Аналоги глюкозозависимого инсулинотропного полипептида (gip), модифицированные по n-концу

























Формула / Реферат
1. Соединение формулы (I)

где А1 представляет собой Сра, His, 4Hppa, 3-Pal, 4-Pal, (Х4,Х5,Х6,Х7,Х8)Phe, Taz, 3Thi, 7HO-Tic, Tyr(Ac), Tyr(Me), 3Br-Tyr, 3,5Br-Tyr, 3Cl-Tyr, 3F-Tyr, hTyr, 3I-Tyr, 3,5I-Tyr, 2,6Me-Tyr, 3МеО-Tyr, 3NO2-Tyr или 3ОН-Tyr;
А2 представляет собой Ala, Aib, D-Ala или Gly;
А3 представляет собой Glu, 4Hyp или hPro;
А7 представляет собой Ile или Acc;
А11 представляет собой Ser, Acc или Aib;
А13 представляет собой Ala или Aib;
А14 представляет собой Met, Acc или Nle;
А29 представляет собой Gln или отсутствует;
А30 представляет собой Lys или отсутствует;
А31 представляет собой Gly или отсутствует;
А32 представляет собой Lys или отсутствует;
А33 представляет собой Lys или отсутствует;
А34 представляет собой Asn или отсутствует;
А35 представляет собой Asp или отсутствует;
А36 представляет собой Trp или отсутствует;
А37 представляет собой Lys или отсутствует;
А38 представляет собой His или отсутствует;
А39 представляет собой Asn или отсутствует;
А40 представляет собой Ile, Acc или отсутствует;
А41 представляет собой Thr, Acc или отсутствует;
А42 представляет собой Gln или отсутствует;
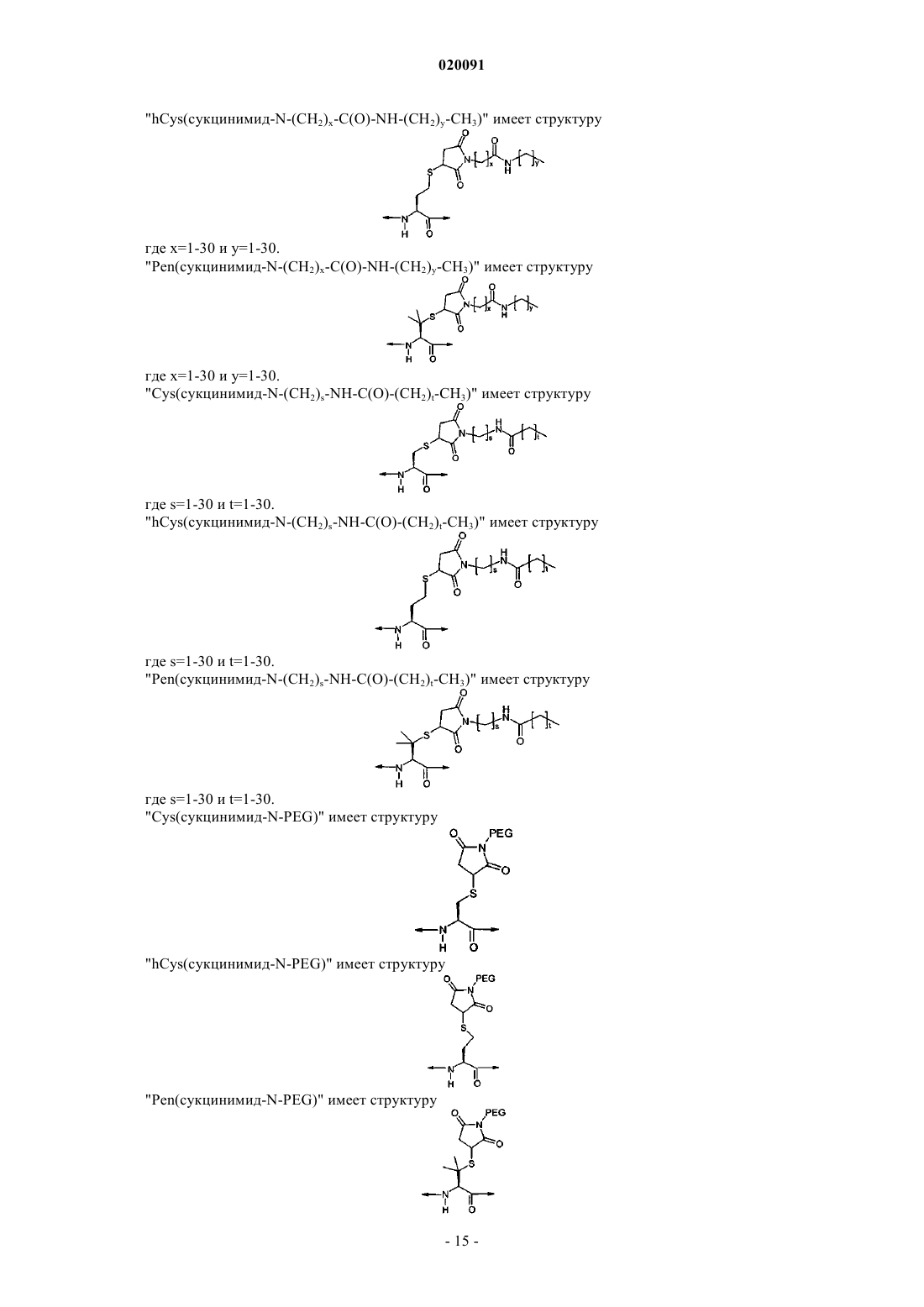
А43 представляет собой His, Cys, Cys(сукцинимид-N-алкил), Cys(сукцинимид-N-(СН2)x-C(О)-NH-(СН2)y-СН3), Cys(сукцинимид-N-(CH2)s-NH-C(O)-(CH2)t-CH3), Lys43(N-C(O)-(СН2)10-СН3) или отсутствует;
R1 представляет собой ОН, NH2, (C1-C30)алкокси или NH-X2-CH2-Z0, где X2 представляет собой (С0-С30) углеводородный остаток и Z0 представляет собой Н, ОН, СО2Н или CONH2;
каждый из R2, R3 независимо выбирают из группы, состоящей из Н, (C1-C30)алкила, (C1-C30)гетероалкила, (C1-C30)ацила, (С2-С30)алкенила, (С2-С30)алкинила, арил(C1-C30)алкила, арил(С1-С30)ацила, замещенного (C1-C30)алкила, замещенного (С1-С30)гетероалкила, замещенного (C1-C30)ацила, замещенного (С2-С30)алкенила, замещенного (С2-С30)алкинила, замещенного арил(С1-С30)алкила и замещенного арил(C1-C30)ацила; при условии, что когда R2 представляет собой (C1-C30)ацил, арил(C1-C30)ацил, замещенный (C1-C30)ацил или замещенный арил(C1-C30)ацил, тогда R3 представляет собой Н, (C1-C30)алкил, (C1-C30)гетероалкил, (С2-С30)алкенил, (С2-С30)алкинил, арил(C1-C30)алкил, замещенный (C1-С30)алкил, замещенный (C1-C30)гетероалкил, замещенный (С2-С30)алкенил, замещенный (С2-С30)алкинил или замещенный арил(С1-С30)алкил;
s, t, х и у, каждый независимо, представляют собой для каждого случая целое число от 1 до 30 включительно;
X4, X5, X6, X7 и X8, каждый независимо, представляют собой для каждого случая Н, F, CF3, Cl, Br, I, (C1-10)алкил, замещенный (C1-10)алкил, арил, замещенный арил, ОН, NH2, -CH2NH2, NO2 или CN;
при условии, что когда А1 представляет собой 4Нрра, тогда R2 и R3 отсутствуют;
при дополнительном условии, что соединение содержит более чем одну аминокислотную замену или модификацию в положениях 1, 2 и 3; и
при дополнительном условии, что если аминокислота в положении 1 модифицирована, она не модифицируется посредством:
(a) N-концевого алкилирования;
(b) N-концевого ацетилирования;
(c) N-концевого ацилирования;
(d) присоединения N-концевой изопропильной группы или
(e) присоединения N-концевой пироглутаминовой кислоты; и
при дополнительном условии, что соединение содержит по меньшей мере одну аминокислотную замену или модификацию в положениях с 4 по 43,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором, когда присутствует Acc,
А7 представляет собой А5с или А6с; или
А11 представляет собой А5с; или
А14 представляет собой А5с; или
А40 представляет собой А5с; или
А41 представляет собой А5с,
или его фармацевтическая соль.
3. Соединение по п.2, которое представляет собой



или его фармацевтически приемлемая соль.
4. Соединение по п.2,
в котором А1 представляет собой 4Нрра;
А43 отсутствует и
по меньшей мере один из А2, А3, А7, А11 и А14 не является аминокислотным остатком из соответствующего положения в нативном GIP,
или его фармацевтическая соль.
5. Соединение по п.4, которое представляет собой

или его фармацевтически приемлемая соль.
6. Соединение по п.2,
в котором А1 представляет собой Tyr(Ac), Tyr(Me), 3Br-Tyr, 3,5Br-Tyr, 3Cl-Tyr, 3F-Tyr, hTyr, 3I-Tyr, 3,5I-Tyr, 2,6Me-Tyr, 3MeO-Tyr, 3NO2-Tyr или 3ОН-Tyr;
А2 представляет собой Aib, D-Ala или Gly и
по меньшей мере один из А3, А11, А13, А14, А40, А41 и А43 не является аминокислотным остатком из соответствующего положения в нативном GIP,
или его фармацевтическая соль.
7. Соединение по п.6,
в котором А2 представляет собой Aib, D-Ala или Gly и
по меньшей мере два из А3, А11, А13, А14, А40, А41 и А43 не являются аминокислотными остатками из соответствующих положений в нативном GIP,
или его фармацевтическая соль.
8. Соединение по п.7, которое представляет собой


или его фармацевтически приемлемая соль.
9. Соединение по п.2,
в котором А1 представляет собой 3Br-Phe, 3Cl-Phe, 4CN-Phe, 3F-Phe, 4F-Phe, 3,4F-Phe, 3,4,5F-Phe, 3,5F-Phe, 4NH2-Phe, 4NH2CH2-Phe или 3ОН-Phe;
A2 представляет собой Aib, D-Ala или Gly;
А11 представляет собой A5c и
по меньшей мере один из А14 и А41 не является аминокислотным остатком из соответствующего положения в нативном GIP,
или его фармацевтическая соль.
10. Соединение по п.9, в котором А2 представляет собой Aib, или его фармацевтическая соль.
11. Соединение по п.10, которое представляет собой

или его фармацевтически приемлемая соль.
12. Соединение по п.2, в котором каждая аминокислота в положениях 1, 2 и 3 является замененной или модифицированной, или его фармацевтическая соль.
13. Соединение по п.1, которое выбирают из группы, состоящей из


или его фармацевтически приемлемая соль.
14. Соединение по п.9, которое выбирают из группы, состоящей из

или его фармацевтически приемлемые соли.
