Способ получения ингибитора протеазы вич из(2-s)-4-пиколил-2-пиперазин-трет-бутилкарбоксамида
Номер патента: 178
Опубликовано: 24.12.1998
Авторы: Аскин Дэвид, Ридер Пол, Енг Кан К., Воланте Ральф П.















Формула / Реферат
1. Способ получения ингибитора протеазы ВИЧ структурной формулы

включающий следующие стадии:
(а) нагревание, по крайней мере, в течение одного часа одного эквивалента (S)-4-(3-пиколил)-2-трет-бутилкарбоксамидпиперазина (1)

с приблизительно одним эквивалентом соединения

в интервале температур между около 25 и около 150°С, при этом указанная смесь необязательно содержит приемлемый растворитель;
(б) снятие защиты у полученного соединения 3 путем обработки кислотой

(в) нейтрализация кислоты.
2. Способ по п.1, отличающийся тем, что нагревание осуществляют в интервале температур от около 50 до около 120°С.
3. Способ по п.1, отличающийся тем, что нагревание осуществляют в интервале температур от около 65 до около 85°С.
4. Способ по п.1, отличающийся тем, что приемлемым растворителем является сложный эфир, спирт, углеводород, простой эфир или формамид или их смесь.
5. Способ по п.1, отличающийся тем, что приемлемым растворителем является спирт.
6. Способ по п.1, отличающийся тем, что приемлемым растворителем является метанол или изопропанол.
7. Способ по п.1, отличающийся тем, что снятие защиты (б) осуществляют с помощью газообразного НСl.
8. Способ ингибитора протеазы ВИЧ по п.1, включающий стадии:
(а) нагревание, по крайней мере, в течение одного часа смеси одного эквивалента (S)-4-(3-пиколил)-2-трет-бутилкарбоксамидпиперазина (1)

с приблизительно одним эквивалентом

в интервале температур между от около 65 до около 85°С, при этом указанная смесь необязательно содержит в качестве растворителя метанол или изопропанол или их смесь;
(б) охлаждение смеси до около 0°С;
(в) удаление защиты полученного соединения 3 с помощью газообразного НСl;
(г) нейтрализация с помощью NаОН.
9. (S)-4-(3-Пиколил)-2-трет-бутилкарбоксамидпиперазин (1)

Текст
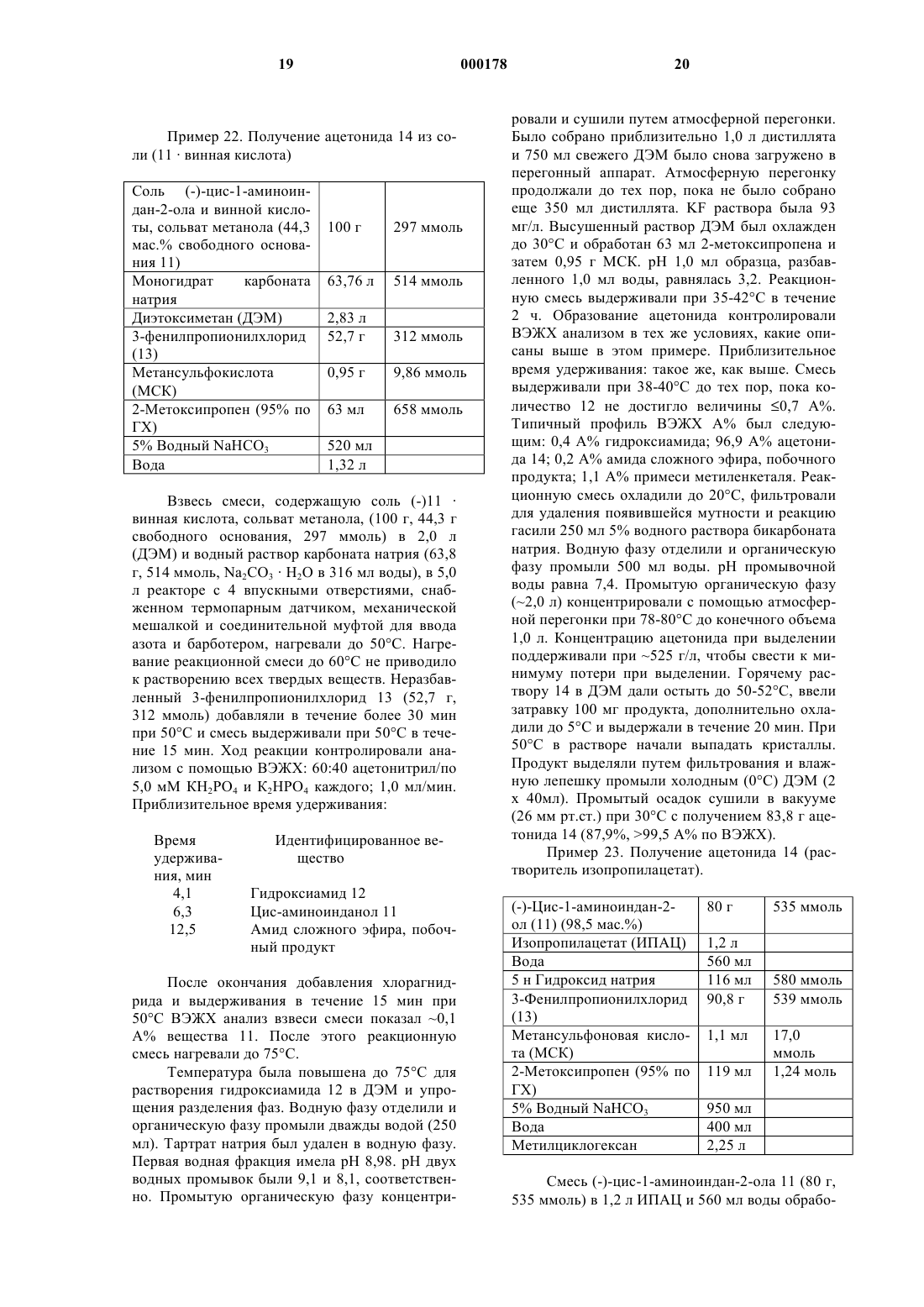
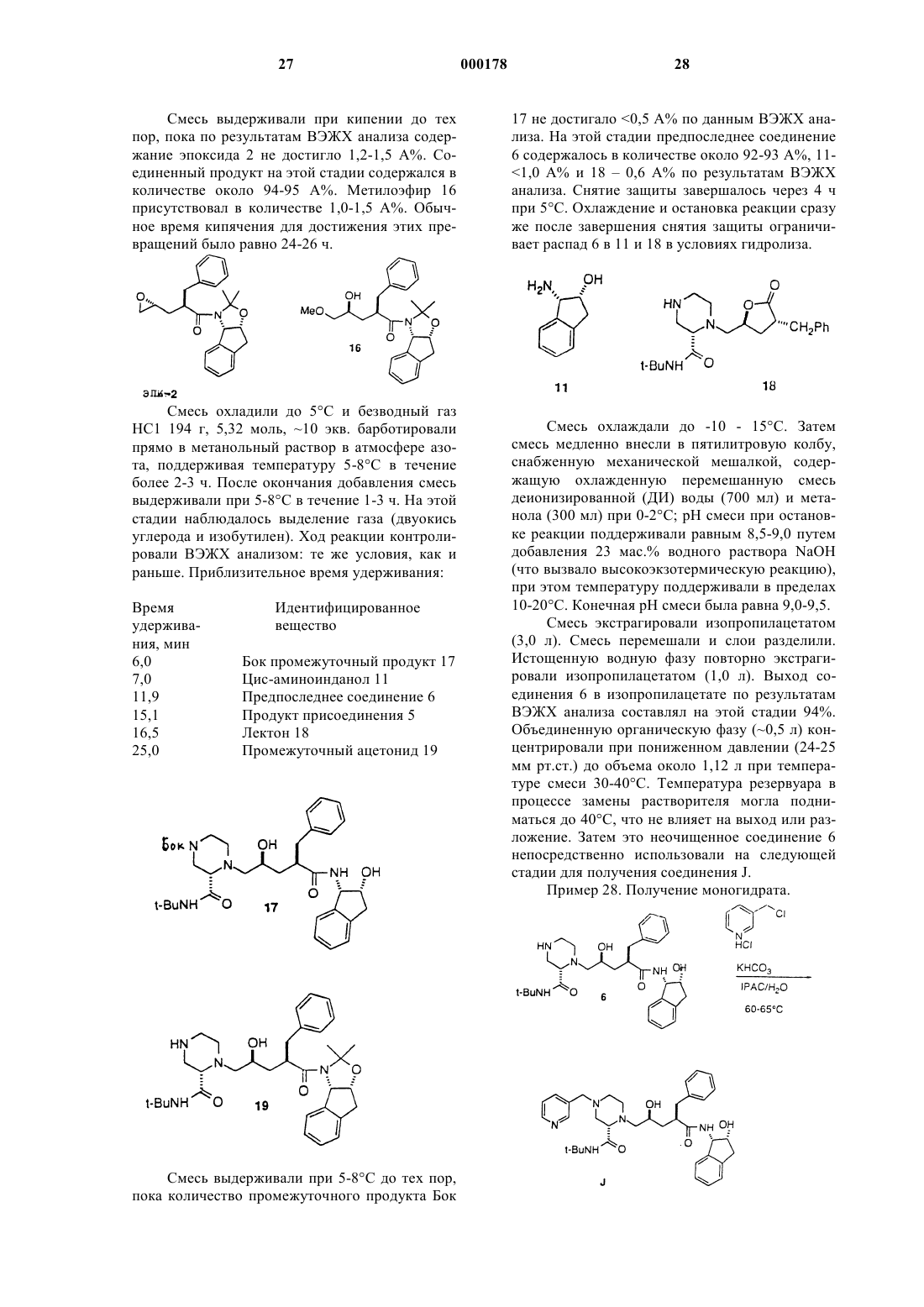
1 Данное изобретение относится к новому промежуточному продукту и способу синтеза соединений, которые ингибируют протеазу, кодированную вирусом иммунодефицита человека(ВИЧ), и, в частности, некоторых аналогов олигопептида, таких как Соединение J в нижеприводимых примерах. Эти соединения важны для профилактики заражения ВИЧ, лечения ВИЧинфекции и лечения возникающего при этом синдрома приобретенного иммунодефицита(СПИД). Эти соединения также могут использоваться для ингибирования ренина и других протеаз. Описанное здесь изобретение относится к способу получения ингибитора протеазы ВИЧ J из 4-пиколилпиперазинкарбоксамида 1 двухстадийным методом. Пиперазин 1 конденсируют с эпоксидом 2 с получением продукта связывания 3. Удаление ацетонидной защитной группы 3 прямо приводит к ингибитору протеазы ВИЧ-1 Ретровирус, названный вирусом иммунодефицита человека (ВИЧ), является этиологическим фактором сложного заболевания, которое включает прогрессивное разрушение иммунной системы (синдром приобретенного иммунодефицита; СПИД) и деградацию центральной и периферической нервной системы. Этот вирус первоначально был известен как LAV, HTLV-III или ARV. Общим признаком репликации ретровируса является обширная посттрансляционная переработка предшественника - полипротеинов с помощью кодируемой вирусом протеазы с образованием зрелых вирусных белков, необхо 000178 2 димых для сборки и функционирования вируса. Ингибирование этой переработки препятствует продуцированию среднеинфекционного вируса. Например, Kohl N.E. et al., Proc. Nat'l Acad. Sci.,85, 4686 (1988) показали, что генетическая инактивация ВИЧ-кодированной протеазы приводит к продукции незрелых, неинфекционных вирусных частиц. Эти результаты показывают,что ингибирование ВИЧ-протеазы представляет собой практически осуществимый метод лечения СПИДа и профилактики или лечения ВИЧинфекции. В нуклеотидной последовательности ВИЧ показано наличие пол гена в одной открытой рамке считывания [Ratner L. et al., Nature, 313,277 (1985)]. Гомология аминокислотной последовательности служит доказательством того,что пол последовательность кодирует обратную транскриптазу, эндонуклеазу и ВИЧ-протеазуet al., Nature, 329, 351 (1987)]. Соединения конечного продукта, включая некоторые аналоги олигопептидов, которые могут быть получены из новых промежуточных продуктов и по способам данного изобретения, являются ингибиторами ВИЧ-протеазы, и они описаны в ЕРО 541168, которая опубликована 12 мая 1993 г. Например, смотри Соединение J, указанные здесь. Ранее синтез Соединения J и родственных соединений осуществлялся с помощью 12 стадийного способа. Этот способ проиллюстрирован в ЕРО 541168. В предыдущих методах ингибитор J ВИЧ-протеазы получали путем связывания промежуточного эпоксида 2 с Бокзащищенным пиперазинкарбоксамидом 4 и образованием Бок-защищенного связанного промежуточного продукта 5. Снятие защиты у соединения 5 приводило к образованию предпоследнего Соединения 6, которое подвергали пиколилированию с образованием J. Неудобство этого способа состоит в том, что для превращения эпоксида 2 в J необходимо три химических стадии. Так, после снятия защиты необходима отдельная стадия пиколилирования для осуществления преобразования в J. Так как более реакционно-способное 4 положение пиперазинкарбоксамида должно быть защищено перед связыванием, наиболее эффективным методом защиты хирального 2-пиперазин-трет-бутилкарбоксамида было бы введение в это положение 3-пиколильной группы. Однако не ожидалось, что взаимодействие 1 с эпоксидом 2 будет эффективным, так как пиперазин 1 содержит три основные аминогруппы, способные вступить в реакцию с эпоксидом 2. Однако Бокзащищенный пиперазин 4 содержит только одну основную аминогруппу, и поэтому ожидалось,что связывание соединений 2 и 4 будет прямым. в интервале температур от около 25 до около 150 С, причем иногда указанная смесь необязательно содержит соответствующий растворитель;(б) снятия защиты путем обработки кислотой, и(в) нейтрализации кислоты с получением целевого соединения. В одном варианте, настоящее изобретение относится к способу синтеза соединения структуры В способе по данному изобретению исключена одна стадия, что является удобным упрощением и без того сложного синтеза Соединения J. Данный способ короче и высокоэффективен. Настоящее изобретение относится к альтернативному конвергентному синтезу, из которого исключена дополнительная синтетическая стадия и который дает более высокий выход. Получаемое Соединение J может использоваться в качестве ингибитора ВИЧ-протеазы, ренина и других протеаз. Настоящее изобретение относится к способу синтеза соединения структуры(а) нагревания в течение, по крайней мере,одного часа смеси одного эквивалента с приблизительно одним эквивалентом включающему стадии:(а) нагревания в течение, по крайней мере,одного часа смеси одного эквивалента в интервале температур от около 65 до около 85 С, причем указанная смесь необязательно 5 содержит в качестве растворителя метанол или изопропанол или их смесь;(б) охлаждения смеси до около 0 С;(г) нейтрализации с помощью NaOH, что приводит к получению целевого соединения. Данное изобретение включает также соединение Связывание может быть выполнено путем нагревания пиперазина 1 и эпоксида 2 в неразбавленном виде или в различных растворителях. Реакция связывания может быть проведена при температуре в интервале от 25 до 150 С, предпочтительно в интервале от 50 до 120 С, наиболее предпочтительными температурами являются от 65 до 85 С. Желательными растворителями для этой стадии являются сложные эфиры,такие как этилацетат, изопропилацетат, нбутилацетат; ацетонитрил; спирты, такие как метанол, этанол, н-пропанол, н-бутанол, третбутанол, трет-амиловый спирт и изопропанол; углеводороды, такие как циклогексан и толуол; простые эфиры, например ТГФ, ДМЭ; и формамиды, такие как ДМФ. Предпочтительными растворителями являются спирты, наиболее предпочтительны метанол и изопропанол. Снятие защиты у промежуточных продуктов 3 с образованием J осуществляется с помощью стандартных методов, т.