Новые разветвленные замещенные аминопроизводные 3-амино-1-фенил-1h[1,2,4]триазола, способы их получения и содержащие их фармацевтические композиции
Номер патента: 4957
Опубликовано: 28.10.2004
Авторы: Галли Даниэль, Роже Пьер, Жеслен Мишель, Маффран Жан-Пьер


















Формула / Реферат
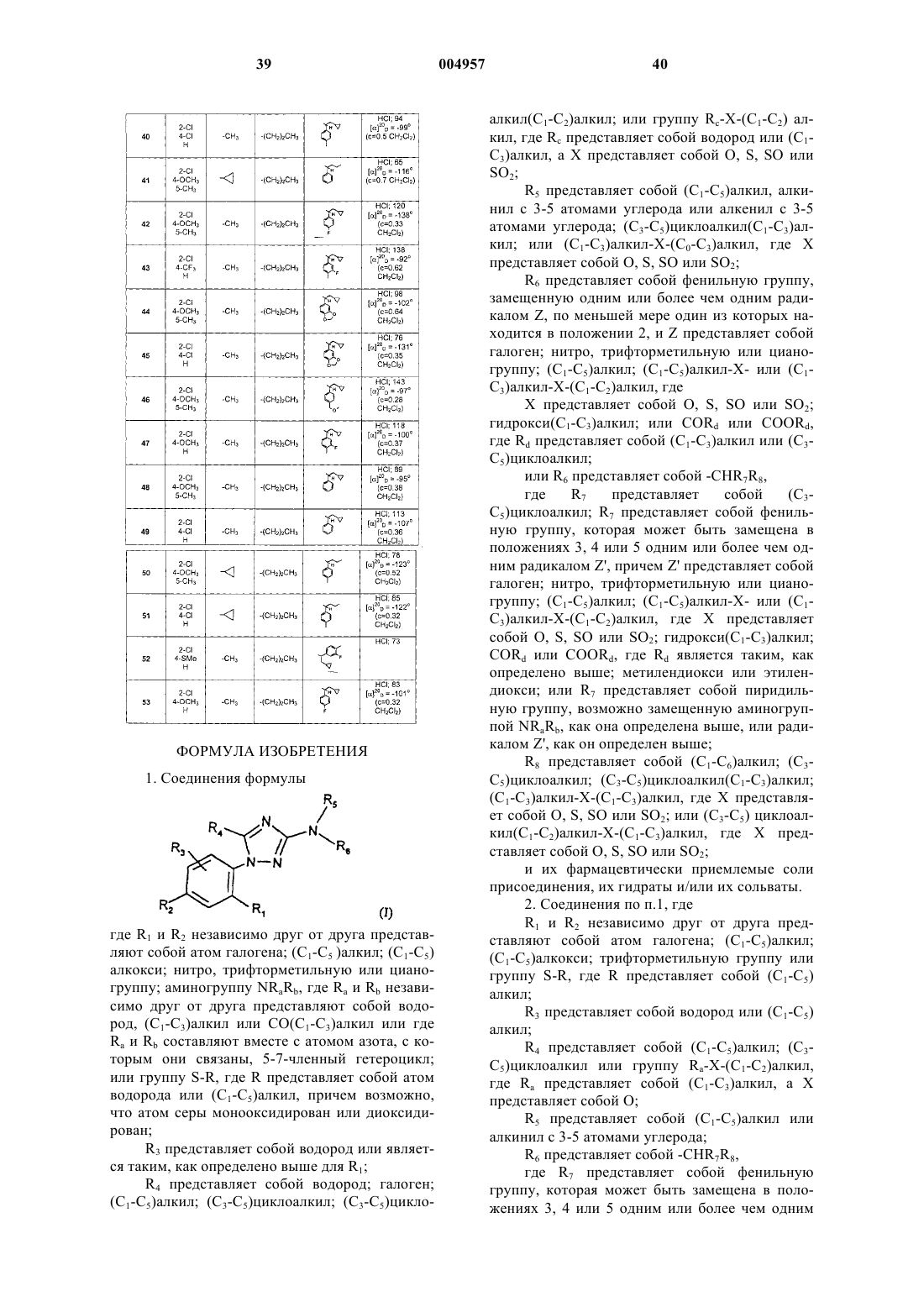
1. Соединения формулы

где R1 и R2 независимо друг от друга представляют собой атом галогена; (C1-C5 )алкил; (C1-C5)алкокси; нитро, трифторметильную или цианогруппу; аминогруппу NRaRb, где Ra и Rb независимо друг от друга представляют собой водород, (C1-C3)алкил или CO(C1-C3)алкил или где Ra и Rb составляют вместе с атомом азота, с которым они связаны, 5-7-членный гетероцикл; или группу S-R, где R представляет собой атом водорода или (C1-C5)алкил, причем возможно, что атом серы монооксидирован или диоксидирован;
R3 представляет собой водород или является таким, как определено выше для R1;
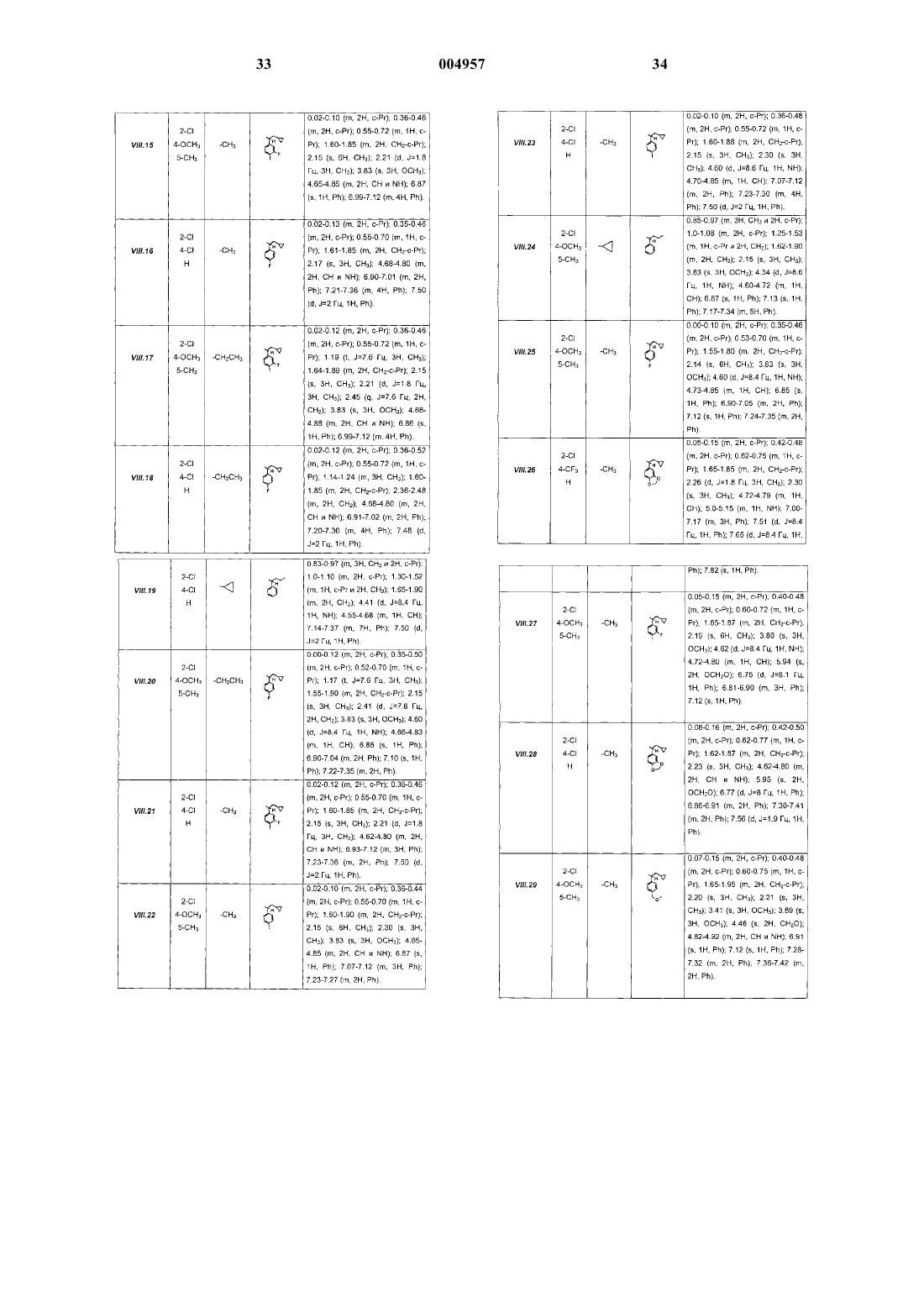
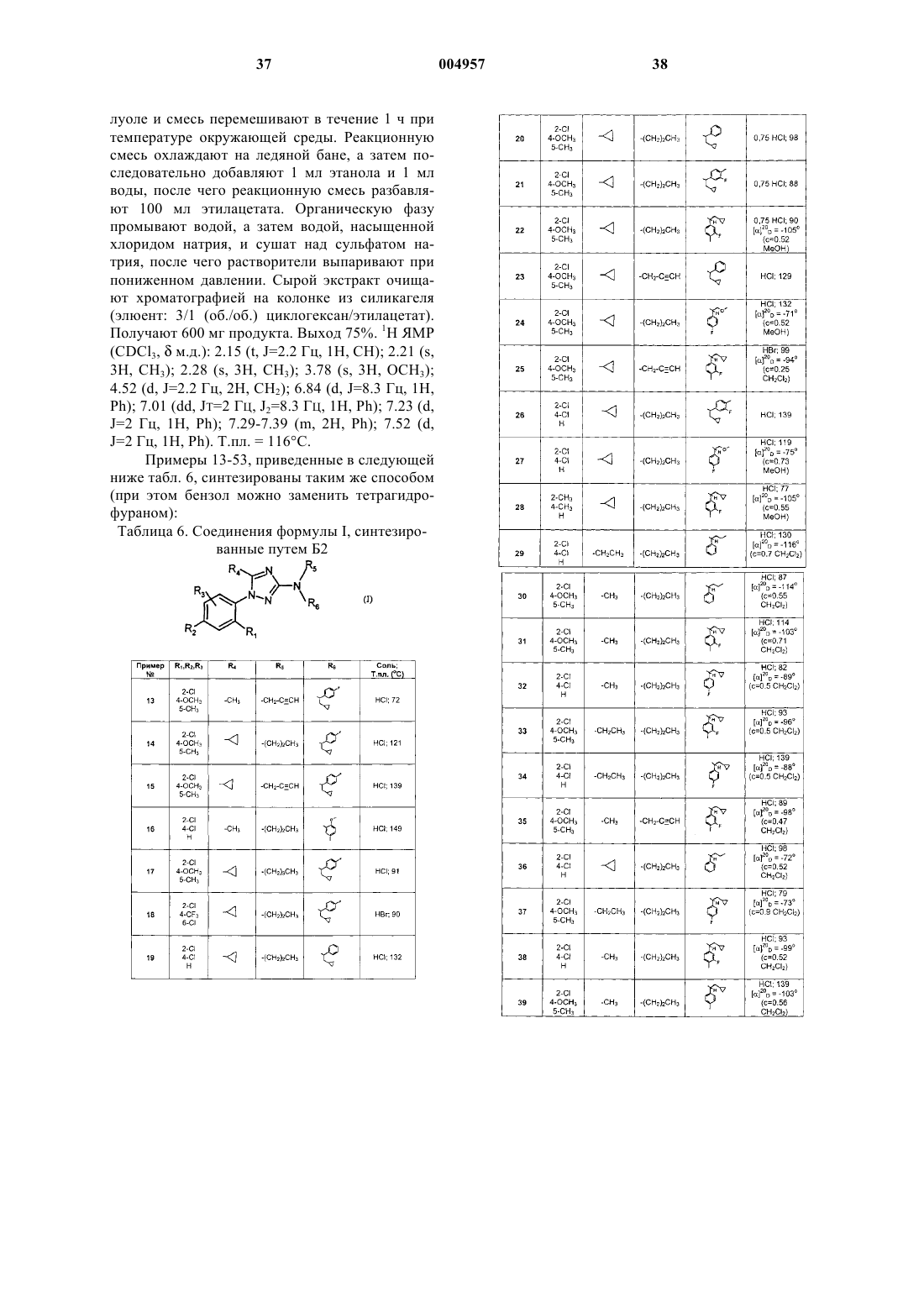
R4 представляет собой водород; галоген; (C1-C5)алкил; (C3-C5)циклоалкил; (C3-C5)циклоалкил(C1-C2)алкил; или группу Rc-X-(C1-C2) алкил, где Rc представляет собой водород или (C1-C3)алкил, а X представляет собой O, S, SO или SO2;
R5 представляет собой (C1-C5)алкил, алкинил с 3-5 атомами углерода или алкенил с 3-5 атомами углерода; (C3-C5)циклоалкил(C1-C3)алкил; или (C1-C3)алкил-X-(C0-C3)алкил, где X представляет собой O, S, SO или SO2;
R6 представляет собой фенильную группу, замещенную одним или более чем одним радикалом Z, по меньшей мере один из которых находится в положении 2, и Z представляет собой галоген; нитро, трифторметильную или цианогруппу; (C1-C5)алкил; (C1-C5)алкил-X- или (C1-C3)алкил-X-(C1-C2)алкил, где
X представляет собой O, S, SO или SO2; гидрокси(C1-C3)алкил; или CORd или COORd, где Rd представляет собой (C1-C3)алкил или (C3-C5)циклоалкил;
или R6 представляет собой -CHR7R8, где R7 представляет собой (C3-C5)циклоалкил; R7 представляет собой фенильную группу, которая может быть замещена в положениях 3, 4 или 5 одним или более чем одним радикалом Z', причем Z' представляет собой галоген; нитро, трифторметильную или цианогруппу; (C1-C5)алкил; (C1-C5)алкил-X- или (C1-C3)алкил-X-(C1-C2)алкил, где X представляет собой O, S, SO или SO2; гидрокси(C1-C3)алкил; CORd или COORd, где Rd является таким, как определено выше; метилендиокси или этилендиокси; или R7 представляет собой пиридильную группу, возможно замещенную аминогруппой NRaRb, как она определена выше, или радикалом Z', как он определен выше;
R8 представляет собой (C1-C6)алкил; (C3-C5)циклоалкил; (C3-C5)циклоалкил(C1-C3)алкил; (C1-C3)алкил-X-(C1-C3)алкил, где X представляет собой O, S, SO или SO2; или (C3-C5)циклоалкил(C1-C2)алкил-X-(C1-C3)алкил, где X представляет собой O, S, SO или SO2;
и их фармацевтически приемлемые соли присоединения, их гидраты и/или их сольваты.
