Синтез пептидов
Номер патента: 8305
Опубликовано: 27.04.2007
Авторы: Брахт Франц-Петер, Хаберль Удо, Франк Ханс-Георг, Рибка Андреас






















Формула / Реферат
1. Применение активированной твердой фазы, содержащей твердый носитель, металл-хелатирующие лиганды, ковалентно связанные с твердым носителем, ионы металла Мn+ с n=1-3, координационно связанные с упомянутыми металл-хелатирующими лигандами, причем указанная активированная твердая фаза обеспечивает координационные участки для координационного и обратимого присоединения якорной части пептида, для твердофазного синтеза пептидов на указанной активированной твердой фазе, при этом пептид представляет собой "растущий пептид" и подвергается процедурам удлинения пептида.
2. Применение по п.1, при котором используют твердый носитель на основе диоксида кремния, стекла, или целлюлозы, или полимера, выбранного из группы, состоящей из полистирольных смол, меламиновых смол и поливиниловых спиртов.
3. Применение по любому из пп.1-2, при котором каждый металл-хелатирующий лиганд содержит по крайней мере один атом азота, кислорода, фосфора или серы, который способен образовывать координационную связь лиганд-металл.
4. Применение по любому из пп.1-3, при котором каждый металл-хелатирующий лиганд содержит по крайней мере одну функциональную группу, выбранную из группы, состоящей из амино, гетероциклического азота, карбокси, гидроксила и меркапто.
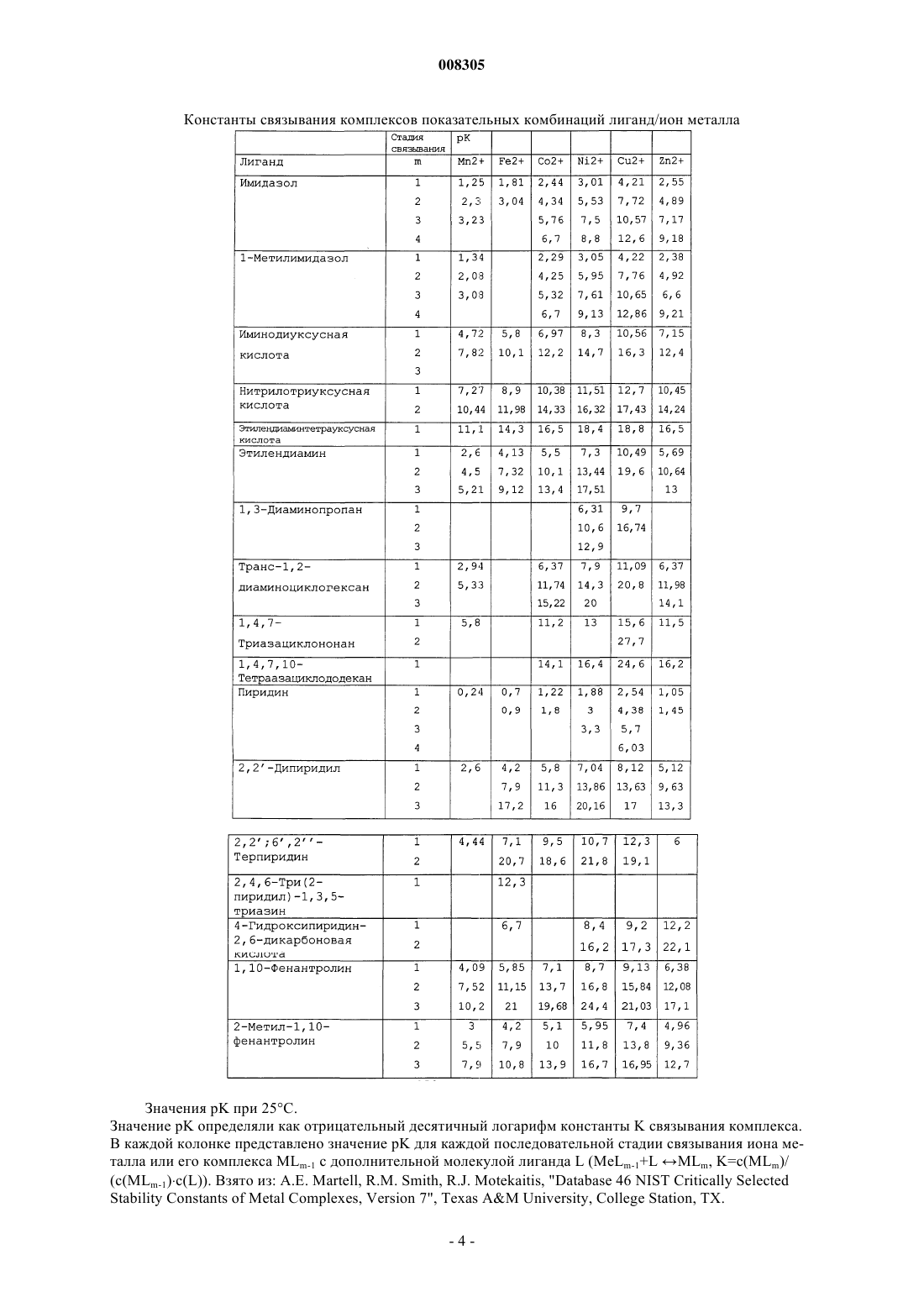
5. Применение по любому из пп.1-4, при котором каждый металл-хелатирующий лиганд, ковалентно связанный с твердым носителем, содержит по крайней мере один фрагмент, выбранный из группы, состоящей из трифенилфосфиновых фрагментов, аминопуриновых фрагментов, предпочтительно 6-аминопуриновых фрагментов, фталоцианиновых фрагментов, 1,10-фенантролиновых фрагментов, предпочтительно 5-амино-1,10-фенантролиновых фрагментов, терпиридиновых фрагментов, предпочтительно 4'-амино-[2,2'; 6',2'']терпиридиновых фрагментов, триазациклононановых фрагментов, предпочтительно [1,4,7]-триазациклононановых фрагментов, и тетраазациклододеканиловых фрагментов, предпочтительно [1,4,7,10]-тетраазациклододеканиловых фрагментов.
6. Применение по любому из пп.1-5, при котором металл Мn+ выбирают из группы, состоящей из ионов Mn2+, Cu2+, Ni2+, Co2+, Zn2+, Mg2+, Ca2+, Fe2+, Fe3+ и лантанида, особо предпочтительным Мn+ является Cu2+, Ni2+, Co2+, Zn2+, Mg2+.
7. Применение по п.1, при котором якорную часть пептида отделяют от указанной активированной твердой фазы добавлением конкурентного лиганда.
8. Применение по п.7, при котором конкурентный лиганд содержит по крайней мере один фрагмент, способный образовывать комплекс с ионами металлов, предпочтительно азотсодержащий фрагмент, выбранный из группы, состоящей из имидазольных, N-метилимидазольных, аминопуриновых, фенантролиновых, бипиридиновых, терпиридиновых, триазациклононановых, тетраазациклододекановых фрагментов, фрагментов иминодиуксусной кислоты, фрагментов нитрилотриуксусной кислоты и фрагментов этилендиаминтетрауксусной кислоты.
9. Применение по п.8, при котором моно- или олигомерные аминокислоты присоединяют к С- или N-концу растущего пептида по протоколу последовательных реакций типа протокола Меррифилда.
10. Применение по любому из пп.7-9, при котором якорная часть пептида содержит по крайней мере один фрагмент, образующий комплекс с ионом металла, причем каждый из указанных фрагментов включает по крайней мере одну группу, содержащую азот, кислород, фосфор или серу, которая способна координировать ионы металла активированной твердой фазы.
11. Применение по любому из пп.7-10, при котором азотсодержащую группу, которая способна координировать ионы металла активированной твердой фазы, выбирают из группы, состоящей из амино, гидроксильных, карбоксильных, меркапто, имидазолильных, N-метилимидазолильных, аминопуринильных фрагментов, фенантролильных фрагментов, пиридильных фрагментов, бипиридильных фрагментов, терпиридинильных фрагментов, триазациклононанонильных фрагментов, тетраазациклододеканильных фрагментов, фрагментов иминодиуксусной кислоты, фрагментов нитрилотриуксусной кислоты и фрагментов этилендиаминотетрауксусной кислоты.
12. Применение по любому из пп.7-11, при котором якорную часть пептидной цепи помещают на С-конце и/или по крайней мере в одной аминокислотной боковой цепи пептида.
13. Применение по п.12, при котором по крайней мере одну аминокислоту якорной части на С-конце пептида удлиняют на одну или более аминокислот, что делает возможным определение с помощью систем детектирования.
14. Применение по любому из пп.7-13, при котором якорную часть пептида удлиняют на ее N-конце аминокислотной последовательностью, обеспечивающей участок узнавания для специфической протеазы.
15. Применение по любому из пп.7-14, при котором после отщепления пептид повторно присоединяют к активированной твердой фазе разбавлением реакционной смеси, содержащей конкурентный лиганд, согласно протоколу последовательных реакций типа протокола Меррифилда.
16. Применение по п.1, которое включает стадию рефолдинга неправильно скрученных структур и/или деагрегации межмолекулярных агрегатов необязательно защищенного пептида, при котором якорная часть пептида координационно и обратимо присоединена к активированной твердой фазе, и повторного создания правильно скрученной структуры пептида, включающее стадии:
a) воздействия на пептид по крайней мере одного хаотропного или денатурирующего реагента и
b) последующего воздействия ряда растворителей, чтобы постепенно уменьшить хаотропность и обеспечить воспроизводимые условия рефолдинга и повторного создания вторичной и третичной структуры.
17. Применение по п.16, при котором вторичную и третичную структуру пептида поддерживают ковалентными связями между реакционноспособными боковыми цепями указанного пептида путем обработки пептида подходящими реагентами, включающей образование указанных ковалентных связей до отщепления пептида от активированной твердой фазы.
18. Применение по п.17, при котором ковалентные связи представляют собой дисульфидные связи, амидные связи или стабильные ароматические или алифатические гидразоны.
19. Применение по пп.7, 16 и 17 для синтеза пептида с последовательностью
![]()
где X - d-аланин и bAla - бета-аланин,
включающее образование дисульфидной связи между остатками цистеина, в результате чего образуется

где X и bАlа являются такими, как описано выше.
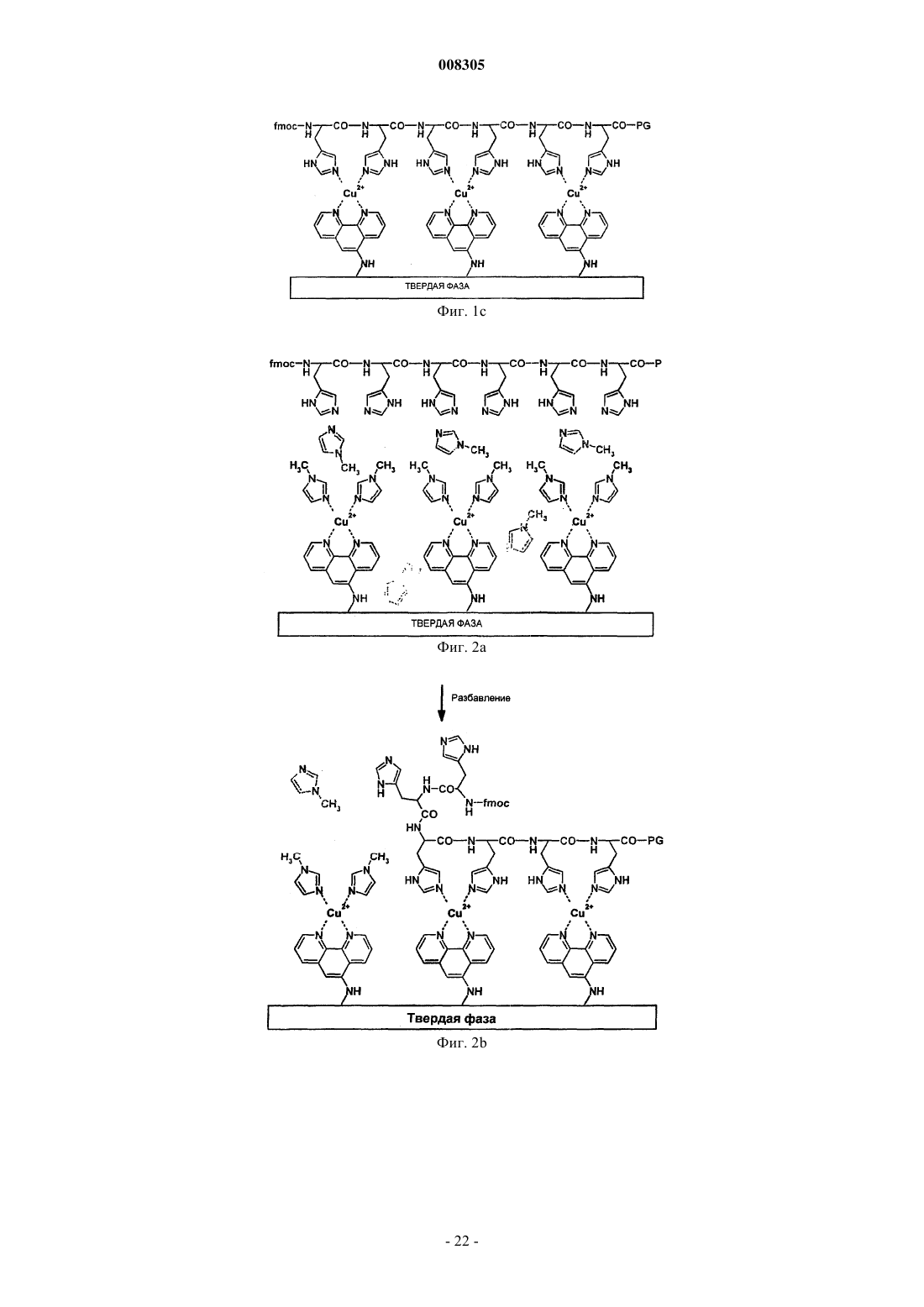
20. Способ твердофазного синтеза пептидов посредством нековалентного присоединения растущей пептидной цепи к активированной твердой фазе, при котором активный компонент соответствующей твердой фазы образован металл-хелатными комплексами со свободными координационными участками для нековалентного присоединения растущей пептидной цепи к активированной твердой фазе через хелатирующие группы, присутствующие на якорной части растущей пептидной цепи, при этом металл-хелатные комплексы образованы комплексами между ионом металла и металл-хелатирующим лигандом, который непосредственно или через линкерную молекулу ковалентно связан с твердой фазой.
21. Способ по п.20, при котором полностью образованный комплекс металла для повторяющихся циклов синтеза в процессе синтеза пептидов включает твердую фазу, металл-хелатирующий лиганд, ион металла, который размещен между металл-хелатирующим лигандом и хелатирующими группами якорной части пептидной цепи, и хелатирующие группы, присутствующие в N- или С-концевых положениях и/или в боковых цепях моно- или олигомерных аминокислот, которые образуют якорную часть пептидной цепи, которую наращивают ступенчатым образом вследствие повторяющихся циклов синтеза.
22. Способ по любому одному из пп.20-21, при котором нековалентное и координационное присоединение иона металла к металл-хелатирующему лиганду характеризуется большей силой, чем сила присоединения к хелатирующим группам.
