Очищенная ксантозин-n7-метилтрансфераза из растения кофе, кодирующие ее нуклеиновые кислоты, трансгенные клетки растения кофе, трансгенные растения кофе и кофейные зерна, не содержащие кофеина, и способы уменьшения синтеза кофеина в клетках растения кофе
Номер патента: 3835
Опубликовано: 30.10.2003
Авторы: Стайлис Джон И., Неупане Каби Радж, Моисиади Истефо















Формула / Реферат
1. Очищенная ксантозин-N7-метилтрансфераза из растения кофе Coffea arabica с выведенной аминокислотной последовательностью


содержащая триптические фрагменты, имеющие аминокислотные последовательности
(i) Ile Asn Tyr Ala Ser Gly Ala Ser Gly Ile Leu Asp Gln - Gly, или
(ii) Ile Asn Tyr Ala Ser Gly Ala Ser Gly Ile Leu Asp Gln Thr, или
(iii) Gln Tyr Val Pro Cys Tyr Phe (Thr/Asp) Phe Ile Asp (Asp) (Gln) Asp.
2. Выделенная последовательность кДНК, кодирующая ксантозин-N7-метилтрансферазу из растения кофе Coffea arabica, охарактеризованную в п.1, имеющая следующую структуру:


а также варианты указанной кДНК, полученные на основе вырожденности генетического кода.
3. Способ получения кофейных зерен, не содержащих кофеина, включающий
трансформирование растений кофе последовательностью ДНК, которая является антисмысловой по отношению к последовательности кДНК или ее вариантам, полученным на основе вырожденности генетического кода, охарактеризованным в п.2; и
получение плодов от трансформированных растений кофе.
4. Растение кофе, трансформированное последовательностью кДНК по п.2, кодирующей РНК, которая является антисмысловой по отношению к последовательности мРНК, кодирующей ксантозин-N7-метилтрансферазу по п.1, причем РНК имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозин-N7-метилтрансферазы.
5. Растение кофе, трансформированное последовательностью кДНК по п.2, кодирующей РНК, которая является смысловой мРНК, кодирующей ксантозин-N7-метилтрансферазу по п.1, причем РНК имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозин-N7-метилтрансферазы.
6. Растение кофе по п.4, где последовательность кДНК связана с промотором транскрипции в антисмысловой ориентации.
7. Растение кофе по п.5, где последовательность кДНК связана с промотором транскрипции в смысловой ориентации.
8. Кофейные зерна от растений кофе по любому из пп.4-7.
9. Трансформирующий вектор, включающий промотор транскрипции, связанный с последовательностью кДНК, охарактеризованной в п.2.
10. Трансформирующий вектор по п.9, где последовательность кДНК связана с промотором транскрипции в смысловой ориентации.
11. Трансформирующий вектор по п.9, где последовательность кДНК связана с промотором транскрипции в антисмысловой ориентации.
12. Трансформирующий вектор по п.9, где промотором является 35S промотор вируса мозаики цветной капусты.
13. Трансформирующий вектор по п.9, где вектором является модифицированная плазмида pBI-121.
14. Клетка растения кофе, трансформированная последовательностью кДНК по п.2, кодирующей РНК, которая является антисмысловой по отношению к последовательности мРНК, кодирующей ксантозин-N7-метилтрансферазу по п.1, причем РНК имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозин-N7-метилтрансферазы.
15. Клетка растения кофе, трансформированная последовательностью кДНК по п.2, кодирующей РНК, которая является смысловой мРНК, кодирующей ксантозин-N7-метилтрансферазу по п.1, причем РНК имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозин-N7-метилтрансферазы.
16. Клетка растения кофе по п.14, где последовательность кДНК связана с промотором транскрипции в смысловой ориентации.
17. Клетка растения кофе по п.15, где последовательность кДНК связана с промотором транскрипции в антисмысловой ориентации.
18. Трансформированная клетка растения кофе, полученная путем введения трансформирующего вектора по п.9 в клетку растения кофе.
19. Трансформированная клетка растения кофе по п.18, где последовательность кДНК связана с промотором транскрипции в смысловой ориентации.
20. Трансформированная клетка растения кофе по п.18, где последовательность кДНК связана с промотором транскрипции в антисмысловой ориентации.
21. Трансформированная клетка растения кофе по любому из пп.14-20, где клетка синтезирует меньшее количество кофеина по сравнению с клеткой растения кофе, не трансформированной последовательностью кДНК.
22. Растение кофе, выращенное из трансформированных клеток растения кофе по любому из пп. с 14 по 21.
23. Кофейные зерна, полученные от растения кофе по п.22.
24. Способ уменьшения синтеза кофеина клетками растения кофе, включающий следующие стадии:
получение трансформирующего вектора, содержащего последовательность кДНК по п.2, кодирующую РНК, которая имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозин-N7-метилтрансферазы, охарактеризованной в п.1, причем последовательность кДНК связана с промотором транскрипции в антисмысловой ориентации; и
введение трансформирующего вектора в клетку растения кофе, в результате чего последовательность кДНК вводится в геном клетки растения кофе для получения трансформированной клетки, причем трансформированные клетки синтезируют меньшее количество кофеина по сравнению с клетками растения кофе, не трансформированными последовательностью кДНК.
25. Способ уменьшения синтеза кофеина клетками растения кофе, включающий следующие стадии:
получение трансформирующего вектора, содержащего последовательность кДНК по п.2, кодирующую РНК, которая имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозин-N7-метилтрансферазы, охарактеризованной в п.1, причем последовательность кДНК связана с промотором транскрипции в смысловой ориентации; и
введение трансформирующего вектора в клетку растения кофе, в результате чего последовательность кДНК вводится в геном клетки растения кофе для получения трансформированной клетки, причем трансформированные клетки синтезируют меньшее количество кофеина по сравнению с клетками растения кофе, не трансформированными последовательностью кДНК.
Текст
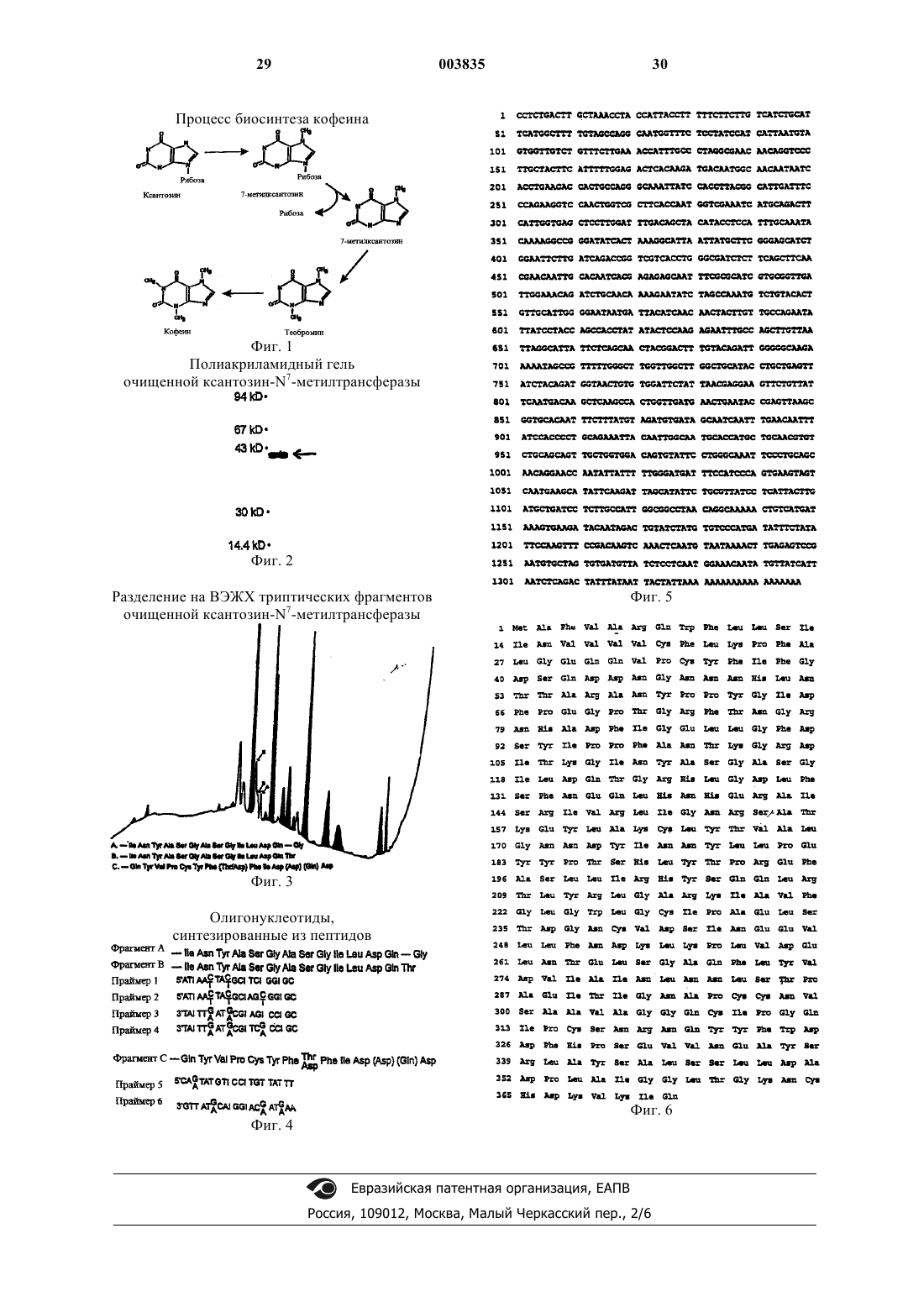
1 Данная заявка относится к очищенной ксантозин-N7-метилтрансферазе,последовательностям рекомбинантной ДНК, клеткам,трансформированным с их помощью, и способам получения напитков и продуктов питания,не содержащих кофеина. Более подробно, данная заявка относится к очищенной ксантозинN7-метилтрансферазе, которая является одним из ключевых ферментов, участвующих в синтезе кофеина в растении кофе, и последовательностям рекомбинантной ДНК, подавляющим экспрессию кофеина в растениях кофе и их плодах. Настоящее изобретение дает стабильные линии растений кофе, не содержащих кофеина, плоды которых после обжаривания и перемалывания могут использоваться для приготовления кофе,не содержащего кофеина. Предполагается, что данное изобретение можно использовать для подавления синтеза кофеина в растениях чая(Camellia sinensis) и коле (Cola acuminata), а также родственных алкалоидов в шоколадном дереве (Theobroma cacao). Предпосылки изобретения Кофе готовят из обжаренных и перемолотых зерен растений рода Кофе (Coffea), обычно из вида С. arabica. Растения кофе производят алкалоид кофеин, который присутствует в их высушенных плодах, кофейных зернах. Т.к. многие любители кофе предпочитают кофе без кофеина, были разработаны многочисленные способы удаления кофеина из кофейных зерен. Все эти способы приводят к удалению из зерен кроме кофеина и других веществ, что приводит к ухудшению вкуса кофе, приготовленного из обработанных такими способами кофейных зерен. Хотя известно несколько сортов растений кофе, не содержащих кофеина, и родственных им видов, встречающихся в природе (Mascarocoffea spp. и Coffea bengalensis), они не имеют коммерческого значения. (Charrier и Berthaud,"Variation Of Caffeine Content In The Coffea Genus", Cafe' Cacao The', 14:251-264 (1975. Поэтому существует необходимость в способе получения кофейных зерен, не содержащих кофеина, результатом которого является удаление кофеина, а не других веществ. Кофеин - это природный пуриновый алкалоид, синтезируемый в числе других растениями кофе и чая. Считается, что синтез кофеина защищает растения от насекомых. Растения кофе синтезируют кофеин из нуклеозида ксантозина в ходе четырех последовательных реакций,показанных на фиг. 1. См. Suzuki, Т., Ashihara,H. и Waller, G.R., Phytochemistry 31:2575 (1992). Первая стадия синтеза - это метилирование нуклеозида ксантозина S-аденозилметионином,которое катализируется ферментом ксантозинN7-метилтрансферазой (ХМТ). Продукт реакции 7-метилксантозин гидролизуется до 7 метилксантина (при этом удаляется рибоза) и подвергается дальнейшему метилированию с получением теобромина и кофеина. Можно 2 ожидать, что прервав эту последовательность реакций, можно предотвратить синтез кофеина. Следовательно, стратегия избирательного удаления кофеина из кофейных зерен заключается в предотвращении синтеза специфических ферментов в процессе биосинтеза кофеина. Настоящее изобретение относится к генетическому изменению растений кофе с целью исключить синтез ХМТ. В предпочтительной реализации синтез ХМТ подавляется путем трансформации растений кофе с помощью последовательности ДНК, кодирующей транскрипцию матричной РНК (мРНК), которая является антисмысловой по отношению к мРНК, кодирующей ХМТ. Изобретение можно использовать для получения напитков и продуктов питания, не содержащих кофеина, включая чай, какао и другие напитки и продукты на основе какао. Сущность изобретения Очищенная ксантозин-N7-метилтрансфераза, кодирующие ее последовательности ДНК и рекомбинантные молекулы ДНК для трансформации растений кофе с целью подавления синтеза кофеина. Фермент ксантозин-N7 метилтрансфераза (ХМТ) катализирует первую стадию синтеза кофеина в растениях кофе. Дается основная последовательность ДНК и выведенная аминокислотная последовательность ХМТ. Растения кофе трансформируют с помощью молекулы ДНК, кодирующей РНК, которая является антисмысловой по отношению к мРНК, кодирующей ксантозин -N7-метилтрансферазу. Антисмысловая РНК связывается с мРНК ХМТ, что приводит к инактивации мРНК,кодирующей фермент первой стадии биосинтеза кофеина. В результате этого трансформированные растения кофе становятся не способными синтезировать кофеин, причем на другие аспекты их метаболизма влияния не оказывается. Краткое описание чертежей Фиг. 1 представляет схематичное изображение процесса биосинтеза кофеина в растенииCoffea arabica. Фиг. 2 представляет фотографию окрашенного серебром полиакриламидного геля(ПААГ) после анализа очищенной ксантозинN7-метилтрансферазы с помощью гельэлектрофореза в присутствии додецилсульфата натрия (ДСН). Фиг. 3 представляет хроматограмму элюции триптических фрагментов очищенной ксантозин-N7-метилтрансферазы после разделения с помощью ВЭЖХ. Фиг. 4 представляет олигонуклеотидные праймеры, которые используются для скрининга библиотеки кДНК с целью поиска кДНК, кодирующей ксантозин-N7-метилтрансферазу. Фиг. 5 представляет основную последовательность кДНК, кодирующую кcaнтoзин-N7 мeтилтpaнcфepaзy. 