15. Соединение по п.10, которое представляет собой (3Cl-Tyr1,Aib2,А5с11,Nle14)hGIP(1-42)-ОН (SEQ ID NO:17), или его фармацевтически приемлемая соль.
16. Соединение по п.12, которое представляет собой (4Нрра1,Aib2,hPro3,Nle14)hGIP(1-30)-NH2 (SEQ ID NO:12), или его фармацевтическая соль.
17. Соединение по п.1 или 2, в котором пептидная связь между А1 и А2 заменена псевдопептидной связью, или его фармацевтическая соль.
18. Соединение по п.17, в котором А1-А2 представляет собой A1-y-(CH2-NH)А2, или его фармацевтическая соль.
19. Соединение по п.18, которое представляет собой [Tyr1-y(CH2-NH)Gly2,A5c11,41]hGIP(1-42)-ОН (SEQ ID NO:43), или его фармацевтическая соль.
20. Соединение по любому из пп.1-19, дополнительно содержащее ковалентно связанный остаток PEG, или его фармацевтически приемлемая соль.
21. Соединение по п.20, в котором указанный остаток PEG ковалентно связывается с соединением через Cys(малеимидный) или Pen(малеимидный) линкер с образованием Cys(сукцинимид-N-PEG), или его фармацевтически приемлемая соль.
22. Соединение по п.21, в котором пегилирование осуществляют по любому из положений аминокислотных остатков 16, 30 и 31-43, при этом Cys(сукцинимид-N-PEG) располагается в любом из положений аминокислотных остатков 16, 30 и 31-43, или его фармацевтически приемлемая соль.
23. Соединение по п.22, в котором пегилирование осуществляют по любому из положений аминокислотных остатков 32, 33 и 43, при этом Cys(сукцинимид-N-PEG) располагается в любом из положений аминокислотных остатков 32, 33 и 43, или его фармацевтически приемлемая соль.
24. Соединение по п.23, в котором указанный остаток PEG имеет среднюю молекулярную массу от примерно 2000 до примерно 80000, или его фармацевтически приемлемая соль.
25. Соединение по п.24, в котором указанный PEG выбирают из группы, состоящей из 5K PEG, 10K PEG, 20K PEG, 30K PEG, 40K PEG, 50K PEG, 60K PEG, с образованием Cys(сукцинимид-N-5K PEG), Cys(сукцинимид-N-10K PEG), Cys(сукцинимид-N-20K PEG), Cys(сукцинимид-N-30K PEG), Cys(сукцинимид-N-40K PEG), Cys(сукцинимид-N-50K PEG) или Cys(сукцинимид-N-60K PEG), или его фармацевтически приемлемая соль.
26. Соединение по любому из пп.21-25, в котором указанный сукцинимид-N-PEG является линейным, или его фармацевтически приемлемая соль.
27. Соединение по п.26, в котором указанный линейный сукцинимид-N-PEG представляет собой сукцинимид-N-(СН2)2-С(О)NH-(CH2)3-PEG, или его фармацевтически приемлемая соль.
28. Соединение по любому из пп.21-25, в котором указанный сукцинимид-N-PEG является разветвленным, или его фармацевтически приемлемая соль.
29. Соединение по п.28, в котором указанный разветвленный сукцинимид-N-PEG представляет собой сукцинимид-N-(СН2)2-C(О)NH-(СН2)3-O-CH2-CH-PEG-CH2-PEG, или его фармацевтически приемлемая соль.
30. Соединение по любому из пп.25-29, которое представляет собой
[3Cl-Tyr1,Aib2,A5c11,Nle14,Cys43(сукцинимид-N-20K PEG)]hGIP(1-43)-NH2 (SEQ ID NO:80) или
[3Cl-Tyr1,Aib2,A5c11,Nle14,Cys43(сукцинимид-N-30K PEG)]hGIP(1-43)-NH2 (SEQ ID NO:81);
или его фармацевтически приемлемая соль.
31. Фармацевтическая композиция, содержащая эффективное количество соединения по любому из пп.1-30.
32. Фармацевтическая композиция по п.31, дополнительно содержащая фармацевтически приемлемый носитель.
33. Способ агонистического воздействия на рецептор GIP у субъекта, нуждающегося в этом, включающий введение указанному субъекту терапевтически эффективного количества соединения по любому из пп.1-30 или фармацевтической композиции по п.31 или 32.
34. Способ антагонистического воздействия на рецептор GIP у субъекта, нуждающегося в этом, включающий введение указанному субъекту терапевтически эффективного количества соединения по любому из пп.1-30 или фармацевтической композиции по п.31 или 32.
35. Способ лечения состояний или заболеваний, опосредуемых связыванием с GIP-рецептором, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп.1-30 или фармацевтической композиции по п.31 или 32.
36. Способ по п.35, в котором указанное состояние или заболевание, опосредуемое связыванием с GIP-рецептором, выбрано из группы, состоящей из диабета типа I, диабета типа II, ожирения, резистентности к инсулину, непереносимости глюкозы, жировой дистрофии печени, глюкагона, секреторных расстройств дыхательных путей, расстройства метаболизма, артрита, остеопороза, заболевания центральной нервной системы, рестеноза, нейродегенеративного заболевания, почечной недостаточности, ишемической болезни сердца, нефротического синдрома, цирроза печени, отека легких, гипертонии и расстройств, где желательным является уменьшение приема пищи и/или уменьшение массы тела.
37. Способ лечения диабета, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп.1-30 или фармацевтической композиции по п.31 или 32.
38. Способ по п.37, в котором указанный диабет представляет собой диабет типа II.
39. Способ лечения расстройств, связанных с диабетом, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп.1-30 или фармацевтической композиции по п.31 или 32.
40. Способ по п.39, в котором указанное расстройство, связанное с диабетом, выбрано из группы, состоящей из гипергликемии, гиперинсулинемии, нарушенной переносимости глюкозы, нарушенной гликемии натощак, дислипидемии, гипертриглицеридемии и резистентности к инсулину.
41. Способ лечения или предотвращения вторичных причин диабета, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп.1-30 или фармацевтической композиции по п.31 или 32.
42. Способ по п.41, в котором указанная вторичная причина выбрана из группы, состоящей из избытка глюкокортикоидов, избытка гормона роста, феохромоцитомы и лекарственного диабета.
43. Способ лечения ожирения, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп.1-30 или фармацевтической композиции по п.31 или 32.
44. Способ стимулирования секреции инсулина у субъекта, нуждающегося в этом, посредством введения указанному субъекту терапевтически эффективного количества соединения по любому из пп.1-30 или фармацевтической композиции по п.31 или 32.
45. Применение соединения по любому из пп.1-30 для получения лекарственного средства для связывания с GIP-рецептором для предотвращения или лечения заболеваний или состояний, связанных с нарушением связывания аналогов GIP-рецепторов.
46. Применение по п.45 для получения лекарственного средства для предотвращения или лечения апоптоза β-клеток поджелудочной железы.
47. Применение по п.45 для получения лекарственного средства для усиления глюкозозависимой пролиферации β-клеток поджелудочной железы.
Текст
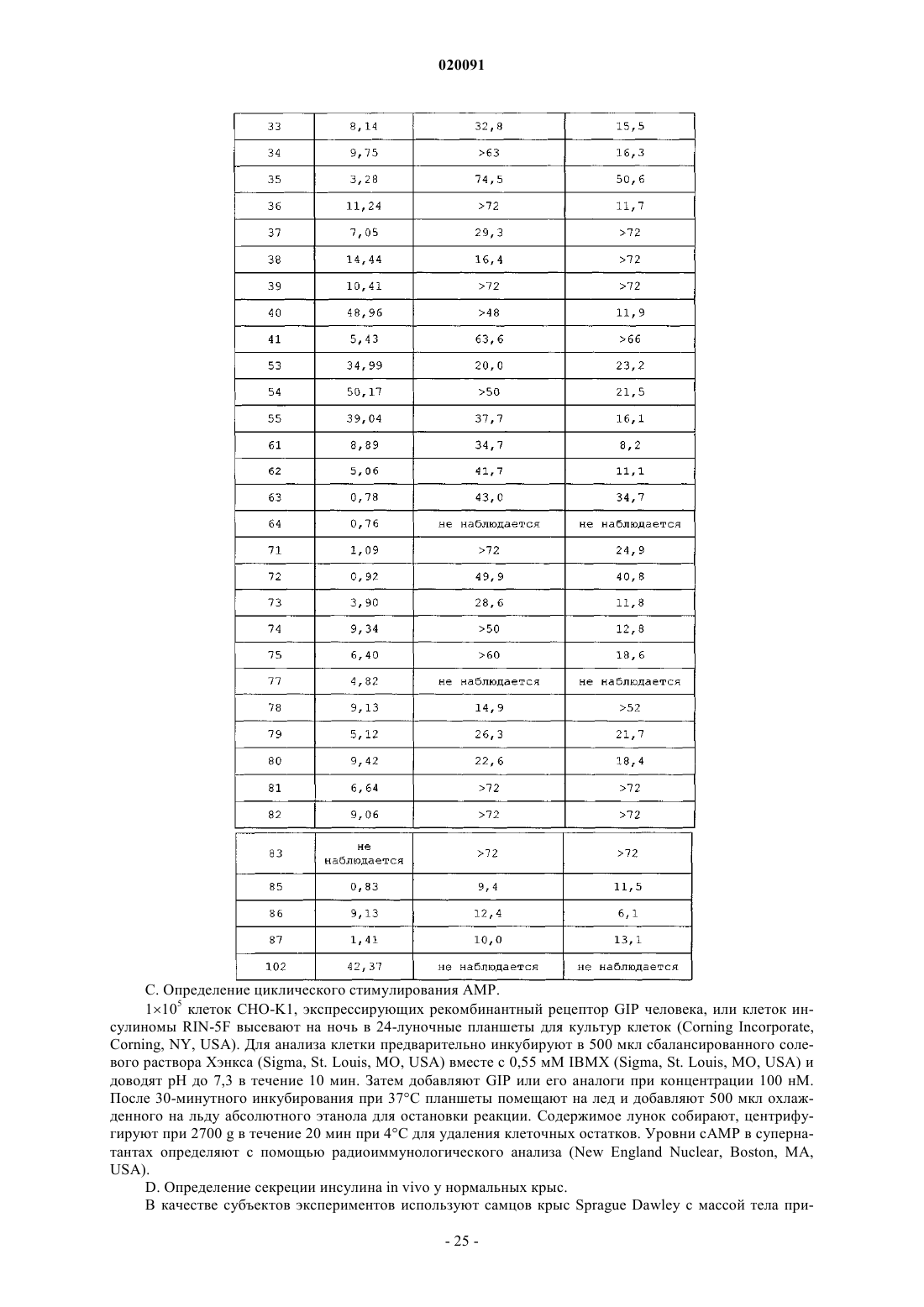
АНАЛОГИ ГЛЮКОЗОЗАВИСИМОГО ИНСУЛИНОТРОПНОГО ПОЛИПЕПТИДА (GIP),МОДИФИЦИРОВАННЫЕ ПО N-КОНЦУ В изобретении предлагается новый ряд аналогов соединений глюкозозависимого инсулинотропного полипептида, фармацевтические композиции, содержащие указанные соединения, и применение указанных соединений в качестве агонистов или антагонистовGIP-рецепторов для лечения состояний, опосредуемых GIP-рецепторами, таких как инсулиннезависимый сахарный диабет и ожирение. Дун Чжэн Синь (US) Медведев В.Н. (RU) Область техники, к которой относится изобретение Настоящее изобретение относится к области новых аналогов соединений глюкозозависимого инсулинотропного полипептида, к фармацевтическим композициям, содержащим указанные соединения, и к применению указанных соединений в качестве агонистов или антагонистов GIP-рецепторов для лечения состояний, опосредуемых GIP-рецепторами, таких как инсулиннезависимый сахарный диабет и ожирение. Уровень техники Глюкозозависимый инсулинотропный полипептид ("GIP", известный так же, как "желудочный ингибиторный полипептид"; SEQ ID NO:1) представляет собой пептид из 42 остатков, секретируемый энтероэндоринными K-клетками тонкого кишечника в кровоток в ответ на пероральное поглощение питательных веществ. GIP ингибирует секрецию желудочного сока и, как показано, является сильнодействующим стимулятором секреции инсулина из -клеток поджелудочной железы после перорального поглощения глюкозы ("инкретиновый эффект") (Creutzefldt, W., et al., 1979, Diabetologia, 16:75-85). Высвобождение инсулина, индуцируемое поглощением глюкозы и других питательных веществ,вызывается как гормональными, так и нервными факторами (Creutzfeldt, W., et al., 1985, Diabetologia,28:565-573). В качестве инкретинов предложены несколько желудочно-кишечных регуляторных пептидов, и среди этих кандидатов только GIP и глюкагоноподобный пептид 1 ("GLP-1"), видимо, удовлетворяют требованиям, чтобы рассматривать их как физиологические стимуляторы постпрандиального выделения инсулина (Nauck, et al., 1989, J. Clin. Endocrinol. Metab., 69:654-662). Показано, что сочетанные воздействия GIP и GLP-1 являются достаточными для объяснения всего инкретинового воздействия энтероинсулярной оси (Fehmann, H.