Глюкопиранозидные конъюгаты 2-(4-гидроксифенил)-1-[4-(2-амин-1-илэтокси)бензил]-1h-индол-5-олов
Номер патента: 5371
Опубликовано: 24.02.2005
Авторы: Миллер Кристофер Пол, Чан Анита Вай-Йин, Коллини Майкл Дэвид, Рубежов Аркадий Зиновий, Трэн Бэч Динх






Формула / Реферат
1. Соединение формулы I, имеющее структуру

где R1 и R2 независимо представляют собой водород, алкильную цепь из 1-6 атомов углерода, бензил, ацил из 2-7 атомов углерода, бензоил,

X представляет собой водород, алкил из 1-6 атомов углерода, CN, галоген, трифторметил или тиоалкил из 1-6 атомов углерода;
n=1-3;
при условии, что по меньшей мере один из R1 или R2 представляет собой

или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором X представляет собой алкил из 1-4 атомов углерода.
3. Соединение по п.1, в котором X представляет собой метил.
4. Соединение по любому из пп.1-3, в котором n равно 2 или 3.
5. Соединение по любому из пп.1-4, в котором R2 представляет собой водород.
6. Соединение по любому из пп.1-5, в котором R1 представляет собой

7. Соединение по любому из пп.1-6, которое имеет D-конфигурацию.
8. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-триацетил-{1-O-[4-(2-пиперидин-1-илэтокси)бензил]-2-[4-(2,2-диметилпропионилокси)фенил]-3-метил-1H-индол-5-ил}-глюкуроновой кислоты или его фармацевтически приемлемую соль.
9. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-триацетил-{1-O-[4-(2-азепан-1-илэтокси)бензил]-2-[4-(2,2-диметилпропионилокси)фенил]-3-метил-1H-индол-5-ил}глюкуроновой кислоты или его фармацевтически приемлемую соль.
10. Соединение по п.1, которое представляет собой триэтиламмониевую соль 5-O-глюкуронида 1-[4-(2-пиперидин-1-илэтокси)бензил]-2-(4-гидроксифенил)-3-метил-1H-индол-5-ола.
11. Соединение по п.1, которое представляет собой 5-O-глюкуронид 1-[4-(2-азепан-1-илэтокси)бензил]-2-(4-гидроксифенил)-3-метил-1H-индол-5-ола или его фармацевтически приемлемую соль.
12. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-O-триацетил-1-O-(4-{5-бензилокси-3-метил-1-[4-(2-пиперидин-1-илэтокси)бензил]-1H-индол-2-ил}фенил)-бета-D-глюкуроновой кислоты или его фармацевтически приемлемую соль.
13. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-O-триацетил-1-O-(4-{5-бензилокси-3-метил-1-[4-(2-азепан-1-илэтокси)бензил]-1H-индол-2-ил}фенил)-бета-D-глюкуроновой кислоты или его фармацевтически приемлемую соль.
14. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-O-триацетил-1-O-(4-{5-гидрокси-3-метил-1-[4-(2-пиперидин-1-илэтокси)бензил]-1H-индол-2-ил}фенил)-бета-D-глюкуроновой кислоты или его фармацевтически приемлемую соль.
15. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-O-триацетил-1-O-(4-{5-гидрокси-3-метил-1-[4-(2-азепан-1-илэтокси)бензил]-1H-индол-2-ил}фенил)-бета-D-глюкуроновой кислоты или его фармацевтически приемлемую соль.
16. Соединение по п.1, которое представляет собой 4-O-глюкуронид 2-(4-гидроксифенил)-3-метил-1-[4-(2-пиперидин-1-илэтокси)бензил]-1H-индол-5-ола или его фармацевтически приемлемую соль.
17. Соединение по п.1, которое представляет собой триэтиламмониевую соль 4-O-глюкуронида 2-(4-гидроксифенил)-3-метил-1-[4-(2-пиперидин-1-илэтокси)бензил]-1H-индол-5-ола.
18. Соединение по п.1, которое представляет собой 4-O-глюкуронид 1-[4-(2-азепан-1-илэтокси)бензил]-2-(4-гидроксифенил)-3-метил-1H-индол-5-ола или его фармацевтически приемлемую соль.
19. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-O-триацетил-1-O-[4-[1-[4-[2-гексагидро-1H-азепин-1-илэтокси]бензил]-3-метил-5-[(2,3,4-O-триацетил-6-O-метил-бета-D-глюкопирануронилозил)окси]-1H-индол-2-ил]фенил]-бета-D-глюкопиранозидуроновой кислоты или его фармацевтически приемлемую соль.
20. Соединение по п.1, которое представляет собой 4-[5-(бета-D-глюкопирануронозилокси)-1-[4-[2-(гексагидро-1H-азепин-1-ил)этокси]бензил]-3-метил-1H-индол-2-ил]фенил-бета-D-глюкопиранозидуроновую кислоту.
21. Способ лечения или ингибирования сердечно-сосудистого заболевания, понижения уровней липидов в крови или повышения уровней ЛВП у млекопитающего, которое в этом нуждается, который включает введение указанному млекопитающему соединения формулы I, заявленного в любом из пп.1-20.
22. Способ лечения или ингибирования рака молочной железы у млекопитающего, которое в этом нуждается, который включает введение указанному млекопитающему соединения формулы I, заявленного в любом из пп.1-20.
23. Способ лечения или ингибирования утраты костной ткани или остеопороза у млекопитающего, которое в этом нуждается, который включает введение указанному млекопитающему соединения формулы I, заявленного в любом из пп.1-20.
24. Способ проведения гормональной заместительной терапии у млекопитающего, которое в этом нуждается, который включает введение указанному млекопитающему соединения формулы I, заявленного в любом из пп.1-20.
25. Способ усиления познавательной функции или лечения или ингибирования деменций или болезни Альцгеймера у млекопитающего, которое в этом нуждается, который включает введение указанному млекопитающему соединения формулы I, заявленного в любом из пп.1-20.
26. Фармацевтическая композиция, которая включает соединение формулы I, заявленное в любом из пп.1-20, и фармацевтический носитель.
27. Способ получения соединения формулы I, который включает превращение соединения формулы Ia

где X и n определены в п.1, а один из R1 и R2 представляет собой

другой представляет собой защитную группу, в условиях, подходящих для удаления защитной группы, с получением соответствующего соединения формулы I, где один из R1 и R2 представляет собой

а другой из R1 и R2 представляет собой водород; указанное соединение может быть гидролизовано одновременно со снятием защиты или последовательно с получением соответствующего соединения формулы I, где один из R1 и R2 представляет собой водород, а другой представляет собой

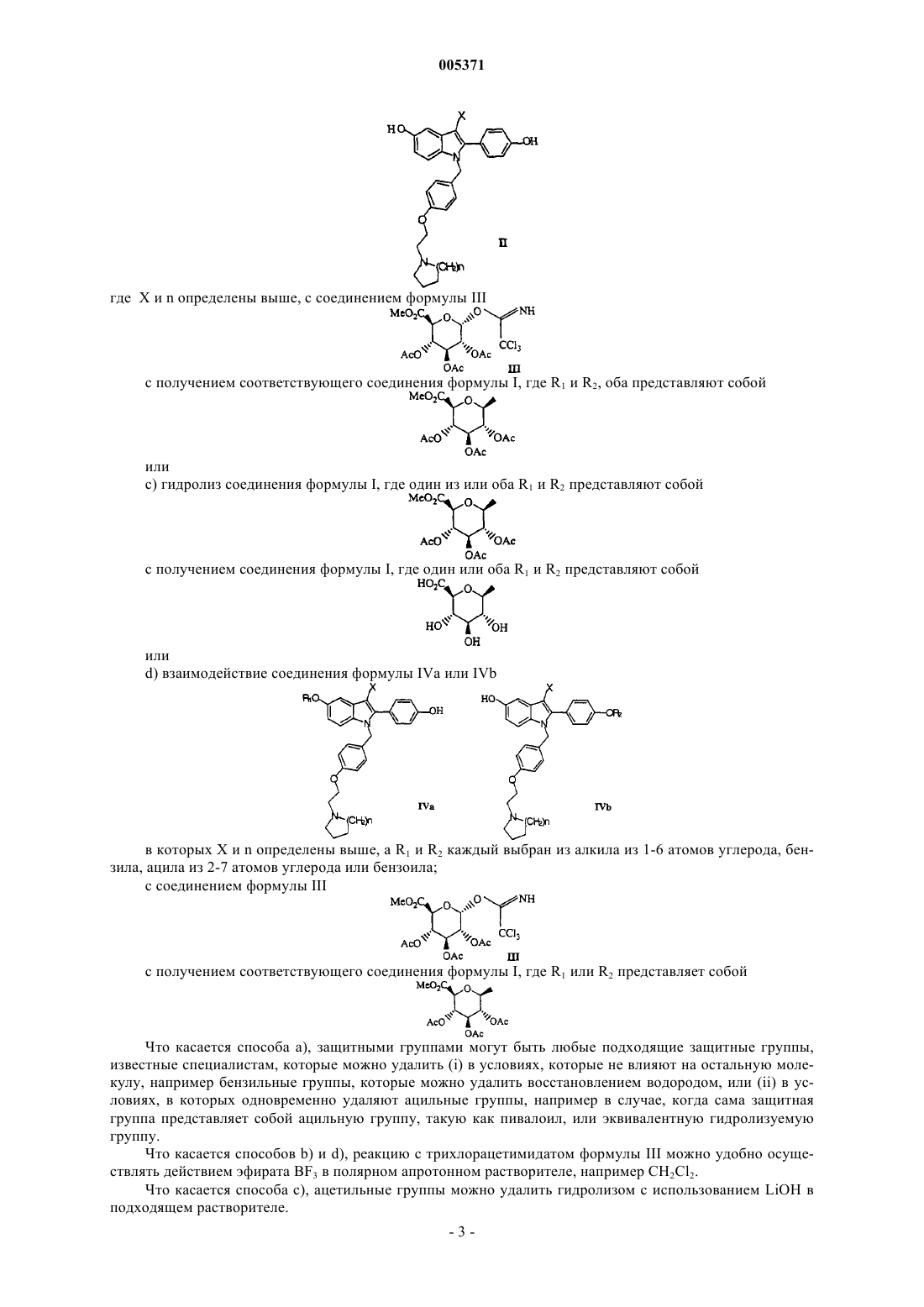
28. Способ получения соединения формулы I, который включает взаимодействие соединения формулы II

где X и n определены в п.1, с соединением формулы III

с получением соответствующего соединения формулы I, где R1 и R2, оба представляют собой

29. Способ получения соединения формулы I, который включает гидролиз соединения формулы I, где один или оба R1 и R2 представляют собой

с получением соединения формулы I, где один или оба R1 и R2 представляют собой

30. Способ получения соединения формулы I, который включает взаимодействие соединения формулы IVa или IVb

в которых X и n определены в п.1, а R1 и R2 каждый выбран из алкила из 1-6 атомов углерода, бензила, ацила из 2-7 атомов углерода или бензоила,
с соединением формулы III

с получением соответствующего соединения формулы I, где R1 или R2 представляет собой

