Замещенные пиридины в качестве избирательных ингибиторов циклооксигеназы-2
Номер патента: 1444
Опубликовано: 23.04.2001
Авторы: Дюб Дэниел, Фресен Ричард, Вонг Заойин, Готье Жак Ив, Фортин Риджен

























Формула / Реферат
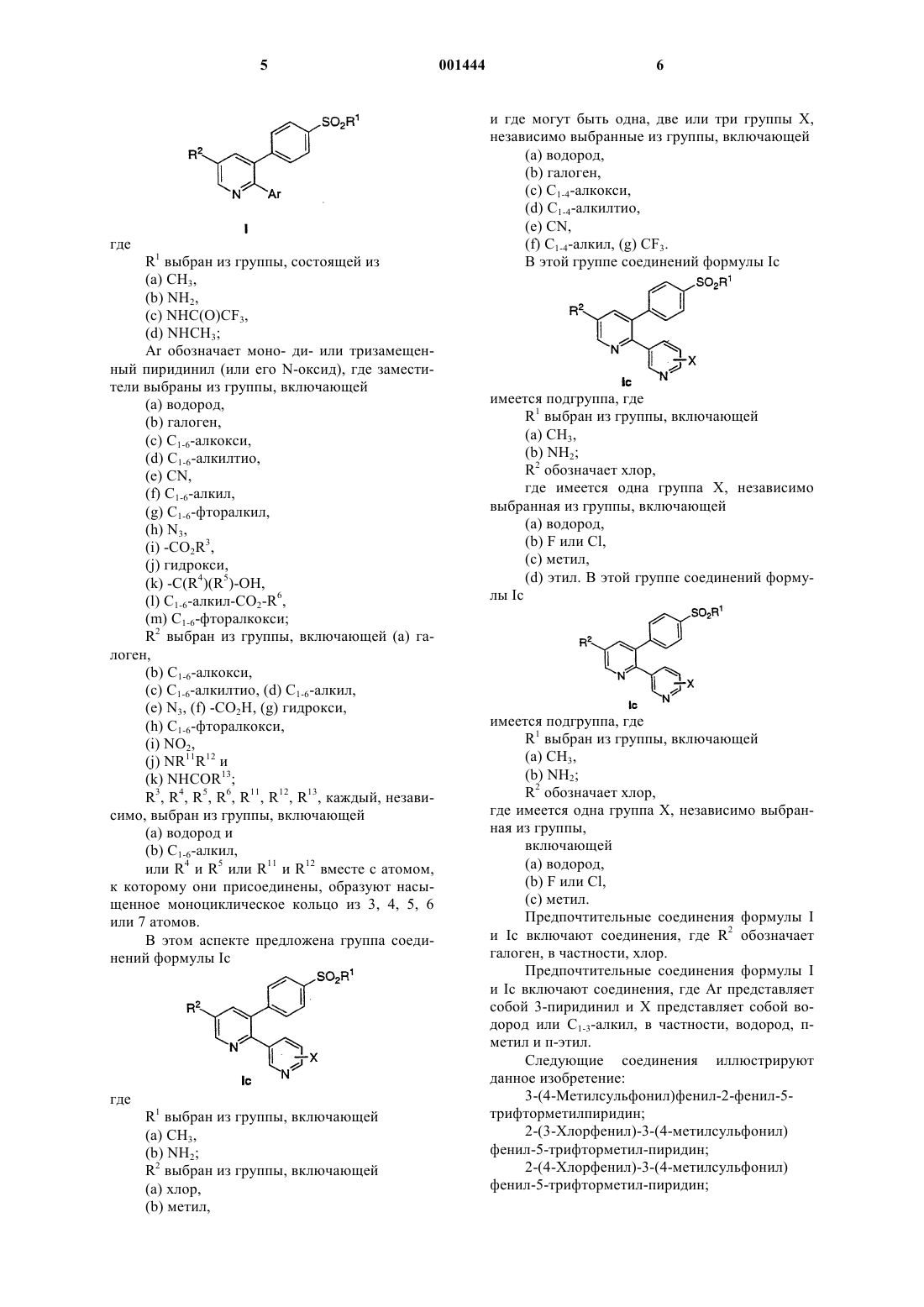
1. Соединение формулы I

или его фармацевтически приемлемая соль, где R1 выбран из группы, включающей:
(а) СН3,
(b) NН2,
(c) NНС(O)СF3,
(d) NНСН3;
Аr обозначает моно-, ди- или тризамещенный фенил или пиридинил (или его N-оксид), где заместители выбраны из группы, включающей:
(a) водород,
(b) галоген,
(c) C1-6-алкокси,
(d) C1-6-алкилтио,
(е) CN,
(f) C1-6-алкил,
(g) C1-6-фторалкил,
(h) N3,
(i) -CO2R3,
(j) гидрокси,
(k) -C(R4)(R5)-OH,
(l) -С1-6-алкил-СО2R6,
(m) C1-6-фторалкокси;
R2 выбран из группы, включающей:
(a) галоген,
(b) C1-6-алкокси,
(c) C1-6-алкилтио,
(d) CN,
(e) C1-6-алкил,
(f) C1-6-фторалкил,
(g) N3,
(h) -CO2R7,
(i) гидрокси,
(j) -C(R8)(R9)-OH,
(k) -С1-6-алкил-СO2-R10,
(l) C1-6-фторалкокси,
(m) NO2,
(n) NR11R12 и
(о) NHCOR13;
R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13 выбраны, каждый независимо, из группы, включающей:
(a) водород и
(b) C1-6-алкил,
или R4 и R5, R8 и R9 или R11 и R12 вместе с атомом, к которому они присоединены, образуют насыщенное моноциклическое кольцо из 3, 4, 5, 6 или 7 атомов, при условии, что, когда Аr представляет собой фторфенил, R2 выбран из (а), (b), (с) или (е)-(о) из определения R2, данного выше.
2. Соединение по п.1, где Аr представляет собой моно-, ди- или тризамещенный 2-пиридинил.
3. Соединение по п.1, где Аr представляет собой моно-, ди- или тризамещенный 3-пиридинил.
4. Соединение по п.1, где R1 представляет собой СН3 или NH2.
5. Соединение по п.1, где Аr обозначает моно-, ди- или тризамещенный 2-пиридинил или 3-пиридинил и заместители выбраны из группы, включающей:
(a) водород,
(b) галоген,
(c) C1-3-алкокси,
(d) C1-3-алкилтио,
(e) C1-3-алкил,
(f) СF3 и
(g) CN.
6. Соединение по п.1, где R1 обозначает СН3 или NH2 и Аr обозначает моно-, ди- или тризамещенный 2-пиридинил или 3-пиридинил и заместители выбраны из группы, включающей:
(а) водород,
(b) галоген,
(c) C1-3-алкокси,
(d) C13-алкилтио,
(e) C1-3-алкил,
(f) СF3, и
(g) CN.
7. Соединение по п.1, где R2 обозначает
(a) галоген,
(b) C1-4-алкокси,
(c) CN,
(d) C1-3-алкил,
(e) C1-3-фторалкил,
(f) -СО2Н,
(g) -С1-3-алкил-СО2Н,
(h) C1-3-фторалкокси или
(i) NO2.
8. Соединение по п.1, где R2 обозначает галоген, СН3 или СF3.
9. Соединение по п.1, где R1 обозначает СН3 или NH2;
R2 обозначает галоген, СН3 или СF3; и
Аr обозначает моно-, ди- или тризамещенный 2-пиридинил или 3-пиридинил и заместители выбраны из группы, включающей:
(a) водород,
(b) галоген,
(c) C1-3-алкокси,
(d) C1-3-aлкилтио,
(e) C1-3-алкил,
(f) СF3 и
(g) CN.
10. Соединение по п.1, где R1 обозначает СН3 или NH2;
R2 обозначает галоген, СН3 или СF3 и
Аr обозначает моно- или дизамещенный 3-пиридинил и заместители выбраны из группы, включающей:
(a) водород,
(b) галоген,
(c) C1-3-алкокси,
(d) C1-3-алкилтио,
(e) C1-3-алкил,
(f) СF3 и
(g) CN.
11. Соединение по п.1, где
R1 обозначает СН3 или NH2;
R2 обозначает галоген, СН3 или СF3 и
Аr обозначает моно- или дизамещенный фенил, и заместители выбраны из группы, включающей:
(a) водород,
(b) галоген,
(c) C1-3-алкокси,
(d) C1-3-алкилтио,
(e) C1-3-алкил,
(f) СF3 и
(g) CN.
12. Соединение по п.1 формулы Iа

или его фармацевтически приемлемая соль, где R2 и Аr выбраны вместе следующим образом:
| R2 | Аr | |
| (1) | СF3 | Ph |
| (2) | СF3 | 3-СlС6Н4 |
| (3) | СF3 | 4-СlС6Н4 |
| (4) | СF3 | 4-FC6H4 |
| (5) | СF3 | 2-(СМе2OН)С6Н4 |
| (6) | СF3 | 3-(СМе20Н)С6Н4 |
| (7) | СF3 | 3-руr |
| (8) | СF3 | 5-(2-Ме)руr |
| (9) | СF3 | 5-(3-Вr)руr |
| (10) | СF3 | 5-(3-Сl)руr |
| (11) | СF3 | 5-(2-OMe)pyr |
| (12) | СF3 | 2-(5-Вr)руr |
| (13) | Me | Ph |
| (14) | Me | 4-СlС6Н4 |
| (15) | Me | 3-руr |
| (16) | Cl | Ph |
| (17) | Cl | 4-СlС6Н4 |
| (18) | Cl | 2-(СМе2OН)С6Н4 |
| (19) | Cl | 3-(СМе2OН)С6Н4 |
| (20) | Cl | 2-руr |
| (21) | Cl | 3-руr |
| (22) | Cl | 4-руr |
| (23) | Cl | 5-(2-Ме)руr |
| (24) | Cl | 5-(3-Вr)руr |
| (25) | Cl | 5-(3-Сl)руr |
| (26) | Cl | 5-(2-ОМе)руr |
| (27) | Cl | 2-(5-Вr)руr |
| (28) | F | Ph |
| (29) | F | 3-руr |
| (30) | F | 5-(2-Ме)руr |
| (31) | Br | Ph |
| (32) | Br | 3-руr |
| (33) | Br | 5-(2-Ме)руr |
| (34) | NO2 | Ph |
| (35) | NO2 | 3-руr |
| (36) | NO2 | 5-(2-Ме)руr |
| (37) | OMe | Ph |
| (38) | OMe | 3-руr |
| (39) | OMe | 5-(2-Ме)руr |
| (40) | NHCOMe | Ph |
| (41) | NHCOMe | 3-руr |
| (42) | NHCOMe | 5-(2-Ме)руr |
| (43) | CO2Me | 4-ClС6Н4 |
| (44) | CO2H | 4-ClС6Н4 |
| (45) | CN | 4-ClС6Н4 |
13. Соединение по п.12, где фармацевтически приемлемая соль является солью лимонной, бромисто-водородной, хлористо-водородной, малеиновой, метансульфоновой, фосфорной, серной или винной кислоты.
14. Соединение по п.13, где фармацевтически приемлемая соль является кислотно-аддитивной солью хлористо-водородной или метансульфоновой кислоты.
15. Соединение по п.1 формулы Ib

или его фармацевтически приемлемая соль, где R2 и Аr выбраны вместе следующим образом:
| R2 | Ar | |
| (48) | CF3 | Ph |
| (49) | CF3 | 4-СlС6Н4 |
| (5) | СF3 | 4-FC6H4 |
| (51) | CF3 | 3-pyr |
| (52) | Me | Ph |
| (53) | Me | 4-СlС6Н4 |
| (54) | Cl | 3-pyr |
| (55) | Cl | 5-(2-Me)pyr |
| (56) | CN | 4-СlС6Н4 |
| (71) | Cl | 5-(2-ethyl)pyr |
| (72) | Cl | 5-(2-Et)pyr-MeSO3H |
| (73) | Cl | 5-(2-c-Pr)pyr |
| (74) | Cl | 3-(2,6-Me2)pyr |
16. Соединение по п.15, где фармацевтически приемлемая соль является солью лимонной, бромисто-водородной, хлористо-водородной, малеиновой, метансульфоновой, фосфорной, серной или винной кислоты.
17. Соединение по п.16, где фармацевтически приемлемая соль является кислотно-аддитивной солью хлористо-водородной или метансульфоновой кислоты.
18. Фармацевтическая композиция для лечения воспалительного заболевания, чувствительного к лечению нестероидным противовоспалительным агентом, содержащая нетоксичное терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
19. Фармацевтическая композиция для лечения опосредованных циклооксигеназой заболеваний, успешно излечиваемых активным агентом, который избирательно ингибирует ЦОГ-2 относительно ЦОГ-1, содержащая нетоксичное терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
20. Способ лечения воспалительного заболевания, чувствительного к лечению нестероидным противовоспалительным агентом, предусматривающий введение пациенту, нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения по п.1 и фармацевтически приемлемого носителя.
21. Способ лечения опосредованных циклооксигеназой заболеваний, успешно излечиваемых активным агентом, который избирательно ингибирует ЦОГ-2 относительно ЦОГ-1, предусматривающий введение пациенту, нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения по п. 1 и фармацевтически приемлемого носителя.
22. Соединение по п.1, где R2 выбран из группы, включающей:
(a) галоген,
(b) C1-6-алкокси,
(c) C1-6-алкилтио,
(d) C1-6-алкил,
(e) N3,
(f) -CO2H,
(g) гидрокси,
(h) C1-6-фторалкокси,
(i) NO2,
(j) NR11R12 и
(k) NHCOR13;
R3, R4, R5, R6, R11, R12, R13, каждый независимо, выбран из группы, включающей:
(а) водород и
(b) C1-6-алкил,
или R4 и R5 или R11 и R12 вместе с атомом, к которому они присоединены, образуют насыщенное моноциклическое кольцо из 3, 4, 5, 6 или 7 атомов.
23. Соединение по п. 22 формулы Ic

где R1 выбран из группы, включающей:
(a) СН3,
(b) NH2;
R2 выбран из группы, включающей:
(a) хлор,
(b) метил,
и где могут быть одна, две или три группы X, независимо выбранные из группы, включающей:
(a) водород,
(b) галоген,
(c) C1-4-алкокси,
(d) C1-4-алкилтио,
(e) CN,
(f) C1-4-алкил,
(g) СF3.
24. Соединение по п.23, где R2 обозначает хлор, где имеется одна группа X, независимо выбранная из группы, включающей:
(a) водород;
(b) F или Сl,
(c) метил,
(d) этил.
25. Соединение по п.24, где имеется одна группа X, независимо выбранная из группы, включающей:
(a) водород,
(b) F или Сl,
(c) метил.
26. Соединение по п.25, где R1 обозначает метил.
27. Соединение по п.1 формулы Iс

где R1 обозначает СН3 или NH2;
R2 обозначает
(a) галоген,
(b) C1-6-алкокси,
(c) C1-6-алкилтио,
(d) C1-6-алкил,
(e) N3,
(f) -CO2H,
(g) гидрокси,
(h) C1-6-фторалкокси,
(i) NO2,
(j) NR11R12,
(k) NHCOR13 и
X обозначает водород.
28. Соединение по п.1, которое представляет собой 5-хлор-3-(4-метилсульфонил)фенил-2-(3-пиридинил)пиридин или его фармацевтически приемлемую соль.
29. Соединение по п.1, которое представляет собой гидрометансульфонат 5-хлор-3-(4-метилсульфонил)фенил-2-(3-пиридинил) пиридина.
30. Соединение по п.1, которое представляет собой гидрохлорид 5-хлор-3-(4-метилсульфонил)фенил-2-(3-пиридинил)пиридина.
31. Соединение по п.1, которое представляет собой 5-хлор-3-(4-метилсульфонил)фенил-2-(2-этил-5-пиридинил)пиридин или его фармацевтически приемлемую соль.
32. Соединение по п.1, которое представляет собой гидрометансульфонат 5-хлор-3-(4-метилсульфонил)фенил-1-(2-этил-5-пиридинил) пиридина.
33. Соединение по п.1 формулы Iс

где R1 обозначает СН3 или NH2;
R2 обозначает
(a) галоген,
(b) C1-3-алкокси,
(c) C1-3-алкилтио,
(d) C1-3-алкил,
(e) N3,
(f) -СО2Н,
(g) гидрокси,
(h) C1-3-фторалкокси,
(i) NO2,
(j) NR11R12,
(k) NHCOR13 и
X обозначает метил, этил, н-пропил, изопропил или циклопропил.
34. Соединение по п.33, где Х обозначает метил.
35. Соединение по п.33, которое представляет собой 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридин или его фармацевтически приемлемую соль.
36. Соединение по п.33, которое представляет собой гидрохлорид 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина.
