Замещенные пиразолы, композиции их содержащие, способ получения и применение
Номер патента: 19454
Опубликовано: 31.03.2014
Авторы: Може Жак, Бьергард Кирстен, Додсон Марк, Наир Анил, Патек Марсель, Табар Мишель






















Формула / Реферат
1. Соединение формулы (I)
в которой:
1) Ar-L-A означает
где каждую группу X1, X2, Х3 и Х4 независимо выбирают из N и C-R'5, где R'5 имеет то же значение, что и R5;
2) А представляет собой фенил, возможно замещенный одним или несколькими заместителями, выбранными из трифторметила, галогена, метокси или метоксикарбонила; Ar представляет собой фенил;
3) L представляет собой NH-CO-NH или O-CO-NH;
4) R1 обозначает Н;
5) X представляет собой О или NH;
6) R3 представляет собой Н или метил;
7) R4a представляет собой Н или (С1-С4)алкил;
8) R4b представляет собой Н или (С1-С4)алкил;
9) R5 представляет собой Н или галоген;
указанное соединение находится в форме:
1) не хиральной; или
2) рацемической; или
3) обогащенной энантиомером,
и, возможно, преобразовано в фармацевтически приемлемую соль.
2. Соединение по п.1, отличающееся тем, что R4a и R4b означают Н.
3. Соединение по п.1, отличающееся тем, что R4a означает Н и R4b означает (С1-С4)алкил.
4. Соединение по п.1, отличающееся тем, что R4a означает (С1-С4)алкил и R4b означает Н.
5. Соединение по любому из пп.1-4, отличающееся тем, что R3 означает Н и X означает NH.
6. Соединение по любому из пп.1-4, отличающееся тем, что R3 означает метил и X означает О.
7. Соединение по любому из пп.1-6, отличающееся тем, что R5 означает Н.
8. Соединение по любому из пп.1-7, отличающееся тем, что L означает NHCONH.
9. Соединение по п.1, отличающееся тем, что оно представляет собой
трифторацетат 4-{[({[3-фенил]карбамоил}окси)бензил]амино}-1Н-пиразол-3-карбоксамида;
хлоргидрат 4-{[3-({[2-фтор-5-(трифторметил)фенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамида;
трифторацетат 4-{[3-({[2-фтор-5-(трифторметил)фенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамида;
трифторацетат 4-[(3-{[(2-фторфенил)карбамоил]амино}бензил)амино]-1Н-пиразол-3-карбоксамида;
трифторацетат 4-[(3-{[(2-метоксифенил)карбамоил]амино}бензил)амино]-1Н-пиразол-3-карбоксамида;
трифторацетат 4-{[3-({[2-фтор-3-(трифторметил)фенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамида;
трифторацетат 4-[(3-{[(3-метоксифенил)карбамоил]амино}бензил)амино]-1Н-пиразол-3-карбоксамида;
трифторацетат 4-{[3-({[3-фтор-5-(трифторметил)фенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамида;
трифторацетат метил 3-{[(3-{[(3-карбамоил-1Н-пиразол-4-ил)амино]метил}фенил)карбамоил]амино}бензоата;
трифторацетат 4-{[3-({[4-(трифторметил)фенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамида;
трифторацетат 4-{[3-({[3-(трифторметил)фенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамида;
трифторацетат 4-{[3-({[2-(трифторметил)фенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамида;
трифторацетат 4-[(3-{[(3,5-диметоксифенил)карбамоил]амино}бензил)амино]-1Н-пиразол-3-карбоксамида;
трифторацетат 4-[(3-{[(4-метоксифенил)карбамоил]амино}бензил)амино]-1Н-пиразол-3-карбоксамида;
трифторацетат 4-[(3-{[(4-фторфенил)карбамоил]амино}бензил)амино]-1Н-пиразол-3-карбоксамида;
трифторацетат 4-{[3-({[4-хлор-3-(трифторметил)фенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамида;
трифторацетат 4-{[3-({[2-хлор-4-(трифторметил)фенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамида;
трифторацетат 4-{[3-({[(3,5-бис-(трифторметил)фенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамида;
трифторацетат 4-[(3-{[(3-фторфенил)карбамоил]амино}бензил)амино]-1Н-пиразол-3-карбоксамида;
трифторацетат 4-[(3-{[(2,5-диметоксифенил)карбамоил]амино}бензил)амино]-1Н-пиразол-3-карбоксамида;
трифторацетат 4-[(3-{[(2,5-дифторфенил)карбамоил]амино}бензил)амино]-1Н-пиразол-3-карбоксамида;
трифторацетат 4-{[3-({[2-фтор-5-(трифторметил)фенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-метилкарбоксилат.
10. Соединение по п.1, отличающееся тем, что оно представляет собой
трифторацетат 4-(1-{3-[3-(2-фтор-5-трифторметилфенил)уреидо]фенилэтиламино}-1Н-пиразол-3-карбоксамида;
4-({3-[3-(2-фтор-5-трифторметилфенил)уреидо]бензил}метиламино)-1Н-пиразол-3-карбоксамид;
4-(этил-{3-[3-(2-фтор-5-трифторметилфенил)уреидо]бензил}амино)-1Н-пиразол-3-карбоксамид;
4-метил-({3-[3-(2-хлор-5-трифторметилфенил)уреидо]бензил}амино)-1Н-пиразол-3-карбоксамид;
4-{[3-({[3-хлор-4-фторфенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамид;
4-{[3-({[3,4-дихлорфенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамид;
4-{[3-({[3-хлор-5-трифторметилфенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамид;
4-{[3-({[3-трифторметил-4-хлорфенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамид;
4-{[3-({[2-хлор-5-трифторметилфенил]карбамоил}амино)бензил]амино}-1Н-пиразол-3-карбоксамид.
11. Фармацевтическая композиция для лечения рака, содержащая соединение по любому из пп.1-10 в комбинации с фармацевтически приемлемым реципиентом.
12. Применение соединения по любому из пп.1-10 в качестве ингибирующего агента одной или нескольких реакций, катализируемых киназой.
13. Применение соединения по п.12, при котором киназу выбирают из KDR и Tie2.
14. Применение соединения по любому из пп.1-10 в производстве лекарственного средства для лечения рака.
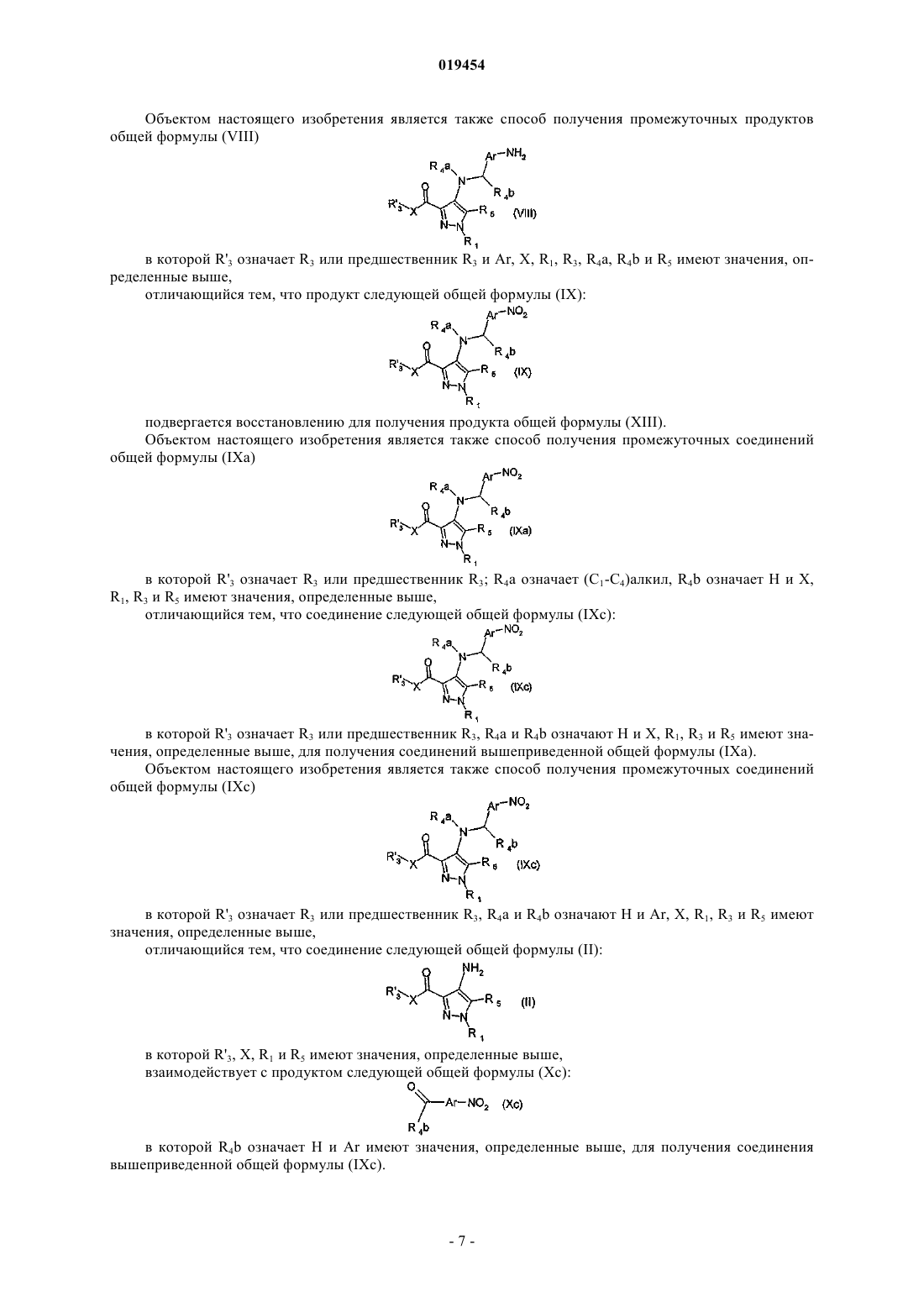
15. Способ получения соединений общей формулы (Ib)
в которой R1, R3, R4b, R5, X, Ar, L и А имеют значения, определенные в п.1, и R4a означает Н,
отличающийся тем, что соединение общей формулы (II)
в которой R'3 означает R3 и X, R1, R3 и R5 имеют значения, определенные в п.1,
взаимодействует с соединением формулы (III)
в которой R4b, Ar, L и А имеют значения, определенные в п.1, для получения соединения общей формулы (Ib).
