Способ получения замещенных 2-аминотиазолонов
Номер патента: 18028
Опубликовано: 30.04.2013
Авторы: Фаул Маргарет, Мониз Джордж А., Фризл Мэтью Дж., Мартинелли Майкл, Бернард Чарльз, Клингенсмит Лиэн, Хансен Карл Б., Лэрроу Джей














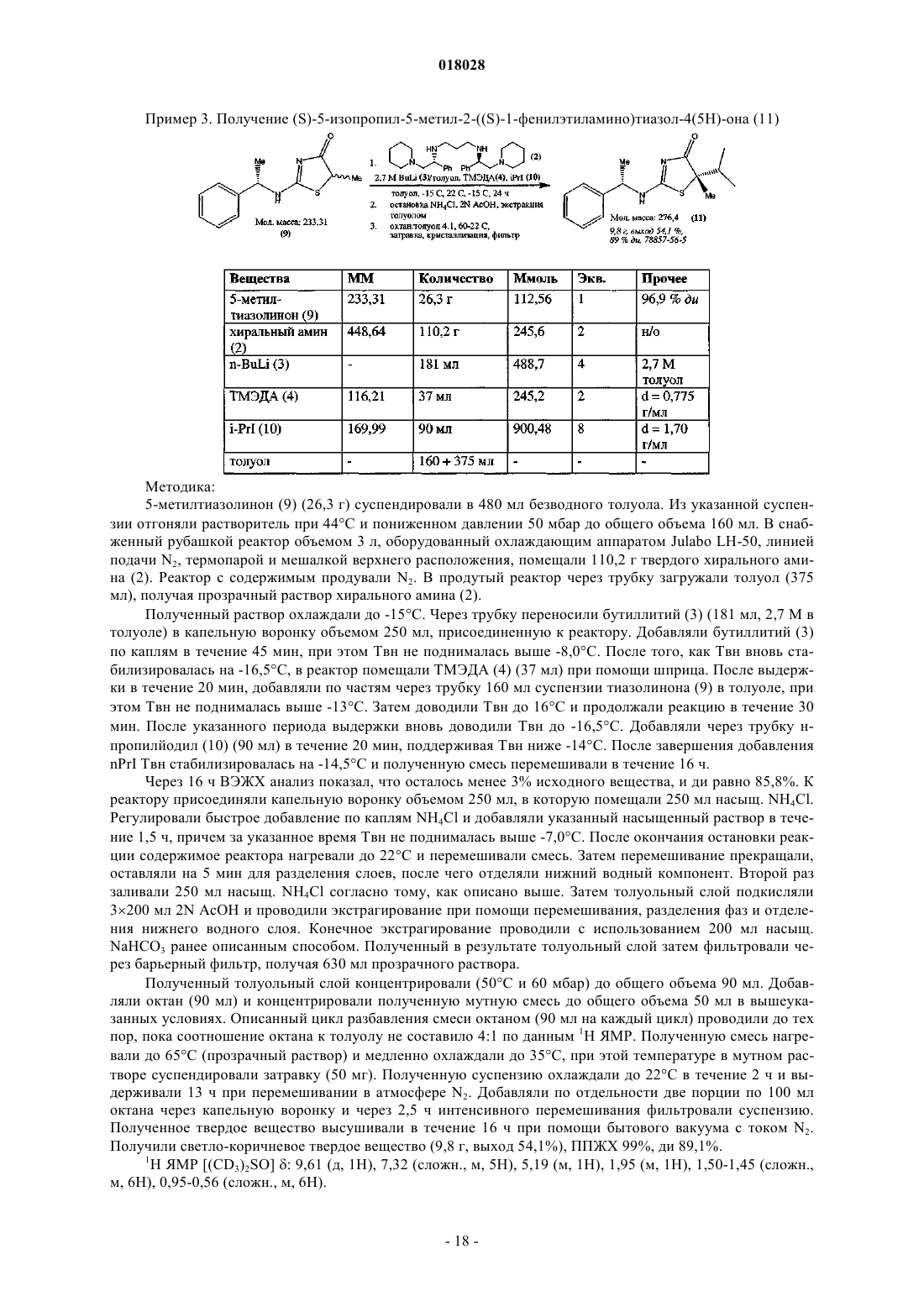
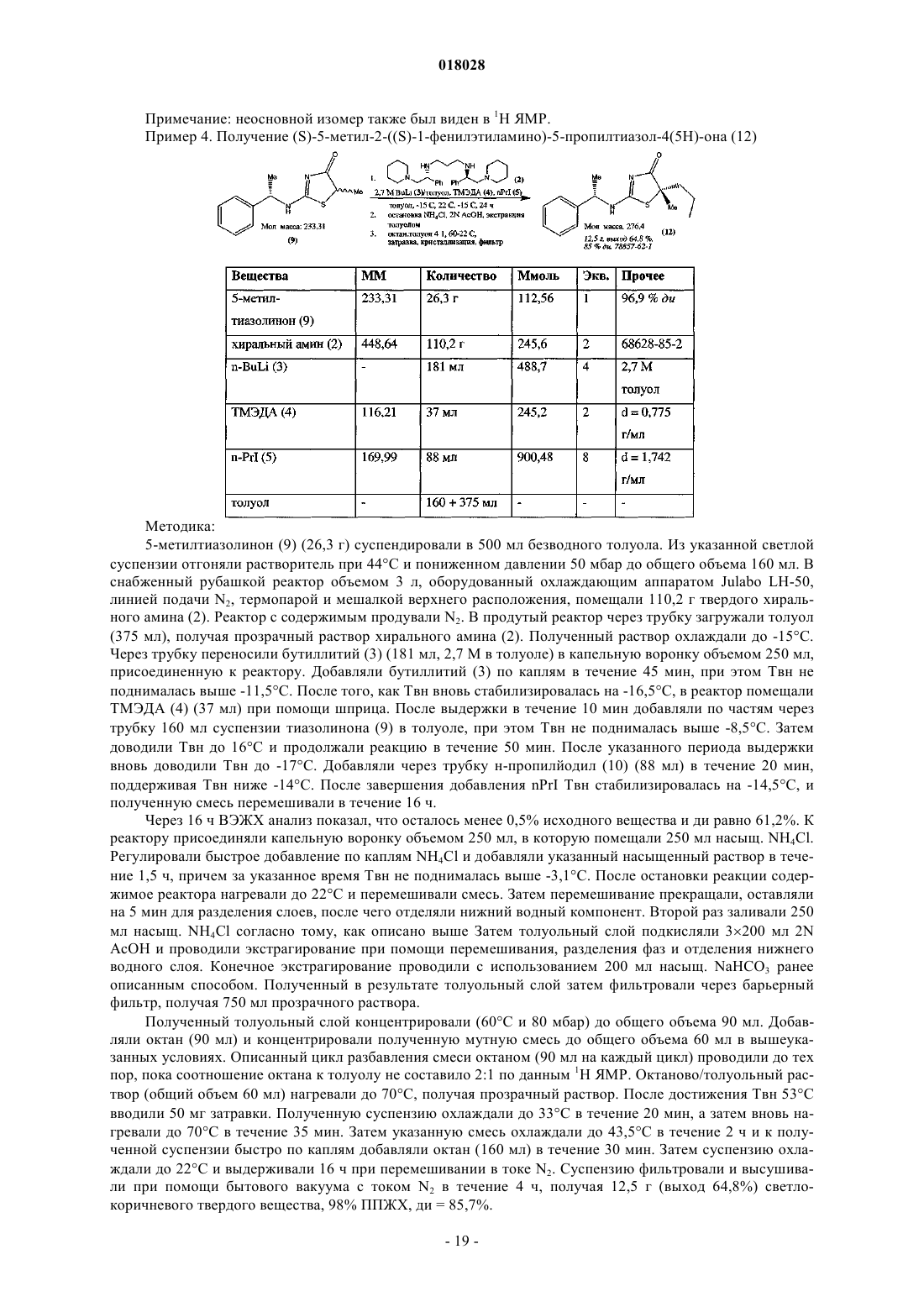










Формула / Реферат
1. Способ получения соединения формулы (I) или стереоизомера, геометрического изомера, оптического изомера или фармацевтически приемлемой соли указанного соединения

включающий приведение в контакт соединения формулы (II)

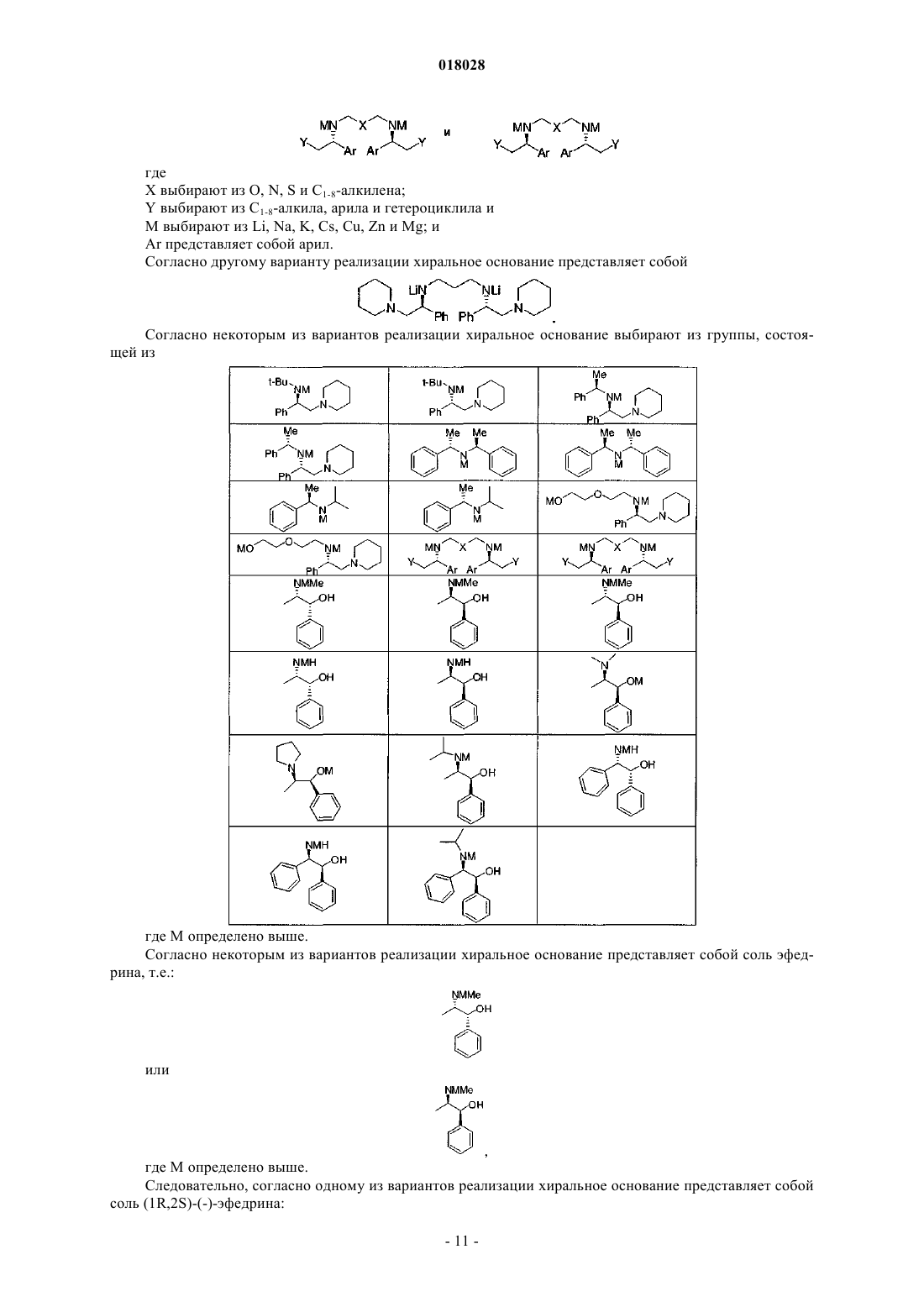
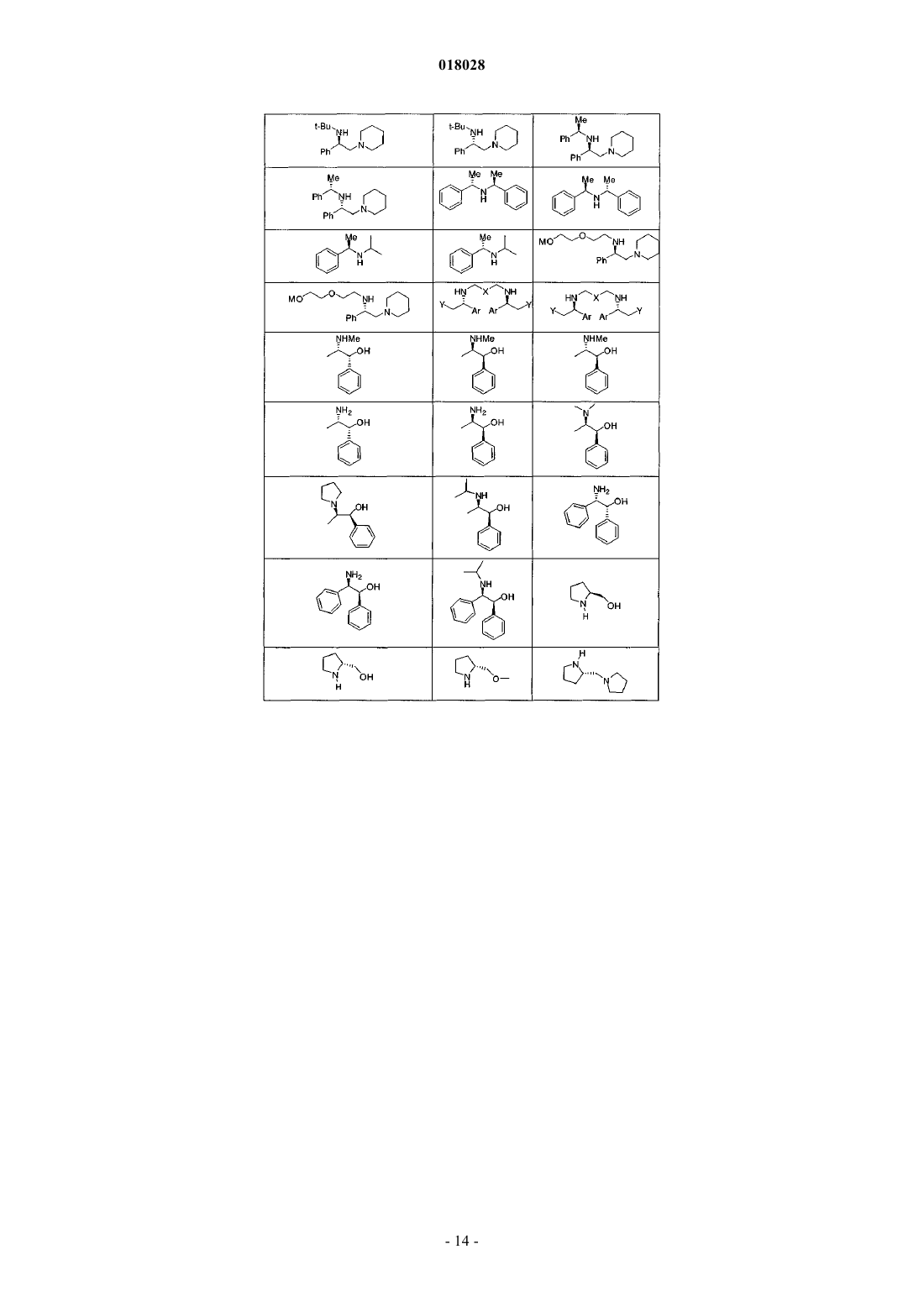
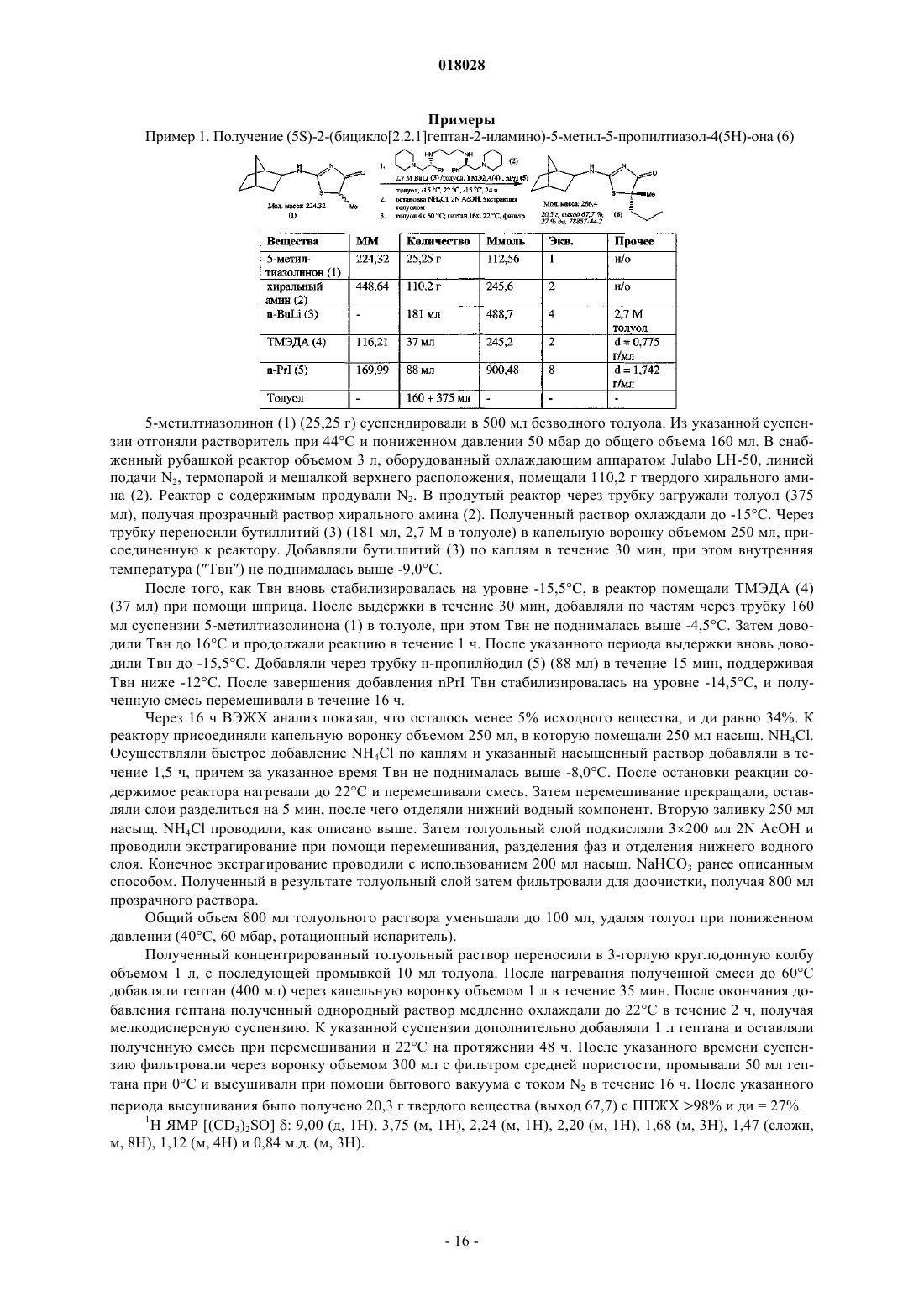
с хиральным основанием формулы