23. Споёюс по любому из пп.20-22, при котором структура комплекса с металлом состоит из
a) металл-хелатирующих лигандов, которые ковалентно присоединены к твердой фазе и способны образовывать комплекс с ионами металлов через атомы N, О, Р и/или S,
b) ионов металла (Me[n+]); n от 1 до 3, предпочтительно 2, которые образуют комплекс с металл-хелатирующими лигандами, при этом остаются доступными свободные координационные участки,
c) одиночных или олигомерных природных или неприродных аминокислот, содержащих природные или химически модифицированные боковые цепи или N-, или С-концевые модификации, к которым присоединены хелатирующие группы и которые, таким образом, способны образовывать комплекс со свободными координационными участками, предоставленными частично насыщенными комплексами металл-хелатирующих лигандов с ионами Меn+ на активированной твердой фазе, описанной в а) и b) и пп.1-4, при этом хелатирующие группы способны образовывать комплекс с ионами металлов через атомы N, О, Р и/или S и при этом по крайней мере одна, предпочтительно 1-3 хелатирующие группы, могут присутствовать в боковой цепи для того, чтобы образовать стабильную якорную часть для присоединения растущей пептидной цепи.
24. Способ по любому из пп.20-23, при котором твердая фаза характеризуется присутствием функциональных химических групп, которые выбирают из аминогрупп, групп, содержащих гетероциклический азот, карбоксигрупп, гидроксильных, тиоловых групп или других функциональных остатков, для которых, самих по себе, известны реакции связывания, таким образом, указанные функциональные химические группы и реакции связывания используют, чтобы ковалентно связать твердую фазу с металл-хелатирующим лигандом по пп.20-23.
25. Способ по любому из пп.20-24, при котором металл-хелатирующие лиганды содержат функциональную группу, которая делает возможным химическое связывание с функциональными химическими группами на поверхности твердой фазы по п.24, и при котором те же металл-хелатирующие лиганды, а также хелатирующие группы аминокислотных боковых цепей по п.23 содержат одну или более, предпочтительно 1-3, функциональных групп, которые способны образовывать комплекс с ионами металлов по п.5 (Меn+) и которые выбирают из аминогрупп, групп, содержащих гетероциклический азот, азагрупп, карбоксигрупп, фрагментов, содержащих серу или фосфор.
26. Способ по любому из пп.20-25, при котором ионы металлов (Меn+) образуют комплекс с металл-хелатирующими лигандами на твердой фазе; причем указанные ионы металлов выбирают из ионов Mn2+, Ni2+, Cu2+, Co2+ или Zn2+, Ca2+, Fe2+, Fe3+ или ионов лантанида.
27. Способ по любому из пп.20-26, при котором последовательные реакции используют, чтобы присоединить дополнительные моно- или олигомерные аминокислоты к С- или N-концу полностью образованного комплекса с металлом по п.21; при этом соответственно защищенные аминокислотные производные или олигомерные фрагменты, которые присоединяют в каждом цикле, можно легко выбрать или составить из любых природных или неприродных аминокислот.
28. Способ по любому из пп.20-27, при котором конкурентный хелатирующий реагент добавляют к смеси реагентов стадии конденсации протокола реакций типа протокола Меррифилда для того, чтобы конкурентно отщепить растущую пептидную цепь от твердой фазы во время указанной стадии; при этом подходящие конкурентные хелатирующие реагенты имеют приблизительно такое же сродство к свободным координационным участкам на активированной твердой фазе, как индивидуальные хелатирующие группы боковых цепей моно- или олигомерных аминокислот, используемых для присоединения растущей пептидной цепи к твердой фазе, причем конкурентные хелатирующие реагенты являются растворимыми в смеси реагентов стадии конденсации и не взаимодействуют или иным образом не влияют на ингредиенты смеси реагентов стадии конденсации.
29. Способ по п.28, при котором реакционную смесь стадии конденсации, содержащую конкурентный хелатирующий реагент, разбавляют перед следующими стадиями промывания для того, чтобы повторно присоединить моно- или олигомерные аминокислоты, образующие якорную часть пептидной цепи, к активированной твердой фазе перед последующими стадиями, такими как полоскание или промывание.
30. Способ по любому из пп.20-29, при котором отщепленный и деблокированный неочищенный продукт синтеза пептидов, несущий моно- и олигомерные аминокислоты с боковыми цепями или N-, или С-концевыми фрагментами с хелатирующими группами по п.23, способен образовывать комплекс металла с твердой фазой по любому из пп.20-26 и может быть далее обработан путем:
a) очистки путем воздействия на раствор неочищенного продукта активированной твердой фазой по пп.20-26 в условиях, при которых происходит повторное присоединение требуемого продукта к твердой фазе, и путем отмывания примесей, таких как остатки защитных групп и поглотители или нежелательные побочные продукты, избытком растворителя при избирательном сохранении на активированной твердой фазе продукта, образовавшего комплекс, и далее
b) удаления возможных нежелательных неправильно скрученных структур продукта и межмолекулярных агрегатов молекул продукта при воздействии на связанный продукт хаотропными или денатурирующими реагентами, например мочевиной, детергентами, такими как додецилсульфат натрия, высокими концентрациями солей, меркаптоэтанолом или другими реагентами, в результате чего связанный продукт переходит в денатурированное состояние, которое характеризуется разрушением вторичной и третичной структуры, тогда как связь с твердой фазой сохраняется.
31. Способ по п.30, при котором очищенный, связанный и денатурированный продукт подвергают воздействию ряда растворителей, причем ряд растворителей подобран и оптимизирован для соответствующего продукта, чтобы постепенно уменьшить хаотропность и создать воспроизводимые условия рефолдинга молекул связанного продукта, а также контролируемого повторного появления вторичной и третичной структуры при сохранении молекул продукта связанными с твердой фазой.
32. Способ по п.30 и/или 31, при котором ковалентные связи между боковыми цепями аминокислот, предпочтительно замыкание дисульфидных связей из свободных сульфгидрильных групп, образование амидных связей или образование стабильных ароматических или алифатических гидразонов осуществляют пропусканием смесей реагентов через повторно скрученный продукт, который связан с твердой фазой.
33. Способ по п.24, при котором твердую фазу выбирают из диоксида кремния, целлюлозы или из полимеров, предпочтительно из полистирольной смолы, поперечно-сшитых с дивинилбензолом, хлортритильной смолы, из модифицированных, предпочтительно карбоксилированных, частиц меламина, или модифицированного, предпочтительно карбоксилированного, полимерного носителя на основе поливинилового спирта.
34. Способ по п.24 и/или 33, при котором матрикс смолы содержит ферромагнитные частицы, что позволяет применять технологии магнитной сепарации.
35. Способ по любому из пп.20-27, при котором моно- или олигомерные аминокислоты содержат имидазольные боковые цепи, предпочтительно не менее шести остатков гистидина, двух или более остатков гистидина, более предпочтительно 6-10 остатков гистидина.
36. Способ по пп.23 и 35, при котором используемые моно- или олигомерные аминокислоты удлиняют на N-конце короткими аминокислотными последовательностями, обеспечивающими участок узнавания для специфической протеазы.
37. Способ по любому из пп.23, 35 и/или 36, при котором модифицированные или немодифицированные (по п.35) моно- или олигомерные аминокислоты удлиняют на С-конце на одну или более аминокислот, что позволяет проводить определения системами детектирования, такими как биотинилированные аминокислоты, которые присоединяют, чтобы иметь возможность определять посредством авидин-подобных взаимодействий.
38. Способ по любому из пп.28, и/или 29, и/или 35-37, при котором конкурентный хелатирующий реагент содержит структурные фрагменты, содержащие металл-хелатирующие фрагменты, имеющшх электронные пары для координационных фрагментов, полученных из имидазолила, N-метилимидазолила, иминодиуксусной кислоты, нитрилотриуксусной кислоты, этилендиаминтетрауксусной кислоты, аминопурина, фенантролина, бипиридила, терпиридинила, триазациклононана или тетра-азациклододекана.
39. Способ по любому из пп.20-26, при котором хелатирующий лиганд содержит структурные фрагменты, имеющие электронные пары для координационных связей, такие как трифенилфосфин, 6-аминопурин, фталоцианин.
40. Способ по п.39, при котором лиганд представляет собой 5-амино-1,10-фенантролин, или амино-терпиридин, или триазациклононан, или тетраазациклододекан, или их производные.
41. Способ по любому из пп.20-40, при котором хелатирующие группы боковых цепей аминокислот выбирают из имидазоло, аминофрагментов, гидроксильного, карбоксильного, тиолового фрагментов, фрагментов нитрилотриуксусной кислоты, фрагментов иминодиуксусной кислоты, фенантролинового фрагмента, пиридинового, бипиридинового, терпиридинового, триазациклононанового, тетраазациклододеканового или пуринового фрагментов или производных перечисленных фрагментов, которые способны образовывать комплексы с металлом по любому из пп.20-26.
42. Способ по любому из пп.20-41, который полностью автоматизирован, совместим с роботами для синтеза и при котором разделение жидкости и твердой фазы во время циклов синтеза осуществляют, по существу, известными способами, предпочтительно просеиванием, разделением по размеру частиц, центрифугированием или технологией магнитной сепарации.
Текст
008305 Химический синтез пептидов хорошо изучен. По существу, можно различать два разных метода. Синтез в растворе часто занимает очень много времени, поэтому его не используют для научных исследований, тогда как синтез на твердом носителе делает возможной быструю оптимизацию реакционных циклов. Методы, подходящие для твердофазного синтеза пептидов (SPPS), основаны на технологии синтеза Меррифилда (Merrifield, R.B., J. Amer. Chem. Soc. 85, 1963, 2149), в соответствии с которой синтезируют пептиды с определенными последовательностями на нерастворимом твердом носителе, преимущество которой заключается в том, что отделение растворимых реагентов легко можно осуществлять простой фильтрацией. SPPS был крайне быстро усовершенствован в виде вариантов (обозначенных в описании как синтез по протоколу типа Меррифилда), и способ включает в себя следующие стадии: 1) на первой стадии подбирают подходящую матрицу твердого носителя (смолу), к которому аминокислотное производное ковалентно присоединяют по С-концу. Обычно первую С-концевую аминокислоту синтезируемой последовательности присоединяют к твердому носителю с помощью линкера. Аминогруппы и функциональные группы боковой цепи первой аминокислоты защищают защитными группами, известными из уровня техники, обычно совместимыми с Fmoc- или Вос-химией; 2) на второй стадии группу, защищающую N-конец первой аминокислоты, избирательно удаляют; 3) на 3-й стадии следующее аминокислотное производное, соответствующее 2-й аминокислоте в синтезируемой последовательности, присоединяют к свободной концевой аминогруппе первой аминокислоты, тогда как она сама имеет аминогруппу и функциональную группу боковой цепи, защищенные подходящими защитными группами; 4) на 4-й стадии группу, защищающую N-конец, удаляют из только что присоединенного аминокислотного производного и повторяют стадию 3 для следующей аминокислоты. Таким образом, твердофазный синтез пептидов включает циклический процесс, посредством которого определенная последовательность аминокислот может быть создана на твердом носителе. В конце процесса пептид отщепляют от твердого носителя, например, обработкой трифторуксусной кислотой (включая различные поглотители). Одновременно защитные группы, которые могут присутствовать на боковых цепях аминокислот,защищая имидазол-, меркапто-, карбокси-, амино-, спирт- или другие функционально подходящие группы, могут быть удалены. Это приводит к конечному пептиду, который часто требует очистки на подходящем хроматографическом оборудовании, обычно системе ЖХ/МС, которая позволяет одновременно разделять и анализировать компоненты. Описанные и другие стадии рассматривают подробно в стандартных руководствах по синтезу пептидов. Тогда как синтез небольших пептидов (вплоть до 30-членных) обычно не составляет никаких проблем с SPPS-методиками, существуют ограничения в отношении пептидов размером от 40 до 120 (или даже более) аминокислот. Их свойства значительно больше соответствуют термину небольшой белок,чем термину большой пептид: а) конструируемые пептиды указанного размера обычно имеют не только первичную структуру(последовательность), а также вторичную структуру (например, спираль, бета-слой) и третичную структуру (например, лейцин-замок, соединение доменов дисульфидной мостиковой связью). Тогда как различные варианты метода SPPS предназначены исключительно для построения первичной структуры, они не предусматривают никакого инструмента, чтобы контролировать внутримолекулярное образование вторичной и третичной структур подходящим способом. Отсутствие контрольных механизмов фолдинга более крупных пептидов усугубляется еще тем фактом, что одновременное отделение всех продуктов от смолы и удаление защитных групп приводит к очень высокой локальной концентрации продукта. При описанных условиях происходит не только внутримолекулярный фолдинг. Во многих случаях небольшие белки, отщепленные от смолы, стремятся к взаимодействию друг с другом, а не к внутримолекулярному скручиванию. Подобные проблемы имеют место, когда лиофилизированные фракции, содержащие очищенный продукт, элюированные из системы ЖХ, разбавляют растворителем. Таким образом, внутримолекулярный фолдинг и межмолекулярные взаимодействия конкурируют, и не существует никакого способа, чтобы адекватно контролировать описанный процесс. Это часто приводит к образованию функционально бесполезных мультимолекулярных агрегатов, которые не проявляют никакой желаемой биологической активности;b) в случае синтеза больших пептидов оказывается предпочтительным использовать соответственно модифицированные (защищенные, частично защищенные или незащищенные) пептидные фрагменты вместо отдельных аминокислотных остатков для каждого цикла синтеза. Обычно небольшие фрагменты соединяют или лигированием, или методами конденсации фрагментов для того, чтобы минимизировать количество стадий синтеза. Это обусловлено тем фактом, что циклы обычно не дают количественных выходов, а слишком много циклов часто заканчивается неприемлемым снижением выхода. Кроме того,проблемы с конденсацией отдельных аминокислот легко можно предотвратить с помощью конденсации целого фрагмента вместо отдельной аминокислоты. В случае конденсации фрагментов возникает проблема, обусловленная необходимостью применять более длительные периоды взаимодействия во время каждого цикла конденсации. Низкая реакционная скорость ограничена диффузией и обусловлена отсутствием диффузии фиксированной пептидной цепи и субоптимальным диффузионным поведением фрагментов в порах окружающей смолы. Продолжительное время реакции часто приводит к значительной-1 008305 степени рацемизации по С-концевой аминокислоте конденсируемых фрагментов, что определяет необходимость поддерживать время реакции настолько коротким, насколько возможно. Наилучшим способом преодоления описанной проблемы является конденсация в растворе. Классический SPPS не обеспечивает способ доставки обоих участников реакции в растворе и возможность повторно присоединять их во время следующей стадии цикла при повторении этой процедуры на каждой стадии синтеза; с) часто бывает трудно контролировать требуемую плотность исходных остатков на полимерном носителе, а нагрузка смолы первой аминокислотой часто происходит вместе с рацемизацией. Хотя большинство предварительно нагруженных смол поставляются коммерчески, коммерческие смолы часто не охватывают все нагрузки, а наилучшую величину для данной задачи пептидного синтеза нередко оказывается трудно подобрать. Сродство к металлу до сих пор находит только незначительное применение в синтезе пептидов, что документально подтверждают в публикации Comely et al. (Journal of the Chemical Society, 2001, 25262531). В упомянутой работе образуют комплекс аминокислоты с хромом, используя ароматические электро-пи-системы, например, которая присутствует в боковой цепи модифицированного фенилаланина. Такие комплексы образуются в растворе и предварительно полученный комплекс с хромом затем прикрепляют к твердой фазе и успешно проводят один цикл синтеза. Хотя принципиальная пригодность прикрепления комплекса с металлом к твердой фазе была продемонстрирована, процедура отличается от следующих аспектов представляемого изобретения.Comely et al. сначала присоединяют ион металла к растворимой части системы, а затем прикрепляют комплекс к твердой фазе на второй стадии, тогда как в представляемом изобретении сначала проводят присоединение ионов металла к твердой фазе, а затем прикрепляют растущую пептидную цепь к твердой фазе. Это устраняет один из недостатков системы, предлагаемой Comely, т.