Амиды, ингибирующие секрецию аполипопротеина в (аро в ) и/или микросомного протеина переноса триглицеридов (мтр)





























Формула / Реферат
1. Соединение формулы

или его стереоизомеры, фармацевтически приемлемые соли или гидраты, где G выбирается из
(а) фенила или гетероциклического кольца, где указанное гетероциклическое кольцо содержит в общей сложности от 3 до 14 атомов в кольце, причем указанное гетероциклическое кольцо включает в общей сложности от 1 до 4 гетероатомов в кольце, выбираемых независимо из кислорода, азота и серы, причем отдельные кольца из указанных гетероциклических колец могут быть независимо насыщенными, частично насыщенными или ароматическими, и где каждое из указанных фенильных или гетероциклических колец может необязательно иметь от 1 до 4 заместителей, выбранных независимо из галогена, гидрокси, циано, нитро, оксо, тиоксо, аминосульфонила, фенила, фенокси, фенилтио, бензила, бензоила, бензилокси, (С1-С10)алкила, (C1-C4)перфторалкила, (C1-C10)алкокси, (C1-С4)перфторалкокси, (C1-С10)алкоксикарбонила, (C1-С10)алкилтио, (C1-С10)алкиламино, ди(C1-С10)алкиламино, (C1-С10)алкиламинокарбонила, ди(C1-С10)алкиламинокарбонила, (C1-С10)ацила, (C1-С10)перфторацила, (C1-C10)ацилокси, (C1-C6)ациламино и (С1-C6)перфторациламино;
(b) -CH2CN,

(d) (C2-C12)алкила или (C2-C12)перфторалкила, причем каждый из указанных (C2-C12)алкила и (С2-С12)перфторалкила необязательно замещен 1-3 заместителями, независимо выбранными из
(1) фенила, галогена, нитро, циано, гидрокси, -NR1R2, -OCORJ, (C1-C4)алкокси, (C1-C4)перфторалкокси, (C1-C4)тиоалкокси или (C1-C4)перфтортиоалкокси,
где R1 и R2 в группе -N-R1R2, каждый независимо, выбирается из водорода, формила, фенила, бензила, бензоила, (С3-С8)циклоалкила, (C3-C8)циалоалкенила, (C1-C4)алкила, (C1-С4)перфторалкила, (C1-C10)алкоксикарбонила, (C1-C6)ацила, (C1-С6)перфторацила, аминокарбонила, (C1-C10)алкиламинокарбонила, ди(C1-C10)алкиламинокарбонила, аминосульфонила, (C1-С4)алкиламиносульфонила, ди(С1-С4)алкиламиносульфонила, (C1-C4)перфторалкиламиносульфонила, (C1-C4)алкилсульфонила и (C1-C4)перфторалкилсульфонила, или
где R1 и R2, взятые вместе с атомом азота, с которым они связаны, образуют насыщенное, частично насыщенное или ароматическое гетероциклическое кольцо, причем указанное гетероциклическое кольцо содержит в общей сложности от 3 до 14 атомов в кольце и необязательно включает в кольцо дополнительно от 1 до 4 гетероатомов, независимо выбранных из кислорода, азота и серы, причем указанное гетероциклическое кольцо может необязательно иметь от 1 до 4 заместителей, независимо выбранных из галогена, гидрокси, циано, нитро, оксо, тиоксо, аминосульфонила, фенила, фенокси, фенилтио, бензила, бензоила, бензилокси, (С1-С10)алкила, (C1-C4)перфторалкила, (C1-С10)алкокси, (C1-C4)перфторалкокси, (C1-С10)алкоксикарбонила, (C1-С10)алкилтио, (C1-С10)алкиламино, ди(С1-С10)алкиламино, (C1-C10)алкиламинокарбонила, ди(С1-С10)алкиламинокарбонила, (C1-С10)ацила, (C1-С10)перфторацила, (C1-С10)ациламино и (C1-С10)ацилокси,
где R3 в группе -OCOR3 выбирается из -NR1R2, фенила, (С1-С10)алкила, (С1-С4)перфторалкила, (C1-C6)алкокси и (C1-С6)перфторалкокси,
(2) (С3-С8)циклоалкила или (С3-С8)циклоалкенила, где каждый из указанных (С3-С8)циклоалкила и (С3-С8)циклоалкенила может необязательно иметь от 1 до 4 заместителей, выбранных независимо из галогена, гидрокси, циано, нитро, оксо, тиоксо, аминосульфонила, фенила, фенокси, фенилтио, бензила, бензоила, бензилокси, (C1-С10)алкила, (С1-С4)перфторалкила, (C1-С10)алкокси, (C1-С4)перфторалкокси, (C1-С10)алкоксикарбонила, (С1-C10)алкилтио, (C1-С10)алкиламино, ди(С1-С10)алкиламино, (C1-C10)алкиламинокарбонила, ди(С1-С10)алкиламинокарбонила, (С1-С10)ацила, (C1-С10)перфторацила, (C1-С10)ациламино, (C1-С10)перфторациламино, (С1-С10)ацилокси и
(3) насыщенного, частично насыщенного или ароматического гетероциклического кольца, содержащего в общей сложности от 3 до 14 атомов в кольце, причем указанное гетероциклическое кольцо включает в общей сложности от 1 до 4 гетероатомов в кольце, независимо выбранных из кислорода, азота и серы, причем указанное гетероциклическое кольцо может необязательно иметь от 1 до 4 заместителей, независимо выбранных из галогена, гидрокси, циано, нитро, оксо, тиоксо, аминосульфонила, фенила, фенокси, фенилтио, бензила, бензоила, бензилокси, (C1-С10)алкила, (С1-С4)перфторалкила, (С1-С10)алкокси, (С1-С4)перфторалкокси, (С1-С10)алкоксикарбонила, (C1-С10)алкилтио, (С1-С10)алкиламино, ди(С1-С10) алкиламино, (С1-С10)алкиламинокарбонила, ди(С1-С10)алкиламинокарбонила, (С1-С10)ацила, (С1-С10)перфторацила, (С1-С10)ациламино, (С1-С10)перфторациламино, (С1-С10)ацилокси
при условии, что (С1-С12)алкил не включает незамещенный алкил;
(e) (С3-С8)циклоалкила или (С3-C8)циклоалкенила, причем каждый из указанных (С3-С8) циклоалкила и (С3-С8)циклоалкенила может необязательно иметь от 1 до 4 заместителей, независимо выбранных из галогена, гидрокси, циано, нитро, оксо, тиоксо, аминосульфонила, фенила, фенокси, фенилтио, бензила, бензоила, бензилокси, (С1-С10)алкила, (С1-С4)перфторалкила, (С1-С10)алкокси, (С1-С4)перфторалкокси, (С1-С10)алкоксикарбонила, (С1-С10)алкилтио, (С1-С10)алкиламино, ди(С1-С10)алкиламино, (С1-С10)алкиламинокарбонила, ди(С1-С10)алкиламинокарбонила, (С1-С10)ацила, (С1-С10)перфторацила, (С1-С10)ациламино, (С1-С10)перфторациламино, (С1-С10)ацилокси и
(f) -(CH2)nCOR4, где R4 в группе -(CH2)nCOR2 выбирается из гидрокси, фениыр, -NR1R2, (С1-С4)алкила, (С1-С4)перфторалкила, (С1-С4)алкокси, (С1-С4)перфторалкокси, (С3-С8)циклоалкила и (С3-С8)циклоалкенила,
где n является целым числом от 1 до 4.
2. Соединение по п.1 и его стереоизомеры, фармацевтически приемлемые соли и гидраты, отличающееся тем, что G выбирается из
(а) фенила или гетероциклического кольца, причем указанное гетероциклическое кольцо содержит в общей сложности от 3 до 7 атомов в кольце, причем указанное гетероциклическое кольцо включает в общей сложности от 1 до 4 гетероатомов в кольце, независимо выбранных из кислорода, азота и серы, причем указанное гетероциклическое кольцо может быть насыщенным, частично насыщенным или ароматическим, и причем каждое из указанных фенильного и гетероциклического колец может необязательно иметь от 1 до 4 заместителей, независимо выбранных из галогена, гидрокси, фенила, бензила, бензоила, бензилокси, (C1-С10)алкила, (C1-C4)перфторалкила, (C1-C10)алкокси, (C1-C4)перфторалкокси, (C1-С10)алкоксикарбонила, (C1-С10)алкилтио, (C1-С10)алкиламино, ди(С1-С10)алкиламино, (C1-C10)алкиламинокарбонила, ди(С1-С10)алкиламинокарбонила, (C1-C10)ацила, (C1-С10)перфторацила, (C1-С10)ациламино, (C1-C6) перфторациламино, (C1-C10)ацилокси;
(b) (С2-С12)алкила, причем указанный (С2-С12)алкил необязательно замещен 1-3 заместителями, выбранными из
(1) фенила, галогена, циано, гидрокси, -NR1R2, -OCOR3, (C1-C4)алкокси или (C1-C4)перфторалкокси,
где R3 в группе -OCOR3 выбирается из -NR1R2, (С1-С4)алкила и (С1-С4)перфторалкила,
(2) (С3-С6)циклоалкила или (С3-С6)циклоалкенила, причем каждый из указанных (C3-C6)циклоалкила и (С3-С6)циклоалкенила может необязательно иметь от 1 до 4 заместителей, независимо выбранных из гидрокси, (C1-C4)алкила, (C1-C4)алкокси и (C1-С4)алкоксикарбонила и
(3) насыщенного, частично насыщенного или ароматического гетероциклического кольца, содержащего в общей сложности от 3 до 6 атомов в кольце, причем указанное, гетероциклическое кольцо включает в общей сложности от 1 до 4 гетероатомов в кольце, выбранных независимо из кислорода, азота и серы, причем указанное гетероциклическое кольцо может необязательно иметь от 1 до 4 заместителей, независимо выбранных из галогена, гидрокси, фенила, бензила, бензоила, бензилокси, (C1-С10)алкила, (C1-С4)перфторалкила, (C1-C10)алкокси, (C1-C10)алкоксикарбонила, (C1-С10)алкилтио, (С1-С10)алкиламино, ди(C1-С10)алкиламино, (C1-С10)алкиламинокарбонила, ди(С1-С10)алкиламинокарбонила, (С1-С4)перфторалкокси, (C1-С10)ацила, (C1-C10)ациламино, (С1-С10)перфторациламино, (C1-С10)ацилокси;
при условии, что (C2-C12)алкил не включает незамещенный аллил,
(c) (С3-С6)циклоалкила или (C3-C6)циклоалкенила, причем каждый из указанных (С3-С6)циклоалкила или (С3-С6)циклоалкенила может необязательно иметь от 1 до 4 заместителей, независимо выбранных из гидрокси, (C1-C4)алкила, (C1-C4)алкокси, (C1-С10)ациламино, (C1-С10)перфторациламино и (C1-C4)алкоксикарбонила; и
(d) -(CH2)nCOR4, где R4 в группе -(СН2)nCOR4 выбирается из гидрокси, фенила, -NR1R2, (С1-С4)алкила, (C1-C4)перфторалкила, (C1-С4)алкокси, (C1-C4)перфторалкокси, (С3-Сб)циклоалкила и (С3-С6)циклоалкенила,
где n является целым числом от 1 до 4.
3. Соединение по п.2 и его стереоизомеры, фармацевтически приемлемые соли и гидраты, отличающееся тем, что G представляет собой (C1-C12)алкил, причем указанный (C2-C12)алкил необязательно замещен группой, выбранной из фенила, галогена, циано, гидрокси, (С1-С4)алкокси или насыщенное, частично насыщенное или ароматическое гетероциклическое кольцо, выбранное из тиенила, пиразолила, пирролидинила, пирролила, фуранила, тиазолила, изоксазолила, имидазолила, триазолила, тетрагидропиранила, пиридила и пиримидила, причем каждое из указанных гетероциклических колец может необязательно иметь от 1 до 3 заместителей, независимо выбранных из галогена, (C1-C4)ацила, (C1-C4)перфторацила, (C1-C4)алкила, (C1-C4)перфторалкила, (C1-C4)алкокси, (С1-С4)алкиламинокарбонила и (C1-C4)ациламино;
при условии, что (С2-С12)алкил не включает незамещенный аллил.
4. Соединение по п.2 и его стереоизомеры, фармацевтически приемлемые соли и гидраты, отличающееся тем, что G представляет собой -(CH2)nNRlR2, a n является целым числом от 2 до 4.
5. Соединение по п.2 и его стереоизомеры, фармацевтически приемлемые соли и гидраты, отличающееся тем, что G представляет собой -(CH2)nCOR4, a n равно 1 или 2.
6. Соединение по п.2, отличающееся тем, что G представляет собой -(CH2)2ОСОСН3, -(СН2)2OCON(CH3)2,

-(СН2)4СН3, -(СН2)2OСН3, -(СН2)ОСН2СН3, -(СН2)3ОСН3, -(CH2)2CN,

-(СН2)2NНS(O)2СН3, -(СН2)2NHCHO, -(СН2)2NHCOCH2CH3, -(CH2)2NHCOCF3, -(CH2)2NHCONHCH3, -(СН2)2NHCOOCH3, -(СН2)2NHCOCH3, -(CH2)2NH2, -(CH2)2CON(CH3)2, -(CH2)2CON(CH2CH3)2, -(CH2)2CON(CH3)2, -CH2COOH.
7. Способ ингибирования или снижения секреции аполипопротеина В у млекопитающих, нуждающихся в этом, заключающийся во введении ингибирующего или снижающего секрецию Аро В количества соединения по п.1 или его стереоизомера, фармацевтически приемлемой соли или гидрата.
8. Способ лечения состояний, включающих атеросклероз, панкреатит, ожирение, гиперхолестеринемию, гипертриглицеридемию, гиперлипидемию или диабет, заключающийся во введении млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по п.1 или его стереоизомера, фармацевтически приемлемой сюыш или гидрата.
9. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по п.1 или его стереоизомера, фармацевтически приемлемой соли или гидрата в сочетании с фармацевтически приемлемым носителем или растворителем (разбавителем).
10. Фармацевтическая композиция для лечения состояний, включающих атеросклероз, панкреатит, ожирение, гиперхолестеринемию, гипертриглицеридемию, гиперлипидемию или диабет, у млекопитающего, которая включает терапевтически эффективное количество соединения по п.1 или его стереоизомера, фармацевтически приемлемой соли или гидрата в сочетании с фармацевтически приемлемым носителем или растворителем.
11. Фармацевтическая композиция, содержащая
a) терапевтически эффективное количество первого соединения, которое является соединением по п.1 или его стереоизомером, фармацевтически приемлемой солью или гидратом;
b) терапевтически эффективное количество второго соединения, которое выбирается из ингибитора абсорбции холестерина, ингибитора белка переноса эфиров холестерина, ингибитора гидроксиметилглютарил-коэнзим А-редуктазы, ингибитора гидроксиметилглютарил-коэнзим А-синтазы, ингибитора экспрессии гена гидроксиметилглютарил-коэнзим А-редуктазы, ниацина, антиоксиданта, ингибитора ацил-коэнзим А-холестеринацилтрансферазы или ингибитора скваленсинтетазы; и
с) фармацевтически приемлемый носитель или растворитель.
12. Фармацевтическая композиция по п.10, отличающаяся тем, что второе соединение выбирается из ловастатина, симвастатина, правастатина, флувастатина, аторвастатина или ривастатина.
13. Способ лечения состояний, включающих атеросклероз, панкреатит, ожирение, гиперхолестеринемию, гипертриглицеридемию, гиперлипидемию или диабет, заключающийся во введении млекопитающему, нуждающемуся в таком лечении:
a) терапевтически эффективного количества первого соединения, которое является соединением по п.1 или его стереоизомером, фармацевтически приемлемой солью или гидратом;
b) терапевтически эффективного количества второго соединения, которое выбирается из ингибитора абсорбции холестерина, ингибитора белка переноса эфиров холестерина, ингибитора гидроксиметилглютарил-коэнзим А-редуктазы, ингибитора гидроксиметилглютарил-коэнзим А-синтазы, ингибитора экспрессии гена гидроксиметилглютарил-коэнзим А-редуктазы, ниацина, антиоксиданта, ингибитора ацил-коэнзим А-холестеринацилтрансферазы или ингибитора скваленсинтетазы.
14. Способ по п.11, отличающийся тем, что второе соединение выбирается из ловастатина, симвастатина, правастатина, флувастатина, аторвастатина или ривастатина.
15. Соединение по п.1, отличающееся тем, что соединение выбирается из
{6-[(4'-трифторметилбифенил-2-карбонил)амино]-3,4-дигидро-1Н-изохинолин-2-ил}уксусной кислоты;
4'-трифторметилбифенил-2-карбоновой кислоты (n-пентил-1,2,3,4-тетрагидроизохинолин-6-ил)амида;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(3-метоксипропил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(2-метоксиэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(2-этоксиэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(2-цианоэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
уксусной кислоты 2-{6-[(4'-трифторметилбифенил-2-карбонил)амино]-3,4-дигидро-1Н-изохинолин-2-ил}-этиловый эфир и
диметилкарбаминовой кислоты 2-{6-[(4'-трифторметилбифенил-2-карбонил)амино]-3,4-дигидро-1Н-изохинолин-2-ил}-этиловый эфир.
16. Соединение по п.1, отличающееся тем, что соединение выбирается из
4'-трифторметилбифенил-2-карбоновой кислоты [2-(2-аминоэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(2-ацетилцианоэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(2-диметилкарбамоилэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
4'-трифторметилбифенил-2-карбоновой кислоты (2-диметилкарбамоилметил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
4'-трифторметилбифенил-2-карбоновой кислоты (2-диэтилкарбамоилметил)-1,2,3,4-тетрагидроизохинолин-6-ил)амида;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(2-метансульфониламиноэтил)-1,2, 3, 4-тетрагидроизохинолин-6-ил]амида;
4'-трифторметилбифенил-2-карболовой кислоты {2-[2-(2,2,2-трифторацетиламино)-этил]-1,2,3,4-тетрагидроизохинолин-6-ил}амида;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(2-пропиониламиноэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
{2-[6-(4'-трифторметилбифенил-2-карбонил)амино]-3,4-дигидро-1Н-изохинолин-2-ил}-этил карбаминовой кислоты метиловый эфир;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(2-фopмилaминoэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида и
4'-трифторметилбифенил-2-карбоновой кислоты {2-[2-(3-метилуреидо)-этил]-1,2,3,4-тетрагидроизохинолин-6-ил}амида;
17. Соединение по п.1, отличающееся тем, что соединение выбирается из
4'-трифторметилбифенил-2-карбоновой кислоты { 2-[2-(1-метил-1Н-пиррол-2-ил)этил]-1,2,3,4-тетрагидроизохинолин-6-ил}-амида;
4'-трифторметилбифенил-2-карбоновой кислоты {2-[2-(2Н-[1,2,4]триазол-3-ил)этил]-1,2,3,4-тетрагидроизохинолин-6-ил}амида;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(2,2-дифенилэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(2-пиридин-2-ил-этил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
4'-трифторметилбифенил-2-карбоновой кислоты (2-фенилэтил-1,2,3,4-тетрагидроизохинолин-6-ил)амида;
4'-трифторметилбифенил-2-карбоновой кислоты [ 2-пиперидин-4-ил-1,2,3,4-тетрагидроизохинолин-6-ил]амида;
4'-трифторметилбифенил-2-карбоновой кислоты [2-(1-трифторметилацетил-пиперидин-4-ил)-1,2,3,4-тетрагидроизохинолин-6-ил]амида.
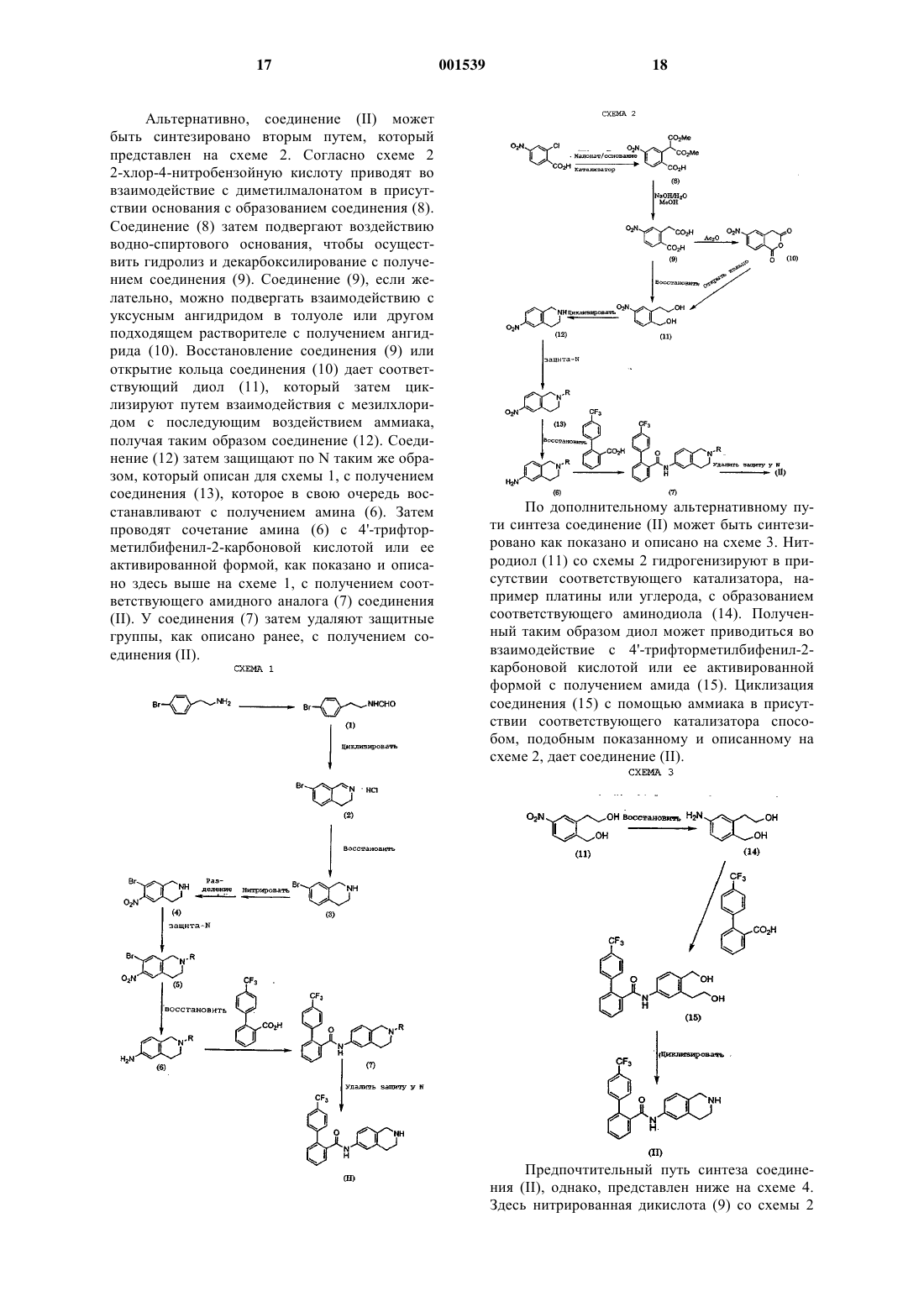
18. Способ получения соединения формулы

