Замещенные соединения миноциклина, фармацевтическая композиция и способ лечения чувствительного к тетрациклину состояния млекопитающего
Номер патента: 9175
Опубликовано: 28.12.2007
Авторы: Макинтайр Лора, Коуза Дэррелл, Хокинс Пол, Лю Гуй, Стэплетон Карен, Верма Атул, Ренни Глен, Баузер Тодд, Фречетт Роджер, Шихэн Пол, Виски Питер, Уорчол Тэд, Измаил Мохамед, Дьюморней Джимми, Бхатиа Бина, Нельсон Марк Л.



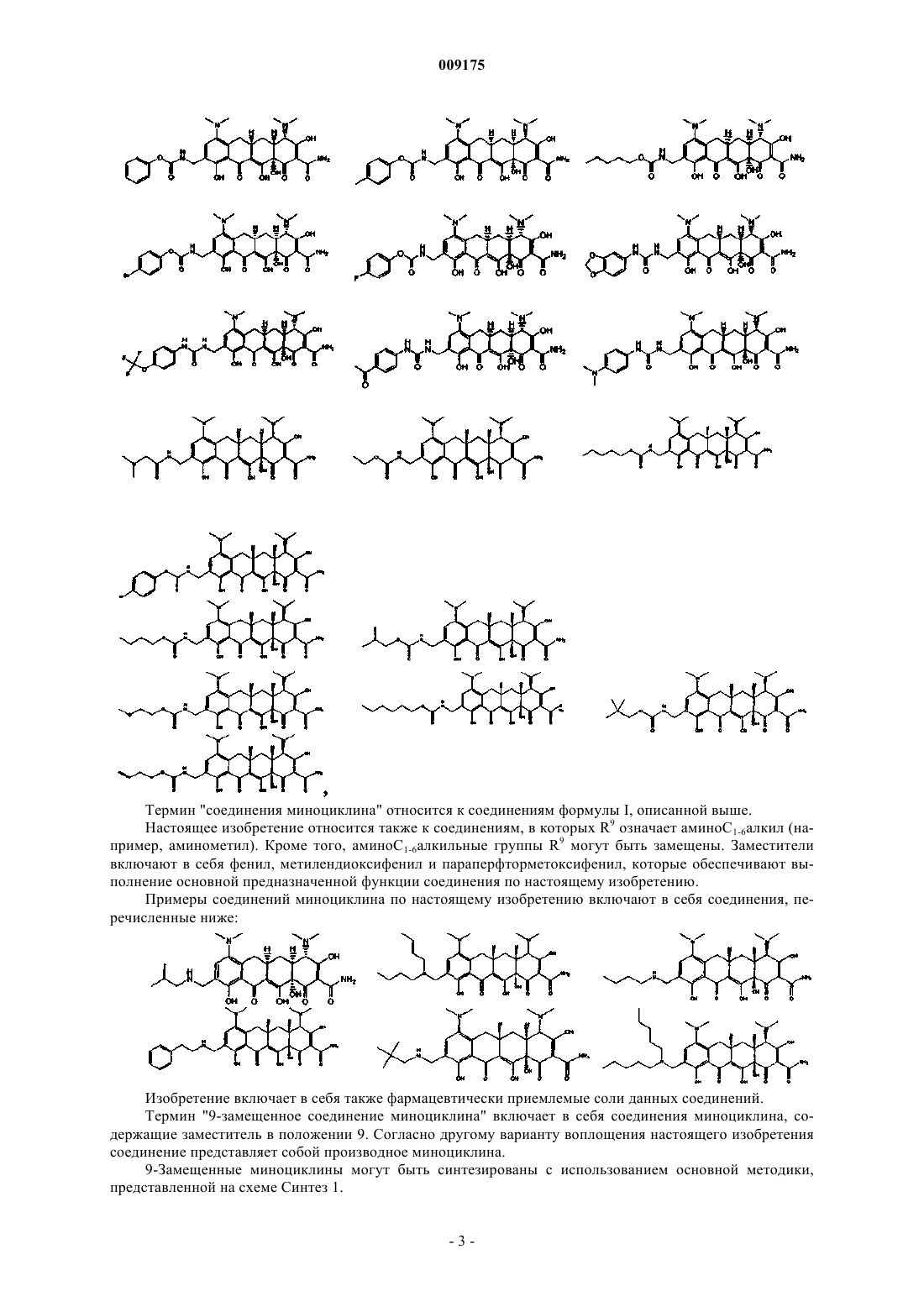


Формула / Реферат
1. Соединение миноциклина общей формулы I

где R4', R4'', R7' и R7'', каждый независимо означает С1-6алкил;
R9 представляет собой С1-6алкил, который замещен аминогруппой, С1-6алкиламиногруппой, диС1-6
алкиламиногруппой или С1-6алкиламиногруппой, в которой С1-6алкил замещен фенилом, метилендиоксифенилом или параперфторметоксифенилом, или группа R9 имеет следующие значения:


и их фармацевтически приемлемые соли или сложные эфиры.
2. Соединение миноциклина по п.1, отличающееся тем, что R9 означает метил, который замещен незамещенной или замещенной С1-6алкиламиногруппой или диС1-6алкиламиногруппой.
3. Соединение миноциклина по п.2, отличающееся тем, что R9 означает метил, который замещен незамещенной С1-6алкиламиногруппой.
4. Соединение миноциклина по п.3, отличающееся тем, что указанная незамещенная С1-6алкиламиногруппа представляет собой пентиламиногруппу.
5. Соединение по п.1, отличающееся тем, что оно выбрано из группы, включающей

или их фармацевтически приемлемые соли или сложные эфиры.
6. Соединение по п.1, представляющее собой

или его фармацевтически приемлемые соли или сложные эфиры.
7. Способ лечения чувствительного к тетрациклину состояния субъекта-млекопитающего, отличающийся тем, что осуществляют лечение указанного субъекта посредством введения соединения миноциклина по любому из пп.1-6.
8. Способ по п.7, отличающийся тем, что указанным чувствительным к тетрациклину состоянием является бактериальная инфекция.
9. Способ по п.8, отличающийся тем, что указанная бактериальная инфекция связана с Е. coli.
10. Способ по п.8, отличающийся тем, что указанная бактериальная инфекция связана с S. aureus.
11. Способ по п.8, отличающийся тем, что указанная бактериальная инфекция связана с Е. faecalis.
12. Способ по п.8, отличающийся тем, что указанная бактериальная инфекция устойчива к другим тетрациклиновым антибиотикам.
13. Способ по п.7, отличающийся тем, что указанное соединение миноциклина вводят в смеси с фармацевтически приемлемым носителем.
14. Способ по п.7, отличающийся тем, что указанным субъектом является человек.
15. Фармацевтическая композиция для воздействия на бактерии, чувствительные к тетрациклину, отличающаяся тем, что она содержит терапевтически эффективное количество соединения миноциклина по любому из пп.1-6 и фармацевтически приемлемый носитель.