2. Соединения по п.1, где
R1 и R2 независимо друг от друга представляют собой атом галогена; (C1-C5)алкил; (C1-C5)алкокси; трифторметильную группу или группу S-R, где R представляет собой (C1-C5)алкил;
R3 представляет собой водород или (C1-C5)алкил;
R4 представляет собой (C1-C5)алкил; (C3-C5)циклоалкил или группу Ra-X-(C1-C2)алкил, где Ra представляет собой (C1-C3)алкил, а X представляет собой O;
R5 представляет собой (C1-C5)алкил или алкинил с 3-5 атомами углерода;
R6 представляет собой -CHR7R8, где R7 представляет собой фенильную группу, которая может быть замещена в положениях 3, 4 или 5 одним или более чем одним радикалом Z', причем Z' представляет собой галоген; (C1-C5)алкил; (C1-C5)алкил-X- или (C1-C3)алкил-X-(C1-C2)алкил, где X представляет собой O; или метилендиоксигруппу;
R8 представляет собой (C1-C6)алкил; (C3-C5)циклоалкил(C1-C3)алкил; или (C1-C3)алкил-X-(C1-C3)алкил, где X представляет собой O;
и их фармацевтически приемлемые соли присоединения, их гидраты и/или их сольваты.
3. Соединения по любому из пп.1 и 2, где R5 представляет собой пропильную или пропаргильную группу.
4. Соединения по любому из пп.1-3 в энантиомерной форме.
5. Соединения по любому из пп.1-4, выбранные из
5-циклопропил-N-[2-циклопропил-1-(4-фторфенил)этил]-1-(2,4-дихлорфенил)-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
N-[2-циклопропил-1-(4-фторфенил)этил]-1-(2,4-дихлорфенил)-5-(метоксиметил)-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
N-[2-циклопропил-1-(4-фторфенил)этил]-1-[2,6-дихлор-4-(трифторметил)фенил]-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-N-[2-циклопропил-1-(4-фторфенил)этил]-5-метил-N-(2-пропинил)-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[2-циклопропил-1-(4-фторфенил)этил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[2-циклопропил-1-(4-фторфенил)этил]-N-(2-пропинил)-1H-1,2,4-триазол-3-амина гидрохлорида,
5-циклопропил-N-[2-циклопропил-1-(4-фторфенил)этил]-1-[2,6-дихлор-4-(трифторметил)фенил]-N-пропил-1H-1,2,4-триазол-3-амина гидробромида,
5-циклопропил-N-(2-циклопропил-1-фенилэтил)-1-(2,4-дихлорфенил)-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-(2-циклопропил-1-фенилэтил)-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-(2-циклопропил-1-фенилэтил)-N-(2-пропинил)-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[(1R)-1-(4-фторфенил)-2-метоксиэтил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-N-(2-пропинил)-1H-1,2,4-триазол-3-амина гидробромида,
5-циклопропил-N-[2-циклопропил-1-(3-фтор-4-метилфенил)этил]-1-(2,4-дихлорфенил)-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
5-циклопропил-1-(2,4-дихлорфенил)-N-[(1R)-1-(4-фторфенил)-2-метоксиэтил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
5-циклопропил-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-1-(2,4-диметилфенил)-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2,4-дихлорфенил)-5-этил-N-[(1S)-1-фенилбутил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-5-метил-N-[(1S)-1-фенилбутил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
N-[(1S)-2-циклопропил-1-(4-фторфенил)этил]-1-(2,4-дихлорфенил)-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-этил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
N-[(1S)-2-циклопропил-1-(4-фторфенил)этил]-1-(2,4-дихлорфенил)-5-этил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-(2-пропинил)-1H-1,2,4-триазол-3-амина гидрохлорида,
5-циклопропил-1-(2,4-дихлорфенил)-N-[(1S)-1-фенилбутил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(4-фторфенил)этил]-5-этил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-1-(2,4-дихлорфенил)-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(4-метилфенил)этил]-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
N-[(1S)-2-циклопропил-1-(4-метилфенил)этил]-1-(2,4-дихлорфенил)-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[(1S)-1-фенилбутил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-(4-фторфенил)этил]-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-[2-хлор-4-(трифторметил)фенил]-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
N-[(1S)-1-(1,3-бензодиоксол-5-ил)-2-циклопропилэтил]-1-(2-хлор-4-метокси-5-метилфенил)-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
N-[(1S)-1-(1,3-бензодиоксол-5-ил)-2-циклопропилэтил]-1-(2,4-дихлорфенил)-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-N-{(1S)-2-циклопропил-1-[(4-метоксиметил)фенил]этил}-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метоксифенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-N-[(1S)-2-циклопропил-1-фенил)этил]-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
N-[(1S)-(2-циклопропил-1-фенил)этил]-1-(2,4-дихлорфенил)-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[(1S)-1-(4-метилфенил)бутил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
5-циклопропил-1-(2,4-дихлорфенил)-N-[(1S)-1-(4-метилфенил)бутил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,
1-[2-хлор-4-(метилсульфанил)фенил]-N-[2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-пропил-1 H-1,2,4-триазол-3-амина гидрохлорида,
1-(2-хлор-4-метоксифенил)-N-[(1S)-2-циклопропил-1-(4-фторфенил)этил]-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида и
соответствующих оснований, других фармацевтически приемлемых солей присоединения, их сольватов и/или их гидратов.
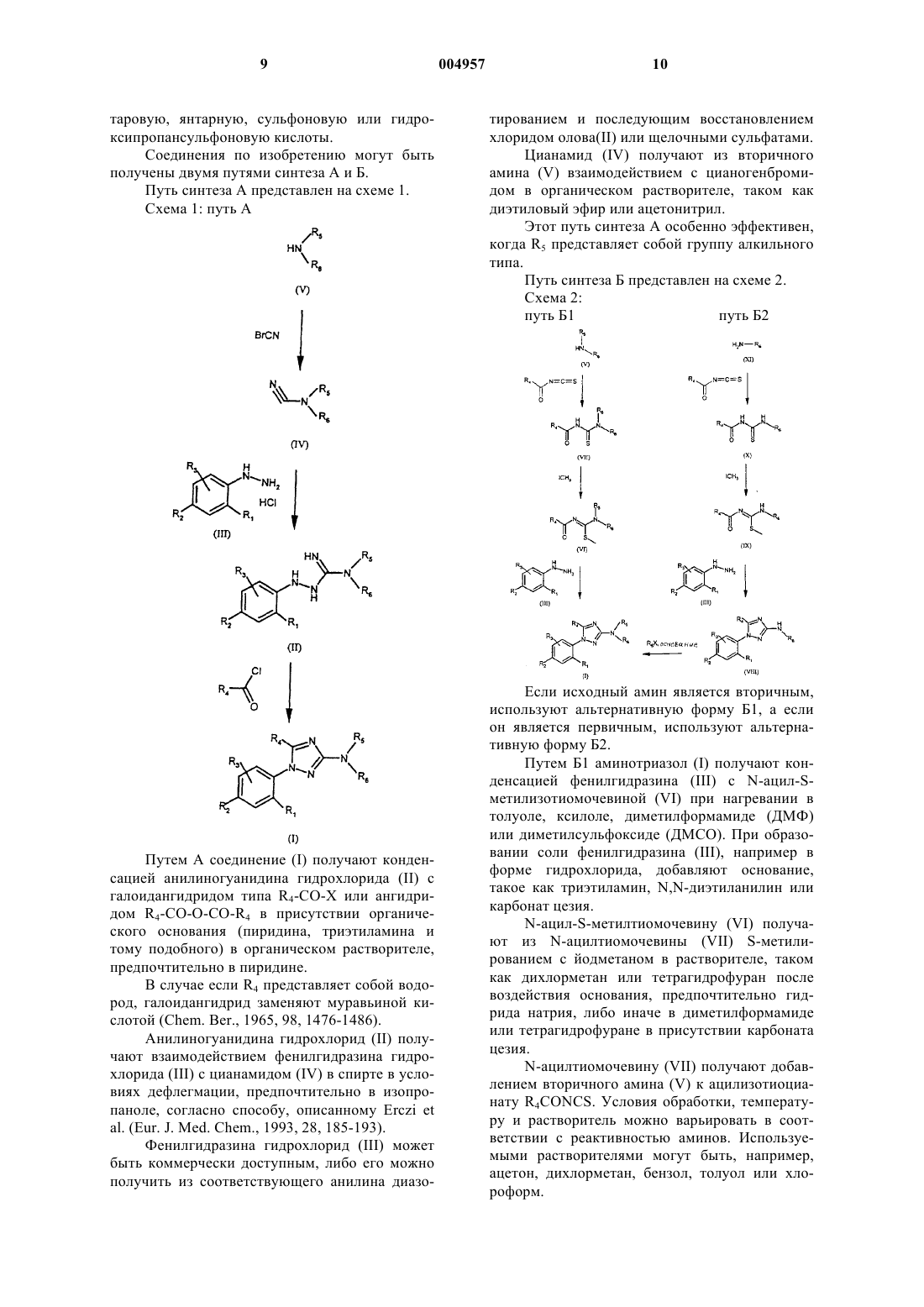
6. Способ получения соединений формулы (I) по п.1, отличающийся тем, что соединение формулы

где R1, R2, R3, R5 и R6 являются такими, как определено для (I), подвергают взаимодействию с соединением формулы R4COX (где X представляет собой галоген) с получением соединения формулы (I).
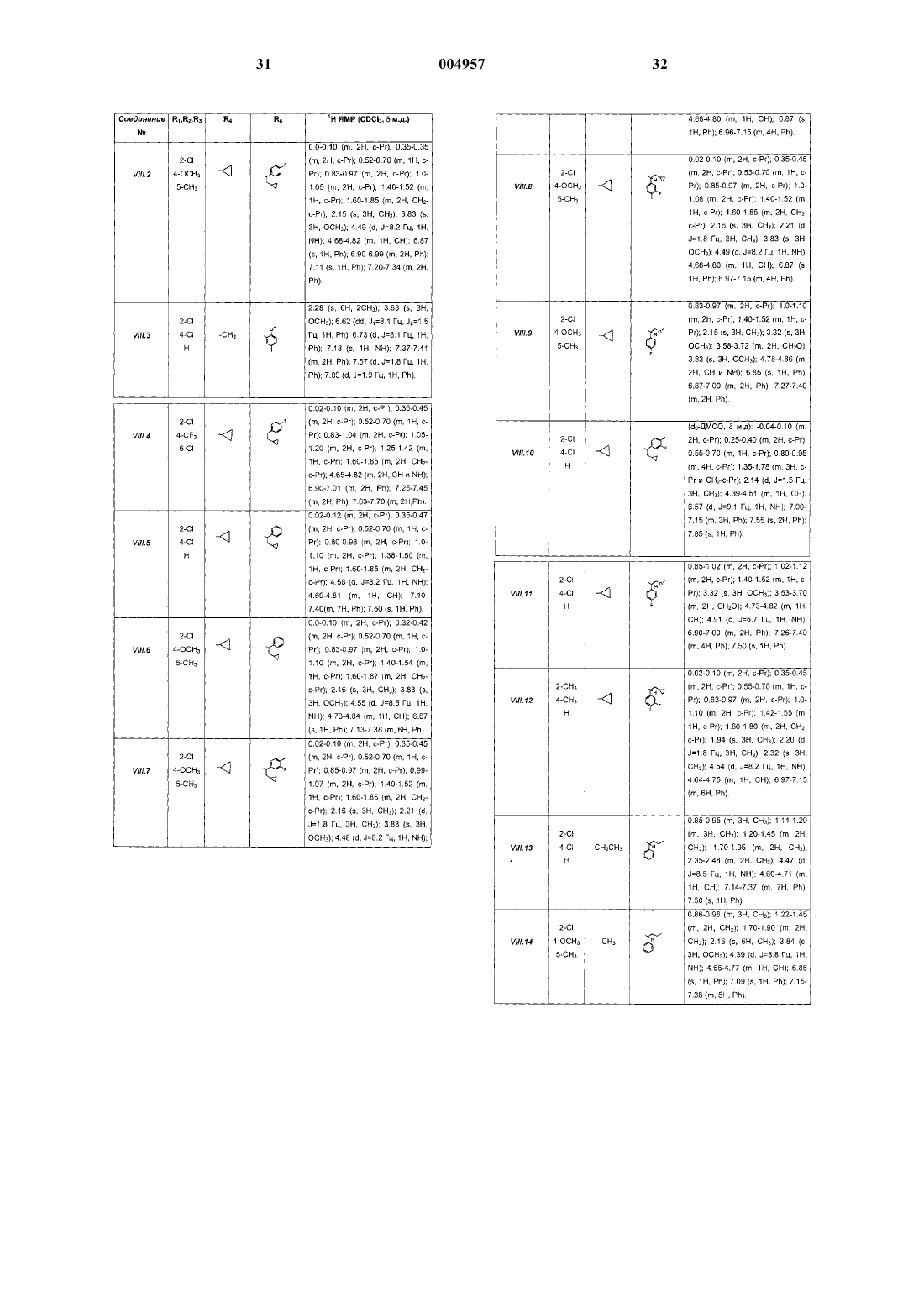
7. Способ получения соединений формулы (I) по п.1, отличающийся тем, что соединение формулы (VIII)

где R1, R2, R3, R4 и R6 являются такими, как определено для (I), подвергают реакции алкилирования с соединением формулы R5X (где X представляет собой галоген) с получением соединения формулы (I).