который включает стадии
а) циклизации дикислоты структурной формулы

или ее активированной формы с бензиламином с получением дионового производного структурной формулы

b) восстановления продукта со стадии (а) с получением изохинолинового производного структурной формулы

с) восстановление продукта со стадии (b) с получением аминопроизводного структурной формулы

d) сочетания продукта со стадии (с) с соединением 4'-трифторметилбифенил-2-карбоновой кислотой или ее активированной формой с получением амидного производного структурной формулы

е) удаление защиты амидного производного со стадии (d) с получением указанного аминопроизводного структурной формулы

f) выделение аминопроизводного со стадии (е) в виде свободного основания или его соли присоединения кислоты.
19. Способ получения соединения формулы

который включает стадии
а) удаление защиты амидного производного структурной формулы

с получением аминопроизводного формулы

b) выделения аминопроизводного со стадии (а) в виде свободного основания или его соли присоединения кислоты.
20. Тозилатная соль присоединения кислоты соединения

21. Соединение, имеющее структурную формулу

или его соль присоединения кислоты, где R выбирается из NO2 и NН2.
Текст
1 Настоящее изобретение относится к соединениям, которые являются ингибиторами микросомного протеина переноса триглицеридов (МТР) и/или аполипопротеина В (Аро В) и которые, соответственно, пригодны для профилактики и лечения атеросклероза и его клинических осложнений, для снижения уровня сывороточных липидов и профилактики и лечения связанных с этим заболеваний. Изобретение также относится к фармацевтическим композициям,содержащим эти соединения, и к способам лечения атеросклероза, ожирения и связанных с этим заболеваний и/или патологических состояний с помощью указанных соединений, или самих по себе, или в комбинации с другими лекарственными веществами, включая средства,снижающие уровень липидов. Кроме того, изобретение относится к некоторым способам получения соединений согласно изобретению. Микросомальный белок переноса триглицеридов катализирует транспорт триглицерида,холестерильного сложного эфира и фосфолипидов, и вовлекается в качестве посредника в образовании содержащих Аро В липопротеинов,биомолекул, которые способствуют образованию атеросклеротических повреждений. В частности, субклеточное (полости микросомной частицы) и тканевое распределение (печень и кишечник) МТР привело к предположению, что он играет какую-то роль в агрегате липопротеинов плазмы, так как они являются местами образований липопротеинов плазмы. Способность МТР катализировать транспорт триглицеридов между мембранами согласуется с этим предположением и наводит на мысль о том, что МТР может катализировать транспорт триглицеридов из места их синтеза в мембране эндоплазматического ретикулума в образующиеся липопротеиновые частицы в просветах эндоплазматического ретикулума. Соединения, которые ингибируют МТР и/или секрецию Аро В, соответственно, применимы при лечении атеросклероза и других патологических состояний, связанных с ними. Такие соединения также применимы при лечении других заболеваний или состояний, при которых, путем ингибирования секреции МТР и/или Аро В могут быть снижены уровни холестерина и триглицеридов в сыворотке. Такие патологические состояния могут включать, например,гиперхолестеринемию, гипертриглицеридемию,панкреатит и ожирение; и гиперхолестеринемию, гипертриглицеридемию и гиперлипидемию, связанные с панкреатитом, ожирением и диабетом. Для детального обсуждения, см. например, Wetterau et al., Science, 258, 999-1001,(1992), Wetterau et al., Biochem. Biophys. Acta.,875, 610-617 (1986), 0584446 A2 и European patent application publication0643057 Al, в последней из которых раскрываются некоторые соединения общих формул которые применяются в качестве ингибиторов МТР. Данное изобретение относится к соединениям, которые являются ингибиторами, представленными структурной формулой (I), включая стереоизомеры и их фармацевтически приемлемлые соли и гидраты(a) фенила или гетероциклического кольца,где указанное гетероциклическое кольцо содержит в общей сложности от 3 до 14 атомов, причем указанное гетероциклическое кольцо включает в общей сложности от 1 до 4 гетероатомов в кольце, выбираемых независимо из кислорода,азота и серы, причем отдельное кольцо из указанных гетероциклических колец может быть независимо насыщенным, частично насыщенным или ароматическим, где каждое из указанных фенильных или гетероциклических колец может необязательно иметь от 1 до 4 заместителей, выбранных независимо из галогена, гидрокси, циано, нитро, оксо, тиоксо, аминосульфонила, фенила, фенокси, фенилтио, бензила, бензоила, бензилокси, (C1-C10)алкила, (C1-C4) перфторалкила, (C1-C10)алкокси, (С 1-С 4) перфторалкокси, (C1-С 10)алкоксикарбонила, (C1-C10) алкилтио, (C1-C10)алкиламино, ди(С 1-С 10) алкиламино, (С 1-С 10)алкиламинокарбонила, ди(С 1 С 10) алкиламинокарбонила, ди(С 1-С 10) алкиламино (С 1-С 10)алкокси, (С 1-С 10)ацила, (C1-C10) перфторацила, (С 1-С 10)ацилокси, (С 1-С 10) ацилокси (С 1-C10)алкила, (С 1-C6)ациламино и (С 1 С 6)перфторациламино;(d) (C2-C12)алкила или (С 2-С 12)перфторалкила, где каждый из указанных (C2-C12)алкил и (C2-C12)перфторалкил необязательно замещен 1-3 заместителями, независимо выбранными из(1) фенила, галогена, нитро, циано, гидрокси, -NR1R2, -OCOR3, (С 1-C4)алкокси, (С 1-C4) перфторалкокси, (С 1-C4)тиоалкокси или (С 1-С 4) перфтортиоалкокси, 3 где R1 и R2 в группе -N-R1R2, каждый, независимо выбираются из водорода, формила,фенила, бензила, бензоила, (С 3-С 8)циклоалкила,(С 3-C8)циклоалкенила, (С 1-C4)алкила, (C1-С 4) перфторалкила, (C1-С 10)алкоксикарбонила, (C1C6)ацила, (C1-С 6)перфторацила, аминокарбонила, (C1-С 10)алкиламинокарбонила, ди(С 1-С 10) алкиламинокарбонила, аминосульфонила, (C1 С 4)алкиламиносульфонила, ди (C1-C4) алкиламиносульфонила, (C1-С 4)перфторалкиламиносульфонила, (C1-C4)алкилсульфонила, ди(С 1-С 4) алкилсульфонила и (С 1-С 4)перфторалкилсульфонила,или где R1 и R2, взятые вместе с атомом азота, с которым они связаны, образуют насыщенное, частично насыщенное или ароматическое гетероциклическое кольцо, причем указанное гетероциклическое кольцо содержит в общей сложности от 3 до 14 атомов в кольце и необязательно включает в кольцо дополнительно от 1 до 4 гетероатомов, выбранных из кислорода, азота и серы, причем указанное гетероциклическое кольцо может необязательно иметь от 1 до 4 заместителей, независимо выбранных из галогена, гидрокси, циано, нитро, оксо, тиоксо, аминосульфонила, фенила, фенокси, фенилтио, бензила, бензоила, бензилокси, (C1-С 10) алкила, (C1-С 4)перфторалкила, (C1-С 10)алкокси,(C1-C4)перфторалкокси, (C1-С 10)алкоксикарбонила, (C1-С 10)алкилтио, (C1-С 10)алкиламино,ди(С 1-С 10)алкиламино, (C1-С 10)алкиламинокарбонила, ди(С 1-С 10)алкиламинокарбонила, ди(C1 С 10)алкиламино (C1-С 10)алкокси, (С 1-С 10)ацила,(С 1-С 10)перфторацила, (C1-С 10)ациламино, (C1 С 10)ацилокси и (C1-С 10)ацилокси (C1-С 10)алкила,где R3 выбирается из -NR1R2, фенила, (C1 С 10)алкила, (С 1-С 4)перфторалкила, (C1-C6)алкокси и (C1-C6) перфторалкокси,(2) (С 3-С 8)циклоалкила или (С 3-С 8)циклоалкенила, где каждый из указанных (С 3 С 8)циклоалкила и (С 3-С 8)циклоалкенила может необязательно иметь от 1 до 4 заместителей,выбранных независимо из галогена, гидрокси,циано, нитро, оксо, тиоксо, аминосульфонила,фенила, фенокси, фенилтио, бензила, бензоила,бензилокси, (C1-С 10)алкила, (C1-C4)перфторалкила, (C1-С 10)алкокси, (C1-С 4)перфторалкокси,(C1-С 10)алкоксикарбонила,(C1-С 10)алкилтио,(C1-С 10)алкиламино, ди(C1-С 10)алкиламино, (C1 С 10)алкиламинокарбонила, ди (C1-С 10)алкиламинокарбонила, ди (C1-С 10)алкиламино(C1-С 10) алкокси, (C1-С 10)ацила, (C1-С 10)перфторацила,(C1-С 10)ациламино, (C1-С 10)перфторациламино,(C1-С 10)ацилокси и (C1-С 10)ацилокси (С 1-С 10) алкила и(3) насыщенного, частично насыщенного или ароматически гетероциклического кольца,содержащего в общей сложности от 3 до 14 атомов в кольце, причем указанное гетероциклическое кольцо включает в общей сложности от 1 до 4 гетероатомов в кольце, независимо выбранных из кислорода, азота и серы, причем 001539 4 указанное гетероциклическое кольцо может необязательно иметь от 1 до 4 заместителей,независимо выбранных из галогена, гидрокси,циано, нитро, оксо, тиоксо, аминосульфонила,фенила, фенокси, фенилтио, бензила, бензоила,бензилокси, (C1-С 10)алкила, (C1-С 4)перфторалкила, (C1-С 10)алкокси, (C1-C4)перфторалкокси,(C1-С 10)алкоксикарбонила,(C1-С 10)алкилтио,(C1-С 10)алкиламино, ди(С 1-С 10)алкиламино, (C1 С 10)алкиламинокарбонила, ди(С 1-С 10)алкиламинокарбонила, ди(C1-С 10)алкиламино(C1-С 10) алкокси, (C1-С 10)ацила, (C1-С 10)перфторацила, (C1 С 10)ациламино, (C1-С 10)перфторациламино, (C1 С 10)ацилокси и (C1-С 10)ацилокси (C1-С 10)алкила,при условии, что (C1-C12) алкил не включает незамещенный алкил;(e) (С 3-С 8) циклоалкила или (С 3 С 8)циклоалкенила, причем каждый из указанных (С 3-C8) циклоалкила и (С 3-C8) циклоалкенила может необязательно иметь от 1 до 4 заместителей, независимо выбранных из галогена,гидрокси, циано, нитро, оксо, тиоксо, аминосульфонила, фенила, фенокси, фенилтио, бензила, бензоила, бензилокси, (C1-С 10) алкила, (С 1 С 4)перфторалкила, (C1-С 10) алкокси, (C1-С 4) перфторалкокси, (C1-С 10)алкоксикарбонила, (C1 С 10)алкилтио, (C1-С 10)алкиламино, ди(C1-С 10) алкиламино, (C1-С 10)алкиламинокарбонила, ди(f) -(CH2)nCOR4, где R4 выбирается из гидрокси, фенила, -NR1R2, (C1-C4)алкила, (C1-C4) перфторалкила,(C1-C4) алкокси,(C1 С 4)перфторалкокси, (С 3-С 8)циклоалкила и (С 3 С 8)циклоалкенила,где n является целым числом от 1 до 4. Предпочтительная подгруппа соединений формулы (1) и их стереоизомеров, фармацевтически приемлемых солей и гидратов включает соединения, в которых G выбран из(a) фенила или гетероциклического кольца,причем указанное гетероциклическое кольцо содержит в общей сложности от 3 до 7 атомов в кольце, причем указанное гетероциклическое кольцо включает в общей сложности от 1 до 4 гетероатомов в кольце, независимо выбранных из кислорода, азота и серы, причем указанное гетероциклическое кольцо может быть насыщено, частично насыщенным или ароматическим,и причем каждое из указанных фенильного и гетероциклического колец может необязательно иметь от 1 до 4 заместителей, независимо выбранных из галогена, гидрокси, фенила, бензила, бензоила, бензилокси, (C1-С 10)алкила, (C1C4) перфторалкила, (C1-С 10)алкокси, (C1-C4) перфторалкокси, (C1-С 10)алкоксикарбонила, (C1 С 10)алкилтио, (C1-С 10)алкиламино, ди(С 1-С 10) алкиламино, (C1-С 10)алкиламинокарбонила, ди(b) (C2-C12)алкила, где указанный (C2-C12) алкил необязательно замещен 1-3 заместителями, выбранными из(1) фенила, галогена, циано, гидрокси,-NR1R2, -OCOR3, (C1-C4)алкокси или (C1-C4) перфторалкокси,где R3 выбирается из -NR1R2, (C1-C4) алкила и (C1-С 4)перфторалкила,(2) (С 3-С 6)циклоалкила или (С 3-Сб) циклоалкенила, причем каждый из указанных (С 3-С 6) циклоалкила и (С 3-С 6)циклоалкенила может необязательно иметь от 1 до 4 заместителей,независимо выбранных из гидрокси, (C1-C4) алкила, (C1-C4) алкокси и (C1-С 4) алкоксикарбонила и(3) насыщенного, частично насыщенного или ароматического гетероциклического кольца, содержащего в общей сложности от 3 до 6 атомов в кольце, причем указанное гетероциклическое кольцо включает в общей сложности от 1 до 4 гетероатомов в кольце, выбранных независимо из кислорода, азота и серы, причем указанное гетероциклическое кольцо может необязательно иметь от 1 до 4 заместителей,независимо выбранных из галогена, гидрокси,фенила, бензила, бензоила, бензилокси, (C1-С 10) алкила, (C1-С 4) перфторалкила, (C1-С 10) алкокси,(C1-С 10) алкоксикарбонила, (C1-С 10) алкилтио,(C1-С 10) алкиламино, ди(С 1-С 10) алкиламино,(C1-С 10) алкиламинокарбонила, ди(С 1-С 10) алкиламинокарбонила, ди(С 1-С 10) алкиламино (C1 С 10)алкокси, (C1-C4)перфторалкокси, (C1-С 10) ацила, (C1-C10)ациламино, (C1-C4) перфторациламино, (C1-С 10)ацилокси и (C1-С 10)ацилокси(C1-С 10) алкила; при условии, что (C2-C12)алкил не включает незамещенный аллил,(c) (С 3-С 6)циклоалкила или (С 3-С 6) циклоалкенила, причем каждый из указанных (С 3-С 6) циклоалкила или (С 3-Сб) циклоалкенила может необязательно иметь от 1 до 4 заместителей,независимо выбранных из гидрокси, (C1-C4) алкила, (C1-C4) алкокси, (C1-С 10)ациламино, (C1 С 10) перфторациламино и (C1-C4) алкоксикарбонила; и(d) -(СН 2)nCOR4, где R4 выбирается из гидрокси, фенила, -NR1R2, (C1-C4)алкила, (С 1-С 4) перфторалкила, (C1-C4)алкокси, (С 1-С 4) перфторалкокси, (С 3-С 6) циклоалкила и (С 3-С 6) циклоалкенила,где n является целым числом от 1 до 4. Еще более предпочтительными из соединений формулы (I), включая их стереоизомеры,фармацевтически приемлемые соли и гидраты,являются те соединения подгруппы, в которых 6 бранной из фенила, галогена, циано, гидрокси,(C1-C4)алкокси или насыщенного, частично насыщенного или ароматического гетероциклического кольца, выбираемого из тиенила, пиразолила, пирролидинила, пирролила, фуранила,тиазолила, изоксазолила, имидазолила, тиазолила, тетрагидропиранила, пиридила и пиримидила, причем каждое из указанных гетероциклических колец может необязательно иметь от 1 до 3 заместителей, независимо выбранных из галогена, (C1-C4)ацила, (С 1-С 4)перфторацила, (C1-C4) алкила, (C1-C4)перфторалкила, (C1-C4)алкокси,(C1-C4)алкиламинокарбонила и(C1C4)ациламино; при условии, что (C2-C12)алкил не включает незамещенный аллил;(b) -(CH2)nNR1R2, где n является целым числом от 2 до 4; и(c) -(CH2)nCOR4, где n равно 1 или 2. Следующие соединения формулы (I),включая их стереоизомеры и фармацевтически приемлемые соли и гидраты, перечисленные ниже вместе с их соответствующими химическими названиями по номенклатуре МСТПХ 4'-трифторметилбифенил-2-карбоновой кислоты [2-(1-трифторметилацетилпиперидин 4-ил-этил)-1,2,3,4-тетрагидроизохинолин-6-ил]амид. Следующие определения выбранных функциональных групп использованы в данном описании и прилагаемой формуле изобретения и предлагаются в качестве иллюстрации, но не ограничивают объем изобретения. Термин ацил относится к углеродному заместителю с прямой или разветвленной цепью, связанному с карбонильной группой. Примерами таких радикалов являются ацетил, пропионил, бутирил и изобутирил и тому подобное. Термин алкил включает углеводородные радикалы с прямой и разветвленной цепью,имеющие необязательную ненасыщенность в виде двойной или тройной связи между атомами углерода. Представителями таких радикалов являются метил, этил, пропил, пропилен, пропинил, изопропил, изопропилен, бутил, изобутил, изобутилен, трет-бутил, пентил, гексил и так далее. Термин алкокси включает углеводородный радикал с прямой или разветвленной цепью, связанный с атомом кислорода. Иллюстрациями таких радикалов являются метокси, этокси, пропокоси, изопропокси, бутокси, изобутокси, пентокси, гексокси, гептокси и тому подобное. Ссылка на термин галоген включает фтор, хлор, бром и йод, если не указано иначе. Термин перфтор, когда он используется в соединении с конкретным углеводородным радикалом, означает включение заместителя, в котором его отдельные углеродные атомы могут быть замещенными для этого одним или более,а предпочтительно от 1 до 9, атомами фтора. Примерами таких радикалов являются трифторметил, пентафторэтил, гептафторпропил и тому подобное. Иллюстративные значения для термина 9 Иллюстративные значения для термина(C1-С 10)алкилтио включает соответствующие серосодержащие производные от термина алкокси, которому дано определение здесь выше,включающие метилтио, этилтио, пропилтио,изопропилтио, бутилтио, изобутилтио, пентилтио, гексилтио, гептилтио и тому подобное. Иллюстративные значения для термина(C1-С 10)алкиламино включают метиламино,этиламино, пропиламино, изопропиламино, бутиламино, изобутиламино и так далее. Иллюстративные значения для термина ди(C1-С 10)алкиламино включают диметиламино, диэтиламино, дипропиламино, диизопропиламино и тому подобное, а также N-метил-N'этиламино, N-этил-N'-пропиламино, N-пропилN'-изопропиламино и тому подобное. Иллюстративные значения для термина(C1-С 10)ацилокси включают ацетилокси, пропионилокси, бутирилокси и тому подобное, а также включают такие радикалы, которые включают циклический заместитель, такой как бензоилокси. Иллюстративные значения для термина(С 3-C8)циклоалкил включают циклопропил,циклобутил, циклопентил, циклогексил и тому подобное. Иллюстративные значения для термина(C1-С 10)циклоалкенил включают циклопропил, циклобутенил, циклогексенил, циклогептенил и тому подобное. Термин гетероциклическое кольцо, который используется в данном описании и прилагаемой формуле изобретения, обозначает гетероциклические радикалы, которые могут быть по природе моноциклическими и полициклическими. Примерами систем моноциклических гетероциклических колец являются такие радикалы, как фуранил, тиенил, пиразолил, пирролидинил, пирролил, тиазолил, изоксазолил,имидазолил, триазолил, тетрагидропиранил,пиридил, пиримидил и так далее. Примерами систем полициклических гетероциклических колец являются такие радикалы, как индолил,бензофуранил, бензимидазолил, хинолинил,акридинил, фталазинил и тому подобное. Кроме того, если карбоциклическое или гетероциклическое кольцо может быть связано или соединено с определенным субстратом,включая соединение (I), через различные атомы кольца без указания конкретной точки присоединения, то предполагаются все возможные точки, или через атом углерода, или трехвалентный атом азота. Например, термин пиридил означает 2-, 3- или 4-пиридил, термин тиенил означает 2- или 3-тиенил и так далее. Термин лечение, как он используется здесь, обозначает как профилактическое, облегчающее, а также устраняющее заболевание лечение. Данное изобретение заявляет также фармацевтические композиции для лечения патоло 001539 10 гических состояний, включающих атеросклероз,панкреатит, ожирение, гиперхолестеринемию,гипертриглицеридемию и диабет, которые содержат терапевтически эффективное количество