5-н-гептил-2-тиенил)-1 метилэтиламин. Пример 67. 1(R)-2-(3-метил-2-тиенил)-1 метилэтиламин. Пример 68. 1(R)-2-(4-метил-2-тиенил)-1 метилэтиламин. Пример 69. 1(R)-2-(3-хлор-2-тиенил)-1 метилэтиламин, []D=-6,1 (с=1, метанол). Пример 70. 1(R)-2-(4-хлор-2-тиенил)-1 метилэтиламин. Пример 71. 1(R)-2-(3-хлор-5-фенил-2 тиенил)-1-метилэтиламин. Пример 72. 1(R)-2-(5-бром-2-хлор-2-тиенил)-1-метилэтиламин. Пример 73. 1(R)-2-(4-метил-5-хлор-2 тиенил)-1-метилэтиламин. Пример 74. 1(R)-2-(2,5-дихлор-3-тиенил)1-метилэтиламин. Пример 75. Смесь 13,88 г (29,7 ммоля) продукта примера 37 и 4,27 г (41 ммоля) формамидинацетата в 200 мл н-ВuОАс кипятят с обратным холодильником в течение 1 ч. В этот момент реакция заканчивается, и продукты можно определить способом высокоэффективной жидкостной хроматографии. После охлаждения реакционную смесь промывают 5% (по весу) NаНСО 3 и насыщенным солевым раствором, сушат над Na2SO4 и фильтруют. Фильтрат обрабатывают 13,8 г (59,4 ммоля) (1R)-(-)-10 камфорсульфоновой кислоты, для отделения липкого твердого вещества. Суспензию подвергают кипячению с обратным холодильником для получения более гранулированного осадка. После охлаждения твердое вещество собирают,промывают EtOAc и сушат в вакууме, получая 10,41 г соли ди-(1R)-(-)-камфорсульфоновой кислоты и [1S-[1a,2b,3b,4a,(S)-4-[7-2-(3 хлор-2-тиенил)-1-этилэтил] амино]-3 Н-имидазо 42 Следующие аналитические данные получены на примере образца соли ди[(1R)-(-)камфорсульфоната [1S-[1a,2b,3b-4a,(S)-4-[72-(3-хлор-2-тиенил)-1-этилэтил]амино]-3 Нимидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамида в виде грязнобелого порошка. Продукт, если требуется,можно перекристаллизовать (СН 3 СN) с высоким выходом и повысить чистоту. Следующие аналитические данные получены на примере образца соли ди[(1R)-(-)камфорсульфоната [1S-[1a,2b,3b,4 а,(S)-4-[72-(3-хлор-2-тиенил)-1-этилэтил]амина]-3 Нимидазо-[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентакарбоксамида: Дифференциальный сканирующий калориметр: т.пл. 188 С. Элементный анализ. Рассчитано: С 53,51; Н 6,42; N 7,43; Cl 3,76; S 10,20. Найдено: С 53,41; Н 6,47; N 7,34; Cl 4,03; S 10,07. Пример 76. В трехгорлую круглодонную колбу на 5 л,снабженную пробками с мешалкой, датчиком температуры с диаграммным самописцем и обратным холодильником (охлаждаемым посредством syltherm XLT при температуре 2,6 С),загружают под током азота диметилацетондикарбоксилат (696,6 г, 4 моля), триметилортоформиат (424,5 г, 4 моля) и уксусный ангидрид(816,72 г, 8 молей) . Полученный янтарный гомогенный раствор быстро перемешивают (400 об/мин) и нагревают до кипения с обратным холодильником при температуре 115 С в течение 40 мин, используя нагревающий кожух. Регулируя нагревание, в течение следующего часа поддерживают умеренное кипение с обратным холодильником (точка кипения смеси постепенно снижается до 95 С с течением реакции). Затем полученную оранжевую гомогенную смесь перегоняют при максимальной температуре 115 С в вакууме (20 мм рт.ст.), используя насадку Кляйзена. Собирают около 1000 мл дистиллята (содержащего АсОН и AсОМе). Темнооранжевый остаток охлаждают на ледяной бане при температуре 6 С и затем добавляют по капле гидроксид аммония (1 л, 8 молей) для регулирования температуры экзотермической реакции ниже 25 С, общее время=1,5 ч. Желтую суспензию подкисляют НСl (рН 2,0) (750 мл, 9 молей) и полученное рыжевато-коричневое твердое вещество отфильтровывают, промывают 1 л МeОН и сушат при отсасывании до постоянного веса. Таким образом получают первый сбор метил-4,6-дигидроксиникотината (351 г, 51%) 97% чистоты. Второй сбор метил-4,6-дигидроксиникотината (35 г) можно получить, уменьшая в два раза объем метанольного фильтрата. Масс-спектр: 169 (М, 97%) 137 (100%). Этот продукт хроматографируют двумя способами. 43 Первый способ высокоэффективной жидкостной хроматографии: используют колонку типа Alltec Аbsobosphere-SСХ, 5 , 250x4,6 мм; подвижную фазу А: 50 мM NaH2PO4, pH=3,5 с Н 3 РО 4, В: СH3 СN, А:В=95:5; скорость течения 1,0 мл/мин; детектирование: УФ поглощение при 210 нм; время удерживания 3-9 мин. Определено, что чистота продукта составляет 97,8%. Второй способ высокоэффективной жидкостной хроматографии: используют колонку типа Sulpeco Sulpecosil-SAX, 5 , 250 х 4,6 мм; подвижную фазу А: 200 мМ KH2PO4, с 0,1%TEA, рН=6,0 с Н 3 РO4, В: СH3 СN, А:В=85:15; скорость течения 1,0 мл/мин; детектирование: УФ поглощение при 210 нм; время удерживания 4,9 мин. Пример 77. В трехгорлую колбу на 2 л, снабженную датчиком температуры с диаграммным самописцем, механической мешалкой, прибором Дина-Старка и обратным холодильником, загружают метил-4,6-дигидроксиникотинат (100 г,97% чистоты, 0,59 моля) и фосфорную кислоту(85%, 300 мл). Суспензию нагревают при помощи нагревательного кожуха и при температуре 120 С получают красно-бурый раствор (кожух при температуре 290 С, прошедшее время 1 ч). При температуре 140 С сразу появляется белый осадок 4,6-дигидроксиникотиновой кислоты(хотя метанол образуется, но метанола в дистилляте не наблюдают, что предполагает образование фосфатного эфира), и с этого момента начинается значительное пенообразова-ние. Это пенообразование регулируют путем добавления поверхностно-активного вещества, такого как силиконовое масло 550 (5-6 капель). Содержащуюся