Соединения пиперазина, способ их получения и фармацевтические композиции, которые их содержат
Номер патента: 9648
Опубликовано: 28.02.2008
Авторы: Миллан Марк, Дессинже Эме, Гумен Бертран, Маннури-Ла-Кур Клотильда, Пеглион Жан-Луи

























Формула / Реферат
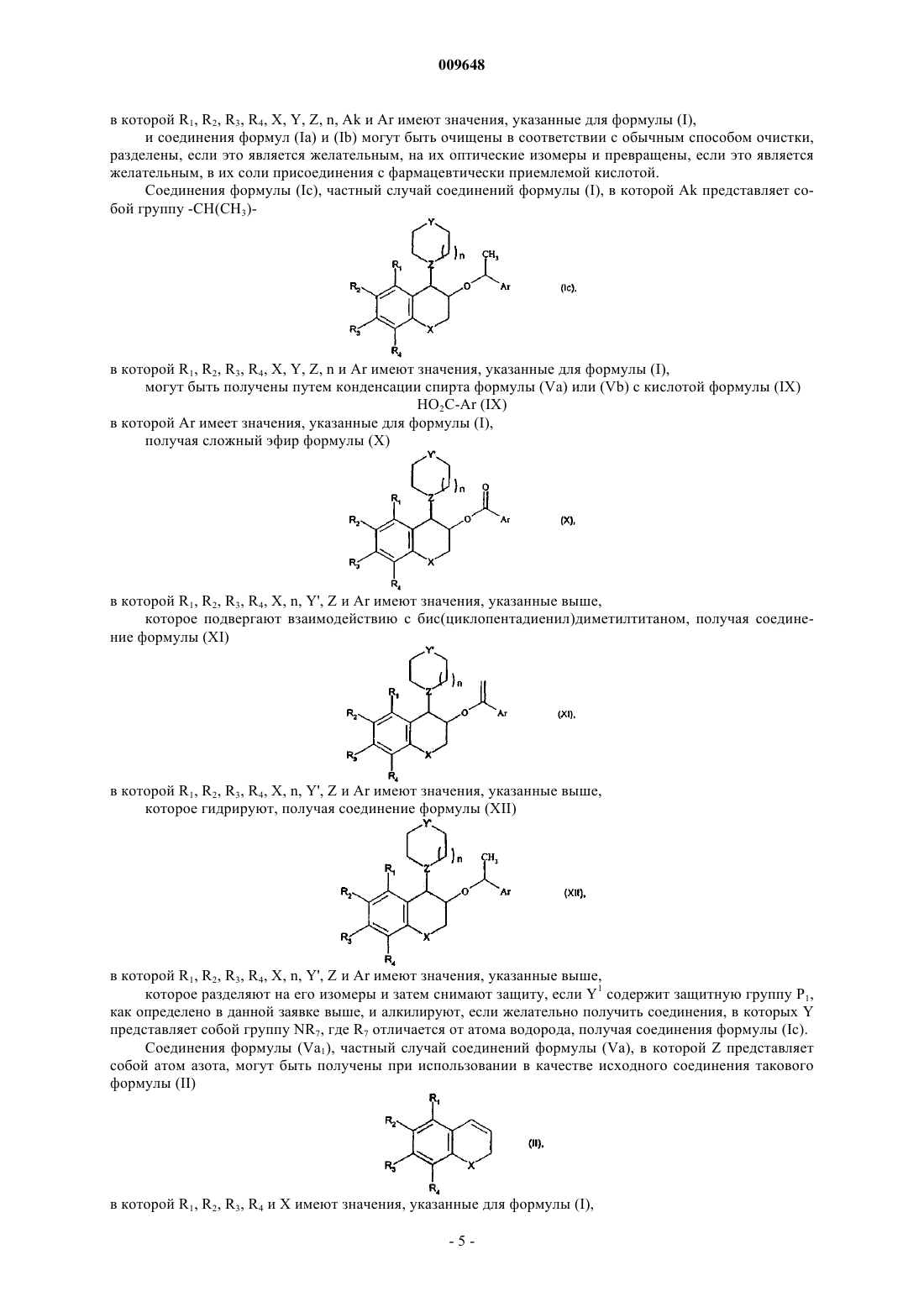
1. Соединение формулы (I)

в которой
R1, R2, R3 и R4, которые могут быть одинаковыми или разными, каждый представляет собой атом или группу, выбранную из Н, галогена, линейного или разветвленного C1-С6-алкила, линейного или разветвленного C1-С6-алкокси, фенила и циано,
X представляет собой связь, атом кислорода или группу, выбранную из -(СН2)m-, -ОСН2- и -NR5-,
m представляет собой 1 или 2,
R5 представляет собой атом водорода или группу, выбранную из линейного или разветвленного C1-С6-алкила, COR6 и CO2R6,
R6 представляет собой линейную или разветвленную C1-С6-алкильную группу,
Y представляет собой атом кислорода или группу, выбранную из NR7 и CHR8,
R7 представляет собой атом водорода или группу, выбранную из COR9 и линейного или разветвленного C1-С6-алкила, где алкильная группа необязательно замещена 5-оксо-4,5-дигидро-1H-1,2,4-триазол-3-ильной или 2,3-дигидро-1,4-бенздиоксин-2-ильной группой,
R9 представляет собой группу, выбранную из линейного или разветвленного C1-С6-алкила, арила и гетероарила,
R8 представляет собой атом водорода или аминогруппу, необязательно замещенную одной или двумя линейными или разветвленными С1-С6-алкильными группами,
Z представляет собой атом азота или СН группу,
n представляет собой 1 или 2,
Ak представляет собой линейную или разветвленную С1-С6-алкиленовую цепь,
Ar представляет собой арильную или гетероарильную группу,
его оптические изомеры, а также его соли присоединения с фармацевтически приемлемой кислотой,
при этом понятно, что
арильная группа обозначает фенил, бифенилил или нафтил, каждая из этих групп необязательно замещена одной или несколькими одинаковыми или разными группами, выбранными из галогена, линейного или разветвленного С1-С6-алкила, линейного или разветвленного C1-С6-алкокси, гидрокси, циано, линейного или разветвленного C1-С6-тригалоалкила и линейного или разветвленного C1-С6-тригалоалкокси,
и гетероарильная группа обозначает ароматическую моно- или бициклическую 5-12-членную группу, содержащую 1, 2 или 3 гетероатома, выбранных из кислорода, азота и серы, при этом понятно, что гетероарильная группа необязательно может быть замещена одной или несколькими одинаковыми или разными группами, выбранными из галогена, линейного или разветвленного C1-С6-алкила, линейного или разветвленного C1-С6-алкокси, гидрокси, циано и линейного или разветвленного C1-С6-тригалоалкила.
2. Соединение формулы (I) по п.1, в котором Y представляет собой NH.
3. Соединение формулы (I) по п.1 или 2, в котором Z представляет собой атом азота.
4. Соединение формулы (I) по любому из пп.1-3, в котором n представляет собой 1.
5. Соединение формулы (I) по любому из пп.1-4, в котором Ar представляет собой арильную группу.
6. Соединение формулы (I) по любому из пп.1-5, в котором X представляет собой связь, атом кислорода или группу, выбранную из -ОСН2- и -(СН2)m-, где m представляет собой 1 или 2.
7. Соединение формулы (I) по п.1, выбранное из
транс-1-{2-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден-1-ил}пиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,
транс-1-{3-[(3,5-дибромбензил)окси]-3,4-дигидро-2H-хромен-4-ил}пиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,
транс-1-{6-[(3,5-дибромбензил)окси]-6,7,8,9-тетрагидро-5H-бензо[7]аннулен-5-ил}пиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,
транс-1-{2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-ил}пиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,
транс-1-{2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-ил}-1,4-диазепана, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,
1-{(1S,2R)-2-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден-1-ил}пиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,
1-{(1S,2R)-2-[(3,5-дифторбензил)окси]-2,3-дигидро-1H-инден-1-ил}пиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,
1-{(1S,2R)-2-[(3,5-диметилбензил)окси]-2,3-дигидро-1H-инден-1-ил}пиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,
транс-1-{3-[(3,5-дихлорбензил)окси]-3,4-дигидро-2H-хромен-4-ил}пиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,
транс-1-{3-[3-фтор-5-(трифторметил)бензилокси]-3,4-дигидро-2H-хромен-4-ил}пиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой и
транс-1-{3-(3-хлор-5-фторбензилокси)-3,4-дигидро-2Н-хромен-4-ил}пиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой.
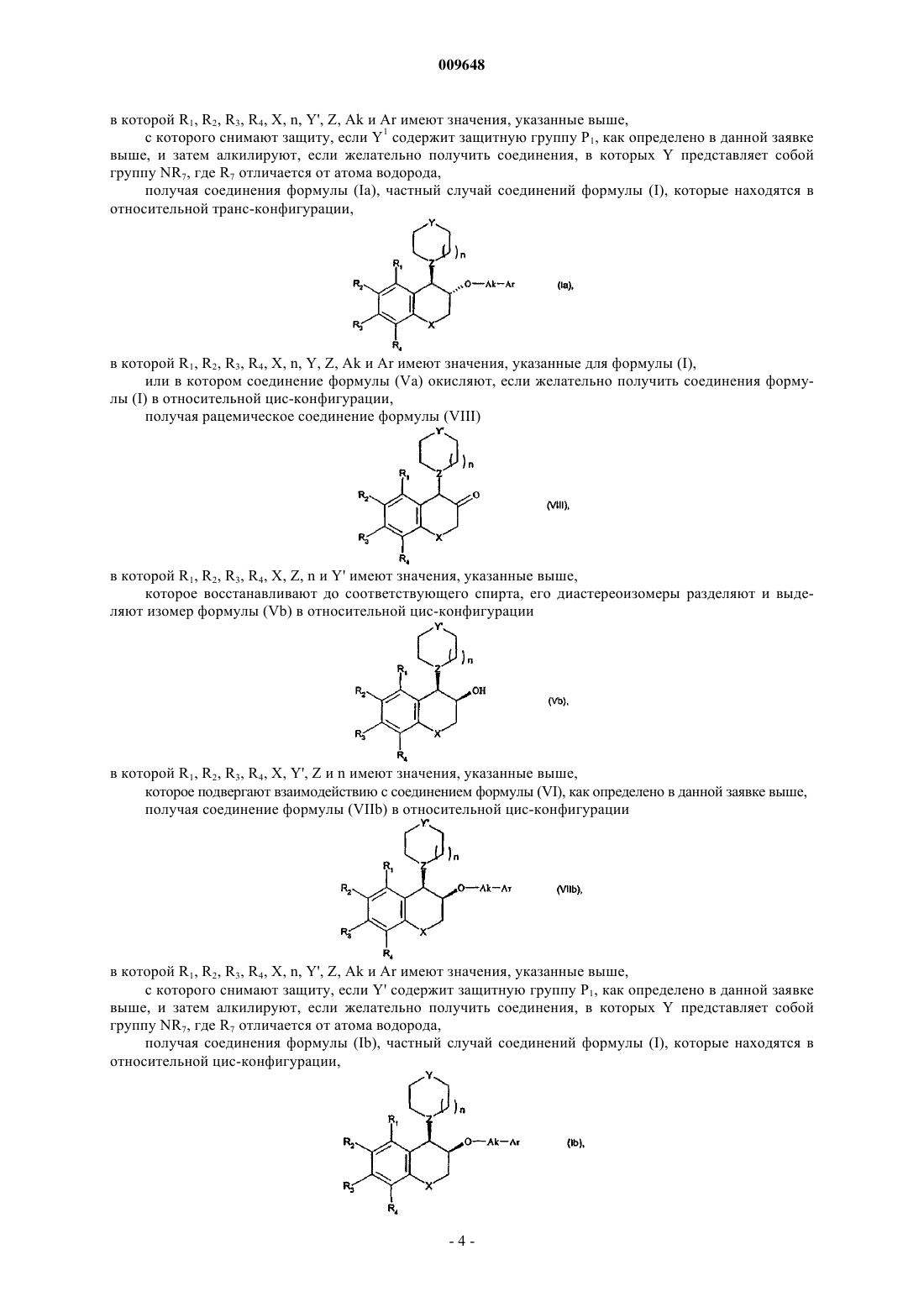
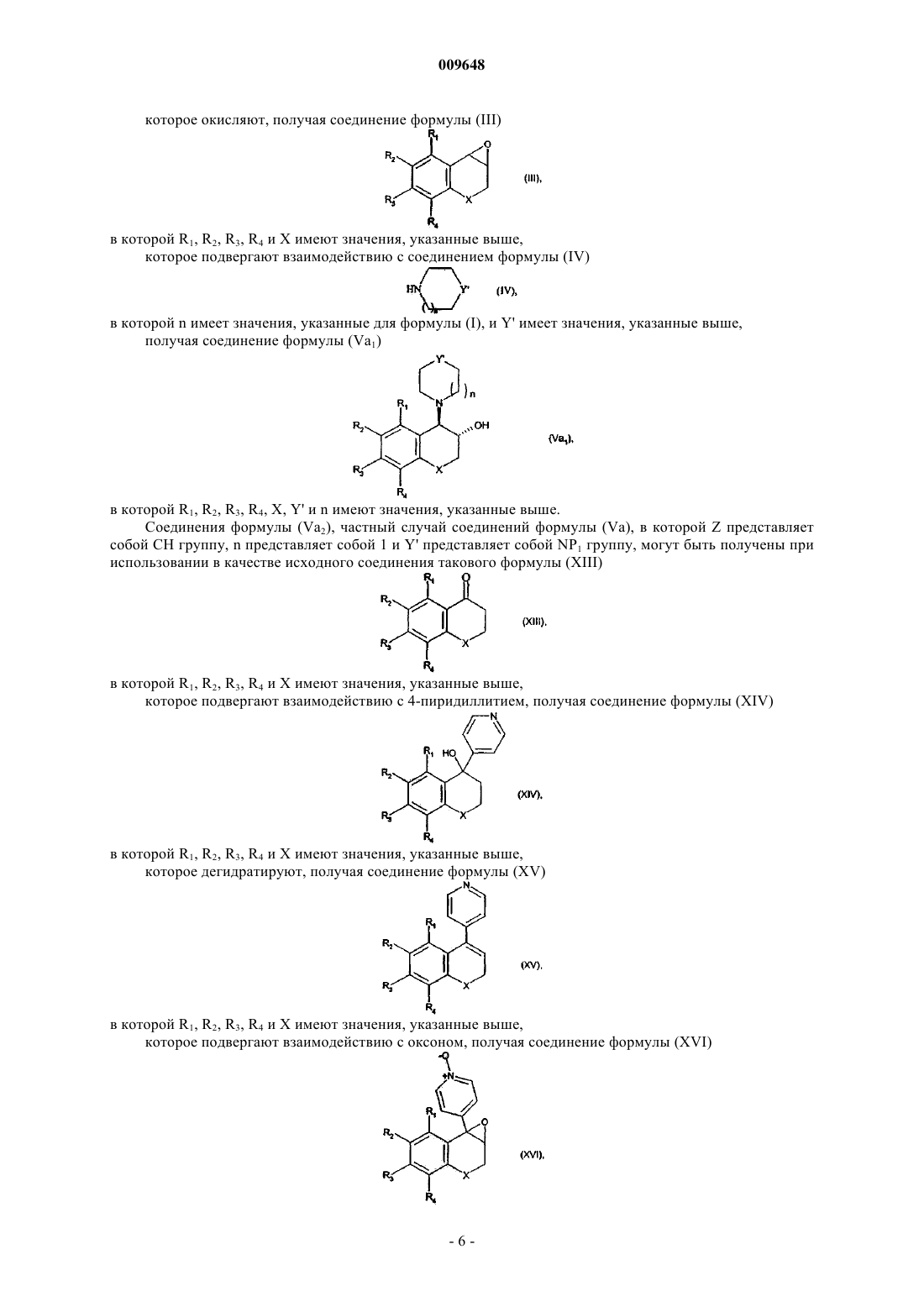
8. Способ получения соединений формулы (I) по п.1 из соединения формулы (Va) в относительной транс-конфигурации

в которой
R1 R2, R3, R4, X, n и Z имеют значения, указанные в п.1, a Y' представляет собой атом кислорода или группу, выбранную из NP1 и CHR'8, в которой R'8 представляет собой атом водорода или группу NHP1 и P1 представляет собой защитную группу для функциональной аминогруппы,
в котором соединение формулы (Va) подвергают взаимодействию, если желательно получить соединения формулы (I) в относительной транс-конфигурации, с соединением формулы
G-Ak-Ar (VI)
в которой Ak и Ar имеют значения, указанные для формулы (I), и G представляет собой уходящую группу, такую как, например, атом галогена или n-толуолсульфонатная, трифторметансульфонатная или метансульфонатная группа,
с получением соединения формулы (VIIa) в относительной транс-конфигурации

в котором R1, R2, R3, R4, X, n, Y', Z, Ak и Ar имеют значения, указанные выше,
с последующим снятием защиты, если Y' содержит защитную группу P1, как определено выше, и затем алкилированием, если желательно получить соединения, в которых Y представляет собой группу NR7, где R7 отличен от атома водорода,
с получением соединения формулы (Ia), частного случая соединения формулы (I), находящегося в относительной транс-конфигурации

в которой R1, R2, R3, R4, X, n, Y, Z, Ak и Ar имеют значения, указанные для формулы (I),
и полученные соединения формулы (Ia) могут быть очищены в соответствии с обычным способом очистки и при желании разделены, если это является желательным, на их оптические изомеры и превращены в их соли присоединения фармацевтически приемлемой кислоты.
