Лекарственная форма, способы ее получения и способ лечения




















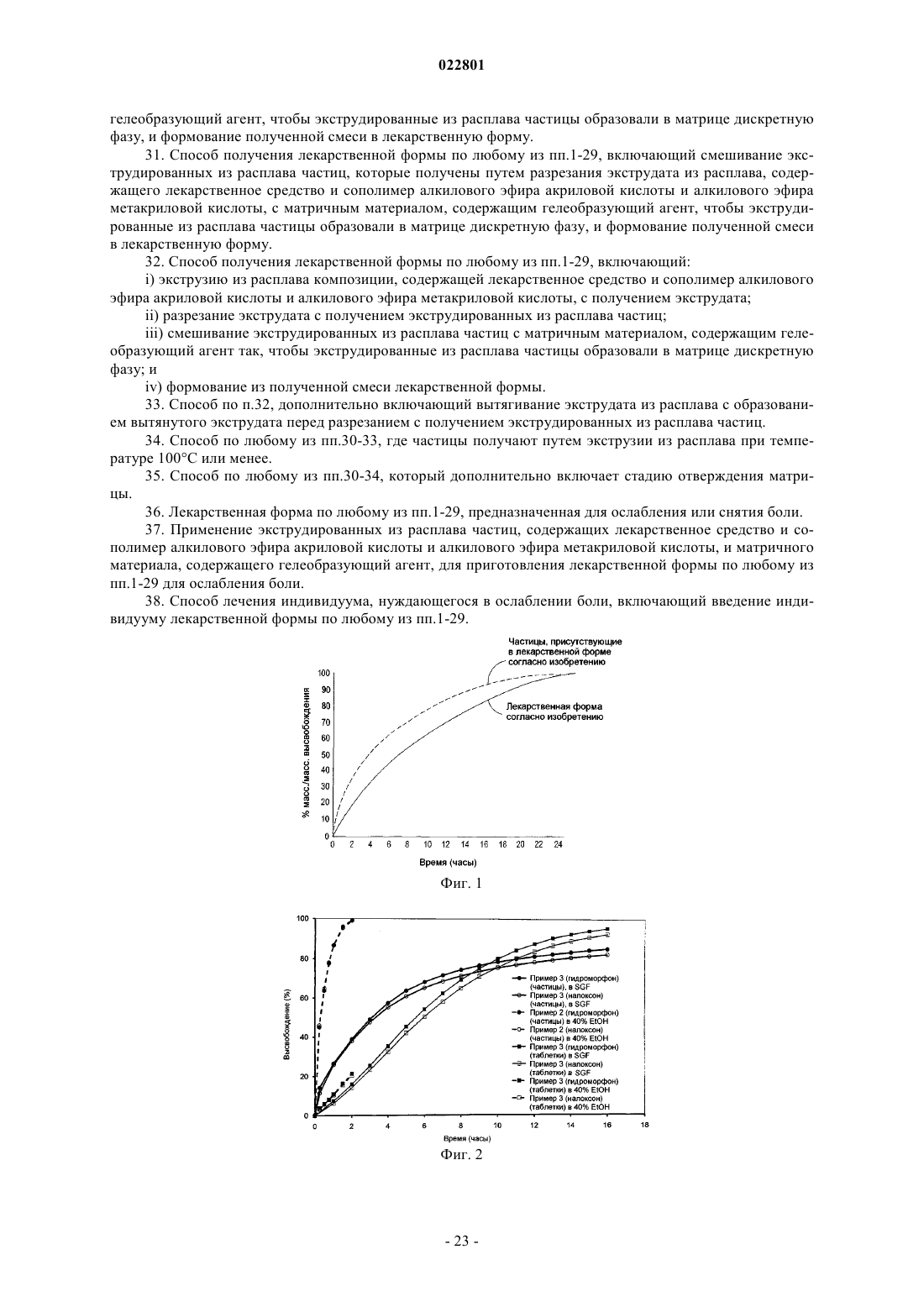
Формула / Реферат
1. Лекарственная форма, содержащая экструдированные из расплава частицы, включающие лекарственное средство, и матрицу, представляющую собой непрерывную фазу, включающую гелеобразующий агент, где экструдированные из расплава частицы присутствуют в матрице в дискретной фазе; и экструдированные из расплава частицы содержат сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты.
2. Лекарственная форма по п.1, где сополимер выбран из сополимеров торговых марок Eudragit® RL 100, Eudragit® RL PO, Eudragit® RS 100, Eudragit® RS PO, Eudragit® NE 40 D и Eudragit® NE 30 D.
3. Лекарственная форма по п.1 или 2, где экструдированные из расплава частицы имеют удлиненную форму.
4. Лекарственная форма по любому из пп.1-3, которая содержит 15-80 мас.% экструдированных из расплава частиц от общей массы лекарственной формы.
5. Лекарственная форма по любому из пп.1-4, которая содержит 20-85 мас.% матрицы от общей массы лекарственной формы.
6. Лекарственная форма по любому из предыдущих пунктов, полученная в форме таблетки.
7. Лекарственная форма по любому из предыдущих пунктов, которая является резистентной к модификации.
8. Лекарственная форма по любому из предыдущих пунктов, где количество лекарственного средства, высвобождаемого из лекарственной формы в течение 0,5 ч, при измерении на приборе USP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов (SGF) в присутствии 40% этанола при 37°С составляет примерно ±20% от количества лекарственного средства, высвобождаемого из лекарственной формы в течение 0,5 ч, при измерении на приборе USP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов (SGF) в отсутствие этанола (0%) при 37°С.
9. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы имеют предел прочности при разрыве по меньшей мере 350 H.
10. Лекарственная форма по любому из предыдущих пунктов, где частицы, экструдированные из расплава, имеют диаметр и/или длину менее чем примерно 1000 мкм.
11. Лекарственная форма по любому из предыдущих пунктов, где лекарственным средством является лекарственное средство, которое представляет собой объект для злоупотребления.
12. Лекарственная форма по любому из предыдущих пунктов, где лекарственным средством являются агонист опиоида; транквилизатор; средство, подавляющее ЦНС; средство, стимулирующее ЦНС; или снотворное средство седативного действия.
13. Лекарственная форма по любому из предыдущих пунктов, где лекарственным средством является агонист опиоида.
14. Лекарственная форма по п.13, где агонист опиоида выбран из группы, состоящей из оксикодона, оксиморфона, гидрокодона, гидроморфона, морфина, кодеина, бупренорфина, фентанила, трамадола, тапентадола и их фармацевтически приемлемых солей.
15. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы содержат 3-50 мас.% лекарственного средства от общей массы частиц.
16. Лекарственная форма по любому из предыдущих пунктов, которая включает один или несколько дополнительных активных ингредиентов.
17. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы содержат 10-50 мас.% указанного сополимера от общей массы частиц.
18. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы дополнительно содержат скорость-регулирующий или скорость-модифицирующий агент.
19. Лекарственная форма по п.18, где скорость-регулирующий или скорость-модифицирующий агент представляет собой алкилцеллюлозу.
20. Лекарственная форма по п.19, где алкилцеллюлоза представляет собой этилцеллюлозу.
21. Лекарственная форма по любому из пп.18-20, где экструдированные из расплава частицы содержат 20-50 мас.% скорость-регулирующего или скорость-модифицирующего агента от общей массы частиц.
22. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы дополнительно содержат замасливатель.
23. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы дополнительно содержат пластификатор.
24. Лекарственная форма по п.1, где экструдированные из расплава частицы содержат оксикодон или гидроморфон, сополимер этилакрилата и метилметакрилата, этилцеллюлозу в качестве скорость-регулирующего или скорость-модифицирующего агента, стеариловый спирт и/или триэтилцитрат в качестве пластификатора, глицерилдибегенат в качестве замасливателя и, необязательно, антагонист опиоида.
25 Лекарственная форма по п.24, где оксикодон или гидроморфон представлены в виде их гидрохлоридной соли.
26. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы содержат агонист опиоида, а также антагонист опиоида.
27. Лекарственная форма по любому из пп.1-26, где гелеобразующий агент выбран из полиэтиленоксида, поливинилового спирта, гидроксипропилметилцеллюлозы, карбомеров, полиуроновых кислот или их смесей.
28. Лекарственная форма по любому из пп.1-27, где гелеобразующий агент является отвержденным.
29. Лекарственная форма по п.28, которая может быть подвергнута уплощению без разрушения до толщины примерно на 60% меньше толщины указанной лекарственной формы до ее уплощения.
30. Способ получения лекарственной формы по любому из пп.1-29, включающий смешивание экструдированных из расплава частиц, содержащих лекарственное средство и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, с матричным материалом, содержащим гелеобразующий агент, чтобы экструдированные из расплава частицы образовали в матрице дискретную фазу, и формование полученной смеси в лекарственную форму.
31. Способ получения лекарственной формы по любому из пп.1-29, включающий смешивание экструдированных из расплава частиц, которые получены путем разрезания экструдата из расплава, содержащего лекарственное средство и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, с матричным материалом, содержащим гелеобразующий агент, чтобы экструдированные из расплава частицы образовали в матрице дискретную фазу, и формование полученной смеси в лекарственную форму.
32. Способ получения лекарственной формы по любому из пп.1-29, включающий:
i) экструзию из расплава композиции, содержащей лекарственное средство и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, с получением экструдата;
ii) разрезание экструдата с получением экструдированных из расплава частиц;
iii) смешивание экструдированных из расплава частиц с матричным материалом, содержащим гелеобразующий агент так, чтобы экструдированные из расплава частицы образовали в матрице дискретную фазу; и
iv) формование из полученной смеси лекарственной формы.
33. Способ по п.32, дополнительно включающий вытягивание экструдата из расплава с образованием вытянутого экструдата перед разрезанием с получением экструдированных из расплава частиц.
34. Способ по любому из пп.30-33, где частицы получают путем экструзии из расплава при температуре 100°С или менее.
35. Способ по любому из пп.30-34, который дополнительно включает стадию отверждения матрицы.
36. Лекарственная форма по любому из пп.1-29, предназначенная для ослабления или снятия боли.
37. Применение экструдированных из расплава частиц, содержащих лекарственное средство и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, и матричного материала, содержащего гелеобразующий агент, для приготовления лекарственной формы по любому из пп.1-29 для ослабления боли.
38. Способ лечения индивидуума, нуждающегося в ослаблении боли, включающий введение индивидууму лекарственной формы по любому из пп.1-29.