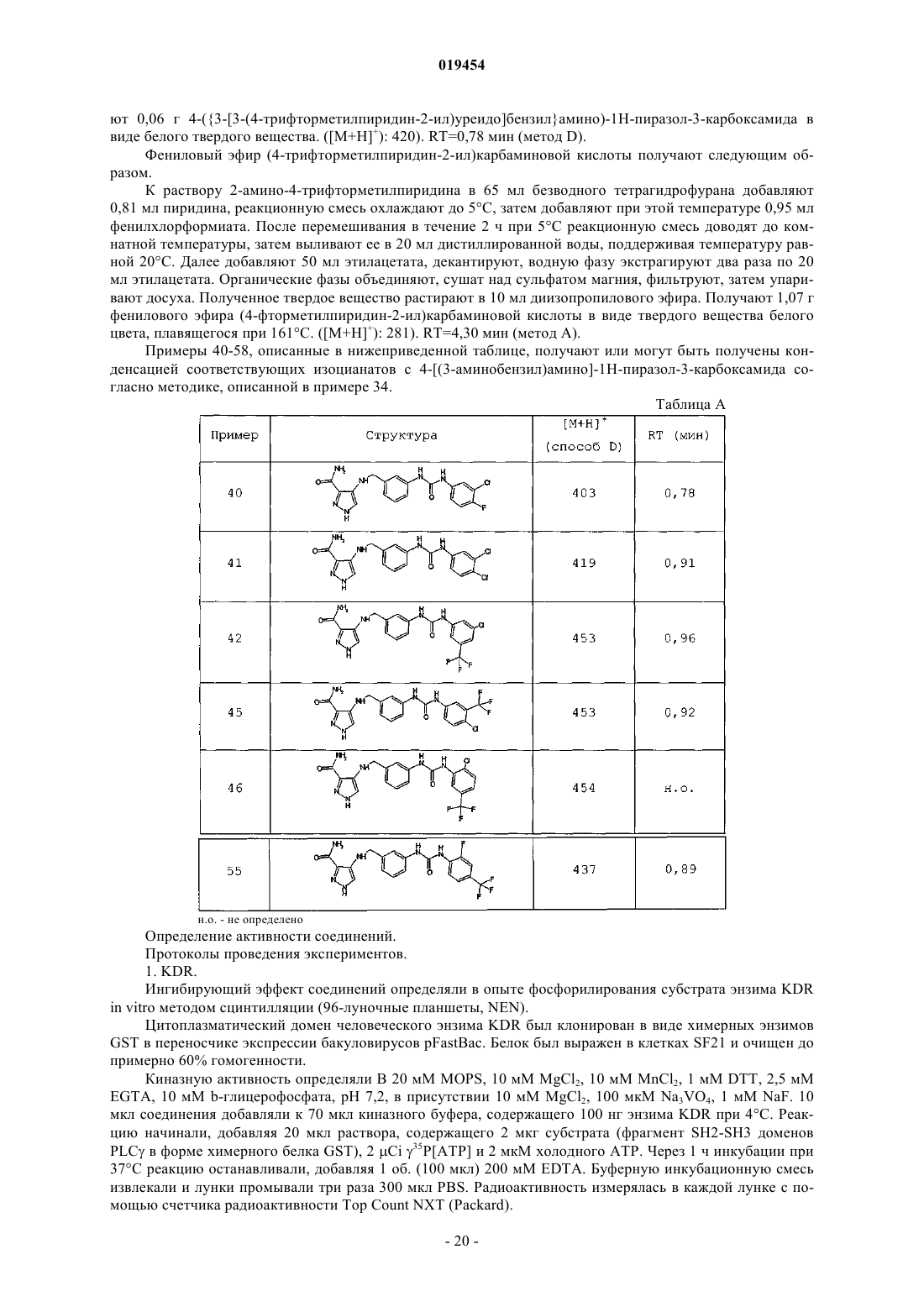
16. Способ получения соединений следующей общей формулы (I):
в которой R1, R3, R4a, R4b, R5, X, Ar и А имеют значения, определенные в п.1, и L означает NHCONH,
отличающийся тем, что соединение общей формулы (VIII)
в которой R'3 означает R3 и X, Ar, R1, R3, R4a, R4b и R5 имеют значения, определенные в п.1,
взаимодействует с соединением общей формулы (VII)
в которой А имеет значение, определенное в п.1, для получения соединения общей формулы (I)
17. Соединения общей формулы (VIII) в качестве промежуточных соединений, где Ar, R3, X, R1, R4a, R4b и R5 имеют значения, определенные в п.16.
Текст
Изобретение относится к замещенным пиразолам общей формулы (I), композициям, их содержащим, и их применению в качестве лекарственных средств для лечения рака. Настоящее изобретение относится особенно к новым химическим соединениям, в частности к замещенным пиразолам, композициям их содержащим, и их применению в качестве лекарственных средств. Более конкретно и согласно первому аспекту настоящее изобретение относится к новым специфическим замещенным пиразолам, обладающим противораковой активностью через модуляцию активности протеинов, в частности протеина киназ. Протеины киназ входят в семейство энзимов, катализирующих фосфорилирование гидроксильных групп специфических остатков протеинов, таких как остатки тирозина, серина или треонина. Такие реакции фосфорилирования могут в широком смысле модифицировать функцию протеинов; так, протеины киназ играют важную роль в регулировании большого разнообразия клеточных процессов, включающих,в частности, метаболизм, клеточную пролиферацию, дифференциацию клеток, миграцию клеток или выживаемость клеток. Среди различных клеточных функций, в которые вовлечена активность протеина киназы, некоторые процессы представляются привлекательными для лечения раковых заболеваний, а также других болезней. Таким образом, одним из объектов настоящего изобретения является предложение композиций, обладающих противораковой активностью, в особенности при воздействии на киназы. Среди киназ, в отношении которых исследуется модуляция активности, можно назвать KDR, Tie2, VEGFR-1, PDGFR,FGFR, FLT1. Киназы KDR и Tie2 являются предпочтительными. Упомянутые соединения отвечают следующей общей формуле (I): где каждую группу X1, X2, X3 и Х 4 независимо выбирают из N и C-R'5, где R'5 имеет то же значение,что и R5; 2) А выбирают из фенила, возможно, замещенного одним или несколькими заместителями, выбранными из группы, состоящей из трифторметила, галогена, метокси и метоксикарбонила; Ar представляет собой фенил; 3) L выбирают из группы, состоящей из NH-CO-NH и O-CO-NH; 4) R1 обозначает Н; 5) X выбирают из группы, состоящей из О и NH; 6) R3 выбирают из группы, состоящей из Н, метила; 7) R4a выбирают из группы, состоящей из Н или (С 1-С 4)алкила; 8) R4b выбирают из группы, состоящей из Н или (С 1-С 4)алкила; 9) R5 выбирают из группы, состоящей из H и галогена. Когда R1 представляет собой Н, две таутомерные формы, представленные ниже, принадлежат к настоящему изобретениюR3 преимущественно означает метил и X преимущественно означает О. В продуктах формулы (I) R5 преимущественно представляет собой Н. Заместитель Ar согласно изобретению может быть выбран из фенила, пиридила и пирролила, замещенного R'5, где R'5 имеет то же значение, что и R5. где каждая из групп X1, X2, Х 3 и Х 4 независимо выбрана из N и C-R'5, где R'5 имеет то же значение,что и R5. Более конкретно, группа R'5 может быть выбрана из группы, состоящей из Н, F и Cl. Заместитель Ar преимущественно означает фенил, где R'5 является Н. Заместитель А по изобретению представляет собой фенил, возможно, замещенный. Из соединений формулы (I), объектов настоящего изобретения, можно назвать, в частности, первую группу продуктов, для которых R4a и R4b представляют собой Н и А, Ar, L, R1, X, R3 и R5 имеют значение, определенное выше. Из соединений первой группы можно, в частности, назвать первую подгруппу соединений, для которых: 1) A и Ar означают фенилы, возможно замещенные; 2) L выбирают из группы, состоящей из NH-CO-NH и O-CO-NH; 3) X означает NH и R3 означает Н или X означает О и R3 означает метил; 4) R1, R4a, R4b и R5 означают Н. Из этой первой подгруппы можно также назвать подгруппу соединений, для которых: 1) A и Ar означают фенилы, возможно замещенные; 2) L означает NH-CO-NH; 3) X означает NH и R3 означает Н; 4) R1, R4a, R4b и R5 означают Н. Из соединений формулы (I), объектов настоящего изобретения, можно, в частности, назвать вторую группу соединений, для которых R4a означает Н, R4b означает (C1-C4)алкил и А, Ar, L, R1, X, R3 и R5 имеют значения, определенные выше. Из соединений второй группы можно, в частности, назвать первую подгруппу соединений, для которых: 1) A и Ar означают фенилы, возможно замещенные; 2) L означает NH-CO-NH; 3) X означает NH и R3 означает Н; 4) R1, R4a и R5 означают Н; 5) R4b означает метил. Из соединений формулы (I), объектов настоящего изобретения, можно, в частности, назвать третью группу соединений, для которых R4a означает (C1-C4)алкил, R4b означает Н и A, Ar, L, R1, X, R3 и R5 имеют значения, определенные выше. Из соединений третьей группы можно, в частности, назвать первую подгруппу соединений, для которых: 1) A и Ar означают фенилы, возможно замещенные; 2) L означает NH-CO-NH; 3) X означает NH и R3 означает Н; 4) R1, R4b и R5 означают Н; 5) R4a означает метил. Из соединений третьей группы можно, в частности, назвать вторую подгруппу соединений, для которых: 1) A и Ar означают собой фенилы, возможно замещенные; 2) L означает NH-CO-NH; 3) X означает NH и R3 означает Н; 4) R1, R4b и R5 означают Н; 5) R4a означает этил. Группа А может быть выбрана из фенила, замещенного по меньшей мере одной группой, выбранной из трифторметила, галогена, метокси и метоксикарбонила. Настоящее изобретение содержит также объекты, соответствующие комбинациям вышеназванных подгрупп. Соединения по изобретению могут быть в форме: 1) нехиральной; или 2) рацемической; или 3) обогащенной стереоизомером; или 4) обогащенной энантиомером; и быть, возможно, в форме соли. Настоящее изобретение относится также к фармацевтическим композициям, содержащим соедине-2 019454 ние по изобретению, в сочетании с фармацевтически приемлемым эксципиентом в соответствии с выбранным способом введения. Фармацевтическая композиция может быть представлена в твердом, жидком виде или в виде липосом. Среди твердых композиций можно назвать порошки, желатиновые капсулы, таблетки. К оральным формам можно также присоединить твердые формы, защищенные от кислой среды желудка. Основы,используемые для твердых форм, состоят, в частности, из минеральных основ, таких как фосфаты, карбонаты, или органических основ, таких как лактоза, целлюлозы, амидон или полимеры. Жидкие формы состоят из растворов, суспензий или дисперсий. В качестве диспергирующей основы они содержат воду либо органический растворитель (этанол, NMP или другие) или смеси поверхностно-активных веществ и растворителей либо комплексообразующих веществ и растворителей. Жидкие формы предпочтительно пригодны для инъекции и поэтому имеют рецептурный состав,подходящий для такого применения. Подходящие способы введения путем инъекции включают внутривенный, внутрибрюшной, внутримышечный и подкожный способы, причем внутривенный способ является предпочтительным. Вводимые дозы веществ согласно изобретению подбирает врач в зависимости от способа введения пациенту и от состояния последнего. Соединения по изобретению применимы в качестве ингибиторов одной или нескольких реакций,катализируемых киназой. KDR и/или Tie2 являются киназами, для которых соединения по изобретению особенно подходят в качестве ингибиторов. Причины, по которым выбирают указанные киназы, приведены ниже.Growth Factor Receptor 2), в основном выражен в эндотелиальных клетках. Этот рецептор связывает проангиогенный фактор VEGF и служит, таким образом, медиатором трансдукционного сигнала через активацию межклеточного домена киназы. Прямое ингибирование активности киназы VEGF-R2 позволяет ослабить явление ангиогенеза в присутствии экзогенного фактора VEGF (Vascular Endothelial GrowthFactor: сосудистый эндотелиальный фактор роста) (Strawn et al., Cancer Research, 1996, vol. 56, p. 35403545). Данный механизм был подтвержден с помощью мутантов VEGF-R2 (Millauer et al., Cancer Research, 1996, vol. 56, p. 1615-1620). По-видимому, рецептор VEGF-R2 не имеет другой функции у взрослых, чем та, которая связана с ангиогенной активностью VEGF. Недавно полученные результаты привели к мысли о том, что кроме этой основной функции в динамическом ангиогенном процессе экспрессияVEGF способствует выживанию опухолевых клеток после химио- и радиотерапии, при этом подчеркивается потенциальная синергия ингибиторов KDR с другими агентами (Lee et al. Cancer Research, 2000, vol. 60, p. 5565-5570).Tie2 (Tek) является членом семейства рецепторов тирозин-киназы, специфичного для эндотелиальных клеток. Tie2 является первым рецептором с активностью тирозин-киназы, которая известна одновременно как агонист (ангиопоэтин 1 или Ang1), который стимулирует самофосфорилирование рецептора и клеточную сигнализацию [S. Davis et al. (1996) Cell, 87, 1161-1169] и как антагонист (ангиопоэтин 2 или Ang2) [Р.С. Maisonpierre et al. (1997) Science, 277, 55-60]. Ангиопоэтин 1 может проявлять синергический эффект с VEGF на последних стадиях нео-ангиогенеза [Asahara T. Circ. Res. (1998) 233-240]. Опыты knock-out и трансгенные работы с экспрессией Tie2 или Ang1 проводились на животных с повреждениями сосудов [D.J. Dumont et al. (1994) Genes Dev., 8, 1897-1909, C. Suri (1996) Cell, 87, 1171-1180]. Связь Ang1 со своим рецептором приводит к самофосфорилированию домена киназы Tie2, что существенно для неоваскуляризации, а также для рекрутирования и взаимодействия сосудов с перицитами и гладкомышечными клетками; эти явления способствуют созреванию и стабильности вновь образовавшихся сосудов [Р.С. Maisonpierre et al. (1997) Science, 277, 55-60], Lin et al. (1997) J. Clin. Invest., 100, 8,2072-2078 и Lin P. (1998) PNAS, 95, 8829-8834, продемонстрировали ингибирование роста и опухолевую васкуляризацию так же, как уменьшение метастаз легких при аденовирусном заражении или впрыскиваниях внеклеточного домена Tie2 (Tek) в модели ксенотрансплантатов опухоли груди и меланомы. По нижеследующим причинам ингибиторы Tie2 могут быть использованы в ситуациях, когда неоваскуляризация, или ангиогенез, происходит несоответствующим образом, т.