Везикулярные составы, содержащие производные органических кислот, и процесс их приготовления
Номер патента: 18246
Опубликовано: 28.06.2013
Авторы: Анес Элза Мария Рибеиро Сантос, Валенте Эмилия Алисе Дос Реис Торроаес, Симоес Марта Филипа Джесус Де Фреитас, Константино Луис Филипе Висенте










Формула / Реферат
1. Везикулярный состав, содержащий противомикобактериальное пролекарство, представляющее собой сложноэфирное производное слабой органической кислоты, имеющее общую формулу
где R1 является пиразиновой кислотой, бензольной кислотой или коричной кислотой;
R2 выбирают из группы октила, децила, додецила, тетрадецила, гексадецила,
при этом пролекарство имеет логарифм коэффициента распределения октанол/вода (log P) более 3,0,
с липосомным везикулярным переносчиком, включающим как минимум один липид, гидрогенизированный или нет, или смесь липидов, выбранных из фосфатидилхолина, фосфатидилглицерина, димиристоилфосфатидилхолина, димиристоилфосфатидилглицерина, дипальмитоилфосфатидилхолина, дипальмитоилфосфатидилглицерина, диэстеароилфосфатидилхолина, диэстеароилфосфатидилглицерина, диолеилфосфатидилхолина, диолеилфосфатидилглицерина, холестерина или его производных, сфингомиелина, арахидоновой кислоты, сфингозина, ганглиозидов, церамидов, фосфатидилинозитола и фосфатидной кислоты.
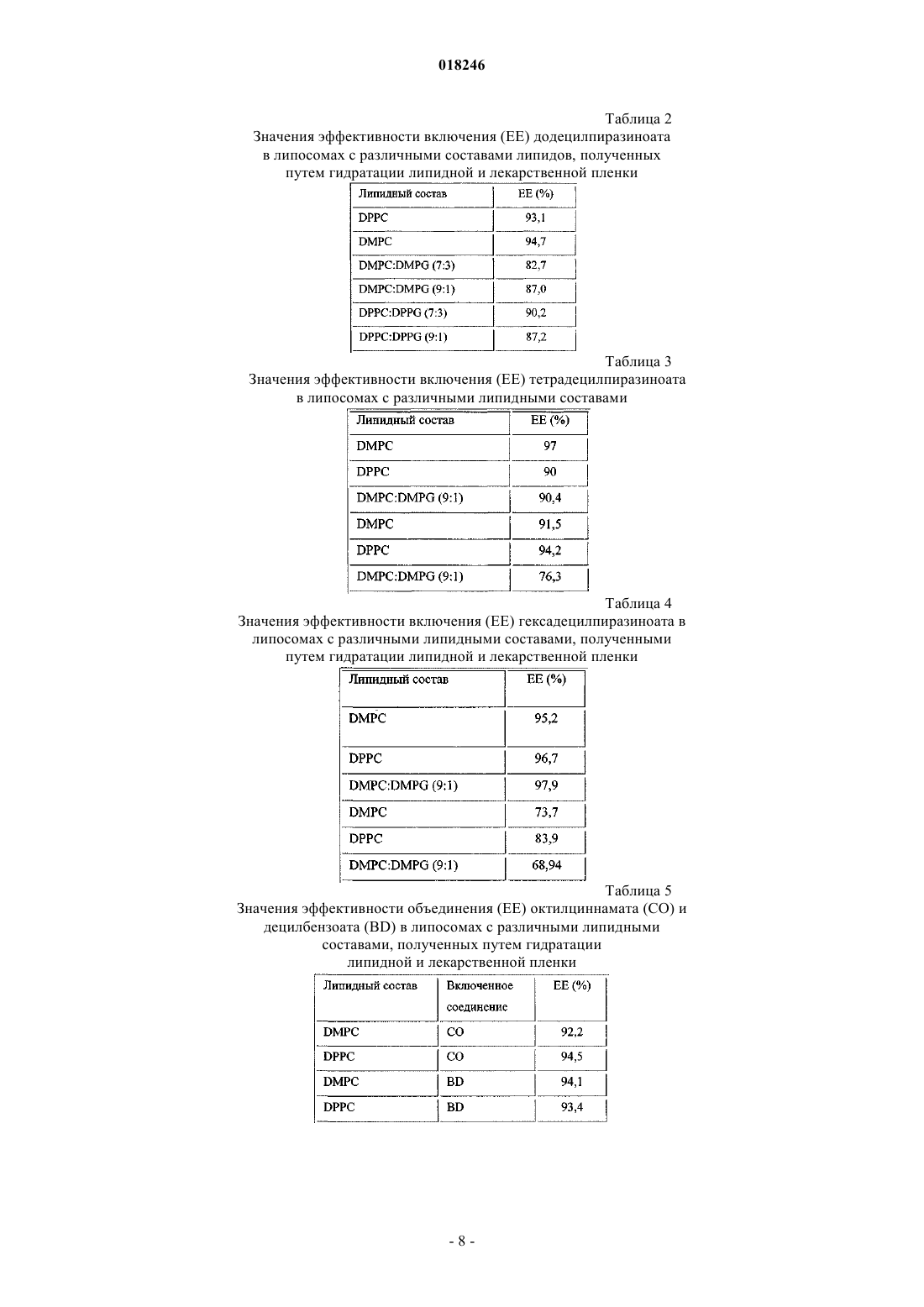
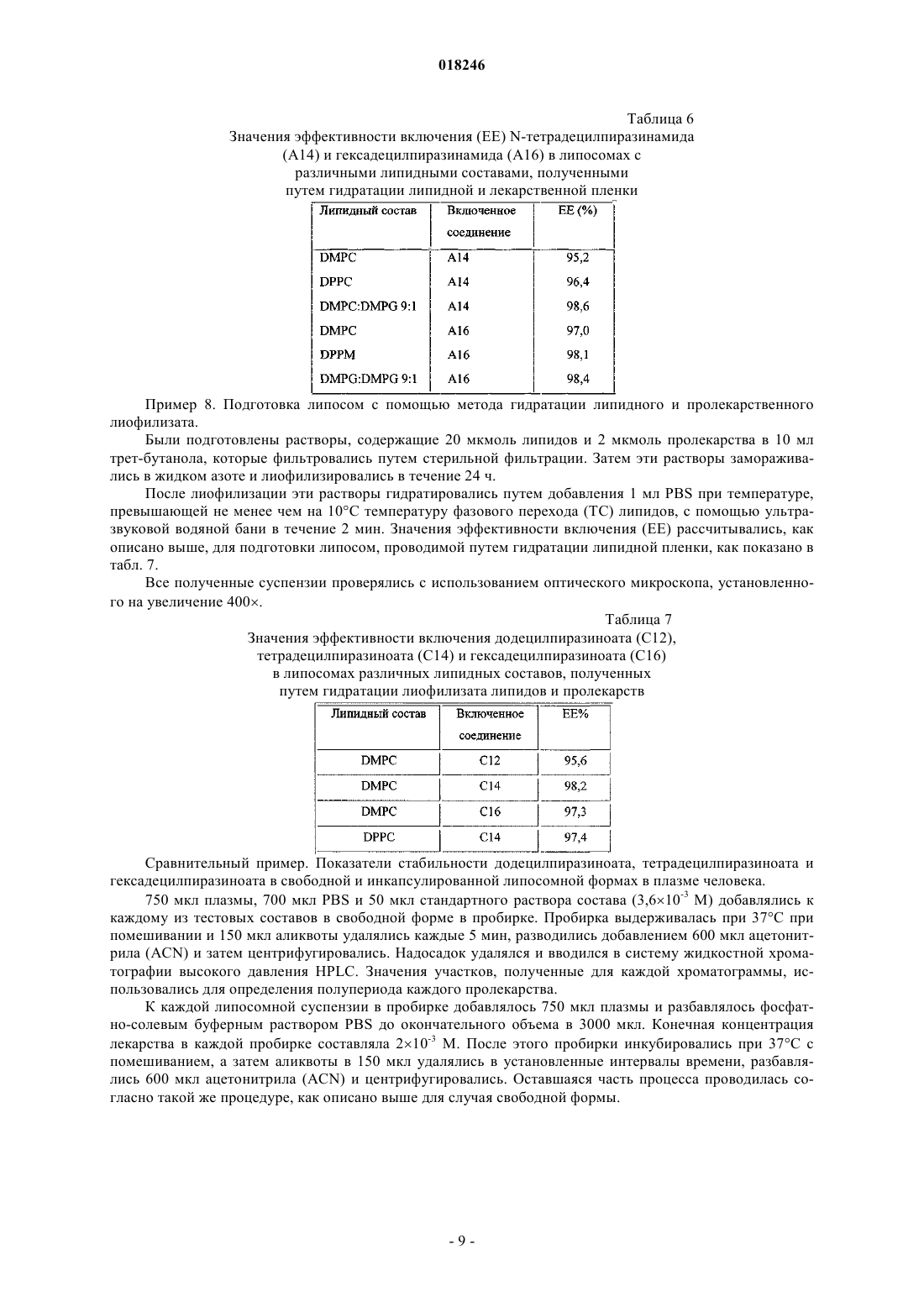
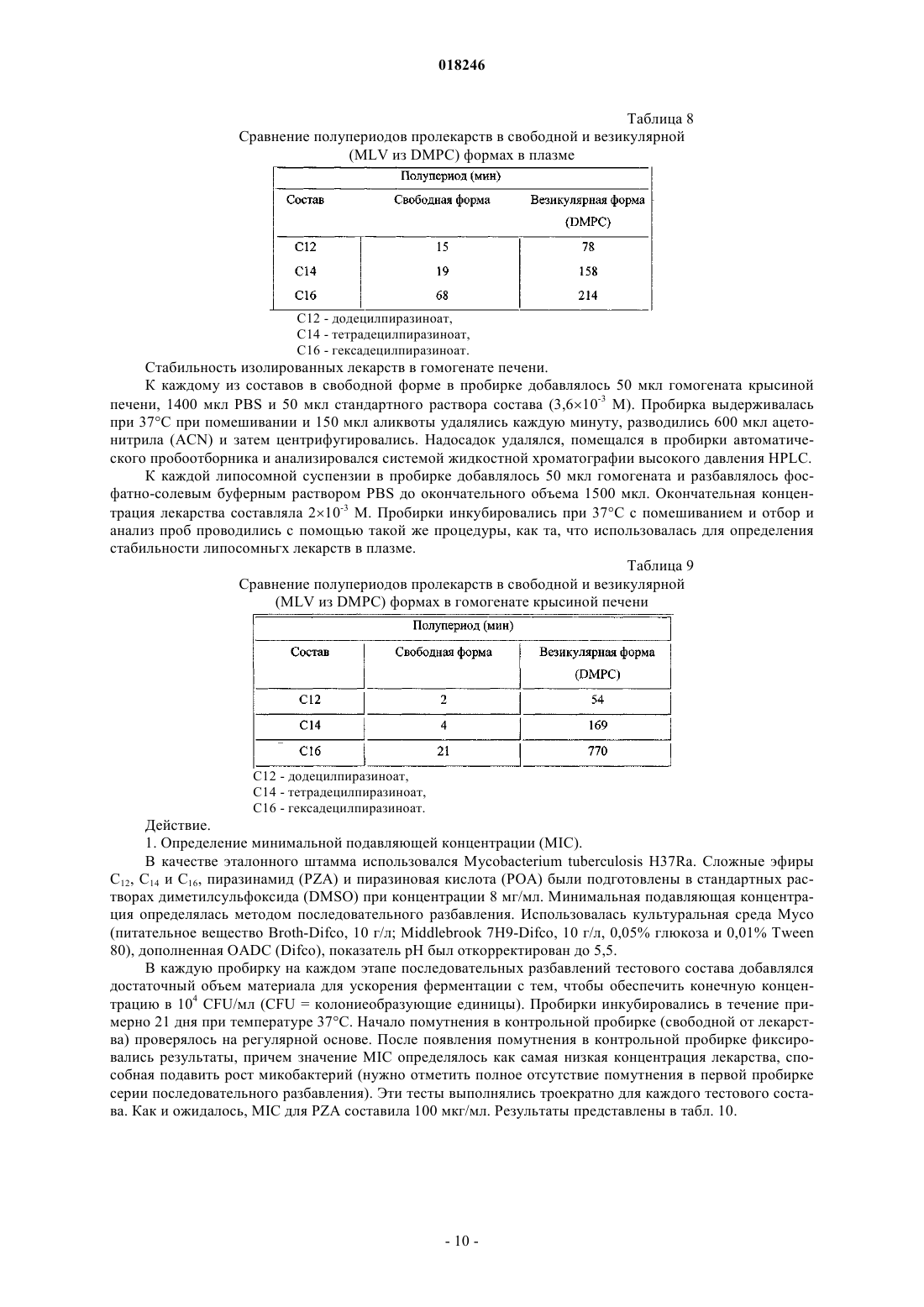
2. Везикулярный состав по п.1, характеризующийся тем, что пролекарство является додецилпиразиноатом.
3. Везикулярный состав по п.1, характеризующийся тем, что пролекарство является тетрадецилпиразиноатом.
4. Везикулярный состав по п.1, характеризующийся тем, что пролекарство является гексадецилпиразиноатом.
5. Везикулярный состав по п.1, характеризующийся тем, что пролекарство является децилбензоатом.
6. Везикулярный состав по п.1, характеризующийся тем, что пролекарство является октилциннаматом.
7. Везикулярный состав по пп.1-6, характеризующийся тем, что липосомный везикулярный состав имеет лиофилизированные или гидратизированные формы.
8. Везикулярный состав по пп.1-6, характеризующийся тем, что везикулярный носитель содержит дополнительные нейтральные или заряженные молекулы, не имеющие характер липидов или поверхностно-активного вещества.
9. Способ приготовления липосомного везикулярного состава по пп.1-8, характеризующийся тем, что содержит следующие этапы:
а) этерификация слабой органической кислоты, выбранной из пиразиновой, бензольной или коричной кислот;
б) подготовка раствора, содержащего липиды и пролекарство, полученное на этапе а), в подходящем растворителе;
в) удаление растворителя путем испарения или лиофилизации;
г) гидратация полученного продукта.
10. Лекарственный препарат для лечения туберкулезной инфекции, содержащий липосомный везикулярный состав по пп.1-8.
11. Лекарственный препарат по п.10 в форме для ингаляции, внутривенного, внутримышечного или подкожного введения.
Текст
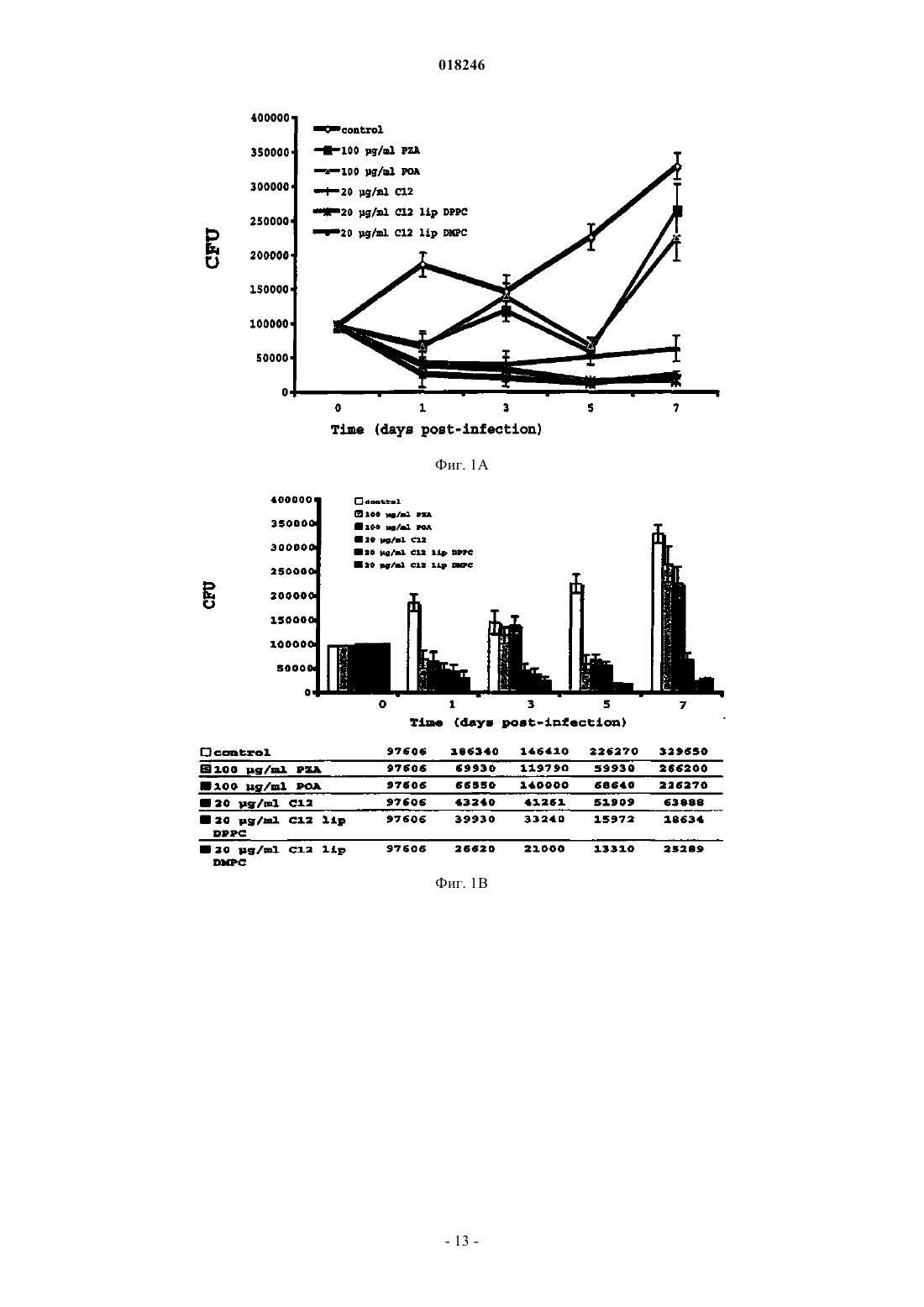
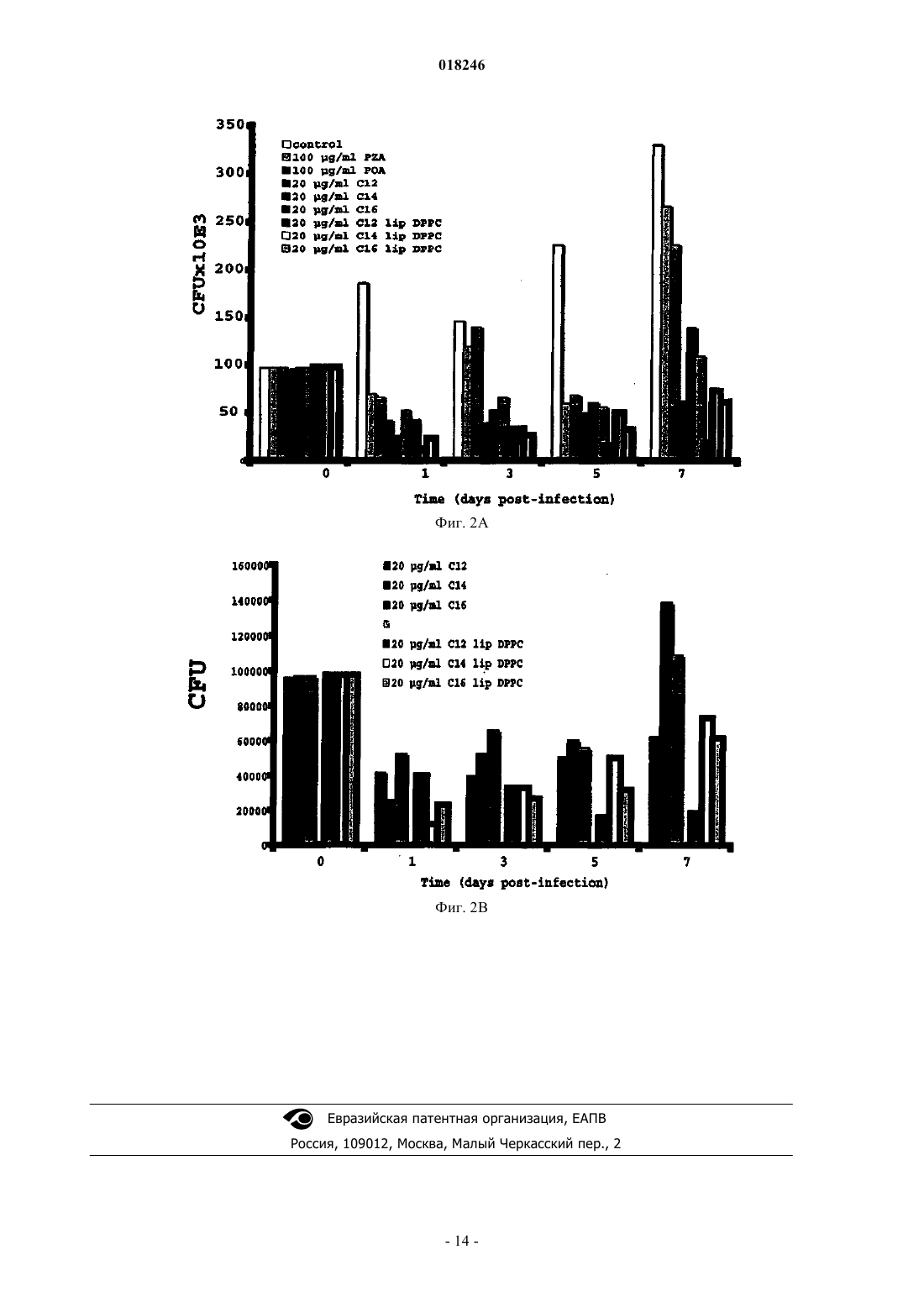
ВЕЗИКУЛЯРНЫЕ СОСТАВЫ, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ОРГАНИЧЕСКИХ КИСЛОТ, И ПРОЦЕСС ИХ ПРИГОТОВЛЕНИЯ Изобретение относится к везикулярному липосомному составу,содержащему противомикобактериальное пролекарство, представляющее собой сложноэфирное производное слабой органической кислоты, характеризующемуся тем, что он состоит из сочетания сложноэфирного производного слабой органической кислоты с общей формулой Константино Луис Филипе Висенте,Анес Элза Мария Рибеиро Сантос,Симоес Марта Филипа Джесус де Фреитас, Валенте Эмилия Алисе Дос Реис Торроаес (PT) Гаврилова Е.А. (RU) где R1 является пиразиновой, бензольной или коричной кислотой; R2 выбирают из группы октила,децила, додецила, тетрадецила, гексадецила, с липосомным везикулярным переносчиком, который защищает пролекарство от распада в плазме. Изобретение касается также процесса приготовления липосомных составов, новых пролекарств и фармацевтических композиций, пригодных для лечения туберкулеза и других видов микобактериоза.(71)(73) Заявитель и патентовладелец: УНИВЕРСИДАДЕ ДЕ ЛИСБОА (PT) Сфера, к которой относится изобретение Данное изобретение относится к везикулярным липосомным составам, содержащим противомикобактериальные сложноэфирные пролекарства органических кислот, процессу их приготовления и их лекарственным препаратам. Такие составы эффективно защищают пролекарство от распада в плазме и пригодны для лечения туберкулеза и других видов микобактериоза. Предпосылки создания изобретения Ряд органических кислот известны своим противомикобактериальным действием. Часто возникают проблемы с усваиванием этих соединений на клеточном уровне или их проникновением в клетку, что значительно ограничивает их применение в лечебных целях. Широко известным примером является пиразиновая кислота, активное вещество пиразинамида и парааминосалициловой кислоты. Недавно в заявке на патент WO 2004/062607 была продемонстрирована фармакологическая активность широкого спектра других слабых органических кислот, таких как бензойная кислота и салициловая кислота, в отношении штаммов Mycobacterium tuberculosis, что придает особое значение необходимости преодолеть указанные выше недостатки органических кислот. Альтернативным методом, позволяющим преодолеть фармакинетические проблемы, связанные с органическими кислотами, является использование пролекарств фармакологически активных соединений. Пролекарство означает новую молекулу, полученную в результате связывания фармакологически активной молекулы со второй химической структурной единицей. Получаемое в результате соединение демонстрирует физико-химические свойства, отличные от свойств исходной молекулы. Данное пролекарство может быть активным само по себе или может быть неактивным и становиться активным после ферментативного или химического разрушения с получением активной молекулы. В форме пролекарства это соединение может поглощаться, преодолевать различные препятствия и проникать в микобактерии. После проникновения внутрь оно активируется с выделением органической кислоты, которая затем может действовать в месте приложения действия. Чтобы быть эффективными, пролекарства должны быть в состоянии выдерживать процесс абсорбции и транспортировки и