Антагонисты мсн-рецепторов
Номер патента: 15127
Опубликовано: 30.06.2011
Авторы: Лу Цзяньлян, Арнольд Маклин Брайан, Гардинир Кевин Мэттью, Грин Стивен Джеймс, Дао Ен, Гармен Дэвид Джозеф, Хембр Эрик Джеймс





























Формула / Реферат
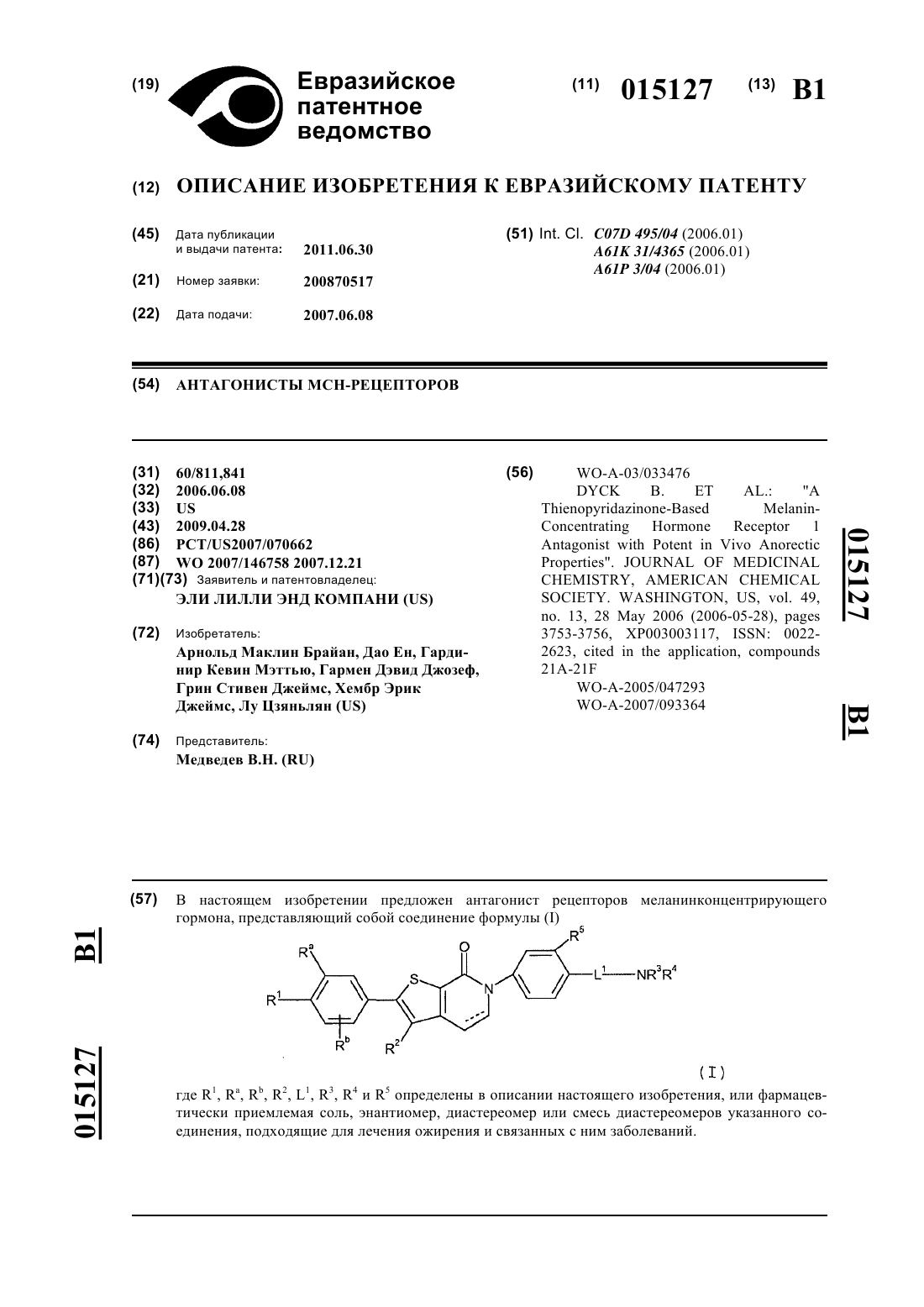
1. Соединение формулы

где ----- представляет необязательную связь, которая образует двойную связь;
R1 независимо выбран из группы, состоящей из водорода, C1-C4-алкила, галогена, гидрокси, C1-C4-галогеналкила, C1-C4-алкокси, C1-C4-галогеналкокси, -О-C3-C4-циклоалкила, -SO2C1-C4-алкила и -NR9R9';
Ra и Rb представляют собой независимо водород, фтор, хлор или метокси;
R2 представляет собой водород или C1-C2-алкил;
L1 выбран из группы, состоящей из связи, -ОСН2СН2-, -ОСН2СН2СН2-, -CF2CH2CH2-, CHFCH2CH2-,
-СН(ОН)СН2СН2-, -NHC(O)CH2-, ОСН2СН=СН2, -NHC(O)CH2CH2, -C(O)CH2CH2-, -C(O)NHCH2CH2-,
-NH(CO)CH2CH2CH2- и -C(O)NHCH2CH2CH2;
R3 и R4независимо выбраны из группы, состоящей из водорода, C1-C4-алкила, или R3и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 4-7-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1в положении a, b, g или d по отношению к атому азота в составе NR3R4 образует 4-7-членную азотсодержащую гетероциклическую группу с L1, при этом каждая 4-7-членная азотсодержащая гетероциклическая группа, образованная объединением R3и R4 или L1и одним из R3 и R4, необязательно замещена одной или двумя группами, выбранными независимо из оксо, гидрокси, -OR6, галогена, C1-C4-алкила, -C(O)C1-C4-алкила, C3-C6-циклоалкила и -NR6R6';
R5 представляет собой водород, хлор, фтор, циано, метил, трифторметокси или метокси;
R6 и R6' независимо выбраны из группы, состоящей из водорода и C1-C4-алкила;
R9 и R9' независимо выбраны из группы, состоящей из водорода и C1-C3-алкила;
или фармацевтически приемлемая соль или энантиомер, диастереомер или смесь диастереомеров указанного соединения.
2. Соединение по п.1, отличающееся тем, что
----- представляет необязательную связь, которая образует двойную связь;
R1 независимо выбран из группы, состоящей из водорода, C1-C3-алкила, галогена, C1-C3-алкокси, -OC3-C4-циклоалкила и C1-C3-галогеналкила;
Ra и Rb представляют собой независимо водород или хлор;
R2 представляет собой водород;
L1 выбран из группы, состоящей из связи, -ОСН2СН2-, CF2CH2CH2-, -CHFCH2CH2-, СН(ОН)СН2СН2-,
-ОСН2СН2СН2-, -ОСН2СН=СН2-, -NHC(O)CH2-, -NHC(O)CH2CH2-, -С(O)СН2СН2-, -С(O)NHCH2CH2- и
-C(O)NHCH2CH2CH2,
R3 и R4независимо выбраны из группы, состоящей из водорода, C1-C4-алкила, или R3и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 4-7-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1в положении a, b, g или d по отношению к атому азота в составе NR3R4 образует 4-7-членный азотсодержащий гетероцикл с L1, при этом каждый из 4-7-членных азотсодержащих гетероциклов, образованных объединением R3и R4 или L1и одним из R3 и R4, выбран из группы, состоящей из азетидинила, морфолино, пирролидинила, имидазолила, пиперазинила и пиперидинила, и каждый необязательно замещен одной или двумя группами, выбранными независимо из группы, состоящей из оксо, галогена, гидрокси, -OR6, -C1-C4-алкила, C(O)C1-C4-алкила и -NR6R6';
R5 выбран из группы, состоящей из -ОМе, хлора, фтора и циано;
R6 и R6' независимо выбраны из группы, состоящей из водорода и -C1-C4-алкила;
R9 и R9' представляют собой независимо водород или метил.
3. Соединение по п.1, отличающееся тем, что
R1 представляет собой метил, хлор, метокси, фтор, трифторметил или циклопропокси;
Ra и Rb представляют собой независимо водород, хлор, фтор или метокси;
R2 представляет собой водород;
L1 представляет собой связь;
R3 и R4независимо выбраны из группы, состоящей из водорода, C1-C4-алкила, или R3и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 4-7-членный азотсодержащий гетероцикл, при этом указанный гетероцикл выбран из группы, состоящей из пирролидинила, морфолино, пиперидинила, пиперазинила, имидазолила и азетидинила, и каждый необязательно замещен одной или двумя группами, выбранными независимо из гидрокси, метила, фтора, -N-метиламина, N,N-диметиламина, оксо, циклопропила и циклобутила;
R5 представляет собой водород, -ОСН3, циано, фтор или хлор.
4. Соединение по п.1, отличающееся тем, что
R1 представляет собой метил, хлор, метокси, фтор, трифторметил или циклопропокси;
Ra и Rb представляют собой независимо водород, фтор или метокси;
R2 представляет собой водород;
L1 выбран из группы, состоящей из связи, -ОСН2СН2-, -NHC(O)CH2CH2-, -NHC(O)СН2-, -С(O)СН2СН2- и
-C(O)NHCH2CH2-;
R3 и R4независимо выбраны из группы, состоящей из водорода, C1-C4-алкила, или R3и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 4-7-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1в положении a, b или g по отношению к атому азота в составе NR3R4образует 4-7-членный азотсодержащий гетероцикл с L1, при этом каждый из 4-7-членных азотсодержащих гетероциклов, образованных объединением R3 и R4или L1 и одним из R3и R4, выбран из группы, состоящей из пирролидинила, морфолино, пиперидинила, пиперазинила, имидазолила и азетидинила, и каждый необязательно замещен одной или двумя группами, выбранными независимо из гидрокси, метила, фтора, -N-метиламина, N,N-диметиламина, оксо, циклопропила и циклобутила;
R5 представляет собой водород, -ОСН3, циано, фтор или хлор.
5. Соединение по п.1, отличающееся тем, что
R1 представляет собой хлор, метокси или трифторметокси;
каждый из Ra и Rb представляет собой водород;
R2 представляет собой водород;
L1 выбран из группы, состоящей из -CF2CH2CH2-, -CHFCH2CH2- и -СН(ОН)СН2СН2-;
R3 и R4независимо выбраны из группы, состоящей из водорода, C1-C4-алкила, или R3и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 4-7-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1в положении a, b или g по отношению к атому азота в составе NR3R4образует 4-7-членный азотсодержащий гетероцикл с L1, при этом каждый из 4-7-членных азотсодержащих гетероциклов, образованных объединением R3 и R4 или L1и одним из R3 и R4, выбран из группы, состоящей из пирролидинила, пиперидинила и пиперазинила, при этом каждый из 4-7-членных азотсодержащих гетероциклов, образованных R3и R4 или L1и одним из R3 и R4, необязательно замещен одной или двумя группами, выбранными независимо из метила, фтора, -N-метиламина, N,N-диметиламина и циклобутила;
R5 выбран из группы, состоящей из водорода, метокси, хлора и фтора.
6. Соединение по п.1, отличающееся тем, что
R1 представляет собой хлор, трифторметил или метокси;
Ra и Rb выбраны независимо из водорода, фтора и хлора;
R2 представляет собой водород;
L1 представляет собой связь, -ОСН2СН2- или -ОСН2СН2СН2-;
R3 и R4совместно с атомом азота, с которым они связаны, образуют 4-7-членный азотсодержащий гетероцикл, выбранный из группы, состоящей из пирролидинила, морфолино, пиперидинила, пиперазинила, имидазолила и азетидинила, при этом каждый из указанных 4-7-членных азотсодержащих гетероциклов необязательно замещен одной или двумя группами, выбранными из гидрокси, метила, фтора, -N-метиламина, N,N-диметиламина, оксо, циклопропила и циклобутила;
R5 представляет собой водород, -ОСН3, циано, фтор или хлор.
7. Соединение по п.1, отличающееся тем, что
R1 представляет собой хлор, метокси, трифторметил или трифторметокси;
каждый из Ra и Rb представляет собой водород;
R2 представляет собой водород;
L1 выбран из группы, состоящей из -NHC(O)CH2-, -NHC(О)СН2СН2-, -C(O)CH2CH2-, C(O)NHCH2CH2и
-С(О)NHCH2CH2CH2-;
R3 и R4независимо выбраны из группы, состоящей из водорода, C1-C4-алкила, или R3и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 4-7-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1в положении a, b или g по отношению к атому азота в составе NR3R4образует 4-7-членный азотсодержащий гетероцикл с L1, при этом каждый из 4-7-членных азотсодержащих гетероциклов, образованных объединением R3 и R4 или L1и одним из R3 и R4, выбран из группы, состоящей из пирролидинила, морфолино, пиперидинила, пиперазинила, имидазолила и азетидинила, и необязательно замещен одной или двумя группами, выбранными независимо из гидрокси, метила, фтора, -N-метиламина, N,N-диметиламина, циклобутила и оксо;
R5 выбран из группы, состоящей из водорода, метокси, циано и хлора.
8. Соединение по п.1, отличающееся тем, что
R1 представляет собой хлор, фтор, метокси, трифторметил или трифторметокси;
каждый из Ra и Rb представляет собой водород;
R2 представляет собой водород;
L1 представляет собой связь;
R3 и R4совместно с атомом азота, с которым они связаны, образуют 4-7-членный азотсодержащий гетероцикл, выбранный из группы, состоящей из пирролидинила, пиперидинила, диазепанила и морфолино; при этом каждый из указанных 4-7-членных азотсодержащих гетероциклов необязательно замещен группой, выбранной из группы, состоящей из -ОН, -NHCH3, -N(CH3)2, -СН3, циклобутила и фтора;
R5 представляет собой водород, метил, метокси, циано или хлор.
9. Соединение по п.1, отличающееся тем, что
R1 представляет собой хлор, фтор, метокси, трифторметил или трифторметокси;
каждый из Ra и Rb представляет собой водород;
R2 представляет собой водород;
L1 представляет собой -ОСН2СН2- или -ОСН2СН2СН2-;
R3 и R4совместно с атомом азота, с которым они связаны, образуют 4-7-членный азотсодержащий гетероцикл, выбранный из группы, состоящей из пирролидинила, пиперидинила, диазепанила и морфолино, при этом каждый из указанных 4-7-членных азотсодержащих гетероциклов необязательно замещен группой, выбранной из группы, состоящей из -ОН, -NHCH3, -N(CH3)2, -CH3, циклобутила и фтора;
R5 представляет собой водород, метил, метокси или циано.
10. Соединение по п.1, отличающееся тем, что
R1 представляет собой хлор, фтор, метокси, трифторметил или трифторметокси;
Ra и Rb независимо выбраны из водорода, фтора и хлора;
R2 представляет собой водород;
L1 выбран из группы, состоящей из -NHC(O)CH2-, NHC(O)CH2CH2, -C(O)NHCH2CH2-, NHC(О)СН2СН2СН2и -С(О)NHCH2CH2CH2-;
R3 и R4независимо выбраны из группы, состоящей из водорода, C1-C4-алкила, или R3и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 4-7-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1в положении a, b или g по отношению к атому азота в составе NR3R4образует 4-7-членный азотсодержащий гетероцикл с L1, при этом каждый из 4-7-членных азотсодержащих гетероциклов, образованных объединением R3 и R4 или L1и одним из R3 и R4, выбран из группы, состоящей из пирролидинила, пиперидинила и морфолино, и каждый из 4-7-членных азотсодержащих гетероциклов, образованных объединением R3и R4 или L1и одним из R3 и R4, необязательно замещен группой, выбранной из группы, состоящей из -ОН, -NHCH3, -N(CH3)2, -СН3, циклобутила и фтора;
R5 представляет собой водород, хлор, фтор или метокси.
11. Соединение по п.1, выбранное из группы, состоящей из
гидрохлорида 2-(4-метоксифенил)-6-[3-метокси-4-(2-пирролидин-1-илэтокси)фенил]-5,6-дигидро-4Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 6-[3-метокси-4-(2-морфолин-4-илэтокси)фенил]-2-(4-метоксифенил)-5,6-дигидро-4Н-тиено[2,3-c]пиридин-7-она,
2-(2,4-дихлорфенил)-6-[3-метокси-4-(2-пирролидин-1-илэтокси)фенил]-5,6-дигидро-4Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 6-[3-метокси-4-(2-пирролидин-1-илэтокси)фенил]-2-(4-трифторметилфенил)-5,6-дигидро-4Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида N-{4-[2-(4-хлорфенил)-7-оксо-4,7-дигидро-5Н-тиено[2,3-c]пиридин-6-ил]-2-метоксифенил}-2-пирролидин-1-илацетамида,
гидрохлорида N-{4-[2-(4-хлорфенил)-7-оксо-7Н-тиено[2,3-с]пиридин-6-ил]-2-метоксифенил}-2-пирролидин-1-илацетамида,
гидрохлорида 2-(4-хлорфенил)-6-[4-((R)-3-гидроксипирролидин-1-ил)-3-метоксифенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-((R)-пиперидин-3-илокси)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-(3-фтор-4-пиперазин-1-илфенил)-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-((S)-пиперидин-3-илокси)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-((R)-пирролидин-3-илокси)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-(пиперидин-4-илокси)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-((R)-1-метилпиперидин-3-илокси)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-((S)-1-метилпиперидин-3-илокси)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-(1-метилазетидин-3-илокси)фенил]-5,6-дигидро-4Н-тиено[2,3-с]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-фтор-4-(1-метилазетидин-3-илокси)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-(1-метилазетидин-3-илокси)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-(2-пирролидин-1-илэтокси)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-(3-метил-3H-имидазол-4-илметокси)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[4-((S)-3-диметиламинопирролидин-1-ил)-3-метоксифенил]-6Н-тиено[2,3-с]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-((S)-3-метиламинопирролидин-1-ил)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[4-((R)-3-диметиламинопирролидин-1-ил)-3-метоксифенил]-6Н-тиено[2,3-с]пиридин-7-она,
2-(4-хлорфенил)-6-[3-метокси-4-((S)-4-метил-5-оксоморфолин-3-илметокси)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[4-(1-метилпирролидин-3-карбонил)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[4-(1-метилазетидин-3-карбонил)фенил]-6Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида (±)-транс-2-(4-хлорфенил)-6-[4-(4-гидрокси-1-метилпирролидин-3-илокси)-3-метоксифенил]-6Н-тиено[2,3-с]пиридин-7-она,
гидрохлорида 2-(4-хлорфенил)-6-[3-метокси-4-(4-метилпиперазин-1-ил)фенил]-5,6-дигидро-4Н-тиено[2,3-c]пиридин-7-она,
гидрохлорида (±)-транс-2-(4-хлорфенил)-6-[4-(4-гидроксипирролидин-3-илокси)-3-метоксифенил]-6Н-тиено[2,3-с]пиридин-7-она,
гидрохлорида {4-[2-(4-хлорфенил)-7-оксо-7Н-тиено[2,3-с]пиридин-6-ил]фенил}амида (±)-1-метилпирролидин-3-карбоновой кислоты,
гидрохлорида транс-2-(4-хлорфенил)-6-[4-(4-гидрокси-1-метилпирролидин-3-илокси)-3-метоксифенил]-6Н-тиено[2,3-с]пиридин-7-она, изомер 1,
гидрохлорида транс-2-(4-хлорфенил)-6-[4-(4-гидрокси-1-метилпирролидин-3-илокси)-3-метоксифенил]-6Н-тиено[2,3-с]пиридин-7-она, изомер 2.