е., как правило, в злокачественных опухолях, а также в особых случаях рака, такого как саркома Капоши или ранняя гемоангиома,ревматоидный артрит, остеоартрит и/или ассоциированные боли, при болезнях воспалительного характера кишечника, таких как геморрогический ректоколит или болезнь Крона, при патологиях глаза, таких как возрастная макулодистрофия, диабетические ретинопатии, при хроническом воспалении, псориазе. Ангиогенез является процессом зарождения новых капиллярных сосудов из уже существующих. Опухолевый ангиогенез (образование новых кровеносных сосудов), ответственных за рост опухоли, является также одним из существенных факторов метастатической диссеминации (Oncongene, 2003, May 19, 22 (20):3172-9, Nat Med. 1995 Jan., 1(1):27-31.). Такая неоваскуляризация происходит за счет миграции, затем пролиферации и дифференциации эндотелиальных клеток под действием ангиогенных факторов, секретируемых раковыми клетками и клетками стромы (Recent Prog Horm Res., 2000, 55, 15-35, 35-6). Система ангиопоэтин 1/рецептор Tie2 играет решающую роль в созревании сосудов, допуская рекрутирование пери-эндотелиальных клеток для стабилизации сосуда (Cell., 1996, Dec., 27, 87(7):1161-9,Recent Prog Horm Res., 2004, 59:51-71). Таким образом, было показано, что введение рекомбинантной растворимой формы внеклеточного домена рецептора Tie2 (exTek) ингибирует опухолевый ангиогенез в муриновых опухолевых моделях, а также метастатическое развитие (Proc Natl Acad Sci USA, 1998, Jul. 21, 95(15), 8829-34; Cancer immunol Immunother, 2004, Jul., 53(7):600-8). В культуре эндотелиальных клеток стимуляция Tie2 активирует путь PI3 киназы, р 42/р 44 пути, участвующие в пролиферации и клеточной миграции; синтез PAF (Cell Signal., 2006, Apr. 14; ahead of print) путь, включенный в провоспалительную активность. Стимуляция Tie2 усиливает путь Akt и ингибирует апоптоз (Exp Cell Res., 2004,Aug. 1, 298(1):167-77) путь трансдукции, известный своим значением для клеточной выживаемости. Добавление Extek (растворимый рецептор Tie2) ингибирует образование псевдоканальцев эндотелиальных клеток на Matrigel (Cancer Immunol. Immunother., 2004, Jul., 53(7):600-8). Данные исследования указывают на то, что система Tie2/ангиопоэтин необходима на первых стадиях образования васкулярных почек в зрелых тканях и что функция рецептора Tie2 состоит в возрастании выживаемости эндотелиальных клеток в процессе образования кровеносных сосудов. Кроме того, ангиопоэтин-1 стимулирует пролиферацию лимфатических эндотелиальных клеток так же, как лимфангиогенез (развитие лимфатических неососудов) - особенно подходящий путь для метастатического развития (Blood., 2005, Jun. 15,105(12):4649-56). Процессы ангиогенеза играют, таким образом, решающую роль в росте многочисленных солидных опухолей. Кроме того, было показано, что вероятность появления метастаз очень сильно увеличивается при увеличении васкуляризации первичной опухоли (Br J. Cancer., 2002, May 20, 86(10):1566-77). В более ранних работах была также установлена потенциальная роль про-ангиогенных агентов в лейкемиях и лимфомах. Действительно, в общем виде было сообщено, что клеточные клоны в этих патологиях могут быть либо разрушены естественным образом иммунной системой, либо переведены в ангиогенный фенотип, который способствует их выживаемости и затем их пролиферации. Такое изменение фенотипа вызвано экспрессией ангиогенных факторов, в частности макрофагами и/или мобилизацией этих факторов из внеклеточной матрицы (Thomas D.A., Giles F.J., Cortes J., Albitar M., Kantarjian H.M.,Acta Haematol. (2001), vol. 207, p. 106-190). Существует связь между процессом ангиогенеза костного мозга и "экстрамедуллярной болезнью" вCML (chronic myelomonocytic leukemia). В различных исследовательских работах показано, что ингибирование ангиогенеза могло бы быть представлено как лучшее лечение против этой патологии (Leuk Res.,2006, Jan., 30(1):54-9; Histol Histopathol., 2004, oct., 19(4):1245-60. Кроме того, убедительно показано, что активация системы Tie2/ангилпоэтин вовлечена в развитие ангиогенеза костного мозга у пациентов, пораженных множественной миеломой (Blood., 2003, Jul. 15, 102(2):638-45). Ревматоидный артрит (RA) является болезнью с неизвестной этиологией. Притом что он затрагивает много органов, наиболее тяжелой формой RA является прогрессивное синовиальное воспаление суставов, ведущее к деструкции. По-видимому, ангиогенез сильно влияет на развитие этой патологии. Таким образом, было показано, что активация Tie2 регулирует ангиогенез в синовиальных тканях, способствуя развитию ревматоидного артрита (Arthritis Rheum., 2003, Sep., 48(9):2461-71). Была также показана экспрессия ангиопоэтина 1 и Tie2 в синовиальных тканях пациентов, пораженных остеоартритом, связанным с активной неоваскуляризацией (Shahrara S. et al. Arthritis Res., 2002,4(3. Таким образом, показано, что блокируя активацию Tie2 путем использования аденовируса, вырабатываемого exTEK (растворимый рецептор Tie2), добиваются ингибирования ангиогенеза, развития артроза и защиты от разрушения костей в модели мыши, у которой артроз вызывался коллагеном (ArthritisRheum., 2005, May, 52(5):1346-8). ВЗК (воспалительное заболевание кишечника) IBD имеет две формы хронических воспалительных заболеваний кишечника: язвенный колит UC (ulcerative colitis) и болезнь Крона (CD). ВЗК характеризуются иммунитетной дисфункцией, которая выражается в выработке несоответствующих вызывающих воспаление цитокинов, стимулирующих образование локальной микроваскулярной системы. Следствием такого ангиогенеза воспалительного характера является интестинальная ишемия, вызванная сужением сосудов (Inflamm Bowel Dis., 2006, Jun., 12(6):515-23). Патологии глаза в связи с явлениями неоваскуляризации, такие как возрастная макулярная дегенерация, являются основной причиной большинства случаев слепоты в индустриально развитых странах. Сигналы молекул, контролирующих явления неоваскуляризации в глазе, таких как VEGF или ангиопоэтины, являются лучшей мишенью в таких патологиях (Campochiaro P.A. Expert Opin Biol Ther., 2004,Sep., 4(9. Таким образом, показано, что блокируя активацию Tie2 путем использования аденовируса,вырабатывающего exTEK (растворимый рецептор Tie2), ингибировали ретинальную хороидную неоваскуляризацию, которая чаще всего является причиной потери зрения (Hum Gene Ther., 2001, Jul. 1,12(10):1311-21). Продукт по изобретению можно использовать для производства лекарственного средства, подходящего для лечения патологического состояния, в частности рака. Благодаря слабой токсичности и фармакологическим и биологическим свойствам соединения согласно настоящему изобретению находят свое применение для лечения любой карциномы со значительной степенью васкуляризации или индуцирующей метастазы или, наконец, для лечения патологий типа лимфомы и лейкемии. Упомянутые соединения предоставляют лечение на выбор либо отдельно, либо в сочетании с соответствующей химиотерапией или радиотерапией и/или в сочетании с другими соединениями, обладающими противоангиогенной активностью, такими как ингибиторы VEGF или FGF. Так, продукты общей формулы (I) особенно подходят для лечения и предотвращения патологического состояния, отличающегося тем, что продукт вводят отдельно или в сочетании с другими действующими веществами, а именно противораковыми, такими как цитотоксические, цитостатические, противоангиогенные или противометастазные вещества. Так, соединения по настоящему изобретению могут быть введены отдельно или в смеси с другими противораковыми соединениями. Среди возможных сочетаний можно назвать алкилирующие агенты и, в частности, циклофосфамид, мелфалан, ифосфамид, хлорамбуцил, бусулфан, тиотепа, преднимустин, кармустин, ломустин, семустин, стептозотоцин, декарбазин, темозоломид, прокарбазин и гексаметилмеламин; производные платины, такие как, в частности, цисплатин, карбоплатин или оксалиплатин; антибиотики, такие как, в частности, блеомицин, митомицин, диактиномицин; агенты для разрушения микроканальцев, такие как, в частности, винбластин, винкристин, виндезин,винорелбин, таксоиды (паклитаксел и доцетаксел); антрациклины, такие как, в частности, доксорубицин, даунорубицин, идарубицин, эпирубицин, митоксантрон, лозоксантрон; ингибиторы топоизомераз групп I и II, такие как этопозид, тенипозид, амсакрин, иринотекан, топотекан и томудекс; фторпиримидины, такие как 5-фторурацил, UFT, флоксуридин; аналоги цитидина, такие как 5-азацитидин, цитарабин, гемцитарадин, 6-меркаптопурин, 6 тиогуанин; аналоги аденозина, такие как пентостатин, цитарабин или флударабина фосфат; метотрексат и фолиновая кислота; энзимы и различные соединения, такие как L-аспарагиназа, гидроксимочевина, транс-ретиноевая кислота, сурамин, дексразоксан, амифостин, герцептин, а также эстрогенные и андрогенные гормоны; антиваскулярные агенты, такие как производные комбретастатина, например СА 4 Р, халконы или колфицин, например ZD6126, и их пролекарственные соединения; антиангиогенные агенты, такие как бевацизумаб, сорафениб или сунитиниба малат; терапевтические агенты ингибирования других тирозинкиназ, такие как иматиниб, гефитиниб и эрлотиниб. Если соединения по настоящему изобретению применяются в сочетании с другим лечением или с лечением радиацией, то такие виды лечения могут осуществляться одновременно, раздельно, последовательно. Лечение будет выбрано лечащим врачом в зависимости от болезни. Определения. Термин "галоген" относится к элементу, выбранному из F, Cl, Br и I. Термин "алкил" относится к насыщенному углеводородному заместителю, линейному или разветвленному, содержащему от 1 до 6 атомов углерода. Заместители метил, этил, пропил, 1-метилэтил, бутил,1-метилпропил, 2-метилпропил, 1,1-диметилэтил, пентил,1-метилбутил, 2-метилбутил, 3-метилбутил,1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, гексил, 1-метилпентил, 2 метилпентил, 1-этилбутил, 2-этилбутил, 3,3-диметилбутил являются примерами алкильного заместителя. Объектом настоящего изобретения является получение соединений следующей общей формулы (I): в которой R1, R3, R4b, R5, X, Ar, L и А имеют значения, определенные выше,отличающееся тем, что соединение следующей общей формулы (II): в которой R'3 означает R3 или предшественник R3 и X, R1, R3 и R5 имеют значения, определенные выше,взаимодействует с соединением следующей общей формулы (III): в которой R4b, Ar, L и А имеют значения, определенные выше, для получения соединения общей формулы (I). Кроме того, объектом настоящего изобретения являются продукты общей формулы (II) в качестве промежуточных продуктов, для которых R'3, X, R1 и R5 имеют значения, определенные выше, а также продукты общей формулы (III), для которых Ar, L и А имеют значения, определенные выше. Объектом настоящего изобретения является также способ получения промежуточных продуктов следующей общей формулы (II): в которой R'3, X, R1 и R5 имеют значения, определенные выше,отличающийся тем, что соединение следующей общей формулы (IV): в которой R1 и R5 имеют значения, определенные выше,взаимодействует с соединением следующей общей формулы (V): в которой Gp означает защитную группу; X и R'3 имеют значения, определенные выше, для