е. в полученном хромсодержащем комплексе на твердой фазе координационная связь между боковыми цепями аминокислоты и хромом оказывается сильнее, чем связь между хромом и твердым носителем. Таким образом, элюция комплекса конкурентами элюирует пептидный комплекс всегда с хромом, присоединенным к пептиду в стехиометрических количествах. Это не подходит ни для анализа и разделения, ни для предполагаемого фармацевтического применения пептида. Образованный комплекс данного изобретения характеризуется сильным хелатообразованием иона металла в твердой фазе и легко обратимым образованием комплекса с пептидной цепью. Процесс завершают элюцией пептида без значительных количеств ионов металла, присоединенных к нему.Comely et al. используют очень необычные комплексы, образованные ароматическими системами,тогда как в данном изобретении применяют образующие комплексы фрагменты, которые координационно связывают ион металла через атомы N, P, S или О. Упомянутые атомы могут быть частью стандартных органических групп. Такие группы можно создавать и оптимизировать для хелатообразования и прочности связывания, используя все разнообразие методов органической химии, а не ограничиваться ароматическими системами. Таким образом, представляемый принцип изобретения предусматривает более значительную гибкость и приспособляемость, чем система Comely. Система, предлагаемая Comely, является невыгодной в том смысле, что она требует применения атмосферы инертного газа, чтобы защитить некоторые из используемых реагентов. Даже при таких условиях выход не превышает 90% на каждую стадию, это означает, что нельзя ожидать, что более длинные пептиды, чем максимально декамеры, будут получены с подходящим выходом. Данное изобретение и используемая химия комплексов являются совместимыми со стандартной fmoc-химией и не требуют применения специальной атмосферы или оборудования. Comely применяет жесткие условия, которые являются медленными и деструктивными в отношении смолы, чтобы освободить пептид (48 ч окисления образующего основу полимера на воздухе при белом свете в ДХМ). В то время как никакое другое применение комплексов металлов до сих пор не найдено в области химического синтеза пептидов, сродство металлов известно в области рекомбинантного получения биомолекул. Однако его используют исключительно в хроматографическом методе как инструмент для очистки биологических молекул из неочищенных биологических смесей или биологических жидкостей в одну стадию. Имеются патенты относительно таких стратегий, использующих производные агарозы,содержащие иминодиацетат и нитрилотриацетат (ЕР-А-253303; US-A-4877830). Гранулированная агароза, подходящая для хроматографической очистки рекомбинантных продуктов, поставляется коммерчески. Кроме того, пептиды, содержащие боковые цепи со сродством к металлу, конструировали с намерением использовать комплексы переходного металла, фиксированные боковой цепью, как люминисцентные меченные реагенты для данного пептида (например, WO-A-9603651A1; ЕРА-А-0178450). Все упомянутые описания документально подтверждают пригодность способов с использованием хелата металла для анализа и/или очистки биомолекул. Однако они не заключают в себе сущности изобретения и его применения, которые описывают ниже. Техническая проблема, которую необходимо решить, заключается в создании подходящего способа твердофазного синтеза, очистки и рефолдинга/деагрегации небольших и больших пептидов, включая пептиды свыше 120 аминокислот. Чтобы решить проблемы SPPS, связанные с большими пептидами,пептиды следует обратимо присоединять к твердому носителю. Другой аспект изобретения заключается-2 008305 в повторном скручивании и отделении синтезированных пептидов, имеющих нежелательные, неправильно скрученные структуры и/или межмолекулярные агрегаты. Упомянутые проблемы можно разрешить согласно способу п.1. Предлагается применение активированной твердой фазы, содержащей твердый носитель, металлхелатирующие лиганды, ковалентно связанные с твердым носителем, ионы металла Мn+ с n=1-3, координационно связанные с металл-хелатирующими лигандами, причем указанная активированная твердая фаза обеспечивает координационные участки для координационного и обратимого присоединения якорной части пептида, для твердофазного синтеза пептидов или очистки пептидов. Активированную твердую фазу еще называют смола со сродством к металлу. В предпочтительном воплощении пептид представляет собой растущий пептид и его подвергают процедурам удлинения пептида. Предпочтительно, растущий пептид состоит по крайней мере из одной аминокислоты. В другом предпочтительном воплощении моно- или олигомерные аминокислоты присоединяют к карбокси- или аминоконцу (С- или N-конец) растущего пептида по протоколу последовательных реакций типа протокола Меррифилда, предпочтительно на основе fmoc-химии. В другом предпочтительном воплощении моно- или олигомерные аминокислоты содержат природные и/или неприродные аминокислоты. Кроме того, легко можно выбрать соответственно защищенные аминокислотные производные или олигомерные фрагменты, присоединяемые в каждом цикле по протоколу последовательных реакций типа протокола Меррифилда. Предпочтительный твердый носитель является носителем на основе диоксида кремния, стекла или целлюлозы или полимера, выбранного из группы, состоящей из полистирольных смол, меламиновых смол и смол на основе поливинилового спирта. Другие подходящие носители данного изобретения представляют собой, например, полистиролы,имеющие функциональные группы, в которых указанные функциональные группы могут быть ковалентно связаны с подходящими метал-хелатирующими лигандами. Примерами таких функциональных групп являются группы хлортритила, амино, гетероциклического азота, карбокси, гидрокси, меркапто и винила. Для твердофазного синтеза пептидов свободные карбоксильные группы или другие реакционноспособные группы твердого носителя должны быть защищены таким образом, чтобы они не могли мешать процедурам удлинения пептида. В предпочтительном воплощении твердый носитель содержит ферромагнитные частицы. В другом предпочтительном воплощении разделение жидкой и активированной твердой фазы во время циклов синтеза осуществляют, например, просеиванием, разделением по размеру, центрифугированием или по способу магнитной сепарации. Реакционноспособные функциональные группы можно вводить в твердый носитель в результате реакции с предварительно полученными фрагментами твердого носителя или, в случае полимеров, также в результате сополимеризации с соответственно модифицированными сополимерами. В предпочтительном воплощении каждый лиганд, образующий комплекс с металлом, содержит по крайней мере один атом азота, кислорода, фосфора или серы, который способен образовать координационную связь металл-лиганд. В другом предпочтительном воплощении каждый металл-хелатирующий лиганд, содержит по крайней мере одну функциональную группу, выбранную из группы, состоящей из амино, гетероциклического азота, карбокси, гидроксила и меркапто. Металл-хелатирующие лиганды ковалентно связаны с твердым носителем или непосредственно или через группы линкера. Подходящие линкерные группы представляют собой, например, группы амино,карбокси, метилена, окси, метилендиокси, полиметилендиокси, этилендиокси и полиэтилендиокси. Для твердофазного синтеза пептидов свободные карбоксильные группы металл-хелатирующих лигандов должны быть защищены таким образом, чтобы они не могли мешать процедурам удлинения пептида. На фиг. 1 а изображен пример твердого носителя, модифицированного 5-амино-1,10 фенантролином. Специалисты в данной области легко смогут идентифицировать многочисленные органические фрагменты, которые способны координационно связывать ионы металла, например, из базы данных, которую можно использовать, чтобы найти подходящие металл-хелатирующие фрагменты для металлхелатирующих лигандов согласно данному изобретению. В рамках данного изобретения существующие сведения можно использовать, чтобы идентифицировать подходящие фрагменты, которые соответствуют сфере действия изобретения. Пример базы данных, содержащей соответствующие сведения, представлен с показательными данными для групп, подходящих для осуществления данного изобретения, в таблице.-3 008305 Константы связывания комплексов показательных комбинаций лиганд/ион металла Значения pK при 25C. Значение pK определяли как отрицательный десятичный логарифм константы K связывания комплекса. В каждой колонке представлено значение pK для каждой последовательной стадии связывания иона металла или его комплекса MLm-1 с дополнительной молекулой лиганда L (MeLm-1+L MLm, K=c(MLm)/-4 008305 Таким образом, данные, касающиеся образования комплекса ионов различных металлов с лигандными молекулами, известны и могут быть обнаружены в основной литературе и базах данных. Данные в таблице взяты из базы данных Мартелла (Martell) и сотрудников (2003) и представляют показательный обзор относительно взаимодействия соответствующих ионов металлов с лигандами. Такие базы данных можно использовать, чтобы присоединять ион металла к твердой фазе. Упомянутые лиганды также могут функционировать в боковой цепи одиночных или олигомерных, природных или неприродных аминокислот как хелатирующие группы, которые связывают ненасыщенный ион металла, как описывают ниже. В другом предпочтительном воплощении каждый металл-хелатирующий лиганд, ковалентно связанный с твердым носителем, содержит по крайней мере один фрагмент, выбранный из группы, состоящей из трифенилфосфиновых фрагментов, аминопуриновых фрагментов, предпочтительно 6 аминопуриновых фрагментов, фталоцианиновых фрагментов, 1,10-фенантролиновых фрагментов, предпочтительно 5-амино-1,10-фенантролиновых фрагментов, терпиридиновых фрагментов, предпочтительно 4'-амино-[2,2'; 6',2]терпиридиновых фрагментов, триазациклононановых фрагментов, предпочтительно [1,4,7]-триазациклононановых фрагментов, и тетраазациклододеканиловых фрагментов, предпочтительно [1,4,7,10]-тетраазациклододекановых фрагментов. Предпочтительно металл Мn+ выбирают из группы, состоящей из Mn2+, Cu2+, Ni2+, Co2+, Zn2+, Mg2+,2+Ca , Fe2+, Fe3+ и ионов лантанида, особо предпочтительными Мn+ являются Cu2+, Ni2+, Co2+ и Zn2+ На фиг. 1b представлен пример активированной твердой фазы, содержащей твердый носитель, ковалентно связанный с 5-амино-1,10-фенантролиновым лигандом, образующим комплекс с металлом, и ионы связанного хелатом металла, Cu2+. На фиг. 1 с представлен пример присоединенного пептида, содержащего пептидный остаток и якорную часть олигогистидинового фрагмента, в котором указанная якорная часть ковалентно присоединена к активированной твердой фазе, включающей твердый носитель, ковалентно связанный 5-амино-1,10 фенантролиновым металл-хелатирующим лигандом, и ионы связанного хелатом Cu2+. Покрывающий слой прикрепленных пептидов и прикрепленных (моно- или олигомерных) аминокислот, последние называют исходным звеном для твердофазного синтеза пептидов, на поверхности активированного твердого носителя очень легко можно контролировать. При условии, что координационные участки активированной твердой фазы равномерно распределены по поверхности, плотность присоединенных пептидов и исходных звеньев (покрывающий слой) можно корректировать для определенных задач синтеза, очистки или рефолдинга пептидов и определять по количеству исходных звеньев,добавленных к смоле во время 1-й стадии присоединения. Покрывающий слой поверхности можно рассчитать из известной величины области поверхности и количества пептидов (определенных на моль) и/или диаметров. Так как реакции комплексообразования обычно являются быстрыми, присоединение может быть завершено в присутствии органического или водного растворителя в течение нескольких минут. Представлен способ, при котором якорную часть пептида, которая координационно связана с координационными участками активированной твердой фазы, содержащей твердый носитель, металлхелатирующие лиганды, ковалентно связанные с твердым носителем, ионы металла Мn+ с n=1-3 в указанных металл-хелатирующих лигандах, отделяют от упомянутой активированной твердой фазы добавлением конкурентного лиганда. Согласно изобретению конкурентный реагент можно добавлять к прикрепленному пептиду для того, чтобы конкурентно отделить якорную часть пептида от активированной твердой фазы. Предпочтительно, конкурентный реагент добавляют к смеси реагентов стадии связывания по протоколу реакций типа протокола Меррифилда. Подходящий конкурентный хелатирующий реагент имеет приблизительно такое же или более слабое сродство к свободным координационным участкам на активированной твердой фазе как каждый индивидуальный хелатирующий ион металла фрагмент якорной части пептида к указанным координационным участкам. В предпочтительном воплощении конкурентный реагент растворим в смеси реагентов стадии связывания и не взаимодействует с ингредиентами из смесей реагентов. На фиг. 2 а представлен принцип отделения с примером, в котором якорная часть пептида представляет собой олигогистидиновый остаток. В отличие от отщепления повторное присоединение якорной части пептида к активированной твердой фазе оказывается возможным в результате разбавления смеси, содержащей активированную твердую фазу, конкурентный реагент и неприсоединенный пептид. В предпочтительном воплощении в пределах протокола реакций типа протокола Меррифилда повторное присоединение проводят перед следующими стадиями промывания. На фиг. 2b представлен принцип повторного присоединения с примером, в котором якорная часть пептида представляет собой олигогистидиновый остаток. Пример влияния концентраций конкурентного реагента на связывание данного пептида с активированной твердой фазой представлен на фиг. 3 а, пример, когда отщепление возрастающими концентрациями конкурента применяют для элюции координационно связанного пептида. Описанный процесс является обратимым.