Способ получения плеуромутилинов





Формула / Реферат
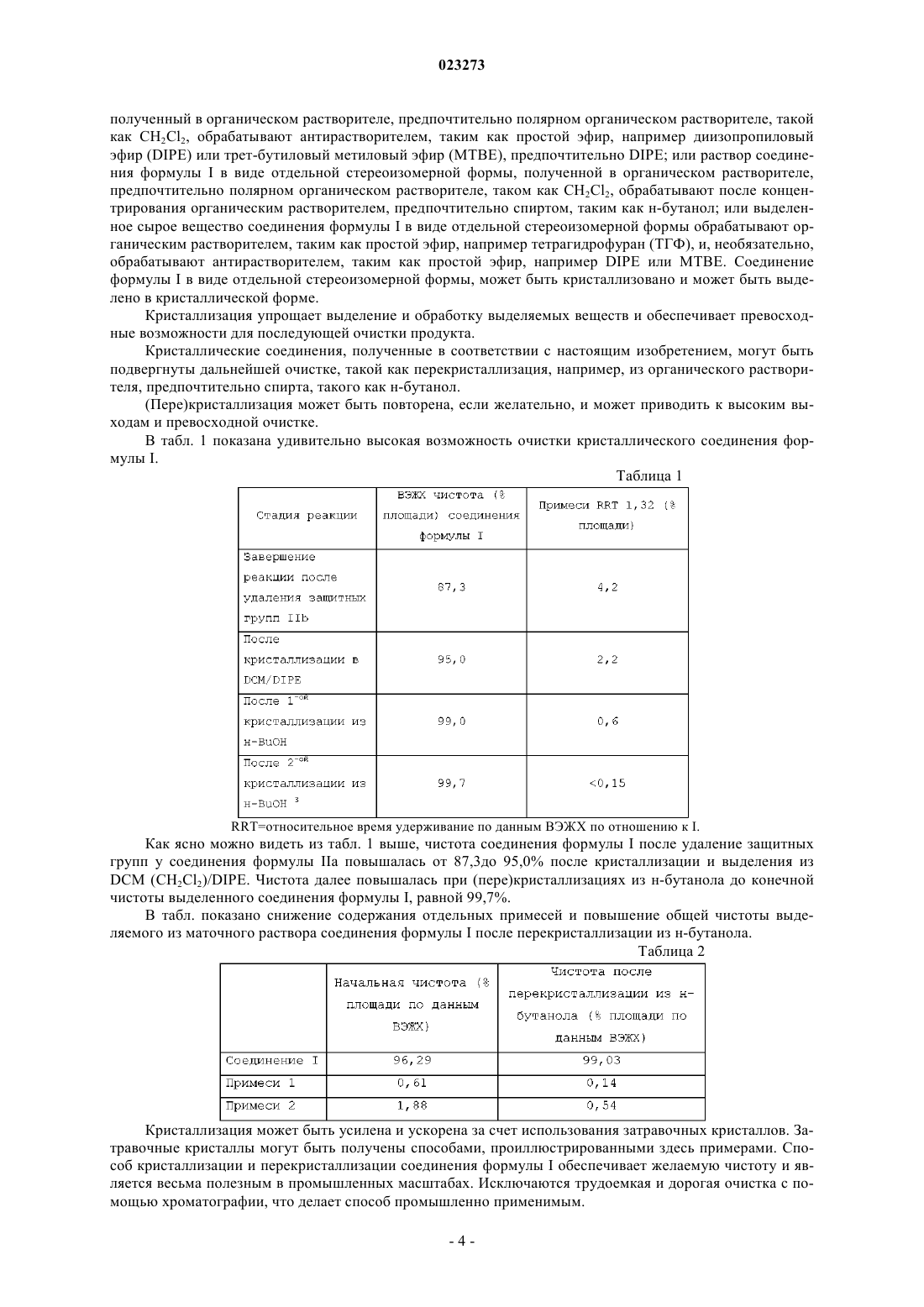
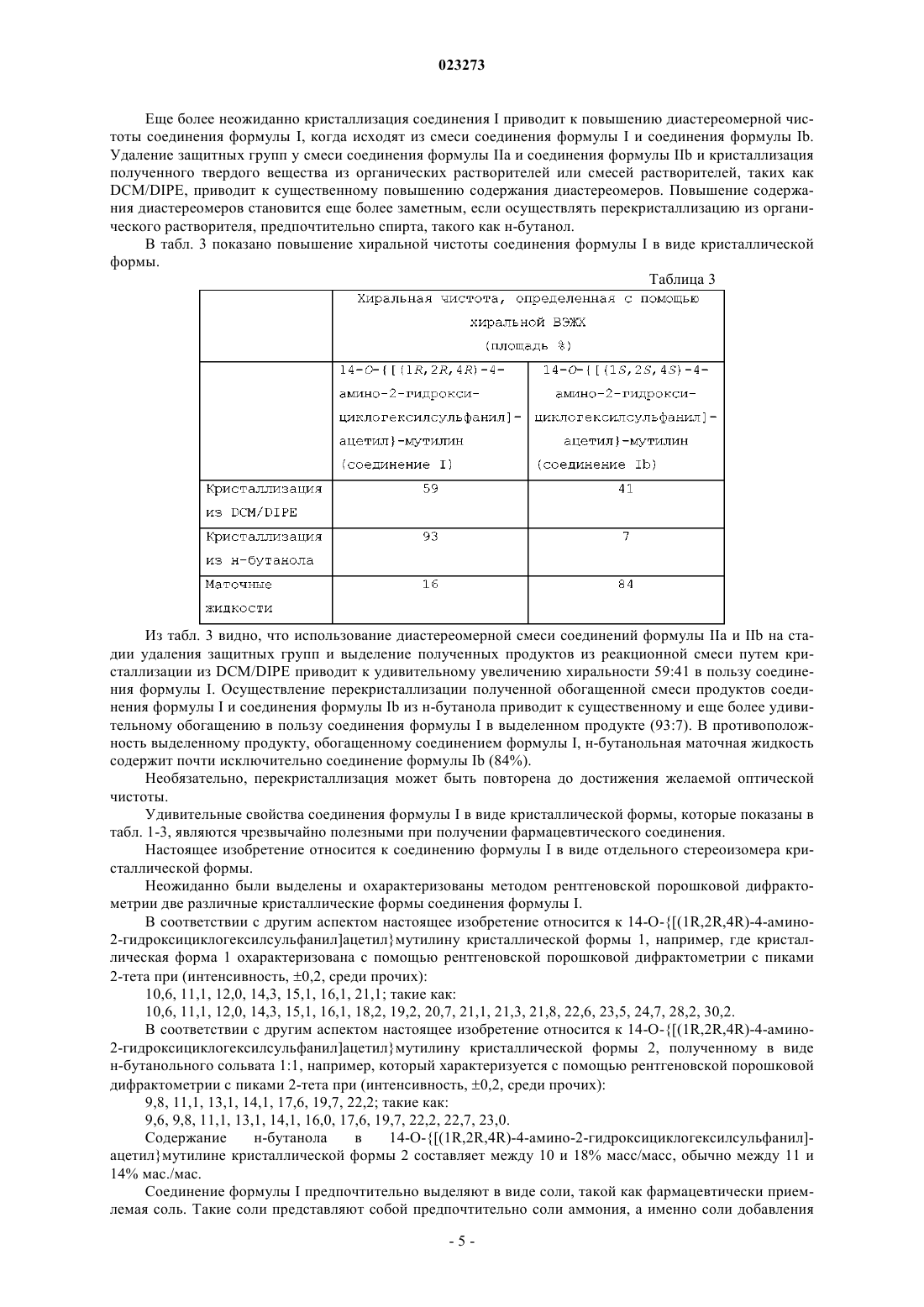
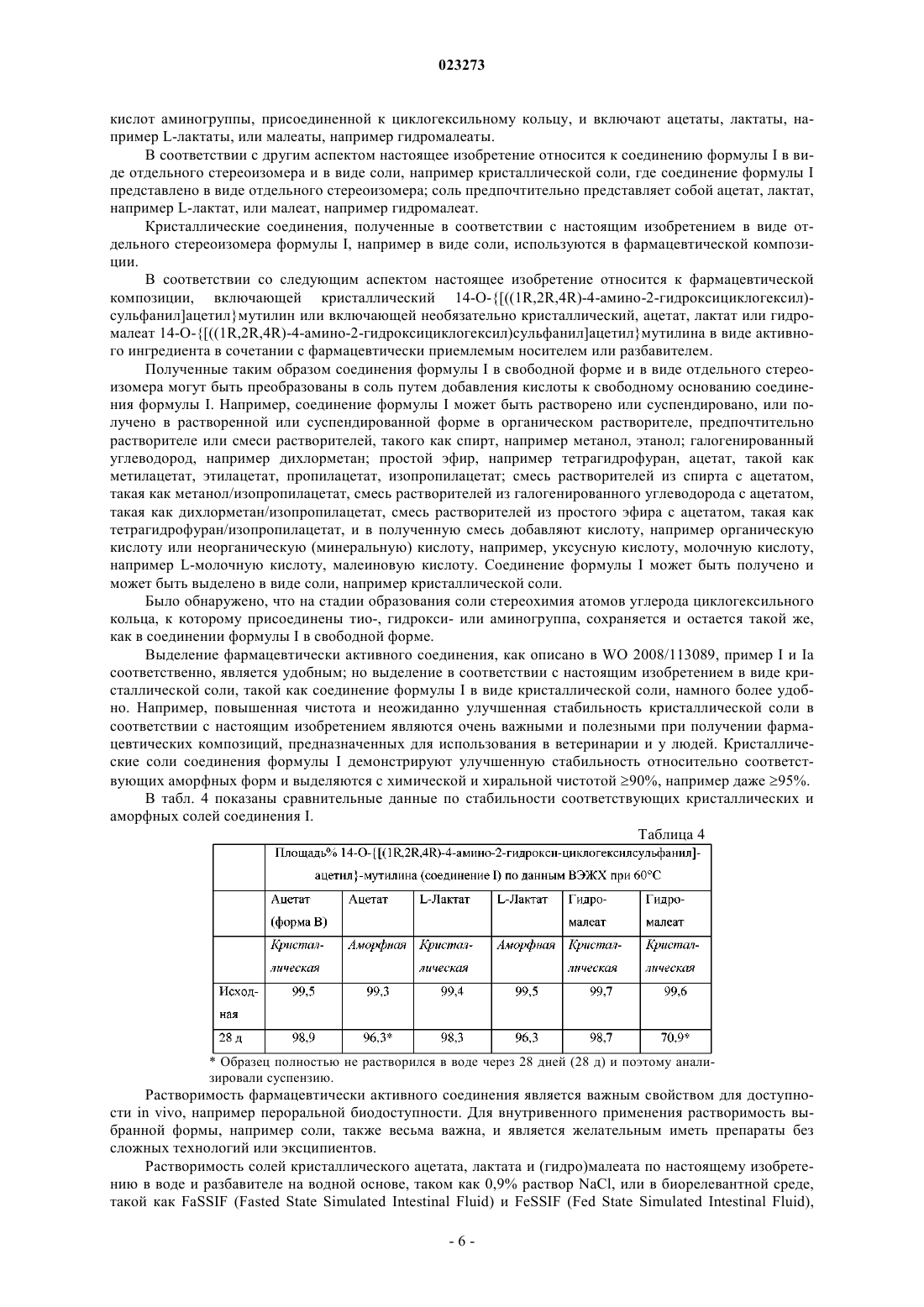
1. Соединение формулы I

в виде отдельного стереоизомера кристаллической формы.
2. Соединение по п.1, которое представляет собой 14-O-{[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетил}мутилин кристаллической формы 1, характеризующееся пиками рентгеновской порошковой дифрактометрии 2-тета 10,6, 11,1, 12,0, 14,3, 15,1, 16,1, 21,1.
3. Соединение по п.1, которое представляет собой 14-O-{[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетил}мутилин кристаллической формы 2 в виде сольвата с н-бутанолом, характеризующееся пиками рентгеновской порошковой дифрактометрии 2-тета 9,8, 11,1, 13,1, 14,1, 17,6, 19,7, 22,2.
4. Соединение формулы I, как определено в п.1, в виде фармацевтически приемлемой кристаллической соли.
5. Кристаллическая соль по п.4, которая представляет собой ацетат, лактат или гидромалеат.
6. Соединение по любому из пп.4 или 5, которое выбрано из группы, состоящей из
ацетата 14-O-{[{1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетил}мутилина кристаллической формы А;
ацетата 14-O-{[{1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетил}мутилина кристаллической формы В;
L-лактата 14-O-{[{1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетил}мутилина кристаллической формы 1 и
гидромалеата 14-O-{[{1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетил}мутилина кристаллической формы 1.