воду, которая образуется в результате дегидратации фосфорной кислоты, затем удаляют (примерно 60 мл) до тех пор, пока внутренняя температура не достигнет 210 С 5 С(кожух при температуре 290 С, общее прошедшее время 2,5 ч). Без дегидратации фосфорной кислоты декарбоксилирование не происходит. После перемешивания в течение 4-5 ч при этой температуре высокоэффективная жидкостная хроматография подтверждает исчезновение 4,6 дигидроксиникотиновой кислоты и метил-4,6 дигидроксиникотината. Затем реакционной смеси дают охладиться до температуры ниже 100 С, в этот момент добавляют уксусную кислоту (300 мл) и поддерживают температуру при 90 С. К этой смеси добавляют с постоянной скоростью (5-6 мл/мин) азотную кислоту (25 мл,624 ммоля), пока высокоэффективная жидкостная хроматoгpaфия не покажет отсутствие 2,4 дигидроксипиридина. Затем к реакционной смеси добавляют воду (300 мл) и прекращают нагревание. При температуре ниже 80 С начинает появляться темно-коричневое твердое вещество,ему дают осадиться при перемешивании в течение 2-3 ч. Фильтрование через стеклянную(средней пористости) дает темно-коричневое твердое соединение, которое затем промывают 250 мл изопропилово-го спирта. Полученный осадок сушат на воздухе в течение 1 ч и затем помещают на 2 дня в сушильную печь под вакуум (508 мм рт.ст. с продувом воздуха) при 50 С. 2,4-дигидрокси-3-нитропи-ридин (60 г) получают с 60% выходом. Дифференциальный сканирующий калориметр: т.пл. 183,85 С. Элементный анализ для C5H4N2O4. Рассчитано: С 38,47; Н 2,58; N 17,95; Найдено: С 38,42; Н 2,62; N 17,69; ИК-спектр 3194,9 (ОН), 1689,2 (С=0),1616,5 (С=С); Масс-спектр (М+Н), 157. Этот продукт хроматографируют двумя способами: Первый способ высокоэффективной жидкостной хроматографии: используют колонку типа Alltec Absobosphere-SCX, 5 , 250 х 4,6 мм; подвижную фазу А: 50 MM NaH2PO4, pH=3,5 с Н 3 РO4, В: СН 3 СN, А:В=95:5; скорость течения 1,0 мл/мин; детектирование: УФ поглощение при 210 нм; время удерживания 4,2 мин. Определено, что чистота продукта составляет 99,92%. Второй способ высокоэффективной жидкостной хроматографии: используют колонку типа Sulpeco Sulpecosil-SAX, 5 , 250 х 4,6 мм; подвижную фазу А: 200 мМ КН 2 РO4, с 0,1%TEA, pH=6,0 с Н 3 РO4, В: СН 3 СN, А:В=85:15; скорость течения 1,0 мл/мин; детектирование: УФ поглощение при 210 нм; время удерживания 10 мин. Пример 78. В трехгорлую колбу на 22 л, снабженную датчиком температуры с диаграммным самописцем, механической мешалкой, прибором Дина-Старка (Dean-Stark) и обратным холодильником, загружают 4,6-дигидроксиникотиновую кислоту (88%, 2 кг, 12,81 моля), фосфорную кислоту (85%, 6 л, 103,15 моля). Отходящие из обратного холодильника газы очищают посредством барботирования через 50% водный NaОH. Эту суспензию нагревают до удаления достаточного количества воды (примерно 1,2 л), то есть дегидратации фосфорной кислоты. Без дегидратации фосфорной кислоты декарбоксилирование не происходит. Для достижения внутренней температуры 210C5C,температуры кожуха составляет 290 С на протяжении 3,5 ч. После перемешивания в течение 4-5 ч при этой температуре высокоэффективная жидкостная хроматография подтверждает исчезновение исходного материала. Затем реакционной смеси дают охладиться до температуры ниже 100 С, в этот момент добавляют уксусную кислоту (6 л, 104,81 моля) и поддерживают температуру при 90 С. К этой смеси добавляют с постоянной скоростью (5-6 мл/мин) азотную 45 кислоту (485 мл, 12,11 моля), пока высокоэффективная жидкостная хроматография не покажет отсутствие 4,6- дигидроксипиридина. Затем к реакционной смеси добавляют воду (6 л, 333,3 моля) и прекращают нагревание. При температуре ниже 80 С начинает появляться желтое твердое вещество, ему дают осадиться при перемешивании в течение ночи. Фильтрование через воронку со стеклянным фильтром(средней пористости) дает желтое твердое вещество, которое затем промывают 2,5 л изопропилового спирта. Полученный осадок сушат на воздухе в течение 48 ч и затем помещают на 3 дня в сушильную печь под вакуум (508 мм рт.ст. с продувом воздуха) при 50 С. 2,4-дигидрокси 3-нитропиридин (1,050 кг) получают с 60% выодом. Осторожно: этот материал проявляет огромную экзотермическую реакцию после начала плавления при 262,62 С, таким образом, авторы рекомендуют не нагревать это вещество до температуры в пределах 100 С от точки его плавления. Дифференциальный сканирующий калориметр: т.пл 183,85 С. Элементный анализ для C5H4N2O4. Рассчитано: С 38,47; Н 2,58; N 17,95; Найдено: С 38,42; Н 2,62; N 17,69; ИК-спектр 3194,9 (ОН), 1689,2 (С=0),1616,5 (С=С); Масс-спектр (М+Н), 157. Этот продукт хроматографируют двумя способами: Способ А высокоэффективной жидкостной хроматографии: используют колонку типа AlltecAbsobosphere-SCX, 5 , 250 х 4,6 мм; подвижную фазу А: 50 мм NаН 2 РO4, рН=3,5 с Н 3 РO4, В: СН 3 СN, А:В=95:5; скорость течения 1,0 мл/мин; детектирование: УФ поглощение при 210 нм; время удерживания 4,2 мин. Определено, что чистота продукта составляет 99,92%. Способ В высокоэффективой жидкостной хроматографии: используют колонку типа Sulpeco Sulpecosil-SAX, 5 , 250 х 4,6 мм; подвижную фазу А: 200 мм KH2PO4, с 0,1% TEA,pH=6,0 с Н 3 РO4, В: СН 3 СN, А:В=85:15; скорость течения 1,0 мл/мин; детектирование: УФ поглощение при 210 нм; время удерживания 10 мин. Пример 79. 