8. Способ получения соединений формулы (I) по п.1 и соединений формулы (VIII), отличающийся тем, что соединения формулы (VI) или (IX)

где R4, R5 и R6 являются такими, как определено для (I), подвергают взаимодействию с соединением формулы (III)

где R1, R2 и R3 являются такими, как определено для (I), с получением соответственно соединений формулы (I) или формулы (VIII).
9. Фармацевтическая композиция, отличающаяся тем, что она содержит в качестве действующего начала соединение по любому из пп.1-5 в комбинации с одним или более чем одним подходящим эксципиентом.
10. Применение соединения по любому из пп.1-5 в приготовлении лекарств, предназначенных для предупреждения и/или лечения кортикотропин-рилизинг-фактор (КРФ)-зависимых заболеваний.
Текст
1 Объектами настоящего изобретения являются новые разветвленные замещенные аминопроизводные 3-амино-1-фенил-1 Н-[1,2,4]триазола, способы их получения и содержащие их фармацевтические композиции. Эти новые производные триазола обладают антагонистической активностью по отношению к КРФ и могут, следовательно, быть действующими началами фармацевтических композиций. Кортикотропин-рилизинг-фактор(КРФ) представляет собой пептид, последовательность которого из 41 аминокислоты была определенаVale W. et al. в 1981 году (Science, 1981, 213,1394-1397). КРФ является главным эндогенным фактором, вовлеченным в регуляцию системы гипоталамус-гипофиз-надпочечники (высвобождение адренокортикотропного гормона(АКТГ и ее патологий, а также в депрессивные синдромы, которые являются результатом ее дисфункции. КРФ также вызывает секрецию эндорфина, -липотропина и кортикостерона. Таким образом, КРФ является физиологическим регулятором секреции адренокортикотропного гормона (АКТГ) и кортизола под воздействием АКТГ на уровне надпочечников и вообще пептидов, производных от проопиомеланокортина(ПОМК). Кроме локализации в гипоталамусе,КРФ широко распространен в центральной нервной системе, а также в тканях вне нервной системы, таких как надпочечниковые железы и семенники. Присутствие КРФ было продемонстрировано также во время воспалительного процесса. В ряде экспериментов на животных было показано, что центральное введение КРФ вызывает различные анксиогенные эффекты, такие как модификация поведения в целом, например неофобия, снижение половой восприимчивости и снижение потребления пищи и медленноволнового сна у крыс. Интрацеребровентрикулярная инъекция КРФ также повышает возбуждение норадренергических нейронов голубого пятна, которое часто связано у животных с состоянием тревоги. У крысы центральное и периферическое введение КРФ или родственных пептидов (например урокортина, саувагина) индуцирует в дополнение к центральным эффектам, таким как повышение настороженности и эмоциональной реактивности по отношению к окружающим, изменения в опорожнении желудка, в секреции кислоты, во времени прохождения содержимого через кишечник и в экскреции фекалий, а также эффекты напряжения. КРФ также вовлечен в комплексную регуляцию воспалительных ответов, с одной стороны, играя провоспалительную роль в некоторых животных моделях (дегрануляция тучных клеток,приводящая к высвобождению воспалительных молекул, таких как гистамин, простагландины и тому подобные), и, с другой стороны, как инги 004957 2 битор эффектов, вызываемых повышением проницаемости сосудов в результате воспаления. Использование пептида-антагониста, спирального КРФ (9-41) (Н-КРФ) или специфичных антител (Rivier J. et al., Science, 1984,224, 889-891) дало возможность подтвердить роль этого пептида во всех этих эффектах. Эти эксперименты также подтвердили важную роль КРФ у человека в интеграции комплексных реакций, наблюдаемых во время физиологического, психологического или иммунологического стресса, одновременно на нейроэндокринном,висцеральном и поведенческом уровнях (MorleySmith M. A. et al., Horm. Res., 1989, 31, 66-71). Кроме того, клинические данные подтверждают действенное вовлечение КРФ во множество расстройств, являющихся результатом состояния стресса (Gulley L. R. et al., J. Clin. Psychiatry,1993, 54, 1 (suppl.), 16-19), например:- существование теста на КРФ (внутривенное (в.в.) введение) у человека дало возможность продемонстрировать модификацию АКТГ-ответа у пациентов с депрессией (Breier- обнаружение эндогенной гиперсекреции КРФ при некоторых патологиях, например при высоком уровне КРФ в спинно-мозговой жидкости у не подвергавшихся лечению лекарственными средствами пациентов, которые страдают депрессией или поражены болезнью типа деменции Альцгеймера (Nemeroff С. В. et al.,Science 1984, 226, 4680, 1342-1343; Regul. Pept.,1989, 25, 123-130), или пониженной плотности рецепторов КРФ в коре головного мозга жертв суицида (Nemeroff et al., Arch. Gen. Psychiatry,1988, 45, 577-579);- предполагается также дисфункция КРФзависимых нейронов при тяжелых патологиях,которыми являются болезни Альцгеймера и Паркинсона, хорея Хантингтона и боковой амиотрофический склероз (De Souza E. В., Hospital Practice, 1988, 23, 59). Центральное введение КРФ у многих видов животных приводит к поведенческим эффектам, подобным эффектам, наблюдаемым у человека в стрессовых ситуациях. Если эти эффекты повторяются с течением времени, то они могут привести к различным патологиям, таким как утомление, гипертензия, сердечные расстройства и расстройства давления, изменение опорожнения желудка и экскреции фекалий (колит, раздраженный кишечник), изменение секреции кислоты, гипергликемия, задержка роста,анорексия, неофобия, мигрени, репродуктивные расстройства, иммуносупрессия (воспалительные процессы, множественные инфекции и рак) и различные нейропсихиатрические расстройства (депрессия, нервная анорексия или нервная булимия и тревога). 3 Инъекция интрацеребровентрикулярным путем известного пептида-антагониста, Н-КРФ(9-41), предупреждает эффекты, получаемые либо при введении экзогенного КРФ, либо при использовании агентов, индуцирующих стресс(эфира, лишения свободы, шума, электрошока,отлучения от этанола или операции), которые сами по себе способны индуцировать повышение уровня эндогенного КРФ. Эти результаты подтверждены исследованием многих пептидных молекул-антагонистов, которые являются структурно родственными КРФ и которые обладают пролонгированной продолжительностью действия относительно Н-КРФ (9-41) (Rivier J.et al., J. Med. Chem., 1993, 36, 2851-2859; Menzaghi F. et al., J. Pharmacol. Exp. Ther., 1994, 269,2, 564-572; Hernandez J. F. et al., J. Med. Chem.,1993, 36, 2860-2867). Такие пептидные соединения-антагонисты КРФ раскрыты, например, в патентах US 5109111, US 5132111 и US 5245009 и в патентных заявках WO 92/22576 и WO 96/19499. Кроме того, предшествующие исследования показали, что трициклические антидепрессанты могут модулировать уровень КРФ и число рецепторов КРФ в головном мозге (Grigiriadis D. E. et al., Neuropsychopharmacology, 1989,2, 53-60). Подобным образом, бензодиазепиновые анксиолитики способны ингибировать эффект КРФ (Britton К. Т. et al., Psychopharmacology, 1988, 94, 306) без полного объяснения механизма действия этих веществ. Эти результаты подтверждают, если необходимо, возрастающую потребность в непептидных молекулахантагонистах рецепторов КРФ. Важно также подчеркнуть три возможных последствия состояний хронического стресса,которые представляют собой иммунодепрессию,нарушение способности к воспроизведению потомства и развитие диабета. КРФ оказывает такие воздействия, взаимодействуя со специфичными мембранными рецепторами, которые были определены в гипофизе и в головном мозге множества видов(мышь, крыса и человек), а также в сердце, в скелетных мышцах (крыса, мышь) и в миометрии и плаценте во время беременности. Соединения 3-амино-1-фенил-1 Н-[1,2,4] триазола широко не представлены. Можно упомянуть, в частности, соединения, несущие фенил в положении 5, раскрытые Ebenreth A. et al.G. and Janaky J. (Arch. Pharm., 1989, 322(10),583-7, патентыHU 44522, HU 195791, 1986),которые заявляют о противовоспалительныхпротиворевматических свойствах. В двух японских патентах (JP 02091061 и JP 2729810, 1988),Inamori et al. заявляют получение 3-амино-1 фенил-1H-[1,2,4]триазолов в качестве инсектицидов. 4 В патентной заявке корпорации Neurocrine,опубликованной под номером WO 96/39400,соединения 3-амино-5-фенил-1 Н-[1,2,4]триазола раскрыты как антагонисты рецепторов КРФ. Теперь согласно настоящему изобретению обнаружено, что некоторые производные 3 амино-1-фенил-1H-[1,2,4]триазола, которые являются объектом настоящего изобретения, обладают великолепным сродством к рецепторам КРФ. К тому же, благодаря своей структуре эти молекулы обладают хорошей диспергируемостью и/или растворимостью в растворителях или растворах, обычно используемых в терапии,которые способствуют их фармакологической активности, а также дают возможность легко изготавливать пероральные и парентеральные фармацевтические лекарственные формы. Объектом настоящего изобретения являются соединения в рацемической или энантиомерной форме формулы где R1 и R2 независимо друг от друга представляют собой атом галогена; (С 1-С 5)алкил; (С 1 С 5)алкокси; нитро, трифторметильную или цианогруппу; аминогруппу NRaRb, где Ra и Rb независимо друг от друга представляют собой водород, (С 1-С 3)алкил или СО(С 1-С 3)алкил или гдеRa и Rb составляют вместе с атомом азота, с которым они связаны, 5-7-членный гетероцикл; или группу S-R, где R представляет собой атом водорода или (С 1-С 5)алкил, причем возможно,что атом серы монооксидирован или диоксидирован;R3 представляет собой водород или является таким, как определено выше для R1;(С 1-С 5)алкил; (С 3-С 5)циклоалкил; (С 3-С 5)циклоалкил(С 1-С 2)алкил; или группу Rc-X-(С 1-С 2)алкил, где Rc представляет собой водород или (С 1 С 3)алкил, а X представляет собой О, S, SO илиR5 представляет собой (С 1-С 5)алкил, алкинил с 3-5 атомами углерода или алкенил с 3-5 атомами углерода; (С 3-С 5)циклоалкил(С 1-С 3) алкил; или (С 1-С 3)алкил-Х-(С 0-С 3)алкил, где X представляет собой О, S, SO или SO2;R6 представляет собой фенильную группу,замещенную одним или более чем одним радикалом Z, по меньшей мере один из которых находится в положении 2, и Z представляет собой галоген; нитро, трифторметильную или цианогруппу; (С 1-С 5)алкил; (С 1-С 5)алкил-Х- или (С 1 С 3)алкил-Х-(С 1-С 2)алкил, где X представляет собой О, S, SO или SO2; гидрокси(С 1-С 3)алкил; 5 или CORd, или COORd, где Rd представляет собой (С 1-С 3)алкил или (С 3-С 5)циклоалкил; или R6 представляет собой -CHR7R8, где R7 представляет собой (С 3-С 5)циклоалкил; фенильную группу, которая может быть замещена в положениях 3, 4 или 5 одним или более чем одним радикалом Z', причем Z' представляет собой галоген; нитро, трифторметильную или цианогруппу; (С 1-С 5)алкил; (С 1-С 5)алкил-Х- или (С 1 С 3)алкил-Х-(С 1-С 2)алкил, где X представляет собой О, S, SO или SO2; гидрокси(С 1-С 3)алкил;CORd или COORd, где Rd является таким, как определено выше; метилендиокси или этилендиокси; или пиридильную группу, возможно замещенную аминогруппой NRaRb, как она определена выше, или радикалом Z', как он определен выше; R8 представляет собой (С 1 С 6)алкил; (С 3-С 5)циклоалкил; (С 3-С 5)циклоалкилX представляет собой О, S, SO или SO2; и их фармацевтически приемлемые соли присоединения, их гидраты и/или их сольваты. В настоящем описании под термином галоген подразумевается атом фтора, хлора,брома или йода. Алкильные группы или алкоксигруппы являются линейными или разветвленными. Согласно другому своему аспекту, данное изобретение относится к соединениям формулы(I) в рацемической или энантиомерной форме,где R1 и R2 каждый независимо друг от