37. Соединение по п.1, выбранное из группы, включающей
3-(4-метилсульфонил)фенил-2-фенил-5-трифторметилпиридин;
2-(3-хлорфенил)-3-(4-метилсульфонил) фенил-5-трифторметилпиридин;
2-(4-хлорфенил)-3-(4-метилсульфонил) фенил-5-трифторметилпиридин;
2-(4-фторфенил)-3-(4-метилсульфонил) фенил-5-трифторметилпиридин;
5-метил-3-(4-метилсульфонил)фенил-2-фенилпиридин;
2-(4-хлорфенил)-5-метил-3-(4-метилсульфонил)фенилпиридин;
5-хлор-2-(4-хлорфенил)-3-(4-метилсульфонил)фенилпиридин;
метиловый эфир 2-(4-хлорфенил)-3-(4-метилсульфонил)фенилпиридинил-5-карбоновой кислоты;
2-(4-хлорфенил)-3-(4-метилсульфонил)фенилпиридинил-5-карбоновая кислота;
5-циано-2-(4-хлорфенил)-3-(4-метилсульфонил)фенилпиридин.
38. Соединение по п.1, выбранное из группы, включающей
3-(4-метилсульфонил)фенил-2-(3-пиридинил)-5-трифторметилпиридин;
5-метил-3-(4-метилсульфонил)фенил-2-(3-пиридинил)пиридин;
5-хлор-3-(4-метилсульфонил)фенил-2-(2-пиридинил)пиридин;
5-хлор-3-(4-метилсульфонил)фенил-2-(3-пиридинил)пиридин;
5-хлор-3-(4-метилсульфонил)фенил-2-(4-пиридинил)пиридин;
5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридин;
гидрометансульфонат 5-хлор-3-(4-метилсульфонил)фенил-2-(3-пиридинил)пиридина;
гидрохлорид 5-хлор-3-(4-метилсульфонил)фенил-2-(3-пиридинил)пиридина;
гидрохлорид 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина;
5-хлор-3-(4-метилсульфонил)фенил-2-(2-этил-5-пиридинил)пиридин и
гидрометансульфонат 5-хлор-3-(4-метилсульфонил)фенил-2-(2-этил-5-пиридинил)пиридина.
39. Соединение формулы I, определенной в п.1, или его фармацевтически приемлемая соль, где
R1 обозначает СН3;
R2 выбран из C1-6-алкила, галогена, СF3 или CN; и
Аr обозначает монозамещенный фенил или незамещенный или монoзамещенный пиридинил, в котором монозаместитель выбран из C1-6-алкила, галогена или С3-6-циклоалкила.
40. Соединение по п.39, где Аr представляет собой монозамещенный фенил.
41. Соединение по п.39, где Аr представляет собой незамещенный или монозамещенный пиридинил.
42. Применение соединения по пп.1, 28, 29, 30, 31, 32, 35, 36, 37, 38, 39, 40 или 41 при приготовлении лекарственного средства для лечения воспалительного заболевания.
43. Фармацевтическая композиция для лечения опосредованных циклооксигеназой-2 заболеваний, содержащая соединение по пп.1, 28, 29, 30, 31, 32, 35, 36, 37, 38, 39, 40 или 41 и фармацевтически приемлемый носитель.
44. Фармацевтическая композиция, содержащая соединение по любому из пп. 1-16 и 22-41 и фармацевтически приемлемый носитель.
45. Противовоспалительная фармацевтическая композиция, содержащая приемлемое противовоспалительное количество соединения формулы I, определенного в любом из пп. 1-11, 22-26 или 29-32, или его фармацевтически приемлемой соли вместе с фармацевтически приемлемым носителем.
46. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-17 или 22-37 в качестве ингибитора ЦОГ-2, избирательного для ЦОГ-2 относительно ЦОГ-1.
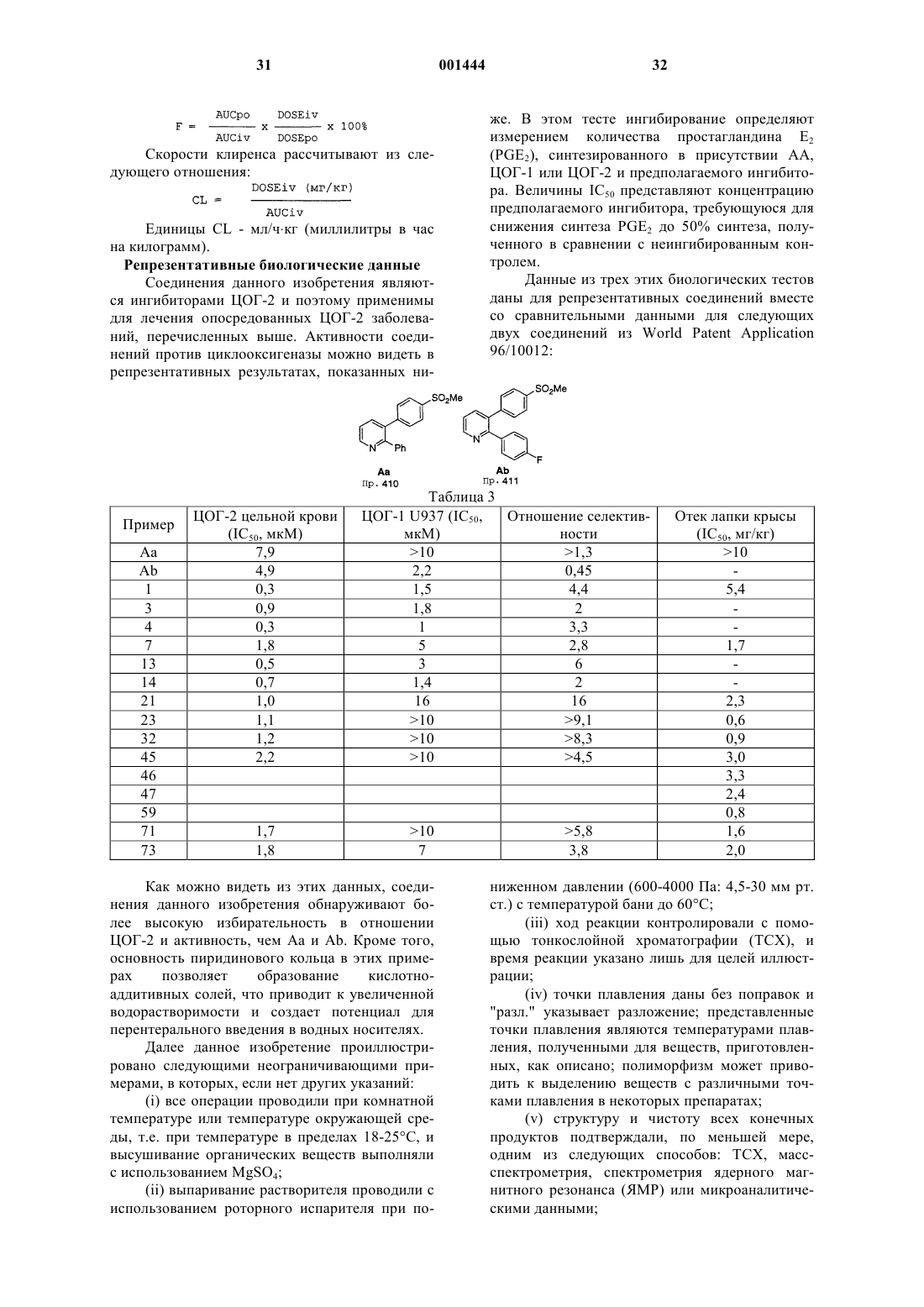
Текст
1 Предпосылки изобретения Данное изобретение относится к способам лечения опосредованных циклооксигеназой заболеваний и к некоторым фармацевтическим композициям. Нестероидные противовоспалительные лекарственные средства оказывают большую часть их противовоспалительной, анальгезирующей и жаропонижающей активности и ингибируют вызываемые гормоном сокращения матки и некоторые типы ракового роста через ингибирование простагландин G/H-синтазы,известной также как циклооксигеназа. Сначала была известна только одна форма циклооксигеназы,соответствующая циклооксигеназе-1(ЦОГ-1), или конститутивному ферменту, обнаруженному первоначально в бычьих семенных пузырьках. Совсем недавно ген для второй индуцируемой формы циклооксигеназы, циклооксигеназы-2 (ЦОГ-2), был клонирован, секвенирован и охарактеризован первоначально из курицы, мыши и человека. Этот фермент отличается от ЦОГ-1, которая была клонирована, секвенирована и охарактеризована из различных источников, в том числе, из овцы, мыши и человека. Вторая форма циклооксигеназы, ЦОГ-2,быстро и легко индуцируется рядом агентов, в том числе, митогенами, эндотоксином, гормонами, цитокинами и факторами роста. Так как простагландины играют как физиологическую,так и патологическую роль, авторы пришли к выводу, что конститутивный фермент, ЦОГ-1,ответственен, в основном, за эндогенное базальное высвобождение простагландинов и,следовательно, имеет важное значение для их физиологических функций, таких как поддержание желудочно-кишечной целостности и почечного кровотока. В противоположность этому, авторы пришли к выводу, что индуцируемая форма, ЦОГ-2, ответственна, в основном, за патологические действия простагландинов, когда индукция этого фермента имеет место в ответ на такие агенты, как воспалительные агенты, гормоны, факторы роста и цитокины. Таким образом, избирательный ингибитор ЦОГ-2 будет иметь сходные противовоспалительные,жаропонижающие и анальгезирующие свойства в сравнении с общепринятым нестероидным противовоспалительным лекарственным средством и, кроме того, будет ингибировать индуцируемые гормонами сокращения матки и иметь потенциальные противораковые эффекты,но будет обладать пониженной способностью индуцировать некоторые относящиеся к механизму действия побочные эффекты. В частности, такое соединение будет обладать пониженным потенциалом в отношении желудочнокишечной токсичности, пониженным потенциалом в отношении почечных побочных эффектов, уменьшенным действием на время кровотечения и, возможно, ослабленной способностью 2 индуцировать приступы астмы у чувствительных к аспирину субъектов. Кроме того, такое соединение будет также ингибировать индуцируемое простаноидами сокращение гладких мышц путем предотвращения синтеза контрактильных простаноидов и,следовательно, может быть использовано при лечении дисменореи, преждевременных родов,астмы и связанных с эозинофилами заболеваний. Оно будет также применимо при лечении болезни Альцгеймера, для уменьшения рарефикации костей, в частности, у женщин в постклимактерическом периоде (т.е. для лечения остеопороза) и для лечения глаукомы. Возможные области применения избирательных ингибиторов циклооксигеназы-2 обсуждаются в следующих статьях: 1. John Vane, "Towards a better aspirin" inSelective Inhibition and Progress to Date", in Medicinal Research Reviews, Vol. 16, pp. 181-206,1996. В WO 96/10012 (DuPont Merck, April 4,1996) описаны соединения, представленные формулой А, как используемые при лечении опосредованных ЦОГ-2 заболеваний благодаря их избирательному ингибированию ЦОГ-2 относительно ЦОГ-1. В настоящее время авторы обнаружили, что подгруппа соединений, представленных А, в которых -J-K-L- обозначает NCHCH-, Х обозначает связь, R1 обозначает ароматический радикал и R3 и R4 оба не являются водородом, проявляет неожиданно превосходящую избирательность в отношении ингибирования ЦОГ-2 относительно ЦОГ-1 и/или превосходящую активность в сравнении с наиболее близкими соединениями,описанными в 96/10012. Эта подгруппа соединений является объектом данного изобретения и представлена формулой I Из более 175 характерных соединений,описанных в WO 96/10012, только 4 являются пиридинами, и ни один из этих пиридинов не содержит заместитель (R3 или R4 в А) в пиридиновом кольце. 3 В WO 96/16934 (Searle, June 6, 1996) описаны соединения, представленные структурой В, в качестве используемых для лечения воспаления и связанных заболеваний. Химически эти соединения отличаются от соединений данного изобретения тем, что центральное из трех ароматических колец является предпочтительнее бензолом, чем пиридином. Сущность изобретения Данное изобретение относится к новому соединению формулы I, а также к способу лечения опосредованных ЦОГ-2 заболеваний, предусматривающий введение пациенту, нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения формулы I Данное изобретение относится также к некоторым фармацевтическим композициям для лечения опосредованных ЦОГ-2 заболеваний,содержащим соединения формулы I. Подробное описание изобретения Данное изобретение относится к новому соединению формулы I, а также к способу лечения опосредованных циклооксигеназой-2 заболеваний, заключающемуся во введении пациенту, нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения формулы I:(a) СН 3,(b) NH2,(c) NНС(O)СF3,(d) NНСН 3; Аr обозначает моно-, ди- или тризамещенный фенил или пиридинил (или их N-оксид), где заместители выбраны из группы, включающей(b) C1-6-алкил,или R4 и R5, R8 и R9 или R11 и R12, вместе с атомом, к которому они присоединены, образуют насыщенное моноциклическое кольцо из 3,4, 5, 6 или 7 атомов. Подходящие алкильные группы включают группы метил, этил, н-пропил, изопропил и бутил, пентил и гексил. Предпочтительной алкильной группой является метильная группа. Как правило, R4 и R5, R8 и R9 и R11 и R12 не являются остатками вышеуказанных моноциклических колец. Когда "алкил" является частью составного термина, такого как алкокси, алкилтио, фторалкил, фторалкокси, значение алкил относится также к составному термину. Предпочтительной подгруппой формулы I является такая, где Аr является моно- или дизамещенным пиридинилом. В этой подгруппе особенно предпочтительны 3-пиридинильные изомеры, такие как изомеры формулы Iс. Когда Аr представляет собой дизамещенный фенил, особенно предпочтительно, чтобы один или оба заместителя были водородом. Другой предпочтительной подгруппой формулы I является такая, где Аr является моноили дизамещенным фенилом. Когда Аr представляет собой дизамещенный фенил, особенно предпочтительно, чтобы один из заместителей был водородом или фтором, а второй был водородом, фтором, хлором,метилом, метокси или трифторметилом. Другой предпочтительной подгруппой формулы I является подгруппа, в которой R1 обозначает СН 3 или NH2. В общем, СН 3 предпочтителен для специфичности ЦОГ-2, a NH2 предпочтителен для активности. Другой предпочтительной подгруппой формулы I является подгруппа, в которой R2 обозначает галоген, СН 3 или СF3. Другой предпочтительной подгруппой формулы I является подгруппа, в которой заместители Аr выбраны из группы, включающей(g) CN. В одном аспекте данное изобретение относится к соединениям формулы I:(a) СН 3,(b) NH2,(c) NНС(O)СF3,(d) NHCH3; Аr обозначает моно- ди- или тризамещенный пиридинил (или его N-оксид), где заместители выбраны из группы, включающей(b) C1-6-алкил,или R4 и R5 или R11 и R12 вместе с атомом,к которому они присоединены, образуют насыщенное моноциклическое кольцо из 3, 4, 5, 6 или 7 атомов. В этом аспекте предложена группа соединений формулы Iс 6 и где могут быть одна, две или три группы X,независимо выбранные из группы, включающей(a) водород,(b) галоген,(c) C1-4-алкокси,(d) C1-4-алкилтио,(e) CN,(f) C1-4-алкил, (g) СF3. В этой группе соединений формулы IсR2 обозначает хлор,где имеется одна группа X, независимо выбранная из группы, включающей(a) водород,(b) F или Сl,(c) метил,(d) этил. В этой группе соединений формулы IсR2 обозначает хлор,где имеется одна группа X, независимо выбранная из группы,включающей(a) водород,(b) F или Сl,(c) метил. Предпочтительные соединения формулы I и Iс включают соединения, где R2 обозначает галоген, в частности, хлор. Предпочтительные соединения формулы I и Iс включают соединения, где Аr представляет собой 3-пиридинил и Х представляет собой водород или C1-3-алкил, в частности, водород, пметил и п-этил. Следующие соединения иллюстрируют данное изобретение: 3-(4-Метилсульфонил)фенил-2-фенил-5 трифторметилпиридин; 2-(3-Хлорфенил)-3-(4-метилсульфонил) фенил-5-трифторметил-пиридин; 2-(4-Хлорфенил)-3-(4-метилсульфонил) фенил-5-трифторметил-пиридин; 7 2-(4-Фторфенил)-3-(4-метилсульфонил) фенил-5-трифторметил-пиридин; 3-(4-Метилсульфонил)фенил-2-(3 пиридинил)-5-трифторметил-пиридин; 5-Метил-3-(4-метилсульфонил)фенил-2 фенилпиридин; 2-(4-Хлорфенил)-5-метил-3-(4-метилсульфонил)фенилпиридин; 5-Метил-3-(4-метилсульфонил)фенил-2-(3 пиридинил)пиридин; 5-Хлор-2-(4-хлорфенил)-3-(4-метилсульфонил)фенилпиридин; 5-Хлор-3-(4-метилсульфонил)фенил-2-(2 пиридинил)пиридин; 5-Хлор-3-(4-метилсульфонил)фенил-2-(3 пиридинил)пиридин; 5-Хлор-3-(4-метилсульфонил)фенил-2-(4 пиридинил)пиридин; 5-Хлор-3-(4-метилсульфонил)фенил-2-(2 метил-5-пиридинил)пиридин; Сложный метиловый эфир 2-(4-хлорфенил)-3-(4-метилсульфонил)фенилпиридинил-5 карбоновой кислоты; 2-(4-Хлорфенил)-3-(4-метилсульфонил) фенилпиридинил-5-карбоновая кислота; 5-Циано-2-(4-хлорфенил)-3-(4-метилсульфонил)фенилпиридин; Гидрометансульфонат 5-хлор-3-(4-метилсульфонил)фенил-2-(3-пиридил)пиридина; Гидрохлорид 5-хлор-3-(4-метилсульфонил) фенил-2-(3-пиридинил)пиридина; Гидрохлорид 5-хлор-3-(4-метилсульфонил) фенил-2-(2-метил-5-пиридинил)пиридина; 5-Хлор-3-(4-метилсульфонил)фенил-2-(2 этил-5-пиридинил)пиридин и Гидрометансульфонат 5-хлор-3-(4-метилсульфонил)фенил-2-(2-этил-5-пиридинил)пиридина. Предпочтительные соединения формулы I и формулы Iс включают соединения, где R1 представляет собой метил или NH2, в частности,метил. В другом аспекте данное изобретение относится к фармацевтической композиции для лечения воспалительного заболевания, чувствительного к лечению нестероидным противовоспалительным агентом, содержащей нетоксичное терапевтически эффективное количество соединения формулы I и фармацевтически приемлемый носитель. В другом аспекте данное изобретение относится также к фармацевтической композиции для лечения опосредованных циклооксигеназой заболеваний, успешно поддающихся лечению активным агентом, который избирательно ингибирует ЦОГ-2 в сравнении с ЦОГ-1, содержащей нетоксичное терапевтически эффективное количество соединения формулы I и фармацевтически приемлемый носитель. В другом аспекте данное изобретение относится также к способу лечения воспалительного заболевания, чувствительного к лечению 8 нестероидным противовоспалительным агентом, который заключается во введении пациенту, нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения формулы I и фармацевтически приемлемого носителя. В другом аспекте данное изобретение относится также к способу лечения опосредованных циклооксигеназой заболеваний, успешно излечиваемых активным агентом, который избирательно ингибирует ЦОГ-2 в сравнении с ЦОГ-1, заключающемуся во введении пациенту,нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения формулы I. В другом аспекте данное изобретение относится также к применению соединения формулы I или фармацевтической композиции при получении лекарственного средства для лечения воспалительного заболевания, чувствительного к лечению нестероидным противовоспалительным агентом. Данное изобретение иллюстрируется примерами 1-56. Следующие аббревиатуры имеют указанные значения:HBSS = сбалансированный солевой раствор Хенкса;NSAID = нестероидное противовоспалительное лекарственное средство (НСПВЛС);tid = ter in die = три раза в день. Для целей данного описания "алкил" обозначает линейные, разветвленные и циклические структуры с указанным числом атомов углерода. Примеры алкильных групп включают метил, этил, пропил, втор- и трет-бутил, бутил,пентил, гексил, 1,1-диметилэтил, циклопропил,циклобутил, циклогексилметил и т.