Текст
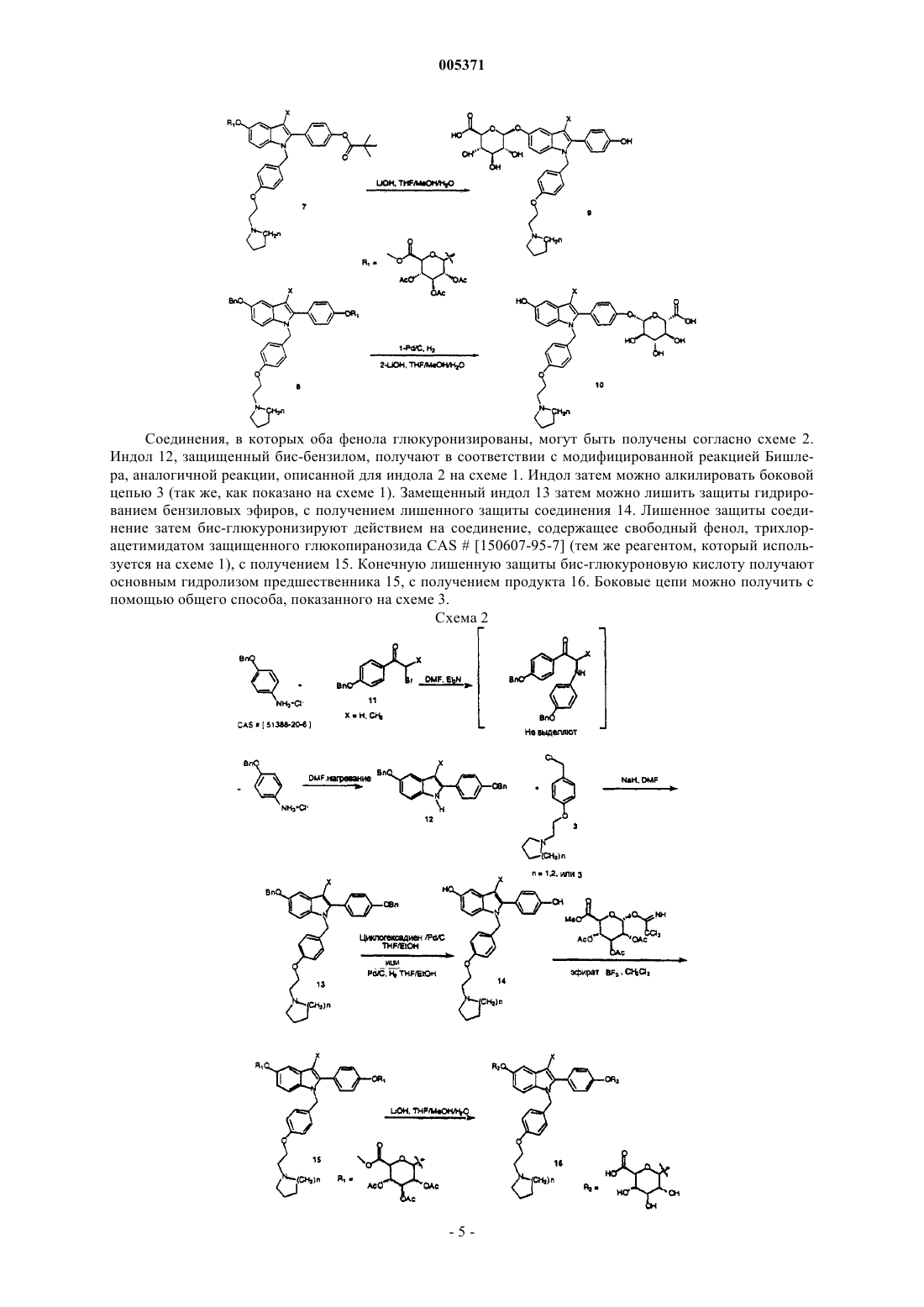
005371 Настоящее изобретение относится к глюкопиранозидным конъюгатам 2-(4-гидроксифенил)-1-[4-(2-амин-1 илэтокси)бензил]-1 Н-индол-5-олов, которые могут использоваться в качестве гистоселективных эстрогенных агентов. Также описаны способы получения указанных соединений и содержащие их фармацевтические композиции. Применение гормональной заместительной терапии для профилактики остеопороза у женщин в постменопаузе хорошо известно. Обычная схема предусматривает дополнительное снабжение организма эстрогенами, с использованием композиций, содержащих эстрон, эстриол, этинилэстрадиол или конъюгированные эстрогены, выделенные из натуральных источников (т.е. PREMARIN; конъюгированные эстрогены лошади). Некоторым пациентам указанная терапия может быть противопоказана из-за пролиферативных эффектов эcтрогенов, применяемых без противопоставления (эстрогенов, которые применяют без комбинации с прогестинами), в отношении ткани матки. Указанная пролиферация связана с повышенным риском эндометриоза и/или рака эндометрия. Действия непротивопоставленных эстрогенов на ткань молочной железы менее ясны, но вызывают некоторые опасения. Потребность в эстрогенах, которые могут поддерживать сохранение костной ткани при минимальных пролиферативных эффектах в отношении матки и молочной железы,является очевидной. Некоторые нестероидные антиэстрогены, как было показано, поддерживают массу костной ткани у крыс с удаленными яичниками, а также в клинических испытаниях у людей. Тамоксифен(продается как NOVALDEX, тамоксифена цитрат), например, является полезным паллиативным средством для лечения рака молочной железы и, как было показано, оказывает действие, подобное агонисту эстрогенов, на костную ткань у людей. Однако он также является частичным агонистом в отношении матки, и это вызывает некоторые опасения. EVISTA (ралоксифен), бензотиофеновый антиэстроген, как было показано,стимулирует рост матки у крыс с удаленными яичниками в меньшей степени, чем тамоксифен, в то же время поддерживая способность сохранять костную ткань. Полезный обзор гистоселективных эстрогенов представлен в статье "Tissue-Selective Actions Of Estrogen Analogs", Bone Vol. 17, No. 4, October 1995, 181S-190S. О применении индолов в качестве антагонистов эстрогенов сообщает Von Angerer, см. J. Med. Chem. 1990,33, 2635-2640; J. Med. Chem. 1987, 30, 131-136. См. также Ger. Offen., DE 3821148 A1 891228 и WO 96/03375.WO A 95 17383 (Kar Bio АВ) описывает индольные антиэстрогены с длинными прямыми цепями. Другой родственный патент WO A 93 10741 описывает 5-гидроксииндол с описанием группы соединений,включающей другие боковые цепи. WO 93/23374 (Otsuka Pharmaceuticals, Japan) описывает соединения,которые отличаются от настоящего изобретения, где OR2 в настоящей формуле I, приведенной ниже, определен как тиоалкил, и в указанной ссылке не описаны соединения, имеющие боковые цепи из азота индода с такой же структурой, которая описана в настоящем изобретении. Там, где заявленная боковая цепь аналогична приведенной в настоящем описании, соединения представляют собой амиды: ацилированные индолы не заявляются в настоящем изобретении. Имеется сообщение о конъюгатах глюкуроновой кислоты и селективного модулятора эстрогеновых рецепторов ралоксифена (бензотиофена) (ЕР 683170 A1 951122). Описание изобретения Настоящее изобретение относится к соединениям формулы I, имеющим структуру где R1 и R2 независимо представляют собой водород, алкильную цепь из 1-6 атомов углерода, бензил,ацил из 2-7 атомов углерода, бензоил, Х представляет собой водород, алкил из 1-6 атомов углерода, CN, галоген, трифторметил или тиоалкил из 1-6 атомов углерода;n=1-3; при условии, что по меньшей мере один из R1 и R2 представляет собой или к их фармацевтически приемлемым солям, которые могут использоваться в качестве гистоселективных эстрогенов.-1 005371 Термин "алкил" включает заместители как с прямыми, так и разветвленными цепями. Примеры включают алкил из 1-4 атомов углерода, такой как метил, этил, н-пропил, изопропил, бутил и трет-бутил."Галоген" означает бром, хлор, фтор и йод. Термин "ацил" включает алканоильные группы, особенно те,в которых алкильная часть представляет собой алкил, как описано выше, например ацетил. Примерами Х являются алкильные группы из 1-4 атомов углерода, например метил. Примерами n являются 2 и 3. Примерами R1 и R2 независимо являются водород, бензил и пивалоил, другой из R1 и R2 является одним из Настоящее изобретение описывает как L-, так и D-форму глюкопиранозидов, при этом D-форма является предпочтительной. Фармацевтически приемлемые соли включают соли, образованные по реакции присоединения с неорганическими или органическими кислотами. Могут использоваться неорганические кислоты, такие как хлористо-водородная кислота, бромисто-водородная кислота, йодисто-водородная кислота, серная кислота,фосфорная кислота и азотная кислота. Также могут использоваться органические кислоты, такие как уксусная кислота, пропионовая кислота, лимонная кислота, малеиновая кислота, яблочная кислота, винная кислота, фталевая кислота, янтарная кислота, метансульфоновая кислота, толуолсульфоновая кислота, нафталинсульфоновая кислота, камфарсульфоновая кислота и бензолсульфоновая кислота. Фармацевтически приемлемые соли по настоящему изобретению также включают четвертичные аммониевые соли, которые могут быть получены кватернизацией основного амина соединений по настоящему изобретению электрофильным органическим галогенидом, мезилатом, тозилатом и т.п. Соединения по настоящему изобретению,которые содержат карбоновую кислоту, могут образовывать фармацевтически приемлемые основно-аддитивные соли действием на нейтральное исходное вещество подходящим неорганическим основанием, таким как гидроксиды или карбонаты щелочных металлов, таких как литий, натрий, калий, цезий, магний, кальций или барий. Или на кислоту можно действовать органическим основанием (таким, как различные органические первичные(включая аммоний), вторичные или третичные амины), с получением солей аммония. Настоящее изобретение относится также к способам получения соединений формулы I. Соответственно, предложен способ получения соединения формулы I, который включает один из следующих: а) взаимодействие соединения формулы Iа другой представляет собой защитную группу в условиях, подходящих для удаления защитной группы, с получением соответствующего соединения формулы I, где один из R1 и R2 представляет собой а другой из R1 и R2 представляет собой водород; указанное соединение может быть гидролизовано одновременно со снятием защиты или последовательно с получением соответствующего соединения формулы I, где один из R1 и R2 представляет собой водород, а другой представляет собойb) взаимодействие соединения формулы II с получением соответствующего соединения формулы I, где R1 и R2, оба представляют собой или с) гидролиз соединения формулы I, где один из или оба R1 и R2 представляют собой с получением соединения формулы I, где один или оба R1 и R2 представляют собойd) взаимодействие соединения формулы IVa или IVb в которых Х и n определены выше, а R1 и R2 каждый выбран из алкила из 1-6 атомов углерода, бензила, ацила из 2-7 атомов углерода или бензоила; с соединением формулы III с получением соответствующего соединения формулы I, где R1 или R2 представляет собой Что касается способа а), защитными группами могут быть любые подходящие защитные группы,известные специалистам, которые можно удалить (i) в условиях, которые не влияют на остальную молекулу, например бензильные группы, которые можно удалить восстановлением водородом, или (ii) в условиях, в которых одновременно удаляют ацильные группы, например в случае, когда сама защитная группа представляет собой ацильную группу, такую как пивалоил, или эквивалентную гидролизуемую группу. Что касается способов b) и d), реакцию с трихлорацетимидатом формулы III можно удобно осуществлять действием эфирата ВF3 в полярном апротонном растворителе, например СН 2 Сl2. Что касается способа с), ацетильные группы можно удалить гидролизом с использованием LiOH в подходящем растворителе.-3 005371 Соединения по настоящему изобретению можно удобно синтезировать в соответствии с основными способами, представленными на схеме 1. Ортогонально защищенный индол получают в соответствии с модифицированной реакцией Бишлера, при которой гидроксипропиофенон или ацетофенон 1, защищенный бром-4-пивалоилом, взаимодействует с 4-бензилоксианилином в DMF в присутствии триэтиламина. Реакцию контролируют с помощью ТСХ по расходованию исходных веществ. Вещество, замещенное анилином, не нуждается в выделении, но, вместо этого, в той же самой колбе на него действуют дополнительными 1,25-1,5 эквивалентами гидрохлорида 4-бензилоксианилина и нагревают до 120-160 С до полного расходования предыдущего промежуточного продукта. На защищенный индол 2 затем действуют подходя-щим основанием,таким как гидрид натрия в DMF, а затем он взаимодействует с подходящим бензилхлори-дом типа 3. Индол 4 затем можно лишить монозащиты или гидрированием бензильной группы в 5 положении индола, или гидролизом пивалоильной группы в положении 4' 2-фенильной группы индола. Удаляемая груп-па определяется положением желаемого конъюгата глюкуроновой кислоты. Удаление бензильной группы или сложного эфира пивалоила дает соединения типа 5 или 6, соответственно. Взаимодействие соединения 5 или 6 с трихлорацетимидатом CAS[150607-95-7] защищенного глюкопиранозида в присутствии эфирата ВF3 в полярном апротонном растворителе, таком как дихлорметан, дает глюкопиранозидированные соединения 7 или 8. Заявители установили, что взаимодействие 5 или 6 с трихлорацетимидатом, описанное выше, в СН 2 Сl2 с использованием молекулярных сит 3 позволяет осуществить синтез глюкопиранозидов с очень хорошим выходом. Соединения образуются исключительно в виде -глюкопиранозидов (экваториально замещенных). Затем на пивалоилированное соединение 7 можно действовать LiOH в смеси ТНF/H2 О/МеОН (или в смеси диоксан/МеОН/Н 2 О), чтобы осуществить полное снятие защиты с соединения, с получением, после соответствующей обработки, моноглюкуроновой кислоты 9. Монобензиловый эфир 8 можно гидрировать переносом водорода между циклогексадиеном и Pd/C, a затем гидролизовать с помощью LiOH в смеси THF/H2O/MeOH(или в смеси диоксан/МеОН/Н 2 О), с получением моноглюкуроновой кислоты 10. Более предпочтительными условиями для гидрирования в более крупных масштабах являются условия, при которых используется катализатор 10% Pd/C, H2 и система растворителей, состоящая из THF/EtOH. Схема 1 Соединения, в которых оба фенола глюкуронизированы, могут быть получены согласно схеме 2. Индол 12, защищенный бис-бензилом, получают в соответствии с модифицированной реакцией Бишлера, аналогичной реакции, описанной для индола 2 на схеме 1. Индол затем можно алкилировать боковой цепью 3 (так же, как показано на схеме 1). Замещенный индол 13 затем можно лишить защиты гидрированием бензиловых эфиров, с получением лишенного защиты соединения 14. Лишенное защиты соединение затем бис-глюкуронизируют действием на соединение, содержащее свободный фенол, трихлорацетимидатом защищенного глюкопиранозида CAS[150607-95-7] (тем же реагентом, который используется на схеме 1), с получением 15. Конечную лишенную защиты бис-глюкуроновую кислоту получают основным гидролизом предшественника 15, с получением продукта 16. Боковые цепи можно получить с помощью общего способа, показанного на схеме 3. Схема 2 Эстрогенная антагонистическая активность соединений, представленных в настоящем изобретении,была показана в ходе стандартной фармакологической тест-процедуры, с использованием клеток MCF-7,трансфектированных рецептором эстрогена. При пероральном введении соединения по настоящему изобретению действуют, по меньшей мере частично, как пролекарства для соответствующих гидроксилированных соединений, описанных в ЕР 802183. Соответственно, соединения по настоящему изобретению представляют собой гистоселективные эстрогены, что означает, что в определенной ткани, содержащей рецепторы эстрогенов, указанные соединения будут действовать как агонисты эстрогенов, а в другой ткани,содержащей рецепторы эстрогенов, указанные соединения будут действовать как антагонисты. Процедура, которую использовали для демонстрации эстрогенной антагонистической активности MCF-7 в отношении клеток карциномы, кратко описана ниже, а полученные результаты представлены в таблице ниже. Приготовление клеток. Клетки