2,4-Дигидрокси-3-нитропиридин (590 г,3,78 моля) загружают в трехгорлую колбу на 12 л, снабженную датчиком температуры с диаграммным самописцем, дополнительной воронкой, насадкой Клайзена, холодильником и механической мешалкой. Отходящие из холодильника газы очищают посредством барботирования через 50% водный NaOH. Начинают перемешивание твердого вещества. Добавляют РОСl3 (1058 мл) и полученное полутвердое вещество нагревают до 45 С для получения пере 000884 46 мешиваемой суспензии. К этой смеси за один час добавляют DIPEA (988 мл). Эту экзотермическую реакцию регулируют скоростью добавления диизопропилэтиламина (DIPEA). По завершении добавления реакцию нагревают до 1005 С. Превращение 2,4-дигидрокси-3 нитропиридина через промежуточное монохлорсоединение завершается через 4 ч (высокоэффективная жидкостная хроматография; способ В). Затем реакционную смесь охлаждают до 20 С и гасят, выливая в воду со льдом при интенсивном перемешивании. Осаждается рыжевато-коричневое твердое вещество, льду дают расплавиться и полученную смесь фильтруют через воронку со стеклянным фильтром. Продукт сушат на воздухе в течение ночи и затем переносят в вакуумную печь и сушат до постоянного веса (737 мм рт.ст, 20 С). 2,4-дихлор-2 нитропиридин получают в виде грязно-белого твердого вещества (699 г, 96% выход). Если цвет или чистота продукта, полученного описанным выше способом, являются неудовлетворительными, продукт можно перекристаллизовать следующим образом: твердое вещество переносят в колбу, снабженную механической мешалкой и устройством Дина-Старка. Затем добавляют гептан (5 мл/г) и смесь нагревают до кипения с обратным холодильником. Оставшуюся воду удаляют азеотропной перегонкой. Раствор фильтруют через целит и фильтрату дают охладиться при перемешивании, затем рыжевато-коричневое твердое вещество собирают путем фильтрования и сушат на воздухе до постоянного веса. Элементный анализ для С 5 Н 2N2O2 Сl2. Рассчитано: С 31,26; Н 1,05; N 14,59; Cl 36,44. Найдено: С 31,31; Н 1,26; N 14,41. Масс-спектр 192 (М+, 90%). Этот продукт хроматографируют двумя способами: Способ А высокоэффективной жидкостной хроматографии осуществляют так, как описано в примере 76, определено, что чистота продукта составляет 99,33%. Способ В высокоэффективной жидкостной хроматографии: используют колонку типа MicrosorbMV-C18, 5 , 250 х 4,6 мм; подвижную фазу А: Н 2 О с 1% АсОН, В: СН 3 СN, А:В=70:30; скорость течения 1,0 мл/мин; детектирование: УФ поглощение при 220 нм; время удерживания 5,3 мин. Пример 80. 2,4-Дигидроксинитропиридин (85 мл, 0,914 моля) загружают в колбу на 1 л, затем загружают РОСl3 при перемешивании, через 30 мин добавляют DMA. Экзотермическая реакция протекает при температуре 56 С. После того, как экзотермическая реакция утихает, реакционную смесь нагревают до 90 С в течение 1 ч и затем нагревают в течение 4 ч до 105-115 С. Через 5 ч смесь гасят, выливая на лед при перемешивании 47 и фильтруют. Выпадающее серое твердое вещество промывают 2 х 100 мл холодной водой, затем сушат на воздухе на фильтре в течение ночи. Выход серого твердого вещества составляет 57 г. Твердое вещество перекристаллизовывают из 500 мл гептана, обрабатывают 2 г угля и фильтруют теплым через целит. Объем фильтрата уменьшают до 75 мл и фильтруют, получая твердое вещество, которое промывают гептаном и сушат на фильтре. Получают 47 г продукта. Объем фильтрата уменьшают и отфильтровывают осаждающееся твердое вещество, получая дополнительно 2,8 г продукта (2 урожая 85%). Пример 81. 2,4-Дигидрокси-3-нитропиридин (23,7 г,0,152 моля) загружают в колбу на 500 мл, затем загружают POCl3 (42,5 мл, 0,457 моля) при перемешивании, через 45 мин добавляют DIPEA(39,7 мл, 0,228 моля), что дает экзотермическую реакцию с повышением температуры до 67 С. После того, как экзотермическая реакция утихает, смесь нагревают до 90 С. По достижении степени определяемой чистоты 98,6% реакционную смесь резко охлаждают, добавляя 250 г льда/50 мл H2O. Твердое вещество отфильтровывают, промывают 2 х 100 мл деионизованнойH2O и оставляют сушить на фильтре в течение 72 ч. Выход продукта, который является светлорыжевато-коричневым по цвету, составляет 25,1 г (85%). Соединения, полученные из промежуточных продуктов, приготовленных согласно способу настоящего изобретения, полезны в качестве гипотензивных агентов для лечения высокого кровяного давления; они также повышают коронарный кровоток и, следовательно, полезны при лечении ишемии миокарда; они также действуют как кардиозащитные агенты, полезные для профилактики или восстановления поражения миокарда вследствие ишемии миокарда; и они также действуют как антилиполитические агенты, полезные для лечения гиперлипидемии и гиперхолестеринемии. Соединения, полученные из промежуточных продуктов, приготовленных согласно способу настоящего изобретения, демонстрируют активность в стандартных исследованиях связывания A1/A2 рецепторами для определения активности агонистов рецепторов аденозина у млекопитающих. Примеры процедур исследования, полезных в определении свойства связывания этими рецепторами данных соединений,описаны ниже. А. Определение in vitro свойства связывания рецепторами аденозина. Способность связывания рецепторами A1 определяют посредством конкурентного