12. Соединение N-{4-[2-(4-хлорфенил)-7-оксо-4,7-дигидро-5Н-тиено[2,3-c]пиридин-6-ил]-2-метоксифенил}-2-пирролидин-1-илацетамид, хлористо-водородная соль

13. Соединение N-{4-[2-(4-хлорфенил)-7-оксо-7Н-тиено[2,3-с]пиридин-6-ил]-2-метоксифенил}-2-пирролидин-1-илацетамид, хлористо-водородная соль

14. Фармацевтическая композиция, содержащая соединение по любому из пп.1-13 и фармацевтически приемлемый носитель и/или разбавитель.
15. Применение соединения по любому из пп.1-13 для получения лекарственного средства для лечения ожирения.
Текст
В настоящем изобретении предложен антагонист рецепторов меланинконцентрирующего гормона, представляющий собой соединение формулы (I) где R1, Ra, Rb, R2, L1, R3, R4 и R5 определены в описании настоящего изобретения, или фармацевтически приемлемая соль, энантиомер, диастереомер или смесь диастереомеров указанного соединения, подходящие для лечения ожирения и связанных с ним заболеваний.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) 015127 Область изобретения Настоящее изобретение относится к области медицины, в частности к лечению ожирения и связанных с ним заболеваний. Уровень техники Меланинконцентрирующий гормон (МСН) представляет собой нейропептид, содержащий остатки 19 аминокислот, вырабатываемый в латеральной области гипоталамуса и неопределенной зоне (zonaincerta). Имеется большое количество данных, свидетельствующих о том, что МСН обладает активностью в отношении возбуждения аппетита. Сообщалось, что мыши с генотипом MCHR1-/- отличаются худобой и ускоренным обменом веществ, что указывает на то, что, по меньшей мере, некоторые из метаболических эффектов МСН опосредуются через изоформу R1. В публикации международной заявки WO 03/033476 А 1 предложены пиримидиноны, подходящие для применения в качестве антагонистов рецепторов меланинконцентрирующего гормона. В публикации международной заявки WO 2005/047293 А 1 предложены соединения, которые, как указано, являются подходящими в качестве антагонистов МСН. В работе Дика и др. (Dyck, B. et al., Journal of MedicinalHormone Receptor 1 Antagonist with Potent In Vivo Anorectic Properties" предложены тиенопиридазиноны,которые, как указано, пригодны для применения в качестве антагонистов МСН. В настоящее время требуются высокоактивные избирательные и терапевтически эффективные средства, позволяющие более эффективно контролировать пищевые привычки и свести к минимуму риск развития ожирения, а также подходящие для лечения и/или облегчения нарушений, обусловленных ожирением и связанными с ним заболеваниями. В настоящем изобретении предложены особенно предпочтительные соединения, обладающие высокой активностью и избирательностью, которые эффективны invivo в качестве антагонистов МСН и подходят для лечения ожирения и связанных с ним заболеваний. Краткое описание изобретения В настоящем изобретении предложено соединение формулы (I) гдепредставляет необязательную связь, которая образует двойную связь;R1 независимо выбран из группы, состоящей из водорода, C1-C4-алкила, галогена, гидрокси, C1-C4 галогеналкила, C1-C4-алкокси, C1-C4-галогеналкокси, -O-C3-C4-циклоалкила, -SO2C1-C4-алкила и -NR9R9';Ra и Rb представляют собой независимо водород, фтор, хлор или метокси;R2 представляет собой водород или C1-C2-алкил;R3 и R4 независимо выбраны из группы, состоящей из водорода, C1-C4-алкила, или R3 и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 4-7-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1 в положении , ,илипо отношению к атому азота в составе NR3R4 образует 4-7-членную азотсодержащую гетероциклическую группу с L1, при этом каждая 4-7-членная азотсодержащая гетероциклическая группа, образованная объединением R3 и R4 или L1 и одним из R3 и R4, необязательно замещена одной или двумя группами, выбранными независимо из оксо, гидрокси, -OR6, галогена, C1-C4-алкила, -C(O)C1-C4-алкила, C3-C6-циклоалкила иR6 и R6' независимо выбраны из группы, состоящей из водорода и C1-C4-алкила;R9 и R9' независимо выбраны из группы, состоящей из водорода и C1-C3-алкила,или фармацевтически приемлемая соль или энантиомер, диастереомер или смесь диастереомеров указанного соединения. В настоящем изобретении также предложена фармацевтическая композиция, содержащая соединение формулы (I). В настоящем изобретении предложено применение соединения формулы (I) для лечения ожирения и связанных с ним заболеваний.-1 015127 Подробное описание изобретения Для целей настоящего изобретения в настоящем описании и прилагаемой формуле изобретения используются следующие термины и определения, если не указано иное. Общепринятые химические термины, используемые в настоящем изобретении при описании соединений, употребляются в их обычном значении. Например, термин "C1-C4-алкил" относится к линейной или разветвленной алифатической цепи, содержащей от 1 до 4 атомов углерода, и ее изомерам, включающим без ограничения метил, этил, пропил, изопропил и н-бутил. Аналогичным образом, термин "C1C4-алкил" включает C1-C3-алкил, C1-C2-алкил, C2-C3-алкил и C2-C4-алкил, каждый из которых содержит указанное количество атомов углерода. Термин "C3-C6-циклоалкил" относится к насыщенному карбоциклу, содержащему от 3 до 6 атомов углерода, включая циклопропил, циклобутил, циклопентил и циклогексил. Аналогичным образом, термин "C3-C4-циклоалкил" относится к группе, включающей циклопропил и циклобутил. Термин "C3-C6 галогеналкил" включает термин "C3-C5-галогеналкил" и т.д. Термин "галоген" относится к галогену, т.е. атому хлора, брома, йода и фтора. Термин "C1-C4-галогеналкил" относится к C1-C4-алкильной группе, содержащей в качестве заместителей один, два, три или более атомов галогена, как указано в настоящем описании или возможно с химической точки зрения. Примеры C1-C4-галогеналкила включают без ограничения трифторметил, хлорэтил и 2-хлорпропил. Аналогичным образом, "C2-C3-галогеналкил" представляет собой метильную или этильную группу, содержащую в качестве заместителей от одного до максимально возможного количества атомов галогена, предпочтительно хлора или фтора. Для специалиста в данной области техники очевидно, что C1-C4-галогеналкил включает C1-C3-галогеналкил и C2-C3-галогеналкил."C1-C4-алкоксигруппа" представляет собой C1-C4-алкильный (или другой указанный) фрагмент,присоединенный через атом кислорода. Примеры алкоксигрупп включают без ограничения метокси(-ОМе), этокси (-OEt), пропокси (-OPr), изопропокси (-OiPr), бутокси (-OBu) и др. Аналогичным образом,термин "C1-C3-алкокси" включает метокси (-ОМе), этокси (-OEt), пропокси (-OPr), изопропокси (-OiPr). Аналогично, C1-C2-алкокси включает ОМе и OEt группы. Термин "C1-C4-галогеналкокси" включает C1-C4-алкокси, где один или несколько атомов водорода в алкиле замещены атомами галогена. Примеры галогеналкоксигрупп включают дифторметокси, трифторметокси, 2-галогенэтокси, 2,2,2-трифторэтокси, 4,4,4-трифторбутокси и другие аналогичные группы, содержащие указанное количество атомов углерода. Например, C1-C2-галогеналкокси включает группыOCF3, OCH2CH2F и другие группы, содержащие один или два атома углерода и подходящее количество атомов галогена. Согласно настоящему изобретению термин "C1-C4-алкил" также включает конкретный алкил, который при присоединении может приводить к появлению хиральности. Полученные хиральные соединения также являются объектами настоящего изобретения. Термины , , или относятся соответственно к положению 1, 2, 3 или 4 по отношению к атому азота в составе группы NR3R4 в соединении формулы (I) при отсчете в направлении против часовой стрелки. Термины , , или обозначают положение в соединении формулы (I), где один изR3 и R4 совместно с атомом в цепи L1 (линкере L1) образует гетероциклическое кольцо. Для специалиста в данной области техники очевидно, что при объединении R3 и R4 или объединении R3 или R4 с L1 с образованием 4-7-членного азотсодержащего гетероцикла, как указано в настоящем описании, должно происходить отщепление одного или двух атомов водорода из СН или СН 2 группы в составе одной или обеих объединяющихся групп. Кроме того, согласно настоящему изобретению подразумевается, что если один из R3 и R4 совместно с L1 образует (4-7-членный) азотсодержащий гетероцикл, то другой из R3 и R4 представляет собой либо атом водорода, либо необязательный заместитель в указанном гетероцикле, при этом необязательные заместители определены ниже или являются такими, как указано для конкретных групп в соединениях формулы (I). Термин "азотсодержащий гетероциклический" означает насыщенный, частично ненасыщенный,полностью ненасыщенный или ароматический 4-, 5-, 6- или 7-членный (или другой указанный) фрагмент, который может дополнительно содержать гетероатомы, выбранные из азота и кислорода. Типичные гетероциклические группы включают азетидинил, морфолинил, пиперидинил, пиперазинил, диазепанил и пирролидинил. Соответственно используемый в настоящем описании термин "4-7-членная азотсодержащая гетероциклическая группа" включает по отдельности и/или в совокупности 4-6-, 5-6-, 5-7- и 4-7-членные азотсодержащие гетероциклические группы. Используемый в настоящем описании термин "оксо" означает атом кислорода, присоединенный к атому углерода, входящему в состав кольца или цепи, с образованием карбонильной группы. В настоящем изобретении предложены химически стабильные соединения, при этом специалисту в данной области техники известна конкретная комбинация заместителей в рамках настоящего изобретения, обеспечивающая химическую стабильность, в том числе присоединение или отщепление атомов водорода, приводящее к образованию указанного в настоящем описании и/или предполагаемого химически стабильного соединения.-2 015127 Термин "подходящий растворитель" относится к любому растворителю или смеси растворителей,которые инертны в условиях протекающей реакции, обеспечивают достаточную солюбилизацию взаимодействующих веществ и создают среду для протекания требуемой реакции. Используемый в настоящем описании термин "пациент" относится к людям, домашним животным(например, собакам, кошкам и др.) и домашнему скоту. Термины "лечение", "лечить" и "лечащий" включают облегчение, прекращение, ограничение, замедление и обратное прогрессирование или снижение степени тяжести патологических симптомов ожирения и связанных с ним заболеваний. Используемый в настоящем описании термин "терапевтически эффективное количество" означает количество соединения согласно настоящему изобретению, которое способно обеспечивать излечение симптомов различных патологических состояний, указанных в настоящем описании. Термин "фармацевтически приемлемый" в настоящем описании используется в качестве прилагательного и означает "по существу, безвредный" для пациента, принимающего препарат. Используемые в настоящем описании термины "заболевания, связанные с ожирением" или "родственные заболевания" относятся к симптомам, заболеваниям или состояниям, которые обусловлены ожирением, обостряются при ожирении, спровоцированы ожирением или сопутствуют ожирению. Указанные заболевания, состояния и/или симптомы включают без ограничения расстройства пищевого поведения (булимию, нервную анорексию и др.), диабет, осложнения при диабете, диабетическую ретинопатию, депрессию, тревожность, гипертензию, внутримозговое кровоизлияние, застойную сердечную недостаточность, атеросклероз, ревматоидный артрит, удар, гиперлипидемию, гипертригликемию, гипергликемию и гиперлипопротеинемию. Фармацевтически приемлемые соли и способы их получения хорошо известны специалисту в данной области техники, см., например, P. Stahl, et al. Handbook of Pharmaceutical Salts: Properties, Selectionsand Use (VCHA/Wiley-VCH, 200); S.M. Berge, et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, vol. 66, No. 1, January 1977. Предпочтительные соединения согласно изобретению Некоторые соединения согласно настоящему изобретению представляют особенный интерес и являются предпочтительными. Далее приведено несколько групп предпочтительных соединений. Очевидно, что каждую из таких групп можно комбинировать с другими группами, представленными в настоящем описании, с образованием дополнительных групп предпочтительных соединений, которые также находятся в рамках настоящего изобретения. Предпочтительные группы R1 независимо выбраны из группы, состоящей из хлора, фтора, метила,трифторметила, трифторметокси, пропокси, -N(СН 3), -SO2CH3 и циклопропокси. Более предпочтительноR1 выбран из группы, состоящей из фтора, хлора, циклопропокси, трифторметокси и метокси. Наиболее предпочтительно R1 представляет собой хлор или метокси. Предпочтительно Ra и Rb представляют собой независимо водород, фтор или метокси.R2 предпочтительно представляет собой водород. Предпочтительно L1 выбран из группы, состоящей из связи, -ОСН 2 СН 2-, -CF2CH2CH2-,-CHFCH2CH2-, СН(ОН)СН 2 СН 2-, -ОСН 2 СН 2 СН 2-, -ОСН 2 СН=СН 2-, -NHC(O)CH2-, -NHC(O)CH2CH2-,-С(О)СН 2 СН 2-, C(O)NHCH2CH2- и -C(O)NHCH2CH2CH2. Предпочтительно R3 и R4 совместно с атомом азота, к которому они присоединены, образуют необязательно замещенный 4-7-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1 в положении ,илипо отношению к атому азота в составе NR3R4 образует 4-7-членный азотсодержащий гетероцикл, необязательно замещенный, при этом указанный гетероцикл выбран из группы, состоящей из азетидинила, морфолино, пирролидинила, имидазолила, пиперазинила, диазепанила и пиперидинила, и каждый из 4-7-членных азотсодержащих гетероциклов, образованных R3 и R4 или же L1 и любым из R3 и R4, необязательно замещен одной или двумя группами, выбранными независимо из метила, оксо, гидрокси, галогена, амино, N-метиламина и N,N-диметиламина. Предпочтительно группа R5 представляет собой водород, хлор, фтор, трифторметокси, метокси или циано. Более предпочтительно R5 представляет собой водород, фтор, хлор или метокси. Наиболее предпочтительно R5 представляет собой метокси, хлор или фтор. Предпочтительно R6 или R6' независимо выбраны из водорода и C1-C2-алкила. Более предпочтительно группы R6 и R6' независимо выбраны из водорода и метила. Предпочтительно R9 и R9' независимо выбраны из группы, состоящей из водорода и метила. Согласно настоящему изобретению предпочтительным является соединение формулы (I), где представляет необязательную связь, которая может образовывать двойную связь;R1 независимо выбран из группы, состоящей из водорода, C1-C3-алкила, галогена, C1-C3-алкокси,-OC3-C4-циклоалкила и C1-C3-галогеналкила;Ra и Rb представляют собой независимо водород или хлор;R3 и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 47-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1 в положении , ,илипо отношению к атому азота в составе NR3R4 образует 4-7-членный азотсодержащий гетероцикл с L1, при этом каждый из 4-7-членных азотсодержащих гетероциклов, образованных R3 и R4 или L1 и одним из R3 и R4, выбран из группы, состоящей из азетидинила, морфолино, пирролидинила, имидазолила, пиперазинила и пиперидинила, и каждый из указанных азотсодержащих гетероциклов необязательно замещен одной или двумя группами, выбранными независимо из оксо, галогена, гидрокси, -OR6, -C1-C4-алкила,С(O)C1-C4-алкила и -NR6R6';R6 и R6' независимо выбраны из группы, состоящей из водорода и -C1-C4-алкила;R9 и R9' представляют собой независимо водород или метил. Также предпочтительным является соединение формулы (I), гдеRa и Rb представляют собой независимо водород, хлор, фтор или метокси;R3 и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 47-членный азотсодержащий гетероцикл, при этом указанный гетероцикл выбран из группы, состоящей из пирролидинила, морфолино, пиперидинила, пиперазинила, имидазолила и азетидинила, и каждый из указанных гетероциклов необязательно замещен одной или двумя группами, выбранными независимо из