соединения формулы (I), представленной и получившей определение выше, включая его стереоизомеры, фармацевтически приемлемые соли и гидраты, в сочетании с фармацевтически приемлемым носителем или растворителем. Соединение согласно изобретению ингибируют или снижают секрецию Аро В, вероятно путем подавления МТР, хотя возможно в это вовлечены еще и другие механизмы. Эти соединения применимы при лечении любого из болезненных состояний, при которых повышены уровни Аро В, холестерина и/или триглицеридов в сыворотке. Соответственно, данное изобретение представляет способ подавления или снижения секреции Аро В у млекопитающих,нуждающихся в этом, который включает введение ингибирующего или снижающего секрецию Аро В количества соединения формулы (I) или его стереоизомера, фармацевтически приемлемой соли или гидрата. Изобретение, также относится к способу лечения состояний, включающих атеросклероз, панкреатит, ожирение,гиперхолестеринемию, гипертриглицеридемию,гиперлипидемию и диабет, заключающемуся в том, что млекопитающему, особенно человеку,нуждающемуся в таком лечении, вводят терапевтически эффективное количество соединения (I) или его стереоизомера, фармацевтически приемлемой соли или гидрата. Предпочтительной подгруппой состояний, описанных здесь выше, предпредставляет атеросклероз, ожирение,гиперхолестеринемия, гипертриглицеридемия, гиперлипидемия и диабет. Соединения согласно изобретению могут использоваться в сочетании с другими фармацевтическими средствами, включающими другие снижающие уровень липидов вещества. Такие средства включают, например, ингибиторы биосинтеза холестерина и ингибиторы абсорбции холестерина, особенно ингибиторы ГМГКоА-редуктазы (HMG-CoA) и ингибиторы ГМГ-КоА-синтетазы; ингибиторы экпрессии гена ГМГ-КоА-редуктазы; ингибиторы СЕТР,вещества, усиливающие секрецию желчных кислот; фибраты ингибиторы абсорбции холестерина; ингибиторы АСАТ, ингибиторы скваленсинтетазы, ионообменные смолы, антиоксиданты и ниацин. При лечении комбинированными терапевтическими средствами соединения данного изобретения и другие лекарственные терапевтические средства можно вводить млекопитающим (например, людям) обычными способами. Конкретно ингибиторы абсорбции холестерина и ингибиторы биосинтеза холестерина подробно описаны ниже. Дополнительные ингибиторы абсорбции холестерина известны спе 11 циалистам и описаны, например, в РСТ WO 94/00480. Согласно изобретению по комбинированной терапии может использоваться любой ингибитор ГМГ-КоА-редуктазы. Термин ингибитор ГМГ-КоА-редуктазы относится к соединению,которое ингибирует биотрансформацию гидроксиметилглютарил-коэнзима А в мевалоновую кислоту, которое катализируется ферментом ГМГ-КоА-редуктазой. Такое ингибирование может быть легко определено специалистом стандартными методами (например, Methods ofEnzymology, 1981; 71:455-509 и источники, процитированные здесь). Ряд этих соединений описан и приведен в виде ссылок ниже. В патенте США 4231938 (описание которого включено сюда в виде ссылки) раскрыты некоторые соединения, выделенные после культивирования микроорганизма, принадлежащего к роду Aspergillus, такие как ловастатин. В патенте США 4444784 (описание которого включено сюда в виде ссылки) также раскрыты синтетические производные вышеупомянутых соединений,такие как симвастатин. Кроме того, в патенте США 4739073 (описание которого включено сюда в виде ссылки) раскрываются некоторые замещенные индолы, такие как флувастатин. Затем в патенте США 4346227 (описание которого включено сюда в виде ссылки) раскрываются производные ML-236B, такие как правастатин. Кроме того, в патенте США 4346227 (описание которого включено сюда в виде ссылки) сообщается о некоторых пиридилдигидроксигептеновых кислотах, таких как ривастатин. В патенте США.4647576 (описание которого включено сюда в виде ссылки) также раскрываются некоторые 6-[2-(замещенныйпиррол-1-ил)-алкил]-пиран-2-оны, такие как аторвастатин. Другие ингибиторы ГМГ-КоАредуктазы будут известны специалистам. В качестве второго соединения в комбинированной терапии согласно изобретению может использоваться любой ингибитор ГМГ-КоАсинтазы. Термин ингибитор ГМГ-КоА-синтезы относится к соединению, которое ингибирует биосинтез гидроксиметилглутарил-коэнзима-А из ацетил-коэнзима А и ацетоацетил-коэнзима А, катализируемый ферментом ГМГ-КоАсинтазой. Такое подавление может быть легко определено специалистом стандартными методами (например, Methods of Enzymology, 1975,35: 155-160 и Methods of Enzymology, 1985, 110: 19-26 и источники процитированные здесь). Ряд этих соединений описан и приводится здесь ниже. В патенте США 5120729 (описание которого включено сюда в виде ссылки) раскрываются некоторые беталактамные производные. В патенте США 5064856 (описание которого включено сюда в виде ссылки) описаны определенные спиролактоновые производные,полученные путем культивирования микроорганизма MF5253. В патенте США 4847271(описание которого включено сюда в виде ссылки) раскрыты некоторые оксэтановые соединения, такие как производные 11-(3-гидроксиметил-4-оксо-2-оксэтаил)-3,5,7-триметил 2,4-ундекадиеновой кислоты. Опытным специалистам будут известны и другие ингибиторы ГМГ-КоА-синтазы. В качестве второго соединения в аспекте комбинированной терапии этого изобретения может использоваться любое соединение, которое снижает экспрессию гена ГМГ-КоАредуктазы. Эти вещества могут быть ингибиторами транскрипции ГМГ-редуктазы, которые блокируют транскрипцию ДНК, или ингибиторами трансляции, которые препятствуют трансляцию мРНК, кодирующей ГМГ-КоА-редуктазу в белок. Такие ингибиторы могут или непосредственно действовать на транскрипцию или трансляцию, или могут подвергаться биотрансформации в соединения, которые обладают вышеупомянутыми свойствами с помощью одного или более ферментов в каскаде биосинтеза холестерина, или могут приводить к накоплению изопренового метаболита, который обладает вышеупомянутой активностью. Такая регуляция легко определяется специалистами в данной области стандартными методами (Methods ofEnzymology, 1985, 110: 9-19). Некоторые такие соединения описаны и процитированы ниже,однако, специалистам будут известны и другие ингибиторы экспрессии гена ГМГ-КоАредуктазы. В патенте США 5041432 (описание которого включено сюда в виде ссылки) раскрыты некоторые производные 15 замещенного ланостерина. Другие оксигенированные стерины, которые угнетают биосинтез ГМГ-КоА-редуктазы, описаны E.I.Mercer (Prog.Lip.Res., 1993; 32: 357-416). В качестве второго соединения в комбинированной терапии согласно изобретению может быть любое соединение, обладающее активностью ингибитора СЕТР. Термин ингибитор СЕТР относится к соединениям, которые ингибируют опосредуемый белком переноса эфиров холестерина (БПХЭ = СЕТР) транспорт различных сложных эфиров холестерина и триглицеридов от липопротеина высокой плотности(ЛПНП) и липопротеину очень низкой плотности (ЛПОНП). Ряд этих соединений описан и приводится ниже, однако и другие ингибиторы БПХЭ известны специалистам. В патенте США 5512548 (описание которого включено сюда в виде ссылки) раскрываются некоторые полипептидые производные, обладающие активностью как ингибиторы БПХЭ, тогда как некоторые ингибирующие БПХЭ розенонолактоновые производные и фосфатосодержащие аналоги эфира холестерина раскрыты в J.Antibiot., 1996; 49(8): 815-816 и Bioorg. Med. Chem. Lett.; 1996; 6: 1951-1954, соответственно. 13 Вторым соединением в комбинированной терапии согласно изобретению может быть любой ингибитор АСАТ. Термин ингибитор АСАТ относится к соединениям, которые ингибируют внутриклеточную эстерификацию пищевого холестерола с помощью фермента ацил-КоА: холестеринацилтрансферазы(АХАТ). Такое ингибирование может быть легко определено специалистом стандартными методами, такими как метод, описанный Heider etal. In Journal of Lipid Research., 1983; 24: 1127. Ряд этих соединений описан ниже, однако, специалистам известны и другие ингибиторы АХАТ. В патенте США 5510379 (описание которого включено сюда в виде ссылки) раскрываются некоторые карбоксисульфонаты,тогда как в WO 96/26948 и WO 96/10559, в обоих, раскрываются производные мочевины, обладающие ингибирующей АХАТ активностью. Вторым соединением в комбинированной терапии согласно изобретению может служить любое соединение, обладающее активностью ингибитора скваленсинтетазы. Термин ингибитор скваленсинтетазы относится к соединениям, которые ингибируют конденсацию двух молекул фамезилпирофосфата с образованием сквалена, реакцию, которая катализируется ферментом скваленсинтетазой. Такое ингибирование легко определит специалист по стандартной методике (Methods of Enzymology, 1969; 15: 393-454 и Methods of Enzymology, 1985; 110: 359-373 и источники, процитированные здесь). Было осуществлено краткое обобщение по ингибиторам скваленсинтетазы (Curr. Op. Ther.Patents (1993) 861-4). В Публикации европейской патентной заявки 0567026 А 1 раскрываются некоторые производные 4,1-бензоксазепина в качестве ингибиторов скваленсинтетазы и их использование при лечении гиперхолестеринемии и в качестве фунгицидов. В публикации европейской заявки 0645378 А 1 раскрыты некоторые семи- и восьмичленные гетероциклы в качестве ингибиторов скваленсинтетазы и их использование при лечении и профилактике гиперхолестеринемии и грибковых инфекций. В публикации европейской патентной заявки 0645377 А 1 раскрыты некоторые бензоксазепиновые производные в качестве ингибиторов скваленсинтетазы, применимых для лечения гиперхолестеринемии или коронаросклероза. В публикации европейской патентной заявки 0611749 А 1 раскрыты некоторые замещенные производные amiс кислоты, применимые для лечения артериосклероза. В публикации европейской патентной заявки 0705607 А 2 раскрываются конденсированные семи- или восьмичленные гетероциклические соединения,применимые в качестве антигипертриглицеридемических средств. В публикации РСТ WO 96/09827 описаны некоторые комбинации ингибиторов абсорбции холестерина и ингибиторов биосинтеза холестерина, включающие бензокса 001539 14 зепиновые производные и бензотиазепиновые производные. В публикации европейской патентной заявки 0071725 А 1 раскрыт способ получения некоторых оптически активных соединений, включающих бензоксазепиновые производные, обладающие активностью по снижению уровней холестерина и триглицеридов в плазме. Специалистам понятно, что некоторые соединения согласно изобретению могут содержать асимметрически замещенный атом углерода и, соответственно, могут существовать, и/или могут быть выделены, в оптически активной и рацемической формах. Кроме того, некоторые соединения могут проявлять полиморфизм. Соответственно данное изобретение охватывает любые и все рацемические, оптически ативные,полиморфные и стереоизомерные формы или их смеси, и эта форма или формы обладают свойствами, полезными для лечения патологических состояний, указанных выше, причем хорошо известно, как получить оптически активные формы (например, путем разделения рацемической формы с помощью методов перекристаллизации, путем синтеза из оптически активных исходных материалов, путем хирального синтеза или путем хроматографического разделения с использованием хиральной стационарной фазы) и как определить эффективность в отношении лечения . патологических состояний, указанных здесь, путем стандартных испытаний, описанных ниже. Далее были использованы некоторые общие химические и методические аббревиатуры и акронимы, которые включают: Me (метил); Et(1,1'-диметиламинопиридин), Ms (метансульфонат, мезил); ТФУ (трифторуксусная кислота); Ас (ацетил); ОФ (обращенная фаза); ВЭЖХ (высокоэффективная жидкостная хроматография); ТСХ (тонкослойная хроматография). Соединения формулы (1) в большинстве случаев обычно синтезируются с использованием способов, аналогичных способам получения подобных соединений, известным специалистам в области химии. Такие способы получения соединения формулы (1), которые детально охарактеризованы здесь выше, представлены как дополнительные признаки изобретения и иллюстрируются следующими процедурами, при которых значения характерных радикалов являются такими же, как и получившие определение выше, если не указано иначе. Методы включают сочетания соединения формулы (II) 15 которое увеличивает западную часть молекулы, т.е. часть, состоящую из формулы (II) с водородом, удаленным у атома азота тетрагидроизохинолинового кольца, с реагентом, который добавляет восточную, т.е. часть -G. Реагенты,которые дают восточную часть обычно коммерчески доступны и хорошо представлены в химической литературе. Соединение формулы (II) имеет химическое название 4'-трифторметилбифенил-2-карбоновой кислоты (1,2,3,4 тетрагидроизохинолин-6-ил)-амид и здесь называется просто соединение (II) для удобства. Западная часть молекулы, которая представляет соединения формулы (I), обычно известна как часть 6-[(4'-трифторметил) бифенил-2 илкарбониламино]-3,4-дигидро-1 Н-изохинолин 2-ил, хотя менее часто, когда называется в качестве заместителя в соединении, эта кольцевая система может быть названа как 6-замещенная 3,4-дигидро-1 Н-изохинолин-2-ил часть. Дополнительно данное изобретение представляет соль соединения (II) с присоединением кислоты. Ядро из изохинолинового кольца соединения (II) содержит изолированный основной центр и поэтому может образовывать соль с присоединением кислоты с различными органическими и неорганическими сопряженными кислотами. В целях данного изобретения выражение соль с присоединением кислоты предназначено для того, чтобы включать, но не ограничиваться такими солями, как гидрохлоридная,гидробромидная, сульфатная, фосфатная, гидрофосфатная, дигидрофосфатная, ацетатная,сукцинатная, цитратная, метансульфонатная(мезилат) и п-толуолсульфонатная (тозилат) соли, а также любые их гидратированные и сольватированные формы. Соединение формулы (II) может быть синтезировано, как представлено на схемах 1-4 и детальные описания эксперимента каждого из этих методов представлены последовательно в примерах 1-4 ниже. Дополнительный признак данного изобретения представляет собой предпочтительный способ, включающий некоторые промежуточные соединения, относящиеся к этому, для получения соединения (II), и этот способ представлен ниже на схеме 4. Обращаясь к схеме 1, 1,2-(4-бромфенил) этиламина гидробромид приводится в реакцию с этилформиатом в присутствии основания с образованием N-[2-(4-бромфенил)этил] формамида (1). Формамидное производное, полученное таким образом, затем циклизируется в желаемое дигидроизохинолиновое производное (2) путем взаимодействия с пентоксидом фосфора в полифосфорной кислоте. При взаимодействии циклизированного продукта с газообразным галогенводородом (например, с хлористым водородом) образуется галогенводородная соль 7 бром-3,4-дигидроизохинолина. Полученную таким образом галогенводородную соль затем восстанавливают с получением 7-бром-1,2,3,4 001539 16 тетрагидроизохинолина (3). Восстановленный продукт затем нитруют путем взаимодействия с нитратом калия в концентрированной серной кислоте, и зонально изомерночистые нитрированные фракции разделяются с получением 7 бром-6-нитро-1,2,3,4-тетрагидроизохинолина(4). Затем защищают нитросоединение (4) путем функционализации атома азота тетрагидроизохинолинового кольца с помощью соответствующей защищающей группы. Примерами таких защищающих групп для атома азота являются бензильная и БОК-группа, соответственно,однако, могут быть также использованы другие защищающие группы в зависимости от других отдельных функциональных групп, присутствующих в молекуле, и условий предполагаемого препаративного метода. В отношении общего описания защитных групп и их использования смотрите: T.W.Greene, Protective Groups in Organic Synthesis, John WileySons, New York,1991. Полученный тетрагидроизохинолиновый продукт (5) с защищенным N галогенируют в присутствии палладия на карбонате кальция с образованием соответствующего 6 аминопроизводного (6). Полученный таким способом амин подвергают сочетанию с 4'трифторметилбифенил-2-карбоновой кислотой или ее активированной формой с получением соответствующего амидного производного (7). Примеры активированных форм карбоновых кислот, а также 4'-трифторметилбифенил-2 карбоновой кислоты, включают соответствующие галогенангидриды кислот, активированные производные, которые могут быть получены путем реакции свободной кислоты с соответствующим коммерчески доступным карбодиимидом, например, 1-(3-диметиламинопропил)-3 этилкарбодиимида гидрохлоридом (EDC) или 1,1'-карбонилдиимидазолом (CDI), а также другие активированные формы, известные специалисту. Если желательно, EDC может быть преимущественно связанным с полимером, как описано в патенте США 5416193 (описание которого включено сюда в виде ссылки). Кроме того, реакцию с активированной карбоновой кислотой обычно проводят в присутствии соответствующего основания, например амина, который может быть связан с полимером, таким как связанный с полимером морфолинополистирол. Вышеприведенный способ активации карбоновых кислот для сочетания с соответствующими субстратами не должен рассматриваться как ограниченный случаем 4'-трифторметилбифенил-2-карбоновой кислоты, но может быть применен также для любого остатка карбоновой кислоты, описанного в данной спецификации и прилагаемой формуле изобретения. Обычное удаление защиты у атома азота функционализированного тетрагидроизохиноли-нового кольца (7) затем дает соединение (II),(1,2,3,4-тетрагидроизохинолин-6-ил)-амид 4'трифторметилбифенил-2-карбоновой кислоты. 