е. обработкой сильными кислотами, такими как газообразная НС 1 в спиртовых растворителях или водной НС 1, причем метод с использованием газообразной НС 1 наиболее предпочтителен. Продукты этого изобретения могут использоваться для ВИЧ-протеазы, профилактики или лечения инфекции вирусом иммунодефицита человека и лечения сопровождающих ее патологических состояний, таких как СПИД. Лечение СПИДа или профилактика или лечение ВИЧ-инфекции определяются как включающие,но не ограниченные ими, случаи применений для лечения самых различных состояний при ВИЧ-инфекции: СПИД, ARC (родственный комплекс СПИД), как симптоматические, так и асимптоматические и фактические или потенциальные ВИЧ-контакты. Например, конечные продукты, которые могут быть получены в данном процессе, и промежуточные продукты по этому изобретению могут использоваться при 6 лечении ВИЧ-инфекции после предполагаемого контакта с ВИЧ, например, при переливании крови, пересадке органов, замещении жидкостей тела, укусах, поломках иглы при уколах или контактах с кровью пациента во время операции. Ингибиторы ВИЧ-протеазы из числа конечных продуктов могут использоваться также при получении и осуществлении скрининговых исследований противовирусных соединений. Например, соединения из числа конечных продуктов могут использоваться для выделения мутантных ферментов, которые являются превосходными скрининговыми инструментами для поиска более мощных противовирусных соединений. Кроме того, такие соединения могут использоваться при установлении и определении участка связывания у других противовирусных соединений, действующих на ВИЧпротеазу, например с помощью конкурентного ингибирования. Таким образом, соединения из числа конечных продуктов, которые получают в этом процессе, и промежуточные продукты этого изобретения являются коммерческими продуктами, которые продаются для этих целей. Конечный продукт - Соединение J, являющееся ингибитором ВИЧ-протеазы, имеет структуру: пригодны также его фармацевтически приемлемые соли или гидраты. Соединение J имеет название:N-(1(S)-2,3-дигидро-2(R)-гидрокси-1 Нинденил)-4(S)-гидрокси-2(R)-фенилметил-5-[4(3-пиридилметил)-2(S)-трет-бутилкарбамоил) пиперазинил]пентанамид. Соединения-ингибиторы ВИЧ-протеазы,которые могут быть получены по способам данного изобретения, описаны в ЕРО 541164. Эти соединения, ингибирующие ВИЧ-протеазу, могут быть введены пациентам, нуждающимся в таком лечении, в фармацевтических составах,включающих фармацевтический носитель и терапевтически эффективные количества соединения или его фармацевтически приемлемой соли. В ЕРО 541164 описаны приемлемые фар 7 мацевтические составы, пути введения, солевые формы и соли и дозы для этих соединений. Соединения по настоящему изобретению,которые могут иметь асимметрические центры и встречаются в виде рацематов, рацемических смесей и в виде индивидуальных диастереомеров или энантиомеров со всеми изомерными формами, входят в объем настоящего изобретения. Когда какая-нибудь переменная (например арил) встречается более одного раза в какомлибо структурном элементе, ее определение в каждом случае не зависит от ее определения в любом другом случае. Кроме того, сочетания заместителей и/или переменных допустимы только в таком виде, когда такие сочетания приводят к стабильным соединениям. Далее подробно описываются показательные экспериментальные методы использования нового способа. Эти методы являются только иллюстративными примерами и ими не ограничивают новый способ по изобретению. Пример 1. Пиразин-2-трет-бутилкарбоксамид 8EtOAc (KF = 56 мкг/мл) ДМФ 120 мл 1-Пропанол 30 л Карбоновую кислоту 7 суспендировали в 27 л EtOAc и 120 мл ДМФ при механическом перемешивании в N2 в 3-горлой колбе объемом 72 л и суспензию охлаждают до 2 С. Добавили оксалилхлорид, поддерживая температуру между 5 и 8 С. Добавление завершают в течение 5 ч. В процессе экзотермического добавления выделяется СО и СО 2. Образовавшаяся НСl главным образом остается в растворе. Имеется осадок,который, по-видимому, является НСl солью хлорангидрида пиразиновой кислоты. Анализ образования этого хлорангидрида осуществляют путем гашения безводной реакционной пробы с помощью трет-бутиламина. После завершения реакции остается 0,7% кислоты 7. Анализ полноты образования хлорангидрида является важным, так как незавершенная реакция приводит к образованию примеси бистрет-бутилоксамида. Реакцию можно контролировать с помощью ВЭЖХ: колонка 25 см Dupont ZorbaxRXC8 со скоростью потока 1 мл/мин и регистрацией при 250 нм; линейный градиент от 98% 8 0,1% водного раствора Н 3 РО 4 и 2% СН 3 СN до 50% водного раствора Н 3 РО 4 и 50% СН 3 СN за 30 мин. Время удерживания: кислота 7 = 10,7 мин, амид 8 = 28,1 мин. Реакционную смесь выдерживали при 5 С в течение 1 ч. Полученную взвесь охлаждали до 0 С и добавляли трет-бутиламин с такой скоростью, чтобы поддерживать внутреннюю температуру ниже 20 С. Для добавления требуется 6 ч, так как реакция была очень экзотермична. Небольшую часть образующегося гидрохлорида третбутиламмония удаляют из реакционной смеси в виде пушистого белого твердого вещества. Смесь выдерживают при 18 С дополнительно 30 мин. Осевшие соли аммония удаляют путем фильтрования. Лепешку на фильтре промывают 12 л EtOAc. Объединенные органические фазы промывают 6 л 3% NaHCO3 и 2 х 2 л насыщенного водного раствора NaCl. Органическую фазу обрабатывают 200 г угля Дарко G 60 и фильтруют через солка-флок (Solka Flok) и лепешку промывают 4 л EtOAc. Обработка углем позволяет эффективно устранить слабофиолетовое окрашивание продукта.Pacтвор 8 в EtOAc концентрируют при 10 мбар до 25% исходного объема. Добавляют 30 л 1-пропанола и перегонку продолжают до достижения конечного объема 20 л. К этому моменту количество EtOAc было ниже предела,обнаруживаемого в 1 Н ЯМР (1%). Внутренняя температура в этом измененном растворителе 30 С. Pacтвор 8 в 1-пропанол/EtOAc стабилен к кипячению при атмосферном давлении в течение нескольких дней. При выпаривании пробы получают твердое вещество рыжевато-коричневого цвета с т.пл. 87-88 С. 13 С ЯМР (75 МГц, СDСl3, м.д.) 161,8; 146,8; 145,0; 143,8; 142,1; 51,0; 28,5. Пример 2. рац-2-трет-Бутил-карбоксамидпиперазин 9 Материалы. Раствор пиразин-2-трет-бутилкарбоксамида 82,4 кг (13,4 моль) в 12 л раствора 1 пропанола с 20%-ным Рd(ОН)2/С, 16 вес.%, воды 144 г. Раствор пиразин-2-трет-бутилкарбоксамида 8 в 1-пропаноле помещали в автоклав объемом 5 гал. Добавили катализатор и смесь гидрировали при 65 С при 40 ф/кв.