гидрокси, метила, фтора, -N-метиламина, N,N-диметиламина, оксо, циклопропила и циклобутила;R5 представляет собой водород, -ОСН 3, циано, фтор или хлор. Также предпочтительным является соединение формулы (I), гдеRa и Rb представляют собой независимо водород, фтор или метокси;R3 и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 47-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1 в положении ,илипо отношению к атому азота в составе NR3R4 образует 4-7-членный азотсодержащий гетероцикл с L1, при этом каждый из 4-7-членных азотсодержащих гетероциклов, образованных R3 и R4 или L1 и одним из R3 и R4, выбран из группы, состоящей из пирролидинила, морфолино, пиперидинила, пиперазинила, имидазолила и азетидинила, и каждый из указанных азотсодержащих гетероциклов необязательно замещен одной или двумя группами, выбранными независимо из гидрокси, метила, фтора, -N-метиламина, N,Nдиметиламина, оксо, циклопропила и циклобутила;R5 представляет собой водород, -ОСН 3, циано, фтор или хлор. Также предпочтительным является соединение формулы (I), гдеR3 и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 47-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1 в положении ,илипо отношению к атому азота в составе NR3R4 образует 4-7-членный азотсодержащий гетероцикл с L1, при этом каждый из 4-7-членных азотсодержащих гетероциклов, образованных R3 и R4 или L1 и одним из R3 и R4, выбран из группы, состоящей из пирролидинила, пиперидинила и пиперазинила, и каждый из 4-7 членных азотсодержащих гетероциклов, образованных R3 и R4 или L1 и одним из R3 и R4, необязательно замещен одной или двумя группами, выбранными независимо из метила, фтора, -N-метиламина, N,Nдиметиламина и циклобутила;R5 выбран из группы, состоящей из водорода, метокси, хлора и фтора. Также предпочтительным является соединение формулы (I), гдеRa и Rb выбраны независимо из водорода, фтора и хлора;R3 и R4 совместно с атомом азота, с которым они связаны, образуют 4-7-членный азотсодержащий гетероцикл, выбранный из группы, состоящей из пирролидинила, морфолино, пиперидинила, пиперазинила, имидазолила и азетидинила, при этом каждый из указанных 4-7-членных азотсодержащих гетероциклов необязательно замещен одной или двумя группами, выбранными из гидрокси, метила, фтора,-4 015127R5 представляет собой водород, -ОСН 3, циано, фтор или хлор. Также предпочтительным является соединение формулы (I), гдеR3 и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 47-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1 в положении ,илипо отношению к атому азота в составе NR3R4 образует 4-7-членный азотсодержащий гетероцикл с L1, при этом каждый из 4-7-членных азотсодержащих гетероциклов, образованных R3 и R4 или L1 и одним из R3 и R4, выбран из группы, состоящей из пирролидинила, морфолино, пиперидинила, пиперазинила, имидазолила и азетидинила, и необязательно замещен одной или двумя группами, выбранными независимо из гидрокси, метила, фтора, -N-метиламина, N,N-диметиламина, циклобутила и оксо;R5 выбран из группы, состоящей из водорода, метокси, циано и хлора. Также предпочтительным является соединение формулы (I), гдеR3 и R4 совместно образуют 4-7-членный азотсодержащий гетероцикл, выбранный из группы, состоящей из пирролидинила, пиперидинила, диазепанила и морфолино, при этом каждый из указанных 47-членных азотсодержащих гетероциклов необязательно замещен одной группой, выбранной из -ОН,-NHCH3, -N(CH3)2, -CH3, циклобутила и фтора; иR5 представляет собой водород, метил, метокси, циано или хлор. Также предпочтительным является соединение формулы (I), гдеR3 и R4 совместно образуют 4-7-членный азотсодержащий гетероцикл, выбранный из группы, состоящей из пирролидинила, пиперидинила, диазепанила и морфолино, при этом каждый из указанных 47-членных азотсодержащих гетероциклов необязательно замещен одной группой, выбранной из -ОН,-NHCH3, -N(СН 3)2, -СН 3, циклобутила и фтора; иR5 представляет собой водород, метил, метокси или циано. Также предпочтительным является соединение формулы (I), гдеRa и Rb независимо выбраны из водорода, фтора и хлора;R3 и R4 совместно с атомом азота, с которым они связаны, образуют необязательно замещенный 47-членный азотсодержащий гетероцикл, или один из R3 и R4 совместно с L1 в положении ,илипо отношению к атому азота в составе NR3R4 образует 4-7-членный азотсодержащий гетероцикл с L1, при этом каждый из 4-7-членных азотсодержащих гетероциклов, образованных R3 и R4 или L1 и одним из R3 и R4, выбран из группы, состоящей из пирролидинила, пиперидинила и морфолино, и каждый из 4-7 членных азотсодержащих гетероциклов, образованных R3 и R4 или L1 и одним из R3 и R4, необязательно замещен группой, выбранной из -ОН, -NHCH3, -N(CH3)2, -CH3, циклобутила и фтора; иR5 представляет собой водород, хлор, фтор или метокси. Получение соединений согласно настоящему изобретению Соединения согласно настоящему изобретению (т.е. соединения формулы (I могут быть получены в соответствии с различными методиками, известными в данной области техники, в том числе методиками, описание которых приведено ниже. Продукты, полученные на каждой из стадий в соответствии со схемой, приведенной ниже, могут быть выделены с помощью традиционных способов, включая экстракцию, выпаривание, осаждение, хроматографию, фильтрование, растирание в порошок, кристаллизацию и др. На приведенных ниже схемах все заместители определены ранее, если не указано иное, а подходящие реагенты хорошо известны и широко применяются в данной области техники. Получение промежуточного соединения формулы (5) может быть осуществлено с помощью реакций, указанных на схеме 1. Согласно схеме 1 на стадии 1 карбамат формулы (I) превращают в лактам формулы (3) с помощью ацилирования по Фриделю-Крафтсу. Например, карбамат (1) растворяют в избытке оксихлорида фосфора и обрабатывают пентоксидом фосфора при примерно 100-130 С. Альтернативным образом, на стадии 2 лактам формулы (3) может быть получен за счет расширения цикла в кетоне формулы (2) путем обработки гидроксиламином и избытком ацетата натрия в спиртовом растворителе, таком как МеОН или EtOH. Промежуточное соединение, представляющее собой имин,выделяют путем фильтрования и обрабатывают сильной кислотой, такой как полифосфорная кислота,при примерно 100-150 С с образованием лактама (3). Бромирование тиофенового кольца с образованием бромтиофена (4) проводят путем обработки бромом в подходящем растворителе, таком как уксусная кислота, вода или четыреххлористый углерод. На стадии 4 в бромтиофен формулы (4) вводят функциональную группу с образованием арилтиофена формулы (5) с помощью подходящей катализируемой металлом реакции кросс-сочетания, хорошо известной специалистам в данной области техники. Например, бромтиофен (4) обрабатывают арилбороновой кислотой в растворителе, таком как ацетонитрил, ДМФ, толуол, вода и др. В реакции арилирования участвуют основание, такое как карбонат калия, и палладиевый катализатор, такой как Pd(OAc)2, Pd(PPh3)4 или Pd(PPh3)2Cl2 и др., как правило, при добавлении фосфинового лиганда, такого как PPh3. Очевидно, что соединения формулы (1) и (2) могут быть легко получены по способам, аналогичным приведенным в настоящем описании, согласно методикам, хорошо известным в данной области техники. Например, соединения формулы (1) получают путем восстановления тиофен-3-ацетонитрила до амина и последующим взаимодействием с этилхлорформиатом. Кетон формулы (2) без труда может быть получен согласно Aparajithan, K., et al. J. Heterocyclic Chem. 1966, 3, 466. Схема 2 Получение соединений формулы (8) может быть проведено в соответствии со способами, представленными на схеме 2. Подходящее соединение формулы (9) представляет собой соединение, в котором Х=Cl или Br, a R2 определено при описании соединений формулы (I). Подходящее соединение формулы(8) представляет собой соединение, в котором R1, Ra, Rb и R2 определены при описании соединений формулы (I). Согласно схеме 2 на стадии 1 ацилазид формулы (6) подвергают циклизации при нагревании с об-6 015127 разованием тиенопиридинона формулы (7). Например, ацилазид (6) растворяют в диоксане и добавляют по каплям в предварительно нагретую колбу (230 С), содержащую Dowtherm А. В бромтиенопиридинон формулы (7) вводят функциональную группу с образованием арилтиенопиридинона с помощью катализируемой металлом реакции кросс-сочетания, как описано выше для схемы 1, стадии 4. Согласно схеме 2 соединения формулы (8) также могут быть получены по способам, представленным на стадиях 3-5. На стадии 3 5-галогентиофен формулы (9) превращают в арилтиофен формулы (10) с помощью катализируемой металлом реакции кросс-сочетания с участием арилбороновой кислоты. Например, 5-галогентиофен формулы (9), где X=Cl, растворяют в растворителе, таком как этанол, и обрабатывают арилбороновой кислотой в присутствии основания, такого как карбонат натрия, калия или цезия. Добавляют палладиевый катализатор, такой как [1,3-бис-(2,6-диизопропилфенил)имидазол-2-илиден)(3 хлорпиридил)]палладий(II) дихлорид, и реакцию проводят в интервале температур от примерно комнатной температуры до примерно температуры дефлегмации выбранного растворителя. На стадии 4 ацеталь формулы (10) превращают в альдегид формулы (11) в кислой среде, при этом подходящие среды хорошо известны в данной области техники. Предпочтительные условия предполагают применение трифторуксусной кислоты. Согласно схеме 2 на стадии 5 альдегид формулы (11) подвергают циклизации за счет внутримолекулярной конденсации в кислой среде с образованием тиенопиридинона формулы (8). Предпочтительные условия предполагают применение в качестве растворителя трифторметансульфоновой кислоты в интервале температур примерно от 50 до 150 С в течение примерно от 1 до 5 ч. Продукт выделяют, выливая реакционную смесь в холодную воду с последующим фильтрованием. Очевидно, что соединения формулы (6) и (9) могут быть получены согласно способам и методикам,приведенным в настоящем описании или известным в данной области техники. Например, соединения формулы (6) получают путем превращения соответствующей кислоты (Gronowitz, S.; Ander, I. ChemicaScripta 1980, 15, 145) в хлорангидрид и последующего взаимодействия с азидом натрия с образованием ацилазида формулы (6). Соединения формулы (9) получают путем ацилирования 5-галогентиофен-2 карбоновой кислоты 2,2-диэтоксиэтиламином. Схема 3 Получение соединений формулы (I) может быть проведено согласно способам, представленным на схеме 3. Подходящее соединение формулы (8 а) представляет собой соединение, в котором R1, Ra, Rb и R2 такие, как определены для формулы (I), a подходящее соединение формулы (12) представляет собой соединение, в котором L1, R3, R4 и R5 такие, как определены для формулы (I), а X представляет собой атом брома или йода. Соединение формулы (12 а) представляет собой соединение, в котором L1a представляет собой частный случай L1, когда L1 представляет собой связь или L1 содержит подходящий терминальный первичный или вторичный амин, спирт или первичный амид (H2NC(O)-), участвующие в реакции сочетания. Например, на стадии 1 соединение формулы (8 а) взаимодействует с соединением формулы (12) в условиях каталитической реакции кросс-сочетания, такой как арилирование амида по Бухвальду (Yin, J.;Buchwald, S.J. J. Am. Chem. Soc. 2002, 124 (21), 6043-6048). Реакцию сочетания проводят с использованием основания, например Cs2CO3, палладиевого реагента, например Pd2dba3, и фосфинового лиганда, например Xantphos, в апротонном растворителе, таком как диоксан, толуол или бензол. Реакцию, как правило, проводят в интервале температур от примерно комнатной температуры до примерно температуры дефлегмации выбранного растворителя. Альтернативным образом реакцию проводят в присутствии меди. Например, соединение формулы(8 а) растворяют в толуоле или диоксане и обрабатывают соединением формулы (12) (1 экв.), K2CO3(2 экв.), N,N'-диметилэтан-1,2-диамином (0,2 экв.) и CuI (от 0,1 до 0,25 экв.). Реакционную смесь перемешивают при температуре примерно от 80 до 110 С. Согласно схеме 3 соединения формулы (I) также могут быть получены по способам согласно стадиям 2 и 3. На стадии 2 лактам или пиридинон формулы (8 а) превращают в бромфениламид формулы (13) в результате реакции сочетания с 4-бромфенилбороновой кислотой. Предпочтительные условия предпола-7 015127 гают использование ацетата меди в присутствии молекулярных сит 4 А в инертном растворителе, таком как дихлорметан, при температуре в интервале примерно от комнатной до температуры дефлегмации растворителя. На стадии 3 бромфениламид формулы (13) превращают в соединения формулы (I) с помощью проводимой в присутствии меди или палладия реакции кросс-сочетания со вторичным или первичным амином, спиртом, первичным амидом (H2NC(O)-), входящим в состав L1a; указанную реакцию проводят по способам, хорошо известным в данной области техники. Например, первичный амид подвергают взаимодействию с соединением формулы (13) в присутствии йодида меди или карбоната цезия в инертном растворителе, таком как диоксан, при температуре примерно от 80 С до температуры дефлегмации растворителя. Для специалиста в данной области техники очевидно, что соединения формулы (12) или (12 а) легко могут быть получены с помощью способов, хорошо известных и широко применяемых в данной области техники. Например, соединения формулы (12) получают путем алкилирования фенола алкилгалогенидом или по реакции Мицунобу, в ходе которой фенол взаимодействует со спиртом. Амидный мостик получают путем ацилирования бензойной кислоты алкиламином. Бензойную кислоту получают путем превращения фенола в трифлат с последующим карбонилированием. Очевидно, что характер и последовательность реакций зависят от природы L1. Очевидно также, что стадии, необходимые для получения соединения формулы (12), могут проводиться в любом порядке, в том числе и после взаимодействия интермедиата соединения формулы (12) с соединением формулы (I) с последующим проведением реакций карбонилирования, алкилирования, ацилирования, арилирования и др., приводящих к образованию соединения формулы (I). Схема 4 Получение соединений формулы (17) может быть осуществлено в соответствии со способами,представленными на схеме 4. Согласно схеме 4 на стадии 1 метилтиофенкарбоновую кислоту формулы (14) ацилируют с помощью анилина формулы (15) с образованием метилтиофениламида формулы (16). Условия проведения реакции ацилирования хорошо известны в данной области техники. Предпочтительные условия предполагают использование оксалилхлорида с образованием ацилхлорида с последующим взаимодействием ацилхлорида с указанным анилином. Метилтиофениламид формулы (16) обрабатывают 2,0-2,3 экв. сильного основания, такого как н-BuLi, трет-BuLi или LDA, при температуре примерно -70 С или ниже. Полученный раствор далее обрабатывают ДМФ, постепенно нагревают до комнатной температуры и перемешивают в течение примерно 0,5-2 ч с образованием тиенопиридинона формулы (17). Для специалиста в данной области техники очевидно, что соединения формулы (14) и (15) легко могут быть получены способами, известными в данной области техники. Например, карбоновая кислота формулы (14) может быть получена в результате реакции сочетания 2-бром-4-метилтиофена с арилбороновой кислотой с последующим карбоксилированием. Соединения формулы (15) получают по реакции ароматического нуклеофильного замещения п-галогеннитробензола, такого как п-хлор- или п-фторнитробензола, со спиртом или амином. Очевидно, что ацилированию может быть подвергнута лишь часть соединения формулы (15), например анилинсодержащая защищенная силильным фрагментом гидроксильная группа, с последующим удалением защитной группы и превращением полупродукта в соединения формулы (17). Получение тиофенлактамов формулы (21) может быть проведено в соответствии со способами,представленными на схеме 5. Подходящее соединение формулы (18) представляет собой соединение, в котором R2 является таким, как определено для формулы (I), а X представляет собой Br или I. Согласно схеме 5 на стадии 1 тиофенлактон формулы (18) превращают в амид формулы (19) с помощью типичной реакции Вайнреба (Basha, Anwer; Lipton, M.; Weinreb, Steven M. Tetrahedron Letters,1977, 48, 4171). Например, амин формулы (15) растворяют в апротонном растворителе, таком как CH2Cl2 или толуол, и обрабатывают 2-2,5 М раствором Me3Al в гексане. Полученный раствор перемешивают при температуре примерно от 0 С до комнатной температуры в течение примерно от 5 до 60 мин, после чего обрабатывают лактономформулы (18). Полученный раствор перемешивают при температуре примерно от комнатной до 110 С в течение примерно то 3 до 24 ч с образованием амида (19). Согласно схеме 5 на стадии 2 реакция циклизации, приводящая к образованию лактама формулы(20), может быть проведена по меньшей мере двумя способами, описание которых приведено ниже. Согласно первому варианту спирт формулы (19) превращают в уходящую группу, предпочтительно мезилат, путем взаимодействия с метансульфонилхлоридом в присутствии подходящего основания, такого как триэтиламин. Промежуточное соединение, представляющее собой мезилат, выделяют путем обработки водой, сразу после этого растворяют в безводном полярном растворителе, таком как ДМФ, и обрабатывают основанием, таким как гидрид натрия (1,5 экв.), при температуре от 0 до 25 С. Второй вариант предполагает проведение реакции Мицунобу (Maligres, Р.