Текст
ЛЕКАРСТВЕННАЯ ФОРМА, СПОСОБЫ ЕЕ ПОЛУЧЕНИЯ И СПОСОБ ЛЕЧЕНИЯ Изобретение относится к лекарственной форме, а в частности, к лекарственной форме,резистентной к модификации и включающей экструдированные из расплава частицы, содержащие лекарственное средство и матрицу, где указанные экструдированные из расплава частицы присутствуют в указанной матрице в дискретной фазе. Область, к которой относится изобретение Настоящее изобретение относится к лекарственным формам, а в частности, к лекарственным формам, которые являются резистентными к модификации и включают частицы, продуцированные путем экструзии из расплава и содержащие лекарственное средство и матрицу; а также к способам получения указанных лекарственных форм. Настоящее изобретение также относится к применению указанных лекарственных форм в медицине, например для устранения боли. Предшествующий уровень техники В общих чертах, желательно получить такие лекарственные препараты, которые позволили бы максимизировать вероятность того, что они будут использованы по назначению. Очевидно, что такой фармацевтический препарат, в свою очередь, будет давать максимально желаемый фармакологический эффект. Еще более важным является тот факт, что получение фармацевтических средств в защищенной от модификаций форме дает гарантию того, что такое фармацевтическое средство будет трудно доступно для злоупотребления. Совершенно очевидно, что фармацевтические препараты, содержащие лекарственные средства определенного типа, являются более привлекательными для злоупотребления, чем лекарственные средства другого типа. Так, например, лекарственные формы (например, таблетки), содержащие агонист опиоидов; транквилизатор; средство, подавляющее ЦНС; средство, стимулирующее ЦНС; или снотворное средство седативного действия, часто являются объектами злоупотребления, а особенно те лекарственные формы, которые содержат агонист опиоидов. Опиоидные аналгетики являются ценными фармацевтическими средствами, которые применяются для ослабления и снятия боли. Лица с наркотической зависимостью обычно пытаются модифицировать лекарственные формы, содержащие опиоидные аналгетики, а в частности, лекарственные формы с регулируемым высвобождением, а затем вводят их таким способом, чтобы за короткий период времени достигалась их высокая концентрация in vivo, вызывающая состояние эйфории. Опиоидсодержащие таблетки регулируемого высвобождения могут быть, например, измельчены в целях получения опиоидов согласно изобретению, доступных для быстрого высвобождения после их перорального или интраназального введения. Другой формой модификации лекарственного препарата является экстракция опиоида из опиоидсодержащих препаратов, главным образом, с использованием этанола, хотя могут быть также использованы и другие растворители, например вода или ацетон. Полученный раствор может быть затем введен в неочищенной форме путем инъекции. Кроме того, лица с наркотической зависимостью иногда не соблюдают инструкции по применению опиоидсодержащих лекарственных форм, и одновременно с введением лекарственного средства они принимают алкоголь для усиления высвобождения лекарственного средства. У лица с наркотической зависимостью такое введение может приводить к более быстрому высвобождению опиоида, чем это предусмотрено производителем. Для минимизации возможности злоупотребления лекарственным средством было предложено приготавливать опиоидные аналгетики в форме таблеток, содержащих высокомолекулярный полиэтиленоксид (ПЭО). ПЭО должен служить для регуляции скорости высвобождения опиоида из лекарственной формы и для предотвращения ее разрушения. Если такую лекарственную форму подвергнуть экстракции этанолом, то ПЭО будет также придавать раствору такую вязкость, при которой данный раствор невозможно будет ввести в шприц и в организм посредством инъекции. Однако количество ПЭО, а в частности его содержание по отношению к лекарственному средству и другим наполнителям (если они присутствуют) в данной лекарственной форме, которое необходимо для регуляции скорости высвобождения лекарственного средства и его резистентности к разрушению, является ограниченным. В частности, получение высококонцентрированных лекарственных форм (то есть лекарственных форм, содержащих относительно большие количества лекарственного средства) представляет определенные трудности, поскольку на практике трудно обеспечить такое количество ПЭО,которое необходимо для регуляции скорости высвобождения лекарственного средства в кровоток и для гарантии его устойчивости к измельчению. В этом случае лекарственная форма (например, таблетка) будет слишком большой и тяжелой, а поэтому непригодной для простого введения. Поэтому получение лекарственных форм, которые обладают пролонгированным высвобождением лекарственного средства, а в частности высвобождением в течение 24 ч, и которые также являются резистентными к модификации,представляет определенные трудности. Специалистам также известны макрочастицы, которые могут быть получены путем экструзии из расплава и которые содержат опиоидные аналгетики. Такие макрочастицы описаны, например, вWO2005/079760. Некоторые из полимеров, введенные в такие макрочастицы для облегчения экструзии из раствора, могут сообщать этим макрочастицам определенную степень резистентности к модификации. Действительно, известно, что чем выше количество таких полимеров, присутствующих в указанных макрочастицах, тем более устойчивыми являются эти частицы к модификации. Однако, с другой стороны, вышеописанные макрочастицы все же сохраняют некоторую чувствительность к экстракции спиртом, которая может быть использована в целях злоупотребления. Например,известно, что такие макрочастицы высвобождают в 2-3 раза больше опиоидов в присутствии спирта, чем в его отсутствие. Очевидно, что это свойство обусловлено высвобождением лекарственного средства с поверхностей, образующихся при разрезании экструдата, получаемого из расплава в процессе гранулирования, проводимого в процессе продуцирования макрочастиц. Указанное свойство является крайне нежелательным из-за относительно высокой вероятности злоупотребления полученным продуктом. В соответствии с этим совершенно очевидно, что необходимо получить альтернативные лекарственные формы, а в частности, лекарственные формы, которые было бы трудно модифицировать и которые обладали бы резистентностью к модификации, а также к экстракции растворителем (например, этанолом). Такие лекарственные препараты должны преимущественно иметь такие формы, размеры и массы, которые были бы подходящими для их перорального введения. Совершенно очевидно, что такие лекарственные препараты могут быть также легко приготовлены экономически эффективным способом. Было неожиданно обнаружено, что если частицы, экструдированные из раствора и содержащие лекарственное средство, ввести в матрицу и в смесь в целях получения лекарственной формы (например,таблетки), то такая лекарственная форма будет обладать превосходной резистентностью к экстракции спиртом (то есть будет лучше защищена от модификации), а также резистентностью к дроблению. Матрица, в которой присутствуют частицы, будет обеспечивать резистентность к экстракции спиртом в результате образования геля или вязкого раствора под воздействием спирта, что будет затруднять введение таких частиц с помощью шприца или путем инъекции. Состав и размер частиц, содержащих лекарственные средства, будут обладать резистентностью к их измельчению, даже если указанной матрицей является измельчаемый материал; при этом все полученные продукты будут представлять собой трудно разделимые частицы, которые будут слишком мелкими для последующего измельчения, но слишком крупными для интразального введения. Матрица может также включать отверждающийся полимер, и в этом случае такая матрица позволяет получить преимущественно крупные лекарственные формы с устойчивостью к измельчению. Описание сущности изобретения Таким образом, в своем первом аспекте настоящее изобретение относится к лекарственной форме,содержащей экструдированные из расплава частицы, включающие лекарственное средство, и матрицу,где указанные экструдированные из расплава частицы присутствуют в указанной матрице в дискретной фазе, при этом указанная матрица представляет собой непрерывную фазу, включающую гелеобразующий агент; и указанные экструдированные из расплава частицы содержат сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты. В предпочтительных лекарственных формах согласно изобретению указанные частицы имеют удлиненную форму. Особенно предпочтительными частицами являются частицы, полученные путем вытягивания и разрезания экструдата из расплава. Более предпочтительные лекарственные формы согласно изобретению содержат 15-80 мас.% указанных частиц по общей массе лекарственной формы. Другие предпочтительные лекарственные формы содержат 20-85 мас.% указанной матрицы по общей массе лекарственной формы. В других предпочтительных лекарственных формах указанная матрица содержит гелеобразующий агент, образующий отвержденный гель. В другом своем аспекте настоящее изобретение относится к способу получения лекарственной формы, включающей лекарственное средство, где указанный способ включает смешивание частиц, полученных путем экструзии из расплава и содержащих лекарственное средство и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, с матричным материалом, содержащим гелеобразующий агент, так, чтобы указанные частицы образовывали в указанной матрице дискретную фазу, и получение из указанной смеси лекарственной формы. В другом своем аспекте настоящее изобретение относится к способу получения лекарственной формы, содержащей лекарственное средство, где указанный способ включает смешивание частиц, полученных путем экструзии из расплава, где указанные частицы получены путем разрезания экструдата (необязательно при его вытягивании), полученного из расплава и содержащего лекарственное средство и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, с матричным материалом, содержащим гелеобразующий агент так, чтобы указанные частицы образовывали в указанной матрице дискретную фазу, и получение из указанной смеси лекарственной формы. В другом своем аспекте настоящее изобретение относится к способу получения лекарственной формы, содержащей лекарственное средство, где указанный способ включает:i) экструзию из расплава композиции, содержащей указанное лекарственное средство и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, с получением экструдата из расплава;ii) разрезание (необязательно при вытягивании) указанного экструдата с получением экструдированных из расплава частиц;iii) смешивание указанных частиц с матричным материалом, содержащим гелеобразующий агент,так, чтобы указанные частицы образовывали в указанной матрице дискретную фазу; иiv) формование из указанной смеси лекарственной формы. В другом своем аспекте, настоящее изобретение относится к лекарственной форме, полученной, как описано выше, в целях ее использования для ослабления или снятия боли. В другом своем аспекте настоящее изобретение относится к применению частиц, полученных путем экструзии из расплава и содержащих лекарственное средство (например, лекарственное средство,которое может быть объектом злоупотребления) и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, и матричного материала, содержащего гелеобразующий агент,в целях изготовления вышеописанной лекарственной формы, которая может быть использована для ослабления боли. Альтернативно, настоящее изобретение также относится к способу лечения индивидуума, нуждающегося в ослаблении боли, где указанный способ включает введение указанному индивидууму лекарственной формы, содержащей лекарственное средство (например, лекарственное средство, которое может быть объектом для злоупотребления), описанное выше. В предпочтительных вариантах настоящего изобретения указанная лекарственная форма является резистентной к модификации. В других предпочтительных вариантах настоящего изобретения указанное лекарственное средство представляет собой лекарственное средство, которое может быть объектом для злоупотребления. В других предпочтительных вариантах настоящего изобретения указанными частицами, присутствующими в указанной лекарственной форме, являются микрочастицы. Подробное описание изобретения Используемый здесь термин "лекарственная форма" означает фармацевтическую молекулу, которая состоит из лекарственного средства, фактически вводимого пациенту или принимаемого самим пациентом. Репрезентативным примером лекарственной формы является таблетка. Другой лекарственной формой является капсула. А предпочтительными лекарственными формами согласно изобретению являются таблетки. Предпочтительные лекарственные формы предназначены для перорального введения. Используемый здесь термин "резистентный к модификации" относится к лекарственным формам,которые являются резистентными к экстракции спиртом. Поэтому такие лекарственные формы могут стать объектами для злоупотребления. Предпочтительными резистентными к модификации лекарственными формами согласно изобретению являются лекарственные формы, резистентные к экстракции спиртом и к измельчению. Предпочтительными лекарственными формами согласно изобретению являются такие лекарственные формы, в которых количество лекарственного средства, высвобождаемого из лекарственной формы в течение 0,5 ч, при измерении на приборе USP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов (SGF) в присутствии 40% этанола при 37 С составляет примерно 20% (например, примерно 10%, еще более предпочтительно примерно 5%) от количества лекарственного средства, высвобождаемого из лекарственной формы в течение 0,5 ч, при измерении на приборе USP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов (SGF) в отсутствии этанола (0%) при 37 С. В особенно предпочтительных лекарственных формах количество лекарственного средства, высвобождаемого из лекарственной формы в течение 0,5 ч,при измерении в вышеупомянутом тесте в SGF в присутствии 40% этанола, меньше, чем количество лекарственного средства, высвобождаемого в SGF в отсутствии этанола (0%), или приблизительно равно такому количеству. Еще более предпочтительно количество лекарственного средства, высвобождаемого в течение 0,5 ч, при измерении в вышеупомянутом тесте в SGF в присутствии 40% этанола составляет 90% или менее, более предпочтительно 80% или менее, например 70% или менее от количества лекарственного средства, высвобождаемого в SGF в отсутствии этанола (0%). Особенно предпочтительные лекарственные формы согласно изобретению могут быть сделаны более плоскими (например, с помощью молотка) без их разрушения, и толщина такой формы может составлять примерно менее 60%, предпочтительно примерно менее чем 50%, а более предпочтительно примерно менее чем 40% от толщины этой лекарственной формы до ее уплощения. Особенно предпочтительные лекарственные формы могут быть сделаны более плоскими (например, с помощью молотка) без их разрушения, и толщина такой формы может составлять примерно в пределах от 10 до 99%, примерно в пределах от 20 до 80% или примерно в пределах от 40 до 60% от толщины этой лекарственной формы до ее уплощения. Особенно предпочтительные лекарственные формы согласно изобретению могут иметь предел прочности при разрыве по меньшей мере 350 Н, предпочтительно 500 Н, например 400495 Н, как было определено в тесте в соответствии с методикой, описанной в примерах. Лекарственные формы согласно изобретению содержат частицы, составляющие их дискретную фазу. Используемый