-5 008305 В другом представляемом способе конкурентный лиганд содержит по крайней мере один фрагмент,способный образовывать комплекс с ионами металлов, предпочтительно азотсодержащий фрагмент, выбранный из группы, состоящей из имидазола, N-метилимидазола, аминопурина, фенантролина, бипиридина, терпиридина, триазациклононана и тетраазациклододекана, фрагментов иминодиуксусной кислоты, фрагментов нитрилотриуксусной кислоты и фрагментов этилендиаминтетрауксусной кислоты. В другом предпочтительном воплощении конкурентные лиганды содержат структурные фрагменты, имеющие электронные пары для координационных связей, такие как трифенилфосфиновые фрагменты, 6-аминопуриновые фрагменты или фталоцианиновые фрагменты. Конкретными примерами конкурентных лигандов являются глутатион, этилендиаминтетрауксусная кислота, имидазол, N-метилимидазол, фенантролины, предпочтительно 5-амино-1,10-фенантролины,аминотерпиридины, триазациклононаны и тетраазациклододеканы. В предпочтительном воплощении указанного способа пептид представляет собой растущий пептид, связанный через якорную часть с ионом металла, который присоединен к металл-хелатирующему лиганду, связанному с твердым носителем, и подвергается процедурам удлинения пептида. Термин растущий пептид имеет отношение к последовательному созданию пептидной цепи обычно с использованием аминокислоты с защищенными N-концом и боковой цепью, например, как в случае всех схем пептидного синтеза с применением fmoc. Предпочтительно моно- или олигомерные аминокислоты присоединяют на С- или N-конце растущего пептида по протоколу последовательных реакций типа протокола Меррифилда. Как упоминали выше, растущий пептид, содержащий по крайней мере одну моно- или олигомерную аминокислоту, ковалентно связанную с ионом металла якорной части, рассматривают как исходное звено для твердофазного синтеза пептидов. Примером такого исходного звена является единственный остаток глицина, ковалентно связанный через его карбоксильную группу с фрагментом, образующим комплекс с ионом металла. Предпочтительно исходное звено содержит по крайней мере одну боковую цепь и/или С-концевую модификацию, которая способна координировать ионы металла активированной твердой фазы. Указанные аминокислоты могут быть природными или неприродными. Каждая прикрепленная моно- или олигомерная аминокислота, используемая как исходное звено для твердофазного синтеза пептидов, должна быть совместимой с протоколами пептидного синтеза. Предпочтительно, якорная часть пептида содержит по крайней мере один фрагмент, образующий комплекс с ионом металла, каждый указанный фрагмент заключает в себе по крайней мере одну группу,содержащую азот, кислород, фосфор или серу, которая способна координировать ионы металла активированной твердой фазы. В предпочтительном воплощении каждый фрагмент, образующий комплекс с ионом металла, содержит от 1 до 10 упомянутых групп, содержащих азот, кислород, фосфор или серу. Более предпочтительный каждый фрагмент, образующий комплекс с ионом металла, содержит от 1 до 10 групп, содержащих амино, гетероциклический азот, аза, карбокси, серу и фосфор. Образующие комплекс с ионом металла фрагменты якорной части могут быть модифицированы при условии, что полученное производное является совместимым со способами пептидного синтеза. На фиг. 1 с, 2 а и 2b представляют олигогистидиновый фрагмент, аминоконец которого защищен в соответствии с Fmoc-химией. В другом воплощении изобретения координационная связь металл-хелатирующих лигандов с ионами металла активированной твердой фазы является более сильной, чем координационная связь якорной части пептида с указанными ионами металла. Более предпочтительную азотсодержащую группу, способную координировать ионы металла активированной твердой фазы, выбирают из группы, состоящей из амино, гидроксила, карбоксила, меркапто,имидазолила, N-метилимидазолила, аминопиринила, фрагментов фенантролила, фрагментов пиридила,фрагментов бипиридила, терпиридиниловых фрагментов, триазациклононанониловых фрагментов, тетраазациклододеканиловых фрагментов, фрагментов иминодиуксусной кислоты, фрагментов нитрилотриуксусной кислоты и фрагментов этилендиаминтетрауксусной кислоты. Особо предпочтительный указанный фрагмент содержит от 2 до 10 природных или неприродных аминокислот. Подходящие боковые цепи указанной аминокислоты, содержащие фрагменты, образующие комплекс с ионом металла, предпочтительно можно выбрать из имидазоло-, амино-, гидроксильного,карбоксильного, меркапто-, фенантролинового, пиридинового, бипиридинового, терпиридинового, триазациклононанового, тетраазациклододеканового и пуринового фрагментов и их производных, которые еще способны образовывать комплексы с металлом. Подходящие органические фрагменты, способные образовывать координационные связи с ионами металлов, можно найти в базах данных, которые иллюстративно представлены в таблице. Для осуществления процессов удлинения пептида свободные карбоксильные группы должны быть соответственно защищены. Предпочтительные моно- или олигомерные аминокислоты якорной части содержат возможно защищенные на N-конце имидазольные боковые цепи. В другом предпочтительном воплощении указанные моно- или олигомерные аминокислоты представляют собой олигогистидин или короткую (1-6 остатков)-6 008305 последовательность неприродных аминокислот, несущих фенантролиновые фрагменты в своих боковых цепях, по крайней мере с одной дополнительной аминокислотой, причем дополнительная аминокислота не взаимодействует с твердым носителем, например глицин. Предпочтительно, указанные олигогистидиновые фрагменты содержат по крайней мере 2 остатка гистидина; более предпочтительно 6-10 остатков гистидина. В другом предпочтительном воплощении указанные олигогистидиновые фрагменты содержат последовательность по крайней мере из 2 последовательных остатков L- или D-гистидина, более предпочтительно 6 остатков L- или D-гистидина. В другом предпочтительном воплощении указанные моно- или олигомерные аминокислоты якорной части содержат по крайней мере один 5-амино-1,10-фенантролиновый фрагмент. Оказалось возможно повторно отщеплять от активированной твердой фазы и повторно присоединять к активированной твердой фазе растущую пептидную цепь. В предпочтительном воплощении отщепление осуществляют во время стадии конденсации синтеза пептидов, тогда как повторное присоединение индуцируют разбавлением реакционной смеси до стадий промывки, предшествующих последующему удалению защиты. Используя описанные стадии, оказалось можно применять переходный жидкофазный синтез и использовать его преимущества особенно для синтеза больших пептидов способами конденсации фрагментов. Предпочтительно указанную якорную часть пептидной цепи помещают на С-конце и/или по крайней мере в одной боковой цепи аминокислот пептида. В другом предпочтительном воплощении указанную якорную часть пептида можно удлинять определенными группами и/или последовательностями природных и/или неприродных аминокислот, что делает возможным дальнейшую обработку и детектирование пептидов. В предпочтительном воплощении по крайней мере одну аминокислоту якорной части на С-конце пептида удлиняют одной или более аминокислотами, что позволяет проводить определение системами обнаружения. В предпочтительном воплощении к растущему пептиду добавляют последовательность, которая обеспечивает прямое распознавание конечной пептидной цепи антителами. Примером такой tagпоследовательности является myc-tag. Кроме того, добавление биотинилированной аминокислоты (например, биотинилированный D-, L-лизин) предпочтительно на С-конце олигомерного исходного звена позволит проводить прямое количественное определение конечного продукта, основанное на специфическом взаимодействии между биотином и авидинподобными молекулами. В другом предпочтительном воплощении якорную часть пептида удлиняют на N-конце аминокислотной последовательностью, обеспечивая участок узнавания для специфической протеазы. В еще одном предпочтительном воплощении исходное звено удлиняют короткой аминокислотной последовательностью, которая кодирует участки узнавания для протеазы. Это делает возможным селективное удаление исходного звена после процесса синтеза и процесса скручивания. В предпочтительном воплощении пептид повторно присоединяют к активированной твердой фазе разбавлением реакционной смеси в ходе последовательных реакций типа протокола Меррифилда, содержащей конкурентный лиганд. Подходящие конкурентные лиганды имеют одинаковое сродство к активированной твердой фазе(смола со сродством к металлу) по сравнению со сродством отдельных остатков предпочтительно олигомерных азотсодержащих групп, образующих комплекс с ионами металла фрагментов якорной части пептида (исходное звено), который следует отделить. В случае олигогистидина в виде якорной части пептида соответствующие конкурентные лиганды представляют собой имидазол (в водных растворителях) илиN-метилимидазол (в органических растворителях). Отделение осуществляют добавлением большого избытка конкурентного лиганда (обычно 102-106 молярного избытка конкурентного лиганда относительно присоединенного лиганда) по сравнению с присоединенным пептидом в растворителе. Например, в водных растворителях концентрация от 100 до 250 мМ имидазола (конкурентный лиганд) оказывается подходящей для того, чтобы достигнуть полного отщепления. При таких же условиях повторного присоединения достигают разведением растворителя, содержащего конкурентный лиганд, предпочтительно в 1020 раз. Это приводит к падению концентрации конкурентного лиганда и обеспечивает повторное присоединение полидентатной якорной части пептида к носителю. Молярное соотношение конкурентного лиганда к якорным частям можно легко определить для пар якорной части и активированной твердой фазы в соответствующем растворителе хроматографическими способами, элюируя якорную часть из смолы со сродством к металлу. В другом предпочтительном воплощении присоединение якорной части к активированной твердой фазе при синтезе пептида (например, олигогистидина) осуществляют в присутствии разбавленного щелочного или нейтрального раствора исходного звена. Кроме того, представляют способ очистки возможно защищенных пептидов, содержащих якорную часть, указанную якорную часть координационно связывают с координационными участками активированной твердой фазы, содержащей твердый носитель, металл-хелатирующие лиганды, ковалентно связанные с твердым носителем, ионы металла Мn+ с n=1-3 в указанных металл-хелатирующих лигандах,-7 008305 промывают, чтобы смыть примеси, такие как остатки защитных групп и поглотители или нежелательные побочные продукты пептидного синтеза. В другом предпочтительном воплощении изобретения используют хелатные группы, присоединенные на конечных стадиях синтеза к пептиду (на аминоконце цепи, например, производное Glyфенантролина). В упомянутом случае очистку неочищенного продукта от загрязняющих побочных продуктов осуществляют методом хроматографии, используя координационное присоединение пептида к активированной твердой фазе. Описанный метод можно применять, используя обычное блокирование конца свободных несвязанных аминогрупп в каждом цикле синтеза. При применении описанного метода очистку неочищенного продукта можно успешно провести за одну хроматографическую стадию. Отщепление можно осуществить добавлением подходящего конкурентного реагента, который описан выше, или увеличением кислотности растворителя до критической степени, предпочтительно, по крайней мере, до величины рН ниже 6, более предпочтительно до рН 5 или ниже, особенно в водном растворе, что является другим элегантным способом отщепления присоединенного пептида от смолы со сродством к металлу. Представлен другой способ, основанный на вышеупомянутом координационном и обратимом присоединении пептида к активированной твердой фазе. Способ рефолдинга неправильно скрученных структур и/или деагрегации межмолекулярных агрегатов возможно защищенного пептида, в котором якорная часть пептида координационно и обратимо присоединена к активированной твердой фазе, и повторное создание правильно скрученной пептидной структуры, включает стадии:a) обработки пептида по крайней мере одним хаотропным или денатурирующим реагеном иb) последующей обработки рядом растворителей, чтобы постепенно снизить хаотропность и обеспечить соответствующие условия рефолдинга и повторного создания вторичной и третичной структуры. В предпочтительном воплощении хаотропный или денатурирующий реагент выбирают из группы,состоящей из мочевины, детергентов, таких как додецилсульфат натрия, высокие концентрации солей и меркаптоэтанол или их смеси в подходящем растворителе. Вместо ковалентного присоединения к соответствующей смоле, в данном изобретении предусмотрена возможность отщепления цепи растущего пептида во время стадии конденсации, хотя его можно присоединять снова перед следующими стадиями. Кроме того, тот же самый принцип обратимого прикрепления пептида к активированной твердой фазе можно использовать, чтобы контролировать фолдинг очищенного продукта. С этой целью продукт очищают и повторно присоединяют к активированной твердой фазе, при этом соотношение молекул присоединенного пептида (определенное в молях) к области поверхности смолы со сродством к металлу выбирают таким образом, чтобы присоединенные пептиды не могли взаимодействовать друг с другом. Это способствует внутримолекулярному фолдингу, а не межмолекулярным взаимодействиям (агрегации). Очищенный и присоединенный продукт затем обрабатывают рядом растворителей, которые делают возможным постепенный фолдинг молекулы и поддерживают правильную внутримолекулярную конфигурацию пептидных цепей. Стадии фолдинга могут завершаться образованием, например, дисульфидных связей в правильном положении. В конце описанной процедуры стабилизированный продукт высвобождают из активированной твердой фазы добавлением конкурентного лиганда или увеличением кислотности растворителя. Рефолдинг присоединенного пептида проводят согласно протоколу рефолдинга. По указанному протоколу продукт растворяют в подходящем растворителе. Обычно наиболее предпочтительным растворителем является элюционный растворитель из ЖХ-хроматографической системы, хотя продукт можно лиофилизировать и перерастворить в подходящем растворителе. Из полученного раствора продукт повторно координационно присоединяют к активированной твердой фазе. Процедуру можно осуществлять, используя смолы на основе незащищенной иминодиуксусной кислоты или тринитрилоуксусных кислот. Это оказывается возможным, так как обычно для рефолдинга не применяют реагенты, которые активируют свободные карбоксильные группы. Предпочитают очень низкую плотность присоединения (покрывающий слой поверхности), чтобы избежать того, чтобы молекулы продукта приходили в близкий контакт друг с другом. После завершения присоединения ряд растворителей постепенно пропускают через смолу. В принципе, первые растворители являются высоко денатурирующими и/или хаотропными для того, чтобы привести все молекулы продукта в сравнимое состояние фолдинга. Постепенно следующие стадии и растворители приближают физиологические условия с тем, чтобы поддержать молекулярный фолдинг. В конце процедуры молекулы продукта оказываются в требуемом, предпочтительно водном, конечном растворителе, возможно скрепленные, например, дисульфидной связью, и могут высвобождаться из активированной твердой фазы. В другом предпочтительном воплощении вторичную и третичную структуру пептида поддерживают ковалентными связями между реакционноспособными боковыми группами цепей указанного пептида в результате обработки пептида соответствующими реагентами, включающей образование указанных ковалентных связей до отщепления пептида от активированной твердой фазы. Предпочтительно ковалентные связи, образованные между реакционноспособными боковыми цепями повторно скрученных пептидов, являются дисульфидными связями, амидными связями или стабильными ароматическими или алифатическими гидразонами.