3 Фиг. 6 представляет выведенную аминокислотную последовательность ксантозин-N7 метилтрансферазы. Подробное описание изобретения Для лучшего понимания настоящего изобретения предлагается следующее подробное описание, в котором используются термины: Нуклеотид - Мономерная единица ДНК или РНК, состоящая из углеводного остатка(пентозы), фосфатной группы и азотистого гетероциклического основания. Основание связано с углеводным остатком через гликозидный атом углерода (1'-атом углерода пентозы), и эта комбинация основания и углевода называется нуклеозидом. Основание характеризует нуклеотид. Четыре основания ДНК - это аденин ("А"), гуанин ("G"), цитозин ("С") и тимин ("Т"). Четыре основания РНК - это А, О, С и урацил ("U"). Последовательность ДНК - Линейная цепь нуклеотидов, связанных друг с другом фосфодиэфирными связями между 3'- и 5'-атомами углерода соседних пентоз. Кодон - Последовательность ДНК из трех нуклеотидов (триплет), который кодирует через РНК аминокислоту, сигнал начала трансляции или сигнал окончания трансляции. Например,триплеты нуклеотидов ТТА, TTG, СТТ, СТС,СТА и CTG кодируют аминокислоту лейцин(Leu), TAG, TAA и TGA являются сигналами окончания трансляции, ATG является сигналом начала трансляции и также кодирует аминокислоту метионин (Met). Полипептид - Линейная последовательность аминокислот, связанных между собой пептидными связями между амино- и карбоксильными группами соседних аминокислот. Геном - полная ДНК клетки или вируса. Он включает структурный ген, кодирующий полипептиды вещества, а также сайты промотора, начала и окончания транскрипции и трансляции. Ген - Последовательность ДНК, кодирующая через ее матричную РНК (мРНК) последовательность аминокислот, характерных для специфического полипептида. Транскрипция - Процесс получения мРНК из гена или последовательности ДНК. Трансляция - Процесс получения полипептида из мРНК. Экспрессия - Процесс получения полипептида под воздействием гена или последовательности ДНК. Комбинация транскрипции и трансляции. Плазмида - внехромосомная двухцепочечная последовательность ДНК, включающая интактный "репликон", такой, что плазмида реплицируется в клетке-хозяине. Когда плазмиду помещают в одноклеточный организм, свойства этого организма могут измениться из-за плазмидной ДНК. Например, плазмида, несущая ген устойчивости к тетрациклину (TETR), превращает клетку, ранее чувствительную к тетрацик 003835"трансформантом". Фаг или Бактериофаг - бактериальные вирусы, многие из которых состоят из последовательностей ДНК, заключенных в белковую оболочку ("капсид"). Клонирующий вектор - Плазмида, фаговая ДНК, космида или другая последовательность ДНК, способная реплицироваться в клеткехозяине, характеризующаяся одним или несколькими сайтами узнавания эндонуклеаз, в которых такие последовательности ДНК могут быть разрезаны определенным образом без потери основной биологической функции ДНК,например, репликации, синтеза белков оболочки или потери промоторных или связывающих сайтов, и которые содержат маркер, подходящий для идентификации трансформированных клеток, например, по их устойчивости к тетрациклину или ампициллину. Клонирующий вектор часто называют вектором. Клонирование - Процесс получения популяции организмов или последовательностей ДНК, взятых от одного из таких организмов,или последовательность бесполой репродукции. Молекула рекомбинантной ДНК или гибридная ДНК - молекула, состоящая из фрагментов ДНК из различных геномов, которые соединены конец в конец вне живых клеток и способны поддерживаться в живых клетках. кДНК - Цепь ДНК, комплементарная мРНК, кодирующая специфический полипептид. Хотя стратегия получения кофе, не содержащего кофеина, может быть использована и для других ферментов, участвующих в процессе биосинтеза кофеина в растениях кофе и других растениях, производящих кофеин, в настоящем изобретении подавляют экспрессию первого уникального фермента в процессе биосинтеза кофеина(ХМТ). Хотя роль ХМТ в синтезе кофеина была показана с помощью радиоактивного мечения предшественников, до сих пор этот фермент не был очищен, и не была определена его аминокислотная последовательность. Поэтому настоящее изобретение включает очищенную ХМТ. Кроме того, изобретение включает аминокислотную последовательность триптических фрагментов, выделенных из очищенной ХМТ. Были синтезированы кДНК-зонды на основе фрагментов аминокислотных последовательностей, полученных из образцов очищенного фермента, и часть гена была амплифицирована с помощью ПЦР. ПЦР-продукты использовали для скрининга библиотеки кДНК, синтезированной на мРНК из молодых листьев, для того,чтобы идентифицировать транскрипты, кодирующие ХМТ. Позитивные транскрипты секвенировали, и было получено около 90% гена,кодирующего ХМТ. 5 ДНК, кодирующую ХМТ, вводили в трансформирующий вектор pBI-121, который включал ген устойчивости к канамицину. Успешное включение этого вектора в клетки растений определялось по приобретению ими устойчивости к антибиотикам. Такие конструкции использовали для трансформации соматических эмбрионов растений кофе в тканевой культуре. Затем из трансформированных эмбрионов выращивали новые растения кофе, которые не производили кофеин. Натуральный кофе, не содержащий кофеина, готовили из обжаренных перемолотых плодов этих новых растений. Более конкретно, свежую ткань из молодых листьев растений С. arabica вымачивали и экстрагировали из нее белок. Очищенные на колонке экстракты анализировали на ферментативную активность путем контроля метилирования ксантозина, используя в качестве субстрата меченый С 14 S-аденозилметионин. Продукт реакции идентифицировали как 7 метилксантозин путем сравнения миграции меченого продукта реакции с миграцией 3 метилксантина,7-метилксантина,8 метилксантина, 7-метилксантозина, ксантина и ксантозина в каждой из четырех различных хроматографических систем. Чистоту белковых изолятов определяли с помощью электрофореза в ПААГ в присутствии ДСН и двухмерного гель-электрофореза. Окрашивание серебром гелей после проведения одномерного электрофореза в ПААГ в присутствии ДСН показало присутствие дублета с ферментативной активностью ХМТ и с молекулярным весом 36-37 килодальтон (кДа), как показано на фиг. 2. Затем каждый белок разделяли с помощью изоэлектрического фокусирования. Полученные данные показали наличие изоферментов ХМТ, что могло быть результатом либо посттрансляционной модификации белка, либо присутствия семейства генов, кодирующих ферменты ХМТ. Дублет, наблюдаемый в ПААГ с ДСН, использовали для секвенирования белка. Очищенную ХМТ подвергали частичному расщеплению трипсином для получения фрагментов для дальнейшего анализа; с помощью ВЭЖХ были получены три отдельных пика. Секвенирование проводили в лаборатории структуры белка Калифорнийского университета (Protein StructureLaboratory of the University of California, Davis) с использованием автоматической деградации по Эдману (Edman, P. and Begg, G., Eur. J. Biochem., 1:80). Были получены две уникальные последовательности, которые использовали для конструирования праймеров для синтеза зондов. Из листьев растений кофе эктрагировали РНК. Затем из этой РНК выделяли мРНК, содержащие поли(А+)-последовательности. Из поли(А+)мРНК получали библиотеку кДНК, используя обратную транскриптазу. Двухцепочечные ДНК получали, используя ДНК-полимеразуI, и выделяли осаждением. кДНК фракционировали и встраивали в фаг для амплификации. Проводили скрининг библиотеки кДНК с помощью синтезированного ПЦР зонда, полученного с использованием праймеров на основе последовательности ДНК, выведенной из аминокислотной последовательности очищенной ХМТ. Был идентифицирован клон, продуцирующий кДНК, содержащую все последовательности, кодирующие ХМТ. кДНК, соответствующая гену, кодирующему ХМТ, используется для трансформации эмбрионов растений кофе. Плазмида pBI-121 используется в качестве трансформирующего вектора. Последовательности, соответствующие ДНК, кодирующей ХМТ, вставляют в плазмиду в инвертированной ориентации рядом с 35S промотором вируса мозаики цветной капусты. РНК, транскрибированная с такой ДНК, будет комплементарна мРНК, кодирующей аминокислотную последовательность ХМТ. Полные конструкции амплифицируют в бактериальных клетках-хозяевах. Клетки-хозяева разрушают, и амплифицированный вектор присоединяют к коллоидным частицам золота. Частицы золота с присоединенными векторами внедряют в протопласты растений кофе путем обстрела клеток этими частицами с большой скоростью, как описано в патенте US 5107065. Успешно трансформированные молодые растения идентифицировали по их устойчивости к антибиотикам. Трансформированные растения не производили кофеин. Примеры А. Очистка ксантозин-N7-метилтрансферазы из листьев растения С. arabica L. cv Guatemalan. Ткань молодых листьев длиной