друг представляет собой атом галогена; (С 1 С 5)алкил; (С 1-С 5)алкокси; трифторметил или группу S-R, где R представляет собой (С 1 С 5)алкил;R3 представляет собой водород или (С 1-С 5) алкил;R6 представляет собой -CHR7R8,где R7 представляет собой фенильную группу, которая может быть замещена в положениях 3, 4 или 5 одним или более чем одним радикалом Z', причем Z' представляет собой галоген; (С 1-С 5)алкил; (С 1-С 5)алкил-Х- или (С 1 С 3)алкил-Х-(С 1-С 2)алкил, где X представляет собой О; или метилендиоксигруппу;R8 представляет собой (С 1-С 6)алкил; (С 3 С 5)циклоалкил(С 1-С 3)алкил; или (С 1-С 3)алкилХ-( С 1-С 3)алкил, где X представляет собой О; и их фармацевтически приемлемым солям присоединения, их гидратам и/или их сольватам. Согласно еще одному своему аспекту,данное изобретение относится к соединениям формулы (I) в рацемической или энантиомерной 6 форме, где R5 представляет собой пропильную или пропаргильную группу, и к их фармацевтически приемлемым солям присоединения, к их гидратам и/или к их сольватам. Более конкретно данное изобретение относится к вышеуказанным соединениям в энантиомерной форме. Согласно еще одному из своих аспектов,данное изобретение относится к следующим соединениям: 5-циклопропил-N-[2-циклопропил-1-(4-фторфенил)этил]-1-(2,4-дихлорфенил)-N-пропил-1H1,2,4-триазол-3-амина гидрохлорид (пример 7),N-[2-циклопропил-1-(4-фторфенил)этил]-1(2,4-дихлорфенил)-5-(метоксиметил)-N-пропил 1H-1,2,4-триазол-3-амина гидрохлорид (пример 9),N-[2-циклопропил-1-(4-фторфенил)этил]-1[2,6-дихлор-4-(трифторметил)фенил]-5-метил-Nпропил-1H-1,2,4-триазол-3-амина гидрохлорид(пример 50),5-циклопропил-1-(2,4-дихлорфенил)-N-[(1S)1-(4-метилфенил)бутил]-N-пропил-1 Н-1,2,4-триазол-3-амина гидрохлорид (пример 51),1-[2-хлор-4-(метилсульфанил)фенил]-N-[2 циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорид (пример 52),1-(2-хлор-4-метоксифенил)-N-[(1S)-2-циклопропил-1-(4-фторфенил)этил]-5-метил-N-пропил 1 Н-1,2,4-триазол-3-амина гидрохлорид (пример 53),к соответствующим основаниям, к другим фармацевтически приемлемым солям присоединения, к их сольватам и/или к их гидратам. Соединения по изобретению в свободной форме обычно проявляют слабоосновные свойства. Соли соединений формулы (I) с фармацевтически приемлемыми кислотами являются предпочтительными солями, но те соли, которые дают возможность выделять соединения формулы (I), в частности очищать их или получать чистые энантиомеры или диастереоизомеры, также являются объектом данного изобретения. Среди фармацевтически приемлемых кислот для получения солей присоединения соединений формулы (I) можно упомянуть соляную, бромоводородную, фосфорную, фумаровую, лимонную, щавелевую, серную, аскорбиновую, винную, малеиновую, миндальную, метансульфоновую, молочную, глюконовую, глу 9 таровую, янтарную, сульфоновую или гидроксипропансульфоновую кислоты. Соединения по изобретению могут быть получены двумя путями синтеза А и Б. Путь синтеза А представлен на схеме 1. Схема 1: путь А Путем А соединение (I) получают конденсацией анилиногуанидина гидрохлорида (II) с галоидангидридом типа R4-CO-X или ангидридом R4-CO-O-CO-R4 в присутствии органического основания (пиридина, триэтиламина и тому подобного) в органическом растворителе,предпочтительно в пиридине. В случае если R4 представляет собой водород, галоидангидрид заменяют муравьиной кислотой (Chem. Ber., 1965, 98, 1476-1486). Анилиногуанидина гидрохлорид (II) получают взаимодействием фенилгидразина гидрохлорида (III) с цианамидом (IV) в спирте в условиях дефлегмации, предпочтительно в изопропаноле, согласно способу, описанному Erczi etal. (Eur. J. Med. Chem., 1993, 28, 185-193). Фенилгидразина гидрохлорид (III) может быть коммерчески доступным, либо его можно получить из соответствующего анилина диазо 004957 10 тированием и последующим восстановлением хлоридом олова(II) или щелочными сульфатами. Цианамид (IV) получают из вторичного амина (V) взаимодействием с цианогенбромидом в органическом растворителе, таком как диэтиловый эфир или ацетонитрил. Этот путь синтеза А особенно эффективен,когда R5 представляет собой группу алкильного типа. Путь синтеза Б представлен на схеме 2. Схема 2: путь Б 1 путь Б 2 Если исходный амин является вторичным,используют альтернативную форму Б 1, а если он является первичным, используют альтернативную форму Б 2. Путем Б 1 аминотриазол (I) получают конденсацией фенилгидразина (III) с N-ацил-Sметилизотиомочевиной (VI) при нагревании в толуоле, ксилоле, диметилформамиде (ДМФ) или диметилсульфоксиде (ДМСО). При образовании соли фенилгидразина (III), например в форме гидрохлорида, добавляют основание,такое как триэтиламин, N,N-диэтиланилин или карбонат цезия.N-ацил-S-метилтиомочевину (VI) получают из N-ацилтиомочевины (VII) S-метилированием с йодметаном в растворителе, таком как дихлорметан или тетрагидрофуран после воздействия основания, предпочтительно гидрида натрия, либо иначе в диметилформамиде или тетрагидрофуране в присутствии карбоната цезия.N-ацилтиомочевину (VII) получают добавлением вторичного амина (V) к ацилизотиоцианату R4CONCS. Условия обработки, температуру и растворитель можно варьировать в соответствии с реактивностью аминов. Используемыми растворителями могут быть, например,ацетон, дихлорметан, бензол, толуол или хлороформ. 11 Ацилизотиоцианат R4CONCS получают заранее взаимодействием между ацилгалогенидом R4COCl и щелочным тиоцианатом (например тиоцианатом аммония), как правило, в ацетоне. Путем Б 2 аминотриазол (I) получают изNH аминотриазола (VIII) реакцией алкилирования. Условия реакции варьируют в соответствии с реактивностью вторичного амина и галогенида. Используемыми системами могут быть гидрид натрия/ДМФ, гидрид калия/бензол/краунэфир, трет-бутилат щелочного металла/ДМСО. Условия синтеза для получения NH аминотриазола (VIII) из первичного амина (XI) через промежуточную N-алкилтиомочевину (X), а затем N-ацил-S-метилтиомочевину (IX) подобны описанным для пути Б 1. Соединения формулы (I) образуют соли,обычно в форме гидрохлорида. Эти гидрохлориды могут находиться в форме кристаллических или аморфных твердых веществ, получаемых осаждением из растворителя (или смеси растворителей), такого как пентан, диизопропиловый эфир или диэтиловый эфир. Используемые первичные амины (XI) и вторичные амины (V), если они имеют асимметрический атом углерода (R6=CH(R8)R7), могут быть в рацемической или энантиомерной форме. Из обычных способов синтеза первичных аминов использовали, в частности, два способа: один из аминокислоты, другой из замещенного фенилкетона (1). В последнем случае синтез проводят согласно приведенной ниже схеме 3. Замещенный фенилкетон (1) превращают в оксим (2), а затем в бензилоксим (3). Этот бензилоксим (3) восстанавливают либо алюмогидридом лития, чтобы получить рацемический амин,либо хиральным комплексом, таким как хиральный комплекс оксазаборолидинборан, чтобы получить амин в форме энантиомера. Схема 3 Приведенные выше соединения формулы(I) также включают соединения, в которых один или более чем один атом водорода или углерода заменен его радиоактивным изотопом, например тритием или углеродом-14. Такие меченые соединения находят применение в исследовательской, метаболической или фармакокинетической работе или, альтернативно, в биохимических анализах в качестве лигандов рецепторов. Соединения по настоящему изобретению стали предметом биохимических и фармакологических исследований. Они обладают весьма предпочтительными фармакологическими свой 004957 12 ствами. Соединения по изобретению вытесняют при концентрациях менее чем 10 мкМ связывание йодированного КРФ (кортикотропинрилизинг-фактора) или родственных пептидов(уротензина, саувагина), например 125I-Tyr-КРФ,с рецепторами, находящимися на мозговых мембранах или на культивируемых клетках,согласно способу, описанному Е. В. De Souza (J.Neurosci., 1987, 7, 1, 88-100). Антагонистическая активность соединений по изобретению продемонстрирована их способностью ингибировать некоторые виды активности, связанные с КРФ. В частности, соединения формулы (I) способны ингибировать секрецию кортикотропина (АКТГ), индуцированную посредством КРФ. Исследование на секрецию АКТГ, индуцированную посредством КРФ, было проведено in vivo на крысах, находящихся в сознании, согласно адаптированному способу С. Rivier et al., Endocrinology, 1982, 110(1), 272-278. КРФ представляет собой нейропептид, который регулирует активность системы гипоталамус-гипофиз-надпочечники. Этот фактор ответственен за эндокринные и поведенческие реакции, связанные со стрессом. Фактически показано, что КРФ может модулировать поведение, а также некоторые функции автономной нервной системы (G. F.Brown, L. A. Fisher, Fed. Proc, 1985, 44, 243). Более конкретно, КРФ индуцирует секрецию кортикотропина (АКТГ), -эндорфинов и других пептидов, производных от проопиомеланокортина (A. Tazi et al., Regul. Peptides, 1987, 18,37; M. R. Brown et al., Regul. Peptides, 1986, 16,321; С L. Williams et al., Am. J. Physiol., 1987, G 582, 253). Таким образом, соединения по изобретению можно применять для регуляции секреции этих эндогенных веществ. Более конкретно, их можно применять в качестве действующего начала в лекарствах для снижения реакции на стресс (поведения, эмоциональных состояний,желудочно-кишечных и сердечно-сосудистых расстройств или расстройств иммунной системы) и, более широко, при патологиях, в которые вовлечен КРФ, например при психиатрических расстройствах, тревоге, депрессии, нервной анорексии и нервной булимии, эпилепсии, расстройствах половой активности и фертильности,болезни Альцгеймера или других. Соединения по изобретению очень стабильны и поэтому особенно подходят в качестве действующего начала лекарств. Изобретение также направлено на фармацевтические композиции, содержащие в качестве действующего начала соединение формулы(I) или одну из его фармацевтически приемлемых солей, возможно в комбинации с одним или более чем одним подходящим инертным эксципиентом. 