9. Способ получения соединений формулы (I) по п.1 окислением соединения формулы (Va), и, если желательно получить соединения (I) в относительной цис-конфигурации,
полученное в результате окисления рацемическое соединение формулы (VIII)

в которой R1, R2, R3, R4, X, Z, n и Y' имеют значения, указанные выше,
восстанавливают до соответствующего спирта, его диастереоизомеры разделяют с выделением изомера формулы (Vb), в относительной цис-конфигурации

в которой R1, R2, R3, R4, X, Y', Z и n имеют значения, указанные выше,
которое подвергают взаимодействию с соединением формулы (VI)
G-Ak-Ar (VI)
в которой Ak и Ar имеют значения, указанные для формулы (I), и G представляет собой уходящую группу, такую как, например, атом галогена или n-толуолсульфонатную, трифторметансульфонатную или метансульфонатную группу,
получая соединение формулы (VIIb) в относительной цис-конфигурации

в котором R1, R2, R3, R4, X, n, Y', Z, Ak и Ar имеют значения, указанные выше,
с последующим снятием защиты, если Y' содержит защитную группу Р1, как определено выше, и алкилированием, если желательно получить соединения, в которых Y представляет собой группу NR7, где R7 отличен от атома водорода,
с получением соединения формулы (Ib), частного случая соединения формулы (I), находящегося в относительной цис-конфигурации,

в которой R1, R2, R3, R4, X, Y, Z, n, Ak и Ar имеют значения, указанные для формулы (I),
и полученные соединения формулы (Ib) могут быть очищены в соответствии с обычным способом очистки и при желании разделены, если это является желательным, на их оптические изомеры и превращены в их соли присоединения фармацевтически приемлемой кислоты.
10. Фармацевтическая композиция, содержащая в качестве активного компонента соединение в соответствии с любым из пп.1-7 в сочетании с одними или несколькими фармацевтически приемлемыми, инертными, нетоксичными носителями.
11. Применение соединений формулы (I) в соответствии с любым из пп.1-7 для приготовления лекарственных средств для применения в качестве ингибиторов обратного поглощения серотонина.
12. Применение соединений формулы (I) в соответствии с любым из пп.1-7 для приготовления лекарственных средств для применения в качестве ингибиторов обратного поглощения серотонина и антагонистов NK1.
13. Применение соединений формулы (I) в соответствии с любым из пп.1-7 для приготовления лекарственных средств для применения для лечения депрессивных состояний, состояний беспокойства, импульсивных расстройств, агрессивного поведения, злоупотребления лекарственными средствами, ожирения и расстройства аппетита, боли и воспаления, деменции, психотических состояний, нарушений хронобиологического ритма, тошноты или желудочно-кишечных расстройств.
Текст
009648 Настоящее изобретение относится к соединениям пиперазина, к способу их получения и фармацевтическим композициям, которые их содержат, а также к их применению в качестве ингибиторов обратного поглощения серотонина. На основании этого, они являются полезными для лечения депрессивных состояний (Goodnick иGoldstein, J. Psychopharmacol. 1998, 12 (Suppl. В): S55-S87; Cheer и Goa, Drugs 2001, 61: 81-110; MacQueen и др., CNS Drug Rev. 2001, 7: 1-24; Wagstaff и др., Drugs 2002, 62: 655-703), состояний беспокойства, таких как генерализованный страх, приступы паники и фобии (Feighner, J. Clin. Psychiatry 1999, 60Anesth. 2004, 15: 154-158); и в отношении тех объектов, расстройств поведения и нейронной дегенерации, которые ассоциированы с деменцией и другими расстройствами, связанными со старением (Lyketos и др., Am. J. Psychiatry 2000, 157: 1686-1689; Lanctot и др., J. Neuropsyhiatry Clin. Neurosci. 2001, 13: 5-21;Lanctot и др., Neuropsychopharmacology 2002, 27: 646-654; Pollock и др., Am. J. Psychiatry 2002, 159: 460-465). Кроме того, большинство соединений в соответствии с настоящим изобретением также являются активными в качестве антагонистов нейрокинина NK1. Благодаря этому они также являются полезными для лечения депрессивных состояний (Rupniak и др., Behav. Pharmacol. 2001, 12: 497-508; Rupniak и др.,Neuropharmacology 2003, 44: 516-523; Kramer и др., Neuropsychopharmacology 2004, 29: 385-392; Dableh и др., Eur. J. Pharmacol. 2005, 507: 99-105), состояний беспокойства, таких как генерализованный страх, приступы паники и фобии (Rupniak и др., Behav. Pharmacol. 2001, 12: 497-508; Santarelli и др., Proc. Natl. Acad.Neurosci. 2003, 18: 1828-1836; Guest и др., Brain Res. 2004, 1002: 1-10; Czeh и др., Neuropsychopharmacology 2005, 30: 67-79), импульсивных состояний, таких как обсессивно-компульсивные расстройства поведения (Cilman и др., Br. J. Pharmacol. 1995, 114: 1310-1316; Tschope и др., Br. J. Pharmacol. 1992, 107: 750755; Rupniak и др., Behav. Pharmacol. 2001, 12: 497-508; Millan и др., Neuropharmacology 2002, 42: 677684), агрессивных состояний (Siegel и Schubert, Rev. Neurosci. 1995, 6: 47-61; De Felipe и др., Nature 1998,392: 394-397; Rupniak и др., Behav. Pharmacol. 2001,12: 497-508), а также злоупотребления лекарственными средствами (Murtra и др., Nature 2000, 405: 180-183; Ripley и др., Neuropharmacology 2002, 43: 12581268; Gadd и др., J. Neurosci. 2003, 23: 8271-8280), психотических состояний (Zachrisson и др., Eur.Neuropsychopharmacol. 2000, 10: 355-363) и экстрапирамидальных моторных эффектов, вызванных антипсихотическими средствами (Anderson и др., J. Pharmacol. Exp. Ther. 1995, 274: 928-936, Steinberg и др., J.Res. 1995, 28: 61-71; Daniels и др., Neurosci. Lett. 2003, 338: 111-114; Kramer и др., Science 1998, 281: 1640-1644; Kramer и др., Neuropsychopharmacology 2004, 29: 385-392), нарушений хронобиологических ритмов, таких как циркадные ритмы (Shibata и др., Brain Res. 1992, 597: 257-263; Challet и др., Brain Res. 1998, 800: 32-39; Challet и др., Neuropharmacology 2001, 40: 408-415; Gannon и др., Neuropharmacology, в печати), боли (Seguin и др., Pain 1995, 61: 325-343; De Felipe и др., Nature 1998, 392: 394-397; Sanger, Br. J.Patel и Lindley, Expert Opin. Pharmacother. 2003, 4: 2279-2296; Sanger, Br. J. Pharmacol. 2004, 141: 13031312); и в отношении тех объектов, расстройств поведения и нейронной дегенерации, которые ассоциированы с деменцией и другими расстройствами, связанными со старением (Raffa, Neurosci. Biobehav.-1 009648 Соединения, которые являются активными как на рецепторах NK1, так и на сайтах обратного поглощения серотонина (5-НТ), должны обладать комплементарными и синергетическими механизмами для контроля импульсивного, агрессивного, болевого и, прежде всего, депрессивного состояний. Более того, было показано, что блокирование рецепторов NK1 усиливает влияние 5-НТ ингибиторов обратного поглощения серотонина на серотонинэргическую трансмиссию: вследствие этого факта такие соединения могли бы оказывать более быстрые и более сильные антидепрессантные эффекты (Guiard и др., J.Neurochem. 2004, 89: 54-63; Froger и др., J. Neurosci. 2001, 21: 8188-8197). Быстрые анксиолитические эффекты антагонистов NK1, кроме того, могут быть комплементарными к анксиолитическим эффектам ингибиторов обратного поглощения 5-НТ, которые экспрессируются после длительного лечения. В отношении анксиогенных эффектов, вызываемых 5-НТ в начале лечения (Bagdy и др., Int. J. Neuropsychopharmacol. 2001, 4: 399-408), то такие предотвращались бы антагонистическими свойствами NK1 (Ballard и др., Eur.J. Pharmacol. 2001, 412: 255-264; Rupniak и др., Neuropharmacology 2003, 44: 516-523). Поскольку другие нежелательные эффекты, ассоциированные с блокированием обратного поглощения 5-НТ, связаны с такими эффектами, как рвотные эффекты (Goldstein и Goodnick, J. Psychopharmacol. 1998, 12 (Suppl. B):S55-S87; Edwards и Anderson, Drugs 1999, 57: 507-533; Waugh и Goa, CNS Drugs 2003, 17: 343-362) и сексуальные дисфункции (Goldstein и Goodnick, J. Psychopharmacol. 1998, 12 (Suppl. B): S55-S87; Montgomery и др., J. Affect. Disord. 2002, 69: 119-140; Hirschfeld, J. Clin. Psychiatry 2003, 64 (Suppl. 18): 20-24), то антагонисты NK1 также могли бы противодействовать таким эффектам. Таким образом, соединения, которые представляют собой как антагонисты NK1, так и ингибиторы обратного поглощения серотонина, будут иметь терапевтические преимущества по сравнению с соединениями, которые взаимодействуют только с одной или другой из этих двух мишеней. В частности, настоящее изобретение относится к соединениям формулы (I)R1, R2, R3 и R4, которые могут быть одинаковыми или разными, каждый представляет собой атом или группу, выбранную из Н, галогена, линейного или разветвленного C1-С 6-алкила, линейного или разветвленного C1-C6-алкокси, фенила и циано,X представляет собой связь, атом кислорода или группу, выбранную из (СН 2)m-, -ОСН 2-и -NR5-,m представляет собой 1 или 2,R5 представляет собой атом водорода или группу, выбранную из линейного или разветвленного C1 С 6-алкила, COR6 и CO2R6,R6 представляет собой линейную или разветвленную С 1-С 6-алкильную группу,Y представляет собой атом кислорода или группу, выбранную из NR7 и CHR8,R7 представляет собой атом водорода или группу, выбранную из COR9 и линейного или разветвленного C1-С 6-алкила, где алкильная группа необязательно замещена 5-оксо-4,5-дигидро-1H-1,2,4-триазол-3 ильной или 2,3-дигидро-1,4-бенздиоксин-2-ильной группой,R9 представляет собой группу, выбранную из линейного или разветвленного C1-С 6-алкила, арила и гетероарила,R8 представляет собой атом водорода или аминогруппу, необязательно замещенную одной или двумя линейными или разветвленными C1-С 6-алкильными группами,Z представляет собой атом азота или СН группу,n представляет собой 1 или 2,Ak представляет собой линейную или разветвленную С 1-С 6-алкиленовую цепь,Ar представляет собой арильную или гетероарильную группу,к их оптическим изомерам, а также к их солям присоединения с фармацевтически приемлемой кислотой. Под оптическими изомерами понимают энантиомеры и диастереоизомеры. Среди фармацевтически приемлемых кислот можно отметить, но не ограничиваясь только ими, соляную кислоту, бромисто-водородную кислоту, серную кислоту, фосфорную кислоту, уксусную кислоту,трифторуксусную кислоту, молочную кислоту, пировиноградную кислоту, малоновую кислоту, янтарную кислоту, глутаровую кислоту, фумаровую кислоту, винную кислоту, малеиновую кислоту, лимонную кислоту, аскорбиновую кислоту, щавелевую кислоту, метансульфокислоту, бензолсульфокислоту,камфорную кислоту, дибензоилвинную кислоту. Под арильной группой понимают фенил, бифенилил или нафтил, каждая из этих групп необязательно замещена одной или несколькими одинаковыми или разными группами, выбранными из галогена, линейного или разветвленного С 1-С 6-алкила, линейного или разветвленного C1-С 6-алкокси, гидрокси, циано,линейного или разветвленного C1-С 6-тригалоалкила и линейного или разветвленного C1-С 6-тригалоалкокси.-2 009648 Под гетероарильной группой понимают ароматическую моно- или бициклическую 5-12-членную группу, содержащую 1, 2 или 3 гетероатома, выбранных из кислорода, азота и серы, при этом понятно,что гетероарильная группа необязательно может быть замещена одной или несколькими одинаковыми или разными группами, выбранными из галогена, линейного или разветвленного C1-С 6-алкила, линейного или разветвленного С 1-С 6-алкокси, гидрокси, циано и линейного или разветвленного С 1-С 6-тригалоалкила.X предпочтительно представляет собой связь, атом кислорода или группу, выбранную из -ОСН 2- и-(СН 2)m-, где m представляет собой 1 или 2,Y предпочтительно представляет собой NH,Z предпочтительно представляет собой атом азота,n предпочтительно представляет собой 1,Ar предпочтительно представляет собой арильную группу. Предпочтительными соединениями в соответствии с изобретением являются транс-1-2-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин, его энантиомеры, а также его соли присоединения с фармацевтически приемлемой кислотой,транс-1-3-[(3,5-дибромбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин, его энантиомеры, а также его соли присоединения с фармацевтически приемлемой кислотой,транс-1-6-[(3,5-дибромбензил)окси]-6,7,8,9-тетрагидро-5H-бензо[7]аннулен-5-илпиперазин, его энантиомеры, а также его соли присоединения с фармацевтически приемлемой кислотой,транс-1-2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-илпиперазин, его энантиомеры, а также его соли присоединения с фармацевтически приемлемой кислотой,транс-1-2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-ил-1,4-диазепан, его энантиомеры,а также его соли присоединения с фармацевтически приемлемой кислотой,1-(1S,2R)-2-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин, его энантиомеры, а также его соли присоединения с фармацевтически приемлемой кислотой,1-[(1S,2R)-2-[(3,5-дифторбензил)окси]-2,3-дигидро-1H-инден-1-ил]пиперазин, его энантиомеры, а также его соли присоединения с фармацевтически приемлемой кислотой,1-[(1S,2R)-2-[(3,5-диметилбензил)окси]-2,3-дигидро-1H-инден-1-ил]пиперазин, его энантиомеры, а также его соли присоединения с фармацевтически приемлемой кислотой,транс-1-3-[(3,5-дихлорбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин, его энантиомеры, а также его соли присоединения с фармацевтически приемлемой кислотой,транс-1-3-[3-фтор-5-(трифторметил)бензилокси]-3,4-дигидро-2H-хромен-4-илпиперазин, его энантиомеры, а также его соли присоединения с фармацевтически приемлемой кислотой, и транс-1-3-(3-хлор-5-фторбензилокси)-3,4-дигидро-2 Н-хромен-4-илпиперазин, его энантиомеры, а также его соли присоединения с фармацевтически приемлемой кислотой. Изобретение также относится к способу получения соединений формулы (I), при использовании в качестве исходного соединения такового формулы (Va), в относительной транс-конфигурации в которой R1, R2, R3, R4, X, n и Z имеют значения, указанные выше, и Y' представляет собой атом кислорода или группу, выбранную из NP1 и CHR'8, в которой R'8 представляет собой атом водорода или группуNHP1, и P1 представляет собой защитную группу для функциональной аминогруппы,в котором соединение формулы (Va) подвергают взаимодействию, если желательно получить соединения формулы (I) в относительной транс-конфигурации, с соединением формулы (VI)G-Ak-Ar (VI) в которой Ak и Ar имеют значения, указанные для формулы (I), и G представляет собой уходящую группу, такую как, например, атом галогена или n-толуолсульфонатную, трифторметансульфонатную или метансульфонатную группу,получая соединение формулы (VIIa) в относительной транс-конфигурации-3 009648 в которой R1, R2, R3, R4, X, n, Y', Z, Ak и Ar имеют значения, указанные выше,с которого снимают защиту, если Y1 содержит защитную группу P1, как определено в данной заявке выше, и затем алкилируют, если желательно получить соединения, в которых Y представляет собой группу NR7, где R7 отличается от атома водорода,получая соединения формулы (Ia), частный случай соединений формулы (I), которые находятся в относительной транс-конфигурации, в которой R1, R2, R3, R4, X, n, Y, Z, Ak и Ar имеют значения, указанные для формулы (I),или в котором соединение формулы (Va) окисляют, если желательно получить соединения формулы (I) в относительной цис-конфигурации,получая рацемическое соединение формулы (VIII) в которой R1, R2, R3, R4, X, Z, n и Y' имеют значения, указанные выше,которое восстанавливают