или солью данного основания, полученной присоединением кислоты, в присутствии депротонирующего агента и алкилирующего агента R3-LG;
при этом
Z представляет собой S или О;
R1 выбирают из

R2 представляет собой метил;
R3 выбирают из пропила и изопропила;
LG представляет собой йод.
2. Способ по п.1, отличающийся тем, что дополнительно включает следующие стадии:
(b) приведение в контакт продукта стадии (а) с кислотой HB с получением соли формулы (I')

при этом В представляет собой органический или неорганический анион; и
(с) взаимодействие соли формулы (I') с основанием с получением соединения формулы (I).
3. Способ по п.1, отличающийся тем, что Z представляет собой S.
4. Способ по п.3, отличающийся тем, что R1 представляет собой

5. Способ по п.4, отличающийся тем, что R2 представляет собой метил и R3 представляет собой изопропил.
6. Способ по п.1, отличающийся тем, что диастереомерный избыток соединения формулы I составляет по меньшей мере 85%.
7. Способ получения соединения формулы (III)

включающий:
(а) приведение в контакт соединения формулы (IV)

с хиральным основанием формулы

или солью данного основания, полученной присоединением кислоты,
в присутствии депротонирующего агента; и
(b) взаимодействие продукта стадии (а) с изопропилйодилом.
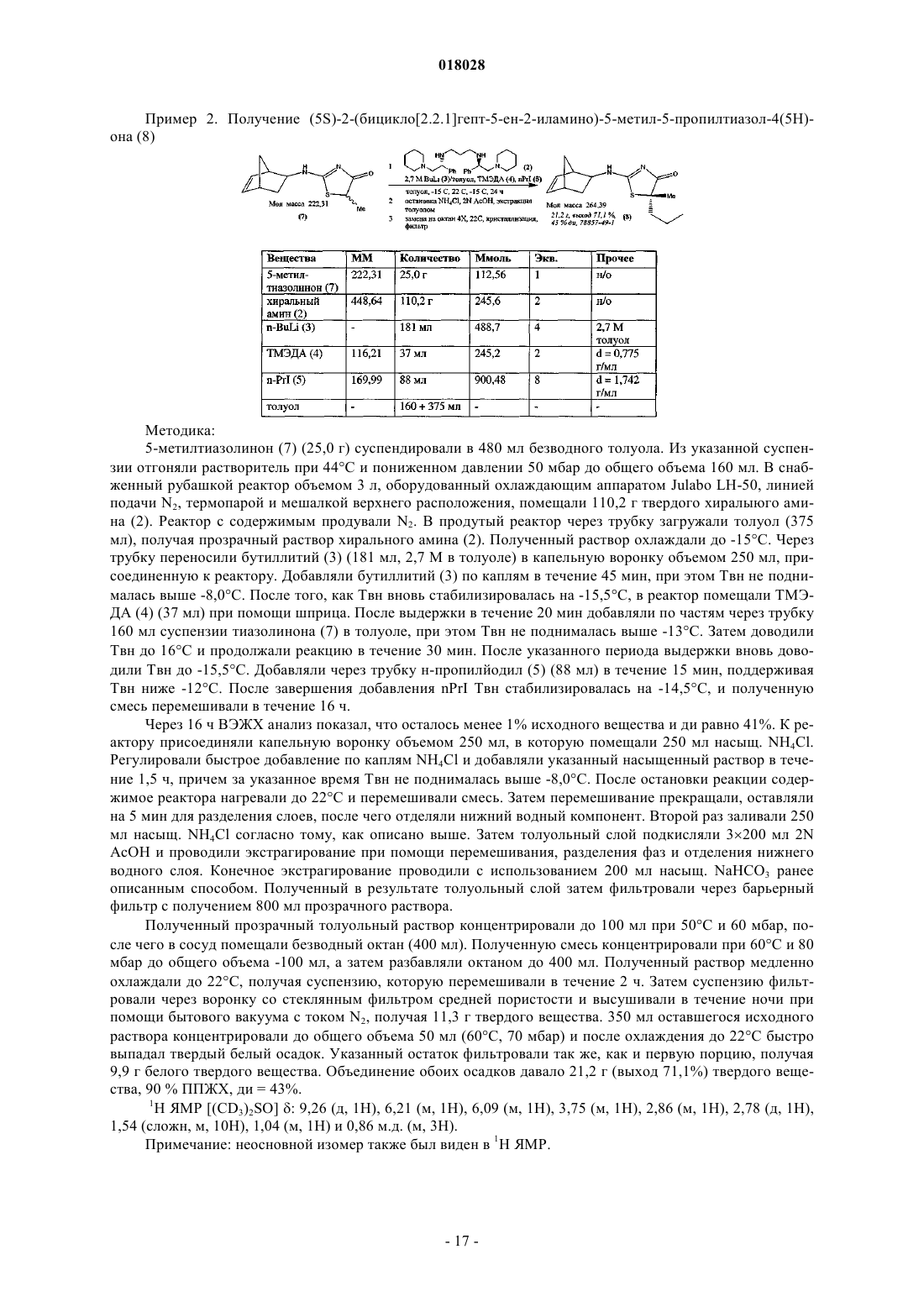
Текст
СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 2-АМИНОТИАЗОЛОНОВ Настоящее изобретение относится к способам получения соединений, ингибирующих фермент 11 гидроксистероиддегидрогеназу типа 1 (11- ГСД 1). Один из указанных способов включает:(а) приведение в контакт соединения формулы (II) с хиральным основанием, депротонирующим агентом и алкилирующим агентом R3-LG, (b) приведение в контакт продукта стадии (а) с кислотой,с образованием соли и (с) взаимодействие полученной соли с основанием, с образованием соединения формулы (I) Уровень техники изобретения Настоящее изобретение относится к способам получения соединений, ингибирующих фермент 11-гидроксистероиддегидрогеназу типа 1 (11- ГСД 1). Гидроксистероиддегидрогеназы (ГСД) регулируют заполнение и активацию рецепторов стероидных гормонов посредством превращения стероидных гормонов в неактивные метаболиты. Обзор современного состояния в этой области можно найти в Nobel et al., Eur. J. Biochem. 2001, 268:4113-4125. Существует множество классов ГСД. 11-бета-гидроксистероиддегидрогеназы (11.бета.-ГСД) катализируют взаимопревращение активных глюкокортикоидов (таких как кортизол и кортикостерон) и их инертных форм (таких как кортизон и 11-дегидрокортикостерон). Изоформа 11-бетагидроксистероиддегидрогеназы типа 1 (11.бета.-ГСД 1) экспрессирована в печени, жировой ткани, мозге,легких и другой глюкокортикоидной ткани, и является возможной мишенью терапии, направленной на многочисленные заболевания, которые можно облегчить путем уменьшения действия глюкокортикоидов, такие как диабет, ожирение и возрастная когнитивная дисфункция (Seckl, et al., Endocrinology, 2001,142:1371-1376). Различные изозимы 17-бета-гидроксистероиддегидрогеназ (17.бета.-ГСД) связываются с рецепторами андрогенов или рецепторами эстрогенов и катализируют взаимопревращение различных половых гормонов, включая эстрадиол/эстрон и тестостерон/андростендион. К настоящему моменту у человека было идентифицировано шесть изозимов, которые экспрессированы в различных тканях человека, включая ткани эндометрия,ткани груди,ткани толстой кишки и яички. 17-бетагидроксистероиддегидрогеназа типа 2 (17.бета.-ГСД 2) экспрессирована в эндометрии человека и известно, что активность указанного фермента связана с раком шейки матки Kitawaki et al., J. Clin. Endocrin.Metab., 2000, 85:1371-3292-3296. 17-бета-гидроксистероиддегидрогеназа типа 3 (17.бета.-ГСД 3) экспрессирована в яичках, и ее модулирование может быть полезным при лечении нарушений, связанных с андрогенами. Андрогены и эстрогены активны в 17.бета.-гидроксиконфигурациях, тогда как их 17-кетопроизводные не связываются с рецепторами андрогенов и эстрогенов и поэтому неактивны. Превращения между активными и неактивными формами (эстрадиол/эстрон и тестостерон/андростендион) половых гормонов катализируются членами группы ферментов 17.бета.-ГСД. 17.бета.-ГСД 1 катализирует образование эстрадиола в ткани груди, что важно для роста злокачественных опухолей груди (Labrie etal., Mol. Cell. Endocrinol. 1991, 78:C113-C118). Предполагают, что 17.бета.-ГСД 4 играет аналогичную роль при раке толстой кишки (English et al., J. Clin. Endocrinol. Metab. 1999, 84:2080-2085). 17.бета.-ГСД 3 экспрессирована почти исключительно в яичках и превращает андростендион в тестостерон. Недостаточность указанного фермента во время развития плода приводит к мужскому псевдогермафродитизму(Geissler et al., Nat. Genet. 1994, 7:34-39). Как 17.бета.-ГСД 3, так и различные изозимы 3.альфа.-ГСД вовлечены в сложные метаболические пути, которые приводят к переключениям андрогенов между неактивными и активными формами (Penning et al., Biochem. J. 2000, 351:67-77). Следовательно, модулирование определенных ГСД может обладать потенциально благоприятными эффектами при лечении нарушений, связанных с андрогенами и эстрогенами. 20-альфа-гидроксистероиддегидрогеназы (20.альфа.-ГСД) катализируют взаимопревращения прогестинов (такие как между прогестероном и 20.альфа.-гидроксипрогестероном). Другие субстраты 20.альфа.-ГСД включают 17.альфа.-гидроксипрегненолон или 17.альфа.-гидроксипрогестерон, давая в результате 20.альфа.-ОН стероиды. Были идентифицированы некоторые изоформы 20.альфа.-ГСД,20.альфа.-ГСД экспрессированы в различных тканях, включая плаценту, яичники, яички и надпочечники(Peltoketo, et al., J. Mol. Endocrinol. 1999, 23:1-11). 3-альфа-гидроксистероиддегидрогеназы (3.альфа.-ГСД) катализируют взаимопревращения андрогенов дигидротестостерона (ДГТ) и 5.альфа.-андростан-3.альфа.,17.бета.-диола и взаимопревращение андрогенов дегидроэпиандростерона (ДГЭА) и андростендиона, и, следовательно, играют важную роль в метаболизме андрогенов (Ge et al., Biology of Reproduction 1999, 60:855-860). 1. Глюкокортикоиды, диабет и производство глюкозы печенью. Более полувека тому назад стало известно, что глюкокортикоиды играют основную роль при диабете. Например, удаление гипофиза или надпочечника у страдающего диабетом животного облегчает наиболее тяжелые симптомы диабета и понижает концентрацию глюкозы в крови (Long, С. D. and Leukins,F. D. W. (1936) J. Exp. Med. 63: 465-490; Houssay, B. A. (1942) Endocrinology 30: 884-892). Также твердо установлено, что глюкокортикоиды делают возможным воздействие глюкагона на печень. Роль 11.бета.ГСД 1 как важного регулятора местного глюкокортикоидного действия, и, следовательно, производства глюкозы печенью, хорошо обоснована (см., например, Jamieson et al. (2000) J. Endocrinol. 165: 685-692). У здоровых людей-добровольцев, получавших неспецифический ингибитор 11.бета.ГСД 1 карбеноксолон (carbenoxolone), повышалась восприимчивость печени к инсулину (Walker,В. R. et al. (1995) J. Clin. Endocrinol. Metab. 80: 3155-3159). Кроме того, был установлен предполагаемый механизм в различных экспериментах на мышах и крысах. Указанные исследования показали, что были снижены уровни мРНК и активности двух ключевых ферментов в производстве глюкозы печенью, а именно: лимитирующего скорость фермента в глюкогенезе, фосфоенолпируваткарбоксикиназы(ФЕПКК), и глюкозо-6-фосфатазы (Г-6-Фазы), фермента, катализирующего последнюю общую стадию глюконеогенеза и гликогенолиза. Наконец, у мышей с выключенным (knocked-out) геном 11.бета.ГСД 1 снижались уровни глюкозы в крови и производства глюкозы печенью. Данные, полученные на указанной модели, также подтверждают, что ингибирование 11.бета.ГСД 1 не вызывает гипогликемии, как и было предсказано на основании того факта, что базальные уровни ФЕПКК и Г-6-Фазы регулируются независимо от глюкокортикоидов (Kotelevtsev, Y. et al., (1997) Ргос. Natl. Acad. Sci. USA 94: 14924-14929). В FR 2384498 описаны соединения, обладающие высоким гипогликемическим эффектом. Следовательно, лечение гипергликемии при помощи указанных соединений может приводить к гипогликемии. 2. Возможное уменьшение ожирения и связанных с ожирением сердечно-сосудистых факторов риска. Ожирение является важным фактором при синдроме X, а также в большинстве случаев ( 80%) диабета типа 2, и, по-видимому, первоочередную важность имеет брюшной жир (omental fat). Абдоминальное ожирение тесно связано с нарушением толерантности к глюкозе, гиперинсулинемией, гипертриглициридемией, и другими факторами так называемого синдрома X (например, повышенным кровяным давлением, пониженными уровнями липопротеина высокой плотности (ХВП) и повышенными уровнями липопротеина сверхнизкой плотности (ЛПОНП (MontagueO'Rahilly, Diabetes 49: 883-888,2000). Показано, что ингибирование фермента 11.бета.ГСД 1 в преадипоцитах (стромальных клетках) уменьшает скорость дифференциации указанных клеток в адипоциты. Предсказано, что это приведет к пониженному распространению (возможно, уменьшению) отложений брюшного жира, т.е., уменьшенному центральному ожирению (Bujalska, I. J., S. Kumar and P. M. Stewart (1997) Lancet 349: 1210-1213). Полагают, что ингибирование 11.бета.ГСД 1 в зрелых адипоцитах ослабляет секрецию ингибитора активатора плазминогена 1 (ИАП-1) - независимого фактора риска сердечно-сосудистых заболеваний(Halleux, С. М. et al. (1999) J. Gin. Endocrinol. Metab. 84: 4097-4105). Кроме того, существует четкая корреляция между активностью глюкокортикоида и фактором риска сердечно-сосудистых заболеваний,позволяющая предположить, что уменьшение действий глюкокортикоидов было бы благотворным(Walker, В. R. et al. (1998) Hypertension 31: 891-895; Fraser, R. et al. (1999) Hypertension 33: 1364-1368). Удаление надпочечников уменьшает действие голодания как на увеличение потребления пищи, так и на экспрессию гипоталамического нейропептида Y. Это подтверждает роль глюкокортикоидов в стимуляции потребления пищи и позволяет предположить, что ингибирование 11.бета.ГСД 1 в мозге может увеличить насыщение и, следовательно, уменьшить потребление пищи (Woods, S. С. et al. (1998) Science,280: 1378-1383). 3. Возможное благоприятное действие на поджелудочную железу Ингибирование 11.бета.ГСД 1 в изолированных бета.-клетках поджелудочной железы мыши улучшает стимулируемую глюкозой секрецию инсулина (Davani, В. et al. (2000) J. Biol. Chem. 2000 Nov. 10; 275(45): 34841-4). Ранее было известно, что глюкокортикоиды уменьшают выделение инсулина поджелудочной железой in vivo (Billaudel, В. and В. С. J. Sutter (1979) Horm. Metab. Res. 11: 555-560). Следовательно, предполагают, что ингибирование 11.бета.ГСД 1 приведет к другим положительным результатам в отношении лечения диабета помимо воздействия на печень и жир. 4. Возможные благотворные действия на когнитивную функцию и деменцию. Стресс и глюкокортикоиды влияют на когнитивную функцию (de Quervain, D. J. R, В. Roozendaal,and J. L. McGaugh (1998) Nature 394: 787-790). Фермент 11.бета.ГСД 1 контролирует уровень действия глюкокортикоидов в мозге и, следовательно, вносит вклад в нейротоксичность (Rajan, V., С. R. W. Edwards, and J. R. Seckl, J. (1996) Neuroscience 16: 65-70; Seckl, J. R., Front. (2000) Neuroendocrinol. 18: 4999). Неопубликованные результаты показывают значительное улучшение памяти крыс, получавших неспецифический ингибитор 11.бета.ГСД 1 (J. Seckl, личное сообщение). Исходя из вышесказанного и из известных действий глюкокортикоидов в мозге, можно также предположить, что ингибирование 11.бета.ГСД 1 в мозге может привести к снижению тревоги (Tronche, F. et al. (1999) Nature Genetics 23: 99-103). Следовательно, в совокупности, существует гипотеза, что ингибирование 11.бета.ГСД 1 в мозге человека могло бы предотвращать реактивацию кортизона в кортизол и защищать от опосредуемых глюкокортикоидами вредных воздействий на выживаемость нейронов и другие аспекты функции нейронов,включая нарушения когнитивной функции, депрессию и повышенный аппетит. 5. Возможное применение иммуномодуляции при помощи ингибиторов 11.бета.ГСД 1 Согласно общепринятым представлениям, глюкокортикоиды подавляют иммунную систему. Но в действительности существует динамическое взаимодействие между иммунной системой и осью ГГН(гипоталамо-гипофизарно-надпочечниковой) (Rook, G. A. W. (1999) Baillier's Clin. Endocrinol. Metab. 13: 576-581). Равновесие между клеточной реакцией и гуморальными реакциями модулируется глюкокортикоидами. Высокая активность глюкокортикоидов, такая как при стрессовом состоянии, ассоциируется с гуморальной реакцией. Следовательно, ингибирование фермента 11.бета.ГСД 1 можно предложить в качестве средства переключения реакции в направлении клеточной реакции. При некоторых болезненных состояниях, включая туберкулез, проказу и псориаз, иммунная реакция обычно смещена в сторону гуморальной реакции, тогда как в действительности соответствующая реакция должна быть клеточной. Временное ингибирование 11.бета.ГСД 1, местное или системное, можно применять, чтобы подтолкнуть иммунную систему к соответствующей реакции (Mason, D. (1991) Immunology Today 12: 57-60; Rook et al., supra). Аналогичное применение ингибирования 11.бета.ГСД 1, в данном случае временное, при необходимости, может усиливать иммунную реакцию совместно с иммунизацией, для уверенности, что будет получена клеточная реакция. 6. Снижение внутриглазного давления Последние данные позволяют предположить, что уровни рецепторов глюкокортикоидов и ферментов 11.бета.ГСД определяют предрасположенность к глаукоме (Stokes, J. et al. (2000) Invest. Ophthalmol. 41: 1629-1638). Кроме того, ингибирование 11.бета.ГСД 1 в последнее время обсуждается в качестве нового подхода к понижению внутриглазного давления (Walker E. A. et al., poster P3-698 at the EndocrineSociety meeting Jun. 12-15, 1999, San Diego). Показано, что прием внутрь карбеноксолона, неспецифического ингибитора 11.бета.ГСД 1, уменьшал внутриглазное давление на 20% у нормальных субъектов. Есть мнение, что экспрессия 11.бета.ГСД 1 ограничена базальными клетками эпителия роговицы и непигментированными эпителиальными клетками роговицы (область выработки глазной жидкости), ресничной мышцей и сфинктерной и расширяющей мышцами радужной оболочки. Напротив, отдаленный изоэнзим 11.бета.ГСД 2 высоко экспрессирован в непигментированном ресничном эпителии и эндотелии роговицы. Ни один из указанных ферментов не обнаружен в трабекулярной сети, области дренажа. Следовательно, полагают, что 11.бета.ГСД 1 играет роль в выработке глазной жидкости, а не в ее дренаже, но в настоящее время неизвестно, происходит ли это благодаря влиянию на активацию рецепторов глюкокортикоидов, на активацию рецепторов минералокортикоидов или на активацию обоих указанных рецепторов. 7. Уменьшенный остеопороз Глюкокортикоиды играют важную роль в развитии и функционировании скелета, но вредны в избытке. Обусловленная глюкокортикоидами потеря костной массы вызвана, по меньшей мере, частично,ингибированием образования кости, включающим подавление пролиферации остеобластов и синтеза коллагена (Kim, С. Н., Cheng, S. L. and Kim, G. S. (1999) J. Endocrinol. 162: 371-379). Отрицательное действие на образование костных узелков (bone nodules) можно заблокировать при помощи неселективного ингибитора карбеноксолона, что позволяет предположить важную роль 11.бета.ГСД 1 в действии глюкокортикоидов (Bellows, С. G., Ciaccia, A. and Heersche, J. N. М. (1998) Bone 23: 119-125). Другие данные позволяют предположить роль 11.бета.ГСД 1 в обеспечении достаточно высоких уровней активных глюкокортикоидов в остеокластах, и, следовательно, в усилении резорбции кости (Cooper, M. S. et al. (2000)Bone 27: 375-381). Вместе взятые указанные данные позволяют предположить, что ингибирование 11.бета.ГСД 1 может оказывать благотворное действие при остеопорозе посредством нескольких механизмов, задействованных параллельно. 8. Уменьшение гипертонии Желчные кислоты ингибируют 11.бета.-гидроксистероиддегидрогеназу типа 2. Это приводит к сдвигу общего баланса организма в сторону кортизола по отношению к кортизону, как показали исследования соотношения метаболитов в моче (Quattropani, С, Vogt, В., Odermatt, A., Dick, В., Frey, В. М.,Frey, F. J. (2001) J Clin Invest. November; 108(9): 1299-305. "Reduced activity of 11beta-hydroxysteroid dehydrogenase in patients with cholestasis"). Предполагают, что уменьшение активности 11bГСД 1 в печени при помощи селективного ингибитора повернет этот дисбаланс в обратную сторону и окажет резкое противодействие таким симптомам как гипертония, пока ожидается хирургическое вмешательство для устранения закупорки желчного протока. В WO 99/65884 описаны замещенные по углероду аминотиазольные ингибиторы циклинзависимых киназ. Указанные соединения могут, например, применяться против рака, воспаления и артрита. В Патенте США 5856347 описан антибактериальный препарат или бактерицидное средство, включающее производное 2-аминотиазола и/или соль указанного соединения. Кроме того, в Патенте США 5403857 описаны производные бензолсульфонамида, имеющие ингибиторную активность в отношении 5 липоксигеназы. Кроме того, тетрагидротиазоло[5,4-с]пиридины описаны в Analgesic tetrahydrothiazolo[5,4-c]pyridines. Fr. Addn. (1969), 18 pp, Addn. to Fr. 1498465. CODEN: FAXXA3; FR 94123 19690704(1967), 39 pp. CODEN: NAXXAN NL 6610324 19670124 CAN 68:49593, AN 1968: 49593 CAPLUS. Однако ни в одном из указанных документов не описан способ получения соединений согласно настоящему изобретению. 9. Лечение ран Кортизол осуществляет широкий диапазон метаболических функций и прочих функций. Примером множественного действия глюкокортикоидов могут служить пациенты с длительным повышенным содержанием глюкокортикоидов в плазме, так называемым синдромом Кушинга. У пациентов с синдромом Кушинга наблюдается длительное повышенное содержание глюкокортикоидов в плазме и развивается ослабленная толерантность к глюкозе, диабет 2 типа, центральное ожирение и остеопороз. У указанных пациентов также наблюдается ухудшенное заживление ран и непрочная кожа (Ganong, W. F. Re-3 018028view of Medical Physiology. Eighteenth edition ed. Stamford, Conn.: AppletonLange; 1997). Было показано, что глюкокортикоиды увеличивают риск инфицирования и замедленного заживления открытых ран (Anstead, G. M. Steroids, retinoids, and wound healing. Adv Wound Care 1998; 11(6): 27785). Пациенты, получавшие глюкокортикоиды, имели в 2-5 раз больший риск осложнений после хирургического вмешательства (Diethelm, A. G. Surgical management of complications of steroid therapy. AnnSurg 1977;185(3):251-63). В Европейской патентной заявкеEP 0902288 описан способ диагностики состояния заживления ран у пациента, включающий определение уровней кортизола в указанной ране. Авторы предположили,что повышенные уровни кортизола в раневой жидкости, по сравнению с нормальными уровнями в плазме здоровых индивидуумов, коррелируют с большими незаживающими ранами (Hutchinson, Т. С, Swaniker, H. Р., Wound diagnosis by quantitating cortisol in wound fluids. European patent application No. EP 0902288, published 17.03.1999). У человека 11.бета.-ГСД катализирует превращение кортизола в кортизон и обратно. Параллельной функцией 11.