здесь термин "частица" означает дискретную массу вещества, которая является твердой, например, при 20 С, или при комнатной температуре, или при температуре окружающей среды. Предпочтительно, чтобы такая частица была твердой при 20 С. Частицы, присутствующие в лекарственных формах согласно изобретению, получают из экструдата расплава, содержащего лекарственное средство, а более предпочтительно из вытянутого экструдата расплава, содержащего лекарственное средство. Таким образом, частицы, присутствующие в лекарственных формах согласно изобретению, предпочтительно получают путем экструзии из расплава композиции,содержащей лекарственное средство, с образованием экструдата расплава, последующего вытягивания указанного экструдата и разрезания вытянутого экструдата. Поэтому предпочтительные частицы, при-3 022801 сутствующие в виде дискретной фазы лекарственных форм согласно изобретению, отличаются от стандартных макрочастиц, которые обычно имеют размер примерно 1 мм (длина)1 мм (диаметр), и эти частицы, поскольку они образованы из вытянутого экструдата расплава, имеют значительно меньший диаметр, чем стандартные макрочастицы. Такие частицы могут быть похожи на волокна или они могут рассматриваться как волокна. Такая форма этих частиц преимущественно усиливает их сопротивление разрушению и дает возможность включать эти частицы в матрицу. При этом предпочтительно, чтобы частицы, присутствующие в лекарственных формах как дискретная фаза, также не были подвержены силам сжатия. Предпочтительными частицами, присутствующими в лекарственных формах согласно изобретению, являются микрочастицы. Используемый здесь термин "микрочастицы" означает частицы, имеющие среднюю длину и средний диаметр 1000 мкм или менее. "Длина" частиц означает размерность частиц,параллельную направлению экструзии. "Диаметр" частиц является наибольшей размерностью, перпендикулярной направлению экструзии. Предпочтительные частицы, присутствующие в лекарственных формах согласно изобретению,обычно имеют цилиндрическую форму. Поэтому диаметр таких частиц представляет собой диаметр их поперечного сечения. Особенно предпочтительные частицы, например микрочастицы, имеют средний диаметр менее чем примерно 1000 мкм, более предпочтительно менее чем примерно 800 мкм, а еще более предпочтительно менее чем примерно 650 мкм. Особенно предпочтительные микрочастицы имеют средний диаметр менее чем 700 мкм, в частности менее чем 600 мкм, а еще более конкретно менее чем 500 мкм, например менее чем 400 мкм. Поэтому особенно предпочтительные частицы имеют средний диаметр в пределах 200-1000 мкм,более предпочтительно 400-800 мкм, еще более предпочтительно 450-700 мкм, а еще более предпочтительно 500-650 мкм, например примерно 500-600 мкм. Другие предпочтительные частицы имеют средний диаметр в пределах примерно от 300 до 400 мкм, примерно от 400 до 500 мкм или примерно от 500 до 600 мкм. Еще меньший диаметр частиц, например микрочастиц согласно изобретению по сравнению с диаметром стандартных макрочастиц, предпочтительно достигается путем вытягивания экструдата из расплава перед гранулированием. Поэтому минимальный средний диаметр частиц, например микрочастиц, определяется, главным образом, степенью его возможного вытягивания экструдата без его разрушения, и такой диаметр может составлять, например, примерно 500 мкм, примерно 400 мкм, примерно 300 мкм или примерно 200 мкм в зависимости от состава экструдата из расплава. Предпочтительные частицы, присутствующие в лекарственных формах согласно изобретению,имеют среднюю длину менее чем примерно 1000 мкм, предпочтительно менее чем примерно 800 мкм,более предпочтительно менее чем примерно 650 мкм, например примерно 600 мкм, примерно 500 мкм,примерно 400 мкм или примерно 300 мкм. Особенно предпочтительные частицы имеют среднюю длину менее чем 700 мкм, в частности менее чем 650 мкм, более предпочтительно 550 мкм, например менее чем 450 мкм. Поэтому особенно предпочтительные частицы имеют среднюю длину в пределах 200-1000 мкм, более предпочтительно 400-800 мкм, еще более предпочтительно 450-700 мкм, а еще более предпочтительно 500-650 мкм, например примерно 500-600 мкм. Минимальную среднюю длину микрочастиц определяют в стадии разрезания, и такая длинаможет составлять, например, 500 мкм, 400 мкм, 300 мкм или 200 мкм. Размер частиц может быть определен любым стандартным методом, известным специалистам, например по рассеянию лазерного излучения, с помощью анализа на ситах, с помощью оптического микроскопа или с помощью визуализирующего анализа. Частицы, присутствующие в лекарственных формах согласно изобретению, являются резистентными к разрушению. Таким образом, предпочтительно частицы могут быть сделаны более плоскими (например, с помощью молотка) без их разрушения, и толщина таких частиц может составлять примерно менее 60%, предпочтительно примерно менее чем 50%, а более предпочтительно примерно менее чем 40% от толщины этих частиц до их уплощения. Особенно предпочтительные частицы могут быть сделаны более плоскими (например, с помощью молотка) без их разрушения, и толщина таких частиц может составлять примерно от 10 до 99%, примерно от 20 до 80% или примерно от 40 до 60% от толщины этих частиц до их уплощения. Некоторые частицы, используемые в настоящем изобретении, могут иметь предел прочности при разрыве по меньшей мере 350 Н, предпочтительно 500 Н, например 400-495 Н, как было протестировано в соответствии с процедурой, описанной в примерах. Частицы, присутствующие в лекарственных формах согласно изобретению, предпочтительно содержат лекарственное средство, которое является объектом для злоупотребления. Такое лекарственное средство, которое является объектом для злоупотребления, предпочтительно представляет собой агонист опиоидов; транквилизатор; средство, подавляющее ЦНС; средство, стимулирующее ЦНС; или снотворное средство седативного действия. Особенно предпочтительное лекарственное средство, которое является объектом для злоупотребления, представляет собой агонист опиоидов. В особенно предпочтительных лекарственных формах согласно изобретению указанным лекарственным средством является агонист опиоидов, выбранный из группы, состоящей из оксикодона, оксиморфона, гидрокодона, гидроморфона, морфина, кодеина, бупренорфина, фентанила, трамадола, тапентадола и их фармацевтически приемлемых солей. Еще более предпочтительным лекарственным средством согласно изобретению является агонист опиоидов, выбранный из группы, состоящей из оксикодона,оксиморфона, гидрокодона, гидроморфона, морфина, кодеина и их фармацевтически приемлемых солей. В некоторых вариантах изобретения предпочтительным агонистом опиоидов является оксикодон. В других предпочтительных вариантах изобретения предпочтительным агонистом опиоидов является гидроморфон. Специалист в данной области может легко выбрать подходящие фармацевтически приемлемые соли. Фармацевтически приемлемыми солями являются, но не ограничиваются ими, соли металлов, такие как соль натрия, соль калия и соль цезия; соли щелочно-земельных металлов, такие как соль кальция и соль магния; соли органических аминов, такие как соль триэтиламина, соль пиридина, соль пиколина,соль этаноламина, соль триэтаноламина, соль дициклогексиламина и соль N,N'-дибензилэтилендиамина; соли неорганических кислот, такие как гидрохлорид, гидробромид, сульфат и фосфат; соли органических кислот, такие как формиат, ацетат, трифторацетат, малеат и тартрат; сульфонаты, такие как метансульфонат, бензолсульфонат и п-толуолсульфонат; соли аминокислот, такие как аргинат, аспаргинат и глутамат. В основном предпочтительными являются соли неорганических кислот. Предпочтительными агонистами опиоидов являются гидрохлорид оксикодона и гидрохлорид гидроморфона. Лекарственное средство, включенное в препарат лекарственных форм согласно изобретению, предпочтительно имеет средний размер частиц менее чем 500 мкм, более предпочтительно менее чем 300 мкм, а еще более предпочтительно менее чем 200 или 100 мкм. Нижний предел среднего размера частиц не имеет каких-либо ограничений и может составлять, например, 50 мкм. Размер частиц лекарственных средств может быть определен любым стандартным методом, известным специалистам, например методом рассеяния лазерного излучения, с помощью анализа на ситах, с помощью оптического микроскопа или с помощью визуализирующего анализа. Вообще говоря, предпочтительно, чтобы наибольшая размерность частиц лекарственного средства была меньше, чем размер частицы (например, меньше, чем наименьшая размерность частицы). Также очевидно, что в основном предпочтительнее использовать частицы лекарственного средства и/или не расплавляющиеся и/или не размягчающиеся наполнители,имеющие частицы, размер которых меньше половины диаметра вытянутого экструдата из расплава. Это служит гарантией того, что такие частицы, содержащие лекарственное средство, будут образовывать дискретную фазу в непрерывной матрице. Если это достижимо, то экструдат может быть вытянут с минимальным риском разрыва. Частицы, присутствующие в лекарственных формах согласно изобретению, предпочтительно содержат 3-50 мас.% лекарственного средства, более предпочтительно 5-40 мас.% лекарственного средства,еще более предпочтительно 7,5-35 мас.% лекарственного средства, например 10-20 мас.% лекарственного средства, где мас.% приводятся по отношению к общей массе частицы. Лекарственные формы согласно изобретению могут также содержать один или несколько дополнительных активных ингредиентов. Дополнительным активным ингредиентом может быть лекарственное средство, которое является объектом для злоупотребления, или другое фармацевтическое средство. Дополнительные активные ингредиенты могут присутствовать в вышеописанных частицах ("интрагранулярные") или в матрице ("экстрагранулярные"). Если дополнительный активный ингредиент присутствует в частице, то он может присутствовать либо в виде комбинации с одним или несколькими активными ингредиентами, присутствующими в тех же самых частицах, либо он может присутствовать в дискретной популяции отдельных частиц и может быть отделен от какого-либо другого активного ингредиента, присутствующего в лекарственной форме. Предпочтительными частицами, присутствующими в лекарственных формах согласно изобретению, являются частицы, имеющие подходящий предел прочности при растяжении, определяемый тестметодом, который в настоящее время применяется специалистами. Другими предпочтительными частицами являются частицы, имеющие модуль Юнга, определяемый специалистами с проведением соответствующих испытаний. Другими предпочтительными частицами являются микрочастицы, имеющие приемлемое относительное удлинение при разрыве. Частицы, присутствующие в лекарственных формах согласно изобретению, предпочтительно содержат полимер, который сообщает резистентность к разрушению, а особенно предпочтительно каучукоподобный полимер или полимер с пластическими свойствами. Это означает, что если лицо, злоупотребляющее лекарственными средствами, имеет намерение разрушить такую лекарственную форму, то присутствие указанного полимера будет препятствовать высвобождению лекарственного средства из микрочастиц. Кроме того, благодаря достаточно малому размеру частиц, присутствующих в матрице,они не могут быть отделены от измельченной матрицы данным лицом. Предпочтительными полимерами,которые сообщают резистентность к разрушению, являются полимер Eudragit NE, или NM полимер, или полимер, обладающий пластическими свойствами, такой как полимер Eudragit RS или RL полимер. В предпочтительных лекарственных формах согласно изобретению полимерами, сообщающими резистентность к разрушению, например каучукоподобными полимерами, являются акриловый полимер,метакриловый полимер или их смеси. Таким образом, лекарственные формы согласно изобретению предпочтительно содержат полимер, выбранный из акрилового полимера, метакрилового полимера или их смесей. Помимо повышения резистентности к разрушению эти полимеры также облегчают экструзию из расплава, а также регуляцию скорости высвобождения лекарственного средства из частиц. Репрезентативными примерами акриловых и метакриловых полимеров являются сополимеры акриловой и метакриловой кислоты, сополимеры метилметакрилата, этоксиэтилметакрилата, цианоэтилметакрилата, полиакриловой кислоты, полиметакриловой кислоты, сополимер метакриловой кислоты и алкиламида, полиметилметакрилата, полиметакрилата, сополимер полиметилметакрилата, полиакриламида, сополимер аминоалкилметакрилата, сополимер ангидрида метакриловой кислоты и сополимеры глицидилметакрилата. Особенно предпочтительные частицы, присутствующие в лекарственной форме согласно изобретению, содержат сополимер алкиловых эфиров акриловой кислоты, алкиловых эфиров метакриловой кислоты и их смесей. Предпочтительными алкиловыми эфирами являются метиловые и этиловые эфиры. Особенно предпочтительным сополимером является сополимер этилакрилата и метилметакрилата. Полимер, сообщающий резистентность к разрушению, например каучукоподобный полимер, может быть нейтральным (то есть не несет заряда) или заряженным. В нейтральных полимерах боковые цепи(например, алкильная группа боковых цепей алкилового сложного эфира) обычно не являются функционализированными. В заряженных полимерах боковая цепь (например, алкильная группа боковых цепей алкилового сложного эфира) обычно функционализирована, например, группой четвертичного аммония,такой как триметиламмоний. Триметиламмониевые группы предпочтительно присутствуют в виде солей и имеют тенденцию к сообщению полимерам водопроницаемости. Предпочтительным полимером, сообщающим резистентность к разрушению, является нейтральный полимер (то есть, незаряженный). Особенно предпочтительный полимер, сообщающий резистентность к разрушению, например каучукоподобный полимер, такой как сополимер этилакрилата и метилметакрилата, имеет среднюю молекулярную массу в пределах от 50000 до 200000, а более предпочтительно от 60000 до 150000, например от 70000 до 100000. Средняя молекулярная масса представляет собой среднечисленную молекулярную массу. Акриловые и метакриловые полимеры, используемые в частицах, присутствующих в лекарственных формах согласно изобретению, являются коммерчески доступными и поставляются, например, фирмой Evonik. Репрезентативными примерами подходящих полимеров являются полимеры, имеющиеся в продаже под торговым знаком Eudragit, а в частности Eudragit RL 100, Eudragit RL РО, Eudragit RS 100, Eudragit RS PO, Eudragit NE 40 D, Eudragit NE 30 D. Особенно предпочтительными являются полимеры Eudragit NE 40 D, Eudragit NE 30 D и Eudragit NM 30 D, которые представляют собой нейтральные сополимеры этилакрилата и метилметакрилата, имеющие среднюю молекулярную массу примерно 150000. Частицы, присутствующие в лекарственных формах согласно изобретению, предпочтительно содержат 10-50 мас.%, более предпочтительно 20-40 мас.%, а еще более предпочтительно 25-35 мас.