Замещенные соединения бензо [e][1,4]оксазино[3,2-g]изоиндола и фармацевтические композиции, которые их содержат
Номер патента: 9697
Опубликовано: 28.02.2008
Авторы: Кэньяр Даньель-Анри, Хикман Джон, Ренар Пьер, Лепифр Франк, Краус-Бертье Лоранс, Пьерр Ален, Кудер Жерар







Формула / Реферат
1. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I)

в которой
W1 представляет собой вместе с атомами углерода, к которому он присоединен, фенильную группу или пиридильную группу,
Z представляет собой водород,
R1 представляет собой группу, выбранную из водорода и группы -C(O)-R5, в которой R5 представляет собой группу, выбранную из водорода и таких групп, как линейный или разветвленный С1-С6-алкокси и арилокси, где под "арилом" подразумевают такую группу, как фенил,
R2 представляет собой атом водорода или группу формулы -CH2CH2O-R8, в которой
R8 представляет собой группу, выбранную из водорода и -S(O)tR6, где R6 представляет собой линейный или разветвленный C1-С6-алкил и t представляет собой число 2,
R3 и R4 каждый представляет собой линейный или разветвленный C1-С6-алкил,
n представляет собой целое число от 1 до 6 включительно,
а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
2. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по п.1, отличающиеся тем, что они представляют собой соединения формулы (IA)

в которой R1, R2, R3, R4, W1, Z и n имеют значения, указанные для формулы (I), их соли присоединения с фармацевтически приемлемой кислотой или основанием.
3. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по п.1 или 2, отличающиеся тем, что они представляют собой соединения формулы (IB)

в которой R1, R2, R3, R4, Z и n имеют значения, указанные выше, их соли присоединения с фармацевтически приемлемой кислотой или основанием.
4. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по п.1 или 2, отличающиеся тем, что они представляют собой соединения формулы (IC)