производить активное вещество после активации при помощи микобактериальных ферментов. Препарат пиразинамид является типичным примером пролекарства органической кислоты. Это соединение делает возможным внутриклеточное высвобождение пиразиновой кислоты после активации при помощи микобактерий. Однако, поскольку активация пиразинамида осуществляется только при помощи одного фермента, а именно пиразинамидазы, зачастую возникает резистентность в результате формирования мутаций микобактерий, что приводит к серьезным проблемам в ходе лечения. Для предотвращения возникновения проблем, связанных с резистентностью микроорганизмов к лекарственному средству, как это описано выше касательно пиразинамида, в качестве пролекарств были предложены сложные эфиры пиразиновой кислоты. Некоторые описания изобретения к патенту отражают общий интерес к использованию этих соединений при лечении туберкулеза и других видов микобактериоза. Патент США 4962111, выданный Велчу Дж.Т. и Цинамону М.Х. (Welch, J.Т.Cynamon, М.Н.),ссылается на сложные эфиры пиразиновой кислоты как на противотуберкулезные вещества. Заявляется использование сложных эфиров пиразиновой кислоты с короткой цепью в качестве противотуберкулезных веществ. Однако заявленные соединения демонстрируют очень низкую стабильность в плазмеantimycobacterial activity of pyrazoinic acid esters", Journal of Medicinal Chemistry, 1996, 39: 3394-3400) и не могут использоваться, так как они подвергаются гидролизу до достижения своих соответствующих мест приложения действия. Эти авторы утверждают, что стабильность сложных эфиров пиразиновой кислоты в плазме снижается экспоненциально с сокращением длины цепи алкокси. Кроме того, в патенте США 5643912, выданном 11 января 1996 г. тем же изобретателям, описывается использование аналогичных соединений для борьбы с инфекциями, вызванными Mycobacteriumavium. Однако, так как эти соединения лабильны под воздействием плазмы, их терапевтическое использование невозможно. Патент США 6120758 (Siddiqui Mukhtar et al.) описывает смесь консервантов для локально применяемых продуктов, включая водно-липосомные суспензии, служащую для предотвращения роста бактерий, дрожжей и плесени и не вызывающую раздражения кожи. В этих системах сложные эфиры парагидробензольной кислоты (парагидроксибензольной кислоты) декларируются как консерванты. Эти сложные эфиры не являются пролекарствами и не демонстрируют противомикобактериального действия. Липосомные составы описаны как основная форма продукта, используемая для локально применяемых продуктов, и не составлены для предотвращения гидролиза в плазме пролекарств слабых кислот в парентеросолюбильной форме или форме для ингаляции. Другие сложные эфиры, которые могут присутствовать в качестве смягчающих веществ в локально применяемых продуктах в виде эмульсия типа "вода-вмасле", включают в себя октилсалицитат, октилпальмитат и C12-C15-алкилбензоат. В WO 01/01949 раскрывается состав для очистки, например эмульсия, содержащая в качестве компонента сложный эфир, который, среди прочих, является смесью гексилдецилбензоата и бутилоктилбензоата. Эфиры в этих составах используются в качестве очищающих веществ, не оказывают терапевтического воздействия и не являются пролекарствами. Липосомные формы не раскрываются. Также в патенте США 2003/003069, выданном на имя Джона К. Карсона (John С. Carson et al.), сложные эфиры, такие как октилциннамат, используются в качестве очищающих веществ в многофазных поверхностноактивных составах, таких как пены для ванн, очистители для кожи и шампуни. В патенте США 4556495 раскрывается микроэмульсионная система, состоящая из алифатических карбоксилатов, таких как п-децилбензоат, п-додецилбензоат, п-тетрадецилбензоат или п-гексадецилбензоат, и вспомогательного поверхностно-активного вещества для восстановления масла из подповерхностной формации. И все же в этом патенте указанные п-децилбензоат, п-додецилбензоат,п-тетрадецилбензоат или п-гексадецилбензоат являются солями бензойной кислоты (карбоксилатами), а не сложными эфирами бензойной кислоты. Заявка на патент WO 2004/062607 также ссылается на метод лечения туберкулеза, в котором используются слабые органические кислоты или их предшественники. Однако проблема низкой стабильности сложных эфиров органических кислот в плазме остается нерешенной. Преимущество этих соединений, казалось бы, очевидно, так как эстеразы в изобилии присутствуют в микобактериях, и, следовательно, активация пролекарства может быть легко осуществлена in situ. Однако это нельзя подтвердить результатами исследований, так как эстеразы, которые присутствуют в человеческой плазме, подвергают пролекарство гидролизу до того, как оно достигнет целевых клеток, что не позволяет ему подействовать. Подробно описанные в литературе опыты, в которых липосомы и прочие везикулярные препараты вводились крысам, показали, что эти везикулы в первую очередь накапливаются в органах, содержащих большое количество клеток ретикулоэндотелиальной системы (РЭС), а именно в печени, селезенке, костном мозге и в легких. Это связано с тем, что имеет место фагоцитоз, вызванный местными макрофагами. Макрофаги - это клетки РЭС, образующие первую линию защиты организма и осуществляющие функции фагоцистоза, а также уничтожения инородных тел. Так как липосомные системы подвергаются фагоцитозу, вызванному макрофагами, их можно использовать в качестве носителя лекарственного средства к инфицированным макрофагам, увеличивая,таким образом, концентрацию лекарственного средства на тех целевых участках, которые больше всего нуждаются в лечении. Проблема с данными инфекциями заключается в том, что микобактерии могут препятствовать действию бактерицидных механизмов макрофагов, оставаясь внутри клеток в латентной форме длительное время и затем вызывая повторное инфицирование. Следовательно, успешное создание терапевтически активной внутриклеточной концентрации лекарственного препарата, такой как та, что ассоциируется с липосомными формулами лекарственных средств, является решающим в лечении этих микобактериозов. Основным резервуаром этиологического агента туберкулеза (М.tuberculosis) является человек(Homo sapiens). Но в некоторых случаях крупный рогатый скот (М.bovis) также может представлять собой важный резервуар инфекции. Некоторые