бета.-ГСД у грызунов является взаимопревращение кортикостерона и 11 дегидрокортикостерона (Frey, F. J., Escher, G., Frey, В. М. Pharmacology of 11 beta-hydroxysteroid dehydrogenase. Steroids 1994; 59(2): 74-9). Были описаны два изоэнзима 11.бета.ТСД, 11.бета.-ГСД 1 и 11.бета.ГСД 2, отличающиеся между собой по действию и распределению в тканях (Albiston, A. L., Obeyesekere,V. R., Smith, R. E., Krozowski, Z. S. Cloning and tissue distribution of the human 11 beta-hydroxysteroid dehydrogenase type 2 enzyme. Mol Cell Endocrinol 1994; 105(2):R11-7). Подобно ГР, 11.бета.-ГСД 1 экспрессирована в многочисленных тканях, таких как печень, жировая ткань, кора надпочечников, половые железы, легкие, гипофиз, мозг, глаз, и т.д. (Monder С., White Р. С. 11 beta-hydroxysteroid dehydrogenase. Vitamtissues. Invest Ophthalmol Vis Sci 2000; 41(7):1629-38). Функцией 11.бета.-ГСД 1 является тонкая настройка местного действия глюкокортикоидов. Была показана активность 11.бета.-ГСД в коже человека и грызунов, в фибробластах человека и ткани кожной кисты крыс (Hammami, M. M., Siiteri, P. K. Regulation of 11 beta-hydroxysteroid dehydrogenase activity in human skin fibroblasts: enzymatic modulation of glucocorticoid action. J Clin Endocrinol Metab 1991;73(2):326-34); Cooper, M. S., Moore, J., Filer, A., Buckley, С D.,Hewison, M., Stewart, P. M. 11beta-hydroxysteroid dehydrogenase in human fibroblasts: expression and regulation depends on tissue of origin. ENDO 2003 Abstracts 2003; Teelucksingh, S., Mackie, A. D., Burt, D.,Mclntyre, M. A., Brett, L., Edwards, C. R. Potentiation of hydrocortisone activity in skin by glycyrrhetinic acid.Cell Biochem 1998; 189(l-2):47-54). Способ заживления ран состоит из нескольких стадий, включая воспаление, пролиферацию фибробластов, секрецию межуточных веществ, выработку коллагена, ангиогенез, стягивание раны и эпителизацию. Его можно разделить на три фазы: воспалительную, пролиферативную и ремоделирующую фазы (см. в Anstead et al., выше). У пациентов, перенесших операцию и получавших глюкокортикоиды, возрастает риск инфицирования и замедленного заживления открытых ран. В моделях на животных было показано, что ограничивающий стресс замедляет заживление повреждений кожи и увеличивает восприимчивость к бактериальной инфекции в процессе заживления ран. Указанные эффекты были обращены путем лечения антагонистом рецепторов глюкокортикоидов RU486 (Mercado, A. M., Quan, N., Padgett, D. A., Sheridan, J. F., Marucha, P. T. Restraint stress alters the expression of interleukin-1 and keratinocyte growth factor at the woundBehav Immun 2002; 16(l):74-84). Глюкокортикоиды приводят к указанным эффектам, подавляя воспаление, уменьшая прочность раны, ингибируя стягивание раны и задерживая эпителизацию (Anstead et al.,выше). Глюкокортикоиды влияют на заживление ран, воздействуя на производство или действие цитокинов и факторов роста, таких как ИФР, ТФР-.бета., ЭФР, ФРК и ФРТ (Beer, Н. D., Fassler, R., Werner, S.reverses the glucocorticoid-induced wound-healing deficit in rats: possible regulation in macrophages by platelet-derived growth factor. Proc Natl Acad Sci USA 1989; 86(7):2229-33). Также было показано, что глюкокортикоиды уменьшают синтез коллагена в коже крыс и мышей in vivo и в фибробластах крысы и человека (Oishi, Y., Fu, Z. W., Ohnuki, Y., Kato, H., Noguchi, T. Molecular basis of the alteration in skin collagencollagenase, and tissue inhibitors of metalloproteinases. Br J Dermatol 2002;147(5):859-68). В публикации заявки на патент США 2006/0142357 и WO 2005/116002 описаны ингибиторы 11-ГСД 1 следующей общей структуры и некоторые способы получения указанных соединений: Очевидно, что указанный тип ингибиторов 11 ГСД 1 весьма важен с медицинской точки зрения. Следовательно, существует потребность в эффективном способе синтеза указанных соединений, особенно высокочистых оптических изомеров указанных соединений, для крупномасштабного получения, подходящего для промышленного производства. Краткое описание изобретения Согласно одному из вариантов реализации настоящего изобретения предложен способ производства соединений формулы I или таутомера, стереоизомера, геометрического изомера, оптического изомера,гидрата, сольвата, пролекарства или фармацевтически приемлемой соли этого соединенияR1 выбирают из группы, состоящей из C1-8-алкила, C2-8-алкенила, C3-10-циклоалкила, C3-10 циклоалкенила, C3-10-циклоалкил-C1-8-алкила, C3-10-циклоалкенил-C1-8-алкила, арила, арил-C1-8-алкила,гетероциклила, гетероциклил-C1-8-алкила и галогеналкила. В определении R1 любой арильный, циклоалкильный или гетероциклический остаток, возможно, независимо замещен одним или более C1-8-алкилом,арилом, галогеном, гало-C1-C8-алкилом, НО-C1-C8-алкилом, R4R5N-C1-C8-алкилом, C1-C8-алкил-OR6,-OR6, (C3-C10)-циклоалкилом или C1-C8-алкилсульфонилом.R2 и R3 независимо выбирают из C1-8-алкила, C1-8-алкоксила, C3-10-циклоалкила, гетероциклила, C3-10 циклоалкил-C1-8-алкила, CN-C1-8-алкила, арила, арил-C1-8-алкила, гетероциклил-C1-8-алкила и галогеналкила. В определениях R2 и R3, любой арильный, циклоалкильный или гетероциклический остаток, возможно, независимо замещен одним или более C1-8-алкилом, арилом, галогеном, гало-C1-C8-алкилом, HOC1-C8-алкилом, R4R5N-C1-C8-алкилом, C1-C8-алкил-OR6, -OR6, (C3-C10)-циклоалкилом или C1-C8 алкилсульфонилом.R4 и R5 независимо выбирают из группы, состоящей из водорода, C1-C8-алкила, C1-C8-алкоксила,6 6-NR R , -S-(С 1-С 8)алкила, арила и гетероциклила. В определениях R4 и R5, любой алкил, алкоксил, гетероциклил или арил имеет от одного до трех заместителей, выбираемых из галогена, незамещенного C1-C8-алкила,незамещенного C1-C8-алкоксила, незамещенного C1-C8-тиоалкоксила и незамещенного арил(С 1-С 4)алкила.R6 независимо выбирают из группы, состоящей из водорода, C1-C8-алкила, арил-C1-C8-алкила, C1C8- алкоксила, -S-(C1-C8)алкила, гетероциклила и арила. В определении R6, любой алкил, гетероциклил или арил могут нести от одного до трех заместителей, выбираемых из галогена, незамещенного C1-C8 алкила, незамещенного C1-C8-алкоксила, незамещенного C1-C8-тиоалкоксила и незамещенного арил(С 1 С 4)алкила. Указанный способ включает следующие стадии:(а) приведение в контакт соединения формулы II с (i) хиральным основанием в присутствии амина и алкилирующего агента R3-LG; где LG представляет собой уходящую группу;(b) приведение в контакт продукта стадии (а) с кислотой HB для получения соли формулы I', где В представляет собой органический или неорганический анион; и(с) взаимодействие соли формулы I' с основанием с получением соединения формулы I. Согласно другому варианту реализации настоящего изобретения предложен другой способ получения соединения формулы I или таутомера, стереоизомера, геометрического изомера, оптического изомера, гидрата, сольвата, пролекарства или фармацевтически приемлемой соли этого соединения: Указанный способ включает приведение в контакт соединения формулы II с хиральным основанием в присутствии депротонирующего агента и алкилирующего агента R3-LG.R1 выбирают из группы, состоящей из C1-8-алкила, C2-8-алкенила, C3-10-циклоалкила, C3-10 циклоалкенила, C3-10-циклоалкил-C1-8-алкила, C3-10-циклоалкенил-C1-8-алкила, арила, арил-C1-8-алкила,гетероциклила, гетероциклил-C1-8-алкила и галогеналкила; причем любой арильный, циклоалкильный или гетероциклический остаток, возможно, независимо имеет в качестве заместителей один или болееR2 и R3 независимо выбирают из C1-8-алкила, C1-8-алкоксила, C3-10-циклоалкила, гетероциклила, C3-10 циклоалкил-C1-8-алкила, CN-C1-8-алкила, арила, арил-C1-8-алкила, гетероциклил-C1-8-алкила и галоалкила; причем любой арильный, циклоалкильный или гетероциклический остаток, возможно, независимо имеет в качестве заместителей один или более C1-8-алкилов, арилов, галогенов, гало-C1-C8-алкилов, HO-C1-C8 алкилов,R4R5N-C1-C8-алкилов,C1-C8-алкил-OR6,-OR6,(C3-C10)циклоалкилов илиR4 и R5 независимо выбирают из водорода, C1-C8-алкила, C1-C8-алкоксила, -NR6R6, -S-(C1C8)алкила, арила и гетероциклила; причем в определении R4 и R5 любой алкил, алкоксил, гетероциклил или арил могут иметь от одного до трех заместителей, выбираемых из галогена, незамещенного C1-C8 алкила, незамещенного C1-C8-алкоксила, незамещенного C1-C8-тиоалкоксила и незамещенного арил(C1C4)алкила.R6 независимо выбирают из водорода, C1-C8-алкила, арил-C1-C8-алкила, C1-C8-алкоксила, -S-(C1C8)алкила, гетероциклила и арила; причем в определении R6 любой алкил, гетероциклил или арил могут иметь от одного до трех заместителей, выбираемых из галогена, незамещенного C1-C8-алкила, незамещенного C1-C8-алкоксила, незамещенного C1-C8-тиоалкоксила и незамещенного арил(C1-C4)алкила.LG представляет собой уходящую группу. Другой вариант реализации настоящего изобретения представляет собой способ получения соединения формулы III: Согласно одному из вариантов реализации, указанный способ включает стадии:(а) приведение во взаимодействие соединения формулы IV(b) взаимодействие продукта стадии (а) с изопропилйодидом. Согласно одному из вариантов реализации указанный способ дополнительно включает следующие стадии:(c) приведение в контакт продукта стадии (b) с MeSO3H с получением мезилатной соли и(d) взаимодействие мезилатной соли, полученной на стадии (с), с NaOH с получением соединения формулы III. Согласно еще одному варианту реализации указанный способ дополнительно включает стадию выделения мезилатной соли после стадии (с) и перед стадией (d). Другой вариант реализации настоящего изобретения представляет собой способ получения соединения формулы V(а) приведение в контакт соединения формулы (VI)(b) взаимодействие продукта стадии (а) с н-пропилйодидом. Подробное описание Ниже определены различные термины, используемые отдельно и в сочетаниях в способе, описанном в настоящей заявке. Выражение включающий означает включающий, без ограничения. Следовательно, могут присутствовать другие, не упомянутые, вещества, добавки, носители или стадии. Термин арил в настоящей заявке включает ароматические кольца (моноциклические или бициклические), содержащие от 6 до 10 атомов углерода, такие как фенил (Ph), нафтил и инданил (т.е., 2,3 дигидроинденил). Арильная группа может иметь в качестве заместителя C1-6-алкил. Примерами замещенных арильных групп являются бензил и 2-метилфенил. Термин гетероарил представляет собой моноциклическую, би- или трициклическую ароматическую кольцевую систему (только одно кольцо обязательно должно быть ароматическим), содержащую от 5 до 14 атомов в кольце (моно- или бициклическом, из которых один или несколько атомов кольца отличны от углерода, такие как азот, сера, кислород и селен, входящие в кольцевую систему. Согласно некоторым из вариантов реализации кольцо содержит от 5 до 10 атомов, то есть, 5, 6, 7, 8, 9 или 10. Примерами таких гетероарильных колец являются пиррол, имидазол, тиофен, фуран, тиазол, изотиазол, тиадиазол, оксазол, изоксазол, оксадиазол, пиридин, пиразин, пиримидин, пиридазин, пиразол, триазол, тетразол, хроман, изохроман, хинолин, хиноксалин, изохинолин, фталазин, циннолин, хиназолин, индол, изоиндол, бензотиофен, бензофуран, изобензофуран, бензоксазол, 2,1,3-бензоксадиазол, бензопиразол, бензотиазол, 2,1,3-бензотиазол, 2,1,3-бензоселенадиазол, бензимидазол, индазол, бензодиоксан, индан, 1,5 нафтиридин, 1,8-нафтиридин, акридин, феназин и ксантен. Термины гетероциклический и гетероциклил относятся к ненасыщенным, а также частично и полностью насыщенным моно-, би- и трициклическим кольцам, содержащим от 4 до 14 атомов, содержащим один или несколько гетероатомов (например, кислород, серу или азот) в составе кольца, при этом остальные атомы представляют собой углерод, так, например, вышеуказанные гетероарильные группы, а также соответствующие частично насыщенные или полностью насыщенные гетероциклические кольца. Примерами насыщенных гетероциклических колец являются азетидин, пирролидин, пиперидин, пиперазин, морфолин, тиоморфолин, 1,4-оксазепан, азепан, фталимид, индолин, изоиндолин, 1,2,3,4 тетрагидрохинолин, 1,2,3,4-тетрагидроизохинолин, 3,4-дигидро-2H-1,4-бензоксазин, гексагидроазепин,3,4-дигидро-2(1H)изохинолин, 2,3-дигидро-1 Н-индол, 1,3-дигидро-2 Н-изоиндол, азокан, 1-окса-4 азаспиро[4.5]дец-4-ен, декагидроизохинолин и 1,4-диазепан. Кроме того, гетероциклил или гетероциклический остаток может, возможно, иметь в качестве заместителей одну или несколько оксогрупп.C1-8-алкил представляет собой линейную или разветвленную алкильную группу, содержащую 1-8 атомов углерода. Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, вторбутил, трет-бутил, пентил, изопентил, гексил, изогексил, н-гептил и н-октил. В качестве частей диапазона C1-8-алкил включены все подгруппы указанного диапазона, такие как C1-7-алкил, C1-6-алкил, C1-5 алкил, C1-8-алкил, C2-8-алкил, C2-7-алкил, C2-6-алкил, C2-5-алкил, C3-7-алкил, C4-6-алкил, и т.д.C1-8-алкоксил представляет собой линейную или разветвленную алкоксильную группу, содержащую 1-8 атомов углерода. Примеры алкоксильных групп включают метокси, этокси, пропокси, изопропокси, бутокси, втор-бутокси, трет-бутокси, пентилокси, изопентилокси, гексилокси, изогексилокси, нгептилокси и н-октилокси. В качестве частей диапазона C1-6-алкоксил включены все подгруппы указанного диапазона, такие как C1-7-алкокси, C1-6-алкокси, C1-5-алкокси, C1-4-алкокси, C2-8-алкокси, C2-7 алкокси, C2-6-алкокси, C2-5-алкокси, C3-7-алкокси, C4-6-алкокси, и т.д. С 2-8-алкенил представляет собой линейную или разветвленную алкенильную группу, содержащую 2-8 атомов углерода. Типичные алкенильные группы включают винил, 1-пропенил, 2-пропенил, изопропенил, 1-бутенил, 2-бутенил, 1-пентенил, 2-пентенил, 1-гексенил, 2-гексенил, 1-гептенил и 1-октенил. В качестве частей диапазона C2-8-алкенил включены все подгруппы указанного диапазона, такие как C2-7 алкенил, C2-6-алкенил, C2-6-алкенил, C2-4-алкенил, C3-8-алкенил, C3-7-алкенил, C3-6-алкенил, С 3-5-алкенил,С 4-7-алкенил, C5-6-алкенил, и т.д.C3-10-циклоалкил представляет собой моноциклическую, бициклическую или трициклическую алкильную группу, содержащую от 3 до 10 атомов углерода, возможно, имеющую заместители. Примеры циклоалкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил,циклооктил, циклононил, циклодецил, бицикло[2.2.1]гепт-2-ил, трицикло[3.3.1.0-3,7-]нон-3-ил,(1R,2R,3R,5S)-2,6,6-триметилбицикло[3.1.1]гепт-3-ил, (1S,2S,3S,5R)-2,6,6-триметилбицикло[3.1.1]гепт-3 ил, 1-адамантил, норадамантил и 2,2,3,3-тетраметилциклопропил. В качестве частей диапазона C3-10 циклоалкил включены все подгруппы, такие как C3-9-циклоалкил, C3-8-циклоалкил, C3-7-циклоалкил, C3-6 циклоалкил, C3-5-циклоалкил, C4-10-циклоалкил, C5-10-циклоалкил, C6-10-циклоалкил, C7-10-циклоалкил, С 8-9 циклоалкил, и т.д. Кроме того, циклоалкильный остаток может иметь в качестве заместителей одну или несколько оксогрупп.C3-10-циклоалкенил представляет собой циклическую, бициклическую или трициклическую алкенильную группу, содержащую в сумме 3-10 атомов углерода, возможно, имеющую в качестве заместителя алкил. Примеры циклоалкенильных групп включают циклопропенил, циклобутенил, циклопентенил,циклогексенил, циклогептенил, циклооктенил, циклононенил, циклодеценил и бицикло[2.2.1]гепт-5-ен 2-ил. В качестве частей диапазона C3-10-циклоалкенил включены все подгруппы, такие как С 3-9 циклоалкенил, C3-8-циклоалкенил, C3-7-циклоалкенил, C3-6-циклоалкенил, С 3-5-циклоалкенил, C4-10 циклоалкенил, C5-10-циклоалкенил, C6-10-циклоалкенил, С 7-10-циклоалкенил, C8-9-циклоалкенил, и т.д. Кроме того, циклоалкенильный остаток может, возможно, иметь в качестве заместителей одну или несколько оксогрупп. Термины галоген и гало в настоящей заявке включают фтор, хлор, бром и йод. Термин -гетеро(С 1-С 8)алкил относится к остатку, в котором гетероатом, выбираемый из азота, серы и кислорода, возможно, несущих заместители, является точкой присоединения к главной молекуле и присоединен к цепи C1-C8-алкила. Настоящее изобретение предусматривает только такие комбинации заместителей и переменных,которые приводят к образованию стабильных соединений. Термин стабильный в настоящей заявке относится к соединениям, обладающим достаточной стабильностью для обеспечения возможности производства и сохраняющим целостность соединения в течение достаточного периода времени для применения в целях, подробно описанных в настоящей заявке (например, терапевтическое введение субъекту для лечения заболевания, ингибирования 11-.бета.-ГСД 1, заболевания, опосредуемого 11-.бета.