MCF-7 пересевали 2 раза в неделю на ростовую среду [среду D-MEM/F-12, содержавшую 10% (об./об.) инактивированной нагреванием фетальной бычьей сыворотки, 1% (об./об.) пенициллинастрептомицина и 2 мМ glutaMax-1]. Клетки выращивали в вентилируемых колбах в термостатах с внутренней температурой 37 С и атмосферой с 5% СО 2 и 95% влажностью. За 1 день до обработки клетки высевали на 96-луночные планшеты с ростовой средой в количестве 25000 на лунку и инкубировали при 37 С в течение ночи. Условия тест-процедуры. Клетки инфицировали в течение 2 ч при 37 С 5-ЕRЕ-tk-люциферазой аденовируса в разведении 1:10 в количестве 50 мкл на лунку, в экспериментальной среде [среда D-MEM/F-12 без Фенолового красного, содержащая 10% (об./об.) инактивированной нагреванием и истощенной активированным углем фетальной бычьей сыворотки, 1% (об./об.) пенициллина-стрептомицина, 2 мМ glutaMax-1, 1 мМ пирувата натрия]. Лунки однократно промывали 150 мкл экспериментальной среды. И, наконец, клетки обрабатывали в течение 24 ч при 37 С, с кратностью повторения 8 лунок на одну обработку, носителем в количестве 150 мкл на лунку ( или равно 0,1% об./об. DMSO) или соединением, которое было разведеноили равно 1000-кратному в экспериментальной среде. Эксперименты, касающиеся реакции доза-ответ, выполняли с использованием активных соединений при log-повышении концентрации с 10-14 до 10-5 М, по агонистическому или антагонистическому варианту. С помощью указанных кривых доза-ответ получали величины EC50 и IC50, соответственно. Последняя лунка в каждой группе обработки содержала 5 мкл 3 х 10-5 ICI-182780 (конечная концентрация 10-6 М) в качестве контроля антагониста ER. После обработки клетки лизировали в шейкере в течение 15 мин с использованием 1 х лизирующего клеточные культуры реагента (Promega Corporation) в количестве 25 мкл на лунку. Клеточные лизаты (20 мкл) переносили в 96-луночный люминометрический планшет и измеряли активность люциферазы на люминометре MicroLumat LB 96 Р (EG и G Bethold) с использованием субстрата для люциферазы (PromegaCorporation) в количестве 100 мкл на лунку. Перед инъекцией субстрата для каждой лунки проводили фоновое измерение в течение 1 с. После инъекции субстрата активность люциферазы измеряли в течение 10 с после 1-секундной задержки. Данные переносили с люминометра на персональный компьютер Макинтош и анализировали с помощью программы JMP (институт SAS); указанная программа вычитает фоновые значения из измерения люциферазы для каждой лунки, а затем определяет среднюю величину и стандартное отклонение для каждой обработки. Анализ результатов. Данные по люциферазе трансформировали с помощью логарифмов и для устранения выпадающих трансформированных наблюдений использовали Huber M-estimator. Для анализа трансформированных и взвешенных данных с помощью анализа ANOVA (критерий Дюннетта) использовали программу JMP. Результаты по обработке соединением сравнивали с контрольными результатами по носителю (0,1 нМ 17-эстрадиол) по антагонистическому варианту. Для первоначального эксперимента с однократной дозой, когда результаты по обработке соединением достоверно отличались от соответствующего контроля(р 0,05), тогда результаты выражали в процентах от контроля 17-эстрадиола [т.е. соединениеконтроль с носителем)/(контроль с 17-эстрадиолом - контроль с носителемх 100]. Величины EC50 и/илиIC50 по нелинейным кривым доза-ответ получали также с помощью программы JMP. Конъюгаты с моноглюкуроновой кислотой испытывали в анализе с MCF-7 как по агонистическому,так и по антагонистическому варианту (совместные дозы с 10 пМ 17-эстрадиола). Все четыре испытанных соединения показали антиэстрогенную активность в указанной клеточной системе, и ни одно из соединений не показало достоверной агонистической активности в отношении указанных клеток. Это является желательным результатом, поскольку клеточная линия MCF-7 получена из ткани молочной железы человека, а полученные результаты показывают, что указанные соединения противодействовали пролиферативным эффектам эстрогенной активности в указанных клетках. Положительным признаком является также то, что указанные соединения демонстрируют способность проникать в клетки и аффинность связывания с рецепторами (поскольку клетки MCF-7 экспрессируют рецепторы эстрогенов). Данные для соединений представлены в таблице ниже. СоединениеIC50 для MCF-7 29 (схема 5) 230 нМ 30 (схема 5) 210 нМ 37 (схема 6) 1200 нМ 38 (схема 6) 1100 нМ Как заявлено выше, соединения по настоящему изобретению представляют собой гистоселективные эстрогены, действуя как агонисты эстрогенов на определенную ткань и как антагонисты на другую ткань. В частности, соединения по настоящему изобретению действуют как агонисты рецепторов эстрогенов в том, что касается обеспечения защиты от остеопороза, понижения уровней липидов и повышения уровней ЛВП. Соединения по настоящему изобретению действуют как антагонисты рецепторов эстрогенов в том, что касается ингибирования роста матки (как потенциального побочного эффекта введения эстрогенных соединений), обеспечивают защиту от рака молочной железы, обеспечивают контрацепцию,ингибируют деменцию и обеспечивают усиление познавательной функции. Соответственно, соединения по настоящему изобретению могут применяться для лечения определенных состояний, вызываемых дефицитом эстрогенов, таких как утрата костной ткани или остеопороз, который может быть результатом дисбаланса образования новых костных тканей и резорбции более старых тканей у пациента, что приводит в итоге к остеопорозу. Истощение костной ткани приводит к появлению особой категории пациентов, особенно женщин в постменопаузе, женщин, которые перенесли двустороннюю овариэктомию, пациентов, получающих или получавших продолжительную кортикостероидную терапию, пациентов с дисгенезией половых органов и пациентов, страдающих синдромом Кушинга. Указанные соединения могут также предназначаться пациентам, имеющим необходимость в замене кости, включая зубы и кости области рта, переломы костей, дефектные костные структуры, пациентам с операциями на костях и/или имплантацией протезов. Помимо проблем, описанных выше, указанные соединения можно использовать для лечения остеоартрита, гипокальциемии, гиперкальциемии, болезни Педжета, остеомаляции, множественной миеломы и других форм злокачественных опухолей, оказывающих вредное влияние на костные ткани. Другие состояния, вызываемые дефицитом эстрогенов, которые можно лечить соединениями по настоящему изобретению, включают гипертрофию предстательной железы, атрофию влагалища и кожи, угри, сердечно-сосудистое заболевание, контрацепцию у женщин в пременопаузе, а также гормональную заместительную терапию у женщин в постменопаузе или имеющих другие состояния дефицита эстрогенов, когда восполнение эстрогенов будет оказывать благоприятное действие. Поскольку соединения по настоящему изобретению действуют также как антагонисты эстрогенов,они являются особенно пригодными для антиэстрогенной терапии, особенно для лечения облысения по мужскому типу, дисфункционального маточного кровотечения, полипов эндометрия, доброкачественного заболевания молочной железы, лейомиома матки, аденомиоза, для лечения новообразований, таких как рак яичника, рак молочной железы, рак эндометрия, меланома, рак предстательной железы, злокачественные опухоли толстого кишечника и злокачественные опухоли ЦНС, для лечения эндометриоза,синдрома поликистозных яичников, бесплодия, болезни Альцгеймера, угасания познавательной функции и других заболеваний ЦНС, а также для обеспечения контрацепции. Соединения по настоящему изобретению являются также пригодными для лечения болезненных состояний, при которых аменорея является благоприятной, таких как лейкоз, удаление эндометрия, хроническое заболевание почек или печени или болезни или нарушения свертываемости крови. Кроме того, соединения 27, 28, 33 и 34 являются промежуточными соединениями, пригодными для получения соединений 29, 30, 35, 36, 37 и 38, как показано на схемах 5 и 6 (ниже). Эффективное введение указанных соединений можно осуществлять в дозах приблизительно от 0,1 до 1000 мг в день. Предпочтительно, доза составляет приблизительно от 10 до 600 мг в день, более предпочтительно от 50 до 600 мг в день, однократно или двумя или более разделенными дозами. Указанные дозы можно вводить любым способом, пригодным для направления активных соединений в кровоток-7 005371 реципиента, включая пероральный, через имплантаты, парентеральный (включая внутривенные, интраперитонеальные и подкожные инъекции), ректальный, вагинальный и чрескожный. Для целей настоящего описания под чрескожным введением подразумеваются все способы введения через поверхность тела и внутренние выстилки различных трактов организма, включая ткани эпителия и слизистых оболочек. Указанные виды введения можно осуществлять с использованием настоящих соединений или их фармацевтически приемлемых солей, в лосьонах, кремах, пенах, пластырях, суспензиях, растворах и суппозиториях (ректальных и вагинальных). Пероральные композиции, содержащие активные соединения по настоящему изобретению, могут включать любые обычные пероральные лекарственные формы, включая таблетки, капсулы, трансбуккальные формы, лепешки, пастилки и жидкости для перорального введения, такие как суспензии или растворы. Капсулы могут содержать смеси активного соединения (соединений) с инертными наполнителями и/или разбавителями, такими как фармацевтически приемлемые крахмалы (например, кукурузный,картофельный крахмал или крахмал из тапиоки), сахара, искусственные подсластители, порошкообразные целлюлозы, такие как кристаллическая и микрокристаллическая целлюлозы, мука, желатины, камеди и т.п. Пригодные таблеточные композиции можно изготавливать обычными способами прессования,влажного гранулирования или сухого гранулирования и использовать фармацевтически приемлемые разбавители, связующие, замасливатели, разрыхлители, суспендирующие или стабилизирующие агенты,включая, без ограничения, стеарат магния, стеариновую кислоту, тальк, лаурилсульфат натрия, микрокристаллическую целлюлозу, кальциевую соль карбоксиметилцеллюлозы, поливинилпирролидон, желатин, альгиновую кислоту, аравийскую камедь, ксантановую камедь, цитрат натрия, комплексные силикаты, карбонат кальция, глицин, декстрин, сахарозу, сорбит, гидрофосфат кальция, сульфат кальция, лактозу, каолин, маннит, хлорид натрия, тальк, сухие крахмалы и порошкообразный сахар. Другие композиции по настоящему изобретению могут включать обычные композиции с замедленным или отсроченным высвобождением активного соединения для того, чтобы изменить всасывание активного соединения (соединений). Суппозитории можно изготавливать из традиционных материалов, включая масло какао, с добавлением или без добавления восков для изменения температуры плавления суппозитория, и глицерин. Можно использовать также водорастворимые основы для суппозиториев, такие как полиэтиленгликоли различных молекулярных масс. Понятно, что доза, схема и способ введения указанных соединений будут изменяться в зависимости от заболевания, пациента и мнения лечащего врача. Предпочтительно, чтобы введение одного или более соединений начиналось с малой дозы и повышалось до тех пор, пока не будет достигнут желаемый результат. Следующие методики содержат примеры, характеризующие настоящее изобретение. Все реакции выполнялись в атмосфере азота. Хроматографию выполняли с использованием силикагеля 230-400 меш. Тонкослойную хроматографию осуществляли на пластинках с силикагелем. Спектры 1H ЯМР получали на установках Bruker AM-400, GE QE 300, Bruker DPX-300 или DPX-301; и химические сдвиги выражали в миллионных долях. Величины температур плавления определялись с помощью аппарата Thomas-Hoover и не корректировались. ИК-спектры определяли с помощью спектрофотометров с дифракционной решеткой Perkin-Elmer, Mattson 5020FT-IR или Perkin-Elmer 784. Масс-спектры определяли с помощью масс-спектрометров Kratos MS 50 или Finnigan 8230. LC/MS выполняли на системеMicromass, Model Platform LC с системой HP 1100 LC и диодно-матричным детектором. Использовали колонку 2,0 х 50 мм С 18 3 мкм. Использовали следующую подвижную фазу: А=950 (10 мМ NH4OAc): 50t=15, 0% А, 100% В. ВЭЖХ осуществляли на системе Waters 60F Pump с колонкой 4,6 мм х 15 см LUNA 3 мкм, С 18 и детекторе 996 Diode Array (при 300 нм или 220 нм). Расход=1,0 мл/мин. Температура=30 С. Подвижная фаза была следующей: А=950 H2O: 50 СН 3 СN, 20 мМ К 2 НРO4/Н 3 РO4; В=300 Н 2O: 700 СН 3 СN,20 мМ К 2 