менее 5 мм(эквивалентных стадии В 3 (Frischknecht, P.M.,Ulmer-Dufek, J. And Baumann, T.W. (1986) Phytochemistry 25:613), получали с деревьев, выращенных на исследовательской станции (Waimanalo) Гавайского Университета, Оаху, Гавайи. Листья немедленно помещали в жидкий азот и хранили до использования при температуре -70 С. Все необходимые процедуры выполняли при температуре 4 С, если не указано иначе. Ткань листьев (150 г) растирали пестиком в ступке в жидком азоте и еще в замороженном виде помещали в бытовую кофемолку и перемалывали около 30 с с небольшим кусочком сухого льда. Измельченную ткань листьев помещали в стакан, содержащий 1,5 л ледяного 80%-ного ацетона, 5 мМ тиомочевины и 12,5 мМ -меркаптоэтанола. После перемешивания на магнитной мешалке в течение 45 мин ткань отфильтровывали под вакуумом на воронке Бюхнера через фильтровальную бумагу Whatman1. Затем ткань промывали 2,5 л ледяного 80%-ного ацетона, содержащего тиомочевину и 7 на воздухе в течение 20 мин и лиофилизировали в течение 48 ч. Полученный ацетоновый порошок смешивали в миксере с 400 мл экстрагирующего буфера (ЭБ) (0,1 М PIPES [рН 7,0], 0,5 мМ Nа 2 ЭДТА,0,5 мМ Nа 2 ЭГТА, 5% аскорбиновой кислоты, 5 мМ дититреитола [ДТТ], 5 мМ тиомочевины, 12 мМ L-цистеина НСl, 1% полиэтиленгликоля(ПЭГ) 20000, 0,1 мМ фенилметилсульфонилфторида [PMSF] и 20 г поливинилполипирролидона [ПВПП]). Смесь перемешивали в течение 10 мин со средней скоростью, переносили в центрифужные стаканы на 250 мл и центрифугировали при 23000 об./мин в течение 30 мин на центрифуге GSA (Dupont-Sorvall). К 350 мл полученного супернатанта в течение 30 мин при перемешивании порциями добавляли 78,86 г сульфата аммония (СА) до получения 40%-ного насыщенного раствора,причем сосуд помещали в баню со льдом. Затем смесь снова переносили в центрифужные стаканы на 250 мл и центрифугировали в течение 30 мин при 23000 об./мин, как описано выше. 350 мл полученного супернатанта наносили на хроматографическую колонку Macro-Prep с метилгидрофобным взаимодействием (Bio-Rad) объемом 40 мл при скорости потока 2,5 мл/мин. Все фракции проверяли на наличие белка, используя поглощение при 280 нм. Колонку сначала уравновешивали буфером, содержащим 1,7 М СА,20 мМ бис-трис-пропана, рН 6,8, и 5 мМ ДТТ до установления базовой линии около нуля. Затем через колонку пропускали буфер, содержащий 10 мМ Трис, рН 7,0, 5 мМ ДТТ, 1 мМ MgCl2. Первые 15 мл элюата отбрасывали, а оставшийся элюат (200 мл) пропускали самотеком через колонку объемом 100 мл, заполненную аффинным гелем Affi-Gel Blue (100-200 меш, Bio-Rad),с матрицей которого ковалентно связан краситель Cibacron blue F3GA. Гель предварительно уравновешивали буфером, содержащим 10 мМ Трис, рН 7,0, 5 мМ ДТТ, 1 мМ MgCl2. Колонку промывали этим буфером до тех пор, пока базовая линия не устанавливалась около нуля, и связавшиеся белки элюировали буфером, содержащим 10 мМ Трис, рН 7,0, 5 мМ ДТТ и 1,5 мМ хлорида натрия. К 142 мл элюата из колонки с гелем AffiGel Blue прибавляли небольшими порциями 31,8 г СА до концентрации 1,7 М при перемешивании в течение 30 мин, при этом сосуд с элюатом помещали в ледяную баню. Затем смесь центрифугировали в центрифужных стаканах на 250 мл при 23000 об./мин в течение 30 мин, как описано выше, и супернатант наносили на колонку с фенил-сефарозой (FPLC PhenylSepharose column XK 26/20, Pharmacia) при 23 С. Колонку уравновешивали буфером, содержащим 20 мМ бис-трис-пропана, рН 6,8, 5 мМ ДТТ и 1,7 М СА. После установления базовой линии около нуля, из колонки в течение 40 мин элюировали белки в обратном градиенте 8 концентрации СА от 1,7 до 0 М при скорости потока 5 мл/мин, собирая 1-минутные фракции. Элюент с 0 М СА содержал 10 мМ Трис (рН 7,0), 5 мМ ДТТ и 1 мМ MgCl2. Анализ активности собранных фракций показал, что большая часть ферментативной активности ксантозин-N7-метилтрансферазы была сосредоточена во фракциях 49-54. Эти фракции объединяли, и полученное количество объемом 30 мл пропускали самотеком через 6 мл колонку с АТР-агарозой (ATP-agarose (SigmaChemicals A2767 при 4 С. Колонку промывали уравновешивающим буфером, содержащим 10 мМ Трис, рН 7,0, 5 мМ ДТТ и 1 мМ MgCl2. После установления базовой линии колонку промывали 20 мл уравновешивающего буфера, содержащего 100 мкМ ксантозина, а затем еще 40 мл уравновешивающего буфера. Оба элюата объединяли и наносили на колонку Mono-P HR 5/20 FPLS (Pharmacia), уравновешенную буфером, содержащим 25 мМ бис-Трис, рН 6,0, и 9% бетаина при 23 С. После установления базовой линии, колонку промывали 100 мл полибуфера 74 [Polybuffer 74 (10 мл:90 мл Н 2 О, рН 4,0),Pharmacia] и 9% бетаина со скоростью 1 мл/мин. Пробирки коллектора содержали 100 мкл 0,5 М трицина, рН 7,0, и 50 мМ ДТТ для получения конечной концентрации в 1 мл 50 мМ трицина,рН 7,0, и 5 мМ ДТТ во фракциях, собранных за 1 мин. Это эффективно стабилизировало конечную величину рН среды для белков, элюированных при слабо кислом рН с колонки Mono-P. Наибольшую активность ксантозин-N7 метилтрансферазы определили в пробирках,содержащих фракции 15 и 16, после градиентного элюирования с колонки с рН 5,42 и 5,35 соответственно. Важно не замораживать образцы белков на любой стадии очистки, т.к. это отрицательно влияет на активность ксантозинN7-метилтрансферазы. В. Определение ферментативной активности. 100 мкл стандартной аналитической смеси содержало 50 мМ трицина (рН 7,0), 1200 мкМ ксантозина, 5 мМ ДТТ, 7,5 мкМ S-аденозил-L[метил-14 С]-метионина (SAM) (60 мКи/ммоль;DuPont NEN), и 1 мМ Na2 ЭДTA. Реакционную смесь (50 мкл без фермента) выдерживали 10 мин при 25 С, затем инициировали реакцию добавлением 50 мкл раствора фермента и оставляли на 1 ч при 25 С. В конце инкубации отбирали три аликвоты реакционной смеси по 30 мкл и прекращали реакцию путем добавления 8 мкл 0,6 М раствора хлорной кислоты (НСlO4). Аналогичные процедуры выполняли с контрольными пробами (с нулевым временем реакции) для получения истинного значения ферментативной активности. Смесь центрифугировали на микроцентрифуге в течение 5 мин и к 19 мкл супернатанта добавляли 1,0 мкл 33 мМ 7 метилксантозина. Эти смеси наносили на хроматографическую бумагу Whatman1 и хро 9 матографировали в растворе н-бутанолуксусная кислота-вода (n-ВuОН-НОАс-Н 2 О)(4:1:1). Положение 7-метилксантозы определяли по голубой флуоресценции в коротковолновой части УФ-спектра. Этот участок вырезали из хроматограмм и определяли радиоактивность в сцинтилляционном счетчике, используя 3 мл сцинтилляционной жидкости (Scinti-verse scintillation fluid (Fisher Scientific. Точность расчетов составляла 74,7%. Значения фона и неспецифической радиации, определенные для 7 метилксантозина в контрольных опытах, вычитались. С. Идентификация продукта реакции. Сайт метилирования ксантинового кольца идентифицировали путем гидролиза сахара из продукта реакции метилирования ксантозина и разделения в четырех различных хроматографических системах. Продукт из двух 100-мкл реакций, проведенных, как описано выше, и содержащий 6 мкл 33 мМ 7-метилксантозина в качестве носителя, наносили на хроматографическую бумагу Whatman1. Хроматографию проводили в растворе n-BuOH-HOAc-Н 2 О(4:1:1). Участок хроматограммы, соответствующий метилированному ксантозину определяли, как описано выше, разрезали на маленькие кусочки, помещали в стерильную пробирку и выдерживали в 35 мл деионизированной воды при 37 С в течение ночи при встряхивании. Экстракт фильтровали через два слоя ткани,0,22 мкм фильтр и лиофилизировали. Высушенный экстракт ресуспендировали в 1,0 мл деионизированной воды, помещали в стеклянный флакон для гидролиза и лиофилизировали. Затем образец ресуспендировали в 400 мкл 1,0 М НСl и выдерживали в течение 1 ч при 100 С. Гидролизат лиофилизировали, ресуспендировали в 400 мкл 3 мМ 7-метилксантина и снова лиофилизировали. Затем этот гидролизат ресуспендировали в 40 мкл деионизированной воды и 10 мкл подвергали хроматографии в каждой из четырех различных систем. Для сравнения на каждую хроматограмму наносили 1 метилксантин, 3-метилксантин, 7-метилксантин,8-метилксантин, 7-метилксантозин, ксантин и ксантозин. Использовали следующие хроматографические системы: бумажную хроматографию на Whatman1 в n-BuOH-HOAc-H2O(4:1:1) и тонкослойную хроматографию на пластинах С 8 (Whatman KC18F) в системе изоамиловый спирт-вода-ацетонитрил (41:4:5), или этанол-вода (4:1), или тpeт-BuOH-HOAc-H2O(4:1:1). После высушивания на хроматограммы распыляли En3Hance (DuPont NEN), снова высушивали и экспонировали в течение 30 дней с предварительно облученной рентгеновской пленкой (Fuji RXGCU) при -70 С.D. Идентификация белков с помощью гель-электрофореза. Экстракты, полученные, как описано выше, использовали для одномерного (1D) элек 003835 10 трофореза (ПААГ с ДСН) в минигелях (основной гель: 12,5% акриламида, 0,8% метиленбисакриламида; концентрирующий гель: 7,5% акриламида, 0,21% метилен-бис-акриламида),смешивая их с буфером Лэммли для образцов(Laemmli, U.K., Nature 227:680 (1970, и для двухмерного (2D) миниизоэлектрофокусирования (ИЭФ)/электрофореза (ПААГ с ДСН) по модифицированному методу O'Farrell и др.(O'Farrell, P.Z., Goodman. H.M., O'Farrell Р.