% полимера, сообщающего резистентность к разрушению, например каучукоподобного полимера, такого как акриловый полимер, метакриловый полимер или их смесь, по общей массе частиц. Лекарственное средство, присутствующее в полимере, сообщающем резистентность к разрушению,например каучукоподобном полимере, таком как акриловый полимер, метакриловый полимер или их смесь, может быть растворимым. Однако предпочтительно, чтобы лекарственное средство, присутствующее в таком полимере, не было растворимым. Частицы, присутствующие в лекарственных формах согласно изобретению, предпочтительно содержат скорость-регулирующий или скорость-модифицирующий агент. Используемый здесь термин"скорость-регулирующий или скорость-модифицирующий агент" означает компонент частиц, который был включен в эти частицы в целях изменения скорости высвобождения лекарственного средства из этих частиц. Предпочтительными скорость-регулирующими или скорость-модифицирующими агентами, используемыми в указанных частицах, являются агенты регулируемым, а в частности пролонгированным высвобождением. Предпочтительными скорость-регулирующими или скорость-модифицирующими агентами, используемыми в настоящем изобретении, являются гидрофобные материалы, а в частности гидрофобные полимеры. Другими предпочтительными скорость-регулирующими или скорость-модифицирующими агентами являются вещества, не растворяющиеся в воде, а в частности не растворимые в воде полимеры. Скорость-регулирующим или скорость-модифицирующим агентом может быть, но необязательно,полимер, сообщающий резистентность к разрушению. Особенно предпочтительными скорость-регулирующими или скорость-модифицирующими агентами, используемыми в указанных частицах, являются алкилцеллюлозы. Такими алкилцеллюлозами являются природные и синтетические алкилцеллюлозы. Подходящими также являются водорастворимые и не растворимые в воде производные целлюлозы. Репрезентативными примерами алкилцеллюлоз и гидроксиалкилцеллюлоз являются водорастворимая метилцеллюлоза, гидроксипропилцеллюлоза и гидроксипропилметилцеллюлоза. Примером не растворимой в воде алкилцеллюлозы является этилцеллюлоза. Особенно предпочтительной алкилцеллюлозой, используемой в качестве скорость-регулирующего или скорость-модифицирующего агента в частицах, присутствующих в лекарственных формах согласно изобретению, является этилцеллюлоза. Подходящие алкилцеллюлозы, используемые в частицах, являются коммерчески доступными. Примерами коммерчески доступных алкилцеллюлоз, которые могут присутствовать, являются этилцеллюлоза N10 и N45, а также водная дисперсия Surerelease Е-7-1940. Особенно предпочтительной является этилцеллюлоза N10 и N45. Такие целлюлозы могут быть закуплены у различных поставщиков. Алкилцеллюлоза может быть получена в форме гранул, порошка или тонкодисперсного порошка. Все эти формы являются коммерчески доступными. Другими скорость-регулирующими или скорость-модифицирующими агентами, которые могут быть подходящими для их использования в частицах лекарственных форм согласно изобретению, являются нерастворимые гидрофильные капиллярные агенты, такие как микрокристаллическая целлюлоза,натрийсодержащая кроскармелоза, кросповидон и гликолят натрия-крахмала; гелеобразующие агенты,которые гидратируются с образованием гелей, используемых для регуляции перемещения воды, такие как высокомолекулярная гидроксипропилметилцеллюлоза эталонной чистоты (с высокой вязкостью)(НРМС), полиэтиленоксид, пектин, камедь рожкового дерева и ксантановая камедь; высокомолекулярные полиэтиленгликоли, такие как ПЭГ 6000; и водопроницаемый метакрилат аммония (также называемый сополимером метакрилата аммония), такой как Eudragit RL PO. Частицы, присутствующие в лекарственных формах согласно изобретению, предпочтительно содержат 20-50 мас.% скорость-регулирующего или скорость-модифицирующего агента, более предпочтительно 25-45 мас.% скорость-регулирующего или скорость-модифицирующего агента, а еще более предпочтительно 30-40 мас.% скорость-регулирующего или скорость-модифицирующего агента по общей массе частицы. Предпочтительные частицы, присутствующие в лекарственных формах согласно изобретению, могут также содержать замасливатель. Замасливатели представляют собой технологические добавки, которые способствуют снижению степени трения между полимерной смесью или полимерными комбинациями и, например, внутренними поверхностями экструдера. Репрезентативными примерами замасливателей являются стеариновая кислота, глицерилбегенат (например, в форме глицерилдибегената), стеарат магния, стеарат кальция, тальк и диоксид кремния (плавленый кварц). Присутствие замасливателя в экструзионной смеси приводит к улучшению смешивания, замеса и перемещения и к снижению силы сцепления. Экструзия в присутствии мягкой смазки при низких или умеренных температурах приводит к улучшению воспроизводимости партий продукта и к снижению деформации как продукта, так и промышленного оборудования. Предпочтительным замасливателем, используемым в частицах, является глицерилдибегенат. Количество замасливателя, присутствующего в частицах, предпочтительно составляет в пределах 125 мас.%, более предпочтительно в пределах 2-15 мас.%, а еще более предпочтительно в пределах 3-10 мас.% по общей массе частиц. Предпочтительные частицы, присутствующие в лекарственных формах согласно изобретению, могут также содержать пластификатор. Пластификаторы облегчают экструзию, а также снижают степень сцепления посредством внутреннего замасливания любых полимеров, присутствующих в частицах. Репрезентативными примерами пластификаторов являются не растворимые в воде твердые вещества (например, цетиловый спирт, стеариловый спирт и цетостеариловый спирт), водорастворимые твердые вещества (например, сорбит, сахароза, полиэтиленгликоль) и жидкости (например, дибутилсебакат, трибутилцитрат, ацетилтрибутилцитрат, триэтилцитрат, ацетилтриэтилцитрат, триацетин, дибутилфталат, диэтилфталат, пропиленгликоль и полисорбат 80). Предпочтительным твердым пластификатором является стеариловый спирт. Предпочтительными также являются жидкие пластификаторы. Предпочтительным жидким пластификатором является триэтилцитрат. Количество пластификатора, присутствующего в лекарственных формах согласно изобретению,предпочтительно составляет в пределах 1-30 мас.%, более предпочтительно 5-20 мас.%, а еще более предпочтительно 10-15 мас.% по общей массе частицы. Пластификаторы могут иногда действовать как замасливатели, а замасливатели могут иногда действовать как пластификаторы. Если частицы, присутствующие в лекарственных формах согласно изобретению, содержат агонист опиоида, то указанная лекарственная форма может также содержать антагонист опиоида. При этом может присутствовать любой подходящий антагонист опиоида, например налтрексон или налоксон или их фармацевтически приемлемые соли. Особенно предпочтительным является налоксон, включая его соли. Антагонист опиоида может присутствовать в частицах или в матрице. Альтернативно, антагонист опиоида может быть введен в вышеописанные лекарственные средства в отдельных частицах. Предпочтительный состав таких частиц аналогичен составу частиц, содержащих лекарственные средства. Особенно предпочтительные лекарственные средства согласно изобретению содержат налоксон, а в частности гидрохлорид налоксона, и частицы, содержащие агонист опиоида, выбранный из оксикодона или одной из его фармацевтически приемлемых солей и гидроморфона или одной из его фармацевтически приемлемых солей. Особенно предпочтительными агонистами опиоида являются гидрохлорид оксикодона и гидрохлорид гидроксиморфона. Отношение агониста опиоида к антагонисту опиоида в лекарственных формах согласно изобретению предпочтительно составляет 1:1 до 3:1 по массе, например примерно 2:1 по массе. Так, например,если агонистом опиоида является гидроморфон-HCl, а антагонистом опиоида является налоксон-HCl, то отношение агонист:антагонист может составлять 1:1 до 3:1 по массе, например примерно 2:1 по массе. Так, например, если агонистом опиоида является оксикодон-HCl, а антагонистом опиоида является налоксон-HCl, то отношение агонист:антагонист может составлять 1:1 до 3:1 по массе, а предпочтительно примерно 2:1 по массе. Если антагонист опиоида присутствует в лекарственных формах согласно изобретению, то общее количество агониста опиоида и антагониста опиоида, присутствующих в частицах, предпочтительно составляет в пределах 5-40 мас.%, более предпочтительно, 10-30 мас.%, а еще более предпочтительно 20-25 мас.% по общей массе частицы. Если агонистом опиоида является гидроморфон или его соль (например,HCl-соль) и если присутствует антагонист (например, налоксон-HCl), то количество гидроморфона или его соли, присутствующих в лекарственной форме, предпочтительно составляет 2-80 или 5-80 мг, например 5, 10, 20, 40 или 80 мг, а еще более предпочтительно количество гидроморфона или его соли составляет 2-32 мг, например 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 24, 28 или 32 мг. Если агонистом опиоида является оксикодон или его соль (например, HCl-соль) и если присутствует антагонист (например, налоксон-HCl),то количество оксикодона или его соли, присутствующих в лекарственной форме, предпочтительно составляет 2-32 или 5-80 мг, например 2, 4, 8, 16 или 32 мг, а еще более предпочтительно количество оксикодона или его соли составляет 5-80 мг, например 5, 10, 20, 30, 40, 50, 60, 70 или 80 мг. Подходящее процентное содержание каждого из вышеописанных предпочтительных компонентов частиц, присутствующих в лекарственных формах согласно изобретению, приводится в нижеследующей таблице, где указанные проценты даны по отношению к общей массе частиц. В этой таблице процентное содержание компонентов может быть равно любому числу в пределах указанных интервалов, а также любому числу в других предпочтительных интервалах. Частицы, присутствующие в лекарственных формах согласно изобретению, могут также содержать другие наполнители, известные специалистам, например разбавители, связующие агенты, стимуляторы грануляции, красители, отдушки, агенты, повышающие скольжение, и другие агенты, модифицирующие высвобождение. Специалист в данной области может легко выбрать и другие подходящие наполнители,а также определить количество каждого из этих наполнителей. Конкретные примеры фармацевтически приемлемых носителей и наполнителей, которые могут быть использованы для приготовления лекарственных форм согласно изобретению, описаны в руководстве Handbook of Pharmaceutical Excipients,American Pharmaceutical Association (1986), содержание которого вводится в настоящее описание посредством ссылки. В качестве наполнителей могут быть использованы лактоза, глюкоза или сахароза, крахмалы и их гидролизаты, микрокристаллическая целлюлоза, целлатоза, спирты ряда сахаров, такие как сорбит или маннит, соли кальция, растворяющиеся в большинстве растворителей, такие как бифосфат кальция, дифосфат или трифосфат кальция. Повидон может быть использован в качестве стимулятора грануляции. В качестве агентов, придающих текучесть, могут быть предпочтительно использованы высокодисперсная двуокись кремния, тальк, кукурузный крахмал, оксид магния и стеарат магния или кальция. Особенно предпочтительные частицы, присутствующие в лекарственных формах согласно изобретению, включают оксикодон или гидроморфон предпочтительно в виде их гидрохлоридных солей, сополимер этилакрилата и метилметакрилата, предпочтительно Eudragit NE 30 D или NE 40 D, этилцеллюлозу в качестве скорость-регулирующего или скорость-модифицирующего агента, стеариловый спирт и/или триэтилцитрат в качестве пластификатора, глицерилдибегенат в качестве замасливателя и, необязательно, антагонист опиоида. Если присутствует антагонист опиоида, то им предпочтительно является налоксон, а в частности его гидрохлоридная соль. Частицы, присутствующие в лекарственных формах согласно изобретению, предпочтительно получают путем смешивания компонентов, экструзии смеси из расплава и последующего вытягивания и разрезания экструдата, например, с получением продукта с предварительно определенными диаметром и длиной. Смешивание может быть осуществлено любым стандартным методом, например путем объединения компонентов и/или их грануляции до достижения гомогенности. В предпочтительном методе смешивание компонентов осуществляют путем грануляции. Гранулят предпочтительно сушат. Затем этот гранулят может быть подвергнут экструзии, вытягиванию и разрезанию, как описано выше. Так,например, лекарственное средство может быть гранулировано вместе с другими компонентами с получением гранул, содержащих лекарственное средство, после чего эти гранулы сушат, а затем высушенные гранулы подвергают экструзии и разрезают. Альтернативно, компоненты, не содержащие лекарственного средства, могут быть гранулированы с получением плацебо-гранул, которые затем сушат, и подвергают сухому смешиванию с лекарственным средством, после чего полученную сухую смесь подвергают экструзии и разрезают. Последний способ является предпочтительным способом получения частиц, содержащих активные агенты, чувствительные к воздействию воды, например гидроморфон или его фармацевтически приемлемые соли, используемые в качестве лекарственного средства. В особенно предпочтительном способе жидкий пластификатор смешивают со скоростьрегулирующим или со скорость-модифицирующим полимером (например, этилцеллюлозой) в количестве 5-25 мас.% (по массе скорость-регулирующего полимера) и оставляют, например, на 5-12 ч. Это позволяет пластификтору глубоко проникать в полимерную структуру, что приводит к снижению температуры перехода в стеклообразное состояние (Tg) и в конечном счете к повышению резистентности скорость-регулирующего или скорость-модифицирующего полимера к разрушению. Затем пластифицированный скорость-регулирующий или скорость-модифицирующий полимер смешивают, например гранулируют с другими компонентами, например с агонистом опиоида, полимером, сообщающим резистентность к разрушению (например, с каучукоподобным полимером и/или с полимером, обладающим пластифицирующими свойствами), с замасливателем и с пластификатором. Смешивание может быть осуществлено в любом подходящем смесителе. Экструзия может быть осуществлена на любом подходящем оборудовании для экструзии, например на экструдере с диском для плавления, а предпочтительно на двухшнековом экструдере, который имеет шнеки, вращающиеся в одном направлении или в противоположных направлениях. Обычно смесь (например, в виде порошка или сухих гранул) подают с помощью подающего устройства в первый отсек барабана, обычно при относительно низкой температуре (например, 10-20 С) для обеспечения постоянного потока, подаваемого в барабанные отсеки с более высокой температурой. Подающее устройство обеспечивает равномерный поток смеси в экструдер. Такая равномерность является желательной, поскольку неравномерная и варьирующаяся скорость подачи может приводить к образованию частиц с измененными физическими свойствами, такими как плотность и пористость. Предпочтительным является экструдер с двумя шнеками, а более предпочтительно со шнеками,вращающимися в противоположном направлении и обеспечивающими подачу, смешивание, прессование, нагревание и размягчение смеси. В зависимости от выбора компонентов смеси и условий экструзии такая смесь может быть расплавлена, а также подвергнута размягчению. Шнеки, которые участвуют в значительной части такого процесса экструзии, изготавливают из различных более мелких элементов,выбранных из ряда элементов для шнеков и элементов для смесителей. Время смешивания и замеса может быть в значительной степени изменено путем изменения типа, длины и конфигурации элементов шнека и, возможно, элементов смесителя. Короткое время пребывания и умеренные или низкие усилия сдвига обеспечивают безопасность обработки продукта и его стабильность, даже если присутствующие лекарственные средства являются термочувствительными. Примерами подходящих экструдеров являются экструдеры, изготавливаемые Leistritz, Brabender, Randcastle и Kurimoto Co. Ltd. Скорость вращения шнеков может оказывать определенное влияние на качество продуцируемых частиц. Высокая скорость вращения без соответствующей компенсации за счет скорости подачи смеси может приводить к продуцированию высокопористых частиц с варьирующейся скоростью высвобождения лекарственного средства. С другой стороны, медленное вращение шнеков вручную может индуцировать нежелательное продолжительное время пребывания частиц в экструдере. Для удаления воздуха,попадаемого в размягченную смесь, желательно, чтобы в барабан экструдера подавался вакуум, в результате чего будут продуцироваться плотные частицы с низкой пористостью. Помимо скорости вращения шнеков другими главными и важными параметрами являются крутящий момент шнека, внутренняя температура барабана, а также давление и температура в экструзионной головке. Предпочтительно экструзию осуществляют при температуре 100 С или менее, например при 80100 С. Экструзионная головка обычно предназначена для продуцирования полимерных нитей фиксированного диаметра. Число, форма и диаметр отверстий экструдера могут быть изменены для удовлетворения предварительно определенным техническим условиям. Однако в основном диаметр экструдата составляет 1,0-1,2 мм, то есть он аналогичен диаметру стандартного экструдата. Поэтому преимущество предпочтительного варианта осуществления изобретения заключается в том, что для экструзии может быть использовано такое же стандартное оборудование, которое используется для получения стандартных макрочастиц. Это означает, что в данном случае не требуется снижения размера отверстий головки(которое может приводить к повышению вероятности забивки головки) или повышения давления, необходимого для достижения экструзии. Экструзия также представляет собой хорошо разработанный способ продуцирования частиц, применяемый в фармацевтике, и такой способ хорошо известен специалистам в данной области. Специалистам в данной области также хорошо известно, что для получения продуктов с нужными свойствами, в процессе проведения экструзии различные параметры, такие как скорость подачи, скорость вращения шнеков, температура обогрева различных зон экструдера (если они имеются), содержание воды и т.