Текст
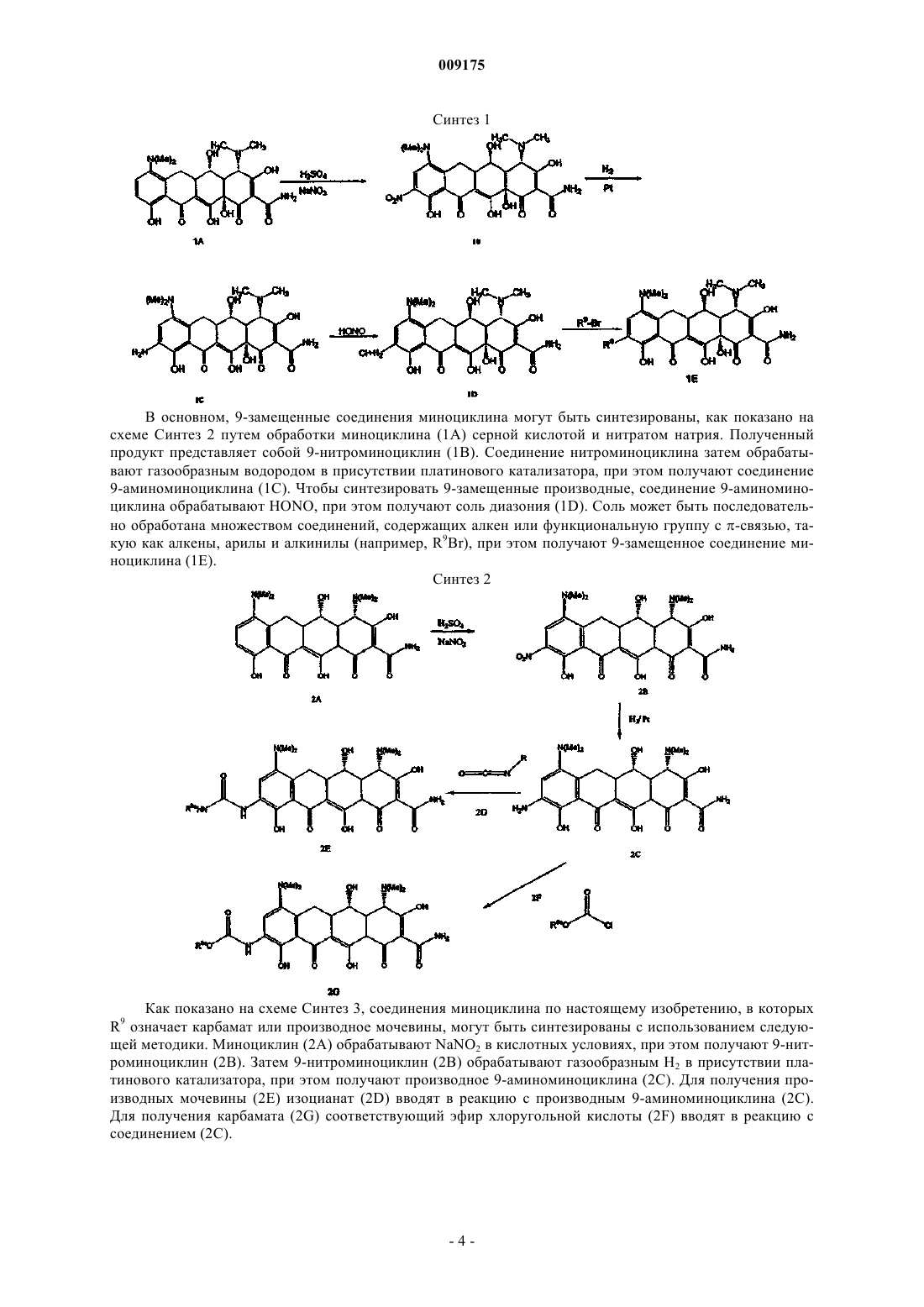
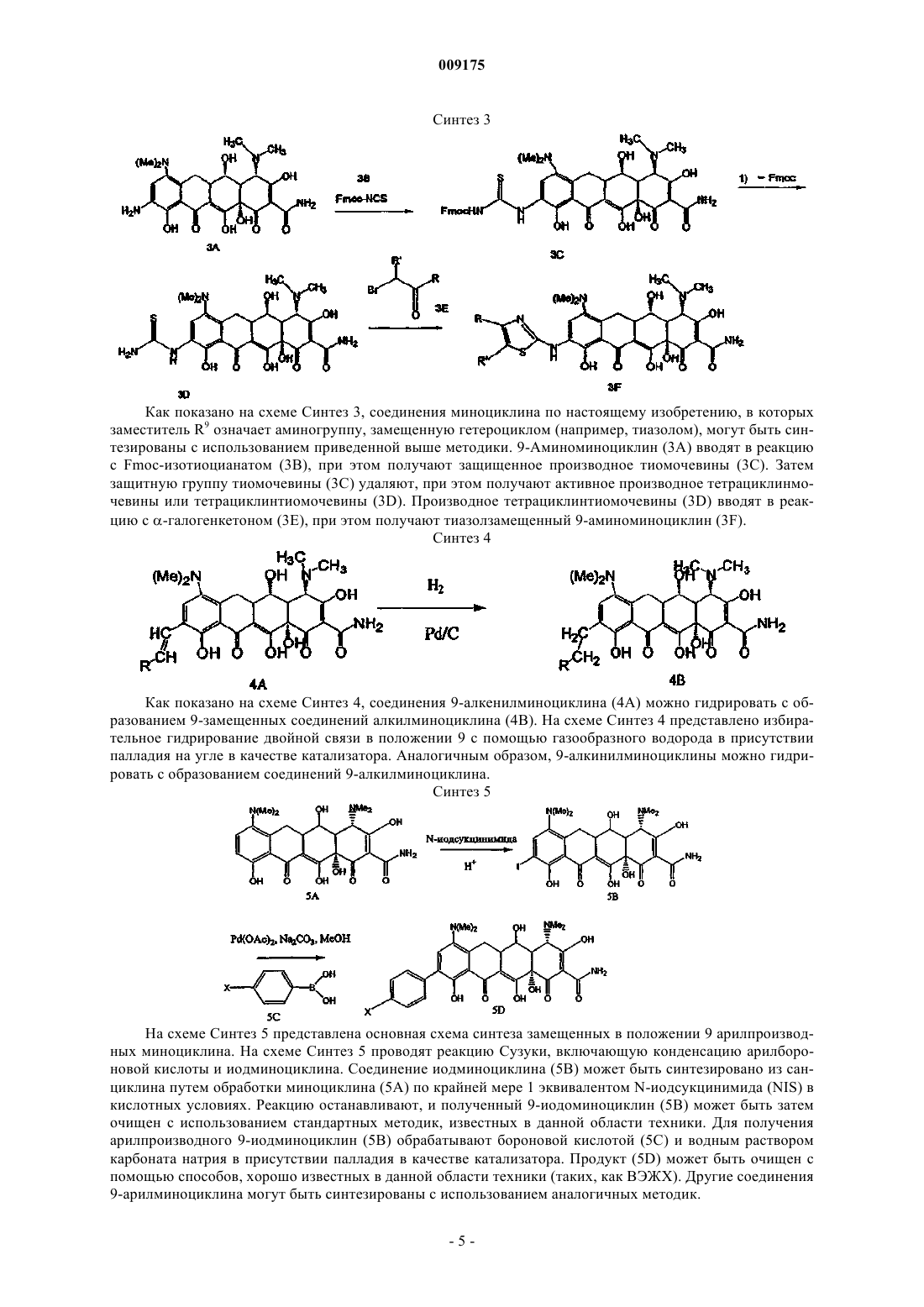
009175 Область техники, к которой относится изобретение Изобретение относится к области медицинской химии, более конкретно к химии антибиотиков, и в частности касается новых замещенных соединений миноциклина, в особенности 9-замещенных соединений миноциклина и их использования для лечения состояний у млекопитающего, чувствительных к действию тетрациклина. Уровень техники Разработка тетрациклиновых антибиотиков является прямым результатом систематического анализа бактериального состава образцов почв, собранных в различных частях земного шара, с целью обнаружения микроорганизмов, способных продуцировать бактерицидные и/или бактериостатические композиции. Первым из таких новых соединений в 1948 г. был обнаружен антибиотик под названием хлортетрациклин. Двумя годами позже был обнаружен окситетрациклин. Изучение химической структуры таких соединений подтвердило сходство их свойств и в 1952 г. явилось аналитической основой для получения третьего представителя этой группы - тетрациклина. Новое семейство соединений тетрациклина, не содержащих присоединенной к кольцу метильной группы, которая присутствовала в предыдущих препаратах тетрациклина,было получено в 1957 г. и получило широкое распространение в 1967 г., а миноциклин используется с 1972 г. Позднее исследования были сфокусированы на разработке новых композиций на основе тетрациклиновых антибиотиков, эффективных при лечении различных состояний с использованием различных способов введения. Были исследованы также новые аналоги тетрациклина с равным или более эффективным действием по сравнению с первыми соединениями тетрациклина. Примеры включают патенты США 2980584, 2990331,3062717, 3165531, 3454697, 3557280, 3674859, 3957980, 4018889, 4024272 и 4126680. В этих патентах представлен широкий спектр фармацевтически активных композиций на основе тетрациклина и аналогов тетрациклина. Позднее, сразу же после их открытия и внедрения было показано, что тетрациклины обладают чрезвычайно высокой фармакологической эффективностью в отношении риккетсий, ряда грамположительных и грамотрицательных бактерий и агентов, вызывающих венерическую лимфогранулему, конъюктивиты с включениями и пситтакоз. Так тетрациклины стали известны как антибиотики "широкого спектра". В связи с последующим исследованием их антимикробной активности in vitro, эффективности по отношению к экспериментальным инфекциям и фармакологических свойств класс тетрациклинов быстро получил широкое распространение в терапии. Однако указанное широко распространенное использование тетрациклинов как для лечения серьезных, так и неопасных заболеваний и расстройств привело к развитию устойчивости к этим антибиотикам даже среди высокочувствительных бактериальных штаммов, как симбиотических, так и патогенных (например, Pneumococci и Salmonella). Появление устойчивых к тетрациклину организмов привело к общему отказу от использования композиций на основе тетрациклина и аналогов тетрациклина. Сущность изобретения Изобретение включает в себя соединения миноциклина общей формулы (I) где R4', R4, R7' и R7, каждый независимо означает С 1-6 алкил; и их фармацевтически приемлемые соли или сложные эфиры. Изобретение относится также к следующим соединениям: или их фармацевтически приемлемым солям или сложным эфирам. Изобретение относится также к способам использования соединений миноциклина по изобретению для лечения субъектов, страдающих от состояний, которые могут быть излечены этими соединениями миноциклина. Изобретение включает в себя также фармацевтические композиции, содержащие соединения миноциклина по настоящему изобретению, и фармацевтически приемлемый носитель. Настоящее изобретение относится также к применению соединения миноциклина по настоящему изобретению для получения лекарственного средства, например лекарственного средства для лечения состояний, чувствительных к тетрациклину. Сведения, подтверждающие возможность осуществления изобретения Настоящее изобретение относится к новым 9-замещенным соединениям миноциклина. Эти соединения миноциклина могут быть использованы для лечения многочисленных чувствительных к тетрациклину состояний, таких как бактериальные инфекции и новообразования, наряду с другими известными областями применений соединений миноциклина и тетрациклина, в основном, в качестве блокаторов оттока тетрациклина и модуляции экспрессии генов. Изобретение включает в себя соединения миноциклина общей формулы (I) где R4', R4, R7' и R7, каждый независимо означает С 1-6 алкил; Термин "соединенияминоциклина" относится к соединениям формулы I, описанной выше. Настоящее изобретение относится также к соединениям, в которых R9 означает аминоС 1-6 алкил (например, аминометил). Кроме того, аминоС 1-6 алкильные группы R9 могут быть замещены. Заместители включают в себя фенил, метилендиоксифенил и параперфторметоксифенил, которые обеспечивают выполнение основной предназначенной функции соединения по настоящему изобретению. Примеры соединений миноциклина по настоящему изобретению включают в себя соединения, перечисленные ниже: Изобретение включает в себя также фармацевтически приемлемые соли данных соединений. Термин "9-замещенное соединение миноциклина" включает в себя соединения миноциклина, содержащие заместитель в положении 9. Согласно другому варианту воплощения настоящего изобретения соединение представляет собой производное миноциклина. 9-Замещенные миноциклины могут быть синтезированы с использованием основной методики,представленной на схеме Синтез 1. В основном, 9-замещенные соединения миноциклина могут быть синтезированы, как показано на схеме Синтез 2 путем обработки миноциклина (1 А) серной кислотой и нитратом натрия. Полученный продукт представляет собой 9-нитроминоциклин (1 В). Соединение нитроминоциклина затем обрабатывают газообразным водородом в присутствии платинового катализатора, при этом получают соединение 9-аминоминоциклина (1 С). Чтобы синтезировать 9-замещенные производные, соединение 9-аминоминоциклина обрабатывают HONO, при этом получают соль диазония (1D). Соль может быть последовательно обработана множеством соединений, содержащих алкен или функциональную группу с -связью, такую как алкены, арилы и алкинилы (например, R9Br), при этом получают 9-замещенное соединение миноциклина (1 Е). Синтез 2 Как показано на схеме Синтез 3, соединения миноциклина по настоящему изобретению, в которыхR9 означает карбамат или производное мочевины, могут быть синтезированы с использованием следующей методики. Миноциклин (2 А) обрабатывают NaNO2 в кислотных условиях, при этом получают 9-нитроминоциклин (2 В). Затем 9-нитроминоциклин (2 В) обрабатывают газообразным Н 2 в присутствии платинового катализатора, при этом получают производное 9-аминоминоциклина (2 С). Для получения производных мочевины (2 Е) изоцианат (2D) вводят в реакцию с производным 9-аминоминоциклина (2 С). Для получения карбамата (2G) соответствующий эфир хлоругольной кислоты (2F) вводят в реакцию с соединением (2 С). Как показано на схеме Синтез 3, соединения миноциклина по настоящему изобретению, в которых заместитель R9 означает аминогруппу, замещенную гетероциклом (например, тиазолом), могут быть синтезированы с использованием приведенной выше методики. 