получения соединения общей формулы (II). Объектом настоящего изобретения является также получение продуктов общей формулы (I)NHCONH,отличающееся тем, что соединение следующей общей формулы (VIII): в которой R'3 означает R3 или предшественник R3 и X, R1, R3, R4a, R4b и R5 имеют значения, определенные выше,взаимодействует с соединением следующей формулы (VII): в которой А имеют значения, определенные выше, для получения соединения следующей общей формулы (I'): в которой предшественник R'3 преобразуется в R3 для получения соединения общей формулы (I). Объектом настоящего изобретения является также способ получения промежуточных продуктов общей формулы (VIII) в которой R'3 означает R3 или предшественник R3 и Ar, X, R1, R3, R4a, R4b и R5 имеют значения, определенные выше,отличающийся тем, что продукт следующей общей формулы (IX): подвергается восстановлению для получения продукта общей формулы (XIII). Объектом настоящего изобретения является также способ получения промежуточных соединений общей формулы (IXa) в которой R'3 означает R3 или предшественник R3; R4a означает (С 1-С 4)алкил, R4b означает Н и X,R1, R3 и R5 имеют значения, определенные выше,отличающийся тем, что соединение следующей общей формулы (IXc): в которой R'3 означает R3 или предшественник R3, R4a и R4b означают Н и X, R1, R3 и R5 имеют значения, определенные выше, для получения соединений вышеприведенной общей формулы (IXa). Объектом настоящего изобретения является также способ получения промежуточных соединений общей формулы (IXc) в которой R'3 означает R3 или предшественник R3, R4a и R4b означают Н и Ar, X, R1, R3 и R5 имеют значения, определенные выше,отличающийся тем, что соединение следующей общей формулы (II): в которой R'3, X, R1 и R5 имеют значения, определенные выше,взаимодействует с продуктом следующей общей формулы (Хс): в которой R4b означает Н и Ar имеют значения, определенные выше, для получения соединения вышеприведенной общей формулы (IXc). Объектом настоящего изобретения является также способ получения промежуточных соединений общей формулы (IXb) в которой R'3 означает R3 или предшественник R3, R4a означает Н, R4b означает (С 1-С 4)алкил и Ar,X, R1, R3 и R5 имеют значения, определенные выше,отличающийся тем, что соединение следующей общей формулы (II): в которой R'3, X, R1 и R5 имеют значения, определенные выше, взаимодействует с соединением следующей общей формулы (Xb): в которой R4b означает (С 1-С 4)алкил и Ar имеют значения, определенные выше, подвергается затем восстановлению для получения соединения вышеприведенной общей формулы (IXb). Используемые исходные соединения коммерчески доступны или получаются способами, известными специалисту в данной области. Под защитной группой Gp подразумевается группа, позволяющая, с одной стороны, защитить такую реакционноспособную функцию, как ОН или амин во время синтеза, и, с другой стороны, регенерировать реакционноспособную функцию в конце синтеза. Примеры защитных групп, а также способы защиты и восстановления приведены в "Protective Groups in Organic Synthesis", Green et al., 2nd Edition (JohnWileySons, Inc., New York). Под предшественником R3 подразумевается группа, позволяющая в конце реакции, или синтеза,получить группу R3. Речь идет, например, о группе -СН 2-смола Ринка, когда X означает NH, или же о группе 2,4-диметоксибензил. Соединения по изобретению могут быть получены классическими способами органической химии. Нижеприведенная схема 1 иллюстрирует способ, используемый для получения примеров 1-31. Поэтому она не ограничивает рамки настоящего изобретения в отношении способов получения заявляемых соединений. Схема 1 1. Получение промежуточной смолы (ii). Смола Rink (v) набухает в ДМФ. ДМФ отфильтровывают, затем заменяют на 50%-ный раствор пиперидина в ДМФ. Через 1/2 ч перемешивания при комнатной температуре смесь фильтруют, затем смолу последовательно промывают ДМФ, метанолом и ДМФ. Затем раствор 3 экв. 4-нитро-3-пиразолкарбоновой кислоты (iv), 3 экв. HOBt (гидроксибензотриазол) и 3 экв. диизопропилкарбодиимида (DIC) в ДМФ добавляют в смолу. Смесь перемешивают в течение одной ночи при комнатной температуре, затем реакционную смесь отделяют фильтрацией. Смолу промывают 3 раза ДМФ, 2 раза метанолом и 5 раз ДМФ. Затем смолу обрабатывают молярным раствором хлорида олова (SnCl2) в течение одной ночи при комнатной температуре. Реакционную смесь отделяют фильтрацией. Смолу промывают 5 раз ДМФ, 2 раза метанолом и 3 раза ДХМ (дихлорметан) и два раза эфиром, затем сушат в вакууме, получают смолу (ii). 2. Получение продуктов (i') восстановительным аминированием. Смола (ii) набухает в дихлорэтане (ДХЭ) в емкости. 3 экв. раствора альдегида (iii) в ДМФ добавляют к 5 экв. цианборгидрида натрия в метаноле, содержащего 10% уксусной кислоты. Смесь нагревают при 80 С в течение 2 ч. После охлаждения реакционную среду фильтруют и смолу последовательно промывают 2 раза метанолом, 3 раза ДХМ (дихлорметан), 2 раза метанолом и 3 раза ДХМ. Конечные продукты получают разделением раствором трифторуксусная кислота/ДХМ (50/50) при комнатной темпера-8 019454 туре в течение 2 ч. Продукт отделяют фильтрацией и выпариванием растворителя. Продукт (i') очищают либо нормально-фазовой жидкостной хроматографией, либо с помощью препаративного LC/MS анализа. Оборудование и методы. Аналитический LC/MS, метод А. Анализ осуществляют на масс-спектрометре Waters модели ZQ, в режиме распыления с положительной и отрицательной ионизацией (диапазон от 10 до 1200 а.е.м.), связанном с жидкостным хроматографом HPLC Agilent HP 1100. Разделение происходит на колонке Waters Xbridge C18 (350 мм, 2,5 мкм диаметр частиц) с поддерживаемой температурой, равной 60 С, с использованием градиентного элюирования смесью ацетонитрил/вода, содержащей 0,1% (об./об.) муравьиной кислоты, расход 1,1 мл/мин. Проход через градиент от 5 до 100% ацетонитрила совершается за 5 мин, градиент удерживается при 100% 1/2 мин, затем доходит до 5% за 1 мин. Общая продолжительность разделения составляет 7 мин. Помимо анализа методом масс-спектроскопии осуществляют детектирование УФ (диодная матрица) при длинах волн от 210 до 400 нм, а также измерение с применением детектора по светорассеянию испаренного образца ELSD (evaporative light scattering) с помощью прибора Sedere Sedex 85. Аналитический LC/MS, метод В. Анализ осуществляют на масс-спектрометре Waters, модель ZQ, методом распыления с положительной и отрицательной ионизацией (диапазон от 10 до 1200 а.е.м.), связанном с прибором Waters Alliance HT. Разделение происходит на колонке Waters Atlantis dC18 (2,150 мм, 5 мкм диаметр частиц) с поддерживаемой температурой, равной 25 С, с использованием градиентного элюирования смесью ацетонитрил/вода, содержащей 0,1% (об./об.) трифторуксусной кислоты, расход 0,5 мл/мин. Проход через градиент от 5 до 85% ацетонитрила совершается за 5 мин, затем градиент удерживается при 90% в течение 1 мин. Общая продолжительность разделения составляет 7 мин. Помимо анализа методом массспектроскопии осуществляют детектирование УФ (диодная матрица) при длинах волн от 210 до 400 нМ. Препаративный LC/MS метод С. Продукты очищают с помощью препаративного метода LC/MS с использованием системы WatersFractionLynx, состоящей из насоса Waters модели 600 для градиентного элюирования, насоса Waters модели 515 для регенерации, насоса Waters Reagent Manager, автоинжектора модели 2700, 2 прерывателейTheodyne модели LabPro, детектора Waters с диодной матрицей, масс-спектрометра Waters модели ZMD и коллектора фракций модели 204. Прибор управляется системой программного обеспечения WatersFractionLynx. На выходе из разделительной колонки 1/1000 потока отводится разделителем LC PakingAccurate; данный поток смешивается с метанолом (расход 0,5 мл/мин) и отправляется к детекторам: 3/4 отправляется на диодную матрицу и 1/4 - на масс-спектрометр; остальная часть потока (999/1000) отправляется на коллектор фракций. Продукт собирается, если наблюдается пик массы на FractionLynx, в противном случае поток непосредственно выводится. Молекулярные формулы продуктов вводятся в систему программного обеспечения FractionLynx и продукт собирается, когда улавливаются пики массыRC10.10. Массу полученного продукта определяют, измеряя массу емкости после выпаривания растворителя. Информация об используемых колонках и градиентах дана для каждого примера в следующей части. Аналитический LC/MS, метод D. Анализ осуществляют на масс-спектрометре Waters SQD методом распыления с положительной и отрицательной ионизацией (диапазон от 10 до 1200 а.е.м.), связанном с прибором Waters Alliance HT. Разделение происходит на колонке ВЕН (2,150 мм, 1,7 мкм диаметр частиц) с поддерживаемой температурой, равной 25 С, с использованием градиентного элюирования смесью ацетонитрил/вода, содержащей 0,1% (об./об.) трифторуксусной кислоты, расход 1 мл/мин. Проход через градиент от 5 до 100% ацетонитрила совершается за 2 мин. Помимо анализа методом масс-спектроскопии осуществляют детектирование УФ (диодная матрица) при длинах волн от 210 до 400 нм. Пример 1. Трифторацетат 4-[([3-фенил]карбамоилокси)бензил]амино-1 Н-пиразол-3 карбоксамида Получение смолы (ii). 30 г смолы Ринка (PolymerLab; 0,99 ммоль/г) набухает в 150 мл ДМФ. Через 10 мин перемешивания ДМФ отфильтровывают и заменяют на 150 мл раствора пиперидина в ДМФ (50/50 об./об.). Смесь выдерживают в течение 1 ч при перемешивании, затем фильтруют. Смолу последовательно промывают 5 раз 150 мл ДМФ, 2 раза 150 мл метанола и 3 раза 150 мл ДМФ. Далее раствор 14,1 г 4-нитро-3 пиразолкарбоновой кислоты (90 ммоль; 3 экв.) и 13,8 г HOBt (90 ммоль; 3 экв.) в 150 мл ДМФ добавляют к смоле, после чего сразу же добавляют 13,8 мл DIC (90 ммоль; 3 экв.). Смесь выдерживают в течение 16 ч при перемешивании при комнатной температуре. Раствор фильтруют, смолу последовательно промывают 5 раз 150 мл ДМФ, 2 раза 150 мл метанола и 3 раза 150 мл ДМФ, затем 150 мл раствора 1 М SnCl2(33 г в 150 мл). Перемешивание при комнатной температуре поддерживают в течение 24 ч, затем смесь фильтруют и смолу промывают 5 раз 150 мл ДМФ, 2 раза 150 мл метанола, 3 раза 150 мл ДХМ и 2 раза 150 мл этиловым эфиром. После сушки в вакууме отделяют 31 г промежуточной смолы (ii). Получение примера 1. 