п.,могут варьироваться. Вышеупомянутые параметры зависят от конкретного типа применяемого экструдера. В процессе экструзии температура зон обогрева, в которых компоненты препарата согласно изобретению расплавляются, может составлять от 40 до 120 С, предпочтительно от 50 до 100 С, более предпочтительно от 50 до 90 С, еще более предпочтительно от 50 до 70 С, а наиболее предпочтительно от 50 до 65 С, в частности, если используются двухшнековые экструдеры со шнеками, вращающимися в противоположном направлении. Специалистам в данной области хорошо известно, что не каждая зона обогрева должна нагреваться. А в частности, в зоне, расположенной за подающим устройством, в которой компоненты смешиваются, может оказаться необходимым охлаждение приблизительно при 25 С. Скорость вращения шнеков может варьироваться от 50 до 500 об/мин, предпочтительно 100-250 об/мин, более предпочтительно 100-200 об/мин, а наиболее предпочтительно примерно 150 об/мин, в частности, если используются двухшнековые экструдеры со шнеками, вращающимися в противоположном направлении. При этом могут быть выбраны необходимые конфигурация и диаметр сопла. Диаметр сопла обычно используемых экструдеров в основном составляет 1-10 мм, предпочтительно 2-8 мм, а наиболее предпочтительно 3-5 мм. Отношение длины к диаметру шнеков экструдера, который может быть использован для получения описанных здесь лекарственных форм, обычно составляет примерно 40:1. В общих чертах, температуры зон обогрева выбирают так, чтобы не происходило повышения температуры до значений, которые могут приводить к разрушению фармацевтически активных соединений. Скорость подачи и скорость вращения шнеков выбирают так, чтобы осуществлялось пролонгированное,независимое и постоянное высвобождение фармацевтически активных соединений из препаратов, получаемых путем экструзии. Если, например, увеличить скорость подачи, то соответственно может быть увеличена скорость вращения шнеков, что будет обеспечивать одинаковое время запаздывания. Специалистам в данной области известно, что вышеупомянутые параметры зависят от конкретных условий продуцирования (типа экструдера, конфигурации шнеков, числа компонентов и т.п.) и могут быть адаптированы так, чтобы полученный экструдат имел нужные свойства. Затем экструдат из расплава предпочтительно вытягивают. Это предпочтительно осуществляют до тех пор, пока еще данный экструдат сохраняет свою эластичность. Вытягивание предпочтительно осуществляют с использованием конвейерной ленты, по которой экструдат подается в гранулирующее устройство и/или на прижимные ролики гранулирующего устройства. Особенно предпочтительно, чтобы скорость движения конвейерной ленты и скорость прижимных роликов были скоординированы и скорректированы для достижения желаемого вытягивания. Обычно конвейерную ленту и/или прижимные ролики устанавливают на более высокую скорость продуцирования экструдата, чем это обычно присходит в экструдере. Однако специалист в данной области может легко выбрать соответствующие параметры для экструдера, конвейерной ленты, прижимных роликов и т.п. в целях достижения желаемого вытягивания. В процессе вытягивания диаметр экструдата из расплава уменьшается, а длина экструдата соответственно увеличивается. При этом предпочтительно, чтобы вытягивание приводило к снижению диаметра экструдата из расплава на 80-30% от исходного диаметра, более предпочтительно на 75-40%, еще более предпочтительно на 70-45%, а наиболее предпочтительно на 60-50%, например приблизительно на 50% от его исходного диаметра. Максимальным количеством, на которое может быть вытянут экструдат из расплава, может быть такое количество, которое позволяет уменьшить диаметр экструдата примерно на 40-30% (например, на 30%) от его исходного диаметра, в зависимости от состава экструдата, полученного из расплава, и/или от размера частиц присутствующего в нем лекарственного средства. Особенно предпочтительно, чтобы средний диаметр вытянутого экструдата из расплава составлял менее чем примерно 1000 мкм, более предпочтительно менее чем примерно 800 мкм, еще более предпочтительно менее чем примерно 650 мкм, например менее чем примерно 600 или 500 мкм, а еще более предпочтительно менее чем примерно 450 мкм, например примерно 300-400 мкм. Поэтому минимальный средний диаметр частиц определяется, главным образом, степенью возможного вытягивания экструдата без разрушения, и такой диаметр может составлять, например, примерно 500, 400, 300 или 200 мкм. И в этом случае такой диаметр зависит от точного состава экструдата. Поэтому средний диаметр вытянутого экструдата из расплава предпочтительно составляет в пределах 200-1000 мкм, более предпочтительно 400-800 мкм, еще более предпочтительно 450-700 мкм, а еще более предпочтительно 500-650 мкм, на- 10022801 пример примерно 500-600 мкм. В других предпочтительных вариантах средний диаметр вытянутого экструдата из расплава составляет в пределах 300-600 мкм, например 400-500 мкм. Диаметр вытянутого экструдата из расплава предпочтительно остается постоянным в течение всего периода времени. Таким образом, вышеупомянутые средние диаметры частиц предпочтительно составляют 20%, а более предпочтительно 10%, например 5%. Измерение диаметра с помощью лазера может быть осуществлено, но необязательно, в период времени между подачей по конвейерной ленте и пропусканием через гранулятор для непрерывного мониторинга диаметра экструдата. Информация, полученная по системе мониторинга, может быть использована для регуляции скорости конвейерной ленты и/или прижимных роликов. Измерение диаметра с помощью лазера может быть также применено для определения среднего диаметра экструдата. Для продуцирования частиц, присутствующих в лекарственной форме согласно изобретению, вытянутый экструдат из расплава разрезают. Разрезание может быть осуществлено любым стандартным методом, известным специалистам. Так, например, вытянутый экструдат может быть подан на гранулятор с помощью прижимных роликов. Затем в этом грануляторе поданный экструдат разрезают, например, с помощью дисковых ножей, на отрезки с заранее определенной длиной, которая в среднем составляет примерно менее чем 1000 мкм, предпочтительно менее чем примерно 800 мкм, более предпочтительно менее чем примерно 650 мкм, например примерно 600 или 500 мкм. В других предпочтительных вариантах поданный экструдат разрезают на отрезки со средней длиной примерно 300-600 мкм, например примерно 400, 450 или 500 мкм. Скорость подачи, например скорость конвейерной ленты с экструдатом из расплава и скорость вращения ножей гранулятора, определяют, главным образом, длину частиц. Минимальная средняя длина частиц может составлять, например, 400-200 мкм (например, 200 мкм). Поэтому средняя длина частиц предпочтительно составляет в пределах 200-1000 мкм, например примерно 300 мкм, примерно 400 мкм или примерно 500 мкм, более предпочтительно 400-800 мкм, например примерно 400-500 мкм, еще более предпочтительно 450-700 мкм, например примерно 450-600 мкм, а еще более предпочтительно 500-650 мкм, например примерно 500-600 мкм. В лекарственных формах согласно изобретению вышеописанные частицы включены в матрицу. Используемый здесь термин "матрица" означает непрерывную фазу, присутствующую в лекарственной форме. Матрица лекарственных форм согласно изобретению предпочтительно содержит один или несколько гелеобразующих агентов и/или силикон. Предпочтительные силиконы описаны ниже. Предпочтительно, чтобы матрица лекарственных форм содержала один или несколько гелеобразующих агентов. Предпочтительными гелеобразующими агентами являются полимеры. Средние молекулярные массы полимеров, присутствующих в матрице, являются среднечисленными, если это не оговорено особо. Используемый здесь термин "гелеобразующий агент" означает соединение, которое после его контактирования с растворителем (например, с водой) абсорбирует растворитель и набухает, в результате чего образуется вязкое или полувязкое вещество. Это вещество может замедлять высвобождение лекарственного средства из включающих его частиц в водную и в водно-спиртовую среду. После полной гидратации обычно образуется густой вязкий раствор или густая вязкая дисперсия, значительно снижающие и/или минимизирующие количество свободного растворителя, который может содержать определенное количество солюбилизированного лекарственного средства и может быть забран в шприц. Образованный таким образом гель может также снижать общее количество лекарственного средства, экстрагируемого растворителем путем удерживания лекарственного средства в гелевой структуре. Таким образом, гелеобразующий агент может играть важную роль в сообщении лекарственным формам согласно изобретению резистентности к модификации. Предпочтительными гелеобразующими агентами, которые могут быть использованы в лекарственных формах согласно изобретению, являются фармацевтически приемлемые полимеры, обычно гидрофильные полимеры, такие как гидрогели. Предпочтительные полимеры, используемые в качестве гелеобразующего агента, приобретают высокую степень вязкости после их контактирования с подходящим растворителем. Если лицо, злоупотребляющее лекарственными средствами, попытается раздробить и растворить содержимое лекарственной формы в водном и/или водно-спиртовом носителе и ввести его внутривенно, то высокая вязкость указанного агента будет усиливать образование высоковязких гелей. Репрезентативными примерами полимеров, которые могут быть использованы в качестве гелеобразующего агента, являются полиэтиленоксид, поливиниловый спирт, гидроксипропилметилцеллюлоза,карбомеры, поли(уроновые) кислоты и их смеси. Предпочтительные свойства каждого из этих полимеров описаны ниже. Особенно предпочтительными силиконами и гелеобразующими агентами (например, полимерами),используемыми в лекарственных формах согласно изобретению, являются силиконы и агенты, которые могут отверждаться. Используемый здесь термин "отверждаемый" относится к агентам, а обычно к полимерам, которые могут подвергаться сшиванию, например, путем нагревания. Сшивание, которое осуществляют в процессе отверждения, служит для повышения жесткости или прочности агента, например полимера, и тем самым сообщает данной лекарственной форме резистентность к разрушению. Такие лекарственные формы являются особенно предпочтительными, поскольку они образуются по двум меха- 11022801 низмам, обеспечивающим их резистентность к разрушению, а именно посредством отверждения матрицы, а также отверждения частиц. Репрезентативными примерами полимеров, которые могут быть использованы в качестве отверждаемых агентов, являются полиэтиленоксид, поливиниловый спирт, карбомеры, поли(уроновые) кислоты, силиконы и их смеси. Особенно предпочтительным отверждаемым гелеобразующим агентом является полиэтиленоксид. Другим предпочтительным отверждаемым агентом является силикон. Полиэтиленоксид. Матрица лекарственных форм согласно изобретению может содержать полиэтиленоксид (ПЭО). ПЭО, присутствующий в лекарственных формах согласно изобретению, предпочтительно представляет собой гомополимер. Особенно предпочтительным ПЭО является гомополимер, имеющий повторяющиеся оксиэтиленовые группы, то есть -(О-СН 2-СН 2)n-, где n может быть равно примерно 2000-180000. Особенно предпочтительно, чтобы ПЭО имел среднюю молекулярную массу, по меньшей мере,примерно 1000000, например, в соответствии с реологическими измерениями. Еще более предпочтительно, чтобы ПЭО имел среднюю молекулярную массу примерно 2000000-7000000, например примерно 3000000-4000000. Предпочтительно, чтобы ПЭО имел вязкость 400-5000 сП и был получен в виде 2%-ного водного раствора при 25 С, более предпочтительно, чтобы ПЭО имел вязкость 400-800 сП и был получен в виде 2%-ного водного раствора при 25 С, а еще более предпочтительно, чтобы ПЭО имел вязкость 2000-4000 сП и был получен в виде 2%-ного водного раствора при 25 С. Предпочтительно также, чтобы ПЭО имел вязкость 1500-12000 сП и был получен в виде 1%-ного водного раствора при 25 С, более предпочтительно, чтобы ПЭО имел вязкость 1650-5500 сП и был получен в виде 1%-ного водного раствора при 25 С,еще более предпочтительно, чтобы ПЭО имел вязкость 5500-7500 сП и был получен в виде 1%-ного водного раствора при 25 С, а более предпочтительно, чтобы ПЭО имел вязкость 7500-10000 сП и был получен в виде 1%-ного водного раствора при 25 С. Особенно предпочтительно, чтобы ПЭО, присутствующий в лекарственных формах согласно изобретению, представлял собой полимер, имеющий среднюю молекулярную массу и вязкость, указанные в нижеследующей таблице. Так, например, предпочтительный ПЭО, используемый в лекарственных формах согласно изобретению, имеет среднюю молекулярную массу 4 000000 и вязкость 1650-5500 сП и представляет собой 1%-ный водный раствор при 25 С. Другой предпочтительный ПЭО, используемый в лекарственных формах согласно изобретению, имеет среднюю молекулярную массу 5000000 и вязкость 5550-7500 сП и представляет собой 1%-ный водный раствор при 25 С. Еще один предпочтительный ПЭО, используемый в лекарственных формах согласно изобретению, имеет среднюю молекулярную массу 7000000 и вязкость 7500-10000 сП и представляет собой 1%-ный водный раствор при 25 С. В некоторых вариантах изобретения матрица может содержать смесь ПЭО, имеющих различные молекулярные массы. Так, например, предпочтительно, чтобы некоторые лекарственные формы включали ПЭО, имеющий среднюю молекулярную массу, составляющую