исследования, основанного на вытеснении лиганда 3H-СНА (циклогексиладенозин) [Research Biochemicals Inc., Natick, Mass.] с рецептора, используя мембранный препарат из целого головного мозга крысы согласно процедуре R.F.Bruns 48 и др., Mol.Pharmacol., 29:331 (1986). Неспецифическое связывание исследуют в присутствии 1 мМ теофилина. Способность связывания рецепторами A2 определяют аналогичным способом, основанным на вытеснении лиганда 3H-CGS 21680, известного специфического агониста аденозина, с рецептора, используя мембраны из полосатого тела (striatum) головного мозга крысы. Неспецифическое связывание исследуют в присутствии 20 M 2-хлораденозина. Опыты проводят в стеклянных пробирках при 25 С, дублируя эксперименты. При добавлении мембран пробирки сразу вращают и инкубируют при 25 С в течение 60 мин (A1 исследование) или 90 мин (А 2 исследование) на роторном шейкере. Пробирки вращают до половины времени инкубации и затем ближе к концу. Испытание заканчивают быстрой фильтрацией через 2,4 см GF/B фильтры, применяя коллектор Brandel Cell Harvestor. Пробирки трижды промывают холодным 50 мМ Трис-НСl (рН 7,7 или 7,4), фильтрацию заканчивают за 15 с. Увлажненные фильтровальные круги помещают в стеклянные пузырьки для сцинтилляционных исследований, заполненные 10 мл Aquasol II(New England Nuclear). Пузырьки оставляют встряхиваться в течение ночи на роторном шейкере и помещают в жидкостной сцинтилляционный анализатор на двухминутный подсчет. Используя компьютерную программу по подгонке кривых RS/1, Bolt, Beranek and Newman,Boston, MA), получают величины IC50 для связывания рецепторов, то есть концентрацию, при которой соединение формулы I замещает радиоактивно-меченый стандарт. Б. Определение in vitro вазорелаксации в выделенных артериях свиней. Коронарные артерии свиней получают с местной бойни, аккуратно рассекают и очищают от жира, крови и присоединенной ткани. Отрезают кольца шириной приблизительно 2-3 мм и переносят их в бани для тканей с водяными рубашками (10 мл), заполненные теплым (37 С),насыщенным кислородом (O2/СO2=95:5) буфером Кребса (Krebs-Henseleit buffer), закрепляют на L-образных крючках между стержнями из нержавеющей стали и силовым датчиком. Состав буфера Кребса следующий (мМ) : NaCl 118; КСl 4,7; CaCl2 2,5; MgSO4 1,2; КН 2 РO4 1,2;NaHCO3 25,0 и глюкоза 10,0. Кольца уравновешивают в течение 90 мин с частыми изменениями буфера при остаточном напряжении 5 г. Чтобы гарантировать создание оптимального напряжения, артериальные кольца дважды наполняют 36 мМ КС 1 и один раз 10 M PGF2a,перед действием 3 М PGF2a. Когда изометрическое напряжение достигает стационарного состояния, в бани добавляют накапливающиеся дозы агонистов аденозина данного изобретения ческой шкале). Напряжение, достигаемое при 3 М PGF2a считают эквивалентным 100%; все другие значения выражают в процентах от этой максимальной величины. Значения IC50 для релаксации, то есть концентрацию, при которой соединение формулы I вызывает 50% уменьшение напряжения, определяют, используя указанную выше компьютерную программу по подгонке кривых. В. Определение in vivo среднего артериального кровяного давления (MAP) и частоты сердечных сокращений (HR) у находящейся под наркозом крысы с нормальным кровяным давлением и крысы со спонтанно повышающимся давлением 1. Крыса под наркозом. Крысам с нормальным кровяным давлением дают наркоз - пентобарбитал натрия (50 мг/кг, внутрибрюшинно) и помещают их на нагретый хирургический стол. В бедренную артерию и вену вводят канюли для возможности измерения артериального давления и облегчения внутривенного введения исследуемых соединений. После хирургического вмешательства животным дают отрелаксировать в течение 10 мин. Непрерывно измеряют и регистрируют среднее артериальное давление и следят за частотой сердечных сокращений, используя пульсацию артериального давления для запуска кардиотахометра. После установления и регистрации исходных параметров вводят внутривенно увеличивающиеся дозы (1, 3, 10, 30, 100, 300 и 1000 цг/кг) подлежащего тестированию соединения формулы I. Наблюдают максимальные изменения сердечно-сосудистых параметров после каждой дозы агониста аденозина. Одной крысе вводят только одно соединение. Способность данных соединений снижать частоту серАктивность связываПр. ния рецепторов Вазорелаксация коронарной артерии свиньи/ 50 дечных сокращений и среднее значение артериального давления оценивают, определяя дозу агента, необходимую для снижения частоты сердечных сокращений или артериального давления на 25% (ED25). 