100 мг смолы (ii) набухает в 0,3 мл ДХЭ, затем добавляют раствор 72 мг фенилкарбамино-3 формилфенилового эфира (0,3 ммоль; 3 экв.) в 0,2 мл ДМФ и 33 мг цианборгидрида натрия (0,5 ммоль; 5 экв.). Смесь нагревают при 80 С в течение 1 ч, затем фильтруют после охлаждения до комнатной температуры. Смолу затем последовательно промывают 2 раза 1 мл МеОН, 5 раз 1 мл ДМФ, 3 раза 1 мл МеОН и 5 раз 1 мл ДХМ. Продукт расщепляют путем обработки смолы 1 мл раствора ТФУ/ДХМ в соотношении 50/50. Раствор упаривают и полученный сырой продукт непосредственно очищают препаративной ВЭЖХ. Получают 2,4 мг ожидаемого продукта 1 (выход=5%). ([М+Н]+: 352). RT=2,49 мин (метод А). Пример 2 а. Трифторацетат 4-[3-([2-фтор-5-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Н-пиразол-3-карбоксамида. Пример 2b. Хлоргидрат 4-[3-([2-фтор-5-(трифторметил)фенил]карбамоиламино)бензил]амино 1 Н-пиразол-3-карбоксамида Получение 1-(2-фтор-5-трифторметилфенил)-3-(3-формилфенил)мочевины. Смесь 484 мг 3-аминобензальдегида (4 ммоль) (полимеризованная форма) и 290 мкл 2-фтор-5 трифторметилфенилизоцианата в 4 мл ДХЭ обрабатывают в микроволновой печи СЕМ Discover при 100 С в течение 10 мин (мощность 90). После охлаждения смесь вносят в 100 мл насыщенного раствора гидрогеносульфата калия и экстрагируют дважды 50 мл этилацетата. Соединенные органические фазы промывают водой, сушат над сульфатом натрия и упаривают. Отделяют 740 мг ожидаемого альдегида(выход 57%) с чистотой LC/MS 82%. Сырой продукт непосредственно используют на следующих стадиях. ([М+Н]+): 327. RT=4,28 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 2 а. Пример 2 а получают способом, описанным для примера 1, исходя из 1 г смолы (ii), 520 мг 1-(2 фтор-5-трифторметилфенил-3-(3-формилфенил)мочевины (1,6 ммоль; 2 экв.) и 264 мг цианборгидрида натрия (4 ммоль; 5 экв.). После очистки препаративной ВЭЖХ получают продукт 2 а. (EIMS ([М+Н]+): 437. RT=3,45 мин (метод А). Получение примера 2b. Пример 2b получают способом, описанным для примера 1, исходя из 1 г смолы (ii), 520 мг 1-(2 фтор-5-трифторметилфенил-3-(3-формилфенил)мочевины (1,6 ммоль; 2 экв.) и 264 мг цианборгидрида натрия (4 ммоль; 5 экв.). После фильтрации и упаривания выделяют 301 мг сырого продукта (чистотаLC/MS 83%). Сырой продукт очищают на колонке с силикагелью, используя в качестве элюента смесь ДХМ/МеОН (90/10). Выделяют 151 мг бледно-желтого твердого вещества (выход 43%). Продукт растворяют в 2 мл МеОН и превращают его в соль хлоргидрата добавлением раствора 4 N HCl в диоксане. После упаривания выделяют продукт 2b в виде бледно-желтого твердого вещества. (EIMS ([М+Н]+): 437.(полимеризованная форма) и 548 мг 2-фторфенилизоцианата в 4 мл ДХЭ. После охлаждения смесь вносят в 100 мл насыщенного раствора гидрогеносульфата калия и экстрагируют дважды 50 мл этилацетата. Соединенные органические фазы промывают водой, сушат над сульфатом натрия и упаривают. Выделяют 864 мг ожидаемого альдегида в форме камеди с чистотой LC/MS 77%. Кристаллическую фракцию получают после растирания в этиловом эфире. Выделяют 165 мг твердого вещества ([М+Н]+): 259.RT=4,28 мин (градиентное элюирование ацетонитрил/вода 5/85% метод В). Получение примера 3. Пример 3 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 104 мг 1-(2 фторфенил)-3-(3-формилфенил)мочевины (0,4 ммоль, 2 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 33 мг продукта 3 (выход=43%). EIMS ([M+Н]+): 369. RT=3,70 мин (градиентное элюирование ацетонитрил/вода от 5 до 85% - метод А). Пример 4. Трифторацетат 4-[(3-[(2-метоксифенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3 карбоксамида(полимеризованная форма) и 597 мг 2-метоксифенилизоцианата (4 ммоль) в 4 мл ДХЭ. 300 мг ожидаемого сырого альдегида выделяют и непосредственно используют на следующей стадии. ([М+Н]+): 271.RT=5,14 мин (градиентное элюирование ацетонитрил/вода от 3 до 90% - метод В). Получение примера 4. Пример 4 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 162 мг 1-(2 метоксифенил)-3-(3-формилфенил)мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 10,4 мг продукта 4 (выход=11%). EIMS(полимеризованная форма) и 820 мг 2-фтор-3-фторметилфенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 300 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии.([М+Н]+): 327. RT=4,21 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 5. Пример 5 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 195 мг 1-(2 фтор-3-фторметилфенил)-3-(3-формилфенил)мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 31,4 мг продукта 5 (выход=29%). EIMS ([M+Н]+): 437. RT=3,33 мин (градиентное элюирование ацетонитрил/вода от 5 до 100% метод А). Пример 6. Трифторацетат 4-[3-[(3-метоксифенил)карбамоил]аминобензиламино]-1 Н-пиразол-3 карбоксамида(полимеризованная форма) и 597 мг 3-триметоксифенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 598 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([М+Н]+): 271.RT=5,05 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 6. Пример 6 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 162 мг 1-(3 метоксифенил)-3-(3-формилфенил)мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 17 мг продукта 6 (выход=17%). EIMS(полимеризованная форма) и 597 мг 3-фтор-5-трифторметилфенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 924 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии.([М+Н]+): 327. RT=4,48 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение продукта примера 7. Продукт примера 7 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 196 мг 1-(3-фтор-5-триформетилфенил)-3-(3-формилфенил)мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 33,6 мг продукта 7 (выход=31%). EIMS ([M+Н]+): 437. RT=3,55 мин (градиентное элюирование ацетонитрил/вода от 5 до 100% метод А). Пример 9. Трифторацетат 3-[(3-[(3-карбамоил-1 Н-пиразол-4-ил)амино]метилфенил)карбамоил]аминометилбензоата(полимеризованная форма) и 812 мг сложного метилового эфира 3-изоцианбензойной кислоты (4 ммоль) в 4 мл ДХЭ. Выделяют 695 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([М+Н]+): 299. RT=2,94 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% метод В). Получение примера 9. Продукт примера 9 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 180 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 25,3 мг продукта 9 (выход=24%). EIMS ([М+Н]+): 409. RT=2,67 мин (градиентное элюирование ацетонитрил/вода от 5 до 100% - метод А). Пример 10. Трифторацетат 4-[3-([4-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Нпиразол-3-карбоксамида(полимеризованная форма) и 748 мг 4-трифторметилфенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 896 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([М+Н]+): 309. RT=4,14 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 10. Пример 10 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 185 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 20,9 мг продукта 10 (выход=19%). EIMS ([M+Н]+): 419. RT=3,33 мин (градиентное элюирование ацетонитрил/вода от 5 до 100% - метод А). Пример 11. Трифторацетат 4-[3-([3-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Нпиразол-3-карбоксамида(полимеризованная форма) и 748 мг 3-трифторметилфенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 744 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([М+Н]+): 309. RT=4,04 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 11. Пример 11 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 185 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 25,9 мг продукта 11 (выход=24%). EIMS ([М+Н]+): 419. RT=3,25 мин (градиентное элюирование ацетонитрил/вода от 5 до 100% - метод А). Пример 12. Трифторацетат 4-[3-([2-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Нпиразол-3-карбоксамида(полимеризованная форма) и 748 мг 2-трифторметилфенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 744 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([М+Н]+): 309. RT=5,51 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 12. Пример 12 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 185 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 17,5 мг продукта 12 (выход=16%). EIMS ([М+Н]+): 419. RT=2,64 мин (градиентное элюирование ацетонитрил/вода от 5 до 100% - метод А). Пример 13. Трифторацетат 4-[(3-[(3,5-диметоксифенил)карбамоил]аминобензил)амино]-1 Нпиразол-3-карбоксамида(полимеризованная форма) и 717 мг 3,5-диметоксифенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 893 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([М+Н]+): 301. RT=2,99 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 13. Пример 13 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 180 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 14,9 мг продукта 13 (выход=14%). EIMS ([М+Н]+): 411. RT=4,61 мин (градиентное элюирование ацетонитрил/вода от 5 до 85% - метод А). Пример 15. Трифторацетат 4-[(3-[(4-метоксифенил)карбамоил]аминобензил)амино]-1 Н-пиразол 3-карбоксамида(полимеризованная форма) и 597 мг 4-метоксифенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 747 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([М+Н]+): 271.RT=2,53 мин (градиентное элюирование ацетонитрил/вода от 3 до 90% - метод В). Получение примера 15. Пример 15 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 162 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 2,6 мг продукта 15 (выход=3%). EIMS ([М+Н]+): 381. RT=4,3 мин (градиентное элюирование ацетонитрил/вода от 5 до 85% - метод В). Пример 16. Трифторацетат 4-[(3-[(4-фторфенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3 карбоксамида(полимеризованная форма) и 548 мг 4-фторфенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 760 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([M+Н]+): 259.RT=2,86 мин (градиентное элюирование ацетонитрил/вода 30-90% - метод В). Получение примера 16. Пример 16 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 155 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 0,6 мг продукта 16 (выход=1%). EIMS ([М+Н]+): 369. RT=4,44 мин (градиентное элюирование ацетонитрил/вода от 5 до 85% - метод В). Пример 17. Трифторацетат 4-[3-([4-хлор-3-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Н-пиразол-3-карбоксамида(полимеризованная форма) и 886 мг 4-хлор-3-трифторфенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 117 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([М+Н]+): 343. RT=4,66 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 17. Пример 17 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 206 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 7,3 мг продукта 17 (выход=6%). EIMS ([М+Н]+): 453. RT=5,38 мин (градиентное элюирование ацетонитрил/вода от 5 до 85% - метод В). Пример 19. Трифторацетат 4-[3-([2-хлор-4-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Н-пиразол-3-карбоксамида(полимеризованная форма) и 886 мг 2-хлор-4-трифторметилфенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 1 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии.([М+Н]+): 343. RT=4,66 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 19. Пример 19 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 206 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 24,3 мг продукта 19 (выход=22%). EIMS ([M+Н]+): 453. RT=5,36 мин (градиентное элюирование ацетонитрил/вода от 5 до 85% - метод В). Пример 25. Трифторацетат 4-([3-([3,5-бис-(трифторметил)фенил]карбамоиламино)бензил]амино)1 Н-пиразол-3-карбоксамида(полимеризованная форма) и 1,02 г 3,5-бис-трифторметилфенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 489 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии.([M+Н]+): 377. RT=5,05 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 25. Пример 25 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 225 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 3,3 мг продукта 25 (выход=4%). EIMS ([М+Н]+): 365. RT=4,5 мин (градиентное элюирование ацетонитрил/вода от 5 до 85% - метод В). Пример 26. Трифторацетат 4-[(3-[(3-фторфенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3 карбоксамида(полимеризованная форма) и 548 мг 3-фторфенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 723 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([М+Н]+): 259.RT=4,04 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 26. Пример 26 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 155 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 23,5 мг продукта 26 (выход=25%). EIMS ([М+Н]+): 369. RT=4,7 мин (градиентное элюирование ацетонитрил/вода от 5 до 85% - метод В).(полимеризованная форма) и 717 мг 2,5-диметоксифенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 853 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([М+Н]+): 301. RT=4,14 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 28. Пример 28 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 170 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 11,8 мг продукта 28 (выход=12%). EIMS ([М+Н]+): 411. RT=4,72 мин (градиентное элюирование ацетонитрил/вода от 5 до 85% - метод В). Пример 30. Трифторацетат 4-[(3-[(2,5-дифторфенил)карбамоил]аминобензил)амино]-1 Н-пиразол 3-карбоксамида(полимеризованная форма) и 620 мг 2,5-дифторфенилизоцианата (4 ммоль) в 4 мл ДХЭ. Выделяют 952 мг ожидаемого сырого альдегида и непосредственно используют на следующей стадии. ([M+Н]+): 277.RT=4,13 мин (градиентное элюирование ацетонитрил/вода от 30 до 90% - метод В). Получение примера 30. Пример 30 получают способом, описанным для примера 1, исходя из 200 мг смолы (ii), 167 мг мочевины (0,6 ммоль, 3 экв.) и 66 мг цианборгидрида натрия (1 ммоль; 5 экв.). После очистки препаративной ВЭЖХ выделяют 14,1 мг продукта 30 (выход=14%). EIMS ([M+Н]+): 387. RT=4,82 мин (градиентное элюирование ацетонитрил/вода от 5 до 85% - метод В). Пример 32. Трифторацетат 4-[3-([2-фтор-5-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Н-пиразол-3-карбоксамида метила Соединение 32 получают прямым восстановительным аминированием метилового эфира 4-амино 3-пиразолкарбоновой кислоты. Раствор 44 мг метилового эфира 4-амино-3-пиразолкарбоновой кислоты(0,31 ммоль) и 100 мг 1-(2-фтор-5-трифторметилфенил)-3-(3-формилфенил)мочевины (см. пример 2) в смеси 0,6 мл ДХЭ и 0,5 мл ДМФ обрабатывают раствором 59 мг цианборгидрида натрия в 0,5 мл метанола и 0,05 мл уксусной кислоты. Смесь перемешивают в течение 2 ч при 80 С, затем охлаждают и выливают в 20 мл воды. Смесь экстрагируют два раза 20 мл этилацетата. Объединенные органические фазы промывают водой, сушат над сульфатом натрия и упаривают в вакууме. После очистки препаративной ВЭЖХ выделяют 20,8 мг продукта 32 (выход=12%). EIMS ([M+Н]+): 452. RT=5,52 мин (градиентное элюирование ацетонитрил/вода от 5 до 85% - способ В). Пример 33. Трифторацетат 4-1-3-[3-(2-фтор-5-трифторметилфенил)уреидо]фенилэтиламино-1 Нпиразол-3-карбоксамида (1:1)(1,34 ммоль) в 1 мл ТГФ оставляют при перемешивании при комнатной температуре на 1 ч, затем упаривают. Твердое вещество извлекают эфиром и фильтруют. Выделяют 307 мг ожидаемого кетона (выход=69%) с чистотой LC/MS 87%. Сырой продукт непосредственно используют на следующей стадии.([M+Н]+): 341. RT=6,06 мин (метод А). Получение примера 33. 450 мг смолы I (0,45 ммоль) набухает в 2 мл ДХЭ, затем добавляют раствор 307 мг 1-(3 ацетилфенил)-3-(2-фтор-5-трифторметилфенил)мочевины (0,9 ммоль; 2 экв.) в 2 мл ДМФ и затем 149 мг цианборгидрида натрия (2,25 ммоль; 5 экв.). Смесь обрабатывают в микроволновой печи СЕМ Discover при 100 С в течение 10 мин (мощность 90). Смолу далее последовательно промывают два раза 2 мл МеОН, 3 раза 2 мл дихлорметана, 2 раза 2 мл МеОН и 3 раза 2 мл дихлорметана. Продукт разделяют путем обработки смолы 4 мл раствора трифторуксусная кислота/дихлорметан в соотношении 50/50. Раствор упаривают и полученный сырой продукт непосредственно очищают препаративной ВЭЖХ. После лиофилизации получают 13,5 мг ожидаемого продукта (белое твердое вещество, выход=5%). ([M+Н]+): 451. К раствору 0,25 г 4-[(3-аминобензил)метиламино]-1 Н-пиразол-3-карбоксамида в 25 мл безводного ТГФ добавляют при 20 С раствор 0,14 мл 1-фтор-2-изоцианато-4-трифторметилбензола. Реакционную смесь перемешивают в течение 12 ч при 20 С, затем разбавляют 100 мл этилацетата. Органическую фазу промывают 50 мл дистиллированной воды, затем декантируют, сушат над сульфатом магния и упаривают досуха при пониженном давлении. Полученный осадок хроматографируют на колонке с силикагелем(15 г силикагелевого патрона Merck с размером частиц от 15 до 45 мкм, диаметр колонки 2,2 см, фракции 5 мл, расход 10 мл/мин, элюент дихлорметан 95 метанол 5 - по объему). Фракции от 35 до 95 объединяют, упаривают досуха при пониженном давлении. Получают 0,02 г 4-(3-[3-(2-фтор-5 трифторметилфенил)уреидо]бензилметиламино)-1 Н-пиразол-3-карбоксамида в виде твердого вещества.([M+Н]+): 451. RT=3,55 мин (метод А). 4-[(3-Аминобензил)метиламино]-1 Н-пиразол-3-карбоксамид получают следующим образом. К раствору 1,26 г 4-[метил(3-нитробензил)амино]-1 Н-пиразол-3-карбоксамида в 90 мл абсолютного этанола добавляют малыми порциями 3,6 г хлорида олова дигидрата при 20 С. Реакционную смесь перемешивают в течение 15 ч при 20 С, упаривают досуха при пониженном давлении. Осадок извлекают 500 мл смеси метиленхлорид 90/метанол 10 об. и 500 мл водного насыщенного раствора гидрогенокарбоната калия. Эту среду перемешивают при 20 С в течение 2 ч, затем фильтруют. Полученное твердое вещество после фильтрации экстрагируют дважды смесью 100 мл метиленхлорида 90 метанол 10 об., жидкие фазы объединяют, декантируют. Водную фазу экстрагируют два раза 200 мл метиленхлорида, органические фазы объединяют, промывают 300 мл водного насыщенного раствора хлорида натрия, сушат над сульфатом натрия, фильтруют и упаривают досуха при пониженном давлении. Получают 0,6 г 4-[(3 аминобензил)метиламино]-1 Н-пиразол-3-карбоксамида в виде крема безе. ([М+Н]+): 246. RT=0,3 мин(метод А). 4-[Метил-(3-нитробензил)амино]-1 Н-пиразол-3-карбоксамид получают следующим образом. К раствору 2,49 г 2,4-диметоксибензиламида 4-[метил-(3-нитробензил)амино]-1 Н-пиразол-3 карбоновой кислоты в 30 мл толуола добавляют 4,45 г моногидрата паратолуолсульфоновой кислоты,затем реакционную среду нагревают при температуре образования флегмы толуола в течение 17 ч. После охлаждения реакционной среды до комнатной температуры добавляют 15 мл метанола, затем 700 мл этилацетата и, наконец, 300 мл дистиллированной воды. Доводят рН этой среды до значения, равного 11,путем добавления 100 мл водного раствора 1 N гидроксида натрия. Данный раствор фильтруют, твердое вещество экстрагируют дважды 30 мл этилацетата, жидкие фазы объединяют, затем декантируют. Водную фазу экстрагируют дважды 200 мл метиленхлорида, органические фазы объединяют, промывают 300 мл водного насыщенного раствора хлорида натрия, сушат над сульфатом магния, фильтруют и упаривают досуха при пониженном давлении. Получают 1,41 г 4-[метил-(3-нитробензил)амино]-1 Нпиразол-3-карбоксамида в виде твердого бежевого вещества, плавящегося при 166 С. ([М+Н]+): 276.RT=2,61 мин (метод А). 2,4-Диметоксибензиламид 4-[метил-(3-нитробензил)амино]-1H-пиразол-3-карбоновой кислоты получают следующим образом. Раствор 1,61 г 2,4-диметоксибензиламид 4-(3-нитробензиламино)-1 Н-пиразол-3-карбоновой кисло- 17019454 ты, 1,25 г параформальдегида и 0,94 г безводного сульфата магния в 70 мл ледяной уксусной кислоты перемешивают при комнатной температуре в течение 4 ч. Далее к данному раствору добавляют небольшими порциями 1,23 г цианборгидрида натрия. Реакционную среду перемешивают в течение 2 ч при комнатной температуре, затем выливают на 300 мл водного раствора натрия 5 N и 120 г 120 г измельченного льда, доводят значение рН до 11. Далее добавляют 300 мл насыщенного раствора хлорида натрия. Данный раствор фильтруют, твердое вещество промывают три раза 60 мл дистиллированной воды. Собранный, таким образом, осадок сушат на воздухе. Получают 1,51 г 2,4-диметоксибензиламид 4-[метил(3-нитробензил)амино]-1 Н-пиразол-3-карбоновой кислоты в виде бледно-желтого твердого вещества.([М+Н]+): 426. RT=3,87 (метод А). 2,4-Диметоксибензиламид 4-(3-нитробензиламино)-1 Н-пиразол-3-карбоновой кислоты получают следующим образом. К раствору 15,43 г 2,4-диметоксибензиламида 4-амино-1 Н-пиразол-3-карбоновой кислоты хлоргидрата и 7,01 г диизопропилэтиламина в 490 мл безводного тетрагидрофурана добавляют 8,2 г 3 нитробензальдегида и 5,9 г безводного сульфата магния. Реакционную среду кипятят с обратным холодильником в течение 1 ч 30 мин, оставляют до тех пор, пока температура не снизится до 20 С, затем охлаждают до 5 С посредством водно-ледяной бани. К полученной суспензии в виде крема небольшими порциями добавляют 15,5 г цианборгидрида натрия. Реакционную смесь перемешивают 5 мин при 5 С,затем оставляют до тех пор, пока температура не повысится до комнатной. Оставляют при перемешивании при комнатной температуре в течение 20 ч. Полученный мутный раствор оранжево-каштанового цвета выливают в 1500 мл дистиллированной воды. К полученному прозрачному раствору молочнокаштанового цвета добавляют 1000 мл дихлорметана. После перемешивания и декантации хлорметиленовой фазы водную фазу снова экстрагируют дважды 500 мл дихлорметана. Органические фазы объединяют, затем промывают 500 мл водного насыщенного раствора хлорида натрия, сушат над сульфатом магния, фильтруют через бумажный фильтр, затем сушат досуха в ротационном испарителе (температура 40 С, Р: 15 мбар). Получают 27,19 г клейкого твердого вещества желто-каштанового цвета, который перекристаллизовывают из 360 мл ацетонитрила при кипячении с обратным холодильником. Получают первую порцию массой 7,7 г 2,4-диметоксибензиламида 4-(3-нитробензиламино)-1 Н-пиразол-3 карбоновой кислоты в виде желтого твердого вещества. Ацетонитрильный фильтрат выделяют, затем сушат досуха в ротационном испарителе (температура 40 С, Р: 15 мбар). Получают 19,7 г клейкой массы цвета охры, которую растирают при 20 С в 50 мл ацетонитрила в течение 1 ч. Полученную суспензию фильтруют на фильтре VF n3, промывают дважды 15 мл ацетонитрила. После сушки на воздухе, а затем в сушильном шкафу Heraeus (температура 40 С, Р: 0,2 мбар) получают вторую порцию массой 6,59 г 2,4-диметоксибензиламида 4-(3-нитробензиламино)-1 Нпиразол-3-карбоновой кислоты в виде твердого желтого вещества. ([М+Н]+): 412. RT=3,93 мин (метод А). 