Антагонисты гонадотропин-высвобождающего фактора.
Номер патента: 829
Опубликовано: 24.04.2000
Авторы: Виврэтт Мэттью Дж., Джиротра Нариндар Н., Лин Питер, Уолш Томас Ф., Фишер Майкл Х., Чу Лин, Гаулет Марк





























Формула / Реферат
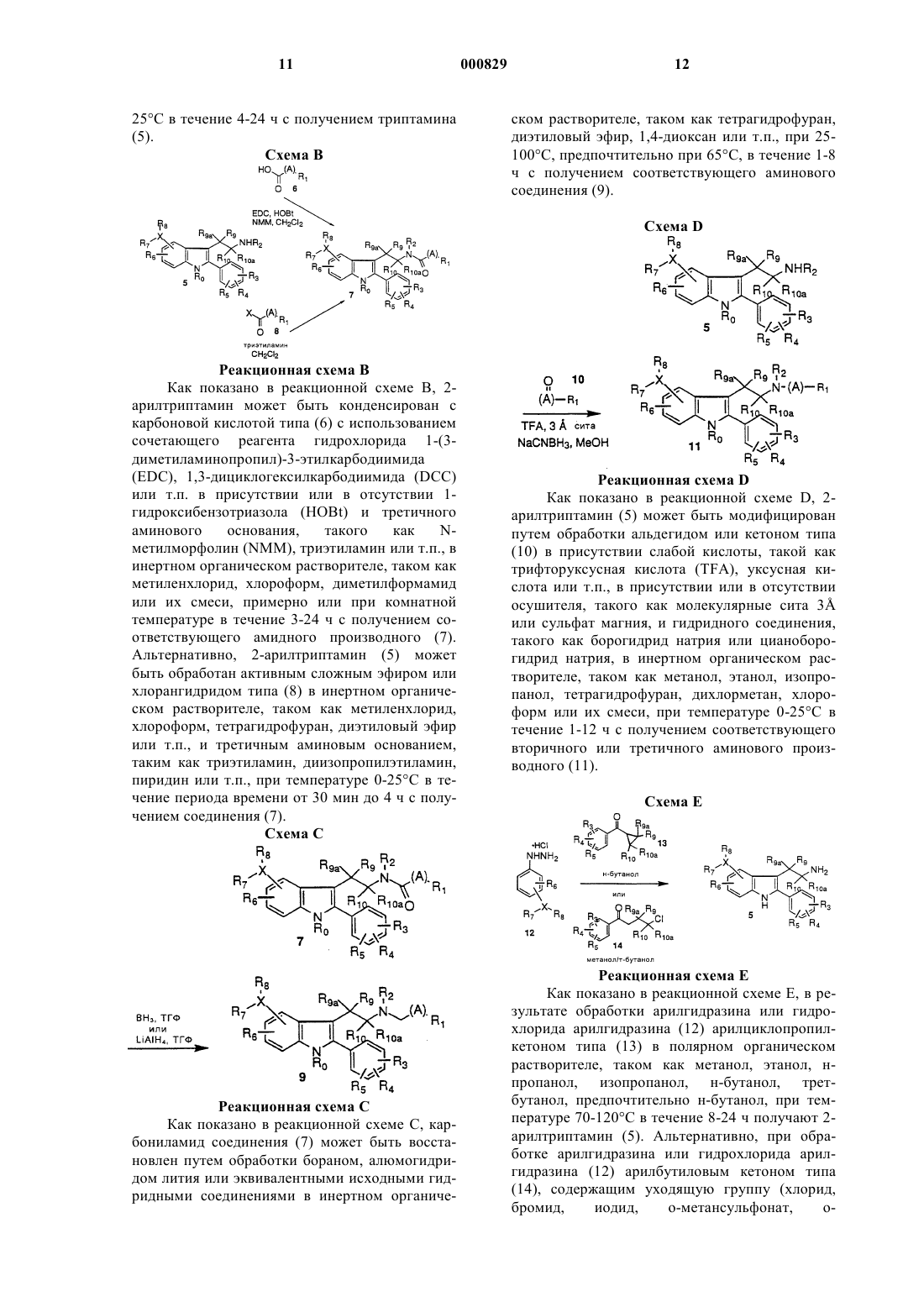
1. Соединение формулы

где А представляет C1-С6 алкил, замещенный C1-С6 алкил, С3-С7-циклоалкил, замещенный С3-С7 циклоалкил, С3-С6 алкенил, замещенный С3-С6 алкенил, С3-С6 алкинил, замещенный С3-С6 алкинил, C1-С6 алкокси или С0-С5 алкил-S(О)n-С0-С5 алкил, С0-С5 алкил-O-С0-С5алкил, С0-С5 алкил-NR18-С0-С5 алкил, где R18 и С0-С5 алкил, взятые вместе, могут образовывать кольцо,  или простую связь;
или простую связь;
R0 представляет водород, C1-С6 алкил, замещенный C1-С6 алкил, где заместители определены ниже; арил, замещенный арил, аралкил или замещенный аралкил, где заместители являются такими, как они определены ниже для R3, R4 и R5;
R1 представляет


R2 представляет водород, C1-С6алкил, замещенный C1-С6алкил, аралкил, замещенный аралкил, арил, замещенный арил, алкил-OR11, C1-C6(NR11R12), C1-C6(CONR11R12) или C(NR11R12)NH;
R2 и А, взятые вместе, образуют кольцо из 5-7 атомов;
R3, R4 и R5 независимо представляют водород, C1-С6 алкил, замещенный C1-С6 алкил, C1-С6 алкенил, замещенный C1-С6 алкенил, CN, нитро, C1-С3 перфторалкил, C1-С3 перфторалкокси, арил, замещенный арил, аралкил, замещенный аралкил, R11О(СН2)p-, R11C(O)O(CH2)p-, R11ОС(О)(СН2)p-, -(CH2)pS(О)nR17, -(СН2)pС(О) NR11R12 или галоген, где R17 представляет водород, C1-С6 алкил, C1-С3 перфторалкил, арил или замещенный арил;
R3 и R4, взятые вместе, образуют карбоциклическое кольцо из 3-7 атомов углерода или гетероциклическое кольцо, содержащее 1-3 гетероатомов, выбранных из N, О и S;
R6 представляет водород, C1-С6 алкил, замещенный C1-С6 алкил, арил, замещенный арил, C1-С3 перфторалкил, CN, NO2, галоген, R11O(CH2)p-, NR12C(O)R11, NR12C(О)NR11R12 или SOnR11;
R7 представляет водород, C1-С6 алкил или замещенный C1-С6 алкил, при условии, что, если Х является водородом или галогеном, то R7 отсутствует;
R8 представляет водород, С(O)ОR9, C(O)NR11R12, NR11R12, C(O)R11, NR12C(O)R11, NR12C(O)NR11R12, NR12S(O)2R11, NR12S(O)2 NR11R12, OC(O)R11, OC(O)NR11R12, OR11, SOnR11, S(O)nNR11R12, C1-С6 алкил или замещенный C1-С6 алкил, при условии, что если Х является водородом или галогеном, то R8 отсутствует; или
R7 и R8, взятые вместе, образуют карбоциклическое кольцо из 3-7 атомов;
R9 и R9a независимо представляют водород, C1-С6алкил, замещенный C1-С6алкил, арил, замещенный арил, аралкил или замещенный аралкил, если mь0; или
R9 и R9a, взятые вместе, образуют карбоциклическое кольцо, содержащее 3-7 атомов углерода, или  если mь0; или
если mь0; или
R9 и А, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода и один или несколько гетероатомов, если mь0; или
R10 и R10a независимо представляют водород, C1-С6 алкил, замещенный С1-С6 алкил, арил, замещенный арил, аралкил или замещенный аралкил; или
R10 и R10a, взятые вместе, образуют карбоциклическое кольцо, содержащее 3-7 атомов углерода, или
R9 и R10, взятые вместе, образуют карбоциклическое кольцо, содержащее 3-7 атомов углерода, или гетероциклическое кольцо, содержащее один или несколько гетероатомов, если mь0 или
R9 и R2, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода, и один или несколько гетероатомов, если mь0; или
R10 и R2, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода, и один или несколько гетероатомов;
R10 и А, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода, и один или несколько гетероатомов; или
R11 и R12 независимо представляют водород, С1-С6 алкил, замещенный C1-С6 алкил, арил, замещенный арил, аралкил, замещенный аралкил, карбоциклическое кольцо, содержащее 3-7 атомов, или замещенное карбоциклическое кольцо, содержащее 3-7 атомов;
R11 и R12, взятые вместе, могут образовывать необязательно замещенное кольцо с 3-7 атомами;
R13 представляет водород, ОН, NR7R8, NR11SO2(C1-С6 алкил), NR11SO2 (замещенный C1-С6 алкил), NR11SO2 (арил), NR11SO2 (замещенный арил), NR11SO2 (С1-С3 перфторалкил), SO2NR11 (C1-С6 алкил), SO2NR11 (замещенный C1-С6алкил), SO2NR11 (арил), SO2NR11 (замещенный арил), SO2NR11 (C1-С3 перфторалкил), SO2NR11(С(О)C1-С6алкил), SO2NR11(С(О) замещенный C1-С6 алкил), SO2NR11(С(О) арил), SO2NR11(C(O) замещенный арил), S(О)n(C1-С6 алкил), S(O)n (замещенный C1-С6 алкил), S(О)n(арил), S(О)n(замещенный арил), C1-С3 перфторалкил, C1-С3 перфторалкокси, C1-С6 алкокси, замещенный C1-С6 алкокси, СООН, галоген, NO2 или CN;
R14 и R15 независимо представляют водород, C1-С6 алкил, замещенный C1-С6 алкил, С2-С6 алкенил, замещенный С2-С6 алкенил, CN, нитро, С1-С3 перфторалкил, C1-С3 перфторалкокси, арил, замещенный арил, аралкил, замещенный аралкил, R11O(CH2)p-, R11C(O)O(CH2)p-, R11OC(O)(СН2)p-, -(CH2)pS(O)nR17, -(СН2)pС(O)NR11R12 или галоген, где R17 представляет водород, C1-С6 алкил, C1-С3 перфторалкил, арил или замещенный арил;
R16 представляет водород, C1-С6 алкил, замещенный C1-С6 алкил или N(R11R12);
R18 представляет водород, C1-С6 алкил, замещенный C1-С6 алкил, С(O)ОR9, C(O)NR11R12, C(O)R11, S(O)nR11;
R19 любой радикал, определенный выше для R13 или R14;
Х представляет водород, галоген, N, О, S(O)n, С(О), (CR11R12)p, С2-С6 алкенил, замещенный С2-С6 алкенил, С2-С6 алкинил, или замещенный С2-С6 алкинил, причем, если Х представляет водород или галоген, то R7 и R8 отсутствуют; а если Х представляет О, S(O)n, С(О) или CR11R12, то может присутствовать только R7 или R8;
Z представляет О, S или NR11;
m=0-3;
n=0-2;
р=0-4; и
где заместители для алкила, алкенила и алкинила выбирают из группы, включающей C1-С6 алкил, С3-С7 циклоалкил, арил, замещенный арил, аралкил, замещенный аралкил, гидрокси, оксо, циано, C1-С6 алкокси, фтор, C(O)OR11, арилС1-С3 алкокси, замещенный арилС1-С3 алкокси, а заместители для арила являются такими, как они были определены для R3, R4 и R5;
или их фармацевтически приемлемые аддитивные соли и/или гидраты, либо геометрические или оптические изомеры, если они являются необходимыми, или их рацемическая смесь.
2. Соединение по п.1 формулы

где R1, R3, R4, R5 и А являются такими, как указано в нижеследующей таблице

3. Соединение по п.1 формулы

где R1, R3, R4, R5 и А являются такими, как указано в нижеследующей таблице



4. Соединение по п.1 формулы

где R1, R3, R4, R5 и X-R7R8 являются такими, как указано в нижеследующей таблице

5. Соединение по п.1 формулы

где A, R1, R6 и X-R7R8 являются такими, как указано в нижеcледующей таблице


(*) - R6 = 5-NO2
(**)-R6=Br
6. Соединение по п.1 формулы

где R1, R7, и R8 являются такими, как указано в нижеследующей таблице




7. Соединение по п.1 формулы

где R1 и А являются такими, как указано в нижеследующей таблице


8. Соединение по п.1 формулы

где R1 и X-R7R8 являются такими, как указано в нижеследующей таблице



9. Соединение по п.1 формулы

где R1, R2 и А являются такими, как указано в нижеследующей таблице

10. Соединение по п.1 формулы

где A, R1, R2 и X-R7R8 являются такими, как указано в нижеследующей таблице



11. Соединение по п.1 формулы

где R1, X-R7R8, R9, R9a, R10 и R10a являются такими, как указано в нижеследующей таблице


12. Соединение по п.1 формулы

где R1 и X-R7R8 являются такими, как указано в нижеследующей таблице

13. Соединение по п.1 формулы

где X-R7R8 является таким, как указано в нижеследующей таблице


14. Соединение по п.1 формулы

где A, R1, R2, X-R7R8, R9, R9a, R10 и R10a являются такими, как указано в нижеследующей таблице