п. Подобным образом алкокси и алкилтио обозначают линейные, разветвленные и циклические структуры с указанным числом атомов углерода. Для целей данного описания "фторалкил" обозначает алкильные группы с указанным числом атомов углерода, в которых один водород заменен фтором. Примерами являются -СF3,-CH2CH2F, -СН 2 СF3, c-Pr-F5, c-Hex-F11 и т.п. Подобным образом фторалкокси обозначает линейные, разветвленные и циклические структуры с указанным числом атомов углерода. Для целей данного описания в ситуациях,когда термин встречается два или более раз,определение этого термина в каждом случае не зависит от определения в каждом другом случае. Для целей данного описания галоген обозначает F, Сl, Вr или I. В другом варианте данное изобретение относится к фармацевтическим композициям для ингибирования ЦОГ-2 и для лечения опосредованных ЦОГ-2 заболеваний, описанных здесь,содержащим фармацевтически приемлемый носитель и нетоксичное терапевтически эффек 001444 10 тивное количество соединения формулы I, описанной выше. Еще в одном варианте данное изобретение относится к способу ингибирования циклооксигеназы и лечения опосредованных циклооксигеназой заболеваний, успешно излечиваемых активным агентом, который избирательно ингибирует ЦОГ-2 в сравнении с ЦОГ-1, как описано здесь, заключающемуся во введении пациенту, нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения формулы I, как описано здесь. Оптические изомеры - Диастереомеры - Геометрические изомеры Некоторые из описанных здесь соединений содержат один или несколько асимметричных центров и, следовательно, могут давать диастереомеры и оптические изомеры. Данное изобретение включает в себя такие возможные диастереомеры, а также их рацемические и разделенные, энантиомерно чистые формы и их фармацевтически приемлемые соли. Некоторые из описанных здесь соединений содержат этиленовые двойные связи и, если нет других указаний, включают как Е, так и Z геометрические изомеры. Соли Фармацевтические композиции данного изобретения содержат соединение формулы I в качестве активного ингредиента или его фармацевтически приемлемую соль и могут также содержать фармацевтически приемлемый носитель и, необязательно, другие терапевтические ингредиенты. Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований, в том числе неорганических оснований и органических оснований. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, соли трехвалентного марганца, соли двухвалентного марганца, калия, натрия,цинка и т.п. Особенно предпочтительны соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований,включают соли первичных, вторичных и третичных аминов, замещенных аминов, в том числе встречающихся в природе замещенных аминов, циклических аминов, и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, N-метилглюкамин, глюкамин, глюкозамин, гистидин, гидрабамин, N-(2-гидроксиэтил)пиперидин, N-(2 гидроксиэтил)пирролидин, изопропиламин, лизин, метил глюк амин, морфолин, пиперазин,пиперидин, полиаминовые смолы, прокаин, пу 11 рины, теобромин, триэтиламин, триметиламин,трипропиламин, трометамин и т.п. Когда соединение данного изобретения является основным, соли могут быть получены из фармацевтически приемлемых нетоксичных кислот, в том числе неорганических и органических кислот. Такие кислоты включают уксусную, адипиновую, аспарагиновую, 1,5-нафталиндисульфоновую, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, 1,2 этандисульфоновую, этансульфоновую, этилендиаминтетрауксусную, фумаровую, глюкогептоновую, глюконовую, глутаминовую, йодоводородную, бромоводородную, хлороводородную, изетионовую, молочную, малеиновую,яблочную, миндальную, метансульфоновую,муциновую, 2-нафталинсульфоновую, азотную,щавелевую, памовую, пантотеновую, фосфорную, пивалиновую, пропионовую, салициловую, стеариновую, янтарную, серную, винную,п-толуолсульфоновую,ундекановую,10 ундеценовую кислоту и т.п. Особенно предпочтительны лимонная, бромоводородная, хлороводородная, малеиновая, метансульфоновая, фосфорная,серная и винная кислоты. Должно быть понятно, что при обсуждении способов лечения, которое представлено далее, ссылки на соединения формулы I включают также фармацевтически приемлемые соли. Полезность Соединение формулы I может использоваться для ослабления боли, жара и воспаления при различных состояниях, включающих ревматическую атаку, симптомы, связанные с гриппом или другими вирусными инфекциями, простуду, боль в нижней части спины и шее, дисменорею, головную боль, зубную боль, растяжения, миозит, невралгию, синовит, артрит, в том числе ревматоидный артрит, дегенеративные заболевания суставов (остеоартрит), подагру и анкилозирующий спондилоартрит, бурсит,ожоги, повреждения, сопровождающие хирургические и зубные процедуры. Кроме того, такое соединение может ингибировать клеточные неопластические превращения и метастатический рост опухоли и, следовательно, может использоваться при лечении рака. Соединение I может быть также использовано при лечении и/или предотвращении опосредованных циклооксигеназой пролиферативных нарушений, таких, какие могут иметь место в случае диабетической ретинопатии и опухолевого ангиогенеза. Соединение I будет также ингибировать индуцируемое простаноидом сокращение гладких мышц путем предотвращения синтеза сокращающихся простаноидов и, следовательно,может быть использовано при лечении дисменореи, преждевременных родов, астмы и связанных с эозинофилами нарушений. Оно также может использоваться при лечении болезни Альцгеймера, для снижения потери костной 12 ткани у женщин в постклимактерическом периоде (то есть при лечении остеопороза) и при лечении глаукомы. Благодаря его высокой ингибирующей ЦОГ-2 активности и/или его специфичности в отношении ингибирования ЦОГ-2 в сравнении с ЦОГ-1, соединение I будет полезным в качестве альтернативы для общепринятых НСПВЛС (нестероидных противовоспалительных лекарственных средств) (NSAID), в частности, когда такие нестероидные противовоспалительные лекарственные средства могут быть противопоказаны, например, в случае пациентов с пептическими язвами, гастритом, регионарным энтеритом, язвенным колитом, дивертикулитом или с рецидивирующим анамнезом желудочнокишечных повреждений; в случае ЖК (желудочно-кишечного) кровотечения, нарушений свертывания крови, в том числе анемии, такой как гипопротромбинемия, гемофилия, или других связанных с кровотечением проблем; в случае заболевания почек; перед хирургическим вмешательством или при приеме антикоагулянтов. Фармацевтические композиции Для лечения любого из этих опосредованных циклооксигеназой заболеваний соединениеI может быть введено перорально, местно, парентерально, ингаляцией или ректально в виде стандартных лекарственных препаратов, содержащих общепринятые нетоксичные фармацевтически приемлемые носители, добавки и разбавители. Термин "парентеральный" в применении здесь включает подкожные инъекции, способы внутривенной, внутримышечной, внутригрудинной инъекции или инфузии. Кроме лечения таких теплокровных животных, как мыши,крысы, лошади, крупный рогатый скот, овцы,собаки, кошки и т.д., соединение данного изобретения эффективно при лечении человека. Соединения данного изобретения, в частности,являются хорошо подходящими для лошадей. Как указано выше, фармацевтические композиции для лечения опосредованных ЦОГ-2 заболеваний, описанных выше, могут необязательно содержать один или несколько ингредиентов, перечисленных выше. Фармацевтические композиции, содержащие активный ингредиент, могут быть в форме,пригодной для перорального применения, например, в виде таблеток, пастилок, лепешек,водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул или сиропов или эликсиров. Композиции, предназначенные для перорального применения, могут быть приготовлены согласно любому способу, известному в данной области для производства фармацевтических композиций, и такие композиции могут содержать один или несколько агентов, выбранных из группы, включающей подсластители, ароматизаторы, красители и консерванты, для обеспече 13 ния фармацевтически элегантных и приятных на вкус препаратов. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями,которые используются для получения таблеток. Эти наполнители могут представлять собой,например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза,фосфат кальция или фосфат натрия; гранулирующие или разрыхляющие агенты, например,кукурузный крахмал или альгиновую кислоту; связывающие агенты, например, крахмал, желатин или аравийскую камедь; и смачивающие агенты, например, стеарат магния, стеариновую кислоту или тальк. Таблетки могут быть непокрытыми или могут быть покрыты известными способами для замедления разрушения и всасывания в желудочно-кишечном тракте и, следовательно, обеспечения замедленного действия на протяжении более продолжительного периода времени. Например, может быть использован такой материал для пролонгирования действия,как глицерилмоностерат или глицерилдистеарат. Они могут быть также покрыты по способу,описанному в патентах США 4 256 108; 4 166 452 и 4 265 874, для получения осмотических терапевтических таблеток для регулируемого высвобождения. Препараты для перорального использования могут быть также представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция,фосфатом кальция или каолином, или мягких желатиновых капсул, в которых активные ингредиенты смешаны с водой или смешивающимися растворителями, такими как пропиленгликоль, ПЭГ и этанол, или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом. Водные суспензии содержат активный материал в смеси с наполнителями, пригодными для получения водных суспензий. Такими наполнителями являются суспендирующие агенты, например, натрий-карбоксиметилцеллюлоза,метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон,трагакантовая камедь и аравийская камедь; диспергирующие или смачивающие агенты могут быть встречающимся в природе фосфатидом,например, лецитином, или продуктами конденсации алкиленоксида с жирными кислотами,например, полиоксиэтиленстеаратом, или продуктами конденсации этиленоксида с алифатическими спиртами с длинной цепочкой, например, гептадекаэтиленоксицетанолом, или продуктами конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и гексита, такими как моноолеат полиоксиэтиленсорбита, или продуктами конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и 14 ангидридов гексита, такими как моноолеат полиэтиленсорбитана. Водные суспензии могут также содержать один или несколько консервантов, например, этил- или н-пропил-п-гидроксибензоат, бензиловый спирт, один или несколько красителей, один или несколько ароматизаторов и один или несколько подсластителей, таких как сахарин или аспартам. Масляные суспензии могут быть приготовлены суспендированием активного ингредиента в растительном масле, например, арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или минеральном масле,таком как жидкий парафин. Масляные суспензии могут содержать загустители, например,пчелиный воск, твердый парафин или цетиловый спирт. Подсластители, такие как описанные выше, и ароматизаторы могут быть добавлены для получения вкусного перорального препарата. Эти композиции могут сохраняться при добавлении антиоксиданта, такого как аскорбиновая кислота. Диспергируемые порошки и гранулы, пригодные для приготовления водной суспензии путем добавления воды, содержат активный ингредиент в смеси с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или несколькими консервантами. Пригодные диспергирующие или увлажняющие агенты и суспендирующие агенты уже описаны выше. Могут также присутствовать дополнительные наполнители, например, подсластители,ароматизаторы и красители. Фармацевтические композиции данного изобретения могут быть также в виде эмульсий типа масло-в-воде. Масляной фазой может быть растительное масло, например, оливковое масло или арахисовое масло, или минеральное масло,например, жидкий парафин, или их смеси. Пригодными эмульгирующими агентами могут быть встречающиеся в природе фосфатиды, например, лецитин сои, и сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и ангидридов гексита, например, моноолеат сорбитана, и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, например, моноолеат полиоксиэтиленсорбитана. Эмульсии могут также содержать подсластители и ароматизаторы. Сиропы и эликсиры могут быть приготовлены с подсластителями, например, глицерином, пропиленгликолем, сорбитом или сахарозой. Такие препараты могут также содержать уменьшающее раздражение средство, консервант и ароматизаторы и красители. Фармацевтические композиции могут быть в виде стерильной инъецируемой водной или масляной суспензии. Такая суспензия может быть приготовлена в соответствии с известными в данной области способами с использованием пригодных диспергирующих или увлажняющих агентов и суспендирующих агентов, которые упо 15 минались выше. Стерильный инъекционный препарат может быть также стерильным инъекционным раствором или суспензией в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые могут быть использованы, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Могут также использоваться сорастворители, такие как этанол, пропиленгликоль или полиэтиленгликоли. Кроме того, в качестве растворителя или суспендирующей среды удобно использовать нелетучие масла. Для этой цели может использоваться любое мягкое нелетучее масло, включающее синтетические моно- или диглицериды. Кроме того, при приготовлении инъекционных растворов могут найти применение жирные кислоты, такие как олеиновая кислота. Соединение формулы I может быть также введено в виде суппозиториев для ректального введения лекарственного средства. Эти композиции могут быть приготовлены смешиванием лекарственного средства с подходящим нераздражающим наполнителем, который является твердым при обычной температуре, но жидким при ректальной температуре и, следовательно,будет расплавляться в прямой кишке с высвобождением лекарственного средства. Такими материалами являются масло какао и полиэтиленгликоли. Для наружного применения используют кремы, мази, гели, растворы или суспензии и т.