Текст
в виде отдельного стереоизомера в кристаллической форме и его солям, которые полезны в качестве антимикробного агента.(71)(73) Заявитель и патентовладелец: НАБРИВА ТЕРАПЬЮТИКС АГ (AT) Настоящее изобретение относится к кристаллическому 14-O-[(4-амино-2 гидроксициклогексил)сульфанил]ацетилмутилину, новым способам его получения и его кристаллическим солям. Плеуромутилин, соединение формулы представляет собой встречающийся в природе антибиотик, например, вырабатываемый базидиальными грибами Pleurotus mutilus и P.passeckerianus, см., например, The Merck Index, 12th edition, item 7694. Ряд последующих плеуромутилинов, имеющих основную кольцевую структуру плеуромутилина и замещенных по первичной гидроксигруппе, были разработаны, например, в качестве противомикробных препаратов. Ввиду их выраженной противомикробной активности было обнаружено, что особый интерес представляет группа производных плеуромутилинов, аминогидроксизамещенные циклогексилсульфанилацетилмутилины, как описано в WO 2008/113089. Как описано в WO 2008/11089, 14-O-[(4-амино-2 гидроксициклогексил)сульфанил]ацетилмутилины являются особенно полезными соединениями ввиду проявляемой ими активности против грамположительных и грамотрицательных патогенов, например,связанных с инфекциями дыхательных путей и кожи и кожных структур. Для получения по существу чистых изомеров/диастереомеров данной группы соединений имеется потребность в способе получения,применяемом в промышленном масштабе, который позволил бы избегнуть применения дорогих исходных продуктов, экологически опасных реагентов и растворителей или отнимающих много времени и трудоемких стадий очистки. Способ получения, описанный в WO 2008/113089, включает хроматографическую очистку соединений, полученных в соответствии с отдельными стадиями синтеза, а целевые диастереомеры разделяют с помощью хиральной хроматографии ВЭЖХ, что не может быть использовано в промышленном масштабе. Неожиданно были обнаружены кристаллические промежуточные соединения, которые, с одной стороны, обладают неожиданной возможностью химической очистки, что является важным для способов получения чистых аминогидроксизамещенных циклогексилсульфанилацетилмутилинов без хроматографической очистки и стадий разделения. Следует отметить, что 14-O-[(4-амино-2-гидроксициклогексил)сульфанил]ацетилмутилины являются потенциальными новыми лекарственными веществами на медицинском рынке с нормативными требованиями, определенными в соответствующих нормативах ICH (Международная конференция по гармонизации технических требований к регистрации лекарственных препаратов для использования у человека - International Conference on Harmonization of Technical Requirements for Registration ofPharmaceuticals for Human Use). Нормативы ICH по содержанию примесей в новых лекарственных субстанциях (Q3A(R2 включают следующие пороги. Как можно видеть из порогов ICH, указанных выше, желательно содержание всех индивидуальных неидентифицируемых примесей ниже 0,10% площади и примесей с идентифицируемыми структурами ниже 0,15% соответственно. Способы, представленные в соответствии с настоящим изобретением, позволяют получать API (активные фармацевтические ингредиенты) в пределах желаемых характеристик и с соблюдением требований ICH. С другой стороны, даже более неожиданно, кристаллические промежуточные соединения приводят к значительному хиральному обогащению, что имеет огромное преимущество при получении чистых стереоизомеров, исходя из дешевых рацемических материалов или менее хирально чистых исходных материалов. Описанные способы не включали какую-либо хроматографическую очистку ни на нормальной, ни на хиральной фазе, в противоположность способам синтеза, описанным в WO 2008/113089, где описано,например,в примере 1,стадия В,что 14-О-[(4-амино-2 гидроксициклогексил)сульфанил]ацетилмутилины были выделены после нормально-фазовой хроматографии как диастереомерные смеси в виде бесцветных аморфных пен. В WO 2008/113089 было описано достижение хиральной чистоты диастереомеров, например в примере 1 А, подвергая смесь хиральной хроматографии, после чего разделенные чистые диастереомеры были выделены в виде бесцветных аморфных пен. Хиральная хроматография, однако, не является настолько технологичной, чтобы могла быть использована в промышленном масштабе, и, более того, кристаллические соли 14-O-[(4-амино-2-1 023273 гидроксициклогексил)сульфанил]ацетилмутилины не были получены в соответствии сWO 2008/113089. В противоположность этому, в соответствии с настоящим изобретением были обнаружены кристаллические фармацевтически приемлемые соли 14-O-[(4-амино-2-гидроксициклогексил)сульфанил]ацетилмутилинов, обладающие неожиданными и превосходными способами по сравнению с аморфными солями предыдущего уровня техники, описанные в WO 2008/113089; например, неожиданно химическая стабильность кристаллический соли по настоящему изобретению является лучшей по сравнению с формами аморфных солей; а также и в дополнение, кристаллические соли по настоящему изобретению показывают неожиданно низкую гигроскопичность. Также были разработаны способы получения таких кристаллических солей, где соли могут быть получены в виде отдельных стереоизомерных форм,исходя из 14-O-[(4-амино-2 гидроксициклогексил)сульфанил]ацетилмутилинов, и способы получения стереохимически чистых 14-O-[(4-амино-2-гидроксициклогексил)сульфанил]ацетилмутилинов кристаллической формы в качестве основы для кристаллических солей были также найдены. В одном аспекте настоящее изобретение относится к способу получения соединения формулы I в виде отдельного стереоизомера кристаллической формы, включающему удаление аминозащитной группы либо в соединении формулы IIa либо в смеси соединения формулы IIa и соединения формулы IIb где R представляет собой аминозащитную группу,и выделение соединения формулы I, получаемого в виде отдельного диастереомера кристаллической формы либо непосредственно из реакционной смеси, либо путем перекристаллизации из органического растворителя. В соответствии с другим аспектом настоящее изобретение относится к соединению формулы I,описанному выше, в виде отдельного стереоизомера кристаллической формы. Соединения формулы IIa являются новыми и также составляют часть настоящего изобретения. В соответствии с другим аспектом настоящее изобретение относится к соединению формулы IIa. В соединении формулы I или IIa соответственно атомы углерода циклогексильного кольца, к которому присоединены гидроксигруппа, аминогруппа и сульфанил-ацетил-мутилиновая группа, все имеютR конфигурацию, и, таким образом, соединение формулы I или IIa представляет собой необязательно аминозащищенный 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилин. В отличие от этого в соединении формулы Ib или IIb атомы углерода циклогексильного кольца, к которому присоединены гидроксигруппа, амино группа и сульфанил-ацетил-мутилиновая группа, все имеют S конфигурацию, и, таким образом, соединение формулы IIb представляет собой необязательно аминозащищенный 14-O-[(1S,2S,4S)-4-амино-2-2 023273 гидроксициклогексилсульфанил]ацетилмутилин. Аминозащитная группа включает защитные группы, известные специалистам в данной области, которые могут быть удалены с помощью кислотных, основных, гидрирующих, окислительных или восстановительных способов, например, путем гидрогенолиза, обработки кислотой, основанием, гидридом,сульфидом. Подходящие аминозащитные группы описаны, например, в обзоре Т.W. Greene,P.G.M. Wuts, Protective Groups in Organic Synthesis, Wiley-Interscience, 4th edition, 2007, в частности p. 696868. Аминозащитные группы, которые могут быть использованы в способе в соответствии с настоящим изобретением, включают, например бензилоксикарбонильную (Cbz) группу, которая может быть удалена, например, путем гидрогенолиза; п-метоксибензилкарбонильную (Moz или MeOZ) группу, которая может быть удалена, например,путем гидрогенолиза; трет-бутилоксикарбонильную (Вос) группу, которая может быть удалена, например, путем обработки сильной кислотой, такой как HCl, H3PO4 или CF3COOH; трифторацетильную группу, которая может быть удалена, например, путем обработки основанием,таким как NaOH, K2CO3, Cs2CO3; 9-флуоренилметилоксикарбонильную (Fmoc) группу, которая может быть удалена, например, путем обработки основанием, таким как пиперидин; бензильную (Bn) группу, которая может быть удалена, например, путем гидрогенолиза; п-метоксибензильную (РМВ) группу, которая может быть удалена, например, путем гидрогенолиза; 3,4-диметоксибензильную (DMPM) группу, которая может быть удалена, например, путем гидрогенолиза; п-метоксифенильную (РМР) группу, которая может быть удалена, например, путем обработки нитратом аммония церия(IV) (CAN); тозильную (Tos) группу, которая может быть удалена, например, путем обработки концентрированной кислотой, такой как HBr, H2SO4, или путем обработки сильными восстанавливающими агентами,такими как натрий в жидком аммиаке, натрий нафталин; группы, которые образуют с амином сульфонамиды, иные, чем Tos-амиды, например включая 2-нитробензолсульфонамид (нозил) или о-нитрофенилсульфенил (Nps), которые могут быть удалены,например, путем обработки йодидом самария, гидридом трибутилолово; бензилиденовую группу, которая может быть удалена, например, путем обработки трифторметансульфоновой кислотой, трифторуксусной кислотой, диметилсульфидом; трифенилметильную (тритильную, Tr) группу, диметокситритильную (DMT), которая может быть удалена, например, путем обработки; кислотой, такой как трифторуксусная кислота; предпочтительно трифторацетильную или трет-бутилоксикарбонильную (Вос). В случае трифторацетила для осуществления удаления защитной группы у соединения формулы IIa и IIb для получения соединения формулы I в виде отдельного стереоизомера может быть использовано основание, например неорганическое, такое как NaOH, KOH, Cs2CO3 и K2CO3, или органическое основание, такое как этаноламин. В предпочтительном варианте осуществления изобретения используется неорганическое основание, такое как NaOH и K2CO3. В случае трет-бутилоксикарбонила (Вос) для осуществления удаления защитной группы у соединения формулы IIa и IIb для получения соединения формулы I в виде отдельного стереоизомера может быть использована кислота, например неорганическая, такая как минеральная кислота, или органическая кислота. В предпочтительном варианте осуществления изобретения используется органическая кислота,такая как трифторуксусная кислота (ТФУ), или минеральная кислота, такая как ортофосфорная кислота. Альтернативно, атом азота, к которому присоединен R, образует гетероциклическое кольцо, например атом азота, присоединенный к циклогексильной группе, является частью фталимидо кольца; удаляемый, например, путем обработки гидразином. В этом случае атом водорода у атома азота отсутствует(см. пример 17). Удаление защитных групп у соединения формулы IIa приводит к соединению формулы I в виде единственного продукта. В случае удаления защитных групп у смеси соединения формулы На и соединения формулы IIb полученные продукты формул Ia и Ib подвергают кристаллизации/перекристаллизации и соединение формулы I получают выделением из смеси. Было обнаружено, что в процессе проведения реакции удаления аминозащитной группы стереохимия атомов углерода циклогексильной группы с присоединенными тио-, гидрокси- и аминогруппой соответственно сохраняется такой же, как в аминозащищенных соединениях формул IIa и IIb, используемых в качестве исходных продуктов. Неожиданно было обнаружено, что соединение формулы I может быть кристаллизовано после выделения или даже непосредственно в реакционной смеси, полученной после удаления защитной группы у аминогруппы. Например, раствор соединения формулы I в виде отдельной стереоизомерной формы,-3 023273 полученный в органическом растворителе, предпочтительно полярном органическом растворителе, такой как CH2Cl2, обрабатывают антирастворителем, таким как простой эфир, например диизопропиловый эфир (DIPE) или трет-бутиловый метиловый эфир (МТВЕ), предпочтительно DIPE; или раствор соединения формулы I в виде отдельной стереоизомерной формы, полученной в органическом растворителе,предпочтительно полярном органическом растворителе, таком как CH2Cl2, обрабатывают после концентрирования органическим растворителем, предпочтительно спиртом, таким как н-бутанол; или выделенное сырое вещество соединения формулы I в виде отдельной стереоизомерной формы обрабатывают органическим растворителем, таким как простой эфир, например тетрагидрофуран (ТГФ), и, необязательно,обрабатывают антирастворителем, таким как простой эфир, например DIPE или MTBE. Соединение формулы I в виде отдельной стереоизомерной формы, может быть кристаллизовано и может быть выделено в кристаллической форме. Кристаллизация упрощает выделение и обработку выделяемых веществ и обеспечивает превосходные возможности для последующей очистки продукта. Кристаллические соединения, полученные в соответствии с настоящим изобретением, могут быть подвергнуты дальнейшей очистке, такой как перекристаллизация, например, из органического растворителя, предпочтительно спирта, такого как н-бутанол.