15. Соединение по п.1, которое представляет собой
a) 1-[2-[2-(3,4-диметоксифенил)-1H-индол-3-ил]этиламино]-3-(пиридин-4-илокси)пропан-2-ол;
b) [2-[2-(3,5-диметилфенил)-5-метансольфонил-1Н-индол-3-ил]этил]-(4-пиридин-4-ил-бутил)амин; и
c) 3-[2-(3,5-диметилфенил)-3-[2-(5-пиридин-4-ил-пентил-амино)этил]-1Н-индол-5-ил]-1,1-диметилмочевина.
16. Фармацевтическая композиция, которая содержит эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
17. Способ антагонизирования гонадотропин-высвобождающего гормона у индивидуума, нуждающегося в этом, предусматривающий введение эффективного количества соединения по п.1 индивидууму, страдающему расстройством, ассоциированным с гонадотропин-высвобождающим гормоном.
18. Способ по п.17, где расстройством, ассоциированным с гонадотропин-высвобождающим гормоном, является состояние, связанное с половым гормоном.
19. Способ по п.17, где расстройством, ассоциированным с гонадотропин-высвобождающим гормоном, является рак, зависимый от половых гормонов, доброкачественная гипертрофия предстательной железы или миома матки.
20. Способ по п.19, где зависимым от половых гормонов раком является рак предстательной железы, рак матки, рак молочной железы и гонадотропные аденомы гипофиза.
21. Способ по п.18, где состояние, зависимое от половых гормонов, выбирают из группы, включающей эндометриоз, поликистоз яичника, фиброз матки и преждевременное половое развитие.
22. Способ предупреждения беременности у индивидуума, нуждающегося в этом, предусматривающий введение этому индивидууму эффективного количества соединения по п.1.
23. Способ лечения системной красной волчанки у индивидуума, нуждающегося в таком лечении, предусматривающий введение этому индивидууму эффективного количества соединения по п.1.
24. Способ лечения синдрома раздражения кишечника у индивидуума, нуждающегося в таком лечении, предусматривающий введение этому индивидууму эффективного количества соединения по п.1.
25. Способ лечения предменструального синдрома у индивидуума, нуждающегося в таком лечении, предусматривающий введение этому индивидууму эффективного количества соединения по п.1.
26. Способ лечения гирсутизма у индивидуума, нуждающегося в таком лечении, предусматривающий введение этому индивидууму эффективного количества соединения по п.1.
27. Способ лечения недостаточного роста или дефицита гормона роста у индивидуума, нуждающегося в таком лечении, предусматривающий введение этому индивидууму эффективного количества соединения, стимулирующего эндогенное продуцирование гормона роста, и эффективное количество соединения по п.1.
28. Способ лечения расстройства сна, такого как, приступы апноэ во сне, у индивидуума, нуждающегося в таком лечении, предусматривающий введение этому индивидууму эффективного количества соединения по п.1.
29. Фармацевтическая композиция, которая содержит инертный носитель и эффективное количество соединения, стимулирующего эндогенное продуцирование или высвобождение гормона роста, в комбинации с соединением по п.1.
30. Фармацевтическая композиция, изготовленная путем смешивания соединения по п.1 и фармацевтически приемлемого носителя.
31. Способ изготовления фармацевтической композиции, предусматривающий смешивание соединения по п.1 и фармацевтически приемлемого носителя.
Текст
(GnRH), называемый также фактором высвобождения лютеинизирующего гормона (LHRH),представляет собой декапептид, который играет ключевую роль в репродукции человека. Этот гормон высвобождается из гипоталамуса и воздействует на гипофиз, стимулируя биосинтез и секрецию лютеинизирующего гормона (LH) и фоллирулостимулирующего гормона (FSH). LH,высвобождаемый из гипофиза, является ответственным, в первую очередь, за регуляцию продуцирования половых стероидных гормонов у обоих полов, а FSH регулирует сперматогенез у мужчин и развитие фоллокулов у женщин. Было показано, что GnRH-агонисты и антагонисты являются эффективными при лечении некоторых состояний, которые требуют ингибирования высвобождения LH/FSH. В частности, было показано, что терапия на основе GnRH является эффективной в лечении эндометриоза, фибромы матки, поликистоза яичников, преждевременного полового созревания и некоторых опухолей,ассоциируемых с половыми стероидными гормонами, главным образом рака предстательной железы, рака молочной железы и рака яичника.GnRH-агонисты и антагонисты были также использованы в различных методах оплодотворения и были исследованы как возможные контрацептивы как для мужчин, так и для женщин. Было также обнаружено, что они могут быть использованы для лечения гонадотропных аденом гипофиза, расстройств сна, таких как приступы апноэ (остановки дыхания) во время сна,синдрома раздражения кишечника, предменструального синдрома, доброкачественной гиперплазии предстательной железы, гирсутизма как следствие терапии путем введения гормона роста при дефиците гормона роста у детей и мышиных моделей волчанки. Соединения настоящего изобретения могут быть также использованы в комбинации с бифосфонатными агентами (бифосфоновыми кислотами) или другими агентами, такими как стимуляторы секреции гормона роста, например МК-0677, для лечения и предупреждения нарушений кальциевого обмена, фосфатного обмена и костного метаболизма, а в частности для предупреждения разрежения кости в процессе терапии с использованием GnRH-антагониста, а также в комбинации с эстрогенами, прогестеронами, антиэстрогенами, антипрогестинами и/или андрогенами для предупреждения или лечения разрежения кости или гипогонадотропных симптомов, таких как, приступообразное ощущение жара ("приливы") во время терапии с использованием GnRHантогониста. Кроме того, соединения настоящего изобретения могут быть введены одновременно с ингибитором 5-редуктазы 2, таким как финастерид или эпристерид; ингибитором 5 редуктазы 1, таким как 4,7-диметил-4-аза-5 000829 холестан-3-он, 3-оксо-4-аза-4,7-диметил-16(4-хлорфенокси)-5-андростан и 3-оксо-4-аза 4,7-диметил-16-(фенокси)-3-андростан, описанным в WO 93/23420 и WO 95/11254; двойными ингибиторами 5-редуктазы 1 и 5 редуктазы 2, такими как 3-оксо-4-аза-17-(2,5 трифторметилфенил-карбамоил)-5-андростан,описанными в WO 95/07927; антиандрогенами,такими как флутамид, казодекс и ацетат ципротерона; и альфа-1-блокаторами, такими как празозин, теразозин, доксазозин, тамсулозин и альфузозин. Кроме того, соединения настоящего изобретения могут быть использованы в комбинации с гормоном роста; фактором, высвобождения гормона роста или стимулятором гормона роста для замедления преждевременного полового созревания у детей с дефицитом гормона роста, что позволяет им набирать рост до образования эпифиза, и прекращения роста с наступлением половой зрелости. Существующие в настоящее время GnRHантагонисты представляют собой GnRHподобные декапептиды, которые, из-за их очень низкой пероральной активности, обычно вводят внутривенно или подкожно. Они имеют аминокислотные замещения, обычно, в положениях 1,2, 3, 6 и 10. Непептидные GnRH-антагонисты имеют,возможно, преимущества в отношении пероральной активности. Непептидные GnRHантагонисты были описаны в Европейской заявке 0219292, в работе De, В et al., J. Med. Chem.,32, 2036-2038 (1989), в WO 95/28405, WO 95/29900 и ЕР 0679642 (все Takeda ChemicalIndustries, Ltd.) В нижеследующих патентах и патентных заявках описаны замещенные индолы, известные специалистам. В патенте США 5030640 описаны альфа-гетероциклические этаноламиноалкилиндолы, которые являются сильнодействующими -агонистами. В патенте США 4544663 описаны индоламиновые производные,которые, как предполагается, могут быть использованы в качестве противозачаточных средств, применяемых мужчинами. В WO 90/05721 описаны альфа-амино-индол-3 уксусные кислоты, которые могут быть использованы в качестве средств против диабета, ожирения и атеросклероза. В патенте Франции 2181559 раскрываются индоловые производные,обладающие седативной, нейролептической,анальгезирующей,гипотензивной,антисеротониновой и адренолитической активностью. В патенте Бельгии 879381 описаны 3 аминоалкил-1 Н-индол-5-тиоамидные и карбоксамидные производные, используемые в качестве сердечно-сосудистых средств для лечения гипертензии, болезни Рейно и мигрени. 3 Краткое описание изобретения Настоящее изобретение относится к соединениям, которые являются непептидными антагонистами GnRH и которые могут быть использованы для лечения ряда состояний, ассоциированных с половыми гормонами как у женщин, так и мужчин; к способам их получения; а также к способам и фармацевтическим композициям, содержащим указанные соединения, и используемым для введения млекопитающим. Поскольку соединения настоящего изобретения являются антагонистами гормона GnRH,то они могут быть использованы для лечения ряда ассоциированных с половыми гормонами состояний как у женщин, так и мужчин. Такими состояниями являются эндометриоз, фиброма матки, поликистоз яичников, гирсутизм, преждевременное половое созревание и некоторые гормональнозависимые опухоли, главным образом рак предстательной железы, рак молочной железы и рак яичника, гонадотропные аденомы гипофиза, расстройство сна, такое как приступы апноэ (остановки дыхания) во время сна, синдром раздражения кишечника, предменструальный синдром, доброкачественная гиперплазия предстательной железы. Указанные соединения могут быть также использованы для лечения состояний, связанных с дефицитом гормона роста, и при низком росте, а также для лечения системной красной волчанки. Кроме того, соединения настоящего изобретения могут быть использованы для in vitro-оплодотворения и в качестве противозачаточных средств. Эти соединения могут быть также использованы в комбинации с андрогенами, эстрогенами, прогестеронами, антиэстрогенами и антипрогестогенами для лечения эндометриоза и фибромы, и для контрацепции. Они могут быть также использованы в комбинации с тестостероном и другими андрогенами или антипрогестогенами в качестве контрацептивов для мужчин. Эти соединения могут быть также использованы в комбинации с ингибитором ангиотензинконвертирующего фермента, таким как эналаприл или каптоприл; антагонистом рецептора ангиотензина II, таким как лосартан; или с ингибитором ренина для лечения фибромы матки. Кроме того, соединения настоящего изобретения могут быть использованы в комбинации с бисфосфонатами (бисфосфоновыми кислотами) и другими агентами, для лечения и предупреждения нарушений кальциевого обмена, фосфатного обмена и костного метаболизма,а в частности, для предупреждения разрежения кости в процессе терапии с использованиемGnRH-антагониста и в комбинации с эстрогенами, прогестеронами и/или андрогенами для предупреждения или лечения разрежения кости или гипогонадотропных симптомов, таких как приступообразное ощущение жара ("приливы") во 4 время терапии с использованием GnRHантагониста. Кроме того, соединения настоящего изобретения могут быть введены одновременно с ингибитором 5-редуктазы 2, таким как финастерид или эпристерид; ингибитором 5 редуктазы 1, таким как 4,7-диметил-4-аза-5 холестан-3-он, 3-оксо-4-аза-4,7-диметил-16(4-хлорфенокси)-5-андростан и 3-оксо-4-аза 4,7-диметил-16-(фенокси)-5-андростан, описанным в WO 93/23420 и WO 95/11254; двойными ингибиторами 5-редуктазы 1 и 5 редуктазы 1, такими как 3-оксо-4-аза-17-(2,5 трифторметилфенил-карбамоил)-5-андростан,описанными в WO 95/07927; антиандрогенами,такими как флутамид, казодекс и ацетат ципротерона, и альфа-1-блокаторами, такими как празозин, теразозин, доксазозин, тамсулозин и альфузозин. Кроме того, соединения настоящего изобретения могут быть использованы в комбинации с гормоном роста; фактором высвобождения гормона роста или стимулятором гормона роста для замедления преждевременного полового созревания у детей с дефицитом гормона роста, что позволяет им набирать рост до образования эпифиза и прекращения роста с наступлением половой зрелости. Подробное описание изобретения Настоящее изобретение относится к соединениям общей формулыR0 представляет водород, C1-С 6 алкил, замещенный C1-С 6 алкил, где заместители определены ниже; арил, замещенный арил, аралкил или замещенный аралкил, где заместители являются такими, как они определены ниже дляR2 и А, взятые вместе, образуют кольцо из 5-7 атомов;R3, R4 и R5 независимо представляют водород, C1-С 6 алкил, замещенный C1-С 6 алкил, С 2 С 6 алкенил, замещенный С 2-С 6 алкенил, CN, нитро, C1-С 3 перфторалкил, C1-С 3 перфторалкокси,арил, замещенный арил, аралкил, замещенный аралкил,R11O(CH2)р-,R11C(O)O(CH2)p-,R11OC(O)(CH2)p-,-(CH2)pS(O)nR17,-(CH2)pC(O)NR11R12 или галоген, где R17 предC1 ставляет водород,C1-С 6 алкил,С 3 перфторалкил, арил или замещенный арил;R3 и R4, взятые вместе, образуют карбоциклическое кольцо из 3-7 атомов углерода или гетероциклическое кольцо, содержащее 1-3 гетероатомов, выбранных из N, О и S;R7 представляет водород, С 1-С 6 алкил или замещенный C1-С 6 алкил, при условии, что, если Х является водородом или галогеном, то R7 отсутствует;R8 представляет водород, С(O)ОR9,C(O)NR11R12, NR11R12, C(O)R11, NR12C(O)R11,NR12S(O)2R11,NR12C(O)NR11R12,NR12S(O)2NR11R12, OC(O)R11, ОС(O)NR11R12,OR11, SOnR11, S(O)nNR11R12, C1-С 6 алкил или замешенный C1-С 6 алкил, при условии, что если Х является водородом или галогеном, то R8 отсутствует; илиR7 и R8, взятые вместе, образуют карбоциклическое кольцо из 3-7 атомов;R9 и R9a независимо представляют водород, C1-С 6 алкил, замещенный C1-С 6 алкил, арил,замещенный арил, аралкил или замещенный аралкил, если m0; илиR9 и А, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода и один или несколько гетероатомов,если m0; илиR10 и R10a независимо представляют водород, C1-С 6 алкил, замещенный C1-С 6 алкил, арил,замещенный арил, аралкил или замещенный аралкил; илиR9 и R10, взятые вместе, образуют карбоциклическое кольцо, содержащее 3-7 атомов углерода, или гетероциклическое кольцо, содержащее один или несколько гетероатомов,если m0; илиR9 и R2, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода, и один или несколько гетероатомов,если m0; илиR10 и R2, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода, и один или несколько гетероатомов;R10 и А, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода, и один или несколько гетероатомов,илиR11 и R12 независимо представляют водород, C1-С 6 алкил, замещенный C1-С 6 алкил, арил,замещенный арил, аралкил, замещенный аралкил, карбоциклическое кольцо, содержащее 3-7 атомов, или замещенное карбоциклическое кольцо, содержащее 3-7 атомов;R11 и R12, взятые вместе, могут образовывать необязательно замещенное кольцо с 3-7 атомами;R14 и R15 независимо представляют водород, -С 6 алкил, замещенный C1-С 6 алкил, С 2 С 6 алкенил, замещенный С 2-С 6 алкенил, CN, нитро, C1-С 3 перфторалкил, C1-С 3 перфторалкокси,арил, замещенный арил, аралкил, замещенный аралкил,R11 О(СН 2)p-,R11C(O)O(CH2)p-,R11OC(O)(CH2)p-,-(CH2)pS(O)nR17,-(CH2)pC(O)NR11R12 или галоген, где R17 предC1-С 3 ставляет водород,C1-С 6 алкил,перфторалкил, арил или замещенный арил;R19 любой радикал, определенный выше для R13 или R14; Х представляет водород, галоген, N, О,S(O)n, С(О), (CR11R12)p, С 2-С 6 алкенил, замещенный С 2-С 6 алкенил, С 2-С 6 алкинил или замещенный С 2-С 6 алкинил, причем, если Х представляет водород или галоген, то R7 и R8 отсутствуют; а если Х представляет О, S(O)n, С(О) или CR11R12,то может присутствовать только R7 или R8;n = 0 - 2; р = 0 - 4; и где заместители для алкильных, алкенильных и алкинильных групп выбирают из группы, включающей C1-С 6 алкил, С 3-С 7 циклоалкил, арил,замещенный арил, аралкил, замещенный аралкил, гидрокси, оксо, циано, C1-С 6 алкокси, фтор,C(O)OR11,арилС 1-С 3 алкокси,замещенный арилС 1-С 3 алкокси, а заместители для арила являются такими как они были определены для R3,R4 и R5; или их фармацевтически приемлемые аддитивные соли и/или гидраты, либо геометрические или оптические изомеры, если они являются подходящими, или их рацемическая смесь. Если это не оговорено особо, то в описании изобретения и в формуле изобретения будут использованы следующие определения. Если какая-либо группа (например, арил,гетероцикл, R1 и т.п.) встречается более одного раза в любом радикале или в формуле I, то его определение в каждом случае является независимым от определения в любом другом случае. Кроме того, комбинации заместителей и/или радикалов являются допустимыми, только если эти комбинации приводят к получению стабильных соединений. Используемый термин "алкил" означает разветвленные или прямые насыщенные алифатические углеводородные группы, имеющие конкретно определенное число атомов углерода,например метил (Me), этил (Et), пропил, бутил,пентил, гексил, гептил, октил, нонанил, децил,ундецил, додецил, и их изомеры, такие как изопропил (i-Pr), изобутил (i-Bu), втор-бутил (s-Bu),трет-бутил (t-Bu), изопентан, изогексан и т.п. Термин "арил" включает фенил и нафтил. Предпочтительным арилом является фенил. Термин "галоген" или "галогеновая группа" включает фтор, хлор, бром и йод. Используемый в настоящем описании термин "композиция" означает продукт, содержащий конкретные ингредиенты в конкретных количествах, а также любой продукт, который получают непосредственно или опосредованно 8 из комбинации конкретных ингредиентов в конкретных количествах. Подразумевается, что связывающая группа А может быть соединена с любыми имеющимися атомами углерода или гетероатомами гетероароматических групп R1, включая оба кольца бензоконденсированных гетероциклических групп и, аналогично, R13, R14 и R15 могут быть связаны с любыми имеющимися атомами углерода гетероароматических групп R1. Кроме того, каждому специалисту хорошо известно, что многие вышеуказанные гетероциклические группы могут существовать в более чем одной таутомерной форме. При этом,имеется в виду, что все указанные таутомеры входят в объем настоящего изобретения. Настоящее изобретение также включает оптические изомерные формы, а именно, смеси энантиомеров или диастереомеров, например,рацематы, а также отдельные энантиомеры или диастереомеры настоящего изобретения. Эти отдельные энантиомеры обычно обозначаются в соответствии с их оптическим вращением, а именно, символами (+) и (-) и (L) и (D), (i) и (d) или их комбинацией. Эти изомеры могут быть также обозначены в соответствии с их абсолютной пространственной конфигурацией символами (S) и (R), которые означают левовращающий и правовращающий изомер соответственно. Отдельные оптические изомеры могут быть получены с использованием стандартных методов разделения, например, путем обработки соответствующей оптически активной кислотой,разделения диастереомеров, а затем выделения нужного изомера. Кроме того, отдельные оптические изомеры могут быть получены методами асимметрического синтеза. Кроме того, данная химическая формула или название охватывают фармацевтически приемлемые аддитивные соли этих соединений и их сольваты, такие как гидраты. Хотя соединения настоящего изобретения сами по себе являются эффективными, однако,для большей стабильности, для облегчения их кристаллизации, повышения растворимости и придания других желаемых свойств они могут быть изготовлены и введены в виде их фармацевтически приемлемых аддитивных солей. Соединения настоящего изобретения могут быть введены в форме фармацевтически приемлемых солей. Используемый в настоящем описании термин "фармацевтически приемлемая соль" включает все приемлемые соли. Примерами таких солей являются соли, которые могут быть образованы хлористоводородной, азотной,серной, фосфорной, муравьиной, уксусной,трифторуксусной, пропионовой, малеиновой,янтарной, малоновой, метансульфоновой кислотой и т.п., и которые могут быть использованы в качестве лекарственной формы для модификации свойств растворимости или гидролизных свойств, либо они могут быть использованы в 9 препаратах пролонгированного действия или пролекарственных препаратах. В зависимости от конкретных функциональных групп, присутствующих всоединении настоящего изобретения, фармацевтически приемлемые соли соединений настоящего изобретения включают соли,образованные катионами, такими как натрий,калий, алюминий, кальций, литий, магний,цинк; а также основаниями, такими как аммиак,этилендиамин, N-метилглутамин, лизин, аргинин, орнитин, холин, N,N'-дибензилэтилендиамин, хлорпрокаин, диэтаноламин, прокаин,N-бензилфенэтиламин, диэтиламин, пиперазин,трис(гидроксиметил)аминометан и гидроксид тетраметиламмония. Эти соли могут быть получены стандартными способами, например посредством реакции свободного основания с подходящим органическим или неорганическим основанием, либо альтернативно, посредством реакции свободного основания с подходящей органической или неорганической кислотой. Кроме того, в случае присутствия кислотной (-СООН) или спиртовой группы могут быть использованы фармацевтически приемлемые сложные эфиры, например метил, этил, бутил,ацетат, малеат, пивалоилоксиметил и т.п., которые, как известно, модифицируют свойства растворимости или гидролизные свойства для изготовления препаратов с пролонгированным высвобождением лекарственного средства или пролекарственных препаратов. Соединения настоящего изобретения могут иметь хиральные центры, отличающиеся от тех центров, стереохимия которых описана формулой I, а поэтому они могут существовать в виде рацематов, рацемических смесей и в виде отдельных энантиомеров или диастереомеров, где все указанные изомерные формы, а также их смеси входят в объем настоящего изобретения. Кроме того, некоторые из кристаллических форм соединений настоящего изобретения могут существовать как полиморфы, и, как таковые, они входят в объем настоящего изобретения. Более того, некоторые соединения настоящего изобретения могут образовывать сольваты с водой или с обычно используемыми органическими растворителями. Эти сольваты также входят в объем настоящего изобретения. Соединения настоящего изобретения были получены в соответствии с нижеследующими реакционными схемами. Все заместители являются такими, как они были определены выше,если это не оговорено особо. Реакционная схема А Как показано в схеме А, после обработки триптамина (1) N-карбоксифталимидом в инертном органическом растворителе, таком как тетрагидрофуран, при температуре 20-65 С, а предпочтительно 65 С, в течение 12-48 ч получают соответствующееN-фталимидотриптаминовое производное (2). N-фталимидотриптамин (2) может быть затем модифицирован путем обработки бромирующим агентом,таким как пербромид гидробромида пиридиния,гидротрибромид пирролидона или т.п., в инертном органическом растворителе, таком как тетрагидрофуран, метиленхлорид, хлороформ или их смеси, при 0-25 С в течение периода времени от 30 мин до 4 ч с получением 2 бромтриптамина (3). Бромид (3) может быть подвергнут реакции с арилбороновой кислотойScr. 1986, 26, 311-314), с палладиевым (O) катализатором, слабой кислотой, такой как водный карбонат натрия или т.п., и с хлоридным соединением, таким как хлорид лития, в инертном растворителе, таком как толуол, бензол, этанол,пропанол или их смеси, при температуре 25100 С, предпочтительно 80 С, в течение 1-6 ч с получением 2-арилтриптаминового производного (4). И наконец, фталимидогруппа может быть удалена путем обработки соединения (4) водным гидразином в инертном растворителе, таком как метанол или этанол, при температуре 0 11 25 С в течение 4-24 ч с получением триптамина 12 ском растворителе, таком как тетрагидрофуран,диэтиловый эфир, 1,4-диоксан или т.