д., содержащие соединение формулы I. (Для целей данной заявки местное применение включает также использование жидкости для полоскания рта и горла). Препараты для местного применения обычно могут содержать фармацевтический носитель, сорастворитель, эмульгатор,усилитель проницаемости, консервант и мягчитель. Диапазоны доз При лечении вышеуказанных состояний применимы уровни доз порядка от около 0,01 мг до около 140 мг/кг массы тела в день или, альтернативно, от около 0,5 мг до около 7 г на пациента в день. Например, воспаление можно эффективно лечить введением от около 0,01 до 50 мг соединения на кг массы тела в день или,альтернативно, от около 0,5 мг до около 3,5 г на пациента в день. Количество активного ингредиента, которое может быть объединено с материаламиносителями для получения единичной дозированной формы, будет изменяться в зависимости от подлежащего лечению хозяина и от конкретного способа введения. Например, препарат,предназначенный для перорального введения человеку, может содержать от 0,5 мг до 5 г активного агента, объединенного с подходящим и удобным количеством материала-носителя, которое может изменяться от около 5 до около 16 95% всей композиции. Единичные дозированные формы обычно содержат от около 1 мг до около 500 мг активного ингредиента, обычно 25, 50, 100, 200, 300, 400, 500, 600, 800 или 1000 мг. Однако должно быть понятно, что уровень конкретной дозы для любого конкретного пациента будет зависеть от множества факторов, в том числе от возраста, массы тела, общего состояния здоровья, пола, пищевого рациона, времени введения, способа введения, скорости выведения, комбинации лекарственных средств и тяжести конкретного заболевания, подлежащего терапии. Комбинации с другими лекарствами Подобным образом соединение формулы I будет применимо в качестве частичного или полного заменителя общепринятых НСПВЛС в препаратах, в которых они в настоящее время вводятся совместно с другими агентами или ингредиентами. Таким образом, в дополнительных аспектах данное изобретение включает фармацевтические композиции для лечения опосредованных ЦОГ-2 заболеваний, таких как описанные выше, содержащие нетоксичное терапевтически эффективное количество соединения формулы I, описанные выше, и один или несколько ингредиентов, таких как другое болеутоляющее средство, в том числе ацетаминофен или фенацетин; потенциирующее средство,включающее кофеин; Н 2-антагонист, гидроксид алюминия или магния, симетикон, противозастойное средство, в том числе фенилэфрин, фенилпропаноламин, псевдофедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин,пропилгекседрин или леводезоксиэфедрин; противокашлевое средство, в том числе кодеин,гидрокодон, карамифен, карбетапентан или декстраметорфан; простагландин, в том числе мизопростол, энпростил, риопростил, орнопростол или розапростол; мочегонное средство; седативное или неседативное антигистаминное средство. Кроме того, данное изобретение включает способ лечения опосредованных циклооксигеназой заболеваний, предусматривающий введение пациенту, нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения формулы I, вводимого необязательно с одним или несколькими ингредиентами, только что перечисленными выше. Способы синтеза Соединения формулы I данного изобретения могут быть получены согласно путям синтеза, представленным в схемах 1-2, и при помощи описанных в изобретении способов. Схема 1 Пиридины формулы Iа и Ib могут быть получены путем многостадийной последовательности реакций из требуемого 2-аминопиридинаII. Исходное бромирование II бромом в уксусной кислоте дает бромид III. Катализируемое 17 палладием связывание III с 4-(метилтио)фенилбороновой кислотой в присутствии подходящего основания, такого как карбонат натрия, дает сульфид IV, который может быть окислен при помощи одного или нескольких окислителей,таких как ММРР, оксон, или OsO4/NMO до соответствующего сульфона V. АминопиридинV может быть превращен в галогенид VI при помощи одного из нескольких способов. Например, обработка V нитритом натрия в присутствии НСl и брома дает бромид VI (X=Br). Альтернативно, обработка V нитритом натрия и НСl с последующим взаимодействием с РОСl3 дает соответствующий хлорид VI (Х=Сl). Второе катализируемое палладием связывание VI с подходящим образом замещенным металлированным ароматическим соединением, таким как арилбороновая кислота или арилстаннан, дает пиридин формулы Iа. Подходящая модификация заместителя R2 в Iа дает дополнительные примеры Iа. Например, когда R2=Me, окисление таким окислителем, как КМnО 4, дает соответствующую кислоту (R2 Iа= СO2 Н), которая затем может быть превращена в сложный метиловый эфир (R2 Ia=CO2Me) с применением такого реагента, как диазометан. Альтернативно, обработка этой кислоты хлорсульфонилизоцианатом и ДМФ дает нитрил (R2 Ia=CN). Пиридинметилсульфоны Iа могут быть превращены в соответствующие пиридинсульфонамиды Ib с применением процедур, описанных в литературе (Huang Схема 2 2-Галогенпиридины VI схемы 1 могут быть получены многостадийным процессом из соответствующих 2-гидроксипиридинов VII. Сначала, обработка VII бромом в уксусной кислоте дает бромид VIII. Последующее взаимодействие VIII с бензилбромидом в присутствии основания, такого как карбонат серебра, дает бензиловый эфир IX, который может быть пре 001444 18 вращен в сульфон Х через последовательность реакций, подобных описанным для превращения бромида III в V в схеме 1. Бензильная защитная группа может быть удалена обработкойIX кислотой, такой как трифторуксусная кислота, с получением гидроксипиридина X. Нагревание Х с РОВr3 или РОСl3 дает соответствующие 2-галогенпиридины VI (X=Br, Cl) Схемы 1. Характерные соединения Таблицы 1 и 2 иллюстрируют соединения формулы Iа и Ib, которые являются характерными соединениями данного изобретения. Таблица 1 Тесты для определения биологической активности Соединение формулы I может быть испытано при помощи следующих тестов для определения их ингибирующей ЦОГ-2 активности. Ингибирование циклооксигеназной активности Соединения тестируют в качестве ингибиторов циклооксигеназной активности в циклооксигеназных тестах на целых клетках. Оба эти теста определяют синтез простагландина Е 2 в ответ на АА (арахидоновую кислоту) при помощи радиоиммуноанализа. Клетками, используемыми для этих тестов, являются линии клеток 143 остеосаркомы человека (которые специфически экспрессируют ЦОГ-2) и линии клеток U-937 человека (которые специфически экспрессируют ЦОГ-1). В этих тестах 100% активностью считают различие между синтезом простагландина Е 2 в отсутствие и в присутствии арахидоната. Тесты на целых клетках Для циклооксигеназных тестов клетки остеосаркомы культивируют в 1 мл среды в 24 луночных мультипланшетах Nunclon) до конфлюентности (1-2 х 105 клеток/лунку). КлеткиU-937 выращивают в роллерных колбах и ресуспендируют до конечной плотности 1,5 х 106 клеток/мл в 24-луночных мультипланшетахNunclon). После промывания и ресуспендирования клеток остеосаркомы и U-937 в 1 мл HBSS добавляют 1 мкл раствора в ДМСО тестсоединения или ДМСО-носителя и пробы тщательно смешивают. Все тесты выполняют, повторяя 3 раза. Затем пробы инкубируют в течение 5 или 15 мин при 37 С перед добавлением АА. АА (не содержащую пероксида, CaymanChemical) готовят в виде 10 мМ исходного раствора в этаноле и затем разбавляют в 10 раз вHBSS. К клеткам добавляют аликвоту 10 мкл этого разбавленного раствора с получением конечной концентрации АА 10 мкМ. Контрольные пробы инкубируют с этанолом-носителем вместо АА. Пробы опять тщательно смешивают и инкубируют еще в течение 10 мин при 37 С. Для клеток остеосаркомы реакции останавливают после этого добавлением 100 мкл 1N НСl с перемешиванием и быстрым удалением раствора из монослоев клеток. Для клеток U-937 реакции останавливают добавлением 100 мкл 1N НСl с перемешиванием. Затем пробы нейтрализуют добавлением 100 мкл 1N NaOH и уровниPGE2 измеряют радиоиммуноанализом. Тесты на целых клетках для ЦОГ-2 и ЦОГ-1 с применением линий трансфицированных клеток ЯКХ Линии клеток яичника китайского хомячка(ЯКХ), которые были стабильно трансфицированы эукариотическим экспрессирующим вектором pCDNAIII, содержащим кДНК либо ЦОГ 21 1, либо ЦОГ-2 человека, используют для этого теста. Эти клеточные линии называются, соответственно, ЯКХ[хЦОГ-1] и ЯКХ[хЦОГ-2]. Для циклооксигеназных тестов клетки ЯКХ[хЦОГ 1] из суспензионных культур и клетки ЯКХ[хЦОГ-2], полученные трипсинизацией прикрепленных культур, собирают центрифугированием (300 х g, 10 мин) и промывают один раз в HBSS, содержащий 15 мМ HEPES, рН 7,4,и ресуспендируют в HBSS, 15 мМ HEPES, рН 7,4, при концентрации клеток 1,5 х 106 клеток/мл. Тестируемые лекарственные средства растворяют в ДМСО в 66,7 раз для наивысшей концентрации лекарственного средства. Соединения обычно тестируют при 8 концентрациях,повторяя два раза, с применением 3-кратных серийных разведений в ДМСО наивысшей концентрации лекарственного средства. Клетки (0,3 х 106 клеток в 200 мкл) проинкубировали с 3 мкл тестируемого лекарственного средства или ДМСО-носителя в течение 15 мин при 37 С. Рабочие растворы не содержащей пероксида АА(5,5 мкМ и 110 мкМ АА для тестов ЯКХ[хЦОГ 1] и ЯКХ[хЦОГ-2], соответственно) получают 10-кратным разведением концентрированного раствора АА в этаноле в HBSS, содержащем 15 мМ HEPES, рН 7,4. Затем клетки стимулируют в присутствии или в отсутствие лекарственного средства раствором AA/HBSS с получением конечной концентрации 0,5 мкМ АА в тесте ЯКХ[хЦОГ-1] и конечной концентрации 10 мкМ в тесте ЯКХ[хЦОГ-2]. Реакцию останавливают добавлением 10 мкл 1N НСl с последующей нейтрализацией 20 мкл 0,5 N NaOH. Пробы центрифугируют при 300 х g при 4 С в течение 10 мин и пробу осветленного супернатанта подходящим образом разбавляют для определения уровней PGE2 с использованием иммуноферментного анализа для PGE2 (Correlate PGE2 enzyme immunoassay kit, Assay Designs, Inc.). Циклооксигеназную активность в отсутствие тестсоединений определяют как разность уровнейPGE2 тестсоединениями рассчитывают в виде процента активности в присутствии лекарственного средства относительно активности в положительных контрольных пробах. Тесты активности ЦОГ-1 микросом из клетокU-937 Клетки U-937 осаждают центрифугированием при 500 х g в течение 5 мин и промывают один раз забуференным фосфатом солевым раствором и повторно осаждают. Клетки ресуспендируют в буфере для гомогенизации, состоящем из 0,1 М Трис-HCl, рН 7,4, 10 мМ ЭДТА, 2 мкг/мл лейпептина, 2 мкг/мл соевого ингибитора трипсина, 2 мкг/мл апротинина и 1 мМ фенилметилсульфонилфторида. Клеточную суспензию обрабатывают ультразвуком 4 раза в течение 10 с и центрифугируют при 10000 х g в 22 течение 10 мин при 4 С. Супернатант центрифугируют при 100000 х g в течение 1 ч при 4 С. Осадок микросом 100000 х g ресуспендируют в 0,1 М Трис-HCl, рН 7,4, 10 мМ ЭДТА до приблизительно 7 мг белка/мл и хранят при -80 С. Препараты микросом оттаивают непосредственно перед использованием, подвергают кратковременной обработке ультразвуком и затем разбавляют до концентрации белка 125 мкг/мл в 0,1 М Трис-НСl-буфере, рН 7,4, содержащем 10 мМ ЭДТА, 0,5 мМ фенол, 1 мМ восстановленный глутатион и 1 мкМ гематин. Тесты выполняют, повторяя два раза, в конечном объеме 250 мкл. Сначала 5 мкл носителя ДМСО или лекарственного средства в ДМСО добавляют к 20 мкл 0,1 М Трис-НСl-буфера, рН 7,4, содержащего 10 мМ ЭДТА, в лунках 96-луночного полипропиленового титрационного планшета с глубокими лунками. Затем добавляют 200 мкл препарата микросом и прeинкубируют в течение 15 мин при комнатной температуре перед добавлением 25 мкл 1 М арахидоновой кислоты в 0,1 М Трис-HCl и 10 мМ ЭДТА, рН 7,4. Пробы инкубируют в течение 40 мин при комнатной температуре и реакцию останавливают добавлением 25 мкл 1N НСl. Пробы нейтрализуют 25 мкл 1N NaOH перед определением количестваPGE2 при помощи радиоиммуноанализа (тестнаборы Dupont-NEN или Amersham). Циклооксигеназную активность определяют в виде разности между уровнями PGE2 в пробах, инкубированных в присутствии арахидоновой кислоты и этанольного носителя. Тест активности очищенной ЦОГ-2 человека Активность фермента измеряют с применением хромогенного теста, основанного на окислении N,N,N',N'-тетраметил-п-фенилендиамина (TMPD) во время восстановления PGG2 доProc. Natl. Acad. Sci. 91, 11202-11206). Рекомбинантную ЦОГ-2 человека очищают из клеток Sf9, как описано ранее (Percival etal (1994) Arch. Biochem. Biophys. 15, 111118).Тест-смесь (180 мкл) содержит 100 мМ фосфат натрия, рН 6,5, 2 мМ генапол Х-100, 1 мкМ гематин, 1 мг/мл желатин, 80-100 единиц очищенного фермента (одна единица фермента определена как количество фермента, требующееся для получения изменения OD 0,001/мин при 610 нм) и 4 мкл тест-соединения в ДМСО. Смесь прeинкубируют при комнатной температуре (22 С) в течение 15 мин перед инициированием ферментативной реакции путем добавления 20 мкл обработанного ультразвуком раствора 1 мМ АА и 1 мМ TMPD в тест-буфере(без фермента или гематина). Активность фермента измеряют оценкой начальной скорости окисления TMPD на протяжении первых 36 с реакции. Неспецифическую скорость окисления наблюдают в отсутствие фермента (0,007-0,010OD/мин) и вычитают перед расчетом % ингибирования. Величины IC50 определяют методом 23 наименьших квадратов для нелинейного регрессионного анализа с 4 параметрами графика выраженной в логарифмических единицах концентрации против % ингибирования. Анализ цельной человека Обоснование Цельная кровь человека обеспечивает богатую белком и клетками среду, пригодную для исследования биохимической эффективности противовоспалительных соединений, таких как избирательные ингибиторы ЦОГ-2. Исследования показали, что кровь здорового человека не содержит фермента ЦОГ-2. Это согласуется с наблюдением, что ингибиторы ЦОГ-2 не влияют на образование PGE2 в нормальной крови. Эти ингибиторы активны только после инкубирования цельной крови человека с LPS (липололисахаридом), который индуцирует ЦОГ-2. Этот тест может быть использован для оценки ингибирующего действия избирательных ингибиторов ЦОГ-2 на образование PGE2. Тромбоциты в цельной крови также содержат большое количество фермента ЦОГ-1. Сразу же после свертывания крови тромбоциты активируются по опосредованному тромбином механизму. Эта реакция приводит к продуцированию тромбоксана В 2 (ТхВ 2) через активацию ЦОГ-1. Следовательно, действие тест-соединений на уровни ТхВ 2 после свертывания крови может быть определено и использовано как показатель активности ЦОГ-1. Таким образом, степень избирательности тест-соединением может быть определена измерением уровней PGE2 после индукции LPS (ЦОГ-2) и TxB2 после свертывания крови (ЦОГ-1) в одном и том же тесте. Метод Стадия А. ЦОГ-2 (индуцированное LPS продуцирование PGE2). Свежую кровь собирают в гепаринизированные пробирки венопункцией у волонтеров(мужчин и женщин). Эти субъекты не имеют видимых воспалительных состояний и они не принимали каких-либо НСПВЛС в течение, по меньшей мере, 7 дней перед взятием крови. Сразу же получают плазму крови из аликвоты 2 мл крови для применения в качестве слепого контроля (фоновых уровней PGE2). Остальную кровь инкубируют с LPS (100 мкг/мл, конечная концентрация, Sigma Chem. L-2630 из Е. coli; разбавленный в 0,1% БСА (в забуференном фосфатом солевом растворе) в течение 5 мин при комнатной температуре. Аликвоты 500 мкл крови инкубируют либо с 2 мкл носителя(ДМСО), либо с 2 мкл тест-соединения при конечных концентрациях, варьирующих от 10 нМ до 30 мкМ, в течение 24 ч при 37 С. В конце инкубирования кровь центрифугируют при 12000 х g в течение 5 мин для получения плазмы. Аликвоту 100 мкл плазмы смешивают с 400 мкл метанола для осаждения белка. Получают супернатант и его анализируют на PGE2 при помощи набора для радиоиммуноанализа (Am 001444ersham, RPA530) после превращения PGE2 в его метилоксиматное производное в соответствии с процедурой изготовителя. Стадия В. ЦОГ-1 (индуцированное свертыванием продуцирование ТхВ 2). Свежую кровь собирают в вакутейнеры, не содержащие антикоагулянтов. Аликвоты 500 мкл сразу же переносят в силиконизированные микроцентрифужные пробирки, предварительно загруженные (2 мкл) либо ДМСО, либо тестсоединением при конечных концентрациях от 10 нМ до 30 мкМ. Пробирки перемешивают вортексом и инкубируют при 37 С в течение 1 ч, давая крови свертываться. В конце инкубирования сыворотку получают центрифугированием (12000 х g в течение 5 мин). Аликвоту 100 мкл сыворотки смешивают с 400 мкл метанола для осаждения белка. Получают супернатант и его анализируют на ТхВ 2 с применением набора для ферментного иммуноанализа (Cayman,519031) в соответствии с инструкцией изготовителя. Тест отека лапки крысы Протокол Самцов крыс Sprague-Dawley (150-200 г) подвергают голоданию в течение ночи и вводят им перорально либо носитель (1% метоцел или 5% Твин 80), либо тест-соединение. Спустя 1 ч проводят стойким маркером линию на уровне выше голеностопного сустава на одной задней лапке для определения зоны лапки, подлежащей наблюдению. Объем лапки (V0) измеряют при помощи плетизмометра (Ugo-Basile, Italy), основанного на принципе вытеснения воды. Затем животных инъецируют субплантарно (через подошву) 50 мкл 1% раствора каррагенана в солевом растворе (FMC Corp. Maine) в лапку при помощи инсулинового шприца с иглой 25 G(т.