-8 008305 Соответствующие реагенты для образования, например, дисульфидных мостиковых связей, можно выбрать из различных окислительно-восстановительных реагентов, таких как иод, ферроцианаты, кислород и перекиси. Также можно выбрать другие реагенты, которые подходят для образования ковалентных связей в общепринятых реакциях в растворе. Представлен способ, при котором вторичную и третичную структуры правильно скрученных пептидов фиксируют образованием ковалентных связей между боковыми цепями аминокислот, предпочтительно образованием дисульфидных связей из свободных меркаптогрупп, образованием амидных связей,или образование стабильных ароматических или алифатических гидразонов достигают пропусканием смесей реагентов через повторно скрученный продукт, который присоединяют к активированной твердой фазе. Кроме того, представлен специальный способ синтеза пептида с последовательностью с X=d-аланин и Ala=бета-аланин, включающий образование дисульфидной связи между остатками цистеина с получением таким образом в которой X и Аlа являются такими, как описано выше. Специальный способ синтеза пептида с последовательностью (I), которую синтезируют на активированной твердой фазе, используя описанный выше способ последовательных реакций по протоколу типа Меррифилда, включает по крайней мере одну стадию очистки и рефолдинга и образования дисульфидной связи между остатками цистеина. Такой же способ применяют в отношении производного пептида с аминокислотными делециями или аминокислотными замещениями, так как еще два остатка цистеина имеются на расстоянии, которое допускает образование дисульфидной связи. Кроме того, представляют пептиды, содержащие якорную часть, предпочтительно состоящую из неприродных аминокислот, для координационного и обратимого присоединения пептида к поверхности активированной твердой фазы, якорная часть которых, размещенная на N- или С-конце и/или в боковой цепи пептида, содержит по крайней мере один фрагмент, образующий комплекс с ионом металла, каждый указанный фрагмент содержит по крайней мере одну азотсодержащую группу, за исключением пептидов, в которых указанный фрагмент, образующий комплекс с ионом металла, является аминокислотной последовательностью из от 2 до 5 остатков гистидина. Предпочтительно группы, образующие комплекс с ионом металла, предпочтительно азотсодержащие группы предлагаемых пептидов, фиксируют на N- или С-конце или части боковой цепи по крайней мере одной аминокислоты и их выбирают из группы, состоящей из амино, гидроксильного, карбоксильного, меркапто, имидазольного, N-метилимидазольного, аминопуринильного фрагментов, фенантролильных фрагментов, пиридильных фрагментов, бипиридильных фрагментов, терпиридинильных фрагментов, триазациклононанонильных фрагментов и тетраазациклододеканильных фрагментов или их комбинаций. В другом предпочтительном воплощении аминокислотный фрагмент содержит отдельные или олигомерные природные или неприродные аминокислоты. В предпочтительном воплощении якорная часть содержит по крайней мере один имидазолильный фрагмент. Более предпочтительно, якорная часть пептида содержит от 1 до 10 имидазолильных фрагментов. В другом предпочтительном воплощении аминокислота содержит по крайней мере два, более предпочтительно от 6 до 10 фрагментов гистидинила. В предпочтительном воплощении аминокислотный фрагмент содержит по крайней мере один фрагмент имидазолила. Более предпочтительно, якорная часть содержит от 1 до 10 фрагментов имидазолила. Другое преимущество изобретения заключается в том, что смолы со сродством к металлу можно повторно использовать после синтеза пептидов. Повторное применение может быть осуществлено в результате удаления катиона и повторной загрузки смолы тем же самым или другим подходящим катионом. Так как прочность связывания различных катионов отличается друг от друга, способ модифицирования процедур присоединения и повторного присоединения и стабильности полученных комплексов подбирают для другого катиона как мостикового лиганда между поверхностью смолы и исходным звеном синтеза. Изобретение в дальнейшем иллюстрируют следующими примерами, не ограничивающими изобретение. Примеры Сокращения вещества ДМФА - N,N-диметилформамид (чистый для синтеза пептидов) ДХМ - дихлорметан (чистый для пептидного синтеза) НОВТ - 1-гидроксибензотриазол (безводный)NaOH - раствор гидроксида натрия ТГФ - тетрагидрофуран ДМСО-d6 - диметилсульфоксид, дейтерированный Ш широкий (сигнал) с - синглет д - дуплет т - триплет кв - квадруплет п - квинтет м - мультиплетNi-NTA - смола Superflow (Novagen) Пример 1. Синтез Fmoc-Gly-His4-Gly-OMe [TAG1]. 2 ммоль первой аминокислоты и 4 ммоль DIPEA растворяли в 10 мл сухого ДХМ. Полученный раствор добавляли к 1,0 г сухой 2-хлортритилхлоридной смолы (200-400 меш) и смесь оставляли для взаимодействия в течение 60 мин на вращающей мешалке. В конце реакции смолу дважды оставляли для взаимодействия с 20 мл смеси ДХМ/MeOH/DIPEA в течение 3 мин, дважды промывали 20 мл ДХМ и дважды ДМФА. Смолу затем дважды обрабатывали 20 мл смеси пиперидин/ДМФА (1:3) в течение 3/20 мин и промывали 6 раз 20 мл ДМФА. 5 ммоль аминокислоты, 7,5 ммоль НОВТ и 10 ммоль DIPEA растворяли в 15 мл ДМФА. Через 5 мин добавляли 5 ммоль РуВОР и 10 ммоль DIPEA и полученный раствор вливали в смолу. После 60 мин перемешивания на вращающей мешалке смолу промывали 6 раз 20 мл ДМФА, дважды обрабатывали 20 мл смеси пиперидин/ДМФА (1:3) в течение 20/20 мин и промывали 6 раз 20 мл ДМФА. В случае Nконцевой аминокислоты смолу не обрабатывали смесью пиперидин/ДМФА. После присоединения последней аминокислоты смолу промывали 6 раз 20 мл ДМФА, дважды 20 мл ДХМ, а затем оставляли для взаимодействия с 50 мл смеси TFE/ДХМ (2/8) в течение 60 мин. Смолу отфильтровывали, растворители удаляли в вакууме, а неочищенный пептидный фрагмент использовали без дальнейшей очистки. Неочищенный пептид, 10 ммоль Сl-НОВТ, 10 ммоль DIEA и 20 ммоль DIC растворяли в минимальном количестве сухого ДХМ. Раствор оставляли для взаимодействия в течение 10 мин и добавляли раствор 10 ммоль DIEA и 10 ммоль Gly-OMe. После 2 ч перемешивания на вращающей мешалке добавляли 100 мл ДХМ, а раствор экстрагировали 3 раза порциями по 200 мл бисульфата натрия (1-2 моль/л), 3 раза порциями по 200 мл концентрированного хлорида натрия и 3 раза порциями по 200 мл бикарбоната натрия (1-2 моль/л). Органический слой высушивали над сульфатом натрия, растворитель удаляли, а неочищенный пептид использовали без дальнейшей очистки. Неочищенный пептид растворяли в 50 мл смеси TFA/TIS/H2O) (95/2,5/2,5) и оставляли для взаимодействия в течение 60 мин на вращающей мешалке. TFA удаляли при одновременном выпаривании с ДХМ. Неочищенный пептид растворяли в ДМСО и 1000 мкл очищали на системе ЖХ-МС Gilson NebulaLCMS System, используя колонку Kromasil RP C18. Линейный градиент составлял от 5% водной TFA(0,1%) до 50% ацетонитрила (содержащего 0,085% TFA) в течение 30 мин. Скорость потока составляла 20 мл/мин и оптическую плотность контролировали при 214 нм. Пример 2. Синтез Fmoc-Gly-His6-Gly-OMe [TAG3]. 2 ммоль первой аминокислоты и 4 ммоль DIPEA растворяли в 10 мл сухого ДХМ. Полученный раствор добавляли к 1,0 г сухой 2-хлортритилхлоридной смолы (200-400 меш) и смесь оставляли для взаимодействия в течение 60 мин на вращающей мешалке. В конце реакции смолу дважды оставляли для взаимодействия с 20 мл смеси ДХМ/MeOH/DIPEA в течение 3 мин, дважды промывали 20 мл ДХМ и дважды ДМФА. Смолу затем дважды обрабатывали 20 мл смеси пиперидин/ДМФА (1:3) в течение 3/20 мин и промывали 6 раз 20 мл ДМФА. 5 ммоль аминокислоты, 7,5 ммоль НОВТ и 10 ммоль DIPEA растворяли в 15 мл ДМФА. Через 5 мин добавляли 5 ммоль РуВОР и 10 ммоль DIPEA и полученный раствор вливали в смолу. После 60 мин перемешивания на вращающей мешалке смолу промывали 6 раз 20 мл ДМФА, дважды обрабатывали 20 мл смеси пиперидин/ДМФА (1:3) в течение 20/20 мин и промывали 6 раз 20 мл ДМФА. В случае Nконцевой аминокислоты смолу не обрабатывали смесью пиперидин/ДМФА.- 10008305 После присоединения последней аминокислоты смолу промывали 6 раз 20 мл ДМФА, дважды 20 мл ДХМ, а затем оставляли для взаимодействия с 50 мл смеси TFE/ДХМ (2/8) в течение 60 мин. Смолу отфильтровывали, растворители удаляли в вакууме, а неочищенный пептидный фрагмент использовали без дальнейшей очистки. Неочищенный пептид, 10 ммоль Сl-НОВТ, 10 ммоль DIEA и 20 ммоль DIC растворяли в минимальном количестве сухого ДХМ. Раствор оставляли для взаимодействия в течение 10 мин и добавляли раствор 10 ммоль DIEA и 10 ммоль Gly-OMe. После 2 ч перемешивания на вращающей мешалке добавляли 100 мл ДХМ, а раствор экстрагировали 3 раза порциями по 200 мл бисульфата натрия (1-2 моль/л), 3 раза порциями по 200 мл концентрированного хлорида натрия и 3 раза порциями по 200 мл бикарбоната натрия (1-2 моль/л). Органический слой высушивали над сульфатом натрия, растворитель удаляли, а неочищенный пептид использовали без дальнейшей очистки. Неочищенный пептид растворяли в 50 мл смеси TFA/TIS/H2O (95/2,5/2,5) и оставляли для взаимодействия в течение 60 мин на вращающей мешалке. TFA удаляли одновременным выпариванием с ДХМ. Неочищенный пептид растворяли в МЕОН и 1000 мкл очищали на системе ЖХ-МС Gilson Nebula LCMSSystem, используя колонку Kromasil RP C18. Линейный градиент составлял от 5% водной TFA (0,1%) до 80% ацетонитрила (содержащего 0,085% TFA) в течение 50 мин. Скорость потока составляла 20 мл/мин и оптическую плотность контролировали при 214 нм. Пример 3. Синтез Fmos-Gly-His3-Gly-His3-Gly-OMe [TAG4]. 2 ммоль первой аминокислоты и 4 ммоль DIPEA растворяли в 10 мл сухого ДХМ. Полученный раствор добавляли к 1,0 г сухой 2-хлортритилхлоридной смолы (200-400 меш) и смесь оставляли для взаимодействия в течение 60 мин на вращающей мешалке. В конце реакции смолу дважды оставляли для взаимодействия с 20 мл смеси ДХМ/MeOH/DIPEA в течение 3 мин, дважды промывали 20 мл ДХМ и дважды ДМФА. Смолу затем дважды обрабатывали 20 мл смеси пиперидин/ДМФА (1:3) в течение 3/20 мин и промывали 6 раз 20 мл ДМФА. 5 ммоль аминокислоты, 7,5 ммоль НОВТ и 10 ммоль DIPEA растворяли в 15 мл ДМФА. Через 5 мин добавляли 5 ммоль РуВОР и 10 ммоль DIPEA, а полученный раствор вливали в смолу. После 60 мин перемешивания на вращающей мешалке, смолу промывали 6 раз 20 мл ДМФА, дважды обрабатывали 20 мл смеси пиперидин/ДМФА (1:3) в течение 20/20 мин и промывали 6 раз 20 мл ДМФА. В случае Nконцевой аминокислоты смолу не обрабатывали смесью пиперидин/ДМФА. После присоединения последней аминокислоты смолу промывали 6 раз 20 мл ДМФА, дважды 20 мл ДХМ, а затем оставляли для взаимодействия с 50 мл смеси TFE/ДХМ (2/8) в течение 60 мин. Смолу отфильтровывали, растворители удаляли в вакууме, а неочищенный пептидный фрагмент использовали без дальнейшей очистки. Неочищенный пептид, 10 ммоль Сl-НОВТ, 10 ммоль DIEA и 20 ммоль DIC растворяли в минимальном количестве сухого ДХМ. Раствор оставляли для взаимодействия в течение 10 мин и добавляли раствор 10 ммоль DIEA и 10 ммоль Gly-OMe. После 2 ч перемешивания на вращающей мешалке добавляли 100 мл ДХМ, а раствор экстрагировали 3 раза порциями по 200 мл бисульфата натрия (1-2 моль/л), 3 раза порциями по 200 мл концентрированного хлорида натрия и 3 раза порциями по 200 мл бикарбоната натрия (1-2 моль/л). Органический слой высушивали над сульфатом натрия, растворитель удаляли, а неочищенный пептид использовали без дальнейшей очистки. Неочищенный пептид растворяли в 50 мл смеси TFA/TIS/H2O (95/2,5/2,5) и оставляли для взаимодействия в течение 60 мин на вращающей мешалке. TFA удаляли одновременным выпариванием с ДХМ. Неочищенный пептид растворяли в ДМСО и 1000 мкл очищали на системе ЖХ-МС Gilson Nebula LCMSSystem, используя колонку Kromasil RP C18. Линейный градиент составлял от 5% водной TFA (0,1%) до 50% ацетонитрила (содержащего 0,085% TFA) в течение 30 мин. Скорость потока составляла 20 мл/мин и оптическую плотность контролировали при 214 нм. Пример 4. Синтез Fmos-Gly-5-амино-1,10-фенантролина [TAG5]. 2 мл DIEA, 6 ммоль Cl-HOBt и 12 ммоль DIC добавляли к раствору 2,56 ммоль Fmoc-Gly-OH в минимальном количестве ДМФА. Полученный раствор перемешивали на вращающей мешалке в течение 5 мин и добавляли раствор 0,5 г 5-амино-1,10-фенантролина в минимальном количестве ДМФА. Смесь инкубировали в течение 12 ч и разбавляли 5-кратным объемом этилацетата. Раствор три раза промывали бикарбонатом натрия. Осадки неочищенного продукта отфильтровывали и очищали методом колоночной хроматографии или ЖХ-МС. Неочищенный пептид растворяли в ДМСО и 1000 мкл очищали на системе ЖХ-МС Gilson Nebula LCMS System, используя колонку Kromasil RP C18. Линейный градиент составлял от 5% водной TFA (0,1%) до 50% ацетонитрила (содержащего 0,085% TFA) в течение 50 мин. Скорость потока составляла 20 мл/мин и оптическую плотность контролировали при 214 нм. Пример 5. Синтез (бис-трет-бутоксикарбонилметиламино)уксусной кислоты [TAG8b]. а) Синтез бензиловогo эфира (бис-трет-бутоксикарбонилметиламино)уксусной кислоты [TAG8a] 20 ммоль п-тозилата бензилового эфира глицина, 40 ммоль трет-бутилового эфира бромуксусной кислоты и 60 ммоль DIEA растворяли в 35 мл сухого ДМФА. Реакционную смесь перемешивали на вращающей мешалке в течение 4 дней. Выпавшие в осадок соли отфильтровывали, а раствор растворяли в 250 мл этилацетата. Органический слой экстрагировали следующими растворами: 2 х 200 мл 1N NaOH,- 11008305 3 х 1N NaOH/солевой раствор 1:1. Раствор высушивали над Na2SO4, растворитель удаляли перегонкой в вакууме, а неочищенный продукт очищали колоночной флэш-хроматографией (этилацетат/гексан 1:4). 