-ГСД 1). В настоящей заявке термин пролекарство обозначает производное соединения, которое способно гидролизоваться, окисляться или иным образом реагировать в биологических условиях (in vitro или invivo), давая активное соединение. Примеры пролекарств включают, без ограничения, производные или метаболиты производных соединения, содержащие способные к биологическому гидролизу группы, такие как биогидролизуемые амиды, биогидролизуемые сложные эфиры, биогидролизуемые карбаматы,биогидролизуемые карбонаты, биогидролизуемые уреиды и биогидролизуемые фосфатные аналоги (например, монофосфат, дифосфат или трифосфат). Предпочтительно пролекарства соединений, содержащих карбоксильные функциональные группы, представляют собой сложные эфиры карбоновой кислоты и низших алкилов. Сложные эфиры карбоксилатов обычно получают этерификацией любого из остатков карбоновой кислоты, присутствующих в молекуле. Пролекарства обычно можно получить хорошо известными способами, такими как способы, описанные в Burger's Medicinal Chemistry and Drug Discovery 6th ed. (Donald J. Abraham ed., 2001, Wiley) и Design and Application of Prodrugs (H. Bundgaard ed., 1985,Harwood Academic Publishers Gmfh). Таутомер представляет собой один из двух или нескольких структурных изомеров, находящихся в равновесии и легко превращающихся из одной изомерной формы в другую. В данном случае, таутомеры приведенной ниже структуры включены в объем настоящего изобретения. В настоящей заявке гидрат представляет собой такую форму соединения, в которой молекулы воды объединены в некотором соотношении в качестве неотъемлемой составной части кристаллической структуры указанного соединения. В настоящей заявке сольват представляет собой такую форму соединения, в которой молекулы растворителя объединены в некотором соотношении в качестве неотъемлемой составной части кристаллической структуры указанного соединения. В настоящей заявке термин геометрические изомеры относится к соединениям, обладающим одинаковой молекулярной формулой, атомы в которых находятся в различных не эквивалентных положениях друг относительно друга. В настоящей заявке термин оптические изомеры относится к соединениям, содержащим хиральные атомы, которые способны вращать плоскополяризованный свет, и обычно обозначаются с использованием общепринятой R/S конфигурации. Термин оптический изомер включает энантиомеры и диастереомеры, а также соединения, которые можно отличить друг от друга при помощи обозначений (D) и (L). Фармацевтически приемлемый означает в настоящей заявке вещество, подходящее для приготовления фармацевтической композиции, которое, в целом, безопасно, нетоксично, не является ни биологически, ни иным образом нежелательным, и включает как вещество, подходящее для ветеринарного применения, так и вещество, подходящее для фармацевтического применения на людях. Фармацевтически приемлемые соли обозначают соли, которые фармацевтически приемлемы, как определено выше, и обладают желаемой фармакологической активностью. Такие соли включают соли присоединения кислоты, образованные органическими и неорганическими кислотами, такими как хлороводород, бромоводород, йодоводород, серная кислота, фосфорная кислота, уксусная кислота, гликолевая кислота, малеиновая кислота, малоновая кислота, щавелевая кислота, метансульфоновая кислота, трифторуксусная кислота, фумаровая кислота, янтарная кислота, винная кислота, лимонная кислота, бензойная кислота, аскорбиновая кислота, и тому подобное. Соли присоединения основания могут быть образованы органическими и неорганическими основаниями, такими как натрий, аммиак, калий, кальций,этаноламин, диэтаноламин, N-метилглюкамин, холин, и тому подобное. В настоящее изобретение включены фармацевтически приемлемые соли или соединения любой из приведенных формул. В зависимости от структуры выражение фармацевтически приемлемая соль в настоящей заявке относится к фармацевтически приемлемой соли указанного соединения и органической или неорганической кислоты или основания. Типичные фармацевтически приемлемые соли включают, например, соли щелочных металлов, соли щелочно-земельных металлов, соли аммония, растворимые и не растворимые в воде соли, такие как ацетат, амсонат (4,4-диаминостильбен-2,2-дисульфонат), бензолсульфонат, бензоат,бикарбонат, бисульфат, битартрат, борат, бромид, бутират, кальций, эдетат кальция, камсилат(camsylate), карбонат, хлорид, цитрат, клавуланат (clavulariate), дигидрохлорид, эдетат, эдисилат (edisylate), эстолат (estolate), эсилат (esylate), фумарат, глюцептат (gluceptate), глюконат, глутамат, гликоллиларсанилат (glycollylarsanilate), гексафторфосфат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат (mucinate), напсилат (napsylate), нитрат, аммониевая соль N-метилглюкамина, З-гидрокси-2-нафтоат, олеат, оксалат, пальмитат, памоат (1,1-метен-бис-2 гидрокси-3-нафтоат, эмбонат (einbonate, пантотенат, фосфат/дифосфат, пикрат, полигалактуронат, пропионат, пара-толуолсульфонат, салицилат, стеарат, субацетат, сукцинат, сульфат, сульфосалицилат, мурамат, таннат, тартрат, теоклат (teoclate), тозилат, триэтиодид (triethiodide) и валерат. Кроме того, фармацевтически приемлемые соли могут содержать в своей структуре несколько заряженных атомов. В указанном случае фармацевтически приемлемая соль может содержать многочисленные противоионы. Следовательно, фармацевтически приемлемая соль может содержать один или несколько заряженных атомов и/или один или несколько противоионов. В настоящем описании и формуле изобретения использованы следующие сокращения, значения которых приведены ниже: ТМЭДА обозначает N,N,N',N'-тетраметилэтилендиамин. ТМПДА обозначает N,N,N',N'-тетраметилпропилендиамин. ТМБДА обозначает N,N,N',N'-тетраметилбутилендиамин.Ph обозначает фенил. ди обозначает диастереомерный избыток. МТБЭ обозначает метил-трет-бутиловый простой эфир. ИПС обозначает изопропиловый спирт. ДХМ обозначает дихлорметан. МСК обозначает метансульфоновую кислоту (MeSO3H). Твн обозначает внутреннюю температуру реакционной смеси. ППЖХ обозначает площадь пика в % по данным ВЭЖХ ТГА обозначает термогравиметрический анализ Химические вещества, применяемые в способах синтеза, определенных в настоящей заявке, включают, например, растворители, реактивы и катализаторы. Описанные выше способы могут также дополнительно включать стадии как до, так и после стадий, конкретно описанных в настоящей заявке, для добавления или удаления подходящих защитных групп с целью, в конечном счете, сделать возможным синтез соединений. Кроме того, различные стадии синтеза можно осуществлять в изменяемой последо-9 018028 вательности или порядке для получения выбранных соединений. Химические превращения при синтезе и методике работы с защитными группами (введение защиты и снятие защиты), подходящие для синтеза применимых соединений, известны в данной области и включают, например, те, которые описаны в R.Reagents for Organic Synthesis, John Wiley and Sons (1995) и в последующих изданиях. Некоторые варианты реализации настоящего изобретения включают способы получения соединения общей формулы I, как описано выше, посредством асимметрического алкилирования соединения формулы II Соединение формулы (II) получают согласно следующему общему способу синтеза: где 5 Х означает пятикратное количество. Если подходящая мочевина, тиомочевина, альфа-бромкарбоновая кислота или сложный эфир отсутствуют в продаже, соответствующее исходное вещество можно получить согласно способу, описанному в публикации заявки на патент США 2006/0142357. Согласно одному из вариантов реализации Z представляет собой S, обозначая тиазолиноны. Переменная Z также может представлять собой О, обозначая оксазолиноны. Согласно другому варианту реализации R1 выбирают из группы, состоящей из Согласно одному из вариантов реализации R2 и R3 независимо выбирают из метила, изопропила и н-пропила. Согласно другому варианту реализации хиральное основание выбирают из следующей группы оснований: Согласно другому варианту реализации, хиральное основание выбирают изAr представляет собой арил. Согласно другому варианту реализации хиральное основание представляет собой Согласно некоторым из вариантов реализации хиральное основание выбирают из группы, состоящей из где M определено выше. Согласно некоторым из вариантов реализации хиральное основание представляет собой соль эфедрина, т.е.: где М определено выше. Следовательно, согласно одному из вариантов реализации хиральное основание представляет собой соль (1R,2S)-(-)-эфедрина: Согласно другому варианту реализации хиральное основание представляет собой соль (1S,2R)-(-)эфедрина Примером М во всех указанных вариантах реализации является ион лития. Согласно другому варианту реализации уходящую группу LG в R3LG выбирают из группы, состоящей из Cl, Br, I, -OS(O)2CH3, -OS(O)2C4F9, -OS(O)2CF3 и -OS(O)2(4-СН 3-фенил). Согласно другому варианту реализации амин выбирают из триэтиламина, триметиламина, триметиламина,триизопропиламина,N,N,N',N'-тетраметилэтилендиамина(ТМЭДА),N,N,N',N'тетраметилпропилендиамина (ТМПДА), и N,N,N'N'-тетраметилбутилендиамина (ТМБДА). Примером амина в указанном случае является ТМЭДА. Согласно другому варианту реализации растворитель, применяемый на стадии (а), выбирают из группы, состоящей из бензола, толуола, трифтортолуола, ксилола, хлорбензола, диалкиловых простых эфиров, ТГФ, диоксана, ДМФА, галогенуглеводородных растворителей, сложноэфирных растворителей и смесей указанных растворителей. Примером растворителя в указанном случае является толуол. Согласно одному из вариантов реализации соединение формулы II вначале приводят в контакт с хиральным основанием, а после этого с алкилирующим агентом R3-LG. Согласно другому варианту реализации соединение формулы II вначале приводят в контакт с алкилирующим агентом R3-LG, а после этого с хиральным основанием. Согласно одному из вариантов реализации кислоту на стадии (b) выбирают из группы, состоящей из HCl, H2SO4, CH3C(O)OH, CF3C(O)OH, MeSO3H, и C6H5SO3H. Согласно другому варианту реализации кислота на стадии (b) представляет собой MeSO3H. Согласно одному из вариантов реализации основание на стадии (с) выбирают из группы, состоящей из LiOH, NaOH, KOH и ацетата натрия. Согласно другому варианту реализации основание на стадии (с) представляет собой NaOH. Согласно одному из вариантов реализации величина диастереомерного избытка (ди) продукта составляет по меньшей мере 85, 90, 95 или 98%. Принимая во внимание вышеизложенные соображения и приведенные ниже конкретные примеры,квалифицированный специалист учтет, что данный выбор хирального основания, амина, растворителя,кислоты или основания может определять хиральность конечного продукта и/или ди конечного продукта. Осуществление указанного выбора находится в компетенции квалифицированного специалиста. Другой вариант реализации настоящего изобретения представляет собой способ получения соединения формулы III из соединения формулы IV как, в общем, описано выше. Согласно указанному варианту реализации способ включает стадии (а) приведение в контакт соединения IV с хиральным основанием следующей формулы в присутствии ТМЭДА и затем (b) взаимодействие продукта стадии (а) с изопропилйодидом. Согласно одному из вариантов реализации указанный способ дополнительно включает стадии (с) приведение в контакт продукта стадии (b) с MeSO3H с образованием мезилатной соли и (d) взаимодействие мезилатной соли, полученной на стадии (с), с NaOH с образованием соединения формулы III. Согласно одному из вариантов реализации продукт стадии (b) имеет величину ди по меньшей мере 90, 95 или 98%. Согласно одному из вариантов реализации продукт стадии (d) имеет величину ди по меньшей мере 99%. Согласно еще одному из вариантов реализации, настоящее изобретение представляет собой дополнительный способ получения соединения формулы III Согласно указанному варианту реализации способ включает(а) приведение в контакт соединения формулы (IV) с хиральным основанием в присутствии депротонирующего реагента и(b) взаимодействие продукта стадии (а) с изопропилйодидом. Термин хиральное основание здесь и далее подразумевает хиральную молекулу, являющуюся основанием. Термин хиральное основание дополнительно включает хиральное основание, полученное в результате депротонирования нейтрального или свободного основания. Так, хиральное основание, содержащее ион М, как определено выше, формально относится к соли свободного основания. Свободно основание отличают, например, группы -ОН и -NH или -NH2, означающие, что хиральные основания не являются депротонированными. Иллюстративные примеры хиральных оснований включают следующие: Согласно некоторым вариантам реализации хиральное основание представляет собой Согласно вышеописанному варианту реализации способ можно осуществлять в присутствии депротонирующего агента. Специалистам в области органического синтеза известно множество депротонирующих агентов. Например, депротонирующие агенты включают, без ограничения, металлоорганические соединения, такие как соединения алкиллития. Типичными примерами соединений алкиллития являются метиллитий, н-бутиллитий, трет-бутиллитий и гексиллитий. Другие депротонирующие агенты включают гидриды металлов, такие как, например, гидрид лития, гидрид натрия и гидрид калия. Далее настоящее изобретение будет описано применительно к следующим примерам. Указанные примеры не следует рассматривать как ограничивающие объем настоящего изобретения, они должны служить только иллюстративным материалом. 5-метилтиазолинон (1) (25,25 г) суспендировали в 500 мл безводного толуола. Из указанной суспензии отгоняли растворитель при 44C и пониженном давлении 50 мбар до общего объема 160 мл. В снабженный рубашкой реактор объемом 3 л, оборудованный охлаждающим аппаратом Julabo LH-50, линией подачи N2, термопарой и мешалкой верхнего расположения, помещали 110,2 г твердого хирального амина (2). Реактор с содержимым продували N2. В продутый реактор через трубку загружали толуол (375 мл), получая прозрачный раствор хирального амина (2). Полученный раствор охлаждали до -15C. Через трубку переносили бутиллитий (3) (181 мл, 2,7 М в толуоле) в капельную воронку объемом 250 мл, присоединенную к реактору. Добавляли бутиллитий (3) по каплям в течение 30 мин, при этом внутренняя температура (Твн) не поднималась выше -9,0C. После того, как Твн вновь стабилизировалась на уровне -15,5C, в реактор помещали ТМЭДА (4)(37 мл) при помощи шприца. После выдержки в течение 30 мин, добавляли по частям через трубку 160 мл суспензии 5-метилтиазолинона (1) в толуоле, при этом Твн не поднималась выше -4,5C. Затем доводили Твн до 16C и продолжали реакцию в течение 1 ч. После указанного периода выдержки вновь доводили Твн до -15,5C. Добавляли через трубку н-пропилйодил (5) (88 мл) в течение 15 мин, поддерживая Твн ниже -12C. После завершения добавления nPrI Твн стабилизировалась на уровне -14,5C, и полученную смесь перемешивали в течение 16 ч. Через 16 ч ВЭЖХ анализ показал, что осталось менее 5% исходного вещества, и ди равно 34%. К реактору присоединяли капельную воронку объемом 250 мл, в которую помещали 250 мл насыщ. NH4Cl. Осуществляли быстрое добавление NH4Cl по каплям и указанный насыщенный раствор добавляли в течение 1,5 ч, причем за указанное время Твн не поднималась выше -8,0C. После остановки реакции содержимое реактора нагревали до 22C и перемешивали смесь. Затем перемешивание прекращали, оставляли слои разделиться на 5 мин, после чего отделяли нижний водный компонент. Вторую заливку 250 мл насыщ. NH4Cl проводили, как описано выше. Затем толуольный слой подкисляли 3200 мл 2N АсОН и проводили экстрагирование при помощи перемешивания, разделения фаз и отделения нижнего водного слоя. Конечное экстрагирование проводили с использованием 200 мл насыщ. NaHCO3 ранее описанным способом. Полученный в результате толуольный слой затем фильтровали для доочистки, получая 800 мл прозрачного раствора. Общий объем 800 мл толуольного раствора уменьшали до 100 мл, удаляя толуол при пониженном давлении (40C, 60 мбар, ротационный испаритель). Полученный концентрированный толуольный раствор переносили в 3-горлую круглодонную колбу объемом 1 л, с последующей промывкой 10 мл толуола. После нагревания полученной смеси до 60C добавляли гептан (400 мл) через капельную воронку объемом 1 л в течение 35 мин. После окончания добавления гептана полученный однородный раствор медленно охлаждали до 22C в течение 2 ч, получая мелкодисперсную суспензию. К указанной суспензии дополнительно добавляли 1 л гептана и оставляли полученную смесь при перемешивании и 22C на протяжении 48 ч. После указанного времени суспензию фильтровали через воронку объемом 300 мл с фильтром средней пористости, промывали 50 мл гептана при 0C и высушивали при помощи бытового вакуума с током N2 в течение 16 ч. После указанного периода высушивания было получено 20,3 г твердого вещества (выход 67,7) с ППЖХ 98% и ди = 27%. 1 Методика: 5-метилтиазолинон (7) (25,0 г) суспендировали в 480 мл безводного толуола. Из указанной суспензии отгоняли растворитель при 44C и пониженном давлении 50 мбар до общего объема 160 мл. В снабженный рубашкой реактор объемом 3 л, оборудованный охлаждающим аппаратом Julabo LH-50, линией подачи N2, термопарой и мешалкой верхнего расположения, помещали 110,2 г твердого хиралыюго амина (2). Реактор с содержимым продували N2. В продутый реактор через трубку загружали толуол (375 мл), получая прозрачный раствор хирального амина (2). Полученный раствор охлаждали до -15C. Через трубку переносили бутиллитий (3) (181 мл, 2,7 М в толуоле) в капельную воронку объемом 250 мл, присоединенную к реактору. Добавляли бутиллитий (3) по каплям в течение 45 мин, при этом Твн не поднималась выше -8,0C. После того, как Твн вновь стабилизировалась на -15,5C, в реактор помещали ТМЭДА (4) (37 мл) при помощи шприца. После выдержки в течение 20 мин добавляли по частям через трубку 160 мл суспензии тиазолинона (7) в толуоле, при этом Твн не поднималась выше -13C. Затем доводили Твн до 16C и продолжали реакцию в течение 30 мин. После указанного периода выдержки вновь доводили Твн до -15,5C. Добавляли через трубку н-пропилйодил (5) (88 мл) в течение 15 мин, поддерживая Твн ниже -12C. После завершения добавления nPrI Твн стабилизировалась на -14,5C, и полученную смесь перемешивали в течение 16 ч. Через 16 ч ВЭЖХ анализ показал, что осталось менее 1% исходного вещества и ди равно 41%. К реактору присоединяли капельную воронку объемом 250 мл, в которую помещали 250 мл насыщ. NH4Cl. Регулировали быстрое добавление по каплям NH4Cl и добавляли указанный насыщенный раствор в течение 1,5 ч, причем за указанное время Твн не поднималась выше -8,0C. После остановки реакции содержимое реактора нагревали до 22C и перемешивали смесь. Затем перемешивание прекращали, оставляли на 5 мин для разделения слоев, после чего отделяли нижний водный компонент. Второй раз заливали 250 мл насыщ. NH4Cl согласно тому, как описано выше. Затем толуольный слой подкисляли 3200 мл 2N АсОН и проводили экстрагирование при помощи перемешивания, разделения фаз и отделения нижнего водного слоя. Конечное экстрагирование проводили с использованием 200 мл насыщ. NaHCO3 ранее описанным способом. Полученный в результате толуольный слой затем фильтровали через барьерный фильтр с получением 800 мл прозрачного раствора. Полученный прозрачный толуольный раствор концентрировали до 100 мл при 50C и 60 мбар, после чего в сосуд помещали безводный октан (400 мл). Полученную смесь концентрировали при 60C и 80 мбар до общего объема -100 мл, а затем разбавляли октаном до 400 мл. Полученный раствор медленно охлаждали до 22C, получая суспензию, которую перемешивали в течение 2 ч. Затем суспензию фильтровали через воронку со стеклянным фильтром средней пористости и высушивали в течение ночи при помощи бытового вакуума с током N2, получая 11,3 г твердого вещества. 350 мл оставшегося исходного раствора концентрировали до общего объема 50 мл (60C, 70 мбар) и после охлаждения до 22C быстро выпадал твердый белый осадок. Указанный остаток фильтровали так же, как и первую порцию, получая 9,9 г белого твердого вещества. Объединение обоих осадков давало 21,2 г (выход 71,1%) твердого вещества, 90 % ППЖХ, ди = 43%. 1 Н ЯМР [(CD3)2SO] : 9,26 (д, 1H), 6,21 (м, 1 Н), 6,09 (м, 1 Н), 3,75 (м, 1H), 2,86 (м, 1H), 2,78 (д, 1H),1,54 (сложн, м, 10 Н), 1,04 (м, 1H) и 0,86 м.