дюйм (3 атм) Н 2. За 24 ч реакции поглощалось теоретическое количество водорода и показатель GC указывал на наличие 1% вещества 8. Смесь охладили, продули газом N2 и катализатор удалили 9 путем фильтрования через солка-флок. Катализатор промыли 2 л теплого 1-пропанола. Обнаружено, что использование теплого 1 пропанола в процессе отмывки лепешки на фильтре улучшало фильтрацию и понижало потери продукта на фильтровальной лепешке. Реакцию контролировали с помощью GC: колонка 30 м Megabore, от 100 до 160 С при 10 С/мин, выдерживание 5 мин, затем при 10 С/мин до 250 С, время удерживания: 8 = 7,0 мин, 9 = 9,4 мин. Реакцию можно также контролировать с помощью ТСХ с использованиемEtOAc/MeOH (50:50) в качестве растворителя и нингидрина в качестве проявителя. Выпаривание пробы показало, что выход после амидирования и гидрирования составляет 88% и что концентрация соединения 9 равна 133 г/л. Выпаривание пробы дает соединение 9 в виде белого твердого вещества с т.пл. 150151 С; 13 С ЯМР (75 МГц, D2O, м.д.) 173,5; 59,8; 52,0; 48,7; 45,0; 44,8; 28,7. Пример 3. Соль(S)-(+)-10-камфарасульфо- 10,0 кг (43,2 кислота (CSA) моль) 1-пропанол 12 л Ацетонитрил 39 л Вода 2,4 л Раствор амина 9 в 1-пропаноле загружали в 100-литровую колбу с присоединенным концентратором. Раствор концентрировали при 10 мбар и температуре 25 С до объема около 12 л. В это время продукт осаждался из раствора, но снова переходил в раствор при нагревании смеси до 50 С. Анализ гомогенной пробы показал, что концентрация соединения 9 была 341 г/л. Концентрацию определяли с помощью ВЭЖХ: колонка 25 см Dupont Zorbax RXC8 со скоростью потока 1,5 мл/мин и регистрацией при 210 нм,изократическая смесь (98/2) СН 3 СN/0,1% водный раствор Н 3 РО 4. Время удерживания 9: 2,5 мин. Добавили ацетонитрил (39 л) и воду (2,4 л) и получили прозрачный светло-коричневый раствор. Определение содержания воды с помощью титрования KF и отношения CH3CN/1-пропанол с помощью интеграции в 1 Н ЯМР показало, что отношение CH3CN/1-пропанол/H2O было равно(S)-10-Камфарсульфокислоту загружали в течение более 30 мин 4 порциями при 20 С. После добавления CSA температура поднималась до 40 С. Через несколько минут образовался густой белый осадок. Белую взвесь нагревали до 76 С до растворения всех твердых веществ, затем в течение 8 ч бледно-коричневому раствору дали охладиться до 21 С. Продукт выпадал в осадок при 62 С. Этот продукт фильтровали, не выдерживая при 21 С,и лепешку на фильтре промыли 5 л смеси растворителей СН 3 СN/1-пропанол/Н 2 О 26/8/1,6. Его высушили при 35 С в вакуумной печи с продувкой N2, получая 5,6 кг (39%) вещества 10 в виде белого твердого кристаллического продукта с т.пл. 288-290 С (с разложением) []25D = 18,9 (с = 0,37, Н 2 О). 13 С ЯМР (75 МГц, D2O,м.д.) 222,0; 164,0; 59,3; 54,9; 53,3; 49,0; 48,1; 43,6; 43,5; 43,1; 40,6; 40,4; 28,5; 27,2; 25,4; 19,9; 19,8. Количество вещества, исключая ошибки,составило 95% согласно следующему тесту хиральной ВЭЖХ: пробу 10 (33 мг) суспендировали в 4 мл EtOH и 1 мл Et2N. Добавили Бок 2 О (11 мг) и реакционную смесь выдерживали в течение 1 ч. Растворитель полностью удалили в вакууме и остаток растворили в около 1 мл EtOAc и фильтровали через пастеровскую пипетку сSiO2, используя в качестве элюента EtOAc. Фракции упаренного продукта вновь растворили в гексане при около 1 мг/мл. Энантиомеры разделили на колонке Daicel Chiracell AS с системой растворителей гексан/IPA (97:3) при скорости потока 1 мл/мин и регистрации при 228 нм. Время удерживания: S антипод = 7,4 мин, R Взвесь бис (+) CSA соли 2(S)-третбутилкарбоксамидпиперазина в 30 мл 1 пропанола промыли дважды 25% растворомNaOH и дважды насыщенным водным раствором сульфата натрия. Гомогенную 1 11 пропанольную фазу концентрировали при пониженном давлении и получили 1,30 г (94%) 2(S)-трет-бутилкарбоксамидпиперазина в виде белого твердого продукта. Сырой 2(S)-третбутилкарбоксамидпиперазин суспендировали в 20 мл толуола и после добавления альдегида добавили изопропоксид титана (IУ). Реакционная смесь потемнела, и после перемешивания при 20 С в течение 2 ч добавили цианборгидрид натрия. После перемешивания в течение 18 ч добавили 50 мл EtOH и осадок удалили путем фильтрования и промывания EtOAc. Объединенные органические фазы упаривали и получили желтое масло (2,3 г), которое очищали с помощью хроматографии на SiO2 (EtOAc/МеОН 50/50). При выпаривании фракций, содержащих продукт,получили 1,20 г 2(S)-третбутилкарбоксамид-4-(3-пиридилметил)пиперазина (40%). Полученное соединение существует в виде бледно-желтого масла и может кристаллизоваться в виде твердой соли малеиновой кислоты. 1 Смесь соединения 1 (7,9 г, 28,5 ммоль) и эпоксида 2 (10 г, 26,5 ммоль) в изопропиловом спирте (88 мл) нагревали до точки кипения 82 С и выдерживали в течение 72 ч до завершения образования соединения 3. Раствор 3 охладили до 0 С и обработали безводной газообразной НСl и смесь выдерживали при 0-5 С в течение 3 ч. Гидролиз гасили путем медленного добавления 50% NaOH, доводя рН смеси до 12, в то же время поддерживали температуру ниже 25 С. Затем смесь разделяли с помощью изопропилацетата (200 мл) и воды (50 мл). Смесь перемешивали и слои разделяли и водную фазу снова экстрагировали изопропилацетатом (50 мл). Изопропилацетатный раствор J концентрировали приблизительно до 100 г/л и добавляли воду до насыщения горячего раствора изопропилацетата. В смесь внесли затравку, охладили и получили J (15,1 г, 90%) из эпоксида 2. Пример 6. Преобразование оксида индена в цис-1-амино-2-инданол (см. табл.1) К одному мл оксида индена (8,33 ммоль),растворенного в 10 мл ацетонитрила, добавили 0,15 мл воды (8,33 ммоль). Смесь охладили до 0-5 С на ледяной бане. Добавили по каплям концентрированную серную кислоту, поддерживая температуру смеси ниже 10 С. После добавления всей кислоты температуре дали достичь 20-25 С. Прозрачный раствор выдерживали в течение 30 мин. К этой смеси добавили 2 мл воды и раствор нагревали в течение 30 мин. Когда метилоксазолин полностью преобразовался в цисаминоинданол, реакционную смесь охладили до комнатной температуры. Добавили раствор 5 н КОН (3 мл, 15 ммоль). Это составляет 90% теоретического для серной кислоты. По лакмусу раствор оставался кислым. Если рН поднималась выше, происходило повторное ацилирование 2 и выход аминоинданола снижался. Белое твердое вещество(К 2SO4) удалили путем фильтрования. Добавили при перемешивании 15 мл смолы Dowex (увлажненную ацетонитрилом). Перемешанную смолу выдерживали в течение 15 мин и брали пробу для ЖХ (dilx 50). Когда пик ЖХ для аминоинданола исчезал, смолу собирали с помощью фильтрации, промывали ацетонитрилом и затем метанолом. Влажную смолу обрабатывали раствором 50 мл 1 H NН 3 в метаноле и взвесь перемешивали при комнатной температуре в течение 30 мин. 13 Смолу снова собирали путем фильтрации и метанол/NН 3 сохраняли. Добавляли другую загрузку 1 н NH3/МеОН (20 мл) и смолу снова переводили во взвесь. После удаления из смолы метанола/NН 3 растворы аминоинданола объединяли и концентрировали для удаления NН 3. Анализ конечного МеОН раствора указывал на наличие 1,0 г (выход 81%) цис-1-амино-2 инданола, подходящего для разделяющего агента винной кислоты. Пример 7. Получение рацемического оксида индена. Инден (95%, 122 мл) растворяли в метаноле (812 мл) и ацетонитриле (348 мл), затем фильтровали. Фильтрат разбавляли 0,05 М двухосновным фосфатом натрия (116 мл), затем рН доводили до 10,5 с помощью 1 М водного раствора гидроксида натрия. Водный пероксид водорода (35%, 105 мл) разбавляли водой (53 мл) и добавляли в течение 3 ч, поддерживая температуру при 25 С и при рН 10,5 с помощью 1 М водного раствора гидроксида натрия (всего 120 мл) . Через 6 ч добавили 1 М водный раствор метабисульфита натрия (26 мл), поддерживая рН около 8,3 путем добавления 1 М водногоNaOH (39 