9-Аминоминоциклин (3 А) вводят в реакцию с Fmoc-изотиоцианатом (3 В), при этом получают защищенное производное тиомочевины (3 С). Затем защитную группу тиомочевины (3 С) удаляют, при этом получают активное производное тетрациклинмочевины или тетрациклинтиомочевины (3D). Производное тетрациклинтиомочевины (3D) вводят в реакцию c -галогенкетоном (3 Е), при этом получают тиазолзамещенный 9-аминоминоциклин (3F). Синтез 4 Как показано на схеме Синтез 4, соединения 9-алкенилминоциклина (4 А) можно гидрировать с образованием 9-замещенных соединений алкилминоциклина (4 В). На схеме Синтез 4 представлено избирательное гидрирование двойной связи в положении 9 с помощью газообразного водорода в присутствии палладия на угле в качестве катализатора. Аналогичным образом, 9-алкинилминоциклины можно гидрировать с образованием соединений 9-алкилминоциклина. Синтез 5 На схеме Синтез 5 представлена основная схема синтеза замещенных в положении 9 арилпроизводных миноциклина. На схеме Синтез 5 проводят реакцию Сузуки, включающую конденсацию арилбороновой кислоты и иодминоциклина. Соединение иодминоциклина (5 В) может быть синтезировано из санциклина путем обработки миноциклина (5 А) по крайней мере 1 эквивалентом N-иодсукцинимида (NIS) в кислотных условиях. Реакцию останавливают, и полученный 9-иодоминоциклин (5 В) может быть затем очищен с использованием стандартных методик, известных в данной области техники. Для получения арилпроизводного 9-иодминоциклин (5 В) обрабатывают бороновой кислотой (5 С) и водным раствором карбоната натрия в присутствии палладия в качестве катализатора. Продукт (5D) может быть очищен с помощью способов, хорошо известных в данной области техники (таких, как ВЭЖХ). Другие соединения 9-арилминоциклина могут быть синтезированы с использованием аналогичных методик.-5 009175 9-Замещенные соединения миноциклина по настоящему изобретению могут также быть синтезированы с использованием реакции кросс-сочетания Стилла. Реакцию кросс-сочетания Стилла можно проводить с использованием соответствующего соединения олова (например, R-SnBu3) и галогенированного соединения тетрациклина (например, 9-иодминоциклина). Соединения олова и соединение иодминоциклина могут быть обработаны палладиевым катализатором (например, Pd(PPh3)2Cl2 или Pd(AsPh3)2Cl2) и,по выбору, дополнительно солью меди, например CuI. Полученное соединение может быть очищено методом, известным в данной области техники. Синтез 6 Соединения по настоящему изобретению могут быть синтезированы также с использованием реакций кросс-сочетания типа Heck. Как представлено на схеме Синтез 6, кросс-сочетание типа Heck можно проводить с использованием галогенированного соединения тетрациклина (например, 9-иодминоциклина, 6 А), реакционноспособного алкена (6 В) или алкина (6D) и соответствующего катализатора на основе палладия или другого переходного металла. Полученные соединения 9-замещенного алкенилминоциклина (6 С) или 9-замещенного алкинилминоциклина (6 Е) могут быть очищены с использованием способов, известных в данной области техники. Термин "С 1-6 алкил" включает в себя насыщенные алифатические группы, включая линейные алкильные группы (метил, этил, пропил, бутил, пентил, гексил), разветвленные алкильные группы (изопропил,трет-бутил, изобутил и т.п.), циклоалкильные (алициклические) группы (циклопропил, циклопентил,циклогексил,). Термин "C1-C6" включает в себя алкильные группы, содержащие от 1 до 6 атомов углерода. Термин "амин" или "амино" включает в себя соединения, в которых атом азота ковалентно связан по крайней мере с 1 атомом углерода или гетероатомом. Термин "С 1-6 алкиламино" включает в себя группы и соединения, в которых атом азота связан по крайней мере с одной дополнительной С 1-6 алкильной группой. Термин "диС 1-6 алкиламино" включает в себя группы, в которых атом азота связан по крайней мере с двумя дополнительными С 1-6 алкильными группами. Следует отметить, что структуры некоторых соединений миноциклина по настоящему изобретению содержат асимметрические атомы углерода. Соответственно, следует понимать, что изомеры, образующиеся в связи с такой асимметрией (например, все энантиомеры и диастереомеры), включены в объем настоящего изобретения, если не указано иное. Такие изомеры могут быть получены в практически полностью очищенной форме с использованием классических методов выделения и методов стереохимического синтеза. Кроме того, структуры и другие соединения и остатки, описанные в настоящем описании, также включают все их таутомеры. Изобретение относится также к способу лечения чувствительных к тетрациклину состояний у субъектов путем введения субъекту эффективного количества соединения миноциклина по настоящему изобретению (например, соединения формулы (I) или представленного в таблице ниже), способного вылечить указанное чувствительное к тетрациклину состояние. Термин "чувствительное к соединению тетрациклина состояние" включает в себя состояние, которое можно вылечить, когда можно провести его профилактику или уменьшить интенсивность его симптомов посредством введения соединения миноциклина по настоящему изобретению. Чувствительные к соединению тетрациклина состояния включают в себя бактериальные инфекции (включая состояния,устойчивые к другим соединениям тетрациклина), онкологические заболевания, диабет и другие состояния, для которых показано, что в этом случае соединения тетрациклина активны (см., например, патенты США 5789395, 5834450 и 5532227). Соединения по настоящему изобретению могут быть использованы для профилактики или борьбы с наиболее распространенными заболеваниями млекопитающих и животных, такими как диарея, инфекции мочевых путей, инфекции кожи и структуры кожи, инфекции уха, горла и носа, инфекции в ранах, маститы и т.