по меньшей мере 1000000 (например,2000000-7000000, как описано выше), в соответствии с реологическими измерениями, а также ПЭО,имеющий среднюю молекулярную массу менее чем 1000000 (например, 200000-800000) в соответствии с реологическими измерениями. Такие лекарственные формы могут обладать преимущественными свойствами, такими как резистентность к разрушению и модифицированное и/или регулируемое, например пролонгированное, высвобождение лекарственного средства. ПЭО, подходящий для применения в лекарственных формах согласно изобретению, является коммерчески доступным и поставляется фирмой Dow. Так, например, в лекарственных формах согласно изобретению могут быть использованы Polyox WSR N-12K, Polyox N-60K, Polyox WSR 301 NF или Polyox WSR 303NF. Поливиниловый спирт. Матрица лекарственных форм согласно изобретению может содержать поливиниловый спирт. Поливиниловый спирт предпочтительно имеет среднюю молекулярную массу примерно 20000-200000. Вязкость поливинилового спирта, полученного в виде 4%-ного водного раствора при 25 С, предпочтительно составляет примерно 4-65 сП. Поливиниловый спирт, используемый в матрице, предпочтительно представляет собой водорастворимый полимер. Предпочтительный поливиниловый спирт имеет формулу -(С 2 Н 4 О)n-, где n может составлять примерно 500-5000. Репрезентативными примерами коммерчески доступных полимеров на ос- 12022801 нове поливинилового спирта, которые могут быть использованы в матрице лекарственных форм согласно изобретению, являются PVA, USP, поставляемые корпорацией Spectrum Chemical Manufacturing Corporation. Гидроксипропилметилцеллюлоза. Матрица лекарственных форм согласно изобретению может содержать полимер гидроксипропилметилцеллюлозы. Вязкость гидроксипропилметилцеллюлозы, полученной в виде 2%-ного водного раствора при 25 С, предпочтительно составляет примерно 1000-150000 сП, более предпочтительно примрено 3000-120000 сП, например 3000-5600 сП, 11250-21000 сП или 80000-120000 сП. Гидроксипропилметилцеллюлоза, присутствующая в матрице лекарственных форм согласно изобретению, предпочтительно представляет собой водорастворимый полимер. Примерами коммерчески доступных полимеров гидроксипропилметилцеллюлозы, которые могут быть использованы в лекарственных формах, являются Methocel K4M, Methocel K15M и Methocel K100M, поставляемые компанией The Dow Chemical Company. Карбомеры. Матрица лекарственных форм согласно изобретению может содержать карбомер. Карбомеры предпочтительно имеют молекулярную массу в пределах от 700000 и примерно до 4000000000. Вязкость карбомера, полученного в виде 1%-ного водного раствора при 25 С и при нейтральном рН, предпочтительно составляет в пределах примерно 4000-39400 сП. Примерами коммерчески доступных карбомеров, которые могут присутствовать в матрице лекарственных форм согласно изобретению, являются Carbopol 934P NF, Carbopol 974P NF и Carbopol 971P NF, поставляемые Lubrizol. Полиуроновые кислоты. Матрица лекарственных форм согласно изобретению может содержать полиуроновую кислоту, а предпочтительно, водорастворимую полиуроновую кислоту. Примерами водорастворимых солей полиуроновой кислоты, которые могут быть использованы в матрице, являются соли щелочного металла и альгиновой кислоты и соли щелочного металла и пектиновой кислоты. В предпочтительных матрицах водорастворимой солью полиуроновой кислоты является соль альгиновой кислоты, которая фактическипредставляет собой смесь двух полиуроновых кислот, а именно маннуороновой кислоты и гулуроновой кислоты. Примерами солей щелочного металла и альгиновой кислоты, которые могут быть использованы в матрицах лекарственных форм согласно изобретению, являются альгинат натрия, альгинат калия и альгинат аммония. При этом может быть использована смесь одинаковых или различных солей альгиновых кислот с одинаковыми или различными вязкостями. Силиконы. Матрица лекарственных форм согласно изобретению может содержать силикон, а предпочтительно силикон, который может отверждаться при температуре менее чем 100 С. Особенно предпочтительными силиконами являются силиконы, содержащие полидиорганосилоксаны, имеющие связанные с кремнием ненасыщенные органические группы, например виниловые группы, которые могут реагировать с полидиорганосилоксанами, имеющими атомы водорода, связанные с атомами кремния. Подходящие силиконы описаны в заявке ЕР-А-0425154, полное содержание которой вводится в настоящее описание посредством ссылки. Матрица лекарственных форм согласно изобретению может содержать, но необязательно, замасливатель. Предпочтительными замасливателями являются замасливатели, описанные выше в разделе, относящемся к составу частиц. Количество замасливателя, присутствующего в матрице, предпочтительно составляет в пределах 1-10 мас.%, а предпочтительно 2-5 мас.% по общей массе матрицы. Матрица лекарственных форм согласно изобретению может дополнительно содержать другие наполнители, известные специалистам, например разбавители; связующие вещества; стимуляторы грануляции; красители; ароматизаторы; агенты, повышающие скольжение; агенты, регулирующие влажность; и дезинтеграторы. Специалист в данной области может самостоятельно легко определить соответствующее количество каждого из указанных наполнителей. Предпочтительные лекарственные формы согласно изобретению содержат 15-80 мас.%, более предпочтительно 20-60 мас.%, еще более предпочтительно 30-55 мас.%, еще более предпочтительно 3545 мас.% частиц по общей молекулярной массе лекарственной формы. Предпочтительные лекарственные формы согласно изобретению содержат 20-85 мас.%, более предпочтительно 30-70 мас.%, еще более предпочтительно 45-65 мас.%, еще более предпочтительно 50-60 мас.% матрицы по общей массе лекарственной формы. Преимущество лекарственных форм согласно изобретению заключается в том, что одни и те же частицы могут быть смешаны с матричным материалом в различных количествах с получением лекарственных форм различной прочности. Более того, поскольку лекарственные формы согласно изобретению обладают превосходной устойчивостью к модификации, то могут быть получены лекарственные формы с высокой прочностью, которая будет обеспечивать высвобождение лекарственного средства в течение продолжительного периода времени. Преимущество таких лекарственных форм состоит в том, что они могут быть введены один или два раза в день. Лекарственные формы согласно изобретению могут быть получены любым стандартным методом. Однако предпочтительным способом получения лекарственных форм является прессование. Таким образом, частицы, определенные как описано выше, предпочтительно смешивают, например объединяют и/или гранулируют (например, подвергают мокрому гранулированию), с матричным материалом, а затем полученную смесь (например, состав или гранулят) спрессовывают предпочтительно в формах с получением таблеток. Также следует отметить, что описанные здесь частицы могут быть включены в матрицу другими способами, такими как грануляция из расплава (например, с использованием жирных спиртов,и/или водорастворимых восков, и/или не растворимых в воде восков) или грануляция с высокими сдвиговыми усилиями, с последующим прессованием. Если матрица лекарственной формы содержит отверждаемый агент, например ПЭО, то указанный способ получения также предпочтительно включает стадию нагревания лекарственной формы, содержащей отверждаемый материал, например ПЭО, до температуры по меньшей мере примерно 60 С, предпочтительно по меньшей мере примерно 65 С, а более предпочтительно по меньшей мере примерно 70 С, например 50-100 С. В особенно предпочтительных способах лекарственную форму нагревают при температуре примерно от 60 до 90 С, предпочтительно примерно от 65 до 85 С или примерно от 70 до 80 С. Такой процесс нагревания или "отверждения" сообщает отверждаемому агенту, например ПЭО,резистентность к разрушению. Стадию нагревания или отверждения предпочтительно осуществляют в течение периода времени, достаточного для достижения нужной резистентности к разрушению. Это может быть осуществлено, например по меньшей мере за 1 мин. В предпочтительных способах получения лекарственных форм согласно изобретению отверждение осуществляют по меньшей мере примерно за 5 мин, предпочтительно по меньшей мере примерно за 15 мин, более предпочтительно по меньшей мере примерно за 30 мин, еще более предпочтительно по меньшей мере примерно за 60 мин, например по меньшей мере примерно за 90 мин. В особенно предпочтительных способах отверждение осуществляют примерно в течение периода времени от 1 мин до 24 ч, предпочтительно примерно в течение периода времени от 5 мин до 12 ч, еще более предпочтительно примерно в течение периода времени от 30 мин до 6 ч, например примерно от 1 ч до 3 ч. Отверждение предпочтительно осуществляют после получения лекарственной формы. Лекарственные формы согласно изобретению могут иметь, но необязательно, покрытие, например косметическое покрытие. Такое покрытие предпочтительно наносят после получения лекарственной формы. Покрытие может быть нанесено до или после отверждения. Предпочтительными покрытиями являются покрытия Opadry, закупаемые у Colorcon. Другими предпочтительными покрытиями являются покрытия Opaglos, также закупаемые у Colorcon. Специалист в данной области может самостоятельно легко определить соответствующее количество лекарственного средства, включенного в лекарственную форму. Так, например, в случае аналгетиков общее количество лекарственного средства, присутствующего в лекарственной форме, является достаточным для достижения аналгезирующего действия. Общее количество лекарственного средства, вводимого пациенту в нужной дозе, может варьироваться в зависимости от различных факторов, включая природу лекарственного средства, массу пациента, остроту боли, природу других вводимых терапевтических средств и т.п. Как упоминалось выше, преимущество лекарственных форм согласно изобретению заключается в том, что могут быть легко получены лекарственные формы с различной прочностью. В качестве общего руководства можно принять во внимание, что общее количество лекарственного средства, присутствующего в лекарственных формах согласно изобретению, может составлять в пределах 1-500 мг,более предпочтительно 2-200 мг, а еще более предпочтительно 5-100 мг, например примерно 10-50 мг. Так, например, если лекарственным средством является гидроморфон-НС 1, то общее количество лекарственного средства в лекарственной форме может составлять 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 24, 28 или 32 мг. Если лекарственным средством является оксикодон-HCl, то общее количество лекарственного средства в лекарственной форме может составлять 5, 10, 20, 30, 40, 50, 60, 70 или 80 мг. Предпочтительные лекарственные формы согласно изобретению высвобождают лекарственное средство в режиме регулируемого высвобождения, например, при проглатывании и под действием желудочного сока, а затем кишечного сока. Точный профиль высвобождения может быть изменен, например,при изменении состава частиц, состава матрицы и/или отношений частица:матрица. Предпочтительными лекарственными формами согласно изобретению являются лекарственные формы пролонгированного высвобождения. Используемые здесь термины "лекарственная форма длительного высвобождения", "лекарственная форма замедленного высвобождения" и "лекарственная форма пролонгированного высвобождения" являются синонимами и означают лекарственную форму, которая обеспечивает непрерывное высвобождение лекарственного средства в течение по меньшей мере 6 ч, например по меньшей мере 12 ч, при измерении на приборе USP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов (SGF) в отсутствие этанола (0%) при 37 С. Так,например, по меньшей мере 5%, например, по меньшей мере 10% лекарственного средства (по общей массе исходного лекарственного средства, присутствующего в лекарственной форме) может высвобождаться в течение первого часа (то есть за период времени от 0 до 1 ч) растворения. Предпочтительные лекарственные формы согласно изобретению обеспечивают непрерывное высвобождение лекарственного средства в течение по меньшей мере 8-12 ч при измерении на приборе USP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов (SGF) в отсутствие этанола(0%) при 37 С. Другие особенно предпочтительные лекарственные формы согласно изобретению обеспечивают непрерывное высвобождение лекарственного средства в течение по меньшей мере 12-24 ч при измерении на приборе USP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов (SGF) в отсутствие этанола (0%) при 37 С. В других предпочтительных лекарственных формах согласно изобретению количество лекарственного средства, высвобождаемого из лекарственной формы пролонгированного высвобождения в течение 1 ч при измерении на приборе USP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов (SGF) в отсутствие этанола (0%) при 37 С, составляет менее чем 20%,предпочтительно менее чем 10%, более предпочтительно менее чем 8%, например менее чем 5% (по общей массе исходного лекарственного средства, присутствующего в лекарственной форме). Другими словами предпочтительно, чтобы лекарственные формы согласно изобретению первоначально не имели высокой скорости высвобождения. То есть лекарственные формы согласно изобретению должны обеспечивать регулируемый профиль высвобождения лекарственного средства. Более предпочтительные лекарственные формы согласно изобретению при измерении на прибореUSP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов(SGF) в отсутствие этанола (0%) при 37 С обеспечивают высвобождение in vitro примерно 0-30 мас.% по общей массе лекарственной формы (например, 10-20 мас.% по общей массе лекарственной формы) в течение 1 ч и более чем 80 мас.% лекарственного средства по общей массе лекарственной формы (например, 85-99 мас.% по общей массе лекарственной формы) в течение 12 ч. Другие предпочтительные лекарственные формы согласно изобретению при измерении на прибореUSP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов(SGF) в отсутствие этанола (0%) при 37 С обеспечивают высвобождение in vitro примерно 0-30 мас.% лекарственного средства по общей массе лекарственной формы (например, 10-20 мас.% по общей массе лекарственной формы) в течение 1 ч и более чем 80 мас.% лекарственного средства по общей массе лекарственной формы (например, 85-99 мас.% по общей массе лекарственной формы) в течение 24 ч. В случае этих лекарственных форм предпочтительно 40-70 мас.% лекарственного средства по общей массе лекарственной формы (например, 50-60 мас.% по общей массе лекарственной формы) высвобождается через 12 ч. Если лекарственным средством, присутствующим в лекарственной форме, является гидроморфон или его соль (например, гидроморфон-HCl), то скорость растворения лекарственной формы in vitro при измерении лопастным методом USP (как описано в Фармакопее XXI (1985 при 100 об/мин в 900 мл водного буфера (рН 1,6-7,2) при 37 С составляет 12,5-42,5 мас.% гидроморфона, высвобождаемого через 1 ч, 25-55 мас.% гидроморфона, высвобождаемого через 2 ч, 45-75 мас.% гидроморфона, высвобождаемого через 4 ч, и 55-85 мас.% гидроморфона, высвобождаемого через 6 ч; скорость высвобождения invitro не зависит от рН, который составляет 1,6-7,2; а максимальный уровень гидроморфона в плазме invivo достигается через 2-4 ч после введения лекарственной формы. Скорость растворения предпочтительно составляет 17,5-37,5 мас.% гидроморфона, высвобождаемого через 1 ч, 30-50 мас.% гидроморфона, высвобождаемого через 2 ч, 50-70 мас.% гидроморфона, высвобождаемого через 4 ч, и 60-80 мас.