2. Крыса со спонтанно повышающимся давлением (SHR). Исследуют гипотензивную активность орально принимаемых соединений формулы I у крыс со спонтанно повышающимся давлением в сознательном состоянии. Этим крысам дают наркоз - пентобарбитал натрия (50 мг/кг, внутрибрюшинно). В брюшную полость крыс имплантируют через серединный разрез приемник дистанционного действия. Канюлю приемника вставляют в брюшную аорту для возможности прямого измерения артериального давления у крыс со спонтанно повышающимся давлением в сознательном состоянии. Приемник закрепляют на стенке брюшины. После восстановления от хирургического вмешательства (минимум семь дней) этих крыс (SHR) помещают на пластину приемника и активируют приемник/передатчик. В течение 1,5 ч регистрируют систолическое,диастолическое и среднее артериальное давление и частоту сердечных сокращений у неограниченной крысы в сознательном состоянии для получения стабильной линии отсчета. Затем каждой крысе дают разовую дозу подлежащего тестированию соединения формулы I или наполнителя и в течение 20 ч отслеживают и регистрируют изменения артериального давления и частоты сердечных сокращений. В таблице II представлены результаты определения биологической активности для типичных соединений и для соединений примера 6, стадии 1, входящих в область соединений,описанных формулой I. Таблица II Кровяное давление/Частота сердечных сокращений Крыса под наркозом Когда кровоток к сердцу прерывается на короткий период времени (от 2 до 5 мин) с последующим восстановлением кровотока (повторная перфузия), сердце становится защищенным против развития повреждений, если кровоток прерывается на более длительные периоды времени (например, 30 мин). Соединения формулы I демонстрируют активность в тестах, используемых для определения способности соединений симулировать кардиозащитную активность по предварительной подготовке миокарда. Ниже описаны примеры тестовых процедур, которые полезны в определении кардиозащитной активности соединений формулы I. Определение кардиозащитной активности на крысах 1. Основная хирургическая подготовка. Взрослым крысам Sprague-Dawley дают наркоз из инактина (Inactin) (100 мг/кг, внутрибрюшинно). В трахею вводят трубку и обеспечивают вентиляцию легких под положительным давлением через небольшой респиратор для животных. В брюшную вену и артерию вводят катетеры для введения подлежащих исследованию соединений настоящего изобретения и измерения кровяного давления, соответственно. На левой стороне грудной клетки выше грудных мышц делают разрез, мышцы оттягивают, открывая четвертое межреберное пространство. Раскрывают грудную полость и открывают сердце. Кусок 4-0 пролиновой нити для смешивания ран протягивают через вентрикулярную стенку рядом с левой основной коронарной артерией и используют для прерывания кровотока через коронарную артерию посредством натяжения скользящего узла. На поверхности сердца размещают импульсный датчик потока (pulsedDoppler flow probe) (прибор, который измеряет кровоток) для подтверждения, что коронарная артерия идентифицирована должным образом. В левый желудочек также помещают катетер для контроля функции левого желудочка во время эксперимента. 2. Предварительная подготовка и процедура исследования Для предварительной подготовки сердца перекрывают коронарную артерию (прерывают кровоток) на две минуты. Эту процедуру прерывания/повторной перфузии повторяют дважды. Через пять минут после завершения последнего этапа предварительной подготовки артерию перекрывают снова на 30 мин с последующей перфузией в течение трех часов. После это 53 го вместо осуществления процедуры прерывания/повторной перфузии испытывают соединение формулы I, это соединение вводят за 30 мин до 30-минутного прерывания кровотока. По окончании 3-часового периода перфузии артерию снова перекрывают, вводят в левый вентрикулярный катетер 1 мл красителя (PatentBlue dye) и останавливают сердце посредством внутривенного введения хлорида калия. Данная процедура позволяет красителю перфузировать нормальные области сердца, тогда как часть сердца, подверженная ишемии, не захватывает краситель (это опасная область, "область риска"). Сердце быстро удаляют для исследования величины инфаркта. Величину инфаркта определяют, делая на сердце от верхушки до основания 4-5 срезов толщиной 1-2 мм. Срезы инкубируют в растворе 1% трифенилтетразолия в течение 15 мин. Этот краситель реагирует с жизнеспособной тканью и вызывает появление в ней кирпично-красного цвета. Подверженная инфаркту ткань не реагирует с красителем и выглядит тускло-белой. Срезы ткани помещают в систему анализа видеоизображения и определяют величину инфаркта посредством планиметрии. Исследованный эффект соединения настоящего изобретения на величину инфаркта миокарда оценивают и используют для количественной оценки степени кардиозащитной активности. Результаты представлены в виде процента области риска, подверженной инфаркту. Результаты проверки типичных соединений формулы I указанными выше способами приведены ниже в таблице III. Группа животных Контрольная 1 Предварительно подготовленная 2 Соединение при низкой дозе 3 Соединение при высокой дозе 4 Животные, не прошедшие предварительную подготовку и не обработанные соединениями. 2 Животные, прошедшие предварительную подготовку посредством процедуры прерывания/повторной перфузии. 3 Животные, которым сделана внутривенная инъекция 1 г/кг с последующим внутривенным вливанием 0,1 г/кг/мин соединения примера 39 за 30 мин до 30-минутного прерывания кровотока. 