(Пере)кристаллизация может быть повторена, если желательно, и может приводить к высоким выходам и превосходной очистке. В табл. 1 показана удивительно высокая возможность очистки кристаллического соединения формулы I. Таблица 1RRT=относительное время удерживание по данным ВЭЖХ по отношению к I. Как ясно можно видеть из табл. 1 выше, чистота соединения формулы I после удаление защитных групп у соединения формулы IIa повышалась от 87,3 до 95,0% после кристаллизации и выделения изDCM (CH2Cl2)/DIPE. Чистота далее повышалась при (пере)кристаллизациях из н-бутанола до конечной чистоты выделенного соединения формулы I, равной 99,7%. В табл. показано снижение содержания отдельных примесей и повышение общей чистоты выделяемого из маточного раствора соединения формулы I после перекристаллизации из н-бутанола. Таблица 2 Кристаллизация может быть усилена и ускорена за счет использования затравочных кристаллов. Затравочные кристаллы могут быть получены способами, проиллюстрированными здесь примерами. Способ кристаллизации и перекристаллизации соединения формулы I обеспечивает желаемую чистоту и является весьма полезным в промышленных масштабах. Исключаются трудоемкая и дорогая очистка с помощью хроматографии, что делает способ промышленно применимым. Еще более неожиданно кристаллизация соединения I приводит к повышению диастереомерной чистоты соединения формулы I, когда исходят из смеси соединения формулы I и соединения формулы Ib. Удаление защитных групп у смеси соединения формулы IIa и соединения формулы IIb и кристаллизация полученного твердого вещества из органических растворителей или смесей растворителей, таких какDCM/DIPE, приводит к существенному повышению содержания диастереомеров. Повышение содержания диастереомеров становится еще более заметным, если осуществлять перекристаллизацию из органического растворителя, предпочтительно спирта, такого как н-бутанол. В табл. 3 показано повышение хиральной чистоты соединения формулы I в виде кристаллической формы. Таблица 3 Из табл. 3 видно, что использование диастереомерной смеси соединений формулы IIa и IIb на стадии удаления защитных групп и выделение полученных продуктов из реакционной смеси путем кристаллизации из DCM/DIPE приводит к удивительному увеличению хиральности 59:41 в пользу соединения формулы I. Осуществление перекристаллизации полученной обогащенной смеси продуктов соединения формулы I и соединения формулы Ib из н-бутанола приводит к существенному и еще более удивительному обогащению в пользу соединения формулы I в выделенном продукте (93:7). В противоположность выделенному продукту, обогащенному соединением формулы I, н-бутанольная маточная жидкость содержит почти исключительно соединение формулы Ib (84%). Необязательно, перекристаллизация может быть повторена до достижения желаемой оптической чистоты. Удивительные свойства соединения формулы I в виде кристаллической формы, которые показаны в табл. 1-3, являются чрезвычайно полезными при получении фармацевтического соединения. Настоящее изобретение относится к соединению формулы I в виде отдельного стереоизомера кристаллической формы. Неожиданно были выделены и охарактеризованы методом рентгеновской порошковой дифрактометрии две различные кристаллические формы соединения формулы I. В соответствии с другим аспектом настоящее изобретение относится к 14-O-[(1R,2R,4R)-4-амино 2-гидроксициклогексилсульфанил]ацетилмутилину кристаллической формы 1, например, где кристаллическая форма 1 охарактеризована с помощью рентгеновской порошковой дифрактометрии с пиками 2-тета при (интенсивность, 0,2, среди прочих): 10,6, 11,1, 12,0, 14,3, 15,1, 16,1, 21,1; такие как: 10,6, 11,1, 12,0, 14,3, 15,1, 16,1, 18,2, 19,2, 20,7, 21,1, 21,3, 21,8, 22,6, 23,5, 24,7, 28,2, 30,2. В соответствии с другим аспектом настоящее изобретение относится к 14-O-[(1R,2R,4R)-4-амино 2-гидроксициклогексилсульфанил]ацетилмутилину кристаллической формы 2, полученному в виде н-бутанольного сольвата 1:1, например, который характеризуется с помощью рентгеновской порошковой дифрактометрии с пиками 2-тета при (интенсивность, 0,2, среди прочих): 9,8, 11,1, 13,1, 14,1, 17,6, 19,7, 22,2; такие как: 9,6, 9,8, 11,1, 13,1, 14,1, 16,0, 17,6, 19,7, 22,2, 22,7, 23,0. Содержание н-бутанола в 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилине кристаллической формы 2 составляет между 10 и 18% масс/масс, обычно между 11 и 14% мас./мас. Соединение формулы I предпочтительно выделяют в виде соли, такой как фармацевтически приемлемая соль. Такие соли представляют собой предпочтительно соли аммония, а именно соли добавления кислот аминогруппы, присоединенной к циклогексильному кольцу, и включают ацетаты, лактаты, например L-лактаты, или малеаты, например гидромалеаты. В соответствии с другим аспектом настоящее изобретение относится к соединению формулы I в виде отдельного стереоизомера и в виде соли, например кристаллической соли, где соединение формулы I представлено в виде отдельного стереоизомера; соль предпочтительно представляет собой ацетат, лактат,например L-лактат, или малеат, например гидромалеат. Кристаллические соединения, полученные в соответствии с настоящим изобретением в виде отдельного стереоизомера формулы I, например в виде соли, используются в фармацевтической композиции. В соответствии со следующим аспектом настоящее изобретение относится к фармацевтической композиции, включающей кристаллический 14-O-[1R,2R,4R)-4-амино-2-гидроксициклогексил)сульфанил]ацетилмутилин или включающей необязательно кристаллический, ацетат, лактат или гидромалеат 14-O-[1R,2R,4R)-4-амино-2-гидроксициклогексил)сульфанил]ацетилмутилина в виде активного ингредиента в сочетании с фармацевтически приемлемым носителем или разбавителем. Полученные таким образом соединения формулы I в свободной форме и в виде отдельного стереоизомера могут быть преобразованы в соль путем добавления кислоты к свободному основанию соединения формулы I. Например, соединение формулы I может быть растворено или суспендировано, или получено в растворенной или суспендированной форме в органическом растворителе, предпочтительно растворителе или смеси растворителей, такого как спирт, например метанол, этанол; галогенированный углеводород, например дихлорметан; простой эфир, например тетрагидрофуран, ацетат, такой как метилацетат, этилацетат, пропилацетат, изопропилацетат; смесь растворителей из спирта с ацетатом,такая как метанол/изопропилацетат, смесь растворителей из галогенированного углеводорода с ацетатом,такая как дихлорметан/изопропилацетат, смесь растворителей из простого эфира с ацетатом, такая как тетрагидрофуран/изопропилацетат, и в полученную смесь добавляют кислоту, например органическую кислоту или неорганическую (минеральную) кислоту, например, уксусную кислоту, молочную кислоту,например L-молочную кислоту, малеиновую кислоту. Соединение формулы I может быть получено и может быть выделено в виде соли, например кристаллической соли. Было обнаружено, что на стадии образования соли стереохимия атомов углерода циклогексильного кольца, к которому присоединены тио-, гидрокси- или аминогруппа, сохраняется и остается такой же,как в соединении формулы I в свободной форме. Выделение фармацевтически активного соединения, как описано в WO 2008/113089, пример I и Ia соответственно, является удобным; но выделение в соответствии с настоящим изобретением в виде кристаллической соли, такой как соединение формулы I в виде кристаллической соли, намного более удобно. Например, повышенная чистота и неожиданно улучшенная стабильность кристаллической соли в соответствии с настоящим изобретением являются очень важными и полезными при получении фармацевтических композиций, предназначенных для использования в ветеринарии и у людей. Кристаллические соли соединения формулы I демонстрируют улучшенную стабильность относительно соответствующих аморфных форм и выделяются с химической и хиральной чистотой 90%, например даже 95%. В табл. 4 показаны сравнительные данные по стабильности соответствующих кристаллических и аморфных солей соединения I. Таблица 4 Образец полностью не растворился в воде через 28 дней (28 д) и поэтому анализировали суспензию. Растворимость фармацевтически активного соединения является важным свойством для доступности in vivo, например пероральной биодоступности. Для внутривенного применения растворимость выбранной формы, например соли, также весьма важна, и является желательным иметь препараты без сложных технологий или эксципиентов. Растворимость солей кристаллического ацетата, лактата и (гидро)малеата по настоящему изобретению в воде и разбавителе на водной основе, таком как 0,9% раствор NaCl, или в биорелевантной среде,такой как FaSSIF (Fasted State Simulated Intestinal Fluid) и FeSSIF (Fed State Simulated Intestinal Fluid),-6 023273 является неожиданно высокой, что делает кристаллические соли по настоящему изобретению еще более фармацевтически целесообразными. Противомикробная,например антибактериальная,активность 14-O-[(4-амино-2 гидроксициклогексил)сульфанил]ацетилмутилинов была описана в WO 2008/113089. Например, соединения по настоящему изобретению, например соединение формулы I, показывает противомикробную,например антибактериальную, активность против грамположительных бактерий, таких как коагулазо положительные стафилококки, например золотистый стафилококк и стрептококк, например пневмококк,например проявляя МИК 0,4 мкг/мл против золотистого стафилококка АТСС 49951 и пневмококка АТСС 49619. Минимальная ингибирующая концентрация (МИК) была определена в соответствии с рекомендациями CLSI. В соответствии со следующим аспектом настоящее изобретение относится к кристаллическому 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилину в виде ацетата кристаллической формы А, например, который характеризуется с помощью рентгеновской порошковой дифрактометрии пиками 2-тета при (интенсивность, 0,2, среди прочих): 7,0, 7,7, 11,6, 12,1, 12,6, 13,5, 13,7, 15,4, 15,7, 16,9, 17,3, 19,0, 19,9, 21,1, 23,4, 24,2, 24,4; такие как: 7,0, 7,7, 11,6, 12,1, 12,6, 13,5, 13,7, 14,1, 15,4, 15,7, 16,5, 16,9, 17,3, 19,0, 19,6, 19,9, 20,1, 21,1, 22,2,22,5, 23,4, 24,2, 24,4, 26,7, 29,1, 29,6, 31,0. В соответствии со следующим аспектом настоящее изобретение относится к кристаллическому 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилину в виде ацетата кристаллической формы В, например, который характеризуется с помощью рентгеновской порошковой дифрактометрии пиками 2-тета при (интенсивность, 0,2, среди прочих): 10,3, 10,7, 12,7, 14,3, 15,5, 16,0, 17,2, 19,5, 20,6, 22,9; такие как: 9,0, 10,3, 10,7, 12,7, 14,3, 15,5, 16,0, 17,2, 19,5, 20,6, 21,7, 22,3, 22,7, 22,9, 24,4. В соответствии со следующим аспектом настоящее изобретение относится к кристаллическому 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилину в виде L-лактата кристаллической формы 1, например, который характеризуется с помощью рентгеновской порошковой дифрактометрии пиками 2-тета при (интенсивность, 0,2, среди прочих): 7,0, 11,6, 12,0, 12,5, 13,4, 13,6, 13,9, 15,3, 16,8, 18,8, 19,5, 19,8, 20,9, 23,3, 23,9, 24,2; такие как: 7,0, 7,6, 11,6, 12,0, 12,5, 13,4, 13,6, 13,9, 15,3, 15,5, 16,8, 17,2, 18,8, 19,5, 19,8, 20,0, 20,9, 22,0, 22,4,22,7, 23,3, 23,9, 24,2; 25,3, 28,9, 29,4, 30,8. В соответствии со следующим аспектом настоящее изобретение относится к кристаллическому 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина в виде гидромалеата кристаллической формы 1, например, который характеризуется с помощью рентгеновской порошковой дифрактометрии пиками 2-тета при (интенсивность, 0,2, среди прочих): 7,0, 11,3, 11,7, 12,5, 13,5, 13,8, 15,3, 16,7, 18,3, 19,4, 19,7, 21,1, 22,2, 23,8, 23,9; такие как: 7,0, 11,3, 11,7, 12,5, 13,3, 13,5, 13,8, 14,1, 15,3, 16,7, 17,2, 18,0, 18,3, 19,4, 19,7, 20,4, 21,1, 21,9, 22,2,22,8, 23,8, 23,9, 24,9, 27,1, 27,8, 28,7, 29,3, 30,6, 30,8. Соединение формулы I в виде (кристаллического) свободного основания проявляет такой же уровень фармацевтической активности, такой как противомикробная активность, что и соединение по настоящему изобретению в виде (кристаллической) соли. Соединение формулы IIa в виде отдельного стереоизомера или смесь соединения формулы IIa и соединения формулы IIb могут быть получены, по мере необходимости, например, в соответствии, например, по аналогии с общеизвестным способом. Предпочтительно соединение формулы IIa в виде отдельного стереоизомера или смесь соединения формулы IIa и соединения формулы IIb получают путем конденсации аминогидроксимеркаптоциклогексанового соединения формулы IIIa или смеси соединения формулы IIIa и соединения формулы IIIb, где аминогруппа защищена аминозащитной группой, с активированным 14-O-ацетилмутилином. В соответствии со следующим аспектом настоящее изобретение относится к способу в соответствии с настоящим изобретением, где соединение формулы IIa или смесь соединения формулы IIa и соединения формулы IIb получают путем конденсации либо соединения формулы IIIa либо смеси соединения формулы IIIa и соединения формулы IIIb где АКТ представляет собой активирующую группу, необязательно мезил, безил, тозил, или-О-АКТ представляет собой галоген; необязательно 14-О-АКТ-ацетил-мутилин представляет собой соединение формулы и выделение из реакционной смеси полученных либо соединения формулы IIa, либо смеси соединения формулы На и соединения формулы IIb. Соединения формулы IIIa, где R имеет значения, указанные выше, являются новыми и также составляют часть настоящего изобретения. В соответствии с другим аспектом настоящее изобретение относится к соединению формулы IIIa в виде отдельной стереоизомерной формы. В соединении формулы IIIa атомы углерода циклогексильного кольца, к которому присоединены гидроксигруппа, аминогруппа и тиогруппа, все имеют R конфигурацию, и поэтому