Е., et al. J. Het. Chem. 2003, 40(2), 229-241). Например, спирт формулы (19) растворяют в подходящем безводном растворителе,таком как ТГФ, CH2Cl2 или толуол, и обрабатывают триалкил- или триарилфосфином, таким как Ме 3 Р,Bu3P или Ph3P, и диалкилазодикарбоксилатом, таким как DEAD (диэтилазодикарбоксилат) или DIAD(диизопропилазодикарбоксилат), при температуре примерно от 0 С до комнатной температуры. Согласно схеме 5 на стадии 3 в лактам формулы (20) вводят функциональную группу с помощью катализируемой металлом реакции кросс-сочетания, условия проведения которой указаны при описании схемы 1, стадии 4. Очевидно, что соединения формулы (18) легко могут быть получены способами, аналогичными приведенным в настоящем описании, с использованием методик, которые хорошо известны и широко применяются в данной области техники. Например, 2-тиофен-3-илэтанол может быть превращен в хлорформиат с использованием трифосгена с последующей циклизацией, приводящей к образованию лактона(тиофенлактона), проводимой с использованием кислоты Льюиса, такой как AlCl3. Полученный тиофенлактон далее подвергают галогенированию, например, путем обработки йодом и бис(трифторацетокси)йодбензолом с образованием соединения формулы (18). При галогенировании тиофенлактона, приводящего к образованию соединения (18), образуется смесь 2- и 3-галогенотиофенлактонов. Фракция, содержащая целевой 2-галогенотиофенлактон (18, R2=H), может быть выделена методом хроматографии (что подтверждается результатами 1 Н ЯМР). Соединения формулы (18), в которых R2 представляет собой C1-C4-алкил, могут быть получены с использованием вышеуказанной фракции, содержащей 3-галогенотиофенлактон. 3-Галогенотиофенлактон алкилируют с помощью подходящим образом замещенного алкилсодержащего соединения путем проведения реакции связывания, например реакции Сузуки (с алкилбороновой кислотой), с образованием 3-алкилтиофенлактона. Полученный 3-алкилтиофенлактон далее галогенируют с образованием целевого соединения формулы Получение соединений формулы (17) может быть проведено в соответствии со способами, представленными на схеме 6. Подходящее соединение формул (22), (15) и (17) представляет собой соединение, в котором все заместители являются такими, как указано для формулы (I). Согласно схеме 6 на стадии 1 аминотиофен формулы (22) превращают в бромтиофен формулы (23) по реакции, аналогичной реакции Зандмейера. Предпочтительные условия предполагают использованиеCuBr2 и трет-бутилнитрила в присутствии инертного растворителя, такого как ацетонитрил, при температуре в интервале примерно от комнатной до температуры дефлегмации растворителя. На стадии 2 бромтиофен формулы (23) подвергают взаимодействию с (триметилсилил)ацетиленом в ходе протекающей в присутствии палладия реакции кросс-сочетания, приводящей к образованию этинилтиофена формулы (24). Например, бромтиофен (23) обрабатывают (триметилсилил)ацетиленом в инертном растворителе, например ацетонитриле, ДМФ или толуоле, при добавлении основания, такого как диизопропиламин, и палладиевого катализатора, такого как Pd(OAc)2, Pd(PPh3)4 или Pd(PPh3)2Cl2 и др., как правило, при добавлении фосфинового лиганда, такого как PPh3. Предпочтительные условия предполагают использование ДМФ и Pd(PPh3)2Cl2 при добавлении CuI при температуре примерно от 50 до 150 С. Наиболее предпочтительно реакцию проводят в микроволновом реакторе в течение примерно 30 мин. Согласно схеме 6 на стадии 3 этинилтиофен формулы (24) подвергают взаимодействию с анилином формулы (15) с образованием амида и последующим проведением циклизации in situ с образованием тиенопиридинона формулы (17). Типичные условия представляют собой условия проведения реакции Вайнреба, указанные при описании схемы 5, стадии 1, и предполагают использование Me3Al в инертном растворителе, таком как толуол. Очевидно, что стадии, необходимые для получения соединений формулы (I), (17) и (21), указанные на представленных ранее схемах, зависят от конкретного синтезируемого соединения, исходного соединения и относительной подвижности замещенных фрагментов. Также подразумевается, что для осуществления указанных выше реакций может потребоваться проведение ряда стадий введения и удаления защитных групп. Например, не требуется предварительное получение промежуточных соединений формулы (12) и (15) до проведения различных стадий сочетания или ацилирования, указанных в настоящем описании. Указанные промежуточные соединения также могут выступать в качестве защитных групп для защиты аминной или гидроксильной функциональных групп, которые после удаления указанных защитных групп вступают в реакцию с образованием соединений, предложенных в настоящем изобретении. Выбор и применение подходящих защитных групп хорошо известны в данной области техники (см., например, Protecting Groups in Organic Synthesis, Theodora Greene (Wiley-Interscience. Демонстрация действия. Все лиганды, радиолиганды, растворители и реагенты, используемые при проведении указанных исследований, могут быть без труда приобретены или получены специалистом в данной области техники. Полноразмерную кДНК для человеческого MCHR1 клонировали из кДНК мозга взрослого человека, полученной из библиотеки ДНК (Edge Biosystems, кат. 38356) с помощью стандартного метода полимеразной цепной реакции (PCR) с использованием следующих праймеров: смысловой, 5'-GCCACCATGGACCT GGAAGCCTCGCTGC-3'; антисмысловой, 5'-TGGTGCCCTGACTTGGAGGTGTGC-3'.PCR (полимеразную цепную реакцию) проводили в конечном объеме, равном 50 мкл, содержащем 5 мкл 10 исходного раствора PCR буфера, 1 мкл 10 мМ смеси dNTP (конечная концентрация 200 мкМ),2 мкл 50 мМ MgSO4 (конечная концентрация 2 мМ), 0,5 мкл 20 мкМ раствора каждого из праймеров (конечная концентрация 0,2 мкМ), 5 мкл матричной кДНК, содержащей 0,5 нг ДНК, 0,5 мкл ДНК- 10015127 полимеразы Platinum Taq High Fidelity DNA polymerase (Gibco Life Technologies) и 36 мкл H2O. PCR амплификацию проводили на термоциклере Perkin Elmer 9600. Проводили денатурацию в течение 90 с при 94 С и повторяли последовательность амплификаций, включающую 94 С в течение 25 с, 55 С в течение 25 с и 72 С в течение 2 мин, 30 раз с последующим проведением конечной стадии элонгации при 72 С в течение 10 мин. Получение требуемого PCR продукта (1,1 Кб) подтверждали методом электрофореза на геле агарозы и экстрагировали диск из геля с помощью Geneclean (Bio101), следуя инструкциям производителя. После проведения экстракции клонировали фрагмент кДНК в pCR2.1-ТОРО плазмиду (Invitrogen Corp.) для подтверждения идентичности и последовательности. Для получения клеточных линий, стабильно экспрессирующих MCHR1, субклонировали инсерт вAV12 клетки, которые были предварительно трансфицированы с помощью смешанного G белка G15. Трансфицированные клетки выбирали с помощью G418 (800 мкг/мл) в течение 10-14 дней и проводили выделение отдельных колоний из культуральных планшетов. Далее выбирали колонии, устойчивые кG418, для экспрессии MCHR1 путем измерения МСН-стимулированных кратковременных Са 2+-токов во флуорометрическом планшет-ридере (FLIPR, Molecular Devices). Как правило, индивидуальные клоны высевали в 96-луночные планшеты при концентрации, составляющей 60000 клеток на лунку, в 100 мкл питательной среды (Дульбекко модифицированная среда Игла (DMEM), 5% фетальной телячьей сыворотки, 2 мМ L-глутамина, 10 мМ HEPES, 1 мМ пирувата натрия, 0,5 мг/мл Zeocin и 0,5 мг/мл Geneticin). После выдержки в течение 24 ч при 37 С среду удаляли и заменяли на 50 мкл буфера, содержащего краситель (сбалансированный солевой раствор Хэнкса (HBSS),содержащий 25 мМ HEPES, 0,04% Pluronate 127 и 8 мкМ Fluo3, оба компонента из Molecular Probes. После проведения инкубации в течение 60 мин с содержащим краситель буфером при комнатной температуре буфер аспирировали и заменяли на 100 мкл HEPES/HBBS. Планшет с клетками и планшет с соединениями, содержащий 2 мкМ МСН в буфере, помещали в FLIPR и считывали основные показания в течение 10 с. Затем FLIPR проводил отбор 100 мкл 2 мкМ МСН (для создания конечной концентрации в пробе 1 мкМ МСН) в планшет с клетками и проводил считывание в течение 105 с для регистрации полного пика кальциевого потока, возникающего в ответ на присутствие агониста (1 мкМ МСН). Для корректировки вариаций количества клеток клонов в различных лунках МСН-отклик нормализуют относительно отклика, индуцированного эпинефрином. При исследовании связывания 125I-МСН и функционального связывания GTP35S использовали мембраны, выделенные из клона, обозначенного, как клон 43. Как правило, клетки из 20 конфлюэнтных колб Т 225 обрабатывали, промывая монослои холодным фосфатным буферным раствором (PBS), туда же переносили клетки и повторно суспендировали клеточную массу в (10 мл/г пасты) мембранном буфере с рН 7,4 (250 мМ сахарозы, 50 мМ HEPES, рН 7,5, 1 мМ MgCl2 и ингибиторы протеазы (1 таблетка Complete-EDTA (Roche Diagnostics), на 100 мл мембранного буфера). Клетки гомогенизировали с помощью гомогенизатора с приводом от двигателя Teflon-glass Potter-Elvehjem с помощью 5-10 ударов, а затем центрифугировали при 260g в течение 15 мин при 4 С. Надосадочную жидкость собирали, осадок повторно суспендировали в мембранном буфере, повторно гомогенизировали и еще раз центрифугировали при 260g в течение 15 мин при 4 С в общей сложности 3 раза. После этого осадок можно было отбросить. Объединенные надосадочные жидкости центрифугировали при 30000g в течение 60 мин при 4 С. Мембранный осадок подвергали повторному суспендированию в мембранном буфере, при этом полученная концентрация белка составляла 3-5 мг/мл (Pierce BCA анализ с применением бычьего сывороточного альбумина в качестве стандарта). Аликвоты хранили при -80 С. Связывание соединений с MCHR1 оценивали с помощью конкурентно-связывающего анализа с использованием 125I-МСН, исследуемого соединения и мембран клона 43. Исследования проводили в 96 луночных планшетах с прозрачным белым дном Costar 3632 в суммарном объеме 200 мкл, содержащем 25 мМ HEPES, рН 7,4, 10 мМ CaCl2, 2 мг/мл бычьего сывороточного альбумина, 0,5% диметилсульфоксида (ДМСО), от 4 до 12 мкг мембран клона 43, 200 пМ 125I-МСН (NEN), 2,5 мг/мл гранул пшеничных зародышей для агглютининового сцинтилляционного бесконтактного анализа (WGA-SPA гранулы, Amersham Inc., в настоящее время GE Healthcare) и изменяющуюся дозу исследуемого соединения. Неспецифическое связывание оценивали в присутствии 0,1 мкМ немеченого МСН. Связанные 125I-МСН определяли, помещая запаянные планшеты в Microbeta Trilux (Perkin Elmer Life and Analytical Sciences Inc.) и подсчитывая после 12-часовой выдержки. Значения IC50 (определяемые, как концентрация исследуемого соединения, необходимая для уменьшения специфического связывания 125I-МСН на 50%) определяли путем приведения данных концентрация-отклик в соответствие с 4-параметровой моделью (максимальный отклик, минимальный отклик, коэффициент Хилла, IC50) с помощью Excel (Microsoft Corp.). Значения Ki рассчитывали из значений IC50 с помощью приближения Ченга-Прусоффа (Cheng-Prusoff), как описано в работе Cheng et al. (Relationshipenzymatic reaction, Biochem. Pharmacol., 22: 3099-3108 (1973. Используемые в качестве примеров соеди- 11015127 нения демонстрировали Ki1 мкМ в условиях проводимого исследования связывания. В частности, соединение согласно примеру 58 демонстрировало среднее значение Ki MCHR1, равное примерно 3 нМ. Функциональный антагонизм МСН активности оценивали путем измерения способности тестируемого соединения ингибировать МСН-стимулированное связывание GTP35S с мембранами клона 43. Исследования проводили в планшетах Costar 3632 с прозрачным белым дном в суммарном объеме 200 мкл,содержащем 50 мМ Hepes, рН 7,4, 5 мМ MgCl2, 10 мкг/мл сапонина, 1,0 мг/мл бычьего сывороточного альбумина, 100 мМ NaCl, 3 мкМ GDP, 0,3 нМ GTP35S, 10 нМ МСН (примерно равного ЕС 90), 0,4 мг/мл мембран клона 43, 5,0 мг/мл гранул пшеничных зародышей для агглютининового сцинтилляционного бесконтактного анализа (WGA-SPA beads, Amersham Inc., в настоящее время GE Healthcare) и изменяющееся количество исследуемого соединения. Планшеты запаивали и выдерживали в течение 16-18 ч при 4 С. После выдерживания в течение 1 ч, для того чтобы температура планшетов сравнялась с температурой окружающей среды, определяли связанные GTP35S путем подсчета в Microbeta Trilux (Perkin ElmerLife и Analytical Sciences Inc.). Значения IC50 (определяемые как концентрация исследуемого соединения, необходимая для уменьшения стимулируемого МСН связывания GTP35S на 50%) определяли путем приведения данных концентрация-отклик в соответствие с 4-параметровой моделью (максимальный отклик, минимальный отклик, коэффициент Хилла, IC50) с помощью Excel (Microsoft). После подтверждения наличия конкурентного антагонизма с помощью анализа Шилда (Schild) рассчитывали значения Kb из значений IC50 для каждого антагониста и ЕС 50 для МСН (определяемые независимо) с помощью модификации приближения Ченга-Прусоффа, как описано Leff и Dougal (Trends Pharmacol. Sci. (1993) 14: 110-112). Используемые в качестве примеров соединения демонстрировали значения Kb1 мкМ в условиях функционального анализа, приведенных в настоящем описании. В частности, соединение согласно примеру 69 демонстрировало значение Kb MCHR1, равное примерно 20 нМ. Для демонстрации эффективности in vivo соединений согласно настоящему изобретению указанные соединения вводили перорально через зонд самцам крыс Лонга-Эванса (Harlan, IN), страдающим от ожирения, вызванного высококалорийной диетой, с массой от 450 до 500 г. Наполнитель содержал 10% гуммиарабика и 0,15% раствора сахарина в воде. Животных содержали по отдельности в помещении с регулируемой температурой (24 С) при 12 часовой смене светлого и темного периодов (темный период 10:00/22:00). Вода и еда (Teklad 95217,Harlan, WI) были доступны ad libitum (без ограничений). Соединения вводили перорально один раз в день перед наступлением темного периода в течение 3 дней. Ежедневно в течение трех дней измеряли потребление пищи и изменение массы тела. Соединение согласно примеру 25 обеспечивало среднее снижение массы тела в размере примерно 6 г 10 мг/кг относительно крыс контрольной группы, принимавших один