виды микобактериоза характерны для животных и могут поражать только людей с ослабленной иммунной системой. Инфекции, вызванные М.avium у людей с ослабленной иммунной системой, являются единственным наиболее важным примером такого типа микобактериоза в плане охраны здоровья населения. Краткое изложение сущности изобретения Во время синтеза сложных эфиров с длинной цепью для их заключения в липосомы авторы неожиданно обнаружили, что они не только демонстрируют высокую противомикобактериальную активность,но также являются чрезвычайно устойчивыми к гидролизу в плазме и, в частности, после их внедрения в липосомы. И действительно, заключение пролекарства в везикулы, такие как липосомы, защищает соединения от гидролиза в плазме, одновременно сохраняя их активность. Авторы изобретения пришли к выводу, что путем объединения пролекарства подходящей структуры с липосомными везикулярными препаратами можно получить лекарственное средство, высвобождающее слабую кислоту в месте приложения действия, защищая, таким образом, пролекарство от гидролиза эстеаратами сыворотки. В контексте данного изобретения термин "везикулярный" означает замкнутую структуру, содержащую внутреннюю полость, обычно заполненную жидкостью, например липосомы. Изучив гидролиз ряда сложных эфиров органических кислот, авторы также обнаружили, что сложные эфиры бензойной кислоты с длинными алкоксиловыми цепями особенно устойчивы к гидролизу в плазме. Это показано, что, действительно, возможно увеличить устойчивость пролекарств на основе органических кислот к гидролизу в плазме, что, возможно, позволит решить вышеуказанную проблему. Таким образом, данное изобретение касается новых везикулярных рецептур, содержащих пролекарства слабых органических кислот в сочетании с липосомным везикулярным носителем, а также процесса их приготовления, новых пролекарств слабых органических кислот и лекарственных средств по указанным выше рецептурам. Липосомы - это везикулярные системы, состоящие из липидных микрочастиц или наночастиц. Включение пролекарств в липосомы защищает пролекарства во время их циркуляции в организме. Эти системы имеют дополнительное преимущество естественной подверженности фагоцитозу, который осуществляется клетками ретикулоэндотелиальной системы, а также того, что они способствуют накоплению больших концентраций пролекарства в органах, которые содержат большое количество клеток этой системы. Пролекарства, предусмотренные данным изобретением, получаются из органических кислот,имеющих общую формулу где R1 является пиразиновой бензольной или коричной кислотой. Обычно эти кислоты имеют pKa от 1 до 5. Как известно специалистам, многочисленные типы пролекарств карбоновых кислот после активации способны высвобождать карбоновую кислоту. Примером могут служить производные сложных эфиров, амиды и ацилоксиалкиловые сложные эфиры. Подходящими пролекарствами, которые используют для новых везикулярных рецептур, являются сложные эфиры слабых кислот, используемые в везикулярных рецептурах данного изобретения, имеющих общую формулу где R1 описано выше в формулах (I) и (II); аR2 выбирают из группы, содержащей октил, децил, додецил, тетрадецил, гексадецил. Пролекарства для везикулярных рецептур данного изобретения предпочтительно являются теми, у которых количество атомов углеродов в алкоксиловых цепях приводит к значению логарифма коэффициента распределения октанола и воды в пролекарстве больше или равному 3. Логарифм коэффициента распределения может быть без труда рассчитан специалистами. Коэффициент распределения является важным фактором поддержания ассоциации пролекарства с липофильными частицами везикула, предотвращая его рассеивание в водной среде. Конкретные новые пролекарства, не имеющие приведенных выше недостатков, такие как додецилпиразиноат, тетрадецилпиразиноат, гексадецилпиразиноат, децилбензоат, также являются объектом данного изобретения. Октилциннамат является еще одним предпочтительным пролекарством, не имеющим указанных выше недостатков. Благодаря своей липофильной природе пролекарства органических кислот являются особенно подходящими для высокопроизводительного включения в липосомы - коллоидную систему, обычно используемую для транспортировки лекарственного препарата, преимущество которой заключается в отсутствии необходимости выделять включенный в нее препарат. Это очень важно, особенно для промышленного изготовления. Пролекарства, описываемые в данном изобретении, также могут использоваться в других коллоидных системах транспортировки лекарственных препаратов, таких как макромолекулярные комплексы,нанокапсулы, микросферы и мицеллы. Эти коллоидные системы имеют разрушаемые естественными факторами и нетоксичные частицы диаметром от 50 нм до 2 мкм. В данном изобретении предусмотрена липосомная транспортная система. Липосомы образуются посредством дисперсии фосфолипидов в водной среде. Фосфолипиды имеют полярную часть - "голову",и неполярную часть - "хвост". Благодаря своему полярному характеру, голова имеет сродство к воде и другим полярным веществам, в то время как неполярный хвост имеет сродство к неполярным частям,таким как, например, хвосты других фосфолипидов, находящихся в непосредственной близости. Эта характеристика означает, что при дисперсии в избытке воды фосфолипиды расположатся бислоями. Полярные головы будут обращены во вне, где они могут контактировать с молекулами воды, а хвосты будут обращены внутрь, прислоняясь друг к другу. Липосомы являются везикулами, которые образуются одним или несколькими бислоями фосфолипидов, окружающих внутреннее водное пространство. В зависимости от метода изготовления липосомы могут значительно отличаться своим размером и количеством слоев. В целом, их можно разделить на однослойные везикулы, когда у них имеется только один бислой фосфолипидов, и многослойные фосфолипиды, если у них несколько бислоев фосфолипидов. Оба эти типа везикул можно классифицировать в соответствии с их диаметром, а именно как малые(0,025-0,1 мкм) и большие (0,1 мкм). Липосомы также можно классифицировать по процедуре их получения, и это приводит к включению нескольких подгрупп для каждого типа везикул. Наиболее распространенными липосомами являются многослойные везикулы (MLV), состоящие из нескольких фосфолипидных слоев, окружающих