НРO4/Н 3 РO4. Использовали следующий градиент: t=0, 100% В, 0% С; t=55, 0% В, 100% С. Элементный анализ получали с помощью элементного анализатора Perkin-Elmer 2400. Соединения, для которых приведены данные CHN, их значения находятся в пределах 0,4% теоретической величины для данной формулы, если не указано иное. Следующие соединения [примеры 1-3] получали в соответствии со схемой 4, как показано ниже. Схема 4-8 005371 Пример 1. Получение тетраацетилглюкуронового сложного эфира В. К суспензии D-глюкуронолактона А (100 г, 56,8 ммоль, продается компанией Aldrich Chemical) в МеОН (750 мл) добавляли гранулы NaOH (0,4 г, 0,01 ммоль) под Аr. Суспензия медленно превращалась в прозрачный раствор желтого цвета в течение 2 ч при перемешивании. Через 16 ч реакционную смесь концентрировали в вакууме с получением пены коричневого цвета. Неочищенный продукт растворяли в пиридине (200 мл), охлаждали на бане со льдом, а затем добавляли порциями по 50 мл уксусный ангидрид(375 мл). Во время добавления реакционная смесь становилась темно-коричневой. После добавления реакционную смесь продували Аr и хранили в холодильнике. Через 36 ч на дне колбы образовался коричневатый осадок. Реакционную смесь фильтровали и промывали теплым EtOH (100 мл), высушивали на воздухе в течение ночи с получением В 78,5 г, 37% твердого вещества не совсем белого цвета. Rf=0,65 (EtOAc); 1H ЯМР (CDCl3) 5,77 (д, 1 Н, J=7,7 Гц), 5,12-5,35 (м, 3 Н), 4,18 (д, 1 Н, J=9,3 Гц), 3,75 (с, 3 Н), 2,04-2,12 (м, 12 Н). Пример 2. Получение гидроксиглюкуронового сложного эфира С. К раствору тетраацетилглюкуронового сложного эфира В в DMF (90 мл) добавляли твердый ацетат гидразина (3 г, 32,6 ммоль) при комнатной температуре под Аr. Суспензию нагревали до 65 С под Аr. Через 1 ч реакционную смесь охлаждали до комнатной температуры, выливали в Н 2O (200 мл) и EtOAc(200 мл). Разделяли два слоя. Органический слой экстрагировали Н 2O (2 х 150 мл), высушивали надNа 2SO4, фильтровали, концентрировали в вакууме. Очистка на SiO2 с элюированием смесью (2:1) гексаны:ЕtOАс давала 4,1 г, 46% выход гидроксиглюкуронового сложного эфира С в виде сиропа желтого цвета. Rf=0,61 (EtOAc); 1H ЯМР (СDСl3) 4,58-5,63 (м, 4 Н), 3,75 (с, 3 Н), 1,97-2,22 (м, 9 Н). Пример 3. Получение трихлорацетимидата D. К раствору гидроксиглюкуронового сложного эфира С (11,1 г, 33,3 ммоль) в CH2Cl2 (100 мл) добавляли карбонат цезия (1,5 г, 4,7 ммоль) и трихлорацетонитрил (10 мл, 63 ммоль) при комнатной температуре под Аr. После перемешивания в течение 4 дней к суспензии добавляли Н 2 О (200 мл) и СНСl3 (100 мл). Разделяли два слоя. Органический слой экстрагировали Н 2 О (2 х 100 мл), высушивали над Na2SO4,фильтровали, концентрировали в вакууме. Очистка на SiO2 с элюированием смесью (1:1) гексаны:EtOAc давала 8,9 г, 58% выход трихлорацетимидата D в виде твердого вещества не совсем белого цвета. Rf=0,67(дд, 1 Н, J=3,54, 10,2 Гц), 4,50 (д, 1 Н, J=10,2 Гц), 3,68 (с, 3 Н), 2,02-2,12 (м, 9 Н). Следующие 5-глюкопиранозидные конъюгаты и промежуточные соединения [примеры 4-21] получали в соответствии со схемами 5 и 6, как показано ниже. Схема 5 Пример 4. 2,2-Диметил-4-(1-oкco-2-бромпропил)фениловый эфир пропановой кислоты (19). К разбавленному раствору 4'-пивалоилпропиофенона (22,4 г, 95,6 ммоль) в эфире (1,2 л) при 0 С медленно добавляли Вr2 (16,7 г, 105,2 ммоль). Реакция протекала при 0 С в течение 20 мин, после чего реакционную смесь оставляли стоять при комнатной температуре на 45 мин. По мере протекания реакции при комнатной температуре наблюдалось постепенное исчезновение окраски брома. У продукта была та же величина Rf, что и у исходного вещества на ТСХ в системе 10% EtOAc/Hex. Реакцию гасили 10% раствором Nа 2SО 3, промывали водой, насыщенным солевым раствором и высушивали над MgSO4. Полученный продукт представлял собой восковидное твердое вещество и использовался на следующем этапе без очистки (выход=30 г, 100%). Т.пл.=40-45 С. Пример 5. 4-(5-Бензилокси-3-метил-1 Н-индол-2-ил)фениловый эфир 2,2-диметилпропионовой кислоты (20). Раствор в DMF (350 мл), содержащий бромкетон 19 (44,82 г, 143 ммоль), гидрохлорид парабензилоксианилина (37,1 г, 157,4 ммоль, 1,1 экв.), Et3N (31,85 г, 314,8 ммоль, 2,2 экв.), нагревали до 130 С в течение 1,5 ч и кипятили с обратным холодильником, после чего выполняли ТСХ (15% EtOAc/Hex). Промежуточный продукт -анилинпропиофенон наблюдался как более полярное пятнo, чем -бром-4'пивалоилпропиофенон. После израсходования всех исходных веществ на реакционный раствор при 130 С действовали дополнительным количеством парабензилоксианилина (42 г, 177 ммоль). Реакционную смесь затем нагревали при 130 С в течение еще 3,5 ч. Реакционную смесь оставляли стоять до достижения ею комнатной температуры, промывали водой (800 мл), экстрагировали EtOAc (3 х 300 мл), промывали насыщенным солевым раствором и высушивали над MgSO4. Органический слой концентрировали с получением неочищенного продукта, который растирали с МеОН (3 раза) с получением твердого вещества белого цвета (38 г, 64%). Т.пл. 156-158 С; 1H ЯМР (DMSO) 11,0 (с, 1 Н), 7,67 (д, 2 Н, J=8,6 Гц),7,49 (д, 2 Н, J=7,1 Гц), 7,43-7,29 (м, 3 Н), 7,28-7,19 (м, 3 Н), 7,12 (д, 1 Н, J=2,3 Гц), 6,83 (дд, 1 Н, J=8,7 Гц, 2,3 Гц), 5,13 (с, 2 Н), 2,37 (с, 3 Н), 1,33 (с, 9 Н); МС 414 (М+Н)+; ИК (KВr) 3380, 2970, 1745 см-1. Пример 6. 4-5-Бензилокси-3-метил-1-[4-(2-пиперидин-1-илэтокси)бензил]-1 Н-индол-2-илфениловый эфир 2,2-диметил-пропионовой кислоты (23). К суспензии NaH (11,7 г, 60% дисперсия в минеральном масле, 0,292 моль, 2,5 экв.) в DMF (500 мл) при -25 С по каплям добавляли раствор 1-незамещенного предшественника индола 20 (48,3 г, 0,117 моль,1 экв.) в DMF (500 мл) в течение 50 мин, и реакция продолжалась при -25 С в течение еще 1 ч. К аниону индола затем добавляли по каплям раствор, состоящий из боковой цепи 21 (51,0 г, 0,176 моль, 1,5 экв.) вDMF (500 мл) в течение периода 55 мин, во время которого поддерживали температуру раствора -25 С. Реакционную смесь перемешивали в течение еще 1 ч при -25 С и в течение 24 ч при комнатной температуре. Реакционную смесь разбавляли EtOAc (1,2 л), промывали насыщенным солевым раствором (8 х 300 мл),- 10005371 высушивали над MgSO4 и концентрировали с получением 92,8 г масла коричневого цвета, которое растворяли в минимальном количестве СН 2 Сl2 и подвергали хроматографии на силикагеле (1 кг) с CH2Cl2,затем CH2Cl2/MeOH 98:2 и, наконец, CH2Cl2/MeOH 96:4. Фракция, элюированная CH2Cl2, содержала непрореагировавшее исходное вещество (14,5 г), и элюат из фракций СН 2 Сl2/МеОН концентрировали с получением желаемого продукта (36,7 г, 70%). 1H ЯМР (DMSO) 7,52-7,45 (м, 2 Н), 7,43-7,36 (м, 4 Н), 7,357,31 (м, 1 Н), 7,28-7,20 (м, 3 Н), 7,15 (д, 1 Н, J=2,3 Гц), 6,84 (дд, 1 Н, J=8,9 Гц, 2,4 Гц), 6,73 (с, 4 Н), 5,18 (с,2 Н), 5,13 (с, 2 Н), 3,94 (т, 2 Н, J=5,9 Гц), 2,56 (т, 2 Н, J=5,9 Гц), 2,41-2,35 (м, 4 Н), 2,18 (с, 3 Н), 1,51-1,42 (м,4 Н), 1,32 (с, 11 Н); ИК (KВr) 3410 (H2O), 2930, 1750; МС 631. Пример 7. 4-5-Бензилокси-3-метил-1-[4-(2-гексаметиленамин-1-илэтокси)бензил]-1 Н-индол-2-ил фениловый эфир 2,2-диметилпропионовой кислоты НСl (24). Индол 20 (2,0 г, 4,84 ммоль) растворяли в DMF и охлаждали до 0 С и действовали на него NaH(0,13 г, 5,32 ммоль, в виде 60% дисперсии в минеральном масле). На бензилхлорид 22 (2,0 г, 6,57 ммоль) в DMF в отдельной колбе действовали NaH (0,17 г, 7,22 ммоль, в виде 60% дисперсии в минеральном масле) при 0 С. Раствор, содержавший бензилхлорид, затем переносили c помощью шприца в раствор,содержащий анион индола. Температуру реакционной смеси поддерживали на уровне 0 С в течение 0,5 ч, а затем позволяли ей нагреться до комнатной температуры и перемешивали в течение еще 5 ч. Реакционную смесь распределяли между водой и EtOAc, промывали EtOAc слой насыщенным солевым раствором и высушивали над MgSO4. Раствор концентрировали и подвергали хроматографии на SiO2 (MeOH/CH2Cl2; 5:95) с получением желаемого продукта в виде пены (0,41 г). На полученную пену действовали 1 Н раствором НСl в Et2O, с получением продукта в виде твердого вещества светло-желтого цвета. Т.пл.=222-225 С; 1H ЯМР (DMSO) 10,17 (ушир. с, 1 Н), 7,95-7,22 (м, 10 Н), 7,16 (д, 1 Н, J=2,3 Гц), 6,87-6,76(7,0 г). Смесь затем гидрировали при комнатной температуре в аппарате Парра при давлении 50 фунт/дюйм 2(345 кПа) в течение 30 ч. Реакционную смесь затем фильтровали через Siluflock и к фильтрату добавляли 0,62 г аскорбиновой кислоты для минимизации любого возможного окисления. Осадок на фильтре Siluflock промывали THF и объединенные фильтраты концентрировали с получением твердого вещества белого цвета, которое промывали гексанами и высушивали с получением 44,5 г 25 (84%) в виде твердого вещества белого цвета. 1H ЯМР (DMSO) 8,76 (с, 1 Н), 7,40 (д, 2 Н, J=8,0 Гц), 7,22 (д, 2 Н, J=7,8 Гц), 7,14 (д, 1 Н,J=8,7 Гц), 6,84 (д, 1 Н, J=1,6 Гц), 6,76-6,72 (м, 4H), 6,63 (дд, 1 Н, J=8,7 Гц, 1,4 Гц), 5,14 (с, 2 Н), 3,94 (т, 2 Н,J=5,8 Гц), 2,57 (т, 2 Н, J=6,0 Гц), 2,42-2,33 (м, 4 Н), 2,14 (с, 3 Н), 1-50-1,39 (м, 4 Н), 1,32 (с, 11 Н); ИК (Кbr) 3400, 2900, 1750 см-1; МС 540. Пример 9. 4-5-Гидрокси-3-метил-1-[4-(2-азепан-1-илэтокси)бензил]-1 Н-индол-2-илфениловый эфир 2,2-диметилпропионовой кислоты (26). Соединение 26 получали в соответствии с примером 25, в виде пены. ИК (KВr) 3410, 2920, 1750 см-1; МС 555. Пример 10. Метиловый эфир 2,3,4-триацетил-1-O-[4-(2-пиперидин-1-илэтокси)бензил]-2-[4-(2,2 диметилпропионилокси)фенил]-3-метил-1 Н-индол-5-илглюкуроновой кислоты (27). К смеси 5-гидроксииндольного соединения 25 (31,2 г, 0,058 моль, 1 экв.), трихлорацетимидата D[150607-95-7] (33,2 г, 0,069 моль, 1,2 экв.) и молекулярных сит (3 , 28 г) в СН 2 Сl2 (0,5 л) по каплям добавляли BF3OEt2 (16,5 г, 0,116 моль, 2 экв.) при 0 С. Смесь перемешивали при комнатной тeмпературе в течение 24 ч. Смесь фильтровали, гасили концентрированным водным NaHCO3 (1 л), органический слой промывали водой, насыщенным солевым раствором, высушивали над Na2SO4 и выпаривали с получением 50,7 г пены желтого цвета. Указанное вещество подвергали хроматографии на силикагеле (2 кг, колонка d=15 см, элюент EtOAc/Et3N 9:1, 100 мл/3 мин, 125 мл фракции) с получением желаемого продукта 27 в виде пены. 1 Н ЯМР (DMSO) 7,42 (д, 2 Н, J=8,5 Гц), 7,31 (д, 1 Н, J=8,9 Гц), 7,23 (д, 2 Н, J=8,5 Гц), 7,18(д, 1 Н, J=2,2 Гц), 6,82 (дд, 1 Н, J=8,8 Гц, 2,3 Гц), 6,79-6,68 (м, 4 Н), 5,59 (д, 1 Н, J=8,0 Гц), 5,47 (т, 1 Н, J=9,6 Гц), 5,20 (с, 2 Н), 5,15-5,03 (м, 2 Н), 4,67 (д, 1 Н, J=9,9 Гц), 3,94 (т, 2 Н, J=5,9 Гц), 3,65 (с, 3 Н), 2,58 (т, 2 Н,J=5,7 Гц), 2,43-2,35 (м, 4 Н), 2,18 (с, 3 Н), 2,06 (с, 3 Н), 2,01 (с, 3 Н), 2,00 (с, 3 Н), 1,51-1,42 (м, 4 Н), 1,32 (с,11 Н); ИК (KВr) 2910, 1752 см-1; МС 857 (М+Н+). Пример 11. Метиловый эфир 2,3,4-триацетил-1-O-[4-(2-азепан-1-илэтокси)бензил]-2-[4-(2,2-диметилпропионилокси)фенил]-3-метил-1 Н-индол-5-илглюкуроновой кислоты (28). Соединение 28 получали в соответствии с методикой получения соединения 27, приведенной выше. Очистка на SiO2 с элюированием (20:1) СНСl3:СН 3 ОН дала 1,2 г, 82%, гликозида 6 в виде пены коричневого цвета. Rf=0,21 (20:1 СНСl3:СН 3 ОН); 1H ЯМР (СDСl3) 6,6-7,4 (м, 11 Н), 5,0-5,4 (м, 6 Н), 4,0-4,2 (м, 3 Н),3,8 (с, 3 Н), 2,8-3,2 (м, 6 Н), 2,2 (с, 3 Н), 2,0-2,2 (м, 9 Н), 1,4-1,8 (м, 8 Н), 1,4 (с, 9 Н); 13 С ЯМР (СDСl3) 176,9,170,1, 170,0, 169,9, 169,6, 169,3, 169,2, 168,5, 167,0, 157,6, 151,1 150,8 138,1, 133,7, 131,3, 130,4, 129,1,128,9, 127,1, 121,5, 114,6, 113,7, 110,7 109,2, 107,2, 101,1, 90,2, 72,6, 72,1, 71,7, 71,0, 69,7, 69,3, 67,8, 65,5,- 11005371 56,0, 55,5, 52,8, 52,6, 47,1, 39,1, 27,0, 26,9, 26,6, 20,6, 20,5, 20,4, 9,3; LC/MS (ESI) время удерживания=15,9, М+Н+=871,4. Пример 12. Триэтиламмониевая соль 5-O-глюкуронида 1-[4-(2-пиперидин-1-илэтокси)бензил]-2-(4 гидроксифенил)-3-метил-1 Н-индол-5-ола (29). Раствор, состоящий из предшественника индола 27 (29,8 г, 0,0385 моль) в смеси диоксан/МеОН/ Н 2O (200 мл/100 мл/100 мл) и LiOHH2O (12,95 г, 0,31 моль, 8 экв.) перемешивали при 60 С в течение 2 ч. Добавляли АсОН (18,5 мл) и смесь выпаривали с получением 53,6 г неочищенного твердого вещества. Полученный материал суспендировали в воде (500 мл) в течение 2 ч, фильтровали, промывали водой и высушивали с получением 24,1 г неочищенного продукта. Указанный материал растворяли в смесиMeOH/Et3N (100 мл/5,3 мл) и использовали эфир (2 л) для осаждения соли в виде твердого вещества белого цвета, которое фильтровали, промывали эфиром и высушивали в вакууме с получением соединения 29 (27 г, 99%). 