д. (м, 3H). Примечание: неосновной изомер также был виден в 1 Н ЯМР. Методика: 5-метилтиазолинон (9) (26,3 г) суспендировали в 480 мл безводного толуола. Из указанной суспензии отгоняли растворитель при 44C и пониженном давлении 50 мбар до общего объема 160 мл. В снабженный рубашкой реактор объемом 3 л, оборудованный охлаждающим аппаратом Julabo LH-50, линией подачи N2, термопарой и мешалкой верхнего расположения, помещали 110,2 г твердого хирального амина (2). Реактор с содержимым продували N2. В продутый реактор через трубку загружали толуол (375 мл), получая прозрачный раствор хирального амина (2). Полученный раствор охлаждали до -15C. Через трубку переносили бутиллитий (3) (181 мл, 2,7 М в толуоле) в капельную воронку объемом 250 мл, присоединенную к реактору. Добавляли бутиллитий (3) по каплям в течение 45 мин, при этом Твн не поднималась выше -8,0C. После того, как Твн вновь стабилизировалась на -16,5C, в реактор помещали ТМЭДА (4) (37 мл) при помощи шприца. После выдержки в течение 20 мин, добавляли по частям через трубку 160 мл суспензии тиазолинона (9) в толуоле, при этом Твн не поднималась выше -13C. Затем доводили Твн до 16C и продолжали реакцию в течение 30 мин. После указанного периода выдержки вновь доводили Твн до -16,5C. Добавляли через трубку нпропилйодил (10) (90 мл) в течение 20 мин, поддерживая Твн ниже -14C. После завершения добавленияnPrI Твн стабилизировалась на -14,5C и полученную смесь перемешивали в течение 16 ч. Через 16 ч ВЭЖХ анализ показал, что осталось менее 3% исходного вещества, и ди равно 85,8%. К реактору присоединяли капельную воронку объемом 250 мл, в которую помещали 250 мл насыщ. NH4Cl. Регулировали быстрое добавление по каплям NH4Cl и добавляли указанный насыщенный раствор в течение 1,5 ч, причем за указанное время Твн не поднималась выше -7,0C. После окончания остановки реакции содержимое реактора нагревали до 22C и перемешивали смесь. Затем перемешивание прекращали,оставляли на 5 мин для разделения слоев, после чего отделяли нижний водный компонент. Второй раз заливали 250 мл насыщ. NH4Cl согласно тому, как описано выше. Затем толуольный слой подкисляли 3200 мл 2N АсОН и проводили экстрагирование при помощи перемешивания, разделения фаз и отделения нижнего водного слоя. Конечное экстрагирование проводили с использованием 200 мл насыщ.NaHCO3 ранее описанным способом. Полученный в результате толуольный слой затем фильтровали через барьерный фильтр, получая 630 мл прозрачного раствора. Полученный толуольный слой концентрировали (50C и 60 мбар) до общего объема 90 мл. Добавляли октан (90 мл) и концентрировали полученную мутную смесь до общего объема 50 мл в вышеуказанных условиях. Описанный цикл разбавления смеси октаном (90 мл на каждый цикл) проводили до тех пор, пока соотношение октана к толуолу не составило 4:1 по данным 1 Н ЯМР. Полученную смесь нагревали до 65C (прозрачный раствор) и медленно охлаждали до 35C, при этой температуре в мутном растворе суспендировали затравку (50 мг). Полученную суспензию охлаждали до 22C в течение 2 ч и выдерживали 13 ч при перемешивании в атмосфере N2. Добавляли по отдельности две порции по 100 мл октана через капельную воронку и через 2,5 ч интенсивного перемешивания фильтровали суспензию. Полученное твердое вещество высушивали в течение 16 ч при помощи бытового вакуума с током N2. Получили светло-коричневое твердое вещество (9,8 г, выход 54,1%), ППЖХ 99%, ди 89,1%. 1- 18018028 Примечание: неосновной изомер также был виден в 1 Н ЯМР. Пример 4. Получение (S)-5-метил-2-S)-1-фенилэтиламино)-5-пропилтиазол-4(5 Н)-она (12) Методика: 5-метилтиазолинон (9) (26,3 г) суспендировали в 500 мл безводного толуола. Из указанной светлой суспензии отгоняли растворитель при 44C и пониженном давлении 50 мбар до общего объема 160 мл. В снабженный рубашкой реактор объемом 3 л, оборудованный охлаждающим аппаратом Julabo LH-50,линией подачи N2, термопарой и мешалкой верхнего расположения, помещали 110,2 г твердого хирального амина (2). Реактор с содержимым продували N2. В продутый реактор через трубку загружали толуол(375 мл), получая прозрачный раствор хирального амина (2). Полученный раствор охлаждали до -15C. Через трубку переносили бутиллитий (3) (181 мл, 2,7 М в толуоле) в капельную воронку объемом 250 мл,присоединенную к реактору. Добавляли бутиллитий (3) по каплям в течение 45 мин, при этом Твн не поднималась выше -11,5C. После того, как Твн вновь стабилизировалась на -16,5C, в реактор помещали ТМЭДА (4) (37 мл) при помощи шприца. После выдержки в течение 10 мин добавляли по частям через трубку 160 мл суспензии тиазолинона (9) в толуоле, при этом Твн не поднималась выше -8,5C. Затем доводили Твн до 16C и продолжали реакцию в течение 50 мин. После указанного периода выдержки вновь доводили Твн до -17C. Добавляли через трубку н-пропилйодил (10) (88 мл) в течение 20 мин,поддерживая Твн ниже -14C. После завершения добавления nPrI Твн стабилизировалась на -14,5C, и полученную смесь перемешивали в течение 16 ч. Через 16 ч ВЭЖХ анализ показал, что осталось менее 0,5% исходного вещества и ди равно 61,2%. К реактору присоединяли капельную воронку объемом 250 мл, в которую помещали 250 мл насыщ. NH4Cl. Регулировали быстрое добавление по каплям NH4Cl и добавляли указанный насыщенный раствор в течение 1,5 ч, причем за указанное время Твн не поднималась выше -3,1C. После остановки реакции содержимое реактора нагревали до 22C и перемешивали смесь. Затем перемешивание прекращали, оставляли на 5 мин для разделения слоев, после чего отделяли нижний водный компонент. Второй раз заливали 250 мл насыщ. NH4Cl согласно тому, как описано выше Затем толуольный слой подкисляли 3200 мл 2N АсОН и проводили экстрагирование при помощи перемешивания, разделения фаз и отделения нижнего водного слоя. Конечное экстрагирование проводили с использованием 200 мл насыщ. NaHCO3 ранее описанным способом. Полученный в результате толуольный слой затем фильтровали через барьерный фильтр, получая 750 мл прозрачного раствора. Полученный толуольный слой концентрировали (60C и 80 мбар) до общего объема 90 мл. Добавляли октан (90 мл) и концентрировали полученную мутную смесь до общего объема 60 мл в вышеуказанных условиях. Описанный цикл разбавления смеси октаном (90 мл на каждый цикл) проводили до тех пор, пока соотношение октана к толуолу не составило 2:1 по данным 1 Н ЯМР. Октаново/толуольный раствор (общий объем 60 мл) нагревали до 70C, получая прозрачный раствор. После достижения Твн 53C вводили 50 мг затравки. Полученную суспензию охлаждали до 33C в течение 20 мин, а затем вновь нагревали до 70C в течение 35 мин. Затем указанную смесь охлаждали до 43,5C в течение 2 ч и к полученной суспензии быстро по каплям добавляли октан (160 мл) в течение 30 мин. Затем суспензию охлаждали до 22C и выдерживали 16 ч при перемешивании в токе N2. Суспензию фильтровали и высушивали при помощи бытового вакуума с током N2 в течение 4 ч, получая 12,5 г (выход 64,8%) светлокоричневого твердого вещества, 98% ППЖХ, ди = 85,7%. Н ЯМР [(CD3)2SO] : 9,60 (д, 1H), 7,33 (сложн, м, 5 Н), 5,19 (м, 1H), 1,68 (м, 2 Н), 1,46-1,43 (сложн,м, 7 Н), 1,06 (м, 1 Н), 0,86 (сложн, м, 3H). Примечание: неосновной изомер также был виден в 1 Н ЯМР. Пример 5. Получение (5S)-2-(бицикло[2.2.1]гептан-2-иламино)-5-метил-5-пропилтиазол-4(5 Н)-она Методика: В 1-горлой круглодонной колбе объемом 250 мл суспендировали 20,4 г неочищенного 5-Me/nPr тиазолинона (6) в 100 мл сухого изопропанола при 22C. К полученной суспензии добавляли метансульфоновую кислоту (14) (5,2 мл, 1,05 экв.), которая по окончании добавления полностью растворяла твердое вещество, образуя однородный раствор. Полученную смесь нагревали в течение 25 мин до 50C, выдерживали в течение 1 ч, затем охлаждали до 22C и выдерживали 16 ч в атмосфере N2. После указанного периода раствор, по-прежнему однородный, переносили в 3-горлую круглодонную колбу объемом 500 мл, снабженную мешалкой верхнего расположения и капельной воронкой объемом 500 мл. Добавляли порциями гептан (285 мл), после чего смесь с температурой 22C охлаждали на ледяной бане. Через 10 мин (Твн = 8,2C) добавляли 100 мг затравки в виде 12 Х суспензии в гептане. Полученную смесь выдерживали 16 ч и оставляли медленно нагреться до 22C, получая густую белую суспензию. Указанную суспензию фильтровали и высушивали (бытовой вакуум/ток N2) в течение 5 ч, получая 10,6 г соли МСК(13), 95,1% ди, в маточном растворе ди = -42,7%. 1 Н ЯМР [(CD3)2SO] : 9,36 (д, 1H), 3,75 (м, 1H), 2,34 (с, 3H), 2,24 (м, 1H), 2,20 (м, 1H), 1,68 (м, 3H),1,47 (сложн, м, 8 Н), 1,12 (м, 4 Н) и 0,84 м.д. (м, 3H). Примечание: неосновной изомер также был виден в 1 Н ЯМР. В колбу Эрленмейера объемом 250 мл помещали 10,6 г соли МСК 5-Me/nPr тиазолинона (13). Затем растворяли указанное твердое вещество в 100 мл сухого ДХМ, получая прозрачный 10 Х раствор. В полученный раствор добавляли NaOH (1N, 50 мл) и энергично перемешивали в течение 20 мин. После прекращения перемешивания переносили полученную двухфазную систему в делительную воронку объемом 250 мл, и удаляли верхний водный слой. Выполняли три промывки органического слоя водой (75 мл каждая), величина pH конечного водного слоя составляла 6,5-7,0. Слой ДХМ фильтровали через барьерный фильтр в круглодонную колбу объемом 250 мл (общий объем 100 мл) и концентрировали до общего объема 20 мл (40C, 60 мбар). В полученный раствор добавляли изопропанол (100 мл) и концентрировали до общего объема 20 мл. Затем в колбу помещали дополнительно 20 мл ИПС, чтобы получить 3,75 Х раствор свободного основания в ИПС. После охлаждения полученной смеси от температуры испарительной бани 40C выпадал белый твердый осадок. Полученный осадок отфильтровывали и высушивали,получая 5,0 г продукта, выход 64,7%, 99,4% ППЖХ и 95,9% ди. 1 Н ЯМР [(CD3)2SO] : 9,00 (д, 1 Н), 3,75 (м, 1H), 2,24 (м, 1H), 2,10 (м, 1H), 1,68 (м, 3H), 1,47 (сложи м, 8 Н), 1,12 (м, 4 Н), и 0,84 м.д. (м, 3H). Пример 6. Получение (5S)-2-(бицикло[2.2.1]гепт-5-ен-2-иламино)-5-метил-5-пропилтиазол-4(5 Н)она в высоком диастереомерном избытке Методика: В 1-горлой круглодонной колбе объемом 250 мл суспендировали 20,4 г неочищенного 5-Me/nPr тиазолинона (8) в 100 мл сухого изопропанола при 22C. К полученной густой жемчужно-белой суспензии добавляли метансульфоновую кислоту (14) (5,5 мл, 1,05 экв.), которая по окончании добавления полностью растворяла твердое вещество, образуя однородный раствор. Полученную смесь нагревали в течение 15 мин до 50C, выдерживали в течение 35 мин, затем охлаждали до 22C и выдерживали 16 ч в атмосфере N2. После указанного периода раствор, по-прежнему однородный, переносили в 3-горлую круглодонную колбу объемом 500 мл, снабженную мешалкой верхнего расположения и капельной воронкой объемом 500 мл. Добавляли гептан (268 мл, 2 Х избыток по отношению к 134 мл общего объема раствора в ИПС) порциями в течение 15 мин, после чего смесь с температурой 22C охлаждали на ледяной бане. Через 50 мин полученной смеси давали нагреться до 22C и выдерживали образовавшуюся густую белую суспензию в течение 16 ч. Указанную суспензию фильтровали и высушивали (бытовой вакуум/ток N2) в течение 5 ч, получая 17,5 г соли МСК (15), выход 73,0%, чистота 99,1%, 82,2% ди. 1H ЯМР [(CD3)2SO] : 9,40 (д, 1H), 6,21 (м, 1H), 6,09 (м, 1H), 3,74 (м, 1 Н), 2,87 (м, 1H), 2,81 (м, 1H),2,31 (с, 3H), 1,54 (сложн, м, 10 Н), 1,04 (м, 1H), и 0,86 м.д. (м, 3H). В колбу Эрленмейера объемом 500 мл помещали 17,5 г соли МСК 5-Me/nPr тиазолинона (15). Затем указанное твердое вещество суспендировали в 175 мл сухого ДХМ, получая суспензию. В полученную суспензию добавляли гидроксид натрия (1 М, 88 мл) и энергично перемешивали полученную двухфазную смесь в течение 16 ч. После указанного периода полученную двухфазную систему переносили в делительную воронку объемом 1 л и оставляли на 5 мин для разделения фаз. Щелочной водный слой отделяли от органической фазы, после чего проводили промывку слоя ДХМ 3 130 мл H2O. Величина pH конечного водного слоя составляла 7,0. Слой ДХМ фильтровали через барьерный фильтр и концентрировали до общего объема 20 мл. В полученный раствор добавляли ИПС (3,75 Х, общий объем 65 мл) и всю полученную смесь концентрировали до общего объема 20 мл (40C, 60 мбар). Затем в колбу помещали дополнительно 105 мл ИПС и концентрировали полученный раствор до общего объема 65 мл (3,75 Х ИПС). Затем полученную смесь нагревали до 70C, а потом медленно охлаждали до 0C. Когда Твн достигала 66C, добавляли воду (52 мл) порциями в течение 5 мин. При Твн = 30,0C образовывалась белая суспензия. Полученную белую суспензию перемешивали при 22C в течение 16 ч в атмосфере N2. После указанного периода суспензию охлаждали до 0C, отфильтровывали и промывали 70 мл раствора 60:40 Н 2 О:ИПС. Полученное твердое вещество высушивали на фритте средней пористости в течение 4 ч в токеH ЯМР [(CD3)2SO] : 9,26 (д, 1H), 6,21 (м, 1H), 6,09 (м, 1 Н), 3,75 (м, 1H), 2,86 (с, 1H), 2,80 (с, 1H),1,54 (сложн, м, 10 Н), 1,04 (м, 1H) и 0,86 м.д. (м, 3H). Примечание: неосновной изомер также был виден в 1H ЯМР. Пример 7. Получение (5S)-2-(бицикло[2.2.1]гептан-2-иламино)-5-изопропил-5-метилтиазол-4(5 Н)она (19) в высоком диастереомерном избытке. Стадия 1. Реактор объемом 20 л оборудовали, как описано в разделе Оборудование (выше) и продували азотом. В реактор загружали (S)-экзо-2-норборнилтиомочевину (16) (801,4 г), а после этого 3,0 л абсолютного этанола. Начинали перемешивание (142 об./мин), после чего добавляли 2-бромпропионовую кислоту (17) (509 мл) при помощи мерного цилиндра. Мерный цилиндр ополаскивали 400 мл абсолютного этанола и переносили промывную жидкость в реактор. Затем добавляли ацетат натрия (965,7 г), за которым следовала последняя загрузка 1,4 л абсолютного этанола. Реакционную смесь нагревали до 80C и выдерживали при указанной температуре в течение 3 ч, после чего охлаждали до 22C. Добавляли деионизированную воду (13 л) и наблюдали слабый экзотермический эффект. Давали полученной смеси вернуться к температуре 22C и выдерживали в течение 12 ч. Полученную суспензию фильтровали через воронку со стеклянным фильтром средней пористости. Примечание: на данной стадии неочищенный материал объединяли при фильтрации с неочищенным продуктом параллельной реакции с 234 г тиомочевины. Это было необходимо из-за того, что максимальная вместимость реактора была 20 л. Оставшееся в реакторе твердое вещество смывали в воронку деионизированной водой (2 л) и промывали осадок на фильтре 1 л деионизированной воды. Полученное твердое вещество высушивали на воздухе на фильтре в течение 3 ч, а затем переносили на лотки для сушки и высушивали при 50C и 15 торр до тех пор, пока анализ ТТА не показывал содержание воды менее 3,0%. Регистрировали сухую массу (1311 г) и переносили твердое вещество в чистый реактор объемом 20 л, снабженный рубашкой. Добавляли МТБЭ (5,9 л) и начинали перемешивание (120 об./мин). Полученную суспензию нагревали до 50C и выдерживали при указанной температуре в течение 2 ч. Затем смесь охлаждали до 22C и фильтровали через воронку со стеклянным фильтром средней пористости. Собранное твердое вещество дважды промывали МТБЭ (по 500 мл на каждую промывку) и высушивали на воздухе в воронке в течение 1 ч. Переносили материал на сушильные лотки и высушивали при 50C и 15 торр до тех пор, пока анализ ТТА не показывал содержание воды менее 1,0%. Высушенное твердое вещество упаковывали (выделено 1240 г, выход 91% , 98 А %). Стадия 2. Асимметрическое алкилирование Реактор объемом 20 л продували азотом. В реактор загружали (R,R)-хиральный амин (2) (1761,2 г). Вслед за этим реактор продували азотом в течение 15 мин. Затем загружали безводный толуол (6,0 л) и начинали перемешивание. Оставляли реакционную смесь перемешиваться в токе азота еще в течение 15 мин, после чего охлаждали реакционную смесь до -5C. Раствор BuLi (3) (2,9 л) наливали в капельную воронку объемом 3 л. При достижении внутренней температуры -5C начинали добавлять по каплям nBuLi, следя за тем, чтобы внутренняя температура не поднималась выше 0C. Затем реакционную смесь охлаждали до -15C и выдерживали 15 мин. Затем через трубку добавляли ТМЭДА (4) (592 мл). 5 метилтиазолинон (18) (400 г) суспендировали в безводном толуоле (1,6 л) в отдельной 3-горлой круглодонной колбе объемом 5 л, в токе азота, в течение 15 мин. Полученную суспензию по частям переносили в реактор через трубку, регулируя скорость добавления и температуру рубашки так, чтобы поддерживать внутреннюю температуру ниже 0C. Круглодонную колбу, использованную для приготовления суспензии субстрата, ополаскивали толуолом (2450 мл) и промывную жидкость переносили в реактор. Нагревали реакционную смесь до 22C и выдерживали при указанной температуре в течение 30 мин. Затем смесь вновь охлаждали до -15C. Добавляли через трубку изопропилйодид (10) (1,42 л), регулируя скорость так, чтобы поддерживать температуру ниже -12,5C, при необходимости регулируя температуру рубашки, чтобы контролировать наблюдавшийся экзотермический эффект. Через 30 мин после окончания добавления изопропилйодида отбирали аналитический образец (следуя протоколу обработки образцов, описанному в аналитическом разделе). Выдерживали реакционную смесь при -15C до достижения конверсии 93%, а затем останавливали реакцию, добавляя по каплям насыщенный раствор NH4Cl, также регулируя скорость подачи и температуру рубашки, чтобы контролировать наблюдавшийся экзотермический эффект. Нагревали реакционную смесь до комнатной температуры и останавливали перемешивание. Оставляли фазы разделиться (не менее 20 мин) и сливали нижний водный слой. Добавляли 3,0 л насыщенного раствора NH4Cl и перемешивали полученную смесь в течение 20 мин. Оставляли фазы разделиться и сливали нижний водный слой. В реактор добавляли раствор уксусной кислоты (2 М, 3,3 л) и перемешивали полученную смесь в течение 20 мин. Оставляли для разделения фаз и сливали нижний водный слой. Повторяли описанную промывку уксусной кислотой. В реактор помещали раствор соли (3,3 л) и перемешивали полученную смесь в течение 20 мин. Оставляли фазы разделиться и сливали нижний водный слой. Медленно добавляли в реактор насыщенный раствор NaHCO3 (3,3 л) при перемешивании в течение 20 мин. Оставляли фазы разделиться (не менее 20 мин) и сливали нижний водный слой. Проводили вторую промывку NaHCO3 (3,3 л) и сливали нижний водный слой. Вновь заливали в реактор раствор соли (3,3 л) и перемешивали полученную смесь в тече- 22018028 ние 20 мин. Оставляли фазы разделиться и сливали нижний водный слой. Образец 200 мл неочищенного толуольного раствора упаривали вакуумной перегонкой до конечного объема 30 мл. Затем выдерживали полученную суспензию при температуре 60C. Добавляли к указанной суспензии 100 мл гептана, поддерживая температуру выше 55C. По окончании добавления гептана охлаждали полученную суспензию до 5C в течение часа. Выдерживали указанную порцию при 5C в течение 90 мин. Затем твердое вещество отфильтровывали через стеклянный фильтр средней пористости и промывали осадок на фильтре минимальным количеством (15 мл) холодного гептана (5C). Полученное твердое вещество высушивали в вакуумной печи при 55C в течение 16 ч. Выделили 5,25 г твердого вещества (выход 67,7%). Стадия 3 (образование соли MCK) Трехгорлую круглодонную колбу объемом 2 л продували N2. Загружали в колбу неочищенный продукт алкилирования (19) (84% ди; 225 г), а после этого изопропиловый спирт (1125 мл, 5 объемов). Организовывали перемешивание и затем добавляли через капельную воронку метансульфоновую кислоту(14) (57,5 мл). Нагревали реакционную смесь до 50C и выдерживали в течение 1 ч. Затем содержимое реактора охлаждали до 18-25C и выдерживали в течение 1,5 ч. Затем выделяли твердый продукт фильтрованием на воронке Бюхнера. Для того, чтобы смыть остатки твердого вещества из круглодонной колбы объемом 2 л, использовали дополнительную порцию изопропилового спирта (338 мл, 1,5 объемов). Оставляли влажное отфильтрованное вещество подсохнуть в воронке по меньшей мере в течение 1 ч. Затем твердое вещество переносили в лоток для сушки и помещали в вакуумную печь при 50C на 16 ч. Получили 272,2 г (неисправленный выход 88,9%, 95,98% ди) сухого соединения (20). Стадия 4 (получение свободного основания) К 40 г суспензии соли МСК (20) в ДХМ (10 Х, 400 мл) в 3-горлой круглодонной колбе объемом 1000 мл, снабженной механической мешалкой и вводом азота, добавляли 5 Х 1N NaOH (200 мл). Полученную смесь перемешивали в течение 1 ч и переносили в делительную воронку. Оставляли слои отстояться в течение 15 мин, а затем разделяли. Затем органический слой промывали 5 объемами деионизированной воды до достижения нейтрального pH водного слоя. Перед проведением обмена растворителей органический слой (ДХМ) фильтровали через стеклянный фильтр средней пористости. Проводили перегонку при атмосферном давлении для уменьшения объема ДХМ до значения 3,75 Х(150 мл). На этом этапе в колбу вводили ИПС (3,75 Х; 150 мл) и возобновляли перегонку при атмосферном давлении до тех пор, пока вновь не получали объем смеси 3,75 Х (150 мл). Добавляли в колбу еще 3,75 Х (150 мл) ИПС и продолжали перегонку до получения конечного объема смеси 3,75 Х (150 мл). Температура смеси на данной стадии равнялась температуре кипения ИПС (-82C). В горячий раствор(755C) продукта в ИПС добавляли воду (3X; 120 мл) с такой скоростью, чтобы поддерживать температуру выше 70C. Охлаждали полученную смесь до 5C на протяжении 1 ч и выдерживали в течение 75 мин. Отфильтровывали твердое вещество и промывали минимальным количеством (2 Х) холодной (5C) смеси ИПС/вода (40/60). Полученное твердое вещество высушивали в вакуумной печи при 55C в течение 17 ч. Выделили 27,97 г продукта (19) (неисправленный выход 95,1%; 99,76% ди). Пример 8. Получение (5S)-2-(бицикло[2.2.1]гепт-5-ен-2-иламино)-5-изопропил-5-метилтиазол 4(5 Н)-она (23) в высоком диастереомерном избытке.