внутреннее водное пространство. Эти системы обычно имеют диаметр от 100 нм до 4 мкм, но их размер можно контролировать, например пропуская их под давлением через отверстия определенного размера. Когда система MLV подвергается разрушению ультразвуком при помощи зонда, образуются более мелкие везикулы, известные как малые однослойные везикулы (SUV). Эти везикулы содержат только один бислой фосфолипидов, окружающий внутреннее водное пространство. При подготовке липосом, предусмотренных данным изобретением, обычно используются фосфолипиды, холестерин и его производные, а также прочие липидные или нелипидные молекулы. Примеры включают в себя следующие липиды, гидрогенизированные или нет, по отдельности или в смеси: фосфатидилхолин (PC), фосфатидилглицерин (PG), димиристоилфосфатидилхолин (DMPC), димиристоилфосфатидилглицерин (DMPG), дипальмитоилфосфатидилхолин (DPPC), дипальмитоилфосфатидилглицерин(DPPG), диэстеароилфосфатидилхолин (DSPC), диэстеароилфосфатидилглицерин (DSPG), диолеилфосфатидилхолин (DOPC), диолеилфосфатидилглицерин (DOPG), холестерин или его производные (Chol),сфингомиелин (SM), арахидоновая кислота, сфингозин, ганглиозиды, церамиды, фосфатидилинозитол(PI) и фосфатидная кислота (РА). К липосомам могут быть добавлены липидные или нелипидные вещества для содействия ассоциации липосом с целевыми клетками, интернализации липосом макрофагами или даже увеличения полупериода циркуляции. Некоторые примеры таких соединений включают антитела, церамиды, полиэтиленгликоли и полиэтиленгликоли-липид коньюгаты. Пролекарства карбоновой кислоты могут быть приготовлены из тех же самых соединений, используя известные специалистам методы. Производные сложных эфиров могут быть синтезированы, к примеру, путем проведения реакции слабой кислоты с тионилхлоридом или другим подходящим реагентом для получения соответствующего ацилгалоида и последующей реакции этого ацилгалоида со спиртом или фенолом для получения пролекарства. Эти производные сложных эфиров также могут быть получены в достаточном количестве из других функциональных групп, например, путем реакции кислоты со спиртом в кислой среде. Эти альтернативные реакции широко известны и могут быть без труда проведены без необходимости дополнительного тестирования. Согласно данному изобретению предпочтительный метод включения пролекарств органических кислот в липосомы состоит из лиофилизации раствора, содержащего пролекарство и липидные компоненты, и последующей гидратации лиофилизата. Этот метод позволяет получать стерильные липосомы, быстро увеличивать их количество до масштабов промышленного производства и с легкостью готовить препарат, который можно будет хранить в виде лиофилизата длительное время до использования, что упрощает как хранение, так и транспортировку. Или же липосомы могут быть без проблем получены путем гидратации пленки, содержащей фосфолипиды и пролекарство. Вкратце, процесс приготовления липосомного состава в соответствии с данным изобретением предпочтительно состоит из следующих этапов: а) этерификация слабой органической кислоты, выбранной из пиразиновой, бензольной или коричной кислот; б) подготовка раствора, содержащего липиды и пролекарство, полученное на этапе а), в подходящем растворителе; в) удаление растворителя путем испарения или лиофилизации; г) гидратация полученного продукта. Тем не менее, можно использовать любой известный специалистам процесс приготовления липосомных везикулярных систем, и это не вызовет отступления от сути и формы данного изобретения. Везикулярные системы, включающие в себя пролекарство, предусмотренное данным изобретением,могут вводиться в организм различными способами, включая внутривенное, внутримышечное, интраперитонеальное введение, а также вдыхание. Была протестирована активность пролекарств органических кислот, предусмотренных данным изобретением, на микобактериях и обнаружено, что они характеризуются более высокой активностью invitro, чем первоначальные органические кислоты. Была также проведена проверка экспериментальным путем, которая показала, что включение пролекарств в везикулы защищает их от распада в плазме, а также от распада, вызванного печеночными эстеразами. Используя макрофаги, инфицированные М.tuberculosis, обнаружено, что пролекарства, предусмотренные данным изобретением, в своей липосомной везикулярной форме были активны в борьбе с внутриклеточными микобактериями. Принимая во внимание естественный фагоцитоз липосом, осуществляемый клетками SRE, везикулярные пролекарства являются особенно подходящими для лечения туберкулеза и прочих микобактериозов. Сочетание пролекарств органических кислот с везикулярным носителем повышает стабильность указанных пролекарств в плазме и меняет фармакокинетику указанных соединений, обеспечивая получение составов с характеристиками, повышающими активность этих соединений. Так как молекулы, о которых идет речь, демонстрируют активность, направленную против микобактерий, этот состав можно использовать в лечении туберкулеза и прочих инфекций. Следовательно, следующим объектом данного изобретения являются фармацевтические составы,состоящие из определенного количества везикулярных композиций, как указано выше, достаточного для получения терапевтически эффективного количества слабых органических кислот, имеющих формулы(I) или (II), описанные выше. Согласно данному изобретению предпочтительным фармацевтическим со-4 018246 ставом является состав для ингаляций, внутривенного, внутримышечного или подкожного введения. Таким образом, в качестве отдельного объекта данное изобретение касается рецептуры состава для использования в качестве лекарственного средства. Предусмотренная данным изобретением рецептура используется для приготовления лекарственного препарата, предназначенного для лечения заболеваний,связанных с микобактериальными инфекциями. В частности, для лечения инфекции, вызванной М.tuberculosis, инфекции, вызванной М.avium, или инфекций, вызванных другими микобактериями. Метод лечения туберкулезной инфекции или других видов микобактериоза у животных предполагает введение животным определенного количества препарата, предусмотренного данным изобретением,достаточного для выработки терапевтически эффективного количества слабой органической кислоты с общей формулой (I) или (II) для