С., et al., 1989, FEBS Lett, 252:109-112). Как хорошо известно специалистам в данной области, известные и потенциальные применения GIP являются разнообразными и многочисленными. Таким образом, введение соединений по настоящему изобретению для целей выяснения воздействия агониста может иметь такие же воздействия и применения, как и сам GIP. Эти разнообразные применения GIP могут быть перечислены как следующие: лечение заболевания, выбранного из группы, состоящей из диабета типа I, диабета типа II (Visboll, Т., 2004,Dan. Med. Bull, 51:364-70), резистентности к инсулину (WO 2005/082928), ожирения (Green, В.D., et al.,2004, Current Pharmaceutical Design, 10:3651-3662), метаболического расстройства (Gault, V.A., et al.,2003, Biochem. Biophys. Res. Commun., 308:207-213), заболевания центральной нервной системы, нейродегенеративного заболевания, ишемической болезни сердца, гипогликемии, и таких расстройств, когда желательными являются уменьшение приема пищи и потеря массы. GIP не только резко увеличивает секрецию инсулина в островковых клетках поджелудочной железы, но он также стимулирует продуцирование инсулина посредством усиления транскрипции и трансляции проинсулина (Wang, et al., 1996,Mol. Cell. Endocrinol., 116:81-87) и усиливает рост и выживаемость -клеток поджелудочной железы(Trumper, et al., 2003, Diabetes, 52:741-750). В дополнение к воздействию на поджелудочную железу для усиления секреции инсулина GIP также оказывает воздействия на целевые ткани инсулина, непосредственно понижая уровень глюкозы в плазме крови: усиление потребления глюкозы при адипозе (Eckel, etal., 1979, Diabetes, 28: 1141-1142) и в мышцах (O'Harte, et al., 1998, J. Endocrinol., 156:237-243) и ингибирование продуцирования глюкозы в печени (Elahi, D., et al., 1986, Can. J. Physiol. Pharmacol., 65:A18). В дополнение к этому, антагонист рецептора GIP в соответствии с настоящим изобретением ингибирует, блокирует или уменьшает поглощение глюкозы из кишечника животного. В соответствии с этим наблюдением терапевтические композиции, содержащие антагонисты GIP, могут быть использованы у пациентов с инсулиннезависимым сахарным диабетом для улучшения переносимости перорального приема глюкозы у млекопитающих, таких как люди, для предотвращения, ингибирования или уменьшения ожирения посредством ингибирования, блокирования или уменьшения поглощения глюкозы из кишечника млекопитающего. Использование немодифицированного GIP в качестве терапевтического средства, однако, ограничивается коротким временем полужизни in vivo, примерно 2 мин (Said and Mutt, 1970, Science, 169:12171218). В сыворотке оба инкретина, как GIP, так и GLP-1, разрушаются под действием дипептидилпептидазы IV ("DPPIV"). Улучшение стабильности GIP по отношению к протеолизу не только поддерживает активность GIP на его рецепторе, но, что более важно, предотвращает образование фрагментов GIP, некоторые из которых действуют как антагонисты рецепторов GIP (Gault, et al., 2002, J. Endocrinol, 175:525533). Известные из литературы модификации включают защиту N-конца GIP от протеолиза под действием DPPIV посредством модифицирования N-концевого тирозина (O'Harte, et al., 2002, Diabetologia, 45: 1281-1291), мутации аланина в положении 2 (Hinke, et al., 2002, Diabetes, 51:656-661), мутации глутаминовой кислоты в положении 3 (Gault, et al., 2003, Biochem. Biophys. Res. Commun., 308:207-213) и мутации аланина в положении 13 (Gault, et al., 2003, Cell Biol. International, 27:41-46). Поданы следующие заявки на патенты, относящиеся к воздействиям аналогов GIP на функционирование различных целевых органов и к их потенциальному применению в качестве терапевтических агентов. РСТ публикация WO 00/58360 описывает пептидильные аналоги GIP, которые стимулируют высво-1 020091 бождение инсулина. В частности, это изобретение описывает конкретные пептидильные аналоги, содержащие по меньшей мере 15 аминокислотных остатков от N-концевой части GIP(1-42), например аналогGIP, содержащий ровно одну аминокислотную замену или модификацию в положениях 1, 2 и 3, такую как [Pro3]GIP(1-42). РСТ публикация WO 98/24464 описывает антагонист GIP, состоящий, по существу, из полипептида,содержащего 24 аминокислоты, соответствующих положениям 7-30 последовательности GIP, способ лечения инсулиннезависимого сахарного диабета и способ улучшения переносимости глюкозы у пациента с инсулиннезависимым сахарным диабетом. РСТ публикация WO 03/082898 описывает С-концевые процессированные фрагменты и аналогиGIP с модифицированным N-концом, а также различные аналоги GIP с восстановленной пептидной связью или с изменениями аминокислот вблизи DPPIV-специфичного сайта расщепления. Это изобретение дополнительно описывает аналоги с различными линкерами между потенциальными сайтами связыванияGIP с рецепторами. Соединения этого изобретения, как предполагается, должны быть пригодными для лечения состояний, опосредуемых GIP-рецепторами, таких как инсулиннезависимый сахарный диабет и ожирение. Имеется необходимость в улучшенных аналогах GIP, которые являются стабильными в виде препаратов и имеют продолжительное время полужизни in vivo в плазме, возникающее в результате понижения чувствительности к протеолизу и уменьшения выделения из организма, в то же время сохраняя сродство связывания с рецептором GIP, чтобы вызвать соответствующие агонистические или антагонистические воздействия. Кроме того, среди других терапевтических воздействий соединений по настоящему изобретению, как иллюстрируется в настоящем описании, более точный контроль уровней глюкозы в плазме крови может предотвратить долговременные диабетические осложнения, обеспечивая тем самым улучшение качества жизни для пациентов. Сущность изобретения В одном из аспектов настоящее изобретение относится к пептидным вариантам GIP следующей формулы (I):R1 представляет собой ОН, NH2, (C1-C30)алкокси или NH-X2-CH2-Z0, где X2 представляет собой (С 0 С 30) углеводородный остаток и Z0 представляет собой Н, ОН, СО 2 Н или CONH2; каждый из R2, R3, R4 и R5 независимо выбирают из группы, состоящей из Н, (C1-C30)алкила, (C1C30)гетероалкила, (С 1-С 30)ацила, (С 2-С 30)алкенила, (С 2-С 30)алкинила, арил(C1-C30)алкила, арил(C1C30)ацила, замещенного (C1-C30)алкила, замещенного (С 1-С 30)гетероалкила, замещенного (C1-C30)ацила,замещенного (С 2-С 30)алкенила, замещенного (С 2-С 30)алкинила, замещенного арил(С 1-С 30)алкила и замещенного арил(C1-C30)ацила; при условии, что когда R2 представляет собой (C1-C30)ацил, арил(C1C30)ацил, замещенный (C1-C30)ацил или замещенный арил(C1-C30)ацил, тогда R3 представляет собой Н,(C1-C30)алкил, (C1-C30)гетероалкил, (С 2-С 30)алкенил, (С 2-С 30)алкинил, арил(C1-C30)алкил, замещенныйn представляет собой, независимо для каждого случая, целое число от 1 до 5 включительно;s, t, х и у, каждый независимо, представляют собой для каждого случая целое число от 1 до 30 включительно;X4, X5, X6, X7 и X8, каждый независимо, представляют собой для каждого случая Н, F, CF3, Cl, Br, I,(C1-10)алкил, замещенный (C1-10)алкил, арил, замещенный арил, ОН, NH2, -CH2NH2, NO2 или CN; при условии, что когда А 1 представляет собой 4 Нрра, тогда R2 и R3 отсутствуют; при дополнительном условии, что более чем одна аминокислота в положениях 1, 2 и 3 соединения заменена или модифицирована; при дополнительном условии, что, если аминокислота в положении 1 модифицирована, она не модифицируется посредством:(e) присоединения N-концевой пироглутаминовой кислоты. Подмножество (А) соединений, охватываемых указанной выше формулой (I), составляют такие соединения, в которых А 1 представляет собой Сра, His, 4Hppa, 2Pal, 3Pal, 4Pal, 3Br-Phe, 4CF3-Phe, 3Cl-Phe, 4CN-Phe, 3FPhe, 4F-Phe, 3,4F-Phe, 3,5F-Phe, 3,4,5F-Phe, 4Me-Phe, 4NH2-Phe, 4NH2CH2-Phe, 3OH-Phe, Taz, 3Thi, 7HOTic, Tyr(Ac), Tyr(Me), -Tyr, 3Br-Tyr, 3,5Br-Tyr, 3Cl-Tyr, 2F-Tyr, 3F-Tyr, hTyr, 3I-Tyr, 3,5I-Tyr, Me-Tyr,2,6Me-Tyr, 3 МеО-Tyr, 3NH2-Tyr, 3NO2-Tyr, 3 ОН-Tyr или 3(HO-CH2)Tyr; А 2 представляет собой Ala, Aib, Gly; А 3 представляет собой Glu, 4Hyp или hPro; А 4 представляет собой Gly; А 5 представляет собой Thr; А 6 представляет собой Phe; А 7 представляет собой Ile, А 5 с или А 6 с; А 8 представляет собой Ser; А 9 представляет собой Asp; А 10 представляет собой Tyr;-4 020091 А 11 представляет собой Ser, A5c или Aib; А 12 представляет собой Ile; А 13 представляет собой Ala или Aib; А 14 представляет собой Met, A5c или Nle; А 15 представляет собой Asp; А 16 представляет собой Lys; А 17 представляет собой Ile; А 18 представляет собой His; А 19 представляет собой Gln; А 20 представляет собой Gln; А 21 представляет собой Asp; А 22 представляет собой Phe; А 23 представляет собой Val; А 24 представляет собой Asn; А 25 представляет собой Trp; А 26 представляет собой Leu; А 27 представляет собой Leu; А 28 представляет собой Ala; А 29 представляет собой Gln; А 30 представляет собой Lys; А 31 представляет собой Gly или отсутствует; А 32 представляет собой Lys или отсутствует; А 33 представляет собой Lys или отсутствует; А 34 представляет собой Asn или отсутствует; А 35 представляет собой Asp или отсутствует; А 36 представляет собой Trp или отсутствует; А 37 представляет собой Lys или отсутствует; А 38 представляет собой His или отсутствует; А 39 представляет собой Asn или отсутствует; А 40 представляет собой Ile, А 5 с или отсутствует; А 41 представляет собой Thr, A5c или отсутствует; А 42 представляет собой Gln или отсутствует; А 43 представляет собой His, Cys(сукцинимид-N-(СН 2)11-СН 3), Orn(N-C(О)-(СН 2)10-СН 3) или отсутствует; при условии, что соединение содержит по меньшей мере одну аминокислотную замену или модификацию в положениях 4-43. Подмножество соединений предыдущего подмножества (А) составляют такие соединения, в которых А 1 представляет собой 4 Нрра; А 43 отсутствует и по меньшей мере один из А 2, А 3, А 7, А 11 и А 14 не является аминокислотным остатком из соответствующего положения в нативном GIP. Другое подмножество (В) соединений предыдущего подмножества (А) составляют такие соединения, в которых А 1 представляет собой Tyr(Ac), Tyr(Me), -Tyr, 3Br-Tyr, 3,5Br-Tyr, 3Cl-Tyr, 2F-Tyr, 3FTyr, hTyr, 