е. 500 мкг каррагенана на лапку). Спустя 3 ч измеряют объем лапки (V3) и рассчитывают увеличения в объеме лапки (V3-V0). Животных умерщвляют асфиксией при помощи СO2 и оценивают отсутствие или наличие повреждений желудка. Данные сравнивают с величинами для контроля-носителя и рассчитывают процентное ингибирование. Все группы обработки шифруют для исключения искажения (вызываемого ошибкой наблюдателя). Индуцируемая НСПВЛС гастропатия у крыс Обоснование Основным побочным действием общепринятых НСПВЛС является их способность производить желудочные повреждения у людей. Считается, что это действие обусловлено ингибированием ЦОГ-1 в желудочно-кишечном тракте. Крысы особенно чувствительны к действиям НСПВЛС. В самом деле, крысиные модели использовали обычно в прошлом для оценки желудочно-кишечных побочных эффектов существующих общепринятых НСПВЛС. В данном тесте индуцируемое НСПВЛС желудочно-кишечное повреждение наблюдают путем 25 измерения фекальной экскреции 51 Сr после системной инъекции 51 Сr-меченых эритроцитов. Фекальная 51 Сr-экскреция представляет собой хорошо отработанный и чувствительный способ для обнаружения желудочно-кишечной целостности у животных и людей. Способы Самцам крыс Sprague-Dawley (150-200 г) вводят перорально тест-соединение либо один раз (острое дозирование), либо b.i.d. (два раза в день) в течение 5 дней (хроническое дозирование). Сразу же после введения последней дозы крыс инъецируют через хвостовую вену 0,5 мл 51 Сr-меченых эритроцитов от крысы-донора. Животных помещают индивидуально в клетки для исследования метаболизма с кормом и водой ad libitum. Фекалии собирают в течение периода 48 ч и 51 Сr-экскрецию рассчитывают в виде процента от общей инъецированной дозы. 51 Сr-меченые эритроциты получают при помощи следующих процедур. 10 мл крови собирают в гепаринизированные пробирки через vena cava(полую вену) от крысы-донора. Плазму удаляют центрифугированием и снова восполняют равным объемом HBSS. Эритроциты инкубируют с 400 мкКи 51 хромата натрия в течение 30 мин при 37 С. В конце инкубирования эритроциты промывают дважды 20 мл HBSS для удаления свободного 51 хромата натрия. Наконец, эритроциты воссоздают (реконституируют) в 10 млHBSS и 0,5 мл этого раствора (около 20 мкКи) инъецируют на крысу. Гастропатия с потерей белка у беличьих обезьян Обоснование Гастропатия с потерей белка (проявляющаяся в виде появления циркулирующих клеток и белков плазмы в желудочно-кишечном тракте) является существенной и ограничивающей дозу ответной реакцией на стандартные нестероидные противовоспалительные лекарственные средства (НСПВЛС). Она может быть количественно оценена внутривенным введением раствора 51 СrCl3. Этот изотопный ион может авидно связываться с клеткой и сывороточными глобинами и эндоплазматическим ретикулумом клетки. Измерения радиоактивности, появляющейся в фекалиях, собираемых в течение 24 ч после введения изотопа, обеспечивает, следовательно, чувствительный и количественный показатель гастропатии с потерей белка. Способы Группы самцов беличьих обезьян (0,8-1,4 кг) обрабатывают путем кормления через желудочный зонд либо 1% метоцелом, либо 5% Твином 80 в Н 2O-носителях, (3 мл/кг b.i.d.), либо тест-соединениями при дозах от 1 до 100 мг/кгb.i.d. в течение 5 дней. 51 Сr (5 мкКи/кг в 1 мл/кг забуференного фосфатом солевого раствора(PBS вводят внутривенно спустя 1 ч после последней дозы лекарственного средства/носителя и фекалии собирают в течение 24 ч в клетке для 26 исследования метаболизма и оценивают по гамма-счету 51 Сr. Венозную кровь собирают спустя 1 ч и 8 ч после последней дозы лекарственного средства и концентрации лекарственного средства в плазме измеряют при помощи ОФВЭЖХ. Индуцируемая LPS гипертермия у находящихся в сознании крыс Самцов крыс Sprague-Dawley (150-200 г) подвергали голоданию в течение 16-18 ч перед использованием. Приблизительно в 9 ч 30 мин утра животных помещали временно в фиксирующие камеры из плексигласа и их фоновую ректальную температуру регистрировали при помощи гибкого температурного зонда (YSIseries 400), соединенного с цифровым термометром (Model 08502, Cole Parmer). Одни и те же зонд и термометр использовали для всех животных для уменьшения экспериментальной ошибки. После измерений температуры животных возвращали в их клетки. При времени ноль,крыс инъецировали внутрибрюшинно либо солевым раствором, либо LPS (липополисахаридом) (2 мг/кг, Sigma Chem.) и ректальную температуру повторно измеряли при 5, 6 и 7 ч. после инъекции LPS. После измерения при 5 ч,когда увеличение температуры достигало плато,инъецированным LPS крысам давали перорально либо носитель (1% метоцел), либо тестсоединение для определения, может ли это соединение обращать гипертермию. Процент обращения гипертермии рассчитывали с использованием ректальной температуры, полученной при 7 ч. в контрольной (обработанной носителем) группе, в качестве сравнительной (обращение ноль) точки. Полное обращение гипертермии до величины фона перед LPS принимали за 100%. Индуцируемая LPS гипертермия у находящихся в сознании беличьих обезьян Температурные зонды хирургически имплантировали под кожу живота в группе беличьих обезьян (Saimiri sciureus) (1,0-1,7 кг). Это позволяет наблюдать температуру тела у находящихся в сознании, не подвергнутых фиксации обезьян при помощи телеметрической воспринимающей системы (Data Sciences International, Minnesota). Животных подвергали голоданию и помещали в индивидуальные клетки для акклиматизации за 13-14 ч. до использования. На боковой стороне клеток устанавливали электронные приемники, которые принимали сигналы от имплантированных температурных зондов. Приблизительно в 9 ч 00 мин утра в день эксперимента обезьян временно фиксировали в стульях для дрессировки и давали им болюсную I.V. (внутривенную) инъекцию LPS(6 мг/кг, растворенные в стерильном солевом растворе). Животных возвращали в их клетки и температуру тела регистрировали непрерывно каждые 5 мин. Через два часа после инъекцииLPS, когда температура тела увеличивалась на 27 1,5-2 С, обезьянам давали перорально дозу либо носителя (1% метоцел), либо тест-соединения (3 мг/кг). Спустя 100 минут определяли разность между температурой тела и фоновой величиной. Процентное ингибирование рассчитывали, принимая величину в контрольной группе за 0% ингибирования. Острая воспалительная гипералгезия, индуцированная каррагенаном у крыс Эксперименты проводили, используя самцов крыс Sprague-Dawley (90-110 г). Гипералгезию к механическому сжатию задней лапки индуцировали интраплантарно (в подошву) инъекцией каррагенана (4,5 мг в одну заднюю лапку) за 3 ч до использования. Контрольные животные получали равный объем солевого раствора (0,15 мл в подошву). Тест-соединение(0,5% метоцел) вводили перорально (2 мл/кг) через 2 ч после каррагенана. Голосовую ответную реакцию на сжатие задней лапки измеряли через 1 ч при помощи алгезиметра Ugo Basile. Статистический анализ индуцируемой каррагенаном гипералгезии выполняли при помощи однофакторного дисперсионного анализаANOVA (BMDP Statistical Software Inc.). Гипералгезию определяли вычитанием голосового порога у инъецированных солевым раствором крыс из голосового порога, который получали у животных,инъецированных каррагенаном. Оценки гипералгезии для получивших лекарственное средство крыс выражали в виде процента этого ответа. Затем величины ID50 (дозы,дающей 50% максимального наблюдаемого ответа) рассчитывали при помощи нелинейного регрессионного анализа средних величин с применением метода наименьших квадратов с использованием GraFit (Erithacus Software). Индуцируемый адъювантом артрит у крыс Семьдесят 6,5-7,5-недельных самок крыс(группа негативного контроля, в которой не индуцировали артрит, группа контроля с носителем, группа позитивного контроля, которой вводили индометацин с суточной дозой 1 мг/кг,и четыре группы, которым вводили тестсоединение при общих суточных дозах 0,10-3,0 мг/кг), так чтобы веса тел были равными в каждой группе. Шесть групп из 10 крыс каждая получали инъекции в заднюю лапку 0,5 мг Mycobacterium butyricum в 0,1 мл легкого минерального масла (адъюванта), а группа негативного контроля из 10 крыс не получала инъекций адъювантом. Веса тел, объемы контралатеральных лапок (определяемые плетизмографией с вытеснением ртути) и латеральные радиографы(рентгенограммы) (полученные под анестезией с применением Кетамина и Ксилазина) определяли до инъекции адъюванта (день -1) и спустя 21 день после инъекции адъюванта и первичные 28 объемы лапки определяли до инъекции адъюванта (день -1) и в дни 4 и 21 после инъекции адъюванта. Крыс анестезировали внутримышечной инъекцией 0,03-0,1 мл комбинации Кетамина (87 мг/кг) и Ксилазина (13 мг/кг) для радиографов и инъекций адъюванта. Радио графы получали для обеих задних лапок в дни 0 и 21 с использованием Faxitron (45 kvp, 30 с) и пленки Kodak X-OMAT TL и проявляли в автоматизированном процессоре. Радиографы (рентгенограммы) оценивались на изменения в мягких и твердых тканях исследователем, который не знал экспериментальных вариантов. Следующие радиографические изменения распределялись по степеням в виде цифр в соответствии с тяжестью: увеличенный объем мягкой ткани (0-4), сужение или расширение полостей суставов (0-5), субхондральная эрозия (0-3),периостеальная реакция (0-4), остеолиз (0-4),подвывих (0-3) и дегенеративные изменения сустава (0-3). Для установления цифровой оценки тяжести для каждого изменения на рентгенограмме использовали специфические критерии. Максимальная возможная оценка на лапку была 26. Тест-соединение при общих суточных дозах 0,1, 0,3, 1 и 3 мг/кг/день, индометацин при общей суточной дозе 1 мг/кг/день или носитель(0,5% метоцел в стерильной воде) вводили перорально b.i.d. (два раза в день), начиная после инъекции адъюванта и продолжая в течение 21 дня. Соединения готовили еженедельно, держали в холодильнике в темноте до использования и перемешивали вортексом непосредственно перед введением. Двухфакторный ("вариант обработки" и"время") дисперсионный анализ с повторяемыми измерениями во "времени" применяли к % изменениям веса тела и объемов лапок (стоп) и к упорядоченным данным общих оценок радиографов. Проводили post hoc-тест Dunnett для сравнения действия вариантов обработок с носителем. Однофакторный дисперсионный анализ применяли для веса тимуса и селезенки с последующим тестом Dunnett для сравнения действия обработок с носителем. Кривые дозаответ для % ингибирования объемов лапок в дни 4, 14 и 21 строили с использованием логистической функции с 4 параметрами при помощи нелинейной регрессии методом наименьших квадратов. ID50 определяли как дозу, соответствующую 50% уменьшению от носителя, и получали интерполяцией из полученного уравнения с 4 параметрами. Фармакокинетика в крысах Пероральная фармакокинетика в крысах Животных содержат, кормят и обеспечивают уход в соответствии с Правилами Канадского Совета по уходу за животными. Самцов крыс Sprague-Dawley (325-375 г) подвергают голоданию в течение ночи для каждого исследования уровня в крови при пероральном введении. 29 Крыс помещают в фиксирующую камеру по одной и камеру прочно фиксируют. Нулевое значение пробы крови получают отсечением небольшого (1 мм или менее) кусочка от кончика хвоста. Затем хвост гладят прочным, но мягким движением сверху вниз для "выдаивания" крови. Приблизительно 1 мл крови собирают в гепаринизированную пробирку-вакутейнер. Соединения готовят, как положено, в стандартном объеме для дозирования 10 мл/кг и вводят перорально проведением иглы для кормления через зонд 16 G, 3" в желудок. Последующие взятия крови выполняют так же, как взятие крови для нулевого значения, за исключением того, что нет необходимости опять отсекать кончик хвоста. Хвост очищают кусочком марли и "выдаивают"/гладят, как описано выше, в соответственно помеченные пробирки. Сразу же после взятия крови кровь центрифугируют, отделяют, помещают в четко помеченные флаконы и хранят в холодильнике до анализа. Типичные временные точки для определения уровней в крови крысы после РОдозирования: 0,15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч. После 4-часовой точки взятия крови крысам дают корм ad libitum. Воду предоставляют все время в процессе исследования. Носители: В определениях уровней в крови крыс при РО-дозировании могут быть использованы следующие носители:Methocel 0,5%-1%: 10 мл/кг; Твин 80: 10 мл/кг. Соединения для РО-уровней в крови могут находиться в форме суспензий. Для лучшего растворения раствор может быть помещен в прибор для обработки ультразвуком приблизительно на 5 мин. Для анализа аликвоты разбавляют равным объемом ацетонитрила и центрифугируют для удаления осадка белка. Супернатант инъецируют непосредственно на колонку С-18 ВЭЖХ с УФ-детектированием. Количественное определение выполняют относительно чистой пробы крови, в которую введено известное количество лекарственного средства. Биодоступность (F) оценивают по сравнительной площади под кривой (AUC) i.v. против р.