мл). Добавили воду (700 мл) и смесь экстрагировали хлористым метиленом (580 мл и 300 мл). Объединенные органические экстракты, содержащие оксид индена (117 г), концентрировали до объема 600 мл. Пример 8. Получение оксида (1S,2R)индена. Субстрат, оксид (IS, 2R)-индена получали по методу, описанному D.J. O'Donnell, et al., J.Organic Chemistry, 43, 4540 (1978). Пример 9. Получение цис-1-амино-2 инданола. Оксид индена (117 г), разбавленный до общего объема 600 мл в метиленхлориде, разбавляли ацетонитрилом (600 мл) и охлаждали до -20 С. Затем добавляли метансульфоновую кислоту (114 мл). Смесь нагревали до 25 С и выдерживали в течение 2 ч. Добавляли воду(600 мл) и смесь нагревали при 45 С в течение 5 ч. Органическую фазу отделяли, а водную фазу продолжали нагревать при кипении в течение 4 ч до концентрации приблизительно 200 г/л. рН раствора доводили до 12,5 с помощью 50%-ного водного гидроксида натрия и затем охлаждали до 5 С и фильтровали, высушивали в вакууме с получением цис-1-амино-2-инданола. Пример 10. Получение 1S-амино-2Rинданола. Оксид (1,S,2R)-индена (85%, энантиомерный избыток) (250 г, 0,185 моль) растворили в хлорбензоле (300 мл) и гептане (1200 мл) и медленно добавили к раствору метансульфокислоты (250 мл, 0,375 моль) в ацетонитриле(1250 мл) при температуре ниже -10 С. Реакционную смесь нагрели до 22 С и выдерживали в течение 1,0 ч. К смеси добавили воду и концен 000178 14 трировали перегонкой до достижения внутренней температуры 100 С. Реакционную смесь грели при 100 С в течение 2-3 ч, затем охладили до комнатной температуры. Добавили хлорбензол (1000 мл), смесь перемешивали, органическую фазу отделили. Оставшуюся водную фазу,содержащую 1S-амино-2R-инданол (85%, энантиомерный избыток, 165 г, 60%), довели до рН 12 с помощью 50% водного гидроксида натрия и продукт собирали фильтрацией и высушивали в вакууме при 40 С с получением 1S-амино-2Rинданола (85% энантиомерный избыток, 160 г). Пример 11. Получение 1S-амино-2Rинданола. Оксид (1S, 2R)-индена (85%, энантиомерный избыток) (250 г, 0,185 моль) растворили в хлорбензоле (300 мл) и гептане (1200 мл) и медленно добавили к раствору дымящей серной кислоты (21% SO3, 184 мл) в ацетонитриле(1250 мл) при температуре ниже -10 С. Реакционную смесь нагрели до 22 С и выдерживали в течение 1,0 ч. К смеси добавили воду и концентрировали перегонкой до достижения внутренней температуры 100 С. Реакционную смесь нагревали при 100 С в течение 2-3 ч, затем охладили до комнатной температуры. Добавили хлорбензол (1000 мл), смесь перемешивали, органическую фазу отделили. Оставшуюся водную фазу, содержащую 1S-амино-2R-инданол(85% энантиомерный избыток, 205 г, 74%) разбавляли равным объемом ацетонитрила. рН доводили до 12,5 50% водным раствором гидроксида натрия и органическую фазу отделили. Оставшуюся водную фазу экстрагировали дополнительной порцией ацетонитрила. Объединенные ацетонитрильные экстракты концентрировали в вакууме с получением 1S-амино-2Rинданола (85% энантиомерный избыток, 205 г). Альтернативно, оставшуюся водную фазу,содержащую 1S-амино-2R-инданол (85% энантиомерный избыток, 205 г 74%) разбавили равным объемом бутанола и рН довели до 12,5 с помощью 50%-ного водного раствора гидроксида натрия и органическую фазу отделили. Органическую фазу промыли хлорбензолом. Добавили L-винную кислоту и воду удалили перегонкой для кристаллизации соли винной кислоты и аминоинданола. Пример 12. Применение бензонитрила. Оксид индена (5 г) растворили в бензонитриле (50 мл) при 25 С и добавили серную кислоту (98%, 2,25 мл). Смесь разбавили 5 М водным раствором гидроксида натрия (50 мл) и экстрагировали метиленхлоридом. Органический экстракт концентрировали в вакууме с получением 5,03 г оксазолина. Пример 13. Разделение цис-1-амино-2 инданола. Цис-1-амино-2-инданол (100 г) растворили в метаноле (1500 мл) и добавили раствор Lвинной кислоты (110 г) в метаноле (1500 мл). Смесь нагрели до 60 С и охладили до 20 С, 15 фильтровали и высушили в вакууме с получением соли L-винной кислоты 1S-амино-2Rинданола в виде сольвата метанола (88 г). Пример 14. Получение 1S-амино-2Rинданола. Метанол сольват L-винной соли 1S-амино 2R-инданола (88 г) растворили в воде (180 мл) и нагревали до 55-60 С. Раствор осветлили фильтрацией и рН довели до 12,5 с помощью 50%-ного водного раствора гидроксида натрия. Смесь охлаждали до 0-5 С свыше 2 ч, затем выдерживали при этой температуре в течение 1 ч,фильтровали, промывали холодной водой и высушивали в вакууме при 40 С с получением 1Sамино, 2R-инданола (100% энантиомерный избыток, чистота 99%, 37 г). Пример 15. Преобразование 1,2-инданола в цис-1-амино-2-инданол (см. табл.2) К 300 мл индандиола, растворенного в 3 мл ацетонитрила, содержащего 0,04 мл воды, по каплям добавили при 0-10 С 0,22 мл концентрированной Н 2SО 4. После окончания добавления ледяную баню удалили и смесь нагревали до комнатной температуры. После выдерживания в течение 30 мин из прозрачного раствора брали пробу для анализа Ic (dilx 500). Когда весь гликоль был поглощен, раствор затем обработали водой и нагрели до кипения на паровой бане для гидролиза оксазолина. Когда анализ Ic указал на завершение гидролиза, добавили 1,6 мл 5 н КОН для нейтрализации серной кислоты. Сульфат калия удалили из раствора фильтрацией. Фильтрат проанализировали на цисаминоинданол. Он содержался в количестве 196 мг (66% от теории, что с поправкой на непрореагировавший исходный материал составляет 75%). Раствор пропустили через 10 мл Dowex 50 х 4 (Н+). Отработавшую колонку проверили на наличие продукта. Весь аминоинданол был абсорбирован. После промывания смолы метанолом продукт элюировали раствором 1 М в NН 3(сухой). Аммиачный метанол концентрировали для удаления NН 3, и конечный раствор аминоинданола, готовый для растворения, был проанализирован (175 мг или 59% от теории, если не вводить поправку на непрореагировавший гликоль). Пример 16 Получение инданольных реагентов.-транс-2-бром-1-инданол получали по методу S.M. Sutter et al., J. Am.(+)-транс-2-бром-1-инданол и цис- и транс-1,2 индандиолы получали по методу М. Imuta et al.,J. Org. Chem., 43, 4540 (1978). Пример 17. Получение цис-1-амино-2 инданола из транс-2-бром-1-инданола. Транс-2-бром-1-инданол (10 г, 46,9 ммоль,растворенный в 100 мл ацетонитрила, содержащего 0,8 мл воды) охладили до -5 С и добавили концентрированную серную кислоту (5,2 мл). Смесь выдерживали 1 ч, затем добавили 5 М водный гидроксид калия для доведения рН до 11. Реакционную смесь фильтровали, удаляя соли сернокислого калия. рН фильтрата водного ацетонитрила устанавливали меньше 2 с помощью серной кислоты и нагревали до 80-100 С,удаляя ацетонитрил с помощью перегонки, и получили водный раствор цис-1-аминоинданола. Раствор концентрировали до объема 20 мл, затем рН довели до 12,5 с помощью гидроксида калия. Кристаллический продукт собрали фильтрацией и сушили в вакууме с получением цис-1-амино-2-инданола (4,25 г). Пример 18. Получение цис-1S-амино-2Rинданода из цис-(1S, 2R)-индандиола. Цис-(1S,2R)-индандиол (1 г) растворили в ацетонитриле (10 мл), охладили до 0 С и добавили концентрированную серную кислоту (1,0 мл). Смесь выдерживали в течение 40 мин с нагреванием до 20 С. Добавили воду (0,8 мл) и смесь нагревали до кипения. Добавили 5 М водный раствор гидроксида калия для достижения рН выше 11 и образовавшийся твердый продукт(сернокислый калий) удалили с помощью фильтрования с получением водного раствора цис-1S-амино-2R-инданола (0,79 г, выход 66%). Пример 19. Получение