3I-Tyr, 3,5I-Tyr, Me-Tyr, 2,6Me-Tyr, 3 МеО-Tyr, 3NH2-Tyr, 3NO2-Tyr, 3 ОН-Tyr или 3(HOCH2)Tyr; А 2 представляет собой А 5 с, А 6 с, Aib, D-Ala, Gly или Ser и по меньшей мере один из А 3, А 11,А 13, А 14, А 40, А 41 и А 43 не является аминокислотным остатком из соответствующего положения в нативном GIP. Подмножество соединений предыдущего подмножества (В) составляют такие соединения, в которых А 2 представляет собой Aib, D-Ala или Gly и по меньшей мере два из А 3, А 11, А 13, А 14, А 40, А 41 и А 43 не являются аминокислотными остатками из соответствующих положений в нативном GIP. Другое подмножество (С) соединений предыдущего подмножества (А) составляют такие соединения, в которых А 1 представляет собой 3Br-Phe, 3Cl-Phe, 4CN-Phe, 3F-Phe, 4F-Phe, 3,4F-Phe, 3,4,5F-Phe,3,5F-Phe, 4NH2-Phe, 4NH2CH2-Phe или 3 ОН-Phe; А 2 представляет собой А 5 с, А 6 с, Aib, D-Ala, Gly илиSer; А 11 представляет собой А 5 с и по меньшей мере один из А 14 и А 41 не является аминокислотным остатком из соответствующего положения в нативном GIP. Подмножество соединений предыдущего подмножества (С) представляет собой такое подмножество, в котором А 2 представляет собой Aib. Другой аспект настоящего изобретения относится к пептидным вариантам GIP, охватываемым указанной выше формулой (I), где пептидная связь между А 1 и А 2 заменяется псевдопептидной связью, где Предпочтительные соединения по настоящему изобретению представляют собой В соответствии с другим аспектом настоящего изобретения соединение в соответствии с настоящим изобретением, как описывается выше и заявляется в прилагаемой формуле изобретения, может дополнительно содержать ковалентно связанный остаток PEG, при этом указанный остаток PEG ковалентно связывается с соединением через Cys(малеимидный), hCys(малеимидный) или Pen(малеимидный) линкер, с образованием Cys(сукцинимид-N-PEG), hCys(сукцинимид-N-PEG) или Pen(сукцинимид-N-PEG), где"сукцинимид-N-PEG" является либо линейным, либо разветвленным, как определено ниже. Такой остаток PEG имеет среднюю молекулярную массу от примерно 2000 до примерно 80000, а предпочтительно такой остаток PEG выбран из группы, состоящей из 5K PEG, 10K PEG, 20K PEG, 30K PEG, 40K PEG,50K PEG и 60K PEG, с образованием Cys(сукцинимид-N-5K PEG), Cys(сукцинимид-N-10K PEG),-8 020091Pen(сукцинимид-N-60K PEG). Пегилирование осуществляют по любому из положений аминокислотных остатков 16, 30 и 31-43, а предпочтительно по любому из положений аминокислотных остатков 32, 33 и 43, при этомCys(сукцинимид-N-PEG), hCys(сукцинимид-N-PEG) или Pen(сукцинимид-N-PEG) располагается в любом из таких положений аминокислотных остатков. Кроме того, приведенная выше формула (I) может быть расширена с получением сайтов пегилирования в положениях А 44-А 47. С-концы таких пегилированных соединений по настоящему изобретению могут быть амидированы, например, (4 Нрра 1,Aib2,А 5 с 7,Nle14)hGIP(1-30)-NH2 (SEQ ID NO:4) или могут оставаться как свободная кислота, например (4Hppa1,Aib2,A5c7,Nle14)hGIP(1-30)-OH (SEQ ID NO:96). Предпочтительные соединения таких пегилированных соединений представляют собой Еще более предпочтительные соединения по настоящему изобретению представляют собой или их фармацевтически приемлемые соли. Еще более предпочтительные соединения по настоящему изобретению представляют собой или их фармацевтически приемлемые соли. И даже еще более предпочтительное соединение по настоящему изобретению представляет собой пример 14: (3Cl-Tyr1,Aib2,А 5 С 11,Nle14)hGIP(1-42)-OH (SEQ ID NO:17) или его фармацевтически приемлемая соль. Подробное описание изобретения Настоящее изобретение использует следующие понятные всем сокращения:Boc: трет-бутилоксикарбонил,BSA: бычий сывороточный альбумин,DCM: дихлорметан,DIPEA: диизопропилэтиламин,DMF: диметилформамид,ESI: электрораспылительная ионизация,Fmoc: 9-флоуренилметилоксикарбонил,HBTU: 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат,НОВТ: 1-гидроксибензотриазол,HPLC: высокоэффективная жидкостная хроматография,IBMX: изобутилметилксантин,LC-MS: жидкостная хроматография - масс-спектрометрия,NMP: N-метилпирролидон,5K PEG: полиэтиленгликоль, который может содержать другие функциональные группы или остатки, такие как линкер, и который является либо линейным, либо разветвленным, как определено ниже, со средней общей молекулярной массой примерно 5000,10K PEG: полиэтиленгликоль, который может содержать другие функциональные группы или остатки, такие как линкер, и который является либо линейным, либо разветвленным, как определено ниже,со средней общей молекулярной массой примерно 10000,20K PEG: полиэтиленгликоль, который может содержать другие функциональные группы или остатки, такие как линкер, и который является либо линейным, либо разветвленным, как определено ниже,со средней общей молекулярной массой примерно 20000,30K PEG: полиэтиленгликоль, который может содержать другие функциональные группы или остатки, такие как линкер, и который является либо линейным, либо разветвленным, как определено ниже,со средней общей молекулярной массой примерно 30000,40K PEG: полиэтиленгликоль, который может содержать другие функциональные группы или остатки, такие как линкер, и который является либо линейным, либо разветвленным, как определено ниже,со средней общей молекулярной массой примерно 40000,50K PEG: полиэтиленгликоль, который может содержать другие функциональные группы или остатки, такие как линкер, и который является либо линейным, либо разветвленным, как определено ниже,со средней общей молекулярной массой примерно 50000,60K PEG: полиэтиленгликоль, который может содержать другие функциональные группы или остатки, такие как линкер, и который является либо линейным, либо разветвленным, как определено ниже,со средней общей молекулярной массой примерно 60000,tBu: трет-бутил,TIS: триизопропилсилан,Trt: тритил,TFA: трифторуксусная кислота,TFFH: тетраметилфторформамидиний гексафторфосфат,Z: бензилоксикарбонил,Греческая буква пси используется в настоящем описании для обозначения того, что пептидная связь заменяется псевдопептидной связью. В наименовании последовательности аминокислот формат членапредставляет собой А 1(Х-Х')А 2, где А 1 представляет собой аминоацильный радикал, у которого карбонильная группа модифицирована как X, и А 2 представляет собой аминоацильный радикал, у которого -аминогруппа модифицирована как X'. X и X' показаны как строчные символы элементов,разделенных связью, например Tyr(CH2-NH)Gly. "Cys(сукцинимид-N-алкил)" имеет структуру За исключением N-концевой аминокислоты, все сокращения аминокислот в настоящем описании(например, Ala) проставлены для структуры -NH-C(R)(R')-CO-, где R и R', каждый независимо, представляют собой водород или боковую цепь аминокислоты (например, R=СН 3 и R1=Н для Ala) или R и R' могут соединяться с образованием кольцевой системы. Для N-концевой аминокислоты сокращение проставлено для структуры (R2R3)N-C(R) (R')-CO-, где R2 и R3 являются такими, как определено в указанной выше формуле (I). Термин "(C1-С 30)углеводородный остаток" охватывает алкил, алкенил и алкинил, и в случае алкенила и алкинила имеются в виду С 2-С 30 остатки. Пептид по настоящему изобретению также обозначается в настоящем описании с помощью другого формата, например (A5c2)hGIP(1-42)-ОН (SEQ ID NO:3), при этом замененные аминокислоты из природной последовательности помещаются в скобках (например, А 5 с 2 для Ala2 в hGIP). Числа между скобками относятся к количеству аминокислот, присутствующих в пептиде (например, hGIP(1-42)-ОН (SEQ IDNO:1) представляет собой аминокислоты 1-42 последовательности пептидов для hGIP). ОбозначениеID NO:1) или hGIP(1-42)-ОН (SEQ ID NO:1) означает, что С-конец представляет собой свободную кислоту."Ацил" относится к R"-C(O)-, где R" представляет собой Н, алкил, замещенный алкил, гетероалкил,замещенный гетероалкил, алкенил, замещенный алкенил, арил, алкиларил или замещенный алкиларил."Алкил" относится к углеводородной группе, содержащей один или несколько атомов углерода, где множество атомов углерода, если они присутствуют, соединяются с помощью одинарных связей. Алкильная углеводородная группа может представлять собой прямую цепь или содержать одну или несколько ветвей или циклических групп."Замещенный алкил" относится к алкилу, где один или несколько атомов водорода углеводородной группы заменяются одним или несколькими заместителями, выбранными из группы, состоящей из галогена (то есть фтора, хлора, брома и йода), -ОН, -CN, -SH, -NH2, -NHCH3, -NO2, -С 1-20 алкила, замещенного галогенами, -CF3, -ОСН 3, -OCF3 и -(СН 2)0-20-СООН. В различных вариантах осуществления присутствуют 1, 2, 3 или 4 заместителей. Присутствие -(СН 2)0-20-COOH приводит к получению алкиловой кислоты. Примеры алкиловых кислот, содержащих -(СН 2)0-20-СООН или состоящих из них, включают 2 норборнануксусную кислоту, трет-масляную кислоту и 3-циклопентилпропионовую кислоту."Гетероалкил" относится к алкилу, где один или несколько атомов углерода в углеводородной группе заменяются одной или несколькими из следующих групп: амино, амидо, -O-, -S- или карбонилом. В различных вариантах осуществления присутствуют 1 или 2 гетероатома."Замещенный гетероалкил" относится к гетероалкилу, где один или несколько атомов водорода углеводородной группы заменяются одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -ОН, -CN, -SH, -NH2, -NHCH3, -NO2, -С 1-20 алкила, замещенного галогенами, -CF3,-ОСН 3, -OCF3 и -(СН 2)0-20-СООН. В различных вариантах осуществления присутствуют 1, 2, 3 или 4 заместителей."