лечения указанной выше инфекции. Указанный препарат вводится путем вдыхания, внутривенно, внутримышечно или подкожно. Предпочтительно вводить препарат, предусмотренный изобретением, млекопитающему, которое в нем нуждается. Еще более предпочтительным является то, чтобы таким млекопитающим был человек. Метод использования в соответствии с данным изобретением состоит из введения указанного препарата в форме лекарственного препарата или в сочетании, как описано в данном документе. Термин "лечение", используемый во всех спецификациях и в пунктах патентной формулы, охватывает все различные формы или способы лечения, известные соответствующим специалистам, и, в частности, включает в себя профилактику, замедление развития и лечение с целью излечения. Доза in vitro MIC (анализ питательной среды) составляла от 10 до 40 и 20 мкм /мл (анализ инфицированных макрофагов). Терапевтически эффективное количество in vivo может быть различным в зависимости от состава и способа введения. Для данного изобретения показательным является то, что активность состава С 12 (фиг. 1 а и 1b), в соответствии с изобретением, как в свободной, так и в липосомно-инкапсулированной форме продемонстрировала 5-10-кратное увеличение нейтрализирующей активности in vivo по сравнению как с контрольными образцами, так и обработкой PZA и РОА. Инкапсуляция С 12 в липосомах увеличивает бактерицидный эффект примерно на 50% относительно того же состава в свободной форме. Краткое описание чертежей Теперь данное изобретение будет описано, ссылаясь на прилагаемые чертежи, на которых показано: на фиг. 1 А - график, на котором явно видно, что состав С 12 как в свободной, так и в липосомноинкапсулированной формах демонстрирует 5-10-кратное увеличение нейтрализирующей активности invivo по сравнению как с пиразинамидом (PZA), так и с пиразиновой кислотой (РОА), на этой фигуре также показано, что липосомные формы более активны, чем свободная форма; на фиг. 1 В - гистограмма, на которой представлены те же самые результаты, что и на фиг. 1 А; на фиг. 2 А - результаты соединений в свободной и липосомной формах относительно контрольных образцов и образцов, обработанных PZA или РОА; на фиг. 2 В - бактерицидный эффект трех новых составов данного изобретения, а именно: додецилпиразиноата, тетрадецилпиразиноата и гексадецилпиразиноата, в свободной и липосомной формах, на этой фигуре также показано, что все три липосомные формы более активны, чем свободная форма. Подробное описание изобретения Следующие примеры иллюстрируют синтез пролекарств слабых кислот, подготовку и определение параметров липосомных препаратов, содержащих пролекарства слабых кислот, стабильность составов в плазме и почечном гомогенате, а также активность пролекарств в свободной и везикулярной формах. Пример 1. Синтез додецилпиразиноата. 25 мл тионилхлорида добавляли в 26,5 ммоль пиразиновой кислоты (3,3 г) и нагревали этот раствор с обратным потоком в течение 2 ч. Вначале наблюдался розовый цвет, который постепенно становился все темнее. Излишек тионилхлорида был выпарен и был получен сублимированный хлорид пиразиновой кислоты в виде острых белых кристаллов. Эти кристаллы были немедленно растворены в 13 мл дихлорметана, после чего смесь была помещена в ванну со льдом, и в нее медленно были добавлены 26,5 ммоль додеканола и 3,70 мл дистиллированного триэтиламина. В ванной со льдом реакция протекала примерно в течение 0,5 ч, после чего реакция протекала при комнатной температуре. Затем реакционная смесь была нагрета и подвергалась дефлегмации в течение 1 ч, затем была оставлена на ночь (примерно на 12 ч) при комнатной температуре. Затем смесь еще раз нагревали и подвергали дефлегмации еще в течение 40 мин, после чего была проведена тонкослойная хроматография (TLC) с использованием гексана:этилацетата (5:1) в качестве элюента. Затем реакционная смесь была отфильтрована и фильтрат был последовательно промыт 20 мл дистиллированной воды и 20 мл насыщенного раствора гидрокарбоната натрия. Раствор был обработан безводным сульфатом магния и растворитель был выпарен. Соединение дважды очищалось методом колоночной хроматографии с использованием гексана:этилацетата (5:1) в качестве элюента. После определения и подтверждения структуры методом ядерного магнитного резонанса (ЯМР) и инфракрасной спектроскопии (ИК) был получен чистый продукт в виде восковидного белого твердого вещества с точкой плавления 33-34 С и выходом в размере 46%. Vmax (см-1) = 1723. Характеристики, полученные методом ЯМР, приведены в табл. 1. Пример 2. Синтез тетрадецилпиразиноата. Был использован тот же процесс, что и при синтезе додецилпиразиноата, описанный в примере 1, за исключением того, что использовалось 26,5 ммоль 1-тетрадеканола. Соединение очищалось методом колоночной хроматографии с использованием гексана:этилацетата (1:1) в качестве элюента. Был получен очищенный продукт в виде восковидного белого твердого вещества с точкой плавления 43-44 С и конечным выходом в размере 42%. Vmax (см-1) = 1722. Характеристики, полученные методом ЯМР, приведены в табл. 1. Пример 3. Синтез гексадецилпиразиноата. Был использован тот же процесс, что и при синтезе додецилпиразиноата, описанный в примере 1, за исключением того, что использовалось 26,5 ммоль 1-гексадеканола. Соединение очищалось методом колоночной хроматографии с использованием гексана:этилацетата (1:1) в качестве элюента. Был получен очищенный продукт в виде восковидного белого твердого вещества с точкой плавления = 53-54 С и конечным выходом в размере 41%. Vmax (см-1) = 1723. Характеристики, полученные методом ЯМР, приведены в табл. 1. Пример 4. Синтез децилбензоата. 