п., при 25100 С, предпочтительно при 65 С, в течение 1-8 ч с получением соответствующего аминового соединения (9). Схема D Реакционная схема В Как показано в реакционной схеме В, 2 арилтриптамин может быть конденсирован с карбоновой кислотой типа (6) с использованием сочетающего реагента гидрохлорида 1-(3 диметиламинопропил)-3-этилкарбодиимидаNметилморфолин (NММ), триэтиламин или т.п., в инертном органическом растворителе, таком как метиленхлорид, хлороформ, диметилформамид или их смеси, примерно или при комнатной температуре в течение 3-24 ч с получением соответствующего амидного производного (7). Альтернативно, 2-арилтриптамин (5) может быть обработан активным сложным эфиром или хлорангидридом типа (8) в инертном органическом растворителе, таком как метиленхлорид,хлороформ, тетрагидрофуран, диэтиловый эфир или т.п., и третичным аминовым основанием,таким как триэтиламин, диизопропилэтиламин,пиридин или т.п., при температуре 0-25 С в течение периода времени от 30 мин до 4 ч с получением соединения (7). Схема С Реакционная схема С Как показано в реакционной схеме С, карбониламид соединения (7) может быть восстановлен путем обработки бораном, алюмогидридом лития или эквивалентными исходными гидридными соединениями в инертном органиче Реакционная схема D Как показано в реакционной схеме D, 2 арилтриптамин (5) может быть модифицирован путем обработки альдегидом или кетоном типа(10) в присутствии слабой кислоты, такой как трифторуксусная кислота (TFA), уксусная кислота или т.п., в присутствии или в отсутствии осушителя, такого как молекулярные сита 3 или сульфат магния, и гидридного соединения,такого как борогидрид натрия или цианоборогидрид натрия, в инертном органическом растворителе, таком как метанол, этанол, изопропанол, тетрагидрофуран, дихлорметан, хлороформ или их смеси, при температуре 0-25 С в течение 1-12 ч с получением соответствующего вторичного или третичного аминового производного (11). Схема Е Реакционная схема Е Как показано в реакционной схеме Е, в результате обработки арилгидразина или гидрохлорида арилгидразина (12) арилциклопропилкетоном типа (13) в полярном органическом растворителе, таком как метанол, этанол, нпропанол, изопропанол, н-бутанол, третбутанол, предпочтительно н-бутанол, при температуре 70-120 С в течение 8-24 ч получают 2 арилтриптамин (5). Альтернативно, при обработке арилгидразина или гидрохлорида арилгидразина (12) арилбутиловым кетоном типа(14), содержащим уходящую группу (хлорид,бромид,иодид,o-метансульфонат,o 13 трифторметансульфонат или т.п.) в 4 положении, в полярном растворителе, таком как, метанол, этанол, н-пропанол, изопропанол,н-бутанол, трет-бутанол или их смеси, при комнатной температуре в течение периода времени от 30 мин до 2 ч с последующим нагреванием до температуры 65-100 С в течение 4-24 ч получают 2-арилтриптамин (5). Схема F 14 при температуре 25-65 С в течение 3-24 ч получают продукт присоединения ацилхлорида (18). Неочищенный продукт (18) может быть подвергнут реакции с амином типа (19) в инертном органическом растворителе, таком как диэтилэфир, тетрагидрофуран, метиленхлорид, хлороформ или т.п., в присутствии аминового основания, такого как триэтиламин, диизопропилэтиламин или пиридин, при температуре 0-25 С в течение периода времени от 30 мин до 4 ч с получением амидного производного (20). Амид(20) может быть затем модифицирован путем обработки восстановителем, таким как боран или алюмогидрид лития, в инертном органическом растворителе, таком как тетрагидрофуран,при повышенной температуре, предпочтительно при нагревании с обратным холодильником в течение 1-5 ч с получением соединения (21). Схема Н Реакционная схема F Как показано в реакционной схеме F, иодоанилины типа (15) могут быть подвергнуты реакции с арилацетиленами, соответствующим палладиевым (О) катализатором, таким как тетракис(трифенилфосфин) палладий; галогенидом меди (I), таким как бромид меди, в инертном органическом растворителе, таком как триэтиламин, при температуре 50-88 С в течение периода времени от 30 мин до 5 ч с получением диарилацетилена (16). Ацетилен (16) может быть затем модифицирован путем обработки палладиевым (II) катализатором, таким как хлорид палладия (II) или ацетат палладия (II), в инертном органическом растворителе, таком как ацетонитрил, при температуре 50-82 С в течение периода времени от 30 мин до 6 ч с получением 2-арилиндола (17). Схема G(22b) могут быть восстановлены с получением вторичных аминовых аналогов (7) путем обработки водородом (1 атм) и соответствующим катализатором, таким как палладий-на-угле,гидроксид палладия-на-угле или т.п., в инертном органическом растворителе, таком как тетрагидрофуран, этилацетат, метанол, этанол или их смеси, к которому добавляют слабую кислоту, такую как 30% водная уксусная кислота, в течение периода времени от 10 мин до 3 ч или до тех пор, пока не будет удалена арильная группа, в результате чего получают вторичный амин. Реакционная схема I Как показано в реакционной схеме I, в результате обработки нитроиндола типа (24) водородом (1 атм) и соответствующим катализатором, таким как никелевый катализатор Ренея(Raney Ni), в инертном органическом растворителе, таком как этанол, метанол или т.п., при комнатной температуре в течение 2-12 ч получают соответствующее аминоиндоловое производное (25). Схема J Реакционная схема J Как показано в реакционной схеме J, амино- или гидроксииндол (25) может быть модифицирован путем ацилирования в различных условиях. Например, в результате обработки соединения (25) хлорангидридом, ангидридом кислоты или активным сложным эфиром, и аминовым основанием, таким как диизопропилэтиламин, пиридин или т.п., в инертном органическом растворителе, таком как метиленхлорид, хлороформ, тетрагидрофуран или их смеси,при температуре от 0 С до комнатной температуры в течение 1-12 ч получают соответствующие амидное или сложноэфирное производные(26). Альтернативно, (25) может быть подвергнуто взаимодействию с карбоновой кислотой в присутствии одного или нескольких обычно используемых дегидратирующих агентов. Так,например, обработка аминоиндола (25) соответствующей карбоновой кислотой и гидрохлори 000829 16 дом 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC), 1,3-дициклогексилкарбодиимидом (DCC) или т.п., в отсутствии или в присутствии 1-гидроксибензотриазола (HOBt) и третичного аминового основания, такого как Nметилморфолин (NММ), триэтиламин или т.п., в инертном органическом растворителе, таком как метиленхлорид, хлороформ, диметилформамид или их смеси, при комнатной температуре или примерно при комнатной температуре в течение 3-24 ч приводит к получению соответствующего амидного или сложноэфирного производного Реакционная схема К Как показано в реакционной схеме К, производное мочевины или карбаматное производное (25) может быть получено путем обработки карбамоилхлоридом типа (27 а) или, альтернативно, изоцианатным реагентом типа (27b) и аминовым основанием, таким как пиридин, триэтиламин,диизопропилэтиламин,Nметилморфолин или т.п., в инертном органическом растворителе, таком как метиленхлорид,хлороформ, диметилформамид, тетрагидрофуран или их смеси, при температуре 0-65 С в течение 1-72 ч с получением соединения (28). Соединение (25) может быть также модифицировано путем обработки би(электрофильным) реагентом, таким как фосген, трифосген, 1,1'карбонилдиимидазол,N,N'-дисукцинимидилкарбонат или т.п., с добавлением или без добавления аминового основания, такого как пиридин, триэтиламин, диизопропилэтиламин, Nметилморфолин, в инертном органическом растворителе, таком как метиленхлорид, хлороформ или т. п., при температуре от -20 С до 0 С в течение периода времени от 20 мин до 2 ч. По истечении этого времени реакционную смесь обрабатывают соответствующим моно- или дизамещенным амином при от -20 до -25 С в течение 1-5 ч с получением мочевинного или карбаматного аналога (28).(25) может быть модифицирован путем обработки соответствующим сульфонилхлоридом типа (29) или сульфамилхлоридом типа (30) в присутствии аминового основания, такого как пиридин, триэтиламин, диизопропилэтиламин,N-метилморфолин или т.п., в инертном органическом растворителе, таком как метиленхлорид,хлороформ, дихлорэтан или их смеси, при температуре -20 -25 С в течение периода времени от 20 мин до 2 ч с получением соответствующего Реакционная схема М Как показано в реакционной схеме М, 2 арилтриптамин (33) может быть модифицирован путем обработки эпоксидом, таким как соединение (34), в инертном органическом растворителе, таком как метанол, этанол, изопропанол,бутанол, трет-бутанол или их смеси, при температуре 65-110 С в течение 8-20 ч с получением соответствующего аминоспиртового производного (35). Реакционная схема N Как показано в реакционной схеме N,амидные производные содержащего кислоту индолового производного, такого как (36), могут быть получены путем обработки соответствующим амином (R11R12NH) и подходящим сочетающим агентом, таким как гексафторфосфат бензотриазол-1-илокси-трис(пирролидино) фосфония (РуВОР), гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)фосфония(HOBt) и третичного аминового основания, такого как N-метилморфолин (NММ), триэтиламин или т.п., в инертном органическом растворителе, таком как метиленхлорид, хлороформ,тетрагидрофуран, диметилформамид или их смеси, при комнатной температуре или примерно при комнатной температуре в течение 3-24 ч с получением соответствующего амидного производного (37). Соединения настоящего изобретения могут быть использованы для лечения различных ассоциированных с половыми гормонами состояний у мужчин и женщин. Эффективность этих соединений может быть подтверждена их способностью действовать в качестве антагонистов нейропептидного гормона GnRH, что было продемонстрировано активностью этих соединений в нижеследующих in vitro-анализах. Анализ на связывание с рецептором GnRH гипофиза крысы Неочищенные плазматические мембраны,полученные из тканей гипофиза крысы, инкубировали в буфере Трис-НСl (50 мМ, рН 7,5), содержащем альбумин бычьей сыворотки (0,1%),[I-125] D-t-Вu-Ser6-Рго 9-этиламид-GnRH и нужную концентрацию испытуемого соединения. Аналитические смеси инкубировали при 4 С в течение 90-120 мин с последующей быстрой фильтрацией и неоднократным промыванием через стекловолоконный фильтр. Радиоактивность связанных с мембранами радиоактивных лигандов определяли на гамма-счетчике. Исходя из этих данных, вычисляли IC50 радиоактивного лиганда, связывающегося с рецепто 19 рами GnRH в присутствии испытуемого соединения. Анализ на ингибирование высвобожденияLH Активные соединения, выявленные в анализе на связывание с рецептором GnRH, были затем оценены с помощью in vitro-анализа на высвобождение LH для подтверждения их антагонистической активностиGnRH-индуцированного высвобождения LH). 1. Получение образцов Анализируемые соединения растворяли и разводили в ДМСО. Конечная концентрация ДМСО в среде для инкубирования составляла 0,5%. 2. Анализ Самцы крыс Wistar (150-200 г) были получены из лаборатории Charles River Laboratories(Wilmington, MA). Крыс выдерживали при постоянной температуре (25 С) в условиях смены дня и ночи: 12 ч при свете и 12 ч в темноте. Крысам давали еду и воду ad libitum (по желанию). Затем животных умерщвляли путем обезглавливания, гипофизы удаляли в асептических условиях и помещали в сбалансированный солевой раствор Хэнкса (HBSS) в 50 миллилитровую полипропиленовую центрифужную пробирку. Пробирку для сбора образцов центрифугировали при 250 х g в течение 5 мин и HBSS удаляли путем отсасывания. Гипофизы переносили в одноразовую чашку Петри и измельчали скальпелем. Затем измельченную ткань переносили в 50-миллилитровую одноразовую центрифужную пробирку путем суспендирования фрагментов ткани в трех последовательно добавляемых 10 мл аликвотах HBSS,содержащих 0,2% коллагеназы и 0,2% гиалуронидазы. Диспергирование клеток осуществляли в водяной бане при 37 С, слегка размешивая, в течение 30 мин. По окончании инкубирования,клетки 20-30 раз отсасывали пипеткой, а негидролизованные фрагменты гипофиза оставляли на 3-5 мин для осаждения. Суспендированные клетки удаляли путем отсасывания, а затем подвергали центрифугированию при 1200 х g в течение 5 мин. После этого клетки ресуспендировали в культуральной среде. Негидролизованные фрагменты гипофиза обрабатывали 30 миллилитровыми аликвотами вышеуказанных гидролизирующих ферментов всего за 3 стадии гидролиза с использованием смеси коллагеназы/гиалуронидазы. Полученные клеточные суспензии объединяли, оценивали и разводили до концентрации 3 х 105 кл./мл и 1,0 мл этой суспензии помещали в каждую лунку 24-луночного планшета (Costar, Cambridge, MA.). Клетки поддерживали в атмосфере воздуха 5% СО 2 при влажности 95% и при 37 С в течение 3-4 дней. Культуральная среда состояла из DMEM (модифицированной по способу Дульбекко среды Игла), содержащей 0,37% NаНСО 3, 10% лошадиной сыворотки, 2,5% фетальной телячьей сы 000829 20 воротки, 1% заменимых (не основных) аминокислот, 1% глутамина и 0,1% гентамицина. В день эксперимента клетки три раза промывали за полтора часа до начала эксперимента и еще два раза непосредственно перед началом эксперимента средой DMEM, содержащей 0,37%(100 Х), 1% пенициллина/стрептомицина (10000 ед. пенициллина и 10000 микрограммов стрептомицина на 1 мл) и 25 мМ HEPES, рН 7,4. Высвобождение LH инициировали путем добавления в каждую лунку (в дубликатах) 1 мл свежей среды, содержащей испытуемые соединения в присутствии 2 нМ GnRH. Инкубирование проводили в течение 3 ч при 37 С. После инкубирования среду удаляли и центрифугировали при 2000 х g в течение 15 мин для удаления всего клеточного материала. Надосадочную жидкость удаляли и оценивали на содержание LH методом РИА с "двойным" антителом с использованием материалов, предоставленных Dr. A. F.Parlow (Harbor-UCLA Medical Center, Torrance,CA). Соединения формулы I могут быть использованы в ряде участков организма, подвергаемых неблагоприятному действию GnRH. Они могут быть использованы при состояниях, ассоциированных с половыми гормонами; гормональнозависимом раке; доброкачественной гипертрофии предстательной железы или миоме матки. Введение соединений настоящего изобретения может дать хороший эффект при раке,ассоциированном с половыми гормонами,включая рак предстательной железы, рак матки,рак молочной железы, и гонадотропные аденомы гипофиза. Соединения настоящего изобретения могут быть успешно введены при других состояниях, ассоциированных с половыми гормонами, включая эндометриоз, поликистоз яичников, фиброму матки и преждевременное половое развитие. Эти соединения могут быть также использованы в комбинации с ингибитором ангиотензин-конвертирующего фермента,таким как эналаприл или каптоприл; антагонистом рецептора ангиотензина II, таким как, лосартан; или с ингибитором ренина для лечения фибромы матки. Соединения настоящего изобретения могут быть также использованы в качестве контрацептивов, применяемых как мужчинами, так и женщинами для предохранения от беременности; для in vitro-оплодотворения; при предменструальном синдроме, а также для лечения системной красной волчанки, гирсутизма, синдрома раздражения кишечника, и расстройств сна,таких как приступы апноэ во сне. Кроме того, соединения настоящего изобретения могут быть использованы как вспомогательное средство при гормональной терапии детей с дефицитом гормона роста. Эти соедине 21 ния могут быть введены вместе с гормоном роста или с соединением, стимулирующим эндогенное продуцирование или высвобождение гормона роста. Был разработан ряд соединений,которые стимулируют высвобождение эндогенного гормона роста. Известны пептиды, которые стимулируют высвобождение эндогенного гормона роста, например фактор высвобождения гормона роста; пептиды, высвобождающие гормон роста, GHRP-6 и GHRP-1 (описанные в патенте США 4411890, в публикации патента РСТWO 89/07110 и в публикации патента РСТWO 89/07111) и GHRP-2 (описанный в публикации патента РСТWO 93/04081), а также гексарелин (J. Endocrinol Invest., 15(Suppl.4) 45 (1992. Другие соединения, которые стимулируют высвобождение эндогенного гормона роста, описаны, например, в следующих работах: патент США 3239345; патент США 4036979; патент США 4411890; патент США 5206235; патент США 5283241; патент США 5284841; патент США 5310737; патент США 5317017; патент США 5374721; патент США 5430144; патент США 5434261; патент США 5438136; публ. патента ЕРО 0144230; публ. патента ЕРО 0513974; публ. патента РСТWO 94/07486; публ. патента РСТWO 94/08583; публ. патента РСТWO 94/11012; публ. патента РСТWO 94/13696; публ. патента РСТWO 94/19367; публ. патента РСТWO 95/03289; публ. патента РСТWO 95/03290; публ. патента РСТWO 95/09633; публ. патента РСТWO 95/11029; публ. патента РСТWO 95/12598; публ. патента РСТWO 95/13069; публ. патента РСТWO 95/14666; публ. патента РСТWO 95/16675; публ. патента РСТWO 95/16692; публ. патента РСТWO 95/17422; публ. патента РСТWO 95/17423; Science, 260, 1640-1643 (11 июня,1993); Ann. Rep. Med. Chem., 28, 177-186 (1993);(1994); и Proc. Natl. Acad. Sci. USA, 92, 70017005 (июль 1995). Предпочтительными характерными стимуляторами гормона роста, используемыми в комбинации с соединениями настоящего изобретения, являются следующие соединения: 1) N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро[3 Н-индол-3,4'-пиперидин]-1'-ил)карбонил]-2-(1H-индол-3-ил)этил]-2-амино-2 метилпропанамид; 2) N-[1(R)-[(1,2-Дигидро-1-метанкарбонилспиро[ЗН-индол-3,4'-пиперидин]-1'-ил)карбонил]-2-(1H-индол-З-ил)этил]-2-амино-2-метилпропанамид; 3) N-[1(R)-[(1,2-Дигидро-1-бензолсульфонилспиро[3 Н-индол-3,4'-пиперидин]-1'-ил) карбонил]-2-(1H-индол-3-ил)этил]-2-амино-2 метилпропанамид;N-[1(R)-[(1,2-Дигидро-1-метансульфонил-5-фторспиро-[3 Н-индол-3,4'-пиперидин]1'-ил)карбонил]-2-(5-фтор-1 Н-индол-3-ил)этил]2-амино-2-метилпропанамид; 16) N-[1(R)-[(1,2-Дигидро-1-(2-этоксикарбонил)метилсульфонилспиро[3 Н-индол-3,4'пиперидин)-1'-ил)карбонил]-2-(1H-индол-3-ил) этил]-2-амино-2-метилпропанамид; 17) N-[1(R)-[(1,2-Дигидро-1,1-диоксоспиро[3 Н-бензотиофен-3,4'-пиперидин]-1'-ил)карбонил]-2-(фенилметилокси)этил]-2-амино-2 метилпропанамид; и их фармацевтически приемлемые соли. Соединения настоящего изобретения могут быть использованы в комбинации с бисфосфонатами (бисфосфоновыми кислотами) и другими агентами, такими как стимуляторы секреции гормона роста, например МК-0677, для лечения и предупреждения нарушений кальциевого об 23 мена, фосфатного обмена и костного метаболизма, а в частности, для предупреждения разрежения кости при терапии с использованиемGnRH-антагониста; и в комбинации с эстрогенами, прогестеронами, и/или андрогенами для предупреждения или лечения разрежения кости или гипогонадотропных симптомов, таких как приступообразное ощущение жара во время терапии с использованием GnRH-антагониста. Известно, что бисфосфонаты (бисфосфоновые кислоты) ингибируют разрежение кости,а поэтому они могут быть использованы для лечения костного литиаза, как описано в патенте США 4621077 (Rosini et al.) В литературе описаны различные бифосфоновые кислоты, которые могут быть использованы для лечения и предупреждения заболеваний, связанных с разрежением кости. Характерные примеры таких соединений могут быть найдены в следующих работах: патент США 3251907, патент США 3422137, патент США 3584125, патент США 3940436, патент США 3944599, патент США 3962432, патент США 4054598, патент США 4267108,патент США 4327039, патент США 4407761, патент США 4578376, патент США 4621077, патент США 4624947, патент США 4746654, патент США 4761406, патент США 4922007, патент США 4942157,патент США 5227506, патент США 5270365, публ. патента ЕРО 0252504, и J. Org.Chem., 36, 3843 (1971). Получение бисфосфоновых кислот и галогенбисфосфоновых кислот хорошо известно специалистам. Характерные примеры такого получения могут быть найдены в вышеупомянутых работах, в которых описаны соединения,используемые для лечения нарушений кальциевого или фосфатного обмена, а в частности, используемые в качестве ингибиторов разрежения кости. Предпочтительные бисфосфонаты выбирают из группы следующих соединений: алендроновая кислота, этидроновая кислота, клодроновая кислота, памидроновая кислота, тилудроновая кислота, ризедроновая кислота, 6-амино 1-гидрокси-гексилиден-бисфосфоновая кислота и 1-гидрокси-3(метилпентиламино)-пропилиден-бисфосфоновая кислота; или их любые фармацевтически приемлемые соли. Особенно предпочтительным бисфосфонатом является алендроновая кислота (алендронат) или ее фармацевтически приемлемая соль. Особенно предпочтительным бисфосфонатом является алендронат натрия, включая тригидрат алендроната натрия. Алендронат натрия получил одобрение Регуляторной комиссии на его продажу в США под торговым знаком FOSAMAX. Кроме того, соединения настоящего изобретения могут быть введены одновременно с ингибитором 5-редуктазы 2, таким как фина 000829 стерид или эпристерид; ингибитором 5 редуктазы 1, таким как 4,7-диметил-4-аза-5 холестан-3-он, 3-оксо-4-аза-4,7-диметил-16(4-хлорфенокси)-З-андростан и З-оксо-4-аза 4,7-диметил-16-(фенокси)-5-андростан, описанным в WO 93/23420 и WO 95/11254; двойными ингибиторами 5-редуктазы 1 и 5 редуктазы 2, такими как 3-оксо-4-аза-17-(2,5 трифторметилфенил-карбамоил)-5-андростан,описанными в WO 95/07927; антиандрогенами,такими как флутамид, казодекс и ацетат ципротерона, и альфа-1-блокаторами, такими как, празозин, теразозин, доксазозин, тамсулозин и альфузозин. Кроме того, соединения настоящего изобретения могут быть использованы в комбинации с гормоном роста; фактором, высвобождения гормона роста или стимулятором гормона роста для замедления преждевременного полового созревания у детей с дефицитом гормона роста, что позволяет им набирать рост до образования эпифиза и прекращения роста с наступлением половой зрелости. В случае комбинированного лечения с использованием более чем одного активного агента, где активные агенты применяются в виде отдельных лекарственных препаратов, указанные активные агенты могут быть введены как отдельно, так и в сочетании друг с другом. Кроме того, введение одного агента может быть осуществлено до, одновременно или после введения другого агента. Фармацевтические композиции, содержащие активный ингредиент, могут быть изготовлены в форме, подходящей для перорального введения, например в виде таблеток, таблетоклепешек, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул,эмульсий, жестких и мягких капсул, сиропов или эликсиров. Композиции, предназначенные для перорального введения, могут быть получены любым стандартным методом, обычно используемым специалистами для изготовления фармацевтических композиций, и эти композиции могут содержать один или несколько агентов, выбранных из группы, включающей подслащивающие агенты, ароматизирующие агенты, красители и консерванты, для получения фармацевтических препаратов, имеющих эстетически привлекательный вид и приятные вкусовые качества. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, которые являются подходящими для изготовления таблеток. Такими наполнителями могут быть, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие агенты, например кукурузный крахмал или альгиновая кислота; связующие агенты, например крахмал, желатин или 25 аравийская камедь; и замасливающие агенты,например стеарат магния, стеариновая кислота или тальк. Таблетки могут быть непокрытыми,либо для замедления дезинтеграции и абсорбции в желудочно-кишечном тракте они могут быть покрыты стандартными методами, в результате чего могут быть получены препараты пролонгированного действия. Для этого, например, может быть использован инерционный материал, такой как глицерилмоностеарат или глицерилдистеарат. Эти таблетки могут быть покрыты методами, описанными в патентах США 4256108; 4166452 и 4265874, с получением осмотических терапевтических таблеток для регулируемого высвобождения. Препараты для перорального введения могут быть также изготовлены в виде жестких желатиновых капсул, содержащих активный ингредиент, смешанный с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, содержащих активный ингредиент, смешанный с водной или масляной средой, например с арахисовым маслом, парафиновым маслом или оливковым маслом. Водные суспензии содержат активный ингредиент в смеси с наполнителями, подходящими для изготовления водных суспензий. Такими наполнителями являются суспендирующие агенты, например, натрийсодержащая карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь, и аравийская камедь; диспергирующие или смачивающие агенты, которыми могут быть природный фосфатид, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, образованными жирными кислотами и гекситом, такие как моноолеат полиоксиэтиленсорбита; или продукты конденсации этиленоксида с неполными сложными эфирами,образованными жирными кислотами и ангидридами гексита, например полиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один или несколько консервантов, например этил или н-пропил, п-гидроксибензоат; один или несколько красителей; один или несколько ароматизирующих агентов; и один или несколько подслащивающих агентов, таких как сахароза, сахарин или аспартам. Масляные суспензии могут быть изготовлены путем суспендирования активного ингредиента в растительном масле, например в арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле; либо в минеральном масле, таком как парафиновое масло. Масляные суспензии могут содержать загущающий 26 агент, например пчелиный воск, твердый парафин, или цетиловый спирт. Для получения перорального препарата с приятными вкусовыми качествами могут быть добавлены подслащивающие агенты, такие как указано выше, а также ароматизирующие агенты. Для лучшего сохранения в эти композиции может быть добавлен антиоксидант, такой как аскорбиновая кислота. Диспергируемые порошки и гранулы, подходящие для приготовления водных суспензий путем добавления воды, содержат активный ингредиент в смеси с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или несколькими консервантами. Примеры подходящих диспергирующих или смачивающих агентов и суспендирующих агентов были уже приведены выше. В качестве добавок могут также присутствовать подслащивающие агенты, ароматизирующие агенты и красители. Фармацевтические композиции настоящего изобретения могут быть также изготовлены в виде эмульсий "масло в воде". Масляной фазой может быть растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например парафиновое масло,или их смеси. Подходящими эмульгирующими агентами могут быть природные фосфатиды,например соевые бобы, лецитин, и сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и ангидридов гексита,например сорбитанмоноолеат; а также продукты конденсации указанных неполных сложных эфиров с этиленоксидом, например полиоксиэтиленсорбитанмоноолеат. Эти эмульсии могут также содержать подслащивающие и ароматизирующие агенты. Сиропы и эликсиры могут быть изготовлены с использованием подслащивающих агентов,например, глицерина, пропиленгликоля, сорбита или сахарозы. Такие препараты могут также содержать средство, смягчающее раздражение; консервант; отдушку; и краситель. Фармацевтические композиции могут быть изготовлены в форме стерильных водных или масляных суспензий для инъекций. Эти суспензии могут быть получены стандартными методами с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов,которые были упомянуты выше. Стерильный препарат для инъекций может быть также получен в виде стерильного инъецируемого раствора или суспензии в нетоксичном парентерально приемлемом разбавителе или растворителе, например в виде раствора в 1,3-бутандиоле. Из приемлемых наполнителей и растворителей могут быть использованы вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя и суспендирующей среды обычно используют стерильные жирные масла. В этих целях может быть ис 27 пользовано любое мягкое жирное масло, включая синтетические моно- или диглицериды. Помимо этого,для получения препаратов для инъекций используются также жирные кислоты,такие как, олеиновая кислота. Соединения формулы I могут быть также изготовлены в форме суппозиториев для ректального введения лекарственного средства. Эти композиции могут быть получены путем смешивания лекарственного средства с подходящим не раздражающим наполнителем, который является твердым при обычных температурах, и жидким при температуре прямой кишки, а поэтому при попадании в прямую кишку он расплавляется с высвобождением лекарственного средства. Такими веществами являются какаомасло и полиэтиленгликоли. Для местного применения используют кремы, мази, гели, растворы или суспензии, и т.п., содержащие соединения формулы I. (В этих целях препаратами для наружного применения могут быть растворы для полоскания рта и горла). Соединения настоящего изобретения могут быть введены через нос путем использования носителей, подходящих для интраназального введения, либо они могут быть введены чрескожно с использованием пластырей для наложения на кожу, хорошо известных каждому специалисту. При введении препарата, изготовленного в форме системы для чрескожной доставки лекарственного средства, введение дозы в процессе лечения будет, разумеется, постоянным, а не периодическим. Соединения настоящего изобретения могут быть также введены в виде суппозитория, изготовленного на соответствующей основе, такой как масло-какао, глицеринированный желатин, гидрогенизированные растительные масла, смеси полиэтиленгликолей различной молекулярной массы со сложными эфирами жирных кислот и полиэтиленглиголя. Схему введения с использованием соединений настоящего изобретения выбирают в зависимости от ряда факторов, включая тип, вид,возраст, вес, пол и состояние здоровья пациента; тяжесть заболевания, подвергаемого лечению; способ введения; функцию почек и печени пациента; и конкретно используемое соединение. Любой врач или ветеринар может легко определить и прописать эффективное количество лекарственного средства, необходимое для предупреждения, подавления или прекращения прогрессирования заболевания или обратного развития заболевания. Для оптимальной оценки пределов нужной концентрации лекарственного средства, в целях определения схемы лечения,дающего желаемый эффект без токсического действия, необходимо учитывать кинетику доступности лекарственного средства для данного участка организма пациента. Для этого необходимо принимать в расчет распределение, равно 000829 28 весие и выведение лекарственного средства. Предпочтительные дозы соединения структурной формулы I, используемые в способе настоящего изобретения, составляют в пределах от 0,01 до 1000 мг в день для взрослого индивидуума. Наиболее предпочтительно, если эти дозы составляют от 0,1 до 500 мг/день. Композиции для перорального введения обычно изготавливают в форме таблеток, содержащих от 0,01 до 1000 миллиграммов активного ингредиента, а в частности 0,01; 0,05; 0,1; 0,5; 1,0; 2,5; 5,0; 10,0; 15,0; 25,0; 50,0; 100 и 500 миллиграммов активного ингредиента для симптоматической оценки дозы для пациента, подвергаемого лечению. Эффективное количество лекарственного средства обычно достигается при уровнях доз, составляющих от около 0,0002 мг/кг до около 50 мг/кг веса тела пациента в день. А более конкретно, этот интервал составляет от около 0,001 мг/кг до 1 мг/кг веса тела в день. Преимущественно, активный агент настоящего изобретения может быть введен в виде одноразовой суточной дозы, либо эта суточная доза может быть введена в виде дробных доз два, три или четыре раза в день. Количество активного ингредиента, которое может быть объединено с материалами носителя для получения унифицированной лекарственной формы, варьируется в зависимости от индивидуума, подвергаемого лечению, и от способа введения лекарственного средства. Следует отметить, однако, что конкретный уровень дозы для каждого конкретного пациента будет зависеть от ряда факторов, включая возраст, вес тела, общее состояние здоровья,пол, режим питания, время введения, способ введения, скорость выведения лекарственного средства из организма, комбинации лекарственных средств и тяжесть конкретного заболевания, подвергаемого терапевтическому лечению. Нижеследующие примеры иллюстрируют получение некоторых соединений настоящего изобретения, однако, они не должны рассматриваться как некое ограничение изобретения. Пример 1. 1-[2-[2-(3,4-Диметоксифенил)1H-индол-3-ил]этиламино]-3-(пиридин-4 илокси)пропан-2-ол К раствору 2-[2-(3,4-диметоксифенил)-1Hиндол-3-ил]этиламина (118 мг в 4 мл сухого метанола) добавляли 20 мг 4-оксиранилметоксипиридина и смесь нагревали до 80 С на масляной бане. Через 6 ч смесь концентрировали в вакууме и очищали с помощью флешхроматографии на силикагеле (метиленхлорид : метанол : гидроксид аммония, 90:10:1) с получением целевого соединения (53 мг). m/е = 448(М+Н). Нижеследующие соединения получали способом, аналогичным описанному в примере 1. Стадия 2.1 А. 2-[2-(1H-индол-3-ил)этил]изоиндол-1,3-дион. К перемешанной суспензии 2-(1H-индол-3 ил)этиламина (2,0 г в 20 мл сухого тетрагидрофурана) добавляли N-карбэтоксифталимид (2,85 г) и смесь нагревали с обратным холодильником на масляной бане. Через 48 ч реакционную смесь охлаждали до комнатной температуры,фильтровали и фильтрат концентрировали в вакууме. Полученное твердое вещество суспендировали в смеси гексан/метиленхлорид (2,5:1) и фильтровали. Собранные твердые вещества очищали с помощью флеш-хроматографии (метиленхлорид:метанол 97:3) и получали целевое соединение (3,1 г). Стадия 2.1 В. 2-[2-(2-Бром-1 Н-индол-3-ил) этил]-изоиндол-1,3-дион. К раствору 2-[2-(1H-индол-3-ил)-этил]изоиндол-1,3-диона (1,0 г в смеси 10 мл сухого тетрагидрофурана и 10 мл сухого хлороформа) при 0 С добавляли пербромид бромида пиридиния (1,14 г) и реакционную смесь перемешивали при 0 С. Через 50 мин реакцию гасили путем добавления насыщенного бикарбоната натрия и экстрагировали этилацетатом. Органическую часть промывали насыщенным бикарбонатом натрия (3 х) и 0,3 М бисульфатом натрия (3 х), а затем сушили сульфатом магния. Концентрат очищали с помощью флеш-хроматографии на силикагеле (гексан:этилацетат 3:1) и получали целевое соединение (1,2 г). 30 Стадия 2.1 С. 2-2-[2-(3,5-Диметилфенил)1H-индол-3-ил] этил-изоиндол-1,3-дион. К раствору 2-[2-(2-бром-1H-индол-3-ил)этил]-изоиндол-1,3-диона (150 мг в смеси 5 мл толуола и 5 мл этанола) добавляли 3,5 диметилфенилбороновую кислоту (85 мг), а затем 1,0 мл 1 М карбоната натрия. К перемешанному раствору добавляли хлорид лития (60 мг),а затем тетракис(трифенилфосфин)палладий (28 мг) и смесь нагревали с обратным холодильником на масляной бане. Через 4 ч смесь охлаждали до комнатной температуры и концентрировали в вакууме, после очистки с помощью флешхроматографии на силикагеле(гексан:этилацетат 5:1) получали целевое соединение (146 мг). Стадия 2.1D 2-[2-(3,5-Диметилфенил)-1Hиндол-3-ил] этиламин. К раствору 2-2-[2-(3,5-диметилфенил)1H-индол-3-ил]этил-изоиндол-1,3-диона (87 мг в смеси 4 мл тетрагидрофурана и 4 мл этанола) добавляли 0,6 мл 95% водного раствора гидразина и реакционную смесь перемешивали при комнатной температуре. Через 18 ч смесь концентрировали в вакууме и очищали с помощью флеш-хроматографии на силикагеле (метиленхлорид : метанол : гидроксид аммония 9:6:1) с получением целевого соединения (54 мг). Стадия 2.1E 2-[2-(3,5-Диметилфенил)-1Hиндол-3-ил]этил-(5-пиридин-4-илпентил)амин. К раствору 2-[2-(3,5-диметилфенил)-1Hиндол-3-ил]этиламина (162 мг) и 5-(4-пиридил) пентаналя (100 мг в 2 мл сухого хлороформа) добавляли безводный сульфат магния (735 мг) и смесь перемешивали в течение 15 мин при 0 С. По истечении этого времени добавляли борогидрид натрия (92,7 мг), а затем 3 мл сухого метанола и смесь перемешивали при 0 С. Через 1 ч реакцию гасили путем выливания в воду (25 мл), а затем перемешивали в течение 30 мин и экстрагировали метиленхлоридом (4 х 25 мл). Объединенные органические экстракты сушили карбонатом калия, концентрировали в вакууме и остаток очищали с помощью флешхроматографии на силикагеле (метиленхлорид : метанол 95:5) с получением целевого соединения (172 мг). m/e = 412 (М+Н). Получение синтетических промежуточных соединений Стадия А. 5-(4-пиридил)-4-пентин-1-ол. Гидрохлоридную соль 4-бромпиридина(3,89 г) растворяли в смеси растворителей, содержащей триэтиламин (25 мл) и воду (5 мл). К соли пиридина добавляли безводный хлорид лития (848 мг), порошок бромида меди (1) (30 мг) и 5-пент-4-ин-1-ол (1,68 г), смесь перемешивали и раствор примерно в течение 15 мин слегка продували потоком активного газообразного азота,после чего добавляли тетракис(трифенилфосфин)палладий (231,1 мг). Реакционную смесь нагревали с обратным холодильником в атмосфере азота и выдерживали 31 при нагревании с обратным холодильником в течение 2,5 ч, после чего нагревание прекращали и реакционную смесь оставляли при комнатной температуре. Продукты реакции выделяли путем распределения между эфиром и рассолом. Водный слой экстрагировали эфиром (4 х 50 мл) и объединенные экстракты сушили порошкообразным безводным сульфатом натрия. Экстракт фильтровали и упаривали при пониженном давлении, в результате чего образовывалось темнокоричневое масло. Полученное таким образом коричневое масло подвергали колоночной хроматографии с использованием чистого этилацетата в качестве элюента, в результате чего получали желтое масло (2,1 г), а именно, 5-(4 пиридил)-4-пентин-1-ол, который медленно отверждался и слегка темнел при отстаивании при комнатной температуре. Стадия В. 5-(4-пиридил)-пентан-1-ол. 5-(4-пиридил)-4-пентин-1-ол (1,5 г), полученный в предыдущей стадии, растворяли в метаноле (35 мл) в сосуде для гидрогенизации Парра и добавляли окись платины (IV) [катализатор Адамса] (0,3 г). Сосуд Парра помещали в аппарат для гидрогенизации Парра и раствор гидрировали в течение 5,5 ч при 40 фунт/кв.дюйм (2,812 кг/см 2), после чего исходное соединение было израсходовано, на что указывала ТСХ. Отработанный катализатор удаляли путем фильтрации через слой Целита и этот слой еще раз осторожно промывали метанолом. Объединенные фильтраты упаривали при пониженном давлении на роторном испарителе, а затем маслянистые остатки подвергали колоночной хроматографии на короткой колонке с двуокисью кремния с использованием в качестве элюента чистого этилацетата, в результате чего получали целевое соединение (1,4 г). Стадия С. 5-(4-Пиридил)-пентаналь. Оксалилхлорид (2 мл 2 М раствора в сухом метиленхлориде) помещали в холодную осушенную в печи колбу и охлаждали до -78 С с использованием охлаждающей бани из сухого льда и ацетона, а затем к этому оксалилхлориду в течение 3 мин по одной капле добавляли раствор ДМСО (632 мг) в сухом метиленхлориде (1 мл) и перемешивали еще 3 мин. В реакционную колбу приблизительно в течение 3 мин добавляли раствор [5-(4-пиридил)-4-пентан-1-ола (0,6 г) в сухом метиленхлориде (5 мл) и эту реакционную смесь перемешивали в течение 15 мин. После добавления безводного триэтиламина (2,82 мл) реакционную смесь перемешивали еще 2 ч,и в течение этого времени холодная баня нагревалась до комнатной температуры. Реакционную смесь гасили путем добавления насыщенного солевого раствора, а затем распределяли с метиленхлоридом. Водный слой отбрасывали, а метиленхлоридный экстракт сушили безводным порошком сульфата натрия, фильтровали и упаривали при пониженном давлении, в результате чего образовывался маслянистый остаток. Про 000829 32 дукт (488 мг) выделяли путем колоночной хроматографии на силикагеле с использованием этилацетата в качестве элюента. Пример 2.2. 2-[2-(3,4-Диметоксифенил)-1H-индол-3 ил]этил-[3-(1H-индол-5-ил)пропил]амин. Стадия 2.2 А. N-2-[2-(3,4-Диметоксифенил)-1H-индол-3-ил]этил-3-(1 Н-индол-5-ил) пропионамид. К раствору 3-(1 Н-индол-5-ил)пропионовой кислоты (50 мг в 2,5 мл N,N-диметилформамида) при 0 С добавляли 43 мг 1 гидроксибензотриазола, а затем 71 мг гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида и смесь нагревали до комнатной температуры. Через 23 мин добавляли 155 мг 2-[2(3,4-диметоксифенил)-1H-индол-3-ил]этиламина и смесь перемешивали при комнатной температуре в течение еще 1 ч. Затем реакцию гасили путем добавления воды и смесь экстрагировали этилацетатом. Объединенные органические экстракты промывали водой и солевым раствором,сушили сульфатом натрия и концентрированное масло очищали с помощью флешхроматографии на силикагеле (метиленхлорид : метанол 95:5 с получением целевого соединения (111 мг). Стадия 2.2 В. 2-[2-(3,4-Диметоксифенил)1H-индол-3-ил]этил-[3-(1H-индол-5-ил)пропил]амин. К раствору N-2-[2-(3,4-диметоксифенил)1H-индол-3-ил]этил-3-(1 Н-индол-5-ил)пропионамида (60 мг в 2,0 мл сухого тетрагидрофурана) при 0 С добавляли 26 мг алюмогидрида лития и смесь нагревали до 77 С на масляной бане. Через 5,5 ч смесь охлаждали до 0 С, гасили путем добавления 0,025 мл воды, интенсивно перемешивали в течение 30 мин, после чего суспензию фильтровали через слой сульфата натрия, а фильтрат концентрировали в вакууме. После очистки с помощью флешхроматографии (метиленхлорид : метанол, 92:8) получали целевое соединение (32 мг). m/е = 454(М+Н). Нижеследующие соединения получали способом, аналогичным описанному в примерах 2.1 и 2.2. Пример 3. [3-(3 Н-Бензимидазол-5-ил)аллил]-2-[2-(3,4-диметоксифенил) -1H-индолЗ-ил]этиламин. Стадия 3 А. 2-[2-(3,4-Диметоксифенил)1H-индол-3-ил]-этил-[3-(1-метансульфонил 1 Н-бензоимидазол-5-ил)-аллил] амин. К раствору 3-(1 Н-бензоимидазол-5 ил)проп-2-ен-1-ола (44 мг в смеси 2 мл метиленхлорида и 0,1 мл N,N-диметилформамида) при 0 С последовательно добавляли 346 мг бромида тетрабутиламмония, 0,110 мл диизопропилэтиламина и 100 мг метансульфонового ангидрида и смесь нагревали до комнатной температуры. Через 1 ч смесь добавляли к раствору 2-[2-(3,4-диметоксифенил)-1H-индол-3-ил)этиламина (300 мг в смеси 6 мл метиленхлорида и 1,5 мл N,N-диметилформамида) и перемешивание продолжали в течение 2,5 ч. После этого смесь концентрировали в вакууме и остаток 34 очищали с помощью флеш-хроматографии на силикагеле (метиленхлорид:метанол, 91:9) с получением целевого соединения (24 мг). Стадия 3 В. [3-(3 Н-Бензоимидазол-5-ил)аллил-2-[2-(3,4-диметоксифенил)-1H-индол-3 ил]-этил амин. К раствору 2-[2-(3,4-диметоксифенил)1H-индол-3-ил]этил-[3-(1-метансульфонил-1 Нбензоимидазол-5-ил)-аллил]амина (24 мг в 1,5 мл метанола) при 0 С добавляли 0,225 мл 2 н раствора гидроксида калия и смесь нагревали до комнатной температуры. Через 1,5 ч реакцию гасили путем добавления 0,30 мл 1 н хлористоводородной кислоты, концентрировали в вакууме и очищали с помощью флеш-хроматографии на силикагеле (метиленхлорид : метанол 88:12) с получением целевого соединения (12 мг). m/е= 453 (М+Н). Получение синтетических промежуточных соединений Стадия А. N,N-Метоксиметиламид 1 Нбензоимидазол-5-карбоновой кислоты. К суспензии 1 Н-бензоимидазол-5 карбоновой кислоты (500 мг в 7 мл N,Nдиметилформамида) при 0 С добавляли 500 мг 1-гидроксибензотриазола (HOBt), а затем 828 мг гидрохлорида 1-(3-диметиламинопропил)-3 этилкарбодиимида (EDC) и смесь нагревали до комнатной температуры. Через 42 мин добавляли 1,05 г гидрохлоридаN,Oдиметилгидроксиламина и 1,5 мл триэтиламина и перемешивание продолжали при комнатной температуре. Через 1 ч реакцию гасили путем добавления воды и продукт выделяли путем экстракции этилацетатом. Концентрат очищали с помощью флеш-хроматографии на силикагеле(метиленхлорид:метанол 90:10) и получали целевое соединение (458 мг). Стадия В. 1 Н-бензоимидазол-5-карбоксальдегид. К раствору N,N-метоксиметиламида 1 Нбензоимидазол-5-карбоновой кислоты (458 мг в смеси 5 мл диэтилового эфира и 10 мл тетрагидрофурана) при -78 С добавляли 3,7 мл 1,5 М раствора гидрида диизобутилаллюминия в толуоле и смесь перемешивали при низкой температуре. Через 1 ч реакционную смесь через канюлю вводили в 1 М раствор тартрата калия-натрия при 0 С. Полученную суспензию интенсивно перемешивали в течение 2 ч при комнатной температуре, после чего экстрагировали этилацетатом. Органические экстракты промывали водой, сушили сульфатом натрия и концентрировали в вакууме, в результате чего получали неочищенное целевое соединение (250 мг). Стадия С. Метиловый эфир 3-(1 Нбензоимидазол-5-ил)-акриловой кислоты. К раствору 1 Н-бензоимидазол-5 карбоксальдегида (250 мг в 12 мл сухого тетрагидрофурана) при 0 С добавляли 1,43 г метил(трифенилфосфоранилиден)ацетата и смесь оставляли для нагревания до комнатной темпе 35 ратуры. Через 24 ч смесь концентрировали в вакууме и очищали с помощью флешхроматографии на силикагеле (метиленхлорид: метанол 93:7) с получением целевого соединения (257 мг). Стадия D. 3-(1 Н-бензоимидазол-5-ил)проп-2-ен-1-ол. К раствору метилового эфира 3-(1 Нбензоимидазол-5-ил)акриловой кислоты (50 мг в 1 мл сухого тетрагидрофурана) при температуре-78 С добавляли 1,24 мл 1 М раствора три-вторбутилборогидрида лития в тетрагидрофуране и смесь перемешивали при -78 С в течение 4 ч, а затем нагревали до -40 С еще 4 ч. Реакцию гасили путем добавления водного раствора метанола, концентрировали в вакууме и очищали с помощью флеш-хроматографии на силикагеле(метиленхлорид:метанол 87:13) с получением целевого соединения (44 мг). Нижеследующее соединение получали способом, аналогичным описанному выше.