Алкенил" относится к углеводородной группе, состоящей из двух или более атомов углерода, где присутствуют одна или несколько двойных связей углерод-углерод. Алкенильная углеводородная группа может представлять собой прямую цепь или содержать одну или несколько ветвей или циклических групп."Замещенный алкенил" относится к алкенилу, где один или несколько атомов водорода заменяются одним или несколькими заместителями, выбранными из группы, состоящей из галогена, ОН, -CN, -SH,-NH2, -NHCH3, -NO2, -C1-20 алкила, замещенного галогенами, -CF3, -OCH3, -OCF3 и -(СН 2)0-2O-СООН. В различных вариантах осуществления присутствуют 1, 2, 3 или 4 заместителей."Арил" относится к необязательно замещенной ароматической группе по меньшей мере с одним кольцом, имеющим систему сопряженных пи-электронов, содержащую вплоть до трех сопряженных или конденсированных кольцевых систем. Арил включает карбоциклическую арильную, гетероциклическую арильную и биарильную группы. Предпочтительно арил представляет собой 5- или 6-членное кольцо. Предпочтительные атомы для гетероциклического арила представляют собой один или несколько атомов серы, кислорода и/или азота. Примеры арила включают фенил, 1-нафтил, 2-нафтил, индол, хинолин, 2 имидазол и 9-антрацен. Арильные заместители выбираются из группы, состоящей из -C1-20 алкила, -C120 алкокси, галогена, -ОН, -CN, -SH, -NH2, -NO2, -C1-20 алкила, замещенного галогенами, -CF3, -OCF3 и-(СН 2)0-20-СООН. В различных вариантах осуществления арил содержит 0, 1, 2, 3 или 4 заместителей."Алкиларил" относится к "алкилу", соединенному с "арилом". Синтез. Пептиды по настоящему изобретению могут быть получены с помощью стандартного твердофазного пептидного синтеза; см., например, Stewart, J.M., et al., 1984, Solid Phase Synthesis, Pierce Chemical Co.,2d ed. Если R1 представляет собой NH-X2-CH2-CONH2, то есть Z0=CONH2, синтез пептида начинается сFmoc-HN-X2-CH2-CONH2, который связан с амидной МВНА смолой Rink. Если R1 представляет собойNH-X2-CH2-COOH, то есть Z0=СООН, синтез пептида начинается с Fmoc-HN-X2-CH2-COOH, который связан со смолой Wang. Для этой конкретной стадии используют 2 мол.экв. Fmoc-HN-X2-COOH, HBTU иHOBt и 10 мол.экв. DIPEA. Время конденсации составляет примерно 8 ч. При синтезе аналога GIP по настоящему изобретению, содержащего А 5 с, А 6 с и/или Aib, время конденсации составляет 2 ч для этих остатков и для остатка, следующего непосредственно после них. Заместители R2 и R3 указанной выше общей формулы могут быть присоединены к свободному амину N-концевой аминокислоты А 1 с помощью стандартных способов, известных в данной области. Например, алкильные группы, например (C1-C30)алкил, могут быть присоединены с использованием восстановительного алкилирования. Гидроксиалкильные группы, например (C1-C30)гидроксиалкил, могут быть также присоединены с использованием восстановительного алкилирования, где свободная гидроксигруппа защищена сложным трет-бутиловым эфиром. Ацильные группы, например -С(О)Х 3, могут быть присоединены посредством связывания свободной кислоты, например -Х 3 СООН, со свободным амином N-концевой аминокислоты посредством смешивания готовой смолы с 3 мол.экв. как свободной кислоты, так и диизопропилкарбодиимида в метиленхлориде в течение примерно 1 ч. Если свободная кислота содержит свободную гидроксигруппу, например 3-фтор-4-гидроксифенилуксусную кислоту,тогда конденсация должна осуществляться с дополнительными 3 мол.экв. НОВТ. Следующие далее примеры описывают способы синтеза для получения пептида по настоящему изобретению, эти способы хорошо известны специалистам в данной области. Другие способы также известны специалистам в данной области. Примеры приводятся для целей иллюстрации и не предназначены для ограничения рамок настоящего изобретения каким-либо образом. Пример 1. (4 Нрра 1, Aib2, A5c7,Nle14) hGIP(1-30)-NH2. Указанный в заголовке пептид синтезируется автоматически на пептидном синтезаторе, модель 433 А, Applied Biosystems (Foster City, CA, USA), на основе флуоренилметилоксикарбонильного (Fmoc) метода. Используют Rink Amide MBHA (4-метилбензгидриламиновую) смолу (Nova Biochem, La Jolla,CA, USA) с емкостью 0,72 ммоль/г. Аминокислотные картриджи Fmoc получают от AnaSpec (San Jose,CA, USA). Аминокислоты Fmoc с защитой боковых цепей являются следующими: Fmoc-Lys(Boc)-ОН,Fmoc-Gln(Trt)-ОН, Fmoc-Ala-OH, Fmoc-Leu-OH, Fmoc-Trp(Boc)-OH, Fmoc-Asn(Trt)-ОН, Fmoc-Val-OH,Fmoc-Phe-OH, Fmoc-Asp(OtBu)-OH Fmoc-His(Trt)-OH, Fmoc-Ile-OH, Fmoc-Nle-OH, Fmoc-Ser(tBu)-OH,Fmoc-Tyr(tBu)-OH, Fmoc-A5c-OH, Fmoc-Thr(tBu)-OH, Fmoc-Gly-OH, и Fmoc-Glu(OtBu)-ОН и Fmoc-AibOH. 3-(4-Гидроксифенил)пропионовую кислоту (4 Нрра) покупают от Sigma-Aldrich (St. Louis, MO, USA). Синтез осуществляют в масштабе 0,25 ммоль. Пептидный синтезатор ABI 433A программируют для осуществления следующего цикла реакций: (1) промывка NMP, (2) удаление Fmoc-защитной группы с помощью 20% пиперидина в NMP в течение 10 мин, (3) промывка NMP - в течение циклов промывки и удаления Fmoc, аминокислота Fmoc (4 экв., 1 ммоль) сначала предварительно активируется с помощью 2 мл раствора 0,45 М 2-(1-Н-бензотриазол-1-ил)-1,1,2,3-тетраметилурония гексафторфосфата/1-гидроксибензотриазола (HBTU/HOBT) в DMF; это активирует сложный эфир аминокислоты, 1 мл 2 M диизопропилэтиламина (DIPEA) и 2,5 мл NMP добавляют к смоле, (4) конденсация с предварительно активированной аминокислотой в течение 1 ч. Конденсацию А 5 с со следующим после нее Phe и Aib со следующей после нее 4 Нрра продолжают до истечения 3 ч. Конденсацию со смолой последовательно проводят дважды в соответствии с последовательностью. После окончания сборки пептидной цепи защищенный пептид отщепляют от смолы в смеси TFA, H2O, TIS (15 мл/1,28 мл/1,35 мл) в течение 3 ч. Смесь фильт- 17020091 руют в 140 мл холодного эфира и центрифугируют с получением осадка. Этот сырой продукт растворяют в 20 мл 50% АсОН и разбавляют 180 мл воды. Продукт очищают с помощью препаративной HPLC с обращенной фазой, с использованием колонки (443 см) C18 DYNAMAX-100 A0 (Varian, Walnut Creek, CA,USA). Колонку элюируют линейным градиентом от 20% В до 45% В через 45 мин, где А представляет собой 0,1% TFA в воде, а В представляет собой 0,1% TFA в CH3CN. После проверки с помощью MS иHPLC все чистые фракции собирают и лиофилизируют досуха. Чистота соединения составляет 99,90%. Анализ с помощью масс-спектрометрии с электрораспылительной ионизацией (ESI-MS) дает молекулярную массу 3510,8 в соответствии с вычисленной молекулярной массой 3510,93. Пример 11. (3,5Br-Tyr1,Aib2,13,Nle14)hGIP(1-42)-ОН. Указанный в заголовке пептид синтезируют автоматически на пептидном синтезаторе Applied Biosystems (Foster City, CA, USA) модель 433 на основе флуоренилметилоксикарбонильного (Fmoc) метода. Используют Fmoc-Gln(Trt) на смоле Wang (Novabiochem., La Jolla, CA, USA) с емкостью 0,59 ммоль/г. Аминокислотные картриджи для Fmoc получают от AnaSpec (San Jose, CA, USA). Аминокислоты Fmoc с защитой боковых цепей являются следующими: Fmoc-Thr(tBu)-ОН, Fmoc-Ile-OH, Fmoc-Asn(Trt)-ОН,Fmoc-His(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Gly-OH FmocGln(Trt)-OH, Fmoc-Ala-OH, Fmoc-Leu-OH, Fmoc-Val-OH, Fmoc-Phe-OH, Fmoc-Ile-OH, Fmoc-Nle-OH,Fmoc-Ser(tBu)-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Glu(OtBu)-ОН и Fmoc-Aib-OH. 3,5Br-Tw-OH получают отChem-Impex International (Wooddale, IL, USA). Синтез осуществляют в масштабе 0,25 ммоль. Пептидный синтезатор ABI 433A программируют для осуществления следующего реакционного цикла: (1) промывкаNMP, (2) удаление Fmoc-защитной группы с помощью 20% пиперидина в NMP в течение 10 мин, (3) промывка NMP - в течение цикла удаления Fmoc и после промывки, аминокислота для Fmoc (4 экв., 1 ммоль) сначала предварительно активируется с помощью 2 мл раствора 0,45 М 2-(1-Н-бензотриазол-1 ил)-1,1,2,3-тетраметилурония гексафторфосфата/1-гидроксибензотриазола (HBTU/HOBT) в DMF; это активирует сложный эфир аминокислоты, к смоле добавляют 1 мл 2 М диизопропилэтиламина (DIPEA) и 2,5 мл NMP, (4) конденсация с предварительно активированной аминокислотой в течение 1 ч. Конденсацию Aib со следующим после нее Ile продолжают до истечения 3 ч. Конденсацию 3,5Br-Tyr-ОН осуществляют вручную с помощью PYAOP [(7-азабензотриазол-1-ил)окси-трис-(пирролидино)фосфония гексафторфосфат] в качестве конденсирующего реагента, в присутствии DIPEA и пентафторфенола. Конденсацию со смолой последовательно проводят дважды в соответствии с последовательностью. После окончания сборки пептидной цепи смолу с защищенным пептидом обрабатывают раствором, содержащим Na2CO3/DMF/DBU (1/0,5/0,1), в течение 2 ч, затем отщепляют Fmoc с помощью 25% пиперидина в течение 45 мин. Продукт отщепляют в смеси TFA, H2O, TIS, тиоанизола и фенола (12 мл/0,64/0,64/0,5/0,5) в течение 3 ч, а затем фильтруют в 140 мл холодного эфира. После центрифугирования получают осадок. Этот сырой продукт растворяют в 20 мл 50% АсОН, разбавляют 180 мл воды и очищают с помощью препаративной HPLC с обращенной фазой, с использованием колонки (443 см) C18DYNAMAX-100 A0 (Varian, Walnut Creek, CA, USA). Колонку элюируют с помощью линейного градиента от 20% В до 45% В через 45 мин, где А представляет собой 0,1% TFA в воде и В представляет собой 0,1% TFA в CH3CN. После проверки с помощью MS и HPLC все чистые фракции собирают и лиофилизируют досуха. Чистота составляет 99,90%. Масс-спектрометрический