1H ЯМР (DMSO-d6)6,99 (м, 11H), 5,11 (ушир. с, 1 Н), 5,00 (д, 1 Н, J=1,5 Гц), 3,99 (т, 2 Н),3,66 (д, 1 Н, J=1,5 Гц), 3,26 (м, 2 Н), 2,92 (кв., 6 Н), 2,74 (т, 2 Н), 2,52 (ушир. с, 4 Н), 2,1 (с, 3 Н), 1,50 (ушир. с,4 Н), 1,37 (ушир. с, 2 Н), 1,08 (т, 9 Н). Пример 13. 5-O-Глюкуронид 1-[4-(2-азепан-1-илэтокси)бензил]-2-(4-гидроксифенил)-3-метил-1 Ниндол-5-ола (30). К раствору гликозидного предшественника индола 28 (2,8 г, 3,3 ммоль) в смеси (2:1:1) п-диокcан: МеОН:Н 2 О (52 мл) добавляли твердый LiOHH2O (1,5 г, 36 ммоль) при комнатной температуре под Аr. Суспензию нагревали до 65 С. Через 1,5 ч добавляли ледяную уксусную кислоту (1,5 мл). Реакционную смесь концентрировали в вакууме. Очистка на SiO2 с элюированием смесью (20:1:2) СН 3 СN:HOAc:Н 2O, а затем смесью (10:1:2) СН 3 СN:HOAc:Н 2O дала сироп коричневого цвета. Добавление H2O (30 мл), а затем МеОН (10 мл) к сиропу коричневого цвета дало суспензию светло-коричневого цвета. Указанную суспензию фильтровали и высушивали на воздухе с получением 840 мг, 40%, глюкуронида 30 в виде твердого вещества светло-коричневого цвета. Rf=0,24 (10:1:2 СН 3 СN:НОАс:Н 2O); время удерживания ВЭЖХ=23,2 мин с 95% площади при 220 нм 1H ЯМР (DMSO) 6,8-7,2 (м, 7 Н), 6,7 (с, 4 Н), 5,1 (ушир. с,ОН), 4,8 (д, 1 Н, J=5,5 Гц), 3,9 (т, 2 Н, J=4,5 Гц), 3,1-3,6 (м, 8 Н), 2,77 (т, 2 Н, J=4,5 Гц), 2,5-2,7 (м, 4 Н) , 2,1(31). Смесь замещенного индола 23 (61,8 г, 0,098 моль, 1 экв.) и LiOHH2O (8,5 г, 0,202 моль, 2,1 экв.) в смеси диоксан/МеОН/Н 2 О (300 мл/150 мл/150 мл) перемешивали при 60 С в течение 2 ч. Смесь охлаждали до комнатной температуры и добавляли 12,1 мл АсОН, чтобы довести рН приблизительно до 7. Затем добавляли воду (600 мл) и смесь экстрагировали CH2Cl2 (6 х 150 мл), органическую фазу промывали концентрированным водным Nа 2 СО 3, Н 2 О, насыщенным солевым раствором, высушивали над Na2SO4 и выпаривали с получением соединения 31 в виде пены желтого цвета. 1H ЯМР (СDСl3)7,07 (м, 19 Н),6,10 (с, 2 Н), 5,03 (с, 2 Н), 4,03 (т, 2 Н), 2,8 (т, 2 Н), 2,53 (ушир. с, 4 Н), 2,21 (с, 3 Н), 1,63 (т, 4 Н), 1,43 (ушир.c, 2 Н). Пример 15. 4-5-Бензилокси-3-метил-1-[4-(2-азепан-1-илэтокси)бензил]-1 Н-индол-2-илфенол (32). Соединение 32 синтезировали аналогично соединению 31. 1H ЯМР (DMSO)9,83 (ушир. с, 1 Н),7,49-7,29 (м, 5 Н), 7,21-7,09 (м, 4 Н), 6,89-6,73 (м, 7 Н), 5,15 (с, 2 Н), 5,11 (с, 2 Н), 4,04 (т, 2 Н, J=7,1 Гц), 2,77(т, 2 Н, J=6,0 Гц), 2,65-2,60 (м, 4 Н), 2,15 (с, 3 Н), 1,51 (ушир. с, 8 Н). Пример 16. Метиловый эфир 2,3,4-О-триацетил-1-О-(4-5-бензилокси-3-метил-1-[4-(2-пиперидин-1 илэтокси)бензил]-1 Н-индол-2-илфенил)-бетаглюкуроновой кислоты (33). К смеси индола 31 (35,6 г, 0,065 моль, 1 экв.), трихлорацетимидата D [150607-95-7] (37,4 г, 0,078 моль,1,3 экв.) и молекулярных сит (3 , 28 г) в CH2Cl2 (500 мл) по каплям добавляли BF3 ОEt2 (18,45 г, 0,13 моль,2 экв.) при 0 С. Смесь перемешивали при комнатной температуре в течение 48 ч. Смесь фильтровали, а затем гасили концентрированным водным NaHCO3 (1 л), органический слой промывали водой, насыщенным солевым раствором, высушивали над Na2SО 4 и выпаривали с получением 65,4 г пены желтого цвета. Указанную пену подвергали хроматографии на силикагеле (2 кг, колонка d=15 см), с использованием EtOAc/Et3N (9:1), с получением продукта 33 (34,5 г, 59%) целевого соединения: МС 863 (M+H). Пример 17. Метиловый эфир 2,3,4-О-триацетил-1-О-(4-5-бензилокси-3-метил-1-[4-(2-азепан-1-илэтокси)бензил]-1 Н-индол-2-илфенил)-бета-D-глюкуроновой кислоты (34). Соединение получали в соответствии с методикой получения соединения 33, приведенной выше.(KВr): mах 3435, 2934, 2862, 1756, 1612, 1510, 1372, 1226, 1176, 1041, 828 см-1; LC/MS (ESI) время удерживания=16,1, М+Н+=877. Пример 18. Метиловый эфир 2,3,4-O-триацетил-1-O-(4-5-гидрокси-3-метил-1-[4-(2-пиперидин-1 илэтокси)бензил]-1 Н-индол-2-илфенил)-бета-D-глюкуроновой кислоты (35). На гликозидированный индол 33 (34 г, 0,039 моль) в 400 мл THF/MeOH (1:1) действовали 10% Pd/C(5,4 г) и гидрировали в аппарате Парра при давлении 50 фунт/дюйм 2 (345 кПа) в течение 30 ч. Реакционную смесь фильтровали через Siluflock и к фильтрату добавляли 0,34 г L-аскорбиновой кислоты. Осадок на фильтре Siluflock промывали THF и объединенные фильтраты концентрировали с получением продукта 35 (29,8 г, 98%) в виде твердого вещества желтовато-белого цвета. 1H ЯМР (СDСl3)6,95 (м, 11 Н),5,34 (м, 2 Н), 5,14 (д, 1 Н, J=2 Гц), 5,01 (с, 2 Н), 4,21 (м, 1 Н), 4,06 (т, 2 Н), 2,83 (т, 2 Н), 2,62 (ушир. с, 4 Н),2,13 (с, 3 Н), 1,65 (т, 4 Н), 1,44 (ушир. с, 2 Н). Пример 19. Метиловый эфир 2,3,4-O-триацетил-1-O-(4-5-гидрокси-3-метил-1-[4-(2-азепан-1-илэтокси)бензил]-1 Н-индол-2-илфенил)-бета-D-глюкуроновой кислоты (36). На гликозидированный индол 34 (3,7 г, 4,2 ммоль) в 60 мл THF/MeOH (1:1) действовали 10% Pd/C(1,5 г) и гидрировали в аппарате Парра при давлении 40 фунт/дюйм 2 (276 кПа) в течение 23 ч. Реакционную смесь фильтровали через подушку из целита и осадок на фильтре промывали THF (20 мл) и EtOH(20 мл). Для того чтобы избежать любого окисления воздухом, к профильтрованному раствору добавляли L-аскорбиновую кислоту (0,37 г). Раствор концентрировали и подвергали хроматографии на силикагеле с использованием CHCl3:iPrOH (7:1) с получением продукта 35 (1,6 г, 48%) в виде пены коричневого цвета. Rf=0,20 (7:1 СНСl3:iРrОН); 1H ЯМР (DMSO) 6,5-7,5 (м, 11 Н), 5,7 (д, 1 Н, J=7,9 Гц), 5,5 (т, 1 Н, J=9,6 Гц), 4,9-5,2 (м, 4 Н), 4,7 (д, 1 Н, J=9,9 Гц), 3,6 (с, 3 Н), 3,3 (с, 8 Н), 2,1 (с, 3 Н), 1,9-2,2 (м, 9 Н), 1,5-1,9 (м, 8 Н); 13 С ЯМР (СDСl3) 170,0, 169,0, 167,0, 157,0, 156,5, 150,0, 138,0, 131,7, 131,6, 130,0, 127,3, 116,9, 114,7,112,0, 111,0, 108,0, 104,5, 98,8, 72,6, 71,8, 71,1, 69,1, 64,4, 55,0, 55,2, 53,0, 47,0, 26,7, 23,6, 20,6, 20,5, 9,5; ИК (KBr): max 3427, 3037, 2935, 2612, 1757, 1612, 1510, 1462, 1374, 1227, 1040 см-1; LC/MS (ESI) время удерживания=13,0, М+Н+=787. Пример 20. Триэтиламмониевая соль 4-O-глюкуронида 2-(4-гидроксифенил)-3-метил-1-[4-(2-пиперидин-1-илэтокси)бензил]-1 Н-индол-5-ола (37). Гликозидированный индол 35 (25 г, 0,029 ммоль, 1 экв.) и LiOHH2O (12,3 г, 0,29 моль, 10 экв.) в 300 мл смеси диоксан/МеОН/Н 2 О (2/1/1) перемешивали при 60 С в течение 2 ч. Смесь оставляли остывать до комнатной температуры и добавляли АсОН (13,5 мл). Раствор концентрировали с получением 44,3 г пены желтого цвета. Указанную пену промывали водой, а остаток высушивали и затем растворяли в 160 мл MeOH/Et3N (15/1), и полученный раствор концентрировали, с получением неочищенного продукта, который повторно растворяли в МеОН (100 мл) при 40 С, и из раствора почти немедленно выпадал осадок белого цвета. Осадок фильтровали, промывали МеОН и высушивали в вакууме с получением 9,7 г соединения 37 (78%) в виде твердого вещества белого цвета. 1H ЯМР (DMSO)7,16 (м, 4 Н), 6,86 (д,2 Н, J=2,8 Гц), 6,81 (м, 1 Н), 6,74 (с, 4 Н), 5,15 (с, 2 Н), 4,90 (д, 1 Н, J=2,0 Гц), 3,98 (м, 2 Н), 3,70 (д, 1 Н, J=2,0 Гц), 3,32 (м, 3 Н), 2,78 (т, 2 Н), 2,58 (ушир. с, 4 Н), 2,14 (с, 3 Н), 1,51 (ушир. с, 4 Н), 1,38 (ушир. с, 2 Н). Пример 21. 4-O-глюкуронид 1-[4-(2-азепан-1-илэтокси)бензил]-2-(4-гидроксифенил)-3-метил-1 Ниндол-5-ола (38). К раствору гликозида индола 36 (2,8 г, 3,3 ммоль) в смеси (2:1:1) диоксан:МеОН:Н 2O (52 мл) добавляли твердый LiOHH2O (1,5 г, 36 ммоль) при комнатной температуре под Аr. Суспензию нагревали до 65 С. Через 1,5 ч добавляли ледяную уксусную кислоту (1,5 мл). Реакционную смесь концентрировали в вакууме. Очистка на SiO2 с элюированием смесью (20:1:2) СН 3 СN:НОАс:Н 2O, а затем смесью (10:1:2) СН 3 СN:НОАс:Н 2O дала сироп коричневого цвета. Добавление Н 2 О (30 мл), а затем МеОН (10 мл) к сиропу коричневого цвета дало суспензию светло-коричневого цвета. Указанную суспензию фильтровали и высушивали на воздухе с получением 840 мг, 40%, глюкуронида 38 в виде твердого вещества светлокоричневого цвета. Rf=0,24 (10:1:2 СН 3 СN:НОАс:H2O); время удерживания ВЭЖХ=23,2 мин с 95% площади при 220 нм; 1H ЯМР (DMSO) 6,8-7,2 (м, 7 Н), 6,7 (с, 4 Н), 5,1 (ушир. с, ОН), 4,8 (д, 1 Н, J=5,5 Гц), 3,9(ESI) время удерживания=9,0, М+Н+=647,3. Бис-глюкуронид, соединения 46 и 47 [примеры 22-26] получали в соответствии со схемой 7, как показано ниже. Пример 22. 5-Бензилокси-2-(4-бензилоксифенил)-3-метил-1H-индол (39). На бромкетон CAS[66414-19-5] (50,0 г, 0,16 моль) в 200 мл DMF действовали гидрохлоридом 4 бензилоксианилина САS[51145-58-5] (44 г, 0,22 моль) и реакционную смесь продували азотом в течение приблизительно 10 мин. Добавляли триэтиламин (54,6 мл) и реакционную смесь нагревали до 120 С в течение 2 ч. Анализ с помощью ТСХ (EtOAc/гексаны) показал исчезновение исходных веществ с образованием более полярного пятна. Реакционной смеси давали остыть и добавляли еще 48 г гидрохлорида анилина. Реакционную смесь нагревали до 150 С в течение 2 ч. Добавляли еще 5 г гидрохлорида анилина и реакционную смесь нагревали до 150 С в течение еще 30 мин. Реакционной смеси давали остыть до комнатной температуры, а затем выливали ее приблизительно в 1,5 л воды и экстрагировали 2 л этилацетата. Твердые вещества, если это необходимо, растворяли в дополнительном количестве этилацетата. Слой этилацетата промывали 1 л 1 Н водного раствора NaOH, 1 л воды, насыщенным солевым раствором, затем высушивали над сульфатом магния и фильтровали. Органические слои концентрировали с получением неочищенного твердого вещества, которое перемешивали с 500 мл метанола и фильтровали. Указанное твердое вещество затем перемешивали с 500 мл этилового эфира и фильтровали. Твердое вещество, альтернативно, перемешивали с метанолом и эфиром, пока оно не приобретало белесый цвет. Реакция дала 36 г продукта. Т.пл.=150-152C; 1 Н ЯМР (DMSO)10,88 (с, 1 Н), 7,56 (д, 2 Н, J=8,8 Гц), 7,48 (д, 4 Н,J=7,9 Гц), 7,42-7,29 (м, 6 Н), 7,21 (д, 1 Н, J=7,0 Гц), 7,13 (д, 2 Н, J=8,8 Гц), 7,08 (д, 1 Н, J=2,2 Гц), 6,94 (дд, 1 Н,J=8,8, 2,4 Гц), 5,16 (с, 2 Н), 5,11 (с, 2 Н), 2,33 (с, 3 Н); ИК (KВr) 3470, 2880, 2820, 1620 см-1; MC EI m/z 419. Пример 23. 5-Бензилокси-2-(4-бензилоксифенил)-3-метил-1-[4-(2-азепан-1-илэтокси)бензил]-1 Н-индол (44). К суспензии NаH (20,0 г, 60% дисперсия в минеральном масле, 0,5 моль, 2,5 экв.) при 0 - +10 С добавляли раствор 5-бензилокси-2-(4-бензилоксифенил)-3-метил-1 Н-индола (84 г, 0,2 моль, 1 экв.) в DMF (100 мл) в течение 1 ч. Реакционную смесь перемешивали в течение 30 мин. Добавляли по каплям раствор бензилхлорида(синтез показан на схеме 7, и подробности приведены в следующей экспериментальной части) (67 г, 0,22 моль,1,1 экв.) в DMF (200 мл) при 0 - +10 С в течение 2 ч. Реакционную смесь перемешивали при 25 С в течение 2 ч. В этот момент ТСХ показала отсутствие исходного вещества и наличие, главным образом, продукта (EtOAc/ гексаны 1:5). Реакционную смесь разбавляли водой (1 л), экстрагировали EtOAc (3 х 1 л) и высушивали надMgSО 4. Раствор концентрировали до 150 мл, выливали в МеОН (750 мл) и перемешивали в течение ночи. Осадок фильтровали и высушивали с получением указанного в заголовке соединения (99 г, 76%). Т.пл.=106-107 С; 1H ЯМР (DMSO)7,47 (д, 4 Н, J=8,3 Гц), 7,41-7,36 (м, 4 Н), 7,36-7,30 (м, 2 Н), 7,29 (д, 2 Н, J=8,8 Гц), 7,19 (д, 1 Н,J=8,8 Гц), 7,14-7,10 (м, 3 Н), 6,80 (дд, 1 Н, J=8,8 Гц), 6,73 (с, 4 Н), 5,15 (с, 2 Н), 5,13 (с, 2 Н), 5,11 (с, 2 Н), 3,90 (т,2 Н, J=5,9 Гц), 2,76 (т, 2 Н, J=5,9 Гц), 2,64-2,56 (м, 4 Н), 2,15 (с, 3 Н), 1,58-1,44 (м, 8 Н); MS FAB m/z 651 (М+Н+). Пример 24. 1-[4-(2-Aзепан-1-илэтокси)бензил]-2-(4-гидроксифенил)-3-метил-1 Н-индол-5-ол (45). Раствор, состоящий из индола 44 (17,5 г, 26,9 ммоль) в THF/EtOH (1:1), гидрировали в атмосфере Н 2 с использованием 10% Pd/C в качестве катализатора. В результате хроматографии на силикагелеCH2Cl2/MeOH (градиент с 100/0 до 85/15) получали целевой продукт в виде 8,5 г пены белого цвета, вместе с 2,5 г фракции, содержавшей незначительные примеси. Хотя свободное основание представляет собой вещество, используемое на следующем этапе (бис-глюкуронидизации), с целью характеризации и повышения сроков хранения соединений можно получить кислотно-аддитивную соль с НСl растворением соединения в МеОН и действием на него 1,1 экв. 2 Н водн. раствора НСl. Соединение медленно выпадает в виде кристаллов белого цвета. Физические данные, перечисленные ниже, описывают НСl соль индола 45. Т.пл.