25 ммоль свежеотогнанного триэтиламина добавлялись в раствор 12 ммоль 1-деканола в 25 мл сухого этилового эфира. Затем смесь взбалтывалась с покапельным добавлением 12,8 ммоль хлорида бензоила. Был предусмотрен дефлегматор, и реакция была оставлена проходить при взбалтывании в течение 2 ч. После этого было добавлено 30 мл воды и помешивание продолжалось еще 15 мин. Органическая фаза была промыта водным раствором 5% HCl (230 мл), а после этого - насыщенным раствором гидрокарбоната натрия (230 мл). И, наконец, органическая фаза была высушена при помощи сульфата магния, а растворитель был удален при помощи вакуумного испарения. Децилбензоат был очищен методом силикагелевой колоночной хроматографии с использованием гексана:этилацетата (5:2) в качестве элюента. Конечный продукт был получен в виде бесцветной жидкости, конечный выход которой составил 74%. Пример 5. Синтез октилциннамата. Был использован тот же процесс, что и при синтезе децилбензоата, описанный в примере 4, за исключением того, что имела место реакция 12 ммоль октанола с 12,8 ммоль циннамилхлорида. Конечный продукт был получен в виде масла, конечный выход которого составил 71%. Пример 6. Синтез N-тетрадецилпиразинамида. Был использован процесс, аналогичный тому, что был использован при синтезе тетрадецилпиразиноата и описан в примере 2, за исключением того, что использовалось 26,5 ммоль тетрадециламина. Для очистки продукта методом колоночной хроматографии в качестве элюента использовались гексан:этилацетат (1:1). Был получен чистый продукт в виде белого твердого вещества с точкой плавления 80-81 С и конечным выходом в размере 28%. Таблица 1 Химические сдвиги 1H 13 С NMR C (ppm) сложноэфирных производных пиразиновой кислоты (в CDCl3, эталон (СН 3)4Si) Пример 7. Подготовка липосом путем гидратации липидной и лекарственной пленки. Различные липиды (20 мкмоль) и пролекарство (2 мкмоль) взвешивались, переносились в колбу с круглым дном, растворялись в хлороформе, и органический растворитель испарялся с использованием вращающегося испарителя для формирования липидной пленки. Затем эта пленка высушивалась в вакуумной бомбе для удаления остатков хлороформа и добавлялся 1 мл буфера изотонического фосфата pH 7,4 (PBS) при температуре, по крайней мере на 10 С превышающей температуру фазового перехода (ТС) используемых липидов. Смесь размешивалась в течение дополнительных 5 мин до полной гидратации липидной пленки и через несколько минут покоя снова размешивалась в течение еще 5 мин. С целью определения эффективности включения (EE) были собраны аликвоты липосомной суспензии после гидратации и липосомной суспензии, полученной после центрифугирования, надосадочного удаления и повторного суспендирования осадка в его начальном объеме (до центрифугирования). EE рассчитывалась как соотношение между концентрацией пролекарства в повторно суспендированной липосомной суспензии и концентрацией пролекарства в начальной липосомной суспензии. В табл. 2-6 показаны значения EE, полученные для нескольких составов различных пролекарств. Все подготовленные суспензии проверялись с использованием оптического фазоконтрастного микроскопа, установленного на увеличение 400. Таблица 2 Значения эффективности включения (EE) додецилпиразиноата в липосомах с различными составами липидов, полученных путем гидратации липидной и лекарственной пленки Таблица 3 Значения эффективности включения (EE) тетрадецилпиразиноата в липосомах с различными липидными составами Таблица 4 Значения эффективности включения (EE) гексадецилпиразиноата в липосомах с различными липидными составами, полученными путем гидратации липидной и лекарственной пленки Таблица 5 Значения эффективности объединения (EE) октилциннамата (СО) и децилбензоата (BD) в липосомах с различными липидными составами, полученных путем гидратации липидной и лекарственной пленки Таблица 6 Значения эффективности включения (EE) N-тетрадецилпиразинамида(А 14) и гексадецилпиразинамида (А 16) в липосомах с различными липидными составами, полученными путем гидратации липидной и лекарственной пленки Пример 8. Подготовка липосом с помощью метода гидратации липидного и пролекарственного лиофилизата. Были подготовлены растворы, содержащие 20 мкмоль липидов и 2 мкмоль пролекарства в 10 мл трет-бутанола, которые фильтровались путем стерильной фильтрации. Затем эти растворы замораживались в жидком азоте и лиофилизировались в течение 24 ч. После лиофилизации эти растворы гидратировались путем добавления 1 мл PBS при температуре,превышающей не менее чем на 10 С температуру фазового перехода (ТС) липидов, с помощью ультразвуковой водяной бани в течение 2 мин. Значения эффективности включения (EE) рассчитывались, как описано выше, для подготовки липосом, проводимой путем гидратации липидной пленки, как показано в табл. 7. Все полученные суспензии проверялись с использованием оптического микроскопа, установленного на увеличение 400. Таблица 7 Значения эффективности включения додецилпиразиноата (С 12),тетрадецилпиразиноата (С 14) и гексадецилпиразиноата (С 16) в липосомах различных липидных составов, полученных путем гидратации лиофилизата липидов и пролекарств Сравнительный пример. Показатели стабильности додецилпиразиноата, тетрадецилпиразиноата и гексадецилпиразиноата в свободной и инкапсулированной липосомной формах в плазме человека. 750 мкл плазмы, 700 мкл PBS и 50 мкл стандартного раствора состава (3,610-3 М) добавлялись к каждому из тестовых составов в свободной форме в пробирке. Пробирка выдерживалась при 37 С при помешивании и 150 мкл аликвоты удалялись каждые 5 мин, разводились добавлением 600 мкл ацетонитрила (ACN) и затем центрифугировались. Надосадок удалялся и вводился в систему жидкостной хроматографии высокого давления HPLC. Значения участков, полученные для каждой хроматограммы, использовались для определения полупериода каждого пролекарства. К каждой липосомной суспензии в пробирке добавлялось 750 мкл плазмы и разбавлялось фосфатно-солевым буферным раствором PBS до окончательного объема в 3000 мкл. Конечная концентрация лекарства в каждой пробирке составляла 210-3 М. После этого пробирки инкубировались при 37 С с помешиванием, а затем аликвоты в 150 мкл удалялись в установленные интервалы времени, разбавлялись 600 мкл ацетонитрила (ACN) и центрифугировались. Оставшаяся часть процесса проводилась согласно такой же процедуре, как описано выше для случая свободной формы. Таблица 8 Сравнение полупериодов пролекарств в свободной и везикулярной Стабильность изолированных лекарств в гомогенате печени. К каждому из составов в свободной форме в пробирке добавлялось 50 мкл гомогената крысиной печени, 1400 мкл PBS и 50 мкл стандартного раствора состава (3,610-3 М). Пробирка выдерживалась при 37 С при помешивании и 150 мкл аликвоты удалялись каждую минуту, разводились 600 мкл ацетонитрила (ACN) и затем центрифугировались. Надосадок удалялся, помещался в пробирки автоматического пробоотборника и анализировался системой жидкостной хроматографии высокого давления HPLC. К каждой липосомной суспензии в пробирке добавлялось 50 мкл гомогената и разбавлялось фосфатно-солевым буферным раствором PBS до окончательного объема 1500 мкл. Окончательная концентрация лекарства составляла 210-3 М. Пробирки инкубировались при 37 С с помешиванием и отбор и анализ проб проводились с помощью такой же процедуры, как та, что использовалась для определения стабильности липосомньгх лекарств в плазме. Таблица 9 Сравнение полупериодов пролекарств в свободной и везикулярной Действие. 