13 В каждой дозировочной единице действующее начало формулы (I) присутствует в количествах, подходящих для предусматриваемых суточных доз. Согласно прогнозируемым дозировке и типу введения соответствующим образом регулируют каждую дозировочную единицу, например таблетки, твердые желатиновые капсулы и тому подобное, пакетики, пузырьки,сиропы и тому подобное, капли или трансдермальные или транмукозальные пластыри, так чтобы такая дозировочная единица содержала от 0,5 до 800 мг действующего начала, предпочтительно от 0,5 до 200 мг, которые должны вводить каждый день. Соединения по изобретению можно также применять в комбинации с другим действующим началом, применяемым в требуемой терапии, таким как, например, анксиолитики, антидепрессанты или анорексигенные агенты. Соединения формулы (I) малотоксичны; их токсичность совместима с их применением в качестве лекарства при лечении вышеуказанных расстройств и заболеваний. Соединения формулы (I) можно включать в фармацевтические композиции для введения млекопитающим, в частности человеку, для лечения вышеуказанных заболеваний. Фармацевтические композиции, полученные таким образом, предпочтительно представлять в различных формах, таких как, например,инъекционные растворы или растворы для перорального приема, драже, таблетки или твердые желатиновые капсулы. Фармацевтические композиции, содержащие по меньшей мере одно соединение формулы (I) или одну из его солей в качестве действующего начала, находят применение, в частности, в превентивном или куративном лечении заболеваний, относящихся к стрессу, и, более широко, в лечении всех патологий, в которые вовлечен КРФ, например таких как болезнь Кушинга, нейропсихиатрические расстройства, такие как депрессия, тревога,панические припадки, посттравматический стресс, обсессивно-компульсивные расстройства, расстройства настроения, поведенческие расстройства, агрессивность, анорексия, булимия, гипергликемия, преждевременные роды,беременность с повышенным риском, задержка роста, расстройства сна, эпилепсия и депрессии всех типов; дегенеративные расстройства: болезнь Альцгеймера или болезнь Паркинсона; хорея Хантингтона и боковой амиотрофический склероз; сосудистые, сердечные и мозговые расстройства; расстройства половой активности и фертильности; преждевременные роды, иммунодепрессия, иммуносупрессия, воспалительные процессы, множественные инфекции, интерстициальный цистит, ревматоидный артрит,остеоартрит, увеит, псориаз и диабет; рак; функциональные желудочно-кишечные расстройства и воспаления, которые являются их результатом (раздраженный и воспаленный ки 004957 14 шечник, диарея); расстройства болевой чувствительности, фибромиалгии, связанные или не связанные с расстройствами сна, утомление или мигрень; или симптомы, относящиеся к (алкогольной) зависимости и к отлучению от наркотических средств. Дозировка может варьировать в широких пределах как функция возраста, массы тела и состояния здоровья пациента, природы и тяжести болезни и пути введения. В данном случае дозировка включает в себя введение одной или более чем одной дозы примерно от 0,5 мг до 800 мг действующего начала, предпочтительно от 0,5 до 200 мг в сутки. В фармацевтических композициях по настоящему изобретению для перорального, подъязычного,подкожного,внутримышечного,внутривенного, трансдермального, трансмукозального, местного или ректального введения действующее начало можно вводить животным и людям в стандартных формах введения в смеси с общепринятыми фармацевтическими носителями. Подходящие стандартные формы введения включают формы, предназначенные для перорального пути введения, такие как таблетки, твердые желатиновые капсулы, порошки,гранулы и растворы или суспензии, которые следует принимать перорально, формы для подъязычного и трансбуккального введения,формы для подкожного, внутримышечного,внутривенного, интраназального или внутриглазного введения и формы для ректального введения. Следующие ниже примеры, не означающие ограничение, иллюстрируют изобретение. Способы синтеза различных промежуточных соединений, которые дают возможность получить соединения по изобретению, описаны в получениях. Все эти промежуточные соединения получают способами, хорошо известными специалисту в данной области техники. Точки плавления измерены по методикеMicro-Kofler и выражены в градусах Цельсия. Спектры протонного ядерного магнитного резонанса (1 Н ЯМР) снимали в дейтерированном хлороформе (CDCl3), если не указано иное,при 200 МГц или при 300 МГц. Химические сдвиги выражены в м.д. (миллионные доли), а константы взаимодействия - в Герцах. Энантиомерный избыток (ее) вычисляли из хроматограмм, полученных либо хроматографией ВЭЖХ с хиральной фазой, либо хиральной суперкритической жидкостной хроматографией(СЖХ). Оптическое вращение оптически активных продуктов охарактеризовано их []toD (концентрация с анализируемых растворов выражена в граммах на 100 мл). Использованы следующие сокращения: s синглет; m - мультиплет; d -дублет; t - триплет; 15 Соединения по изобретению по данным элементного анализа согласуются с теорией. Соединения по изобретению, описанные в табл. 1 и 6, также имеют ЯМР-спектры и массспектры, соответствующие их структурам. ПОЛУЧЕНИЕ ИСХОДНЫХ СИНТОНОВ 1) Получение фенилгидразинов формулы III 2-Хлор-4-метокси-5-метилфенилгидразина гидрохлорид, соединение III.1 Раствор 8,6 г (50 ммоль) 2-хлор-4-метокси 5-метиланилина в 75 мл 5 н. соляной кислоты перемешивают при -5 С и добавляют 3,52 г (51 ммоль) нитрита натрия в растворе в 12,5 мл воды. Смесь перемешивают в течение 1 ч при 0 С,а затем добавляют раствор 22,56 г (100 ммоль) хлорида олова (II) в 20 мл 35% соляной кислоты. Эту смесь перемешивают в течение 2 ч с постепенным возвратом к температуре окружающей среды. Образовавшийся осадок отфильтровывают и промывают нормальным раствором соляной кислоты, этанолом и диэтиловым эфиром. После сушки в эксикаторе получено 7,8 г соединения III.1. Т.пл. = 140 С. Выход 70%. 1 Н ЯМР (d6-ДМСО;м.д.): 2.09 (s, 3 Н,СН 3); 3.73 (s, 3 Н, ОСН 3); 7.0 (s, 1 Н, Ph); 7.05 (s,1H, Ph); 7.58 (s, 1H, NH); 10.13 (s, 3 Н, NH3+). 2) Получение вторичных аминов формулы V[2-Циклопропил-1-(4-фторфенил)этил]пропиламин, соединение V.1 Раствор 8,9 г (50 ммоль) 2-циклопропил-1(4-фторфенил)этанона в 100 мл дихлорметана с 16,4 мл (200 ммоль) пропиламина перемешивают при 0 С и медленно добавляют 30 мл нормального раствора тетрахлорида титана в дихлорметане. Эту смесь перемешивают в течение 15 ч при температуре окружающей среды, а затем охлаждают до 0 С и добавляют 100 мл метанола. Добавляют порциями 2,1 г (55 ммоль) боргидрида натрия, и эту смесь перемешивают в течение двух часов при температуре окружающей среды. Эту смесь концентрируют при пониженном давлении примерно до 100 мл, а затем добавляют 100 мл воды. Суспензию фильтруют и фильтрат растворяют в дихлорметане,промывают водой, а затем водой, насыщенной хлоридом натрия, и сушат над сульфатом натрия, после чего растворители выпаривают при пониженном давлении. Получают 9,43 г масляного продукта. Выход 85%. 1 Н ЯМР (CDCl3,м.д.): -0.08-0.12 (m, 2H, с-Рr); 0.28-0.50 (m, 2H,с-Рr); 0.50-0.62 (m, 1H, с-Рr); 0.81-0.88 (m, 3 Н,СН 3); 1.35-1.70 (m, 5H, СН 2, СН 2-с-Рr и NH); 2.30-2.45 (m, 2H, N-CH2); 3.62-3.68 (m, 1H, CH); 6.90-7.05 (m, 2H, Ph); 7.21-7.31 (m, 2H, Ph). 3) Получение первичных аминов формулы XI Первый способ: получение первичных аминов из аминокислот а) (1R)-2-амино-2-(4-фторфенил)этанол 240 мл (240 ммоль) 1 М раствора алюмогидрида лития в тетрагидрофуране перемешивают при кипячении с обратным холодильни 004957 16 ком, а затем добавляют порциями 20 г (118 ммоль) (R)-(4-фторфенил)глицина. После перемешивания при кипячении с обратным холодильником в течение 6 ч и 30 мин реакционную смесь перемешивают при 0 С, а затем медленно добавляют 9,5 мл воды, 9,5 мл 15% раствора гидроксида натрия, а затем 28,5 мл воды. Полученную суспензию фильтруют через целит. Фильтрат концентрируют и растворяют в 1 л дихлорметана. Этот раствор промывают насыщенным раствором хлорида натрия и сушат над безводным сульфатом натрия, а затем растворители выпаривают при пониженном давлении. Кристаллизация из изопропилового эфира дает возможность получить 13,22 г (85,2 ммоль) кристаллического продукта. Выход 72%. Т.пл. = 95 С. 1 Н ЯМР (d6-ДМСО): 1.82 (s, 2H, NH2); 3.35-3.45 (m, 2H, СН 2 О); 3.84 (m, 1H, СН); 4.73(s, 1 Н, ОН); 7.01-7.13 (m, 2 Н, Ph); 7.30-7.41 (m,2 Н, Ph). б) (1R)-1-(4-Фторфенил)-2-метоксиэтиламин, соединение XI.1 3,64 г (91 ммоль) гидрида калия, полученного промыванием 8,1 г маслянистой суспензии пентаном, суспендируют в 70 мл тетрагидрофурана и перемешивают при 10 С. Медленно добавляют раствор 13,22 г (85 ммоль) (1R)-2 амино-2-(4-фторфенил)этанола в 175 мл тетрагидрофурана. После перемешивания в течение 16 ч при температуре окружающей среды добавляют раствор 5,2 мл (83,5 ммоль) йодметана в 105 мл тетрагидрофурана в течение 2 ч. Эту реакционную смесь перемешивают в течение 3 ч при температуре окружающей среды, а затем выливают на 1 л охлажденной во льду соленой воды. Эту смесь экстрагируют 1 л третбутилметилового эфира. Органическую фазу промывают водой, а затем насыщенным раствором хлорида натрия и сушат над безводным сульфатом натрия, после чего растворители выпаривают при пониженном давлении. Получают 11,87 г маслянистого амина. Выход 82%. 1 Н ЯМР (CDCl3): 1.66 (s, 2 Н, NH2); 3.29 (d,1H, CH2); 3.36 (s, 3 Н, ОСН 3); 3.45 (dd, 1 Н, СН 2); 4.16 (т, 1 Н, СН); 6.93-7.05 (m, 2 Н, Ph); 7.24-7.38(т, 2 Н, Ph). Второй способ: получение первичных аминов из фенилкетонов а) Синтез замещенных фенилкетонов Соединение 1 Методика А 2-Циклопропил-1-(3-фтор-4-метилфенил) этан-1-он, соединение 1.1 Раствор 61 г (323 ммоль) 4-бром-3-фтортолуола в 280 мл диэтилового эфира медленно добавляют к 7,8 г (323 ммоль) магниевой стружки, так чтобы вызвать спокойную дефлегмацию. Эту смесь последовательно нагревают с обратным холодильником в течение 2 ч, а затем охлаждают и фильтруют через стекловату. Фильтрат перемешивают при 0 С и добавляют 25 г (308 ммоль) циклопропилацетонитрила, 17 разбавленного 20 мл диэтилового эфира. Эту реакционную смесь перемешивают в течение 3 ч при температуре окружающей среды, а затем охлаждают до 0 С и медленно добавляют 1 н. раствор соляной кислоты до тех пор, пока не достигнут значения рН 1. Эту смесь экстрагируют три раза этилацетатом, объединенные органические фазы промывают водой, а затем водой, насыщенной хлоридом натрия и сушат над сульфатом натрия, растворители выпаривают при пониженном давлении. Получают 53 г сырого продукта, который используют как таковой на следующей стадии. Выход: примерно 85%. 1 Н ЯМР (CDCl3,м.д.): 0.15-0.21 (m, 2H, сРr); 0.55-0.65 (m, 2H, с-Рr); 1.07-1.20 (m, 1 Н, сРr); 2.31 (d, J = 1.9 Гц, 3 Н, СН 3); 2.82 (d, J = 6.7 Гц, 2 Н, СН 2-с-Рr); 7.22-7.30 (m, 1 Н, Ph); 7.547.64 (m, 2 Н, Ph). Следующий кетон синтезирован таким же способом. 2-Циклопропил-1-(4-метилфенил)этан-1-он Методика Б Соединение 1.2 2-Циклопропил-1-(4-метоксиметилфенил) этан-1-он, соединение 1.3 Раствор 32,5 г (162 ммоль) 1-бром-4-метоксиметилфенила в 300 мл тетрагидрофурана перемешивают при -60 С и медленно добавляют 112 мл (179 ммоль) 1,6 М раствора бутиллития. Эту реакционную смесь перемешивают в течение 30 мин при -60 С, а затем медленно добавляют раствор 27,6 г (192 ммоль) 2-циклопропил-N-метокси-N-метилацетамида. Эту реакционную смесь перемешивают, одновременно давая возможность температуре постепенно вернуться к температуре окружающей среды. После перемешивания в течение 4 ч ее охлаждают до 0 С и медленно добавляют 5 мл этанола. Эту смесь экстрагируют этилацетатом, и органическую фазу промывают водой, а затем насыщенным раствором хлорида натрия и сушат над безводным сульфатом натрия, после чего растворители выпаривают при пониженном давлении. Полученный остаток очищают хроматографией на колонке из силикагеля (растворитель: циклогексан, затем 20/1 (об./об.) циклогексан/этилацетат). Получают 21,8 г кетона. Выход 66%. 1 Н ЯМР (CDCl3,м.д.): 0.13-0.21 (m, 2 Н, сРг); 0.53-0.62 (m, 2 Н, с-Рr); 0.84-0.93 (m, 1 Н, сРr); 2.85 (d, J = 6.6 Гц, 2 Н, СН 2-с-Рr); 3.40 (s, 3H,ОСН 3); 4.68 (s, 2H, ОСНз); 7.57 (d, J = 7.5 Гц,2 Н, Ph); 7.92 (d, J = 7.5 Гц, 2 Н, Ph). Следующий кетон синтезирован таким же способом: 2-циклопропил-1-(3,4-метилендиоксифенил)этан-1-он, соединение 1.4 б) Синтез замещенных оксимов Соединение 2 18 Раствор 53 г (275 ммоль) соединения 1.1 в 200 мл пиридина перемешивают при 0 С и медленно добавляют 28,5 г (410 ммоль) гидроксиламина гидрохлорида. Эту смесь перемешивают в течение 12 ч при температуре окружающей среды, а затем концентрируют при пониженном давлении. Остаток растворяют в этилацетате и органическую фазу промывают три раза водой,а затем водой, насыщенной хлоридом натрия, и сушат над сульфатом натрия, после чего растворители выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке из силикагеля (элюент: 20/1 (об./об.) циклогексан/этилацетат). Получают 30 г оксима. Выход 52%. 1 Н ЯМР (d6-ДМСО,м.