анализ с помощью ESI показывает 5150,8. Пример 22. (3,4,5F-Phe1,Aib2,A5c11,Nle14)hGIP(1-42)-ОН.Fmoc-[Aib2, A5c11, Nle14]hGIP (2-42) с защищенными боковыми цепями на смоле Wang синтезируют на пептидном синтезаторе, модель 433 А, Applied Biosystems (Foster City, CA, USA) с использованием флуоренилметилоксикарбонильного (Fmoc) метода. Используют Fmoc-Gln(Trt) на смоле Wang (Novabiochem., San Diego, CA, USA) с емкостью 0,59 ммоль/г. Для Fmoc используют аминокислоты (AnaSpec, SanJose, CA, USA) Fmoc-Ala-OH, Fmoc-Aib-OH, Fmoc-Asn(Trt)-OH, Fmoc-Asp(tBu)-OH, Fmoc-A5c-OH,Fmoc-Gln(Trt)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Gly-OH, Fmoc-His(Trt)-OH, Fmoc-Ile-OH, Fmoc-Leu-OH,Fmoc-Lys(Boc)-OH, Fmoc-Phe-OH, Fmoc-Nle-OH, Fmoc-Ser(tBu)-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Thr(tBu)OH, Fmoc-Trp(Boc)-ОН и Fmoc-Val-OH. Синтез осуществляют в масштабе 0,2 ммоль. Группы Fmoc удаляют посредством обработки 20% пиперидином в N-метилпирролидоне (NMP) в течение 30 мин. На каждой стадии конденсации аминокислота Fmoc (3 экв., 0,3 ммоль) сначала предварительно активируется в 2 мл раствора 0,45 M 2-(1-Н-бензотриазол-1-ил)-1,1,2,3-тетраметилурония гексафторфосфата/1 гидроксибензотриазола (HBTU/HOBT) в NMP. Так получают активированный сложный эфир аминокислоты, к смоле добавляют 1 мл диизопропилэтиламина (DIPEA) и 1 мл NMP. Пептидный синтезатор ABI 433A программируют для осуществления следующего цикла реакций: (1) промывка NMP, (2) удалениеFmoc-защитной группы с помощью 20% пиперидина в NMP в течение 30 мин, (3) промывка NMP, (4) конденсация с предварительно активированной аминокислотой для Fmoc в течение 3 ч, (5) промывкаNMP и (6) конденсация с предварительно активированной аминокислотой Fmoc в течение 3 ч. Один эквивалент TFFH (тетраметилфторформамидиния гексафторфосфата; Perceptive Biosystems, Warrington,UK) добавляют для связывания Fmoc-A5c-ОН и Fmoc-Tyr(tBu)-ОН в положениях 10 и 11. Конденсацию со смолой последовательно проводят дважды в соответствии с последовательностью указанного в заго- 18020091 ловке пептида. После сборки пептидной цепи смолу отмывают полностью посредством использованияNN-диметилформамида (DMF) и дихлорметана (DCM). В конце сборки пептидной цепи на 433 А смолу с пептидом переносят в реакционную емкость на шейкере и Fmoc удаляют с использованием 25% Pip/DMF в течение 30 мин. Смолу промывают DMF.(Oakwood Products; West Columbia, SC, USA) (2,0 ммоль) и DIPEA (2,0 ммоль). Группу Fmoc удаляют,как указано выше. Для отщепления указанного в заголовке пептида смолу обрабатывают смесью TFA, Н 2 О и триизопропилсилана (TIS) (9,5 мл/0,85 мл/0,8 мл) в течение 4 ч. Смолу отфильтровывают и фильтрат выливают в 200 мл эфира. Осадок собирают посредством центрифугирования. Этот сырой продукт растворяют в смеси ацетонитрила и воды и очищают в системе препаративной HPLC с обращенной фазой с помощью колонки (250-21,2 мм) C18 Luna (Phenomenex). Колонку элюируют в течение 80 мин с использованием линейного градиента от 100% А: до 55% А:45% В, где А представляет собой 0,1% TFA в воде и В представляет собой 0,1% TFA в ацетонитриле. Фракции проверяют с помощью аналитической HPLC, и те,которые содержат чистый продукт, собирают и лиофилизируют досуха с получением 156,5 мг (15,5%) белого твердого вещества. Чистоту анализируют с использованием HPLC и находят, что она равна приблизительно 99,90%. Анализ с помощью масс-спектрометрии с электрораспылительной ионизацией (ESIMS) дает молекулярную массу 5041,5. Пример 40. [Tyr1(CH2NH)Gly2,A5c11,14]hGIP(1-42)-ОН. А. Сборка пептидной цепи (Gly2, A5c11,41)hGIP(2-42)ОН. Сборку проводят с использованием метода Fmoc с микроволновым нагреванием на пептидном синтезаторе Liberty (СЕМ; Matthews, NC, USA) в масштабе 0,20 ммоль. Предварительно загруженную FmocGln(Trt) на смоле Wang (0,59 ммоль/г; Novabiochem, San Diego, CA, USA) используют для генерирования пептида с С-концевой кислоты. Смолу (0,423 г) помещают в 50-мл коническую пробирку вместе с 15 мл диметилформамида (DMF) и помещают на место смолы в синтезаторе. Затем смолу количественно переносят в реакционную емкость с помощью автоматизированного способа. Используют стандартный протокол синтеза Liberty для синтеза в масштабе 0,25 ммоль. Этот протокол включает снятие защиты с Nконцевого остатка Fmoc посредством начальной обработки с помощью 7 мл 20% пиперидина, содержащего 0,1 М N-гидроксибензотриазол (НОВТ), в DMF. Начальная стадия снятия защиты осуществляется в течение 30 с с помощью микроволновой мощности (45 Вт, максимальная температура 75 С) и барботирования азота (3 с включено/7 с выключено). Затем реакционную емкость очищают и осуществляют вторую обработку пиперидином, идентичную первой обработке, за исключением того, что она происходит в течение 3 мин. Затем смолу очищают и несколько раз тщательно промывают DMF. Цикл 2 представляет собой связывание Fmoc-A5c-0H, затем добавляют ее 0,2 М исходный раствор в DMF (2,5 мл, 5 экв.), а затем 1,0 мл 0,45 М (4,5 экв.) HBTU [2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат] в DMF. За этим следует добавление 0,5 мл 2 М (10 экв.) DIPEA (диизопропилэтиламин) в NMP (Nметилпирроллидинон). Стадию конденсации осуществляют в течение 5 мин с использованием микроволновой мощности 20 Вт при максимальной температуре 75 С и такой же, как раньше, скорости барботирования азота. После начальной стадии конденсации реакционную емкость очищают от отходов и конденсацию повторяют еще раз. Цикл 3 после Fmoc А 5 с использует агрессивную конденсацию, при которой стадию конденсации осуществляют в течение 10 мин с использованием микроволновой мощности 20 Вт при максимальной температуре 90 С. Все аминокислоты вводят подобно тому, что описано для цикла 2, за исключением того, что агрессивные конденсации применяют к Fmoc A5c-OH на цикле 32 и для следующей после нее Fmoc Tyr(tBu)-OH. Стратегию двойной конденсации используют по всей последовательности. Циклы 2, 4, 20, 21, 26, 27, 31, 36, 37, 38, 39, 41 включают процедуру кэппирования непосредственно после стадии конденсации. Кэппирование осуществляют посредством добавления 7 мл 0,5 М уксусного ангидрида, содержащего 0,015 М НОВТ, в NMP, вместе с 2 мл 2 М раствора DIPEA с использованием многостадийного микроволнового протокола: мощность 50 Вт в течение 30 с (максимальная температура 65 С), затем 30 с отключения микроволновой мощности, затем второй заход в 30 с подключения микроволновой мощности (50 Вт), а затем опять 30 с без микроволновой мощности. Затем смолу очищают и тщательно промывают DMF. Используют следующие аминокислоты (Advanced Chemtech, Louisville, KY, USA): цикл 2: Fmoc-A5c-OH; цикл 3: Fmoc-Ile-OH; цикл 4: Fmoc-Asn (Trt)-ОН; цикл 5: Fmoc-His(Trt)-ОН; цикл 6: Fmoc-Lys(Boc)-ОН; цикл 7: Fmoc-Trp(Boc)-ОН; цикл 8: Fmoc-Asp(OtBu)ОН; цикл 9: Fmoc-Asn (Trt)-ОН; цикл 10: Fmoc-Lys(Boc)-ОН; цикл 11: Fmoc-Lys(Boc)-ОН; цикл 12:Gly-OH, цикл 40: Fmoc-Glu(OtBu)-ОН и цикл 41: Fmoc-Gly-OH. Протокол конденсации для FmocHis(Trt)-ОН представляет собой слегка модифицированную версию стандартного протокола. Микроволновую мощность отключают в течение первых 2 мин, затем следуют 4 мин подачи микроволновой мощности (20 Вт; максимальная температура 50 С). После завершения сборки пептидной цепи смолу обрабатывают 20% пиперидина в DMF в течение 40 мин для снятия защиты с Fmoc на N-конце. В. Синтез Fmoc-Tyr(tBu)-CHO.(1 г, 10 ммоль), НОВТ (1,37 г, 10,1 ммоль) и DIPEA (5,25 мл, 30 ммоль) на ледяной бане в течение 15 мин, к ним добавляют EDC (2,11 г, 11 ммоль). Реакционный раствор перемешивают при комнатной температуре в течение 15 ч. Его разбавляют DCM, который последовательно промывают насыщеннымNaHCO3 (50 мл 3), 10% лимонной кислотой (50 мл 3) и насыщенным раствором соли (50 мл 3). После того как весь слой DCM сушат над MgSO4, его удаляют с получением 4,62 г с MS 503,4 (MW 503,6).N-метокси-N-метил(Fmoc-Tyr(tBu)-карбоксамид восстанавливают до Fmoc-Tyr(tBu)-CHO. Литий-алюминий гидрид (36 мл, 1 М) медленно добавляют к 60 мл охлаждаемого перемешиваемого раствора N-метокси-N-метил(Fmoc-Tyr(tBu-карбоксамида в THF (3,6 г, 7,1 ммоль) через 50 мин. Восстановление заканчивается через 20 мин. Смесь гидролизуют с помощью 100 мл 0,5 н. KHSO4 в течение 0,5 ч. Ее экстрагируют 200 мл простого эфира. Весь органический слой промывают 10% KHSO4 (50 мл 2), насыщенным раствором соли (50 мл 3), затем сушат над MgSO4. Растворитель выпаривают с получением сырого продукта альдегида. Указанный выше сырой Fmoc-Tyr(tBu)-CHO (3,115 ммоль) в 5 мл DMF добавляют к смоле с пептидом (Gly2,A5c11,41)hGIP(2-42) (0,2 ммоль) в 5 мл DMF с 100 мкл АсОН. Смесь обрабатывают NaBH3CN(0,1957 г) в течение 1 ч, затем добавляют вторую порцию NaBH3CN (0,1957 г). Это повторяют еще раз,затем смолу встряхивают в течение ночи. После этого смолу промывают и отщепляют Fmoc с помощью 25% пиперидина в течение 45 мин. Его отщепляют с помощью раствора TFA, H2O, TI и DTT (15 мл/1,28 мл/1,35 мл/0,75 г) в течение 3 ч, а затем фильтруют в 140 мл холодного эфира. После центрифугирования получают осадок. Этот сырой продукт растворяют в 20 мл 50% АсОН, разбавляют 180 мл воды и очищают с помощью препаративной HPLC с обращенной фазой, с использованием колонки (443 см) C18DYNAMAX-100 А 0 (Varian, Walnut Creek, CA, USA). Колонку элюируют линейным градиентом от 20% В до 45% В через 45 мин, где А представляет собой 0,1% TFA в воде и В представляет собой 0,1% TFA вCH3CN. После проверки с помощью MS и HPLC все чистые фракции