(21) (587 г), а после этого 2,97 л абсолютного этанола. Начинали перемешивание, после чего добавляли 2-бромпропионовую кислоту (17) (377 мл) при помощи мерного цилиндра. Затем добавляли ацетат натрия (715 г). Реакционную смесь нагревали до 80C и выдерживали при указанной температуре в течение 4 ч, после чего охлаждали до 22C. Добавляли деионизированную воду (9 л) и наблюдали слабый экзотермический эффект. Давали полученной смеси вернуться к температуре 22C и выдерживали в течение 12 ч. Полученную суспензию фильтровали через воронку со стеклянным фильтром средней пористости. Оставшееся в реакторе твердое вещество смывали в воронку деионизированной водой (1,5 л) и промывали осадок на фильтре 0,5 л деионизированной воды. Полученное твердое вещество высушивали на воздухе на фильтре в течение 3 ч, а затем переносили на лотки для сушки и высушивали при 50C и 3-30 торр до тех пор, пока анализ ТГА не показывал содержание воды менее 3,0%. Регистрировали сухую массу (728,9 г, выход 94%). Стадия 2. Асимметрическое алкилирование(2) (1610 г). Вслед за этим реактор продували азотом в течение 50 мин. Затем загружали безводный толуол (5,42 л) и начинали перемешивание. Оставляли реакционную смесь перемешиваться в токе азота еще в течение 10 мин, после чего охлаждали реакционную смесь до -7,5C в течение 45 мин. Раствор BuLi (3)(2,66 л) наливали в капельную воронку и начинали добавлять по каплям n-BuLi. Во время указанного добавления регулировали скорость добавления и температуру рубашки, чтобы внутренняя температура не поднималась выше 0C. По окончании добавления промывали капельную воронку безводным толуолом (100 мл), который подавали через трубку из герметичной бутыли с толуолом. Реакционную смесь охлаждали до -15 С, а затем добавляли через трубку ТМЭДА (4) (540 мл). 5-Метилтиазолинон (22) (361,6 г) суспендировали в безводном толуоле (1,45 л) в отдельной 3-горлой круглодонной колбе объемом 5 л, в токе азота, в течение 30 мин. Полученную суспензию по частям переносили в реактор через трубку, регулируя скорость добавления и температуру рубашки так, чтобы поддерживать внутреннюю температуру ниже 0C. Круглодонную колбу, использованную для приготовления суспензии субстрата, ополаскивали толуолом (2468 мл) и переносили промывные жидкости в реактор. Нагревали реакционную смесь до 15C в течение 1,5 ч и выдерживали при этой температуре в течение 30 мин (охладитель устанавливали на 22C, макс. темп. = 20C). Затем полученную смесь вновь охлаждали до -15C в течение 1 ч. Добавляли через трубку 2-йодпропан (10) (1,3 л), регулируя скорость так, чтобы поддерживать температуру ниже -12,5C, при необходимости регулируя температуру рубашки, чтобы контролировать наблюдавшийся экзотермический эффект. Выдерживали реакционную смесь при -15C до достижения конверсии 93%, а затем останавливали реакцию, добавляя по каплям насыщенный раствор NH4Cl (3,62 л), также регулируя скорость подачи и температуру рубашки, чтобы контролировать наблюдавшийся экзотермический эффект. Нагревали реакционную смесь до комнатной температуры и останавливали перемешивание. После разделения фаз сливали нижний водный слой. Добавляли 4,82 л насыщенного раствора NH4Cl и перемешивали полученную смесь в течение 20 мин. После разделения фаз сливали нижний водный слой. В реактор добавляли раствор уксусной кислоты (2 М, 3 л) и перемешивали полученную смесь в течение 30 мин. После разделения фаз сливали нижний водный слой. Повторяли описанную промывку уксусной кислотой. Медленно добавляли в реактор насыщенный раствор NaHCO3 (3 л) при перемешивании в течение 20 мин. Оставляли смесь до разделиться фаз (не менее 20 мин) и сливали нижний водный слой. Медленно добавляли в реактор воду (3 л) при перемешивании в течение 20 мин. Оставляли фазы разделиться и сливали нижний водный слой. В толуольном слое проводили замену растворителя на октан, с конечным соотношением раствори- 24018028 телей 20:1, октан:толуол. Перегонку проводили при внутренней температуре в диапазоне от 19 до 54C и давлении в диапазоне от 40 до 275 торр. После достижения выбранного соотношения растворителей,при конечном объеме 3,9 л, полученную суспензию фильтровали через воронку со стеклянным фильтром средней пористости, промывая двумя порциями октана (всего 1400 мл). Полученное твердое вещество высушивали на фильтре в течение 1-1,5 ч, а затем переносили на тарелку для сушки и высушивали в вакуумной печи при 45-55C, 3-30 торр в течение 18-42 ч. Получили 370 г белого твердого вещества, выход 86, 80,5% ди. Стадия 3 (образование соли MCK)(23) (80,5% ди; 303,3 г), а после этого изопропиловый спирт (1820 мл, 6 объемов). Организовывали перемешивание и затем добавляли через капельную воронку метансульфоновую кислоту (14) (78,2 мл). Нагревали реакционную смесь до 50C и выдерживали в течение 1 ч. Затем содержимое реактора охлаждали до 20-24C и выдерживали в течение 1,5 ч. Затем выделяли твердый продукт фильтрованием через воронку объемом 2 л со стеклянным фильтром средней пористости. Для того, чтобы смыть остатки твердого вещества из реактора объемом 5 л, использовали две дополнительные порции изопропилового спирта (2303 мл, всего 2 объема). Оставляли влажное отфильтрованное вещество подсохнуть в воронке,по меньшей мере, в течение 1 ч. Затем твердое вещество переносили в лоток для сушки и помещали в вакуумную печь при 50C, 3-30 торр на 16 ч. Получили 367,4 г (неисправленный выход 88,9, 96,8% ди) сухого соединения. Выделенное твердое вещество (367,4 г) вновь загружали в реактор с последующим добавлением изопропилового спирта (1886 мл). Организовывали перемешивание и нагревали содержимое реактора до 50C в течение 105 мин. Выдерживали полученную смесь при указанной температуре в течение 23 ч. Затем смесь охлаждали до 20-24C в течение 2 ч и выдерживали еще 3 ч. Твердое вещество выделяли фильтрованием через воронку объемом 8 л со стеклянным фильтром средней пористости. Для промывки влажного остатка на фильтре использовали дополнительную порцию изопропилового спирта (2269 мл). Оставляли влажное твердое вещество подсохнуть в воронке по меньшей мере в течение 1 ч. Затем переносили указанное вещество на лоток для сушки и помещали в вакуумную печь при 50C на 16 ч. Получили 357,2 г (выход 97,2, 99,3% ди) сухого соединения. Стадия 4 (получение свободного основания) Реактор объемом 20 л продували азотом. В реактор загружали соль метансульфоновой кислоты (24)(598,6 г) и 5,73 л дихлорметана. Начинали перемешивание и добавляли к полученной суспензии 2,86 л 1N гидроксида натрия в течение 10 мин, что вызывало повышение температуры с 18,1 до 21,6C. Полученную смесь перемешивали в течение 1 ч, затем прекращали перемешивание и оставляли слои отстояться. Сливали нижний органический слой. Затем сливали верхний водный слой (pH = 14). Органический слой возвращали в реактор для промывок водой. В реактор загружали 2,86 л ДИ воды и перемешивали полученную двухфазную смесь в течение 15 мин. Затем прекращали перемешивание и оставляли слои отстояться. Сливали нижний органический слой. Затем сливали верхний водный слой (рН = 10). Повторяли промывку водой еще один раз, получая pH 7. Конечный органический слой фильтровали через воронку со стеклянным фильтром средней пористости и возвращали в чистый реактор объемом 20 л, снабженный перегонным аппаратом. Проводили вакуумную перегонку для уменьшения объема от 7,8 до 4,0 л (6,7 Х). Диапазон температур составлял от 11 до 40C, а диапазон давлений составлял 80-180 торр. При достижении объема 4,0 л добавляли 4,0 л ИПС и повторяли вакуумную перегонку до достижения объема 3,0 л (6,8 объемов), при этом не наблюдали обнаружимых количеств ДХМ. На этом этапе полученный раствор нагревали до 60C в течение 2 ч, после чего добавляли 2420 мл ДИ воды в течение 10 мин, в результате температура понижалась на 8C. Затем переводили охладитель на 35C, и когда внутренняя температура достигала 41C,добавляли еще 580 мл ДИ воды (всего воды = 6,8 объемов, ИПС:вода = 1:1). В течение 1 ч температуру раствора снижали до 0-3C, затем фильтровали раствор через воронку объемом 8 л со стеклянным фильтром средней пористости. Отфильтрованное твердое вещество промывали 880 мл (2 Х) смеси 70:30 вода:ИПС. Полученное вещество переносили на лоток для сушки и помещали в вакуумную печь при 50C, 3-30 торр, на 16 ч. Получили 392,3 г (выход 89,3, 99,3 % дм) белого твердого вещества (23). Пример 9. Получение литиевой соли (R)-пропан-1,3-диил-бисR)-1-фенил-2-(пиперидин-1 ил)этил)амида) (25) Согласно методикам, аналогичным способу, показанному на схеме выше, легко можно получить другие хиральные основания согласно настоящему описанию. Пример 10. Однореакторная реакция алкилирования для получения (5S)-2-(бицикло[2.2.1]гептан-2 иламино)-5-изопропил-5-метилтиазол-4(5 Н)-она В 3-горлую круглодонную колбу объемом 250 мл, снабженную мешалкой верхнего расположения и термопарой, загружали 5-метилтиазолинон (2 г, 8,92 ммоль, 1 экв.). Одно из горл колбы закрывали диафрагмой и вставляли иглу, присоединенную к линии азота и вакуума. Колбу вакуумировали и заполняли азотом. При помощи шприца загружали в колбу толуол (40 мл,20 объемов, Aldrich Sure-Seal). Начинали перемешивание и вводили в диафрагму иглу, через которую подавали положительное давление азота для продувания атмосферы. При помощи шприца добавляли ТМЭДА (2,96 мл, 19,6 ммоль, 2,2 экв.) и продували атмосферу в течение 5 мин. Полученный раствор охлаждали до -15C (5C) и добавляли при помощи шприца n-BuLi (2,6 М в толуоле) (15,1 мл, 39,2 ммоль,4,4 экв.) в течение 35 мин. Температура смеси не превышала -15C (5C). Реакционный сосуд нагревали до 22C (3C) в течение 30 мин и выдерживали 90 мин. В этот момент реакционную смесь охлаждали до 0C (3C) и добавляли 2-йодпропан (7,14 мл, 71,4 ммоль, 8,0 экв.) в течение 15 мин. Наблюдался небольшой скрытый экзотермический эффект 4C. Оставляли реакционную смесь нагреться до 22C в течение 1-2 ч, а затем выдерживали при 22C еще в течение 16 ч. Останавливали реакцию, добавляя насыщенный водный раствор хлорида аммония (16 мл, 8 объема) по каплям при помощи шприца в течение 30 мин. Помещали реакционную смесь в делительную воронку и разделяли два слоя. Обнаружили, что верхний органический слой содержал 1,86 г, аналитический выход (неисправленный) 78%, (5S)-2(бицикло[2.2.1]гептан-2-иламино)-5-изопропил-5-метилтиазол-4(5 Н)-она со стереоселективностью 87:13,и 110 мг, 5,5 % исходного вещества. Описанное направление реакции можно реализовать аналогичным образом как двухреакторную реакцию алкилирования. Пример 11. Однореакторное асимметрическое алкилирование с использованием (+)-эфедрина HCl А. С использованием 2,5 М н-бутиллития в гексане В реактор объемом 5 л, снабженный мешалкой верхнего расположения и крышкой на 5 входов, с присоединенными капельной воронкой, входом для азота и термопарой, загружали 5-метилтиазолинон Продували реактор азотом в течение 45 мин, вдувая азот через впускной переходник и затем выпуская через присоединенный выпускной переходник. Через трубку добавляли Ме-ТГФ (573 мл, 493 г, 6 объемов) и продували реактор еще в течение 30 мин. Удаляли переходник для выпуска азота, так чтобы реактор находился под слоем азота, а затем охлаждали реактор до -15C (3C). В капельную воронку через трубку помещали 2,5 М раствор н-бутиллития в гексане (0,815 л, 2,04 моль, 4,8 экв.). Присоединенный к реактору охладитель устанавливали на -30C, и добавляли в реактор бутиллитий по каплям в течение 2 ч, так чтобы внутренняя температура не превышала -9C. После окончания добавления нагревали реактор до 22C (+3C) в течение 1 ч и выдерживали при указанной температуре в течение 30 мин. В этот момент начинали добавление 2-йодпропана (341 мл, 3,41 моль, 8,0 экв.) порциями из круглодонной колбы с инертной атмосферой. Через 10 мин от начала добавления устанавливали охладитель на 10C для нейтрализации небольшого экзотермического эффекта, в результате температура достигала 26C. Вся процедура добавления заняла 25 мин, и охладитель снова устанавливали на 22C. Полученную реакционную смесь перемешивали при 22C в течение 16 ч, и анализ аликвоты при помощи ВЭЖХ показал конверсию 99% и 77:23 сд (соотношение диастереомеров). Устанавливали охладитель на 10C и добавляли серную кислоту (1,05 М, 907 мл, 9,5 объемов) по каплям через капельную воронку в течение 45 мин. Вновь устанавливали охладитель на 22C и перемешивали полученную смесь в течение 1 ч. Добавляли дихлорметан (478 мл, 5 объемов) и воду (287 мл, 3 объема) и перемешивали в течение 10 мин. После разделения сливали нижний водный слой (1,4 кг), анализировали при помощи ВЭЖХ и обнаружили, что он содержал 45 г эфедрина (53%). Добавляли в реактор раствор натрия бисульфата моногидрата(20 мас./об.%, 907 мл, 9,5 объемов) и перемешивали полученные два слоя в течение 30 мин. Сливали нижний водный слой (1 кг), анализировали при помощи ВЭЖХ и обнаружили, что он содержал 19 г(23%) эфедрина. Органический слой (2,2 кг) слили, проанализировали при помощи ВЭЖХ и обнаружили, что он содержал желаемый (5S)-2-(бицикло[2.2.1]гептан-2-иламино)-5-изопропил-5-метилтиазол-4(5 Н)-он (101 г,89%, 77:23 сд) и (+)-эфедрин (2,9 г, 3%). При необходимости в этом месте можно включить дополнительную промывку раствором натрия бисульфата моногидрата (20 мас./об.%, 907 мл, 9,5 объемов) для удаления избытка эфедрина. Органический слой возвращали в реактор и промывали бикарбонатом натрия (насыщ. водн.) (907 мл, 9,5 объемов), слои сливали и подвергали органический слой стадиям получения соли и свободного основания, аналогично стадиям 3 и 4 примера 7, описанным выше, с выделением продукта. В. С использованием 6.6 М н-гексиллития в гексане [0180] В круглодонную колбу объемом 100 мл, снабженную термопарой, помещали 5-метилтиазолинон (5 г, 22,3 ммоль, 1,0 экв.) и (1S, 2R)-(+)-эфедрина HCl (5,4 г, 26,7 ммоль, 1,2 экв.). Присоединяли диафрагму для запечатывания колбы, затем колбу вакуумировали и заполняли азотом. Добавляли при помощи шприца Ме-ТГФ (30 мл, 6 объемов) и охлаждали колбу до -15C (3C). Добавляли в колбу раствор н-гексиллития в гексане(6,6 М, 16,2 мл, 107 ммоль, 4,8 экв.) по каплям при помощи шприца в течение 20 мин, так чтобы внутренняя температура не превышала -15C (3C). Колбу нагревали до 22C (3C) в течение 30 мин и выдерживали при указанной температуре 45 мин. Добавляли в колбу 2-йодпропан (17,8 мл, 178 ммоль, 8,0 экв.) при 22C (3C) в течение 5 мин и наблюдали небольшой скрытый экзотермический эффект до 26C. Полученную реакционную смесь перемешивали при 22C (3C) в течение 16 ч, а затем добавляли в реакционную смесь серную кислоту (1,05 М, 47 мл, 9,5 объемов) по каплям в течение 90 мин. В процессе указанного добавления внутренняя температура не превышала 26C. Добавляли в реакционную смесь дихлорметан (15 мл, 3 объема) и воду (10 мл, 2 объема) и перемешивали для растворения осадка. Полученные слои переносили в делительную воронку, сливали нижний водный слой (70 г), анализировали при помощи ВЭЖХ и обнаружили, что он содержал эфедрин (3,5 г, 80%). Органический слой промывали раствором натрия бисульфата моногидрата (20 мас./об., 47 мл, 9,5 объемов) и оставляли слои разделиться. Сливали нижний водный слой (56 г), анализировали при помощи ВЭЖХ и обнаружили, что он содержал эфедрин (800 мг, 18%), (5S)-2-(бицикло[2.2.1]гептан-2-иламино)-5-изопропил-5-метилтиазол 4(5 Н)-он и рацемат указанного соединения (28 мг, 0,5%). Сливали органический слой (89 г), анализировали при помощи ВЭЖХ и обнаружили, что он содержал (5S)-2-(бицикло[2.2.1]гептан-2-иламино)-5-изопропил-5-метилтиазол-4(5 Н)-он (5,1 г, 85%, 76:24 сд). При необходимости можно в этом месте включить дополнительную промывку раствором натрия бисульфата моногидрата (20 мас./об, 47 мл, 9,5 объемов) для удаления избытка эфедрина. Органический слой возвращали в делительную воронку и промывали бикарбонатом натрия (насыщ. водн.) (47 мл, 9,5 объемов). Полученные два слоя разделялись медленно, и для улучшения разделения добавляли дополнительно раствор соли (3 объема). Наконец, слои разделяли и сливали. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы (I) или стереоизомера, геометрического изомера, оптического изомера или фармацевтически приемлемой соли указанного соединения включающий приведение в контакт соединения формулы (II) или солью данного основания, полученной присоединением кислоты, в присутствии депротонирующего агента и алкилирующего агента R3-LG; при этомR3 выбирают из пропила и изопропила;LG представляет собой йод. 2. Способ по п.1, отличающийся тем, что дополнительно включает следующие стадии:(b) приведение в контакт продукта стадии (а) с кислотой HB с получением соли формулы (I') при этом В представляет собой органический или неорганический анион; и(с) взаимодействие соли формулы (I') с основанием с получением соединения формулы (I). 3. Способ по п.1, отличающийся тем, что Z представляет собой S. 4. Способ по п.3, отличающийся тем, что R1 представляет собой 5. Способ по п.4, отличающийся тем, что R2 представляет собой метил и R3 представляет собой изопропил. 6. Способ по п.1, отличающийся тем, что диастереомерный избыток соединения формулы I составляет по меньшей мере 85%. 7. Способ получения соединения формулы (III)(а) приведение в контакт соединения формулы (IV) или солью данного основания, полученной присоединением кислоты,в присутствии депротонирующего агента; и(b) взаимодействие продукта стадии (а) с изопропилйодилом.
МПК / Метки
МПК: C07D 277/42
Метки: замещенных, 2-аминотиазолонов, способ, получения
Код ссылки
<a href="https://eas.patents.su/30-18028-sposob-polucheniya-zameshhennyh-2-aminotiazolonov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения замещенных 2-аминотиазолонов</a>
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Мейллян Пьер, Бьенейм Юг, Ансель Жан-Эрик
МПК: B01J 31/24, A61K 31/355, C07C 39/19...
Метки: замещенных, использованием, фенолов, cпособ, витамина, способ, получения
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Способ получения замещенных 4-фенил-4-цианоциклогексановых кислот