до соответствующего спирта, его диастереоизомеры разделяют и выделяют изомер формулы (Vb) в относительной цис-конфигурации в которой R1, R2, R3, R4, X, n, Y', Z, Ak и Ar имеют значения, указанные выше,с которого снимают защиту, если Y' содержит защитную группу P1, как определено в данной заявке выше, и затем алкилируют, если желательно получить соединения, в которых Y представляет собой группу NR7, где R7 отличается от атома водорода,получая соединения формулы (Ib), частный случай соединений формулы (I), которые находятся в относительной цис-конфигурации,-4 009648 в которой R1, R2, R3, R4, X, Y, Z, n, Ak и Ar имеют значения, указанные для формулы (I),и соединения формул (Ia) и (Ib) могут быть очищены в соответствии с обычным способом очистки,разделены, если это является желательным, на их оптические изомеры и превращены, если это является желательным, в их соли присоединения с фармацевтически приемлемой кислотой. Соединения формулы (Ic), частный случай соединений формулы (I), в которой Ak представляет собой группу -СН(СН 3) в которой R1, R2, R3, R4, X, Y, Z, n и Ar имеют значения, указанные для формулы (I),могут быть получены путем конденсации спирта формулы (Va) или (Vb) с кислотой формулы (IX) НО 2 С-Ar (IX) в которой Ar имеет значения, указанные для формулы (I),получая сложный эфир формулы (X) в которой R1, R2, R3, R4, X, n, Y', Z и Ar имеют значения, указанные выше,которое подвергают взаимодействию с бис(циклопентадиенил)диметилтитаном, получая соединение формулы (XI) в которой R1, R2, R3, R4, X, n, Y', Z и Ar имеют значения, указанные выше,которое гидрируют, получая соединение формулы (XII) в которой R1, R2, R3, R4, X, n, Y', Z и Ar имеют значения, указанные выше,которое разделяют на его изомеры и затем снимают защиту, если Y1 содержит защитную группу P1,как определено в данной заявке выше, и алкилируют, если желательно получить соединения, в которых Y представляет собой группу NR7, где R7 отличается от атома водорода, получая соединения формулы (Ic). Соединения формулы (Va1), частный случай соединений формулы (Va), в которой Z представляет собой атом азота, могут быть получены при использовании в качестве исходного соединения такового формулы (II) в которой R1, R2, R3, R4 и X имеют значения, указанные для формулы (I),-5 009648 которое окисляют, получая соединение формулы (III) в которой n имеет значения, указанные для формулы (I), и Y' имеет значения, указанные выше,получая соединение формулы (Va1) в которой R1, R2, R3, R4, X, Y' и n имеют значения, указанные выше. Соединения формулы (Va2), частный случай соединений формулы (Va), в которой Z представляет собой СН группу, n представляет собой 1 и Y' представляет собой NP1 группу, могут быть получены при использовании в качестве исходного соединения такового формулы (XIII) в которой R1, R2, R3, R4 и X имеют значения, указанные выше,которое подвергают взаимодействию с 4-пиридиллитием, получая соединение формулы (XIV) в которой R1, R2, R3, R4 и X имеют значения, указанные выше,которое дегидратируют, получая соединение формулы (XV) в которой R1, R2, R3, R4 и X имеют значения, указанные выше,которое подвергают взаимодействию с оксоном, получая соединение формулы (XVI)-6 009648 в которой R1, R2, R3, R4 и X имеют значения, указанные выше,которое подвергают взаимодействию с восстановителем, получая соединение формулы (XVII) в которой R1, R2, R3, R4 и X имеют значения, указанные выше,которое подвергают реакции каталитической гидрогенизации, получая соединение формулы (XVIII) в которой R1, R2, R3, R4 и X имеют значения, указанные выше,цис- и транс-изомеры которого разделяют, функциональную аминогруппу его транс-изомера защищают, получая соединение формулы (Va2) в относительной транс-конфигурации в которой R1, R2, R3, R4 и X имеют значения, указанные выше. Соединения в соответствии с настоящим изобретением представляют собой ингибиторы обратного поглощения серотонина, также большинство из этих соединений являются антагонистами NK1. Они являются полезными в качестве лекарственных средств для лечения депрессивных состояний, состояний беспокойства, импульсивных расстройств, агрессивного поведения, злоупотребления лекарственными средствами, ожирения и расстройства аппетита, боли и воспаления, деменции, психотических состояний,нарушений хронобиологического ритма, тошноты и желудочно-кишечных расстройств. Настоящее изобретение также относится к фармацевтическим композициям, содержащим в качестве активного компонента соединение формулы (I) или его соль присоединения с фармацевтически приемлемой кислотой, в комбинации с одним или несколькими фармацевтически приемлемыми, инертными,нетоксичными наполнителями или носителями. Среди фармацевтических композиций согласно изобретению особенно можно отметить те, которые являются пригодными для перорального, парентерального (внутривенного, внутримышечного или подкожного), чрескожного, назального, ректального, подъязычного, внутриглазного введения или введения в дыхательные пути, и в особенности таблетки или драже, подъязычные таблетки, желатиновые капсулы,капсулы, суппозитории, кремы, мази, кожные гели, препараты для инъекций или препараты для питья,аэрозоли, глазные и носовые капли. Полезная дозировка может изменяться в зависимости от возраста и веса пациента, пути введения,природы и тяжести расстройства, а также проведения какого-либо ассоциированного лечения и находится в пределах от 0,5 до 500 мг в сутки на одно или больше введений. Последующие примеры иллюстрируют изобретение. Используемые исходные вещества являются известными продуктами или могут быть получены в соответствии с известными способами. Различные получения обеспечивают синтез промежуточных продуктов, которые используются при получении соединений в соответствии с изобретением. Структуры соединений, описанных в примерах, были определены в соответствии с обычными спектрофотометрическими способами (ИК, ядерный магнитный резонанс, масс-спектрометрия). Точки плавления определялись либо на столе Кофлера (BK), либо на горячей пластинке под микроскопом (MK).-7 009648 Получение А. 6-Бром-1,2-дигидронафталин. Этап А. 7-Бром-1,2,3,4-тетрагидронафт-1-ол. К 9,5 г 7-бром-3,4-дигидронафтален-1(2H)-она (42 ммоль), полученного согласно способу, описанному в Synth. Comm. 1994, 2777, растворенного в 100 мл этанола, добавляли, при 0 С и двумя порциями,0,8 г борогидрида натрия (21 ммоль). После этого реакционной смеси позволяли нагреться до температуры окружающей среды в течение 30 мин и затем этанол упаривали. Остаток ресуспендировали в 100 мл толуола и 100 мл воды. После отделения водную фазу экстрагировали 50 мл толуола. Толуольные фазы объединяли, промывали насыщенным водным раствором хлорида натрия и после этого упаривали, получая 7-бром-1,2,3,4-тетрагидронафт-1-ол в виде масла. Этап Б. 6-Бром-1,2-дигидронафталин. Раствор 8,6 г соединения, полученного на вышеописанном этапе (37,9 ммоль) в 200 мл толуола, нагревали до 100 С. При этой температуре по каплям добавляли в течение 1 ч раствор 0,3 г паратолуолсульфоновой кислоты (1,2 ммоль), растворенной в 400 мл толуола. Затем реакционную смесь охлаждали до 25 С и после этого гидролизовали, используя 100 мл воды. Органическую фазу экстрагировали и после этого высушивали, фильтровали и упаривали, получая 6-бром-1,2-дигидронафталин в виде масла. Получение Б. 7-Бром-2,3-дигидро-1-бензоксепин. Этап А. 4-[5-Бром-2-(бут-3-ен-1-илокси)фенил]-4-гидроксибутан-2-он. К 300 мл ацетона добавляли 10 г 5-бром-2-гидроксибензальдегида (49,7 ммоль), 13,7 г карбоната калия (99,5 ммоль) и 10,1 мл 4-бромбут-1-ена (99,5 ммоль); после этого реакционную смесь нагревали в колбе с обратным холодильником в течение 36 ч, затем охлаждали, фильтровали и упаривали насухо,получая 4-[5-бром-2-(бут-3-ен-1-илокси)фенил]-4-гидроксибутан-2-он в виде масла. Этап Б. 4-Бром-1-(бут-3-ен-1-илокси)-2-винилбензол. К 90 мл безводного тетрагидрофурана добавляли 1,6 г 60% гидрида натрия в масле (39,8 ммоль) и после этого, при 0 С и одной порцией, 10,67 г бромида метил(трифенил)фосфония (29,9 ммоль). Смеси позволяли нагреться до температуры окружающей среды и перемешивали в течение 30 мин при 25 С. Затем к реакционной смеси по каплям добавляли раствор 8 г соединения, полученного на вышеописанном этапе (24,9 ммоль) в 30 мл безводного тетрагидрофурана, при температуре окружающей среды. Наблюдали экзотермическую реакцию от 25 до 35 С в течение 45 мин. Дополнительно перемешивали в течение 2 ч при температуре окружающей среды; после этого реакционную смесь фильтровали и фильтрат вливали в смесь 100 мл этилацетата, 200 мл насыщенного водного раствора хлорида натрия и 50 г льда. После экстрагирования этилацетатом объединенные органические фазы промывали водой, высушивали, фильтровали и упаривали насухо. Полученный остаток очищали путем фильтрации через 100 г диоксида кремния (элюент: толуол 100%), получая 4-бром-1-(бут-3-ен-1-илокси)-2-винилбензол в виде масла. Этап В. 7-Бром-2,3-дигидро-1-бензоксепин. 5 г соединения, полученного на вышеописанном этапе (19,8 ммоль), растворяли в 500 мл толуола и затем раствор дегазировали в течение 30 мин с помощью азота. Добавляли 335 мг (0,39 ммоль) [1,3-бис(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(фенилметилен)(трициклогексилфосфин)рутения],или катализатора Грубба II. После этого реакционную смесь нагревали при 50 С в течение 30 мин; затем толуол упаривали и полученный остаток очищали на колонке 70 г диоксида кремния (элюент: циклогексан/толуол: 95/5), получая 7-бром-2,3-дигидро-1-бензоксепин. Пример 1. транс-1-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин дигидрохлорид. Этап А. трет-Бутил транс-4-(2-гидрокси-2,3-дигидро-1 Н-инден-1-ил)пиперазин-1-карбоксилат. 11,6 г трет-бутил пиперазин-1-карбоксилата (62 ммоль) и 8,2 г оксида индена (62 ммоль) растворяли в 30 мл ацетонитрила. После этого реакционную смесь нагревали всю ночь при 60 С и упаривали насухо. Полученный остаток очищали путем флэш-хроматографии на 1 кг диоксида кремния (элюент: дихлорметан/этанол 95/5), получая трет-бутил транс-4-(2-гидрокси-2,3-дигидро-1H-инден-1-ил)пиперазин-1-карбоксилат в виде белой меренги. Этап Б. трет-Бутил транс-4-2-[(3,5-дибромбензил)окси]-2,3-дигидро-1 Н-инден-1-илпиперазин-1 карбоксилат. К 3 г соединения, полученного на вышеописанном этапе (9,42 ммоль), в 30 мл безводного диметилформамида добавляли 452 мг гидрида натрия в виде 60%-ной суспензии в масле (11,3 ммоль, 1,2 экв.). После перемешивания в течение 30 мин при температуре окружающей среды добавляли 3,1 г 3,5-дибромбензилбромида (9,42 ммоль). Наблюдали незначительно экзотермическую реакцию. После этого реакционную смесь перемешивали всю ночь при температуре окружающей среды и затем диметилформамид упаривали. Полученный остаток ресуспендировали в дихлорметане. После промывания водой,высушивания, фильтрования и упаривания получали 6 г остатка, который очищали путем флэш-хроматографии на 500 г диоксида кремния (элюент: дихлорметан/этилацетат 90/10), получая трет-бутил транс-42-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин-1-карбоксилат в виде меренги. Этап В. транс-1-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1 Н-инден-1-илпиперазин дигидрохлорид. 3,5 г соединения, полученного на вышеописанном этапе (6,18 ммоль), растворяли в 250 мл этилацетата и после этого через раствор барботировали газообразный хлористый водород. Температуру повышали до 45 С и после этого перемешивали в течение 2 ч при температуре окружающей среды. После это-8 009648 го реакционную смесь концентрировали на две трети и после этого добавляли 50 мл простого эфира. Полученный осадок отфильтровали и после этого высушивали, получая транс-1-2-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин дигидрохлорид в виде бежевого твердого вещества. Точка плавления (МК): 154-167 С. Пример 2. (-)Изомер транс-1-2-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин дигидрохлорид. После обратного превращения в основание рацемическое соединение из примера 1 разделяли путем препаративной хиральной ВЭЖХ (элюент: изопропанол/ацетонитрил/диэтиламин 100/900/1) на фазеChiralpak AD. Первый из изомеров, разделенных таким образом, превращали в соль с помощью соляной кислоты, получая дигидрохлорид (-)изомера транс-1-2-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден 1-илпиперазина. Точка плавления (МК): 117-125 С. Оптическое вращение : []D=-27,86 (с=1%, МеОН, 20 С, 589 нм). Пример 3. (+)Изомер транс-1-2-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин дигидрохлорид. Второй из изомеров, разделенных в примере 2, превращали в соль с помощью соляной кислоты, получая дигидрохлорид (+)изомера транс-1-2-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазина. Точка плавления (MK): 115-121 С. Оптическое вращение : []D=+27,29 (с=1%, МеОН, 20 С, 589 нм). Пример 4. транс-1-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин дигидрохлорид. Этап А. транс-3-Бромхроман-4-ол. К 15 г 2H-хромена (0,113 моля), растворенного в 330 мл смеси тетрагидрофуран/вода 50/50, добавляли 22,1 г N-бромсукцинимида (0,124 моля, 1,1 экв.) и затем смесь перемешивали в течение 1 ч при температуре окружающей среды. После этого реакционную смесь разводили водой и затем дважды экстрагировали этилацетатом. Объединенные органические фазы промывали водой, высушивали, фильтровали и упаривали насухо, получая транс-3-бромхроман-4-ол в виде светло-желтого твердого вещества. Точка плавления (BK): 96-98 С. Этап Б. 1a,7b-Дигидро-2 Н-оксирен[с]хромен. 4,4 г гранул гидроксида калия (78,5 ммоль) добавляли к 10 г соединения, полученного на вышеописанном этапе (43,6 ммоль), растворенного в 170 мл тетрагидрофурана и 85 мл воды. После перемешивания в течение 2 ч при 25 С реакционную смесь разводили водой и 2 раза экстрагировали простым эфиром. Органические фазы объединяли и промывали водой, высушивали, фильтровали и упаривали насухо,получая 1 а,7b-дигидро-2H-оксирен[с]хромен в виде светло-желтого масла. Этап В. трет-Бутил транс-4-(3-гидрокси-3,4-дигидро-2H-хромен-4-ил)пиперазин-1-карбоксилат. 4,9 г соединения, полученного на вышеописанном этапе (33 ммоль), подвергали реакции с 6,1 г трет-бутил пиперазин-1-карбоксилата (33 ммоль) в соответствии со способом, описанным на этапе А примера 1. Полученное масло очищали путем флэш-хроматографии на 500 г диоксида кремния (элюент: дихлорметан/этанол 95/5), получая ожидаемый продукт в виде масла. Этап Г. трет-Бутил транс-4-3-[(3,5-дибромбензил)окси]-3,4-дигидро-2 Н-хромен-4-илпиперазин-1 карбоксилат. 3 г соединения, полученного на вышеописанном этапе (8,9 ммоль), обрабатывали в соответствии со способом, описанным на этапе Б примера 1. Полученное масло очищали путем флэш-хроматографии на 300 г диоксида кремния, получая ожидаемый продукт в виде белой меренги. Этап Д. транс-1-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2 Н-хромен-4-илпиперазин дигидрохлорид. 