% гидроморфона, высвобождаемого через 6 ч. Наиболее предпочтительная скорость растворения составляет 22,5-32,5 мас.% гидроморфона, высвобождаемого через 1 ч, 3545 мас.% гидроморфона, высвобождаемого через 2 ч, 55-65 мас.% гидроморфона, высвобождаемого через 4 ч, и 65-75 мас.% гидроморфона, высвобождаемого через 6 ч. Используемый в предыдущем параграфе термин "независимо от рН" означает, что различие в любой момент времени между количеством гидроморфона, высвобождаемым при рН 1,6, и количеством гидроморфона, высвобождаемым при любом другой рН вплоть до рН 7,2, включая указанное значение(при измерении in vitro лопастным методом USP (как описано в Фармакопее XXI (1985 при 100 об/мин в 900 мл водного буфера) составляет 10 мас.% или менее от высвобождаемого количества, которое во всех случаях дано как среднее значение по меньшей мере для трех экспериментов. Используемый в предыдущем параграфе термин "максимальный уровень гидроморфона в плазме invivo" означает максимальную среднюю концентрацию гидроморфона, обнаруженную в плазмепо меньшей мере у шести здоровых добровольцев, которым вводили одну дозу препарата при проведении фармакокинетического исследования. Предпочтительно максимальный уровень гидроморфона в плазме in vivo достигается через 2,253,75 ч после введения лекарственной формы. Если лекарственным средством, присутствующим в лекарственной форме, является оксикодон или его соль (например, оксикодон-HCl), то скорость растворения лекарственной формы in vitro при измерении лопастным методом USP (как описано в Фармакопее XXII (1990 при 100 об/мин в 900 мл водного буфера (рН 1,6-7,2) при 37 С составляет 12,5-42,5 мас.% оксикодона, высвобождаемого через 1 ч, 25-56 мас.% оксикодона, высвобождаемого через 2 ч, 45-75 мас.% оксикодона, высвобождаемого через 4 ч, и 55-85 мас.% оксикодона, высвобождаемого через 6 ч; скорость высвобождения in vitro в основном не зависит от рН, а максимальный уровень оксикодона в плазме in vivo достигается через 2-4,5 ч после введения лекарственной формы. Скорость растворения предпочтительно составляет 17,5-38 мас.% оксикодона, высвобождаемого через 1 ч, 30-50 мас.% оксикодона, высвобождаемого через 2 ч, 50-70 мас.% оксикодона, высвобождаемого через 4 ч, и 60-80 мас.% оксикодона, высвобождаемого через 6 ч. Наиболее предпочтительная скорость растворения составляет 17,5-32,5 мас.% оксикодона, высвобождаемого через 1 ч, 35-45 мас.% оксикодона, высвобождаемого через 2 ч, 55-65 мас.% оксикодона, высвобождаемого через 4 ч, и 65-75 мас.% оксикодона, высвобождаемого через 6 ч. Используемый в предыдущем параграфе термин "в основном независимо от рН", если он относится к оксикодону, означает, что различие в любой момент времени между количеством оксикодона, высвобождаемым при рН 1,6, и количеством оксикодона, высвобождаемым при любом другом рН, например рН 7,2, (при измерении in vitro лопастным методом USP (как описано в Фармакопее XXII (1990 при 100 об/мин в 900 мл водного буфера) составляет 10 мас.% или менее от высвобождаемого количества, которое во всех случаях дано как среднее значение по меньшей мере для трех экспериментов. Термин "максимальный уровень оксикодона в плазме in vivo", используемый в предыдущем параграфе, относящемся к оксикодону, означает максимальную среднюю концентрацию оксикодона, обнаруживаемую в плазме по меньшей мере у шести здоровых добровольцев, которым вводили одну дозу препарата при проведении фармакокинетического исследования. Если лекарственная форма содержит оксикодон или его соль (например, оксикодон-HCl) и налоксон (например, налоксон-HCl), то такая лекарственная форма предпочтительно высвобождает 1-40 мас.%, предпочтительно 5-35 мас.%, более предпочтительно 10-30 мас.%, а еще более предпочтительно 15-25 мас.% оксикодона и/или налоксона через 15 мин как было определено барабанным методом USP при рН 1,2 с помощью ВЭЖХ. Предпочтительно лекарственные формы высвобождают 15-20 мас.%, 2025 мас.%, примерно 15 мас.%, примерно 20 мас.% или примерно 25 мас.% оксикодона и/или налоксона через 15 мин, как было определено вышеупомянутым методом. В другом варианте изобретения лекарственные формы, содержащие оксикодон или его соль (например, оксикодон-HCl) и налоксон (например, налоксон-НС 1), высвобождают 25-65 мас.%, предпочтительно 30-60 мас.%, более предпочтительно 35-55 мас.%, а еще более предпочтительно 40-50 мас.% ) оксикодона и/или налоксона через 1 ч, как было определено барабанным методом USP при рН 1,2 с помощью ВЭЖХ. Предпочтительно такие лекарственные формы высвобождают 40-45 мас.%, 45-50 мас.%,примерно 40 мас.%, примерно 45 мас.% или примерно 50 мас.% оксикодона и/или налоксона через 1 ч,как было определено вышеупомянутым методом. В другом варианте изобретения лекарственные формы, содержащие оксикодон или его соль (например, оксикодон-HCl) и налоксон (например, налоксон-HCl), высвобождают 40-80 мас.%, предпочтительно 45-75 мас.%, более предпочтительно 45-70 мас.%, а еще более предпочтительно 45-50 мас.%, 5055 мас.%, 55-60 мас.%, 60-65 мас.% или 65-70 мас.% оксикодона и/или налоксона через 2 ч, как было определено барабанным методом USP при рН 1,2 с помощью ВЭЖХ. Предпочтительно такие лекарственные формы высвобождают примерно 45 мас.%, примерно 50 мас.%, примерно 55 мас.%, примерно 60 мас.%, примерно 65 мас.% или примерно 70 мас.% оксикодона и/или налоксона через 2 ч, как было определено вышеупомянутым методом. В другом варианте изобретения лекарственные формы, содержащие оксикодон или его соль (например, оксикодон-HCl) и налоксон (например, налоксон-HCl), высвобождают 70-100 мас.%, предпочтительно 75-95 мас.%, более предпочтительно 80-95 мас.%, а еще более предпочтительно 80-90 мас.% оксикодона и/или налоксона через 4 ч, как было определено барабанным методом USP при рН 1,2 с помощью ВЭЖХ. Предпочтительно такие лекарственные формы высвобождают 80-85 мас.%, 85-90 мас.%,примерно 80 мас.%, примерно 85 мас.% или примерно 90 мас.% оксикодона и/или налоксона через 4 ч,как было определено вышеупомянутым методом. В другом варианте изобретения лекарственные формы, содержащие оксикодон или его соль (например, оксикодон-HCl) и налоксон (например, налоксон-HCl), высвобождают 70-100 мас.%, предпочтительно 75-100 мас.%, более предпочтительно 80-95 мас.%, а еще более предпочтительно 80-85 мас.%, 8590 мас.% или 90-95 мас.% оксикодона и/или налоксона через 7 ч, как было определено барабанным методом USP при рН 1,2 с помощью ВЭЖХ. Предпочтительно такие лекарственные формы высвобождают примерно 80 мас.%, примерно 85 мас.%, примерно 90 мас.% или примерно 95 мас.% оксикодона и/или налоксона через 7 ч, как было определено вышеупомянутым методом. В другом варианте изобретения лекарственные формы, содержащие оксикодон или его соль (например, оксикодон-HCl) и налоксон (например, налоксон-HCl), высвобождают 85-100 мас.%, предпочтительно 90-100 мас.%, более предпочтительно 95-100 мас.%, а еще более предпочтительно 97-100 мас.% оксикодона и/или налоксона через 12 ч, как было определено барабанным методом USP при рН 1,2 с помощью ВЭЖХ. Другое преимущество лекарственных форм согласно изобретению заключается в том, что лекарственное средство, находящееся в матрице лекарственной формы, является защищенным, то есть оно почти или вообще не подвергается разложению. В предпочтительных лекарственных формах потеря лекарственного средства при разложении составляет менее чем 10 мас.%, предпочтительно менее чем 5 мас.%, а более предпочтительно менее чем 1 мас.% после его длительного хранения при 40 С и при относительной влажности 75% в течение 6 месяцев. Частицы и лекарственные формы согласно изобретению могут быть использованы в медицине, например в качестве аналгетиков. Поэтому такие частицы и лекарственные формы являются особенно подходящими для ослабления или снятия боли. В таких лекарственных формах указанное лекарственное средство предпочтительно является аналгетиком. Подробное описание предпочтительных вариантов изобретения Настоящее изобретение описано на нижеследующих неограничивающих примерах, где на фиг. 1 указана гипотетическая скорость растворения частиц, присутствующих в лекарственных формах согласно изобретению, а также в лекарственной форме per se; на фиг. 2-7 указана скорость растворения in vitro частиц, присутствующих в лекарственных формах согласно изобретению, а также в лекарственной форме per se. Процедуры тестирования. Скорость растворения in vitro. Таблетки тестировали in vitro стандартными методами, например на приборе USP Apparatus 1 (барабанном) или на приборе USP Apparatus 2 (лопастном), например при 50 об/мин, например в 900 мл имитированного желудочного сока без ферментов (SGF) при 37 С, на спектрометре Perkin Elmer UWVISSpectrometer Lambda 20, в УФ соответствующей длины волны, используемом для детектирования присутствующего лекарственного средства. Могут быть также протестированы неотвержденные таблетки,отвержденные таблетки и модифицированные таблетки, то есть уплощенные частицы или таблетки. Модифицированные таблетки/частицы делали плоскими вручную 7 ударами молотка для изменения их физической формы. Размер таблеток/частиц до и после уплощения и профили растворения оценивали на отдельных образцах. Скорость растворения частиц, присутствующих в лекарственной форме согласно изобретению, а также в лекарственной форме per se, указана на фиг. 1. На фиг. 1 показано, что скорость высвобождения лекарственного средства из частиц выше, чем скорость высвобождения лекарственного средства из лекарственной формы. Однако скорость высвобождения лекарственного средства из частиц является недостаточно высокой для достижения эффекта эйфории. Таким образом, даже если лицо, злоупотребляющее лекарственными средствами, попытается раздробить лекарственную форму согласно изобретению,то все равно скорость высвобождения лекарственного средства не будет значительно увеличиваться. Таким образом, у лица, злоупотребляющего лекарственными средствами, пропадет интерес к модификации такой лекарственной формы. Испытания на резистентность к модификации.(i) Возможность дробления. Отвержденные таблетки подвергали испытанию на предел прочности при разрыве путем приложения силы максимум 196 Н на аппарате Schleuniger 2E/106 для оценки резистентности к разрыву. Частицы могут быть подвергнуты тому же самому или аналогичному испытанию на предел прочности при разрыве.(ii) Резистентность к экстракции этанолом. Таблетки тестировали in vitro с использованием этанола/среды SGF при концентрациях этанола 0%,20% и 40% для оценки возможности их экстракции спиртом. Испытания проводили стандартными методами, например на приборе USP Apparatus 1 (барабанном) или на приборе USP Apparatus 2 (лопастном),например при 50 об/мин, например, в 500 мл среды при 37 С, на спектрометре Perkin Elmer UV/VIS Spectrometer Lambda 20, в УФ на соответствующей длине волны, используемом для детектирования присутствующего лекарственного средства. Результаты тестирования образца регистрировали через 0,5 ч и 1 ч. Пример 1. Частицы, имеющие состав, указанный в табл. 1, получали следующим образом. Таблица 1 Препарат этилцеллюлозы/триэтилцитрата получали путем загрузки в смеситель этилцеллюлозы с последующим постепенным добавлением, например путем распыления триэтилцитрата. Смешивание проводили до тех пор, пока смесь не становилась однородной, а затем эту смесь оставляли на ночь для пропитки этилцеллюлозы триэтилцитратом. Гидроморфон-HCl, налоксон-HCl, стеариловый спирт, глицерилдибегенат и вышеупомянутый препарат этилцеллюлозы/триэтилцитрата добавляли в смеситель и смешивали. Полученную смесь гранулировали с использованием водной дисперсии Eudragit NE 40 D. Затем гранулят сушили до достижения постоянной массы. Осушенный гранулят подвергали экструзии. Экструдер с диском для плавления устанавливали на предварительно определенные условия экструзии и осуществляли экструзию. Полученный экструдат имел средний диаметр 1 мм. Затем экструдат вытягивали на конвейерной ленте с прижимными роликами в процессе его подачи в гранулятор. Вытянутый экструдат имел средний диаметр примерно 500 мкм. После этого вытянутый экструдат разрезали на частицы со средней длиной примерно 500 мкм. Таблетки, имеющие состав, указанный в табл. 2 получали, как описано ниже. Таблица 2 Частицы смешивали с матричным материалом и, необязательно, с другими наполнителями. Затем добавляли замасливатель, и смесь перемешивали до образования однородной смеси. После этого смесь спрессовывали в подходящем устройстве с получением таблеток, имеющих предварительно определенную массу и толщину. Затем может быть насенено покрытие, и осуществлено отверждение в одном блоке устройства. Если требуется нанесение покрытия перед отверждением, то таблетку нагревают до предварительно определенной температуры, наносят покрытие путем распыления и сушат, а затем температуру повышают до значения, необходимого для отверждения. Если перед нанесением покрытия требуется отверждение, то таблетку нагревают до нужной температуры в течение определенного периода времени, а затем охлаждают. После этого может быть, но необязательно, нанесено покрытие путем распыления до достижения предварительно определенной массы. Пример 2. Экструдированные из расплава частицы, имеющие состав, указанный ниже в табл. 3, получали: сначала путем формования (грануляции в псевдоожиженном слое) плацебо-гранул, имеющих состав,указанный ниже в табл. 4; затем измельчения указанных плацебо-гранул (на мельнице Ретча с ситами 0,5 мм); после этого смешивания измельченных плацебо-гранул с гидрохлоридом гидроморфона, гидрохлоридом налоксона и стеаратом магния в коническом смесителе соответствующего размера с получением смешанных гранул; и наконец, экструзии из расплава указанных смешанных гранул в экструдере с диском для плавления Leistritz Micro 27 с получением экструдата, который затем вытягивали и, наконец,разрезали на грануляторе с получением частиц, экструдированных из расплава. Полученные частицы имели средний диаметр 0,80 мм и среднюю длину 0,84 мм. Таблица 3 Таблетки, имеющие состав, указанный в табл. 5, получали путем смешивания частиц с гидроксипропилметилцеллюлозой (Methocel K4M) и стеаратом магния с последующим прямым прессованием (на прессе Manesty F3 Betapress) полученной смеси. Таблица 5 Пример 3. Лабораторную партию таблеток, имеющих состав, указанный в табл. 6, изготавливали путем мокрой грануляции частиц примера 2 (см. табл. 3) с различными наполнителями (водой, используемой в качестве жидкого связующего вещества, и гидроксипропилметилцеллюлозой (Methocel K4M) в качестве связующего вещества) в процессоре Kenwood с последующим их прессованием на прессе Manesty F3 Частицы и таблетки тестировали на растворение в лопастном приборе для растворения Ph. Eur при 37 С и при 75 об/мин отдельно в 500 мл имитированного желудочного сока без ферментов (SGF) при рН 1,2 и в 500 мл 40% этанола. Для измерения скорости высвобождения in vitro проводили стандартные ВЭЖХ-анализы, и по полученным результатам строили график, представленный на фиг. 2. Пример 4. Таблетки, имеющие состав, указанный в табл. 7, получали следующим образом: 1. Частицы, полученные, как описано в примере 2, и лактозу загружали в смеситель Kenwood и подвергали сухому смешиванию. 2. Для грануляции смеси по каплям добавляли воду до получения крупных гранул. 3. К сырым гранулам добавляли ГПМЦ (Methocel K100M) при непрерывном перемешивании. 4. Снова добавляли воду до получения порошкообразной смеси. 5. Гранулы сушили в печи Gallenkamp в течение 2 ч при 50-55 С. 6. Осушенные гранулы смешивали со стеаратом магния в смесителе Pharmatech. 