Н.,Cell 12:1133 (1977. Двухмерный электрофорез проводили, осаждая белки 50 объемами 100% этанола в течение 1 ч и далее растворяя их в буфере для образцов для изоэлектрофокусирования (ИЭФ), содержащем 5% амфолинов (1:1,(об./об.), рН 3-10:рН 5-7, LKB-Pharmacia). Соотношение исходного объема белкового экстракта и ИЭФ-буфера для образцов поддерживали, по крайней мере, 1:2 для того, чтобы оставшиеся составляющие буфера после хроматографии не мешали ИЭФ. Равные количества белковых проб (20 мкг) наносили на щелочной конец предварительно сфокусированных гелей (8,8% акриламида, 1,6% метилен-бисакриламида), содержащих 5% амфолинов, как описано выше. Гели фокусировали в течение 10000 В-ч и еще 2 ч при 1000 В. После фокусирования гели разрезали на участки по 5 мм и инкубировали в течение 24 ч в 0,5 мл 100 мМ CaCl2, затем определяли рН сегментов. На основе этого анализа рассчитывали градиент рН ИЭФ-геля, который находился в диапазоне от 4,4 до 6,0. Гели из трубок, приготовленные для гельэлектрофореза с ДСН, промывали сначала водой, а затем 3 раза (каждый раз по 10 мин) горячим буфером Лэммли для образцов. Затем их помещали в верхнюю часть гелей (ПААГ с ДСН) (основной гель: 12,5% акриламида, 0,8% метилен-бисакриламида; концентрирующий гель: 7,5% акриламида, 0,21% метиленбисакриламида), удерживали на месте с помощью 3%-ной агарозы в буфере Лэммли для образцов. Белки визуализировали, окрашивая серебром. В 1D-гелях фракция 16 Моnо-Р, имеющая наибольшую ферментативную активность,показала после окрашивания серебром только присутствие дуплета (фиг. 2). Был определен молекулярный вес этих белков - приблизительно 37,6 и 36,1 кДа. В 2D-гелях каждый белок разделился на два пятна. Изоэлектрическая точка (IP) более кислого белка имела среднее значение по нескольким гелям 5,2, а более основного - 5,3. Их молекулярные веса, однако, теперь имели среднее значение 43,5 кДа, с высшими и низшими пептидами, связанными друг с другом. Следовательно, имеется разница между 1D- и 2D-гелями при определении молекулярного веса. Одинаковая миграция всех этих четырех пептидов в колонках с Mono-P, 1D- и 2D-гелях показывает, что они являются изоферментами,которые могут подвергаться посттрансляционной модификации. С другой стороны, они могут 11 быть продуктами семейства генов, имеющих небольшие различия в структуре, приводящие к существованию различных изоферментов. Е. Секвенирование белков. Оценка количества белка методом ЛоуриRandall, R.J., J. Biol. Chem. 193:265 (1951 для фракции 16 Mono-P показала, что во фракциях объемом 1 мл содержится всего 100 мкг белка. Обычно при таких низких концентрациях белка значения по Лоури могут отличаться от действительной концентрации. Чтобы избежать этого,авторы решили использовать значительную часть этих фракций для секвенирования белка. 900 мкл фракции 16 Mono-P, содержащей 90 мкг белка, помещали в стерильную микроцентрифужную пробирку на 1,5 мл и добавляли 216 мкл 100%-ной трихлоруксусной кислоты (ТХУ). После перемешивания пробирку выдерживали в течение ночи на льду, затем центрифугировали при 14000 об./мин в микроцентрифуге в течение 30 мин при 4 С. Супернатант отсасывали и осадок дважды промывали 1 мл 75%-ного этанола,причем за каждой промывкой следовало центрифугирование. Затем осадок высушивали в вакуумной центрифуге при вращении в течение 1 мин. К осадку прибавляли 20 мкл 2 х буфера Лэммли для образцов, кипятили на водяной бане в течение 5 мин и центрифугировали в течение 1 мин. Затем пробирку охлаждали до 23 С,и все количество наносили на одну дорожку 12,5%-ного 1D-геля. После окончания электрофореза белки визуализировали путем окрашивания 0,1% водным раствором Coomassie R-250,содержащим 50% этанола и 10% уксусной кислоты (вес:об.:об.), и затем отмывали от краски. Дублет с 37,6 и 36,1 кДа, который наблюдали в гелях, окрашенных серебром, также был виден и в гелях, окрашенных Coomassie R-250. Участок геля, содержащий этот дуплет вырезали, и использовали для секвенирования белка методом автоматической деградации по Эдману. Секвенирование белка проводили в лаборатории структуры белков Калифорнийского университета (Protein Structure Laboratory of theUniversity of California, Davis) стандартным методом. Кусочек геля, содержащий дуплет, промывали 4 раза 15 мл воды при осторожном встряхивании в течение 15 мин для удаления уксусной кислоты и ДСН, оставшихся от предыдущих операций. Затем кусочек геля разрезали лезвием бритвы на квадратики площадью 2 кв.мм и помещали в центрифужную пробирку на 1,5 мл. Кусочки геля дегидратировали в вакуумной центрифуге в течение 2 ч до тех пор,пока они не прилипали к пробирке. Затем добавляли 30 мкл гидратирующего буфера (0,1 М Трис-HCl, рН 9,0, 0,05% ДСН) и рН доводили до 8,0, контролируя путем нанесения 0,5 мкл раствора на рН бумагу. Добавляли гидролазу Lys-C(0,2 мкг) из Achomobacter luticus (Wako) и такое количество гидратирующего буфера, чтобы 12 полностью гидратировать кусочки геля и оставить некоторое количество буфера вне геля. Смесь инкубировали в течение ночи при 30 С,после этого супернатант переносили в чистую,стерильную центрифужную пробирку и хранили. К оставшимся кусочкам геля добавляли воду так, чтобы она их покрыла, и выдерживали при 30 С в течение 2 ч. Супернатант переносили в пробирку для хранения, как описано выше. Подобную промывку повторяли еще один раз и объединяли с супернатантами, полученными после первых двух промывок. Затем кусочки геля заливали раствором, содержащим 0,1% трифторуксусной кислоты (ТФУ) в 80%-ном ацетонитриле, и инкубировали 1 ч при 30 С. Супернатант собирали и добавляли к ранее полученным супернатантам. Последнюю промывку повторяли еще один раз, и объединенные супернатанты высушивали в вакуумной центрифуге. Высушенные продукты триптического расщепления растворяли в 25 мкл 6 М гуанидин-HCl, 0,4 М Трис (рН 8,2), рН полученного раствора определяли, нанося 0,5 мкл на рН бумагу. Добавляли 1 мкл 450 мМ ДТТ и гидролизат инкубировали 45 мин при 50 С. После охлаждения до комнатной температуры добавляли 2 мкл 500 мМ йодацетамида и инкубировали еще 15 мин при 23 С. Затем добавляли 72 мкл воды для достижения конечной концентрации 1,5 М гуанидина и 0,1 М Трис. Затем пробы центрифугировали 5 мин при 14000 об./мин в микроцентрифуге и супернатант осторожно переносили в новую центрифужную пробирку. К осадку прибавляли 25 мкл 0,1% ТФУ и интенсивно перемешивали. Затем центрифугировали,как описано выше, и супернатант объединяли с супернатантом, полученным на предыдущей стадии. Фрагменты, полученные в результате триптического расщепления, разделяли на капиллярной колонке С 18 1 мм х 10 см методом высокоэффективной жидкостной хроматографии (ВЭЖХ),применяя линейный градиент от 5% растворителя А (0,1% ТФУ) до 70% растворителя В (0,075% ацетонитрила) за 90 мин со скоростью потока 100 мкл/мин. УФ-детекцию осуществляли при 210 нм со шкалой от 0 до 0,1 А. Наличие индивидуальных пиков отражает присутствие нескольких различных пептидов, как показано на фиг. 3. В качестве контроля использовали часть исходного геля ПААГ с ДСН, не содержащего белка, с которым проводили те же процедуры ферментативного расщепления. Закрашенные пики на фиг. 3 присутствовали на хроматограммах и контроля, и образца. Три пика, отмеченные буквами А, В и С, подвергались автоматической деградации по Эдману. Два пика (А и В) представляли перекрывающиеся уникальные последовательности, представляющие один и тот же фрагмент белка (фиг. 2, фрагменты А и В). Третий пик (С) давал другую уникальную последовательность (фиг. 2, фрагмент С).F. Синтез олигонуклеотидных праймеров ксантозин-N7-метилтрансферазы. Химический синтез 20-членных праймеров для двух аминокислотных последовательностей,проводившийся путем ферментативного расщепления ксантозин-N7-мeтилтpaнcфepaзы, был осуществлен The Midland Certified ReagentCompany. Выбранные участки фрагментов имели минимальную вырожденность нуклеиновых кислот, и в них отсутствовали аминокислоты,имеющие большую избыточность генетического кода. Невозможно было синтезировать более одного праймера для одного и того же фрагмента, чтобы он включал все возможные альтернативные комбинации кодонов. Более того, авторы синтезировали праймеры, комплементарные кодирующей цепи последовательностей ДНК,которые кодируют аминокислотную последовательность. Вырожденность нуклеотидов в третьей позиции преодолели путем использования инозина в этих позициях. Там, где вырожденность нуклеотидов имела двоякий характер,оба нуклеотида включали в синтез праймераG. Экстракция РНК из молодых листьев растений кофе на стадии В 3. Все этапы экстракции осуществляли в стерильных условиях, в отсутствие РНКазы и с использованием воды, обработанной диэтилпирокарбонатом (ДЭПК). Все стадии центрифугирования проводили при 4 С, если не указано иначе. Молодые листья растений кофе на стадии В 3 собирали и хранили, как описано выше. Общую РНК выделяли из 100 г ткани этих молодых листьев путем их растирания в жидком азоте и немедленного переноса в охлажденную бытовую кофемолку. Ткань растирали в порошок с небольшим кусочком сухого льда. Затем к ткани добавляли 200 мл буфера для гомогенизации, содержащего 100 мМ Трис-НСl (рН 9,0),200 мМ NaCl, 15 мМ Na2 ЭДTA, 0,5% саркозила и 100 мМ -меркаптоэтанола, добавленного непосредственно перед использованием. К этому добавляли 200 мл фенола и 40 мл смеси хлороформ:изоамиловый спирт (24:1, об.:об.). Затем ткань гомогенизировали в стеклянном стакане на ледяной бане в течение 2 мин с высокой скоростью в гомогенизаторе Polytron. Сразу после гомогенизации добавляли 14 мл 3 М ацетата натрия (рН 4,0) и перемешивали в гомогенизаторе еще 1 мин. Гомогенат выдерживали 15 мин на льду, затем переносили в два полипропиленовых центрифужных стакана на 250 мл и центрифугировали со скоростью 16000 об./мин в течение 10 мин на центрифуге GSA (Du PontSorvall). Водную фазу (верхний слой) переносили в новые полипропиленовые центрифужные стаканы на 250 мл и добавляли равный объем изопропанола. Эту смесь инкубировали при -20 С в течение ночи и центрифугировали при 10000 14 об./мин 10 минут для выделения осажденной РНК. Осадок РНК промывали 70% этанолом и вновь центрифугировали при 10000 об./мин в течение 5 мин. Этанол декантировали и осадок сушили под вакуумом в течение 5 мин. Затем осадок суспендировали в 15 мл обработанной ДЭПК воды. Суспензию РНК переносили в стерильную центрифужную пробирку на 40 мл с крышкой и нерастворимый материал удаляли центрифугированием в течение 5 мин при 10000 об./мин. Супернатант переносили в новую центрифужную пробирку на 40 мл с крышкой и добавляли 5 мл 8 М LiCl для получения конечной концентрации 