17 Альтернативно, соединение (II) может быть синтезировано вторым путем, который представлен на схеме 2. Согласно схеме 2 2-хлор-4-нитробензойную кислоту приводят во взаимодействие с диметилмалонатом в присутствии основания с образованием соединения (8). Соединение (8) затем подвергают воздействию водно-спиртового основания, чтобы осуществить гидролиз и декарбоксилирование с получением соединения (9). Соединение (9), если желательно, можно подвергать взаимодействию с уксусным ангидридом в толуоле или другом подходящем растворителе с получением ангидрида (10). Восстановление соединения (9) или открытие кольца соединения (10) дает соответствующий диол (11), который затем циклизируют путем взаимодействия с мезилхлоридом с последующим воздействием аммиака,получая таким образом соединение (12). Соединение (12) затем защищают по N таким же образом, который описан для схемы 1, с получением соединения (13), которое в свою очередь восстанавливают с получением амина (6). Затем проводят сочетание амина (6) с 4'-трифторметилбифенил-2-карбоновой кислотой или ее активированной формой, как показано и описано здесь выше на схеме 1, с получением соответствующего амидного аналога (7) соединения(II). У соединения (7) затем удаляют защитные группы, как описано ранее, с получением соединения (II). По дополнительному альтернативному пути синтеза соединение (II) может быть синтезировано как показано и описано на схеме 3. Нитродиол (11) со схемы 2 гидрогенизируют в присутствии соответствующего катализатора, например платины или углерода, с образованием соответствующего аминодиола (14). Полученный таким образом диол может приводиться во взаимодействие с 4'-трифторметилбифенил-2 карбоновой кислотой или ее активированной формой с получением амида (15). Циклизация соединения (15) с помощью аммиака в присутствии соответствующего катализатора способом, подобным показанному и описанному на схеме 2, дает соединение (II). Предпочтительный путь синтеза соединения (II), однако, представлен ниже на схеме 4. Здесь нитрированная дикислота (9) со схемы 2 19 реагирует с бензиламином в кислой среде с получением циклизированного изохинолинового диона (16). Этот тип реакции циклоконденсации обычно осуществляется в протонном растворителе с высокой температурой кипения, таком как уксусная кислота, при температуре, достаточной, чтобы вызвать закрытие кольца, обычно при температуре кипения растворителя. При практическом осуществлении данного способа,предпочтительно, раствор бензиламина и соединения (9) в уксусной кислоте нагревают при температуре кипения в течение срока в 18 ч и образовавшийся изохинолиндион (16) выделяют путем обычной обработки. Альтернативно дикислоту (9) можно превращать в активированную форму, как описано здесь выше, и затем подвергать конденсации с бензиламином. Двухстадийное восстановление дает защищенное у N 6-аминоизохинолиновое производное (18), которое затем функционализируют с помощью 4'трифторметилбифенил-2-карбоновой кислоты или ее активированной формы с получением бензилированного амида (19). На начальной стадии процесса восстановления дион (16) может быть восстановлен до соединения (17) с помощью ряда восстанавливающих веществ,включая диборановые и борогидридные комплексы, такие как диборан/ТГФ, диборан/ДМС,натрия борогидрид/трифторидэтерат бора и тому подобное, в системах апротонных растворителей, таких как ТГФ, алкиловые эфиры, толуол и так далее, при температуре в интервале от примерно 0 С до температуры кипения выбранного растворителя. При предпочтительном способе данного процесса соединение (16) восстанавливают до N-защищенного 6-нитроизохинолинового производного (17) с использованием борогидрида натрия/трифторидэтерата бора в ТГФ при температуре от 0 С до температуры кипения в течение периода, равного примерно 16 ч. На второй стадии восстановления данного процесса соединение (17) восстанавливают до амина (18). Опытный специалист легко представит ряд доступных методов восстановления ароматической нитрогруппы соединения (17),включая метод с Zn/водным раствором НСl,Fe/уксусной кислотой/водой, и разнообразные каталитические методы, включая гидрогенизацию в присутствии палладия, платины, оксида платины, рения и тому подобного, в протонном или апротонном растворителе при давлении водорода в интервале от 6,89103 н/м 2 (1 При практическом осуществлении данного процесса предпочтительно, чтобы восстановление соединения (17) осуществлялось с использованием каталитической гидрогенизации в присутствии оксида платины в ТГФ при давлении водорода в 3,41 атм. (3,44105 н/м 2; 50 фунт/ дюйм 2). Соединение (18) затем подвергают сочетанию с 4'-трифторметилбифенил-2-карбо 001539 20 новой кислотой, как описано на схеме 1 с получением амида (19). Ряд препаративных методов удаления защитных групп у соединения (19) должен быть хорошо известен опытным специалистам. При практическом воплощении данного процесса у соединения (19) удаляют защиту, предпочтительно используя гидроксид палладия на угле в присутствии формиата аммония. Стадия удаления защиты осуществляется при температуре, равной 60 С в течение срока, равного примерно 3 ч, в системе растворителей метанол/ТГФ. Процессы сочетания, выполняемые при синтезе соединений формулы (I) начинаются с соединения (II) и в основном включают общепринятые методы или их незначительные модификации. Обращаясь конкретно к соединениям данного изобретения, использовали в общей сложности десять препаративных путей получения соединений со структурой (I), и каждый из них описан в общих чертах в разделе МетодыA-J ниже. Должно быть понятно, однако, что различные препаративные методы, раскрытые в данном описании были выбраны главным образом на основании удобства, а не в порядке ограничения. Опытный специалист поймет, что возможны многие концептуально осуществимые пути синтеза соединений со структурой (I), влючая различные комбинации способов A-J, и приведенные в качестве примеров схемы, представленные здесь ниже, не предназначены быть единственно возможными препаративными подходами. Экспериментальные детали и некоторые физико-химические данные по каждому из методов синтеза представлены в примерах 514. Для каждого отдельного способа A-J представлен пример синтеза, который следует далее в виде перечисления родственных препаратов для каждого из следующих соответствующих способов. Для соединений со структурой (I),описанных в этом разделе, отмечается, что сво 21 бодное основание обычно выделяют. Для использования при биологическом отборе свободное основание в большинстве случаев превращали в форму гидрохлоридной соли с помощью общепринятых методов. Способ А. Методика способа А представлена следующей схемой и по существу аналогична пути,описанному Abdel-Magid, et al., TetrahedronLett., 1990; 31: 5595. В способе А соответственно замещенный альдегид конденсируется с соединением (II) посредством метода восстановительного аминирования. Хотя с успехом можно применять множество разнообразных восстанавливающих веществ, таких как борогидрид натрия, цианоборогидрид натрия, литийалюмогидрида, триацетоксиборогидрида натрия и других подходящих борогидридных восстанавливающих веществ при практическом осуществлении способа А, обычно предпочтителен триацетоксиборогидрид натрия. Процедура восстановительного аминирования способа А может проводиться или в протонном или в апротонном растворителе, такой как метанол, этанол, ТГФ или ДМФ. Схема реакции Детали эксперимента и физико-химические данные соединений, полученных по способу А, представлены в примере 5 здесь ниже. Этот метод с незначительной модификацией использован для синтеза нескольких родственных производных, и эти соединения вместе с типичными препаратами также включены в пример 5. Способ В. Методика способа В представлена следующей схемой. В способе В реакция циклоконденсации с участием диола (15) и соответственно замещенного амина осуществляется в присутствии соответствующего активирующего вещества. Может использоваться любое подходящее активирующее вещество, которое способно превращать два гидроксильных заместителя у (15) в реактивные удаляемые группы. Например, гидроксильные группы могут быть превращены в их соответственные мезилаты,тозилаты или трифталаты путем реакции с соответствующей кислотой, галогенидом кислоты или ангидридом кислоты. Альтернативно, дигидроксиалкильные заместители соединения(15) могут быть превращены в алкилгалогениды. При практическом осуществлении данного способа обычно предпочтителен мезилхлорид в качестве катализатора циклизации. Реакция циклоконденсации способа В обычно выполняется в апротонном растворителе, таком как ТГФ или метиленхлорид. Детали эксперимента и физикохимические данные соединений, полученных по способу В, представлены в примере 6, ниже. Способ С. Процедура способа С представлена на нижеследующей схеме. В способе С соединение(II) сочетается в присутствии основания с соответствующим несущим G заместителем, активированным удаляемой группой (-Х). Хотя опытному специалисту известно много таких удаляемых групп, для целей данного изобретения обычно предпочтительно, чтобы удаляемая группа была атомом галогена, такого как хлор,бром или йод, наиболее предпочтительно брома. В этой реакции сочетания можно с успехом использовать широкий ряд органических и неорганических оснований. Однако обычно предпочтительно, чтобы использовалось неорганическое основание, такое как карбонат калия. Обычно реакция сочетания способа С проводится в апротонном растворителе, таком как ДМФ или ацетонитрил. Схема реакции В двух примерах, описанных ниже, применялись незначительные модификации, касающиеся растворителя для реакции и основания,которые использовались в вышеприведенной реакции сочетания. Детали эксперимента и физико-химические данные для соединений, полученных по способу С, представлены в примере 7 здесь ниже. Способ D. Процедура способа D представлена на нижеследующей схеме. В способе D проводят сочетание соединения (II) с соответствующим несущим G субстратом путем реакции присоединения по Михаэлю. Такие реакции присоединения известны опытным специалистам, включая их различные преобразования. В отношении детального обсуждения реакции присоединения Михаэля см., например, Н.О.House, Modern Synthetic Reactions, 2nd Ed., W.A.Benjamin, Inc.,Menlo Park, CA, 1972). Эти реакции присоединения обычно проводятся в протонных растворителях, таких как вода, низшие спирты (например, метанол или этанол) или уксусная кислота. В данном примере обычно предпочтительна уксусная кислота. Схема реакцииD, представлены в примере 8 ниже. Способ Е. Процедура способа Е представлена на нижеследующей схеме. По способу Е активированная форма соединения 70 из примера 7 превращается в соответствующее амидное производное путем реакции с соответственно замещенным амином. Опытному специалисту известны разнообразные препаративные методы амидирования остатков карбоновых кислот. Например, кислотный субстрат может быть активирован в форму галогенангидрида, такого как хлорангидрид кислоты. Альтернативный,предпочтительный метод активирования карбоновой кислоты включает образование активированного промежуточного соединения путем реакции свободной карбоновой кислоты с соответствующим карбодиимидом, например 1,1'карбонилимидазола (КДИ) или, предпочтительно, 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (ЭДК). Такие способы описаны детально выше. Схема реакции Детали эксперимента и физикохимические данные соединений, полученных по способу Е, представлены в примере 9 ниже. Способ F. Процедура способа F представлена на нижеследующей схеме. При практическом осуществлении способа F предшественник, включающий восстанавливаемую часть, т.е. G', превращается в соединение (I). Должно быть понятно,что используя этот способ, можно успешно трансформировать широкий ряд восстанавливаемых субстратов и что эта методика никаким образом не ограничивается данным примером,который является иллюстративным для синтеза соединения строения (I) путем восстановления карбонильного соединения. Для целей данного способа соответственно замещенное карбонильное производное (II') может быть синтезировано путем приведения во взаимодействие соединения (II) с карбонильной кислотой или ее активированной формой. В данном случае соединение 4'-трифторметилбифенил-2-карбоновой кислоты[2-(тиофен-2-ил-ацетил)-1,2,3,4 тетрагидроизохинолин-6-ил]-амид (II') превращают в соответствующую восстановленную форму (I) путем взаимодействия с соответствующим восстанавливающим веществом. Восстанавливающие средства, пригодные для этой цели, могут включать, например, борогидрид натрия, литийалюмогидрид и другие подходящие борогидридные соединения, каталитическую гидрогени 001539 24 зацию и так далее. В данном примере обычно предпочтителен борогидрид натрия. Опытный специалист легко определит систему растворителей, совместимую с другими функциональными группами, которые могут присутствовать,и природой подразумеваемой восстанавливаемой части. Такие растворители включают, например, ТГФ, ДМФ, уксусную кислоту, метанол, этанол и тому подобное. Однако в зависимости от реактивных свойств восстанавливаемой части могут быть необходимы некоторые изменения помимо общих указаний, которые хорошо известны опытным специалистам. Схема реакции Связанные с этим детали синтеза и физико-химические данные для соединений, полученных по способу F, включая соединение (II') описаны ниже в примере 10. Способ G. Процедура способа G представлена на нижеследующей общей схеме. По способу G из соединения 67 из примера 6 получают производное путем следующей последовательности синтеза, предпочтительно, ацилированием Nгидроксиэтильного заместителя изохинолинового ядра. Где это применимо, такие реакции получения производных, включая ацилирование, может быть осуществлено в присутствии основания. Например, удобно и можно использовать различные органические основания, такие как триэтиламин, пиридин, N,N'-диметиламинопиридин и тому подобное. Таким же образом с успехом можно использовать некоторые неорганические основания, такие как карбонат натрия, карбонат калия и тому подобное. При практическом осуществлении способа G обычно предпочтительно органическое основание, такое как N,N'-диметиламинопиридин. Как было отмечено ранее, получение других производныхN-гидроксиэтильной части, а не превращения путем ацилирования, описанного в способе G,полностью находится в компетенции опытного специалиста, и указания данного способа не предназначены для ограничения только этим. Схема реакции Конкретные значения для -G будут представлены в примере 11 вместе с изложением деталей синтеза и физико-химических данных,относящихся к этому. Способ Н. 25 Полная процедура способа Н представлена на нижеследующих двух схемах. На стадии 1 способа Н соединения 49 из примера 6 гидролизуют до свободного Nаминоэтильного соединения 94. При практическом осуществлении способа Н наиболее удобно выделять соединение 94 в виде свободного основания, однако, опытный специалист поймет, что если желательно, с этим субстратом может образовываться широкий ряд солей с присоединением кислоты, как описано ниже. Стадия 1. Схема реакции На стадии 2 способа Н N-аминоэтильная часть ядра из изохинолинового кольца соединения 94 превращают в производное в соответствующей последовательности синтеза, предпочтительно амидирования. N-аминоэтильную часть соединения 94 можно амидировать с помощью широкого ряда реагентов, которые могут включать карбоновые кислоты, галогенангидриды кислот, ангидриды кислот, изоцианаты, изотиоцианаты и подобные реагенты, которые хорошо известны опытным специалистам. Процедуры амидирования способа Н могут быть осуществлены в присутствии основания или без него. Кроме того, где нужно, можно использовать активирующее вещество, такое как 1,l'-карбонилдиимидазол (КДИ) или 1-(3 диметиламинопропил)-3-этилкарбодиимида гидрохлорид (ЭДК). Получение других производных N-аминоэтильной части, а не предпочтительных превращений путем амидирования,описанных в способе Н, полностью относится к компетенции опытного специалиста, и указания по данному способу не могут быть ограничивающими только этим. В нескольких примерах были применены незначительные изменения в выборе реагентов для амидирования при синтезе некоторых соединений этим способом, и соответствующие детали эксперимента для каждого из этих изменений представлены в примере 12. Конкретные значения для -G, а также детали синтеза и физико-химические данные, относящиеся к этому, представлены в примере 12,представленном здесь ниже. Способ I. Полная процедура способа 1 представлена следующими двумя схемами. 26 На стадии 1 способа I у соединения 61 из примера 6 удаляют защиту с получением соединения 106. В данном примере для осуществления удаления защиты используется каталитическая гидрогенизация, хотя опытному специалисту изестны и альтернативные пути. Стадия 1. Схема реакции На стадии 1 способа I по атому азота пиперидинового кольца соединения 106 получают производное с соответствующей функциональной группой, предпочтительно алкильной или ацильной функциональной группой. Опытный специалист легко поймет значение того факта,что возможны многие аналогичные трансформации у этого атома азота путем введения других заместителей, а не тех, которые описаны здесь. Стадия 2. Согласно изобретению предпочтительные алкильные или ацильные заместители могут вводиться с помощью ряда известных способов. Процедуры алкилирования могут включать приведения в контакт субстрата из амина с алкилирующим веществом, например, и алкилгалогенидом, или, предпочтительно, алкилирование амина с помощью процедуры восстановительного аминирования, такой, которая описана здесь выше для способа А. Ацилирование амина предпочтительно осуществляется с помощью процесса, аналогичного описанному выше для способа Н. Приведенные в качестве примера, не ограничивающие значения -G, а также детали синтеза и физико-химические данные, относящиеся к этому для способа I будут представлены в примере 13, ниже. Способ J. Процедура способа J представлена ниже. В данном случае цианоэтильная группа соединения 74 из примера 7 циклизируется с помощью гидразин муравьиной кислотой в присутствии основания с получением соединения 111. Известно, что нитрильная часть соединения 74 способна к функционированию в качестве предпоследнего предшественника для других систем гетероциклических колец, и данный пример предлагается только в целях иллюстрации, но не ограничения. Схема реакции Детали процесса получения и его физикохимические данные, для соединения 111 по способу J представлены в примере 14 ниже. Для выделения соединений согласно изобретению могут использоваться обычные методы и/или методики очистки и разделения, известные специалистам. Такие методики включают, например, хорошо известные и признанные виды хроматографии (такие как ВЭЖХ,колоночная хроматография с использованием обычных адсорбентов, таких как силикагель, и тонкослойная хроматография), перекристаллизацию и методики дифференциального (т.