наполнитель. Применение. Соединения согласно настоящему изобретению подходят для применения в качестве антагонистов связывания MCHR1 при лечении патологических состояний у людей и животных (особенно домашних животных), на которые, как было показано, оказывает влияние рецептор MCHR1. За счет ингибирования активности МСН соединения согласно настоящему изобретению обеспечивают подавление аппетита. Таким образом, соединения согласно настоящему изобретению подходят для применения в качестве препаратов, подавляющих аппетит, и/или препаратов, обеспечивающих снижение веса, для лечения ожирения. Таким образом, указанные соединения подходят для лечения состояний, обусловленных ожирением, обостряющихся при ожирении, спровоцированных ожирением или сопутствующих ожирению. При лечении животных, не относящихся к домашним животным, соединения согласно настоящему изобретению можно применять для снижения привеса, и/или улучшения эффективности использования пищи, и/или увеличения массы тела худого животного. Соединения согласно настоящему изобретению можно вводить различными способами, в том числе пероральным, ректальным, трансдермальным, подкожным, внутривенным, внутримышечным и интраназальным. Составы на основе указанных соединений предпочтительно получают до введения, при этом выбор состава в случае каждого конкретного пациента осуществляется лечащим врачом. Соответственно другой аспект настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, разбавитель или наполнитель. Конкретная вводимая доза устанавливается в каждом случае в зависимости от конкретных обстоятельств. Эти обстоятельства включают способ введения, предшествующую историю болезни пациента,патологическое состояние или симптом, подвергаемые лечению, степень тяжести состояния/симптома,подвергаемого лечению, а также возраст и пол реципиента. Однако очевидно, что вводимая терапевтическая доза устанавливается лечащим врачом с учетом соответствующих факторов или ветеринаром в случае реципиентов-животных. В общем случае эффективная минимальная суточная доза соединения согласно настоящему изобретению составляет примерно от 10 до 200 мг в сутки. Как правило, эффективная максимальная доза со- 12015127 ставляет примерно от 200 до 1000 мг в сутки. Точная доза может быть установлена в соответствии со стандартной медицинской практикой определения дозы для реципиента, т.е. путем первоначального введения низкой дозы соединения с последующим постепенным увеличением дозы до тех пор, пока не будет наблюдаться требуемый терапевтический эффект. Фармацевтические композиции согласно настоящему изобретению могут быть адаптированы для указанных выше различных способов введения и могут быть введены пациенту, например, в виде таблеток, капсул, крахмальных капсул, саше, papers, лепешек, пластинок, эликсиров, мазей, трансдермальных пластырей, аэрозолей, средств для ингаляции, суппозиториев, растворов и суспензий. Общее содержание активных ингредиентов в такой композиции составляет от 0,1 до 99,9 мас.% состава (см. Remington'sPharmaceutical Sciences, 18th Edition, Mack Publishing Co. (1990, где в целом обсуждаются составы, способы доставки лекарственных препаратов и др. Примеры Приведенные далее примеры лишь иллюстрируют методики получения и способность заявителей получать соединения согласно настоящему изобретению на основании представленных схем и/или их известных модификаций. Указанные примеры не являются ограничивающими и не исчерпываются соединениями, которые синтезированы или могут быть получены. Используемые в настоящем описании сокращения определены согласно Aldrichimica Acta, vol. 17, No. 1, 1984 и в общем случае известны специалистам в данной области техники или могут быть установлены с минимальными усилиями. Другие сокращения, используемые при описании экспериментов, являются следующими: N-метил-2 пирролидинон (NMP), трет-бутилметиловый эфир (МТВЕ) и комнатная температура (КТ). Названия соединений согласно настоящему изобретению получены с помощью ChemDraw Ultra, версия 7.0.1. Названия солей приводят в виде "свободное основание плюс сопряженная кислота". Препаративный пример 1. Триизопропил(2-метокси-4-нитрофенокси)силан. 4-Нитрогваякол (50,0 г, 295,6 ммоль) растворяли в ДМФ (безводном, 1000 мл) и охлаждали полученный раствор до 0-5 С. Медленно добавляли NaH (60% в минеральном масле, 13,4 г, 335,0 ммоль),поддерживая температуру 10 С. Осуществляли механическое перемешивание полученного желтооранжевого раствора при КТ в течение 30 мин, после чего раствор охлаждали до 0-5 С. Смесь обрабатывали триизопропилсилилтрифлатом (90,0 мл, 334,8 ммоль), поддерживая температуру 10 С. Реакционную смесь перемешивали при КТ (комнатная температура) в течение ночи. Смесь гасили 14% водным раствором NH4Cl (1000 мл) и экстрагировали EtOAc (31000 мл). Полученные органические растворы объединяли, промывали насыщенным солевым раствором (1000 мл) и концентрировали под вакуумом с образованием светло-желтого масла. Полученное масло очищали методом флэш-хроматографии с использованием 100% гексанов, затем 10% EtOAc/гексанов с образованием 95,8 г (99,6%) указанного в заголовке соединения в виде светло-желтого масла.NaH (3,2 г, 80 ммоль) добавляли порциями к раствору 2-пирролидин-1-илэтанола (9,4 мл, 80 ммоль) в ДМФ (180 мл) с температурой 0 С. Реакционную смесь перемешивали в течение 15-20 мин до прекращения выделения газа. С помощью канюли добавляли раствор 1-хлор-2-метокси-4-нитробензола (15,0 г,80 ммоль) в ДМФ (100 мл). Нагревали реакционную смесь до 130 С и перемешивали в течение примерно 18 ч. Охлаждали до КТ и разбавляли EtOAc (300 мл). Промывали водой (2100 мл). Экстрагировали органический слой 1 N HCl (3150 мл). Полученный кислый раствор промывали EtOAc (100 мл), затем подщелачивали путем добавления 1 N NaOH (500 мл) до pH 8-9. Экстрагировали полученный щелочной водный раствор CH2Cl2 (3150 мл). Органический слой сушили над Na2SO4 и концентрировали с образованием 8,5 г (40%) целевого продукта в виде твердого вещества оранжевого цвета.MS/ES m/z 267,3 [М+Н]+. Препаративный пример 3. 3-Метокси-4-триизопропилсиланилоксифениламин. Триизопропил(2-метокси-4-нитрофенокси)силан (95,7 г, 294,0 ммоль) растворяли в EtOH (1800 мл) и добавляли 5% Pd/C (10,0 г). Проводили гидрирование суспензии при КТ водородом при давлении 50 psi (фунтов/кв.дюйм) в течение 8 ч. Суспензию фильтровали через слой целита (Celite) и промывалиEtOH. Фильтрат концентрировали под вакуумом с образованием коричневого масла. Полученное масло очищали методом флэш-хроматографии с использованием градиента от 100% гексанов до 20%EtOAc/гексанов с образованием 67,4 г (77,6%) указанного в заголовке соединения в виде твердого вещества коричневого цвета.MS/ES m/z 296,2 [М+Н]+. Препаративный пример 4. 3-Метокси-4-(2-пирролидин-1-илэтокси)фениламин. Указанное в заголовке соединение получали, по существу, согласно препаративному примеру 3 с использованием 1-[2-(2-метокси-4-нитрофенокси)этил]пирролидина.- 13015127 Препаративный пример 5. 4,5-Дигидротиено[2,3-c]пиран-7-он. 2-Тиофен-3-илэтанол (65,2 мл, 590,5 ммоль) растворяли в CH2Cl2 (безводном, 1480 мл) и охлаждали до 0-5 С, после чего добавляли трифосген (87,7 г, 295,5 ммоль). Полученную смесь перемешивали при 05 С в течение примерно 5 мин, а затем медленно добавляли диизопропилэтиламин (102,8 мл,590,2 ммоль) в течение примерно 1,5 ч, поддерживая температуру 8 С. Осуществляли механическое перемешивание раствора при 0-5 С в течение 4,5 ч, после чего гасили реакцию 1 N HCl (890 мл). Органический раствор отделяли и экстрагировали водный слой CH2Cl2 (2600 мл). Органические растворы объединяли, промывали насыщенным солевым раствором (900 мл) и концентрировали под вакуумом с образованием 125,3 г (выход 78,0%, рассчитанный исходя из чистоты, равной 70%, подтвержденной методом ВЭЖХ) хлорформиата в виде масла. Смешивали AlCl3 (68,0 г, 510,0 ммоль) с толуолом (560 мл) и медленно добавляли в течение примерно 30 мин раствор хлорформиата (масса брутто 125,2 г, масса нетто 87,8 г, 460,5 ммоль) в толуоле(1000 мл), поддерживая температуру равной 40 С. Осуществляли механическое перемешивание смеси при КТ в течение 1 ч. Смесь охлаждали до 0-5 С и медленно гасили насыщенным водным раствором соли Рошеля (тартрат калия-натрия) (800 мл) в течение 30 мин, поддерживая температуру смеси 22 С. Отделяли органический раствор. Водный слой обрабатывали EtOAc (500 мл) и Celite (130 г) и перемешивали смесь при КТ в течение 10-15 мин. Смесь фильтровали и промывали осадок на фильтре EtOAc. Отделяли органический раствор и экстрагировали водную фазу дополнительным количеством EtOAc(2500 мл). Объединяли все полученные органические растворы и промывали насыщенным солевым раствором (1000 мл), затем концентрировали под вакуумом с образованием 111,7 г масла, частично затвердевавшего при хранении. Полученное полутвердое вещество подвергали перекристаллизации изEtOAc/гексана и сушили под вакуумом с образованием 67,2 г (94,6%) указанного в заголовке соединения.MS/ES m/z 155,1 [М+Н]+. Препаративный пример 6 а. 3-Йод-4,5-дигидротиено[2,3-c]пиран-7-он. Препаративный пример 6b. 2-Йод-4,5-дигидротиено[2,3-c]пиран-7-он. 4,5-Дигидротиено[2,3-c]пиран-7-он (1,1 г, 7,14 ммоль) суспендировали в CCl4 (7 мл). Полученную суспензию нагревали до 65 С. Добавляли I2 (1,08, 4,28 ммоль) с последующим добавлением бис(трифторацетокси)йодбензола (1,84 г, 4,28 ммоль, Aldrich). Смесь продолжали нагревать в течение 10 мин, после чего охладили до КТ. Смесь вылили в водный раствор 1 N Na2S2O3 (200 мл) и экстрагировали CH2Cl2 (2200 мл). Объединенные органические слои промывали насыщенным солевым раствором(200 мл). Полученный органический раствор сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексаны) с образованием 684 мг (34%) 3-йод-изомера. Rf=0,37 в смеси 1 EtOAc/3 гексана (видимая УФ-область).ES/MS m/z 281,0 [М+Н]+. 1 Н ЯМР (CDCl3):2,91 (т, 2 Н, J=6,2 Гц), 4,62 (т, 2 Н, J=6,2 Гц), 7,80 (с, 1 Н). Проводили выделение второго компонента с образованием 333 мг (17%) 2-йод-изомера. Rf=0,21 в смеси 1 EtOAc/3 гексана (видимая УФ-область).(428 мг, 7,14 ммоль) в смеси 7:3:1 диметоксиэтан:вода:EtOH (10 мл) и добавляли 2,0 М Na2CO3 (3,5 мл). Через реакционную смесь в течение 10-15 мин барботировали осушенный аргон для удаления кислорода. Добавляли Pd(PPh3)4 (210 мг, 0,18 ммоль) и нагревали смесь с помощью микроволнового излучения до 130 С в течение 45 мин. Выливали смесь в 1 N NaOH (50 мл) и промывали гексаном (50 мл). Водный слой подкисляли 5 N HCl до pH 1 и экстрагировали EtOAc (350 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (50 мл). Концентрировали, после чего добавляли толуол (35 мл) с образованием сырого промежуточного продукта. Добавляли моногидрат п-толуолсульфокислоты (339 мг, 1,8 ммоль) и нагревали до 80 С в течение ночи. Охлаждали до КТ, выливали в водный раствор NaHCO3 (100 мл) и экстрагировали EtOAc (3100 мл). Промывали объединенные экстракты насыщенным солевым раствором (100 мл). Осушали полученный органический раствор,фильтровали и концентрировали. Полученный продукт очищали с помощью хроматографии на силикагеле (0-50% EtOAc/гексаны) с образованием 133 мг (22%) целевого продукта.ES/MS m/z 169,3 [М+Н]+. Препаративный пример 8. 2-Йод-3-метил-4,5-дигидротиено[2,3-c]пиран-7-он. Указанное в заголовке соединение получали, по существу, согласно препаративному примеру 6 а/6b с использованием 3-метил-4,5-дигидротиено[2,3-c]пиран-7-она.CuBr2 (1,0 г, 4,44 ммоль) и трет-бутилнитрит (0,66 мл, 5,55 ммоль) в CH3CN (915 мл) перемешивали при КТ в течение 30 мин и добавляли метиловый эфир 3-амино-5-(4-хлорфенил)тиофен-2-карбоновой кислоты (1,0 г, 3,7 ммоль). Реакционную смесь перемешивали при 70 С в течение 4 ч. Охлаждали реакционную смесь до КТ, выливали в 20% HCl (200 мл) и экстрагировали CH2Cl2 (200 мл). Органический слой экстрагировали 20% HCl, после чего осушали и концентрировали. Полученный сырой продукт очищали методом хроматографии с использованием в качестве элюента 10% EtOAc в гексане с образованием 65 г (53%) указанного в заголовке соединения.LC-MS/ES m/z (35Cl, 81Br) 332,0 [М+]. Препаративный пример 10. Метиловый эфир 5-(4-хлорфенил)-3-винилтиофен-2-карбоновой кислоты. Метиловый эфир 3-бром-5-(4-хлорфенил)тиофен-2-карбоновой кислоты (570 мг, 1,72 ммоль) растворяли в толуоле (10 мл) и добавляли трибутилвинилолово (0,55 мл, 1,89 ммоль) и тетракис(трифенилфосфин)палладий (40 мг, 0,034 ммоль). Реакционную смесь нагревали до 110 С в течение 17 ч. Охлаждали реакционную смесь до комнатной температуры и концентрировали под вакуумом. Полученный продукт очищали методом хроматографии с использованием в качестве элюента 5% этилацетата/гексана с образованием 0,37 г (77%) указанного в заголовке соединения.LC-MS/ES m/z (35Cl) 279,0 [M+H]+. Препаративный пример 11. Метиловый эфир 5-(4-хлорфенил)-3-(2-гидроксиэтил)тиофен-2 карбоновой кислоты. Метиловый эфир 5-(4-хлорфенил)-3-винилтиофен-2-карбоновой кислоты (5,35 г, 19,19 ммоль) растворяли в тетрагидрофуране (220 мл) и охлаждали до 0 С. Медленно добавляли 9 борбицикло[3.3.1]нонан (112 мл, 0,5 М, 56 ммоль) и нагревали реакционную смесь до комнатной температуры при перемешивании в течение 16 ч. Охлаждали реакционную смесь до 0 С и медленно добавляли перекись водорода (74 мл, 721 ммоль) с последующим добавлением 5 N гидроксида натрия (74 мл,370 ммоль). Добавляли воду (14 мл) и проводили экстракцию этилацетатом (3150 мл). Полученный раствор сушили над Na2SO4, фильтровали и выпаривали. Продукт очищали методом хроматографии с использованием в качестве элюента 50% этилацетата/гексан с образованием 5,70 г (100%) указанного в заголовке соединения.LC-MS/ES m/z (35Cl) 296,70 [М+Н]+. Препаративный пример 12. 2-(4-Хлорфенил)-4,5-дигидротиено[2,3-c]пиран-7-он. Метиловый эфир 5-(4-хлорфенил)-3-(2-гидроксиэтил)тиофен-2-карбоновой кислоты (3,4 г,11,46 ммоль) растворяли в толуоле (60 мл), добавляли моногидрат п-толуолсульфокислоты (0,3 г,1,58 ммоль) и нагревали до 80 С в течение одного часа. Охлаждали реакционную смесь до комнатной температуры и разбавляли этилацетатом (50 мл). Полученный раствор промывали 1 N NaOH и дважды экстрагировали водный слой этилацетатом. Полученные органические фракции объединяли, сушили надNa2SO4, фильтровали и концентрировали. Твердое вещество растирали с добавлением простого эфира,затем отфильтровывали и сушили под вакуумом с образованием 1,3 г (43%) указанного в заголовке соединения.LC-MS/ES m/z (35Cl) 265,0 [М+Н]+. Препаративный пример 13. 2-(4-Хлорфенил)-3-метил-4,5-дигидротиено[2,3-c]пиран-7-он. 4-Хлорфенилбороновую кислоту (215 мг, 1,38 ммоль) добавляли к суспензии 2-йод-3-метил-4,5 дигидротиено[2,3-с]пиран-7-она (135 мг, 0,46 ммоль) в CH3CN (2 мл). Добавляли PPh3 (37 мг,0,14 ммоль), а затем 2,0 М Na2CO3 (0,69 мл). Через реакционную смесь в течение 15 мин барботировали осушенный аргон. Добавляли Pd(OAc)2 (12 мг, 0,05 ммоль) и нагревали смесь с помощью микроволнового излучения до 100 С в течение 45 мин. Выливали реакционную смесь в 1 N HCl (50 мл) и экстрагировали EtOAc (350 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (50 мл). Концентрировали, после чего растворяли полученный остаток, представляющий собой сырой продукт, в толуоле (5 мл) и обрабатывали моногидратом п-толуолсульфокислоты (50 мг,0,26 ммоль). Нагревали реакционную смесь до 80 С в течение 8 ч. Охлаждали до КТ, выливали в водный раствор NaHCO3 (50 мл) и экстрагировали EtOAc (350 мл). Промывали объединенные экстракты насыщенным солевым раствором (50 мл) и концентрировали. Полученный продукт очищали с помощью хроматографии на силикагеле (0-50% EtOAc/гексаны) с образованием 111 мг (87%) целевого продукта. Препаративный пример 14. 