2 М LiCl. Пробирку выдерживали в течение ночи при 4 С и выделяли РНК путем центрифугирования в течение 10 мин при 14000 об./мин. Осадок РНК промывали 70% этанолом, центрифугировали 5 мин при 10000 об./мин и быстро сушили под вакуумом. Затем осадок ресуспендировали в 5 мл воды, обработанной ДЭПК, и центрифугировали 5 мин при 10000 об./мин для удаления нерастворимого материала. Супернатант переносили в 4 стерильные микроцентрифужные пробирки на 1,5 мл и хранили на льду. Анализ 10 мкл раствора общей РНК на спектрофотометре Shimadzu UV 160U в диапазоне от 230 до 330 нм показал, что количество полученной РНК составляло 42,8 мг. Пробирки с РНК хранили при -70 С. Н. Выделение поли(А+)мРНК из общей РНК. Обогащенную фракцию поли(А+)РНК(мРНК) получали из общей РНК, используя набор реактивов для выделения мРНК PolyATtractII (Promega Corporation). Аликвоту 600 мкл общей РНК, содержащей 5,1 мг, вносили в пробирку из вышеупомянутого набора, и конечный объем доводили до 2,43 мл водой, свободной от РНКазы. После нагревания при 60 С в течение 10 мин добавляли 10 мкл 50 пмоль/мл биотинилированных олиго(dТ) и 60 мкл 20 х SSCбуфера (175,3 г/л NaCl, 88,2 г/л цитрата натрия,рН 7,0) и медленно охлаждали до комнатной температуры в течение примерно 30 мин. Аликвоту парамагнитных частиц со стрептавидином промывали 3 раза 0,5 х SSC (1,5 мкл на промывку) и ресуспендировали в 0,5 мл 0,5 х SSC. Раствор РНК, содержащий биотинилированные олиго(dТ), смешивали с промытыми парамагнитными частицами стрептавидина. После 10 мин инкубации при комнатной температуре парамагнитные частицы с поглощенной мРНК осаждали на стенках пробирки с помощью магнита. Супернатант удаляли и частицы промывали 4 раза 0,1 х SSC (1,5 мл на промывку). мРНК получали путем суспендирования частиц в 1,0 мл воды, свободной от РНКазы, и удаления воды после того, как частицы осаждались на стенках пробирки. Воду, по 500 мкл за 1 раз, переносили в стерильные микроцентрифужные пробирки на 1,5 мл. После добавления 0,1 объема 3 15 М ацетата натрия (50 мкл на пробирку) выделяли мРНК путем осаждения равным объемом изопропанола (550 мкл на пробирку). Пробирки выдерживали в течение ночи при -20 С и затем центрифугировали при 14000 об./мин при 4 С в течение 30 мин. Осадок промывали 500 мкл 75%-ного ледяного этанола и снова центрифугировали. Этанол декантировали и осадок быстро сушили под вакуумом. мРНК растворяли в 60 мкл стерильной, свободной от нуклеазы, воды,обработанной ДЭПК. Количественное определение мРНК осуществляли в 15 мкл раствора,как описано для общей РНК. Из 5 мг общей РНК было получено приблизительно 9,6 мкг мРНК.I. Конструирование библиотеки кДНК. Первую и вторую цепи кДНК синтезировали с использованием набора реактивов для синтеза ZAP-cDNA (Stratagene). 4 мг мРНК в 25 мкл воды инкубировали при 65 С в течение 5 мин. Добавляли 3 мкл 100 мМ метилртути и инкубировали при комнатной температуре 10 мин. Добавляли 4 мкл 700 мМ -меркаптоэтанола и инкубировали еще 5 мин. К денатурированной мРНК добавляли 5 мкл 10 х буфера для синтеза первой цепи, 5 мкл 10 мМ ДТТ 3 мкл смеси нуклеотидов (по 10 мМ каждогоdATP, dGTP, dTTP и 5-метил-dCTP), 2 мкл 1,4 мг/мл линкерного праймера,(5' GAGAGAGAGAGAGAGAGAGAACTAGTCTCGAGTTTTTTTTTTTTTTTTTT 3'),1 мкл блокирующей РНКазы и 5 мл воды. Реакцию проводили при комнатной температуре в течение 10 мин для отжига праймера с мРНК,затем добавляли 2,5 мкл 20 ед./мкл обратной транскриптазы M-MuLV. 5 мкл этой реакционной смеси переносили в пробирку, содержащую 0,5 мкл 800 Ки/ммоль [-32P]dCTP (DuPontNEN). Обе реакции проводили при 37 С в течение 1 ч. Эту реакционную смесь с радиоактивной меткой замораживали при -20 С для дальнейшего анализа в геле. К 45 мкл основной реакционной смеси прибавляли 40 мкл буфера для синтеза второй цепи, 15 мкл 100 мМ ДТТ, 6 мкл смеси нуклеотидов (10 мМ dATP, dGTP, dTTP и 26 мМP]dCTP. После перемешивания добавляли 4,5 мкл 1 ед./мкл РНКазы Н и 19,2 мкл 5,2 ед./мкл ДНК-полимеразы I из Е. coli и реакцию инкубировали 2,5 ч при 16 С. Затем реакционную смесь экстрагировали смесью фенол : хлороформ (1:1) и фазы разделяли центрифугированием. Водную фазу переносили в новую пробирку и повторно экстрагировали хлороформом. Эту операцию повторяли дважды. Двухцепочечную кДНК выделяли осаждением в течение ночи при -20 С после добавления 33,3 мкл 3 М ацетата натрия и 867 мкл 100%-ного этанола. Осадок отделяли центрифугированием на микроцентрифуге при 4 С в течение 60 мин. Осадок 16 промывали 1 мкл 80%-ного этанола и центрифугировали при комнатной температуре при максимальной скорости на микроцентрифуге. Супернатант удаляли, осадок сушили под вакуумом и растворяли в 45 мкл воды. Отбирали 3 мкл ресуспендированной двухцепочечной кДНК и замораживали при -20 С для дальнейшего анализа с помощью гель-электрофореза. К оставшимся 42 мкл раствора двухцепочечной кДНК добавляли 5 мкл 10 х буфера Кленова (buffer 3), 2,5 мкл 2,5 мМ нуклеотидов(dCTP, dGTP, dATP и dTTP), 0,5 мкл 5 ед./мкл фрагмента Кленова. Реакционную смесь выдерживали 30 мин при 37 С, добавляли 50 мкл воды и экстрагировали равным объемом смеси фенол:хлороформ (1:1), затем хлороформом, как описано выше. После добавления 7 мкл 3 М ацетата натрия и 226 мкл 100%-ного этанола выделяли двухцепочечную кДНК с "тупыми концами" путем осаждения в течение 30 мин на льду и последующего центрифугирования на микроцентрифуге при 4 С в течение 60 мин с максимальной скоростью. Осадок промывали 300 мкл 80%-ного этанола, центрифугировали и сушили, как описано выше. Затем к высушенной кДНК добавляли 7 мкл 0,4 мг/мкл раствораEcoRI-линкеров. Структура линкеров следующая: 5' AATTCGGCACGAG 3'- 3' GCCGTGCTC 5' После интенсивного перемешивания к ресуспендированной кДНК добавляли 1 мкл 10 х лигирующего буфера, 1 мкл 10 мМ АТР и 1 мкл 4 Weiss ед./мкл Т 4 ДНК-лигазы и реакцию инкубировали в течение ночи при 8 С. Затем лигазу инактивировали нагреванием до 70 С в течение 30 мин. 5'-концы EcoRI-линкеров, присоединенные к кДНК, фосфорилировали с помощью полинуклеотидкиназы. В лигирующую смесь добавляли 1 мкл 10 х буфера 3, 2 мкл 10 мМ АТР, 6 мл воды и 1 мл 10 ед./мл Т 4 полинуклеотидкиназы. После 30 мин инкубации киназу инактивировали нагреванием до 70 С в течение 30 мин.XhoI-"липкие концы" создавали на конце кДНК, соответствующем 3'-концу мРНК, путем расщепления XhoI-сайта в линкерном праймере(см. выше). 28 мкл XhoI-буфера и 3 мкл 40 ед./мл рестриктазы XhoI добавляли к кДНК и реакцию инкубировали при 37 С в течение 1,5 ч. Затем кДНК с EсоRI-"липкими концами" на 5'-конце и с XhoI-"липкими концами" на 3'конце (относительно природной мРНК) фракционировали по размеру на вращающейся колонке Sephacryl S-400, как описано ниже. Добавляли 5 мкл 10 х STE буфера (100 мМ Трис(рН 7,0), 5 мМ ЭДТА и 100 мМ NaCl) и кДНК наносили на верхнюю часть 1 мкл-шприца, содержащего Sephacryl S-400. Микроцентрифужную пробирку на 500 мкл помещали в основании шприца, колонку помещали в центрифужную пробирку и центрифугировали 2 мин при 400 х g. Затем добавляли 60 мкл 10 х STE в 17 верхнюю часть шприца, новую микроцентрифужную пробирку помещали в основании шприца и колонку снова центрифугировали, как описано выше. Эту процедуру повторяли до тех пор, пока не было собрано 6 фракций. Примерно 10% каждой фракции подвергали электрофорезу в 1%-ном агарозном геле для определения распределения кДНК по размерам в каждой фракции. Остаток каждой фракции экстрагировали равным объемом смеси фенол:хлороформ, затем хлороформом, как описано выше, и осаждали добавлением 2 объемов 100%-ного этанола. После инкубации в течение ночи при -20 С выделяли кДНК центрифугированием при 14000 об./мин при 4 С в течение 60 мин в микроцентрифуге. кДНК промывали 200 мкл 80%-ного этанола, как описано выше, и сушили. Затем кДНК растворяли в 5 мкл воды и 0,5 мкл раствора отбирали для определения концентрации кДНК методом флюорографии на ДНК флюориметре Hoefer ТKО 100. Оставшиеся 4,5 мл фракции 1, содержащей самые большие молекулы кДНК, содержали 304 нг кДНК. 100 нг кДНК из фракции 1 лигировали в 1 мкг Uni-Zap, вектор на основе бактериофага лямбда ZAP, который был расщеплен рестриктазами EcoRI и XhoI (Stratagene). Фракцию 1 кДНК (2,9 мкл) добавляли к 0,54 мкл 10 х лигирующего буфера, 0,5 мкл 10 мМ АТР, 1 мкл 1 мкг/мкл вектора Uni-Zap XR и 0,5 мкл 4 Weiss ед./мкл Т 4 ДНК-лигазы. Реакцию инкубировали 44 ч при 8 С. Затем аликвоту 1 мкл реакционной смеси прибавляли к одной аликвоте экстракта"Freeze-Thaw" из набора реактивов для упаковывания Gigapack II Gold (Stratagene). Добавляли 15 мкл озвученного экстракта, и содержимое осторожно перемешивали. Упаковывание проводили при комнатной температуре. Через 2 ч к реакционной смеси прибавляли 500 мкл буфераSM (0,01 М Трис-HCl, рН 7,5, 0,01 М MgCl2, 0,1 мМ Na2 ЭДТА) и 20 мкл хлороформа, дебрис удаляли с помощью короткого центрифугирования в микроцентрифуге, упакованные фаги хранили при 4 С.J. Определение титра первичной библиотеки. 1 мкл из 500 мкл первичной библиотеки смешивали с 9 мкл буфера SM для разбавления 1/10. 