собирают и лиофилизируют досуха. Чистота составляет 98,93%. Масс-спектрометрический анализ ESI показывает 4989,7 в соответствии с вычисленной молекулярной массой 4989,67. Другие пептиды по настоящему изобретению могут быть получены специалистом в данной области с использованием процедур синтеза, аналогичных тем, которые описаны в указанных выше примерах. Физические данные для соединений, иллюстрируемых в настоящем описании, представлены в табл. 1. Функциональные анализы. А. Анализ связывания с рецептором hGIP in vitro. Мембраны для анализа связывания с рецептором in vitro готовят посредством гомогенизации клональных клеток СНО-K1, экспрессирующих рекомбинантный рецептор GIP человека, с помощью Brinkman Polytron (настройки 6, 15 с), в 50 мМ охлаждаемого на льду Tris-HCl, а затем подвергают воздейст- 22020091 вию двух центрифугирований при 39000 g в течение 10 мин с повторным суспендированием в свежем буфере между ними. Для анализа аликвоты промытых препаратов мембран инкубируют (100 мин при 25 С вместе с 0,05 нМ [125I]GIP (2200 Кюри/ммоль) в 50 мМ Tris-HCl, 0,1 мг/мл бацитрацина и 0,1%BSA. Конечный объем анализа составляет 0,5 мл. Инкубирования завершают посредством быстрого фильтрования через фильтры GF/C (предварительно пропитанные в 0,5% полиэтиленимина) с использованием фильтрационного коллектора Brandel. Каждую пробирку и фильтр затем промывают три раза с помощью 5-мл аликвоты буфера, охлажденного на льду. Специфичное связывание определяют как общее количество связанного радиоактивного лиганда минус то количество, которое связывается в присутствии 1000 нМ GIP. Данные связывания с рецепторомhGIP in vitro для соединений, иллюстрируемых в настоящем описании, представлены в табл. 2. В. Анализ времени полужизни в плазме крови человека и крысы. Пептид GIP (50 мкл, 1 мг/мл) добавляют к 450 мкл плазмы крови (человека или крысы), встряхивают в течение короткого времени и инкубируют при 37 С. 50 мкл удаляют в различные моменты времени,например через 0, 1, 2, 3, 4, 8, 24, 32, 48, 56, 72 ч, смешивают с 5 мкл муравьиной кислоты и 150 мкл ацетонитрила в пробирке для микроцентрифуги, встряхивают и центрифугируют в течение 10 мин при 10000 об/мин. Супернатант переносят во флакон для инъекций и анализируют с помощью LC-MS. Система LC-MS состоит из масс-спектрометра API4000 с датчиком ESI. Используют режим положительных ионов и детектирование полного сканирования. Разделение с помощью HPLC осуществляют на колонкеLuna 3 мкм С 8 (2), 230 мм с градиентом от 90% А до 90% В через 10 мин при скорости потока 0,3 мл/мин. Буфер А представляет собой 1% муравьиную кислоту в воде, а буфер В представляет собой 1% муравьиную кислоту в ацетонитриле. Данные относительно времени полужизни для плазмы крови человека и крысы для соединений, иллюстрируемых в настоящем описании, представлены в табл. 2. Таблица 2C. Определение циклического стимулирования AMP. 1105 клеток СНО-K1, экспрессирующих рекомбинантный рецептор GIP человека, или клеток инсулиномы RIN-5F высевают на ночь в 24-луночные планшеты для культур клеток (Corning Incorporate,Corning, NY, USA). Для анализа клетки предварительно инкубируют в 500 мкл сбалансированного солевого раствора Хэнкса (Sigma, St. Louis, МО, USA) вместе с 0,55 мМ IBMX (Sigma, St. Louis, МО, USA) и доводят рН до 7,3 в течение 10 мин. Затем добавляют GIP или его аналоги при концентрации 100 нМ. После 30-минутного инкубирования при 37 С планшеты помещают на лед и добавляют 500 мкл охлажденного на льду абсолютного этанола для остановки реакции. Содержимое лунок собирают, центрифугируют при 2700 g в течение 20 мин при 4 С для удаления клеточных остатков. Уровни сАМР в супернатантах определяют с помощью радиоиммунологического анализа (New England Nuclear, Boston, MA,USA).D. Определение секреции инсулина in vivo у нормальных крыс. В качестве субъектов экспериментов используют самцов крыс Sprague Dawley с массой тела при- 25020091 близительно 275-300 г. В день перед лечением правые атриальные канюли имплантируют через югулярную вену под хлоргидратным наркозом. Каждую канюлю заполняют 100 единиц/мл гепаринового солевого раствора и перевязывают. Крыс фиксируют в течение приблизительно 18 ч перед дозированием соединения или носителя (солевой раствор/0,25% BSA). В день эксперимента аликвоты соединения оттаивают, доводят до комнатной температуры и тщательно встряхивают. Тщательно проверяют наличие любого признака соединения, выпадающего из раствора. За 10 мин перед инъекцией соединения/глюкозы 500-мкл образец крови отбирают и заменяют равным объемом гепаринизированного солевого раствора(10 единиц/мл). В момент времени 0 отбирают через канюлю 500-мкл образец крови. Затем либо носитель, либо соответствующую дозу соединения вводят в канюлю и проталкивают ее с помощью раствора глюкозы (1 г/кг) или носителя. Наконец, 500-мкл объем гепаринизированного солевого раствора (10 единиц/мл) используют для проталкивания остальной глюкозы через канюлю. Дополнительные 500-мкл образцы крови отбирают через 2,5, 5, 10 и 20 мин после дозирования глюкозы; после каждого отбора непосредственно следует болюс, внутривенная инъекция 500 мкл гепаринизированного солевого раствора (10 единиц/мл) через канюлю. Плазму собирают из образцов крови посредством центрифугирования и хранят при -20 С до осуществления анализа на содержание инсулина. Численные значения общей секреции инсулина, которые показывают воздействия соединений примеров 14 и 28 in vivo, представлены в табл. 3. Таблица 3 Способы введения. Пептиды по настоящему изобретению могут предусматриваться в форме фармацевтически приемлемых солей. Примеры таких солей включают, но не ограничиваясь этим, соли, образованные с органическими кислотами (например, с уксусной, молочной, малеиновой, лимонной, яблочной, аскорбиновой,янтарной, бензойной, метансульфоновой, толуолсульфоновой или памовой кислотой), с неорганическими кислотами (например, с хлористо-водородной кислотой, серной кислотой или фосфорной кислотой) и с полимерными кислотами (например, с дубильной кислотой, карбоксиметилцеллюлозой, полимолочной,полигликолевой кислотой или с сополимерами полимолочных-гликолевых кислот). Типичный способ получения соли пептида по настоящему изобретению хорошо известен в данной области и может осуществляться с помощью стандартных способов солевого обмена. Соответственно соль TFA и пептида по настоящему изобретению (соль TFA получается в результате очистки пептида посредством использования препаративной HPLC, элюирования с помощью буферных растворов, содержащих TFA) может быть преобразована в другую соль, такую как ацетатная соль, посредством растворения пептида в малом количестве водного раствора 0,25 н. уксусной кислоты. Полученный раствор наносят на полупрепаративную колонку для HPLC (Zorbax, 300 SB, C-8). Колонку элюируют (1) 0,1 н. водным раствором ацетата аммония в течение 0,5 ч, (2) 0,25 н. водным раствором уксусной кислоты в течение 0,5 ч и (3) линейным градиентом (от 20 до 100% раствора В в течение 30 мин) при скорости потока 4 мл/мин (раствор А представляет собой 0,25 н. водный раствор уксусной кислоты; раствор В представляет собой 0,25 н. уксусную кислоту в ацетонитриле/воде, 80:20). Фракции, содержащие пептид, собирают и лиофилизируют досуха. Доза активного ингредиента в композиции по настоящему изобретению может изменяться; однако необходимо, чтобы количество активного ингредиента было таким, чтобы получалась пригодная для использования лекарственная форма. Выбранная дозировка зависит от желаемого терапевтического воздействия, от способа введения и от продолжительности лечения. Как правило, эффективная дозировка для активности по настоящему изобретению находится в пределах от 110-7 до 200 мг/кг/день, предпочтительно от 110-4 до 100 мг/кг/день, которая может быть введена как одна доза или разделена на множество доз. Соединения по настоящему изобретению могут быть введены посредством перорального, парентерального (например, внутримышечной, внутрибрюшинной, внутривенной или подкожной инъекции или импланта), назального, вагинального, ректального, сублингвального или местного способа введения и могут быть составлены вместе с фармацевтически приемлемыми носителями для получения лекарственных форм, соответствующих каждому способу введения. Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли,порошки и гранулы. В таких твердых лекарственных формах активное соединение смешивается по меньшей мере с одним инертным фармацевтически приемлемым носителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы могут также содержать, что является обычной практикой, дополнительные вещества, иные, чем такие инертные разбавители, например смазывающие агенты, такие как стеарат магния. В случае капсул, таблеток и пилюль лекарственные формы могут также содержать буферные агенты. В дополнение к этому, таблетки и пилюли могут быть приготовлены с энтеральными покрытиями. Жидкие лекарственные формы для перорального введения включают, без ограничения, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры и т.п., содержащие инертные разбавители, обычно используемые в данной области, такие как вода. Кроме таких инертных разбавителей, композиции могут также содержать вспомогательные вещества, такие как смачивающие агенты,эмульгирующие и суспендирующие агенты и подслащивающие, ароматизирующие и парфюмерные агенты. Препараты в соответствии с настоящим изобретением для парентерального введения включают, без ограничения, стерильные водные или неводные растворы, суспензии, эмульсии и т.