о. Скорости клиренса рассчитывают из следующего отношения 30 Животных содержат, кормят и обеспечивают уход в соответствии с Правилами Канадского Совета по уходу за животными. Самцов крыс Sprague-Dawley (325-375 г) помещают в пластиковые клетки с размером коробок для туфель с подвешенным дном,крышкой клетки, поилкой для воды и кормом. Соединение готовят, как положено, в стандартном объеме для дозирования 1 мл/кг. У крыс берут пробу крови для нулевой величины и их дозируют при воздействии СО 2 в качестве седативного средства. Крыс, по одной,помещают в заполненную CO2 камеру и вынимают, как только они теряют их рефлекс выпрямления. Затем крысу помещают на фиксирующий столик, носовой конус с подачей СО 2 помещают над мордой и крысу закрепляют на столике эластичными бинтами. При помощи пинцета и ножниц обнажают яремную вену и берут нулевую пробу, после чего отмеренную дозу соединения инъецируют в яремную вену. Легко зажимают пальцем место инъекции и носовой конус удаляют. Замечают время. Это время является временем нулевой точки. 5-Минутное взятие крови выполняют отсечением кусочка (1-2 мм) от кончика хвоста. Затем гладят прочным, но мягким движением сверху вниз для "выдаивания" крови. Приблизительно 1 мл крови собирают в гепаринизированный флакон для сбора крови. Последующие взятия крови выполняют подобным образом, за исключением того, что нет необходимости опять отсекать кончик хвоста. Хвост очищают кусочком марли и берут кровь, как описано выше, в соответствующие помеченные пробирки. Типичные временные точки для определения уровней в крови после I.V.-дозирования: 0,5 мин, 15 мин, 30 мин, 1 ч, 2 ч, 6 ч или 0,5 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч. Носители: В определениях уровней в крови крыс приIV-дозировании могут быть использованы следующие носители: Декстроза: 1 мл/кг 1 мл/кг ограничение до объема дозы 0,1 мл на животное не более 60% в смеси с 40% стерильной воды -1 мл/кг В случае декстрозы, если раствор мутный,может быть добавлен бикарбонат натрия или карбонат натрия. Для анализа аликвоты разбавляют равным объемом ацетонитрила и центрифугируют для удаления осадка белка. Супернатант инъецируют непосредственно на колонку С-18 ВЭЖХ с УФ-детектированием. Количественное определение выполняют относительно чистой пробы крови, в которую введено известное количество лекарственного средства. Биодоступность (F) оценивают по сравнительной площади под кривой (AUC) i.v. против р.о. Скорости клиренса рассчитывают из следующего отношения: Единицы CL - мл/чкг (миллилитры в час на килограмм). Репрезентативные биологические данные Соединения данного изобретения являются ингибиторами ЦОГ-2 и поэтому применимы для лечения опосредованных ЦОГ-2 заболеваний, перечисленных выше. Активности соединений против циклооксигеназы можно видеть в репрезентативных результатах, показанных ни Пример Аа 32 же. В этом тесте ингибирование определяют измерением количества простагландина Е 2(PGE2), синтезированного в присутствии АА,ЦОГ-1 или ЦОГ-2 и предполагаемого ингибитора. Величины IC50 представляют концентрацию предполагаемого ингибитора, требующуюся для снижения синтеза PGE2 до 50% синтеза, полученного в сравнении с неингибированным контролем. Данные из трех этих биологических тестов даны для репрезентативных соединений вместе со сравнительными данными для следующих двух соединений из World Patent Application 96/10012: Как можно видеть из этих данных, соединения данного изобретения обнаруживают более высокую избирательность в отношении ЦОГ-2 и активность, чем Аа и Ab. Кроме того,основность пиридинового кольца в этих примерах позволяет образование кислотноаддитивных солей, что приводит к увеличенной водорастворимости и создает потенциал для перентерального введения в водных носителях. Далее данное изобретение проиллюстрировано следующими неограничивающими примерами, в которых, если нет других указаний:(i) все операции проводили при комнатной температуре или температуре окружающей среды, т.е. при температуре в пределах 18-25 С, и высушивание органических веществ выполняли с использованием MgSO4;(ii) выпаривание растворителя проводили с использованием роторного испарителя при по 5,8 3,8 ниженном давлении (600-4000 Па: 4,5-30 мм рт. ст.) с температурой бани до 60 С;(iii) ход реакции контролировали с помощью тонкослойной хроматографии (ТСХ), и время реакции указано лишь для целей иллюстрации;(iv) точки плавления даны без поправок и"разл." указывает разложение; представленные точки плавления являются температурами плавления, полученными для веществ, приготовленных, как описано; полиморфизм может приводить к выделению веществ с различными точками плавления в некоторых препаратах;(v) структуру и чистоту всех конечных продуктов подтверждали, по меньшей мере,одним из следующих способов: ТСХ, массспектрометрия, спектрометрия ядерного магнитного резонанса (ЯМР) или микроаналитическими данными;(vi) выходы даны только для иллюстрации;(vii) если данные ЯМР представлены, то они даны в форме величин дельтадля основных диагностических протонов, представленных в миллионных долях (ррm), относительно тетраметилсилана (TMS) в качестве внутреннего стандарта, определенных при 300 мГц или 400 мГц с использованием указанного растворителя; общепринятыми аббревиатурами, используемыми для формы сигнала, являются: s,синглет, d, дуплет, t, триплет, m, мультиплет, br,широкий; и т.д., кроме того, '"Аr" обозначает ароматический сигнал;(viii) химические символы имеют их обычные значения; использовались также следующие аббревиатуры: V (объем), W (вес), т. кип. (точка кипения), т. пл. (точка плавления), л (литр, литры),мл (миллилитры), г (граммы), мг (миллиграммы), моль (моли), ммоль (миллимоли), экв. (эквиваленты), к.т. (комнатная температура). Пример 1. 3-(4-Метилсульфонил)фенил-2 фенил-5-трифторметилпиридин. Стадия 1. 2-Амино-3-бром-5-трифторметилпиридин. К раствору 2-амино-5-трифторметилпиридина (9 г) в уксусной кислоте (75 мл) медленно добавляли бром (5,8 мл). Через 1 ч кислоту нейтрализовали осторожным добавлением гидроксида натрия (10N) при 0 С. Полученный оранжевый остаток растворяли в эфире и промывали последовательно насыщенным карбонатом калия, насыщенным Na2SO3 и насыщенным раствором соли, сушили и концентрировали. Оставшееся твердое вещество интенсивно перемешивали в гексане в течение 1 ч с получением, после фильтрования, соединения заголовка в виде белого твердого вещества (10,2 г). Стадия 2. 2-Амино-3-(4-метилтио)фенил-5 трифторметилпиридин. Смесь бромида со стадии 1, 4-метилтиобензолбороновой кислоты (Li, et al. J. Med.Chem. 1995, 38, 4570) (8,5 г), 2 М водного карбоната натрия (60 мл) и палладий-тетракис (трифенилфосфина) (490 мг) в смеси этанол/бензол(100 мл, 1:1) нагревали с обратным холодильником в течение 15 ч. Смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали эфиром. Органические вещества концентрировали и остаток энергично перемешивали в смеси эфир/гексан в течение 1 ч с получением, после фильтрования, соединения заголовка (11,2 г) в виде бежевого твердого продукта. Стадия 3. 2-Амино-3-(4-метилсульфонил) фенил-5-трифторметилпиридин. Смесь 2-амино-3-(4-метилтио)фенил-5 трифторметилпиридина (9,7 г), OsO4 (2 мл 4% раствора в воде) и NMO (13 г) в смеси ацетон/вода (60 мл: 5 мл) перемешивали при комнатной температуре в течение ночи. Затем добавляли насыщенный водный Nа 2SО 3 и полу 001444 34 ченную смесь перемешивали в течение 30 мин. Ацетон выпаривали и полученную смесь экстрагировали эфиром и этилацетатом. Объединенные органические экстракты промывалиNа 2SO3, водой, насыщенным раствором соли и затем концентрировали. Твердый остаток интенсивно перемешивали в гексане и эфире в течение 1 ч. и затем фильтровали с получением соединения заголовка в виде бледно-желтого твердого вещества (9,9 г). Стадия 4. 2-Хлор-3-(4-метилсульфонил) фенил-5-трифторметилпиридин. К раствору 2-амино-3-(4-метилсульфонил) фенил-5-трифторметилпиридина (1,2 г) в смеси вода/концентрированная НСl (9,5 мл : 1 мл) при 0 С добавляли раствор нитрита натрия (262 мг) в 5 мл воды. Смесь нагревали до комнатной температуры и перемешивали в течение ночи. Добавляли еще 30 мг нитрита натрия и после 3 ч гетерогенную смесь фильтровали. Часть твердого вещества (250 мг) и РОСl3 (110 мкл) в ДМФ(2 мл) нагревали при 70 С в течение 60 ч. Смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли, сушили и концентрировали с получением соединения заголовка в виде бледножелтого твердого вещества (270 мг), которое использовали сразу в следующей реакции. Стадия 5. 3-(4-Метилсульфонил)фенил-2 фенил-5-трифторметилпиридин. Смесь 2-хлор-3-(4-метилсульфонил)фенил 5-трифторметил-пиридина (260 мг), бензолбороновой кислоты (113 мг), 2 М водного карбоната натрия (2,1 мл) и палладий-тетракис (трифенилфосфина) (30 мг) в смеси этанол/бензол (8 мл, 1:1) нагревали с обратным холодильником в течение 24 ч. Смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом. Органический слой концентрировали и твердое вещество подвергали флэш-хроматографии (с элюированием смесью гексан/этилацетат, 4:1 об/об) с получением соединения заголовка в виде белого твердого вещества, т. пл. 191-192 С (215 мг). Пример 2. 2-(3-Хлорфенил)-3-(4-метилсульфонил)фенил-5-трифторметилпиридин. Стадия 1. 2-Бром-3-(4-метилсульфонил) фенил-5-трифторметилпиридин. К раствору 2-амино-3-(4-метилсульфонил) фенил-5-трифторметил-пиридина примера 1,стадии 3 (2 г), в 48% НВr (25 мл) при 0 С добавляли порциями бром (3 мл) и затем нитрит натрия (1,1 г). После 2 ч раствор нейтрализовали добавлением гидроксида натрия (10N) и затем экстрагировали этилацетатом. Органический слой промывали насыщенным Na2SO3 и насыщенным раствором соли, сушили и концентрировали. Флэш-хроматография оставшегося продукта (с элюированием смесью гексан/ этилацетат, 7:3 - 3:7 об/об) давала соединение заголовка в виде белого твердого вещества (435 мг). 35 Стадия 2. 2-(3-Хлорфенил)-3-(4 метилсульфонил)фенил-5-трифторметилпиридин. Смесь 2-бром-3-(4-метилсульфонил)фенил-5-трифторметилпиридина (178 мг), 3 хлорфенилбороновой кислоты (110 мг), фосфата калия (225 мг) и палладий-тетракис (трифенилфосфина) (20 мг) в диоксане (10 мл) нагревали с обратным холодильником в течение 24 ч. После охлаждения при комнатной температуре смесь разбавляли водой и экстрагировали эфиром. Органический экстракт сушили и концентрировали и оставшийся продукт подвергали флэшхроматографии (с элюированием смесью гексан/этилацетат, 7:3 об/об). Полученное твердое вещество энергично перемешивали в смеси гексан/эфир в течение 1 ч с получением соединения заголовка в виде бледно-желтого твердого вещества, т. пл. 136-137 С (115 мг). Пример 3. 2-(4-Хлорфенил)-3-(4-метилсульфонил)фенил-5-трифторметилпиридин. Согласно приемам, описанным в примере 1 стадии 5, но с заменой бензолбороновой кислоты 4-хлорфенилбороновой кислотой соединение заголовка получали в виде белого твердого вещества, т. пл. 192-193 С (155 мг). Пример 4. 2-(4-Фторфенил)-3-(4-метилсульфонил)фенил-5-трифторметилпиридин. Согласно приемам, описанным в примере 1 стадии 5, но с заменой бензолбороновой кислоты 4-фторфенилбороновой кислотой соединение заголовка получали в виде белого твердого вещества, т. пл. 163-164 С (152 мг). Пример 7. 3-(4-Метилсульфонил)фенил-2(3-пиридинил)-5-трифторметилпиридин. Смесь 2-бром-3-(4 метилсульфонил)фенил-5 трифторметилпиридина (600 мг) (пример 2, стадия 2), диэтил-3-пиридинилборана (255 мг),карбоната натрия (2 М, 2,2 мл) и дибромида бис(трифенилфосфин)палладия (25 мг) в смеси бензол/этанол (1:1, 32 мл) нагревали с обратным холодильником в течение 24 ч. После охлаждения до комнатной температуры смесь концентрировали, разбавляли водой и экстрагировали этилацетатом. Органический экстракт концентрировали и оставшийся продукт растворяли в смеси 10% HCl/эфир. Органическую фазу удаляли и водную фазу доводили до рН 10 путем добавления насыщенного бикарбоната натрия. Смесь экстрагировали этилацетатом и объединенные органические экстракты концентрировали и подвергали флэш-хроматографии (с элюированием этилацетатом) с получением соединения заголовка в виде белого твердого вещества, т. пл. 171-172 С (180 мг). Пример 13. 5-Метил-3-(4-метилсульфонил) фенил-2-фенилпиридин. Стадия 1. 2-Aмино-3-бром-5-метилпири дин. К раствору 2-амино-5-пиколина (5 г) в уксусной кислоте (40 мл) при комнатной темпера 001444 36 туре медленно добавляли бром (2,6 мл). Через 1 ч кислоту нейтрализовали осторожным добавлением гидроксида натрия (10N) при 0 С. Полученный оранжевый остаток растворяли в эфире и промывали последовательно насыщенным карбонатом калия, насыщенным Nа 2S2 О 3 и насыщенным раствором соли, сушили и концентрировали. Флэш-хроматография (с элюированием смесью гексан/этилацетат, 3:2 об/об) оставшегося твердого вещества давала соединение заголовка в виде бледно-желтого твердого вещества (7,1 г). Стадия 2. 2-Амино-5-метил-3-(4-метилсульфонил)фенилпиридин. Согласно приемам, описанным в примере 1, стадии 2 и 3, но с заменой 2-амино-3-бром-5 трифторметилпиридина 2-амино-3-бром-5-метилпиридином (7,1 г) со стадии 1, соединение заголовка получали в виде бледно-желтого твердого вещества (3,7 г). Стадия 3. 2-Бром-5-метил-3-(4-метилсульфонил)фенилпиридин. Согласно приемам, описанным в примере 2, стадия 1, но с заменой 2-амино-3-(4-метилсульфонил)фенил-5-трифторметилпиридина 2 амино-5-метил-3-(4-метилсульфонил)фенилпиридином (3 г) со стадии 2, соединение заголовка получали в виде белого твердого вещества (2,7 г). Стадия 4. 5-Метил-3-(4-метилсульфонил)фенил-2-фенилпиридин. Согласно приемам, описанным в примере 1, стадия 5, но с заменой 2-хлор-3-(4-метилсульфонил)фенил-5-трифторметилпиридина 2-бром 5-метил-3-(4-метилсульфонил)фенилпиридином(300 мг) со стадии 3 соединение заголовка получали в виде бледно-желтого твердого вещества (270 мг). 1H ЯМР (300 MHz, СDСl3)2,42 (s,3 Н), 3,03 (s, 3 Н), 7,19-7,28 (m, 5H), 7,35 (d, 2H),7,51 (d, 1H), 7,81 (d, 2H), 8,56 (d, 1H). Пример 14. 2-(4-Хлорфенил)-5-метил-3-(4 метилсульфонил)фенилпиридин. Согласно приемам, описанным в примере 3, но с заменой 2-хлор-3-(4-метилсульфонил) фенил-5-трифторметилпиридина 2-бром-5 метил-3-(4-метилсульфонил)фенилпиридином(300 мг) со стадии 3 примера 14 соединение заголовка получали в виде белого твердого вещества, т. пл. 155-156 С (125 мг). Пример 15. 5-Метил-3-(4-метилсульфонил) фенил-2-(3-пиридинил)пиридин. Стадия 1. Три-н-пропокси-3-пиридинилборонат лития. К раствору 3-бромпиридина (39,5 г) в эфире (800 мл) при -90 С (внутренняя температура) добавляли н-BuLi (100 мл, 2,5 М) при такой скорости, чтобы внутренняя температура не превышала -78 С. Полученную смесь перемешивали в течение 1 ч при -78 С и затем добавляли триизопропоксиборат (59 мл) и полученную смесь нагревали до 0 С. Добавляли метанол и смесь выпаривали три раза из метанола и затем 37 два раза из н-пропанола. Остаток откачивали при высоком вакууме в течение 3 дней и полученную пену (76 г смеси 1:1 соединение заголовка/н-пропанол) использовали как таковую на следующей стадии. Стадия 2. 