1 мкл полученного раствора использовали для инфицирования 200 мкл клеток Е. coli XL1Blue MRF', выращенных до плотности, равнойOD600-0,5. Клетки инкубировали 15 мин при 37 С при осторожном перемешивании. Инфицированные клетки затем смешивали с 2,5 мл нагретого до 48 С агара, содержащего 15 мкл 0,5 М IPTG и 50 мкл 250 мг/мл X-gal и высевали на чашки размером 100 х 15 мм со средой NZY(5 г/л NaCl, 2 г/л MgSO47H2O, 5 г/л дрожжевого экстракта, 10 г/л NZ амина, рН 7,5, 15 г/л агара фирмы Difco). Чашки инкубировали в течение ночи при 37 С. Фоновые колонии были голубыми, тогда как рекомбинантные - белыми. Ус 003835 18 реднение по трем чашкам показало, что 1 мкл первичной библиотеки продуцировал 1930 белых рекомбинантных колоний и 65 голубых. Рассчитали, что все 500 мкл первичной библиотеки дали 965000 рекомбинантных колоний. К. Амплификация первичной библиотеки. В 20 стерильных пробирок вносили по 300 мкл клеток Е. coli XL1-Blue MRF', выращенных до плотности равной OD600=0,5. В каждую пробирку добавляли по 12,5 мкл исходного раствора первичной библиотеки и 90 мкл буфера SM и инкубировали 15 мин при 37 С. 2,5 мл нагретого до 48 С агара добавляли в каждую пробирку,и клетки высевали на чашки размером 100 х 15 мм со средой NZY. Чашки выдерживали в течение ночи при 37 С. В каждую чашку добавляли по 5 мл буфера SM, и выдерживали при 4 С еще 8 ч. Затем стерильной пипеткой отбирали буферSM и хранили в стерильной центрифужной пробирке на 250 мл. Каждую чашку промывали примерно 4 мл свежего буфера SM, который добавляли к ранее собранному материалу. К амплифицированной библиотеке добавляли хлороформ до конечной концентрации 5%. Затем библиотеку инкубировали при комнатной температуре в течение 15 мин и центрифугировали 10 мин при 2000 х g для удаления дебриса. Супернатант (114,5 мл) удаляли, а затем переносили его в стерильную полипропиленовую бутылку. Добавляли хлороформ до конечной концентрации 0,3% и амплифицированную библиотеку хранили при 4 С.L. Определение титра амплифицированной библиотеки. 1 мкл разбавленной в 1011 раз в буфере SM амплифицированной библиотеки содержал 192 рекомбинантных колоний при посеве, как описано выше. Для получения 50000 рекомбинантных колоний, 25 мкл разведения в 107 раз использовали для инфицирования 600 мкл клеток Е. coli XL1-Blue MRF', выращенных до плотности, равной OD600=0,5, которые затем инкубировали 15 мин при 37 С. Далее к этим клеткам прибавляли 6,5 мл нагретого до 48 С агара, и библиотеку высевали на чашки размером 100 х 15 мм со средой NZY. Были получены 4 такие чашки, содержащие 200000 рекомбинантных колоний, которые инкубировали в течение ночи при 37 С. Затем чашки охлаждали 4 ч при 4 С и использовали далее для скрининга библиотеки ДНК. М. Амплификация кДНК ксантозин-N7 метилтрансферазы с помощью полимеразной цепной реакции (ПЦР). Синтез первой цепи кДНК осуществляли,как описано выше в протоколе Stratagene. Две уникальные пептидные последовательности,полученные путем триптического расщепления,позволили синтезировать вырожденные праймеры, представленные на фиг. 4. Проводили полимеразную цепную реакцию (ПЦР) (Saiki,R.K., Gelfand, D.H., Stoffel, S., Scharf, S.J., Higu 19chi, R., Horn, G.T., Mullis, K.B. and Erlich, H.A.,Science 239:487 (1988 между парами праймеров (1-6, 2-6, 3-5 или 4-5) с использованием 4 нг кДНК, 1 мкл 20 мкМ праймеров, 0,5 мкл каждого 1 мМ дезоксирибонуклеотидтрифосфата, 1,5 мМ MgCl2, 0,3 мкл Taq ДНК-полимеразы (5000 ед./мл), 2,5 мкл 10 х ПЦР-буфера (10 мМ ТрисHCl, рН 9,0, 0,1% тритона Х-100) и стерильной воды до конечного объема 25 мкл. Условия ПЦР: 94 С - 4 мин (1 цикл); 94 С - 1 мин, 43 С 1 мин; 72 С - 1 мин (35 циклов); 72 С - 5 мин (1 цикл). Реакции проводили в стерильных микроцентрифужных пробирках на 500 мкл, используя термоциклер (Perkin Elmer DNA thermal cycler 480). Только комбинация праймеров 1 и 6 дала единственный продукт при температуре отжига 43 С. Анализ с помощью электрофореза в агарозном геле (SeaPlaque agarose (FMC показал наличие продукта длиной приблизительно 750 пар нуклеотидов (п.н.). В качестве маркера для размеров использовали имеющуюся в продаже 100 п.н. "лестницу" (Promega Corporation).N. Клонирование ПЦР-продукта, специфичного для ксантозин-N7-метилтрансферазы из растений кофе. К 50 мкл ПЦР-реакционной смеси, содержащей фрагмент 750 п.н., полученный с использованием праймеров 1 и 6 (фиг. 4), добавляли 50 мкл хлороформа и 100 мкл стерильной воды. Смесь интенсивно перемешивали и затем центрифугировали в микроцентрифуге при 14000 об./мин в течение 2 мин. Отбирали верхний водный слой, содержащий ДНК, и помещали в стерильную пробирку. Количественный анализ с помощью бромистого этидия показал присутствие примерно 5 нг амплифицированной в результате ПЦР ДНК/мкл. Затем ПЦР-продукт лигировали в вектор pCR II из набора для клонирования ТА (Invitrogen Corporation) в 10 мкл лигирующей реакционной смеси, содержащей 1 мкл 10 х лигирующего буфера, 2 мкл вектора(5 нг/мкл), 1 мкл Т 4 ДНК-лигазы и 3 мкл стерильной воды. Реакцию проводили при 14 С в течение ночи. Затем реакционную смесь центрифугировали 2 мин при 14000 об./мин и помещали на лед. Во флакон со свежеразмороженными компетентными клетками Е. coli XL1-Blue добавляли 2 мкл 0,5 М -меркаптоэтанола и осторожно перемешивали кончиком пипетки,затем добавляли 2 мкл лигирующей реакционной смеси и также осторожно перемешивали кончиком пипетки. Флакон с клетками выдерживали на льду в течение 30 мин и затем подвергали клетки тепловому шоку точно в течение 30 с в 42 С термостате. Затем флакон снова помещали на лед. Через 2 мин в него добавляли 450 мкл стерильной среды SOC (20 г/л триптона, 5 г/л дрожжевого экстракта, 0,5 г/л NaCl, 10 мл/л 250 мМ KСl, 10 мл/л MgCl2, 20 мл/л 1 М глюкозы, рН 7,0). Содержимое флакона пере 003835 20 мешивали на роторном шейкере при 225 об./мин в течение 1 ч и затем помещали на лед. Суспензию трансформированных клеток наносили пипеткой по 50 мкл и/или 200 мкл на одну из двух LB чашек (10 г/л триптона, 5 г/л дрожжевого экстракта, 10 г/л NaCl, 15 г/л агараDifco, рН 7,5), содержащих 50 мкг/мл ампициллина и 40 мкг/мл X-gal. Чашки инкубировали при 37 С в течение 20 ч и затем помещали на 3 ч при 4 С для развития цвета. Шесть белых колоний трансформантов анализировали на наличие и ориентацию ПЦР-фрагмента.O. Кипячение мини-препарата плазмиды. Каждую из колоний трансформантов выращивали в 5 мл стерильного бульона (12 г/л триптона, 24 г/ л дрожжевого экстракта, 4 мл/л глицерина, 100 мл/л 10 х фосфатного буфера ТВ[0,17 М KН 2 РО 4, 0,72 М K2 НРО 4]), дополненного 50 мкг/мл ампициллина. Пробирки выдерживали в течение ночи в роторном шейкере при 37 С. 3 мл каждой колонии (по 1 мл за 1 раз) переносили в центрифужные пробирки на 1,5 мл, и клетки концентрировали центрифугированием при 14000 об./мин в течение 2 мин. Каждый раз супернатант отбрасывали, а осадок клеток максимально высушивали. Затем клетки один раз промывали 1 мл стерильной воды и центрифугировали, как описано выше. Супернатант отбрасывали, и осадок клеток суспендировали в 320 мкл буфера STET (8% сахарозы, 0,5% тритона Х-100, 50 мМ ЭДТА , 10 мМ Трис-HCl,рН 8,0). Далее к этим клеткам добавляли 32 мкл 10 мг/мл раствора лизоцима в буфере ТЕ (10 мл/л 1 М Трис-HCl, рН 8,0, 2 мл/л 0,5 М ЭДТА,рН 8,0) и перемешивали, несколько раз переворачивая пробирку. Пробирки помещали на 5 мин в кипящую водяную баню и затем немедленно на лед. После охлаждения содержимое центрифугировали при 14000 об./мин в течение 30 мин при 4 С. Осадок удаляли из каждой пробирки с помощью стерильной зубочистки. К супернатанту добавляли 170 мкл 7,5 М NH4OAc и 550 мкл ледяного изопропанола и осаждали ДНК при -20 С в течение ночи. После этого пробирки центрифугировали со скоростью 14000 об./мин в течение 30 мин при 4 С, осадок промывали 75%-ным этанолом и сушили 1 мин в вакуумной центрифуге. ДНК ресуспендировали в 50 мкл стерильной воды, содержащей 1 мкл 5 мг/мл РНКазы А.P. Рестрикционное расщепление для удаления вставки из плазмиды pCR II. 25 мкл реакционной смеси получали из 15 мкл мини-препарата плазмидной ДНК, полученной, как описано выше, 2,5 мкл буфера Н (90 мМ Трис-HCl, рН 7,5, 10 мМ MgCl2, 50 мМNaCl), 1 мкл EcoRI (8-12 ед./мкл) и 6,5 мкл стерильной Н 2 О. Смесь инкубировали при встряхивании на водяной бане при 37 С в течение 1 ч и затем кипятили на водяной бане 1 мин. Пробирки центрифугировали при 14000 об./мин в течение 15 с и охлаждали до комнатной темпе 21 ратуры. К 10 мкл каждой смеси добавляли 2 мкл утяжеленного красителя, и продукты расщепления анализировали с помощью 1,5%-ного гельэлектрофореза из особо чистой агарозы (GibcoBRL), используя в качестве маркера размеров 100 п.н. "лестницу" (Promega Corporation). Только одна из шести реакций показала присутствие рестриктной вставки 750 п.н. Колонию бактерий, соответствующую плазмиде с ПЦР-продуктом длиной 750 п.н. для ксантозинN7-метилтрансферазы, вносили в колбу Эрленмейера на 250 мл, содержащую 50 мл стерильной среды LB (10 г/л триптона, 5 г/л дрожжевого экстракта, 10 г/л NaCl, рН 7,5) и 50 мг/мл ампициллина. Колбу инкубировали на роторном шейкере при 30 С в течение ночи. 18 мл полученной культуры клеток концентрировали центрифугированием в микроцентрифужной пробирке на 1,5 мл, как описано выше. Плазмидную ДНК очищали с помощью мининабора реагентов QIAGEN для получения плазмиды (Qiagen Inc.). Промытый осадок бактерий ресуспендировали в 0,3 мл буфера Р 1,содержащего РНКазу. К этой суспензии добавляли 0,3 мл щелочного лизирующего буфера Р 2,осторожно перемешивали, встряхивая пробирку, и инкубировали не более 5 мин при комнатной температуре. Затем добавляли 0,3 мл охлажденного буфера Р 3 и перемешивали, переворачивая пробирку 6 раз. После 10 мин инкубации на льду экстракт центрифугировали при 14000 об./мин в течение 15 мин в микроцентрифуге. Супернатант удаляли и наносили на колонкуQIAGEN-tip 20, предварительно уравновешенную 1 мл буфера QBT, пропущенного самотеком. Супернатант клеточного экстракта также пропускали через колонку самотеком. После того, как поток жидкости через колонку прекращался, ее промывали 4 раза 1 мл буфера QC. ДНК элюировали 0,8 мл буфера QF и осаждали добавлением 0,7 объемов (560 мкл) изопропанола комнатной температуры. Пробирку немедленно центрифугировали при 14000 об./мин в течение 30 мин и осторожно удаляли cупернатант. Осажденную ДНК промывали 1 мл ледяного 70%-ного изопропанола, центрифугировали,как описано выше, и сушили на воздухе 5 мин. Далее ДНК ресуспендировали в 100 мкл стерильной воды. Анализ 1 мкл суспензии ДНК с помощью УФ-спектрометрии, описанный выше,показал, что в 100 мкл содержится 55 мкг очищенной рекомбинантной плазмидной ДНКpCRII. Автоматическое секвенирование последовательности ДНК-вставки в плазмиде pCRII от 5'-конца выполняли с использованием обратного праймера М 13, который присоединялся кpCRII рядом с сайтом, куда был вставлен ПЦРпродукт. Секвенирование проводили в лаборатории биотехнологии Гавайского Университета. Реакционная смесь для секвенирования содержала 1 мкг плазмидной матрицы и 3,2 пмоль 22 праймера М 13. Полученная последовательность показала, что ПЦР-продукт кодировал последовательность ДНК первых 6 аминокислот пептидных фрагментов А и В (фиг. 4), из последовательностей которых были получены вырожденные ДНК-праймеры 1 и 2 (фиг. 4). Кроме того, полученная последовательность также кодировала следующие 7 аминокислот пептидного фрагмента, ДНК-последовательность которого не использовалась при конструировании праймера. Таким образом, фактически была клонирована последовательность ДНК нужного белка.Q. Создание произвольно праймированного зонда для скрининга кДНК с использованием ПЦР-продукта. Два рестрикционных расщепления с помощью рестриктазы EcoRI (по 25 мкл каждое) выполняли в двух аликвотах по 17,5 мкл очищенной плазмиды pCRII, как описано выше. Продукты реакции разделяли в 1%-ном агарозном геле, как описано выше, и вставку длиной 750 п.