Номер патента: 3609
Опубликовано: 26.06.2003
Авторы: Вебб Кевин, Чен Дзианхао, Мендельсон Вилфорд
МПК: C07C 51/08, C07D 303/46
Метки: замещенных, кислот, 4-фенил-4-цианоциклогексановых, способ, получения
Формула / Реферат:
1. Способ получения замещённых 4-фенил-4-цианоциклогексановых кислот формулы I в которой R1 представляет группу -(CR4R5)rR6, в которой алкильные фрагменты могут быть необязательно замещены одним или более галогенами; r = 0-6; R4 и R5 независимо выбраны из водорода или C1-2алкила; R6 представляет C3-6циклоалкил или C4-6циклоалкил, содержащий одну или две ненасыщенные связи, причём циклоалкильные фрагменты могут быть необязательно замещены 1-3...
Соединения и способ получения замещенных 4-фенил-4-цианоциклогексановых кислот

Номер патента: 1783
Опубликовано: 27.08.2001
Авторы: Дидерих Энн Мари, Аллен Эндрю, Мендельсон Уилфорд, Уэбб Кевин, Лиу Ли
МПК: C07C 233/00, A61P 11/06, C07D 303/00...
Метки: способ, соединения, получения, замещенных, 4-фенил-4-цианоциклогексановых, кислот
Формула / Реферат:
1. Способ получения соединения формулы где R1 обозначает -(CR4R5)rR6, гдe алкильные звенья могут быть не замещены или замещены одним или более атомом галогена; r принимает значения от 0 до 6; R4 и R5 независимо выбирают из водорода или C1-2алкила; R6 представляет метил, гидроксил, арил, талогензамещенный арил, арилокси С1-3алкил, галогензамещенный арилокси C1-3алкил, инданил, инденил, С7-11полициклоалкил, тетрагидрофуранил, фуранил,...
Способ получения замещенных бензотиазиноиндолов