7. Полученную смесь спрессовывали в таблетки на прессе Manesty F3 Betapress. Таблица 7 Частицы и таблетки тестировали на растворение в лопастном приборе для растворения Ph. Eur при 37 С и при 75 об/мин в 500 мл SGF при рН 1,2. Для измерения скорости высвобождения in vitro проводили стандартные ВЭЖХ-анализы, и по полученным результатам строили график, представленный на фиг.3. Пример 5. Таблетки, имеющие состав, указанный в табл. 8, получали следующим образом. 1. Частицы, полученные как описано в примере 2, и лактозу загружали с сосуд и подвергали сухому смешиванию. 2. Для увлажнения смеси по каплям добавляли воду до получения крупных гранул. 3. К сырым гранулам добавляли ПЭО при непрерывном перемешивании. 4. Гранулы сушили в печи Gallenkamp в течение 2 ч при 50-55 С. 5. Осушенные гранулы смешивали со стеаратом магния в смесителе Pharmatech. 6. Полученную смесь спрессовывали в таблетки на прессе Manesty F3 Betapress. 7. Полученные таблетки отверждали в печи при 72 С в течение 1 ч. Таблица 8 Частицы и таблетки тестировали на растворение в SGF как описано в примере 4. Эти таблетки дополнительно тестировали на растворение в 40% этаноле как описано в примере 3. Для измерения скорости высвобождения in vitro проводили стандартные ВЭЖХ-анализы, и по полученным результатам строили график, представленный на фиг. 4 и 5. Пример 6. Экструдированные из расплава частицы, имеющие состав, указанный в табл. 9, получали сначала путем формования (грануляции в псевдоожиженном слое) плацебо-гранул, имеющих состав, указанный в табл. 10; затем измельчения указанных плацебо-гранул (на мельнице Ретча с ситами 0,5 мм); после этого смешивания измельченных плацебо-гранул с гидрохлоридом гидроморфона, гидрохлоридом налоксона,стеаратом магния и лаурилсульфатом натрия (2 мг/ед.) в коническом смесителе соответствующего размера с получением смешанных гранул, и наконец, экструзии из расплава указанных смешанных гранул в экструдере с диском для плавления Leistritz Micro 27 с получением экструдата, который затем вытягивали и, наконец, измельчали на грануляторе с получением частиц, экструдированных из расплава. Полученные частицы имели средний диаметр 0,82 мм и среднюю длину 0,81 мм. Таблица 9 Таблетки, имеющие состав, указанный в табл. 11, получали способом, описанным в примере 5, за исключением того, что стадию 7 (отверждение таблетки) не проводили; в стадии 1, частицы, описанные в этом примере, вместо частиц примера 2 подвергали сухому смешиванию с лактозой и с тринатрийцитратом, а в стадии 3 к сырым гранулам вместо ПЭО добавляли альгинат натрия при непрерывном перемешивании. Таблица 11 Пример 7. Таблетки, имеющие состав, указанный в табл. 12, получали способом, описанным в примере 5, за исключением того, что стадию 7 (отверждение таблетки) не проводили; в стадии 1 вместо частиц примера 2 сухому смешиванию с лактозой и со стеаратом магния подвергали частицы, описанные в примере 6; а в стадии 3 к сырым гранулам вместо ПЭО добавляли ксантановую камедь при непрерывном перемешивании. Таблица 12 Частицы и таблетки тестировали на растворение в SGF как описано в примерах 4 и 5. Эти таблетки дополнительно тестировали на растворение в 40%-ном этаноле как описано в примерах 3 и 5. Для измерения скорости высвобождения in vitro проводили стандартные ВЭЖХ-анализы, и по полученным результатам строили график, представленный на фиг. 6 и 7. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Лекарственная форма, содержащая экструдированные из расплава частицы, включающие лекарственное средство, и матрицу, представляющую собой непрерывную фазу, включающую гелеобразующий агент, где экструдированные из расплава частицы присутствуют в матрице в дискретной фазе; и экструдированные из расплава частицы содержат сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты. 2. Лекарственная форма по п.1, где сополимер выбран из сополимеров торговых марок EudragitRL 100, Eudragit RL PO, Eudragit RS 100, Eudragit RS PO, Eudragit NE 40 D и Eudragit NE 30 D. 3. Лекарственная форма по п.1 или 2, где экструдированные из расплава частицы имеют удлиненную форму. 4. Лекарственная форма по любому из пп.1-3, которая содержит 15-80 мас.% экструдированных из расплава частиц от общей массы лекарственной формы. 5. Лекарственная форма по любому из пп.1-4, которая содержит 20-85 мас.% матрицы от общей массы лекарственной формы. 6. Лекарственная форма по любому из предыдущих пунктов, полученная в форме таблетки. 7. Лекарственная форма по любому из предыдущих пунктов, которая является резистентной к модификации. 8. Лекарственная форма по любому из предыдущих пунктов, где количество лекарственного средства, высвобождаемого из лекарственной формы в течение 0,5 ч, при измерении на приборе USP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов (SGF) в присутствии 40% этанола при 37 С составляет примерно 20% от количества лекарственного средства,высвобождаемого из лекарственной формы в течение 0,5 ч, при измерении на приборе USP Apparatus 1 (в барабане) при 100 об/мин в 900 мл имитированного желудочного сока без ферментов (SGF) в отсутствие этанола (0%) при 37 С. 9. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы имеют предел прочности при разрыве по меньшей мере 350 H. 10. Лекарственная форма по любому из предыдущих пунктов, где частицы, экструдированные из расплава, имеют диаметр и/или длину менее чем примерно 1000 мкм. 11. Лекарственная форма по любому из предыдущих пунктов, где лекарственным средством является лекарственное средство, которое представляет собой объект для злоупотребления. 12. Лекарственная форма по любому из предыдущих пунктов, где лекарственным средством являются агонист опиоида; транквилизатор; средство, подавляющее ЦНС; средство, стимулирующее ЦНС; или снотворное средство седативного действия. 13. Лекарственная форма по любому из предыдущих пунктов, где лекарственным средством является агонист опиоида. 14. Лекарственная форма по п.13, где агонист опиоида выбран из группы, состоящей из оксикодона,оксиморфона, гидрокодона, гидроморфона, морфина, кодеина, бупренорфина, фентанила, трамадола,тапентадола и их фармацевтически приемлемых солей. 15. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы содержат 3-50 мас.% лекарственного средства от общей массы частиц. 16. Лекарственная форма по любому из предыдущих пунктов, которая включает один или несколько дополнительных активных ингредиентов. 17. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы содержат 10-50 мас.% указанного сополимера от общей массы частиц. 18. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы дополнительно содержат скорость-регулирующий или скорость-модифицирующий агент. 19. Лекарственная форма по п.18, где скорость-регулирующий или скорость-модифицирующий агент представляет собой алкилцеллюлозу. 20. Лекарственная форма по п.19, где алкилцеллюлоза представляет собой этилцеллюлозу. 21. Лекарственная форма по любому из пп.18-20, где экструдированные из расплава частицы содержат 20-50 мас.% скорость-регулирующего или скорость-модифицирующего агента от общей массы частиц. 22. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы дополнительно содержат замасливатель. 23. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы дополнительно содержат пластификатор. 24. Лекарственная форма по п.1, где экструдированные из расплава частицы содержат оксикодон или гидроморфон, сополимер этилакрилата и метилметакрилата, этилцеллюлозу в качестве скоростьрегулирующего или скорость-модифицирующего агента, стеариловый спирт и/или триэтилцитрат в качестве пластификатора, глицерилдибегенат в качестве замасливателя и, необязательно, антагонист опиоида. 25. Лекарственная форма по п.24, где оксикодон или гидроморфон представлены в виде их гидрохлоридной соли. 26. Лекарственная форма по любому из предыдущих пунктов, где экструдированные из расплава частицы содержат агонист опиоида, а также антагонист опиоида. 27. Лекарственная форма по любому из пп.1-26, где гелеобразующий агент выбран из полиэтиленоксида, поливинилового спирта, гидроксипропилметилцеллюлозы, карбомеров, полиуроновых кислот или их смесей. 28. Лекарственная форма по любому из пп.1-27, где гелеобразующий агент является отвержденным. 29. Лекарственная форма по п.28, которая может быть подвергнута уплощению без разрушения до толщины примерно на 60% меньше толщины указанной лекарственной формы до ее уплощения. 30. Способ получения лекарственной формы по любому из пп.1-29, включающий смешивание экструдированных из расплава частиц, содержащих лекарственное средство и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, с матричным материалом, содержащим гелеобразующий агент, чтобы экструдированные из расплава частицы образовали в матрице дискретную фазу, и формование полученной смеси в лекарственную форму. 31. Способ получения лекарственной формы по любому из пп.1-29, включающий смешивание экструдированных из расплава частиц, которые получены путем разрезания экструдата из расплава, содержащего лекарственное средство и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, с матричным материалом, содержащим гелеобразующий агент, чтобы экструдированные из расплава частицы образовали в матрице дискретную фазу, и формование полученной смеси в лекарственную форму. 32. Способ получения лекарственной формы по любому из пп.1-29, включающий:i) экструзию из расплава композиции, содержащей лекарственное средство и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, с получением экструдата;ii) разрезание экструдата с получением экструдированных из расплава частиц;iii) смешивание экструдированных из расплава частиц с матричным материалом, содержащим гелеобразующий агент так, чтобы экструдированные из расплава частицы образовали в матрице дискретную фазу; иiv) формование из полученной смеси лекарственной формы. 33. Способ по п.32, дополнительно включающий вытягивание экструдата из расплава с образованием вытянутого экструдата перед разрезанием с получением экструдированных из расплава частиц. 34. Способ по любому из пп.30-33, где частицы получают путем экструзии из расплава при температуре 100 С или менее. 35. Способ по любому из пп.30-34, который дополнительно включает стадию отверждения матрицы. 36. Лекарственная форма по любому из пп.1-29, предназначенная для ослабления или снятия боли. 37. Применение экструдированных из расплава частиц, содержащих лекарственное средство и сополимер алкилового эфира акриловой кислоты и алкилового эфира метакриловой кислоты, и матричного материала, содержащего гелеобразующий агент, для приготовления лекарственной формы по любому из пп.1-29 для ослабления боли. 38. Способ лечения индивидуума, нуждающегося в ослаблении боли, включающий введение индивидууму лекарственной формы по любому из пп.1-29.
МПК / Метки
МПК: A61K 45/06, A61K 31/485, A61K 9/16, A61K 9/20
Метки: лекарственная, способ, форма, способы, получения, лечения
Код ссылки
<a href="https://eas.patents.su/26-22801-lekarstvennaya-forma-sposoby-ee-polucheniya-i-sposob-lecheniya.html" rel="bookmark" title="База патентов Евразийского Союза">Лекарственная форма, способы ее получения и способ лечения</a>
Лекарственная форма фентанила для перорального применения, способ ее получения и способы лечения

Номер патента: 10826
Опубликовано: 30.12.2008
Авторы: Хабиб Валид, Моэ Дерек, Агарвал Викас
МПК: A61P 25/04, A61K 9/46, A61K 9/20...
Метки: способ, применения, фентанила, лекарственная, форма, способы, лечения, перорального, получения
Формула / Реферат:
1. Лекарственная форма, содержащая от 90 до 880 мкг фентанила, в расчете на свободное основание фентанила, или эквивалентное количество его соли, шипучую пару в количестве от 5 до 85 мас.% лекарственной формы, средство, регулирующее рН, в количестве от 0,5 до 25 мас.% лекарственной формы и гликолят крахмала в количестве от 0,25 до 20 мас.% лекарственной формы, где указанная лекарственная форма предназначена для доставки указанного фентанила...
Лекарственная форма силимарина и способ ее получения

Номер патента: 6157
Опубликовано: 27.10.2005
Авторы: Даскалов Веселин Евгениев, Тимчев Ангел Радославов, Павлова Виолета Андреева, Славкова Снежана Иванова, Кытовски Бисер Христов, Атанасов Димитр Георгиев, Иванов Тихомир Георгиев
МПК: A61K 36/00, A61K 31/355, A61J 3/10...
Метки: силимарина, получения, форма, способ, лекарственная
Формула / Реферат:
1. Твердая лекарственная форма силимарина, содержащая ядро таблетки из силимарина, поливинилпирролидона К25, пшеничного крахмала, лактозы, глюкозы, сорбита, микрокристаллической целлюлозы, талька, стеарата магния и бикарбоната натрия, покрытое слоем ацетофталатацеллюлозы и оболочкой, отличающаяся тем, что ядро таблетки включает поливинилпирролидон К25 - от 0,0010 до 0,009 г, крахмал - от 0,050 до 0,060 г, лактозу - от 0,050 до 0,060 г, глюкозу -...
Лекарственная форма с замедленным высвобождением, содержащая прамипексол, и способ лечения субъекта

Номер патента: 9663
Опубликовано: 28.02.2008
Авторы: Ганоркар Локсидх Д., Ноак Роберт М., Рео Джозеф П., Ли Эрнест Дж., Эмидон Грегори Э., Скоуг Конни Дж., Хаймлих Джон М.
МПК: A61K 9/28, A61K 9/20, A61K 31/428...
Метки: лечения, форма, замедленным, способ, содержащая, субъекта, лекарственная, высвобождением, прамипексол
Формула / Реферат:
1. Лекарственная форма с замедленным высвобождением в форме таблетки, обеспечивающая пероральную доставку, содержащая растворимую в воде соль прамипексола, диспергированную в матрице, содержащей гидрофильный полимер и крахмал, имеющий предел прочности на разрыв по меньшей мере примерно 0,15 кН/см2 при относительном содержании твердого вещества, типичном для таблетки. 2. Лекарственная форма по п.1, содержащая крахмал, имеющий предел прочности на...
Лекарственная форма для лечения эссенциальной тромбоцитемии

Номер патента: 22715
Опубликовано: 29.02.2016
Автор: Видманн Рудольф
МПК: A61K 31/60, A61K 9/20, A61K 47/10...
Метки: форма, тромбоцитемии, лечения, эссенциальной, лекарственная
Формула / Реферат:
1. Лекарственная форма, представляющая собой таблетку или капсулу, содержащая частицы анагрелида, повидон, кросповидон, микрокристаллическую целлюлозу, стеарат магния и по меньшей мере 60 мг моногидрата лактозы, при этом по меньшей мере 90% указанных частиц анагрелида имеют диаметр менее 10 мкм.2. Лекарственная форма по п.1, содержащая анагрелид в количестве менее 1 мг, предпочтительно менее 0,70 мг, предпочтительно менее 0,65 мг,...
Твердая, полученная прямым прессованием, лекарственная форма и способ ее получения

Номер патента: 223
Опубликовано: 24.12.1998
Авторы: Хантер Эдвард А., Зелезник Юзеф А., Шервуд Боб Е.
МПК: A61K 9/14
Метки: способ, лекарственная, твердая, прямым, форма, получения, прессованием, полученная
Формула / Реферат:
1. Твердая, полученная прямым прессованием, лекарственная форма, представляющая собой гомогенный гранулят, включающий: а) примерно 40-95 маc.% гранулированного ацетаминофена; в) примерно 1-59 маc.% носителя для прямого прессования, представляющего собой микрокристаллическую целлюлозу, обработанную совместно с примерно 0,1-20 маc.% диоксида кремния; и с) примерно 0,01-4,0 маc.% фармацевтически приемлемого смазывающего вещества. 2. Твердая...
Предыдущий патент: Противоэрозионный берегозащитный сотовый матрац
Следующий патент: Соединения, композиции и способы для лечения заболеваний, опосредованных jak-киназой
Случайный патент: Сельскохозяйственные препараты с ацилморфолинами и полярными апротонными сорастворителями