[2-[2-(3,5-Диметилфенил)-5-метансульфонил-1 Н-индол-3-ил]этил]-(4-пиридин-4-илбутил)амин. Стадия 4.1 А. 2-[2-(3,5-Диметилфенил)-5 метансульфонил-1 Н-индол-3-ил]этиламин. Суспензию 4-(метансульфонил)фенилгидразина (3,0 г, 16,1 ммоль) и 3-хлорпропил 3,5 диметилфенилкетона (3,4 г, 16,1 ммоль) в третбутаноле (25 мл) перемешивали при комнатной температуре в течение 20 мин и быстро нагревали на паровой бане. После добавления метанола (250 мл) смесь нагревали с обратным холодильником в течение ночи. После этого смесь концентрировали до небольшого объема и кристаллы отфильтровывали и промывали холодным метанолом. Объединенные фильтраты упаривали, в результате чего образовывался остаток, который распределяли между этиловым эфиром и водой. Затем эфирный слой экстраги 36 ровали водой (2 х), объединенные водные экстракты промывали этиловым эфиром и подщелачивали 5 н гидроксидом натрия. Продукт экстрагировали этилацетатом (3 х). Объединенные органические экстракты промывали водой, сушили сульфатом натрия и упаривали, в результате чего образовывался сироп (3,1 г). Неочищенный продукт очищали с помощью флешхроматографии на силикагеле (метиленхлорид:метанол, 9:1) и получали целевое соединение (1,99 г, 36%), Rf = 0,19 (нингидринположительная реакция). Стадия 4.1 В. N-[2-[2-(3,5-Диметилфенил)5-метансульфонил-1 Н-индол-3-ил]этил]-4 пиридин-4-ил-бутирамид. Смесь 2-[2-(3,5-диметилфенил)-5-метансульфонил-1 Н-индол-3-ил]этиламина (1,29 г 3,77 ммоль), 4-(4-пиридил)бутановой кислоты(10 мл) перемешивали при комнатной температуре в течение 14 ч. После этого раствор подвергали совместной дистилляции с н-бутанолом(метиленхлорид : метанол, 97:3) и получали целевое соединение (1,45 г, 79%). Стадия 4.1 С. [2-[2-(3,5-Диметилфенил)-5 метансульфонил-1 Н-индол-3-ил]этил]-(4 пиридин-4-ил-бутил)амин. К раствору N-[2-[2-(3,5-диметилфенил)-5 метансульфонил-1H-индол-3-ил]этил]-4 пиридин-4-ил-бутирамида (1,45 г, 2,96 ммоль) в сухом тетрагидрофуране (20 мл) по каплям добавляли боран-ТГФ (1,0 М раствор в тетрагидрофуране, 27 мл) и раствор нагревали с обратным холодильником в течение 2 ч. После этого добавляли метанол для гашения избытка борана и полученный раствор упаривали досуха. Остаток растворяли в тетрагидрофуране (20 мл) и добавляли N,N-диметилэтаноламин (8,9 мл,88,86 ммоль). Раствор нагревали с обратным холодильником в течение 2,5 ч, а затем концентрировали досуха. Неочищенный продукт очищали с помощью флеш-хроматографии на силикагеле (метиленхлорид : метанол, 95:5) и получали целевое соединение (1,0 г, 71%). m/е = 476(М+Н). Стадия 4.1D. 4-[2-[2-(3,5-Диметилфенил)5-метансульфонил-1 Н-индол-3-ил]этил]-(4 пиридин-4-ил-бутил)амин (дигидрохлорид). К раствору [2-[2-(3,5-диметилфенил)-5 метансульфонил-1 Н-индол-3-ил]этил]-(4-пиридин-4-ил-бутил)амина (300 мг, 0,63 ммоль) в сухом тетрагидрофуране (20 мл) при 0 С по каплям добавляли хлористый водород в этиловом эфире (1,0 М, 1,4 мл). Через 10 мин избыток хлористого водорода и растворители выпаривали с получением твердой массы. Затем продукт 37 сушили в высоком вакууме при 50 С и получали целевое соединение. Пример 4.2. 3-[2-(3,5-Диметилфенил)-3-[2-(5-пиридин 4-ил-пентиламино)этил]-1 Н-индол-5-ил]-1,1 диметилмочевина. Стадия 4.2 А. 2-[2-(3,5-Диметилфенил)-5 нитро-1 Н-индол-3-ил]этиламина гидрохлоридная соль. 1-(4-Хлор-1-оксо-бутил)-3,5-диметилбензол (2,5 г) растворяли в 12 мл трет-бутанола,перемешивали при комнатной температуре, а затем добавляли 4-нитрофенилгидразин (1,65 г). Реакционную смесь перемешивали при комнатной температуре в течение 20 мин, а затем добавляли воду (12 мл) и метанол (108 мл), нагревали с обратным холодильником и выдерживали при нагревании с обратным холодильником в течение 17 ч. Реакционную смесь охлаждали до комнатной температуры, а летучие растворители удаляли при пониженном давлении на роторном испарителе. Остатки упаривали досуха с использованием потока активного газообразного азота в течение ночи. Сухие твердые остатки растирали с этилацетатом (примерно 100 мл),что инициировало кристаллизацию в результате механического царапанья, после чего образец помещали в холодильник на 8 ч. Полученное таким образом твердое вещество собирали в воронке из спеченного стекла под вакуумом и твердый продукт промывали безводным этилацетатом. Было получено твердое вещество с выходом 1,4 г. Стадия 4.2 В. Трет-бутиловый эфир 2-[2(3,5-диметилфенил)-5-нитро-1 Н-индол-3 ил]этилкарбаминовой кислоты. Гидрохлоридную соль 2-[2-(3,5 диметилфенил)-5-нитро-1 Н-индол-3-ил]этиламина (1 г) суспендировали в сухом метиленхлориде (25 мл), добавляли сухой триэтиламин(0,806 мл) и перемешивали в течение 5 мин при комнатной температуре. К триптамину в течение 5 мин небольшими порциями добавляли [2(трет-бутоксикарбонилоксиимино) фенилацетонитрил] (890 мг) и реакционную смесь оставляли для перемешивания в течение ночи при комнатной температуре. Реакционную смесь распределяли между метиленхлоридом и 5% лимонной кислотой. Метиленхлоридный слой отделяли, промывали солевым раствором и сушили порошкообразным безводным сульфатом натрия. Экстракт фильтровали и упаривали досуха. Продукт выделяли с помощью колоночной хроматографии на силикагеле, используя в качестве элюента этилацетат и гексан (35:65, 000829 38 об./об.). Выход целевого соединения составил 1 г. Стадия 4.2 С. Трет-бутиловый эфир 2- [5 амино-2- (3, 5-диметилфенил)-1H-индол-3-ил] этил карбаминовой кислоты. Трет-бутиловый эфир 2-[2-(3,5 диметилфенил)-5-нитро-1 Н-индол-3-ил]этил карбаминовой кислоты (0,4 г) растворяли в метаноле (25 мл) и добавляли окись платины (IV)(40 мг). Смесь помещали в гидрогенизатор Парра и гидрировали при 45 фунт/кв.дюйм (2,812 кг/см 2) в течение 4 ч; к этому времени исходное соединение было израсходовано и превращено в один продукт. Катализатор удаляли путем фильтрации, фильтраты упаривали при пониженном давлении на роторном испарителе и конечные следовые количества растворителя удаляли в высоком вакууме в течение ночи. Был получен амин (338 мг) в виде порошка. Стадия 4.2D. 2-[2-(3,5-Диметилфенил)-5(3,3-диметилуреидо)-1H-индол-3-ил]этилкарбаминовой кислоты трет-бутиловый эфир. Тонко измельченный трет-бутиловый эфир 2-[5-амино-2-(3,5-диметилфенил)-1H-индол-3 ил]-этил-карбаминовой кислоты (265 мг) суспендировали в сухом тетрагидрофуране (5 мл), а затем добавляли диизопропилэтиламин (0,146 мл) и диметилкарбамоилхлорид (0,077 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2,5 дней. Эту реакционную смесь концентрировали на роторном испарителе, остатки наносили на четыре (20 х 20 см,1000 микрон) препаративные пластины с силикагелем и элюировали с использованием системы растворителей, содержащей метанол и метиленхлорид в соотношении 1:9, об./об. Был выделен продукт (299 мг) в виде густой смолы, который отверждался при продолжительном отстаивании. Стадия 4.2 Е. 3-[3-(2-Аминоэтил)-2-(3,5 диметилфенил)-1H-индол-5-ил]-1,1-диметилмочевина. Трет-бутиловый эфир 2-[2-(3,5 диметилфенил)-5-(3,3-диметилуреидо)-1Hиндол-3-ил]-этил-карбаминовой кислоты (295 мг) растворяли в трехкомпонентной смеси растворителей, содержащей метиленхлорид (6 мл),трифторуксусную кислоту (2 мл) и анизол (2 мл), в течение 2,5 ч. Компоненты летучего растворителя удаляли на роторном испарителе и остатки растворяли в метаноле и очищали на четырех (20 х 20 см, 1000 микрон) препаративных пластинах с силикагелем, используя в качестве элюента систему растворителей, содержащую метанол и метиленхлорид (1:9, об./об.). Был выделен продукт (222 мг) в виде пенистого вещества. Стадия 4.2F. 3-[2-[3,5-Диметилфенил)-3-[2(5-пиридин-4-ил-пентиламино) этил]-1 Н-индол 5-ил]-1,1-диметилмочевина. 3-[3-(2-Аминоэтил)-2-(3,5-диметилфенил)1 Н-индол-5-ил]-1,1-диметилмочевину (25 мг), 39 полученную в предыдущей стадии, растворяли в тетрагидрофуране (1 мл) и дейтерохлороформе(1 мл) приблизительно при 0 С, а затем добавляли 5-(4-пиридил) пентаналь (10,6 мг). Реакционную смесь поддерживали при этой температуре в течение 15 мин, после чего добавляли тонко измельченный в порошок борогидрид натрия, а затем безводный метанол. Реакционную смесь перемешивали в течение 15 мин приблизительно при 0 С, а затем гасили путем добавления 20 капель 2 н хлористоводородной кислоты и воды (1 мл). Летучие вещества удаляли на роторном испарителе, а воду с использованием активного потока газообразного азота. Остатки помещали на четыре (20 х 20 см, 500 микрон) препаративные пластины с силикагелем и элюировали системой растворителей, включающей метанол и метиленхлорид (1:9, об./об.). Таким образом выделяли продукт (14,2 мг) в виде густого масла. Нижеследующие соединения получали способом, аналогичным описанному в примерах 4.1 и 4.2.(50 ммоль) 4 гидразинбензойной кислоты, 10,55 г (50 ммоль) 3-хлорпропил 3,5-диметилфенилкетона и 200 мл абсолютного этанола перемешивали в атмосфере азота и нагревали с обратным холодильником. Через 12 ч смесь охлаждали и фильтровали. Твердый осадок на фильтре промывали дополнительными небольшими объемами этанола. Фильтрат обрабатывали 4 мл концентрированной серной кислоты и перемешивали при нагревании с обратным холодильником в атмосфере азота в течение 4 дней. Охлажденную смесь перемешивали в ледяной бане при добавлении по каплям раствора этоксида натрия (21 мас.% в этаноле) до тех пор, пока смесь не становилась щелочной, на что указывала индикаторная бумага для определения рН. Смесь фильтровали и концентрировали в вакууме при 30 С. Остаток распределяли между диэтиловым эфиром и водой с добавлением определенного количества насыщенного водного раствора хлорида натрия для облегчения разделения слоев. Водную фазу промывали еще 100 мл эфира. Объединенные органические экстракты сушили сульфатом натрия, фильтровали и концентрировали в вакууме. Остаточную смолу очищали с помощью флеш-хроматографии на силикагеле (элюент:метиленхлорид:метанол:гидроксид аммония, 97:3:0,3, а затем 95:5:0,5) и получали целевое соединение (4,8 г). 1 Н-ЯМР (400 МГц,CDCl3) соответствовал предполагаемой структуре. Масс-спектр (РВ-NН 3/Сl): m/е = 337(М+Н). Стадия 5.1 В. Этиловый эфир 2-(3,5 диметилфенил)-3-[2-[4-(пиридин-4-ил)бутиламино] этил]-1 Н-индол-5-карбоновой кислоты. В сухую колбу добавляли 5,0 г (14,9 ммоль) этилового эфира 3-(2-аминоэтил)-2-(3,5 диметилфенил)-1 Н-индол-5-карбоновой кислоты, 1,98 г (13,5 ммоль) 4-(пиридин-4 ил)бутиральдегида (полученного, в основном,как описано в примере 2.1, разбавленного 0,5 мл СDСl3) и 8,12 г (67,7 ммоль) безводного сульфата магния и вводили магнитный стержень для перемешивания. Колбу продували азотом, охлаждали до -10 С и перемешивали при постепенном добавлении шприцем 11,5 мл сухогоCDCl3. Смесь перемешивали в атмосфере азота приблизительно в течение 20 мин. Затем перегородку удаляли и быстро добавляли 670 мг(17,6 ммоль) борогидрида натрия. После этого перегородку сразу же устанавливали и систему снова продували азотом. Смесь перемешивали в атмосфере азота при температуре около -5 С при постепенном добавлении шприцем 10 мл сухого метанола. После выдерживания в течение нескольких минут при этой температуре,реакционную смесь удаляли из охлаждающей бани и распределяли между 80 мл этилацетата и 100 мл воды. Органический слой сушили сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали с помощью флешхроматографии на силикагеле (элюирование градиентом 4-9% метанола в метиленхлориде; повторное элюирование с использованием 515% метанола в метиленхлориде) и получали целевое соединение (3,19 г). 1H-ЯМР (500 МГц,CDCl3) соответствовал предполагаемой структуре. Масс-спектр (РВ-NН 3/Сl): m/е = 470,4(М+Н). Было также выделено еще 1,91 г менее чистого продукта. Стадия 5.1 С. 3-[2-[Бензилоксикарбонил-[4(пиридин-4-ил)бутил]амино]этил]-2-(3,5-диметилфенил)-1 Н-индол-5-карбоновой кислоты сложный этиловый эфир. Раствор 3,19 г (6,83 ммоль) сложного этилового эфира 2-(3,5-диметилфенил)-3-[2-[4(пиридин-4-ил)бутиламино] этил]-1 Н-индол-5 карбоновой кислоты в 25 мл сухого метиленхлорида перемешивали в атмосфере азота и охлаждали до -78 С в бане из сухого льда и ацетона, добавляя при этом 2,38 мл (1,76 г, 13,7 ммоль) N,N-диизопропилэтиламина, с последующим постепенным добавлением порциями 3,4 мл (4,06 г, 23,7 ммоль) бензилхлороформата с помощью шприца. Через 2,5 ч раствор удаляли из охлаждающей бани и оставляли нагреваться до комнатной температуры. После этого раствор распределяли между этилацетатом и 5%-ным водным раствором бисульфата калия. Органическую фазу осушали сульфатом магния, фильтровали и концентрировали в вакууме. После очистки с помощью флеш-хроматографии на силикагеле (элюировали градиентом 0,5-10% метанола в метиленхлориде), получали количественный выход продукта в виде желтой пены. 1 Н-ЯМР (500 МГц) был комплексным из-за присутствия ротамеров, но соответствовал предполагаемой структуре. Масс-спектр (РВ-NН 3/Сl): 42 ремешивали при около 60 С, постепенно добавляя 19 мл воды. Перемешивание продолжали с обратным холодильником в течение ночи. Охлажденную смесь концентрировали в вакууме и получали желтое твердое вещество, которое распределяли между 250 мл смеси этилацетаттетрагидрофуран (1:1) и 250 мл 0,5 н соляной кислоты. Органическую фазу два раза промывали 0,5 н соляной кислотой, а затем сушили сульфатом магния и концентрировали в вакууме. Полученное твердое вещество растирали с диэтиловым эфиром и собирали на фильтре, в результате чего получали (после осушки) 3,46 г желтого твердого вещества, т.пл. 133,5-137,5 С. Гомогенность была подтверждена с помощью ТСХ (95:5:0,5 CH2Cl2-MeOH-AcOH). 1 Н-ЯМР= 576,4 (М+Н). Стадия 5.1E. [2-[5-Диэтилкарбамоил-2(3,5-диметилфенил)-1 Н-индол-3-ил]этил]-[4(пири-дин-4-ил)бутил] карбаминовой кислоты бензиловый эфир. Смесь 846 мг (1,38 ммоль) гидрохлорида 3[2-[бензилоксикарбонил-[4-(пиридин-4-ил) бутил]амино]этил]-2-(3,5-диметилфенил)-1 Ниндол-5-карбоновой кислоты, 862 мг (1,66 ммоль) гексафторфосфата бензотриазол-1 илокси-трис(пирролидино)фосфония (РуВОР) и 8,5 мл безводного метиленхлорида обрабатывали 1,16 мл (839 мг, 8,29 ммоль) триэтиламина, а затем, через несколько минут 0,715 мл (505 мг,6,91 ммоль) диэтиламина. Полученный раствор перемешивали в атмосфере азота при комнатной температуре в течение ночи, а затем распределяли между этилацетатом и насыщенным раствором бикарбоната натрия. Органический слой сушили сульфатом натрия и концентрировали в вакууме. После очистки с помощью флешхроматографии на силикагеле (элюировали градиентом 1-5% метанола в метиленхлориде), получали количественный выход нужного продукта. Гомогенность была подтверждена с помощью ТСХ в 95:5 CH2Cl2-MeOH. 1H-ЯМР 500 МГц являлся комплексным из-за присутствия ротамеров, но соответствовал предполагаемой структуре. Масс-спектр (РВ-NН 3/Сl): m/е = 631,5 (М+Н). Стадия 5.1F. 2-(3,5-Диметилфенил)-3-[2-[4(пиридин-4-ил)бутиламино]этил]-1 Н-индол-5 карбоновой кислоты диэтиламид. Смесь 871 мг (1,38 ммоль) бензилового эфира[2-[5-диэтилкарбамоил-2-(3,5-диметилфенил)-1H-индол-3-ил]этил]-[4-(пиридин-4-ил) бутил]карбаминовой кислоты, 300 мг 20% гидроксида палладия на угле и 40 мл 2 метоксиэтанола перемешивали встряхиванием в присутствии водорода (45 фунт,кв.дюйм = 3,164 кг/см 2) в автоклаве в течение 2,3 ч. Катализатор удаляли путем фильтрации через диатомовую землю и фильтрат концентрировали в вакууме. После очистки остатка с помощью 43 флеш-хроматографии на силикагеле (элюировали градиентом из 99:1:0,1-93:7:0,7 СН 2 Сl2 МеОН-конц. NH4OH) получали количественный выход продукта в виде желтой пены; гомогенность была подтверждена с помощью ТСХm/e = 497,5 (М+Н). Стадия 5.1G. 2-(3,5-Диметилфенил)-3-[2[4-(пиридин-4-ил) бутиламино]этил]-1 Н-индол 5-карбоновой кислоты диэтиламид дигидрохлорид. Раствор 452 мг (0,914 ммоль) диэтиламида 2-(3,5-диметилфенил)-3-[2-[4-(пиридин-4-ил) бутиламино]этил]-1 Н-индол-5-карбоновой кислоты в 45 мл метанола обрабатывали 1,83 мл(3,66 ммоль) 2 н соляной кислоты. Через несколько минут раствор выпаривали досуха. Остаток снова концентрировали из метанола, а затем растирали диэтиловым эфиром. Твердый остаток собирали на фильтре, промывали дополнительно диэтиловым эфиром и сушили, в результате чего получали 455 мг (87%) желтоватого порошка, т.пл. 154-157 С. 1H-ЯМР (500 МГц, ДМСО-d6) соответствовал предполагаемой структуре. Пример 5.2. Методом, аналогичным методу, описанному в примере 5, были получены следующие соединения:CDCl3) при 0 С добавляли 240 мг сульфата магния, а затем 30,6 мг 4-(3-метилизоксазол-5 ил)бутиральдегида и смесь перемешивали при низкой температуре. Через 15 мин добавляли холодный раствор борогидрида натрия (30,2 мг в 1,5 мл сухого метанола) и смесь перемешивали еще 15 мин. По истечении этого времени реакцию гасили путем добавления воды, экстрагировали этилацетатом, а затем метиленхлоридом, и объединенные органические экстракты сушили сульфатом натрия. После очистки концентрата с помощью препаративной ТСХ на силикагеле (метиленхлорид: метанол, 9:1) получали целевое соединение (28 мг). m/е = 557(М+Н). Получение синтетических промежуточных соединений 4-(3-метилизоксазол-6-ил)бутиральдегид. Стадия А. Трет-бутилгекс-5-инилоксидиметилсилан. К раствору гекс-5-ин-1-ола (1,96 г в 40 мл безводного метиленхлорида) при 0 С добавляли 3,48 мл триэтиламина, а затем 3,31 г третбутилдиметилсилилхлорида и смесь перемешивали, медленно нагревая до комнатной температуры. Через 60 ч смесь фильтровали для удаления твердых веществ и фильтрат концентрировали в вакууме. После очистки с помощью флеш-хроматографии на силикагеле(этилацетат:гексан, 1:9, а затем 1:3) получали целевое соединение (1,62 г). Стадия В. 5-[4-(Трет-бутилдиметилсиланилокси)бутил]-3-метилизооксазол. К раствору трет-бутилгекс-5-инилоксидиметилсилана (1,0 г в 20 мл сухого толуола) добавляли 530 мг нитроэтана, а затем 1,31 мл триэтиламина и 1,1 г 4-хлорфенилизоцианата и смесь перемешивали при комнатной температуре. Через 1 ч содержимое нагревали с обратным холодильником на масляной бане еще 20 ч, а затем охлаждали до комнатной температуры и фильтровали для удаления твердых веществ. После концентрирования фильтрата в вакууме и очистки с помощью флеш-хроматографии на силикагеле (этилацетат: гексан, 1:9, а затем 1:2) получали целевое соединение (388 мг). 46 Стадия С. 4-(3-Метилизоксазол-5-ил)бутан-1-ол. К раствору 5-[4-(трет-бутилдиметилсиланилокси) бутил]-3-метилизоксазола (350 мг в 5 мл безводного тетрагидрофурана) при 0 С добавляли 1,62 мл 1 М раствора фторида тетрабутиламмония в тетрагидрофуране и смесь перемешивали, нагревая до комнатной температуры. Через 16 ч смесь концентрировали в вакууме и остаток очищали с помощью флешхроматографии на силикагеле (этилацетат: гексан, 1:3, а затем 1:1) получали целевое соединение (147 мг). Стадия D. 4-(3-Метилизоксазол-5-ил) бутиральдегид. К раствору оксалилхлорида (0,40 мл 2 М раствора в метиленхлориде в 2 мл безводного метиленхлорида) при 78 С добавляли раствор метилсульфоксида (126 мг в 1 мл метиленхлорида) и смесь перемешивали в течение 3 мин при низкой температуре. Затем добавляли раствор 4-(3-метилизоксазол-5-ил)-бутан-1-ола (100 мг в 1 мл метиленхлорида) и смесь оставляли на 15 мин для прохождения реакции, после чего добавляли 0,67 мл триэтиламина и полученную смесь оставляли нагреваться до комнатной температуры. Через 30 мин добавляли солевой раствор и смесь экстрагировали метиленхлоридом. Органическую часть сушили сульфатом натрия и концентрировали в вакууме. После очистки с помощью флеш-хроматографии на силикагеле(этилацетат:гексан, 1:1) получали целевое соединение (48 мг). Способом, аналогичным описанному в примере 6, были получены следующие соединения:N,N-диэтил-2-(4-гидразинофенил)изобутирамида в 92 мл абсолютного этанола перемешивали с обратным холодильником в атмосфере азота в течение 43 ч. Затем раствор охлаждали и концентрировали в вакууме. После очистки с помощью флеш-хроматографии на силикагеле(элюировали градиентом 0-5% метанола в метиленхлориде, а затем метиленхлорид-метанолгидроксид аммония 95:5:0,5, и метиленхлоридметанол гидроксид аммония 92,5:7,5:0,75) полу 000829 48 чали 873 мг (9,4%) продукта кирпичного цвета в виде густой пены, который имел достаточную чистоту, о чем свидетельствовала ТСХ в 95:5:0,5 CH2Cl2-MeOH-конц.NН 4 ОН. 1H-ЯMP(500 МГц, СDСl3) соответствовал предполагаемой структуре. Масс-спектр (РВ-NН 3/Сl): m/е = 406 (М+Н). Стадия 7.1 В. 2-[2-(3,5-Диметилфенил)-3[2-[4-(пиридин-4-ил)бутиламино]этил]-1 Ниндол-5-ил]-N,N-диэтилизобутирамид. Смесь 93,3 мг (0,23 ммоль) 2-[3-(2 аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5 ил]-N,N-диэтилизобутирамида, 37,7 мг (0,253 ммоль) 4-(пиридин-4-ил)бутиральдегида и 138 мг (1,15 ммоль) сульфата магния продували азотом и охлаждали в бане из метанола со льдом при около -10 С, постепенно добавляя шприцом 0,50 мл CDCl3. Смесь перемешивали при этой температуре в атмосфере азота в течение 1 ч. Мембрану удаляли на время, достаточное для того, чтобы добавить 11,3 мг (0,30 ммоль) борогидрида натрия, и раствор снова продували азотом. Смесь перемешивали при -10 С-5 С, постепенно добавляя 0,50 мл безводного метанола и продолжали перемешивать при этой температуре. Через 35 мин смесь распределяли между 5 мл этилацетата и 5 мл воды. Этилацетатный слой промывали солевым раствором, а затем осушали сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали в две стадии с помощью препаративной ТСХ на конусообразных пластинах силикагеля GF Analtech(проявленных сначала в 90:10:1 метиленхлоридметанол-гидроксид аммония, а затем в 90:10 метиленхлорид-метанол). После выделения полос продукта получали 25,7 мг (21%) слегка окрашенного в золотистый цвет стеклообразного остатка, который являлся фактически гомогенным, о чем свидетельствовала ТСХ в 92,5:7,5:0,75 СН 2 Сl2-МеОН-конц. NН 4OН. 1HЯМР (500 МГц, СDСl3) соответствовал предполагаемой структуре. Масс-спектр ОСИ): m/е = 539 (М+Н). Получение синтетических промежуточных соединений. Стадия А. 4-Хлор-N-метокси-N-метилбутирамид. К раствору 4-хлорбутирилхлорида (10,0 г в 200 мл безводного метиленхлорида) добавляли 10,4 г гидрохлоридаN,О-диметилгидроксиламина. Смесь перемешивали в атмосфере азота и поддерживали при температуре ниже 25 С, охлаждая по мере необходимости в бане со льдом, и добавляя по каплям триэтиламин (29,1 мл) в течение примерно 20 мин, в результате чего происходило осаждение. После выдерживания в течение 1,5 ч при комнатной температуре, смесь концентрировали в вакууме. Остаток распределяли между 100 мл диэтилового эфира и 100 мл насыщенного водного раствора бикарбоната натрия. Органический слой промывали еще 100 мл насыщенного бикарбо 49 ната натрия и водные фракции подвергали обратной экстракции эфиром. Объединенные органические фазы сушили сульфатом натрия,фильтровали и концентрировали в вакууме, в результате чего получали 10,5 г (90%) маслообразного продукта, который имел достаточную чистоту, что подтверждалось 1H-ЯМР (CDCl3). Масс-спектр (РВ-NН 3/Сl): m/е = 166 (М+Н). Стадия В. 3-Хлорпропил 3,5-диэтилфенилкетон. Раствор 10,2 мл (13,9 г; 72 ммоль) 5-бромм-ксилола в 200 мл безводного тетрагидрофурана перемешивали в атмосфере азота при -78 С,добавляя по каплям 35,8 мл (84 ммоль) 2,5 М нбутиллития в тетрагидрофуране. После выдерживания в течение 15 мин при -78 С по каплям в течение 25-30 мин добавляли раствор 10,0 г(60 ммоль) 4-хлор-N-метокси-N-метилбутирамида в 30 мл безводного тетрагидрофурана. Полученный раствор поддерживали при-78 С в течение 45 мин, а затем быстро нагревали до комнатной температуры. Реакцию гасили путем добавления 40 мл 2 н соляной кислоты, а затем распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным водным раствором бикарбоната натрия, а затем насыщенным водным раствором хлорида натрия. Органический раствор сушили сульфатом натрия, фильтровали и концентрировали в вакууме. После флеш-хроматографии остатка получали 8,91 г (70 %) маслообразного продукта, который имел достаточную чистоту, что подтверждалось 1 Н-ЯМР (CDCl3). Стадия АА. Этил 2-(4-гидразинофенил) ацетата гидрохлорид и 2-(4-гидразинофенил) уксусной кислоты гидрохлорид. Это соединение (смесь этилового сложного эфира и карбоновой кислоты) получали из 13,4 г (75 ммоль) этил 2(4-аминофенил)ацетата путем диазотирования и восстановления диазониевой соли в соответствии с методом, описанным L.J. Street, et al., J. Med. Chem., 36, 1529(1993). Этот продукт был получен в два сбора. Первый сбор состоял из 6,40 г порошка, т. пл.200 С. ЯМР (400 МГц, ДМСО-d6) подтвердил,что этот продукт соответствовал смеси карбоновой кислоты и этилового сложного эфира в молярном отношении 4:3. Масс-спектр (РВNН 3/Сl): 195 (катион арилгидразония для этилового эфира). Второй сбор состоял из 4,60 г порошка, т. пл.180 С. ЯМР (400 МГц, ДМСОd6) подтвердил, что этот продукт соответствовал смеси карбоновой кислоты и этилового сложного эфира в молярном отношении 7:1. После уточнения состава смеси двух сборов, оцененный общий выход составлял 69%. Поскольку этерификация любой карбоновой кислоты происходит в следующей стадии, то сложный эфир и кислота взаимодействуют с образованием того же самого продукта. 