е.,жидкость-жидкость) экстрагирования. Соединения, описанные здесь, образуют катионные соли, такие как соли с присоединением кислоты, и выражение фармацевтически приемлемые соли предназначено для определения, но не для ограничения ими, таких солей как гидрохлоридные, гидробромидные, сульфатные, гидросульфатные, фосфатные, гидрофосфатные, дигидрофосфатные, ацетатные, сукцинатные, цитратные, метансульфонатные (мезилаты) и п-толуолсульфонатные (тозилаты) соли, а также их гидратные и сольватные формы. Для многих соединений возможны и/или желательны соли с полиприсоединением. Соли с присоединением кислоты соединений согласно изобретению, включая соли соединения (II), могут быть легко получены путем реакции соединений в виде оснований с соответствующими сопрягаемыми кислотами. Когда соль является солью моноосновной кислоты(например, гидрохлорид, гидробромид, птолуолсульфонат, ацетат), однозамещенной формой двухосновной кислоты (например, гидросульфат, сукцинат) или однозамещаемой формой трехосновной кислоты (например, дигидрофосфат, цитрат) применяют, по меньшей мере, один молярный эквивалент, а обычно молярный избыток кислоты. Однако когда желательны такие соли, как сульфат, гемисукцинат,гидрофосфат или фосфат, обычно должно использоваться соответствующий и стохиометрический эквивалент кислоты. Свободное основание и кислота обычно соединяются в сорастворителе, из которого желаемая соль выпадает в осадок или может быть выделена иначе путем концентрации маточной жидкости или путем эффекта осаждения, получаемого путем добавления не растворителя. Соединения согласно изобретению можно применять перорально и соответственно применять в комбинации с фармацевтически приемлемым носителем или растворителем, приемлемым для пероральных дозированных форм. Со 001539 28 ответствующие фармацевтически приемлемые носители включают инертные твердые наполнители или разбавители и стерильные водные или органические растворы. Активное соединение будет присутствовать в таких фармацевтических композициях в количествах, достаточных для обеспечения желательного дозированного количества в интервале, описанном ниже. Так,для перорального введения соединения могут сочетаться с подходящим твердым или жидким носителем или разбавителем для получения капсул, таблеток, порошков, сиропов, растворов,суспензий и тому подобного. Фармацевтические композиции могут, если желательно, содержать дополнительные компоненты, такие как улучшающие запах вещества, подсластители, наполнители и тому подобное. Таблетки, пилюли, капсулы и тому подобное могут содержать связывающее вещество,такое как камедь трагаканта, акации, кукурузный крахмал или желатин; наполнители, такие как дикальцийфосфат; дезинтегрирующее средство, такое как кукурузный крахмал, картофельный крахмал, альгиновая кислота; увеличивающее скольжение вещество, такое как стеарат магния; и подслащивающее вещество, такое как сахароза, лактоза или сахарин. Когда дозированная форма представляет собой капсулы,она может содержать в дополнение к веществам вышеназванных типов жидкий носитель, такой как жирное масло. Различные другие материалы могут присутствовать в качестве покрытий или для изменения физической формы дозированной единицы. Например, таблетки могут быть покрыты щеллаком, сахаром или и тем и другим. Сироп или эликсир может содержать, кроме того, активный ингредиент, сахарозу, как подслащивающее вещество, метил- и пропилпарабены в качестве консервантов, подкрашивающее вещество и улучшающее вкус и запах вещество, такое как вишневый или апельсиновый концентрат. Соединения согласно изобретению можно также вводить парентерально. Парентеральное введение соединений может сочетаться со стерильной водной или органической средой для получения инъекционных растворов или суспензий. Растворы или суспензии этих активных соединений могут быть приготовлены в воде,соответственно смешанной с поверхностноактивным веществом, таким как гидроксипропилцеллюлоза. Если необходимо, водные растворы должны быть соответственно забуферены, и водный растворитель в первую очередь должен быть сделан изотоничным с помощью достаточного количества соли или глюкозы. В этом плане, применяемые стерильные водные среды все легко получаются по стандартным методам, хорошо известным специалистам. Препараты, вводимые парентерально, могут также производиться в виде стерильных компо 29 зиций в твердом виде, которые могут также растворяться в стерильной воде или какой-то другой стерильной инъекционной среде непосредственно перед назначенным применением. Дисперсии могут быть приготовлены в кунжутном или арахисовом масле, этаноле, воде, полиоле(например, глицероле, пропиленгликоле и жидком полиэтиленгликоле), подходящих их смесях, растительных маслах, N-метилглюкамине,поливинилпирролидоне и их смесях, в маслах, а также водных растворах водорастворимых фармацевтически приемлемых солей этих соединений. При обычных условиях хранения и применения эти препараты могут содержать консервант для предотвращения роста микроорганизмов. Полученные таким образом растворы для инъекций можно затем вводить внутривенно,внутрибрюшинно, подкожно и внутримышечно. Фармацевтические формы, пригодные для инъекционного введения включают стерильные водные растворы или дисперсии и стерильные порошки для экстемпорального приготовления стерильных инъекционных растворов или дисперсий. Во всех случаях лекарственная форма должна быть стерильной и должна быть жидкой для такой степени, чтобы была возможность легкого введения через шприц. Она должна быть стабильна в условиях производства и хранения, и должна содержать консервант от загрязнения микроорганизмами, такими как бактерии и грибы. Они должны поддаваться стерилизации, например, путем фильтрации через задерживающий бактерии фильтр, путем включения в композиции стерилизующих веществ или путем облучения или нагревания композиций, когда такое облучение или нагревание и приемлемо и совместимо с изготовлением лекарственной формы. Дополнительные фармацевтические лекарственные формы могут включать, между прочим, суппозитории, подъязычные таблетки, дозированные формы для местного применения и тому подобное, и они могут быть получены методами общепринятыми в данной области технологии. Дозировка соединения согласно изобретению, которое назначено для введения, будет в основном меняться в соответствии с принципами, хорошо известными специалистам, с учетом тяжести патологического состояния, которое нужно лечить, и пути введения. В основном соединение будет вводиться теплокровным животным (таким как человек), так чтобы они получали эффективную дозу, обычно суточную дозу, вводимую однократно или разделенную на части, например, дозу в интервале от примерно 0,1 до примерно 15 мг/кг веса тела, предпочтительно от примерно 1 до примерно 5 мг/кг веса тела. Общая получаемая суточная доза будет в основном находиться в интервале между 1 и 1000 мг, предпочтительно между 3 и 350 мг. Вышеприведенные дозировки являются приме 001539 30 рами для обычного случая; конечно, могут быть отдельные случаи, когда требуются дозировки в более высоком или более низком интервале, и такие отклонения входят в объем этого изобретения. Соединения согласно изобретению являются активными, что определено при следующих биологических испытаниях по отбору. Активность соединения согласно изобретению может быть оценена путем измерения подавления секреции Аро В в клетках HepG2. В клетках HepG2 подращивают в среде Игла, модифицированной Дульбекко с 10% эмбриональной бычьей сыворотки (среда для роста; Gibco) в 96-ячеечных платах в атмосфере с увлажнением, содержащей 5% двуокиси углерода до тех пор, пока у них не будет примерно 70% слияния. Соединение, которое нужно испытать, растворяют до концентрации 10-20 мМ в диметилсульфоксиде и затем разбавляют до 1 мМ в среде для роста. В среде для роста делают серийные разведения 1:1 этого стандартного раствора, и 100 мкл каждого разведения добавляют в отдельные ячейки 96-ячеечных плат для культивирования, содержащие клетки HepG2. Через двадцать четыре часа ростовую среду собирают и исследуют с помощью специфического ТИФА (твердофазного иммуноферментного анализа) на Аро В с различными концентрациями Аро AI в качестве контроля. Ингибиторы идентифицируют как соединения, которые снижают секрецию Аро В в среду без слияния на секрецию Аро AI. ТИФА на Аро В выполняется следующим образом. Моноклональные антитела к человеческому Аро В (Chemicon; Temecula,СА) разводят до 5 мг/мл в физиологическом/ азидном растворе (ФБР + 0,02% азида Na) с фосфатным буфером (8,76 г/л Nacl, 0,385 г/л КН 2 РO4, 1,25 г/л К 2 НРO4) и добавляют по 100 мл в каждую ячейку 96-ячеечной платы (NUNCMaxisorb, Rochester, NY). После инкубации в течение ночи при комнатной температуре раствор антител удаляют и ячейки промывают 3 раза ФБР/азидным раствором. Неспецифические сайты связывания на пластике блокируют путем инкубации ячеек в течение 1-3 ч в растворе 1%(вес/объем) растворе бычьего сывороточного альбумина (БСА), приготовленного в ФБР/ азидном растворе. 100 мкл разных разведении ростовой среды от клеток HepG2 или Аро В в виде отделенного центрифугированием на ультрацентрифуге ЛПНП (разведенного в 0,004% Тви 20/1% БСА в ФБР/азидном растворе) добавляют в каждую ячейку и инкубируют в течение 18 ч. Жидкость из ячеек отсасывают и промывают их 3 раза (0,1% Твин 20 в ФБР) перед добавлением 100 мкл разведения 1/1000 вторых антител, козлиных к человеческому Аро В(Chemicon). После 3 ч инкубации при комнатной температуре этот раствор отсасывают и ячейки снова промывают 3 раза, как и выше. Затем в каждую ячейку добавляют 100 мкл раз 31 ведения 1:1600 (в ФБР/1% БСА/2 мМ МgС 12) кроличьего против козлиного IgG, конъюгированного с щелочной фосфатазой (Sigma; St.Louis, МО) и инкубируют в течение 1 ч при комнатаной температуре. После аспирации ячейки промывают 4 раза, как и выше, и в каждую ячейку добавляют 100 мкл раствора 1 мг/мл п-нитрофенилфосфата (пНФФ; Sigma) в 25 мМ растворе бикарбоната натрия/2 мМ MgCl2, pH 9,5 и затем реакцию заканчивают путем добавления 50 мкл 0,2N NaOH. Поглощение каждой ячейки считывают при 405 нм и вычитается фон при 650 нм. Концентрацию Аро В рассчитывают по стандартной кривой, построенной по чистым стандартным ЛПНП, которые проводят параллельно с таким же исследованием. Результаты Аро А 1 измеряют аналогичным образом, за исключением того, что вместо антител к Аро В используют антитела к Аро A1 (Chemicon) и инкубацию с антигеном проводят при 37 С вместо комнатной температуры. Активность может быть также подтверждена, если испытуемое соединение непосредственно ингибирует активность МТР. Ингибирование активности МТР с помощью соединения может быть оценено количественно путем наблюдения ингибирования переноса радиоактивного меченного триглицерида от донорского носителя к акцепторному носителю в присутствии растворимого человеческого МТР. Процедура получения МТР основана на методе Wetterau and Zilversmit (Biochem. Biophys. Acta (1986) 875:610). В кратком изложении, куски человеческой печени, замороженные при -80 С, оттаивают на льду, измельчают и промывают несколько раз 0,25 М раствором сахарозы с температурой льда, причем все последующие стадии выполняются на льду. 50% гомогенат в 0,25 М сахарозе готовят, используя тефлоновый пестик Potter-Elvehjem. Гомогенат разбавляют 1:1 0,25 М раствором сахарозы и центрифугируют при 10000 х g в течение 20 мин при 4 С. Осадок снова суспендируют в растворе сахарозы и повторно центрифугируют при 10000 х g в течение 20 мин. Супернатанты объединяют, и микросомы осаждают центрифугированием при 105000 х g в течение 75 мин. Супернатант отбрасывают, а осадок микросом суспендируют в минимальном объеме 0,25 М растворе сахарозы, разбавляют до 3 мл на грамм исходного веса печени 0,15 М Трис-HCl с рН 8. Эту суспензию разделяют на 12 частей и центрифугируют при 105000 х g в течение 75 мин. Супернатанты отбрасывают, а микросомный осадок хранят замороженным при -80 С до тех пор, пока не понадобится. Для получения МТР перед выполнением исследования оттаявший осадок суспендируют в 12 мл холодного 50 мМ Трис-HCl, 50 мМ КСl, 5 мМ MgCl2, рН 7,4 и медленно добавляют 1,2 мл 0,54% раствора дезоксихолата (рН 7,4) при перемешивании, чтобы разрушить микросомные мембраны. После 30 32 мин инкубации на льду при легком перемешивании суспензию центрифугируют при 105000 хg в течение 75 мин. Супернатант, содержащий растворенный белок МТР, диализируют в течение 2-3 дней с 4 заменами буфера для исследования (150 мМ Трис-HCl, 40 мМ NaCl, 1 мМ ЭДТУ, 0,02% NaN3, рН 7,4). МТР человеческой печени хранят при 4 С и разводят 1:5 буфером для исследования перед использованием. В препаратах МТР не показано значительной потери активности по переносу при хранении в течение вплоть до 30 дней. Липосомы получают обработкой дисперсии 400 мМ яичного фосфатидилхолина (ФХ;PC), 75 мМ кардиолипина бычьего сердца и 0,82 мМ [14 С]-триолеина (110 Ки/моль) (NEN,Boston, MA) в буфере для исследования в атмосфере азота на ультразвуковой бане при комнатной температуре. В соответствующих количествах доабвляют липиды в хлороформе и сушат в потоке азота перед гидратированием буфером для исследования. Акцепторные липосомы также получают обработкой дисперсии 1,2 мМ ФХ, 2,3 мМ триолеина и 30 рМ [3 Н]-ФХ (50 Ки/моль) (NEN; Boston, MA) в буфере для исследования в атмосфере азота на ультразвуковой бане при комнатной температуре. Донорские и акцепторные липосомы центрифугируют при 160000 х g в течение 2 ч при 7 С. Верхние 80% супернатанта, который содержит однослойные липосомы небольшого размера, осторожно удаляют и хранят при 4 С до тех пор, пока они не потребуются для последующего использования в исследованиях по переносу. Активность МТР измеряют, применяя исследования по переносу, которое начинается путем смешивания донорских и акцепторных частичек вместе с растворенным МТР и соединением, которое нужно испытать. К 100 мкл или 5% БСА (контроль) или 5% БСА, содержащего испытуемое вещество, добавляют 500 мкл буфера для испытаний, 100 мкл донорских липосом, 200 мкл акцепторных липосом и 100 мкл разведенного белка МТР. После инкубации при 37 С в течение 45 мин перенос триглицерида прекращают путем добавления 500 мл 50%(вес/объем) суспензии ДЭАЭ-целлюлозы в буфере для исследования. После 4 мин встряхивания донорские липосомы, связанные с ДЭАЭцеллюлозой, селективно осаждают, применяя низкоскоростное центрифугирование. Образец супернатанта, содержащий акцепторные липосомы, просчитывают, и числа распадов 3 Н и 14 С используют для расчета процента выделения акцепторных липосом и процента переноса триглицеридов, применяя кинетику первого порядка. Подавление переноса триглицеридов испытуемым соединением проявляется как снижение в радиоактивности 14 С по сравнению с контролями, где испытуемое вещество не присутствовало. 33 Активность испытуемого соединения как ингибитора МТР может быть продемонстрирована in vivo в соответствии со следующим исследованием. Мышам-самцам (20-30 г; разных линий) перорально через зонд вводили дозу (0,25 мл/25 г веса тела) испытуемого соединения в виде суспензии в водном растворе 0,5% водном растворе метилцеллюлозы. Дозы растворов соединения вводят или многократно в течение нескольких дней или, альтернативно, один раз за 90 мин перед тем, как мышь забивали, и кровь собирают для получения сыворотки. Сыворотку исследуют на концентрацию триглицеридов и холестерина, используя коммерческие ферментативные исследования (триглицерид G. WakoBoeringer Mannheim, Indianapolis, IN). Соединения, которые являются ингибиторами МТР идентифицируют по их способности снижать уровень триглицеридов в сыворотке по сравнению с контрольными мышами, которым вводили только носитель. Данное изобретение иллюстрируется следующими примерами, причем эти примеры предлагаются с целью иллюстрации и не должны никаким образом истолковываться как ограничивающие. Пример 1. Следующие примеры являются иллюстративными для тех процедур, которые представлены и описаны выше, для последующего получения соединений 1-7 и (II) схемы 1. Числа,представленные в скобках после названия каждого вышеуказанного соединения соответствуют соответствующим номерам соединений на схеме 1.N-[2-(4-Бромфенил)-этил]-формамид (1). Объединяют 500 г (1,78 моль) гидробромида 2-(4-бромфенил)-этиламина, 1 л (12,4 моль) этилформиата и 248 мл (1,78 моль) триэтиламина и нагревают до кипения с обратным холодильником в течение 3 ч. Реакционную смесь приводят во взаимодействие с 1 л (каждого из) деионизированной воды и этилацетата. Органический слой отделяют и промывают 1 л(каждого из) воды и насыщенного раствора соли. Органический слой сушат над безводным сульфатом магния, фильтруют и концентрируют с получением 378 г твердого вещества. МС(Сl): 245 (М + NH4+). 7-Бром-3,4-дигидроизохинолина гидрохлорид (2). В 12 литровой трехгорлой круглодонной колбе нагревают полифосфорную кислоту до 150 С и перемешивают. К перемешиваемой полифосфорной кислоте добавляют 530 г (3,75 моль) пентоксида фосфора тремя частями по примерно 176,7 г в каждой. Когда пентоксид фосфора растворится, добавляют 378 г (1,66 моль) N-[2-(4-бромфенил)этил]формамида. Затем температуру реакционной смеси повышают 34 до 200 С и поддерживают в течение двух часов. В этот момент температуре реакционной смеси дают снизиться до 160 С и выливают на 16 л льда. Смесь перемешивают в течение 0,5 ч,подщелачивают до рН 12 10N раствором гидроксида натрия и затем экстрагируют три раза 3 л метиленхлорида. Объединенные органические слои промывают 1 л насыщенного раствора хлорида натрия, сушат над безводным сульфатом натрия, фильтруют и концентрируют до масла. Масло растворяют в 2,5 л метанола и насыщают безводным газообразным НСl. Полученный раствор концентрируют до объема одного литра и добавляют 1 л диэтилового эфира. Полученный осадок отфильтровывают, промывают диэтиловым эфиром и сушат воздухом с получением 219 г вышеуказанного соединения в виде твердого вещества. МС(Сl): 210 (М+Н+). 