соединение формулыIIIa представляет собой необязательно аминозащищенный (1R,2R,4R)-4-амино-2-гидрокси-1 меркаптоциклогексан; и в соединении формулы IIIb соответственно атомы углерода циклогексильного кольца, к которому присоединены гидроксигруппа, аминогруппа и тиогруппа, все имеют S конфигурацию, и поэтому соединение формулы IIIb представляет собой необязательно аминозащищенный(1S,2S,4S)-4-амино-2-гидрокси-1-меркаптоциклогексан. Реакция конденсации соединения формулы IIIa или смеси соединения формулы IIIa и соединения формулы IIIb соответственно, где R имеет значения, указанные выше, с активированным 14-O-АКТ-ацетил-мутилином, где АКТ имеет значения, указанные выше, для получения соединения формулы IIa или смеси IIa и IIb, где R имеет значения, указанные выше, может быть осуществлена, по мере необходимости, например, соответственно, например, аналогично общеизвестному способу, такому как в стандартных условиях, известных для этих реакций; например, в присутствии основания, например сильного неорганического основания, подобного гидроксиду, такому как NaOH, например, в двухфазной системе, и, если реакцию осуществляют в двухфазной системе, предпочтительно в присутствии катализатора, такого как катализатор фазового переноса, например бензил-трибутиламмоний хлорид. Реакция конденсации также может быть осуществлена в системе отдельного растворителя, например в органическом растворителе, таком как галогенированный углеводород, например хлорбензол, дихлорметан, ароматический растворитель, такой как толуол, нитрил, такой как ацетонитрил или простой эфир, например трет-бутил-метиловый эфир, тетрагидрофуран, в присутствии основания, предпочтительно органического основания, такого как DBU. Предпочтительно реакцию конденсации осуществляют в органическом растворителе, например аполярном растворителе, таком как простой эфир, например трет-бутилметиловый эфир (МТВЕ), или полярном растворителе, таком как галогенированный углеводород, например дихлорметан; предпочтительно в присутствии водного раствора основания, такого как водный NaOH; или в присутствии основания в органическом растворителе, такого как DBU, DBN, предпочтительно в присутствии катализатора фазового переноса в случае использования водного раствор основания, такого как бензилтрибутиламмоний хлорид. Было обнаружено, что в процессе реакции конденсации стереохимия атомов углерода циклогексильной группы, к которой присоединены тио-, гидрокси- и аминогруппа, сохраняется и остается такой же, как в соединении формулы IIIa и соединении формулы IIIb соответственно, используемых в качестве исходного продукта. Соединение формулы IIIa в виде отдельного стереоизомера или смеси соединения формулы IIIa и соединения формулы IIIb соответственно могут быть получены, по мере необходимости, например, соответственно, например, аналогично общеизвестному способу. Предпочтительно соединение формулы IIIa или смесь соединения формулы IIIa и соединения формулы IIIb соответственно получают путем удаления защитной группы тиола в аминогидрокси-меркаптоциклогексане, где обе группы, аминогруппа и тиоловая группа, защищены. В соответствии с другим аспектом настоящее изобретение относится к способу в соответствии с настоящим изобретением, где соединение формулы IIIa или смесь соединения формулы IIIa и соединения формулы IIIb получают путем удаления тиоловой группы, либо в соединении формулы IVaR1 представляет собой защитную группу тиола, и выделение из реакционной смеси полученных либо соединения формулы IIIa, либо смеси соединения формулы IIIa и соединения формулы IIIb соответственно. Защитная группа тиола, например, в значении R1 в соединении формулы IVa и IVb в соответствии с настоящим изобретением включает, например-(C1-6)алкил, где алкил необязательно дополнительно замещен, например далее замещен(С 6-12)арилом, таким как фенил, такой как тритил; например, который может быть удален с помощью сильной кислоты или обработкой AgNO3;-(C1-6)алкилкарбонил, например ацетил, например, который может быть удален обработкой с помощью основания, такого как метоксид натрия, -(С 6-12)арилкарбонил, такой как бензоил, например, который может быть удален путем обработки восстанавливающим агентом, таким как DIBAL, или путем обработки основанием, таким как гидразин, предпочтительно -C(=O)-(C6-12)арил; более предпочтительно бензоил. Подходящие защитные группы для серы, например, описаны в Т.W. Greene, P.G.M. Wuts, ProtectiveGroups in Organic Synthesis, Wiley-Interscience, 4th Edition, 2007, в частности p. 647-695. В соединении формулы IVa атомы углерода циклогексильного кольца, к которому присоединены гидроксигруппа, аминогруппа и тиогруппа, все имеют R конфигурацию, и поэтому соединение формулыIVa представляет собой аминои тиозащищенный(1R,2R,4R)-4-амино-2-гидрокси-1 меркаптоциклогексан; и в соединении формулы IVb атомы углерода циклогексильного кольца, к которому присоединены гидроксигруппа, аминогруппа и тиогруппа, все имеют S конфигурацию, и поэтому соединение формулы IVb представляет собой амино- и тиозащищенный (1S,2S,4S)-4-амино-2-гидрокси 1-меркаптоциклогексан. Было обнаружено, что в процессе реакции удаления защитных групп тиола стереохимия атомов углерода циклогексильной группы, к которой присоединены тио-, гидрокси- и аминогруппа, сохраняется и остается такой же, как в соединении формулы IVa и соединении формулы IVb соответственно, используемых в качестве исходного продукта. Удаление защитных групп у тиоловой группы осуществляют в зависимости от ситуации, например с использованием отщепляющего агента, например гидразингидрата. Реакцию удаления защитных групп осуществляют в органическом растворителе, таком как галогенированный углеводород, например хлорбензол, дихлорметан, ароматический растворитель, такой как толуол, нитрил, такой как ацетонитрил или простой эфир, например трет-бутил-метиловый эфир, тетрагидрофуран. Для минимизации образования дисульфида удаление защитных групп, необязательно, осуществляют в присутствии антиоксиданта или восстанавливающего агента, например дитиотреитола (DTT). Соединение формулы IVa или смесь соединения формулы IVa и соединения формулы IVb соответственно могут быть получены в зависимости от ситуации, например аналогично обычному способу или как здесь описано, например в разделе "Примеры". В соответствии с другим аспектом настоящее изобретение относится к способу получения соединения формулы I в виде отдельного стереоизомера, включающему использование соединения формулы IIa в виде отдельного стереоизомера, или смеси соединения формулыIIa и соединения формулы IIb соответственно, и/или включающему использование соединения формулыIIIa в виде отдельного стереоизомера или смеси соединения формулы IIIa и соединения формулы IIIb соответственно и/или использование соединения формулы IVa в виде отдельного стереоизомера или смеси соединения формулы IVa и соединения формулы IVb соответственно в качестве промежуточного соединения; и к соединению формулы IIa в виде отдельного стереоизомера, и/или соединению формулы IIIa в виде отдельного стереоизомера, и/или соединению формулы IVa в виде отдельного стереоизомера для применения в качестве промежуточного соединения в способе получения соединения формулы I в виде отдельного стереоизомера. В соответствии со следующим аспектом настоящее изобретение относится к способу получения соединения формулы I в виде отдельного стереоизомера, включающему конденсацию соединения формулыIIIa в виде отдельного стереоизомера, или смеси соединения формулы IIIa и соединения формулы IIIb соответственно, где R имеет значения, указанные выше, с активированным 14-O-АКТ-ацетилмутилином, где АКТ является активирующей группой, такой как мезил, безил или тозилгруппа, или-О-АКТ представляет собой галоген, предпочтительно АКТ представляет собой тозильную группу,например, соединение формулы Tos-PLEU, для получения соединения формулы IIa или смесь соединения формулы IIa и соединения формулы IIb соответственно, где R имеет значения, указанные выше; необязательно выделение соединения формулы IIa в виде отдельного стереоизомера или смеси соединения формулы IIa и соединения формулы IIb соответственно, где R имеет значения, указанные выше, полученные из реакционной смеси; удаление функциональной аминогруппы в полученных соединениях формулы IIa в виде отдельного стереоизомера в смеси соединения формулы IIa и соединения формулы IIb соответственно, где R имеет значения, указанные выше; и выделение соединения формулы I в виде отдельного стереоизомера, необязательно в виде соли, из реакционной смеси, необязательно, путем перекристаллизации, и, если желательно преобразование соединения формулы I в виде отдельного стереоизомера, полученного в свободной форме, в соединение формулы I в солевой форме в виде отдельного стереоизомера, или наоборот; где необязательно соединение формулы IIIa в виде отдельного стереоизомера или смесь соединения формулы IIIa и соединения формулы IIIb соответственно, где R имеет значения, указанные выше, получают путем удаления защитных групп у тиоловой функциональной группы в соединении формулы IVa в виде отдельного стереоизомера или в смеси соединения формулы IVa и соединения формулы IVb, где R и R1 определены выше. В качестве аминозащитной группы в соединении формул IIa, IIb, IIIa, IIIb, IVa или IVb могут быть использованы общеизвестные аминозащитные группы, предпочтительно трифторацетильная или третбутоксикарбонильная группа. При необходимости, аминозащитная группа R в соединении формул IIa,IIb, IIIa, IIIb, IVa или IVb может быть изменена таким образом, что атом углерода циклогексильного кольца, к которой присоединена аминогруппа, не меняет своей конфигурации, т.е. не изменяет стереохимию, например, путем удаления защитных групп у аминогруппы, с последующей защитой с отличающейся аминозащитной группой. Способы по настоящему изобретению позволяют выделить соединения формулы I в виде отдельного стереоизомера кристаллической формы. С одной стороны, способ контролирует, когда исходят из хиральных чистых исходных материалов, стереохимию и выходы продуктов в виде отдельных стереоизомеров, в соответствии с чем нет необходимости в усложненных способах разделения смесей диастереомеров и изомеров положения, таких как хроматография, например нормально фазовая или хирально фазовая, например хиральная ВЭЖХ, например, описанные в WO 2008/113089, что является большим преимуществом по отношению к промышленно применимому способу. Обнаружение кристаллического соединения формулы I является ключевым в изобретении и позволяет осуществить идеальный контроль химической чистоты. В связи с наблюдаемыми желаемыми выходами кристаллических форм соединения формулы I способы по настоящему изобретению являются чрезвычайно приемлемыми для получения лекарственного средства, когда подходящий/хороший выход необходим с экономической точки зрения, и контроль чистоты имеет важное значение для конечного качества активного фармацевтического ингредиента (API). С другой стороны, способы в соответствии с настоящим изобретением контролируют даже более неожиданно хиральную чистоту соединения формулы I опять по причине неожиданных свойств кристаллического соединения I. Неожиданные свойства кристаллического соединения I позволяют исходить из намного менее дорогих диастереомерных смесей. Хиральная чистота контролируется по кристаллическому соединению I, таким образом, соединение I выделяют в виде кристаллического твердого продукта,и количество нежелаемого диастереомера в маточной жидкости понижено. Соединения формулы I в кристаллической форме никаким образом не описаны в WO 2008/113089. В примере 1 стадии В в WO 2008/113089 смесь 14-O-[(1R, 2R, 4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина (соединение I по настоящему изобретению) и 14-O-[(1S,2S,4S)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина выделена в виде аморфной пены. В примере 1 А в WO 2008/113089 описано разделение диастереомеров посредством хиральной хроматографии, и разделенные 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилинI по настоящему изобретению) и 14-O-[(1S,2S,4S)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилин (соединение Ib по настоящему изобретению) вновь выделяют в виде аморфной пены. Неожиданные свойства кристаллического соединения I, контролирующие химическую и хиральную чистоту соединения I, являются чрезвычайно ценными при получении фармацевтического активного соединения для введения людям или животным. Настоящее изобретение, кроме того, относится к новым кристаллическим формам 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина в виде ацетата,L-лактата или гидромалеата. 14-O-[(1R,2R,4R)-4-Амино-2-гидроксициклогексилсульфанил]ацетилмутилин может быть преобразован в формы кристаллических солей с использованием способа кристаллизации в органической среде с образующим соль агентом. Способ получения новых кристаллических солей может быть усилен и ускорен за счет использования затравочных кристаллов. Затравочные кристаллы могут быть получены показанными в примерах способами. Было обнаружено, что кристаллические соли по настоящему изобретению сохраняют стереохимию соединения формулы I в свободном виде в виде отдельного стереоизомера. Кристаллические соли в соответствии с настоящим изобретением имеют явные неожиданные преимущества, т.