3-(2-Гидроксиэтил)-5-йодтиофен-2-карбоновой кислоты [3-метокси-4(2-пирролидин-1-илэтокси)фенил]амид 3-Метокси-4-(2-пирролидин-1-илэтокси)фениламин (препаративный пример 4) (266 мг, 1,13 ммоль) растворяли в толуоле (8 мл) и добавляли триметилалюминий (2,0 М раствор в толуоле, 565 мкл,1,13 ммоль). Перемешивали в течение 5-10 мин при КТ и добавляли 2-йод-4,5-дигидротиено[2,3-c]пиран 7-он (315 мг, 1,13 ммоль). Нагревали реакционную смесь до 50 С в течение ночи. Охлаждали до КТ, до- 15015127 бавляли водный раствор соли Рошеля (100 мл) и перемешивали в течение 1 ч. Добавляли 1 N NaOH(50 мл) и экстрагировали EtOAc (3100 мл). Объединяли органические слои и промывали насыщенным солевым раствором (100 мл). Концентрировали органическую часть смеси с образованием сырого продукта в виде твердого вещества. Полученный сырой продукт растирали с диэтиловым эфиром, отфильтровывали и получали 370 мг (63%) продукта в виде твердого вещества белого цвета.MS/ES m/z 517,0 [М+Н]+. Представленные в нижеследующей таблице промежуточные соединения получали, по существу, согласно препаративному примеру 14 с использованием подходящего 4,5-дигидротиено[2,3-c]пиран-7-она и соответствующего фениламина.TEA (576 мкл, 4,14 ммоль) добавляли к суспензии 3-(2-гидроксиэтил)-5-йодтиофен-2-карбоновой кислоты [3-метокси-4-(2-пирролидин-1-илэтокси)фенил]амида (1,64 г, 3,19 ммоль) в 20 мл CH2Cl2, температура которой составляла 0 С. Добавляли метансульфонилхлорид (320 мкл, 4,14 ммоль) и перемешивали в течение ночи. При необходимости добавляли дополнительное количество метансульфонилхлорида, чтобы обеспечить вступление в реакцию всего количества спирта. После завершения реакции образования мезилата, которое устанавливали с помощью MS анализа, смесь выливали в 1 N NaOH (250 мл) и экстрагировали CH2Cl2 (2200 мл). Объединенные органические слои промывали 100 мл насыщенного солевого раствора, сушили над Na2SO4, фильтровали и концентрировали. Полученный сырой мезилат растворяли в ДМФ (20 мл), охлаждали до 0 С и добавляли NaH (192 мг, 4,79 ммоль). Перемешивали смесь в течение ночи, при этом температура смеси постепенно достигала комнатной температуры. Повторяли те же стадии водной обработки, которые проводили при получении мезилата. Полученный сырой продукт очищали с помощью хроматографии на силикагеле с использованием в качестве элюента 010% градиента (2 N NH3 в МеОН)/CHCl3 с образованием 1,07 г (68%) целевого продукта.MS/ES m/z 499,2 [М+Н]+. Препаративный пример 18. 6-(3-Метокси-4-триизопропилсиланилоксифенил)-5,6-дигидро-4 Нтиено[2,3-c]пиридин-7-он. Указанное в заголовке соединение получали, по существу, согласно препаративному примеру 17 с использованием 3-(2-гидроксиэтил)тиофен-2-карбоновой кислоты (3-метокси-4-триизопропилсиланилоксифенил)амида. Указанное в заголовке соединение получали, по существу, в соответствии со способом, описанным в работе Aparajithan, K.; Thompson, А.С.; Sam, J. J. Heterocyclic. Chem. 1966, 3, 466. 4,5-Дигидроциклопента[b]тиофен-6-он (Bonini, В.F.; et al. Eur. J. Org. Chem. 2004, 4442 и ссылки, приведенные в настоящем описании) (0,658 г, 4,77 ммоль) растворяли в МеОН (50 мл) и добавляли гидрохлорид гидроксиламина (0,365 г, 5,25 ммоль) и NaOAc (2,35 г, 28,62 ммоль). Реакционную смесь перемешивали при КТ в течение ночи. Удаляли под вакуумом органический растворитель и обрабатывали полученный остаток EtOAc (60 мл). Раствор фильтровали через силикагель, промывали EtOAc и концентрировали. Остаток обрабатывали РРА (полифосфорной кислотой) (30 г) и нагревали на масляной бане до 130 С при периодическом перемешивании в течение 30 мин. После охлаждения реакционной смеси до КТ смесь выливали в воду со льдом (100 мл). Проводили экстракцию CH2Cl2 (3150 мл). Объединенные органические слои промывали 0,1 М NaOH (100 мл), сушили над Na2SO4 и концентрировали. Продукт очищали методом хроматографии с использованием в качестве элюента 75% EtOAc/гексаны с образованием указанного в заголовке соединения (0,521 г, 71%).- 16015127 Альтернативный способ получения. Стадия 1. Гидрохлорид 2-тиофен-3-илэтиламина. Борметилсульфидный комплекс (30,4 мл, 304,4 ммоль) медленно добавляли к раствору тиофен-3 илацетонитрила (25,0 г, 203,0 ммоль) в тетрагидрофуране (450 мл). Нагревали реакционную смесь до температуры кипения с обратным холодильником и поддерживали при этой температуре в течение 16 ч,после чего охлаждали до КТ. Медленно гасили реакцию метанолом (50 мл) до прекращения пенообразования. К полученной смеси медленно добавляли метанол (100 мл), насыщенный хлороводородом. Смесь перемешивали при КТ в течение 20 мин, после чего концентрировали под вакуумом. Добавляли к смеси метанол (100 мл) и концентрировали под вакуумом. Полученное твердое вещество суспендировали в диэтиловом эфире (200 мл) и отфильтровывали с образованием 31,1 г (94%) сырого продукта, представляющего собой указанное в заголовке соединение.MS/ES m/z 128,3 [М+Н]+. Стадия 2. Этиловый эфир (2-тиофен-3-илэтил)карбаминовой кислоты. Диизопропилэтиламин (54,0 г, 418,0 ммоль) добавляли к суспензии гидрохлорида 2-тиофен-3 илэтиламина в дихлорметане (400 мл) и перемешивали смесь в течение 40 мин при КТ. Охлаждали смесь до 0 С и добавляли по каплям этилхлорформиат (22,7 г, 209,0 ммоль) в течение 15 мин. После завершения добавления реакционную смесь перемешивали при 0 С в течение 1 ч и промывали 10% раствором бисульфата натрия (500 мл). Водную часть экстрагировали дихлорметаном (2100 мл), сушили объединенные органические слои над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали с помощью хроматографии на силикагеле с использованием в качестве элюента 100% дихлорметана с образованием 22,6 г (60%) указанного в заголовке соединения.MS/ES m/z 200,3 [М+Н]+. Стадия 3. 5,6-Дигидро-4 Н-тиено[2,3-c]пиридин-7-он. Пентоксид фосфора (32,1 г, 225,8 ммоль) добавляли к раствору этилового эфира 2-тиофен-3 илэтил)карбаминовой кислоты (22,5 г, 112,9 ммоль) в оксихлориде фосфора (167 мл), реакционную смесь нагревали до 110 С и выдерживали при этой температуре в течение 3 ч 45 мин. Охлаждали реакционную смесь до КТ и концентрировали под вакуумом. Остаток растворяли в дихлорметане (50 мл) и выливали на лед (300 г). Доводили рН смеси до pH 7 с помощью 5 N гидроксида натрия и экстрагировали дихлорметаном (4100 мл). Объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали под вакуумом. Полученный остаток очищали с помощью хроматографии на силикагеле с использованием в качестве элюента смеси от 0 до 70% этилацетата/гексана с образованием 8,38 г (48%) указанного в заголовке соединения.MS/ES m/z 154,3 [М+Н]+. Препаративный пример 20. 2-Бром-5,6-дигидро-4 Н-тиено[2,3-c]пиридин-7-он. 5,6-Дигидро-4 Н-тиено[2,3-c]пиридин-7-он (4,80 г, 31,37 ммоль) растворяли в смеси НОАс (40 мл) и воды (30 мл). Охлаждали до 0 С и добавляли по каплям Br2 (1,8 мл, 34,51 ммоль). Перемешивали реакционную смесь при 0 С в течение 1 ч. Разбавляли реакционную смесь водой (100 мл) и экстрагировалиEtOAc (3100 мл). Объединенные органические слои промывали 5% раствором Na2SO3 (250 мл) и насыщенным NaHCO3 (2100 мл), сушили над Na2SO4, фильтровали и концентрировали с образованием 6,198 г (85%) указанного в заголовке соединения. Добавляли 2-бром-5,6-дигидро-4 Н-тиено[2,3-c]пиридин-7-он (1,024 г, 4,42 ммоль), 4-метоксифенилбороновую кислоту (0,671 г, 4,42 ммоль) и Na2CO3 (0,94 г, 8,83 ммоль) к смеси воды (10 мл), диметоксиэтана (75 мл) и СН 3 ОН (50 мл). Полученную смесь продували азотом в течение 5 мин. ДобавлялиPd(PPh3)4 (0,153 г, 0,1325 ммоль) и осуществляли кипячение полученной смеси с обратным холодильником в течение ночи. Охлаждали реакционную смесь до КТ, разбавляли водой (100 мл), экстрагировалиEtOAc (3100 мл) и концентрировали. Остаток обрабатывали EtOAc (40 мл), выделяли твердое вещество и промывали EtOAc (20 мл) и Et2O (220 мл) с образованием указанного в заголовке соединения (0,950 г). Концентрировали фильтрат и очищали полученный остаток методом хроматографии с образованием дополнительного количества продукта (0,140 г). Суммарный выход составлял 1,090 г (95%).(21 мл), метанола (5 мл) и воды (1 мл). Реакционную смесь нагревали до 90 С и выдерживали при этой температуре в течение 16 ч. После охлаждения реакционной смеси до КТ смесь выливали в воду (75 мл). Полученное твердое вещество отфильтровывали и сушили под вакуумом при 80 С с образованием 0,40 гMS/ES m/z 248,0 [М+Н]+. Представленные в нижеследующей таблице промежуточные соединения получали по реакции сочетания Сузуки, по существу, согласно препаративному примеру 21 (в случае соединений согласно препаративному примеру 23) или 22 (в случае соединений согласно препаративному примеру 24) с использованием подходящей арилбороновой кислоты.(Е)-3-(5-Бромтиофен-3-ид)акриловую кислоту (Gronowitz, S.; Ander, I. Chemica Scripta, 1980, 15,145) (2,04 г, 8,79 ммоль) суспендировали в CH2Cl2 (30 мл), обрабатывали оксалилхлоридом (1,5 мл,17,58 ммоль), после чего добавляли ДМФ (3 капли). Реакционную смесь перемешивали при КТ в течение 30 мин, при этом образовывался прозрачный раствор, который продолжали перемешивать в течение 1,5 ч. Удаляли избыток реагента и растворитель под вакуумом. Полученный остаток растворяли в 1,4 диоксане (10 мл), раствор переносили в капельную воронку и добавляли по каплям к раствору NaN3(1,8 г, 26,37 ммоль) в смеси воды (10 мл) и ацетона (10 мл) при 0 С. В процессе добавления температуру реакционной смеси поддерживали равной менее 5 С. Смесь перемешивали при 0 С в течение 1 ч. Смесь разбавляли водой (15 мл) и экстрагировали EtOAc (330 мл). Органические слои объединяли и концентрировали под вакуумом без нагревания. Полученный остаток растворяли в EtOAc (50 мл), промывали водой (30 мл) и насыщенным солевым раствором (20 мл), затем сушили над Na2SO4, фильтровали и концентрировали под вакуумом без нагревания с образованием сырого промежуточного соединения, представляющего собой ацилазид. Полученный ацилазид растворяли в 1,4-диоксане (10 мл), переносили раствор в капельную воронку,присоединенную к колбе, содержащей Dowtherm A (15 мл), снабженной ловушкой Дина-Старка и холодильником. Смесь, содержащую Dowtherm А, нагревали до 230 С и добавляли по каплям раствор ацилазида. В ходе добавления температура реакционной смеси снижалась до 160 С, после чего вновь достигала 230 С. Низкокипящий растворитель скапливался в ловушке Дина-Старка. Реакционную смесь перемешивали при 230 С в течение 1 ч, охлаждали до КТ и разбавляли гексаном (40 мл). Выпавший осадок отделяли фильтрованием и промывали гексаном (220 мл) с образованием 1,838 г (91%) указанного в заголовке соединения. 1 Н ЯМР (ДМСО-d6):6,64 (д, 1 Н, J=6,8 Гц), 7,29 (д, 1 Н, J=6,8 Гц), 7,57 (с, 1 Н), 11,64 (ушир.с, 1 Н).MS/ES m/z (81Br) 229,8 [М-Н]-. Препаративный пример 26. 5-Хлортиофен-2-карбоновой кислоты (2,2-диэтоксиэтил)амид. 5-Хлортиофен-2-карбоновую кислоту (100 г, 0,60 моль) и дихлорметан (1000 мл) добавляли в трехгорлую круглодонную колбу вместимостью 3 л, снабженную подвесной мешалкой, отверстием для подачи/вывода азота, капельной воронкой и термопарой. Полученный раствор перемешивали в атмосфере азота с одновременным охлаждением до 4 С. С помощью капельной воронки добавляли раствор 2,2 диэтилоксиэтиламина (88,5 мл, 0,60 моль) в дихлорметане (35 мл) в течение 12 мин. К охлажденной смеси добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (123 г, 0,64 моль). Добавляли дополнительное количество дихлорметана (165 мл) и перемешивали реакционную смесь в течение 22 ч при комнатной температуре. Реакцию гасили водой (1000 мл) и отделяли образовавшиеся слои. Проводили обратную экстракцию водного слоя дихлорметаном (500 мл), объединяли органические слои и сушили над сульфатом натрия. После этого проводили очистку путем пропускания через слой силикагеля,используя в качестве элюента дихлорметан, затем 1% МеОН в дихлорметане и далее смесь 10% МеОН и дихлорметана с образованием 108 г (65%) указанного в заголовке соединения в виде твердого вещества белого цвета. 1 Н ЯМР (500 МГц, CDCl3):7,26 (д, J=3,5 Гц, 1 Н), 6,85 (д, J=3,5 Гц, 1 Н), 6,37 (ушир.с, 1 Н), 4,57 (т,J=5,5 Гц, 1 Н), 3,68-3,74 (м, 2 Н), 3,48-3,57 (м, 4 Н), 1,19 (т, J=7,5 Гц, 6 Н).EtOH (1000 мл) добавляли в трехгорлую круглодонную колбу вместимостью 2 л, снабженную подвесной мешалкой, отверстием для подачи/вывода азота, капельной воронкой и термопарой. Полученную суспензию нагревали в течение 35 мин, затем добавляли активированный уголь (5,8 г) и нагревали дополнительно в течение 0,5 ч. Суспензию фильтровали через фильтр из стекловолокна и промывали осадокEtOH (500 мл). Из фильтрата под разрежением удаляли часть растворителя таким образом, чтобы оставалось 15-20% растворителя. К полученному фильтрату добавляли воду (1300 мл) и перемешивали полученную суспензию в течение 1 ч при комнатной температуре, а затем в течение 0,5 ч при 0-5 С. Суспензию фильтровали, осадок промывали водой (1000 мл) и высушивали с образованием 66 г сырого продукта, представляющего собой 5-(4-хлорфенил)тиофен-2-карбоновой кислоты (2,2-диэтилоксиэтил)амид. Полученный сырой 5-(4-хлорфенил)тиофен-2-карбоновой кислоты (2,2-диэтилоксиэтил)амид кипятили с обратным холодильником в гептане (1625 мл) в течение 1 ч, а затем фильтровали через фильтр из стекловолокна. Фильтрат переносили в круглодонную колбу и удаляли часть растворителя таким образом,чтобы оставалось 725 мл растворителя. Смесь перемешивали при охлаждении в течение 40 мин. Полученную суспензию фильтровали, промывали гептаном (100 мл) и высушивали с образованием 41 г (64%) продукта в виде твердого вещества белого цвета. 1 Н ЯМР (500 МГц, CDCl3):7,54 (д, J=9,0 Гц, 2 Н), 7,45 (д, J=4,5 Гц, 1 Н), 7,38 (д, J=9,0 Гц, 2 Н), 7,24(д, J=4,5 Гц, 1 Н), 6,20 (ушир.с, 1 Н), 4,62 (т, J=5,5 Гц, 1 Н), 3,73-3,79 (м, 2 Н), 3,57-3,62 (м, 4 Н), 1,25 (т,J=7,0 Гц, 6 Н). Препаративный пример 28. 5-(4-Хлорфенил)тиофен-2-карбоновой кислоты (2-оксоэтил)амид. В трехгорлую круглодонную колбу вместимостью 250 мл, снабженную подвесной мешалкой, отверстием для подачи/вывода азота и термопарой, добавляли воду (21 мл) и затем трифторуксусную кислоту (100 г). К полученному раствору трифторуксусной кислоты при перемешивании добавляли 5-(4 хлорфенил)тиофен-2-карбоновой кислоты (2,2-диэтилоксиэтил)амид (25 г, 0,07 моль), при этом все указанное количество амида добавляли за один прием. Реакционную смесь перемешивали в течение 4 ч, выливали в воду со льдом (1200 мл) и перемешивали в течение 1,25 ч. Полученную суспензию фильтровали и промывали осадок водой (500 мл) и гептаном (500 мл), а затем сушили с образованием 18,65 г (95%) указанного в заголовке соединения в виде твердого вещества желто-белого цвета. 2-Бром-6 Н-тиено[2,3-c]пиридин-7-он (25,0 г, 108,7 ммоль), 4-хлорфенилбороновую кислоту (18,7 г,119,5 ммоль) и карбонат натрия (23,5 г, 217,3 ммоль) смешивали с этанолом (121 мл), 1,2 диметоксиэтаном (604 мл) и водой (121 мл). Реакционную смесь продували азотом в течение 20 мин. Добавляли тетракис(трифенилфосфин)палладий (3,77 г, 3,26 ммоль). Нагревали реакционную смесь до 85 С и выдерживали при этой температуре в течение ночи. Реакционную смесь охлаждали на воздухе до КТ. С помощью роторного испарителя выпаривали половину объема растворителя. Фильтровали смесь и промывали водой (2400 мл), простым эфиром (400 мл) и этилацетатом (20 мл), а затем высушивали полученное твердое вещество под вакуумом при 40 С с образованием 25,4 г (89%) указанного в заголовке соединения.(2-оксоэтил)амид (1 г, 0,004 моль) добавляли в трехгорлую круглодонную колбу вместимостью 25 мл,снабженную магнитной мешалкой, отверстием для подачи/вывода азота, ловушкой Дина-Старка и термопарой. Реакционную смесь нагревали до 95 С в течение 2 ч, затем охлаждали до 40 С и выливали в холодную воду (20 мл, 1,11 моль). Перемешивали смесь в течение 10 мин. Фильтровали полученную суспензию, промывали осадок водой (100 мл) и высушивали с образованием сырого продукта, представляющего собой 2-(4-хлорфенил)-6 Н-тиено[2,3-c]пиридин-7-он (0,95 г, 0,004 моль), в виде твердого вещества коричневого цвета.