5-Метил-3-(4-метилсульфонил) фенил-2-(3-пиридинил)пиридин. Согласно приемам, описанным в примере 14, но с заменой 4-хлорфенилбороновой кислоты три-н-пропокси-3-пиридинилборонатом лития со стадии 1 получали соединение заголовка в виде белого твердого вещества, т. пл. 166167 С (2,1 г). Пример 17. 5-Хлор-2-(4-хлорфенил)-3-(4 метилсульфонил)фенилпиридин. Стадия 1. 2-Амино-3-бром-5-хлорпиридин. К раствору 2-амино-5-хлорпиридина (10 г) в уксусной кислоте (75 мл) при комнатной температуре медленно добавляли бром (2,6 мл). Через 30 мин кислоту нейтрализовали осторожным добавлением гидроксида натрия (10N) при 0 С. Полученный оранжевый остаток растворяли в этилацетате и промывали последовательно насыщенным карбонатом калия, насыщеннымNа 2S2O3 и насыщенным раствором соли, сушили и концентрировали. Флэш-хроматография (с элюированием смесью гексан/этилацетат, 3:1 об/об) оставшегося твердого вещества давала соединение заголовка в виде бледно-желтого твердого вещества (14,8 г). Стадия 2. 2-Амино-5-хлор-3-(4-метилсульфонил)фенилпиридин. Согласно приемам, описанным в примере 1, стадии 2 и 3, но с заменой 2-амино-3-бром-5 трифторметилпиридина 2-амино-3-бром-5-хлорпиридином (5 г) со стадии 1 соединение заголовка получали в виде белого твердого вещества (5 г). Стадия 3. 2-Бром-5-хлор-3-(4-метилсульфонил)фенилпиридин. К охлажденному (баня со льдом) раствору 2-амино-5-хлор-3-(4-метилсульфонил)фенилпиридина (5 г) со стадии 2 в смеси вода/ концентрированная НСl (50 мл/10 мл) добавляли нитрит натрия (1,5 г) в воде (10 мл). Полученную смесь перемешивали при комнатной температуре в течение 15 ч и полученный осадок удаляли фильтрованием и промывали последовательно водой и ССl4. После сушки на воздухе белое твердое вещество (4,8 г) и РОВr3 (10,5 г) в ДМФ(40 мл) нагревали при 100 С в течение 2 дней. Полученную смесь выливали в смесь воды со льдом и экстрагировали этилацетатом. Водную фазу подщелачивали 1N NaOH и экстрагировали этилацетатом. Объединенные органические экстракты сушили и концентрировали. Флэшхроматография (с элюированием смесью гексан/этилацетат, 1:1 об/об) остатка давала соединение заголовка в виде белого твердого вещества (2,7 г). Стадия 4. 5-Хлор-2-(4-хлорфенил)-3-(4 метилсульфонил)фенилпиридин. 38 Согласно приемам, описанным в примере 3, но с заменой 2-хлор-3-(4-метилсульфонил) фенил-5-трифторметилпиридина 2-бром-5-хлор 3-(4-метилсульфонил)фенилпиридином (300 мг) со стадии 3 получали соединение заголовка в виде белого твердого вещества, т. пл. 187-188 С(200 мг). Пример 20. 5-Хлор-3-(4-метилсульфонил) фенил-2-(2-пиридинил)пиридин. Стадия 1. 3-Бром-5-хлор-2-гидроксипиридин. Смесь 5-хлор-2-гидроксипиридина (100 г) и брома (40,1 мл) в уксусной кислоте (400 мл) перемешивали при комнатной температуре в течение 1 ч. Смесь выливали в 3 л воды и перемешивали в течение 30 мин и затем фильтровали. Оставшееся твердое вещество промывали 2 л холодной воды, сушили на воздухе и затем выпаривали вместе с толуолом три раза и с бензолом два раза. Полученное таким образом твердое вещество (81 г) использовали в следующей реакции. Стадия 2. 2-Бензилокси-3-бром-5-хлорпиридин. Смесь 3-бром-5-хлор-2-гидроксипиридина(81 г), бензилбромида (52 мл) и карбоната серебра (97 г) в бензоле (1 л) нагревали при 70 С в течение 1 ч. Смесь охлаждали до комнатной температуры и затем фильтровали через слой целита. Фильтрат концентрировали и оставшееся твердое вещество нестандартного белого цвета перекристаллизовывали из гексана с получением соединения заголовка в виде белого твердого вещества (102 г). Стадия 3. 2-Бензилокси-5-хлор-3-(4 метилсульфонил)фенилпиридин. Согласно приемам, описанным в примере 1, стадии 2 и 3, но с заменой 2-амино-3-бром-5 трифторметилпиридина 2-бензилокси-3-бром-5 хлорпиридином (81 г) со стадии 2 получали соединение заголовка в виде белого твердого вещества (72 г). Стадия 4. 5-Хлор-2-гидрокси-3-(4-метилсульфонил)фенилпиридин. Раствор 2-бензилокси-5-хлор-3-(4-метилсульфонил)фенилпиридина (72 г) в трифторуксусной кислоте (250 мл) перемешивали при 40 С в течение 15 мин и затем выливали в смесь воды со льдом (1 л). После перемешивания в течение 10 мин белое твердое вещество фильтровали, промывали дважды еще 1 л воды и затем сушили на воздухе с получением соединения заголовка. Стадия 5. 2,5-Дихлор-3-(4-метилсульфонил)фенилпиридин. Неочищенный 5-Хлор-2-гидрокси-3-(4 метилсульфонил)фенилпиридин со стадии 4 нагревали в запаянном сосуде при 150 С с РОСl3 (400 мл) в течение 15 ч. После охлаждения до комнатной температуры избыток РОСl3 удаляли отгонкой под вакуумом. Остаток разбавляли этилацетатом и водой и затем нейтра 39 лизовали гидроксидом натрия (10N) до рН 7. Органический экстракт удаляли, промывали насыщенным раствором соли и концентрировали. Оставшееся твердое вещество перекристаллизовывали из эфира с получением соединения заголовка в виде белого твердого вещества (61 г). Стадия 6. Три-н-пропокси-2-пиридилборонат лития. Согласно приемам, описанным в примере 15, стадия 1, но с заменой 3-бромпиридина 2 бромпиридином получали соединение заголовка в виде твердого вещества нестандартного белого цвета (4,1 г). Стадия 7. 5-Хлор-3-(4-метилсульфонил) фенил-2-(2-пиридинил)пиридин. Смесь 2,5-дихлор-3-(4-метилсульфонил) фенилпиридина (1 г), три-н-пропокси-2-пиридилбороната лития (1,22 г), карбоната натрия (5 мл, 2 М) и дибромида бис(трифенилфосфин) палладия 520 мг) в толуоле (100 мл), изопропаноле (10 мл) и воде (25 мл) нагревали с обратным холодильником в течение 7 ч. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через слой целита. Фильтрат экстрагировали 6N НСl и водный слой промывали этилацетатом. Водную фазу подщелачивали до рН 10 с помощью 10N гидроксида натрия и затем экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли, сушили и концентрировали. Флэш-хроматография (с элюированием смесью гексан/этилацетат, 1:1 об/об) остатка давала соединение заголовка в виде белого твердого вещества, т. пл. 134-135 С, (350 мг). Пример 21. 5-Хлор-3-(4-метилсульфонил) фенил-2-(3-пиридинил)пиридин. Согласно приемам, описанным в примере 20, стадия 7, но с заменой три-н-пропокси-2 пиридинилбороната лития три-н-пропокси-3 пиридинилборонатом лития по примеру 15, стадия 1, получали соединение заголовка в виде белого твердого вещества, т. пл. 168-169 С. Пример 22. 5-Хлор-3(4-метилсульфонил) фенил-2-(4-пиридинил)пиридин. Стадия 1. Триметокси-4-пиридинилборо нат лития. Согласно приемам, описанным в примере 15, стадия 1, но с заменой 3-бромпиридина 4 бромпиридином получали соединение заголовка. Неочищенный продукт использовали перед выпариванием из н-пропанола. Стадия 2. 5-Хлор-3-(4-метилсульфонил) фенил-2-(4-пиридинил)пиридин. Согласно приемам, описанным в примере 20, стадия 7, но с заменой три-н-пропокси-2 пиридинилбороната лития три-н-метокси-4 пиридинилборонатом лития со стадии 1 получали соединение заголовка в виде белого твердого вещества, т. пл. 187-188 С. Пример 23. 5-Хлор-3-(4-метилсульфонил) фенил-2-(2-метил-5-пиридинил)пиридин. 40 Стадия 1. 2-Метилпиридин-5-иловый сложный эфир трифторметансульфоновой кислоты. К смеси 5-гидрокси-2-метилпиридина (2 г) и пиридина (1,9 мл) в дихлорметане (100 мл) при 0 С добавляли ангидрид трифторметансульфоновой кислоты (3,4 мл). Смесь перемешивали при этой температуре в течение 15 мин и затем при комнатной температуре в течение 45 мин. Добавляли ацетат аммония (25%) и органические вещества удаляли и промывали 1N НСl, сушили и концентрировали. Соединение заголовка получали в виде бежевой жидкости (4 г), которую использовали как таковую. Стадия 2. 2-Meтил-5-триметилстаннилпиридин. Смесь 2-метилпиридин-5-илового сложного эфира трифторметан-сульфоновой кислоты(2,1 г), гексаметилдиолова (2,85 г), хлорида лития (1,1 г) и палладий-тетракис (трифенилфосфина) (190 мг) нагревали с обратным холодильником в течение 180 мин и затем охлаждали до комнатной температуры. Смесь фильтровали через слой целита, промывали этилацетатом. Фильтрат промывали дважды 5% фторидом калия, сушили и концентрировали. Флэшхроматография (с элюированием смесью гексан/этилацетат, 6:1 об/об) остатка давала соединение заголовка в виде бледно-желтого масла(1,3 г). Стадия 3. 5-Хлор-3-(4-метилсульфонил) фенил-2-(2-метил-5-пиридинил)пиридин. Смесь 2,5-дихлор-3-(4-метилсульфонил) фенилпиридина по примеру 20, стадия 5 (750 мг), 2-метил-5-триметилстаннилпиридина (1,3 г) и палладий-тетракис (трифенилфосфина) (290 мг) в NMP (10 мл) нагревали при 100 С в течение 15 ч. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через слой целита. Фильтрат промывали водой, дважды 5% фторидом калия и затем экстрагировали 1N НСl. Водную фазу нейтрализовали 10N гидроксидом натрия и затем экстрагировали этилацетатом. Органический слой концентрировали и остаток подвергали флэшхроматографии (с элюированием этилацетатом) с получением соединения заголовка в виде твердого вещества, т. пл. 127-128 С. Пример 43. Сложный метиловый эфир 2(4-хлорфенил)-3-(4-метилсульфонил)фенилпиридинил-5-карбоновой кислоты. К раствору 2-(4-хлорфенил)-5-метил-3-(4 метилсульфонил)фенилпиридина (пример 14,1,9 г) в смеси трет-бутанол/вода (1:2, 60 мл) при 90 С добавляли порциями твердый перманганат калия (2,5 г) в течение 2 ч. Полученную смесь перемешивали при 90 С в течение 15 ч и охлаждали до комнатной температуры. Смесь фильтровали через слой целита и фильтрат подкисляли до рН 2 с помощью 6N НСl. Белый осадок фильтровали и затем часть этого продукта поглощали в смеси ТГФ/дихлорметан. К это 41 му раствору добавляли диазометан в эфире до прекращения выделения пузырьков при добавлении. Полученную смесь концентрировали и подвергали флэш-хроматографии (с элюированием смесью гексан/этилацетат, 1:1 об/об). Соединение заголовка получали в виде белого твердого вещества, т. пл. 216-218 С. Пример 44. 2-(4-хлорфенил)-3-(4-метилсульфонил)фенилпиридинил-5-карбоновая кислота. К раствору сложного метилового эфира 2(4-хлорфенил)-3-(4 метилсульфонил)фенилпиридинил-5 карбоновой кислоты (140 мг) в смеси этанол/вода (1:1, 10 мл) добавляли гидроксид лития (0,35 мл, 3N) и полученную смесь перемешивали при комнатной температуре в течение 45 мин. Добавляли насыщенный бикарбонат натрия и водный слой экстрагировали этилацетатом. Водную фазу обрабатывали 3N НСl до установления рН 2 и затем экстрагировали хлороформом. Органический слой концентрировали с получением соединения заголовка в виде белого твердого вещества, т. пл. 300 С(60 мг). Пример. 45 5-Циано-2-(4-хлорфенил)-3-(4 метилсульфонил)фенилпиридин. К 2-(4-хлорфенил)-3-(4-метилсульфонил) фенилпиридинил-5-карбоновой кислоте (300 мг) в дихлорметане (10 мл) при нагревании с обратным холодильником добавляли хлорсульфонилизоцианат (0,4 мл). Через 90 мин при нагревании с обратным холодильником смесь охлаждали до комнатной температуры и затем медленно добавляли ДМФ (1,5 мл). Через 15 мин добавляли воду и смесь экстрагировали этилацетатом. Органическую фазу промывали водой, насыщенным раствором соли, сушили и концентрировали. Флэш-хроматография (с элюированием смесью этилацетат/гексан, 2:1 об/об) остатка давала соединение заголовка в виде белого твердого вещества (100 мг). 1H ЯМР (300 MHz,CDCl3)3,06 (s, 3 Н), 7,21-7,28 (m, 4 Н), 7,37 (d,2H), 7,90 (d, 2H), 7,96 (d, 1H), 8,94 (d, 1H). Пример 46. Гидрометансульфонат 5-хлор 3-(4-метилсульфонил)фенил-2-(3-пиридинил) пиридина. Раствор 5-хлор-3-(4-метилсульфонил)фенил-2-(3-пиридинил)пиридина (5,31 г, пример 21) в этилацетате (100 мл) обрабатывали по каплям метансульфоновой кислотой (1 мл) в этилацетате (20 мл). Полученный осадок фильтровали и сушили в вакууме с получением соединения заголовка (6,4 г) в виде белого твердого вещества. 1H ЯМР (300 MHz, CD3OD)2,68 (s,3H), 3,15 (s, 3H), 7,60 (d, 2H), 7,96-8,00 (m, 3H),8,14 (d, 1H), 8,47 (dt, 1H), 8,80 (d, 1H), 8,86 (m,2H). Пример 47. Гидрохлорид 5-хлор-3-(4 метилсульфонил)фенил-2-(3-пиридинил)пиридина. 42 Раствор 5-хлор-3-(4-метилсульфонил)фенил-2-(3-пиридинил)пиридина (2,05 г, пример 21) в горячем этилацетате (75 мл) обрабатывали по каплям хлороводородной кислотой (1,5 мл, 4 М в диоксане). Полученный осадок фильтровали и сушили под вакуумом с получением соединения заголовка (2,2 г) в виде белого твердого вещества. 1 Н ЯМР (300 MHz, DMSO-d6)3,24(s, 3H), 7,59 (d, 2H), 7,80 (dd, 1H), 7,91 (d, 2H),8,15 (d, 1H), 8,22 (d, 1H), 8,69 (d, 1H), 8,77 (d,1H), 8,90 (d, 1H). Пример 59. Гидрохлорид 5-хлор-3-(4 метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина. Согласно приемам, описанным в примере 47, но с заменой 5-хлор-3-(4-метилсульфонил) фенил-2-(3-пиридинил)пиридина 5-хлор-3-(4 метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридином (по примеру 23) получали соединение заголовка в виде белого твердого вещества. 1H ЯМР (300 MHz, DMSO-d6):2,68 (s,3H), 3,25 (s, 3H), 7,61 (d, 2H), 7,70 (d, 1H), 7,92Org. Chem. 1988, 53, 386) (4,6 г) получали соединение заголовка в виде желтого твердого вещества. Стадия 2: 5-Хлор-3-(4-метилсульфонил) фенил-2-(2-этил-5-пиридинил)пиридин. Смесь 2,5-дихлор-3-(4-метилсульфонил) фенилпиридина (пример 20, стадия 5) (5,6 г),три-н-пропокси-2-этил-5-пиридилбороната лития (стадия 1) (4,0 г), карбоната натрия (17 мл, 2 М) и дибромида бис(трифенилфосфин)палладия(420 мг) в толуоле (75 мл), этаноле (75 мл) и воде (15 мл) нагревали с обратным холодильником в течение 7 ч. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через слой целита. Фильтрат экстрагировали 6N НСl и водный слой промывали этилацетатом. Водную фазу подщелачивали до рН 10 с помощью 10N гидроксида натрия и затем экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли, сушили и концентрировали. Флэшхроматография (с элюированием смесью гексан/этилацетат, 1:1 об/об) остатка давала соединение заголовка в виде белого твердого вещества (4,0 г). 1H ЯМР (500 MHz, ацетон-d6):1,21 Стадия 1. 5-Бром-2-этилпиридин. 280 мл 5 N гидроксида натрия (1,4 мл, 2,09 экв.) добавляли к раствору 158 г 2,5-дибромпиридина (0,67 моль, 1 экв.) в 1,4 л ТГФ. К полученному раствору добавляли 700 мл 1N триэтилбора в ТГФ (0,70 моль, 1,04 экв.), 195 мг хлорида бис(ацетонитрил)палладия(II) (0,75 ммоль, 0,0011 экв.) и 414 мг 1,1'-бис (дифенилфосфино)ферроцена (0,75 ммоль, 0,0011 экв.). Реакционную смесь медленно нагревали до очень слабого кипения с обратным холодильником в течение 3 ч. Затем ее охлаждали до 0 С и обрабатывали последовательно 140 мл 5N гидроксида натрия (0,70 моль, 1,04 экв.) и 53 мл 30% пероксида водорода (0,70 моль, 1,05 экв.) при такой скорости, чтобы температура не превышала 10 С. Смесь экстрагировали эфиром и эфирные экстракты промывали гидроксидом натрия, водой, насыщенным раствором соли и сушили над MgSO4. Затем эфирный раствор концентрировали и коричневый остаток перегоняли в вакууме (40 С, 1 тор) с получением 112 г соединения заголовка в виде прозрачного масла,слегка загрязненного исходным продуктом и 2,5-диэтилпиридином. Выход 90%. 