е. обладают повышенной стабильностью по сравнению с соответствующими формами аморфных солей, как, например,это видно из табл. 4. Кроме того и довольно неожиданно, гигроскопичность кристаллической соли по настоящему изобретению, предпочтительно соли L-лактата и ацетата кристаллической формы В, является незначительной в соответственных интервалах уровней влажности, т.е. при соответственной влажности между 0-80% поглощение воды ниже 2%, что делает соли в сочетании с превосходной химической стабильностью фармацевтически приемлемыми. Кристаллические соли по настоящему изобретению имеют желаемую и соответствующую химическую и хиральную чистоту, обладают лучшей стабильностью по сравнению с аморфными лиофилизованными формами, удобны при хранении и получении фармацевтических композиций. Форма кристаллической соли соединения формулы I не была описана ранее, например, в WO 2008/113089. В целом, формы кристаллических солей соединения формулы I обладают превосходной чистотой, а наблюдаемая стабильность не только превосходит стабильность форм аморфных солей, но и представляется абсолютно фармацевтически выгодной при нормальных условиях хранения. Как здесь используется, "в виде отдельного стереоизомера" обозначается форма, когда соединение показывает диастереомерный или энантиомерный избыток 90% указанной стереохимии. Особенно предпочтительные соединения по настоящему изобретению включают соединения по примерам 1-17, например, соединение, выбранное из группы, включающей трет-бутил [(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]карбамат,14-O-[(1R,2R,4R)-4-трет-бутоксикарбониламино-2-гидроксициклогексилсульфанил]ацетилмутилин,14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилин,кристаллическая форма 1,14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилин,кристаллическая форма 2,2,2,2-трифтор-N-[(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]ацетамид,14-O-[(1R,2R,4R)-4-[(2,2,2-трифторацетил)амино]-2-гидроксициклогексилсульфанил]ацетилмутилин,14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилин ацетат, кристаллическая форма А,14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилин ацетат, кристаллическая форма В,14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилин L-лактат, кристаллическая форма 1,14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилин гидромалеата, в виде кристаллической формы 1,14-O-[(1R,2R,4R)-4-этоксикарбониламино-2-гидроксициклогексилсульфанил]ацетилмутилин, и 14-O-[(1R,2R,4R)-2-гидрокси-4-(фталимидо-N-ил)циклогексилсульфанил]ацетилмутилин. Краткое описание чертежей На фиг. 1 представлены данные порошковой дифрактограммы 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина, форма 1. На фиг. 2 показана порошковая дифрактограмма 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина, форма 2. На фиг. 3 показана порошковая дифрактограмма 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина в виде ацетата, форма А. На фиг. 4 показана порошковая дифрактограмма 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина в виде ацетата, форма В. На фиг. 5 показана порошковая дифрактограмма 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина в виде L-лактата, форма 1. На фиг. 6 показана порошковая дифрактограмма 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина в виде гидромалеата, форма 1. Способ синтеза соединения формулы I, исходя из хиральных предшественников IIa, IIIa и IVa,представлен на схеме реакции 1. Схема реакции 1 На схеме реакции 1 R представляет собой аминозащитную группу и R1 представляет собой защитную группу серы и определены выше. Способ синтеза соединения формулы I, исходя из смесей IIa и IIb, IIIa и IIIb и IVa и IVb, представлен на схеме реакции 2. На схеме реакции 2 R представляет собой аминозащитную группу и R1 представляет собой защитную группу серы (R и R1 имеют значения, указанные выше). В данном документе, включая примеры и включая схемы реакций 1-6, используются следующие аббревиатуры:C - градусы Цельсия; 1 Н ЯМР - спектроскопия протонного ядерного магнитного резонанса; 13 С ЯМР - спектроскопия углеродного ядерного магнитного резонанса;[]D - угол специфического оптического вращения при 589 нм;"Массовый стрип-анализ", как указано в примерах, определяется следующим образом: содержание аликвоты партии или всей партии определяется путем удаления растворителя и определения содержания с помощью ВЭЖХ или ЯМР, используя внутренний или внешний стандарт и/или вычитая идентифицированные примеси из соединения. В случае аликвоты обратная экстраполяция к общей массе/объем не осуществляется."Линейной промывкой", как указано в примерах, является система промывки с помощью соответствующего растворителя, чтобы минимизировать потери продукта и вводимых веществ.Tos-PLEU представляет собой соединение формулыPLEU представляет собой остаток формулы Соединение, предложенное в настоящем изобретении, указывается здесь также как "соединение(я) по (в соответствии с) настоящему(им) изобретению(ем)", и способ, предложенный в настоящем изобретении, указывается здесь также как "способ(ы) по (в соответствии с) настоящему(им) изобретению(ем)". Соединение по настоящему изобретению может быть получено, в зависимости от ситуации, например, аналогично общеизвестному способу или как здесь описано. В следующих примерах все температуры даны в градусах Цельсия (С) и являются нескорректированными. Используемые здесь аббревиатуры определены выше (после схемы реакции 2). 3,94 кг (1R,2R,4R)-4-[(трет-бутоксикарбонил)-амино]-2-гидроксициклогексилбензолкарботиоата и 37 л CH2Cl2 загружали в сосуд и полученную смесь перемешивали при температуре 15-25 С. К смеси добавляли 0,39 кг 1,4-дитио-DL-треитола (10 мас.%) и промывали с помощью 2 л CH2Cl2. В полученную смесь добавляли 0,84 кг моногидрата гидразина. Полученную смесь перемешивали при температуре 18-22 С в течение 3 ч и протекание реакции отслеживали с помощью ВЭЖХ. После завершения взаимодействия добавляли 39 л 1 М раствора фосфорной кислоты и полученную смесь перемешивали в течение еще 15-30 мин. Две образовавшиеся фазы разделяли и полученную органическую фазу промывали с помощью 39 л 1 М раствора фосфорной кислоты, затем 39 л 1%-ного водного раствора NaCl. Полученный органический слой упаривали в вакууме при температуре 40 С, к упаренному остатку добавляли 20 лCH2Cl2 и полученную смесь опять упаривали. К полученному упаренному остатку добавляли еще 8 лCH2Cl2 и полученную смесь упаривали досуха. Получали 2,89 кг трет-бутил [(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]карбамата в виде твердого вещества белого цвета. 1 22-О-Тозилплеуромутилин является известным в литературе соединением. Тем не менее, далее представлен способ его получения. Раствор 13,0 кг плеуромутилина и 6,57 кг 4-толуолсульфонил хлорида в 42,1 л CH2Cl2 при температуре от 10 до 15 С обрабатывали 9,1 л 5,7 М водного раствора NaOH в течение 20 мин, поддерживая температуру 25 С. Полученную суспензию не совсем белого цвета нагревали при кипячении с обратным холодильником в течение 20 ч и реакцию проводили до полного завершения, что определялось с помощью ВЭЖХ. После завершения взаимодействия полученную смесь охлаждали до температуры от 20 до 30 С, разбавляли 52 л CH2Cl2, перемешивали при температуре от 15 до 25 С в течение 10 мин и полученные слои разделяли. Полученную органическую фазу промывали несколько раз 52 л воды до достижения рН водного слоя 9. Полученный органический слой упаривали до 4 объемов и азеотропно высушивали два раза с помощью 52 л CH2Cl2. К полученному раствору добавляли по каплям 52 л гептана и полученный раствор упаривали при температуре 40 С до приблизительно 4 объемов. К полученному концентрату добавляли 52 л гептана и полученную суспензию перемешивали при температуре от 20 до 25 С в течение от 2 до 2,5 ч, фильтровали, полученную лепешку на фильтре промывали с помощью 39 л гептана и отжимали досуха на фильтре. Твердый продукт сушили в вакууме при температуре 40 С в течение по меньшей мере 12 ч. Получали 16,9 кг 22-O-тозилплеуромутилина в виде твердого вещества белого цвета. 1 4,75 кг тозилата плеуромутилина (Tos-PLEU) и 44,4 л МТВЕ загружали в реакционный сосуд, в полученную смесь добавляли 0,31 кг бензил-три-н-бутиламмоний хлорида и промывали с помощью 2,4 л МТВЕ. В полученную смесь добавляли 20 л 1 М водного раствора NaOH и 2,84 кг трет-бутил[(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]карбамата и полученную смесь перемешивали при температуре от 17 до 23 С в течение 3 ч. После завершения взаимодействия (определено с помощью ВЭЖХ) образовавшиеся два слоя разделяли и нижний водный слой удаляли. Полученную органическую фазу промывали с помощью 19 л 1 М водного раствора NaOH, два раза 20 л 0,1 М фосфорной кислоты, 20 л 10%-ного водного раствора NaHCO3 и два раза 20 л воды. Полученные органические жидкости упаривали, полученный концентрат обрабатывали 7,46 кг 2-пропанола, полученную смесь опять упаривали и сушили в вакууме при температуре 40 С. Получали 6,66 кг 14-O-[(1R,2R,4R)-4-третбутоксикарбониламино-2-гидроксициклогексилсульфанил]ацетилмутилина в виде пены белого цвета. 1 Стадия A. 14-O-[(1R, 2R, 4R)-4-Амино-2-гидроксициклогексилсульфанил]ацетилмутилин кристаллической формы 1. 6,6 кг 14-O-[(1R,2R,4R)-4-трет-бутоксикарбониламино-2-гидроксициклогексилсульфанил]ацетилмутилина и 13,2 л изопропанола загружали в реакционный сосуд и перемешивали при температуре от 20 до 25 С. Добавляли 11,20 кг 85%-ной фосфорной кислоты и полученную смесь нагревали приблизительно при 50 С в течение по меньшей мере 16 ч. Полученную смесь анализировали на завершение взаимодействия с помощью ВЭЖХ. После завершения взаимодействия смесь охлаждали до температуры от 20 до 25 С и добавляли 52 л CH2Cl2. Полученную смесь охлаждали до температуры от 0 до 5 С и добавляли 51 л 30%-ного водного раствора K2CO3 в течение 1 ч при температуре 25 С. Полученную смесь нагревали до комнатной температуры, перемешивали в течение 30 мин и определяли рН водного слоя. В полученную смесь добавляли еще 15 л 30%-ного водного раствора K2CO3 при температуре 25 С, полученную смесь перемешивали при температуре от 15 до 25 С в течение 30 мин и полученные две фазы разделяли. Полученную водную фазу экстрагировали 51 л CH2Cl2 и объединенные органические фазы промывали с помощью 51 л очищенной воды. Полученную смесь упаривали до объема 25 л, добавляли 33,6 кг CH2Cl2 и полученную смесь упаривали до 25 л. К полученному концентрату добавляли 33,6 кг CH2Cl2 и полученную смесь упаривали до 10 л. Полученный концентрированный остаток охлаждали до температуры от 18 до 22 С и добавляли 50 л диизопропилового эфира в течение периода в 1 ч. Полученную взвесь перемешивали при температуре от 15 до 25 С в течение минимум 2 ч, фильтровали и полученное твердое вещество промывали с помощью 10 л диизопропилового эфира и сушили. Получали 3,79 кг 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина кристаллической формы 1. Стадия B. 14-O-[(1R,2R,4R)-4-Амино-2-гидроксициклогексилсульфанил]ацетилмутилин, кристаллическая форма 2. Для последующей очистки, 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилин со стадии А и 18,75 л н-бутанола нагревали при температуре от 88 до 92 С до полного раство- 16023273 рения и перемешивали в течение от 30 до 60 мин. Полученной смеси давали охладиться до температуры от 40 до 45 С в течение по меньшей мере 2 ч и далее перемешивали при данной температуре в течение 2 ч. Полученную смесь фильтровали и полученный осадок промывали с помощью 3,75 л н-бутанола, затем 3,75 л МТВЕ. Указанный способ очистки повторяли и полученный продукт сушили в вакууме при температуре 40 С. Получали 3,27 кг кристаллического 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина кристаллической формы 2 в виде твердого вещества белого цвета. 1 К раствору 900 г 14-O- [(1R,2R,4R)-4-трет-бутоксикарбониламино-2-гидроксициклогексилсульфанил]ацетилмутилина в 9 л CH2Cl2 при температуре от 15 до 25 С добавляли 1,8 л ТФУ при температуре от 15 до 25 С и полученный раствор перемешивали в течение 2 ч. После завершения реакции смесь упаривали в вакууме и полученный концентрированный остаток подвергали азеотропной сушке с использованием всего 9 л CH2Cl2. Полученный концентрат растворяли в 4,5 л CH2Cl2, полученный раствор охлаждали до температуры от 0 до 5 С и рН устанавливали равным рН 11 с помощью 3,6 л водного раствора K2CO3 (2,5 М). Двухфазную полученную смесь нагревали до температуры от 15 до 20 С и перемешивали в течение от 5 до 10 мин. Полученные слои разделяли, полученную водную фазу экстрагировали 1,8 л CH2Cl2, полученные органические фазы объединяли, промывали с помощью 2,3 л H2O, сушили над Na2SO4 и упаривали досуха в вакууме при температуре 40 С. Получали сырой 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилин. Выход: 744 г. Для последующей очистки применяли следующий способ. К 744 г сырого 14-O-[(1R,2R,4R)-амино-2-гидроксициклогексилсульфанил]ацетилмутилина добавляли 2,23 л ТГФ и полученную суспензию перемешивали при температуре от 15 до 25 С в течение 60 мин. В полученную смесь добавляли 7,44 л МТВЕ в течение от 15 до 30 мин, полученную суспензию выдерживали в течение 60 мин и фильтровали в атмосфере азота. Собранные твердые вещества промывали с помощью всего 3 л МТВЕ и отжимали досуха на фильтре в атмосфере азота в течение 1,5 ч. Получали 626 г 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексил-сульфанил]ацетилмутилина кристаллической формы 1. Спектр 1 Н ЯМР подтверждал структуру 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина. ЯМР спектр 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина описан в примере 4. Пример 6. 