п. Кроме того, в изобретение включены способы лечения новообразований с использованием соединений миноциклина по настоящему изобретению (см. статью van der Bozert и соавт., Cancer Res., 48:6686-6690 (1988. В случае некоторых чувствительных к тетрациклину состояний может потребоваться соединение миноциклина по настоящему изобретению, обла-6 009175 дающее незначительной антибактериальной активностью или не проявляющее такую активность. Бактериальные инфекции могут быть вызваны широким спектром грамположительных и грамотрицательных бактерий. Соединения по настоящему изобретению используют в качестве антибиотиков против организмов, устойчивых к другим соединениям тетрациклина. Антибиотическую активность соединений миноциклина по настоящему изобретению можно определить с использованием способа, который описан в примере 2,или с использованием стандартного способа разведения питательной среды in vitro, который описан в документе Waitz J.A., National Commission for Clinical Laboratory Standarts (Стандарты национальной комиссии по клиническим лабораторным методам), Document M7-A2, т. 10,8, стр. 13-20, 2-издание, Villanova, PA (1990). Соединения миноциклина могут быть также использованы для лечения инфекций, которые обычно лечат соединениями тетрациклина, таких как риккетсии, ряд грамположительных и грамотрицательных бактерий,а также агенты, вызывающие венерическую лимфогранулему, конъюктивиты с включениями, пситтакоз. Соединения миноциклина могут быть использованы для лечения инфекций, например, таких как K. pneumoniae,Salmonella, E. hirae, A. baumanii, В. catarrhalis, H. influenzae, P. aeruginosa, E. faecium, E. coli, S. aureus илиE. faecalis. Согласно варианту воплощения настоящего изобретения соединение миноциклина используют для лечения бактериальной инфекции, устойчивой к другим тетрациклиновым антибиотикам. Соединение миноциклина по настоящему изобретению можно вводить в смеси с фармацевтически приемлемым носителем. Термин "эффективное количество" соединения означает количество, необходимое или достаточное для лечения или профилактики состояния, чувствительного к соединению тетрациклина. Эффективное количество может варьировать в зависимости от факторов, таких как размер и масса субъекта, тип заболевания или конкретное замещенное соединение миноциклина. Например, выбор соединения миноциклина может влиять на величину "эффективного количества". Специалист в данной области техники способен оценить вышеупомянутые факторы и сделать выводы относительно эффективного количества соединения миноциклина без дополнительных экспериментов. Изобретение относится также к способу лечения инфекций, вызванных микроорганизмами, и связанных с ними заболеваний. Способы включают введение субъекту эффективного количества по меньшей мере одного соединения миноциклина. Субъектом может являться растение или предпочтительно животное, например млекопитающее, в особенности человек. Согласно терапевтическим способам по настоящему изобретению одно или более соединений миноциклина по настоящему изобретению могут быть введены субъекту отдельно или в более типичном случае соединение по настоящему изобретению вводят в виде компонента в составе фармацевтической композиции в смеси со стандартным наполнителем, то есть с фармацевтически приемлемыми органическими или неорганическими веществами-носителями, подходящими для парентерального, перорального или другого требуемого способа введения и которые не оказывают отрицательного воздействия путем взаимодействия с активными соединениями, а также не оказывают отрицательного действия на реципиента. Согласно варианту воплощения настоящее изобретение относится к фармацевтическим композициям, которые содержат терапевтически эффективное количество соединения миноциклина и необязательно фармацевтически приемлемый носитель. Термин "фармацевтически приемлемый носитель" включает в себя соединения, которые могут быть введены совместно с соединением (соединениями) миноциклина и которые обеспечивают осуществление функций соединений, то есть лечение или профилактику состояния, чувствительного к тетрациклину. Подходящие фармацевтически приемлемые носители включают в себя, без ограничения перечисленным,воду, солевые растворы, спирт, растительные масла, полиэтиленгликоли, желатин, лактозу, амилозу,стеарат магния, тальк, кремниевую кислоту, вязкий парафин, ароматическое масло, моноглицериды жирных кислот и диглицериды, петролейные эфиры жирных кислот, гидроксиметилцеллюлозу, поливинилпирролидон и т.п. Фармацевтические препараты могут быть стерилизованы и, при необходимости,смешаны со вспомогательными агентами, например, включающими замасливатели, консерванты, стабилизаторы, увлажнители, эмульгаторы, соли для регулировки осмотического давления, буферные вещества, красители, вкусовые добавки и/или ароматические вещества и т.п., которые не оказывают отрицательного действия путем взаимодействия с активными соединениями по настоящему изобретению. Соединения миноциклина по настоящему изобретению по своей природе являются основаниями и могут образовывать множество солей с различными неорганическими и органическими кислотами. К кислотам, которые могут быть использованы для получения фармацевтически приемлемых кислотноаддитивных солей с основными по природе соединениями миноциклина по настоящему изобретению,относятся кислоты, которые образуют нетоксичные кислотно-аддитивные соли, то есть соли, содержащие фармацевтически приемлемые анионы, такие как гидрохлорид, гидробромид, гидроиодид, нитрат,сульфат, бисульфат, фосфат, кислый фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, кислый цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкаронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и пальмоат (то есть 1,1'-метилен-бис-(2-гидрокси-3-нафтоат. Несмотря на то, что такие соли должны быть фармацевтически приемлемыми для введения субъекту, например млекопитающему,на практике часто требуется выделять соединение миноциклина по настоящему изобретению из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто превращать последнюю в сво-7 009175 бодное основание путем обработки щелочным реагентом с последующим превращением упомянутого свободного основания в фармацевтически приемлемую кислотно-аддитивную соль. Кислотно-аддитивные соли основных соединений по настоящему изобретению легко получить посредством обработки основного соединения эквивалентным количеством выбранной неорганической или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. После осторожного упаривания растворителя легко получить требуемую твердую соль. Соединения миноциклина по настоящему изобретению, которые имеют кислотную природу, могут образовывать множество солей с основаниями. К химическим основаниям, которые могут быть использованы в качестве реагентов для получения фармацевтически приемлемых основных солей таких соединений миноциклина по изобретению, которые имеют кислотную природу, относятся основания, которые образуют нетоксичные основные соли таких соединений. Такие нетоксичные основные соли включают в себя, без ограничения перечисленным, соли, содержащие фармацевтически приемлемые катионы, такие как катионы щелочных металлов (например, калия и натрия) и щелочно-земельных металлов (например, кальция и магния), соли аммония или водно-растворимые аминоаддитивные соли, такие как N-метилглюкамин(меглюмин), а также соли (низш.)