2,9 г соединения, полученного на вышеописанном этапе (4,97 ммоль), обрабатывали в соответствии со способом, описанным на этапе Б примера 1. Полученный продукт кристаллизовали из простого эфира,фильтровали и высушивали, получая ожидаемый продукт в виде белых кристаллов. Точка плавления (MK): 130-135 С. Пример 5. (-)Изомер транс-1-3-[(3,5-дибромбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин дигидрохлорид. После обратного превращения в основание рацемическое соединение из примера 4 разделяли путем препаративной хиральной ВЭЖХ (элюент: этанол/диэтиламин 1000/1) на фазе Chiralpak AD. Первый из изомеров, разделенных таким образом, превращали в соль с помощью соляной кислоты, получая дигидрохлорид (-)изомера транс-1-3-[(3,5-дибромбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазина. Точка плавления (MK): 128-132 С. Оптическое вращение: []D=-40,36 (с=1%, МеОН, 20 С, 589 нм). Пример 6. (+)Изомер транс-1-3-[(3,5-дибромбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин дигидрохлорид. Второй из изомеров, разделенных в примере 5, превращали в соль с помощью соляной кислоты, получая дигидрохлорид (+)изомера транс-1-3-[3,5-дибромбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазина. Точка плавления (MK): 126-130 С.-9 009648 Оптическое вращение: []D=+40,92 (с=1%, МеОН, 20 С, 589 нм). Пример 7. транс-1-6-[(3,5-Дибромбензил)окси]-6,7,8,9-тетрагидро-5H-бензо-[7]аннулен-5-илпиперазин дигидрохлорид. Этап А. трет-Бутил транс-4-(6-гидрокси-6,7,8,9-тетрагидро-5 Н-бензо[7]аннулен-5-ил)пиперазин-1 карбоксилат. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах А, Б и В примера 4, заменяя 2H-хромен 6,7-дигидро-5H-бензо[7]аннуленом. Этап Б. транс-1-6-[(3,5-Дибромбензил)окси]-6,7,8,9-тетрагидро-5 Н-бензо[7]аннулен-5-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах Б и В примера 1,используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Точка плавления (MK): 126-131 С. Пример 8. транс-1-4-[(3,5-Дибромбензил)окси]-2,3,4,5-тетрагидро-1-бензоксепин-5-илпиперазин дигидрохлорид. Этап А. 1 а,2,3,8b-Тетрагидрооксирен[d][1]бензоксепин. К 0,5 г 2,3-дигидро-1-бензоксепина (3,42 ммоль), полученного согласно способу, описанному в J.Org. Chem., 1969, 34 (1), 207, растворенного в 30 мл смеси этилацетат/вода 50/50, добавляли 1,44 г гидрокарбоната натрия (17,1 ммоль) и после этого в течение 1 ч раствор 2,1 г Oxone (3,42 ммоль) в 15 мл воды. После окончания добавления дополнительно перемешивали в течение часа и после этого органическую фазу отделяли. Водную фазу снова экстрагировали 10 мл этилацетата и объединенные органические фазы промывали водой, высушивали, фильтровали и упаривали насухо, получая ожидаемый продукт в виде масла. Этап Б. трет-Бутил транс-4-(4-гидрокси-2,3,4,5-тетрагидро-1-бензоксепин-5-ил)пиперазин-1-карбоксилат. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе А примера 1, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Этап В. транс-1-4-[(3,5-Дибромбензил)окси]-2,3,4,5-тетрагидро-1-бензоксепин-5-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах Б и В примера 1,используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Точка плавления (MK): 132-139 С. Пример 9. транс-1-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперидин-4-амин дигидрохлорид. Этап А. трет-Бутил транс-[1-(3-гидрокси-3,4-дигидро-2 Н-хромен-4-ил)пиперидин-4-ил]карбамат. К 3 г соединения, полученного на этапе Б примера 4, растворенного в 45 мл ацетонитрила, добавляли 4 г трет-бутил пиперидин-4-илкарбамата (20,2 ммоль). Затем смесь нагревали в колбе с обратным холодильником в течение 12 ч и после этого упаривали насухо. Получали желтое масло, которое очищали путем флэш-хроматографии на диоксиде кремния (элюент: дихлорметан/этанол 95/5), получая ожидаемый продукт в виде светло-желтой меренги. Этап Б. трет-Бутил транс-1-3-[(3,5-дибромбензил)окси]-3,4-дигидро-2 Н-хромен-4-ил-4-пиперидилкарбамат. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе Б примера 1, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Этап В. транс-1-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперидин-4-амин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе В примера 1, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Точка плавления (MK): 160-165 С. Пример 10. транс-4-2-[(3,5-Дибромбензил)окси]-1,4-тетрагидронафт-1-ил]морфолин гидрохлорид. Этап А. 1 а,2,3,7b-Тетрагидронафто[1,2-b]оксирен. 36,5 г 1,2-дигидронафталина (280,3 ммоль) подвергали реакции с Oxone в соответствии со способом, описанным на этапе А примера 8, получая ожидаемый продукт в виде масла. Этап Б. транс-1-Морфолин-4-ил-1,2,3,4-тетрагидронафт-2-ол. К 3,95 г соединения, полученного на вышеописанном этапе (27,02 ммоль), растворенного в 65 мл ацетонитрила, добавляли 2,35 мл морфолина (27,02 ммоль) и после этого 185 мг ZnCl2 (1,35 ммоль). После нагревания в колбе с обратным холодильником в течение 12 ч смесь упаривали насухо и полученный остаток очищали путем флэш-хроматографии на диоксиде кремния (дихлорметан/этанол 95/5), получая ожидаемый продукт в виде светло-коричневого твердого вещества. Точка плавления (ВК): 88-90 С. Этап В. транс-4-2-[(3,5-Дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-илморфолин. 3 г соединения, полученного на вышеописанном этапе (12,85 ммоль), обрабатывали в соответствии со способом, описанным на этапе Б примера 1. Получали масло, которое очищали путем флэш-хромато- 10009648 графии на диоксиде кремния (элюент: дихлорметан), получая ожидаемый продукт в виде масла. Этап Г. транс-4-2-[(3,5-Дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-илморфолин гидрохлорид. К 1,9 г соединения, полученного на вышеописанном этапе (3,95 ммоль), растворенного в 40 мл этилацетата, добавляли 2,96 мл 2 М эфирного раствора HCl (5,92 ммоль). После фильтрования и высушивания кристаллов получали ожидаемый продукт. Точка плавления: (MK)=175-180 С. Пример 11. транс-1-2-[(3,5-Дибромбензил)окси]-5-метокси-2,3-дигидро-1H-инден-1-илпиперазин. Этап А. 4-Метокси-6,6 а-дигидро-1 аН-индено[1,2-b]оксирен. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах А и Б примера 4,используя в качестве исходного соединения 6-метокси-1H-инден. Этап Б. трет-Бутил транс-4-2-[(3,5-дибромбензил)окси]-5-метокси-2,3-дигидро-1 Н-инден-1-илпиперазин-1-карбоксилат. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах А и Б примера 1,используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Этап В. транс-1-2-[(3,5-Дибромбензил)окси]-5-метокси-2,3-дигидро-1 Н-инден-1-илпиперазин. 1,3 г соединения, полученного на вышеописанном этапе (2,17 ммоль), обрабатывали в соответствии со способом, описанным на этапе Б примера 1. После упаривания насухо полученный продукт ресуспендировали в воде, и значение рН водной фазы доводили до 8 путем добавления раствора гидроксида натрия (1 н.), и экстрагировали простым эфиром. Органическую фазу промывали водой, высушивали,фильтровали и упаривали насухо. Полученный остаток кристаллизовали из изопропилового эфира. Кристаллы отфильтровали и высушивали, получая ожидаемый продукт. Точка плавления (MK): 101-104 С. Пример 12. транс-1-2-[(3,5-Дибромбензил)окси]-7-метокси-1,2,3,4-тетрагидронафт-1-илпиперазин дигидрохлорид. Этап А. 2-Бром-7-метокси-1,2,3,4-тетрагидронафт-1-ол. 20 г 6-метокси-1,2-дигидронафталина (126 ммоль) обрабатывали в соответствии со способом, описанным на этапе А примера 4, получая ожидаемый продукт. Этап Б. трет-Бутил транс-4-(2-гидрокси-7-метокси-1,2,3,4-тетрагидронафт-1-ил)пиперазин-1-карбоксилат. К 2 г соединения, полученного на вышеописанном этапе (7,78 ммоль), растворенного в 30 мл тетрагидрофурана, добавляли 1,24 г гидрида натрия в виде 60%-ной суспензии в масле (31,1 ммоль) и перемешивали в течение 4 ч при температуре окружающей среды. Реакционную смесь фильтровали. После этого фильтрат, который содержит 6-метокси-1 а,2,3,7b-тетрагидронафто[1,2-6]оксирен в растворе, подвергали реакции с 1,5 г трет-бутил пиперазин-1-карбоксилата (7,78 ммоль) и 5 мл диметилформамида. Из реакционной смеси отгоняли тетрагидрофуран и затем смесь нагревали при 110 С в течение 24 ч. Смесь охлаждали, вливали в воду и экстрагировали дихлорметаном. Органическую фазу высушивали и после этого фильтровали и упаривали насухо. Полученный остаток очищали путем флэш-хроматографии на диоксиде кремния (элюент: дихлорметан/этанол: 98/2), получая ожидаемый продукт. Этап В. транс-1-2-[(3,5-Дибромбензил)окси]-7-метокси-1,2,3,4-тетрагидронафт-1-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах Б и В примера 1,используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Точка плавления (MK): 175-182 С. Пример 13. транс-1-2-[(3,5-Дибромбензил)окси]-6-метокси-1,2,3,4-тетрагидронафт-1-илпиперазин дигидрохлорид. Этап А. 2-Бром-6-метокси-1,2,3,4-тетрагидронафт-1-ол. 10,8 г 7-метокси-1,2-дигидронафталина (67,4 ммоль) обрабатывали в соответствии со способом,описанным на этапе А примера 4, получая ожидаемый продукт. Этап Б. трет-Бутил транс-4-(2-гидрокси-6-метокси-1,2,3,4-тетрагидронафт-1-ил)пиперазин-1-карбоксилат. К 9,7 г соединения, полученного на вышеописанном этапе (37,8 ммоль), растворенного в 150 мл тетрагидрофурана, добавляли 3 г гидрида натрия в виде 60%-ной суспензии в масле (75,6 ммоль, 2 экв.) и после этого реакционную смесь перемешивали в течение 24 ч при температуре окружающей среды. После фильтрования фильтрат, который содержит 5-метокси-1 а,2,3,7b-тетрагидронафто[1,2-6]оксирен в растворе, подвергали взаимодействию с 8,4 г трет-бутил пиперазин-1-карбоксилата (45,3 ммоль) в 20 мл диметилформамида. После этого тетрагидрофуран отгоняли и затем реакционную смесь нагревали при 110 С в течение 1 ч. После этого реакционную смесь охлаждали, затем вливали в воду и экстрагировали дихлорметаном. Затем органическую фазу высушивали, фильтровали и упаривали насухо, получая остаток, который очищали путем флэш-хроматографии на диоксиде кремния (элюент: дихлорметан/этанол: 98/2), получая ожидаемый продукт. Этап В. транс-1-2-[(3,5-Дибромбензил)окси]-6-метокси-1,2,3,4-тетрагидронафт-1-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах Б и В примера 1,используя в качестве исходного вещества соединение, полученное на вышеописанной стадии.- 11009648 Точка плавления (MK): 137-150 С. Пример 14. транс-1-2-[(3,5-Дибромбензил)окси]-5-фенил-2,3-дигидро-1H-инден-1-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, используя в качестве исходного соединения 6-фенил-1H-инден. Точка плавления (MK): 145-153 С. Пример 15. транс-1-2-[(3,5-Дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-илпиперазин дигидрохлорид. Этап А. транс-2-Бром-1,2,3,4-тетрагидронафт-1-ол. К 13,0 г (100 ммоль) дигидронафталина, растворенного в смеси 100 мл воды и 400 мл тетрагидрофурана, при температуре окружающей среды одной порцией добавляли 19,6 г (110 ммоль) N-бромсукцинимида и перемешивали в течение 3 ч. После этого добавляли 200 мл ледяной воды и 200 мл простого эфира; перемешивали и фазы разделяли. Водную фазу экстрагировали 200 мл простого эфира; затем объединенные органические фазы промывали 200 мл насыщенного водного раствора хлорида натрия, и высушивали, и после этого упаривали, получая ожидаемое соединение. Этап Б. 1a,2,3,7b-Тетрагидронафто[1,2-b]оксирен. К 27,5 г соединения, полученного на вышеописанном этапе, растворенного в 180 мл тетрагидрофурана, по каплям в течение 5 мин при температуре окружающей среды добавляли раствор 10 г (180 ммоль) гидроксида калия в 10 мл воды. После перемешивания в течение 5 дней при температуре окружающей среды добавляли 200 мл воды и экстрагировали простым этиловым эфиром. Объединенные органические фазы промывали и после этого высушивали. После упаривания полученный продукт перегоняли в вакууме (приблизительно 0,1 мм рт.ст.) в установке Kugelrohr. Ожидаемый продукт перегонялся приблизительно при 80 С. Этап В. трет-Бутил 4-[транс-2-гидрокси-1,2,3,4-тетрагидронафт-1-ил]пиперазин-1-карбоксилат. При температуре окружающей среды смешивали вместе 4,4 г (30 ммоль) соединения, полученного на вышеописанном этапе, и 5,6 г (30 ммоль) N-трет-бутилоксикарбонилпиперазина в 60 мл ацетонитрила и после этого нагревали в колбе с обратным холодильником в течение 40 ч. После упаривания и флэш-хроматографии на 500 г диоксида кремния (элюент: дихлорметан/этанол 98/2) собирали ожидаемый продукт. Этап Г. трет-Бутил 4-транс-2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-илпиперазин-1 карбоксилат. При температуре окружающей среды к 0,44 г (11 ммоль) гидрида натрия в виде суспензии в 20 мл тетрагидрофурана добавляли в течение нескольких минут 3,3 г (10 ммоль) соединения, полученного на вышеописанном этапе, растворенного в 20 мл тетрагидрофурана. Смесь нагревали при 50 С в течение 2 ч,после этого добавляли одной порцией 3,3 г (10 ммоль) 3,5-дибромбензилбромида и затем 3,7 г (10 ммоль) йодида тетра-н-бутиламмония и нагревали всю ночь при 70 С. Затем смесь вливали в 100 мл воды и экстрагировали 2 раза, каждый раз 100 мл этилового эфира. Объединенные органические фазы промывали и высушивали. После упаривания и флэш-хроматографии на 300 г диоксида кремния (элюент: дихлорметан 100%, и затем дихлорметан/этилацетат 90/10) собирали ожидаемое соединение. Этап Д. транс-1-2-[(3,5-Дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-илпиперазин дигидрохлорид. Газообразный хлористый водород барботировали через 5,8 г (10 ммоль) вышеописанного соединения, растворенного в 400 мл этилацетата, при температуре окружающей среды, в течение нескольких минут. После этого перемешивали в течение 3 ч при температуре окружающей среды; затем смесь концентрировали в 2 раза (появлялся осадок) и добавляли 100 мл простого эфира; после этого перемешивали всю ночь при температуре окружающей среды. После отфильтровывания, промывания и высушивания осадка собирали ожидаемое соединение в рацемической форме. Точка плавления (MK): 110-125 С. Пример 16. (-)Изомер транс-1-2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-илпиперазин дигидрохлорид. После обратного превращения в основание рацемическое соединение, полученное в примере 15,разделяли путем препаративной хиральной ВЭЖХ на колонке CHIRALPAK AD (элюент: ацетонитрил/изопропанол/диэтиламин 900/100/1). Первый из изомеров, разделенных таким образом, превращали в гидрохлорид, используя этанольную HCl, и после этого осаждали из простого эфира. После фильтрования, промывания и высушивания получали (-)энантиомер в энантиомерном избытке, превышающем 98%. Точка плавления (MK): 113-126 С. Оптическое вращение: []=-14,5 (с=1%, МеОН, 20 С, =589 нм). Пример 17. (+)Изомер транс-1-2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-илпиперазин дигидрохлорид. Второй из изомеров, разделенных в примере 16, превращали в гидрохлорид, получая ожидаемый продукт. Точка плавления (MK): 118-126 С. Оптическое вращение: []=+14,4 (с=1%, МеОН, 20 С, =589 нм).