1b) Синтез (бис-трет-бутоксикарбонилметиламино)уксусной кислоты [TAG8b] 17,5 ммоль бензилового эфира (бис-трет-бутоксикарбонилметиламино)уксусной кислоты растворяли в 75 мл ТГФ и добавляли палладиевый катализатор, 10% уголь 0,70 г. Реакционный сосуд несколько раз заполняли водородом, а реакционную смесь перемешивали в атмосфере водорода до тех пор, пока не прекращалось потребление водорода. Катализатор отфильтровывали через целит, а растворитель удаляли в вакууме. 1 Н-ЯМР (400 МГц, ДМСО-d6) млн.д. 12,2 (ш с, 1 Н), 3,46 (с, 2 Н), 3,44 (с, 4 Н), 1,40 (с, 18 Н); ЖХ-МС(ESI): (М+Н)+=304. Пример 6. Синтез 4-(бис-трет-бутоксикарбонилметилкарбамоил)масляной кислоты [TAG10b]. а) Синтез бензилового эфира 4-(бис-трет-бутоксикарбонилметилкарбамоил)масляной кислоты[TAG10a] 24 ммоль монобензилового эфира пентандиовой кислоты, 30 ммоль оксалилхлорида и две капли ДМФА растворяли в 20 мл сухого ДХМ. Реакционную смесь кипятили с обратным холодильником до тех пор, пока не прекращалось выделение газа. Раствор выпаривали с 10 мл толуола. Неочищенный продукт растворяли в ДХМ и добавляли капля за каплей к раствору 12,0 ммоль трет-бутилового эфира (третбутоксикарбонилметиламино)уксусной кислоты и 26,4 ммоль DIEA в ДХМ при 0 С. Реакционную смесь перемешивали в течение 1 ч при 0 С и в течение ночи при комнатной температуре. Растворитель удаляли в вакууме, а остаток растворяли в 150 мл диэтилового эфира. Органический слой экстрагировали следующими растворами: 100 мл 1N HCl, 100 мл полуконцентрированного раствора бикарбоната натрия и 100 мл солевого раствора. Раствор высушивали над сульфатом натрия, а растворитель удаляли в вакууме. Неочищенный продукт очищали колоночной хроматографией на диоксиде кремния (этилацетат/гексан 1:1). 1b) Синтез 4-(бис-трет-бутоксикарбонилметилкарбамоил)масляной кислоты [TAG10b] 2 ммоль бензилового эфира 4-(бис-трет-бутоксикарбонилметилкарбамоил)масляной кислоты растворяли в 20 мл ТГФ и добавляли палладиевый катализатор, 10% уголь, 0,09 г. Реакционный сосуд несколько раз заполняли водородом, а реакционную смесь перемешивали в атмосфере водорода до тех пор,пока не прекращалось потребление водорода. Катализатор отфильтровывали через целит, а растворитель удаляли в вакууме. 1a) Синтез метилового эфира 4-([1,10]-фенантролин-5-ил-карбамоил)масляной кислоты [TAG12a] В атмосфере аргона 5 ммоль 5-амино-[1,10]-фенантролина растворяли в 25 мл сухого ДМФА. Добавляли 6,5 ммоль DIEA и капля за каплей добавляли 5,5 ммоль метилового эфира 4-хлоркарбонилмасляной кислоты. После перемешивания в течение 1 ч растворитель и избыток основания удаляли перегонкой в вакууме. Неочищенный продукт очищали методом колоночной хроматографии с обращенной фазой (Колонка Merck Lobar RP18, элюент: ацетонитрил/бикарбонат аммония, 5 мас.%, градиент: от 20 до 40%). 1b) Синтез 4-([1,10]-фенантролин-5-ил-карбамоил)масляной кислоты [TAG12b] 1,5 ммоль метилового эфира 4-([1,10]-фенантролин-5-ил-карбамоил)масляной кислоты растворяли в 15 мл 1,4-диоксана и 15 мл дистиллированной воды. Добавляли 1,5 мл 1N раствора гидроксида калия и раствор кипятили в течение 1 ч. Растворитель удаляли перегонкой в вакууме, а неочищенный продукт очищали методом колоночной хроматографии с обращенной фазой (Колонка Merck Lobar RP18, элюент:ацетонитрил/трифторуксусная кислота, 1 мас.%, градиент: от 10 до 20%). 1 Н-ЯМР (400 МГц, CD3OD) в виде соли K: млн. д. 9,11 (дд, J=l,6, 4,3 Гц, 1 Н), 9,04 (дд, J=l,6, 4,4 Гц,1 Н), 8,65 (дд, J=l,5, 8,4 Гц, 1 Н), 8,41 (дд, J=l,6, 8,1 Гц, 1 Н), 8,14 (с, 1 Н), 7,82 (дд, J=4,4, 8,4 Гц, 1 Н), 7,74(М+Н)+=310. Кроме того, метки [TAGs], содержащие производные других дикарбоновых кислот кроме глутаровой кислоты, можно получать аналогичным образом, используя описанные процедуры.[TAG15]. В атмосфере аргона 1 ммоль смолы Ванга (Wang), на которую загружен Fmoc-Gly-OH (0,7 ммоль/г),суспендировали в смеси пиперидин/ДМФА 1:3 и облучали в микроволновой печи (система "Discover" от СЕМ) три раза в течение 30 с. Смолу промывали три раза ДМФА и три раза ДХМ. Смолу суспендировали в 15 мл ДХМ и добавляли 9 ммоль DIEA и 10 ммоль адипоилхлорид. После перемешивания суспензии в течение 30 мин на вращающей мешалке реакционный раствор отфильтровывали, а смолу промывали два раза ДХМ и один раз ДМФА. Смолу суспендировали в 15 мл ДМФА и добавляли раствор 3 ммоль 5 амино-1,10-фенантролина и 5 ммоль DIEA в 20 мл ДМФА. После перемешивания на вращающей мешалке суспензии в течение ночи смолу промывали шесть раз ДМФА, а продукт отделяли от смолы суспендированием в смеси TFA/TIS/H2O, 95:2,5:2,5. Смолу отфильтровывали, а фильтрат несколько раз выпаривали одновременно с хлороформом, чтобы получить продукт. Описанный способ также аналогичным образом можно применять для других пептидов, связанных со смолой и других активированных производных дикарбоновых кислот. Пример 9. 5-амино-1,10-фенантролин/2-хлортритилхлоридная смола. 2 ммоль 5-амино-1,10-фенантролин суспендировали в атмосфере инертного газа в 50 мл сухого ДМФА. К полученной смеси добавляли 5 г 2-хлортритилхлоридной смолы (200-400 меш) и смолу инкубировали при комнатной температуре в течение 2 ч. Смолу отфильтровывали и промывали ДМФА до тех пор, пока не удаляли избыток фенантролина. Чтобы блокировать непрореагировавшие группы на смоле,последнюю инкубировали три раза с 10 мл смеси ДХМ/MeOH/DIEA (80:15:5) в течение 10 мин, а затем промывали 3 раза 10 мл ДМФА. Смолу можно хранить под ДМФА при комнатной температуре. Чтобы загрузить смолу ионами Ni2+, ее инкубировали в течение 30 мин с насыщенным раствором NiCl2 в ДМФА. Смолу промывали ДМФА до тех пор, пока больше не обнаруживали никакого NiCl2. После 5 дополнительных промывок ДМФА применяли следующие процедуры: 4x5' ДМФА/N-метилимидазол (0,25 моль/л) 4 х 15' ДМФА/N-метилимидазол (0,25 моль/л) 1 х 12 ч ДМФА/N-метилимидазол (0,25 моль/л) 2x5' ДМФА 1x1' изопропанол 1 х 15' изопропанол 1x5' ДМФА 1 х 30' ДМФА 1x1' изопропанол 1 х 15' изопропанол 1x5' ДМФА 1 х 30' ДМФА 1x1' изопропанол 1 х 15' изопропанол 1x5' ДМФА 1 х 30' ДМФА Пример 10. 5-Амино-1,10-фенантролин/Novasyn-TG-карбоксисмола. 5 г смолы Novasyn TG уравновешивали 50 мл сухого ДМФА в течение 30 мин. Добавляли 10 ммольDIEA и 10 ммоль HOBt. После 5-минутной инкубации добавляли 2 г 5-амино-1,10-фенантролина и 10 ммоль Pybop, а также 10 ммоль DIEA и смесь перемешивали в течение ночи на вращающей мешалке. На следующее утро смолу промывали 6 раз 50 мл ДМФА каждый раз. Для загрузки смолы никелем, добавляли 10 мл насыщенного раствора хлорида никеля в ДМФА и перемешивали на вращающей мешалке в течение 30 мин. После описанной стадии супернатант удаляли, а смолу промывали порциями по 50 мл ДМФА до тех пор, пока нельзя было обнаружить никакого видимого окрашивания супернатанта. Адсорбированный избыток ионов никеля удаляли следующими процедурами промывания: 4x5' ДМФА/N-метилимидазол (0,25 моль/л) 4 х 15' ДМФА/N-метилимидазол (0,25 моль/л) 1 х 12 ч ДМФА/N-метилимидазол (0,25 моль/л) 2x5' ДМФА 1x1' изопропанол 1 х 15' изопропанол 1x5' ДМФА 1 х 30' ДМФА 1x1' изопропанол 1 х 15' изопропанол 1x5' ДМФА 1 х 30' ДМФА 1x1' изопропанол- 13008305 1 х 15' изопропанол 1x5' ДМФА 1 х 30' ДМФА Пример 11. Синтез 1, 4,7-триазациклононан/2-хлортритилхлоридной смолы. 2,5 г 2-хлортритилхлоридной смолы (200-400 меш) суспендировали в 40 мл сухого ДМФА. Добавляли 500 мг тригидрохлорида 1,4,7-триазациклононана и 2 мл диизопропилэтиламина. Смолу инкубировали при комнатной температуре в течение 8 ч. Смолу фильтровали и промывали ДМФА шесть раз. Чтобы блокировать непрореагировавшие группы на смоле, последнюю инкубировали два раза с 20 мл смеси ДХМ/MeOH/DIEA (80:15:5) в течение 30 мин, а затем промывали 3 раза 20 мл ДМФА. Загрузку смолы никелем можно проводить согласно примеру 10. Смолу хранили под ДМФА в холодильнике. Пример 12. Загрузка смолы метками (загрузочные эксперименты). 1 мл соответствующей смолы промывали 3 раза ДМФА (чистый для пептидного синтеза). 1 мг метки растворяли в 1 мл ДМФА и добавляли 1 каплю DIEA. 50 мкл полученного раствора разбавляли 950 мкл воды и анализировали методом ВЭЖХ. Остаток смеси добавляли к промытой смоле и инкубировали в течение 30 мин. Чтобы проверить присоединение меток к смоле, 50 мкл раствора разбавляли 950 мкл воды и анализировали методом ВЭЖХ. Пример 12 а. Загрузка ТАG1 на Ni-фенантролин-2 Сl-Trt-смолу. При ВЭЖХ супернатантной жидкости больше не обнаруживали никакого определяемого TAG1. Пример 12b. Загрузка TAG3 на Ni-фенантролин-2 Сl-Trt-смолу. При ВЭЖХ супернатантной жидкости больше не обнаруживали никакого определяемого TAG3. Оставшиеся следы с подобным временем удерживания представляли собой минорные примеси, которые не были связаны. Проведенный эксперимент также продемонстрировал потенциал способа для очистки продуктов от загрязнения. Пример 12 с. Загрузка ТАG4 на Ni-фенантролин-2 Сl-Trt-смолу. При ВЭЖХ супернатантной жидкости больше не обнаруживали никакого определяемого TAG3. Пример 12d. Загрузка TAG3 на Ni-фенантролин-Novasyn-TG-смолу. При ВЭЖХ супернатантной жидкости больше не обнаруживали никакого определяемого TAG3. Пример 13. Элюция TAG3 из смол (загрузочные эксперименты). 10 мл насыщенной смолы 3 раза промывали 20 мл смеси 1 и смолу делили на 10 порций. Супернатантную жидкость удаляли, а смолы инкубировали в течение 5 мин со следующими растворами:(TAG3-10) 3,28 мг N-метилимидазола в 2 мл смеси 1 (20 ммоль/л). Что касается экспериментов по элюции более высокими концентрациями имидазола и Nметилимидазола, исследования проводили, используя смолы из пробирок (TAG3-07) и (TAG3-10). Указанные смолы инкубировали с возрастающими концентрациями элюционных реагентов (100 ммоль, 200 ммоль, 500 ммоль), супернатант анализировали, а смолу инкубировали в течение 5 мин со следующей концентрацией элюционного реагента. Результаты. Элюция меток может быть легко осуществлена с помощью хлористо-водородной кислоты. Концентрации свыше 100 ммоль/л имидазола или N-метилимидазола также элюировали эффективно. Пример 14. Связывание и элюция ТАG3 на Ni-NTA-смолу (FPLC-(жидкостная хроматография высокого разрешения) эксперименты). Пептид: TAG3 (Fmoc-(His)6-OMe, M=1191,25 г/моль), чистый для ВЭЖХ Смола: Ni-NTA SuperFlow (Емкость приблизительно 5 мкмоль/мл) Колонка: Biorad, 2,0 мл Оборудование для FPLC: Akta Pharmacia а) Элюция N-метилимидазолом Вводили 1,1 мг (0,92 мкмоль) TAG 3 в 0,75 мл элюента А. Элюент А: 2,2,2-трифторэтанол/Н 2O/диизопропилэтиламин/N-метилимидазол, 1:1:+1%:+5 мМ. Элюент В: TFE/H2O/DIEA/NMI, 1:1:+1%:+500 мМ. Скорость: 1 мл/мин. Программа: Введение (5 мл А)Промывка (10 мл А)Градиент: элюция (0% В 100% В для 10 мл)Сбор (5 мл В)Регенерация (8 мл А). Определенное количество TAG3 связывали и элюировали при концентрациях N-метилимидазола- 14008305 100-125 мМ. Такую FPLC-процедуру также можно использовать для определения пороговых концентраций, выше которых присоединенная молекула эффективно конкурентно вытесняется хелатирующим реагентом. Ниже пороговой концентрации присоединенная молекула остается еще связанной со смолой. В загрузочной процедуре разбавление смеси до точки ниже пороговой концентрации приводит к повторному связыванию со смолой смытой присоединенной молекулы.TAG элюировали при 60% элюента А 2. Пример 15. Цикл синтеза с Fmoc-Gly. Конденсация TAG3 с Fmoc-Gly на TG-смоле. 1 мл TG-смолы, в который вводили TAG3, промывали 3 раза 5 мл ДМФА. Чтобы удалить защиту Nконца, добавляли 5 мл 20% пиперидина и смолу инкубировали в течение 30 мин. Затем смолу промывали 6 раз ДМФА и добавляли предварительно активированный раствор 1 ммоль Fmoc-Gly-OH, 2 ммольDIEA, 2 ммоль HOBt и 2 ммоль DIC в минимальном количестве ДМФА. Через 60 мин смолу промывали 6 раз порциями ДМФА по 5 мл и 6 раз порциями воды по 2 мл. Для отделения пептида от смолы добавляли 1 мл 1N HCl, а смолу инкубировали в течение 30 мин. Результат: Отщепленный пептид анализировали методом ЖХ-МС на колонке (218MS5415) массспектрометра Vydac C18. Использовали градиент от 5 до 95% В в течение периода 30 мин и скорость потока 1 мл/мин(А=вода, 0,1% TFA; В=ацетонитрил, 0,085% TFA). Никаких выделенных веществ не определяли. Только пептид, присутствующий в смеси, показал ожидаемую массу (М=1247, М 2+=625). Кроме HOBt не смогли выявить никаких других существенных примесей. Пример 16. Синтез TAG5-G-A-Fmoc на Ni-NTA-смоле. 2,65 мг TAG5 связывали с Ni-NTA-смолой в колонке Omnifit, 1,7 мл. Смолу переносили в микроволновый реакционный сосуд и промывали 6 раз 5 мл ДМФА каждый раз. Супернатанты отбрасывали, а смолу инкубировали с 5 мл 25% пиперидина в ДМФА, подвергая воздействию микроволновых импульсов, 3 х 30 с, при 50 В каждый в микроволновой печи Discovery (СЕМ GmbH). Смолу промывали 6 раз ДМФА. Получали раствор 1 мМ требуемой аминокислоты (соответственно защищенной) в минимальном количестве растворителя (ДМФА). К полученному раствору добавляли 2 ммоль DIEA, 2 ммоль HOBt и 2 ммоль DIC. После 5-минутной инкубации полученный раствор добавляли к смоле, лишенной защиты, и подвергали воздействию микроволновых импульсов, 3 х 30 с, при 50 В каждый. После присоединения последней аминокислоты, концевую Fmoc-группу не удаляли. Смолу промывали 5 раз водой (рН свыше 7). Наконец, 2 мл 1N HCl добавляли к смоле. Смолу инкубировали в течение 30 мин, супернатант удаляли и сохраняли. Супернатант использовали непосредственно для аналитической ЖХ-МС. Смолу собирали для рециркуляции. При ЖХ-МС установили точную массу пептида в единственном определяемом пептидном пике. Непептидными примесями были HOBt и HOBt-Н 2 О. Пример 17. Очистка иммуномера IL-2. Очистка иммуномера IL-2 (DIM03A) на Ni-NTA в смеси 2,2,2-трифторэтанол/Н 2O/ диизопропилэтиламин/N-метилимидазол. Пептид: Который указан в примере 19 (М=6237 г/моль), неочищенный продукт после расщепления смолы. Смола: Ni-NTA SuperFlow 7 6,7 мг (12,2 мкмоль) в 5 мл элюента А, введенные в 5 фракциях, смотри подписи к фиг. 3 а-с. Колонка: Biorad, 2,0 мл. Элюент A: TFE/H2O/DIEA/NMI, 1:1:+l%+5 мМ Элюент В: TFE/H2O/DIEA/NMI, 1:1:+l% + 500 мМ Скорость: 1 мл/мин. Программа: Введение (5 мл А)Промывка (10 мл А)Градиент: элюция (0% В 100% В в пределах 10 мл)Сбор (5 мл В)Регенерация (8 мл А). Требуемый продукт элюировали в одном пике приблизительно при 125 мМ NMI. Идентичность подтверждали аналитической ЖХ-МС. Пример 18. Постсинтетические модификации пептида. Образование дисульфидной мостиковой связи в пептиде.Acm и Trt являются стандартными защитными группами для SH-группы на остатках цистеина. Пеп- 15008305 тид синтезировали обычным способом синтеза пептида и очищали на системе ЖХ/МС до чистоты свыше 95%. 