4 Животные, которым сделана внутривенная инъекция 10 г/кг с последующим внутривенным вливанием 1 г/кг/мин соединения примера 39 за 30 мин до 30-минутного прерывания кровотока через 2 ч после начала повторной перфузии. Соединения формулы I демонстрируют активность в тестах, используемых для определе 54 ния способности соединений ингибировать липолиз. Типичные тестовые процедуры, полезные для определения антилиполитической активности соединений формулы I, описаны ниже. Определение антилиполитической активности на адипоцитах крыс 1. Выделение адипоцитов из эпидидимального жира (Epididymal Fat Pads). Жировую ткань выделяют из крыс под наркозом и дважды промывают в инкубационной среде (2,09 г бикарбоната натрия и 0,04 г натриевой соли ЭДТА в 1 л буфера Кребса(Krebs. Из каждой крысы (300-350 г) получают приблизительно 4 мл жировой ткани. Жировую ткань (35 мл) разрезают на мелкие кусочки при помощи медицинских ножниц и промывают инкубационной средой (50 мл). Смесь выливают в цилиндр шприца на 50 мл, к которому вместо иглы присоединен короткий кусок пережатой трубки. Водную фазу оставляют дренироваться. Второе промывание инкубационной средой проводят через шприц. Ткань добавляют к 50 мл раствора коллагеназы (90 мг коллагеназы, 500 мг бычьего сывороточного альбумина (BSA) и 1 мл 0,1 М раствора хлорида кальция в 50 мл инкубационной среды) в бутылке на 1 л. Смесь встряхивают при температуре 37 С в течение примерно 60 мин в атмосфере 95% кислорода/5% диоксида углерода, выполняя переваривание ткани. Диспергированные клетки выливают через 2 слоя марли в пластиковый лабораторный стакан на 1 л. Оставшиеся на марле непереваренные комки промывают один раз инкубационной средой (20 мл). Клетки из стакана центрифугируют в 2 пластиковых пробирках в течение 30 с при комнатной температуре и 300 об/мин. Водную фазу удаляют из нижней части свободно заполненного слоя плавающих жировых клеток и отбрасывают. Адипоциты осторожно выливают в пластиковый стакан на 250 мл, содержащий 100 мл раствора для промывания (1 г BSA на 100 мл инкубационной среды). После спокойного перемешивания повторяют стадию центрифугирования. Затем промывают еще раз раствором для промывания. Клетки объединяют и определяют их объем при помощи мерного цилиндра. Адипоциты разбавляют в два раза буфером (120 мл инкубационной среды, 1,2 г BSA, 13 мг пировиноградной кислоты). 2. Исследование липолиза in vitro. Исследование проводят в пластиковой пробирке на 20 мл для сцинтилляционных измерений, общий объем для этого исследования составляет 4,2 мл. Буфер для исследования (2,5 мл), разбавленные адипоциты (1,5 мл) и раствор подлежащего тестированию соединения агониста аденозина (12,3 л; меняющейся концентрации) инкубируют в шейкере в течение 15 мин,затем начинают реакцию с раствором норепинафрина (41,2 л) (10 нМ в несущем растворе,содержащем 100 мл воды, 4 мг BSA, 20 л 0,1 М ЭДТА и аденозиндеаминазу (1 г/мл, 41,2 л. Через 60 мин в шейкере реакцию заканчивают,помещая пробирки на лед. Содержимое каждой пробирки переносят в стеклянную пробирку 12 х 75 мм и центрифугируют при 8-10 С и 3600 об/мин в течение 20 мин. Твердый липидный слой удаляют путем аспирации, а водный слой исследуют на глицерин (400 л образца). Позитивный контроль готовят в отсутствие агониста аденозина, заменяя подлежащий исследованию раствор водой. Результаты исследования соединений формулы I приведены ниже в таблице IV и представлены как % ингибирования производства глицерина 1 М и/или 0,1 M исследуемого соединения относительно позитивного контроля и как величину EC50, то есть концентрацию исследуемого соединения, необходимую для 50% ингибирования производства глицерина. Для сравнения приведены также результаты для соединений из литературы N-циклопентиладенозина (СРА), N-этилкарбоксамидоаденозина Связывание A1 и A2 рецепторами аденозина и активность вазорелаксации для соединений из литературы из таблицы IV, определенные описанными здесь выше способами, приведены ниже в таблице V. Таблица V Связывание A1 и А 2 рецепторами аденозима (С 50) Соединение А 2(нМ) Антилиполитическая активность аденозина опосредована активацией A1 подтипа рецепторов. Селективные агонисты А 2 подтипа рецепторов, такие как CGS 21680, не проявляют антилиполитической активности. Соответственно, тогда как некоторые A1 селективные агонисты могут не обладать требуемой гипотензивой активностью, а А 2 агонисты могут быть неэффективными антилиполитическими агентами, 56 соединения настоящего изобретения, которые являются смешанными агонистами, особо подходят для эффективного лечения обоих обсуждаемых здесь выше факторов риска, то есть,гипертензии и гиперлипидемии. В ходе лечения пациентам, страдающим от гипертензии, ишемии миокарда, или пациентам,нуждающимся в кардиозащитной терапии или антилиполитической терапии, обычно можно вводить соединения формулы I орально или парентерально. Используемый здесь термин "пациенты" включает людей и других млекопитающих. Соединения формулы I, предпочтительно в виде солей, можно приготовить для введения любым общеизвестным способом, и область данного изобретения включает фармацевтические композиции, содержащие, по крайней мере, одно соединение формулы I, приспособленное для применения человеком или в ветеринарии. Такие композиции можно составить обычным образом, используя один или более фармацевтически пригодных носителей или наполнителей. Подходящие носители включают разбавители или наполнители, стерильные водные среды и различные нетоксические органические растворители. Композиции можно составить в виде таблеток, капсул, лепешек, пастилок, твердых свечей, порошков, водных суспензий или растворов, растворов для инъекций, эликсиров,сиропов и подобного, они могут содержать один или более агентов, выбранных из