1H ЯМР сравним с сообщенным J.W. Tilley and S. Zawoiski; J. Org. Chem. 1988, 53, 386-390. Этот продукт пригоден для использования на следующей стадии без дополнительной очистки. Стадия 2. Три-н-пропокси-2-этил-5 пиридилборонат лития. Согласно приемам, описанным в примере 15, стадия 1, но с заменой 3-бромпиридина 5 бром-2-этилпиридином со стадии 1 получали соединение заголовка в виде желтого твердого вещества. Стадия 3. 5-Хлор-3-(4-метилсульфонил) фенил-2-(2-этил-5-пиридинил)пиридин. Смесь 2,5-дихлор-3-(4-метилсульфонил) фенилпиридина (пример 20, стадия 5) (5,6 г),три-н-пропокси-2-этил-5-пиридилбороната лития (стадия 2) (4,0 г), карбоната натрия (17 мл, 2 М) и дибромида бис(трифенилфосфин)палладия(420 мг) в толуоле (75 мл), этаноле (75 мл) и воде (15 мл) нагревали с обратным холодильником в течение 7 ч. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через слой целита. Фильтрат экстрагировали 6N НСl и водный слой промывали этилацетатом. Водную фазу подщелачивали до 44 рН 10 с помощью 10 N гидроксида натрия и затем экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли, сушили и концентрировали. Флэшхроматография (с элюированием смесью гексан/этилацетат, 1:1 об/об) остатка давала соединение заголовка в виде белого твердого вещества (4,0 г). 1H ЯМР (500 MHz, ацетон-d6):1,21 Раствор 5-хлор-3-(4-метилсульфонил) фенил-2-(2-этил-5-пиридинил)пиридина (3,4 г,пример 71) в этилацетате (40 мл) и эфире (100 мл) обрабатывали по каплям метансульфоновой кислотой (873 мг) в эфире (20 мл). Полученный осадок охлаждали до -20 С, фильтровали и сушили под вакуумом с получением соединения заголовка (4,3 г) в виде белого твердого вещества. 1H ЯМР (500 MHz, CD3OD)1,40 (t, 3H),2,68 (s, 3H), 3,07 (q, 2H), 3,15 (s, 3H), 7,60 (d,2H), 7,86 (d, 1H), 7,99 (d, 2H), 8,11 (d, 1H), 8,34 или его фармацевтически приемлемая соль, где(а) СН 3,(b) NН 2,(c) NНС(O)СF3,(d) NНСН 3; Аr обозначает моно-, ди- или тризамещенный фенил или пиридинил (или его N-оксид), где заместители выбраны из группы, включающей(b) C1-6-алкил,или R4 и R5, R8 и R9 или R11 и R12 вместе с атомом, к которому они присоединены, образуют насыщенное моноциклическое кольцо из 3, 4, 5,6 или 7 атомов, при условии, что, когда Аr представляет собой фторфенил, R2 выбран из (а), (b),(с) или (е)-(о) из определения R2, данного выше. 2. Соединение по п.1, где Аr представляет собой моно-, ди- или тризамещенный 2-пиридинил. 3. Соединение по п.1, где Аr представляет собой моно-, ди- или тризамещенный 3-пиридинил. 4. Соединение по п.1, где R1 представляет собой СН 3 или NH2. 5. Соединение по п.1, где Аr обозначает моно-, ди- или тризамещенный 2-пиридинил или 3-пиридинил и заместители выбраны из группы, включающей: 46 и заместители выбраны из группы, включающей:R2 обозначает галоген, СН 3 или СF3; и Аr обозначает моно-, ди- или тризамещенный 2-пиридинил или 3-пиридинил и заместители выбраны из группы, включающей:R2 обозначает галоген, СН 3 или СF3 и Аr обозначает моно- или дизамещенный 3 пиридинил, и заместители выбраны из группы,включающей:R2 обозначает галоген, СН 3 или СF3 и Аr обозначает моно- или дизамещенный фенил и заместители выбраны из группы, включающей: или его фармацевтически приемлемая соль, гдеR2 и Аr выбраны вместе следующим образом:CN 4-ClС 6 Н 4 13. Соединение по п.12, где фармацевтически приемлемая соль является солью лимонной,бромисто-водородной,хлористо-водородной,малеиновой, метансульфоновой, фосфорной,серной или винной кислоты. 14. Соединение по п.13, где фармацевтически приемлемая соль является кислотно или его фармацевтически приемлемая соль, гдеR2 и Аr выбраны вместе следующим образом:Cl 3-(2,6-Me2)pyr 16. Соединение по п.15, где фармацевтически приемлемая соль является солью лимонной,бромисто-водородной,хлористо-водородной,малеиновой, метансульфоновой, фосфорной,серной или винной кислоты. 17. Соединение по п.16, где фармацевтически приемлемая соль является кислотноаддитивной солью хлористо-водородной или метансульфоновой кислоты. 18. Фармацевтическая композиция для лечения воспалительного заболевания, чувствительного к лечению нестероидным противовоспалительным агентом, содержащая нетоксичное терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель. 19. Фармацевтическая композиция для лечения опосредованных циклооксигеназой заболеваний, успешно излечиваемых активным агентом, который избирательно ингибирует ЦОГ-2 относительно ЦОГ-1, содержащая нетоксичное терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель. 20. Способ лечения воспалительного заболевания, чувствительного к лечению нестероидным противовоспалительным агентом, предусматривающий введение пациенту, нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения по п.1 и фармацевтически приемлемого носителя. 21. Способ лечения опосредованных циклооксигеназой заболеваний, успешно излечиваемых активным агентом, который избирательно ингибирует ЦОГ-2 относительно ЦОГ-1,предусматривающий введение пациенту, нуж 49 дающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения по п. 1 и фармацевтически приемлемого носителя. 22. Соединение по п.1, где R2 выбран из группы, включающей:(b) C1-6-алкил,или R4 и R5 или R11 и R12 вместе с атомом, к которому они присоединены, образуют насыщенное моноциклическое кольцо из 3, 4, 5, 6 или 7 атомов. 23. Соединение по п. 22 формулы Ic(a) хлор,(b) метил,и где могут быть одна, две или три группы X,независимо выбранные из группы, включающей:(a) водород,(b) галоген,(c) C1-4-алкокси,(d) C1-4-алкилтио,(e) CN,(f) C1-4-алкил,(g) СF3. 24. Соединение по п.23, где R2 обозначает хлор, где имеется одна группа X, независимо выбранная из группы, включающей:(b) F или Сl,(c) метил,(d) этил. 25. Соединение по п.24, где имеется одна группа X, независимо выбранная из группы,включающей:X обозначает водород. 28. Соединение по п.1, которое представляет собой 5-хлор-3-(4-метилсульфонил)фенил 2-(3-пиридинил)пиридин или его фармацевтически приемлемую соль. 29. Соединение по п.1, которое представляет собой гидрометансульфонат 5-хлор-3-(4 метилсульфонил)фенил-2-(3-пиридинил) пиридина. 30. Соединение по п.1, которое представляет собой гидрохлорид 5-хлор-3-(4-метилсульфонил)фенил-2-(3-пиридинил)пиридина. 31. Соединение по п.1, которое представляет собой 5-хлор-3-(4-метилсульфонил)фенил 2-(2-этил-5-пиридинил)пиридин или его фармацевтически приемлемую соль. 32. Соединение по п.1, которое представляет собой гидрометансульфонат 5-хлор-3-(4 метилсульфонил)фенил-1-(2-этил-5-пиридинил) пиридина. 33. Соединение по п.1 формулы IсX обозначает метил, этил, н-пропил, изопропил или циклопропил. 34. Соединение по п.33, где Х обозначает метил. 35. Соединение по п.33, которое представляет собой 5-хлор-3-(4-метилсульфонил)фенил 2-(2-метил-5-пиридинил)пиридин или его фармацевтически приемлемую соль. 36. Соединение по п.33, которое представляет собой гидрохлорид 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридина. 37. Соединение по п.1, выбранное из группы, включающей 3-(4-метилсульфонил)фенил-2-фенил-5 трифторметилпиридин; 2-(3-хлорфенил)-3-(4-метилсульфонил) фенил-5-трифторметилпиридин; 2-(4-хлорфенил)-3-(4-метилсульфонил) фенил-5-трифторметилпиридин; 2-(4-фторфенил)-3-(4-метилсульфонил) фенил-5-трифторметилпиридин; 5-метил-3-(4-метилсульфонил)фенил-2 фенилпиридин; 2-(4-хлорфенил)-5-метил-3-(4-метилсульфонил)фенилпиридин; 5-хлор-2-(4-хлорфенил)-3-(4-метилсульфонил)фенилпиридин; метиловый эфир 2-(4-хлорфенил)-3-(4 метилсульфонил)фенилпиридинил-5 карбоновой кислоты; 2-(4-хлорфенил)-3-(4-метилсульфонил) фенилпиридинил-5-карбоновая кислота; 5-циано-2-(4-хлорфенил)-3-(4-метилсульфонил)фенилпиридин. 38. Соединение по п.1, выбранное из группы, включающей 3-(4-метилсульфонил)фенил-2-(3-пиридинил)-5-трифторметилпиридин; 5-метил-3-(4-метилсульфонил)фенил-2-(3 пиридинил)пиридин; 5-хлор-3-(4-метилсульфонил)фенил-2-(2 пиридинил)пиридин; 5-хлор-3-(4-метилсульфонил)фенил-2-(3 пиридинил)пиридин; 5-хлор-3-(4-метилсульфонил)фенил-2-(4 пиридинил)пиридин; 5-хлор-3-(4-метилсульфонил)фенил-2-(2 метил-5-пиридинил)пиридин; 52 гидрометансульфонат 5-хлор-3-(4-метилсульфонил)фенил-2-(3-пиридинил)пиридина; гидрохлорид 5-хлор-3-(4-метилсульфонил) фенил-2-(3-пиридинил)пиридина; гидрохлорид 5-хлор-3-(4-метилсульфонил) фенил-2-(2-метил-5-пиридинил)пиридина; 5-хлор-3-(4-метилсульфонил)фенил-2-(2 этил-5-пиридинил)пиридин и гидрометансульфонат 5-хлор-3-(4-метилсульфонил)фенил-2-(2-этил-5-пиридинил)пиридина. 39. Соединение формулы I, определенной в п.1, или его фармацевтически приемлемая соль, гдеR2 выбран из C1-6-алкила, галогена, СF3 или CN; и Аr обозначает монозамещенный фенил или незамещенный или монoзамещенный пиридинил, в котором монозаместитель выбран из C1-6 алкила, галогена или С 3-6-циклоалкила. 40. Соединение по п.39, где Аr представляет собой монозамещенный фенил. 41. Соединение по п.39, где Аr представляет собой незамещенный или монозамещенный пиридинил. 42. Применение соединения по пп.1, 28,29, 30, 31, 32, 35, 36, 37, 38, 39, 40 или 41 при приготовлении лекарственного средства для лечения воспалительного заболевания. 43. Фармацевтическая композиция для лечения, опосредованных циклооксигеназой-2 заболеваний, содержащая соединение по пп.1,28, 29, 30, 31, 32, 35, 36, 37, 38, 39, 40 или 41 и фармацевтически приемлемый носитель. 44. Фармацевтическая композиция, содержащая соединение по любому из пп. 1-16 и 2241 и фармацевтически приемлемый носитель. 45. Противовоспалительная фармацевтическая композиция, содержащая приемлемое противовоспалительное количество соединения формулы I, определенного в любом из пп. 1-11,22-26 или 29-32, или его фармацевтически приемлемой соли вместе с фармацевтически приемлемым носителем. 46. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-17 или 22-37 в качестве ингибитора ЦОГ-2,избирательного для ЦОГ-2 относительно ЦОГ-1.
МПК / Метки
МПК: A61P 29/00, A61K 31/44, C07D 213/34
Метки: качестве, циклооксигеназы-2, ингибиторов, избирательных, замещенные, пиридины
Код ссылки
<a href="https://eas.patents.su/27-1444-zameshhennye-piridiny-v-kachestve-izbiratelnyh-ingibitorov-ciklooksigenazy-2.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные пиридины в качестве избирательных ингибиторов циклооксигеназы-2</a>
(метилсульфонил) фенил-2-(5н)-фураноны в качестве ингибиторов циклооксигеназы-2.

Номер патента: 795
Опубликовано: 24.04.2000
Авторы: Рой Патрик, Гримм Эрик, Беллей Мишель, Ли Чунг-Синг, Прасит Петпибун, Блэк Камерон, Готье Жак И., Лау Чеук-Кун, Леблан Ив, Терьен Мишель
МПК: C07D 307/58, A61K 31/365, C07C 317/24...
Метки: ингибиторов, качестве, циклооксигеназы-2, фенил-2-(5н)-фураноны, метилсульфонил
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль, где: Х выбран из группы, состоящей из (a) СН2, (b) СНОН, (c) СО, (d) О, (e) S и (f) N(R15), при условии, что, когда R3 и R4 иные, чем (1) водород (оба), (2) C1-10-алкил (оба) или (3) соединенные вместе с углеродом, к которому они присоединены, образуют насыщенное моноциклическое углеродное кольцо из 3, 4, 5, 6 или 7 атомов, то X выбран из СО, О, S или N(R15); ...
Замещенные 2-фенилпиридины в качестве гербицидов

Номер патента: 83
Опубликовано: 25.06.1998
Авторы: Клинтц Ральф, Хайстрахер Элизабет, Кениг Хартманн, Вальтер Хельмут, Вестфален Карл-Отто, Шэфер Петер, Хампрехт Герхард, Мисслитц Ульф
МПК: C07D 213/61, A01N 43/40
Метки: гербицидов, 2-фенилпиридины, замещенные, качестве
Формула / Реферат:
1. Замещенные 2-фенилпиридины общей формулы 1 в которой переменные имеют следующее значение: n обозначает 0 или 1; R1, R3 и R4 независимо друг от друга обозначают водород, галоген, С1-C4алкил, C1-C4галогеналкил, гидрокси, C1-C4алкокси, С1-C4галогеналкокси, нитро, амино, С1-C4алкиламино, ди-(С1-C4 алкил)амино, меркапто, C1-C4алкилтио, С1-C4галогеналкилтио, циано, карбоксил, аминокарбонил, (С1-C4 алкиламино)карбонил или...
Замещенные 2-фенилпиридины в качестве гербицидов

Номер патента: 84
Опубликовано: 25.06.1998
Авторы: Вестфален Карл-Отто, Шэфер Петер, Хайстрахер Элизабет, Мисслитц Ульф, Вальтер Хельмут, Клинтц Ральф, Хампрехт Герхард, Харреус Альбрехт, Кениг Хартманн, Гетц Норберт
МПК: C07D 213/61, A01N 43/40
Метки: качестве, гербицидов, замещенные, 2-фенилпиридины
Формула / Реферат:
1. Замещенные 2-фенилпиридины общей формулы I в которой переменные имеют следующие значения: n обозначает 0 или 1; R1, R3 и R4 независимо друг от друга обозначают водород, галоген, C1-C4алкил, C1-C4 галогеналкил, гидрокси, C1-C4алкокси, C1-C4галогеналкокси, нитро, амино, C1-C4 алкиламино, ди-(C1-C4алкил)амино, меркапто, C1-C4алкилтио, C1-C4галогеналкилтио, циано, карбоксил, (C1-C4алкокси)карбонил, аминокарбонил, C1-C4алкиламинокарбонил...
Замещенные 2-фенилпиридины в качестве гербицидов

Номер патента: 85
Опубликовано: 25.06.1998
Авторы: Хайстрахер Элизабет, Клинтц Ральф, Вестфален Карл-Отто, Мисслитц Ульф, Кениг Хартманн, Хампрехт Герхард, Вальтер Хельмут, Шэфер Петер
МПК: A01N 43/40, C07D 213/61
Метки: замещенные, 2-фенилпиридины, качестве, гербицидов
Формула / Реферат:
1. Замещенные 2-фенилпиридины общей формулы I в которой переменные имеют следующее значение: n обозначает 0 или 1; R1, R3 и R4 независимо друг от друга обозначают водород, галоген, C1-C4алкил, C1-C4 галогеналкил, гидрокси, C1-C4алкокси, C1-C4галогеналкокси, нитро, амино, C1-C4 алкиламино, ди-(C1-C4алкил)амино, меркапто, C1-C4алкилтио, C1-C4галогеналкилтио, циано, гидроксикарбонил, (C1-C4алкокси)карбонил, аминокарбонил,...
Сульфонилалканоиламиногидроксиэтиламиносульфонамид в качестве ингибиторов ретровирусной протеазы

Номер патента: 533
Опубликовано: 28.10.1999
Авторы: Макдонэлд Джозеф Дж., Нагараян Сринивазан, Васкез Майкл Л., Девадас Балекудру, Гетмен Даниел П., Фрескос Джон Н., Сикорски Джеймс А., Декрессенцо Гэри А.
МПК: A61K 31/34, C07D 317/62, C07C 311/00...
Метки: сульфонилалканоиламиногидроксиэтиламиносульфонамид, ретровирусной, ингибиторов, качестве, протеазы
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль, пролекарство или сложный эфир, где символы n и t, каждый независимо друг от друга, равны 0,1 или 2; R1 обозначает водород, алкил с 1-5 атомами углерода, алкенил с 2-5 атомами углерода, алкинил с 2-5 атомами углерода, гидроксиалкил с 1-3 атомами углерода, алкоксиалкил, состоящий из алкила с 1-3 атомами и алкокси с 1-3 атомами углерода, цианоалкил, содержащий алкил с 1-3 атомами...
Предыдущий патент: Неводные медицинские средства для очистки толстой кишки
Следующий патент: Способ для удаления краски с подложки
Случайный патент: Цеолит типа lsx