- 12009648 Пример 18. транс-1-2-[(3,5-Дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-ил-1,4-диазепан дигидрохлорид. Этап А. трет-Бутил транс-4-[2-гидрокси-1,2,3,4-тетрагидронафт-1-ил]-1,4-диазепан-1-карбоксилат. При температуре окружающей среды 3,0 г (20,5 ммоль) соединения, полученного на этапе Б примера 15, и 4,1 г (20,5 ммоль) N-трет-бутилоксикарбонилгомопиперазина в 41 мл ацетонитрила смешивали вместе и после этого нагревали в колбе с обратным холодильником в течение 48 ч. После упаривания и флэш-хроматографии на 300 г диоксида кремния (элюент: дихлорметан/этанол 98/2) собирали ожидаемое соединение. Этап Б. трет-Бутил транс-4-2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-ил-1,4-диазепан-1-карбоксилат. При температуре окружающей среды к 0,76 г (19 ммоль) гидрида натрия в виде суспензии в 20 мл тетрагидрофурана добавляли в течение нескольких минут 6,0 г (17,3 ммоль) соединения, полученного на вышеописанном этапе, растворенного в 50 мл тетрагидрофурана. Смесь нагревали при 50 С в течение 2 ч, после этого одной порцией добавляли 5,7 г (17,3 ммоль) 3,5-дибромбензилбромида и затем 6,4 г (17,3 ммоль) йодида тетра-н-бутиламмония и нагревали всю ночь при 70 С. Затем смесь вливали в 200 мл воды и экстрагировали простым этиловым эфиром. Объединенные органические фазы промывали и высушивали. После упаривания и флэш-хроматографии на 500 г диоксида кремния (элюент: дихлорметан 100%, затем дихлорметан/этилацетат 95/5 и после этого дихлорметан/этанол 95/5) собирали ожидаемое соединение. Этап В. транс-1-2-[(3,5-Дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-ил)-1,4-диазепан дигидрохлорид. Газообразный хлористый водород осторожно пропускали через 4,9 г (8,2 ммоль) соединения, полученного на вышеописанном этапе, растворенного в 250 мл этилацетата, при температуре окружающей среды в течение нескольких минут. Перемешивали всю ночь при температуре окружающей среды; затем смесь концентрировали в 2 раза (появлялся осадок) и дополнительно перемешивали в течение 2 ч. После отфильтровывания, промывания и высушивания осадка собирали ожидаемое соединение в рацемической форме. Точка плавления (MK): 112-118 С. Пример 19. (-)Изомер транс-1-2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-ил-1,4-диазепан дигидрохлорид. После обратного превращения в основание рацемическое соединение, полученное в примере 18,разделяли путем препаративной хиральной ВЭЖХ на колонке CHIRALPAK AD (элюент: метанол/диэтиламин 1000/1). Первый из изомеров, разделенных таким образом, превращали в гидрохлорид, используя этанольную HCl. После фильтрования, промывания и высушивания получали (-)энантиомер в энантиомерном избытке, превышающем 98%. Точка плавления (MK): 122-126 С. Пример 20. (+)Изомер транс-1-2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-ил-1,4-диазепан дигидрохлорид. Второй из изомеров, разделенных в примере 19, превращали в гидрохлорид, получая ожидаемый продукт. Точка плавления (MK): 119-140 С. Пример 21. 1-(1S,2R)-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин дигидрохлорид. Этап А. (1S,2R)-1-4-[(4-Метилфенил)сульфонил]пиперазин-1-илиндан-2-ол. К 8 г (1S,2R)-1-аминоиндан-2-ола (53,6 ммоль), растворенного в 80 мл диметилформамида, добавляли 15 мл триэтиламина (107,2 ммоль) и 15,8 г N,N-бис-(2-хлорэтил)-n-толуолсульфонамида (53,6 ммоль). Смесь нагревали при 80 С в течение 24 ч и после этого диметилформамид упаривали. Остаток ресуспендировали в дихлорметане, промывали, высушивали, фильтровали и упаривали. Полученное масло очищали путем флэш-хроматографии на диоксиде кремния (элюент: дихлорметан/этилацетат 95/5), получая ожидаемый продукт в виде меренги. Этап Б. 1-(1S,2R)-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1 Н-инден-1-ил-4-[(4-метилфенил)сульфонил]пиперазин. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе Б примера 1, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Этап В. 1-(1S,2R)-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1 Н-инден-1-илпиперазин. 1,1 г соединения, полученного на вышеописанном этапе (1,77 ммоль), нагревали в течение 2 ч при 90 С в присутствии 10 мл раствора HBr/уксусной кислоты (33 %) и 808 мг 4-гидроксибензойной кислоты(5,85 ммоль). Смесь охлаждали и добавляли NaOH (20%) до установления рН 10; после этого экстрагировали дихлорметаном, высушивали, фильтровали и упаривали. Полученное масло очищали путем флэш-хроматографии на 100 г диоксида кремния (элюент: дихлорметан/этанол/аммиак 90/10/1), получая ожидаемый продукт в виде бесцветного масла. Этап Г. 1-(1S,2R)-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1 Н-инден-1-илпиперазин дигидрохлорид. К 1,2 г соединения, полученного на вышеописанном этапе (2,57 ммоль), растворенного в этилацетате, добавляли при температуре окружающей среды 2 М раствор соляной кислоты в эфире. Смесь переме- 13009648 шивали в течение 30 мин и после этого упаривали насухо. Остаток кристаллизовали из этилацетата,фильтровали и высушивали, получая ожидаемый продукт (цис-соединение) в виде белых кристаллов. Точка плавления (MK): 162-176 С. Оптическое вращение []=-2,9 (с=1%, МеОН, 20 С, =589 нм). Пример 22. 1-(1R,2S)-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной в примере 21, используя в качестве исходного соединения (1R,2S)-1-аминоиндан-2-ол. Точка плавления (MK): 142-150 С. Оптическое вращение: []=+5,8 (с=1%, МеОН, 20 С, =589 нм). Пример 23. цис-1-4-[(3,5-Дибромбензил)окси]-2,3,4,5-тетрагидро-1-бензоксепин-5-илпиперазин дигидрохлорид. Этап А. трет-Бутил 4-(4-оксо-2,3,4,5-тетрагидро-1-бензоксепин-5-ил)пиперазин-1-карбоксилат. При -78 С 0,88 мл оксалилхлорида (10 ммоль) вливали в 25 мл дихлорметана и после этого при этой же температуре медленно добавляли 1,02 мл диметилсульфоксида (14,3 ммоль). Перемешивали в течение 10 мин при -78 С. 2,5 г (7,17 ммоль) соединения, полученного на этапе Б примера 8, растворенного в 15 мл дихлорметана, вливали в реакционную смесь. Перемешивали в течение 15 мин при -78 С и после этого добавляли в течение 20 мин 5 мл (35,9 ммоль) триэтиламина. Затем смесь оставляли нагреться до 0 С и после этого реакционную смесь вливали в 100 мл ледяной воды. Экстрагировали дихлорметаном, высушивали, фильтровали и упаривали насухо, получая ожидаемый продукт в виде масла. Этап Б. трет-Бутил цис-4-(4-гидрокси-2,3,4,5-тетрагидро-1-бензоксепин-5-ил)пиперазин-1-карбоксилат. К 2,4 г (6,93 ммоль) соединения, полученного на вышеописанном этапе, растворенного в 50 мл метанола, добавляли, при 0 С и порциями, 130 мг NaBH4 (3,5 ммоль, 0,5 экв.). Перемешивали в течение 1 ч при 0 С и после этого метанол упаривали. Полученный остаток ресуспендировали в толуоле, промывали,высушивали, фильтровали и упаривали. Полученный остаток очищали путем флэш-хроматографии на диоксиде кремния (элюент: толуол/этанол 95/5), получая ожидаемый продукт в виде масла. Этап В. цис-1-4-[(3,5-Дибромбензил)окси]-2,3,4,5-тетрагидро-1-бензоксепин-5-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах Б и В примера 1,используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Точка плавления (MK): 166-191 С. Пример 24. цис-1-[2-3,5-Дибромбензилокси)-1,2,3,4-тетрагидронафт-1-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной в примере 23, используя в качестве исходного вещества соединение, полученное на этапе В примера 15. Точка плавления (MK): 155-159 С. Пример 25. цис-1-6-[(3,5-Дибромбензил)окси]-6,7,8,9-тетрагидро-5H-бензо[7]аннулен-5-илпиперазин дигидрохлорид. Этап А. трет-Бутил 4-(6-оксо-6,7,8,9-тетрагидро-5 Н-бензо[7]аннулен-5-ил)пиперазин-1-карбоксилат. К 4,4 г (12,6 ммоль) соединения, полученного на этапе А примера 7, растворенного в 44 мл диметилсульфоксида, добавляли при температуре окружающей среды 8,8 г стабилизированной 2-йодоксибензойной кислоты (SIBX) (56,3 ммоль, 4,4 экв.). Перемешивали в течение 2 ч при 25 С и затем смесь вливали в воду. Полученное нерастворимое вещество отфильтровали. Фильтрат экстрагировали этилацетатом, высушивали, фильтровали и упаривали насухо. Полученный остаток очищали путем флэш-хроматографии на диоксиде кремния (элюент: дихлорметан/этилацетат 95/5), получая ожидаемый продукт. Точка плавления (BK): 85-95 С. Этап Б. трет-Бутил 4-(6-гидрокси-6,7,8,9-тетрагидро-5 Н-бензо[7]аннулен-5-ил)пиперазин-1-карбоксилат. 1,5 г (4,3 ммоль) соединения, полученного на вышеописанном этапе, обрабатывали в соответствии со способом, описанным на этапе Б примера 23. Полученное желтое масло очищали путем флэш-хроматографии на диоксиде кремния (элюент: дихлорметан/этилацетат 95/5), получая ожидаемый продукт в виде цис/транс-смеси (соотношение цис/транс=80/20). Этап В. трет-Бутил цис-4-6-[(3,5-дибромбензил)окси]-6,7,8,9-тетрагидро-5 Н-бензо[7]аннулен-5 илпиперазин-1-карбоксилат. 650 мг соединения, полученного на вышеописанном этапе (цис/транс=80/20) (1,87 ммоль), обрабатывали в соответствии со способом, описанным на этапе Б примера 1, получая масло, которое очищали путем флэш-хроматографии на диоксиде кремния (элюент: циклогексан/этилацетат 95/5), получая ожидаемый продукт. Этап Г. цис-1-6-[(3,5-Дибромбензил)окси]-6,7,8,9-тетрагидро-5 Н-бензо[7]аннулен-5-илпиперазин дигидрохлорид. 350 мг соединения, полученного на вышеописанном этапе (0,58 ммоль), обрабатывали в соответствии со способом, описанным на этапе Б примера 1, и после этого полученный продукт кристаллизовали из изопропилового эфира, получая ожидаемый продукт в виде белых кристаллов.- 14009648 Точка плавления (MK): 149-158 С. Пример 26. транс-3-[(3,5-Дибромбензил)окси]-4-пиперазин-1-илхроман-6-карбонитрил дигидрохлорид. Этап А. 6-Бром-1 а,7b-дигидро-2 Н-оксирен[с]хромен. 8,5 г 6-бром-2H-хромена (40,3 ммоль), полученного в соответствии со способом синтеза, описанным в J. Org. Chem., 1998, 63, 864, обрабатывали в соответствии со способом, описанным на этапе А примера 8, получая ожидаемый продукт в виде масла. Этап Б. трет-Бутил транс-4-(6-бром-3-гидрокси-3,4-дигидро-2 Н-хромен-4-ил)пиперазин-1-карбоксилат. 7,6 г соединения, полученного на вышеописанном этапе (33,5 ммоль), обрабатывали в соответствии со способом, описанным на этапе А примера 1. Полученный остаток очищали путем флэш-хроматографии на диоксиде кремния (элюент: толуол/этанол 95/5), получая ожидаемый продукт в виде масла. Этап В. трет-Бутил транс-4-(6-циано-3-гидрокси-3,4-дигидро-2 Н-хромен-4-ил)пиперазин-1-карбоксилат. 1 г соединения, полученного на вышеописанном этапе (2,4 ммоль), растворяли в 10 мл диметилформамида. Раствор дегазировали с помощью аргона и после этого добавляли 112 мг тетракис(трифенилфосфин)палладия(0) (0,09 ммоль) и 170 мг цианида цинка (1,4 ммоль). Нагревали при 80 С в течение 3 дней. После этого реакционную смесь охлаждали и вливали в воду. Смесь экстрагировали этилацетатом, промывали водой, высушивали, фильтровали и упаривали насухо. Полученный остаток очищали путем флэш-хроматографии на диоксиде кремния (элюент: дихлорметан/этилацетат 90/10), получая ожидаемый продукт в виде масла. Этап Г. транс-3-[(3,5-Дибромбензил)окси]-4-пиперазин-1-илхроман-6-карбонитрил дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах Б и В примера 1,используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Точка плавления (MK): 185-200 С. Пример 27. транс-7-[(3,5-Дибромбензил)окси]-8-пиперазин-1-ил-5,6,7,8-тетрагидронафталин-2-карбонитрил дигидрохлорид. Этап А. 6-Бром-1 а,2,3,7b-тетрагидронафто[1,2-b]-оксирен. 7,4 г соединения из получения А (35 ммоль) подвергали реакции с Oxone в соответствии со способом, описанным на этапе А примера 8, получая ожидаемый продукт в виде масла. Этап Б. трет-Бутил транс-4-(7-бром-2-гидрокси-1,2,3,4-тетрагидронафт-1-ил)пиперазин-1-карбоксилат. 6,5 г трет-бутил пиперазин-1-карбоксилата (35 ммоль) и 7,9 г соединения, полученного на вышеописанном этапе (35 ммоль), растворяли в 45 мл диметилформамида и после этого реакционную смесь нагревали при 110 С в течение 24 ч. После упаривания насухо получали остаток, который очищали путем флэш-хроматографии на диоксиде кремния (элюент: толуол/этанол 98/2), получая ожидаемый продукт в виде масла. Этап В. трет-Бутил транс-4-(7-циано-2-гидрокси-1,2,3,4-тетрагидронафт-1-ил)пиперазин-1-карбоксилат. 4 г соединения, полученного на вышеописанном этапе (9,7 ммоль), обрабатывали в соответствии со способом, описанным на этапе Б примера 26. Нагревали при 80 С в течение 2 ч. После этого реакционную смесь охлаждали и вливали в воду. Смесь экстрагировали дихлорметаном, промывали водой, высушивали, фильтровали и упаривали насухо. Полученный остаток очищали путем флэш-хроматографии на диоксиде кремния (элюент: дихлорметан/этанол 98/2), получая ожидаемый продукт в виде масла. Этап Г. транс-7-[(3,5-Дибромбензил)окси]-8-пиперазин-1-ил-5,6,7,8-тетрагидронафталин-2-карбонитрил дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах Б и В примера 1,используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Точка плавления (MK): 169-191 С. Пример 28. транс-4-[(3,5-Дибромбензил)окси]-5-пиперазин-1-ил-2,3,4,5-тетрагидро-1-бензоксепин 7-карбонитрил дигидрохлорид. Этап А. 7-Бром-1 а,2,3,8b-тетрагидрооксирен[d][1]бензоксепин. 4 г соединения из получения В подвергали реакции с Oxone в соответствии со способом, описанным на этапе А примера 8, получая ожидаемый продукт в виде масла. Этап Б. трет-Бутил транс-4-(7-бром-4-гидрокси-2,3,4,5-тетрагидро-1-бензоксепин-5-ил)пиперазин 1-карбоксилат. 3,9 г соединения, полученного на вышеописанном этапе (16,2 ммоль), обрабатывали в соответствии со способом, описанным на этапе А примера 1. Продолжали нагревать дополнительно в течение 3 дней. Таким образом получали остаток, который очищали путем флэш-хроматографии на диоксиде кремния(элюент: дихлорметан/этанол 98/2), получая ожидаемый продукт в виде масла. Этап В. трет-Бутил транс-4-(7-циано-4-гидрокси-2,3,4,5-тетрагидро-1-бензоксепин-5-ил)пиперазин 1-карбоксилат. 5,8 г соединения, полученного на вышеописанном этапе (13,6 ммоль), обрабатывали в соответствии со способом, описанным на этапе Б примера 26. Нагревали при 80 С в течение 20 ч. После этого реакционную смесь охлаждали и вливали в воду. Смесь экстрагировали дихлорметаном, промывали водой, вы- 15009648 сушивали, фильтровали и упаривали насухо. Полученный остаток очищали путем флэш-хроматографии на диоксиде кремния (элюент: толуол/этанол 95/5), получая ожидаемый продукт в виде масла. Этап Г. транс-4-[(3,5-Дибромбензил)окси]-5-пиперазин-1-ил-2,3,4,5-тетрагидро-1-бензоксепин-7 карбонитрил дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах Б и В примера 1,используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Точка плавления (MK): 163-195 С. Пример 29. транс-5-[(4-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин-1-ил) метил]-2,4-дигидро-3H-1,2,4-триазол-3-он дигидрохлорид. Этап А. транс-5-[(4-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин-1-ил)метил]2,4-дигидро-3 Н-1,2,4-триазол-3-он. После обратного превращения в основание соединение, полученное на этапе В примера 1 (2,14 ммоль),растворяли в 30 мл диметилформамида. Добавляли 0,75 мл (4,28 ммоль) диизопропилэтиламина и 314 мг(2,35 ммоль) 5-(хлорметил)-2,4-дигидро-3H-1,2,4-триазол-3-она, полученного согласно способу, описанному в Tetrahedron Letters, 2000, 41, 8661. Смесь перемешивали в течение 12 ч при температуре окружающей среды и после этого упаривали насухо. Полученный остаток ресуспендировали в дихлорметане,промывали, высушивали, фильтровали и упаривали. Полученное масло очищали путем флэш-хроматографии на диоксиде кремния, получая ожидаемый продукт. Этап Б. транс-5-[(4-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1 Н-инден-1-илпиперазин-1-ил)метил]2,4-дигидро-3 Н-1,2,4-триазол-3-он дигидрохлорид. 800 мг соединения, полученного на вышеописанном этапе (1,42 ммоль), растворяли в 50 мл этилацетата. Затем добавляли 2,8 мл (5,68 ммоль) 2 М раствора соляной кислоты в эфире. После перемешивания в течение 30 мин при температуре окружающей среды смесь упаривали насухо и остаток кристаллизовали из ацетонитрила, получая ожидаемый продукт. Точка плавления (MK): 198-202 С. Пример 30. транс-1-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1H-инден-1-ил-4-[(2S)-2,3-дигидро 1,4-бенздиоксин-2-илметил]пиперазин дигидрохлорид. Этап А. трет-Бутил 4-[(2R)-2,3-дигидро-1,4-бенздиоксин-2-илкарбонил]пиперазин-1-карбоксилат. 