Номер патента: 12750
Опубликовано: 30.12.2009
Авторы: Котхмиркар Прабхакар, Рамакришна Венката Сатья Нироги, Ширсатх Викас Шрикришна, Дешпанде Амол Динкар, Джасти Венкатесварлу, Камбхампати Рама Састри
МПК: C07D 513/06
Метки: получения, замещенных, способ, бензотиазиноиндолов
Формула / Реферат:
1. Способ получения замещенных бензотиазиноиндолов общей формулы (I) где R1, R2 и R4 могут быть одинаковыми или различными и, каждый независимо, представляют водород, хлор, фтор, амино, нитро, циано, CHO, (C1-С3)алкил, пергалоген(C1-С3)алкил, (C1-С3)алкокси, арил, аралкил, аралкокси, (С5-С7)гетероциклил, (С5-С7)гетероциклилалкил, (С5-С7)гетероциклилокси, ацил, ацетил, алкиламино, аминоалкил, амид, гидроксиалкил, группу карбоновой кислоты и ее...
Способ получения замещенных производных бензола

Номер патента: 122
Опубликовано: 27.08.1998
Авторы: Ногути Сигеру, Камихара Синдзи, Канаи Казуаки
МПК: B01J 23/44, C07C 233/15
Метки: способ, производных, получения, бензола, замещенных
Формула / Реферат:
1. Способ получения соединения, представленного формулой (2) где R1 и R2 каждый независимо представляет атом водорода, атом галогена, гидроксильную группу или C1-6 алкильную группу, Х представляет аминогруппу, имеющую защитную группу, и n представляет целое число от 0 до 4, отличающийся тем, что он включает гидрирование в присутствии палладиевого катализатора соединения, представленного формулой (1) где X, R1, R2 и n имеют такие же...
Предыдущий патент: Кляммер (варианты) для крепления облицовочных элементов фасада
Следующий патент: Прорезиненный по месту слоистый корд для каркасного армирования шины
Случайный патент: Хирургический светильник с управлением световым излучением