=172-174 С; 1H ЯМР (DMSO) 10,11 (ушир. с, 1 Н), 9,70 (с, 1 Н), 8,71 (с, 1 Н), 7,15 (д, 2 Н,J=8,6 Гц), 7,05 (д, 1 Н, J=8,8 Гц), 6,85 (д, 2 Н, J=8,8 Гц), 6,80-6,77 (м, 5 Н), 6,56 (дд, 1 Н, J=8,8 Гц, 2,2 Гц),5,11 (с, 2 Н), 4,26 (т, 2 Н, J=4,6 Гц), 3,48-3,30 (м, 4 Н), 3,22-3,08 (м, 2 Н), 2,09 (с, 3 Н), 1,83-1,76 (м, 4 Н), 1,671,48 (м, 4 Н); ИК (KВr) 3500 br, 3250 br, 2900, 1610; MS FAB m/z 471 (M+H+). Пример 25. Метиловый эфир 2,3,4-O-триацетил-1-O-[4-[1-[4-[2-гексагидро-1 Н-азепин-1-илэтокси] бензил]-3-метил-5-[(2,3,4-O-триацетил-6-О-метил-бета-D-глюкопирануронилозил)окси]-1 Н-индол-2-ил] фенил]-бета-D-глюкопиранозидуроновой кислоты (46). К смеси, состоящей из бис-фенольного индола 45 (2,5 г, 5,3 ммоль) и глюкуронилимидата D (5,60 г,11,7 ммоль, 2,2 экв.) в CH2Cl2 (25 мл), медленно добавляли BF3OEt2 (1,43 мл, 11,7 ммоль, 2,2 экв.) при интенсивном перемешивании реакционной смеси. После добавления реакционную смесь кипятили с обратным холодильником в течение 2,5 ч. Во время реакции некоторое количество исходного вещества оставалось прилипшим ко дну колбы. К реакционной смеси дополнительно добавляли CH2Cl2 и небольшое количество МеОН и органический слой промывали водой, насыщенным солевым раствором и высушивали над MgSO4. Неочищенное вещество очищали хроматографией на силикагеле с СН 2 Сl2/МеОН (95/5) с получением защищенного бис-глюкуронида 46 (0,95 г). Т.пл.=110-116 С; 1 Н ЯМР (DMSO-d6)7,36 (д, 2 Н,J=8,5 Гц), 7,25 (д, 1 Н, J=8,8 Гц), 7,16-7,10 (м, 3 Н), 6,79 (дд, 1 Н, J=8,9 Гц, 2,0 Гц), 6,75 (ушир. с, 4 Н), 5,74 (д,1 Н, J=7,7 Гц), 5,58 (д, 1 Н, J=8,0 Гц), 5,47 (дт, 2 Н, J=9,5 Гц, 2,3 Гц), 5,18 (ущир. с, 2 Н), 5,15-5,04 (м, 4 Н), 4,73(д, 1 Н, J=9,9 Гц), 4,66 (д, 1 Н, J=10,0 Гц), 3,93 (т, 2 Н, J=5,8 Гц), 3,64 (с, 3 Н), 3,636 (с, 3 Н), 2,81-2,60 (м, 6 Н),2,16 (с, 3 Н), 2,05 (с, 3 Н), 2,03 (с, 3 Н), 2,02 (с, 3 Н), 2,00 (ушир. с, 9 Н), 1,61-1,47 (м, 8 Н); МС 1103,7 (М+Н+). Пример 26. 4-[5-(бета-D-Глюкопирануронозилокси)-1-[4-[2-(гексагидро-1 Н-азепин-1-ил)этокси]бензил]-3-метил-1 Н-индол-2-ил]фенил-бета-D-глюкопиранозидуроновая кислота (47). Защищенный бис-глюкуронид индола 46 (0,89 г, 0,81 ммоль, 1 экв.) растворяли в 24 мл смеси пдиоксан/МеОН (5/1) и медленно добавляли 24 мл водного раствора LiOH (0,31 г, 12,9 ммоль, 12 экв.). Реакционную смесь нагревали до 60 С в течение 2 ч. Реакционную смесь оставляли стоять до достижения ею комнатной температуры, добавляли АсОН (0,97 г, 16,1 ммоль, 20 экв.) и раствор концентрировали при пониженном давлении. Добавляли бензол и указанный процесс повторяли 2 раза для азеотропной отгонки всей остаточной воды из реакционной смеси. Неочищенный остаток очищали с помощью ВЭЖХ с обращенной фазой с получением целевого продукта 47 (0,294 г). 1H ЯМР (DMSO-d6)7,31 (д, 1 Н, J=8,8 Гц), 7,22 (д, 2 Н, J=8,5 Гц), 7,19 (ушир. с, 1 Н), 7,09 (д, 2 Н, J=8,4 Гц), 6,90 (д, 1 Н, J=8,7 Гц), 6,75-6,64 (м,4 Н), 5,45-5,35 (м, 2 Н), 5,28-5,10 (м, 4 Н), 4,99-4,93 (м, 2 Н), 4,87-4,83 (м, 2 Н), 4,10-3,00 (несколько протонов скрыты под пиком H2O), 2,13 (с, 3 Н), 1,70 (ушир. с, 4 Н), 1,56 (ушир. с, 4 Н) ; МС 823 (М+Н+). Получение боковой цепи 22 показано на схеме 8, представленной ниже. Боковую цепь 21 получали аналогичным способом. Схема 8- 15005371 Пример 27. 4-(2-Гексаметиленимин-1-илэтокси)бензиловый альдегид (40). К тщательно перемешиваемой суспензии NaH (65 г, 60% дисперсия в минеральном масле, 1,6 моль,2,2 экв.) в DMF (500 мл) по каплям при 0 С добавляли раствор гидрохлорида п-гидроксибензальдегида(90 г, 0,74 моль, 1,0 экв.). Реакционную смесь перемешивали в течение 30 мин, а затем порциями добавляли 4-[2-(гексаметиленимин)]этилхлорид (153 г, 0,77 моль, 1,0 экв.). Реакционную смесь перемешивали в течение 1 ч. В этот момент ТСХ показала незначительное количество исходного вещества и наличие,главным образом, продукта (EtOAc/гексан 1:1). Реакционную смесь разбавляли водой (1 л), экстрагировали эфиром (5 л). Органический слой высушивали над МgSO4 и концентрировали в роторном испарителе с получением 176,8 г (97%) альдегида 40 в виде масла желтого цвета. 1H ЯМР (CDCl3/TMS):9,87 (с,1 Н), 7,81 (д, 2 Н, J=8,7 Гц), 7,02 (д, 2 Н, J=8,7 Гц), 4,14 (т, 2 Н, J=6,09 Гц), 2,98 (т, 2 Н, J=6,14 Гц), 2,78 (м,4 Н), 1,66-1,61 (м, 8 Н). Пример 28. 4-(2-Гексаметиленимин-1-илэтокси)бензиловый спирт (41). К перемешиваемому раствору альдегида 40 (200 г, 0,72 моль, 1,0 экв.) в метаноле (400 мл) порциями добавляли боргидрид натрия (15,6 г, 0,41 моль, 0,57 экв.) при 0 - +5. Реакционную смесь перемешивали в течение 30 мин. В этот момент ТСХ показала отсутствие исходного вещества и наличие, главным образом, продукта (EtOAc/гексан/триэтиламин 3:7:1). Реакционную смесь разбавляли водой (400 мл),экстрагировали метиленхлоридом (3 х 400 мл) и высушивали над MgSО 4. Раствор концентрировали в роторном испарителе с получением 201 г (100%) спирта 41 в виде густого масла. 1H ЯМР (CDCl3/TMS): 7,27 (д, 2 Н, J=8,5 Гц), 6,87 (д, 2 Н, J=8,5 Гц), 4,60 (с, 2 Н), 4,05 (т, 2 Н, J=6,21 Гц), 2,93 (т, 2 Н, J=6,15 Гц),2,77 (м, 4 Н), 1,7-1,5 (м, 8 Н). Пример 29. Гидрохлорид (4-хлорметилфенокси)этилгексаметиленимин-1-ила (22). К раствору спирта 41 (179 г, 0,72 моль, 1 экв.) в THF (300 мл) по каплям добавляли НСl (26,3 г НСl в 263 мл THF, 0,72 моль, 1,0 экв.) при 0 - +10 С. Образовывался осадок белого цвета. К густой суспензии гидрохлорида 42 добавляли тионилхлорид (80 мл, 1,1 моль, 1,5 экв.) и смесь нагревали до 50 С до прозрачности. Реакционную смесь концентрировали до 350 мл и хранили в холодильнике в течение ночи. Полученное твердое вещество белого цвета фильтровали, промывали холодным THF (100 мл) и высушивали с получением 147 г (67%) хлорида 22. 1H ЯМР (DMSO-d6): 11 (ушир. с, НСl), 7,40 (д, 2 Н, J=8,6 Гц),7,00 (д, 2 Н, J=8,6 Гц), 4,74 (с, 2 Н), 4,44 (т, 2 Н, J=5,25), 3,64-3,39 (м, 4 Н), 3,25-3,17 (м, 2 Н), 1,84-1,54 (м,8 Н). Пример 30. Гидрохлорид (4-хлорметилфенокси)этилпиперидин-1-ила (21). Боковую цепь 21 получали аналогично боковой цепи 22, описанной выше. 1H ЯМР (DMSO-d6): 11 где R1 и R2 независимо представляют собой водород, алкильную цепь из 1-6 атомов углерода, бензил,ацил из 2-7 атомов углерода, бензоил, Х представляет собой водород, алкил из 1-6 атомов углерода, CN, галоген, трифторметил или тиоалкил из 1-6 атомов углерода;n=1-3; при условии, что по меньшей мере один из R1 или R2 представляет собой- 16005371 или его фармацевтически приемлемая соль. 2. Соединение по п.1, в котором Х представляет собой алкил из 1-4 атомов углерода. 3. Соединение по п.1, в котором Х представляет собой метил. 4. Соединение по любому из пп.1-3, в котором n равно 2 или 3. 5. Соединение по любому из пп.1-4, в котором R2 представляет собой водород. 6. Соединение по любому из пп.1-5, в котором R1 представляет собой 7. Соединение по любому из пп.1-6, которое имеет D-конфигурацию. 8. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-триацетил-1-O-[4-(2 пиперидин-1-илэтокси)бензил]-2-[4-(2,2-диметилпропионилокси)фенил]-3-метил-1 Н-индол-5-ил-глюкуроновой кислоты или его фармацевтически приемлемую соль. 9. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-триацетил-1-O-[4-(2 азепан-1-илэтокси)бензил]-2-[4-(2,2-диметилпропионилокси)фенил]-3-метил-1 Н-индол-5-илглюкуроновой кислоты или его фармацевтически приемлемую соль. 10. Соединение по п.1, которое представляет собой триэтиламмониевую соль 5-O-глюкуронида 1[4-(2-пиперидин-1-илэтокси)бензил]-2-(4-гидроксифенил)-3-метил-1 Н-индол-5-ола. 11. Соединение по п.1, которое представляет собой 5-O-глюкуронид 1-[4-(2-азепан-1-илэтокси)бензил]-2-(4-гидроксифенил)-3-метил-1 Н-индол-5-ола или его фармацевтически приемлемую соль. 12. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-O-триацетил-1-O-(4-5 бензилокси-3-метил-1-[4-(2-пиперидин-1-илэтокси)бензил]-1 Н-индол-2-илфенил)-бета-D-глюкуроновой кислоты или его фармацевтически приемлемую соль. 13. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-O-триацетил-1-O-(4-5 бензилокси-3-метил-1-[4-(2-азепан-1-илэтокси)бензил]-1 Н-индол-2-илфенил)-бета-D-глюкуроновой кислоты или его фармацевтически приемлемую соль. 14. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-O-триацетил-1-O-(4-5 гидрокси-3-метил-1-[4-(2-пиперидин-1-илэтокси)бензил]-1 Н-индол-2-илфенил)-бета-D-глюкуроновой кислоты или его фармацевтически приемлемую соль. 15. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-O-триацетил-1-O-(4-5 гидрокси-3-метил-1-[4-(2-азепан-1-илэтокси)бензил]-1 Н-индол-2-илфенил)-бета-D-глюкуроновой кислоты или его фармацевтически приемлемую соль. 16. Соединение по п.1, которое представляет собой 4-O-глюкуронид 2-(4-гидроксифенил)-3-метил 1-[4-(2-пиперидин-1-илэтокси)бензил]-1 Н-индол-5-ола или его фармацевтически приемлемую соль. 17. Соединение по п.1, которое представляет собой триэтиламмониевую соль 4-O-глюкуронида 2(4-гидроксифенил)-3-метил-1-[4-(2-пиперидин-1-илэтокси)бензил]-1 Н-индол-5-ола. 18. Соединение по п.1, которое представляет собой 4-O-глюкуронид 1-[4-(2-азепан-1-илэтокси)бензил]-2-(4-гидроксифенил)-3-метил-1 Н-индол-5-ола или его фармацевтически приемлемую соль. 19. Соединение по п.1, которое представляет собой метиловый эфир 2,3,4-O-триацетил-1-O-[4-[1-[4[2-гексагидро-1 Н-азепин-1-ил)этокси]бензил]-3-метил-5-[(2,3,4-O-триацетил-6-О-метил-бета-D-глюкопирануронилозил)окси]-1 Н-индол-2-ил]фенил]-бета-D-глюкопиранозидуроновой кислоты или его фармацевтически приемлемую соль. 20. Соединение по п.1, которое представляет собой 4-[5-(бета-D-глюкопирануронозилокси)-1-[4-[2(гексагидро-1 Н-азепин-1-илэтокси]бензил]-3-метил-1 Н-индол-2-ил]фенил-бета-D-глюкопиранозидуроновую кислоту. 21. Способ лечения или ингибирования сердечно-сосудистого заболевания, понижения уровней липидов в крови или повышения уровней ЛВП у млекопитающего, которое в этом нуждается, который включает введение указанному млекопитающему соединения формулы I, заявленного в любом из пп.120. 22. Способ лечения или ингибирования рака молочной железы у млекопитающего, которое в этом нуждается, который включает введение указанному млекопитающему соединения формулы I, заявленного в любом из пп.1-20. 23. Способ лечения или ингибирования утраты костной ткани или остеопороза у млекопитающего,которое в этом нуждается, который включает введение указанному млекопитающему соединения формулы I, заявленного в любом из пп.1-20. 24. Способ проведения гормональной заместительной терапии у млекопитающего, которое в этом нуждается, который включает введение указанному млекопитающему соединения формулы I, заявленного в любом из пп.1-20. 25. Способ усиления познавательной функции или лечения или ингибирования деменций или болезни Альцгеймера у млекопитающего, которое в этом нуждается, который включает введение указанному млекопитающему соединения формулы I, заявленного в любом из пп.1-20.- 17005371 26. Фармацевтическая композиция, которая включает соединение формулы I, заявленного в любом из пп.1-20, и фармацевтический носитель. 27. Способ получения соединения формулы I, который включает превращение соединения формулы Iа другой представляет собой защитную группу, в условиях, подходящих для удаления защитной группы, с получением соответствующего соединения формулы I, где один из R1 и R2 представляет собой а другой из R1 и R2 представляет собой водород; указанное соединение может быть гидролизовано одновременно со снятием защиты или последовательно с получением соответствующего соединения формулы I, где один из R1 и R2 представляет собой водород, а другой представляет собой 28. Способ получения соединения формулы I, который включает взаимодействие соединения формулы II с получением соответствующего соединения формулы I, где R1 и R2, оба представляют собой 29. Способ получения соединения формулы I, который включает гидролиз соединения формулы I,где один или оба R1 и R2 представляют собой с получением соединения формулы I, где один или оба R1 и R2 представляют собой 30. Способ получения соединения формулы I, который включает взаимодействие соединения формулы IVa или IVb в которых Х и n определены в п.1, a R1 и R2 каждый выбран из алкила из 1-6 атомов углерода, бензила, ацила из 2-7 атомов углерода или бензоила,с соединением формулы III с получением соответствующего соединения формулы I, где R1 или R2 представляет собой
МПК / Метки
МПК: C07H 17/00, A61K 31/7056, A61P 5/30
Метки: 2-(4-гидроксифенил)-1-[4-(2-амин-1-илэтокси)бензил]-1h-индол-5-олов, конъюгаты, глюкопиранозидные
Код ссылки
<a href="https://eas.patents.su/20-5371-glyukopiranozidnye-konyugaty-2-4-gidroksifenil-1-4-2-amin-1-iletoksibenzil-1h-indol-5-olov.html" rel="bookmark" title="База патентов Евразийского Союза">Глюкопиранозидные конъюгаты 2-(4-гидроксифенил)-1-[4-(2-амин-1-илэтокси)бензил]-1h-индол-5-олов</a>
Производные 2-фенил-1- [4-(2-аминоэтокси)бензил] индола в качестве эстрогенных агентов