н. вырезали в стерильных условиях из двух дорожек геля. Кусочки геля массой 0,65 г переносили в стерильную полипропиленовую пробирку на 40 мл и подвергали очистке с помощью набора реактивов Geneclean II (BIO 101,Inc.). К кусочкам геля добавляли 4,5 объема исходного раствора NaI (2,93 мл), 0,5 объема гельмодификатора ТВЕ (325 мкл) и инкубировали 5 мин при 45 С. Затем в пробирку прибавляли 15 мкл суспензии glassmilk и инкубировали еще 5 мин. Далее комплекс glassmilk/ДНК осаждали центрифугированием в течение 10 с при 1000 об./мин и супернатант удаляли. Осадок glassmilk промывали 3 раза по 1 мл раствором NewWash, и ДНК элюировали 50 мкл стерильной воды. Анализ с помощью бромистого этидия показал, что концентрация ДНК составляла 10 нг/мкл. Произвольно праймированный зонд синтезировали из 30 нг (3 мкл) очищенной ДНК. 3 мкл ДНК добавляли к 27 мкл стерильной воды,и ДНК денатурировали нагреванием в кипящей водяной бане. К денатурированной ДНК добавляли реактивы из набора Prime-a-Gene (PromegadATP (50 мкКи, 3000 Ки/ммоль; DuPont NEN) до конечного объема 50 мкл и инкубировали при комнатной температуре 1 ч. Реакцию прекращали добавлением 2 мкл 0,5 М Na2 ЭДTA (до конечной концентрации 20 мМ) и нагреванием в кипящей водяной бане в течение 2 мин.R. Скрининг амплифицированной библиотеки с помощью произвольно праймированного зонда. Четыре чашки размером 150 х 15 мм со средой NZY, в которых находилось приблизительно по 50000 рекомбинантных клонов на чашку(см. выше об условиях посева и выращивания),охлаждали до 4 С, затем рекомбинантные колонии переносили на 132 мм нейлоновых мембранах Magna (MSI Corporation) на хроматографическую бумагу, пропитанную 5 х SSC-буфером,на 10 с. Мембраны помещали на чашки, содержащие рекомбинантные колонии, на 5 мин, затем убирали и наносили на 2 мин на хроматографическую бумагу, пропитанную раствором 0,5 М NaOH и 1,5 М NaCl, стороной, содержащей фаговые частицы, вверх. После этого мембраны нейтрализовали путем переноса на хроматографическую бумагу, пропитанную буфером 0,5 М Трис-HCl, рН 8,0, и 1,5 М NaCl, на 5 мин. Затем их помещали на 10 с на хроматографическую бумагу, насыщенную 2 х SSCбуфером, 0,2 М Трис-HCl, рН 7,5, и промокали насухо. После сушки на воздухе в течение 1 ч,ДНК сшивали с мембранами путем экспозиции с УФ-излучением с энергией 12000 мкДж с помощью UV Stratalinker 1800 (Stratagene Corporation). Четыре мембраны предварительно гибридизовали при 65 С в течение 2 ч в 100 мл 6 хNaH2PO4H2O, 2,2 г/л Na2 ЭДTA, рН 7,4), 5 х растворе Денхардта (1 г/л фикола, 1 г/л поливинилпирролидона, 1 г/л БСА (фракция V), 0,5% ДСН и 100 мкг/мл денатурированной ДНК спермы сельди в термостате для гибридизации HybridMark II. Гибридизацию проводили при 65 С в течение 12 ч в 10 мл 6 х SSPE-буфера, 0,5% ДСН,100 мкг/мл денатурированной ДНК спермы сельди и 52 мкл 15 х 106 имп/мин/мл произвольно праймированного зонда, описанного выше. После окончания гибридизации зонд удаляли, а мембраны быстро промывали (30 с) 100 мл буфера, содержащего 2 х SSPE и 0,5% ДСН, с температурой 65 С. Затем мембраны снова промывали в течение 30 мин вежей порцией (100 мл) указанного буфера. После этого мембраны промывали еще 2 раза по 30 мин 100 мл буфера,содержащего 0,2 х SSPE и 0,5% ДСН, с температурой 65 С, помещали в целлофановый пакет и экспонировали с предварительно облученной рентгеновской пленкой Fuji RXGCU в течение 24 ч при -70 С. Наблюдали 15 позитивных клонов. Эти клоны отбирали и помещали в 1 мл буфераSM, содержащего 20 мкл хлороформа (исходный фаговый концентрат). 11 из них подвергали скринингу во второй или третий раз, пока не были получены индивидуальные клоны.S. Характеристика клонов кДНК ксантозин-N7-метилтрансферазы. Размеры предполагаемых клонов кДНК ксантозин-N7-метилтрансферазы определяли с помощью ПЦР с использованием праймеров,гомологичных Т 3 и Т 4 промоторам, присутствующих в клонирующем векторе и примыкающих к сайту вставки кДНК. Условия ПЦР были аналогичны описанным выше, за исключением того, что цикл включал 35 циклов по 1 мин при 24 95 С, 1 мин при 50 С и 2 мин при 72 С. Анализ путем электрофореза в агарозном геле проводили, как описано выше. Три самых больших полученных клона подвергали эксцизии in vivo,смешивая в стерильной пробирке 200 мкл исходного раствора индивидуального фага с 200 мкл свежей культуры клеток XL1-Blue MRF,выращенных до плотности OD600=1,0. К этой смеси прибавляли 1 мкл фага-хелпера ExAssist(Stratagene Corporation) (1x106 pfu/мкл) и выдерживали при 37 С 15 мин. Затем добавляли 3 мл среды LB и инкубацию продолжали еще 3 ч при 37 С при встряхивании. Далее культуру нагревали при 70 С в водяной бане в течение 20 мин и центрифугировали при 1000 х g в течение 15 мин. 1 мл супернатанта, содержащего вырезанный pBluescript фагмид, упакованный в виде нитевидных фаговых частиц, переносили в стерильную микроцентрифужную пробирку на 1,5 мл и хранили при 4 С в качестве исходного раствора. 25 мкл этого исходного раствора добавляли в микроцентрифужную пробирку с 200 мкл клеток Е. coli Solar, выращенных до плотностиOD600=1,0. После инкубации при 37 С в течение 15 мин 200 мкл клеток высевали на агаровые чашки размером 100 х 15 мм со средой NZY, содержащей 50 мкг/мл ампициллина. Чашки инкубировали при 37 С в течение ночи до появления колоний. Отдельную колонию переносили в 10 мл стерильной среды LB, содержащей 50 мкг/мл ампициллина и выращивали при 37 С в течение ночи при встряхивании. 10 мл клеточной суспензии концентрировали в стерильной микроцентрифужной пробирке на 1,5 мл, из осадка клеток выделяли плазмиду с использованием набора реактивов QIAGEN для очистки плазмид, как описано выше. Очищенную плазмидную ДНК ресуспендировали в 50 мкл стерильной воды. Автоматическое секвенирование ДНК проводили, смешивая 8 мкл этого образца ДНК (0,8 мкг) с 4 мкл либо Т 3, либо Т 7 праймеров для секвенирования (0,8 пмоль/мкл). Реакции выполняли, как описано выше. Каждый этап секвенирования давал последовательность длиной приблизительно 350 оснований. Последовательность представлена на фиг. 5. Аминокислотная последовательность ксантозин-N7 метилтрансферазы, предсказанная на основании последовательности кДНК, представлена на фиг. 6. Описанные примеры служат только для иллюстрации, и их не следует рассматривать в качестве примеров, ограничивающих объем охраны изобретения, который обозначен в формуле изобретения, приведенной далее. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Очищенная ксантозин-N7-метилтрансфераза из растения кофе Coffea arabica с выведенной аминокислотной последовательностьюPhe Ile Asp (Asp) (Gln) Asp. 2. Выделенная последовательность кДНК,кодирующая ксантозин-N7-метилтрансферазу из растения кофе Coffea arabica, охарактеризованную в п.1, имеющая следующую структуру: а также варианты указанной кДНК, полученные на основе вырожденности генетического кода. 3. Способ получения кофейных зерен, не содержащих кофеина, включающий трансформирование растений кофе последовательностью ДНК, которая является антисмысловой по отношению к последовательности кДНК или ее вариантам, полученным на основе вырожденности генетического кода, охарактеризованным в п.2; и получение плодов от трансформированных растений кофе. 4. Растение кофе, трансформированное последовательностью кДНК по п.2, кодирующей РНК, которая является антисмысловой по отношению к последовательности мРНК, кодирующей ксантозин-N7-метилтрансферазу по п.1,причем РНК имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозинN7-метилтрансферазы. 5. Растение кофе, трансформированное последовательностью кДНК по п.2, кодирующей РНК, которая является смысловой мРНК, кодирующей ксантозин-N7-метилтрансферазу по п.1,причем РНК имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозин-N7-метилтрансферазы. 6. Растение кофе по п.4, где последовательность кДНК связана с промотором транскрипции в антисмысловой ориентации. 27 7. Растение кофе по п.5, где последовательность кДНК связана с промотором транскрипции в смысловой ориентации. 8. Кофейные зерна от растений кофе по любому из пп.4-7. 9. Трансформирующий вектор, включающий промотор транскрипции, связанный с последовательностью кДНК, охарактеризованной в п.2. 10. Трансформирующий вектор по п.9, где последовательность кДНК связана с промотором транскрипции в смысловой ориентации. 11. Трансформирующий вектор по п.9, где последовательность кДНК связана с промотором транскрипции в антисмысловой ориентации. 12. Трансформирующий вектор по п.9, где промотором является 35S промотор вируса мозаики цветной капусты. 13. Трансформирующий вектор по п.9, где вектором является модифицированная плазмидаpBI-121. 