алканоламмония и другие основные соли фармацевтически приемлемых органических аминов. Фармацевтически приемлемые основно-аддитивные соли соединений миноциклина по изобретению, которые имеют кислотную природу, могут быть получены с фармацевтически приемлемыми катионами с использованием стандартных способов. Так, эти соли могут быть легко получены путем обработки соединения миноциклина по изобретению водным раствором требуемого фармацевтически приемлемого катиона и упаривания полученного раствора досуха, предпочтительно при пониженном давлении. В другом варианте раствор замещенного соединения тетрациклина по изобретению в низшем алкиловом спирте может быть смешан с алкоксидом требуемого металла и затем раствор упаривают досуха. Получение других соединений миноциклина по настоящему изобретению, которое подробно не описано выше в экспериментальной части, можно осуществить с использованием комбинации реакций,описанных выше и известных для специалиста в данной области техники. Соединения миноциклина по изобретению и их фармацевтически приемлемые соли могут быть введены с помощью перорального, парентерального или местного способов. В основном, наиболее предпочтительно вводить эти соединения в эффективных дозах в зависимости от массы и состояния субъекта, нуждающегося в лечении, от конкретного выбранного способа введения. Возможны варианты в зависимости от вида субъекта,подлежащего лечению, и от его чувствительности к упомянутому лекарственному средству, а также от типа выбранного фармацевтического состава, от периода времени и интервала, через который осуществляют введение. Фармацевтические композиции по настоящему изобретению могут быть введены отдельно или в комбинации с другими известными композициями для лечения чувствительных к тетрациклину состояний у млекопитающих. Предпочтительные млекопитающие включают в себя домашних животных (например, кошки, собаки, хорьки и т.п.), сельскохозяйственный скот (крупный рогатый скот, овцы, свиньи,лошади, козы и т.п.), лабораторных животных (крысы, мыши, обезьяны и т.п.) и приматов (шимпанзе,человекообразные, гориллы). Термин "в комбинации с" известной композицией включает в себя одновременное введение композиции по изобретению и известной композиции, введение сначала композиции по изобретению с последующим введением известной композиции и введение сначала известной композиции с последующим введением композиции по изобретению. Согласно способам по настоящему изобретению может быть использована любая терапевтическая композиция, известная в данной области техники для лечения чувствительных к тетрациклину состояний. Соединения миноциклина по настоящему изобретению могут быть введены отдельно или в комбинации с фармацевтически приемлемыми носителями или разбавителями любым из ранее упомянутых способов, и введение осуществляют в виде однократной или многократной дозы. Например, новые терапевтические агенты по настоящему изобретению могут быть эффективно введены в виде широкого спектра различных лекарственных форм, то есть они могут быть смешаны с различными фармацевтически приемлемыми инертными носителями в форме таблеток, капсул, лепешек, пастилок, твердых капсул, порошков,спреев, кремов, бальзамов, суппозиториев, желе, гелей, паст, лосьонов, мазей, водных суспензий, растворов для инъекций, эликсиров, сиропов и т.п. Такие носители включают в себя твердые разбавители или наполнители, стерильные водные среды и различные нетоксичные органические растворители и т.п. Более того,в пероральные фармацевтические композиции могут быть добавлены подходящие подсластители и/или ароматизаторы. В основном, в таких формах терапевтически эффективные соединения по настоящему изобретению содержатся при концентрации в диапазоне от приблизительно 5,0 до приблизительно 70 мас.%. Для перорального введения могут быть использованы таблетки, содержащие различные носители,такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, кислый фосфат кальция и глицин, наряду с различными дезинтегрирующими агентами, такими как крахмал (предпочтительно кукурузный, картофельный крахмал или крахмал из тапиоки), альгиновая кислота и определенные комплексные силикаты в смеси со связующими гранул, такими как поливинилпирролидон, сахароза, желатин и аравийская камедь. Кроме того, для формирования таблеток в большинстве случаев используют замасливатели, такие как стеарат магния, лаурилсульфат натрия и тальк. В качестве наполнителей в желатиновых капсулах используют также твердые композиции аналогичного типа, причем предпочтительные ма-8 009175 териалы в этом случае включают также лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Если требуются водные суспензии и/или эликсиры для перорального введения, активный компонент может быть смешан с различными подсластителями или ароматизаторами, красителями или пигментами и при необходимости с эмульгаторами и/или суспендирующими агентами наряду с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин и другие их аналогичные комбинации. Для парентерального введения (включая внутрибрюшинное, подкожное, внутривенное, чрескожное или внутримышечное введение) могут быть использованы растворы терапевтического соединения по изобретению в кунжутном или арахисовом масле или в водном пропиленгликоле. В водных растворах при необходимости следует использовать буферные вещества (предпочтительно при рН выше 8) и жидкий разбавитель, который, в первую очередь, придает изотонические свойства. Такие водные растворы подходят для внутривенной инъекции. Масляные растворы подходят для внутрисуставной, внутримышечной и подкожной инъекции. Получение всех таких растворов в стерильных условиях с использованием стандартных фармацевтических методов известны специалистам в данной области техники. Примеры подходящих препаратов для парентерального применения включают в себя растворы, предпочтительно масляные или водные, а также суспензии, эмульсии или имплантаты, включая суппозитории. Терапевтические соединения могут быть переработаны в стерильную форму, содержащую многократную или однократную дозу, например, в виде дисперсии в жидком носителе, таком как стерильный физиологический солевой раствор или 5%-ный солевой раствор декстрозы, которые обычно используют в составах для инъекции. Кроме того, возможно также введение соединений по настоящему изобретению местным способом при лечении воспалительных процессов на коже. Примеры способов местного введения включают чрескожный, буккальный или подъязычный. Для местного введения подходящим способом может быть смешивание терапевтических соединений в фармацевтически инертном носителе для местного введения,таком как гель, мазь, лосьон или крем. Такие носители для местного введения включают в себя воду,глицерин, спирт, пропиленгликоль, жирные спирты, триглицериды, сложные эфиры жирных кислот или минеральные масла. Другими возможными носителями для местного введения являются жидкий вазелин, изопропилпальмитат, полиэтиленгликоль, 95%-ный этанол, 5%-ный монолаурат полиоксиэтилена в воде, 5%-ный лаурилсульфат натрия в воде и т.п. Кроме того, при необходимости могут быть добавлены материалы, такие как антиоксиданты, увлажнители, стабилизаторы вязкости и т.п. Для энтерального введения подходящими являются, прежде всего, таблетки, драже или капсулы, содержащие тальк и/или углеводный связующий носитель или аналогичный материал, причем в качестве носителя предпочтительно используют лактозу, и/или кукурузный крахмал, и/или картофельный крахмал. Могут быть также использованы сироп, эликсир и т.