цис-1-амино-2 инданола из транс-1,2-индандиола. Транс-1,2-индандиол (1,5 г) растворили в ацетонитриле (2,5 мл), охлажденном до 0 С, и добавили концентрированную серную кислоту(1,1 мл). Смесь ступенчато нагрели до 20 С и выдерживали до 3 ч. Добавили воду (2 мл) и смесь нагревали до кипения. Добавили концентрированный водный раствор гидроксида натрия, чтобы довести рН до 12. Образовавшийся твердый продукт удалили фильтрованием с получением раствора цис-1-амино-2-инданола в водном ацетонитриле (1,02 г, выход 63%) . Пример 20. Получение цис-1-амино-2 инданола из цис-1,2-индандиола. Цис-1,2-индандиол (1,0 г) растворили в ацетонитриле (20 мл), охлажденном до -40 С, и добавили дымящую серную кислоту (21% SO3,0,8 мл). Смесь выдерживали в течение 1 ч при ступенчатом нагревании до 0 С. Добавили воду и смесь нагревали до 80 С в течение 1 ч с получением водного раствора цис-1-амино-2 инданола. Взвесь смеси, содержащей (-)-цис-1 аминоиндан-2-ол (11, 900 г, 6,02 моль) в 40 л ДЭМ и водный раствор карбоната натрия (760 г,6,13 моль Nа 2 СО 3 Н 2 О в 6,4 л воды) в реакторе объемом 100 л с 4 впускными отверстиями,снабженном термопарным датчиком, механической мешалкой и соединительной муфтой для ввода азота и барботером, нагревали до 46-47 С и выдерживали в течение 15 мин. Реакционную смесь нагрели до 46-47 С и выдерживали 15 мин для растворения твердых веществ. Водная фаза имела рН 11,5. Добавили неразбавленный 3-фенилпропионилхлорид 13 (1,05 кг, 6,23 моль) в течение более 2 ч при 47-59 С. В процессе добавления 13 внутренняя температура увеличилась от 47 до 59 С; в процессе добавления хлорангидрида из раствора кристаллизовался гидроксид амида 12. После завершения добавления реакционную смесь выдерживали при 59 С в течение 0,5 ч и затем нагревали до 72 С для растворения твердых веществ. Температуру подняли до 72 С для растворения гидроокиси амида таким образом, чтобы можно было получить гомогенный образец для анализа ВЭЖХ и упростить разделение фаз. Ход реакции контролировали с помощью анализа ВЭЖХ: 60:40 Ацетонитрил/по 5,0 мМ КН 2 РО 4 и К 2 НРО 4, каждого. Приблизительное время удерживания: Время удерживания, мин 4,1 6,3 12,5 Идентифицированное вещество Гидроксиамид 12 Цис-аминоинданол 11 Амид сложного эфира,побочный продукт После завершения добавления хлорангидрида и выдерживания при 72 С 0,5 ч анализ реакционной смеси с помощью ВЭЖХ показал наличие 0,6 А% соединения 11; 0,2 А% побочного продукта, сложного эфира амида, и 98,7 А% гидроксиамида. Гидроксиамид 12 не эффективно отводился при выделении ацетонида 14. 18 Водную фазу отделяли, а органическую фазу дважды промывали 4,5 л воды. Промытую органическую фазу концентрировали и высушивали с помощью атмосферной азеотропной перегонки. Исходный объем 40 л концентрировали до 27 л. Всего 16 л свежего ДЭМ было загружено в перегонный аппарат и всю загрузку концентрировали при 88-89 С до объема 40 л. Высушенную взвесь гидроксиамида 12 в ДЭМ обработали 1,28 л 2-метоксипропена и затем 18,6 г МСК при 30 С. Добавление МСК в отсутствии 2-метоксипропена приводило к образованию сложного эфира амина. Эта примесь снова превращается в гидроксиамид 12 в процессе обработки в щелочной среде к концу образования ацетонида. Было обнаружено, что рН 1,0 мл образца, разведенного в 1 мл воды, была 2,8-3,0. Полученную смесь выдерживали при 39-40 С в течение 3 ч. Образование ацетонида контролировали с помощью ВЭЖХ анализа в тех же условиях, как описано выше в этом примере. Приблизительное время удерживания: Время удерживания, мин 4,1 6,9 9,0 12,5 Идентифицированное вещество Гидроксиамид 12 Примесь метиленкеталя Ацетонид 14 Сложный эфир амида,побочный продукт Смесь выдерживали при 38-40 С до тех пор, пока количество соединения 12 не стало 0,4 А%. Типичный профиль ВЭЖХ А% был следующим: 0,4 А% гидроксиамида 12; 96,9 А% ацетонида 14; 0,2 А% сложного эфира амида побочного продукта; 1,1 А% примеси метиленкеталя. Реакционную смесь охладили до 24 С и реакцию гасили с помощью 10,8 л 5% водного раствора бикарбоната натрия. Водную фазу отделили и органическую фазу промыли дважды 10,8 л воды. рН промывной воды была равна 7,6. Если бы рН была слишком низкой, группа ацетонида могла бы снова подвергнуться гидролизу с получением гидроксиамида 12. Промытую органическую фазу (34,2 л) концентрировали с помощью атмосферной перегонки при 7880 С до конечного объема 3,5 л. Концентрацию ацетонида сделали равной 525 г/л для минимизации потерь при выделении. Горячему раствору 14 в ДЭМ дали охладиться до 57 С, внесли затравку 0,5 г 14, дополнительно охладили до 0 С и выдерживали 0,5 ч. При температуре от 53 до 55 С в растворе начали выпадать кристаллы. Продукт выделяли с помощью фильтрования и влажную лепешку промыли холодным (0 С) ДЭМ (300 мл). Промытую лепешку сушили в вакууме (26 мм рт.ст.) при 30 С с получением 1,74 кг ацетонида 14 (90%, 99,5 А% по ВЭЖХ). Пример 22. Получение ацетонида 14 из соли (11 винная кислота) Соль (-)-цис-1-аминоиндан-2-ола и винной кислоты, сольват метанола (44,3 маc.% свободного основания 11) Моногидрат карбоната натрия Диэтоксиметан (ДЭМ) 3-фенилпропионилхлорид(ДЭМ) и водный раствор карбоната натрия (63,8 г, 514 ммоль, Na2CO3 Н 2 О в 316 мл воды), в 5,0 л реакторе с 4 впускными отверстиями, снабженном термопарным датчиком, механической мешалкой и соединительной муфтой для ввода азота и барботером, нагревали до 50 С. Нагревание реакционной смеси до 60 С не приводило к растворению всех твердых веществ. Неразбавленный 3-фенилпропионилхлорид 13 (52,7 г,312 ммоль) добавляли в течение более 30 мин при 50 С и смесь выдерживали при 50 С в течение 15 мин. Ход реакции контролировали анализом с помощью ВЭЖХ: 60:40 ацетонитрил/по 5,0 мМ КН 2 РО 4 и К 2 НРО 4 каждого; 1,0 мл/мин. Приблизительное время удерживания: Время удерживания, мин 4,1 6,3 12,5 Идентифицированное вещество Гидроксиамид 12 Цис-аминоинданол 11 Амид сложного эфира, побочный продукт После окончания добавления хлорагнидрида и выдерживания в течение 15 мин при 50 С ВЭЖХ анализ взвеси смеси показал 0,1 А% вещества 11. После этого реакционную смесь нагревали до 75 С. Температура была повышена до 75 С для растворения гидроксиамида 12 в ДЭМ и упрощения разделения фаз. Водную фазу отделили и органическую фазу промыли дважды водой (250 мл). Тартрат натрия был удален в водную фазу. Первая водная фракция имела рН 8,98. рН двух водных промывок были 9,1 и 8,1, соответственно. Промытую органическую фазу концентри 20 ровали и сушили путем атмосферной перегонки. Было собрано приблизительно 1,0 л дистиллята и 750 мл свежего ДЭМ было снова загружено в перегонный аппарат. Атмосферную перегонку продолжали до тех пор, пока не было собрано еще 350 мл дистиллята. KF раствора была 93 мг/л. Высушенный раствор ДЭМ был охлажден до 30 С и обработан 63 мл 2-метоксипропена и затем 0,95 г МСК. рН 1,0 мл образца, разбавленного 1,0 мл воды, равнялась 3,2. Реакционную смесь выдерживали при 35-42 С в течение 2 ч. Образование ацетонида контролировали ВЭЖХ анализом в тех же условиях, какие описаны выше в этом примере. Приблизительное время удерживания: такое же, как выше. Смесь выдерживали при 38-40 С до тех пор, пока количество 12 не достигло величины 0,7 А%. Типичный профиль ВЭЖХ А% был следующим: 0,4 А% гидроксиамида; 96,9 А% ацетонида 14; 0,2 А% амида сложного эфира, побочного продукта; 1,1 А% примеси метиленкеталя. Реакционную смесь охладили до 20 С, фильтровали для удаления появившейся мутности и реакцию гасили 250 мл 5% водного раствора бикарбоната