Номер патента: 1448
Опубликовано: 23.04.2001
Авторы: Трэн Бэч Динх, Сантилли Артур Аттилио, Миллер Крис Пол, Коллини Мишель Дэвид
МПК: C07D 209/22, A61K 31/404, A61P 9/00...
Метки: агентов, производные, эстрогенных, 2-фенил-1, качестве, индола, 4-(2-аминоэтокси)бензил
Формула / Реферат:
1. Производные 2-фенил-1-[4-(2-аминоэтокси)бензил] индола общей структурной формулы I или II где R1 выбран из Н, ОН, -О-(С=O)-(C1-C12)алкила (где алкил представляет собой линейную цепь или разветвленную), -О-(C1-C12)алкила (где алкил представляет собой линейную цепь, разветвленную или циклическую цепь), галогена; или -O-(C1-C4)-галогенированного алкила (включая трифторметокси и трихлорметокси); R2, R3, R4, R5 и R6 независимо выбраны из Н, ОН,...
2-(1h-индол-3-ил)-2-оксоацетамиды, обладающие противоопухолевой активностью

Номер патента: 4925
Опубликовано: 28.10.2004
Авторы: Мента Эрнесто, Пескалли Николетта
МПК: A61P 35/00, C07D 405/12, A61K 31/404...
Метки: обладающие, активностью, противоопухолевой, 2-(1h-индол-3-ил)-2-оксоацетамиды
Формула / Реферат:
1. Соединения формулы I где R1, R2 и R5 независимо представляют собой водород или C1-C6алкильную группу; R3 представляет собой водород, C1-C4алкил, аралкил, возможно замещенный фенил; R4 представляет собой водород, прямой или разветвленный C1-C8алкил, C5-C6циклоалкил; аралкил; гетероаралкил; X представляет собой одну или более чем одну группу, максимально четыре, независимо выбранную из водорода; C1-C6алкила; C1-C6галогеналкила;...
Конъюгаты гксф