1. Определение минимальной подавляющей концентрации (MIC). В качестве эталонного штамма использовался Mycobacterium tuberculosis H37Ra. Сложные эфирыC12, C14 и C16, пиразинамид (PZA) и пиразиновая кислота (РОА) были подготовлены в стандартных растворах диметилсульфоксида (DMSO) при концентрации 8 мг/мл. Минимальная подавляющая концентрация определялась методом последовательного разбавления. Использовалась культуральная среда Мусо(питательное вещество Broth-Difco, 10 г/л; Middlebrook 7H9-Difco, 10 г/л, 0,05% глюкоза и 0,01% Tween 80), дополненная OADC (Difco), показатель pH был откорректирован до 5,5. В каждую пробирку на каждом этапе последовательных разбавлений тестового состава добавлялся достаточный объем материала для ускорения ферментации с тем, чтобы обеспечить конечную концентрацию в 104 CFU/мл (CFU = колониеобразующие единицы). Пробирки инкубировались в течение примерно 21 дня при температуре 37 С. Начало помутнения в контрольной пробирке (свободной от лекарства) проверялось на регулярной основе. После появления помутнения в контрольной пробирке фиксировались результаты, причем значение MIC определялось как самая низкая концентрация лекарства, способная подавить рост микобактерий (нужно отметить полное отсутствие помутнения в первой пробирке серии последовательного разбавления). Эти тесты выполнялись троекратно для каждого тестового состава. Как и ожидалось, MIC для PZA составила 100 мкг/мл. Результаты представлены в табл. 10. Таблица 10 Минимальные подавляющие концентрации (MIC) для каждого лекарства Как показано в табл. 10, составы С 12, С 14 и С 15 продемонстрировали, in vitro, в 10 раз более низкие значения MIC по сравнению со значениями для PZA и РОА, проявляя более высокий бактерицидный эффект при прямом контакте с М.tuberculosis при pH на уровне 5,5. 2. Противомикобактериальная активность составов в свободной и инкапсулированной формах с использованием макрофага, инфицированного Mycobacterium tuberculosis. При оценке противомикобактериальной активности применялся традиционный метод (Anes et al.,Nat. Cell Biol., 2003) с использованием клеточной культуры макрофага крысы (клеточная линия J774.A1). Кратковременно макрофаг помешался в среду DMEM с высокой концентрацией глюкозы, дополненную 10% эмбриональной бычьей сывороткой, в 24-ячейковые планшеты культуры ткани при 37 С в атмосфере 5% углекислого газа. Как только было получено примерно 80% слияние, клетки инфицировались М.tuberculosis H37Ra с такой скоростью, которая обеспечила концентрацию инокулята на уровне 106 микобактерий/мл на ячейку. После 3 ч бактериального внедрения ожидалось 1-5 бацилл на инфицированную клетку и всего около 10 Е 4-5 бактерий/мл на ячейку. После этого три раза проводилось промывание инфицированных культур буферным раствором PBS с pH 7 с целью удаления всех бактерий, не интернализированных макрофагом. Свежая среда DMEM добавлялась с тестовым составом и без него. Инкубационный период составлял 7 дней с использованием свежей среды каждые 3 дня. Составы добавлялись после 3 ч интернализации и оставались в контакте с инфицированными клетками, пока не добавлялась свежая среда DMEM. По истечении 3 ч, 1, 3, 5 и 7 дней от начала инфекции, внутриклеточные бактерии восстанавливались для лизирования инфицированного макрофага с помощью 1% водного раствора IGEPAL (Sigma). При этой концентрации макрофаги лизируются, не оказывая эффекта на жизнеспособность микобактерий. После последовательных разбавлений лизата водой выжившие бактерии культивировались в средеMiddlebrook 7H10, дополненной OADC (Difco). После примерно 2 недель инкубации при 37 С подсчитывалось количество формирующих единицу колоний (UFC). Каждый тест проводился троекратно в рамках независимых экспериментов. Как видно на фиг. 1 А, состав С 12 в свободной или липосомно-инкапсулированной форме показал 5-10-кратное увеличение нейтрализирующей активности in vivo по сравнению как с контрольными образцами, так и обработкой PZA и РОА. Этот эффект увеличивался после 7 дней инфекции, когда в первых 3 случаях бактерии восстанавливали свою способность внутриклеточного роста, в то время как в составах, содержащих С 12, наблюдалась латентность. Этот эффект более очевиден на фиг. 1 В, который показывает те же результаты в форме гистограммы. Инкапсуляция С 12 в липосомах увеличивает бактерицидный эффект примерно на 50% относительно того же состава в свободной форме. Однако в случае с составами DPPC и DMPC такие значительные различия не наблюдались. На фиг. 2 А сравниваются результаты, полученные для всех составов, как в свободной, так и в везикулярной формах, по сравнению с контрольным, обработкой PZA и обработкой РОА. Все эти пролекарства как в свободной, так и в везикулярной формах были более бактерицидными, чем эталонное пролекарство. Из всех новых пролекарств С 12 как в свободной, так и в везикулярной формах показало наибольший бактерицидный эффект (фиг. 2 В). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Везикулярный состав, содержащий противомикобактериальное пролекарство, представляющее собой сложноэфирное производное слабой органической кислоты, имеющее общую формулу где R1 является пиразиновой кислотой, бензольной кислотой или коричной кислотой;R2 выбирают из группы октила, децила, додецила, тетрадецила, гексадецила,при этом пролекарство имеет логарифм коэффициента распределения октанол/вода (log P) более 3,0,с липосомным везикулярным переносчиком, включающим как минимум один липид, гидрогенизированный или нет, или смесь липидов, выбранных из фосфатидилхолина, фосфатидилглицерина, димиристоилфосфатидилхолина,димиристоилфосфатидилглицерина,дипальмитоилфосфатидилхолина,дипальмитоилфосфатидилглицерина, диэстеароилфосфатидилхолина, диэстеароилфосфатидилглицерина, диолеилфосфатидилхолина, диолеилфосфатидилглицерина, холестерина или его производных, сфингомиелина, арахидоновой кислоты, сфингозина, ганглиозидов, церамидов, фосфатидилинозитола и фосфатидной кислоты. 