натрия. Водную фазу отделили и органическую фазу промыли 500 мл воды. рН промывочной воды равна 7,4. Промытую органическую фазу(2,0 л) концентрировали с помощью атмосферной перегонки при 78-80 С до конечного объема 1,0 л. Концентрацию ацетонида при выделении поддерживали при 525 г/л, чтобы свести к минимуму потери при выделении. Горячему раствору 14 в ДЭМ дали остыть до 50-52 С, ввели затравку 100 мг продукта, дополнительно охладили до 5 С и выдержали в течение 20 мин. При 50 С в растворе начали выпадать кристаллы. Продукт выделяли путем фильтрования и влажную лепешку промыли холодным (0 С) ДЭМ (2 х 40 мл). Промытый осадок сушили в вакууме Смесь (-)-цис-1-аминоиндан-2-ола 11 (80 г,535 ммоль) в 1,2 л ИПАЦ и 560 мл воды обрабо 21 тали соединением 5 (90,8 г, 539 ммоль), при этом рН поддерживалась в пределах 8,0-10,5 при 70-72 С с помощью 5 н гидроксида натрия(116 мл, 580 ммоль). Ход реакции контролировали ВЭЖХ анализом: 60:40 ацетонитрил/по 5,0 мМ КН 2 РО 4 и К 2 НРО 4, каждый. Приблизительное время удерживания: Время удерживания, мин 4,1 6,3 12,5 Идентифицированное вещество Гидроксиамид 12 Цис-аминоинданол 11 Амид сложного эфира, побочный продукт К концу реакции водные фазы отделяли и органическую фазу промывали водой (400 мл) при 72-73 С. рН водной фазы и промывной воды была равна 8,1 и 7,9, соответственно. Влажную фазу ИПАЦ высушивали с помощью атмосферной перегонки. Всего было загружено 3,0 л ИПАЦ для уменьшения KF раствора до 100 мг/л. Конечный объем составляет 1,60 л. Образовавшуюся взвесь гидроксиамида 12 в ИПАЦ обработали 2-метоксипропеном (119 мл, 1,24 моль) и затем МСК (1,1 мл, 3,2 моль %) при 3538 С в течение 4,5 ч. Образование ацетонида контролировали ВЭЖХ анализом, используя такие же условия, как описано выше. Смесь выдерживали при 38-40 С до тех пор, пока количество 12 не достигло величины 0,4 А%. Реакционную смесь фильтруют для устранения мутного осадка и фильтрат гасят холодным раствором бикарбоната натрия (950 мл) более 15 мин. Водную фазу отделили и органическую фазу промыли водой (400 мл). Раствор бикарбоната натрия охладили до 0-5 С. Обнаружили, что рН водной фазы и промывной воды равно 7,5 и 7,9,соответственно. Осуществляли атмосферную перегонку, при этом растворитель ИПАЦ вытесняли метилциклогексаном. Исходный объем перед атмосферным концентрированием был равен 1,65 л. Всего 1,5 л метилциклогексана было добавлено для полного вытеснения растворителя ИПАЦ метилциклогексаном. Температура раствора к концу замены растворителя была равна 101 С и конечный объем раствора был равен 900 мл. Раствор нагрели до 65-70 С,чтобы растворить твердые вещества, затем охладили до 55 С, внесли затравку и охладили до 0 С. Смесь выдерживали при 0 С в течение 15 мин и продукт выделили путем фильтрования и отмыли холодным метилциклогексаном (200 мл). Промытую лепешку сушили в вакууме (26 мм рт.ст.) при 30 С с получением 151 г ацетонида 14 (87,5%, 99,5 А% по ВЭЖХ). Кристаллический ацетонид 14 (200 г, 0,622 моль, 99,1 маc.%) растворили в 1,25 л высушенного над ситами ТГФ (KF = 11 мг/л) в атмосфере азота при 25 С при механическом перемешивании. Полученная на этом этапе величина KF раствора составляла 40 мг/л. Раствор подвергли трем циклам попеременно вакуум/продувка азотом до полного удаления из раствора растворенного кислорода. К раствору ТГФ добавили аллилбромид. Полученная величина KF составляла 75 мг/л. В этой процедуре было получено типичное полное превращение (99,5%) при уровне KF раствора в пре-ЛДС 200 мг/л и наличии избытка основания 10%. Затем раствор охладили до -20 С. К раствору аллилбромид/14 добавили раствор гексаметилдисилазида лития (ЛДС, 1,32 М) в ТГФ с такой скоростью, чтобы температура реакции поддерживалась при -20 С. Добавление ЛДС заняло 30 мин. Смесь выдержали при 15-20 С и реакцию гасили, когда превращение составило 99%. Анализ реакции осуществляли с помощью ВЭЖХ. Приблизительное время удерживания: побочного продукта - гидроксиацетонида = 5,3 мин, этилбензола = 5,6 мин,ацетонида 14 = 6,6 мин, аллилацетонида 15 = 11,8 мин, эпи-15 = 13,3 мин. За время реакции 1 ч превращение составляло 99,5%. Реакцию гасили путем добавления раствора лимонной кислоты (35,7 г, 0,186 моль) в 186 мл ТГФ. После добавления лимонной кислоты смесь выдерживали при 15 С в течение 30 мин. Смесь концентрировали при пониженном давлении Затем вытесняли растворитель, используя всего 2,7 л изопропилацетата (ИПАЦ), при этом непрерывно поддерживали пониженное давление при перегонке. Замену растворителя остановили, когда по данным 1H ЯМР (смотри аналитическое сообщение по методу ГХ) оставалось 1 моль.% ТГФ. Максимальная температура в процессе перегонки не должна превышать 35 С. Неочищенную смесь в ИПАЦ промыли 1,05 л дистиллированной воды; 1,18 л 0,3 М серной кислоты и 1,18 л 6%-ного водного раствора бикарбоната натрия. Объем органической фазы после этих промывок составлял 1,86 л. рН смеси после этих трех водных промывок была 6,5, 1,3 и 8,5, соответственно. При ВЭЖХ анализе смеси на этом этапе установили,что выход соединения 15 составлял 93-94%. По данным ВЭЖХ (условия те же, что и выше) соотношение желаемого 15:эпи-15 было равно 96:4. С помощью ГХ анализа на этом этапе установлено, что побочный продукт гексаметилдисилазан полностью удалялся при такой обработке. Пример 25. 24 теме может быть достигнуто разделение компонентов. Перемешивание проводили с перерывами и слои разделили. К органической фазе добавляли водный раствор сульфита натрия (80 г,0,635 моль в 400 мл) в течение более 10-15 мин. После добавления сульфита натрия температура смеси поднялась до 26-29 С. Смесь перемешивали в течение 40 мин при 25 С. После промывки сульфитом раствор в основном обесцветился. Слои разделили; величина KF органической фазы на этой стадии была равна 25 г/л. Объем органической фазы был равен 1,97 л. По результатам количественного анализа смеси с помощью ВЭЖХ (та же система, что и выше) полный выход иодгидрина 11 в тесте составил на этой стадии 86% (с поправкой на совместную элюцию диастереомеров). Пример 26. К раствору аллиламида 15 в ИПАЦ из предыдущей стадии при 25 С добавили раствор 36,6 г бикарбоната натрия в 1,03 л дистиллированной воды и двухфазную смесь охладили до 5 С. Добавили твердый N-хлорсукцинимид(141,2 г, 1,06 моль). После добавления NCS выделение тепла не происходило. К этой смеси добавили водный раствор йодистого натрия(158,6 г, 106 моль), при этом температуру реакционной смеси поддерживали при 6-11 С. Добавление заняло 30 мин, и смесь потемнела. Смесь подогрели до 25 С и выдерживали при интенсивном перемешивании. Ход реакции контролировали с помощью ВЭЖХ: та же система,что и выше, приблизительно время удерживания: иодгидринов 16, эпи-16, бис-эпи-16 = 8,1 мин; аллиламид 15 = 11,8 мин. Результаты анализа смеси с помощью ВЭЖХ по истечении 2,25 ч показали, что превращение составляло 99,5%. Приблизительно отношение диастереомеров 16:эпи-16:бис-эпи-16 в неочищенной смеси равно 94:2:4 на этой стадии, когда в этой сис 25 мас.% в МеОН Раствор иодгидрина 16 концентрировали в вакууме (28 мм рт.