50 Стадия ААА. (+/-)-2-(4-Нитрофенил) пропионовой кислоты этиловый сложный эфир. К раствору 9,76 г (50 ммоль) (+/-)-2-(4 нитрофенил)пропионовой кислоты в 150 мл абсолютного этанола добавляли 3,0 мл концентрированной серной кислоты. Полученный раствор перемешивали при нагревании с обратным холодильником в атмосфере азота. Через 6 ч раствор охлаждали и интенсивно перемешивали при постепенном добавлении 250 мл насыщенного водного раствора бикарбоната натрия (осторожно: образуется пена). Смесь распределяли между 750 мл этилацетата и 500 мл воды. Органический слой промывали 100 мл насыщенного водного раствора бикарбоната натрия, а затем 100 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили сульфатом магния, фильтровали и концентрировали в вакууме, в результате чего получали 10,86 г (97%) масла,которое является гомогенным(ТСХ:гексан-этилацетат, 9:1). 1H-ЯМР (400 МГц, СDСl3) соответствовал предполагаемой структуре. Стадия ВВВ. 2-Метил-2-(4-нитрофенил) пропионовой кислоты этиловый эфир. Суспензию 924 мг (23 ммоль) гидрида натрия (60% в масле) в 21 мл сухого N,Nдиметилформамида перемешивали в атмосфере азота в ледяной бане при постепенном добавлении в течение около 10 мин раствора 4,68 г (21 ммоль) этилового эфира(+/-)-2-(4 нитрофенил)пропионовой кислоты в 20,5 мл сухого N,N-диметилформамида. Во время добавления появлялась интенсивная фиолетовая окраска. Затем смесь оставляли для нагревания до комнатной температуры. Приблизительно через 1 ч смесь снова охлаждали в ледяной бане,добавляя по каплям шприцем раствор 1,44 мл(3,28 г, 23 ммоль) иодметана в 5 мл сухого N,Nдиметилформамида, в течение примерно 10 мин,поддерживая внутреннюю температуру при 1015 С. Смесь нагревали до комнатной температуры, после чего цвет этой смеси становился коричневым. Через 1 ч добавляли еще 187 мл(426 мг, 3 ммоль) метилиодида. На следующий день, смесь представляла собой суспензию из некоторого количества сероватого твердого вещества в золотистой жидкости. Эту суспензию интенсивно перемешивали и гасили путем постепенного добавления 10 мл 5%-ного водного раствора бисульфата калия. Смесь распределяли между 400 мл диэтилового эфира и 400 мл воды. Органический слой промывали еще 3 х 400 мл воды, а затем 50 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили сульфатом магния, фильтровали и концентрировали в вакууме. Остаток подвергали флеш-хроматографии на силикагеле (элюируя гексан - EtOAc, 19:1) и получали 4,31 г (87%) масла, которое являлось гомогенным, на что указывала ТСХ в гексан:этилацетат 9:1. 1H-ЯМР(400 МГц, CDCl3) соответствовал предполагаемой структуре. Стадия ССС. 2-Метил-2-(4-нитрофенил) пропионовая кислота. Раствор 11 г (24 ммоль) этилового эфира 2 метил-2-(4-нитрофенил)пропионовой кислоты в 1 л 0,5 м гидроксида калия в метаноле перемешивали в атмосфере азота и нагревали примерно до 50 С, постепенно добавляя при этом 111 мл воды. Полученный раствор перемешивали с обратным холодильником в течение ночи, а затем концентрировали в вакууме. Остаток распределяли между этилацетатом - тетрагидрофураном и 0,5 н соляной кислотой. Водный слой также неоднократно экстрагировали хлороформом. Органические фракции сушили сульфатом магния и концентрировали в вакууме, в результате чего получали 9,4 г (94 %) аморфного желто-коричневого твердого вещества, гомогенность которого была подтверждена ТСХ в 95:5:0,5 CH2Cl2-MeOH-AcOH. 1H-ЯМР (500 МГц, СDСl3) соответствовал предполагаемой структуре. СтадияN,N-Диэтил-2-(4 нитрофенил) изобутирамид. К 8,36 г (40 ммоль) 2-метил-2-(4 нитрофенил) пропионовой кислоты добавляли 18 мл циклогексана и 9 мл тионилхлорида. Смесь перемешивали в атмосфере азота и нагревали с обратным холодильником. Твердый продукт постепенно растворялся, и наблюдалось выделение газа. Через 20 ч раствор охлаждали и выпаривали в потоке азота, а затем сушили в вакууме. Оставшееся светло-оранжевое твердое вещество растворяли в 50 мл безводного тетрагидрофурана и по каплям добавляли к раствору 9,10 мл (6,43 г, 88 ммоль) диэтиламина в 100 мл безводного тетрагидрофурана, перемешанного в бане из льда и метанола при около-10-(-15)С. После завершения добавления, которое сопровождалось осаждением, смесь оставляли для постепенного нагревания до комнатной температуры. Через 2 дня смесь концентрировали в вакууме, а остаток распределяли между 250 мл этилацетата и 200 мл воды. Органическую фазу еще раз промывали водой, затем насыщенным водным раствором бикарбоната натрия и, наконец, рассолом. Этилацетатный раствор сушили сульфатом магния, фильтровали и концентрировали в вакууме. Маслообразный остаток очищали с помощью флешхроматографии на силикагеле (элюировали 6:1,а затем 5:1 этилацетат : гексан ) и получали 9,45 г (85 %) светло-желтого твердого вещества, т. пл. 59,5-61 С, гомогенность которого подтверждалась ТСХ в 2:1 гексан : EtOAc. 1 Н-ЯМР (500 МГц, CDCl3) соответствовал предполагаемой структуре. Масс-спектр (РВ-NН 3/Сl): m/е =265 52 Стадия ЕЕЕ. 2-(4-Аминофенил)-N,Nдиэтилизобутирамид. Смесь 9,38 г (35,5 ммоль) N,N-диэтил-2-(4 нитрофенил)изобутирамида, 400 мг 10% палладия-на-угле и 120 мл абсолютного этанола перемешивали путем встряхивания в присутствии водорода (первоначальное давление водорода составляло 47 фунт/кв.дюйм = 3,3 кг/см 2) в автоклаве в течение 22 ч. Катализатор удаляли путем фильтрации через диатомовую землю в присутствии азота и остаток на фильтре дополнительно промывали этанолом. После концентрирования фильтрата в вакууме получали 8,5 г(100 %) беловатого твердого вещества, т.пл. 8990 С, гомогенность которого подтверждалась ТСХ в 92:2 CH2Cl2-MeOH. 1H-ЯМР (500 МГц,СDСl3) соответствовал предполагаемой структуре. Масс-спектр (РВ-NН 3/Сl): m/e = 235(М+Н) . Стадия FFF. N,N-Диэтил-2-(4-гидразинофенил)изобутирамид. К 8,5 г (35,5 ммоль) 2-(4-аминофенил)N,N-диэтилизобутирамида добавляли 35,5 мл концентрированной соляной кислоты и смесь перемешивали до тех пор, пока не получали гомогенный раствор. Полученный раствор перемешивали при температуре от -10 до -5 С в бане лед - ацетон, добавляя по каплям в течение 25 мин раствор 2,55 г (36,9 ммоль) нитрита натрия в 15,3 мл воды. Затем перемешивание продолжали при этой температуре еще в течение 1 ч. Полученную смесь поддерживали в холодном состоянии и в течение 1 ч небольшими порциями добавляли в раствор 40,1 г (178 ммоль) дигидрата хлорида олова (2) в 28,5 мл концентрированной соляной кислоты в присутствии азота и в бане из льда и ацетона ( -10 С). После завершения добавления, смесь продолжали перемешивать в охлаждающей бане в течение 1 ч. Затем смесь доводили до почти комнатной температуры, энергично размешивая при этом, в результате чего получали гомогенный раствор. Этот раствор распределяли между 500 мл этилацетата и 50 мл воды. Этилацетатный слой промывали еще 50 мл воды, а затем тщательно обрабатывали 500 мл полунасыщенного водного раствора бикарбоната натрия. Смесь осторожно перемешивали, в результате чего наблюдалось заметное выделение газа и осаждение. Густую смесь фильтровали, а затем разделяли на фазы. Этилацетатную фазу промывали 50 мл рассола,а затем сушили сульфатом магния, фильтровали и концентрировали в вакууме при комнатной температуре. В результате получали 7,04 г (80%) немного клейкого оранжевого твердого вещества, которое не совсем точно подтверждалось ТСХ, ЯМР и масс-спектроскопией, но которое было подходящим для использования в следующей стадии. 2-[2-(3,5-Диметилфенил)-3-[2-[4-(пиридин 4-ил)бутиламино]этил]-1 Н-индол-5-ил-3-N,Nдибутилизобутирамид. Стадия 7.2 А. 2-[2-(3,5-Диметилфенил)-3[2-[4-(пиридин-3-ил)бутиламино]этил]-1 Ниндол-5-ил]-2-метилпропионовой кислоты этиловый эфир. Сухую колбу, содержащую 3,00 г (7,93 ммоль) этилового эфира 2-[3-(аминоэтил)-2(3,5-диметилфенил)-1 Н-индол-5-ил]-2-метилпропионовой кислоты (полученного, в основном, как описано в примере 7.1, стадии А), 4,76 г (39,7 ммоль) безводного MgSО 4 и магнитный стержень для перемешивания, снабжали перегородкой и иглой адаптера, ведущей к клапануFirestone. Колбу тщательно продували N2, смесь охлаждали в бане лед - МеОН при температуре от -10 до -5 С и интенсивно размешивали, постепенно в течение 10-15 мин, добавляя шприцем раствор 1,32 г (8,88 ммоль) 4-(пиридин-3 ил)бутиральдегида в 15 мл сухого СDСl3. Полученную смесь перемешивали в присутствии Na при температуре от -10 до -5 С в течение 40-45 мин. Затем перегородку удаляли на время, достаточное для добавления 390 мг (10,3 ммоль) борогидрида натрия. Смесь перемешивали в присутствии N2 при температуре от -10 до -5 С,по каплям в течение нескольких минут добавляя шприцем 10 мл сухого МеОН. Через 30 мин смесь удаляли из охлаждающей бани и распределяли между 90 мл EtOAc и 90 мл Н 2 О. Органический слой промывали 2 х 30 мл рассола,затем сушили безводным Na2SO4. Отфильтрованный раствор концентрировали в вакууме и остаток подвергали флеш-хроматографии на силикагеле (элюируя градиентом 0-10% МеОН в СН 2 Сl2). Фракции, содержащие продукт и небольшое количество не прореагировавшего исходного материала, объединяли и концентрировали в вакууме, в результате чего получали 3,00 г светло-бежевого продукта в виде густой пены,который непосредственно использовали в следующей стадии без дополнительной очистки или характеризации. Стадия 7.2 В. 2-[3-[2-[Бензилоксикарбонил[4-(пиридин-3-ил)бутил]амино]этил]-2-(3,5 диметилфенил)-1 Н-индол-5-ил]-2-метилпропионовой кислоты этиловый эфир. Раствор 3,00 г (теоретический максимум 5,86 ммоль) неочищенного этилового эфира 2[2-(3,5-диметилфенил)-3-[2-[4-(пиридин-3-ил) бутиламино]этил]-1H-индол-5-ил]-2-метилпропионовой кислоты в 30 мл сухого CH2Cl2 перемешивали в атмосфере N2 при охлаждении в бане из сухого льда-ацетона. К этому растворуN,N-диизопропилэтиламина. Затем шприцем по каплям добавляли 956 мкл (1,14 г,6,36 ммоль) бензилхлорформиата в течение 5-10 мин. Через 20 мин раствор удаляли из охлаждающей бани и нагревали до комнатной температуры. Через 2 ч раствор разбавляли 50 млCH2Cl2, переносили в делительную воронку и встряхивали с 80 мл Н 2 О. Органическую фазу сушили сульфатом магния, фильтровали и концентрировали в вакууме. Остаточную смолу подвергали флеш-хроматографии на силикагелеCH2Cl2) и получали 2,81 г (полный выход 55% для Стадии 1 и 2) смолы со слабой золотистожелтой окраской; продукт фактически являлся гомогенным, на что указывала ТСХ в CH2Cl2 МеОН, 95:5. 1 Н-ЯМР (500 МГц, СDСl3) являлся комплексным из-за присутствия ротамеров, но соответствовал предполагаемой структуре. Масс-спектр (ЭСИ): m/е = 646 (М+Н)+. Стадия 7.2 С. 2-[3-[2-[Бензилоксикарбонил[4-(пиридин-3-ил)бутил]амино]этил]-2-(3,5-диметилфенил)-1 Н-индол-5-ил]-2-метилпропионовая кислота. Смесь 2,78 г (4,30 ммоль) этилового эфира 2-[3-[2-[бензилоксикарбонил-[4-(пиридин-3 ил)бутил]амино]этил]-2-(3,5-диметилфенил)1 Н-индол-5-ил]-2-метилпропионовой кислоты в 43,0 моль (21,5 ммоль) 0,5 М КОН в МеОН и 25 мл ТГФ перемешивали в атмосфере N2 и нагревали с обратным холодильником. К полученному раствору постепенно добавляли 18 мл Н 2 О и раствор поддерживали при нагревании с обратным холодильником в течение 39 ч. Затем раствор охлаждали и концентрировали до небольшого объема с сопровождающимся осаждением. Смесь обрабатывали 10,75 мл (21,5 ммоль) 2 н НСl и помешивали несколько минут. Твердое вещество собирали на фильтре и тщательно промывали водой. После вакуумной осушки в потоке N2, твердое вещество растирали и промывали диэтиловым эфиром, а затем сушили в вакууме, в результате чего получали 2,43 г(92%) кремового порошка, т.пл. 152-154 С (частичное разл.), гомогенность которого подтверждалась ТСХ в CH2Cl2 -МеОН 90:10. 1H-ЯМР(500 МГц, ДМСО-d6) соответствовал предполагаемой структуре. Масс-спектр (ЭСИ): m/е = 618 (М+Н)+. Стадия 7.2D. [2-[5-(1-Дибутилкарбамоил 1-метилэтил)-2-(3,5-диметилфенил)-1H-индол 3-ил]этил]-[4-(пиридин-3-ил) бутил] карбаминовой кислоты бензиловый эфир. Раствор 92,7 мг (0,15 ммоль) 2-[3-[2[бензилоксикарбонил-[4-(пиридин-3-ил)бутил] амино]этил]-2-(3,5-диметилфенил)-1 Н-индол-5 ил]-2-метилпропионовой кислоты, 83,2 мг (0,16 ммоль) реагента РуВОР и 0,151 мл (116 мг, 0,9 ммоль) триэтиламина в 0,750 мл сухого CH2Cl2 перемешивали при комнатной температуре в колбе с притертой пробкой в течение 2 дней. 55 Затем раствор распределяли между 10 мл EtOAc и 10 мл 0,5 н НСl. Органическую фазу промывали 10 мл насыщенного водного раствораNаНСО 3, а затем 5 мл насыщенного водного раствора NaCl. Этилацетатную фазу сушили(MgSO4), фильтровали и концентрировали в вакууме при комнатной температуре. Остаток очищали с помощью препаративной ТСХ на шести конусообразных пластинах с силикагелемAnaltech (20 х 20 см), которые были проявлены в СН 2 Сl2-МеОН, 95:5. Полосу продукта от каждой пластины выделяли, объединяли и экстрагировали CH2Cl2 - МеОН, 95:5. Экстракты концентрировали в вакууме и получали 85,2 мг(78%) смолы со слабой золотисто-желтой окраской; продукт являлся фактически гомогенным,на что указывала ТСХ в CH2Cl2 - МеОН, 95:5. 1H-ЯМР (500 МГц, CDCl3) являлся комплексным из-за присутствия ротамеров, но соответствовал предполагаемой структуре. Масс-спектр(ЭСИ): m/е = 729,7 (М+Н)+. Стадия 7.2 Е. 2-[2-(3,5-Диметилфенил)-3[2-[4-(пиридин-3-ил)-бутиламино]этил]-1 Ниндол-5-ил]-N,N-дибутилизобутирамид. Смесь 76,5 мг (0,105 ммоль) бензилового эфира [2-[5-(1-дибутилкарбамоил-1-метилэтил)2-(3,5-диметилфенил)-1H-индол-3-ил]этил]-[4(пиридин-3-ил)бутил]карбаминовой кислоты, 40 мг 10% палладия-на-угле, 4 мл абсолютного этанола и 4 мл EtOAc встряхивали в присутствии H2 (приблизительно 47 фунт/кв.дюйм = 3,305 кг/см 2) в автоклаве в течение 6 ч. Катализатор удаляли путем фильтрации через Целит в атмосфере N2 и фильтрат концентрировали в вакууме при комнатной температуре. Остаток очищали с помощью препаративной ТСХ на 4 конусообразных пластинах с силикагелем Analtech (20 х 20 см), которые были проявлены в СН 2 Сl2-МеОН-конц.NН 4 ОН, 92,5:7,5:0,75. Полосу продукта от каждой пластины выделяли,объединяли и экстрагировали CH2Cl2-MeOHконцент.NH4OH, 92,5:7,5:0,75. Экстракты концентрировали в вакууме и получали 51,4 мг(82%) бледно-желтой густой смолы; продукт являлся гомогенным, на что указывала ТСХ вCH2Cl2-MeOH-конц.NH4OH, 92,5:7,5:0,75. 1HЯМР (500 МГц, CDCl3) соответствовал предполагаемой структуре. Масс-спектр (ЭСИ): m/е = 595,6 (М+Н)+. Способом, аналогичным описанному в примерах 7.1 и 7.2, были получены следующие соединения.CDCl3), а затем 206 мг сульфата магния и смесь перемешивали при низкой температуре. Через 15 мин добавляли 26 мг борогидрида натрия, а затем 1,0 мл метанола и смесь перемешивали еще 30 мин. По истечении этого времени, реакцию гасили путем добавления 2 н соляной кислоты и упаривали досуха. Концентрат очищали с помощью препаративной ТСХ на силикагеле(метиленхлорид: метанол, 9:1) и получали целевое соединение (8,4 мг). Получение синтетических промежуточных соединений 3-Пиридин-3-ил-циклогексанон. Стадия А. 3-Пиридин-3-илциклогекс-2 енон. К раствору 3-этокси-2-циклогексен-1-она(1,4 г в 10 мл сухого тетрагидрофурана) добавляли 1,58 г 4-бромпиридина и смесь интенсивно перемешивали при -78 С. К этой смеси по каплям добавляли трет-бутиллитий (11,8 мл 1,7 М раствора в пентане) и реакционную смесь оставляли при низкой температуре для прохождения реакции. Через 2 ч реакцию гасили путем добавления 2 н соляной кислоты и нагревали с обратным холодильником в течение 14 ч. По истечении этого времени, смесь охлаждали и нейтрализовали до рН 7 путем осторожного добавления 1 н гидроксида натрия. Затем смесь экстрагировали метиленхлоридом и органическую часть сушили сульфатом натрия. После очистки концентрата с помощью флешхроматографии на силикагеле (этилацетат : гексан 1:1, а затем 2:1) получали целевое соединение (1,37 г). Стадия В. 3-Пиридин-3-ил-циклогексанон. К раствору 3-пиридин-3-ил-циклогекс-2 энона (1,25 г в 50 мл этанола) добавляли 164 мг катализатора окиси платины и в полученную смесь подавали водород под давлением 40 фунт/кв.дюйм (2/8 кг/см 2). Через 2 ч катализатор удаляли путем фильтрации и фильтрат концентрировали в вакууме. После очистки остатка с помощью флеш-хроматографии на силикагеле(этилацетат : гексан 1:1) получали целевое соединение (427 г). Методом, аналогичным описанному в примере 8, получали следующие соединения: 2-(3,5-Диметилфенил)-3-[2-(1-метил-4 пиридин-3-ил)бутиламино]этил]-1 Н-индол-5 карбоновой кислоты диэтилизобутирамид. Стадия 9.1 А. 3-(2-(Трет-бутоксикарбониламиноэтил)-2-(3,5-диметиметилфенил)-1 Ниндол-5-карбоновой кислоты этиловый эфир. К раствору этилового эфира 3-(2 аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5 карбоновой кислоты (полученного как описано в примере 5.1; 1,5 г в 30 мл сухого тетрагидрофурана) при 0 С добавляли раствор ди-третбутилдикарбоната (1,9 г в 3 мл тетрагидрофурана), а затем водный раствор карбоната калия (1 г в 10 мл воды), и полученную суспензию интенсивно перемешивали при 0 С. Через 12 мин реакцию гасили путем добавления избыточного количества насыщенного водного раствора хлорида аммония и смесь экстрагировали этилацетатом. Органическую часть сушили сульфатом натрия и концентрировали в вакууме. Полученное твердое вещество последовательно промывали метиленхлоридом, гексаном и этилацетатом, в результате чего получали целевое соединение (1,77 г). Стадия 9.1 В. 3-(2-Трет-бутоксикарбониламиноэтил)-2-(3,5-диметилфенил)-1H-индол-5 карбоновая кислота. К суспензии этилового эфира 3-(2-третбутоксикарбониламиноэтил)-2-(3,5-диметилфенил)-1 Н-индол-5-карбоновой кислоты (830 мг в 50 мл метанола) добавляли 8 мл 1,25 н раствора гидроксида натрия и смесь нагревали до 75 С на масляной бане. Через 5,5 ч добавляли еще 3 мл 1,25 н гидроксида натрия и реакцию оставляли для протекания еще на 2 ч. По истечении этого времени, смесь охлаждали до комнатной температуры и реакцию гасили путем добавления буфера с рН=2. Смесь экстрагировали этилацетатом, промывали насыщенным раствором хлорида аммония, а органическую часть сушили сульфатом натрия. После концентрирования в вакууме получали неочищенную кислоту с количественным выходом. 59 Стадия 9.1 С. 2-[5-Диизобутилкарбамоил 2-(3,5-диметилфенил)-1H-индол-3-ил]этил карбаминовой кислоты трет-бутиловый эфир. К раствору 3-(2-трет-бутоксикарбониламиноэтил)-2-(3,5-диметилфенил)-1 Н-индол-5 карбоновой кислоты (200 мг в 12 мл метиленхлорида) добавляли 104 мг 1 гидроксибензотриазола, а затем 118 мг гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида и смесь перемешивали при комнатной температуре. Через 30 мин добавляли 0,35 мл диизобутиламина и смесь перемешивали при комнатной температуре в течение 14 ч. Затем реакционную смесь концентрировали в вакууме и очищали с помощью флешхроматографии на силикагеле (гексан: этилацетат 2:1) с получением целевого соединения (230 мг). Стадия 9.1D. 3-(2-Аминоэтил)-2-(3,5 диметилфенил)-1 Н-индол-5-карбоновой кислоты диизобутиламид. К раствору трет-бутилового эфира 2-[5 диизобутилкарбамоил-2-(3,5-диметилфенил)1H-индол-3-ил]этилкарбаминовой кислоты(230 мг в 12 мл метиленхлорида) при 0 С добавляли 0,55 мл анизола, а затем 3,4 мл трифторуксусной кислоты и смесь перемешивали при 0 С. Через 1 ч смесь концентрировали в вакууме, а остаточную кислоту удаляли путем азеотропной перегонки с толуолом. Концентрат очищали с помощью флеш-хроматографии на силикагеле(метиленхлорид : метанол : гидроксид аммония,90:8:1) и получали целевое соединение (173 мг). Стадия 9.1 Е. 2-(3, 5-Диметилфенил)-3-[2(1-метил-4-пиридин-3-ил-бутиламино)этил]-1 Ниндол-5-карбоновой кислоты диизобутиламид. К раствору диизобутиламида 3-(2 аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5 карбоновой кислоты (142 мг в сухом метаноле) добавляли 19 мг 5-пиридин-3-ил-пентан-2-она и рН доводили до 6 путем добавления трифторуксусной кислоты. К этому раствору добавляли приблизительно 10 мг молекулярных сит ЗА, а затем 27 мг цианоборогидрида натрия. рН поддерживали при 6, добавляя время от времени в течение 36 ч трифторуксусную кислоту. Реакцию гасили путем добавления водного раствора карбоната калия и смесь экстрагировали этилацетатом. Органическую часть последовательно промывали водным раствором карбоната калия и рассолом, а затем сушили сульфатом натрия. Концентрат очищали с помощью флешхроматографии на силикагеле (метиленхлорид : метанол : гидроксид аммония, 90:8:1) и получали целевое соединение (32 мг) [m/е = 567(М+Н)] + непрореагировавший амин (115 мг). Получение синтетических промежуточных соединений Стадия А. 4-Пиридин-3-ил-бутиральдегид. К раствору оксалилхлорида (0,74 мл в 5 мл сухого метиленхлорида) при -78 С добавляли раствор метилсульфоксида (0,90 мл в 5 мл ме 000829 60 тиленхлорида) и смесь перемешивали при низкой температуре в течение 15 мин. Добавляли раствор 4-пиридин-3-ил-бутан-1-ола (полученного, в основном, как описано в примере 2.1,820 мг в 10 мл метиленхлорида) и реакцию оставляли на 45 мин, после чего добавляли 4 мл триэтиламина и смесь нагревали до комнатной температуры. Через 35 мин добавляли насыщенный бикарбонат натрия и смесь экстрагировали метиленхлоридом. Органическую часть промывали насыщенным бикарбонатом натрия, сушили сульфатом натрия и концентрировали в вакууме с получением неочищенного целевого соединения (813 мг). Стадия В. 5-Пиридин-3-ил-пентан-2-ол. К раствору бромида метилмагния (0,75 мл 3 М раствора (диэтилового эфира) в 3,5 мл сухого тетрагидрофурана) при 0 С добавляли раствор 4-пиридин-3-ил-бутиральдегида (133 мг в 1 мл тетрагидрофурана) и смесь перемешивали при низкой температуре. Через 30 мин реакцию гасили путем добавления насыщенного хлорида аммония и экстрагировали этилацетатом. Органическую часть промывали насыщенным бикарбонатом натрия, сушили сульфатом натрия и концентрировали в вакууме. После очистки с помощью флеш-хроматографии на силикагеле(метиленхлорид : метанол 95:5) получали целевое соединение (71 мг). Стадия С. 5-Пиридин-3-ил-пентан-2-он. К раствору оксалилхлорида (0,060 мл в 1 мл сухого метиленхлорида) при -78 С добавляли раствор метилсульфоксида (0,075 мл в 0,25 мл метиленхлорида) и смесь перемешивали при низкой температуре в течение 18 мин. Затем добавляли раствор 5-пиридин-3-ил-пентан-2-ола(71 мг в 1 мл метиленхлорида) и реакцию оставляли на 40 мин, после чего добавляли 0,35 мл триэтиламина, и смесь нагревали до комнатной температуры. Через 35 мин добавляли воду и смесь экстрагировали метиленхлоридом. Органическую часть промывали насыщенным бикарбонатом натрия, сушили сульфатом натрия и концентрировали в вакууме. Остаток очищали с помощью флеш-хроматографии на силикагеле(метиленхлорид : метанол, 96:4) и получали целевое соединение (61 мг). Пример 9.2.
МПК / Метки
МПК: A61K 31/40, C07D 401/12
Метки: антагонисты, гонадотропин-высвобождающего, фактора
Код ссылки
<a href="https://eas.patents.su/30-829-antagonisty-gonadotropin-vysvobozhdayushhego-faktora.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты гонадотропин-высвобождающего фактора.</a>
Антагонисты гонадотропин-высвобождающего фактора.