7-Бром-1,2,3,4-тетрагидроизохинолин (3). В общей сложности 219 г (0,89 моль) гидрохлорида 7-бром-3,4-дигидроизохинолина и 1,5 л воды соединяют и нагревают до 50 С. Добавляют в общей сложности 33,7 г (0,89 моль) борогидрида натрия частями в течение 0,5 ч, во время чего температура повышается до 62 С. Затем реакционную смесь охлаждают до комнатной температуры и экстрагируют три раза по 1 л метиленхлорида. Объединенные органические слои промывают 1 л насыщенного раствора хлорида натрия, сушат над безводным сульфатом натрия и концентрируют с получением 173 г масла. МС(Сl): 212 (М+Н+). 7-Бром-6-нитро-1,2,3,4-тетрагидроизохинолин (4). В 5-литровой трехгорлой круглодонной колбе осторожно растворяют 173 г (0,813 моль) 7-бром-1,2,3,4-тетрагидроизохинолина в 950 мл концентрированной серной кислоты. Полученный раствор охлаждают до -5 С и по каплям добавляют раствор 82,7 г (0,816 моль) нитрита калия в 1 л концентрированной серной кислоты. После добавления реакционную смесь выдерживают при -5 С в течение 15 мин и выливают на 3 л льда. Полученную смесь подщелачивают до рН 14 50% раствором гидроксида натрия. Основной раствор экстрагируют три раза 1 л метиленхлорида. Объединенные органические слои промывают 1 л (каждого из) воды и насыщенного раствора хлорида натрия. Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют с получением 201 г масла. Масло, предварительно адсорбированное на силикагеле, загружают на колонку из 4 кг силикагеля и элюируют градиентом 15% метанол/ метиленхлориде. Фракции, содержащие продукт, объединяют и концентрируют с получением 115 г твердого вещества. 1(0,447 моль) триэтиламина, 97,5 г (0,447 моль) ди-трет-бутилдикарбоната, 3,2 литра диоксана и 0,5 л воды соединяют и перемешивают при комнатной температуре в течение 1,5 ч. Реакционную смесь концентрируют для удаления диоксана, добавляют 1 л насыщенного раствора бикарбоната натрия и смесь экстрагируют два раза 1 л метиленхлорида. Органический слой промывают насыщенным раствором соли, сушат над сульфатом магния и концентрируют. Полученное твердое вещество перекристаллизовывают из изопропанола с получением 118 г твердого вещества. 1 Н ЯМР (250 МГц, ДМСО) : 7,89(с,1 Н); 7,81(с,1 Н), 4,58(с,2 Н); 3,56 (т,2 Н); 2,81(т,2 Н); 1,42(с,9 Н). Трет-бутиловый эфир 6-амино-3,4-дигидро-1 Н-изохинолин-2-карбоновой кислоты (6). В общей сложности 59 г (0,16 моль) третбутилового эфира 7-бром-6-нитро-3,4-дигидро 1 Н-изохинолин-2-карбоновой кислоты, 10 г 5% палладия на карбонате кальция и 49 г ацетата аммония в 1 л уксусной кислоты гидрогенизируют в шейкере Парра в течение 5 ч. Реакционную смесь фильтруют, концентрируют, подщелачивают до рН 12 4N гидроксидом натрия и экстрагируют метиленхлоридом. Органический слой промывают водой и насыщенным раствором соли, сушат над сульфтом магния и концентрируют с получением 40 г вышеуказанного соединения в виде масла. 1H ЯМР (300 МГц, ДМСО) : 4,87(с,2 Н); 4,27(с,2 Н), 3,44(т,2 Н); 2,57(т,2 Н); 1,39(с,9 Н). Трет-бутиловый эфир 6-[(4'-трифторметилбифенил-2-карбонил)-амино]-3,4-дигидро-1 Низохинрлин-2-карбоновой кислоты (7). Образец 7,6 г (29 ммоль) 4'-трифторметилбифенил-2-карбоновой кислоты, 7,1 г (29 ммоль) трет-бутиловый эфир 6-амино-3,4 дигидро-1 Н-изохинолин-2-карбоновой кислоты,100 мг ДМАФ и 6,1 г (32 ммоль) EDCI приводят во взаимодействие в 130 мл метиленхлорида в течение 12 ч. Реакционную смесь экстрагируют 2 х 150 мл 1N НСl, 2 х 150 мл 1N NaOH, 150 мл воды, концентрированного раствора соли и затем концентрируют с получением 14 г бежевой пены. МС(Сl): 519 (M+Na+). 1 Н ЯМР (300 МГц, CDCl3) : 4,49 (с, 2 Н); 3,60 (т, 2 Н); 2,77 (т,2 Н). 36 2-карбоновой кислоты и 6 мл (78 ммоль) трифторуксусной кислоты смешивают в 60 мл метиленхлорида в течение 5 ч. Добавляют метиленхлорид (40 мл) и органический слой промывают 3 х 50 мл насыщенным растором бикарбоната натрия с последующим промывным насыщенным раствором соли. Органический слой сушат над сульфатом натрия и концентрируют с получением 3,1 г твердого вещества. МС(Сl): 397(М+Н+). Пример 2. Следующие примеры синтеза являются иллюстративными для тех процедур, которые представлены и описаны выше для последующего получения соединений 8 - 13 со схемы 2. Цифры, представленные в скобках после названия каждого соединения в заголовке, соответствуют соответствующим номерам на схеме 2. Диметиловый эфир 2-(карбокси-5-нитрофенил)малоновой кислоты (8). Раствор 2-хлор-4-нитробензойной кислоты(75 г, 372 ммоль) в диметилмалонате (900 мл) продувают азотом в течение 15 мин. Добавляют одной порцией метоксид натрия (48,3 г, 894 ммоль) и содержимое нагревается при выделении тепла до 48 С. Через 15 мин добавляют бромид меди (I) (5,4 г, 37 ммоль) и содержимое нагревают до 70 С в течение 24 ч. Реакционную смесь, где реакция прошла примерно на 70%, что определяется по ЯМР, затем нагревают до 85 С в течение 5 ч до полного израсходования оставшейся 2-хлор-4-нитробензойной кислоты. К охлажденной реакционной смеси добавляют воду (900 мл) с последующим добавлением гексанов (900 мл). Водный слой отделяют, добавляют толуол (900 мл), смесь фильтруют и водный слой отделяют. К водному слою добавляют свежий толуол (1800 мл) и бифазную смесь подкисляют 6N водным раствором НСl (90 мл). Образовавшийся белый осадок и содержимое перемешивают в течение 18 ч. Продукт отфильтровывают и сушат с получением белого твердого вещества (78,1 г, 70%) т. пл. 153 С. 1 Н ЯМР (ДМСО) : 8,37 (д, J=2 Гц, 1 Н); 8,30(д, J=l Гц, 2 Н); 5,82(с, 1 Н); 3,83 (с, 6 Н). 13 С ЯМР (ДМСО) : 168,0, 167,3, 149,4,137,1, 135,8, 132,5, 125,4, 123,7, 54,5, 53,4. Анал. Вычислено для C11H10NO8:C 48,49; Н 3,73; N 4,71. Найдено: С 48,27; Н 3,72; N 4,76. 2-Карбоксиметил-4-нитробензойная кислота (9). К раствору диметилового эфира 2(карбокси-5-нитрофенил)малоновой кислоты(25,0 г, 84 ммоль) в метаноле (200 мл) добавляют гидроксид натрия (5 г, 125 ммоль) в воде(200 мл). Через 3 ч реакция завершается, метанол удаляют под пониженным давлением, содержимое охлаждают до 0 С и подкисляют концентрированной НСl (37 мл). Водный слой дважды экстрагируют этилацетатом (200 мл, затем 37 100 мл), объединенные органические слои сушат сульфатом магния, большую часть растворителя удаляют под пониженным давлением и затем добавляют метиленхлорид (30 мл). Образовавшееся твердое вещество отфильтровывают и сушат с получением 19,3 г продукта в виде белого твердого вещества, т.пл. 180-82 С. ИК(КВr) 3080, 3055, 2983, 1707, 1611,1585, 1516, 1491, 1424, 1358,1298, 1237 см-1. 13 С ЯМР (ДМСО) : 172,3, 167,5; 149,2; 138,8; 137,3; 132,1; 127,2; 122,4; 39,8. Анал. Вычислено для C9H17NO6: С 48,01; Н 3,13; N 6,22. Найдено: С 47,67; Н 3,19; N 6,31. 2-(2-Гидроксиметил-5-нитрофенил)-этанол(11) через альтернативное промежуточное соединение (10). Смесь 2-карбоксиметил-4-нитробензойной кислоты (13 г, 57,7 ммоль), уксусного ангидрида(5,45 мл, 57,7 ммоль) и толуола (130 мл) нагревают до кипения с обратным холодильником в течение 5 ч. Растворитель удаляют под пониженным давлением с получением 6 нитроизохроман-1,3-диона (соединение (10) на схеме 2) в виде желтого твердого вещества(10,51 г, 88%). К раствору 6-нитроизохроман 1,3-диона (2 г, 9,66 ммоль) в ТГФ (40 мл) при 0 С по каплям добавляют боран-тетрагидрофурановый комплекс (35,6 мл, 1 М в ТГФ) в течение 40 мин. Затем содержимое перемешивают в течение 18 ч при 25 С, охлаждают до 0 С, гасят метанолом (30 мл) и перемешивают в течение 1 ч. Растворители удаляют под пониженным давлением, добавляют этилацетат (30 мл) и органическую фазу промывают 10% водным раствором соляной кислоты. Водный кислый раствор промывают обратной струей этилацетатом (30 мл), объединенные органические слои сушат сульфатом магния и выпаривают под пониженным давлением до примерно 2 мл остающегося этил-ацетата. Этот раствор фильтруют через силикатель, промывая его метиленхлоридом (30 мл) для удаления примесей. Силикагель смывают этилацетатом, растворитель удаляют под пониженным давлением с получением твердого вещества, из которого готовят суспензию в метиленхлориде и фильтруют с получением вышеуказанного диола в виде белого твердого вещества, 1,38 г, 73%. 2-(2-Гидроксиметил-5-нитрофенил)-этанол(11). Раствор 2-карбоксиметил-4-нитробензойной кислоты (3,0 г, 13,3 ммоль) в ТГФ (60 мл) приводят во взаимодействие с боран-ТГФ комплексом (53,3 мл, 53,3 ммоль) в течение 15 мин при 0 С. Реакционную смесь перемешивают в течение 18,5 ч, гасят ТГФ/водой (1:1, 30 мл),добавляют воду (20 мл) и слои разделяют. Водный слой промывают обратной струей ТГФ (30 мл), объединенные органические фазы промывают насыщенным раствором соли, сушат сульфатом магния и растворитель удаляют под пониженным давлением с получением продукта в 38 виде белого твердого вещества (2,05 г, 78%) т. пл. 79-81 С. ИК (КВr) 3277, 3192, 2964, 2932, 1614,1525, 1507, 1170, 1134, 1089, 1067 см-1. 13 С ЯМР (ДМСО) : 149,1; 146,6, 139,2; 127,8; 124,3; 121,3; 61,2; 60,6; 34,9. Анал. Вычислено для С 9 Н 11NO4: С 54,82; Н 5,62; N 7,10. Найдено: С 54,54; Н 5,49; N 7,07. 6-Нитро-1,2,3,4-тетрагидроизохинолин(12). К раствору 2-(2-гидроксиметил-5 нитрофенил)-этанола (1,0 г, 5,07 ммоль), триэтиламина (1,8 мл, 12,9 ммоль) в метиленхлориде (20 мл) по каплям добавляют метансульфонилхлорид (0,9 мл, 11,63 ммоль) в течение 10 мин. ТСХ показывает, что реакция завершается через 30 мин. 1 Н ЯМР (300 МГц, CDCl3) : 8,17-11 (м,2 Н); 7,65(д, J=9 Гц, 1 Н); 5,36 (с, 2 Н); 4,49 (т, J=6 Гц, 2 Н); 3,25 (т, J=6 Гц, 2 Н), 3,08 (с, 3 Н), 2,98 (с,3 Н). Реакционную смесь промывают 10% водным раствором НСl, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой сушат сульфатом магния, метиленхлорид удаляют под пониженным давлением и выгоняют с ТГФ (3 х 100 мл). Продукт (1,9 г) непосредственно используют на следующей стадии без дополнительной очистки. К димезилату (1,9 г) в ТГФ (30 мл) при 78 С добавляют аммиак (50 мл). Содержимое нагревают до 24 С в течение 60 ч, избыток аммиака отгоняют и растворитель удаляют под пониженным давлением с получением продукта-сырца (786 мг, 82%). Добавляют толуол, раствор фильтруют через сульфат магния и растворитель удаляют под вакуумом с получением 721 мг (75%) янтарного масла. 1 Н ЯМР (CDCl3) : 7,97 (с, 1 Н); 7,95 (д, J=9 Гц, 1 Н), 7,15 (д, J=9 Гц, 1 Н); 4,07 (с, 2 Н); 3,15 (т,J=6 Гц, 2 Н), 2,89 (т, J=6 Гц, 2 Н), 1,98 (шир.с,1 Н). Трет-бутиловый эфир 6-нитро-3,4-дигидро-1 Н-изохинолин-2-карбоновой кислоты (13). К раствору 6-нитро-1,2,3,4-тетрагидроизохинолина (840 мг, 4,71 ммоль) в метиленхлориде (17 мл), содержащем триэтиламин (0,72 мл,5,17 ммоль) добавляют БОК-ангидрид (1,44 мл,6,26 ммоль). Через 5 ч добавляют насыщенный водный раствор бикарбоната натрия, фазы разделяют, органический слой сушат сульфатом магния и растворитель удаляют под пониженным давлением с получением продукта в виде тусклого белого твердого вещества (1,2 г, 92%). Т. пл. 138-41 С. ИК(КВr) 3056, 3018, 2982, 2935, 1734,1684, 1612, 1522, 1399, 1236 см-1. 1 Н ЯМР (CDCl3) : 8,04 (т, J=5 Гц, 1 Н),8,01 (с, 1 Н), 7,26 (т, J=5 Гц, 1 Н); 4,65 (с, 2 Н); 3,68 (т, J=6 Гц, 2 Н); 2,93 (т, J=6 Гц, 2 Н), 1,49 (с,9 Н). течение 5 ч. Катализатор отфильтровывают,растворитель удаляют под пониженным давлением и остаток подвергают хроматографии на двуокиси кремния с этилацетатом/гексанами с получением 42 мг (57%) вышеуказанного продукта. ИК(КВr) 3005, 2975, 2928, 1685, 1627,1509,1423, 1365, 1166 см-1. 1 Н ЯМР (CDCl3) : 6,90 (д, J=6 Гц, 1 Н); 6,56 (д, J=6 Гц, 1 Н); 6,48 (с, 1 Н); 4,47 (с, 2 Н),3,60 (м, J=6 Гц, 4 Н), 2,73 (т, J=6 Гц, 2 Н), 1,49 (с,9 Н). Затем этот продукт взаимодействует с активированной формой 4'-трифторметилбифенил-2-карбоновой кислотой, которая описана ранее, с получением N-защищенного тетрагидроизохинолина (7), у которого затем удаляют защитные группы с получением соединения (II). Пример 3. Следующие примеры синтеза являются иллюстративными для тех процедур, которые представлены и описаны выше для последующего получения соединений 14, 15 и (II) со схемы 3. Цифры, представленные в скобках после названия каждого соединения в заголовке, соответствуют соответствующим номерам на схеме 3. 2-(5-Амино-2-гидроксиметилфенил)этанол (14).Pt-C (50% увлажнения водой, 200 мг) добавляют в раствор 2-(2-гидроксиметил-5 нитрофенил)-этанола (1,0 г, 5 ммоль) в ТГФ (40 мл) и смесь гидрогенизируют при 3,41 атм. ЯМР показала завершение реакции с образованием 2-(5-амино-2-гидроксиметилфенил)этанола (соединение (14) на схеме 3). 1 Н ЯМР (CD3Cl) : 7,08 (д, J=2 Гц, 1 Н); 6,54-6,50 (м. 2 Н); 4,51 (с, 2 Н), 3,82 (т, J=6 Гц,2 Н), 3,80-2,95 (шир.с, 4 Н), 2,84 (т, J=6 Гц, 2 Н). 4'-Трифторметилбифенил-2-карбонилхлорид. Раствор 4'-(трифторметил-2-бифенилкарбоновую кислоту (9,08 г, 34 ммоль), тионилхлорид (12 мл) и диметилформамид (0,05 мл) нагревают до кипения с обратным холодильником в течение 2 ч, при этом сроке по ЯМР определяется, что реакция завершена. Избыток тионилхлорида отгоняют с заменой толуолом (56 мл). Растворитель удаляют под пониженным давлением с получением хлорангидрида кислоты из заголовка в виде белого твердого вещества (9,46 г, 97%). Н ЯМР (CDCl3) : 8,12 (дд, J=8 Гц, 1 Н); 7,70-7,37 (м, 7 Н). 13 С ЯМР, CD3Cl(СО): 168. 4'-трифторметилбифенил-2-карбоновой кислоты-[3-(2-гидроксиэтил)-4-гидроксиметилфенил)]амид (15). Катализатор из гидрогенизации с Pt-C,описанной выше, отфильтровывают, добавляют триэтиламин (1,4 мл, 10 ммоль) с последующим добавлением раствора 4'-трифторметилбифенил-2-карбонилхлорида (1,44 г, 5 ммоль) в ТГФ (10 мл) в течение срока в 1 ч. Содержимое перемешивают в течение 24 ч, растворитель удаляют под пониженным давлением и добавляют этилацетат (40 мл). Органическую фазу промывают водой (2x40 мл), сушат сульфатом магния и выпаривают под пониженным давлением. Остаток отгоняют с толуолом (3x40 мл) и выпаривают с получением 2,11 г белого твердого вещества, которое снова превращают в пульпу в метиленхлориде (21 мл) на 18 ч, фильтруют и сушат с получением продукта, названного в заголовке в виде твердого белого вещества, 1,71 г (81%). 4'-Трифторметилбифенил-2-карбоновой кислоты-(1,2,3,4)-трифторгидроизохинолин-6 ил)-амид (II). К раствору 4'-трифторметилбифенил-2 карбоновой кислоты-[3-(2-гидроксиэтил)-4 гидроксиметилфенил]-амида (214 мг, 0,51 ммоль) и триэтиламина (0,18 мл) в ТГФ (8,5 мл) при 0 С добавляют метансульфонилхлорид(0,085 мл). ТСХ показывает завершение реакции через 30 мин. Содержимое охлаждают до -78 С,добавляют избыток амиака и содержимое перемещают в течение 18 ч при 25 С. Растворители удаляют под пониженным давлением, добавляют метиленхлорид (10 мл) и 1N водный раствор НСl, и содержимое перемешивают течение 1 ч. Фазы разделяют и водную фазу делают щелочной с помощью водного раствора гидроксида натрия до рН 12. Органическую фазу экстрагируют метиленхлоридом (4x10 мл), растворитель удаляют под пониженным давлением с получением белого твердого вещества (108 мг), которое подвергают хроматографии на двуокиси кремния, элюируя 5% метанол/ метиленхлоридом с 0,5% гидроксида аммония. Продукт получают в виде белого твердого вещества (40 мг,20%). 1(с, 2 Н), 3,52(д, J=7 Гц, 0,5 Н); 3,04 (т, J=6 Гц,2 Н). 2,74 (м. 0,5 Н), 2,66 (т, J=7 Гц, 2 Н), 2,27 (с,1 Н). 13 С ЯМР CD3Cl) : (только алифатические углероды) 47,8, 43,6, 29,1. Пример 4. Следующие примеры синтеза являются иллюстративными для тех процедур, которые представлены и описаны выше для последующего получения соединений 16 - 19 и (II) со 41 схемы 4. Цифры, представленные в скобках после названия каждого соединения в заголовке,соответствуют соответствующим номерам на схеме 4. 2-Бензил-6-нитро-4 Н-изохинолин-1,3-дион(16). К суспензии дикислоты (9) (55 г, 0,244 моль) в уксусной кислоте (550 мл) добавляют бензиламин (28,91 г, 0,27 моль). Реакционную смесь нагревают до 115 С в течение 18 ч, охлаждают до 25 С, добавляют воду (450 мл), содержимое перемешивают в течение 2 ч и продукт отфильтровывают в виде твердого белого вещества. Продукт промывают водой (400 мл) и сушат под пониженным давлением с получением 59,51 г (82%). 1 Н ЯМР (ДМСО) : 8,29-8,20 (м, 3 Н); 7,37,19 (м. 5 Н); 5,03 (с, 2 Н); 4,35 (с, 2 Н). ИК 3076, 2976, 1712, 1669, 1618, 1528,1338, 1308 см-1. Анал. Вычислено для C16H12N2 О 4: С 64,83; Н 4,08; N 9,46. Найдено: С 64,72; Н 3,97; N 9,49. 2-Бензил-6-нитро-1,2,3,4-тетрагидроизохинолин (17). К суспензии борогидрида натрия (2,13 г,56,2 ммоль) в ТГФ (56 мл) при 0 С добавляют по каплям трифторэтерат бора (9 мл, 73 ммоль). После перемешивания содержимого в течение 1 ч, добавляют дион (16) (5,0 г, 16,9 ммоль) в ТГФ(150 мл) при 0 С в течение 1,5-часового периода. Содержимое нагревают до 25 С в течение 30 мин и затем кипятят с обратным холодильником в течение 16 ч. Реакционную смесь после завершения реакции охлаждают до 0 С и осторожно гасят 1N водным раствором гидроксида натрия (75 мл, 75 ммоль) при поддержании температуры на уровне примерно 9 С. Погашенную реакционную смесь перемешивают в течение 30 мин при 0 С, 1 ч при 25 С, при 50 С в течение 18 ч и затем охлаждают до 20 С. Растворители удаляют под пониженным давлением, добавляют этилацетат (100 мл) и слои разделяют. Органический слой промывают насыщенным раствором соли, обрабатывают сульфатом натрия и растворители удаляют под пониженным давлением с получением 4,73 г светло-коричневого масла, которое было достаточно чистым, чтобы использоваться на следующей стадии. 1 Н ЯМР (CDCl3) : 7,98-7,29 (м. 5 Н); 7,387,10 (м, 5 Н); 3,71 (м, 2 Н); 2,99 (м, 2 Н), 2,82(м,2 Н). ИК 3064, 2940, 2794, 1589, 1518, 1492,1347, 1315 см-1. Анал. Вычислено для C16H16N2O2: С 71,50; Н 6,00; N 10,44. Найдено: С 70,76; Н 5,99; N 10,33. 4'-Трифторметилбифенил-2-карбоновой кислоты (2-бензил-1,2,3,4-тетрагидроизохинолин-6-ил)амид (19) через аминное промежуточное соединение (18).