2,4-Диметоксибензиламид 4-амино-1 Н-пиразол-3-карбоновой кислоты получают следующим образом. К суспензии 10,72 г 2,4-диметоксибензиламида 4-нитро-1 Н-пиразол-3-карбоновой кислоты в 600 мл абсолютного этанола добавляют небольшими порциями 27,64 г хлорида олова дигидрата. Реакционную смесь перемешивают в течение 48 ч при 20 С. Полученный прозрачный раствор каштанового цвета сушат досуха в ротационном испарителе. Полученный светлый крем каштанового цвета извлекают 700 мл смеси 90/10 об. метиленхлорид/метанол. К полученному коричневому раствору добавляют 700 мл водного насыщенного раствора гидрогенокарбоната натрия. Полученную суспензию в виде крема перемешивают в течение 1 ч при комнатной температуре. К суспензии добавляют 30 г Clarcel Flo, затем оставляют перемешиваться на 10 мин при комнатной температуре. Фильтруют, осадок на фильтре промывают дважды 250 мл смеси 90/10 об. метиленхлорид/метанол. Твердое вещество отжимают, фильтрат извлекают,затем переливают его в емкость для декантации. Хлорметиленовую фазу декантируют, затем водную фазу снова экстрагируют дважды 250 мл дихлорметана. Органические фазы объединяют, затем сушат над сульфатом магния, фильтруют через бумажный фильтр, затем сушат досуха в ротационном испарителе. Получают 8,53 г 2,4-диметоксибензиламид 4-амино-1 Н-пиразол-3-карбоновой кислоты в виде бледно-розового твердого вещества. ([М+Н]+): 277). RT=2,36 мин (метод А). 2,4-Диметоксибензиламид 4-нитро-1 Н-пиразол-3-карбоновой кислоты получают следующим образом. К раствору 28,76 г 1-(3-диметиламинопропил)-3-этилкарбодиимида хлоргидрата и 20,27 г 1 гидроксибензотриазола в 100 мл диметилформамида добавляют 23 г 2,4-диметоксибензиламина, затем 20,04 г 4-нитро-3-пиразолкарбоновой кислоты (98%). Реакционную смесь перемешивают при комнатной температуре в течение 20 ч. Затем полученный желтый прозрачный раствор выливают в 1000 мл дистиллированной воды. Получают белую суспензию, которую оставляют на 1 ч при комнатной температуре. Суспензию фильтруют, осадок на фильтре промывают 3 раза 250 мл дистиллированной воды. Полученный твердый осадок отжимают, сушат на воздухе, а затем в сушильном шкафу Hraeus под вакуумом(температура 40 С, Р: 0,2 мбар). Получают 40,65 г белого твердого вещества, который растирают в 750 мл изопропанола при кипячении с обратным холодильником в течение 20 мин. Полученную суспензию охлаждают на бане вода+лед в течение 2 ч, затем фильтруют. Осадок на фильтре промывают 2 раза 100 мл изопропанола, затем 2 раза 100 мл изопропилового эфира. Полученное твердое вещество сушат на воздухе, затем в сушильном шкафу Heraeus (температура 40 С, Р: 0,2 мбар). Получают 32,16 г 2,4-диметоксибензиламида 4-нитро-1 Н-пиразол-3-карбоновой кислоты ([М+Н]+): 307). RT=3,12 мин. Пример 35. 4-(Этил 3-[3-(2-фтор-5-трифторметилфенил)уреидо]бензиламино)-1 Н-пиразол-3 карбоксамид 4-(Этил 3-[3-(2-фтор-5-трифторметилфенил)уреидо]бензиламино)-1 Н-пиразол-3-карбоксамид получают конденсацией 1-фтор-2-изоцианато-4-трифторметилбензола с 4-[(3-аминобензил)этиламино]-1 Нпиразол-3-карбоксамид согласно методике, описанной в примере 34, причем указанное производное получают из 4-[(3-нитробензил)этиламино]-1 Н-пиразол-3-карбоксамида согласно методике, описанной также в примере 34. 4-[(3-Нитробензил)этиламино]-1 Н-пиразол-3-карбоксамид получают следующим образом. Раствор 2,2 г 2,4-диметоксибензиламида 4-амино-1 Н-пиразол-3-карбоновой кислоты хлоргидрата,1,3 мл диизопропилэтиламина в 70 мл ТГФ перемешивают в течение 5 мин, затем к данному раствору добавляют 1,2 г 3-нитробензальдегида и 0,85 г сульфата магния. Реакционную смесь кипятят с обратным холодильником в течение 1 ч, затем после охлаждения до 45 С небольшими порциями добавляют 7,4 г триацетоксиборгидрида натрия, затем снова кипятят с обратным холодильником в течение 3 ч. Пока весь исходный продукт не исчезнет, после охлаждения до 45 С небольшими порциями добавляют 7,4 г триацетоксиборгидрида натрия, затем снова кипятят с обратным холодильником в течение 2 ч. После охлаждения реакционной смеси до комнатной температуры ее выливают в 350 мл дистиллированной воды. рН молочного бледно-желтого раствора, полученного таким образом, доводят до значения 8-9 путем добавления 60 мл водного раствора гидроксида натрия 2 N. Добавляют 250 мл дихлорметана, затем после декантации, экстракции водной фазы два раза 150 мл дихлорметана органические фазы объединяют,промывают 250 мл водного насыщенного раствора хлорида натрия, сушат над сульфатом магния, фильтруют и сушат досуха при пониженном давлении. Получают 3,19 г клейкого масла, которое хроматографируют на колонке с силикагелем (90 г силикагелевого патрона Merck с размером частиц от 15 до 45 мкм, диаметр колонки 4,7 см, фракции 15 мл, расход 18 мл/мин, элюент этилацетат 70 циклогексан 30 об.). Фракции 34-110 объединяют, упаривают досуха при пониженном давлении. Получают 2,22 г 4-[(3 нитробензил)этиламино]-1 Н-пиразол-3-карбоксамида в виде крема безе желтого цвета. ([М+Н]+): 453). К раствору 0,12 г 4-(3-аминобензиламино)-1 Н-пиразол-3-карбоксамида в 5 мл безводного ТГФ добавляют при 20 С 88 мкл триэтиламина и 0,18 г фенилового эфира (4-фторметилпиридин-2 ил)карбаминовой кислоты. Реакционную среду нагревают 20 мин в микроволновой печи, затем после охлаждения растворяют в 25 мл этилацетата. Органическую фазу промывают два раза 15 мл дистиллированной воды, затем декантируют, сушат над сульфатом магния и упаривают досуха при пониженном давлении. Полученный осадок хроматографируют на колонке с силикагелем (15 г силикагельного патрона Merck с размером частиц от 15 до 45 мкм, диаметр колонки 2,2 см, фракции 3,5 мл, расход 7 мл/мин,элюент этилацетат). Фракции 36-60 объединяют, упаривают досуха при пониженном давлении. Получа- 19019454 ют 0,06 г 4-(3-[3-(4-трифторметилпиридин-2-ил)уреидо]бензиламино)-1 Н-пиразол-3-карбоксамида в виде белого твердого вещества. ([М+Н]+): 420). RT=0,78 мин (метод D). Фениловый эфир (4-трифторметилпиридин-2-ил)карбаминовой кислоты получают следующим образом. К раствору 2-амино-4-трифторметилпиридина в 65 мл безводного тетрагидрофурана добавляют 0,81 мл пиридина, реакционную смесь охлаждают до 5 С, затем добавляют при этой температуре 0,95 мл фенилхлорформиата. После перемешивания в течение 2 ч при 5 С реакционную смесь доводят до комнатной температуры, затем выливают ее в 20 мл дистиллированной воды, поддерживая температуру равной 20 С. Далее добавляют 50 мл этилацетата, декантируют, водную фазу экстрагируют два раза по 20 мл этилацетата. Органические фазы объединяют, сушат над сульфатом магния, фильтруют, затем упаривают досуха. Полученное твердое вещество растирают в 10 мл диизопропилового эфира. Получают 1,07 г фенилового эфира (4-фторметилпиридин-2-ил)карбаминовой кислоты в виде твердого вещества белого цвета, плавящегося при 161 С. ([М+Н]+): 281). RT=4,30 мин (метод А). Примеры 40-58, описанные в нижеприведенной таблице, получают или могут быть получены конденсацией соответствующих изоцианатов с 4-[(3-аминобензил)амино]-1 Н-пиразол-3-карбоксамида согласно методике, описанной в примере 34. Таблица А Определение активности соединений. Протоколы проведения экспериментов. 1. KDR. Ингибирующий эффект соединений определяли в опыте фосфорилирования субстрата энзима KDRin vitro методом сцинтилляции (96-луночные планшеты, NEN). Цитоплазматический домен человеческого энзима KDR был клонирован в виде химерных энзимовGST в переносчике экспрессии бакуловирусов pFastBac. Белок был выражен в клетках SF21 и очищен до примерно 60% гомогенности. Киназную активность определяли В 20 мМ MOPS, 10 мМ MgCl2, 10 мМ MnCl2, 1 мМ DTT, 2,5 мМEGTA, 10 мМ b-глицерофосфата, рН 7,2, в присутствии 10 мМ MgCl2, 100 мкМ Na3VO4, 1 мМ NaF. 10 мкл соединения добавляли к 70 мкл киназного буфера, содержащего 100 нг энзима KDR при 4 С. Реакцию начинали, добавляя 20 мкл раствора, содержащего 2 мкг субстрата (фрагмент SH2-SH3 доменовPLC в форме химерного белка GST), 2 Ci 35 Р[АТР] и 2 мкМ холодного АТР. Через 1 ч инкубации при 37 С реакцию останавливали, добавляя 1 об. (100 мкл) 200 мМ EDTA. Буферную инкубационную смесь извлекали и лунки промывали три раза 300 мкл PBS. Радиоактивность измерялась в каждой лунке с помощью счетчика радиоактивности Top Count NXT (Packard). Фоновый шум определяли путем измерения радиоактивности в четырех различных лунках, содержащих радиоактивный АТР и один субстрат. Контроль общей активности проводили в четырех различных лунках, содержащих все реактивы(33 Р-[АТР], KDR и субстрат PLC), но в отсутствие соединения. Ингибирование активности KDR соединением по изобретению выражалось в проценте ингибирования контрольной активности, определяемой в отсутствие соединения. Соединение SU 5614 (Calbiochem) (1 мкМ) включалось в каждую лунку в качестве пробы ингибирования. 2. Tie2. Кодовая последовательность человеческого рецептора Tie2, соответственно аминокислотам межклеточного домена 776-1124, была образована путем реакции PCR с использованием кДНК, изолированной от человеческой плаценты, в качестве модели. Данная последовательность была введена в переносчик экспрессии baculovirus pFastBacGT в форме химерного протеина GST. Ингибирующий эффект молекул определяли в опыте фосфорилирования PLC рецептором Tie-2 в присутствии белка GST-Tie2, очищенного до примерно 80% гомогенности. Субстрат состоял из фрагментов SH2-SH3 домена PLC, выраженного в форме химерного белка GST. Киназная активность Tie2 измерялась в буфере MOPS 20 мМ рН 7,2, содержащем 10 мМ MgCl2, 10 мМ MnCl2, 1 мМ DTT, 10 мМ глицерофосфата. В 96-луночный планшет, выдерживаемый на льду, поместили реакционную смесь, содержащую 70 мкл киназного буфера, содержащего 100 нг энзима GST-Tie2 на лунку. Далее добавляли 10 мкл исследуемого вещества, растворенного в ДМСО в максимальной концентрации 10%. Для одной заданной концентрации каждое измерение повторялось четыре раза. Реакцию инициировали, добавляя 20 мкл раствора, содержащего 2 мкг GST-PLC, 2 мкМ холодного АТР и 1 мк CiP33[ATP]. Через 1 ч инкубации при 37 С реакцию останавливали, добавляя 1 об. (100 мкл) EDTA в концентрации 200 мМ. После извлечения буфера инкубации лунки промывали три раза 300 мкл PBS. Радиоактивность измеряли на MicroBeta1450Wallac. Измеряли ингибирование активности Tie2 и оно выражалось в проценте ингибирования относительно контрольной активности, определяемой в отсутствие соединения. Результаты. Соединения примеров по изобретению представляли концентрацию, ингибирующую 50% киназной активности, равную в основном от 0,1 нМ до 2 мкМ на KDR, и/или Tie2 предпочтительно от 0,1 до 500 нМ, более предпочтительно от 0,1 до 50 нМ. Значения в таблице, приведенной ниже, даны в иллюстративном порядке. где каждую группу X1, X2, Х 3 и Х 4 независимо выбирают из N и C-R'5, где R'5 имеет то же значение,что и R5; 2) А представляет собой фенил, возможно замещенный одним или несколькими заместителями, выбранными из трифторметила, галогена, метокси или метоксикарбонила; Ar представляет собой фенил; 3) L представляет собой NH-CO-NH или O-CO-NH; 4) R1 обозначает Н; 5) X представляет собой О или NH; 6) R3 представляет собой Н или метил; 7) R4a представляет собой Н или (С 1-С 4)алкил; 8) R4b представляет собой Н или (С 1-С 4)алкил; 9) R5 представляет собой Н или галоген; указанное соединение находится в форме: 1) не хиральной; или 2) рацемической; или 3) обогащенной энантиомером,и, возможно, преобразовано в фармацевтически приемлемую соль. 2. Соединение по п.1, отличающееся тем, что R4a и R4b означают Н. 3. Соединение по п.1, отличающееся тем, что R4a означает Н и R4b означает (С 1-С 4)алкил. 