п., в которые добавляют подсластитель в качестве носителя. Могут быть получены композиции пролонгированного действия, в которых активный компонент защищен дифференциально разрушаемыми покрытиями, например, с помощью микроинкапсуляции, многокомпонентных покрытий и т.д. Кроме лечения человека, терапевтические способы лечения по настоящему изобретению имеют большое значение в ветеринарной практике, например при лечении сельскохозяйственного скота, такого как крупный рогатый скот, овцы, козы, коровы, свиньи и т.п., домашней птицы, такой как куры, утки,гуси, индейки и т.п., лошади и домашние животные, такие как собаки и кошки. Соединения по изобретению могут быть также использованы для лечения объектов неживотного мира, таких как растения. Следует понимать, что действительные предпочтительные количества активных соединений, используемых для данного лечения, могут различаться в зависимости от конкретного используемого соединения, от конкретных полученных композиций, способа применения, специфического участка введения и т.д. Оптимальная частота введения для данной схемы введения может быть легко определена специалистами в данной области техники с использованием обычного тестирования для определения дозировки, которое осуществляют в соответствии с вышеупомянутыми рекомендациями. В основном, соединения по настоящему изобретению, предназначенные для лечения, могут быть введены субъекту в дозировках, которые использовали в предшествующей практике лечения миноциклином. См., например, издание "Physicians' Desk Reference" (Настольные рекомендации для терапевтов). Например, подходящая эффективная доза одного или более соединений по настоящему изобретению изменяется в диапазоне от 0,01 до 100 мг/кг массы тела реципиента в сутки, предпочтительно в диапазоне от 0,1 до 50 мг/кг массы тела реципиента в сутки, более предпочтительно в диапазоне от 1 до 20 мг/кг массы тела реципиента в сутки. Требуемую дозу вводят 1 раз в сутки или в виде нескольких разделенных доз, например от 2 до 5 разделенных доз, причем введение осуществляют через определенные интервалы в течение суток или по другой соответствующей схеме введения. Следует также понимать, что с целью обеспечения эффективности введенных соединений в нормальных условиях применения следует принимать стандартные известные меры предосторожности при введении миноциклинов. Прежде всего, при использовании для терапевтического лечения человека и животныхin vivo практикующий врач должен принимать все разумные меры предосторожности, чтобы исключить известные обычные отрицательные действия и токсические эффекты. Таким образом, следует надлежащим образом учитывать определяемые обычным способом неблагоприятные реакции с учетом заболеваний желудочно-кишечного тракта и воспалительных процессов, нефротоксичности, гиперчувствительности,изменений состава крови и ухудшения всасываемости, связанного с ионами алюминия, кальция и магния.-9 009175 Согласно другому варианту воплощения изобретения соединения миноциклина по настоящему изобретению не включают соединения, описанные в заявке на выдачу патента США 09/823884, включенной в данное описание в качестве ссылки. Кроме того, настоящее изобретение относится также к использованию соединения миноциклина формулы I или II для получения лекарственных средств. Лекарственное средство может содержать фармацевтически приемлемый носитель и соединение миноциклина в эффективном количестве, например эффективном для лечения чувствительного к тетрациклину состояния. Примеры Соединения по настоящему изобретению могут быть получены, как описано ниже, причем возможны модификации описанных ниже методик, которые представляются очевидными для специалистов в данной области техники. Пример 1. Получение соединений миноциклина по изобретению. Получение 9-иодминоциклина. К 200 мл 97%-ной метансульфоновой кислоты медленно при температуре окружающей среды порциями добавляют бис-гидрохлорид миноциклина (30 г, 56,56 ммоль). Затем раствор темного желто-коричневого цвета перемешивают при температуре окружающей среды и при этом 6 равными порциями в течение 3 ч добавляют N-иодсукцинимид (38 г, 169,7 ммоль). Реакционную смесь анализируют методом ЖХ и реакцию проводят до исчезновения исходного материала. Реакцию медленно останавливают, выливая смесь при интенсивном перемешивании в 2 л охлажденной льдом воды, содержащей тиосульфат натрия (17,88 г, 1134,1 ммоль). Полученную смесь перемешивают при температуре окружающей среды в течение приблизительно 30 мин. Затем водный слой экстрагируют этилацетатом (6200 мл) и водный слой выливают в раствор гидрокарбоната натрия (259,8 г,3,08 моль) в 300 мл н-бутанола. Фазы разделяют и водную фазу экстрагируют н-бутанолом (4250 мл). Органические фазы объединяют, промывают водой (3250 мл) и 1 раз насыщенным солевым раствором(250 мл). Полученную органическую фазу упаривают досуха при пониженном давлении. Остаток суспендируют в метаноле (600 мл) и через полученную смесь пропускают поток безводного газообразного HCl до образования раствора. Этот раствор упаривают досуха при пониженном давлении. Фильтраты упаривают досуха при пониженном давлении. Полученное вещество растирают в 300 мл метил-трет-бутилового эфира и выделяют фильтрованием. Полученный материал снова растворяют в 300 мл метанола и обрабатывают 0,5 г древесного угля, отфильтровывают и фильтраты упаривают досуха при пониженном давлении. Продукт снова растирают в метил-трет-бутиловом эфире, выделяют фильтрованием в вакууме и промывают эфиром, а затем гексаном. Полученное вещество высушивают в вакууме, при этом получают 22,6 г порошка светлого желто-коричневого цвета. Общая методика получения 9-алкинилзамещенных соединений миноциклина. 1 ммоль 9-иодминоциклина, 50 мг тетракис-трифенилфосфинатопалладата, 12 мг ацетата палладия,32 мг иодида меди (I) растворяют/суспендируют в 10 мл ацетонитрила. Затем добавляют от 2 до 5 мл триэтиламина и от 3 до 5 ммоль алкинилпроизводного. Реакционную смесь интенсивно перемешивают при температуре от окружающей среды до 70 С. Продолжительность реакции составляет 2-24 ч. После завершения реакции суспензию темного цвета фильтруют через слой целита и концентрируют. Неочищенный продукт очищают препаративной ВЭЖХ. Объединенные фракции концентрируют и переносят в 1 мл метанола. Затем добавляют 3 мл насыщенного HCl/метанола и продукт высаживают эфиром. Общая методика получения 9-арилзамещенных соединений миноциклина. 