5 г (2R)-2,3-дигидро-1,4-бенздиоксин-2-карбоновой кислоты (27,8 ммоль) растворяли в 300 мл ацетонитрила. Добавляли 6 г N,N'-дициклогексилкарбодиимида (29,1 ммоль) и затем 4,1 г 1-гидроксибензотриазола (30,6 ммоль). В завершение, добавляли 6,2 г трет-бутил пиперазин-1-карбоксилата (33,4 ммоль) и перемешивали в течение 12 ч при температуре окружающей среды. Затем реакционную смесь фильтровали и после этого фильтрат упаривали насухо. Полученное таким образом масло очищали путем флэш-хроматографии на диоксиде кремния (элюент: толуол/этилацетат 80/20), получая ожидаемый продукт в виде масла. Этап Б. трет-Бутил 4-[(2S)-2,3-дигидро-1,4-бенздиоксин-2-илметил]пиперазин-1-карбоксилат. 500 мг соединения, полученного на вышеописанном этапе (1,44 ммоль), растворяли в 5 мл безводного тетрагидрофурана. После этого по каплям добавляли при температуре окружающей среды 4,4 мл(4,3 ммоль) 1 М раствора борана в тетрагидрофуране. Смесь нагревали в колбе с обратным холодильником в течение 2 ч, охлаждали, медленно гидролизовали, используя 5 мл этанола, и упаривали насухо. Полученный остаток кристаллизовали из воды, фильтровали и высушивали, получая ожидаемый продукт. Точка плавления (BK)=97-99 С. Этап В. 1-[(2S)-2,3-Дигидро-1,4-бенздиоксин-2-илметил]пиперазин дигидрохлорид. 7,5 г соединения, полученного на вышеописанном этапе (22,4 ммоль), перемешивали в течение 2 дней при температуре окружающей среды в присутствии 100 мл 2,6 н. раствора соляной кислоты в этаноле. Образованные кристаллы отфильтровали и высушивали, получая ожидаемый продукт в виде белого твердого вещества. Точка плавления (BK)=166-172 С. Этап Г. транс 1-4-[(2S)-2,3-Дигидро-1,4-бенздиоксин-2-илметил]пиперазин-1-илиндан-2-ол. К 1,6 г (6,8 ммоль) 1-[(25)-2,3-дигидро-1,4-бенздиоксин-2-илметил]пиперазина, полученного путем обратного превращения соединения с вышеописанной стадии в основание, растворенного в 10 мл ацетонитрила, добавляли 1,08 г оксида индена (8,16 ммоль). Реакционную смесь нагревали при 80 С в течение 30 ч и после этого упаривали насухо. Полученное таким образом масло очищали путем флэш-хроматографии на диоксиде кремния (элюент: дихлорметан/этанол/аммиак 90/10/1), получая ожидаемое соединение в виде меренги. Этап Д. транс-1-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1 Н-инден-1-ил-4-[(2S)-2,3-дигидро-1,4 бенздиоксин-2-илметил]пиперазин. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе Б примера 1, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Этап Е. транс-1-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1 Н-инден-1-ил-4-[(2S)-2,3-дигидро-1,4 бенздиоксин-2-илметил]пиперазин дигидрохлорид. К 210 мг соединения, полученного на вышеописанном этапе (0,34 ммоль), растворенного в 20 мл- 16009648 этилацетата, добавляли при температуре окружающей среды 0,5 мл (1 ммоль) 2 М раствора соляной кислоты в эфире. После перемешивания в течение 30 мин реакционную смесь упаривали насухо. Полученный остаток кристаллизовали из этилацетата, фильтровали и высушивали, получая ожидаемый продукт в виде белых кристаллов. Точка плавления (MK): 137-145 С. Пример 31. (+)-цис-1-(1S,2R)-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1H-инден-1-ил-4-(пиридин 2-илкарбонил)пиперазин дигидрохлорид. Этап А. цис-1-(1S,2R)-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1 Н-инден-1-ил-4-(пиридин-2-илкарбонил)пиперазин. К 263 мг пиридин-2-карбоновой кислоты (2,14 ммоль), растворенной в 10 мл тетрагидрофурана, добавляли 382 мг карбонилдиимидазола (2,35 ммоль, 1,1 экв.) и затем, после перемешивания в течение 3 ч при температуре окружающей среды, раствор 1 г (2,14 ммоль) соединения, полученного на этапе В примера 21, в 20 мл тетрагидрофурана. Перемешивали в течение 12 ч при 25 С и после этого добавляли воду, экстрагировали дихлорметаном, высушивали, фильтровали и упаривали насухо. Полученное масло очищали путем флэш-хроматографии на диоксиде кремния (элюент: дихлорметан/этанол 95/5), получая ожидаемый продукт в виде масла. Этап Б. цис-1-(1S,2R)-2-[(3,5-Дибромбензил)окси]-2,3-дигидро-1 Н-инден-1-ил-4-(пиридин-2-илкарбонил)пиперазин дигидрохлорид. 900 мг соединения, полученного на вышеописанном этапе (1,57 ммоль), обрабатывали в соответствии со способом, описанным на этапе Е примера 30. Полученное твердое вещество отфильтровали и высушивали, получая ожидаемый продукт в виде белых кристаллов. Точка плавления (MK): 108-122 С. Оптическое вращение: []=+27,3 (с=1%, метанол, 20 С, =589 нм). Пример 32. 1-(транс-2-1-[3,5-бис-(трифторметил)фенил]этокси-1,2,3,4-тетрагидронафт-1-ил)пиперазин дигидрохлорид, диастереоизомер 1. Этап А. трет-Бутил 4-(транс-2-[3,5-бис-(трифторметил)бензоил]окси-1,2,3,4-тетрагидронафт-1 ил)пиперазин-1-карбоксилат. К 11,7 г (35,2 ммоль) соединения, полученного на этапе В примера 15, и 5,9 мл (42,2 ммоль) триэтиламина, растворенного в 300 мл дихлорметана, добавляли при температуре окружающей среды 7,05 мл(38,7 ммоль) бис-(трифторметил)бензоилхлорида в 50 мл дихлорметана, по каплям в течение 1 ч 10 мин,и после этого 0,5 г диметиламинопиридина. После этого реакционную смесь нагревали в колбе с обратным холодильником в течение 5 дней, и после этого дополнительно добавляли 1,3 мл хлорангидрида кислоты, и продолжали нагревать в колбе с обратным холодильником в течение 20 ч. После упаривания среды полученный остаток фильтровали через 200 г диоксида кремния (элюент: дихлорметан), получая ожидаемый продукт. Этап Б. трет-Бутил 4-[транс-2-(1-[3,5-бис-(трифторметил)фенил]винилокси)-1,2,3,4-тетрагидронафт-1-ил]пиперазин-1-карбоксилат. К 13,2 г (23 ммоль) соединения, полученного на вышеописанном этапе, в 92 мл тетрагидрофурана добавляли при температуре окружающей среды 46 мл 1 М раствора дициклопентадиенилдиметилтитана в толуоле и реакционную смесь нагревали при 85 С в течение 20 ч. Дополнительно по каплям добавляли 23 мл 1 М раствора дициклопентадиенилдиметилтитана в толуоле при 85 С в течение 10 мин и после этого продолжали нагревать при этой температуре дополнительно в течение 24 ч. Смесь оставляли охладиться, добавляли 500 мл пентана; смесь фильтровали через целит и промывали пентаном до обесцвечивания фильтрата. После упаривания всех объединенных фильтратов и флэш-хроматографии на 800 г диоксида кремния (элюент: дихлорметан и затем дихлорметан/этилацетат 98/2) получали ожидаемый продукт. Этап В. трет-Бутил 4-(транс-2-1-[3,5-бис-(трифторметил)фенил]этокси-1,2,3,4-тетрагидронафт-1 ил)пиперазин-1-карбоксилат. 8,34 г (14,6 ммоль) соединения, полученного на вышеописанном этапе, растворенного в 150 мл этанола, гидрировали при температуре окружающей среды и атмосферном давлении в течение 7 ч в присутствии 1 г 5% палладия на угле. После фильтрования через целит, промывания этанолом и упаривания получали ожидаемый продукт в виде смеси диастереоизомеров, которая не поддается количественному определению и разделению. Этап Г. 1-(транс-2-1-[3,5-бис-(трифторметил)фенил]этокси-1,2,3,4-тетрагидронафт-1-ил)пиперазин дигидрохлорид, диастереоизомер 1. Газообразный хлористый водород осторожно пропускали через 8,4 г (14,6 ммоль) соединения, полученного на вышеописанном этапе, растворенного в 500 мл этилацетата, при температуре окружающей среды в течение нескольких минут. Перемешивали всю ночь при температуре окружающей среды и после этого упаривали насухо. Остаток растворяли в 100 мл воды и, при интенсивном перемешивании, превращали в основание, используя 8 г карбоната натрия. Пасту, которая выпала в осадок, 2 раза экстрагировали каждый раз с помощью 100 мл дихлорметана. Объединенные органические фазы высушивали и концентрировали и полученный остаток хроматографировали на 700 г диоксида кремния (элюент: дихлорметан/этанол/аммиак 95/5/0,5), получая первый диазастереоизомер, который превращали в дигидро- 17009648 хлорид путем реакции с эфирной HCl. После кристаллизации из пентана получали ожидаемый продукт. Точка плавления (MK): 98-102 С. Пример 33. 1-(транс-2-1-[3,5-бис-(Трифторметил)фенил]этокси-1,2,3,4-тетрагидронафт-1-ил)пиперазин дигидрохлорид, диастереоизомер 2. Второй продукт, элюируемый на этапе Г примера 32, представляет собой второй диастереоизомер,который превращали в дигидрохлорид. Точка плавления (MK): 98-101 С. Пример 34. 1-[(1S,2R)-2-Бензилокси-2,3-дигидро-1H-инден-1-ил]пиперазин дигидрохлорид. Ожидаемый продукт (цис-соединение) получали в соответствии с методикой из примера 21, заменяя 3,5-дибромбензилбромид на этапе Б бензилбромидом. Точка плавления: 104-125 С. Пример 35. транс-1-2-[(3,5-Диметилбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах Г и Д примера 15,используя в качестве исходного вещества соединение, полученное на этапе А примера 1, и 3,5-диметилбензилбромид. Точка плавления: 161-70 С. Пример 36. 1-[(1S,2R)-2-[(3,5-Дифторбензил)окси]-2,3-дигидро-1H-инден-1-ил]пиперазин дигидрохлорид. Ожидаемый продукт (цис-соединение) получали в соответствии с методикой из примера 21, заменяя 3,5-дибромбензилбромид на этапе Б 3,5-дифторбензилбромидом. Точка плавления: 160-190 С. Пример 37. 1-[(1S,2R)-2-Бензилокси-2,3-дигидро-1H-инден-1-ил]-4-метилпиперазин дигидрохлорид. Ожидаемый продукт получали путем N-метилирования соединения из примера 34. Точка плавления: 182-189 С. Пример 38. 1-[(1S,2R)-2-[(3,5-Диметилбензил)окси]-2,3-дигидро-1H-инден-1-ил]пиперазин дигидрохлорид. Этап А. 1-(1S,2R)-2-[(3,5-Диметилбензил)окси]-2,3-дигидро-1 Н-инден-1-ил-4-[(4-метилфенил)сульфонил]пиперазин. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах А и Б примера 21,заменяя 3,5-дибромбензилбромид на этапе Б 3,5-диметилбензилбромидом. Этап Б. 1-(1S,2R)-2-[(3,5-Диметилбензил)окси]-2,3-дигидро-1 Н-инден-1-илпиперазин. 3 г натрия (130 ммоль) добавляли к раствору 3,6 г нафталина (28 ммоль) в 30 мл 1,2-диметоксиэтана. Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч до образования раствора натрий/нафталин/1,2-диметоксиэтан. 14,8 мл (64 ммоль) полученного раствора добавляли при температуре -70 С к раствору 3 г (6,1 ммоль) соединения, полученного на вышеописанном этапе, в 55 мл 1,2-диметоксиэтана. Цвет раствора изменялся с белого на синий. Перемешивали в течение 30 мин при -70 С, затем гидролизовали, используя 100 мл воды. Затем смесь экстрагировали этилацетатом, высушивали, фильтровали и упаривали насухо. Полученный остаток очищали путем флэш-хроматографии на 200 г диоксида кремния (элюент: дихлорметан/этанол/аммиак 90/10/1), получая ожидаемый продукт в виде масла. Этап В. 1-(1S,2R)-2-[(3,5-Диметилбензил)окси]-2,3-дигидро-1 Н-инден-1-илпиперазин дигидрохлорид. 3,6 мл (7,16 ммоль, 2 экв.) 2 М раствора эфирного хлористого водорода добавляли к 1,2 г (3,58 ммоль) соединения, полученного на вышеописанном этапе, в 50 мл ацетонитрила. Кристаллизацию осуществляли путем царапанья и после этого перемешивали в течение 15 мин при температуре окружающей среды. Полученные белые кристаллы высушивали, получая ожидаемый продукт. Точка плавления: 170-193 С. Пример 39. транс-1-2-[(3,5-Дифторбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 1, заменяя 3,5-дибромбензилбромид на этапе Б 3,5-дифторбензилбромидом. Точка плавления: 185-198 С. Пример 40. транс-1-(2-[3,5-бис-(трифторметил)бензил]окси-2,3-дигидро-1H-инден-1-ил)пиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 1, заменяя 3,5-дибромбензилбромид на этапе Б 3,5-бис-(трифторметил)бензилбромидом. Точка плавления: 140-160 С. Пример 41. транс-1-2-[(3,5-Дибромбензил)окси]-6-метокси-2,3-дигидро-1H-инден-1-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 2H-хромен на этапе А 5-метокси-1H-инденом. Точка плавления: 184-195 С. Пример 42. транс-1-2-[(3,5-Дихлорбензил)окси]-6-метокси-2,3-дигидро-1H-инден-1-илпиперазин- 18009648 дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, используя в качестве исходного соединения 5-метокси-1H-инден и 3,5-дихлорбензилбромид. Точка плавления: 169-176 С. Пример 43. транс-1-2-[(3,5-Дихлорбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 1, заменяя 3,5-дибромбензилбромид на этапе Б 3,5-дихлорбензилбромидом. Точка плавления: 115-127 С. Пример 44. транс-1-3-[(3,5-Дихлорбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 3,5-дибромбензилбромид на этапе Г 3,5-дихлорбензилбромидом. Точка плавления: 110-118 С. Пример 45. транс-1-2-[(3,5-бис-(Трифторметил)бензил)окси]-1,2,3,4-тетрагидронафт-1-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 15, заменяя 3,5-дибромбензилбромид на этапе Г 3,5-бис-(трифторметил)бензилбромидом. Точка плавления (MK): 98-101 С. Пример 46. транс-1-3-[3-Фтор-5-(трифторметил)бензилокси]-3,4-дигидро-2H-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 3,5-дибромбензилбромид на этапе Г 3-фтор-5-(трифторметил)бензилбромидом. Точка плавления: 102-113 С. Пример 47. транс-1-3-(3-Хлор-5-фторбензилокси)-3,4-дигидро-2 Н-хромен-4-илпиперазин дигидрохлорид. Этап А. трет-Бутил транс-4-3-[(3-хлор-5-фторбензил)окси]-3,4-дигидро-2 Н-хромен-4-илпиперазин 1-карбоксилат. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах А-Г примера 4,заменяя 3,5-дибромбензилбромид на этапе Г 3-хлор-5-фторбензилбромидом. Этап Б. транс-1-3-(3-Хлор-5-фторбензилокси)-3,4-дигидро-2 Н-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе В примера 1, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Точка плавления: 90-95 С. Пример 47 бис. транс-1-3-(3-Хлор-5-фторбензилокси)-3,4-дигидро-2 Н-хромен-4-илпиперазин диметансульфонат. Ожидаемый продукт получали путем взаимодействия соединения из примера 47 с гидроксидом натрия, затем путем превращения продукта, полученного таким образом, в соль с применением метансульфоновой кислоты. Точка плавления: 161-171 С. Пример 48. цис-4-[2-(3,5-Дибромбензилокси)-1,2,3,4-тетрагидронафт-1-ил]морфолин гидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 23, заменяя на этапе А соединение, полученное на этапе Б примера 8, соединением, полученным на этапе Б примера 10. Точка плавления: 195-198 С. Пример 49. транс-4-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2H-хромен-4-илморфолин гидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах Б-Г примера 10,используя в качестве исходного вещества соединение, полученное на этапе Б примера 4. Точка плавления: 143-148 С. Пример 50. транс-4-2-[(3,5-Дибромбензил)окси]-1,2,3,4-тетрагидро-1-нафтилпиперидин гидрохлорид. Этап А. 1-(4-Пиридил)-1,2,3,4-тетрагидро-1-нафтол. 85 мл 1,5 М раствора н-бутиллития в гексане добавляли по каплям к раствору 20 г 4-бромпиридина в 73 мл простого эфира, охлаждали до -78 С. Перемешивали в течение 30 мин при этой температуре, затем вливали в раствор 1-тетралона в 73 мл простого эфира и после окончания добавления смесь оставляли нагреться до температуры окружающей среды. После перемешивания в течение ночи вливали в насыщенный водный раствор хлорида аммония. После отделения экстрагировали простым эфиром. Эфирные фазы объединяли и экстрагировали 1 н. соляной кислотой. Значение рН объединенных кислых фаз устанавливали равным 8, используя 20%-ный раствор гидроксида натрия, и экстрагировали дихлорметаном. После высушивания выделяли ожидаемый продукт, который очищали путем высокоскоростной хроматографии на диоксиде кремния (элюент: дихлорметан/метанол 95/5), получая ожидаемый продукт. Точка плавления (BK)=160-162 С. Этап Б. 4-(3,4-Дигидро-1-нафтил)пиридин. 