(230 мг) и гидрогенизацию продолжают в течение 72 ч. Смесь после завершения гидрогенизации фильтруют с получением ТГФ раствора промежуточного соединения, 2-бензил-1,2,3,4 тетрагидроизохинолин-6-иламина (18). К раствору (18) в ТГФ добавляют триэтиламин с последующим добавлением по каплям 4'трифторметилбифенил-2-карбонилхлорида (4,88 г, 17 ммоль) из примера 3. Содержимое затем перемешивают в течение 17 ч. Растворитель удаляют под пониженным давлением, добавляют воду (50 мл), содержимое перемешивают в течение 3 ч, продукт отфильтровывают и сушат под пониженным давлением с получением 5,93 г (71%) соединения (19) в виде белого твердого вещества. 4'-Трифторметилбифенил-2-карбоновой кислоты (1,2,3,4-тетрагидроизохинолин-6-ил)амид (II). К гидроксиду палладия на угле (1,5 г, 50% увлажнения водой) добавляют метанол (65 мл) и ТГФ (135 мл) в атмосфере азота с последующим добавлением амина (19) (5,0 г, 10,3 ммоль) из предыдущего примера и формиата аммония(6,48 г, 103 ммоль). Содержимое нагревают до 60 С в течение 3 ч, охлаждают до 25 С и фильтруют. К фильтрату добавляют водный раствор гидроксида натрия (1N, 10 мл), органические растворители удаляют под пониженным давлением и добавляют воду (40 мл). Содержимое перемешивают в течение 2 ч и продукт-сырец отфильтровывают и сушат с получением 4,09 г твердого вещества. Это вещество суспендируют в гексане/метиленхлориде (40 мл в отношении 3:1, соответственно) и перемешивают в течение 2 ч. Чистый продукт отфильтровывают и сушат с получением соединения заголовка в виде белого твердого вещества (3,74 г, 92%). Тозилатная соль 4'-трифторметилбифенил 2-карбоновой кислоты (1,2,3,4-тетрагидроизохинолин-6-ил)амида (II). Образец соединения (II), 1,13 г (2,85 ммоль) растворяют в 3 мл этилацетата и добавляют п-толуолсульфоновую кислоту (436 мг, 2,3 ммоль). Содержимое перемешивают при комнатной температуре в течение 18 ч и полученное белое вещество отфильтровывают и промывают этилацетатом. Твердый продукт сушат под пониженным давлением с получением 1,097 г тозилатной соли, т.пл. 187 С. 1 Н ЯМР (250 МГц, ДМСО) : 10,47 (с, 1 Н); 7,73-6,94 (м, 16 Н); 4,54 (с, 2 Н); 2,47(с, 2 Н), 2,25(с, 3 Н). Следующие примеры синтеза являются иллюстративными для тех процедур, которые представлены и описаны выше в способах A-J 43 для синтеза соединений со строением (I), представленным выше. Пример 5. Соединения 20-39. Следующие соединения получают по методике способа А, описанной выше. Следующий синтез является примером методики способа А. Соединение 20.(2-фенетил-1,2,3,4-тетрагидроизохинолин-6-ил)амид. Образец соединения (II), 400 мг (1,01 ммоль), фенилацетальдегид (121 мг, 1,01 ммоль), триацетоксиборогидрид натрия (320 мг,1,52 ммоль) и уксусную кислоту (61 мг, 1,01 ммоль) перемешивают в 10 мл 1,2-дихлорэтане в течение 12 ч при комнатной температуре. Реакционную смесь разбавляют метиленхлоридом, промывают 1N NaOH, водой и насыщенным раствором соли. Очистку проводят хроматографией на силикагеле с использованием 50% этилацетата/гексанов в качестве элюента. МС(Сl): 501 (М+Н+). 1 Н ЯМР (250 МГц, CDCl3) : 3,65 (с,2 Н); 2,91(м, 4 Н); 2,87(м, 4 Н). Следующие соединения синтезируются способом, аналогичным описанному выше для соединения 20. Соединение 21.H ЯМР (250 МГц, CDCl3) : 3,55 (с, 2 Н); 2,82 (т, 2 Н); 2,49 (м, 2 Н); 1,49 (м, 2 Н); 0,93 (с,9 Н). Следующие соединения синтезируют по модификации способа А. Общее описание синтеза и список соединений, которые получают по этой модификации, являются следующими. Раствор соответственно замещенного альдегида (7,5 мкммоль), соединения (II) (5 мкмоль), уксусной кислоты (7,5 мкмоль) и триацетоксиборогидрида натрия (10 мкмоль) в 300 мкл 1,2-дихлорэтана встряхивают в течение 60 ч при комнатной температуре. Отбирали 7,5 мкл образец и разбавляют 93 мкл метанола для ТСХ и МС анализа. Оставшийся образец выпаривали до сухости под пониженным давлением. Неочищенное твердое вещество растворяют в 500 45 мкл этилацетата и промывают 300 мкл 5% бикарбоната натрия. Органический слой концентрируют до сухости под пониженным давлением. Соединение 29. 4'-трифторметилбифенил-2-карбоновой кислоты 2-[2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этил]-1,2,3,4-тетрагидроизохинолин-6-ил-амид. МС(Сl): 570(М+Н+). Пример 6. Соединения 40-67. Следующие соединения получают по методике способа В, описанном выше. Следующий синтез является примером методики способа В. Соединение 40.(0,25 мл, 1,79 ммоль) в ТГФ (7 мл). Через 30 мин ТСХ показывает, что реакция завершена. Добавляют в общей сложности 1,06 г (14,4 ммоль) н-бутиламина и содержимое перемешивают в течение 18 ч при комнатной температуре. Растворитель удаляют под пониженным давлением, остаток растворяют в метиленхлориде,промывают 1N NaOH и сушат над сульфатом натрия. Очистку остатка, полученного при выпаривании, проводят на силикагеле с использованием градиента 0-8% метанола в метиленхлориде в качестве элюента. МС(Сl): 453 (M+Na+). 1 Н ЯМР (250 МГц, ДМСО) : 3,43(с, 2 Н); 2,70(т, 2 Н); 2,57(т, 2 Н); 2, 39 (т, 2 Н); 1,47(м,2 Н); 1,31(м, 2 Н); 0, 88 (т, 3 Н). Следующие соединения синтезируются способом, аналогичным описанному выше для соединения 40. Соединение 41. МС(Сl): 441 (М+Н+). Пример 7. Соединения 68-75. Следующие соединения получают по методике способа С, описанного здесь выше. Следующий синтез является примером методики способа С. Соединение 68.[2-(2-оксо-2-фенилэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]-амид. Соединение (II) (0,30 г, 0,76 ммоль), 2 бромацетофенон (0,15 г, 0,76 ммоль) и карбонат калия (0,12 г, 0,83 ммоль) соединяют в 20 мл ацетонитрила и нагревают до кипения с обратным холодильником в течение 1 ч, а затем растворитель выпаривают под пониженным давлением. Остаток растворяют в хлороформе и промывают насыщенным раствором бикарбоната натрия. Очистку осушенного органического слоя производят с помощью хроматографии на силикагеле, используя этилацетат в гексане в качестве элюента. МС(Сl): 515(М+Н+). Следующие соединения синтезируют способом, аналогичным описанному здесь выше для соединения 68. Детали получения для соединения 74 и 75, которые синтезируют с небольшими модификациями вышеприведенного способа, представлены под соответствующими им заголовками. Соединение 69.[2-(2-цианоэтил)-1,2,3,4-тетрагидроизохинолин 6-ил]-амид. Соединение (II) (780 мг, 1,97 ммоль), 3 бромпропионитрил (290 мг, 2,17 ммоль) и 4 диметиламинопиридин (264 мг, 2,16 ммоль) соединяют в 10 мл ДМФИ нагревают до 70 С в течение 72 ч. Реакционную смесь разбавляют метиленхлоридом, промывают водой и насыщенным раствором соли, а затем сушат над сульфатом магния. Очистку остатка, полученного при выпаривании, производят с помощью хроматографии на силикагеле, используя градиент 10-100% этилацетата в гексане в качестве элюента. МС(Сl): 450 (М+Н+). Соединение 75.(2-пиридин-2-ил)-1,2,3,4-тетрагидроизохинолин-6-ил-амид. Соединение (II) (0,20 г, 0,50 ммоль), 2 бромпиридин (0,16 г, 1,0 ммоль) и карбонат калия (0,14 г, 1,0 ммоль) соединяют в 5 мл хлорбензола и полученную смесь нагревают до кипения с обратным холодильником в течение 48 ч. Растворитель удаляют под пониженным давлением и остаток очищают с помощью хроматографии на силикагеле, используя 50% этилацетат в гексане в качестве элюента. МС(Сl): 474 (М+Н+). 1 Н ЯМР (250 МГц, CDCl3) : 4,63 (с, 2 Н); 3,79 (т, 2 Н); 2,89(т,2 Н). Пример 8. Соединения 76 и 77. Следующие соединения получают по методике способа D, описанного выше. Следующий синтез является примером методики способа D. Соединение 76.[2-(2-пиридин-2-ил-этил)-1,2,3,4-тетрагидроизохинолин-6-ил]-амид. Соединение (II) (300 мг, 0,76 ммоль), 2 винилпиридин (95 мг, 0,91 ммоль) и ледяную уксусную кислоту (24 мг, 0,40 ммоль) соединяют в 15 мл метанола и нагревают до кипения с обратным холодильником в течение 24 ч. Растворитель удаляют под пониженным давлением и остаток очищают с помощью хроматографии на силикагеле с градиентом 0-3% метанола в этилацетате в качестве элюента. МС(Сl): 502 (М+Н+). 1 Н ЯМР (250 МГц, CDCl3) : 3,67(с,2 Н.); 3,09 (м, 2 Н); 2,92(м, 2 Н); 2,80(м,4 Н). Пример 9. Соединения 78-87. Следующие соединения получают по методике способа Е, описанного выше. Следующий синтез является примером методики способа Е. Соединение 78. 4'-трифторметилбифенил-2-карбоновой кислоты 2-[2-(4-метилпиперазин-1-ил)-2-оксоэтил]1,2,3,4-тетрагидроизохинолин-6-ил-амид. Соединение 70 из примера 7 (60 мг, 0,13 ммоль), 1-метилпиперазин (22 мкл, 0,20 ммоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (28 мг, 0,15 ммоль) соединяют в 4 мл безводного метиленхлорида и перемешивают при комнатной температуре в течение 15 ч. Реакционную смесь разбавляют метиленхлоридом и промывают 1N NaOH с последующим промыванием насыщенным раствором соли и затем сушат над сульфатом натрия. Органический слой выпаривают под пониженным давлением и остаток очищают с помощью колоночной хроматографии на силикагеле, используя градиент 1-8% метанола в метиленхлориде в качестве элюента. МС(Сl): 537 (М+Н+). Следующие соединения синтезируют способом, аналогичным описанному выше для соединения 78. Соединение 79.[2-(2-оксо-2-тиазолидин-3-ил-этил)-1,2,3,4 тетрагидроизохинолин-6-ил]-амид. МС(Сl): 526(М+Н+). Пример 10. Соединения (II') и 88. Следующие соединения получают по методике способа F, описанного выше. В целях иллюстрации включено также получение карбонильного соединения (II'). Соединение (II'). 4'-Трифторметилбифенил-2-карбоновой кислоты[2-(2-(тиофен-2-ил-ацетил)-1,2,3,4 тетрагидроизохинолин-6-ил]-амид. Соединение (II) (3,1 г, 7,8 ммоль), 2 тиофенуксусную кислоту (1,14 г; 8,0 ммоль) и 1(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (1,7 г; 8,8 ммоль) перемешивают в 50 мл метиленхлорида в течение 12 ч при комнатной температуре. Реакцонную смесь разбавляют метиленхлоридом, промывают 1N соляной кислотой, 1N ТаОН, водой и насыщенным раствором соли. Очистку остатка, полученного при выпаривании, осуществляли хроматографией на силикагеле, используя 50% этилацетат в гексанах в качестве элюента с получением вышеуказанного соединения в виде твердого вещества. Соединение 88.[2-(2-тиофен-2-ил-этил)-1,2,3,4-тетрагидроизохинолин-6-ил]-амид. Образец соединения (II') 200 мг (0,38 ммоль) и борогидрид натрия (45 мг, 1,2 ммоль) соединяют в 1 мл пиридина и смесь нагревают до кипения с обратным холодильником в течение 18 ч. Пиридин удаляют под пониженным давлением и остаток смешивают с 10% соляной кислотой в течение 20 ч при 50 С. Реакционную смесь делают основной (рН 12) и экстрагируют метиленхлоридом. Органический слой промывают водой и насыщенным раствором соли, а затем сушат над карбонатом калия. Очистку остатка, полученного при выпаривании, выполняли хроматографией на силикагеле с использованием 0,5% метанола в метиленхлориде в качестве элюента. МС(Сl): 507(М+Н+). 1 Н ЯМР (250 МГц, ДМСl3) : 3,65 (с, 2 Н); 3,11(т, 2 Н); 2,79(м, 6 Н). Пример 11. Соединения 89-93. Следующие соединения получают по методике способа G, описанного выше. Нижесле 001539 56 дующий синтез является примером методики способа G. Соединение 89.G представляет собой -(СН 2)2 ОСОСН 3 2-6-[(4'-трифторметилбифенил-2-карбонил)амино]-3,4-дигидро-1 Н-изохинолин-2-илэтиловый эфир уксусной кислоты. Ацетилхлорид (100 мг, 1,25 ммоль) и 4 диметиламинопиридин (700 мг, 5,7 ммоль) соединяли в 5 мл толуола и смесь охлаждают до 0 С на ледяной бане. К этой смеси добавляют раствор соединения 67 из примера (500 мг, 1,14 ммоль) в 3 мл метиленхлорида. Реакционной смеси дают нагреться до комнатной температуры и перемешивают в атмосфере азота в течение 2 ч. Реакционную смесь промывают 1N соляной кислотой, насыщенным раствором бикарбоната натрия и насыщенным раствором соли и затем сушат над сульфатом магния. Очистку остатка,полученного при выпаривании, выполняют хроматографией на силикегеле, используя 3% метанол в этилацетате в качестве элюента. МС(Сl): 483 (М+Н+). 1 Н ЯМР (400 МГц, ДМСО) : 4,25(дд, 2 Н); 3,62 (с, 2 Н); 2,78(м, 6 Н); 2,06(с, 3 Н). Следующие соединения синтезируют способом, аналогичным описанному выше для соединения 89. Соединение 90.G представляет собой -(СН 2)2OCON(CH)2 2-6-[(4'-трифторметилбифенил-2-карбонил)амино]-3,4-дигидро-1 Н-изохинолин-2-илэтиловый эфир диметилкарбаминовой кислоты МС(Сl): 512(М+Н+). 1G представляет собой -(СН 2)2 ОСООСН 3 метиловый эфир карбоновой кислоты 2-6-[(4'трифторметилбифенил-2-карбонил)-амино]-3,4 дигидро-1 Н-изохинолин-2-ил-этиловый эфир. МС(Сl): 499(М+Н+). 1 Н ЯМР (400 МГц, CDCl3) : 4,32(т,2 Н); 3,77(с,3 Н); 3,63 (с, 2 Н); 2,79(м, 6 Н). Соединение 93. 57 МС(Сl): 545(М+Н+). 1 Н ЯМР (400 МГц, CDCl3) : 4,52(т,2 Н); 3,70(с, 2 Н); 2,93(т, 3 Н); 2,83(с, 4 Н). Пример 12. Соединеиния 94-105. Следующие соединения получают по методике способа Н, описанного выше. Соединение 94. 4'-Трифторметилбифенил-2-карбоновой кислоты[2-(2-аминоэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]-амид. Образец соединения 49 в 8,00 г (16,6 ммоль) из примера 6 нагревают до кипения с обратным холодильником в 100 мл 2N соляной кислоты в течение 24 ч. Реакционную смесь делают основной 1N NaOH и смесь экстрагируют этилацетатом. Остаток, полученный при выпаривании, очищают хроматографией на силикагеле, используя градиент 0-10% метанола в метиленхлориде с 1% гидроксида аммония в качестве элюента, с получением 3,5 г вышеуказанного соединения. МС(Сl): 440(М+Н+). 1[2-(2-метансульфониламиноэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]-амид. К 5 мл раствора соединения 94 (250 мг,0,57 ммоль) в метиленхлориде, предварительно охлажденному до 0 С, добавляют метансульфонилхлорид (65 мг, 0,57 ммоль). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 12 ч. Реакционную смесь промывают насыщенным раствором бикарбоната натрия, экстрагируют этилацетатом и сушат над сульфатом магния. Очистку остатка, полученного при выпаривании, выполняют хроматографией на силикагеле, используя 10% метанол в метиленхлориде с 1% гидроксида аммония в качестве элюента. МС(Сl): 518 (М+Н+). 1 Н ЯМР (400 МГц, CDCl3) : 3,62 (с,2 Н),3,29 (м,2 Н); 2,95 (с, 3 Н); 2,77 (м, 6 Н). Следующие соединения синтезируют способом, аналогичным описанному выше для соединения 95. Соединение 96H ЯМР (400 МГц, CDCl3) : 3,62 (м, 4 Н); 2,84(м,2 Н); 2,75(м, 4 Н). Следующие соединения синтезируют по методике для соединения 95, за исключением того, что соответствующий ангидрид кислоты заменяется на галогенангидрид кислоты. Соединение 101. 59 и нагревают до кипения с обратным холодильником в течение 30 мин. Очистку остатка, полученного при выпаривании, выполняют путем хроматографии на силикагеле, используя 10% метанол в метиленхлориде с 1% гидроксида аммония в качестве элюента. МС(Сl): 468(М+Н+). 1 Н ЯМР (400 МГц, CDCl3) : 3,59(с,2 Н); 3,48(м, 2H); 2,83(м,2H); 2,74 (т, 2 Н); 2,67(т,2 Н). Соединение 104.[2-(2-уреидоэтил)-1,2,3,4-тетрагидроизохинолин-6-ил]-амид. Соединение 94 (250 мг, 0,57 ммоль) соединяют с 1,1'-карбонилдиимидазолом в 6 мл метиленхлорида и перемешивают в течение 4 ч. Реакционную смесь охлаждают до 0 С на ледяной бане и через раствор пропускают газообразный аммиак. Реакционную смесь перемешивают при комнатной температуре в течение 12 ч. Очистку остатка, полученного при выпаривании, выполняют путем хроматографии на силикагеле, используя 10% метанол в метиленхлориде с 1% гидроксида аммония в качестве элюента. МС(Сl): 483(М+Н+). 1 Н ЯМР (400 МГц, CDCl3) : 3,62(м, 1 Н); 3,58(с,2 Н); 3,35(м, 1 Н); 2,82(м, 2 Н); 2,76(м, 2 Н); 2,65(м, 2 Н). Следующее соединение синтезируют способом, аналогичным описанному для соединения 104. Соединение 105.(СН 2)2NHCONHCH3 4'-трифторметилбифенил-2 карбоновой кислоты 2-[2-(3-метилуреидо)этил]-1,2,3,4-тетрагидроизохинолин-6-ил-амид. МС(Сl): 497(М+Н+). 1 Н ЯМР (400 МГц, CDCl3) : 3,59(с,2 Н); 3,35(с,2 Н); 2,82(м, 2 Н); 2,74(т,2 Н); 2,70(с,3 Н); 2,65(т,2 Н). Пример 13. Соединения 106-110. Следующие соединения получают по методике способа I, описанного выше. Соединение 106. 4'-трифторметилбифенил-2-карбоновой кислоты(2-пиперидин-4-ил-1,2,3,4-тетрагидроизохинолин-6-ил)-амид. Образец соединения 61, 20 г (415 ммоль) из примера 6 соединяют с гидроксидом палладия на угле (6,1 г) в 260 мл метанола и 550 мл ТГФ. Добавляют формиат аммония (26 г, 415 ммоль) и реакционную смесь нагревают до 60 С в течение 4 ч. Реакционную смесь фильтруют и осадок на фильтре промывают ТГФ с последующим промыванием метанола. Раствор концентрируют и добавляют 40 мл 1N NaOH и 150 60 мл воды. Смесь перемешивают в течение 2 ч и фильтруют с получением 15 г вышеуказанного соединения. Следующие соединения синтезируют путем функционализации соединения 106 во время процедуры восстановительного аминирования способа А. Соединение 107. 4'-трифторметилбифенил-2-карбоновой кислоты[2-(1-метилпиперидин-4-ил)-1,2,3,4-тетрагидроизохинолин-6-ил]-амид. МС(Сl): 494(М+Н+). 1 Н ЯМР (250 МГц, CDCl3) : 3,70 (с, 2 Н); 2,95(д,2 Н); 2,79(с,4 Н); 2,29(с,3 Н). Следующее соединение синтезируют путем функционализации соединения 106 при процедуре амидирования способа Н. Соединение 108. 4'-трифторметилбифенил-2-карбоновой кислотыH ЯМР (250 МГц, CDCl3) : 3,75(с,2 Н); 2,83(с,6 Н). Пример 14. Следующее соединение синтезируют по методике способа J, описанного выше. Соединение 111. 4'-трифторметилбифенил-2-карбоновой кислоты 2-[2-(2 Н-[1,2,4]триазол-3-ил)этил]-1,2,3,4 тетрагидроизохинолин-6-ил-амид. Образец соединения 74, 300 мг (0,67 ммоль) и метоксид натрия (3,6 мг, 0,067 ммоль) соединяют в 2 мл метанола и перемешивают при комнатной температуре в течение 5 ч. До
МПК / Метки
МПК: C07D 217/04, A61P 3/10, A61K 31/472
Метки: мтр, переноса, триглицеридов, аро, секрецию, амиды, аполипопротеина, микросомного, ингибирующие, протеина
Код ссылки
<a href="https://eas.patents.su/30-1539-amidy-ingibiruyushhie-sekreciyu-apolipoproteina-v-aro-v-i-ili-mikrosomnogo-proteina-perenosa-trigliceridov-mtr.html" rel="bookmark" title="База патентов Евразийского Союза">Амиды, ингибирующие секрецию аполипопротеина в (аро в ) и/или микросомного протеина переноса триглицеридов (мтр)</a>
Амиды карбамоилкарбоновой кислоты.