д.): 0.10-0.20 (m,2 Н, с-Рr); 0.28-0.40 (m, 2 Н, с-Рr); 0.78-0.95 (m,1 Н, с-Рr); 2.21 (d, J = 1.7 Гц, 3 Н, СН 3); 2.63 (d, J= 6.8 Гц, 2 Н, СН 2-с-Рr); 7.20-7.56 (m, 3 Н, Ph); 11.16 (s, 1H, ОН). Следующие оксимы синтезированы таким же способом: 2-циклопропил-1-фенилэтан-1-она оксим,соединение 2.2; 2-циклопропил-1-(4-фторфенил)этан-1-она оксим, соединение 2.3; 2-циклопропил-1-(4-метилфенил)этан-1 она оксим, соединение 2.4; 2-циклопропил-1-(4-метоксиметилфенил) этан-1-она оксим, соединение 2.5; 2-циклопропил-1-(3,4-метилендиоксифенил)этан-1-она оксим, соединение 2.6; 1-фенилбутан-1-она оксим, соединение 2.7; 1-(4-метилфенил)бутан-1-она оксим, соединение 2.8 в). Синтез замещенных O-бензилоксимов, соединение 3. 2-Циклопропил-1-(3-фтор-4-метилфенил) этан-1-она (Е)-О-бензилоксим, соединение 3.1 Раствор 30 г (144 ммоль) соединения 2.1 в 140 мл диметилформамида перемешивают при 0 С и добавляют порциями 8,3 г (180 ммоль) 55% гидрида натрия в масле. Эту смесь перемешивают в течение 30 мин при 0 С, а затем медленно добавляют 20,5 мл (172 ммоль) бензилбромида. Эту реакционную смесь перемешивают в течение 3 ч при температуре окружающей среды, а затем охлаждают до 0 С и добавляют 10 мл этанола, а затем 500 мл воды. Эту смесь экстрагируют этилацетатом и органическую фазу промывают три раза водой, а затем водой, насыщенной хлоридом натрия и сушат над сульфатом натрия, после чего растворители выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке из силикагеля (элюент: 95/5 (об./об.) циклогексан/дихлорметан). Получают 30,2 г (Е)бензилоксима. Выход 70%. 1 Н ЯМР (d6-ДМСО,м.д.): 0.10-0.17 (m,2H, с-Рr); 0.28-0.40 (m, 2H, с-Рr); 0.78-0.90 (m,1 Н, с-Рr); 2.22 (d, J = 1.8 Гц, 3 Н, СН 3); 2.67 (d, J= 6.8 Гц, 2 Н, СН 2-с-Рr); 5.16 (s, 2 Н, О-СН 2-Рh); 7.20-7.43 (m, 8 Н, Ph). Следующие бензилоксимы синтезированы таким же способом: 2-циклопропил-1-фенилэтан-1-она (Е)-Обензилоксим, соединение 3.2; 2-циклопропил-1-(4-фторфенил)этан-1-она(Е)-Обензилоксим, соединение 3.8. г) Синтез рацемических первичных аминов формулы XI. 2-Циклопропил-1-(4-фторфенил)этиламин, соединение XI.2 Суспензию 10,6 г (280 ммоль) алюмогидрида лития в 500 мл тетрагидрофурана перемешивают при температуре окружающей среды и медленно добавляют 40 г (140 ммоль) 2 циклопропил-1-(4-фторфенил)этан-1-она Обензилоксима. Эту смесь перемешивают при кипячении с обратным холодильником в течение 3 ч, а затем охлаждают до 0 С и добавляют по каплям 10,6 мл воды, 10,6 мл 15% раствора гидроксида натрия, а затем 32 мл воды. Полученную суспензию фильтруют через целит и промывают этилацетатом. Объединенные органические фильтраты промывают водой, а затем водой, насыщенной хлоридом натрия, и сушат над сульфатом натрия, после чего растворители выпаривают при пониженном давлении. Сырой экстракт очищают хроматографией на колонке из силикагеля (элюент: 95/5 (об./об.) дихлорметан/метанол). Получают 14,2 г амина в форме масла. Выход 57%. 1 Н ЯМР (CDCl3,м.д): -0.05-0.18 (m, 2 Н,с-Рг); 0.32-0.50 (m, 2 Н, с-Рr); 0.50-0.70 (m, 1 Н, сРr); 1.40-1.70 (m, 2 Н, СН 2-с-Рr); 1.76 (s, 2 Н,NH2); 3.97-4.05 (m, 1 Н, СН); 6.92-7.04 (m, 2 Н,Ph); 7.24-7.34 (m, 2 Н, Ph). Следующие рацемические амины синтезированы таким же способом: 2-циклопропил-1-фенилэтиламин, соединение XI.3,2-циклопропил-1-(3-фтор-4-метилфенил) этиламин, соединение XI.4,д) Синтез хиральных первичных аминов формулы XI.(1S)-2-Циклопропил-1-(3-фтор-4-метилфенил)этиламин, соединение XI.5. Раствор 37,17 г (145 ммоль) (S)-2-амино-3 метил-1,1-дифенилбутан-1-ола в 180 мл тетрагидрофурана перемешивают при -40 С и медленно добавляют 285 мл 1 М раствора комплекса 20 боран-тетрагидрофуран (285 ммоль). Эту смесь перемешивают в течение 2 ч при температуре от-40 С до температуры окружающей среды, а затем охлаждают до -10 С и добавляют раствор 17 г (57 ммоль) соединения 3.1. Реакционную смесь перемешивают в течение 20 ч при температуре окружающей среды, а затем охлаждают до -10 С и добавляют 285 мл 2 н. соляной кислоты. Эту смесь оставляют перемешиваться в течение 20 ч, а затем тетрагидрофуран выпаривают при пониженном давлении. Образуется осадок (S)-2-амино-3-метил-1,1-дифенилбутан 1-ола гидрохлорида, и его отфильтровывают и промывают 1 н. соляной кислотой. Объединенные кислотные фильтраты промывают третбутилметиловым эфиром, затем охлаждают до 0 С и медленно подщелачивают 35% водным раствором гидроксида натрия. После трехкратной экстракции дихлорметаном объединенные органические фазы промывают водой, а затем водой, насыщенной хлоридом натрия, и сушат над сульфатом натрия, после чего растворители выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке из силикагеля (элюент: 95/5 (об./об.) дихлорметан/метанол). Получают 7,2 г амина в масляной форме. Выход 47%, ее 98%.[]22D = + 5.4 (с = 1,16, дихлорметан) 1 Н ЯМР (CDCl3,м.д.): -0.05-0.15 (m, 2 Н,с-Рr); 0.35-0.50 (m, 2 Н, с-Рr); 0.55-0.70 (m, 1 Н, сРr); 1.43-1.70 (m, 2 Н, СН 2-с-Рr); 2.0 (s, 2 Н, NH2); 2.25 (d, J = 1.7 Гц, 3 Н, СН 3); 3.99-4.04 (m, 1 Н,СН); 6.99-7.17 (m, 3 Н, Ph). Следующие хиральные амины синтезированы таким же способом:(1S)-2-циклопропил-1-фенилэтиламин, ее = 96%, соединение XI.6,(1S)-2-циклопропил-1-(4-фторфенил)этиламин, ее = 98%, соединение XI. 7,(1S)-2-циклопропил-1-(4-метилфенил) этиламин, ее = 97,2%, соединение XI.8,(1S)-2-циклопропил-1-(4-метоксиметилфенил)этиламин, ее = 97,8%, соединение XI.9,(1S)-2-циклопропил-1-(3,4-метилендиоксифенил)этиламин, ее = 96,6%, соединение XI.10,(1S)-1-фенилбутиламин, ее 99%, соединение Xl.11,(1S)-1-(4-метилфенил)бутиламин, ее=97,9%,соединение XI. 12. Энантиомерный избыток (ее) этих соединений оценивали с помощью хиральной сверхкритической жидкостной хроматографии их ацетамидного или тиомочевинного производного. Только энантиомерный избыток соединенияXI.11 оценивали непосредственно с помощью ВЭЖХ с хиральной фазой. Для данного соединения образование соли, а затем перекристаллизация с N-ацетил-L-лейцином (Yamamoto Y. etal., Bull. Chem. Soc. Jpn., 1976, 49(11), 32473249) дали возможность увеличить энантиомерный избыток. 21 Получение путем А 1) Получение цианамидов формулы IV(1-Фенилбутил)пропилцианамид, соединение IV. 1 Суспензию 1,4 г (16,4 ммоль) карбоната магния в 20 мл 9/1 (об./об.) смеси диэтиловый эфир/вода перемешивают при 0 С и добавляют 5 г (47 ммоль) цианогенбромида. К этой смеси,перемешиваемой при 0 С, медленно добавляют 9 г (47 ммоль) (1-фенилбутил)пропиламина. После перемешивания в течение 1 ч при температуре окружающей среды к этой реакционной смеси добавляют 50 мл воды, а затем 100 мл диэтилового эфира. Эфирную фазу промывают водой, а затем насыщенным раствором хлорида натрия. Ее сушат над сульфатом натрия, а затем растворители выпаривают при пониженном давлении. Полученный маслянистый остаток перегоняют в реакционной камере при 150 С при приблизительно или равно 0,3 мм рт.ст. Получают 7,6 г бесцветного масла. Выход 74%. 1 Н ЯМР (CDCl3,м.д.): 0.84-0.95 (m, 6 Н,2 СН 3); 1.28-2.10 (m, 6 Н, 3 СН 2); 2.65-2.90 (m, 2 Н,NCH2); 3.77 (t, J = 7.5 Гц, 1 Н, СН); 7.25-7.39 (m,5 Н, Ph). Следующее соединение синтезировано таким же способом:[2-Циклопропил-1-(4-фторфенил)этил]пропилцианамид, соединение IV.2. 1 Н ЯМР (CDCl3,м.д.): 0-0.2 (m, 2H, с-Рr); 0.37-0.55 (m, 2H, с-Рr); 0.60-0.75 (m, 1 Н, с-Рr); 0.85-0.93 (m, 3 Н, СН 3); 1.55-1.75 (m, 3 Н, СН 2 и НСН-с-Рr); 1.95-2.10 (m,1 Н, НСН-с-Pr); 2.722.90 (m, 2H, NCH2); 3.87 (t, J = 7.5 Гц, 1 Н, CH); 6.98-7.09 (m, 2H, Ph); 7.25-7.35 (m, 2H, Ph). 2) Получение анилиногуанидинов формулы II(30 ммоль) соединения IV.1 и 10 мл безводного н-пропанола, перемешивают при 130 С в течение 24 ч. После охлаждения эту реакционную смесь суспендируют в 50 мл диэтилового эфира. Осадок отфильтровывают, а затем растворяют в 50 мл ацетона. Полученную суспензию перемешивают в течение 30 мин при температуре окружающей среды, после чего осадок отфильтровывают и промывают ацетоном. Осадок затем растворяют в условиях подогрева в 10 мл этанола, а затем добавляют 50 мл диэтилового эфира. Образуется белый осадок, который отфильтровывают, промывают эфиром и сушат. Получают 6 г (14 ммоль) белого порошка. Выход 58%. Т.пл. = 225 С. Следующее соединение синтезировано таким же способомN-[2-Циклопропил-1-(4-фторфенил)этил]N'-(2,4-дихлорфениламино)-N-пропилгуанидина гидрохлорид, соединение II.2. 1 Н ЯМР (d6-ДМСО,м.д.): 0.1-0.7 (m, 5H,с-Рr); 0.78 (m, 3 Н, СН 3); 1.4-1.75 (m, 2 Н, СН 2); 1.85-2.15 (m, 2 Н, СН 2); 3.0-3.4 (m, 2 Н, NCH2); 5.41 (t, J = 7.3 Гц, 1 Н, СН); 7.73 (d, 1 Н, Ph); 7.17.6 (m, 6 Н, Ph); 8.1 (s, 2H, NH2); 8.23 (s, 1H, NH); 9.98 (s, 1H, NH). 3) Получение аминотриазолов формулы I путем А Пример 1. 1-(2,4-Дихлорфенил)-5-метилN-(1-фенилбутил)-N-пропил-1H-1,2,4-триазол-3 амин. Суспензию 1,29 г (3 ммоль) соединенияII.1 в 12 мл пиридина перемешивают при 0 С и медленно добавляют 1,07 мл (15 ммоль) ацетилхлорида. Эту реакционную смесь перемешивают в течение 20 ч при температуре окружающей среды, а затем выливают на 100 мл охлажденной во льду воды. После подкисления до рН 1 1 н. соляной кислотой смесь экстрагируют этилацетатом. Органическую фазу последовательно промывают насыщенным раствором однозамещенного карбоната натрия, водой, а затем водой, насыщенной хлоридом натрия. Эту фазу сушат безводным сульфатом натрия, а затем упаривают. Сырой остаток очищают хроматографией на колонке из силикагеля (элюент: 9/1(m, 7 Н, Ph); 7.53 (d, J = 1.9 Гц, 1 Н, Ph). Этот продукт образует соль в форме гидрохлорида; Т.пл. = 142 С (HCl). Примеры 2-9 в следующей ниже табл. 1 синтезированы таким же способом: Таблица 1. Соединения формулы I, синтезированные путем АVII.1. Раствор 2 г (26,4 ммоль) тиоцианата аммония в 27 мл ацетона перемешивают при температуре окружающей среды и медленно добавляют 1,82 мл (24,2 ммоль) ацетилхлорида. После перемешивания в течение 10 мин медленно добавляют раствор 4,87 г (22 ммоль) соединенияV.1 в 44 мл дихлорметана. После перемешивания в течение 30 мин при температуре окружающей среды к этой реакционной смеси добавляют 100 мл дихлорметана и 100 мл воды. Органическую фазу промывают водой, а затем водой, насыщенной хлоридом натрия, и сушат над сульфатом натрия и выпаривают растворители при пониженном давлении. Сырой остаток очищают хроматографией на колонке из силикагеля (элюент: 4/1 (об./об.) циклогексан/этилацетат). Получают 5,5 г соединения VII.1. Выход 77%. 1 Н ЯМР (d6-ДМСО,м.д.): 0.1-0.2 (m, 2 Н,с-Рr); 0.35-0.50 (m, 2 Н, с-Рr); 0.50-0.70 (m, 3 Н,СН 3); 0.75-0.95 (m, 1 Н, с-Рr); 1.25-1.75 (m, 2 Н,СН 2); 1.80-2.10 (m, 5 Н, СН 2-с-Рr и СОСН 3); 3.103.60 (m, 2 Н, NCH2); 5.32 (m, 1 Н, CH); 7.15-7.24N-Ацетил-N'-[2-циклопропил-1-(4-фторфенил)этил]-S-метил-N'-пропилизотиомочевина,соединение VI.1 Раствор 5,5 г (17 ммоль) соединения VII.1 в 170 мл дихлорметана перемешивают при 0 С и добавляют 740 мг (18,5 ммоль) 60% гидрида натрия в масле. После перемешивания в течение 24 10 мин при 0 С к этой реакционной смеси добавляют 2,1 мл (34 ммоль) метилйодида. Эту реакционную смесь перемешивают в течение 3 ч при температуре окружающей среды, а затем охлаждают до 0 С и добавляют 10 мл этанола, а затем 10 мл воды. Органическую фазу промывают водой, а затем водой, насыщенной хлоридом натрия, сушат над сульфатом натрия и выпаривают растворители при пониженном давлении. Бесцветный маслянистый сырой остаток используют как есть на следующей стадии (количественный выход). 1 Н ЯМР (CDCl3,м.д.): 0.1-0.16 (m, 2H, cPr); 0.45-0.52 (m, 2H, с-Рr); 0.62-0.70 (m, 3 Н,СН 3); 0.80-0.95 (m, 1 Н, СН, с-Рr); 1.0-1.2 и 1.451.65 (2m, 2H, СН 2); 1.8-2.05 (m, 2 Н, СН 2-с-Рr); 2.18 (s, 3 Н, СН 3 СО); 2.41 (s, 3 Н, SCH3); 3.04-3.20(m, 2 Н, Ph); 7.30-7.38 (m, 2 Н, Ph). 