ст.) до азеотропически сухой смеси. Всего было собрано 700 мл дистиллята,при этом температура смеси поддерживалась при 22-28 С. Дистиллят вытесняли 500 мл ИПАЦ (KF = 275 мг/л). Раствор охладили до 26 С и в течение более 10 мин добавляли 25% раствораNaOMe/MeOH (168,1 г). После добавления метилата натрия температура упала до 24 С. Смесь стала темнее, и быстро образовалось смолистое твердое вещество, которое снова растворилось. Смесь выдерживают 1 ч при 25 С. Анализ реакции был выполнен с помощью ВЭЖХ (условия такие же, как выше); приблизительное время удерживания: эпоксид эпи-2 = 6,5 мин, эпоксид 2, бис-эпи-2 = 7,1 мин, иодгидрин 16 = 8,1 мин. По результатам ВЭЖХ анализа превращение иодгидрина в эпоксид равно 99%. По истечении 40 мин добавили 4,1 г раствора метоксид натрия/метанол. По результатам ВЭЖХ анализа через 20 мин превращение составило 99,5%. Реакцию гасили путем добавления 366 мл воды при 25 С, затем быстро перемешали (10 мин) и слои разделили. В дальнейшем было обнаружено, что длительное выдерживание реакции и перемешивание/осаждение в промывной воде приводит в этих условиях эксперимента к значительной обратной реакции с 25 образованием иодгидрина. Эта проблема является особенно острой при промывании водой. Для разрешения этой проблемы реакцию проводили при 15 С. Когда превращение достигло величины 99% (через 1 ч после добавленияNaOMe), смесь разбавили ИПАЦ (40% объема смеси) и сначала промыли увеличенным объемом воды (732 мл) при 20 С. Промывки при более низких температурах и при более высоких концентрациях смесей могли привести к преждевременному осаждению соединения 2 во время промывок. Время перемешивания/осаждения свели к минимуму (10 мин/30 мин, соответственно). Таким способом обратную реакцию можно было снизить до величины 1%. Неочищенные смеси,содержащие эпоксид 2/иодгидрин 16 (97:3), брали для выделения продукта - эпоксида, содержащего 0,6% иодгидрина. Эпоксидный продукт, содержащий такой уровень иодгидрина, выделялся без трудностей. Органическую фазу промывали 3%-ным водным раствором сульфата натрия (2 х 750 мл). После промывки объем органической фазы составлял 1,98 л. рН трех порций промывающей воды был равен 10,7; 9,4 и 8,6 соответственно. По результатам ВЭЖХ анализа суммарный выход эпоксида 2 на этой стадии составлял 86%(с поправкой 4% на совместную элюцию бисэпи-2). Раствор эпоксида в ИПАЦ концентрировали при пониженном давлении (28 мм рт.ст.) до объема около 600 мл, при этом температура раствора поддерживалась при 15-22 С. Раствор вытесняли н-РrОН путем добавления 750 мл нРrОН в процессе концентрирования в вакууме до объема приблизительно 500 мл, поддерживая температуру раствора 30 С. При температуре 35 С в процессе концентрирования/замены растворителя из эпоксида 2 может образоваться н-пропиловый эфир в качестве побочного продукта. Результаты анализа состава растворителя с помощью 1 Н ЯМР указывают на наличие остатка ИПАЦ в количестве 1 моль.%. Вязкую суспензию охладили до -10 С более, чем за один час, и выдерживали в течение 45 мин. Твердые вещества собрали фильтрованием и промыли 125 мл холодного н-РrОН. Продукт сушили в вакуумной печи при 25 С. Получили 188,5 г эпоксида 2 (98,9 А%, 97,6 маc.%, 0,8 маc.% эпи 2, 79,3% суммарного выхода из 14). В нормальной фазе ВЭЖХ (смотри инструкцию по применению этой процедуры для анализа) не обнаружено присутствие бис-эпи-2 в выделенных твердых веществах. Пример 27. Получение предпоследнего соединения 6. 23% NaOH 740 мл Изопропилацетат 4,0 л Вода 700 мл с поправкой на чистоту, мас.% Твердый 2(S)-трет-бутилкарбоксамид-4 трет-бутоксикарбонилпиперазин 4 (159 г, 557 ммоль) и эпоксид 2 (200 г, 530 моль) добавили в двухлитровую 3-х горлую колбу, снабженную механической мешалкой, обратным холодильником, нагревательным кожухом, термопарой в тефлоновой капсуле и трубкой для ввода азота. Добавили метанол (756 мл) и полученную взвесь нагрели до температуры кипения. Через 40 мин образовался гомогенный раствор. Температура этого раствора во время кипения была равна 64-65 С. Ход реакции контролировали ВЭЖХ анализом: 60:40, ацетонитрил/10 мМ(КН 2 РО 4/К 2 НРО 4), время удерживания: Время удерИдентифицированное веживания,щество мин 4,8 Пиперазин 4 6, 6 Простой метиловый эфир 16 8,2 Эпоксид эпи-2 8,9 Эпоксид 2 15,2 Соединенный продукт 5 27 Смесь выдерживали при кипении до тех пор, пока по результатам ВЭЖХ анализа содержание эпоксида 2 не достигло 1,2-1,5 А%. Соединенный продукт на этой стадии содержался в количестве около 94-95 А%. Метилоэфир 16 присутствовал в количестве 1,0-1,5 А%. Обычное время кипячения для достижения этих превращений было равно 24-26 ч. Смесь охладили до 5 С и безводный газ НС 1 194 г, 5,32 моль, 10 экв. барботировали прямо в метанольный раствор в атмосфере азота, поддерживая температуру 5-8 С в течение более 2-3 ч. После окончания добавления смесь выдерживали при 5-8 С в течение 1-3 ч. На этой стадии наблюдалось выделение газа (двуокись углерода и изобутилен). Ход реакции контролировали ВЭЖХ анализом: те же условия, как и раньше. Приблизительное время удерживания: Время удерживания, мин 6,0 7,0 11,9 15,1 16,5 25,0 Идентифицированное вещество Бок промежуточный продукт 17 Цис-аминоинданол 11 Предпоследнее соединение 6 Продукт присоединения 5 Лектон 18 Промежуточный ацетонид 19 Смесь выдерживали при 5-8 С до тех пор,пока количество промежуточного продукта Бок 28 17 не достигало 0,5 А% по данным ВЭЖХ анализа. На этой стадии предпоследнее соединение 6 содержалось в количестве около 92-93 А%, 111,0 А% и 180,6 А% по результатам ВЭЖХ анализа. Снятие защиты завершалось через 4 ч при 5 С. Охлаждение и остановка реакции сразу же после завершения снятия защиты ограничивает распад 6 в 11 и 18 в условиях гидролиза. Смесь охлаждали до -10 - 15 С. Затем смесь медленно внесли в пятилитровую колбу,снабженную механической мешалкой, содержащую охлажденную перемешанную смесь деионизированной (ДИ) воды (700 мл) и метанола (300 мл) при 0-2 С; рН смеси при остановке реакции поддерживали равным 8,5-9,0 путем добавления 23 маc.% водного раствора NaOH(что вызвало высокоэкзотермическую реакцию),при этом температуру поддерживали в пределах 10-20 С. Конечная рН смеси была равна 9,0-9,5. Смесь экстрагировали изопропилацетатом(3,0 л). Смесь перемешали и слои разделили. Истощенную водную фазу повторно экстрагировали изопропилацетатом (1,0 л). Выход соединения 6 в изопропилацетате по результатам ВЭЖХ анализа составлял на этой стадии 94%. Объединенную органическую фазу (0,5 л) концентрировали при пониженном давлении (24-25 мм рт.ст.) до объема около 1,12 л при температуре смеси 30-40 С. Температура резервуара в процессе замены растворителя могла подниматься до 40 С, что не влияет на выход или разложение. Затем это неочищенное соединение 6 непосредственно использовали на следующей стадии для получения соединения J. Пример 28. Получение моногидрата. 29 Предпоследнее соединение 6 Бикарбонат калия Вода Пиколилхлорид Изопропилацетат Раствор предпоследнего соединения 6 в изопропилацетате (4,96 л, 52,5 г/л предпоследнего соединения) концентрировали при пониженном давлении до объема 1,18 л (260 г, 499 ммоль). Температуру раствора поддерживали в пределах 35-44 С, при этом давление в вакууме поддерживалось при 25 мм рт.ст. Соединение метанола составляло 1,0 об.%. Полученную суспензию обработали водным раствором бикарбоната калия (152 г в 630 мл воды, 1,59 моль,3 экв.) и нагрели до 60 С. Затем в течение более 4 ч добавляли водный раствор пиколилхлорида (93,8 г в 94 мл воды, 572 ммоль, 1,14 экв.). В смесь ввели затравку моногидрата J после загрузки 75% всего загружаемого пиколилхлорида. Температура смеси находилась в пределах 60-65 С. По окончании добавления взвесь смеси выдержали в течение 20 ч при 60-65 С. По результатам ВЭЖХ анализа реакция завершилась,когда количество предпоследнего соединения оставалось 1,0 А%. Соединение пиколилхлорида составляло 0,5-0,8 А%. Смесь затем разбавили 2,5 л изопропилацетата и 1,34 л воды и нагрели до 78 С. Слои разделили и органическую фазу промыли горячей водой (3 х 1,34 л) при 78 С. При отмывке горячей водой удалялся бис-алкилированный J и его уровень уменьшился до величины 0,1 А% по результатам ВЭЖХ анализа. Органическую фазу медленно охладили до 75 С и в нее внесли затравку моногидрата J (8,0 г) и затем охлаждали до 4 С более 2 ч. Смесь профильтровали, чтобы собрать продукт, и влажный слежавшийся материал промыли холодным изопропилацетатом (2 х 335 мл). Влажный слежавшийся материал высушили в вакууме (28 мм рт.