4. Соединение по п.1, отличающееся тем, что R4a означает (С 1-С 4)алкил и R4b означает Н. 5. Соединение по любому из пп.1-4, отличающееся тем, что R3 означает Н и X означает NH. 6. Соединение по любому из пп.1-4, отличающееся тем, что R3 означает метил и X означает О. 7. Соединение по любому из пп.1-6, отличающееся тем, что R5 означает Н. 8. Соединение по любому из пп.1-7, отличающееся тем, что L означает NHCONH. 9. Соединение по п.1, отличающееся тем, что оно представляет собой трифторацетат 4-[([3-фенил]карбамоилокси)бензил]амино-1 Н-пиразол-3-карбоксамида; хлоргидрат 4-[3-([2-фтор-5-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Н-пиразол 3-карбоксамида; трифторацетат 4-[3-([2-фтор-5-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Нпиразол-3-карбоксамида; трифторацетат 4-[(3-[(2-фторфенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3-карбоксамида; трифторацетат 4-[(3-[(2-метоксифенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3-карбоксамида; трифторацетат 4-[3-([2-фтор-3-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Нпиразол-3-карбоксамида; трифторацетат 4-[(3-[(3-метоксифенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3 карбоксамида; трифторацетат 4-[3-([3-фтор-5-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Нпиразол-3-карбоксамида; трифторацетат метил 3-[(3-[(3-карбамоил-1 Н-пиразол-4-ил)амино]метилфенил)карбамоил]аминобензоата; трифторацетат 4-[3-([4-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Н-пиразол-3 карбоксамида; трифторацетат 4-[3-([3-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Н-пиразол-3 карбоксамида; трифторацетат 4-[3-([2-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Н-пиразол-3 карбоксамида; трифторацетат 4-[(3-[(3,5-диметоксифенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3 карбоксамида; трифторацетат 4-[(3-[(4-метоксифенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3-карбоксамида; трифторацетат 4-[(3-[(4-фторфенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3-карбоксамида; трифторацетат 4-[3-([4-хлор-3-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Нпиразол-3-карбоксамида; трифторацетат 4-[3-([2-хлор-4-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Нпиразол-3-карбоксамида; трифторацетат 4-[3-([(3,5-бис-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Нпиразол-3-карбоксамида; трифторацетат 4-[(3-[(3-фторфенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3-карбоксамида; трифторацетат 4-[(3-[(2,5-диметоксифенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3 карбоксамида; трифторацетат 4-[(3-[(2,5-дифторфенил)карбамоил]аминобензил)амино]-1 Н-пиразол-3 карбоксамида; трифторацетат 4-[3-([2-фтор-5-(трифторметил)фенил]карбамоиламино)бензил]амино-1 Нпиразол-3-метилкарбоксилат. 10. Соединение по п.1, отличающееся тем, что оно представляет собой трифторацетат 4-(1-3-[3-(2-фтор-5-трифторметилфенил)уреидо]фенилэтиламино-1 Н-пиразол-3 карбоксамида; 4-(3-[3-(2-фтор-5-трифторметилфенил)уреидо]бензилметиламино)-1 Н-пиразол-3-карбоксамид; 4-(этил-3-[3-(2-фтор-5-трифторметилфенил)уреидо]бензиламино)-1 Н-пиразол-3-карбоксамид; 4-метил-(3-[3-(2-хлор-5-трифторметилфенил)уреидо]бензиламино)-1 Н-пиразол-3-карбоксамид; 4-[3-([3-хлор-4-фторфенил]карбамоиламино)бензил]амино-1 Н-пиразол-3-карбоксамид; 4-[3-([3,4-дихлорфенил]карбамоиламино)бензил]амино-1 Н-пиразол-3-карбоксамид; 4-[3-([3-хлор-5-трифторметилфенил]карбамоиламино)бензил]амино-1 Н-пиразол-3 карбоксамид; 4-[3-([3-трифторметил-4-хлорфенил]карбамоиламино)бензил]амино-1 Н-пиразол-3 карбоксамид; 4-[3-([2-хлор-5-трифторметилфенил]карбамоиламино)бензил]амино-1 Н-пиразол-3 карбоксамид. 11. Фармацевтическая композиция для лечения рака, содержащая соединение по любому из пп.1-10 в комбинации с фармацевтически приемлемым реципиентом. 12. Применение соединения по любому из пп.1-10 в качестве ингибирующего агента одной или нескольких реакций, катализируемых киназой. 13. Применение соединения по п.12, при котором киназу выбирают из KDR и Tie2. 14. Применение соединения по любому из пп.1-10 в производстве лекарственного средства для лечения рака. 15. Способ получения соединений общей формулы (Ib) в которой R1, R3, R4b, R5, X, Ar, L и А имеют значения, определенные в п.1, и R4a означает Н,отличающийся тем, что соединение общей формулы (II) в которой R4b, Ar, L и А имеют значения, определенные в п.1, для получения соединения общей формулы (Ib). 16. Способ получения соединений следующей общей формулы (I):NHCONH,отличающийся тем, что соединение общей формулы (VIII) в которой R'3 означает R3 и X, Ar, R1, R3, R4a, R4b и R5 имеют значения, определенные в п.1,взаимодействует с соединением общей формулы (VII) в которой А имеет значение, определенное в п.1, для получения соединения общей формулы (I) 17. Соединения общей формулы (VIII) в качестве промежуточных соединений, где Ar, R3, X, R1,R4a, R4b и R5 имеют значения, определенные в п.16.
МПК / Метки
МПК: C07D 231/38, A61K 31/415, A61P 35/00
Метки: применение, получения, композиции, пиразолы, способ, замещенные, содержащие
Код ссылки
<a href="https://eas.patents.su/25-19454-zameshhennye-pirazoly-kompozicii-ih-soderzhashhie-sposob-polucheniya-i-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные пиразолы, композиции их содержащие, способ получения и применение</a>
Замещенные 7-азаиндазолы, содержащие их композиции, способ получения и применение

Номер патента: 19302
Опубликовано: 28.02.2014
Авторы: Патек Марсель, Додсон Марк, Бьергард Кирстен, Акерман-Беррьер Марта, Ронан Батист, Наир Анил, Табар Мишель, Вивьяни Фабрис, Смрчина Мартин, Суай Катрин, Бак Эрик, Леруа Венсан
МПК: C07D 213/85, A61K 31/437, A61P 35/00...
Метки: применение, получения, способ, замещенные, 7-азаиндазолы, содержащие, композиции
Формула / Реферат:
1. Соединение следующей общей формулы (I):в которой:1) А представляет собой фенил, необязательно замещенный одним или двумя заместителями, выбранными из галогена, (С1-С4)-алкила, (C1-C3)-алкилгалогена, О(C1-C4)-алкила, S-(C1-C4)-алкила, О(С1-С4)-алкилгалогена, S-(C1-C4)-алкилгалогена;2) Ar выбирают из группы, состоящей из фенила, тиазолила, тиенила, фурила, пирролила, оксазолила, изоксазолила, изотиазолила, тиадиазолила, пиразолила, имидазолила,...
Замещённые индолы, содержащие их композиции, способ получения и применение

Номер патента: 12613
Опубликовано: 30.10.2009
Авторы: Бак Эрик, Алле Франк, Филош-Ромм Брюно, Ронан Батист, Леталлек Жан-Филипп, Вивьяни Фабрис, Табар Мишель, Суай Катрин
МПК: A61K 31/407, C07D 209/12, A61K 31/404...
Метки: способ, композиции, содержащие, замещённые, индолы, получения, применение
Формула / Реферат:
1. Продукт, отвечающий следующей формуле (I) где a) Ar обозначает фенил; b) А обозначает фенил, пиразол или изоксазол, в случае необходимости замещенный; c) R1 означает Н; d) X означает N или N-оксид или СН; e) L обозначает NH-CO-NH или NH-SO2; f) R5, R6, R7 и R12, каждый независимо, выбирают из группы, состоящей из Н, NO2, O(R2), N(R2)(R3), NHC(О)R2N(R3)(R4); где каждый R2, R3, R4 независимо выбирают из группы, состоящей из Н, (C1-C4)алкила, в...
Замещённые пироллы, содержащие их композиции, способ получения и применение

Номер патента: 13060
Опубликовано: 26.02.2010
Авторы: Суай Катрин, Бак Эрик, Демазо Паскаль, Ронан Батист, Вивьяни Фабрис, Табар Мишель, Летталек Жан-Филипп
МПК: C07D 207/34, A61P 35/00, A61K 31/40...
Метки: композиции, замещённые, содержащие, получения, пироллы, применение, способ
Формула / Реферат:
1. Соединение, отвечающее следующей формуле (I)в которой1) Ar является фенилом;2) A является необязательно замещенным фенилом;3) L является NH-CO-NH;4) Ra является Н;5) R1 является Н;6) R2 независимо выбраны из группы, состоящей из Н, галогена, R'2, O(R'2), NH(R'2), NHCO(R'2), NHCONH(R'2), CONH2; причем каждый из R'2 независимо выбран из группы, состоящей из Н, (С1-С3)алкила, (С1-С3)алкилена, (С3-С5)циклоалкила, замещенного (С1-С3)алкила,...
Замещённые пирролы и имидазолы, содержащие их композиции, способ получения и применение

Номер патента: 12267
Опубликовано: 28.08.2009
Авторы: Суай Катрин, Бак Эрик, Алле Франк, Табар Мишель, Ронан Батист, Вивьяни Фабрис, Эль Амад Юссеф
МПК: A61K 31/40, A61K 31/4164, A61K 35/00...
Метки: содержащие, получения, композиции, имидазолы, способ, пирролы, применение, замещённые
Формула / Реферат:
1. Соединение, отвечающее следующей формуле (I): в которой: 1) А обозначает фенил, необязательно замещенный одним или несколькими одинаковыми или разными заместителями, независимо выбранными из группы, включающей F, Cl, CH3, OCH3, OCF3; 2) Ar обозначает фенил; 3) L обозначает NH-CO-NH; 4) Ra обозначает Н; 5) R1 обозначает Н; 6) X выбирают из CR3 и N; 7) R2 выбирают из Н и галогена, 8) R3 выбирают из группы, состоящей из Н, галогена, NH2,...
Стероиды, замещенные в положении 11, способ их получения, их применение в качестве лекарственного средства и содержащие их фармацевтические композиции

Номер патента: 1868
Опубликовано: 22.10.2001
Авторы: Тетш Жан-Жорж, Ван Де Вельд Патрик, Ник Франсуа, Буали Иамина
МПК: C07J 41/00, A61P 19/10, A61K 31/565...
Метки: средства, применение, получения, стероиды, замещенные, способ, фармацевтические, лекарственного, композиции, качестве, положении, содержащие
Формула / Реферат:
1. Соединения общей формулы в которой n - целое число, равно 2 или 3, либо R1 и R2, идентичные или разные, означают атом водорода или алкил, содержащий от 1 до 4 атомов углерода, либо R1 и R2 образуют вместе с атомом азота, с которым они связаны, моно- или полициклический гетероцикл, имеющий от 5 до 15 звеньев, ароматический или не ароматический, содержащий, при необходимости, от 1 до 3 дополнительных гетероатомов, выбираемых из кислорода,...
Предыдущий патент: Твердая композиция для очистки поверхности, способ ее производства и применение для гигиенической обработки унитаза
Следующий патент: Вязкоупругий разделитель буровых растворов в скважине
Случайный патент: Производные пиразолотриазинов, фармацевтические композиции, содержащие их, способы лечения