0,15 ммоль 9-иодминоциклина, PdOAc (3,2 мг), 229 мкл 2 М Na2CO3 и 2 экв. фенилбороновой кислоты растворяют/суспендируют в 10 мл метанола. Реакционную колбу продувают аргоном и проводят реакцию в течение, минимум, 4 ч или до тех пор, пока данные анализа ВЭЖХ не свидетельствуют о потреблении исходного материала и/или о появлении продуктов. Суспензию фильтруют через целит и подвергают очистке с помощью препаративной ВЭЖХ на колонке с дивинилбензольным сорбентом. Соединение OU (9-(4-трифторметоксифенилуреидо)метилминоциклин). Синтез 7- 10009175 К 3 мл диметилформамида при 25 С добавляют 150 мг (0,25 ммоль) тригидрохлорида 9-метиламиноминоциклина и 67 мл (0,50 ммоль) триэтиламина. Затем при перемешивании добавляют 75 мл (0,50 ммоль) 4-трифторметоксифенилизоцианата и полученную реакционную смесь перемешивают при 25 С в течение 2 ч. Реакционную смесь анализируют методом аналитической ВЭЖХ (колонка Luna C18 с обращенной фазой, 4,650 мм, линейный градиент от 1 до 100% буферного раствора В в течение 5 мин, буферный раствор А: вода, содержащая 0,1% трифторуксусной кислоты, буферный раствор В: ацетонитрил,содержащий 0,1% трифторуксусной кислоты). После завершения реакции ее останавливают 1 мл воды и величину рН доводят приблизительно до 2,0 с помощью конц. HCl. Раствор фильтруют и соединение очищают препаративной ВЭЖХ. Выход соединения OU составляет 64 мг (37%). Чистота соединения OU,определенная методом МС-ЖХ (М+1=690), составляет 95%. Соединение LA (9-(4'-карбоксифенил)миноциклин). Синтез 8 В чистый сухой реакционный сосуд загружают бис-гидрохлорид 9-иодминоциклина (500 мг, 0,762 ммоль),ацетат палладия(II) (17,2 мг, 0,076 ммоль) и 10 мл метанола класса "химический реагент". Раствор немедленно продувают потоком аргона при перемешивании в течение 5 мин. Реакционный сосуд нагревают с обратным холодильником и последовательно добавляют с помощью шприца 2 М раствор карбоната калия (1,91 мл, 3,81 ммоль) и раствор п-карбоксифенилбороновой кислоты (238,3 мг, 1,53 ммоль) в 5 мл ДМФ. Оба указанных раствора предварительно дегазируют аргоном в течение приблизительно 5 мин. Реакционную смесь нагревают в течение 45 мин, ход реакции контролируют методом обращенно-фазной ВЭЖХ. Реакционную смесь отфильтровывают в вакууме через слой диатомовой земли и слой промывают ДМФ. Фильтраты упаривают в вакууме до маслообразного остатка и остаток обрабатывают третбутилметиловым эфиром. Неочищенный материал очищают методом обращенно-фазной ВЭЖХ с использованием дивинилбензольного сорбента, и градиента воды, и метанол/ацетонитрила, содержащих 1,0% трифторуксусной кислоты. Структуру продукта подтверждают масс-спектрометрией: найдено М+1=578,58; а также 1 Н ЯМР-спектром. Пример 2. Анализ in vitro минимальной ингибирующей концентрации (МИК). Следующий анализ используют для определения эффективности соединений миноциклина по отношению к распространенным бактериям. 2 мг каждого соединения растворяют в 100 мкл ДМСО. Затем раствор добавляют в питательную среду Mueller Hinton, сбалансированную по катионам (САМНВ), до конечной концентрации соединения 200 мкг/мл. Растворы соединений миноциклина разбавляют до 50 мкл, при этом концентрация исследуемого соединения составляет 0,098 мкг/мл. Оптическую плотность (ОД) определяют относительно свежеприготовленных культур в логарифмической фазе роста исследуемых штаммов. Готовят разбавления, в которых конечная плотность клеток составляет приблизительно 1106 КОЕ (колониеобразующих единиц)/мл. Если ОД=1, то плотность клеток для различных видов бактерий приблизительно составляет Е. coli 1109 КОЕ/млEnterococcus sp. 2,5109 КОЕ/мл В каждую лунку микропланшета для титрования добавляют по 50 мкл клеточной суспензии. Конечная плотность клеток составляет приблизительно 5105 КОЕ/мл. Эти планшеты инкубируют при 35 С в атмосфере окружающей среды в инкубаторе в течение приблизительно 18 ч. Планшеты считывают с использованием ридера для микропланшет и при необходимости наблюдают визуально. Величину МИК определяют в виде наименьшей концентрации соединения миноциклина, при которой наблюдается ингибирование роста. Соединения по настоящему изобретению проявляют достаточно высокую ингибирующую активность в отношении роста бактерий. В нижеследующей таблице соединения, обладающие удовлетворительным ингибированием роста конкретного организма, обозначены знаком , соединения, обладающие высоким ингибированием роста конкретного организма, обозначены знаком , соединения,обладающие чрезвычайно высоким ингибированием роста конкретного организма, обозначены знаком . Эквивалентные решения Специалисты в данной области техники знают или способны выявить с использованием простых стандартных экспериментов множество эквивалентов специфических методов, описанных в данном контексте. Такие эквивалентные решения включены в объем притязаний настоящего изобретения и определены в следующей ниже формуле изобретения. Содержание всех ссылок, патентов и заявок на выдачу патентов, цитированных в данном описании, полностью включено в данную заявку в качестве ссылок. Для настоящего изобретения и вариантов его воплощения могут быть выбраны соответствующие компоненты, процессы и способы, описанные в указанных патентах, заявках и других документах. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение миноциклина общей формулы I где R4', R4, R7' и R7, каждый независимо означает С 1-6 алкил; и их фармацевтически приемлемые соли или сложные эфиры. 2. Соединение миноциклина по п.1, отличающееся тем, что R9 означает метил, который замещен незамещенной или замещенной С 1-6 алкиламиногруппой или диС 1-6 алкиламиногруппой. 3. Соединение миноциклина по п.2, отличающееся тем, что R9 означает метил, который замещен незамещенной С 1-6 алкиламиногруппой. 4. Соединение миноциклина по п.3, отличающееся тем, что указанная незамещенная С 1-6 алкиламиногруппа представляет собой пентиламиногруппу. 5. Соединение по п.1, отличающееся тем, что оно выбрано из группы, включающей или их фармацевтически приемлемые соли или сложные эфиры. 6. Соединение по п.1, представляющее собой- 27009175 или его фармацевтически приемлемые соли или сложные эфиры. 7. Способ лечения чувствительного к тетрациклину состояния субъекта-млекопитающего, отличающийся тем, что осуществляют лечение указанного субъекта посредством введения соединения миноциклина по любому из пп.1-6. 8. Способ по п.7, отличающийся тем, что указанным чувствительным к тетрациклину состоянием является бактериальная инфекция. 9. Способ по п.8, отличающийся тем, что указанная бактериальная инфекция связана с Е. coli. 10. Способ по п.8, отличающийся тем, что указанная бактериальная инфекция связана с S. aureus. 11. Способ по п.8, отличающийся тем, что указанная бактериальная инфекция связана с Е. faecalis. 12. Способ по п.8, отличающийся тем, что указанная бактериальная инфекция устойчива к другим тетрациклиновым антибиотикам. 13. Способ по п.7, отличающийся тем, что указанное соединение миноциклина вводят в смеси с фармацевтически приемлемым носителем. 14. Способ по п.7, отличающийся тем, что указанным субъектом является человек. 15. Фармацевтическая композиция для воздействия на бактерии, чувствительные к тетрациклину,отличающаяся тем, что она содержит терапевтически эффективное количество соединения миноциклина по любому из пп.1-6 и фармацевтически приемлемый носитель.