1 мл Ni-NTA-смолы, которую загружали пептидом, промывали 3 раза 2 мл МеОН. В течение 2 ч добавляли 50 мкл раствора иода (0,5 моль/л), а смесь инкубировали в течение дополнительных 2 ч при комнатной температуре. Смолу промывали 6 раз МеОН, 3 раза водой (рН 7), а затем инкубировали с 2 мл НСl (1). Смолу отфильтровывали и раствор анализировали в системе ЖХ/МС. Получен единственный пик продукта, который показал точную массу. Пример 19. Синтез Интерлейкин-2-иммуномера, используя стадии рефолдинга способа, представленного в описании. Химическая формула вещества, которое синтезировали:Ala = бета-аланин другие буквы = стандартный аминокислотный код Общая стратегия Синтез проводили на 2-хлортритилхлоридной смоле (200-400 меш) со степенью замещения 0,2 ммоль/г. Первые семь аминокислот конденсировали в виде фрагмента. Присоединение следующих аминокислот достигали посредством одиночной, двойной или тройной конденсации при 10-кратном избытке аминокислот и PyBOP/HOBT/DIPEA как конденсирующих добавок. Удаление защиты с N-конца растущей пептидной цепи проводили двойной обработкой смесью пиперидин/ДМФА (1/3). В трудных случаях проводили третью обработку смесью ДБУ/пиперидин/ДМФА (2/2/96). Количество примененных циклов конденсации и снятия защиты представлено в следующей схеме (Информация относительно трудной конденсации/снятия защиты, полученная при проверке каждого удлинения методом ВЭЖХ-МС). 1, 2, 3 = количество стадий конденсации и удаления защитыX = D-аланин В = аланин Протокол 1. Синтез первого пептидного фрагмента 2 ммоль первой аминокислоты и 4 ммоль DIPEA растворяли в 10 мл сухого ДХМ. Полученный раствор добавляли к 1,0 г сухой 2-хлортритилхлоридной смолы (200-400 меш) и смесь оставляли для взаимодействия в течение 60 мин на вращающей мешалке. В конце реакции смолу дважды оставляли для взаимодействия с 20 мл смеси ДХМ/MeOH/DIPEA в течение 3 мин, дважды промывали 20 мл ДХМ и дважды ДМФА. Смолу затем дважды обрабатывали 20 мл смеси пиперидин/ДМФА (1:3) в течение 3/20 мин и промывали 6 раз 20 мл ДМФА. Присоединение аминокислот 2-7 достигали по следующему способу: 5 ммоль аминокислоты, 7,5 ммоль НОВТ и 10 ммоль DIPEA растворяли в 15 мл ДМФА. Через 5 мин добавляли 5 ммоль РуВОР и 10 ммоль DIPEA и полученный раствор вливали в смолу. После 60 мин перемешивания на вращающей мешалке смолу промывали 6 раз 20 мл ДМФА, дважды обрабатывали 20 мл смеси пиперидин/ДМФА (1:3) в течение 20/20 мин и промывали 6 раз 20 мл ДМФА. В случае Nконцевой аминокислоты, смолу не обрабатывали смесью пиперидин/ДМФА. Протокол 2. Отщепление пептидного фрагмента. После присоединения последней аминокислоты смолу промывали 6 раз 20 мл ДМФА, дважды 20 мл ДХМ, а затем оставляли для взаимодействия с 50 мл TFE/ДХМ (2/8) в течение 60 мин. Смолу отфильтровывали, растворитель удаляли в вакууме, а неочищенный пептидный фрагмент использовали без дальнейшей очистки. Протокол 3. Повторное присоединение пептидного фрагмента. 1 ммоль фрагмента и 2 ммоль DIPEA растворяли в 50 мл сухого ДХМ. Полученный раствор добавляли к 5,0 г сухой 2-хлортритилхлоридной смолы (200-400 меш) и смесь оставляли для взаимодействия в течение 12 ч на вращающей мешалке. В конце реакции смолу дважды оставляли для взаимодействия с 50 мл смеси ДХМ/MeOH/DIPEA в течение 3 мин, дважды промывали 50 мл ДХМ и дважды 50 мл ДМФА. Протокол 4. Конденсация аминокислот 8-57. Высушенную смолу оставляли набухать в 50 мл смеси пиперидин/ДМФА (1/3) в течение 30 мин,обрабатывали 30 мл смеси пиперидин/ДМФА (1/3) в течение 20 мин, обрабатывали 30 мл смеси ДБУ/пиперидин/ДМФА (2/2/96) в течение 20 мин и промывали 6 раз 30 мл ДМФА. 10 ммоль аминокислоты, 15 ммоль НОВТ и 20 ммоль DIPEA растворяли в 30 мл ДМФА. Через 5 мин добавляли 10 ммоль РуВОР и 20 ммоль DIPEA и полученный раствор вливали в смолу. После 60 мин перемешивания на вра- 16008305 щающей мешалке смолу промывали дважды 30 мл ДМФА и конденсацию повторяли один раз или (в трудных случаях) дважды в течение 60 мин. Смолу затем промывали 6 раз 30 мл ДМФА. Смолу можно использовать непосредственно для присоединения следующей аминокислоты или после 2 промывок 30 мл ДХМ высушивать в вакууме и хранить при -80 С. Протокол 5. Отщепление и удаление защиты. После конденсации последней аминокислоты N-концевую защитную группу удаляли двойной обработкой 30 мл смеси пиперидин/ДМФА (1/3) в течение 20 мин и обработкой смесью ДБУ/пиперидин/ДМФА (2/2/96) в течение 20 мин. Смолу промывали 6 раз 30 мл ДМФА, дважды 30 мл ДХМ и обрабатывали 50 мл смеси TFE/ДХМ (2/8) в течение 180 мин. После фильтрования растворитель удаляли в вакууме, а защитную группу удаляли обработкой 30 мл смеси TFA/TIS/EDT/вода (94/1/2,5/2,5) в течение 180 мин в атмосфере инертного газа. Полученный раствор вливали в 300 мл холодного эфира,осадок растворяли в ацетонитриле, а пептид очищали методом ВЭЖХ-ОФ (Kromasil 100 С 4 10 мкм,250 х 4,6 мм) и собранные фракции непосредственно использовали для процедуры рефолдинга. Альтернативно, неочищенный продукт иммуномера IL-2 можно очищать эффективно на аффинной колонке со сродством к металлу. Неочищенный продукт IL-2, 76,7 мг (12,2 мкмоль) вводили приблизительно в 5 мл элюента А за 5 циклов (см. также фиг. 3 а-с). Колонка: Omnifit 1,7 мл. Элюент A: TFE/H2O/DIEA/NMI, 1:1:+1%:+5 мМ. Элюент В: TFE/H2O/DIEA/NMI, 1:1:+1%:+500 мМ. Скорость 1 мл/мин. Программа: Введение (5 мл А)Промывка (16,5 мл А)Градиент: элюция (0% В 100% В в пределах 10 мл)Сбор (5 мл В)Регенерация (8 мл А) Протокол 6. Процедура рефолдинга. Соответствующие элюированные фракции разбавляли до конечного объема 20 мл, используя точно такой растворитель, который присутствует в элюированной фракции. Полученный раствор очищенного продукта инкубировали в течение 30 мин при комнатной температуре с 10 мл Ni-NTA Superflow (Qiagen) при небольшом перемешивании в химическом стакане. Частицы Superflow упаковывали в пустую колонку FPLC и прикрепляли к прибору FPLC. Используя хроматографическую программу этого прибора,растворитель заменяли за 10 мин. Затем градиент водой в течение других 10 мин. Использовали градиент, чтобы заменить растворитель смесью вода/трифторэтанол (1/1). Во время 24-часового периода используемый растворитель окисляли барботированием кислорода через резервуарный сосуд и при использовании атмосферы кислорода в сосуде, чтобы закрыть дисульфидные мостиковые связи. Во время насыщения кислородом поддерживали постоянную скорость 0,1 мл/мин в течение ночи по всей колонке,тогда как элюат постоянно рециркулировал в резервуарном сосуде. В конце 24-часового периода конечный продукт, повторно скрученный и уплотненный в результате образования дисульфидной связи, элюировали применением забуференного фосфатом физиологического раствора (PBS, рН 7,2), содержащего 150 мМ имидазола, или применением 0,1 М ацетатного буфера (рН 4,5). Пример 20. Конденсация (бис-трет-бутоксикарбонилметиламино)уксусной кислоты [TAG8b] с Nконцом пептида. 1 ммоль пептида с последовательностью WETGLRLAPL получали на 2-хлортритилхлоридной смоле (200-400 меш), следуя стандартным процедурам, которые описаны в примере 19, протокол 1. Для конденсации Tag8b к N-концу пептида аналогичным образом применяли процедуру конденсации аминокислот. 5 ммоль Tag8b, 7,5 ммоль НОВТ и 10 ммоль DIPEA растворяли в 15 мл ДМФА. Через 5 мин добавляли 5 ммоль РуВОР и 10 ммоль DIPEA и полученный раствор вливали в смолу. После 60 мин перемешивания на вращающей мешалке смолу промывали 6 раз 20 мл ДМФА, дважды 30 мл ДХМ. Отщепление и снятие защиты пептида осуществляли, следуя стандартным процедурам. Смолу обрабатывали 50 мл смеси TFE/ДХМ (2/8) в течение 180 мин. После фильтрования растворитель удаляли в вакууме, а защитные группы удаляли обработкой 30 мл смеси TFA/TIS/вода (95/2,5/2,5) в течение 180 мин в атмосфере инертного газа. Полученный раствор вливали в 300 мл холодного эфира, осадок растворяли в ацетонитриле, а пептид очищали методом ВЭЖХ-ОФ (Kromasil 100 С 4 10 мкм, 250 х 4,6 мм). На стадии удаления защиты группы трет-бутиловых эфиров Tag отщепляли с тем, чтобы получить пептид[TAG10b] с N-концом пептида. 1 ммоль пептида с последовательностью GDQYIQQAHRSHI был получен на 2 хлортритилхлоридной смоле (200-400 меш), следуя стандартным процедурам, которые описывают в примере 19, протокол 1. Для конденсации Tag10b к N-концу пептида аналогичным образом применяли процедуру конденсации аминокислот. 5 ммоль Tag10b, 7,5 ммоль НОВТ и 10 ммоль DIPEA растворяли в 15 мл ДМФА. Через 5 мин добавляли 5 ммоль РуВОР и 10 ммоль DIPEA и полученный раствор вливали в смолу. После 60 мин перемешивания на вращающей мешалке смолу промывали 6 раз 20 мл ДМФА, дважды 30 мл ДХМ. Отщепление и снятие защиты пептида осуществляли, следуя стандартным процедурам: смолу обра- 17008305 батывали 50 мл смеси TFE/ДХМ (2/8) в течение 180 мин. После фильтрования растворитель удаляли в вакууме, а защитные группы удаляли обработкой 30 мл смеси TFA/TIS/вода (95/2,5/2,5) в течение 180 мин в атмосфере инертного газа. Полученный раствор вливали в 300 мл холодного эфира, осадок растворяли в ацетонитриле, а пептид очищали методом ВЭЖХ-ОФ (Kromasil 100 С 4 10 мкм, 250 х 4,6 мм). На стадии удаления защиты, группы трет-бутиловых эфиров Tag отщепляли с тем, чтобы получить пептид Tag10-GDQYIQQAHRSHI. ЖХ-МС (ESI): (М+Н)+: 1783 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Применение активированной твердой фазы, содержащей твердый носитель, металлхелатирующие лиганды, ковалентно связанные с твердым носителем, ионы металла Мn+ с n=1-3, координационно связанные с упомянутыми металл-хелатирующими лигандами, причем указанная активированная твердая фаза обеспечивает координационные участки для координационного и обратимого присоединения якорной части пептида, для твердофазного синтеза пептидов на указанной активированной твердой фазе, при этом пептид представляет собой растущий пептид и подвергается процедурам удлинения пептида. 2. Применение по п.1, при котором используют твердый носитель на основе диоксида кремния,стекла, или целлюлозы, или полимера, выбранного из группы, состоящей из полистирольных смол, меламиновых смол и поливиниловых спиртов. 3. Применение по любому из пп.1-2, при котором каждый металл-хелатирующий лиганд содержит по крайней мере один атом азота, кислорода, фосфора или серы, который способен образовывать координационную связь лиганд-металл. 4. Применение по любому из пп.1-3, при котором каждый металл-хелатирующий лиганд содержит по крайней мере одну функциональную группу, выбранную из группы, состоящей из амино, гетероциклического азота, карбокси, гидроксила и меркапто. 5. Применение по любому из пп.1-4, при котором каждый металл-хелатирующий лиганд, ковалентно связанный с твердым носителем, содержит по крайней мере один фрагмент, выбранный из группы,состоящей из трифенилфосфиновых фрагментов, аминопуриновых фрагментов, предпочтительно 6 аминопуриновых фрагментов, фталоцианиновых фрагментов, 1,10-фенантролиновых фрагментов, предпочтительно 5-амино-1,10-фенантролиновых фрагментов, терпиридиновых фрагментов, предпочтительно 4'-амино-[2,2'; 6',2]терпиридиновых фрагментов, триазациклононановых фрагментов, предпочтительно [1,4,7]-триазациклононановых фрагментов, и тетраазациклододеканиловых фрагментов, предпочтительно [1,4,7,10]-тетраазациклододеканиловых фрагментов. 6. Применение по любому из пп.1-5, при котором металл Мn+ выбирают из группы, состоящей из ионов Mn2+, Cu2+, Ni2+, Co2+, Zn2+, Mg2+, Ca2+, Fe2+, Fe3+ и лантанида, особо предпочтительным Мn+ является Cu2+, Ni2+, Co2+, Zn2+, Mg2+. 7. Применение по п.1, при котором якорную часть пептида отделяют от указанной активированной твердой фазы добавлением конкурентного лиганда. 8. Применение по п.7, при котором конкурентный лиганд содержит по крайней мере один фрагмент, способный образовывать комплекс с ионами металлов, предпочтительно азотсодержащий фрагмент, выбранный из группы, состоящей из имидазольных, N-метилимидазольных, аминопуриновых, фенантролиновых, бипиридиновых, терпиридиновых, триазациклононановых, тетраазациклододекановых фрагментов, фрагментов иминодиуксусной кислоты, фрагментов нитрилотриуксусной кислоты и фрагментов этилендиаминтетрауксусной кислоты. 9. Применение по п.8, при котором моно- или олигомерные аминокислоты присоединяют к С- илиN-концу растущего пептида по протоколу последовательных реакций типа протокола Меррифилда. 10. Применение по любому из пп.7-9, при котором якорная часть пептида содержит по крайней мере один фрагмент, образующий комплекс с ионом металла, причем каждый из указанных фрагментов включает по крайней мере одну группу, содержащую азот, кислород, фосфор или серу, которая способна координировать ионы металла активированной твердой фазы. 11. Применение по любому из пп.7-10, при котором азотсодержащую группу, которая способна координировать ионы металла активированной твердой фазы, выбирают из группы, состоящей из амино,гидроксильных, карбоксильных, меркапто, имидазолильных, N-метилимидазолильных, аминопуринильных фрагментов, фенантролильных фрагментов, пиридильных фрагментов, бипиридильных фрагментов,терпиридинильных фрагментов, триазациклононанонильных фрагментов, тетраазациклододеканильных фрагментов, фрагментов иминодиуксусной кислоты, фрагментов нитрилотриуксусной кислоты и фрагментов этилендиаминотетрауксусной кислоты. 12. Применение по любому из пп.7-11, при котором якорную часть пептидной цепи помещают на Сконце и/или по крайней мере в одной аминокислотной боковой цепи пептида. 13. Применение по п.12, при котором по крайней мере одну аминокислоту якорной части на Сконце пептида удлиняют на одну или более аминокислот, что делает возможным определение с помощью систем детектирования.- 18008305 14. Применение по любому из пп.7-13, при котором якорную часть пептида удлиняют на ее N-конце аминокислотной последовательностью, обеспечивающей участок узнавания для специфической протеазы. 15. Применение по любому из пп.7-14, при котором после отщепления пептид повторно присоединяют к активированной твердой фазе разбавлением реакционной смеси, содержащей конкурентный лиганд, согласно протоколу последовательных реакций типа протокола Меррифилда. 16. Применение по п.1, которое включает стадию рефолдинга неправильно скрученных структур и/или деагрегации межмолекулярных агрегатов необязательно защищенного пептида, при котором якорная часть пептида координационно и обратимо присоединена к активированной твердой фазе, и повторного создания правильно скрученной структуры пептида, включающее стадии:a) воздействия на пептид по крайней мере одного хаотропного или денатурирующего реагента иb) последующего воздействия ряда растворителей, чтобы постепенно уменьшить хаотропность и обеспечить воспроизводимые условия рефолдинга и повторного создания вторичной и третичной структуры. 17. Применение по п.16, при котором вторичную и третичную структуру пептида поддерживают ковалентными связями между реакционноспособными боковыми цепями указанного пептида путем обработки пептида подходящими реагентами, включающей образование указанных ковалентных связей до отщепления пептида от активированной твердой фазы. 18. Применение по п.17, при котором ковалентные связи представляют собой дисульфидные связи,амидные связи или стабильные ароматические или алифатические гидразоны. 