Номер патента: 828
Опубликовано: 24.04.2000
Авторы: Виврэтт Мэттью Дж., Фишер Майкл Х., Чу Лин, Эштон Уоллес Т., Гаулет Марк
МПК: A61K 31/535, C07D 413/02
Метки: антагонисты, фактора, гонадотропин-высвобождающего
Формула / Реферат:
1. Соединение формулы где А представляет C1-C6 алкил, замещенный C1-C6 алкил, С3-С7 циклоалкил, замещенный С3-С7 циклоалкил, С3-C6 алкенил, замещенный С3-С6 алкенил, С3-С6 алкинил, замещенный С3-С6 алкинил, C1-C6 алкокси или C0-C5алкил-S(O)n-С0-С5 алкил, C0-C5 алкил-О-С0-С5алкил, C0-C5 алкил-NR18-С0-С5 алкил, где R18 и C0-C5 алкил, взятые вместе, могут образовывать кольцо или простую связь; R0 представляет водород, C1-С6 алкил,...
Производные индола как антагонисты рецептора 5-нт

Номер патента: 304
Опубликовано: 29.04.1999
Авторы: Дакворт Дэвид Малькольм, Гэстер Лэрэми Мэри, Дэвис Дэвид Томас, Вайман Пол Эдриан, Джоунз Грэхэм Элджин, Форбес Ян Томсон, Малхоллэнд Кит Раймонд
МПК: C07D 401/12, A61K 31/44
Метки: антагонисты, индола, производные, 5-нт, рецептора
Формула / Реферат:
1. Соединение формулы (I) или его соль где Р1 и Р2 независимо представляет собой фенил, ароматические или частично насыщенные моноциклические или бициклические гетероциклические кольца, содержащие до трех гетероатомов, выбранных из азота, кислорода или серы; А представляет собой связь, цепь из 1-5 атомов, необязательно замещенных С1-6алкилом, или А представляет собой необязательно замещенный фенил или необязательно замещенное 5-7-членное...
Пиразолпиримидины как антагонисты крф рецепторов.