Номер патента: 4685
Опубликовано: 24.06.2004
Автор: Байон Паскаль Себастьян
МПК: A61K 47/48, C07K 14/535
Формула / Реферат:
1. Физиологически активный конъюгат, имеющий формулу где G представляет гранулоцитарный колониестимулирующий фактор, без тех его аминогрупп, которые образуют амидную связь с полиэтиленгликолевой частью в указанном конъюгате, R представляет алкил с прямой или разветвленной цепью, имеющий от одного до шести атомов углерода, n представляет целое число от 420 до 550, а m представляет целое число от 1 до 5. 2. Конъюгат по п.1, где R представляет...
Способ получения 4-замещенных 1н-индол-3-глиоксамидов

Номер патента: 3582
Опубликовано: 26.06.2003
Авторы: Харн Нэнси Кей, Андерсон Бенджамин Алан
МПК: C07D 209/22
Метки: получения, 4-замещенных, 1н-индол-3-глиоксамидов, способ
Формула / Реферат:
1. Способ получения соединения формулы I или его фармацевтически приемлемой соли или пролекарственного производного где R1 выбран из группы, включающей (C7-C20)алкил, где R10 выбран из группы, включающей галоген, (C1-C10)алкил, (C1-C10)алкокси, -S-(C1-C10)алкил и галоген(C1-C10)алкил, и t представляет собой целое число от 0 до 5 включительно; R2 выбран из группы, включающей водород, галоген, (C1-C3) алкил, (C3-C4)циклоалкил,...
Конъюгаты полиол-&beta-интерферон

Номер патента: 3789
Опубликовано: 30.10.2003
Авторы: Соливич Вэйн, Робертс Майкл Дж., Эль Таяр Набиль, Харрис Милтон
МПК: A61P 31/00, A61K 47/48
Метки: полиол-&beta-интерферон, конъюгаты
Формула / Реферат:
1. Конъюгат полиол-b-интерферон, полиольная часть которого ковалентно связана с Cys17 человеческого b-интерферона. 2. Конъюгат полиол-b-интерферон по п.1, в котором указанная полиольная часть является полиалкиленгликольной частью. 3. Конъюгат полиол-b-интерферон по п.2, где указанная полиалкиленгликольная часть является полиэтиленгликольной (ПЭГ) частью. 4. Конъюгат полиол-b-интерферон по любому из пп.1-3, где конъюгат полиол-b-интерферон...
Предыдущий патент: Способ концентрирования отработанной кислоты
Следующий патент: Замещенные производные пурина в качестве ингибиторов клеточной адгезии
Случайный патент: Фармацевтические композиции с ингибиторами dpp iv