2. Везикулярный состав по п.1, характеризующийся тем, что пролекарство является додецилпиразиноатом. 3. Везикулярный состав по п.1, характеризующийся тем, что пролекарство является тетрадецилпиразиноатом. 4. Везикулярный состав по п.1, характеризующийся тем, что пролекарство является гексадецилпиразиноатом. 5. Везикулярный состав по п.1, характеризующийся тем, что пролекарство является децилбензоатом. 6. Везикулярный состав по п.1, характеризующийся тем, что пролекарство является октилциннаматом. 7. Везикулярный состав по пп.1-6, характеризующийся тем, что липосомный везикулярный состав имеет лиофилизированные или гидратизированные формы. 8. Везикулярный состав по пп.1-6, характеризующийся тем, что везикулярный носитель содержит дополнительные нейтральные или заряженные молекулы, не имеющие характер липидов или поверхностно-активного вещества. 9. Способ приготовления липосомного везикулярного состава по пп.1-8, характеризующийся тем,что содержит следующие этапы: а) этерификация слабой органической кислоты, выбранной из пиразиновой, бензольной или коричной кислот; б) подготовка раствора, содержащего липиды и пролекарство, полученное на этапе а), в подходящем растворителе; в) удаление растворителя путем испарения или лиофилизации; г) гидратация полученного продукта. 10. Лекарственный препарат для лечения туберкулезной инфекции, содержащий липосомный везикулярный состав по пп.1-8. 11. Лекарственный препарат по п.10 в форме для ингаляции, внутривенного, внутримышечного или подкожного введения.
МПК / Метки
МПК: A61K 9/127
Метки: составы, содержащие, кислот, производные, приготовления, процесс, органических, везикулярные
Код ссылки
<a href="https://eas.patents.su/15-18246-vezikulyarnye-sostavy-soderzhashhie-proizvodnye-organicheskih-kislot-i-process-ih-prigotovleniya.html" rel="bookmark" title="База патентов Евразийского Союза">Везикулярные составы, содержащие производные органических кислот, и процесс их приготовления</a>
Трициклические соединения для лечения воспалительных и аллергических нарушений, способы их приготовления и содержащие их фармацевтические составы

Номер патента: 10408
Опубликовано: 29.08.2008
Авторы: Анупинди Рагху Рам, Баласубраманиан Гопалан, Гхарат Лаксмикант Атмарам, Лакдавала Афтаб Давудбхай
МПК: A61K 31/381, A61K 31/34, A61K 31/403...
Метки: лечения, способы, составы, приготовления, фармацевтические, аллергических, содержащие, воспалительных, нарушений, соединения, трициклические
Формула / Реферат:
1. Соединение общей формулы (1) где R1 представляет собой водород, С1-С8 алкил, С3-С12 циклоалкил, (С3-С12 циклоалкил)(С1-С8 алкил), (С6-С14 арил)(С1-С8 алкил) или защитную группу, где С1-С8 алкил, С3-С12 циклоалкил, (С3-С12 циклоалкил)(С1-С8 алкил) и (C6-C14 арил)(С1-С8 алкил) возможно имеют от 1 до 3 заместителей, выбранных из группы, включающей С1-С8 алкил, галоген, -ORx, оксо- и -COORx; R2 представляет собой водород, С1-С8 алкил, С3-С15...
Противоопухолевые составы, содержащие производные таксанов

Номер патента: 6878
Опубликовано: 28.04.2006
Автор: Биссери Мари-Кристин
МПК: A61P 35/00, A61K 31/337
Метки: производные, составы, противоопухолевые, содержащие, таксанов
Формула / Реферат:
1. Фармацевтический состав, содержащий соединение формулы 1 или его производное, а также по крайней мере одно из соединений: антибиотик, алкалоид барвинка, платиновый координационный комплекс или ингибитор топоизомеразы. 2. Фармацевтический состав по п.1, отличающий тем, что антибиотик представляет собой доксорубицин. 3. Фармацевтический состав по п.1, отличающийся тем, что алкалоид барвинка представляет собой навелбин. 4. Фармацевтический...
Производные дигидро- и тетрагидрохинолина, способ их получения и содержащие их фармацевтические составы

Номер патента: 3272
Опубликовано: 27.02.2003
Авторы: Локар Бриан, Дорей Жильбер, Казара Патрик, Лестаж Пьер
МПК: A61K 31/4709, C07D 215/06, A61P 25/28...
Метки: производные, тетрагидрохинолина, способ, составы, получения, содержащие, фармацевтические, дигидро
Формула / Реферат:
1. Соединения общей формулы (I) где R1 представляет собой атом водорода или группу где A представляет собой атом водорода или группу -BNZ1Z2, в которой B представляет собой линейную или разветвленную (C1-C6)алкиленовую группу и Z1 и Z2 независимо представляют собой атом водорода или алкильную, (C3-C8)циклоалкильную или необязательно замещенную арильную группу или вместе с соединяющим их атомом азота образуют необязательно замещенную...
Способ получения ангидридов кислот или смешанных ангидридов органических и кислородсодержащих неорганических кислот

Номер патента: 9665
Опубликовано: 28.02.2008
Авторы: Богенштеттер Томас, Клопп Инго, Франке Дирк
МПК: B01J 8/20, C07F 9/141, C07C 45/79...
Метки: кислородсодержащих, получения, органических, неорганических, ангидридов, смешанных, кислот, способ
Формула / Реферат:
1. Способ получения ангидридов кислот или смешанных ангидридов органических и кислородсодержащих неорганических кислот, в котором слежавшийся осадок первого реагента, выбранного из щелочных или аммонийных солей ароматических или гетероароматических карбоновых кислот, осажденных на фильтрующем элементе, промывают раствором второго реагента, выбранного из галогенидов неорганических или органических кислот, растворенных в растворителе, выбранном из...
Сыпучий гранулят на основе органических кислот

Номер патента: 874
Опубликовано: 26.06.2000
Авторы: Мейер Иоахим, Брёкель Ульрих, Кеслер Бруно, Мюшен Ганс, Сальвадор Биати
МПК: A23L 3/3454, A23K 1/00
Метки: кислот, гранулят, основе, сыпучий, органических
Формула / Реферат:
1. Сыпучий гранулят, гранулы которого содержат органическую кислоту, нанесенную на пористый носитель, отличающийся тем, что гранулы покрыты оболочкой, которая состоит из покровного вещества, которое при 20шС растворимо или набухает в воде. 2. Сыпучий гранулят по п.1, отличающийся тем, что оболочка дополнительно содержит на поверхности пудрящее вещество. 3. Сыпучий гранулят по п.1, отличающийся тем, что ядро содержит 30-90 вес.% органической...