3) Получение аминотриазолов формулы I путем Б 1. Пример 10. N-[2-Циклопропил-1-(4-фторфенил)этил]-1-[2,6-дихлор-4-(трифторметил)фенил]-5-метил-N-пропил-1 Н-1,2,4-триазол-3 амин. Смесь, составленную из 1 г (3 ммоль) соединения VI.1, 1 г (4 ммоль) 2,6-дихлор-4 трифторметилфенилгидразина и 8 мл безводного диметилсульфоксида, перемешивают и постепенно нагревают от 100 до 200 С в течение 30 ч, пока исходная изотиомочевина не исчезает. Эту реакционную смесь, предварительно охлажденную, вливают в охлажденную во льду воду и подкисляют нормальным раствором соляной кислоты. Смесь экстрагируют этилацетатом, органический экстракт промывают насыщенным водным раствором гидрокарбоната натрия, затем водой и водой, насыщенной хлоридом натрия. Затем экстракт сушат над безводным сульфатом натрия и растворители выпаривают при пониженном давлении. Сырой остаток очищают хроматографией на колонке из силикагеля (элюент: 9/1 (об./об.) циклогексан/метилацетат). Получают 630 мг смоляного продукта. Выход 44%. 1 Н ЯМР (CDCl3 м.д.): 0.08-0.18 (m, 2H, сРr); 0.37-0.41 (m, 2H, с-Рr); 0.67-0.75 (m, 3 Н, СН 3 и 1 Н, с-Рr); 1.30-1.45 и 1.50-1.65 (2m, 2 Н, СН 2); 1.84-2.03 (m, 2 Н, СН 2-с-Рr); 2.21 (s, 3 Н, СН 3); 2.94-3.02 и 3.10-3.18 (2m, 2 Н, NCH2); 5.43 (t, 7.5 Гц, 1 Н, СН); 6.93-7.0 (m, 2 Н, Ph); 7.36-7.41 (m,2 Н, Ph); 7.71 (s, 2H, Ph). ЭТО соединение образует соль в форме гидрохлорида: Т.пл. = 135 С (НСl). Получение путем Б 2 1) Получение N-ацилтиомочевин формулы X.N-Ацетил-N'-[2-циклопропил-1-(4-фторфенил)этил]тиомочевина, соединение X.1 Раствор 3,38 г (44,4 ммоль) тиоцианата аммония в 44 мл ацетона перемешивают при температуре окружающей среды и добавляют 25 3,07 мл (40,7 ммоль) ацетилхлорида. После перемешивания в течение 5 мин добавляют 88 мл бензола, и эту реакционную смесь нагревают до 60 С. Затем добавляют раствор 6,62 г (37 ммоль) соединения XI.2 в 27 мл бензола. Поддерживают температуру 60 С в течение 5 мин, а затем смесь охлаждают до температуры окружающей среды и разбавляют 100 мл этилацетата. Эту смесь промывают водой, а затем водой,насыщенной хлоридом натрия, сушат над сульфатом натрия и выпаривают растворители при пониженном давлении. Сырой остаток (11 г) очищают хроматографией на колонке из силикагеля (элюент: 4/1 (об./об.) циклогексан/этилацетат). Получают 5,6 г соединения Х.1. Выход 54%. 1 Н ЯМР (CDCl3,м.д.): 0.10-0.25(m, 1 Н, с-Рr); 1.75-1.85 (m, 2 Н, СН 2-с-Рr); 2.11 (s,3 Н, СН 3 СО); 5.38-5.49 (m, 1 Н, СН); 6.95-7.06 (m,2 Н, Ph); 7.22-7.32 (m, 2 Н, Ph); 8.73 (s, 1H, NH); 11.0 (d, 1H, NH). Следующие соединения синтезированы таким же способом:N-Ацетил-N'-(2-метокси-5-метилфенил) тио-мочевина, соединение Х.3. Т.пл. = 152 С. Соединения в следующей ниже табл. 2 также синтезированы таким же способом или с заменой бензола хлороформом: Таблица 2. N-ацилтиомочевины формулы X.IX. 1. Раствор 2,8 г (10 ммоль) соединения Х.1 в 50 мл тетрагидрофурана перемешивают при 0 С и добавляют порциями 440 мг (11 ммоль) 60% гидрида натрия в масле. После перемешивания в течение 20 мин при 0 С добавляют 0,75 мл (12 ммоль) метилйодида. Эту реакционную смесь перемешивают в течение 4 ч при температуре окружающей среды, а затем охлаждают до 0 С и медленно добавляют 5 мл этанола, а затем 5 мл воды. Эту смесь экстрагируют 150 мл этилацетата, и органическую фазу промывают водой, а затем водой, насыщенной хлоридом натрия,сушат над сульфатом натрия и растворители выпаривают при пониженном давлении. Сырой остаток (3,5 г) используют как есть на следующей стадии (количественный выход). 1 Н ЯМР(m, 2 Н, СН 2-с-Рr); 2.15 (s, 3 Н, СН 3 СО); 2.42 (s,3 Н, SCH3); 4.62-4.71 (m, 1 Н, СН); 6.92-7.06 (m,2 Н, Ph); 7.19-7.33 (т, 2 Н, Ph); 11.57 (s, 1H, NH). Продукты, указанные в табл. 3, получены таким же способом. Таблица 3. N-ацил-S-метилтиомочевины формулы IX.S-Метил-N-[(1S)-1-фенилбутил]-N'-пропионил-изотиомочевина, соединение IX.8. Раствор 6,4 г (24,2 ммоль) соединения Х.8 в 120 мл диметилформамида перемешивают при температуре окружающей среды и добавляют 7,9 г (24,2 ммоль) карбоната цезия. Затем медленно добавляют 3 мл (24 ммоль) йодметана. Эту смесь перемешивают при температуре окружающей среды в течение 2 ч, затем добавляют лед и проводят экстракцию этилацетатом. Органическую фазу промывают водой, а затем водой, насыщенной хлоридом натрия, сушат над сульфатом натрия и растворители выпаривают при пониженном давлении. Сырой остаток очищают хроматографией на колонке из силикагеля (элюент: 9/1 (об./об.) циклогексан/этилацетат). Получают 5,15 г соединенияIX.8. Выход 72%. 1 Н ЯМР (CDCl3,м.д.): 0.860.94 (m, 3 Н, СН 3); 1.09-1.17 (m, 3 Н, СН 3); 1.221.45 (m, 2 Н, СН 2); 1.72-1.86 (m, 2 Н, СН 2); 2.38 (s,3 Н, SCH3); 2.35-2.50 (m, 2 Н, СН 2); 4.57-4.63 (m,1 Н, СН); 7.19-7.37 (m, 5 Н, Ph); 11.52 (s, 1H, NH). Продукты, указанные в табл. 4, получены таким же способом. Таблица 4. N-ацил-S-метилтиомочевины формулы IX. 3) Получение NH аминотриазолов формулы VIII. 1-(2-Хлор-4-метокси-5-метилфенил)-N-[2 циклопропил-1-(4-фторфенил)этил]-5-метил 1H-1,2,4-триазол-3-амин, соединение VIII.1. 3 г (11,5 ммоль) 2-хлор-4-метокси-5 метилфенилгидразина гидрохлорида (соединение III.1) добавляют к раствору 3,2 г (10 ммоль) соединения IX.1 в 25 мл толуола с последующим добавлением 3,5 мл (25 ммоль) триэтиламина и молекулярного сита 4 А. Эту реакционную смесь перемешивают в течение ночи при умеренной дефлегмации, а затем охлаждают. Молекулярное сито удаляют фильтрованием, и фильтрат разбавляют 100 мл этилацетата. Органическую фазу промывают 1 н. соляной кислотой, водой, насыщенным водным раствором гидрокарбоната натрия, водой, а затем водой,насыщенной хлоридом натрия. Эту фазу сушат над сульфатом натрия, а затем растворители выпаривают при пониженном давлении. Сырой остаток очищают хроматографией на колонке из силикагеля (элюент: 3/1, затем 2/1, затем 1/1(m, 2 Н, СН 2-с-Рr); 2.17 (s, 6H, 2 СН 3); 3.83 (s, 3 Н,ОСН 3); 4.60 (d, J = 8.2 Гц, 1 Н, NH); 4.73-4.84 (m,1 Н, СН); 6.85 (s, 1H, Ph); 6.90-7.00 (m, 2 Н, Ph); 7.05 (s, 1 Н, Ph); 7.25-7.37 (m, 2 Н, Ph). Соединения VIII, указанные в табл. 5, получены таким же способом. Толуол можно заменить ксилолом, диметилформамидом или диметилсульфоксидом. Если требуется основание,то триэтиламин можно заменить N,Nдиэтиланилином или карбонатом цезия. Таблица 5. NH аминотриазолы формулы VIII.VII 1.36. 470 мг (2,2 ммоль) 2,4-дихлорфенилгидразина гидрохлорида добавляют к раствору 600 мг (1,9 ммоль) N-ацетил-N'-[2-циклопропил 1-(3-фтор-4-метилфенил)этил]-S-метилизотиомочевины в 10 мл диметилформамида с последующим добавлением 800 мг (2,5 ммоль) карбоната цезия и молекулярного сита 4. Эту реакционную смесь перемешивают в течение 4 ч при 140 С, а затем охлаждают. Молекулярное сито удаляют фильтрованием, и фильтрат разбавляют 100 мл этилацетата. Органическую фазу промывают 1 н. соляной кислотой, водой, насыщенным водным раствором гидрокарбоната натрия, водой, а затем водой, насыщенной хлоридом натрия. Эту фазу сушат над сульфатом натрия, а затем растворители выпаривают при пониженном давлении. Сырой остаток очищают хроматографией на колонке из силикагеля (элюент: 3/1 (об./об.) циклогексан/этилацетат). Получают 385 мг N-[2-циклопропил-1-(3-фтор-4-метилфенил)этил-1-(2,4-дихлорфенил)-5-метил-1H-1,2,4 триазол-3-амина и 115 мг соединения VIII.36.(d, J=8.3 Гц, 1 Н, Ph); 7.33 (d, J=2 Гц, 1 Н, Ph). 4) Получение аминотриазолов формулы (I) путем Б 2. Пример 11. [1-(2-Хлор-4-метокси-5-метилфенил)-N-[2-циклопропил-1-(4-фторфенил)этил]5-метил-N-пропил-1H-1,2,4-триазол-3-амин. 240 мг (6 ммоль) гидрида калия (полученного из масляной суспензии промыванием пентаном с последующим высушиванием в атмосфере аргона) суспендируют в 2 мл безводного бензола. Эту смесь перемешивают при 5-10 С и последовательно добавляют 780 мг (1,85 ммоль) соединения VIII.1, растворенного в 6 мл бензола, и 750 мг (2 ммоль) 2,3,11,12-циклогексано 1,4,7,10,13,16-гексаоксациклооктадекана. После перемешивания в течение 1 ч 30 мин при температуре окружающей среды добавляют 0,6 мл (6 ммоль) йодпропана, и эту смесь перемешивают в течение 3 ч при температуре окружающей среды. Эту реакционную смесь охлаждают на ледяной бане и добавляют 1 мл этанола, а затем 1 мл воды, после чего смесь разбавляют 100 мл этилацетата. Органическую фазу промывают водой, а затем водой, насыщенной хлоридом натрия и сушат над сульфатом натрия, после чего растворители выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке из силикагеля (элюент: 4/1 (об./об.) циклогексан/этилацетат). Получают 590 мг смолистого продукта. Выход 75%. 1 Н ЯМР (CDCl3, м.д.): 0.1-0.17 (m, 2H, с-Рr); 0.39-0.45 (m, 2H, сРr); 0.69-0.76 (m, 4 Н, СН 3 и СН, с-Рr); 1.30-1.45 и 1.50-1.65 (2m, 2 Н, СН 2); 1.88-1.98 (m, 2 Н,СН 2); 2.21 (s, 3 Н, СН 3); 2.22 (s, 3 Н, СН 3); 2.943.02 и 3.10-3.20 (2m, 2 Н, NCH2); 3.87 (s, 3 Н,СН 3); 5.47 (t, 1 Н, СН); 6.92 (s, 1 Н, Ph); 6.94-7.02(m, 2 Н, Ph); 7.19 (s, 1 Н, Ph); 7.38-7.44 (m, 2H,Ph). ЭТОТ продукт образует соль гидрохлорид; Т.пл. = 38 С (HCl). Пример 12. [1-(2,4-Дихлорфенил)-N-(2-метокси-5-метилфенил)-5-метил-N-(2-пропинил)1H-1,2,4-триазол-3-амин. 240 мг (6 ммоль) гидрида калия (полученного из масляной суспензии промыванием пентаном с полседующим высушиванием в атмосфере аргона) суспендируют в 2 мл безводного бензола. Эту смесь перемешивают при 10 С и добавляют 730 мг (2 ммоль) соединения VIII.3,растворенного в 8 мл бензола. Добавляют 75 мг(0,2 ммоль) 2,3,11,12-циклогексано-1,4,7,10,13,16-гексаоксациклооктадекана, и смесь перемешивают в течение 2 ч при температуре окружающей среды. Затем добавляют 0,66 мл (6 ммоль) 80% раствора пропаргилбромида в то 37 луоле и смесь перемешивают в течение 1 ч при температуре окружающей среды. Реакционную смесь охлаждают на ледяной бане, а затем последовательно добавляют 1 мл этанола и 1 мл воды, после чего реакционную смесь разбавляют 100 мл этилацетата. Органическую фазу промывают водой, а затем водой, насыщенной хлоридом натрия, и сушат над сульфатом натрия, после чего растворители выпаривают при пониженном давлении. Сырой экстракт очищают хроматографией на колонке из силикагеля(CDCl3,м.д.): 2.15 (t, J=2.2 Гц, 1 Н, СН); 2.21 (s,3 Н, СН 3); 2.28 (s, 3 Н, СН 3); 3.78 (s, 3 Н, ОСН 3); 4.52 (d, J=2.2 Гц, 2 Н, СН 2); 6.84 (d, J=8.3 Гц, 1 Н,Ph); 7.01 (dd, JT=2 ГЦ, J2=8.3 ГЦ, 1 Н, Ph); 7.23 (d,J=2 Гц, 1 Н, Ph); 7.29-7.39 (m, 2H, Ph); 7.52 (d,J=2 Гц, 1 Н, Ph). Т.пл. = 116C. Примеры 13-53, приведенные в следующей ниже табл. 6, синтезированы таким же способом(при этом бензол можно заменить тетрагидрофураном): Таблица 6. Соединения формулы I, синтезированные путем Б 2 где R1 и R2 независимо друг от друга представляют собой атом галогена; (С 1-С 5 )алкил; (С 1-С 5) алкокси; нитро, трифторметильную или цианогруппу; аминогруппу NRaRb, где Ra и Rb независимо друг от друга представляют собой водород, (С 1-С 3)алкил или СО(С 1-С 3)алкил или гдеRa и Rb составляют вместе с атомом азота, с которым они связаны, 5-7-членный гетероцикл; или группу S-R, где R представляет собой атом водорода или (С 1-С 5)алкил, причем возможно,что атом серы монооксидирован или диоксидирован;R3 представляет собой водород или является таким, как определено выше для R1; 40 алкил(С 1-С 2)алкил; или группу Rc-X-(С 1-С 2) алкил, где Rc представляет собой водород или (С 1 С 3)алкил, а X представляет собой О, S, SO илиR5 представляет собой (С 1-С 5)алкил, алкинил с 3-5 атомами углерода или алкенил с 3-5 атомами углерода; (С 3-С 5)циклоалкил(С 1-С 3)алкил; или (С 1-С 3)алкил-Х-(С 0-С 3)алкил, где X представляет собой О, S, SO или SO2;R6 представляет собой фенильную группу,замещенную одним или более чем одним радикалом Z, по меньшей мере один из которых находится в положении 2, и Z представляет собой галоген; нитро, трифторметильную или цианогруппу; (С 1-С 5)алкил; (С 1-С 5)алкил-Х- или (С 1 С 3)алкил-Х-(С 1-С 2)алкил, где(С 3 С 5)циклоалкил; R7 представляет собой фенильную группу, которая может быть замещена в положениях 3, 4 или 5 одним или более чем одним радикалом Z', причем Z' представляет собой галоген; нитро, трифторметильную или цианогруппу; (С 1-С 5)алкил; (С 1-С 5)алкил-X- или (С 1 С 3)алкил-Х-(С 1-С 2)алкил, где X представляет собой О, S, SO или SO2; гидрокси(С 1-С 3)алкил;CORd или COORd, где Rd является таким, как определено выше; метилендиокси или этилендиокси; или R7 представляет собой пиридильную группу, возможно замещенную аминогруппой NRaRb, как она определена выше, или радикалом Z', как он определен выше;(С 1-С 3)алкил-Х-(С 1-С 3)алкил, где X представляет собой О, S, SO или SO2; или (С 3-С 5) циклоалкил(С 1-С 2)алкил-Х-(С 1-С 3)алкил, где X представляет собой О, S, SO или SO2; и их фармацевтически приемлемые соли присоединения, их гидраты и/или их сольваты. 