(0,969 г, 5,08 ммоль), а затем Et3N (1,06 мл, 7,62 ммоль). Реакционную смесь перемешивали при КТ вMS/ES m/z (81Br) 380,9 [M+Na]+. Препаративный пример 31. 4-Бромфениловый эфир толуол-4-сульфокислоты. Указанное в заголовке соединение получали, по существу, согласно препаративному примеру 30 с использованием 4-бромфенола (1,58 г, 9,13 ммоль) с образованием 2,76 г (92%) продукта. 1 Н ЯМР (400 МГц, CDCl3):2,45 (с, 3H), 6,84-6,89 (м, 2 Н), 7,32 (д, J=8,7 Гц, 2 Н), 7,38-7,43 (м, 2 Н),7,68-7,72 (м, 2 Н). Препаративный пример 32. 2-Метокси-4-[2-(4-метоксифенил)-7-оксо-4,7-дигидро-5 Н-тиено[2,3c]пиридин-6-ил]фениловый эфир толуол-4-сульфокислотыPd2(dba)3 (8,6 мг, 0,09 ммоль). Реакционную смесь при перемешивании кипятили с обратным холодильником в течение 16 ч. Смесь разбавляли EtOAc (100 мл), промывали водой (230 мл), сушили надNa2SO4, фильтровали и концентрировали. Полученный сырой продукт очищали методом хроматографии с использованием в качестве элюента 30-50% EtOAc/гексаны с образованием 0,432 г (86%) указанного в заголовке соединения.LC-MS/ES m/z 536,0 [М+Н]+. Представленные в нижеследующей таблице соединения получали, по существу, согласно препаративному примеру 32 с использованием подходящего тиенопиридинона и подходящего арилбромида или гетероарилбромида. Обработка: разбавляли EtOAc, фильтровали через целит (Celite) и концентрировали. Полученный остаток суспендировали в Et2O и фильтровали.(30 мл) и водой (20 мл). Реакционную смесь кипятили с обратным холодильником в течение 10 ч и разбавляли водой (50 мл). Смесь подкисляли 5,0 М HCl (8 мл). Удаляли под вакуумом органический растворитель и разбавляли остаток водой (10 мл). Твердое вещество отделяли фильтрованием и промывали водой (310 мл) и Et2O (215 мл), после чего сушили в вакуум-сушильном шкафу с образованием 0,646 гMS/ES m/z 382,0 [М+Н]+. Препаративный пример 39. 6-(4-Гидроксифенил)-2-(4-метоксифенил)-5,6-дигидро-4 Н-тиено[2,3c]пиридин-7-он. Указанное в заголовке соединение получали, по существу, согласно препаративному примеру 38 с использованием 4-[2-(4-метоксифенил)-7-оксо-4,7-дигидро-5 Н-тиено[2,3-c]пиридин-6-ил]фенилового эфира толуол-4-сульфокислоты (0,256 г, 0,51 ммоль) с образованием 0,135 г (76%) продукта.LC-MS/ES m/z 352,3 [М+Н]+. Пример 1. Гидрохлорид 2-(4-метоксифенил)-6-[3-метокси-4-(2-пирролидин-1-илэтокси)фенил]-5,6 дигидро-4 Н-тиено[2,3-c]пиридин-7-она. 2-(4-Метоксифенил)-5,6-дигидро-4 Н-тиено[2,3-c]пиридин-7-он (0,234 г, 0,90 ммоль), 1-[2-(4-бром-2 метоксифенокси)этил]пирролидин (Bastian, J.A., et al. WO 9725033 Al) (0,271 г, 0,90 ммоль), N,N'диметилэтан-1,2-диамин (16 мг, 0,18 ммоль), K2CO3 (0,249 г, 1,81 ммоль) и CuI (17 мг, 0,09 ммоль) смешивали в толуоле (10 мл) и перемешивали при 110 С в течение 48 ч. Разбавляли EtOAc (100 мл) и трижды промывали водой (50 мл), содержащей NH3 Н 2 О (5 мл), сушили над Na2SO4 и фильтровали. Продукт очищали методом хроматографии с использованием в качестве элюента 7% 2 М NH3 в СН 3 ОН и 93%CH2Cl2 с образованием 0,125 г очищенного продукта, который растворяли в СН 3 ОН (5 мл) и обрабатывали 1,0 М HCl в EtOH (270 мкл, 0,27 ммоль). Перемешивали при КТ в течение 5 мин и концентрировали с образованием указанного в заголовке соединения (0,133 г, 0,26 ммоль, 100%).(0,12 г, 0,31 ммоль) в ДМФ (5 мл) обрабатывали гидрохлоридной солью 4-(2-хлорэтил)морфолина (59 мг,0,31 ммоль) и 60% NaH (38 мг, 0,94 ммоль). Реакционную смесь перемешивали при 85 С в течение ночи. Реакцию гасили водой (5 мл) и разбавляли реакционную смесь EtOAc (150 мл). Проводили разделение фаз и органическую фазу промывали водой (350 мл), сушили над Na2SO4, фильтровали и концентрировали. Полученный сырой продукт очищали методом хроматографии с использованием в качестве элюента 1% NH3 Н 2 О, 10% СН 3 ОН и 90% EtOAc с образованием 31 мг продукта, который растворяли в CH3OH(2 мл) и обрабатывали 1,0 М HCl в EtOAc (65 мкл, 0,065 ммоль) при перемешивании при КТ в течение 5 мин. Удаляли под вакуумом растворитель с образованием 33 мг (20%) указанного в заголовке соединения.(3 мл) и воды (0,5 мл) при КТ. Добавляли 2,4-дихлорфенилбороновую кислоту (151 мг, 0,79 ммоль) с последующим добавлением трифенилфосфина (55 мг, 0,21 ммоль). Добавляли Pd(OAc)2 (16 мг, 0,7 ммоль) и нагревали до 80 С в атмосфере азота в течение 1,5 ч. Смесь охлаждали до КТ и помещали на колонку с силикагелем. Продукт очищали с помощью хроматографии на силикагеле с использованием градиента 030% (2 N NH3 в MeOH)/CHCl3 с образованием 155 мг (42%) целевого конечного продукта.- 21015127 Представленные в нижеследующей таблице соединения получали, по существу, согласно примеру 4 с использованием 4-(N,N-диметиламино)фенилбороновой кислоты, 4-(метансульфонил)фенилбороновой кислоты, 4-хлорфенилбороновой кислоты и п-толилбороновой кислоты соответственно. Гидрохлорид получали путем растворения свободного основания вCH2Cl2 и обработки 2,0 М HCl в диэтиловом эфире. Альтернативно, 2-(4-хлорфенил)-6-[3-метокси-4-(2-пирролидин-1-илэтокси)фенил]-5,6-дигидро-4 Нтиено[2,3-с]пиридин-7-он (пример 7) получали согласно препаративным примерам 40, 41 и 44 с последующим алкилированием согласно примеру 9. Препаративный пример 40. 2-Бром-6-(3-метокси-4-триизопропилсиланилоксифенил)-5,6-дигидро 4 Н-тиено[2,3-c]пиридин-7-он. 6-(3-Метокси-4-триизопропилсиланилоксифенил)-5,6-дигидро-4 Н-тиено[2,3-с]пиридин-7-он (5 г,11,6 ммоль) растворяли в ТГФ (100 мл) в трехгорлой круглодонной колбе. Раствор охлаждали на бане из сухого льда/ацетона. К одной из горловин колбы была присоединена капельная воронка, охлаждаемая с помощью сухого льда/ацетона, содержащая 1,2-дибром-1,1,2,2-тетрафторэтан (2,8 мл, 23,2 ммоль) в 10 мл ТГФ. Через одну из боковых горловин в колбу быстро добавляли трет-BuLi (9,7 мл 1,2 М раствора в пентане, 11,6 ммоль), позволяя раствору стекать внутрь колбы. По прошествии не более 30 с после окончания добавления трет-BuLi открывали капельную воронку, содержащую электрофильный реагент. Перемешивали смесь в течение 5-10 мин, затем выливали в водный раствор NaHCO3 (300 мл) и экстрагировали EtOAc (2300 мл). Объединенные органические слои промывали насыщенным солевым раствором (200 мл). Концентрировали и очищали продукт с помощью хроматографии на силикагеле с использованием градиента 0-25% EtOAc/гексаны с образованием 2,78 г (47%) целевого продукта.(2,87 г, 5,46 ммоль) растворяли в диметоксиэтане (28 мл) с последующим добавлением EtOH (4 мл) и воды (8 мл). Добавляли 4-хлорфенилбороновую кислоту (1,28 г, 8,19 ммоль) и 2 М Na2CO3 (4 мл,8,19 ммоль). Через реакционную смесь в течение 15-20 мин барботировали осушенный аргон. ДобавлялиPd(PPh3)4 (315 мг, 0,27 ммоль) и нагревали смесь до 90 С в атмосфере аргона. Через 3 ч реакционную смесь фильтровали через целит (Celite) и элюировали CH2Cl2. Концентрировали и очищали с помощью хроматографии на силикагеле с использованием градиента 0-25% EtOAc/гексаны с образованием 2,37 гMS/ES m/z (35C1) 542,1 [М+Н]+. Представленные в нижеследующей таблице соединения получали, по существу, согласно препаративному примеру 41 с использованием п-толуолбороновой кислоты и 4-циклопропоксифенилбороновой кислоты (полученной согласно Olofsson, K. et al., WO 2005123673). Варианты: DME/EtOH/2 M Na2CO3 (в отсутствие воды). Действие микроволнового излучения при 100 С в течение 3 ч. Обработка CH2Cl2 и 1 N NaOH. Препаративный пример 44. 2-(4-Хлорфенил)-6-(4-гидрокси-3-метоксифенил)-5,6-дигидро-4 Нтиено[2,3-c]пиридин-7-он. 2-(4-Хлорфенил)-6-(3-метокси-4-триизопропилсиланилоксифенил)-5,6-дигидро-4 Н-тиено[2,3 с]пиридин-7-он (1,68 г, 3,1 ммоль) растворяли в ТГФ (20 мл) и добавляли 1,0 М раствор тетрабутиламмонийфторида в ТГФ (3,1 мл, 3,1 ммоль). Перемешивали при КТ в течение 1 ч. Медленно добавляли 1 NHCl до достижения рН примерно 1, при этом начинал выпадать осадок. Раствор отстаивали при КТ в течение примерно 2-3 ч. Отфильтровывали образовавшийся осадок с образованием 850 мг (выход 71%) целевого продукта.MS/ES m/z (35Cl) 386,0 [М+Н]+. Представленные в нижеследующей таблице соединения получали, по существу, согласно препаративному примеру 44. Суспензию ДМФ (8 мл) и 2-(4-хлорфенил)-6-(4-гидрокси-3-метоксифенил)-5,6-дигидро-4 Нтиено[2,3-c]пирдин-7-она (850 мг, 2,21 ммоль) охлаждали до 0 С. Добавляли NaH (97 мг, 2,43 ммоль) и перемешивали в течение 5 мин. Получали свободное основание гидрохлорида 1-(2 хлорэтил)пирролидина путем промывания в делительной воронке суспензии коммерчески доступной соли (500 мг) в CH2Cl2 (50 мл) 1 N раствором NaOH (50 мл). Объединяли органические слои и промывали насыщенным солевым раствором (50 мл). Сушили над Na2SO4 и концентрировали с образованием 1-(2 хлорэтил)пирролидина. К реакционной смеси добавляли свободное основание (391 мг, 2,94 ммоль) и нагревали до 60 С в течение ночи. Реакционную смесь переносили в делительную воронку, содержащую 1 N NaOH (100 мл). Экстрагировали CH2Cl2 (2100 мл). Объединенные органические слои промывали насыщенным солевым раствором (100 мл). Часть полученного продукта очищали с помощью хроматографии на силикагеле с использованием 0-7% (2 N NH3 в МеОН)/CHCl3 градиента с образованием 330 мг(выход 30%) чистого свободного основания, идентичного соединению согласно примеру 8.MS/ES m/z 483,0 [М+Н]+. Альтернативно, продукт превращали в гидрохлорид путем растворения свободного основания вCH2Cl2 (примерно 10 мг/мл) и добавления 2,0 М HCl в диэтиловом эфире (1,5 экв.). Раствор либо концентрировали, либо осаждали диэтиловым эфиром с последующим фильтрованием с образованием 493 мгMS/ES m/z (35Cl) 483,0 [М+Н]+. Соединения, указанные в нижеследующей таблице, а также соединения согласно примеру 10 и препаративному примеру 48 получали, по существу, путем проведения алкилирования согласно примеру 9 с использованием подходящего фенола. Соединение согласно примеру 10 получали с использованием гидрохлорида N-(2-хлорэтил)морфолина вместо гидрохлорида 1-(2-хлорэтил)пирролидина. Пример 11. 2-(4-Хлорфенил)-6-[3-метокси-4-(2-пирролидин-1-илэтокси)фенил]-3-метил-5,6 дигидро-4 Н-тиено[2,3-c]пиридин-7-он. Указанное в заголовке соединение получали, по существу, согласно препаративному примеру 17 с использованием 5-(4-хлорфенил)-3-(2-гидроксиэтил)-4-метилтиофен-2-карбоновой кислоты [3-метокси 4-(2-пирролидин-1-илэтокси)фенил]амида.MS/ES m/z (35Cl) 497,0 [М+Н]+. Пример 12. Гидрохлорид 2-(4-циклопропоксифенил)-6-[3-метокси-4-(2-пирролидин-1 илэтокси)фенил]-5,6-дигидро-4 Н-тиено[2,3-с]пиридин-7-она. Свободное основание 2-хлорэтилпирролидина получали путем добавления соляно-кислой соли(84 мг, 0,49 ммоль) в делительную воронку, содержащую 1 N NaOH (75 мл) и CH2Cl2 (75 мл). Органический слой промывали насыщенным солевым раствором, после чего сушили над Na2SO4 и концентрировали. В отдельном сосуде, подходящем для использования в микроволновой печи, смешивали 2-(4 циклопропоксифенил)-6-(4-гидрокси-3-метоксифенил)-5,6-дигидро-4 Н-тиено[2,3-c]пиридин-7-он(100 мг, 0,24 ммоль) в ДМФ (1 мл). Смесь осторожно нагревали горячим воздухом до растворения фенола. Добавляли порошкообразный K2CO3 (68 мг, 0,49 ммоль). Перемешивали в течение 5-10 мин при КТ,после чего добавляли вышеуказанный хлорид в виде раствора в ДМФ (500 мкл). Сосуд накрывали крышкой и нагревали смесь с помощью микроволнового излучения до 110 С в течение 2 ч. Реакционную смесь сразу переносили на колонку с силикагелем и очищали с помощью хроматографии на силикагеле с использованием градиента 0-10% (2 N NH3 в МеОН)/CHCl3. Свободное основание продукта растворяли в(156 мг, 0,35 ммоль) растворяли в смеси диметоксиэтан:EtOH: вода, 7:2:1 (2 мл). Добавляли 4 фторфенилбороновую кислоту (63 мг, 0,45 мл) с последующим добавлением 2 М Na2CO3 (225 мкл,0,45 ммоль). Через реакционную смесь в течение 2-3 мин барботировали осушенный аргон, а затем добавляли Pd(PPh3)4 (20 мг, 0,18 ммоль). Сосуд с реакционной смесью герметично закрывали и смесь нагревали с помощью микроволнового излучения до 100 С в течение 40 мин. Реакционную смесь сразу переносили на колонку с силикагелем и очищали с помощью хроматографии на силикагеле с использованием градиента 0-10% (2 N NH3 в МеОН)/CHCl3 с образованием 139 мг (85%) целевого продукта в виде свободного основания. Получали HCl соль путем растворения свободного основания в CH2Cl2 (примерно 0,1-0,3 М). Добавляли 1,5 экв. 4,0 М HCl в виде раствора в диоксане. Полученную соль выделяли либо путем осаждения диэтиловым эфиром с последующим отделением осадка путем фильтрования, либо путем простого концентрирования CH2Cl2 раствора.MS/ES m/z 467,0 [М+Н]+. Представленные в нижеследующей таблице соединения получали, по существу, согласно примеру 13 с использованием подходящей бороновой кислоты и 2-бром-6-[3-метокси-4-(2-пирролидин-1 илэтокси)фенил]-5,6-дигидро-4 Н-тиено[2,3-c]пиридин-7-она.(0,26 мл, 1,53 ммоль) и перемешивали реакционную смесь при КТ в течение ночи. Избыток реагента и растворитель удаляли под вакуумом. Полученный остаток растворяли в CH2Cl2 (50 мл). Промывали насыщенным CuSO4 (230 мл) и сушили над Na2SO4. Фильтровали и концентрировали с образованием 0,649 г (100%) указанного в заголовке соединения.(15 мл) в реакторе Парра под давлением, оборудованном мешалкой с тефлоновым покрытием. Сосуд с реакционной смесью продували азотом (4) и монооксидом углерода (4). В сосуд нагнетали монооксид углерода (100 фунтов на квадратный дюйм, 690 кПа), герметизировали и перемешивали при 100 С в течение 6 ч, поддерживая давление в реакторе равным 100 фунтов на квадратный дюйм. После охлаждения реакционной смеси до комнатной температуры из реакционного сосуда удаляли монооксид углерода. Реакционную смесь фильтровали через силикагель, промывали EtOAc (100 мл) и концентрировали. Полученный сырой продукт очищали методом хроматографии с использованием в качестве элюента 50%MS/ES m/z 424,0 [М+Н]+. Препаративный пример 51. 2-Метокси-4-[2-(4-метоксифенил)-7-оксо-4,7-дигидро-5 Н-тиено[2,3c]пиридин-6-ил]бензойная кислота. Метиловый эфир 2-метокси-4-[2-(4-метоксифенил)-7-оксо-4,7-дигидро-5 Н-тиено[2,3-c]пиридин-6 ил]бензойной кислоты (0,2274 г, 0,54 ммоль) растворяли в диоксане (10 мл) и добавляли моногидрат гидроксида лития (124 мг, 2,69 ммоль) с последующим добавлением воды (5 мл). Реакционную смесь перемешивали при КТ в течение ночи, разбавляли водой (10 мл) и подкисляли 1 N HCl. Твердое вещество отделяли фильтрованием и промывали водой (210 мл) и Et2O (210 мл). Твердое вещество сушили в вакуумной печи с образованием 0,145 г продукта (66%).(5 мл) и добавляли 1 N NaOH (1,13 мл, 1,13 ммоль). Смесь нагревали с обратным холодильником в течение 16 ч, охлаждали до КТ и концентрировали под вакуумом. Остаток суспендировали в воде (25 мл) и подкисляли 10% раствором бисульфата натрия. Смесь экстрагировали этилацетатом (420 мл) с образованием суспензии продукта в органической фазе. Твердое вещество отфильтровывали и сушили под вакуумом с образованием 0,19 г (49%) указанного в заголовке соединения. Фильтрат концентрировали под вакуумом с образованием дополнительно 0,09 г (23%) указанного в заголовке соединения.