19. Применение по пп.7, 16 и 17 для синтеза пептида с последовательностью где X - d-аланин и Ala - бета-аланин,включающее образование дисульфидной связи между остатками цистеина, в результате чего образуется где X и Аlа являются такими, как описано выше. 20. Способ твердофазного синтеза пептидов посредством нековалентного присоединения растущей пептидной цепи к активированной твердой фазе, при котором активный компонент соответствующей твердой фазы образован металл-хелатными комплексами со свободными координационными участками для нековалентного присоединения растущей пептидной цепи к активированной твердой фазе через хелатирующие группы, присутствующие на якорной части растущей пептидной цепи, при этом металлхелатные комплексы образованы комплексами между ионом металла и металл-хелатирующим лигандом,который непосредственно или через линкерную молекулу ковалентно связан с твердой фазой. 21. Способ по п.20, при котором полностью образованный комплекс металла для повторяющихся циклов синтеза в процессе синтеза пептидов включает твердую фазу, металл-хелатирующий лиганд, ион металла, который размещен между металл-хелатирующим лигандом и хелатирующими группами якорной части пептидной цепи, и хелатирующие группы, присутствующие в N- или С-концевых положениях и/или в боковых цепях моно- или олигомерных аминокислот, которые образуют якорную часть пептидной цепи, которую наращивают ступенчатым образом вследствие повторяющихся циклов синтеза. 22. Способ по любому одному из пп.20-21, при котором нековалентное и координационное присоединение иона металла к металл-хелатирующему лиганду характеризуется большей силой, чем сила присоединения к хелатирующим группам. 23. Способ по любому из пп.20-22, при котором структура комплекса с металлом состоит изa) металл-хелатирующих лигандов, которые ковалентно присоединены к твердой фазе и способны образовывать комплекс с ионами металлов через атомы N, О, Р и/или S,b) ионов металла (Me[n+]); n от 1 до 3, предпочтительно 2, которые образуют комплекс с металлхелатирующими лигандами, при этом остаются доступными свободные координационные участки,c) одиночных или олигомерных природных или неприродных аминокислот, содержащих природные или химически модифицированные боковые цепи или N-, или С-концевые модификации, к которым присоединены хелатирующие группы и которые, таким образом, способны образовывать комплекс со свободными координационными участками, предоставленными частично насыщенными комплексами металл-хелатирующих лигандов с ионами Меn+ на активированной твердой фазе, описанной в а) и b) и пп.1-4, при этом хелатирующие группы способны образовывать комплекс с ионами металлов через атомы N, О, Р и/или S и при этом по крайней мере одна, предпочтительно 1-3 хелатирующие группы, могут присутствовать в боковой цепи для того, чтобы образовать стабильную якорную часть для присоединения растущей пептидной цепи. 24. Способ по любому из пп.20-23, при котором твердая фаза характеризуется присутствием функциональных химических групп, которые выбирают из аминогрупп, групп, содержащих гетероциклический азот, карбоксигрупп, гидроксильных, тиоловых групп или других функциональных остатков, для- 19008305 которых, самих по себе, известны реакции связывания, таким образом, указанные функциональные химические группы и реакции связывания используют, чтобы ковалентно связать твердую фазу с металлхелатирующим лигандом по пп.20-23. 25. Способ по любому из пп.20-24, при котором металл-хелатирующие лиганды содержат функциональную группу, которая делает возможным химическое связывание с функциональными химическими группами на поверхности твердой фазы по п.24, и при котором те же металл-хелатирующие лиганды, а также хелатирующие группы аминокислотных боковых цепей по п.23 содержат одну или более, предпочтительно 1-3, функциональных групп, которые способны образовывать комплекс с ионами металлов по п.5 (Меn+) и которые выбирают из аминогрупп, групп, содержащих гетероциклический азот, азагрупп,карбоксигрупп, фрагментов, содержащих серу или фосфор. 26. Способ по любому из пп.20-25, при котором ионы металлов (Меn+) образуют комплекс с металлхелатирующими лигандами на твердой фазе; причем указанные ионы металлов выбирают из ионов Mn2+,Ni2+, Cu2+, Co2+ или Zn2+, Ca2+, Fe2+, Fe3+ или ионов лантанида. 27. Способ по любому из пп.20-26, при котором последовательные реакции используют, чтобы присоединить дополнительные моно- или олигомерные аминокислоты к С- или N-концу полностью образованного комплекса с металлом по п.21; при этом соответственно защищенные аминокислотные производные или олигомерные фрагменты, которые присоединяют в каждом цикле, можно легко выбрать или составить из любых природных или неприродных аминокислот. 28. Способ по любому из пп.20-27, при котором конкурентный хелатирующий реагент добавляют к смеси реагентов стадии конденсации протокола реакций типа протокола Меррифилда для того, чтобы конкурентно отщепить растущую пептидную цепь от твердой фазы во время указанной стадии; при этом подходящие конкурентные хелатирующие реагенты имеют приблизительно такое же сродство к свободным координационным участкам на активированной твердой фазе, как индивидуальные хелатирующие группы боковых цепей моно- или олигомерных аминокислот, используемых для присоединения растущей пептидной цепи к твердой фазе, причем конкурентные хелатирующие реагенты являются растворимыми в смеси реагентов стадии конденсации и не взаимодействуют или иным образом не влияют на ингредиенты смеси реагентов стадии конденсации. 29. Способ по п.28, при котором реакционную смесь стадии конденсации, содержащую конкурентный хелатирующий реагент, разбавляют перед следующими стадиями промывания для того, чтобы повторно присоединить моно- или олигомерные аминокислоты, образующие якорную часть пептидной цепи, к активированной твердой фазе перед последующими стадиями, такими как полоскание или промывание. 30. Способ по любому из пп.20-29, при котором отщепленный и деблокированный неочищенный продукт синтеза пептидов, несущий моно- и олигомерные аминокислоты с боковыми цепями или N-, или С-концевыми фрагментами с хелатирующими группами по п.23, способен образовывать комплекс металла с твердой фазой по любому из пп.20-26 и может быть далее обработан путем:a) очистки путем воздействия на раствор неочищенного продукта активированной твердой фазой по пп.20-26 в условиях, при которых происходит повторное присоединение требуемого продукта к твердой фазе, и путем отмывания примесей, таких как остатки защитных групп и поглотители или нежелательные побочные продукты, избытком растворителя при избирательном сохранении на активированной твердой фазе продукта, образовавшего комплекс, и далееb) удаления возможных нежелательных неправильно скрученных структур продукта и межмолекулярных агрегатов молекул продукта при воздействии на связанный продукт хаотропными или денатурирующими реагентами, например мочевиной, детергентами, такими как додецилсульфат натрия, высокими концентрациями солей, меркаптоэтанолом или другими реагентами, в результате чего связанный продукт переходит в денатурированное состояние, которое характеризуется разрушением вторичной и третичной структуры, тогда как связь с твердой фазой сохраняется. 31. Способ по п.30, при котором очищенный, связанный и денатурированный продукт подвергают воздействию ряда растворителей, причем ряд растворителей подобран и оптимизирован для соответствующего продукта, чтобы постепенно уменьшить хаотропность и создать воспроизводимые условия рефолдинга молекул связанного продукта, а также контролируемого повторного появления вторичной и третичной структуры при сохранении молекул продукта связанными с твердой фазой. 32. Способ по п.30 и/или 31, при котором ковалентные связи между боковыми цепями аминокислот,предпочтительно замыкание дисульфидных связей из свободных сульфгидрильных групп, образование амидных связей или образование стабильных ароматических или алифатических гидразонов осуществляют пропусканием смесей реагентов через повторно скрученный продукт, который связан с твердой фазой. 33. Способ по п.24, при котором твердую фазу выбирают из диоксида кремния, целлюлозы или из полимеров, предпочтительно из полистирольной смолы, поперечно-сшитых с дивинилбензолом, хлортритильной смолы, из модифицированных, предпочтительно карбоксилированных, частиц меламина, или модифицированного, предпочтительно карбоксилированного, полимерного носителя на основе поливинилового спирта.- 20008305 34. Способ по п.24 и/или 33, при котором матрикс смолы содержит ферромагнитные частицы, что позволяет применять технологии магнитной сепарации. 35. Способ по любому из пп.20-27, при котором моно- или олигомерные аминокислоты содержат имидазольные боковые цепи, предпочтительно не менее шести остатков гистидина, двух или более остатков гистидина, более предпочтительно 6-10 остатков гистидина. 36. Способ по пп.23 и 35, при котором используемые моно- или олигомерные аминокислоты удлиняют на N-конце короткими аминокислотными последовательностями, обеспечивающими участок узнавания для специфической протеазы. 37. Способ по любому из пп.23, 35 и/или 36, при котором модифицированные или немодифицированные (по п.35) моно- или олигомерные аминокислоты удлиняют на С-конце на одну или более аминокислот, что позволяет проводить определения системами детектирования, такими как биотинилированные аминокислоты, которые присоединяют, чтобы иметь возможность определять посредством авидинподобных взаимодействий. 38. Способ по любому из пп.28, и/или 29, и/или 35-37, при котором конкурентный хелатирующий реагент содержит структурные фрагменты, содержащие металл-хелатирующие фрагменты, имеющие электронные пары для координационных фрагментов, полученных из имидазолила, Nметилимидазолила, иминодиуксусной кислоты, нитрилотриуксусной кислоты, этилендиаминтетрауксусной кислоты, аминопурина, фенантролина, бипиридила, терпиридинила, триазациклононана или тетраазациклододекана. 39. Способ по любому из пп.20-26, при котором хелатирующий лиганд содержит структурные фрагменты, имеющие электронные пары для координационных связей, такие как трифенилфосфин, 6 аминопурин, фталоцианин. 40. Способ по п.39, при котором лиганд представляет собой 5-амино-1,10-фенантролин, или аминотерпиридин, или триазациклононан, или тетраазациклододекан, или их производные. 41. Способ по любому из пп.20-40, при котором хелатирующие группы боковых цепей аминокислот выбирают из имидазоло, аминофрагментов, гидроксильного, карбоксильного, тиолового фрагментов,фрагментов нитрилотриуксусной кислоты, фрагментов иминодиуксусной кислоты, фенантролинового фрагмента, пиридинового, бипиридинового, терпиридинового, триазациклононанового, тетраазациклододеканового или пуринового фрагментов или производных перечисленных фрагментов, которые способны образовывать комплексы с металлом по любому из пп.20-26. 42. Способ по любому из пп.20-41, который полностью автоматизирован, совместим с роботами для синтеза и при котором разделение жидкости и твердой фазы во время циклов синтеза осуществляют, по существу, известными способами, предпочтительно просеиванием, разделением по размеру частиц, центрифугированием или технологией магнитной сепарации.
МПК / Метки
МПК: C07K 1/22, C07K 1/04, C07K 14/55
Код ссылки
<a href="https://eas.patents.su/25-8305-sintez-peptidov.html" rel="bookmark" title="База патентов Евразийского Союза">Синтез пептидов</a>
Производные пептидов

Номер патента: 1000
Опубликовано: 28.08.2000
Авторы: Накано Масахару, Сакурада Синобу, Нукуи Ерико, Окаяма Тору, Хонго Томоко, Такесима Сатоко, Хонго Казуя, Огава Тадаси, Таке Нобухиро
МПК: A61K 38/06, A61P 29/00, C07K 5/087...
Метки: производные, пептидов
Формула / Реферат:
1. Соединение, имеющее формулу Y-L-Tyr-Q-NR1-CH(R2)-СО-Х где R1 представляет собой атом водорода; R2 представляет собой незамещенную бензильную группу; Q представляет собой D-Arg; Y представляет собой один атом водорода и C1-6-алкильную группу, или же Y представляет один атом водорода и группу формулы HN=C(R3), где R3 обозначает C1-6-алкильную группу; и Х представляет собой NR7-C(R8)(R9)(R10), где R7 обозначает C1-6-алкильную группу, R8...
Производные пептидов

Номер патента: 1001
Опубликовано: 28.08.2000
Авторы: Сакурада Синобу, Таке Нобухиро, Накано Масахару, Огава Тадаси, Такесима Сатоко, Окаяма Тору, Хонго Казуя, Хонго Томоко, Нукуи Ерико
МПК: A61P 29/00, A61K 38/06, C07K 5/087...
Метки: пептидов, производные
Формула / Реферат:
1. Соединение, представляемое следующей формулой: HN=C (R1)-X-Y-NH-CH(R2)-CO-Q, где R1 представляет собой C1-6-алкильную группу; R2 представляет арилзамещенную C1-6-алкильную группу (предпочтительно фенилзамещенный метил); Х представляет L-тирозин (L-Tyr); Y представляет, по меньшей мере, одну из следующих аминокислот: D-лизин, 2-амино-4-метилсульфинилмасляная кислота (=AMSB или MetO), 2-амино-3-гуанидинпропионовая кислота (=AGPR),...
Фармацевтические композиции пептидов, имеющих низкую растворимость в физиологической среде

Номер патента: 2227
Опубликовано: 28.02.2002
Автор: Ричардсон Питер
МПК: A61K 38/09
Метки: пептидов, низкую, имеющих, растворимость, среде, фармацевтические, композиции, физиологической
Формула / Реферат:
1. Фармацевтическая композиция, включающая: а) пептидный агонист ЛГРФ, пептидный антагонист ЛГРФ или пептид ГРФ в качестве активного ингредиента; б) никотинамид и с) физиологический водный раствор. 2. Фармацевтическая композиция по п.1, в которой активный ингредиент представляет пептидный антагонист ЛГРФ. 3. Фармацевтическая композиция по любому из предшествующих пунктов, в которой активный ингредиент представляет антид или человеческий ГРФ. 4....
Способ определения состояния организма посредством измерения пептидов

Номер патента: 1033
Опубликовано: 28.08.2000
Авторы: Шульц-Кнаппе Петер, Опитц Ханс-Георг, Форссманн Вольф-Георг, Шрадер Михель
МПК: G01N 33/68
Метки: пептидов, посредством, организма, состояния, определения, способ, измерения
Формула / Реферат:
1. Способ определения состояния организма, в соответствии с которым отбирают пробу исследуемого организма и определяют качественный и количественный состав низкомолекулярных пептидов в этой пробе, а о состоянии организма судят по результатам сравнения этого состава с эталоном. 2. Способ по п.1, отличающийся тем, что проба представляет собой ткань или жидкость организма, или сам организм, или его комбинации. 3. Способ по п.1 и/или 2,...
Способ превращения пептидов класса эхинокандина в их с4-гомотирозиновые монодезокси-аналоги

Номер патента: 2909
Опубликовано: 31.10.2002
Авторы: Яиванти Кения, Мукхопадхиаи Триптикумар, Кумар Ерра Котесвара Сатия Вийая
МПК: A61P 31/00, C07K 7/56, A61K 38/12...
Метки: способ, с4-гомотирозиновые, пептидов, превращения, эхинокандина, класса, монодезокси-аналоги
Формула / Реферат:
1. Способ превращения пептидов класса эхинокандина формулы в которой W, X, Y, Z, R и R' имеют значения, определенные ниже W Х Y 1. Эхинокандин В ОН ОН OH 2. Пневмокандин А0 ОН ОН OH 3. Пневмокандин A1 Н ОН ...
Предыдущий патент: 8 – замещённые имидазопиридины
Следующий патент: Конструкция сборной винтовой металлической сваи (анкера)
Случайный патент: Устройство для уменьшения ударной нагрузки на конвейерную ленту