14. Клетка растения кофе, трансформированная последовательностью кДНК по п.2, кодирующей РНК, которая является антисмысловой по отношению к последовательности мРНК,кодирующей ксантозин-N7-метилтрансферазу по п.1, причем РНК имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозин-N7-метилтрансферазы. 15. Клетка растения кофе, трансформированная последовательностью кДНК по п.2, кодирующей РНК, которая является смысловой мРНК, кодирующей ксантозин-N7-метилтрансферазу по п.1, причем РНК имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозин-N7-метилтрансферазы. 16. Клетка растения кофе по п.14, где последовательность кДНК связана с промотором транскрипции в смысловой ориентации. 17. Клетка растения кофе по п.15, где последовательность кДНК связана с промотором транскрипции в антисмысловой ориентации. 18. Трансформированная клетка растения кофе, полученная путем введения трансформирующего вектора по п.9 в клетку растения кофе. 19. Трансформированная клетка растения кофе по п.18, где последовательность кДНК связана с промотором транскрипции в смысловой ориентации. 20. Трансформированная клетка растения кофе по п.18, где последовательность кДНК 28 связана с промотором транскрипции в антисмысловой ориентации. 21. Трансформированная клетка растения кофе по любому из пп.14-20, где клетка синтезирует меньшее количество кофеина по сравнению с клеткой растения кофе, не трансформированной последовательностью кДНК. 22. Растение кофе, выращенное из трансформированных клеток растения кофе по любому из пп. с 14 по 21. 23. Кофейные зерна, полученные от растения кофе по п.22. 24. Способ уменьшения синтеза кофеина клетками растения кофе, включающий следующие стадии: получение трансформирующего вектора,содержащего последовательность кДНК по п.2,кодирующую РНК, которая имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозин-N7-метилтрансферазы, охарактеризованной в п.1, причем последовательность кДНК связана с промотором транскрипции в антисмысловой ориентации; и введение трансформирующего вектора в клетку растения кофе, в результате чего последовательность кДНК вводится в геном клетки растения кофе для получения трансформированной клетки, причем трансформированные клетки синтезируют меньшее количество кофеина по сравнению с клетками растения кофе,не трансформированными последовательностью кДНК. 25. Способ уменьшения синтеза кофеина клетками растения кофе, включающий следующие стадии: получение трансформирующего вектора,содержащего последовательность кДНК по п.2,кодирующую РНК, которая имеет длину, достаточную для того, чтобы препятствовать экспрессии ксантозин-N7-метилтрансферазы, охарактеризованной в п.1, причем последовательность кДНК связана с промотором транскрипции в смысловой ориентации; и введение трансформирующего вектора в клетку растения кофе, в результате чего последовательность кДНК вводится в геном клетки растения кофе для получения трансформированной клетки, причем трансформированные клетки синтезируют меньшее количество кофеина по сравнению с клетками растения кофе,не трансформированными последовательностью кДНК. Фиг. 1 Полиакриламидный гель очищенной ксантозин-N7-метилтрансферазы Фиг. 2 Разделение на ВЭЖХ триптических фрагментов очищенной ксантозин-N7-мeтилтpaнcфepaзы
МПК / Метки
Метки: клетки, растения, кофеина, кислоты, способы, клетках, кофейные, очищенная, содержащие, ксантозин-n7-метилтрансфераза, уменьшения, кодирующие, зерна, кофе, синтеза, нуклеиновые, трансгенные
Код ссылки
<a href="https://eas.patents.su/16-3835-ochishhennaya-ksantozin-n7-metiltransferaza-iz-rasteniya-kofe-kodiruyushhie-ee-nukleinovye-kisloty-transgennye-kletki-rasteniya-kofe-transgennye-rasteniya-kofe-i-kofejjnye-zerna-ne.html" rel="bookmark" title="База патентов Евразийского Союза">Очищенная ксантозин-n7-метилтрансфераза из растения кофе, кодирующие ее нуклеиновые кислоты, трансгенные клетки растения кофе, трансгенные растения кофе и кофейные зерна, не содержащие кофеина, и способы уменьшения синтеза кофеина в клетках растения кофе</a>
Мутантный il-6 человека и его внутренний фрагмент, кодирующие их последовательности днк, способы их получения, содержащие их фармацевтические композиции, содержащие их векторы, линии клеток- хозяев испособ лечения il-6 опосредованных заболеваний

Номер патента: 852
Опубликовано: 26.06.2000
Авторы: Розе-Йон Штефан, Элерс Марк, Гротзингер Йоахим
МПК: A61K 38/20, A61P 19/10, A61P 35/00...
Метки: получения, днк, лечения, последовательности, человека, клеток, мутантный, испособ, заболеваний, содержащие, внутренний, фрагмент, кодирующие, линии, композиции, хозяев, фармацевтические, способы, векторы, опосредованных
Формула / Реферат:
1. Мутантный интерлейкин-6 (IL-6) человека, имеющий аминокислотную последовательность содержащую следующие точечные мутации по сравнению с природным IL-6 человека: Pro 54, Glu 159, Pro 162, Leu 170 и Аrg 176. 2. Внутренний фрагмент мутантного IL-6 человека по п.1 формулы, обладающий аналогичной биологической активностью. 3. Последовательность ДНК, кодирующая мутантный IL-6 человека по п.1 формулы. 4. Последовательность ДНК, кодирующая...
Полипептиды, содержащие домены протеина gax, участвующие в подавлении транскрипции и/или взаимодействующие с другими протеинами, соответствующие нуклеиновые кислоты и их применение

Номер патента: 3694
Опубликовано: 28.08.2003
Авторы: Марсиро Кристоф, Мафуди Абдерраим, Бранеллек Дидье, Фурнье Алан
МПК: A61K 38/17, C07K 14/47, C12N 15/12...
Метки: подавлении, взаимодействующие, другими, применение, участвующие, кислоты, нуклеиновые, полипептиды, содержащие, протеина, транскрипции, протеинами, домены, соответствующие
Формула / Реферат:
1. Полипептид, отличающийся тем, что он представляет собой фрагмент протеина GAX (Growth-Arrest-Specific Homeobox) человека, содержащий, по меньшей мере, остатки 1-32 протеина GAX и обладающий репрессорной активностью в отношении транскрипции и/или оказывающий положительное или отрицательное влияние на репликацию ДНК. 2. Полипептид по п.1, отличающийся тем, что он дополнительно содержит остатки 140-230 протеина GAX человека. 3. Полипептид,...
Циклоалкильные соединения, лактамы, лактоны и родственные соединения, содержащие их фармацевтические композиции и способы ингибирования высвобождения и/или синтеза β-амилоидного пептида с помощьюуказанных соединений

Номер патента: 2100
Опубликовано: 24.12.2001
Авторы: Танг Джей С., Генри Стивен С., Фридман Стефен, Торсетт Юджин Д., Дрессман Брюс А., Кви Синтия Л., Джон Варгес, Макданиел Стейси Л., Дрост Джеймс Дж., Нейц Джеффри, Ниссен Джеффри С., Мабри Томас Э., Рил Джон К., Ву Джинг, Скотт Уильям Леонард, Латимер Ли Х., Стаки Расселл Д., Портер Варрен Дж., Одия Джеймс Е., Плейсс Майкл А., Бриттон Томас К.
МПК: C07D 243/10, A61P 25/28, A61K 31/55...
Метки: beta;-амилоидного, соединений, помощьюуказанных, композиции, пептида, родственные, соединения, способы, содержащие, ингибирования, лактамы, фармацевтические, лактоны, циклоалкильные, синтеза, высвобождения
Формула / Реферат:
1. Способ ингибирования высвобождения и/или синтеза b -амилоидного пептида в клетке, который заключается в том, что в такую клетку вводят соединение или смесь соединений в количестве, эффективном для ингибирования высвобождения и/или синтеза b -амилоидного пептида в клетке, и указанные соединения имеют формулу I в которой R1 выбирают из группы, включающей C1-С10алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из...
Линия клеток растения сахарной свеклы и растение сахарной свеклы, устойчивые к имидазолиноновому гербициду, семена указанного растения, способы получения указанной клеточной линии и гибридных растенийсвеклы, устойчивых к данному гербициду, и применение указанного гербицида для борьбы с сорняками, растущими на полях с указанными растениями сахарной свеклы

Номер патента: 3296
Опубликовано: 24.04.2003
Авторы: Райт Терри Р., Пеннер Доналд
Метки: указанного, сахарной, семена, гербициду, гибридных, указанной, клеточной, данному, гербицида, борьбы, растущими, клеток, полях, растения, линия, применение, свеклы, растениями, имидазолиноновому, линии, устойчивых, способы, растение, сорняками, указанными, получения, устойчивые, растенийсвеклы
Формула / Реферат:
1. Линия клеток растения сахарной свеклы с мутированным геном ацетолактатсинтазы, в котором в положении 337 гуанин замещен на аденин, устойчивая к имидазолиноновому гербициду, способная регенерировать в растение сахарной свеклы, причем указанная устойчивость передается при общепринятом кроссбридинге регенерированных растений с растениями, чувствительными к данному гербициду. 2. Линия клеток по п.1, которая получена из чувствительных клеток...
Линия клеток растения сахарной свеклы и растение сахарной свеклы, устойчивые к имидазолиноновым и сульфонилмочевинным гербицидам, семена указанного растения, способы получения указанной клеточной линии и гибридных растений свеклы, устойчивых к данным гербицидам, и применение имидазолинонового гербицида для борьбы с сорняками, растущими на полях с указанными растениями сахарной свеклы

Номер патента: 3297
Опубликовано: 24.04.2003
Авторы: Пеннер Доналд, Райт Терри Р.
Метки: гербицида, борьбы, способы, применение, сахарной, имидазолинонового, линия, гербицидам, гибридных, клеток, линии, устойчивых, получения, указанными, семена, устойчивые, указанной, растение, указанного, полях, растущими, данным, растений, сорняками, имидазолиноновым, клеточной, растения, растениями, свеклы, сульфонилмочевинным
Формула / Реферат:
1. Линия клеток растения сахарной свеклы с мутированным геном ацетолактатсинтазы, в котором в положении 562 цитозин замещен на тимин и в положении 337 гуанин замещен на аденин, устойчивая к имидазолиноновым и сульфонилмочевинным гербицидам, способная регенерировать в растение сахарной свеклы, причем указанная устойчивость передается при общепринятом кроссбридинге регенерированных растений с растениями, чувствительными к данным гербицидам. 2....