(45 мг, 0,39 ммоль), EDCI (1-этил-3-[3-(диметиламино)пропил]карбодиимид хлоргидрат) (81 мг,0,42 ммоль), HOBt (57 мг, 0,42 ммоль) и Et3N (0,15 мл, 1,06 ммоль). Реакционную смесь перемешивали при КТ в течение ночи, разбавляли EtOAc (100 мл), промывали водой (230 мл) и сушили над Na2SO4. Сырой продукт очищали с помощью хроматографии на силикагеле с использованием в качестве элюента 2% NH3-H2O, 50% CH3OH/EtOAc с образованием 9 мг (5%) указанного в заголовке соединения.LC-MS/ES m/z 506,2 [М+Н]+. Пример 18. Гидрохлорид 4-[2-(4-хлорфенил)-7-оксо-4,7-дигидро-5 Н-тиено[2,3-с]пиридин-6-ил]-2 метокси-N-(2-морфолин-4-илэтил)бензамида. Раствор 4-[2-(4-хлорфенил)-7-оксо-4,7-дигидро-5 Н-тиено[2,3-c]пиридин-6-ил]-2-метоксибензойной кислоты (0,10 г, 0,24 ммоль) в тионилхлориде (1 мл) нагревали с обратным холодильником в течение 1 ч. Реакционную смесь охлаждали и концентрировали под вакуумом. Остаток растворяли в дихлорметане(0,5 мл) и добавляли к раствору 4-(2-аминоэтил)морфолина (0,038 г, 0,29 ммоль) и триэтиламина (0,029 г,0,29 ммоль) в дихлорметане (0,5 мл), при этом температура раствора составляла 0 С. Реакционную смесь перемешивали при КТ в течение 16 ч, промывали водой (2 мл) и проводили обратную экстракцию водной части дихлорметаном (22 мл). Объединенные органические растворы сушили (Na2SO4), фильтровали и концентрировали под вакуумом. Полученный продукт очищали с помощью хроматографии на силикагеле с использованием в качестве элюента градиента от чистого дихлорметана до 5% 2 М раствора аммиака в метаноле/дихлорметане с образованием 0,085 г свободного амина. Амин растворяли в дихлорметане (0,5 мл) и добавляли 1 N HCl в этаноле (0,16 мл). Добавляли диэтиловый эфир (2 мл), отфильтровывали образовавшееся твердое вещество и сушили под вакуумом при 80 С с образованием 0,08 г (59%) указанного в заголовке соединения. 2,0 М раствор триметилалюминия в толуоле (0,26 мл, 0,53 ммоль) добавляли к раствору 1-(2 аминоэтил)пирролидина (0,042 г, 0,37 ммоль) в толуоле (7 мл) и перемешивали смесь в течение 5 мин. Добавляли суспензию метилового эфира 4-[2-(4-хлорфенил)-7-оксо-4,7-дигидро-5 Н-тиено[2,3c]пиридин-6-ил]-2-метоксибензойной кислоты (0,15 г, 0,35 ммоль) в толуоле (2 мл), нагревали реакционную смесь до 100 С и выдерживали при этой температуре в течение 3 ч. Охлаждали реакционную смесь до комнатной температуры и медленно гасили реакцию водой до прекращения пенообразования. Смесь разбавляли 1 N NaOH и экстрагировали этилацетатом (310 мл). Объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали под вакуумом с образованием свободного амина(0,117 мл). Добавляли диэтиловый эфир (5 мл), отфильтровывали и сушили полученное твердое вещество белого цвета под вакуумом с образованием 0,053 г (28%) указанного в заголовке соединения.MS/ES m/z (37Cl) 511,3 [М+]. Соединения, указанные в нижеследующей таблице, а также соединения согласно примерам 20 и 21 и препаративным примерам 53 и 54 получали, по существу, согласно примеру 19 с использованием подходящего сложного эфира и амина. Очищены с помощью хроматографии на силикагеле с использованием в качестве элюента градиента от 0 до 75 или 80%EtOAc/гексаны. Пример 22. Гидрохлорид -4-[2-(4-хлорфенил)-7-оксо-4,7-дигидро-5 Н-тиено[2,3-c]пиридин-6-ил]2-метокси-N-морфолин-2-илметилбензамида. Раствор трет-бутилового эфира 4-[2-(4-хлорфенил)-7-оксо-4,7-дигидро-5 Н-тиено[2,3-c]пиридин-6 ил]-2-метоксибензоиламинометилморфолин-4-карбоновой кислоты (0,20 г, 0,33 ммоль) в 4 М HCl и 1,4 диоксане (3 мл) перемешивали при КТ в течение 2 ч. Добавляли диэтиловый эфир (5 мл), фильтровали и сушили под вакуумом при 80 С с образованием 0,153 г (85%) указанного в заголовке соединения.MS/ES m/z (37Cl) 497,0 [М+]. Пример 24. Гидрохлорид 2-хлор-4-[2-(4-хлорфенил)-7-оксо-4,7-дигидро-5 Н-тиено[2,3-c]пиридин-6 ил]-N-(2-пирролидин-1-илэтил)бензамида. Хлорид индия(III) (0,046 г, 0,093 ммоль) добавляли к суспензии метилового эфира 2-хлор-4-[2-(4 хлорфенил)-7-оксо-4,7-дигидро-5 Н-тиено[2,3-c]пиридин-6-ил]бензойной кислоты (0,2 г, 0,46 ммоль) в 1(2-аминоэтил)пирролидине (0,9 мл, 7,1 ммоль), нагревали смесь до 120 С и выдерживали при этой температуре в течение 16 ч. Смесь охлаждали до КТ и разбавляли водой (10 мл). Экстрагировали смесь дихлорметаном (310 мл). Объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали под вакуумом. Остаток очищали с помощью хроматографии на силикагеле с использованием в качестве элюента градиента от дихлорметана до 6% 2 М раствора аммиака в смеси метанол/дихлорметан с образованием 71 мг (30%) свободного амина. Свободный амин растворяли в хлороформе (0,5 мл) и добавляли 1 N HCl в этаноле (0,138 мл). Раствор разбавляли диэтиловым эфиром (5 мл), фильтровали и сушили полученное твердое вещество белого цвета под вакуумом при 80 С с образованием 0,06 г (24%) указанного в заголовке соединения.MS/ES m/z (35Cl35Cl) 514,0 [М+Н]+. Препаративный пример 55. 1-(2-Метокси-4-нитрофенил)-4-метилпиперазин. 1-Хлор-2-метокси-4-нитробензол (1,0 г, 5,33 ммоль) и 1-метилпиперазин (1,33 г, 13,30 ммоль) нагревали до 100 С и выдерживали при этой температуре в запаянной трубке в течение 16 ч. Смесь охлаждали до комнатной температуры и подвергали разделению между дихлорметаном (50 мл) и водой(50 мл). Органический слой отделяли, а водный слой экстрагировали дихлорметаном (220 мл). Объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали под вакуумом с образованием 1,05 г (78%) указанного в заголовке соединения.(50 мл) и добавляли 10% Pd/C (0,20 г). Суспензию гидрировали при комнатной температуре под колоко- 27015127 лом, наполненным водородом, в течение 16 ч. Фильтровали суспензию через слой целита (Celite) и промывали этилацетатом. Концентрировали фильтрат под вакуумом с образованием твердого вещества красноватого цвета (0,66 г, выход 71%).MS/ES m/z 220,0 (М+1)+. Препаративный пример 57. трет-Бутиловый эфир 4-(2-фтор-4-нитрофенил)пиперазин-1-карбоновой кислоты. 3,4-Дифторнитробензол (2,0 г, 12,6 ммоль) растворяли в ацетонитриле (35 мл) и добавляли третбутиловый эфир пиперазин-1-карбоновой кислоты (4,7 г, 25,2 ммоль). Смесь нагревали с обратным холодильником в течение 16 ч. Охлаждали смесь до комнатной температуры и концентрировали под вакуумом. Проводили разделение остатка между дихлорметаном (75 мл) и водой (75 мл), отделяли органическую часть, а водную часть экстрагировали дихлорметаном (225 мл). Объединяли органические растворы, сушили (Na2SO4), фильтровали и концентрировали под вакуумом с образованием 2,83 г (72%) указанного в заголовке соединения.N-трет-Бутил-(S)-3-гидроксипирролидин (35 г, 187 ммоль) растворяли в осушенном ДМФ (500 мл) при 0 С в течение 10 мин. Добавляли небольшими порциями NaH (60% в минеральном масле, 8,98 г,224 ммоль). Реакционную смесь перемешивали в течение 30 мин при 0 С, а затем с помощью полой трубки в атмосфере азота добавляли по каплям раствор 2-хлор-5-нитроанизола (35,1 г, 187 ммоль) в ДМФ (120 мл). Реакционную смесь перемешивали в течение ночи при 130 С. Органический растворитель удаляли под разрежением. Разбавляли остаток водой (1 л) и экстрагировали EtOAc (3200 мл). Объединенные органические слои промывали водой (2200 мл), сушили (Na2SO4), фильтровали и концентрировали под вакуумом. Полученный остаток очищали с помощью хроматографии на силикагеле с использованием в качестве элюента градиента растворителя 0-50% EtOAc в гексанах. Выделяли целевой продукт и растирали с добавлением Et2O в течение ночи. Отфильтровывали осадок методом вакуумфильтрования с образованием 37 г (58%) указанного в заголовке соединения. 1 Н ЯМР (CDCl3):1,43 (с, 9 Н), 2,27-2,05 (м, 2 Н), 3,69-3,48 (м, 4 Н), 4,98 (м, 1 Н), 3,89 (с, 3H), 6,856,83 (д, J=9,2 Гц, 1 Н), 7,73-7,72 (д, J=2,6 Гц, 1 Н), 7,85-7,83 (ушир.д, J=9,0 Гц, 2 Н). Препаративный пример 59. трет-Бутиловый эфир (R)-3-(2-метокси-4-нитрофенокси)пиперидин-1 карбоновой кислоты. Указанное в заголовке соединение получали, по существу, согласно препаративному примеру 58 с использованием трет-бутилового эфира (R)-3-гидроксипиперидин-1-карбоновой кислоты (35,0 г,174 ммоль) с образованием 47,0 г (76%) указанного в заголовке соединения.MS/ES m/z 297,2 [M-tBu+H]+. Препаративный пример 60. трет-Бутиловый эфир 3-(2-метокси-4-нитрофенокси)азетидин-1 карбоновой кислоты. 1-Фтор-2-метокси-4-нитробензол (118 г, 689 ммоль) и трет-бутиловый эфир 3-гидроксиазетидин-1 карбоновой кислоты (125 г, 724 ммоль) растворяли в ТГФ (800 мл) и охлаждали до 0 С. К полученному раствору в атмосфере азота добавляли по каплям 1 М раствор tBuOK (1 л, раствор в ТГФ). По окончании добавления перемешивали темно-коричневый раствор при 0 С в течение 30 мин, после чего разбавляли водой (1 л), добавление которой проводили в течение 10 мин. Смесь перемешивали в течение 5 мин и затем дважды экстрагировали МТВЕ (трет-бутилметиловым эфиром). Органические растворы объединяли и промывали насыщенным солевым раствором (2700 мл), затем сушили и концентрировали. Полученное твердое вещество сушили под вакуумом при 45 С в течение 20 ч с образованием 216 г (95%) указанного в заголовке соединения в виде твердого вещества желтого цвета. 1 Н ЯМР (300 МГц, CDCl3):1,42 (с, 9 Н), 3,80 (с, 3H), 4,04 (м, 2 Н), 4,18 (м, 2 Н), 4,76 (м, 1 Н), 6,17(дд, 1 Н, J=2,4, 8,5), 6,30 (д, 1 Н, J=2,6), 6,47 (д, 1 Н, J=8,5). Нижеприведенные соединения получали, по существу, согласно препаративному примеру 60 с использованием подходящего спирта.(0,99 г, 5,95 ммоль) и N,N-диизопропилэтиламин (1,08 мл, 6,55 ммоль) растворяли в N,Nдиметилформамиде (20 мл) и добавляли О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат (2,49 г, 6,55 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч, разбавляли водой (50 мл) и экстрагировали дихлорметаном (325 мл). Объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали под вакуумом. Продукт очищали методом хроматографии (от 0 до 4% МеОН/CH2Cl2) с образованием 0,77 г (46%) указанного в заголовке соединения.(R)-1-(2-Метокси-4-нитрофенил)-3-триизопропилсиланилоксипирролидин. 1-Хлор-2-метокси-4-нитробензол (10 г, 53,3 ммоль) смешивали с (R)-3-пирролидинолом (9,3 г,106,6 ммоль). Смесь нагревали до 100 С в течение ночи. Смесь охлаждали, растворяли в CH2Cl2 (200 мл) и промывали 1 N NaOH (100 мл). Экстракт промывали насыщенным солевым раствором (350 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали с образованием промежуточного соединения, представляющего собой пирролидинол, в виде влажного твердого вещества краснокоричневого цвета (12,17 г, 95%).MS (ЭС+) 239,1 (М+1)+. Сырой продукт, представляющий собой (R)-1-(2-метокси-4-нитрофенил)пирролидин-3-ол (10,9 г,45,5 ммоль), растворяли в осушенном пиридине (50 мл) и охлаждали до 0 С. Добавляли по каплям хлортриизопропилсилан (19,8 мл, 91 ммоль) и нагревали смесь до 80 С в течение ночи. Удаляли под разрежением пиридин, промывали сырой продукт раствором NaHSO3 и экстрагировали EtOAc (3100 мл). Органические растворы объединяли, сушили и концентрировали с образованием сырого продукта. Продукт очищали на колонке с силикагелем с помощью гексанов (300 мл) и промывали 10% EtOAc в гексанах(800 мл) с образованием указанного в заголовке соединения в виде красноватого масла (17,85 г, 99%).(S)-1-(2-Метокси-4-нитрофенил)-3-триизопропилсиланилоксипирролидин. Указанное в заголовке соединение получали, по существу, согласно препаративному примеру 56 с использованием (S)-3-пирролидинола.MS/ES m/z 395,3 [М+Н]+. Препаративный пример 68. трет-Бутиловый эфир (S)-3-(4-амино-2-метоксифенокси)пирролидин-1 карбоновой кислоты. трет-Бутиловый эфир (S)-3-(2-метокси-4-нитрофенокси)пирролидин-1-карбоновой кислоты (2,0 г,5,91 ммоль) растворяли в этилацетате (50 мл) и добавляли 10% Pd/C (0,20 г). Суспензию гидрировали при комнатной температуре под колоколом, наполненным водородом, в течение 16 ч. Фильтровали суспензию через слой целита (Celite) и промывали этилацетатом. Фильтрат концентрировали под вакуумом с образованием твердого вещества красноватого цвета (1,82 г, выход 100%).MS/ES m/z 253,0 [М-трет-бутил+Н]+. Нижеприведенные соединения получали, по существу, согласно препаративному примеру 68 с той разницей, что в препаративных примерах 72 и 73 в качестве растворителя использовали метанол и в препаративном примере 72 использовали 5% Pd/C.
МПК / Метки
МПК: C07D 495/04, A61K 31/4365, A61P 3/04
Метки: антагонисты, мсн-рецепторов
Код ссылки
<a href="https://eas.patents.su/30-15127-antagonisty-msn-receptorov.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты мсн-рецепторов</a>
Антагонисты мускариновых ацетилхолиновых рецепторов

Номер патента: 9899
Опубликовано: 28.04.2008
Авторы: Буш-Петерсен Якоб, Чжу Чунцзе, Янь Хунсин, Вань Цзехун, Палович Майкл Р.
МПК: C07D 451/02, A61K 31/46
Метки: рецепторов, ацетилхолиновых, антагонисты, мускариновых
Формула / Реферат:
1. Соединение, которое является (эндо)-3-(2-циано-2,2-дифенилэтил)-8,8-диметил-8-азониабицикло [3,2,1]октана бромидом и (эндо)-3-(2-циано-2,2-дифенилэтил)-8,8-диметил-8-азониабицикло[3,2,1]октана йодидом. 2. Соединение по п.1, которое является (эндо)-3-(2-циано-2,2-дифенилэтил)-8,8-диметил-8-азониабицикло[3,2,1]октана бромидом. 3. Соединение по п.1, которое является (эндо)-3-(2-циано-2,2-дифенилэтил)-8,8-диметил-8-азониабицикло[3,2,1]октана...
Антагонисты мускариновых ацетилхолиновых рецепторов

Номер патента: 13435
Опубликовано: 30.04.2010
Авторы: Вань Цзехун, Буш-Петерсен Якоб, Янь Хунсин, Палович Майкл Р., Чжу Чунцзе
МПК: C07D 451/02, A61K 31/46
Метки: ацетилхолиновых, мускариновых, антагонисты, рецепторов
Формула / Реферат:
1. Соединение, имеющее структуру Iв котором обозначенный атом Н находится в экзо-положении;R1-представляет собой анион, связанный с положительным зарядом атома N;R2 и R3 независимо выбраны из арила или гетероарила, где арил представляет собой фенил или нафтил и гетероарил представляет собой 5-10-членную систему ароматических колец, в которой одно или более колец содержит один и более гетероатомов, выбранных из группы, состоящей из N, О или S;R4...
Антагонисты мускариновых ацетилхолиновых рецепторов

Номер патента: 13689
Опубликовано: 30.06.2010
Авторы: Палович Майкл Р., Янь Хунсин, Чжу Чунцзе, Буш-Петерсен Якоб, Вань Цзехун
МПК: A61K 31/46, C07D 451/02
Метки: мускариновых, рецепторов, ацетилхолиновых, антагонисты
Формула / Реферат:
1. Соединение, имеющее структуру IIв котором обозначенный атом Н находится в экзо-положении;R2 и R3 независимо выбраны из арила или гетероарила, где арил представляет собой фенил или нафтил и гетероарил представляет собой 5-10-членную систему ароматических колец, в которой одно или более колец содержит один и более гетероатомов, выбранных из группы, состоящей из N, О или S;R4 представляет собой -CN.2. Соединение по п.1, где R2 и R3 оба являются...
Антагонисты рецепторов эндотелина

Номер патента: 952
Опубликовано: 28.08.2000
Авторы: Луэнго Хуан Игнасио, Эллиотт Джон Дункан
МПК: A61K 31/415, C07D 405/02, A61P 9/02...
Метки: рецепторов, эндотелина, антагонисты
Формула / Реферат:
1. Соединение общей формулы I в которой Z представляет собой радикал в котором R1 и R3 являются водородом, R4 является хлором или н.алкоксигруппой, R5 является группой -О-СН2-Аr, в которой Аr является фенилом, замещенным одним или двумя заместителями, выбранными из СООН, хлора, или Аr является пиридилом, замещенным СООН, R15 является низшим алкилом; Р представляет СООН; R2 представляет группу в которой Z1 и Z2 является...
Пиразолпиримидины как антагонисты крф рецепторов.

Номер патента: 803
Опубликовано: 24.04.2000
Авторы: Маккарти Джеймс Р., Хуанг Чарльз, Моран Тиренс Дж., Вебб Томас Р., Чен Чен, Уилкоуксен Кейт М.
МПК: C07D 487/04, A61K 31/495
Метки: пиразолпиримидины, рецепторов, крф, антагонисты
Формула / Реферат:
1. Соединение формулы I включая его стереоизомерные формы и фармацевтически приемлемые кислотно-аддитивные соли, где R1 представляет собой NR4R5 или OR5; R2 представляет C1-6-алкил, C1-6-алкилокси или C1-6-алкилтио; R3 представляет водород, C1-6-алкил, C1-6-алкилсульфонил, C1-6-алкилсульфокси или C1-6-алкилтио; R4 представляет водород, C1-6-алкил, моно- или ди(С3-6-циклоалкил) метил, С3-6-циклоалкил, С3-6-алкенил, гидрокси-С1-6-алкил,...
Предыдущий патент: Пиридил- и пиримидинилзамещенные производные пиррола, тиофена и фурана в качестве ингибиторов киназ
Следующий патент: Превращение кислородсодержащих соединений в олефины с димеризацией и диспропорционированием
Случайный патент: Балансировочный груз с патроном, содержащим материал для балансировки