МПК / Метки
МПК: A61K 31/65, A61P 31/04, C07C 237/26
Метки: композиция, чувствительного, миноциклина, тетрациклину, млекопитающего, состояния, замещенные, лечения, способ, фармацевтическая, соединения
Код ссылки
<a href="https://eas.patents.su/29-9175-zameshhennye-soedineniya-minociklina-farmacevticheskaya-kompoziciya-i-sposob-lecheniya-chuvstvitelnogo-k-tetraciklinu-sostoyaniya-mlekopitayushhego.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные соединения миноциклина, фармацевтическая композиция и способ лечения чувствительного к тетрациклину состояния млекопитающего</a>
Замещенные соединения тетрациклина (варианты), фармацевтическая композиция и способ лечения чувствительного к тетрациклину состояния субъекта

Номер патента: 6170
Опубликовано: 27.10.2005
Авторы: Банадарадж Юпул, Макинтайр Лора, Виски Питер, Измаил Мохамед, Бхатиа Бина, Хокинс Пол, Баузер Тодд, Ренни Глен, Мессершмит Дэвид, Фречетт Роджер, Нельсон Марк Л., Уорчол Тэд, Коуза Дэррелл, Шихэн Пол, Верма Атул
МПК: C07C 237/26, A61K 31/65
Метки: замещенные, фармацевтическая, субъекта, способ, тетрациклину, лечения, чувствительного, варианты, состояния, композиция, тетрациклина, соединения
Формула / Реферат:
1. Замещенные соединения тетрациклина общей формулы I где X означает CHC(R13Y'Y), CR6'R6, C=CR6'R6, S, NR6 или O; R2, R2', R4' и R4'', каждый независимо, означает водород, C1-C4алкил, C2-C6алкенил, C2-C6алкинил, C1-C4алкокси, C1-C4алкилтио, C1-C4алкилсульфинил, C1-C4алкилсульфонил, C1-C4алкиламино, C5-C10 арилC1-C4алкил, C5-C10арил, гетероциклический остаток, содержащий 3-5 атомов углерода и 1-2 атома N, гетероароматический остаток, включающий...
Пиперазинил-бензилиденил-лактамовые соединения в качестве агонистов и антагонистов 5-нт1а и 5-нт1d рецепторов, фармацевтическая композиция на их основе и способ лечения или профилактики нарушения илисостояния, связанных с недостаточной серотонергической нейротрансмиссией у млекопитающего

Номер патента: 2157
Опубликовано: 24.12.2001
Автор: Ховард Гэрри Ральф
МПК: C07D 233/96, A61P 25/00, A61K 31/4166...
Метки: агонистов, пиперазинил-бензилиденил-лактамовые, млекопитающего, основе, качестве, композиция, фармацевтическая, нарушения, соединения, профилактики, 5-нт1d, рецепторов, лечения, серотонергической, недостаточной, связанных, 5-нт1а, способ, илисостояния, антагонистов, нейротрансмиссией
Формула / Реферат:
1. Пиперазинил-бензилиденил лактамовые соединения формулы где R1 представляет пиперазин-1-ил или пиперазин-1-ил, замещенный в положении 4 (C1-C4) алкилом; R2 представляет водород; R представляет-(СН2)mВ, где m равно 0 или 1 и В представляет водород, фенил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, (C1-C6)алкила или трифторметила; Х представляет водород или галоген; Y представляет...
Соединения пиразинона, фармацевтическая композиция (варианты) и лекарственная форма, способ ингибирования сериновой протеазы, способ снижения тромбообразующей способности поверхности, способ лечения нарушенного протеолиза и тромбоза, связанного с различными заболеваниями, способ снижения свертывания крови у млекопитающего

Номер патента: 4884
Опубликовано: 26.08.2004
Авторы: Томкзук Брюс Э., Лу Тианбао, Маркоутэн Томас П.
МПК: A61K 38/05, A61P 7/02, C07K 5/078...
Метки: соединения, тромбообразующей, лекарственная, тромбоза, нарушенного, различными, форма, способности, пиразинона, протеазы, протеолиза, свертывания, заболеваниями, сериновой, лечения, фармацевтическая, композиция, ингибирования, снижения, млекопитающего, крови, поверхности, способ, варианты, связанного
Формула / Реферат:
1. Соединение формулы I или его сольват, гидрат или фармацевтически приемлемая соль, где W означает R1; R1 означает R2, R2(CH2)tC(R12)2, где t равно 0-3 и оба R12 одинаковые или различные, (R2)(OR12)CH(CH2)p, где p равно 1-4, R2C(R12)2(CH2)t, где t равно 1-3 и оба R12 одинаковые или различные, причем (R12)2 может быть объединен с атомом углерода, к которому он присоединен, с образованием от 3- до 7-членного циклоалкильного кольца; ...
Производные аминогуанидина и алкоксигуанидина (варианты) и способ их получения (варианты), фармацевтическая композиция и способ ингибирования протеолиза у млекопитающего, способ лечения различных заболеваний млекопитающего, способы ингибирования индуцированной тромбином агрегации тромбоцитов и свертывания фибриногена в плазме, тромбина, агрегации тромбоцитов и образования тромбов в крови, устройство для сбора крови, искусственного кровообращения и хранения крови

Номер патента: 3461
Опубликовано: 26.06.2003
Авторы: Сайедем Коллин, Лу Тианбао, Томкзук Брюс Э., Маркоутэн Томас П., Солл Ричард М.
МПК: A61K 31/445, A61P 7/02, C07C 279/00...
Метки: тромбов, способы, заболеваний, тромбина, искусственного, устройство, хранения, лечения, млекопитающего, крови, способ, кровообращения, варианты, фибриногена, протеолиза, свертывания, агрегации, индуцированной, плазме, фармацевтическая, аминогуанидина, различных, алкоксигуанидина, образования, ингибирования, сбора, получения, производные, композиция, тромбоцитов, тромбином
Формула / Реферат:
1. Соединение формулы I или его сольват, гидрат или фармацевтически приемлемая соль, где L - означает -C(O)- или -SO2-; R1 означает группу R2 означает группу или R1 и R2 могут быть объединены с атомом азота, к которому они присоединены, с образованием цикла, содержащего от трех до семи атомов, который необязательно содержит дополнительный атом азота или кислорода, и который по выбору является бензо- или пиридоконденсированным циклом, причем...
Новые замещенные димерные соединения, способ их получения и содержащая их фармацевтическая композиция

Номер патента: 3300
Опубликовано: 24.04.2003
Авторы: Ренар Пьер, Делягранж Филипп, Лефулон Франсуа, Вио Мари-Клод, Иус Саид, Лезьер Даниель, Беннежан Каролин, Декамп-Франсуа Кароль, Гийоме Жераль
МПК: C07C 233/72, C07D 403/12, A61K 31/353...
Метки: композиция, содержащая, соединения, способ, димерные, получения, новые, фармацевтическая, замещенные
Формула / Реферат:
1. Соединения формулы (I) A-G1-Cy-G2-Cy-G3-B (I), где A представляет собой группы формулы -NHC(Q)R1 или -NHC(Q)NHR2, где Q обозначает атом кислорода или серы, R1 обозначает линейную или разветвленную (C1-C6)алкильную группу, замещенную или незамещенную атомом галогена, линейную или разветвленную (C2-C6)алкенильную группу, (C3-C8)циклоалькильную группу, арильную или гетероарильную группу, R2 обозначает линейную или разветвленную...
Предыдущий патент: Система аутентификации
Случайный патент: Производные имидазопиридина как ингибиторы индуцируемой no - синтазы