14-O-[(1R,2R,4R)-4-[(2,2,2-Трифторацетил)амино-2-гидроксициклогексилсульфанил]ацетилмутилин. Способ 1. 45,3 г (1R,2R,4R)-4-[(2,2,2-трифторацетил)амино]-2-гидроксициклогексилбензолкарботиоата и 453 мл CH2Cl2 помещали в колбу и полученную смесь дегазировали при температуре 15-25 С с помощью аргона в течение 25 мин. В полученную смесь добавляли по каплям 12,79 г моногидрата гидразина, затем 90,6 мл CH2Cl2 линейной промывки. Полученную смесь перемешивали при температуре от 20 до 25 С в течение 2 ч и протекание реакции отслеживали с помощью ТСХ до завершения. После завершения взаимодействия полученную смесь охлаждали до температуры от 15 до 20 С и промывали с помощью 158,6 мл 2 М раствора HCl. Объединенные фазы разделяли, полученную водную фазу возвращали в ре- 17023273 акционный сосуд, добавляли 158,6 мл насыщенного водного раствора NaCl и полученную смесь вновь экстрагировали 278,8 мл CH2Cl2. Полученные объединенные органические фазы промывали с помощью 90,6 мл насыщенного водного раствора NaCl и полученный органический слой упаривали до 2 объемов в вакууме при температуре 40 С. К полученному концентрату добавляли 226,5 мл CH2Cl2 и полученную смесь упаривали до 2 объемов. К полученному концентрату добавляли 362,4 мл CH2Cl2. Массовый стрип-анализ полученной смеси осуществляли для определения содержания 2,2,2-трифтор-N-[(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]ацетамида, и был определен выход в 27,1 г. Раствор 2,2,2-трифтор-N-[(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]ацетамида (содержащий 27,1 г), полученный, как описано выше, дегазировали с помощью аргона. Добавляли 56,5 г тозилата плеуромутилина, затем 54,3 мл CH2Cl2 линейной промывки и полученную смесь перемешивали при температуре от 20 до 25 С в течение 15 мин. В полученную смесь в течение 30 мин добавляли 34,0 г DBU, растворенного в 34 мл CH2Cl2 и полученную смесь перемешивали при температуре от 20 до 25 С в течение 1 ч до полного завершения реакции. Полученную смесь промывали с помощью 2222,5 мл 2 М H2SO4,затем 2222,5 мл 5%-ного водного раствора NaHCO3 и полученную смесь упаривали досуха в вакууме при температуре 40 С. Получали 72,7 г 14-O-[(1R,2R,4R)-[(2,2,2-трифторацетил)амино-2-гидроксициклогексилсульфанил]ацетилмутилина в виде пены не совсем белого цвета. 1 Стадия А. 2,2,2-Трифтор-N-[(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]ацетамид. 5,79 г (1R,2R,4R)-4-[(2,2,2-трифторацетил)амино]-2-гидроксициклогексилбензолкарботиоата растворяли в 81 мл дихлорметана, добавляли 1,67 г (1,62 мл) гидразингидрата и полученный раствор перемешивали при комнатной температуре в течение 3,5 ч. В полученную смесь добавляли 40 мл 1 М HCl и двухфазную полученную смесь энергично перемешивали в течение 10 мин. Фазы разделяли и полученную органическую фазу промывали с помощью 4 0 мл 1 М HCl. Полученные объединенные водные слои насыщали NaCl и промывали с помощью 30 мл DCM; полученные объединенные органические фазы сушили над безводным сульфатом натрия и упаривали досуха. Получали 3,41 г 2,2,2-трифтор-N-[(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]ацетамида в виде бесцветных кристаллов. 1H ЯМР (200 МГц, ДМСО-d6, м.д., среди прочих)9,31 (д, J=7,2 Гц, 1H), 5,12 (д, J=5,l Гц, 1 Н), 3,823,6 (м, 1H), 3,24-3,09 (м, 1 Н), 2,62-2,5 (м, 1 Н), 2,40 (с, широкий, 1 Н), 2,03-1,84 (м, 2 Н), 1,74-1,71 (м, 1 Н),1,47-1,21 (м, 3 Н). Стадия В. 14-O-[(1R,2R,4R)-[(2,2,2-Трифторацетил)амино]-2-гидроксициклогексилсульфанил]ацетилмутилин. 5,91 г 2,2,2-трифтор-N-[(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]ацетамида и 11,78 г тозилата плеуромутилина растворяли в 82,5 мл DCM и полученный раствор дегазировали с помощью аргона в течение 20 мин. В полученную смесь в течение 30 мин добавляли 6,91 г DBU в 27,5 мл DCM. После завершения взаимодействия (контроль с помощью ТСХ) в полученную смесь добавляли 25 мл 2 М HCl и полученную смесь энергично перемешивали в течение 10 мин. Полученные фазы разделяли, полученную органическую фазу промывали с помощью 12,5 мл 2 М HCl и далее 25 мл 5%-ным раствором бикарбоната натрия, сушили над безводным сульфатом натрия и фильтровали. Из полученного фильтрата упаривали в вакууме растворитель. 16,8 г 14-O-[(1R,2R,4R)-4-[(2,2,2-трифторацетил)амино]-2-гидроксициклогексилсульфанил]ацетилмутилина получали в виде бесцветной пены (вещество содержало тозилатную соль DBU и остаточный DCM). Спектр 1 Н ЯМР подтверждал структуру 14-O-[(1R,2R,4R)-4-[(2,2,2-трифторацетил)амино]-2 гидроксициклогексилсульфанил]ацетилмутилина. ЯМР спектр 14-O-[(1R,2R,4R)-4-[(2,2,2 трифторацетил)амино]-2-гидроксициклогексилсульфанил]ацетилмутилина описан в примере 6,способ 1. 72,7 г сырого 14-O-[(1R,2R,4R)-4-[(2,2,2-трифторацетил)амино-2-гидроксициклогексилсульфанил]ацетилмутилина, 419, 9 мл метанола и 168 мл воды помещали в колбу и полученную смесь нагревали до температуры от 40 до 45 С. В полученную смесь добавляли 67,3 г K2CO3 и полученную смесь перемешивали при температуре от 40 до 45 С в течение 5 ч. Протекание реакции отслеживали с помощью ВЭЖХ до завершения. После завершения взаимодействия полученную смесь охлаждали до температуры от 20 до 25 С, добавляли 588 мл CH2Cl2 и 588 мл 2 М фосфорной кислоты и полученную смесь перемешивали при температуре от 20 до 25 С в течение 15 мин. Полученную смесь фильтровали бифазно, разделяли и полученный органический слой экстрагировали 588 мл 1 М фосфорной кислоты. Объединенные фазы разделяли и к полученным объединенным водным (продукт) слоям добавляли 588 мл CH2Cl2 и полученную смесь охлаждали до температуры от 10 до 15 С. В полученную смесь добавляли по каплям 6 М NaOH при температуре 25 С до достижения рН 9 (потребовалось 275 мл). Полученную смесь фильтровали бифазно и полученные слои разделяли. Полученный органический (продукт) слой упаривали приблизительно до 5 объемов в вакууме при температуре 40 С, добавляли 176 мл мл CH2Cl2 и полученную смесь упаривали еще раз до 2 объемов в вакууме при температуре 40 С. К полученному концентрату добавляли по каплям 323,5 мл н-бутанола, полученную смесь упаривали до 5 объемов в вакууме при температуре 40 С и полученный концентрат перемешивали при температуре от 20 до 25 С в течение 2 ч. Полученную смесь фильтровали, полученный осадок промывали с помощью 117,6 мл н-бутанола и полученное твердое вещество сушили в течение ночи в вакууме при температуре 40 С, получая 44,2 г 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина кристаллической формы 2. 44,2 г сырого 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина помещали в чистый сосуд и добавляли 221 мл н-бутанола. Полученную смесь нагревали до от 88 до 92 С,перемешивали в течение 40 мин, давали охладиться с постоянной скоростью в течение 3 ч до температуры от 40 до 45 С и перемешивали в течение еще 2 ч. Полученную смесь фильтровали, промывали с помощью 44,2 мл н-бутанола, затем 44,2 мл МТВЕ и сушили в вакууме при температуре 40 С. 37,6 г кристаллического 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина кристаллической формы 2 получали в виде кристаллического твердого продукта белого цвета. Спектр 1 Н ЯМР подтверждал структуру 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина. ЯМР спектр 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина описан в примере 4. Пример 8. трет-Бутил [(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]карбамат и трет-бутил [(1S,3S,4S)-3 гидрокси-4-меркаптоциклогексил]карбамат(1S,2S,4S)-4-[(трет-бутоксикарбонил)амино]-2 гидроксициклогексилбензолкарботиоата и 72 мл CH2Cl2 и полученную смесь перемешивали при температуре 15-25 С. Полученную смесь дегазировали с использованием аргона в течение интервала времени в 20 мин. В полученную смесь добавляли 0,72 г 1,4-дитио-DL-треитола и добавляли по каплям 1,5 г моногидрата гидразина. Протекание реакции отслеживали с помощью ТСХ до завершения. После завершения взаимодействия (2,5 ч) в полученную смесь добавляли 72 мл 1 М раствора фосфорной кислоты и полученную смесь перемешивали в течение 15 мин. Объединенные фазы разделяли, нижнюю органическую фазу промывали с помощью 72 мл 1 М фосфорной кислоты, затем 72 мл 1%-ного раствора NaCl,- 19023273 сушили над безводным сульфатом магния (10 г), фильтровали и твердый продукт промывали с помощью 210 мл CH2Cl2. Полученную смесь упаривали в вакууме при температуре 40 С. Получали 5,01 г смеси трет-бутил [(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]карбамата и трет-бутил [(1S,3S,4S)-3 гидрокси-4-меркаптоциклогексил]карбамата в виде твердого вещества белого цвета. 1 В колбу помещали 9,68 г тозилата плеуромутилина и 101 мл МТВЕ и полученную смесь дегазировали при комнатной температуре с использованием аргона в течение от 5 до 10 мин. В полученную смесь добавляли при перемешивании 0,64 г хлорида бензил-три-н-бутиламмония, 4,72 г смеси трет-бутил[(1R,3R,4R)-3-гидрокси-4-меркаптоциклогексил]карбамата и трет-бутил [(1S,3S,4S)-3-гидрокси-4 меркаптоциклогексил]карбамат и 40 мл 1 М водного раствора NaOH. Полученную смесь перемешивали при температуре 20-25 С и протекание реакции отслеживали с помощью ВЭЖХ до завершения. После завершения взаимодействия (1 ч) полученные слои разделяли и нижний водный слой удаляли. Полученную органическую фазу промывали с помощью 40 мл 1 М водного раствора NaOH, 240 мл 0,1 М фосфорной кислоты, затем 40 мл 10%-ного раствора NaHCO3 и 40 мл Н 2 О. Полученные органические жидкости упаривали и сушили в вакууме при температуре 40 С. Получали 11,88 г смеси 14-O-[(1R,2R,4R)4-трет-бутоксикарбониламино-2-гидроксициклогексилсульфанил]ацетилмутилина и 14-O-[(1S,2S,4S)4-трет-бутоксикарбониламино-2-гидроксициклогексилсульфанил]ацетилмутилина в виде пены белого цвета (без поправки на остатки растворителя) 1 В колбу помещали 10,69 г смеси 14-O-[(1R, 2R, 4R)-4-трет-бутоксикарбониламино-2 гидроксициклогексилсульфанил]ацетилмутилина и 14-O-[(1S,2S,4S)-4-трет-бутоксикарбониламино-2 гидроксициклогексилсульфанил]ацетилмутилина (с поправкой на остатки растворителя) и 24 мл изо- 20023273 пропанола и полученную смесь перемешивали. В полученную смесь добавляли 12 мл 85% фосфорной кислоты и полученную смесь нагревали при 50 С в течение ночи. После завершения взаимодействия,определенного с помощью ВЭЖХ, полученную смесь охлаждали до комнатной температуры и добавляли 119 мл CH2Cl2. Полученную смесь охлаждали до температуры от 0 до 5 С и добавляли по каплям в течение 1 ч 119 мл 30%-ного водного раствора K2CO3. Полученную смесь нагревали до комнатной температуры и давали отстояться. Образовавшиеся фазы разделяли и полученные нижний органический (продукт) слой удаляли. Полученный водный слой (измеренный рН 10) экстрагировали 119 мл CH2Cl2 и полученные объединенные органические фазы промывали с помощью 119 мл Н 2 О. Полученную органическую фазу упаривали до приблизительно 5 объемов, добавляли 59 мл CH2Cl2 и полученную смесь упаривали опять до приблизительно 5 объемов. К полученному концентрату вновь добавляли 59 мл дихлорметана и полученную смесь упаривали до 2 объемов. К полученному концентрату при перемешивании добавляли по каплям 119 мл диизопропилового эфира в течение интервала времени в 1 ч. Получали густой маслянистый осадок, который становился кристаллами белого цвета через приблизительно 1 ч. Полученную смесь перемешивали при температуре от 15 до 25 С в течение 2 ч, фильтровали, полученный осадок промывали с помощью 24 мл диизопропилового эфира и отжимали досуха на фильтре. Получали 8,28 г(без поправки на остатки растворителя) смеси 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина и 14-O-[(1S,2S,4S)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина в виде твердого продукта (соотношение 59:41). В колбу помещали 8,28 г полученной сырой смеси и 41 мл н-бутанола и полученную смесь нагревали при температуре от 88 до 92 С при перемешивании в течение 30 мин. Полученной смеси давали охладиться до комнатной температуры в течение приблизительно 3 ч. При температуре 50 С наблюдалось начало образования осадка и полученную смесь перемешивали при комнатной температуре в течение ночи. Полученную смесь фильтровали и полученный осадок промывали с помощью 16,6 мл н-бутанола,затем 16,6 мл МТВЕ и сушили в вакууме при температуре 40 С. Получали 4,27 г 14-O-[(1R,2R,4R)-4 амино-2-гидроксициклогексилсульфанил]ацетилмутилина в виде кристаллического твердого продукта белого цвета кристаллической формы 2. Оптическая чистота была определена равной 93% с помощью хиральной ВЭЖХ и химическая чистота была 99,14% площади, определенная с помощью ОФ ВЭЖХ. Кроме того, сравнение оптического вращения оригинального образца 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина с оптическим вращением 14-O-[(1R,2R,4R)-4-амино 2-гидроксициклогексилсульфанил]ацетилмутилина, полученного, как описано выше, также подтверждало хиральную чистоту; ([]D (CHCl3)=+24,9 по сравнению с +25,9 для оригинального образца). Полученные после перекристаллизации в н-бутаноле маточные жидкости упаривали досуха с получением пены белого цвета. Получали 3,67 г смеси 14-O-[(1S,2S,4S)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина и 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина (соотношение 8 4,0:16,0; определено с помощью хиральной ВЭЖХ). 