2 г соединения, полученного на вышеописанном этапе, 10 мл воды и 10 мл 95%-ной серной кислоты смешивали вместе, нагревали при 80 С в течение 30 мин, охлаждали до 0 С и значение рН доводили до- 19009648 10, использовали 20%-ный раствор гидроксида натрия. Смесь экстрагировали дихлорметаном, промывали водой, высушивали и упаривали, получая ожидаемый продукт. Этап В. 4-(2,3-Дигидронафто[1,2-b]оксирен-7b(1 аН)-ил)пиридин 1-оксид. При температуре от 20 до 25 С раствор, содержащий 5,4 г продукта, полученного на вышеописанной стадии, 20 г бикарбоната натрия, 35 мл ацетона, 20 мл воды и 200 мл этилацетата вливали в раствор 29,3 г Oxone в 200 мл воды. Перемешивали всю ночь при температуре окружающей среды, разводили водой и экстрагировали этилацетатом. После обычной обработки выделяли ожидаемый продукт. Этап Г. 1-(1-Оксидо-4-пиридил)-1,2,3,4-тетрагидро-2-нафтол. К раствору 450 мг продукта, полученного на вышеописанной стадии, в 10 мл безводного тетрагидрофурана, в присутствии следов бромкрезола зеленого, одной порцией при температуре окружающей среды добавляли 289 мг цианоборогидрида натрия. Добавляли трифторэтерат бора до изменения цвета индикатора на желтый и столько раз, сколько это было необходимо при осуществлении реакции для поддержания значения рН при 4-5. После окончания реакции добавляли концентрированную соляную кислоту до установления рН равным 1 и перемешивали в течение 30 мин при температуре окружающей среды. Значение рН смеси доводили до 8 с помощью раствора гидроксида натрия и экстрагировали этилацетатом и после обработки выделяли ожидаемый продукт (80% транс, 20% цис). Этап Д. 1-(4-Пиперидил)-1,2,3,4-тетрагидро-2-нафтол. 1,78 г продукта, полученного на вышеописанной стадии, 1 г оксида платины, 0,75 мл концентрированной соляной кислоты и 75 мл этанола смешивали в реакторе и гидрировали под давлением 1 бар. После взаимодействия в течение 6 ч при температуре окружающей среды смесь фильтровали; добавляли 8 мл раствора гидроксида натрия, этанол упаривали, для растворения использовали небольшое количество воды и значение рН доводили до 10. После экстрагирования и обычной обработки получали ожидаемый продукт (80% транс, 20% цис). Этап Е. трет-Бутил 4-(2-гидрокси-1,2,3,4-тетрагидро-1-нафтил)-1-пиперидинкарбоксилат. Раствор 2,1 г ди(трет-бутил)дикарбоната в 50 мл дихлорметана добавляли к 2,06 г продукта, полученного на вышеописанной стадии, растворенного в 50 мл дихлорметана. Перемешивали в течение 2 ч при температуре окружающей среды, затем упариванили насухо. После очистки на диоксиде кремния(элюент: дихлорметан/метанол 95/5) выделяли ожидаемый продукт (80% транс, 20% цис). Этап Ж. трет-Бутил 4-2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидро-1-нафтил-1-пиперидинкарбоксилат. 241 мг гидрида натрия (60% в масле) вносили в раствор 1,9 г соединения, полученного на вышеописанном этапе, в 20 мл безводного тетрагидрофурана, охлажденного до 0 С. Перемешивали в течение 15 мин и после этого добавляли, все еще при этой температуре, 20 мг йодида тетрабутиламмония и в завершение 1,9 г 3,5-дибромбензилбромида. Смесь оставляли нагреться до температуры окружающей среды и перемешивали в течение 24 ч. Смесь упаривали насухо, ресуспендировали в воде и дихлорметане и после обычной обработки и хроматографирования на диоксиде кремния (элюент: дихлорметан) выделяли ожидаемый продукт в виде белой меренги (80% транс, 20% цис). Этап 3. транс-4-2-[(3,5-Дибромбензил)окси]-1,2,3,4-тетрагидро-1-нафтилпиперидин гидрохлорид. 2 г соединения, полученного на вышеописанном этапе, в 20 мл этанола, подвергали реакции с 19 мл 3,6 н. раствора этанольного хлористого водорода. Через 24 ч образованный осадок отфильтровали, промывали и высушивали, получая ожидаемый продукт в виде гидрохлорида (цис-соединение находится в фильтрате). Точка плавления: 152-167 С. Пример 51. цис-1-Ацетил-3-[(3,5-дибромбензил)окси]-4-(1-пиперазинил)-1,2,3,4-тетрагидрохинолин дигидрохлорид. Этап А. 1-Ацетил-1,2-дигидрохинолин. 23,43 г борогидрида натрия порциями вносили в раствор 20 г хинолина в 200 мл уксусной кислоты и 77,5 мл уксусного ангидрида, охлажденного до 0 С. Затем смесь нагревали в течение 2 ч при 60 С и перемешивали всю ночь при температуре окружающей среды. Смесь концентрировали, разводили водой,рН доводили до 10 с помощью раствора гидроксида натрия и экстрагировали простым эфиром. Объединенные эфирные фазы промывали 1 н. соляной кислотой и после этого при нейтральном рН и после обычной обработки выделяли ожидаемый продукт. Этап Б. 3-Ацетил-1 а,2,3,7b-тетрагидрооксирен[2,3-с]хинолин. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе А примера 8, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Этап В. трет-Бутил транс-4-[1-ацетил-3-гидрокси-1,2,3,4-тетрагидро-4-хинолил]-1-пиперазинкарбоксилат. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе А примера 1, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Этап Г. цис-1-Ацетил-3-[(3,5-дибромбензил)окси]-4-(1-пиперазинил)-1,2,3,4-тетрагидрохинолин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 23, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии.- 20009648 Точка плавления: 164-167 С. Пример 52. транс-1-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2H-хромен-4-ил-N-метил-4-пиперидинамин дигидрохлорид. Этап А. трет-Бутил транс-1-3-[(3,5-дибромбензил)окси]-3,4-дигидро-2 Н-хромен-4-ил-N-метил-4 пиперидилкарбамат. 0,2 г гидрида натрия (60% в масле) добавляли к 1 г соединения, полученного на этапе Б примера 9,и 0,42 мл метилйодида в 10 мл тетрагидрофурана, поддерживая температуру реакционной смеси при 0 С. После осуществления взаимодействия в течение 15 мин при этой температуре смесь перемешивали в течение 48 ч при температуре окружающей среды, разводили водой и экстрагировали этилацетатом. После обычной обработки выделяли ожидаемый продукт. Этап Б. транс-1-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2 Н-хромен-4-ил-N-метил-4-пиперидинамин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе В примера 1, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Точка плавления: 192-195 С. Пример 53. транс-1-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2H-хромен-4-ил-N,N-диметил-4-пиперидинамин дигидрохлорид. Этап А. транс-1-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2 Н-хромен-4-ил-N-метил-4-пиперидинамин. Ожидаемый продукт получали путем обратного превращения соединения из примера 52 в основание. Этап Б. транс-1-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2 Н-хромен-4-ил-N,N-диметил-4-пиперидинамин. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе А примера 52, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Этап В. транс-1-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2 Н-хромен-4-ил-N,N-диметил-4-пиперидинамин дигидрохлорид. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанном этапе, в соль с помощью соляной кислоты. Точка плавления: 187-190 С. Пример 54. транс-1-3-[(3,5-Диметоксибензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 3,5-дибромбензилбромид на этапе Г 3,5-диметоксибензилбромидом. Точка плавления: 108-115 С. Пример 55. транс-1-3-Бензилокси-3,4-дигидро-2H-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 3,5-дибромбензилбромид на этапе Г бензилбромидом. Точка плавления: 66-80 С. Пример 56. транс-3-[(3-Фторбензил)окси]-3,4-дигидро-2H-хромен-4-ил)пиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 3,5-дибромбензилбромид на этапе Г 3-фторбензилбромидом. Точка плавления: 180-184 С. Пример 57. транс-1-3-[(3-Хлорбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 3,5-дибромбензилбромид на этапе Г 3-хлорбензилбромидом. Точка плавления: 97-107 С. Пример 58. транс-1-3-[(3,4-Дихлорбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 3,5-дибромбензилбромид на этапе Г 3,4-дихлорбензилбромидом. Точка плавления: 114-121 С. Пример 59. транс-1-2-[(3-Хлор-5-фторбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин диметансульфонат. Этап А. транс-1-2-[(3-Хлор-5-фторбензил)окси]-2,3-дигидро-1 Н-инден-1-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапах Б и В примера 1,заменяя 3,5-дибромбензилбромид на этапе Б 3-хлор-5-фторбензилбромидом. Этап Б. транс-1-2-[(3-Хлор-5-фторбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазин диметансульфонат. Ожидаемый продукт получали путем обратного превращения соединения, полученного на вышеописанной стадии, в основание и последующего превращения в соль с помощью метансульфоновой кислоты. Точка плавления: 175-182 С. Пример 60. транс-1-3-[(3-(Трифторметил)бензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин диметансульфонат.- 21009648 Этап А. транс-1-3-[(3-(Трифторметил)бензил)окси]-3,4-дигидро-2 Н-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 3,5-дибромбензилбромид на этапе Г 3-(трифторметил)бензилбромидом. Этап Б. транс-1-3-[(3-(Трифторметил)бензил)окси]-3,4-дигидро-2 Н-хромен-4-илпиперазин диметансульфонат. Ожидаемый продукт получали путем обратного превращения соединения, полученного на вышеописанной стадии, в основание и последующего превращения в соль с помощью метансульфоновой кислоты. Точка плавления: 123-127 С. Пример 61. транс-1-3-[(3-Цианобензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин диметансульфонат. Этап А. транс-1-3-[(3-Цианобензил)окси]-3,4-дигидро-2 Н-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 3,5-дибромбензилбромид на этапе Г 3-цианобензилбромидом. Этап Б. транс-1-3-[(3-Цианобензил)окси]-3,4-дигидро-2 Н-хромен-4-илпиперазин диметансульфонат. Ожидаемый продукт получали путем обратного превращения соединения, полученного на вышеописанной стадии, в основание и последующего превращения в соль с помощью метансульфоновой кислоты. Точка плавления: 118-121 С. Пример 62.(+)Изомер транс-1-3-(3-хлор-5-фторбензилокси)-3,4-дигидро-2 Н-хромен-4 илпиперазин дибензоилтартрат (+). Этап А. (+)Изомер трет-бутил транс-4-3-[(3-хлор-5-фторбензил)окси]-3,4-дигидро-2 Н-хромен-4 илпиперазин-1-карбоксилат. Ожидаемое соединение получали путем разделения рацемической смеси, полученной на этапе А примера 47, с помощью препаративной хиральной ВЭЖХ. Этап Б. (+)Изомер транс-1-3-(3-хлор-5-фторбензилокси)-3,4-дигидро-2 Н-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе В примера 1, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Этап В. (+)Изомер транс-1-3-(3-хлор-5-фторбензилокси)-3,4-дигидро-2 Н-хромен-4-илпиперазин дибензоилтартрат (+). Ожидаемый продукт получали путем обратного превращения соединения, полученного на вышеописанной стадии, в основание и последующего превращения в соль с помощью (+)-дибензоилвинной кислоты. Точка плавления: 100-107 С. Пример 63. (-)Изомер транс-1-3-(3-хлор-5-фторбензилокси)-3,4-дигидро-2 Н-хромен-4-илпиперазин дибензоилтартрат (-). Этап А. (-)Изомер трет-бутил транс-4-3-[(3-хлор-5-фторбензил)окси]-3,4-дигидро-2 Н-хромен-4 илпиперазин-1-карбоксилат. Ожидаемое соединение представляет собой второй из энантиомеров, разделенных на этапе А примера 62. Этап Б. (-)Изомер транс-1-3-(3-хлор-5-фторбензилокси)-3,4-дигидро-2 Н-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой, описанной на этапе В примера 1, используя в качестве исходного вещества соединение, полученное на вышеописанной стадии. Этап В. (-)Изомер транс-1-3-(3-хлор-5-фторбензилокси)-3,4-дигидро-2 Н-хромен-4-илпиперазин дибензоилтартрат (-). Ожидаемый продукт получали путем обратного превращения соединения, полученного на вышеописанной стадии, в основание и последующего превращения в соль с помощью (-)-дибензоилвинной кислоты. Точка плавления: 100-107 С. Пример 64. транс-1-3-[(3,5-Дифторбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин диметансульфонат. Этап А. транс-1-3-[(3,5-Дифторбензил)окси]-3,4-дигидро-2 Н-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 3,5-дибромбензилбромид на этапе Г 3,5-дифторбензилбромидом. Этап Б. транс-1-3-[(3-Дифторбензил)окси]-3,4-дигидро-2 Н-хромен-4-илпиперазин диметансульфонат. Ожидаемый продукт получали путем обратного превращения соединения, полученного на вышеописанной стадии, в основание и последующего превращения в соль с помощью метансульфоновой кислоты. Точка плавления: 178-182 С. Пример 65. транс-4-3-[(3,5-Дибромбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперидин гидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 50, заменяя на этапе А 1-тетралон 2,3-дигидро-4 Н-хромен-4-оном. Точка плавления: 148-167 С.- 22009648 Пример 66. транс-1-3-[(3-(Трифторметокси)бензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазин диметансульфонат. Этап А. транс-1-3-[(3-(Трифторметокси)бензил)окси]-3,4-дигидро-2 Н-хромен-4-илпиперазин дигидрохлорид. Ожидаемый продукт получали в соответствии с методикой из примера 4, заменяя 3,5-дибромбензилбромид на этапе Г 3-(трифторметокси)бензилбромидом. Этап Б. транс-1-3-[(3-(Трифторметокси)бензил)окси]-3,4-дигидро-2 Н-хромен-4-илпиперазин диметансульфонат. Ожидаемый продукт получали путем обратного превращения соединения, полученного на вышеописанной стадии, в основание и последующего превращения в соль с помощью метансульфоновой кислоты. Точка плавления: 132-135 С. Фармакологическое исследование соединений по изобретению Пример 67. Определение аффинности для сайтов обратного поглощения серотонина у крысы. Аффинность соединений для сайта обратного поглощения серотонина (5-НТТ) оценивали с помощью конкурентных экспериментов с [3 Н]-циталопрамом на мембранах лобной коры крыс. Кору гомогенизировали при использовании Polytron в 40 объемах (мас./об.) холодного Трис-HCl (50 мМ, рН 7,4) буфера для инкубации, содержащего 120 мМ NaCl и 5 мМ KCl и после этого смесь сначала центрифугировали. Затем полученный осадок ресуспендировали в том же буфере, инкубировали в течение 10 мин при 37 С, а потом повторно центрифугировали. В дальнейшем мембраны дважды промывали, а осадок ресуспендировали в приемлемом объеме буфера для инкубации. После этого мембраны инкубировали в течение 2 ч при 25 С с исследуемыми соединениями в присутствии 0,7 нМ [3 Н]-циталопрама. Неспецифическое связывание определяли с 10 мкМ флуоксетина. В конце инкубационного периода образцы фильтровали через фильтры типа Unifilter GF/B, которые были предварительно обработаны с помощьюPEI (0,5%), и несколько раз промывали при использовании инкубационного буфера. Оставшуюся на фильтрах радиоактивность подсчитывали после добавления сцинтиляционной жидкости с использованием сцинтиляционного счетчика. Полученные изотермы анализировали с помощью нелинейной регрессионной зависимости для определения значений IC50, которые превращали в Ki при использовании уравнения Ченга-Прусоффа:Ki=IC50/(1+L/kD) где L представляет собой концентрацию радиолиганда, а kD является константой диссоциации [3 Н]-циталопрама на сайте обратного поглощения серотонина (0,7 нМ). Результаты выражали в виде pKi=-log Ki. Полученные результаты для типичных соединений в соответствии с изобретением представлены в следующей таблице. Пример 68. Связывание hNK1. Аффинность соединений в соответствии с изобретением определяли путем конкурентных анализов в присутствии [3 Н]-вещества Р (Sar-9, MetO2-11,2-пропил-3,4-3 Н). IM9 лимфобластоидные клетки человека, которые эндогенно экспрессируют рецепторы NK1, центрифугировали и помещали в буфер для инкубации, содержащий 50 мМ Трис, 150 мМ NaCl, 4 мМ CaCl2, ингибиторы протеиназы при 1/100 е (CocktailSIGMA P8340) и 0,2% БСА. Определяли объем инкубационного буфера так, чтобы получить концентрацию 5106 клеток/мл. Затем препарат клеток инкубировали вместе с 1,5 нМ [3 Н]-вещества Р и исследуемого соединения в течение 90 мин при комнатной температуре. Неспецифическое связывание определяли в присутствии 1 мкМ GR 205171. В конце периода инкубации образцы фильтровали через фильтры типа Unifilter GF/B, которые предварительно обрабатывали PEI (0,1%) и несколько раз промывали с по- 23009648 мощью фильтрационного буфера (50 мМ Трис, 150 мМ NaCl, 4 мМ CaCl2). Оставшуюся на фильтрах радиоактивность подсчитывали после добавления к фильтрам сцинтиляционной жидкости. Полученные результаты анализировали с помощью нелинейной регрессионной зависимости, которая позволяет вычерчивать изотермы и определять значения IC50. Последние затем превращали в константы ингибирования (Ki) при использовании уравнения Ченга-Прусоффа:Ki=IC50/(1+L/KD) где L представляет собой концентрацию [3 Н]-вещества Р, а KD является константой диссоциации [3 Н]вещества Р для рецепторов NK1 человека (0,53 нМ). Результаты выражали в виде pKi(-log Ki). Полученные результаты для типичных соединений в соответствии с изобретением представлены в следующей таблице. Пример 69. Фармацевтическая композиция. Формула для приготовления 1000 таблеток, каждая из которых содержит 10 мг активного компонента: Соединение из примера 1 10 г Гидроксипропилцеллюлоза 2 г Пшеничный крахмал 10 г Лактоза 100 г Стеарат магния 3 г Тальк 3 г ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I)R1, R2, R3 и R4, которые могут быть одинаковыми или разными, каждый представляет собой атом или группу, выбранную из Н, галогена, линейного или разветвленного C1-С 6-алкила, линейного или разветвленного C1-С 6-алкокси, фенила и циано,X представляет собой связь, атом кислорода или группу, выбранную из -(СН 2)m-, -ОСН 2- и -NR5-,m представляет собой 1 или 2,R5 представляет собой атом водорода или группу, выбранную из линейного или разветвленного C1 С 6-алкила, COR6 и CO2R6,R6 представляет собой линейную или разветвленную C1-С 6-алкильную группу,Y представляет собой атом кислорода или группу, выбранную из NR7 и CHR8,R7 представляет собой атом водорода или группу, выбранную из COR9 и линейного или разветв- 24009648 ленного C1-С 6-алкила, где алкильная группа необязательно замещена 5-оксо-4,5-дигидро-1H-1,2,4-триазол-3-ильной или 2,3-дигидро-1,4-бенздиоксин-2-ильной группой,R9 представляет собой группу, выбранную из линейного или разветвленного C1-С 6-алкила, арила и гетероарила,R8 представляет собой атом водорода или аминогруппу, необязательно замещенную одной или двумя линейными или разветвленными С 1-С 6-алкильными группами,Z представляет собой атом азота или СН группу,n представляет собой 1 или 2,Ak представляет собой линейную или разветвленную С 1-С 6-алкиленовую цепь,Ar представляет собой арильную или гетероарильную группу,его оптические изомеры, а также его соли присоединения с фармацевтически приемлемой кислотой,при этом понятно, что арильная группа обозначает фенил, бифенилил или нафтил, каждая из этих групп необязательно замещена одной или несколькими одинаковыми или разными группами, выбранными из галогена, линейного или разветвленного С 1-С 6-алкила, линейного или разветвленного C1-С 6-алкокси, гидрокси, циано, линейного или разветвленного C1-С 6-тригалоалкила и линейного или разветвленного C1-С 6-тригалоалкокси,и гетероарильная группа обозначает ароматическую моно- или бициклическую 5-12-членную группу,содержащую 1, 2 или 3 гетероатома, выбранных из кислорода, азота и серы, при этом понятно, что гетероарильная группа необязательно может быть замещена одной или несколькими одинаковыми или разными группами, выбранными из галогена, линейного или разветвленного C1-С 6-алкила, линейного или разветвленного C1-С 6-алкокси, гидрокси, циано и линейного или разветвленного C1-С 6-тригалоалкила. 2. Соединение формулы (I) по п.1, в котором Y представляет собой NH. 3. Соединение формулы (I) по п.1 или 2, в котором Z представляет собой атом азота. 4. Соединение формулы (I) по любому из пп.1-3, в котором n представляет собой 1. 5. Соединение формулы (I) по любому из пп.1-4, в котором Ar представляет собой арильную группу. 6. Соединение формулы (I) по любому из пп.1-5, в котором X представляет собой связь, атом кислорода или группу, выбранную из -ОСН 2- и -(СН 2)m-, где m представляет собой 1 или 2. 7. Соединение формулы (I) по п.1, выбранное из транс-1-2-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,транс-1-3-[(3,5-дибромбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,транс-1-6-[(3,5-дибромбензил)окси]-6,7,8,9-тетрагидро-5H-бензо[7]аннулен-5-илпиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,транс-1-2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-илпиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,транс-1-2-[(3,5-дибромбензил)окси]-1,2,3,4-тетрагидронафт-1-ил-1,4-диазепана, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,1-(1S,2R)-2-[(3,5-дибромбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,1-(1S,2R)-2-[(3,5-дифторбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,1-(1S,2R)-2-[(3,5-диметилбензил)окси]-2,3-дигидро-1H-инден-1-илпиперазина, его энантиомеров,а также его солей присоединения с фармацевтически приемлемой кислотой,транс-1-3-[(3,5-дихлорбензил)окси]-3,4-дигидро-2H-хромен-4-илпиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой,транс-1-3-[3-фтор-5-(трифторметил)бензилокси]-3,4-дигидро-2H-хромен-4-илпиперазина, его энантиомеров, а также его солей присоединения с фармацевтически приемлемой кислотой и транс-1-3-(3-хлор-5-фторбензилокси)-3,4-дигидро-2 Н-хромен-4-илпиперазина, его энантиомеров,а также его солей присоединения с фармацевтически приемлемой кислотой. 8. Способ получения соединений формулы (I) по п.1 из соединения формулы (Va) в относительной транс-конфигурацииR1 R2, R3, R4, X, n и Z имеют значения, указанные в п.1, a Y' представляет собой атом кислорода или группу, выбранную из NP1 и CHR'8, в которой R'8 представляет собой атом водорода или группу NHP1 иP1 представляет собой защитную группу для функциональной аминогруппы,в котором соединение формулы (Va) подвергают взаимодействию, если желательно получить соединения формулы (I) в относительной транс-конфигурации, с соединением формулыG-Ak-Ar (VI) в которой Ak и Ar имеют значения, указанные для формулы (I), и G представляет собой уходящую группу, такую как, например, атом галогена или n-толуолсульфонатная, трифторметансульфонатная или метансульфонатная группа,с получением соединения формулы (VIIa) в относительной транс-конфигурации в котором R1, R2, R3, R4, X, n, Y', Z, Ak и Ar имеют значения, указанные выше,с последующим снятием защиты, если Y' содержит защитную группу P1, как определено выше, и затем алкилированием, если желательно получить соединения, в которых Y представляет собой группуNR7, где R7 отличен от атома водорода,с получением соединения формулы (Ia), частного случая соединения формулы (I), находящегося в относительной транс-конфигурации в которой R1, R2, R3, R4, X, n, Y, Z, Ak и Ar имеют значения, указанные для формулы (I),и полученные соединения формулы (Ia) могут быть очищены в соответствии с обычным способом очистки и при желании разделены, если это является желательным, на их оптические изомеры и превращены в их соли присоединения фармацевтически приемлемой кислоты. 9. Способ получения соединений формулы (I) по п.1 окислением соединения формулы (Va), и, если желательно получить соединения (I) в относительной цис-конфигурации,полученное в результате окисления рацемическое соединение формулы (VIII) в которой R1, R2, R3, R4, X, Z, n и Y' имеют значения, указанные выше,восстанавливают до соответствующего спирта, его диастереоизомеры разделяют с выделением изомера формулы (Vb), в относительной цис-конфигурацииG-Ak-Ar (VI) в которой Ak и Ar имеют значения, указанные для формулы (I), и G представляет собой уходящую группу, такую как, например, атом галогена или n-толуолсульфонатную, трифторметансульфонатную или метансульфонатную группу,получая соединение формулы (VIIb) в относительной цис-конфигурации в котором R1, R2, R3, R4, X, n, Y', Z, Ak и Ar имеют значения, указанные выше,с последующим снятием защиты, если Y' содержит защитную группу Р 1, как определено выше, и алкилированием, если желательно получить соединения, в которых Y представляет собой группу NR7,где R7 отличен от атома водорода,с получением соединения формулы (Ib), частного случая соединения формулы (I), находящегося в относительной цис-конфигурации, в которой R1, R2, R3, R4, X, Y, Z, n, Ak и Ar имеют значения, указанные для формулы (I),и полученные соединения формулы (Ib) могут быть очищены в соответствии с обычным способом очистки и при желании разделены, если это является желательным, на их оптические изомеры и превращены в их соли присоединения фармацевтически приемлемой кислоты. 10. Фармацевтическая композиция, содержащая в качестве активного компонента соединение в соответствии с любым из пп.1-7 в сочетании с одними или несколькими фармацевтически приемлемыми,инертными, нетоксичными носителями. 11. Применение соединений формулы (I) в соответствии с любым из пп.1-7 для приготовления лекарственных средств для применения в качестве ингибиторов обратного поглощения серотонина. 12. Применение соединений формулы (I) в соответствии с любым из пп.1-7 для приготовления лекарственных средств для применения в качестве ингибиторов обратного поглощения серотонина и антагонистов NK1. 13. Применение соединений формулы (I) в соответствии с любым из пп.1-7 для приготовления лекарственных средств для применения для лечения депрессивных состояний, состояний беспокойства,импульсивных расстройств, агрессивного поведения, злоупотребления лекарственными средствами,ожирения и расстройства аппетита, боли и воспаления, деменции, психотических состояний, нарушений хронобиологического ритма, тошноты или желудочно-кишечных расстройств.
МПК / Метки
МПК: C07D 319/20, A61K 31/496, A61K 31/495, C07D 313/08, C07D 213/54, C07D 405/04, C07D 311/68, C07D 295/033, C07D 249/12, A61K 31/453, C07D 215/42
Метки: которые, фармацевтические, получения, способ, композиции, соединения, пиперазина, содержат
Код ссылки
<a href="https://eas.patents.su/28-9648-soedineniya-piperazina-sposob-ih-polucheniya-i-farmacevticheskie-kompozicii-kotorye-ih-soderzhat.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения пиперазина, способ их получения и фармацевтические композиции, которые их содержат</a>
Замещённые соединения [1,4]бенздиоксино[2,3-e]изоиндола, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 8498
Опубликовано: 29.06.2007
Авторы: Леонс Стефан, Ренар Пьер, Кудер Жерар, Хикман Джон, Сеньар Даньель-Анри, Пьерр Ален, Эрб Натали, Лепифр Франк, Рутье Сильвен
МПК: A61K 31/5365, C07D 487/04, A61P 35/00...
Метки: содержат, 1,4]бенздиоксино[2,3-e]изоиндола, замещённые, фармацевтические, способ, получения, которые, композиции, соединения
Формула / Реферат:
1. Соединения формулы (I) в которой А вместе с атомами углерода, с которыми он связан, представляет собой группу формулы (а) или (b) в которой W1 вместе с атомами углерода, с которыми он связан, представляет собой фенильную группу или пиридильную группу, Z представляет собой группу, выбранную из атомов водорода и галогена и таких групп, как линейный или разветвленный C1-C6-алкил, нитро, циано, гидрокси, линейный или разветвленный...
Соединения бензотиазина и бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 7227
Опубликовано: 25.08.2006
Авторы: Корди Алекс, Десо Патрис, Лестаж Пьер
МПК: A61K 31/549, A61K 31/5415, A61P 25/00...
Метки: композиции, бензотиадиазина, бензотиазина, которые, получения, соединения, фармацевтические, способ, содержат
Формула / Реферат:
1. Соединения формулы (I) где R1 представляет собой арильную или гетероарильную группу, R2 представляет собой атом водорода, атом галогена или гидроксильную группу, А представляет собой CR4R5 группу или NR4 группу, R3 представляет собой атом водорода, линейную или разветвленную С1-С6-алкильную группу или С3-С7-циклоалкильную группу, R4 представляет собой атом водорода или линейную или разветвленную С1-С6-алкильную группу, или А представляет...
Новые соединения пирролидина и тиазолидина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 9292
Опубликовано: 28.12.2007
Авторы: Де Нантей Гийом, Комбетт Мюриелль, Арле Элизабет, Бенуа Ален
МПК: A61K 31/4025, A61K 31/40, A61K 31/426...
Метки: способ, тиазолидина, фармацевтические, получения, содержат, которые, пирролидина, соединения, новые, композиции
Формула / Реферат:
1. Соединение пирролидина или тиазолидина формулы (I) в которой X1 представляет собой атом или группу, выбранную из CR4aR4b, О, S(O)q1 и NR5, где R4a и R4b, которые могут быть одинаковыми или разными, каждый представляет собой атом водорода или С1-С6-алкильную группу, или R4a и R4b вместе с атомом углерода, к которому они присоединены, образуют С3-С7-циклоалкильную группу, q1представляет собой 0 или 2, и R5 представляет собой атом водорода или...
Новые соединения бензотиазина и бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 9522
Опубликовано: 28.02.2008
Авторы: Корди Алекс, Десо Патрик, Лестаж Пьер
МПК: A61K 31/542, A61K 31/5415, A61P 25/28...
Метки: содержат, композиции, способ, которые, новые, бензотиадиазина, соединения, получения, бензотиазина, фармацевтические
Формула / Реферат:
1. Соединения формулы (I) в которой R1 представляет собой гетероциклическую группу, R2 представляет собой атом водорода, А представляет собой атом азота и вместе со смежной группой -CHR3- образует кольцо где m представляет собой 1, а также их аддитивные соли с фармацевтически приемлемой кислотой или основанием, где под гетероциклической группой подразумевается моноциклическая, ароматическая или неароматическая группа, которая содержит от 1 до...
Новые соединения бензотиазина и бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 8797
Опубликовано: 31.08.2007
Авторы: Лестаж Пьер, Корди Алекс, Десо Патрик
МПК: C07D 513/04, A61K 31/549, A61P 25/28...
Метки: соединения, способ, новые, композиции, которые, бензотиадиазина, бензотиазина, получения, фармацевтические, содержат
Формула / Реферат:
1. Соединения формулы (I) где R1 представляет собой атом водорода, атом галогена или линейную или разветвленную C1-С6-алкильную группу, R1a представляет собой атом водорода или линейную или разветвленную C1-С6-алкильную группу, R2 представляет собой атом водорода, атом галогена или гидроксильную группу, А представляет собой CR4R5-группу или NR4-группу, R3 представляет собой атом водорода, линейную или разветвленную C1-С6-алкильную группу или...
Предыдущий патент: Арилсульфонамидные соединения и их применение для лечения ожирения, диабета типа ii
Следующий патент: Установка и способ обработки пара сжиженного природного газа
Случайный патент: Медицинский электрод, пучок электродов и массив из пучков электродов