Номер патента: 515
Опубликовано: 28.10.1999
Авторы: Вагнер Оливер, Айкен Карл, Штратман Зигфрид, Дитрих Клаус, Веттерих Франк, Лоренц Гизела, Аммерманн Эберхард
МПК: A01N 47/12, C07C 271/22
Метки: карбамоилкарбоновой, кислоты, амиды
Формула / Реферат:
1. Амиды карбамоилкарбоновой кислоты общей формулы I где R1 означает С1-С8алкил, С2-С8алкенил или С2-С8алкинил, причем эти радикалы могут быть частично либо полностью галогенированы и/или могут быть замещены одной-тремя группами из числа следующих: циано, С1-С4алкокси, С1-C4галогеналкокси, С1-С4алкилтио, C1-С4алкоксикарбонил, С3-С7циклоалкил, С3-С7циклоалкенил, арил, арилокси и гетероарил, причем циклические и ароматические кольца этих...
Производные 2-хинолона, ингибирующие фарнезилтрансферазу

Номер патента: 719
Опубликовано: 28.02.2000
Авторы: Энд Девид Вилльям, Анжибо Патрик Рене, Санз Жерар Шарль, Вене Марк Гастон
МПК: C07D 401/06, A61K 31/47
Метки: производные, ингибирующие, фарнезилтрансферазу, 2-хинолона
Формула / Реферат:
1. Соединение формулы (I) его стереоизомерная форма, фармацевтически приемлемая соль присоединения кислоты или основания, в которых пунктирная линия обозначает необязательную связь; Х представляет кислород или серу; R1 представляет водород, С1-12алкил, Аr1, Аr2C1-6алкил, хинолинилС1-6алкил, пиридилС1-6алкил, гидроксиС1-6алкил, C1-6-алкилоксиC1-6алкил, моно- или ди(C1-6алкил)аминоC1-6алкил, аминоС1-6алкил, или радикал формулы...
(имидазол-5-ил) метил-2-хинолиноновые производные, ингибирующие фарнезилпротеин-трансферазу.

Номер патента: 710
Опубликовано: 28.02.2000
Авторы: Мюллер Филипп, Вене Марк Гастон, Санз Жерар Шарль, Анжибо Патрик Рене
МПК: C07D 401/06, A61K 31/47
Метки: производные, имидазол-5-ил, метил-2-хинолиноновые, фарнезилпротеин-трансферазу, ингибирующие
Формула / Реферат:
1. Соединение формулы (I) его стереоизомерная форма, фармацевтически приемлемая кислотно-аддитивная или основно-аддитивная соль, где пунктирная линия означает необязательную связь; Х представляет кислород или серу; R1 представляет водород, С1-12алкил, Аr1, Аr2C1-6алкил, хинолинил-C1-6алкил, пиридилС1-6алкил, гидроксиС1-6алкил, C1-6алкилокси-C1-6алкил, моно- или ди(С1-6алкил)аминоС1-6алкил, аминоС1-6алкил или радикал формулы...
Предыдущий патент: Неводные суспензии, снижающие сопротивление течению углеводородов в трубопроводах
Следующий патент: Система передачи на борт воздушного судна вещательных программ в реальном времени
Случайный патент: Способ получения стирола из толуола и метанола