группы, включающей подслащивающие агенты, агенты для вкуса и запаха, красители и консерванты, для обеспечения фармацевтически пригодного препарата. Конкретный носитель и соотношение агонистов аденозина и носителя определяются растворимостью и химическими свойствами соединений, конкретным способом введения и стандартной фармацевтической практикой. В производстве таблеток можно использовать, например, такие наполнители как лактоза, цитрат натрия, карбонат кальция и дикальцийфосфат, и различные дезинтеграторы, такие как крахмал,альгиновая кислота и некоторые комплексные силикаты, вместе со смазывающими веществами, такими как стеарат магния, лаурилсульфат натрия и тальк. Среди предпочтительных фармацевтически пригодных носителей для препаратов в виде капсул находятся лактоза и полиэтиленгликоли с высоким молекулярным весом. Когда составляют водные суспензии для орального приема, носитель может быть эмульгатором или суспендирующим агентом. Наряду с другими материалами можно применять такие разбавители как этанол, пропиленгликоль, глицерин и хлороформ, и их комбинации. Для парентерального введения можно применять растворы или суспензии таких соединений в масле кунжутного или земляного ореха, или растворы в водном пропиленгликоле, 57 а также стерильные водные растворы описанных здесь растворимых фармацевтически пригодных солей. Растворы солей этих соединений особенно подходят для введения путем внутримышечной и подкожной инъекции. Водные растворы, включая растворы солей, растворенных в чистой дистиллированной воде, подходят для введения путем внутривенной инъекции, при условии, что их рН отрегулирован должным образом, и что они имеют подходящий буфер,изотоничны при достаточном количестве соляного раствора или глюкозы и стерилизованы посредством нагревания или микрофильтрации. Схема приема лекарственного препарата такая, которая обеспечивает максимальную терапевтическую реакцию до получения улучшения, а после этого минимальный эффективный уровень, который дает успокоение. Таким образом, именно дозировка является терапевтически эффективным фактором в снижении кровяного давления при лечении гипертензии, в повышении коронарного кровотока при лечении ишемии миокарда, в производстве кардиозащитного действия, то есть уменьшении ишемического поражения или величины инфаркта миокарда вследствие ишемии миокарда, или производстве антилиполитического действия. Вообще, доза для орального приема может составлять примерно от 0,1 до 100 мг/кг (предпочтительно в диапазоне от 1 до 10 мг/кг), а доза для внутривенного введения примерно от 0,01 до 10 мг/кг(предпочтительно в диапазоне от 0,1 до 5 мг/кг),конечно помня, что при выборе подходящей дозы в каждом отдельном случае необходимо учитывать вес, общее состояние здоровья, возраст пациента и другие факторы, которые могут влиять на реакцию на лекарственное средство. Соединения формулы I можно вводить так часто, как необходимо для достижения и поддержания требуемой терапевтической реакции. Некоторые пациенты могут реагировать быстро на относительно большую или маленькую дозу и требовать небольшой дозы для поддержания эффекта или не требовать вовсе. С другой стороны, другим пациентам может требоваться длительное введение лекарства примерно от 1 до 4 раз в день в зависимости от физиологических потребностей конкретного пациента. Обычно лекарственный препарат можно вводить орально примерно от 1 до 4 раз в день. Ожидается, что многим пациентам будет требоваться не более 1-2 доз в день. 58 Также ожидается, что соединения формулы I будут полезны в виде дозированных форм для инъекций, которые можно вводить в случае крайней необходимости пациенту, страдающему от острой гипертензии или ишемии миокарда,или пациенту, нуждающемуся в кардиозащите или антилиполитической терапии. За таким лечением может следовать внутривенное вливание активного соединения, и введенное такому пациенту количество должно быть эффективным и поддерживать требуемую терапевтическую реакцию. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения 2,4-дигидроксипиридина, включающий нагревание соединения формулы А где R является Н, алкилом или аралкилом, и фосфорной кислоты, когда массовое соотношение фосфорной кислоты к воде составляет не менее чем примерно 27:1. 2. Способ по п.1, отличающийся тем, что используют разбавленную фосфорную кислоту и массовое соотношение фосфорной кислоты к воде, равное 27:1, достигается посредством удаления воды из реакционной смеси. 3. Способ по п.2, отличающийся тем, что удаление производят посредством перегонки. 4. Способ по п.1, отличающийся тем, что фосфорную кислоту и соединение формулы А нагревают примерно до 210 С. 5. Способ получения 2,4-дигидрокси-3 нитропиридина, включающий реакцию 2,4 дигидроксипиридина с азотной кислотой. 6. Способ получения 2,4-дигидрокси-3 нитропиридина, включающий реакцию продукта по п.1 без его выделения с азотной кислотой. 7. Способ по п.5, отличающийся тем, что до реакции с азотной кислотой добавляют органическую кислоту. 8. Способ по п.7, отличающийся тем, что органическая кислота является уксусной кислотой. 9. Способ по п.6, отличающийся тем, что до реакции с азотной кислотой добавляют органическую кислоту. 10. Способ по п.9, отличающийся тем, что органическая кислота является уксусной кислотой.