ст., 22 С). Получили 273 г моногидрата J с выходом 79% относительно эпоксида. Вышеприведенное описание способов показывает принципы данного изобретения, а примеры приведены с целью иллюстрации, но следует иметь в виду, что практическое применение этого изобретения охватывает все обычные в таких случаях изменения, адаптирование и модификации, как это следует из совокупности приводимой формулы изобретения и их эквивалентов. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения ингибитора протеазы ВИЧ структурной формулы(а) нагревание, по крайней мере, в течение одного часа одного эквивалента (S)-4-(3 пиколил)-2-трет-бутилкарбоксамидпиперазина с приблизительно одним эквивалентом соединения в интервале температур между около 25 и около 150 С, при этом указанная смесь необязательно содержит приемлемый растворитель;(в) нейтрализация кислоты. 2. Способ по п.1, отличающийся тем, что нагревание осуществляют в интервале температур от около 50 до около 120 С. 3. Способ по п.1, отличающийся тем, что нагревание осуществляют в интервале температур от около 65 до около 85 С. 4. Способ по п.1, отличающийся тем, что приемлемым растворителем является сложный эфир, спирт, углеводород, простой эфир или формамид или их смесь. 5. Способ по п.1, отличающийся тем, что приемлемым растворителем является спирт. 6. Способ по п.1, отличающийся тем, что приемлемым растворителем является метанол или изопропанол. 7. Способ по п.1, отличающийся тем, что снятие защиты (б) осуществляют с помощью газообразного НСl. 8. Способ получения ингибитора протеазы ВИЧ по п.1, включающий стадии:(а) нагревание, по крайней мере, в течение одного часа смеси одного эквивалента (S)-4-(3 пиколил)-2-трет-бутилкарбоксамидпиперазина в интервале температур между от около 65 до около 85 С, при этом указанная смесь необязательно содержит в качестве растворителя метанол или изопропанол или их смесь;(б) охлаждение смеси до около 0 С;(в) удаление защиты полученного соединения 3 с помощью газообразного НСl; Таблица 1 Материалы Оксид индена Ацетонитрил Вода Конц. Н 2SO4 5 н КОН Грамм или мл 1 мл 10 мл 2,15 мл 0,92 мл 3,0 мл 15 мл влажной смолы Материалы 1,2-индандиол Ацетонитрил Вода Серная кислота 5 н КОН Граммы или мл 300 мг 2,5 мл 0,04 мл 0,22 мл 1,6 мл 10 мл 30 мл Моногидрат карбоната натрия Диэтоксиметан (ДЭМ) 3-Фенилпропионилхлорид (13) Метансульфоновая кислота (МСК) 2-Метоксипропен (95% по ГХ) 5% Водный NаНСО 3 Вода Ацетонид (14) (99,1 мас,%) Аллилбромид Гексометилдисилазид метил(ЛДС) (FMC 9404) Лимонная кислота ТГФ, высушенный над ситами Вода 0,3 М Н 2SО 4 6% NaHCO3 ИПАЦ
МПК / Метки
МПК: A61K 31/495, C07D 401/06
Метки: способ, протеазы, вич, ингибитора, из(2-s)-4-пиколил-2-пиперазин-трет-бутилкарбоксамида, получения
Код ссылки
<a href="https://eas.patents.su/18-178-sposob-polucheniya-ingibitora-proteazy-vich-iz2-s-4-pikolil-2-piperazin-tret-butilkarboksamida.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения ингибитора протеазы вич из(2-s)-4-пиколил-2-пиперазин-трет-бутилкарбоксамида</a>
Хелатообразователь железа в качестве ингибитора процесса окисления, опосредованного железом

Номер патента: 176
Опубликовано: 24.12.1998
Авторы: Гибсон Бредфорд В., Рив Джозеф, Хорвиц Маркус А., Хорвиц Лоренс
МПК: A61K 31/55
Метки: железом, качестве, процесса, хелатообразователь, ингибитора, опосредованного, железа, окисления
Формула / Реферат:
1. Композиция для защиты живой ткани млекопитающего от разрушения, вызванного воздействием свободных гидроксильных радикалов, образовавшихся вслед за восстановлением потока жидкости к органу после ограничения поступления к нему кровотока, включающая дезферриэкзохелин и физиологически приемлемый носитель, отличающаяся тем, что в качестве дезферриэкзохелина она содержит эффективное количество, по меньшей мере, одного дезферриэкзохелина формулы ...
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Ансель Жан-Эрик, Бьенейм Юг, Мейллян Пьер
МПК: B01J 31/24, C07C 39/19, A61K 31/355...
Метки: фенолов, способ, витамина, cпособ, замещенных, получения, использованием
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Мезопористый алюмогель и способ его получения

Номер патента: 16
Опубликовано: 30.12.1997
Авторы: Беллусси Джузеппе, Перателло Стефано, Миллини Роберто, Калемма Винченцо
МПК: B01J 21/04, C04B 35/10, C01F 7/02...
Метки: получения, способ, алюмогель, мезопористый
Формула / Реферат:
1. Мезопористый гель, содержащий матрицу оксида алюминия, в которой могут быть гомогенно диспергированы один или более оксидов, выбранных из группы, включающей диоксид кремния, оксид бора, оксид фосфора, оксид металла группы VIII и/или VIB общей формулы МОx, при следующих молярных соотношениях между указанными оксидами и оксидом алюминия: SiO2/Al2O3 = 0-3,0 B2O3/Al2O3 = 0-4,0 P2O5/Al2O3 = 0-0,2 МОx/Аl2O3 = 0-0,2, и имеющий удельную...
Трициклические соединения, способ их получения, способы получения оптически активных или рацемических производных колхицина и тиохолкицина с использованием трициклических соединений и промежуточныепродукты синтеза

Номер патента: 93
Опубликовано: 25.06.1998
Авторы: Тороманофф Эдмон, Шаппер Бернадетт, Миддендорп Мишель, Брион Франсис, Мазюри Алан, Мари Кристиан, Диолез Кристиан, Пронин Дидье
МПК: C07D 317/44, C07C 43/21
Метки: способ, трициклические, получения, активных, колхицина, соединения, соединений, оптически, тиохолкицина, рацемических, производных, промежуточныепродукты, трициклических, способы, синтеза, использованием
Формула / Реферат:
1. Трициклические соединения общей формулы I в которой либо а) оба R1 и R2 представляют собой алкильную группу, a R3 представляет собой атом водорода или группу A-SO2-, либо б) оба R2 и R3 представляют собой атом водорода или алкил, a R1 представляет собой группу A-SO2-, либо в) все три: R1, R2 и R3 представляют собой атом водорода или все три представляют собой алкил, либо г) R1 представляет собой группу А-SO2- или атом водорода, a...
Способ получения микромезопористого геля

Номер патента: 13
Опубликовано: 30.12.1997
Авторы: Миллини Роберто, Беллусси Джузеппе, Перего Карло, Перего Джованни, Паццукони Джаннино, Басси Джанлука
МПК: C01B 33/46, B01J 20/18
Метки: получения, микромезопористого, геля, способ
Формула / Реферат:
1. Способ получения микромезопористого геля, содержащего силикагелевую матрицу с однородным распределением пор, в которой могут быть гомогенно распределены один или более оксидов металлов, выбранных из переходных металлов или из металлов групп IIIA, IVA и VA, включающий: а) проведение гидролиза и гелеобразования тетраалкилортосиликата, взятого в чистом виде или в спиртовом растворе, при температуре 20-80°С с водным раствором гидроксида...
Предыдущий патент: Способ нанесения на съедобный твердый носитель метилового эфира alpha-l-аспартил-l-фенилаланина
Следующий патент: Производные пиразола
Случайный патент: Способ получения 3,4,5-тризамещённых 1-арилпиразолов