1 СпектрH ЯМР подтверждал структуру 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина. ЯМР спектр 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина описан в примере 4. Пример 11. Ацетат 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексил-сульфанил]ацетилмутилина, кристаллическая форма А К суспензии 615 г 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексил-сульфанил]ацетилмутилина в 12,3 л метилацетата при температуре от 50 до 55 С добавляли 62 мл воды и полученный мутный раствор осветляли с помощью фильтровальной бумаги GF. Полученный фильтраты нагревали при температуре от 50 до 55 С (прозрачный раствор), добавляли 123 мл уксусной кислоты, полученную смесь перемешивали при температуре от 50 до 55 С в течение 25 мин, охлаждали до температуры от 15 до 25 С в течение 80 мин и далее охлаждали до температуры от 0 до 5 С в течение 60 мин. Полученной суспензии давали отстояться при температуре от 0 до 5 С в течение 80 мин, фильтровали и лепешку на фильтре промывали с помощью 3,08 л метилацетата. Лепешку на фильтре отжимали досуха на фильтре в атмосфере азота в течение 2 ч. Получали 574,1 г 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина в виде ацетата в виде кристаллического тонкого белого порошка кристаллической формы А. 1 СпектрH ЯМР подтверждал структуру ацетата 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина. ЯМР спектр ацетата 14-O-[(1R,2R,4R)-4-амино-2- 21023273 Способ 1. К суспензии 3260 г 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина в 56,8 л изопропилацетата добавляли 10 г затравочных кристаллов 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина кристаллической формы В. Кристаллизация протекает также и без затравочных кристаллов. Полученную суспензию перемешивали при температуре от 20 до 25 С в течение 10 мин. В полученную смесь добавляли 353 мл уксусной кислоты и полученную смесь перемешивали при температуре от 20 до 25 С в течение 1 ч и исследовали на завершение реакции и полиморфную форму с помощью XRPD. Полученную суспензию перемешивали в течение еще 1 ч при температуре от 20 до 25 С, фильтровали и полученную лепешку на фильтре два раза промывали по 2,84 л изопропилацетата. Полученное твердое вещество сушили в вакууме при температуре 50 С в течение по меньшей мере 12 ч. Получали 3,15 кг ацетата 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина в виде кристаллического белого твердого продукта кристаллической формы В. Способ 2. В 6 мл метанола растворяли 2,00 г 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина. В полученную смесь одной порцией добавляли 0,338 мл уксусной кислоты и полученный раствор перемешивали в течение 15 мин. В полученную смесь добавляли 30 мл изопропилацетата в течение 30 мин и полученную суспензию перемешивали в течение 30 мин. Полученную взвесь нагревали при 30 С, отгоняли 15 объемов и осуществляли 3 отгонки (по 3 объема каждый раз) изопропилацетата. Полученную взвесь фильтровали и полученный осадок белого цвета отделяли, промывали с помощью изопропилацетата и сушили в течение ночи. Получали 1,74 г ацетата [(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина кристаллической формы В в виде кристаллического твердого продукта белого цвета. 1 В реакционный сосуд помещали 22 г 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина и 1,1 л EtOAc. Полученную суспензию нагревали до 50 С и выдерживали до растворения. В полученную смесь добавляли 1 экв. 98% L-молочной кислоты и полученную смесь постепенно охлаждали до температуры 25 С в течение 3 ч. Необязательно добавляли затравочные кристаллы L-лактата 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина. Кристаллизация протекает также и без затравочных кристаллов. Полученную суспензию перемешивали при температуре 20-25 С в течение ночи и затем охлаждали до температуры 5 С в течение 1 ч. Полученный осадок выделяли путем фильтрования и сушили в вакууме при температуре 40 С в течение ночи. Получали 23,7 г В колбу помещали 5,5 г 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина и 110 мл EtOAc. Полученную суспензию нагревали до 80 С и выдерживали до растворения. В полученную смесь помещали 10,8 мл 1 М малеиновой кислоты в ТГФ и полученной смеси давали охладиться до комнатной температуры в течение ночи при перемешивании. Необязательно добавляли затравочные кристаллы гидромалеата 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина. Кристаллизация протекает также и без затравочных кристаллов. 6,16 г 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина в виде кристаллической соли гидромалеата кристаллической формы 1 выделяли путем фильтрования и сушили в течение 6 ч в вакууме. 1H ЯМР (400 МГц, ДМСО-d6, м.д., среди прочих)6,13 (дд, J=11 и 18 Гц, 1 Н), 6,00 (с, 2 Н), 5,54 (д,J=8 Гц, 1 Н), 5,10-5,01 (м, 2 Н), 4,54 (д, J=6 Гц, 1 Н), 3,40 (AB, J=15 Гц, 2 Н), 3,05 (м, 1 Н), 2,56-2,49 (м, 1 Н),2,40 (с, широкий, 1 Н), 1,36 (с, 3 Н), 1,05 (с, 3 Н), 0,81 (д, J=7 Гц, 3 Н), 0,61 (д, J=7 Гц, 3 Н). Способ замещения аминозащитной группой R соединения формулы I показан на схеме реакции 3. Схема реакции 3 На схеме реакции 3 R представляет собой аминозащитную группу, определенную выше. Пример 15. 14-O-[(1R,2R,4R)-4-[(2,2,2-Трифторацетил)амино-2-гидроксициклогексилсульфанил]ацетилмутилин В колбу помещали 1 г 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина и 10 мл DCM при комнатной температуре и полученную смесь перемешивали. В полученную смесь добавляли по каплям 0,41 мл триэтиламина, затем 0,29 мл ангидрида трифторуксусной кислоты и полученную смесь перемешивали до завершения реакции (определялось с помощью ТСХ). Полученную смесь промывали с помощью 10 мл 0,1 М HCl, затем 10 мл 5% NaHCO3 и 10 мл воды и упаривали досуха. Получали 1,20 г 14-O-[(1R,2R,4R)-4-[(2,2,2-трифторацетил)амино-2-гидроксициклогексилсульфанил]ацетилмутилина в виде пены. Спектр 1 Н ЯМР подтверждал структуру 14-O-[(1R,2R,4R)-4-[(2,2,2-трифторацетил)амино-2 гидроксициклогексилсульфанил]ацетилмутилина. ЯМР спектр 14-O-[(1R,2R,4R)-4-[(2,2,2 трифторацетил)-амино-2-гидроксициклогексилсульфанил]ацетилмутилина описан в примере 6. В колбу при комнатной температуре помещали 1 г 14-O-[(1R,2R,4R)-4-амино-2 гидроксициклогексилсульфанил]ацетилмутилина и 10 мл DCM и полученную смесь перемешивали. В полученную смесь добавляли по каплям 0,41 мл триэтиламина, затем 0,2 мл этилхлорформиата и полученную смесь перемешивали до завершения реакции (определялось с помощью ТСХ). Полученную смесь промывали с помощью 10 мл 0,1 М HCl, затем 10 мл 5% NaHCO3 и 10 мл воды и упаривали досуха. Получали 1,05 г 14-O-[(1R,2R,4R)-4-этоксикарбониламино-2-гидроксициклогексилсульфанил]ацетилмутилина в виде пены. 1 1 г 14-O-[(1R,2R,4R)-4-амино-2-гидроксициклогексилсульфанил]ацетилмутилина и 40 мл толуола помещали в колбу при комнатной температуре и полученную смесь перемешивали. В полученную смесь добавляли по каплям 0,41 мл триэтиламина, затем 0,30 г фталевого ангидрида и полученную смесь нагревали при кипячении с обратным холодильником и удаляли воду с помощью насадки Дина-Старка до завершения реакции (определялось с помощью ВЭЖХ). Полученную смесь промывали с помощью 10 мл 0,1 М HCl, затем 10 мл 5% NaHCO3 и 10 мл воды, сушили над сульфатом натрия и упаривали досуха. Получали 0,87 г 14-O-[(1R,2R,4R)-2-гидрокси-4-(фталимидо-Nил)циклогексилсульфанил]ацетилмутилина в виде кристаллов бледного белого цвета. 1H ЯМР (400 МГц, ДМСО-d6, м.д., среди прочих)7,9-7,7 (м, 4 Н), 6,17 (дд, J=17,6 Гц, J=11,2 Гц,1 Н), 5,58 (д, 1 Н, J=7,8 Гц), 5,16-5,06 (м, 3 Н), 4,54 (д, 1 Н, J=6,0 Гц), 4,13-4,01 (м, 1 Н), 3,62-3,29 (м, 4 Н),2,69-2,60 (м, 1 Н), 2,43 (м, 1 Н), 2,30-1,80 (м, 8 Н), 1,80-1,15 (м, 12 Н), 1,1-0,9 (м, 4 Н), 0,83 (д, J=6,6 Гц, 3 Н),0,65 (д, J=5,8 Гц, 3 Н). Способ получения исходного продукта формулы IVa, используемого для получения соединений формулы IIIa, представлен на схеме реакции 4. На схеме реакции 4 R представляет собой аминозащитную группу и R1 представляет собой защитную группу серы и определены выше. Пример 18. В колбу помещали 1000 г рацемической циклогекс-3-ен-1-карбоновой кислоты и добавляли 5 объемов ацетона. Полученную смесь перемешивали, нагревали при температуре от 55 до 60 С и перемешивали в течение 30 мин. В полученную смесь добавляли по каплям в течение приблизительно 25 мин 960,5 г(S)-(-) метилбензиламина в 2 объемах ацетона. Получали прозрачный раствор оранжевого цвета и его медленно охлаждали. Кристаллизация начиналась при температуре 53 С (через 30 мин). Полная кристаллизация протекала через 1 ч при температуре 49 С. Полученную смесь охлаждали до комнатной температуры в течение еще 3 ч на ледяной бане, затем перемешивали при комнатной температуре в течение еще 1,5 ч. Полученный осадок отфильтровывали и промывали с помощью ацетона. Получали-метилбензиламиновую соль циклогекс-3-ен-1-карбоновой кислоты, как показано на схеме реакции выше. Выход (влажный): 1966,9 г; оптическое вращение: 20[]D=+8,05 (с=1, МеОН).B. Разделение солей. В 10-литровый сосуд помещали 1966,9 г (влажной) соли со стадии A и 3,8 объемов ацетона и полученную смесь нагревали при температуре от 55 до 60 С. Когда продукт растворялся, полученную смесь перемешивали в течение еще 15 мин и затем медленно охлаждали до комнатной температуры. Кристаллизация начиналась через 1 ч 10 мин (53 С). Полученную смесь охлаждали до температуры от 20 до 25 С в течение 4,5 ч и перемешивали при комнатной температуре в течение еще 1,5 ч. Полученный осадок отфильтровывали и промывали с помощью ацетона. Получали -метилбензиламиновую соль циклогекс 3-ен-1-карбоновой кислоты, обогащенную R-изомером. Выход (влажный): 1143 г; оптическое вращение: 20[]D=+20,65 (с=1, МеОН). Стадию В повторяли до достижения требуемого оптического вращения (20[]D40). 579,6 г (S)-(-)метилбензиламиновой соли циклогекс-3-ен-1(R)-карбоновой кислоты и 5 объемов МТВЕ помещали в колбу при температуре 20-25 С и полученную смесь перемешивали. В полученную смесь добавляли 10 объемов 1 М HCl, полученную смесь перемешивали в течение 5-10 мин и образовывались два слоя. Полученные слои разделяли и водный слой экстрагировали МТВЕ. Полученные органи- 25023273 ческие слои объединяли и промывали с помощью насыщенного солевого раствора. Полученную органическую фазу сушили над Na2SO4, фильтровали и полученную лепешку на фильтре промывали с помощью МТВЕ. Из полученного фильтрата в вакууме удаляли растворитель. Получали циклогекс-3-ен-1(R)карбоновую кислоту в виде прозрачного масла. Выход: 301,78 г. Оптическое вращение 20[]D=+83,1 (с=1, CHCl3). Циклогекс-3-ен-1(R)-карбоновая кислота может быть получена аналогично способу, описанномуD. Перегруппировка Курциуса для получения трет-бутил циклогекс-3-енил-(R)-карбамата В колбу помещали 305 г циклогекс-3-ен-1(R)-карбоновой кислоты и 10 объемов толуола при температуре 20-25 С и полученную смесь перемешивали. В полученную смесь добавляли по каплям в течение 15 мин 1,1 эквивалентов триэтиламина и полученную смесь перемешивали в течение еще 20 мин. В полученную смесь добавляли по каплям в течение приблизительно 20 мин 1,05 экв. DPPA, и температура повышалась до 95 С (экзотермическая реакция) с интенсивным выделением газа. Полученную смесь перемешивали в течение 15 мин и нагревали при температуре кипения. Протекание реакции отслеживали с помощью 1 Н ЯМР измерений до полного завершения. Полученную смесь охлаждали до температуры 80 С в течение 35 мин и по каплям добавляли в течение 10 мин 5 экв. трет-бутанола, затем 7,65 г CuCl. Полученную смесь нагревали до температуры 100 С и перемешивали в течение еще 40 мин. Протекание реакции отслеживали с помощью 1 Н ЯМР измерений до полного завершения. Полученную смесь охлаждали и в течение 10 мин добавляли 5 объемов насыщенного водного раствора NaHCO3. Полученную смесь перемешивали в течение 20 мин и оставляли на ночь. Полученную смесь фильтровали и оставшийся твердый продукт два раза промывали толуолом. Органические слои разделяли и водный слой промывали два раза толуолом. Все полученные органические слои объединяли, промывали с помощью Н 2 О и растворитель удаляли в вакууме. Получали трет-бутил циклогекс-3-енил-1(R)-карбамат в виде твердого вещества светло-коричневого цвета. Выход сырого продукта: 479,7 г. Полученный сырой трет-бутил циклогекс-3-енил-1(R)-карбамат подвергали хроматографии. Для 160 г сырого продукта колонку наполняли 1,5 кг силикагеля, используя 2,5 л циклогексана, и сверху наполняли песком. Сырой продукт загружали в 0,8 л смеси 5% EtOAc/циклогексан. Через колонку пропускали следующую градиентную систему, каждый раз собирая отдельные фракции: 2% EtOAc/циклогексан (90,8 л фракции),5% EtOAc/циклогексан (70,8 л фракции),10% EtOAc/циклогексан (40,8 л фракции). Общий выход после хроматографии: 81,3% от теоретического. 1 В сосуд помещали 4500 г mCPBA (70%) и 24 л мл CH2Cl2 и полученную смесь охлаждали до температуры 15 С. Добавляли по каплям в течение приблизительно 30 мин 3000 г трет-бутил циклогекс-3 енил-1(R)-карбамата в 4,5 л CH2Cl2, поддерживая температуру от 15 до 25 С. В полученную смесь добавляли 1,5 л CH2Cl2 и полученную смесь перемешивали при температуре от 20 до 25 С в течение 1 ч и нагревали до температуры кипения (40 С) в течение 2 ч. После завершения взаимодействия (1 Н ЯМР контроль) смесь охлаждали до температуры от -5 до 0 С, перемешивали в течение ночи и полученный твердый осадок отфильтровывали и промывали с помощью CH2Cl2. Полученный фильтрат промывали 10%-ным водным раствором тиосульфата натрия для удаления пероксидов, 10%-ным водным растворомNaHCO3 до достижения рН 7 в водной фазе и водой. Полученную органическую фазу упаривали до минимального объема и добавляли 15 л толуола и полученную смесь упаривали опять до минимального объема. Этот способ выделения повторяли еще два раза, получали 2,63 кг (выход 2,05 кг с поправкой на остаточные mCBA и толуол) трет-бутил (1R,3R,6S)-(7-оксабицикло[4.1.0]гепт-3-ил)карбамата в виде раствора в толуоле. 