2. Соединения по п.1, гдеR1 и R2 независимо друг от друга представляют собой атом галогена; (С 1-С 5)алкил;(С 1-С 5)алкокси; трифторметильную группу или группу S-R, где R представляет собой (С 1-С 5) алкил;R3 представляет собой водород или (С 1-С 5) алкил;R6 представляет собой -CHR7R8,где R7 представляет собой фенильную группу, которая может быть замещена в положениях 3, 4 или 5 одним или более чем однимR8 представляет собой (С 1-С 6)алкил; (С 3 С 5)циклоалкил(С 1-С 3)алкил; или (С 1-С 3)алкилХ-(С 1-С 3)алкил, где X представляет собой О; и их фармацевтически приемлемые соли присоединения, их гидраты и/или их сольваты. 3. Соединения по любому из пп.1 и 2, гдеR5 представляет собой пропильную или пропаргильную группу. 4. Соединения по любому из пп.1-3 в энантиомерной форме. 5. Соединения по любому из пп.1-4, выбранные из 5-циклопропил-N-[2-циклопропил-1-(4 фторфенил)этил]-1-(2,4-дихлорфенил)-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,N-[2-циклопропил-1-(4-фторфенил)этил]1-(2,4-дихлорфенил)-5-(метоксиметил)-N-пропил-1 Н-1,2,4-триазол-3-амина гидрохлорида,N-[2-циклопропил-1-(4-фторфенил)этил]1-[2,6-дихлор-4-(трифторметил)фенил]-5-метилN-пропил-1 Н-1,2,4-триазол-3-амина гидрохлорида,1-(2-хлор-4-метокси-5-метилфенил)-N-[2 циклопропил-1-(4-фторфенил)этил]-5-метил-N(2-пропинил)-1 Н-1,2,4-триазол-3-амина гидрохлорида,1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[2-циклопропил-1-(4-фторфенил) этил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[2-циклопропил-1-(4-фторфенил) этил]-N-(2-пропинил)-1 Н-1,2,4-триазол-3-амина гидрохлорида,5-циклопропил-N-[2-циклопропил-1-(4 фторфенил)этил]-1-[2,6-дихлор-4-(трифторметил)фенил]-N-пропил-1H-1,2,4-триазол-3 амина гидробромида,5-циклопропил-N-(2-циклопропил-1-фенилэтил)-1-(2,4-дихлорфенил)-N-пропил-1 Н 1,2,4-триазол-3-амина гидрохлорида,1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-(2-циклопропил-1-фенилэтил)-Nпропил-1 Н-1,2,4-триазол-3-амина гидрохлорида,1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[(1S)-2-циклопропил-1-(3-фтор-4 метилфенил)этил]-N-пропил-1 Н-1,2,4-триазол 3-амина гидрохлорида,1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-(2-циклопропил-1-фенилэтил)-N(2-пропинил)-1 Н-1,2,4-триазол-3-амина гидрохлорида,1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[(1R)-1-(4-фторфенил)-2-метоксиэтил]-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида,1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[(1S)-2-циклопропил-1-(3-фтор-4 004957 1-[2-хлор-4-(трифторметил)фенил]-N[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил) этил]-5-метил-N-пропил-1 Н-1,2,4-триазол-3 амина гидрохлорида,N-[(1S)-1-(1,3-бензодиоксол-5-ил)-2-циклопропилэтил]-1-(2-хлор-4-метокси-5-метилфенил)-5-метил-N-пропил-1 Н-1,2,4-триазол-3 амина гидрохлорида,N-[(1S)-1-(1,3-бензодиоксол-5-ил)-2-циклопропилэтил]-1-(2,4-дихлорфенил)-5-метил-Nпропил-1 Н-1,2,4-триазол-3-амина гидрохлорида,1-(2-хлор-4-метокси-5-метилфенил)-N(1S)-2-циклопропил-1-[(4-метоксиметил)фенил]этил-5-метил-N-пропил-1 Н-1,2,4-триазол 3-амина гидрохлорида,1-(2-хлор-4-метоксифенил)-N-[(1S)-2-циклопропил-1-(3-фтор-4-метилфенил)этил]-5-метил-N-пропил-1 Н-1,2,4-триазол-3-амина гидрохлорида,1-(2-хлор-4-метокси-5-метилфенил)-N[(1S)-2-циклопропил-1-фенил)этил]-5-метил-Nпропил-1 Н-1,2,4-триазол-3-амина гидрохлорида,N-[(1S)-(2-циклопропил-1-фенил)этил]-1(2,4-дихлорфенил)-5-метил-N-пропил-1 Н-1,2,4 триазол-3-амина гидрохлорида,1-(2-хлор-4-метокси-5-метилфенил)-5-циклопропил-N-[(1S)-1-(4-метилфенил)бутил]-Nпропил-1 Н-1,2,4-триазол-3-амина гидрохлорида,5-циклопропил-1-(2,4-дихлорфенил)-N[(1S)-1-(4-метилфенил)бутил]-N-пропил-1 Н 1,2,4-триазол-3-амина гидрохлорида,1-[2-хлор-4-(метилсульфанил)фенил]-N-[2 циклопропил-1-(3-фтор-4-метилфенил)этил]-5 метил-N-пропил-1 Н-1,2,4-триазол-3-амина гидрохлорида,1-(2-хлор-4-метоксифенил)-N-[(1S)-2-циклопропил-1-(4-фторфенил)этил]-5-метил-N-пропил-1H-1,2,4-триазол-3-амина гидрохлорида и соответствующих оснований, других фармацевтически приемлемых солей присоединения, их сольватов и/или их гидратов. 6. Способ получения соединений формулы 44 где R1, R2, R3, R5 и R6 являются такими, как определено для (I), подвергают взаимодействию с соединением формулы R4COX (где X представляет собой галоген) с получением соединения формулы (I). 7. Способ получения соединений формулы(I) по п.1, отличающийся тем, что соединение формулы (VIII) где R1, R2, R3, R4 и R6 являются такими, как определено для (I), подвергают реакции алкилирования с соединением формулы R5X (где X представляет собой галоген) с получением соединения формулы (I). 8. Способ получения соединений формулы(I) по п.1 и соединений формулы (VIII), отличающийся тем, что соединения формулы (VI) или (IX) где R4, R5 и R6 являются такими, как определено для (I), подвергают взаимодействию с соединением формулы (III) где R1, R2 и R3 являются такими, как определено для (I), с получением соответственно соединений формулы (I) или формулы (VIII). 9. Фармацевтическая композиция, отличающаяся тем, что она содержит в качестве действующего начала соединение по любому из пп.1-5 в комбинации с одним или более чем одним подходящим эксципиентом. 10. Применение соединения по любому из пп.1-5 в приготовлении лекарств, предназначенных для предупреждения и/или лечения кортикотропин-рилизинг-фактор
МПК / Метки
МПК: C07D 249/14, A61K 31/4196, A61P 25/00
Метки: способы, 3-амино-1-фенил-1h[1,2,4]триазола, аминопроизводные, содержащие, фармацевтические, получения, замещенные, новые, разветвленные, композиции
Код ссылки
<a href="https://eas.patents.su/23-4957-novye-razvetvlennye-zameshhennye-aminoproizvodnye-3-amino-1-fenil-1h124triazola-sposoby-ih-polucheniya-i-soderzhashhie-ih-farmacevticheskie-kompozicii.html" rel="bookmark" title="База патентов Евразийского Союза">Новые разветвленные замещенные аминопроизводные 3-амино-1-фенил-1h[1,2,4]триазола, способы их получения и содержащие их фармацевтические композиции</a>
Новые замещенные пиридины или пиперидины, способ их получения и содержащие их фармацевтические композиции

Номер патента: 3053
Опубликовано: 26.12.2002
Авторы: Мет-Кон Отто, Лестаж Пьер, Кеньяр Даниель-Анри, Ренар Пьер, Лебрэн Мари-Сесиль, Ю Чу-Йи
МПК: A61K 31/44, A61P 25/28, C07D 211/22...
Метки: получения, способ, пиперидины, содержащие, замещенные, композиции, фармацевтические, новые, пиридины
Формула / Реферат:
1. Соединения формулы (I) где А означает пиридиновую, пиридиниевую или пиперидиновую группу; R2 означает атом водорода и R3 означает гидроксил или R2 и R3 вместе образуют оксогруппу; R4 означает незамещенный или замещенный фенил, незамещенный или замещенный нафтил или незамещенный или замещенный гетероарил; R1 означает атом водорода; или R1 и R4 вместе с несущими их двумя атомами углерода образуют цикл, содержащий 6 атомов углерода; или R1 и R2...
Новые замещенные димерные соединения, способ их получения и фармацевтические композиции, содержащие их

Номер патента: 3391
Опубликовано: 24.04.2003
Авторы: Депре Патрик, Ренар Пьер, Ларрайа Карлос, Беннежан Каролин, Делагранж Филипп, Шев Гвенаэль, Декамп-Франсуа Кароль, Гийоме Жеральд, Йус Сэд, Вио Мари-Клод
МПК: A61P 25/00, C07C 233/25, A61K 31/343...
Метки: содержащие, замещенные, фармацевтические, композиции, соединения, способ, димерные, получения, новые
Формула / Реферат:
1. Соединение формулы (I) A-G1-Cy-G2-Cy'-G3-B (I), где A обозначает -NHC(Q)R2, где Q означает O или S, a R2 означает (C1-C6)алкильную группу; B обозначает -NHC(Q)R2, C(Q)OR2, где Q и R2 имеют вышеуказанные значения; G1 и G3 обозначают возможно разветвленную алкиленовую группу, Cy и Cy' обозначает нафтил, 1,4-бензодиоксин, тетрагидронафтил, бензотиенил, дигидробензотиенил, бензофурил, дигидробензофурил, пирроло[3,2-b]пиридинил, индолил,...
Новые циклические α-амино-γ-гидроксиамидные соединения, способ их получения и фармацевтические композиции, их содержащие

Номер патента: 4955
Опубликовано: 28.10.2004
Авторы: Буланже Мишель, Вержбицкий Мишель, Фуркез Жан-Мари, Юссон-Робер Бернадетта, Левен Нижель, Ножан Оливье
МПК: A61P 3/10, A61K 31/40, C07C 237/14...
Метки: содержащие, получения, фармацевтические, новые, alpha;-амино-γ-гидроксиамидные, соединения, циклические, композиции, способ
Формула / Реферат:
1. Соединения формулы (I) в которой представляет собой насыщенное углеводородное кольцо, имеющее от 4 до 8 атомов углерода в кольце, необязательно замещенное одной или более линейными или разветвленными (C1-C6)алкильными группами, R1 и R4, которые могут быть одинаковыми или различными, каждый представляет собой атом водорода или линейную или разветвленную (C1-C6)ацильную группу, R2 и R3, которые могут быть одинаковыми или различными,...
Новые ингибиторы фарнезилтрансферазы, способы их получения, содержащие их фармацевтические композиции и их применение для получения медикаментов.

Номер патента: 2114
Опубликовано: 24.12.2001
Авторы: Дере Норбер, Коммерсон Алан, Майлие Патрик, Бурза Жан-Доминик, Шев Мишель, Капе Марк, Мартэн Жан-Поль, Суниго-Томсон Фабьенн
МПК: A61K 31/40, A61P 35/00, C07D 209/72...
Метки: медикаментов, новые, содержащие, способы, применение, композиции, получения, фарнезилтрансферазы, фармацевтические, ингибиторы
Формула / Реферат:
1. Соединения формулы I в которой Аr обозначает фенил, замещенный одним или несколькими атомами галогена, радикалом алкил С1-С4, С1-С4 алкокси, которые могут в свою очередь быть замещены атомами галогена или радикалом гидрокси; радикалом 3-N,N-диметиламино или 4-N,N-диметиламино; радикалом 4-метилсульфанил или радикалом циано; 2,3-дигидро-1,4-бензодиоксин-6-ил; дигидробензофуран; дигидробензопиран; нафтил или тетрагидронафтил; инданил; тиенил...
Новые 19-норстероиды, замещенные в положении 11&beta, способ и промежуточные продукты для их получения, применение в качестве лекарственных средств и содержащие их фармацевтические композиции

Номер патента: 3133
Опубликовано: 27.02.2003
Автор: Ник Франсуа
МПК: C07J 41/00, A61P 19/10, A61K 31/566...
Метки: продукты, качестве, композиции, 11&beta, содержащие, получения, промежуточные, 19-норстероиды, положении, лекарственных, замещенные, средств, применение, способ, новые, фармацевтические
Формула / Реферат:
1. Соединения общей формулы (I) в которой R1 обозначает атом водорода иди радикал ацил, R2 обозначает радикал (C1-C4)алкил, X обозначает атом галогена или атом водорода, n равно 3, 4 или 5, R3 и R4 обозначает (C1-C4)алкил или R3 и R4 образуют вместе с атомом азота, с которым они связаны, группу пироолидинил или пиперидинил, R5 обозначает OH и R6 обозначает H, (C1-C4)алкил возможно замещенный одним или тремя атомами галогена или R5 и R6...
Следующий патент: Фунгицидная смесь и способ борьбы с фитопатогенными грибами
Случайный патент: Переработка масличных семян