п. Примеры неводных растворителей или наполнителей включают пропиленгликоль, полиэтиленгликоль, растительные масла,такие как оливковое масло и кукурузное масло, желатин и сложные органические эфиры для инъекций,такие как этилолеат. Такие лекарственные формы могут также содержать вспомогательные вещества,такие как консервирующие, смачивающие, эмульгирующие и диспергирующие агенты. Они могут быть стерилизованы, например, с помощью фильтрования через фильтр, удерживающий бактерии, посредством введения в композиции стерилизующих агентов, посредством облучения композиций или посредством нагревания композиций. Они могут также быть получены в форме стерильных твердых композиций,которые могут растворяться в стерильной воде или в некоторой другой стерильной среде для инъекций непосредственно перед использованием. Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые могут содержать, в дополнение к активному веществу, наполнители, такие как масло какао или воск для суппозиториев. Композиции для назального или сублингвального введения также получают с помощью стандартных наполнителей, хорошо известных в данной области. Кроме того, соединение по настоящему изобретению может быть введено в композиции с замедленным высвобождением, такой как та, которая описывается в следующих далее патентах и заявках на патенты. Патент США 5672659 сообщает о композициях с замедленным высвобождением, содержащих биологически активный агент и сложный полиэфир. Патент США 5595760 сообщает о композициях с замедленным высвобождением, содержащих биологически активный агент в гелеобразной форме. Патент США 5821221 сообщает о полимерных композициях с замедленным высвобождением, содержащих биологически активный агент и хитозан. Патент США 5916883 сообщает о композициях с замедленным высвобождением, содержащих биологически активный агент и циклодекстрин. Публикация РСТWO 99/38536 сообщает о поглощаемых композициях с замедленным высвобождением биологически активного агента. Публикация РСТWO 00/04916 сообщает о способе получения микрочастиц,содержащих терапевтический агент, такой как пептид, в способе типа масло-в-воде. Публикация РСТWO 00/09166 сообщает о комплексах, содержащих терапевтический агент, такой как пептид, и фосфорилированный полимер. Публикация РСТWO 00/25826 сообщает о комплексах, содержащих терапевтический агент, такой как пептид, и полимер, несущий неполимеризуемый лактон. Если не утверждается иного, все технические и научные термины, используемые в настоящем описании, имеют такое же значение, как обычно понимается специалистами в области, к которой принадлежит настоящее изобретение. Также все публикации, заявки на патент, патенты и другие ссылки, упоминаемые в настоящем описании, включаются тем самым в качестве ссылок, каждая во всей своей полноте. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I)R1 представляет собой ОН, NH2, (C1-C30)алкокси или NH-X2-CH2-Z0, где X2 представляет собой (С 0 С 30) углеводородный остаток и Z0 представляет собой Н, ОН, СО 2 Н или CONH2; каждый из R2, R3 независимо выбирают из группы, состоящей из Н, (C1-C30)алкила, (C1C30)гетероалкила, (C1-C30)ацила, (С 2-С 30)алкенила, (С 2-С 30)алкинила, арил(C1-C30)алкила, арил(С 1 С 30)ацила, замещенного (C1-C30)алкила, замещенного (С 1-С 30)гетероалкила, замещенного (C1-C30)ацила,замещенного (С 2-С 30)алкенила, замещенного (С 2-С 30)алкинила, замещенного арил(С 1-С 30)алкила и замещенного арил(C1-C30)ацила; при условии, что когда R2 представляет собой (C1-C30)ацил, арил(C1C30)ацил, замещенный (C1-C30)ацил или замещенный арил(C1-C30)ацил, тогда R3 представляет собой Н,(C1-C30)алкил, (C1-C30)гетероалкил, (С 2-С 30)алкенил, (С 2-С 30)алкинил, арил(C1-C30)алкил, замещенныйs, t, х и у, каждый независимо, представляют собой для каждого случая целое число от 1 до 30 включительно;X4, X5, X6, X7 и X8, каждый независимо, представляют собой для каждого случая Н, F, CF3, Cl, Br, I,(C1-10)алкил, замещенный (C1-10)алкил, арил, замещенный арил, ОН, NH2, -CH2NH2, NO2 или CN; при условии, что когда А 1 представляет собой 4 Нрра, тогда R2 и R3 отсутствуют; при дополнительном условии, что соединение содержит более чем одну аминокислотную замену или модификацию в положениях 1, 2 и 3; и при дополнительном условии, что если аминокислота в положении 1 модифицирована, она не модифицируется посредством:(e) присоединения N-концевой пироглутаминовой кислоты; и при дополнительном условии, что соединение содержит по меньшей мере одну аминокислотную замену или модификацию в положениях с 4 по 43,или его фармацевтически приемлемая соль. 2. Соединение по п.1, в котором, когда присутствует Acc,А 7 представляет собой А 5 с или А 6 с; или А 11 представляет собой А 5 с; или А 14 представляет собой А 5 с; или А 40 представляет собой А 5 с; или А 41 представляет собой А 5 с,или его фармацевтическая соль. 3. Соединение по п.2, которое представляет собой
МПК / Метки
МПК: C07K 14/46, C07K 14/62
Метки: аналоги, модифицированные, n-концу, глюкозозависимого, инсулинотропного, gip, полипептида
Код ссылки
<a href="https://eas.patents.su/30-20091-analogi-glyukozozavisimogo-insulinotropnogo-polipeptida-gip-modificirovannye-po-n-koncu.html" rel="bookmark" title="База патентов Евразийского Союза">Аналоги глюкозозависимого инсулинотропного полипептида (gip), модифицированные по n-концу</a>
Аналоги глюкозозависимого инсулинотропного полипептида

Номер патента: 20019
Опубликовано: 29.08.2014
Автор: Дун Чжэн Синь
МПК: C07K 14/46, C07K 14/62
Метки: инсулинотропного, глюкозозависимого, аналоги, полипептида
Формула / Реферат:
1. Соединение формулы (I)в котором А1 представляет собой Tyr, N-Me-Tyr, 4Hppa или Orn(N-C(O)-(СН2)12-СН3);А2 представляет собой Ala, A5c, А6с, Aib, D-Ala или Gly;А3 представляет собой Glu, Dhp, 3Нур, 4Нур, Pro, hPro или Tic;А4 представляет собой Gly или Aib;А5 представляет собой Thr, A5c или Aib;А6 представляет собой Phe;А7 представляет собой Ile, А5с, А6с или Aib;А8 представляет собой Ser или Aib;А9 представляет собой Asp или Aib;А10...
Процессированные аналоги глюкозозависимого инсулинотропного полипептида

Номер патента: 20018
Опубликовано: 29.08.2014
Авторы: Дун Чжэн Синь, Шен Йилана, Деоливейра Дэниел Б.
МПК: A61K 38/00
Метки: процессированные, полипептида, глюкозозависимого, инсулинотропного, аналоги
Формула / Реферат:
1. Соединение формулы (I)где А1 отсутствует;А2 представляет собой Ala, 4Hppa, Aib или отсутствует;А3 представляет собой Glu, 4Нур, Pro или отсутствует;А4 представляет собой Gly или отсутствует;А5 представляет собой Thr или отсутствует;А6 представляет собой Phe или отсутствует;А7 представляет собой Ilе, А5с, А6с, СН3-(СН2)8-С(О)-А6с, СН3-(СН2)4-С(О)-А6с или отсутствует;А8 представляет собой Ser, Chc-Ser или отсутствует;А9 представляет собой Asp...
Аналоги глюкозазависимого инсулинотропного полипептида

Номер патента: 20005
Опубликовано: 30.07.2014
Автор: Дун Чжэн Синь
МПК: A61K 38/00
Метки: полипептида, инсулинотропного, глюкозазависимого, аналоги
Формула / Реферат:
Данные на сервере публикаций отсутствуют
Штамм трансгенных растительных клеток и трансгенное растение, содержащие кассету экспрессии полипептида-мишени usp рецептора, способы получения их потомства, способы регулирования экспрессии полипептида-мишени в указанном растении с помощью ювенильного гормона или его агонистов, способы выявления и получения лиганда для полипептида usp рецептора и соответствующий лиганд

Номер патента: 3423
Опубликовано: 24.04.2003
Авторы: Кросслэнд Лайл Дин, Гофф Артур Стефен
МПК: C12N 15/82, C12Q 1/68, A01H 5/00...
Метки: потомства, соответствующий, трансгенных, гормона, полипептида-мишени, экспрессии, кассету, растение, помощью, содержащие, регулирования, рецептора, получения, клеток, ювенильного, агонистов, растительных, трансгенное, лиганда, растении, способы, указанном, лиганд, выявления, полипептида, штамм
Формула / Реферат:
1. Штамм трансгенных растительных клеток, содержащих: а) кассету экспрессии USP рецептора, кодирующую полипептид USP рецептора для контроля экспрессии полипептида-мишени, и б) кассету экспрессии полипептида-мишени, включающую ген полипептида-мишени, экспрессия которого опосредуется USP рецептором в ответ на ювенильный гормон или его агонист, и необязательно в) вспомогательную кассету экспрессии рецептора, кодирующую вспомогательный полипептид...
Усеченный по амино-концу хемокин rantes и способы его использования

Номер патента: 3940
Опубликовано: 30.10.2003
Авторы: Вам Дамме Йо, Прост Поль, Стрюйф Софи
МПК: C07K 14/52, A61P 43/00, A61K 38/19...
Метки: усеченный, хемокин, использования, способы, амино-концу, rantes
Формула / Реферат:
1. Усеченный по амино-концу хемокин RANTES, у которого отсутствуют NH2-концевые аминокислоты, соответствующие аминокислотным остаткам 1-2 природного RANTES, имеющий аминокислотную последовательность SEQ ID No:2, который обладает антагонистической активностью в отношении хемокинов MCP-3, MCP-1a , MCP-1b и RANTES. 2. Усеченный по амино-концу RANTES по п.1 в гликозилированной форме. 3. Молекула ДНК, кодирующая усеченный по амино-концу...
Предыдущий патент: Способ получения и рекристаллизации кристаллического гемитартрата бензоимидазол-2-илпиримидинового производного
Следующий патент: Способ и система для промывки электродов
Случайный патент: Новый способ синтеза сложных эфиров n-[(s)-1-карбоксибутил]-(s)-аланина и их применение для синтеза периндоприла