Номер патента: 803
Опубликовано: 24.04.2000
Авторы: Чен Чен, Моран Тиренс Дж., Уилкоуксен Кейт М., Хуанг Чарльз, Маккарти Джеймс Р., Вебб Томас Р.
МПК: C07D 487/04, A61K 31/495
Метки: рецепторов, пиразолпиримидины, антагонисты, крф
Формула / Реферат:
1. Соединение формулы I включая его стереоизомерные формы и фармацевтически приемлемые кислотно-аддитивные соли, где R1 представляет собой NR4R5 или OR5; R2 представляет C1-6-алкил, C1-6-алкилокси или C1-6-алкилтио; R3 представляет водород, C1-6-алкил, C1-6-алкилсульфонил, C1-6-алкилсульфокси или C1-6-алкилтио; R4 представляет водород, C1-6-алкил, моно- или ди(С3-6-циклоалкил) метил, С3-6-циклоалкил, С3-6-алкенил, гидрокси-С1-6-алкил,...
Анализ крови на соотношение активности фактора v для оценки вероятности заболевания тромбоэмболией

Номер патента: 673
Опубликовано: 28.02.2000
Авторы: Ку Дехью Вэйн, Аркел Йэйл С.
МПК: G01N 33/86, C12Q 1/56
Метки: заболевания, анализ, оценки, активности, фактора, соотношение, крови, тромбоэмболией, вероятности
Формула / Реферат:
1. Способ идентификации субъектов с риском тромботических нарушений вследствие дефекта фактора V, включающий a) инкубацию анализируемой плазмы с первым реагентом, содержащим фосфолипидную композицию и контактный активатор; b) добавление второго реагента, содержащего активированный белок С, к первой аликвоте анализируемой плазмы и инкубации первой аликвоты и второй аликвоты; и c) измерение активности фактора V плазмы в первой аликвоте и во...
Cпособ получения фактора ix из биологических источников и фактор ix, полученный этим способом

Номер патента: 222
Опубликовано: 24.12.1998
Авторы: Хоффер Лутц, Морфельд Франк, Йосич Дьюро
Метки: получения, фактора, фактор, полученный, биологических, способом, cпособ, этим, источников
Формула / Реферат:
1. Способ получения фактора IX из биологических источников с помощью хроматографии, отличающийся тем, что перед разделением с помощью аффинной хроматографии указанный источник обрабатывается сульфатом аммония в концентрации от 1,5-2,3 моль/л. 2. Способ по п.1, отличающийся тем, что указанным биологическим источником является плазма крови или плазма, истощенная по фактору VIII и фибриногену путем отделения криопреципитата (криообедненная...
Предыдущий патент: Антагонисты гонадотропин-высвобождающего фактора.
Следующий патент: Наружное зеркало с шаговым и контролируемым поворотом
Случайный патент: Катализатор и способ получения олефинов