МПК / Метки
МПК: C07D 213/04
Метки: способ, 2,4-дигидроксипиридина, получения, 2,4-дигидрокси-3-нитропиридина
Код ссылки
<a href="https://eas.patents.su/30-884-sposob-polucheniya-24-digidroksipiridina-i-24-digidroksi-3-nitropiridina.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения 2,4-дигидроксипиридина и 2,4-дигидрокси-3-нитропиридина</a>
Способ получения аморфной гемисоли кальция [r-(r*,r*)]-2-(4-фторфенил)-бета, дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1н-пиррол-1-гептановой кислоты (аторвастатина) и его гидратов

Номер патента: 625
Опубликовано: 29.12.1999
Авторы: Швайсс Дитер, Минь Линь
МПК: C07D 207/34
Метки: r-(r*,r*)]-2-(4-фторфенил)-бета, получения, аторвастатина, гемисоли, аморфной, дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1н-пиррол-1-гептановой, способ, кислоты, гидратов, кальция
Формула / Реферат:
1. Способ получения аморфной гемисоли кальция [R-(R*,R*)]-2-(4-фторфенил)-b ,d -дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино) кабронил]-1Н-пиррол-1-гептановой кислоты (аторвастатина) и его гидратов, отличающийся тем, что аторвастатин кристаллической формы I со следующими характеристиками по таблицам. Интенсивность и локализация пика всех дифракционных линий при относительной интенсивности выше 20% в случае кристаллической формы I...
Способ очистки 11&beta-21-дигидрокси-2 ‘-метил-5′ &beta н-прегна-1,4-диено – (17,16-ди)-оксазол-3,20-диона

Номер патента: 789
Опубликовано: 24.04.2000
Автор: Форте Луиджи
МПК: C07J 71/00
Метки: метил-5, н-прегна-1,4-диено, 17,16-ди)-оксазол-3,20-диона, способ, очистки, beta, 11&beta-21-дигидрокси-2
Формула / Реферат:
1. Способ очистки соединения 11b -21-дигидрокси-2'-метил-5'b Н-прегна-1,4-диено[17,16-ди-]-оксазолин-3,20-дион формулы который включает: а) адсорбцию вышеназванного соединения, содержащегося в водном растворе, полученном из ферментационных бульонов или из потоков в ходе процесса на адсорбирующей полимерной смоле, имеющей в основе стирольный или акриловый матриксы, б) промыв смолы водой, в) десорбцию соединения формулы I со смолы путем...
Кристаллическая кислая кальциевая соль [r-(r, r)]-2-(4-фторфенил)-бета, дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1h-пиррол-1гептановой кислоты (аторвастатин)

Номер патента: 474
Опубликовано: 26.08.1999
Авторы: Ичикава Шигеру, Дженнингс Рекс Аллен, Накагава Шинсуке, Бриггс Кристофер, Харасава Кикуко, Вейд Роберт, Минохара Казуо
МПК: A61K 31/40, C07D 207/34
Метки: аторвастатин, кислая, кристаллическая, r)]-2-(4-фторфенил)-бета, соль, r-(r, кальциевая, дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1h-пиррол-1гептановой, кислоты
Формула / Реферат:
1. Кристаллическая форма I аторвастатина или его гидрата, имеющая в форме порошка дифракцию рентгеновских лучей, содержащую в качестве основного пика следующую величину 2q , измеренную с использованием излучения СuKa : 21,6. 2. Кристаллическая форма I аторвастатина или его гидрата согласно п.1, характеризующаяся также следующими величинами 2q , измеренными с использованием излучения СuKa : 17,1, 19,5 и 23,7. 3. Кристаллическая форма I...
Форма iii кристаллической кислой кальциевой соли [r-(r*, r*)]-2-(4-фторфенил)-бета-дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)-карбонил]-1н-пиррол-1-гептановой кислоты (аторвастатин)

Номер патента: 505
Опубликовано: 28.10.1999
Автор: Маккензи Энн
МПК: A61K 31/40, C07D 207/34
Метки: кальциевой, r*)]-2-(4-фторфенил)-бета-дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)-карбонил]-1н-пиррол-1-гептановой, кристаллической, аторвастатин, кислоты, соли, r-(r, форма, кислой
Формула / Реферат:
1. Кристаллическая форма III аторвастатина или его гидрата, имеющая в форме порошка дифракцию рентгеновских лучей, содержащую в качестве основного пика следующую величину 2q , измеренную с использованием излучения CuKa :8,5. 2. Кристаллическая форма III аторвастатина или его гидрата согласно п.1, характеризующаяся также следующими величинами 2q , измеренными с использованием излучения CuKa :16,6, 20,0, 20,3 и 24,4. 3. Кристаллическая форма III...
Производные 5-0-дезозаминил-6-0-метилэритронолида а, способ их получения и их применение для получения биологически активных продуктов

Номер патента: 575
Опубликовано: 29.12.1999
Авторы: Бонне Алан, Дельтиль Мишель, Мазюри Алан
МПК: C07H 17/08
Метки: биологически, 5-0-дезозаминил-6-0-метилэритронолида, применение, производные, получения, продуктов, способ, активных
Формула / Реферат:
1. Соединения формулы (I): в которой или R1 представляет радикал алкил, содержащий до 8 атомов углерода, замещенный одним или несколькими радикалами алкила, содержащими до 8 атомов углерода, или одним или несколькими радикалами арила, содержащими до 14 атомов углерода, или R1 представляет радикал арил, содержащий до 14 атомов углерода, который может быть замещен одним или несколькими радикалами алкил, алкенил или алкинил, содержащими до 8...
Предыдущий патент: Роторно-поршневые двигатели внутреннего сгорания.
Следующий патент: Лекарственное средство и способ медикаментозного воздействия на организм
Случайный патент: Упаковка для табачных изделий