1 В сосуд помещали 2630 г (уточненные 2050 г) трет-бутил (1R,3R,6S)-(7-оксабицикло[4.1.0]гепт-3 ил)карбамата в виде раствора в толуоле (масса раствора: 15,44 кг) со стадии В выше и 3,1 л толуола и полученную смесь перемешивали при температуре 15-25 С. В полученную смесь добавляли по каплям 1,44 л тиобензойной кислоты (10%). Температуру поддерживали ниже 30 С. Добавляли еще 1,9 л толуола и 85,3 г моногидрата тетрабутиламмоний хлорида одной порцией, прекращали контроль внешней температуры и полученную смесь подвергали экзотермической реакции. Полученную смесь нагревали при температуре 40-45 С и перемешивали в течение 4 ч. После завершения взаимодействия (ТСХ и 1 Н ЯМР контроль) полученную смесь охлаждали до температуры от 15 до 20 С и промывали два раза 5%-ным водным раствором NaHCO3, затем два раза H2O. Полученный органический слой упаривали в вакууме до минимального объема. Добавляли 10,25 л толуола и полученную смесь опять концентрировали до минимального объема. Этот способ повторяли и определяли полученную сухую массу. Все последующие объемы взвесей соотносятся с этой массой. К полученному сырому концентрированному остатку при перемешивании добавляли 0,5 объемов толуола и полученную смесь перемешивали при температуре от 15 до 25 С в течение 30 мин. В полученную смесь добавляли по каплям 0,5 объемов гептана в течение 15 мин и полученную смесь перемешивали при температуре от 15 до 25 С в течение 40 мин. Полученное твердое вещество фильтровали и промывали с помощью 0,25 объемов смеси толуол-гептан (1:1), затем взвесь промывали с помощью 0,5 объемов толуол-гептан (1:1), затем фильтрат промывали с помощью 0,25 объемов толуол-гептан (1:1). Данный способ уменьшал количество нежелаемых пространственного изомера и тиобензойной кислоты до неопределяемого количества (с помощью 1 Н ЯМР). Полученное твердое вещество выделяли и сушили в вакууме при температуре 30 С. Получали 1090 г (1R,2R,4R)-4-[(трет-бутоксикарбонил)амино]-2-гидроксициклогексилбензолкарботиоата в виде твердого вещества белого цвета. 1H ЯМР (200 МГц, ДМСО-d6, м.д.)7,92-7,87 (м, 2H), 7,71-7,63 (м, 1 Н), 7,58-7,49 (м, 2 Н), 6,85 (д,J=8 Гц, 1H), 5,11 (д, J=5,6 Гц, 1 Н), 3,49-3,25 (м, 3 Н), 2,12-1,95 (м, 2 Н), 1,79-1,69 (м, 1 Н), 1,54-1,14 (м,12 Н). Альтернативный способ (в сокращенном виде) получения исходного продукта формулы IVa, который использовался для получения соединений формулы IIIa, представлен на схеме реакции 5. Схема реакции 5 где R представляет собой аминозащитную группу и R1 представляет собой защитную группу серы и описаны выше. Стадия A. N-(Циклогекс-3-ен-1(R)-ил)-2,2,2-трифторацетамид. 50 г 3-циклогексен-1(R)-карбоновой кислоты и 425 мл хлорбензола помещали в колбу при температуре 20-25 С и полученную смесь перемешивали. В полученную смесь добавляли по каплям 110 мл триэтиламина, затем 25 мл хлорбензола. Полученную смесь нагревали до температуры от 78 до 82 С и добавляли контролируемыми дозами 109,2 г DPPA, поддерживая температуру от 80 до 90 С и равномерное выделение газа и дополняли 20 мл хлорбензола для линейной промывки. Полученную смесь перемешивали при температуре от 78 до 82 С в течение 1 ч до полного завершения, определенного по данным ТСХ. Полученную смесь охлаждали до температуры приблизительно 70 С и добавляли по каплям 226 г трифторуксусной кислоты в 34 мл хлорбензола, поддерживая температуру от 70 до 80 С, затем 1,57 гCuCl и 25 мл хлорбензола для линейной промывки. Полученную смесь перемешивали при температуре от 90 до 95 С в течение 2 ч и реакцию проводили до ее завершения по данным ТСХ. Полученную смесь охлаждали до температуры от 15 до 25 С и добавляли 375 мл 20%-ного водного раствора K2CO3 и полученную смесь перемешивали в течение 15 мин. Полученные слои разделяли и к полученному верхнему органическому слою добавляли 375 мл 20%-ного водного раствора K2CO3. Полученную смесь фильтровали через целит для удаления оставшегося твердого продукта и целит промывали с помощью 50 мл хлорбензола. Полученные слои разделяли. Полученные объединенные низшие водные слои опять экстрагировали с помощью 250 мл хлорбензола и объединенные полученные органические фазы промывали с помощью 500 мл 0,5 М фосфорной кислоты. Полученный водный слой вновь экстрагировали 300 мл хлорбензола и полученные объединенные органические фазы промывали с помощью 500 мл 5%-ного водного раствора NaCl. Осуществляли массовый стрип-анализ для определения содержания N-(циклогекс-3-ен-1(R)-ил)2,2,2-трифторацетамида для использования далее на стадии В эпоксидирования. Полученный хлорбензольный раствор содержал 69,52 г N-(циклогекс-3-ен-1(R)-ил)-2,2,2 трифторацетамида. 1MS (ESI, г/моль): m/z 194 [М+Н]+Стадия B. 2,2,2-Трифтор-N-(1R,3R,6S)-(7-оксабицикло[4.1.0]гепт-3-ил)ацетамид. В сосуд, содержащий охлажденный (10-15 С) раствор 69,5 г N-(циклогекс-3-ен-1(R)-ил)-2,2,2 трифторацетамида со стадии А, порциями помещали 106,5 г м-хлорпербензойной кислоты (70%), поддерживая температуру 30 С и промывали с помощью 69,5 мл хлорбензола. Полученную смесь перемешивали при температуре от 20 до 25 С в течение 1 ч и реакцию проводили до ее завершения по данным ТСХ. После завершения взаимодействия полученную смесь охлаждали до температуры от 0 до -5 С, перемешивали в течение 30 мин и полученный твердый осадок (mCBA) отфильтровали и промывали с помощью 234,8 мл хлорбензола. Полученный фильтрат промывали с помощью 347,6 мл 10%-ного раствора тиосульфата натрия для удаления пероксидов и полученный водный слой вновь экстрагировали 208,6 мл хлорбензола. Полученные объединенные органические слои промывали с помощью 347,6 мл 5%-ного раствора бикарбоната натрия для установления рН 7 водной фазы и полученный водный слой вновь экстрагировали 208,6 мл хлорбензола. Объединенные органические слои промывали с помощью 347,6 мл воды. Для определения содержания 2,2,2-трифтор-N-(1R,3R,6S)-(7-оксабицикло[4.1.0]гепт-3 ил)ацетамида проводили массовый стрип-анализ. Полученный хлорбензольный раствор содержал 58,64 г 2,2,2-трифтор-N-(1R,3R,6S)-(7 оксабицикло[4.1.0]гепт-3-ил)ацетамида, включающий приблизительно 11% анти(транс)эпоксида и органический раствор использовали на стадии С далее (раскрытие кольца). 1MS (ESI, г/моль): m/z 208 [МН]-. Стадия С. (1R,2R,4R)-4-[(2,2,2-Трифторацетил)амино]-2-гидроксициклогексилбензолкарботиоат. Хлорбензольный раствор со стадии В, содержащий 58,64 г 2,2,2-трифтор-N-(1R,3R,6S)-(7- 28023273 оксабицикло[4.1.0]гепт-3-ил)ацетамида, упаривали приблизительно до 5 объемов, основываясь на эпоксиде. Полученный концентрат дегазировали при температуре 15-25 С с помощью аргона в течение 30 мин и температуру устанавливали от 15 до 20 С. В полученную смесь добавляли по каплям 58,1 г тиобензойной кислоты (90%), устанавливая температуру ниже 30 С. В сосуд к полученной смеси добавляли 17,6 мл хлорбензола в качестве линейной промывки и порциями добавляли 2,49 г моногидрата тетрабутиламмоний хлорида при температуре 30 С. Полученную смесь нагревали при температуре от 4045 С, перемешивали и реакцию проводили до завершения по данным ТСХ. После завершения взаимодействия полученную смесь охлаждали до температуры от 0 до 5 С, перемешивали в течение 1 ч, фильтровали и полученную лепешку на фильтре промывали с помощью 258,64 мл хлорбензола. Полученное твердое вещество сушили в вакууме при температуре 40 С. Получали 45,5 г (1R,2R,4R)-4-[(2,2,2 трифторацетил)амино]-2-гидроксициклогексилбензолкарботиоата в виде твердого продукта. 1MS (ESI, г/моль): m/z 348,0 [М+Н]+. Способ получения исходной смеси соединения формулы IVa и соединения формулы IVb, используемой для получения смеси соединения формулы IIIa и соединения формулы IIIb, представлен на схеме реакции 6. Схема реакции 6 где R представляет собой аминозащитную группу и R1 представляет собой защитную группу серы,как определено выше. Пример 20. В 2-литровую колбу при температуре 20-25 С помещали 50,0 г 3-циклогексен-1-карбоновой кислоты и 500 мл толуола, полученную смесь перемешивали и добавляли по каплям 60,7 мл триэтиламина в течение 15 мин, затем в течение 20 мин добавляли по каплям 89,7 мл DPPA (выделение газа, экзотерм до 50 С). В полученную смесь добавляли 50 мл толуола в качестве линейной промывки. Полученную смесь нагревали при кипячении с обратным холодильником и перемешивали до завершения реакции,определяемой с помощью ТСХ и 1 Н ЯМР. Завершение реакции было показано через 1 ч. Полученную смесь охлаждали до температуры 80 С и в течение 10 мин добавляли по каплям 186 мл трет-BuOH, затем 1,26 г CuCl и полученную смесь нагревали при кипячении с обратным холодильником. Реакцию отслеживали с помощью 1 Н ЯМР и было показано ее полное завершением через 1 ч. Полученную смесь охлаждали до температуры от 20 до 25 С и в течение от 5 до 10 мин добавляли 250 мл насыщенного раствораNaHCO3. Полученную смесь перемешивали в течение 30 мин, фильтровали для удаления оставшегося твердого продукта и твердый продукт промывали 25 мл толуола. Слои разделяли и водный слой экстрагировали 2150 мл толуола. Органические слои объединяли, промывали с помощью 150 мл воды и кон- 29
МПК / Метки
МПК: A61P 17/00, C07C 323/52, A61P 31/00, A61K 31/215
Метки: способ, плеуромутилинов, получения
Код ссылки
<a href="https://eas.patents.su/30-23273-sposob-polucheniya-pleuromutilinov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения плеуромутилинов</a>
Способ получения дихлорпропанола, способ получения эпихлоргидрина, способ получения эпоксидных смол и применение оборудования, обладающего коррозионной стойкостью, в способе получения дихлорпропанола

Номер патента: 14241
Опубликовано: 29.10.2010
Авторы: Бальтазар Доминик, Краффт Филипп, Сметс Валентин, Франк Кристиан, Жильбо Патрик
МПК: B01J 19/02, C07C 29/62, C07C 31/36...
Метки: обладающего, эпоксидных, оборудования, эпихлоргидрина, получения, дихлорпропанола, способ, коррозионной, смол, способе, применение, стойкостью
Формула / Реферат:
1.Способ получения дихлорпропанола, содержащий:(a) стадию, на которой глицерин или сложный эфир глицерина или их смесь вводят во взаимодействие с агентом хлорирования, содержащим хлороводород,(b) по меньшей мере одну другую стадию, осуществляемую на оборудовании, выполненном или имеющем покрытие из материалов, обладающих стойкостью по отношению к агенту хлорирования, в условиях осуществления этой стадии,причем другая стадия является стадией...
Способ получения композиции сложного полиэфира, полученная композиция, содержащая ее пленка, раствор для получения композиции и способ его получения

Номер патента: 14016
Опубликовано: 30.08.2010
Авторы: Отто Бригитта, Зайдель Экхард, Хельдманн Карл-Хайнц
МПК: C08K 5/098, C08J 5/18, C08K 5/00...
Метки: содержащая, композиции, полиэфира, сложного, способ, раствор, пленка, получения, полученная, композиция
Формула / Реферат:
1. Способ получения композиции сложного полиэфира, включающий следующие стадии:A) этерификацию дикарбоновой кислоты алкандиолом или переэтерификацию диалкилового эфира дикарбоновой кислоты алкандиолом;B) предварительную поликонденсацию полученного диалкилового эфира дикарбоновой кислоты до форполиконденсата;C) поликонденсацию в расплаве форполиконденсата до сложного полиэфира, причем смесь, которую подвергают поликонденсации, содержит по меньшей...
Способ получения залеплона

Номер патента: 7505
Опубликовано: 27.10.2006
Автор: Радль Станислав
МПК: C07D 239/00, C07D 231/00, C07D 487/04...
Метки: получения, залеплона, способ
Формула / Реферат:
1. Способ получения N-(3-(3-цианопиразоло[1,5-а]пиримидин-7-ил)фенил)-N-этилацетамида, известного под международным непатентованным названием залеплон, формулы I отличающийся тем, что N-[3-(3-диметиламиноакрилоил)фенил]-N-этилацетамид формулы II приводят во взаимодействие с 5-амино-1Н-пиразол-4-карбонитрилом формулы III в среде, содержащей раствор соляной или бромисто-водородной кислоты в С1-С5 спирте, или в С4-С6 алифатическом или...
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Колладан Колетт, Ларкин Джон Патрик, Крок Вероник, Руссель Патрик
МПК: C07D 487/04
Метки: способ, кислоты, октагидро-6, терапевтически, диазепин-1-карбоновой, 1,2, применение, 1,2-а, активных, соединений, получения, производные, 10-диоксо-6н-пиридазино
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...
Цеолитный катализатор l-типа, способ его получения, способ получения ароматических углеводородов, способ получения бензина

Номер патента: 3559
Опубликовано: 26.06.2003
Авторы: Фукунага Тецуя, Иннес Роберт А., Сугимото Митио
МПК: B01J 29/61, C10G 35/095, C07C 5/41...
Метки: углеводородов, цеолитный, получения, бензина, l-типа, катализатор, ароматических, способ
Формула / Реферат:
1. Цеолитный катализатор L-типа, который получают при нанесении на цеолит L-типа платинового компонента, одного или более галогеновых компонентов и одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, при этом наносимое количество одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, находится в интервале от 0,001 до 3 мас.% из расчета на общую массу катализатора, молярное отношение...
Предыдущий патент: Бициклические пиридинилпиразолы
Следующий патент: Солнечный модуль
Случайный патент: Система приведения в действие поршня пресс-подборщика