в которой R1, R2, R3, R4, Z и n имеют значения, указанные выше, их соли присоединения с фармацевтически приемлемой кислотой или основанием.
5. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по любому из пп.1-4, отличающиеся тем, что R1 представляет собой атом водорода или группу -C(O)-R5, где R5 более предпочтительно представляет собой атом водорода, их соли присоединения с фармацевтически приемлемой кислотой или основанием.
6. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по любому из пп.1-5, отличающиеся тем, что R2 представляет собой атом водорода или группу -CH2CH2O-R8, где R8 более предпочтительно представляет собой атом водорода, их соли присоединения с фармацевтически приемлемой кислотой или основанием.
7. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) в соответствии с любым из пп.1-6, отличающиеся тем, что n представляет собой целое число 2, их соли присоединения с фармацевтически приемлемой кислотой или основанием.
8. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по п.1, которые представляют собой
2-[2-(диметиламино)этил]-5-гидроксибензо[а]пирроло[3,4-с]феноксазин-1,3-дион,
2-[2-(диэтиламино)этил]-5-гидроксибензо[а]пирроло[3,4-с]феноксазин-1,3-дион,
2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)-2,3-дигидробензо[а]пирроло[3,4-с]феноксазин-8-карбальдегид-1,3-дион,
2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)бензо[а]пирроло[3,4-с]-феноксазин-1,3-дион,
2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)бензо[е]пиридо[2',3':5,6][1,4]-оксазино[3,2-g]изоиндол-1,3-дион,
их соли присоединения с фармацевтически приемлемой кислотой или основанием.
9. Фармацевтическая композиция, обладающая противоопухолевой активностью, содержащая в качестве активного компонента по крайней мере одно замещенное соединение бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по любому из пп.1-8, самостоятельно или в сочетании с одним или несколькими фармацевтически приемлемыми инертными, нетоксичными наполнителями или носителями.
10. Фармацевтическая композиция по п. 9, пригодная в качестве лекарственного средства для лечения злокачественных опухолей.
Текст
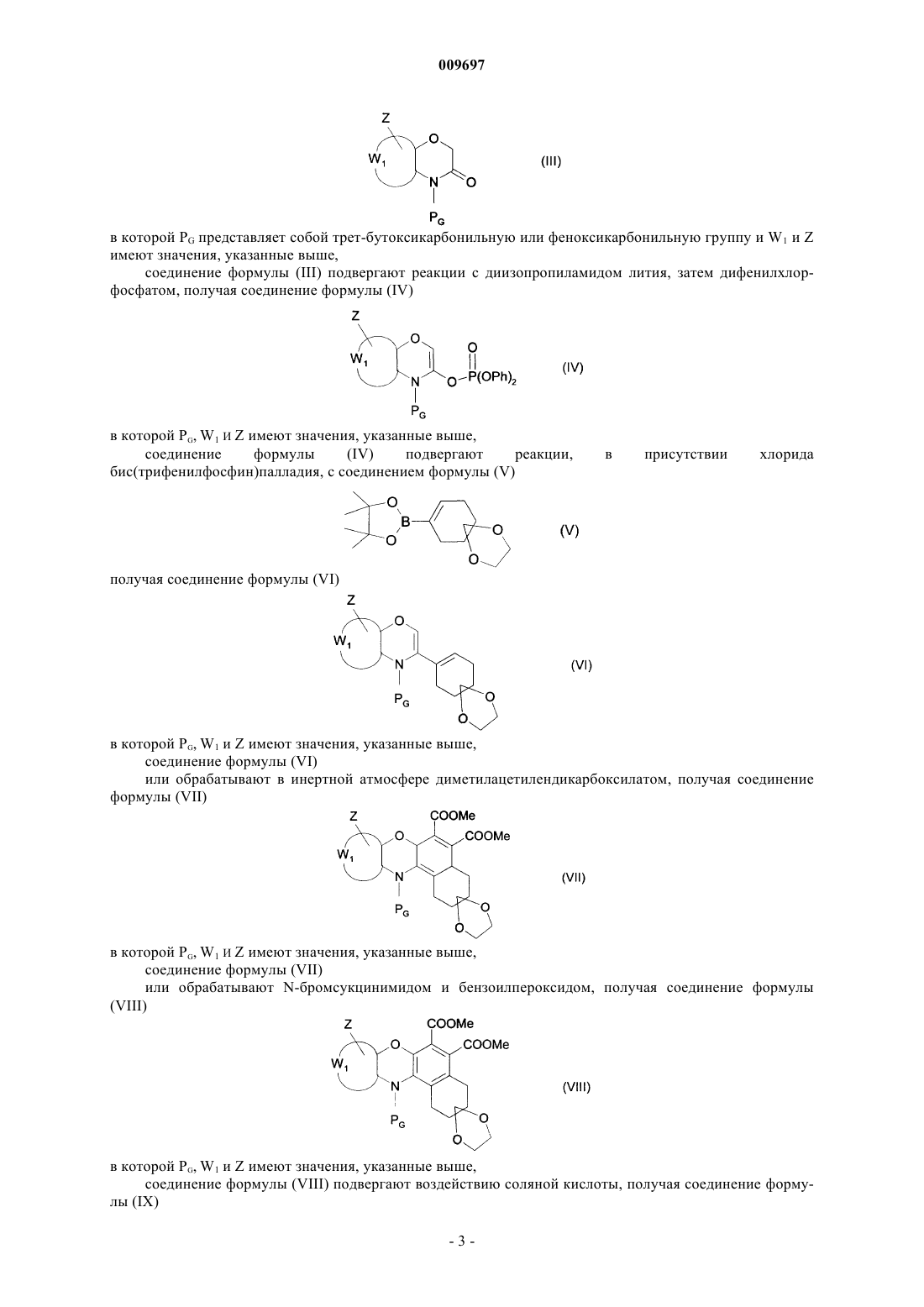
009697 Настоящее изобретение относится к новым соединениям бензо[е][1,4]оксазино[3,2-g]изоиндола, к способу их получения и к фармацевтическим композициям, которые их содержат. Соединения согласно настоящему изобретению являются ценными для терапевтического применения в связи с их противоопухолевой активностью. В литературе, J. Pharm. Sciences, 1974, 63(8), сс. 1314-1316, описан синтез соединений бензоксазинохинолина, обладающих противоопухолевыми свойствами. В патентной заявке ЕР 0841337 заявлены замещенные соединения 7,12-диоксабензо[а]антрацена и описаны их противоопухолевые свойства. Соединения согласно настоящему изобретению являются новыми как по своей структуре, так и для их применения в качестве противоопухолевых средств. Их биодоступность также значительно превышает биодоступность соединений, известных из уровня техники. В частности, настоящее изобретение относится к замещенным соединениям бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I)W1 представляет собой вместе с атомами углерода, к которому он присоединен, фенильную группу или пиридильную группу,Z представляет собой водород,R1 представляет собой группу, выбранную из водорода и группы -C(O)-R5, в которой R5 представляет собой группу, выбранную из водорода и таких групп, как линейный или разветвленный С 1-С 6 алкокси и арилокси, где под арилом подразумевают такую группу, как фенил,R2 представляет собой атом водорода или группу формулы -CH2CH2O-R8, в которой R8 представляет собой группу, выбранную из водорода и -S(O)tR6, где R6 представляет собой линейный или разветвленный С 1-С 6-алкил и t представляет собой число 2,R3 и R4, каждый, представляет собой линейный или разветвленный С 1-С 6-алкил.n представляет собой целое число от 1 до 6 включительно,а также их солям присоединения с фармацевтически приемлемой кислотой или основанием,где под арилом подразумевают такую группу, как фенил, нафтил, дигидронафтил, тетрагидронафтил, инденил или инданил, каждая из этих групп необязательно замещена одной или несколькими одинаковыми или разными группами, выбранными из галогена, линейного или разветвленного C1-C6-алкила,линейного или разветвленного C1-С 6-тригалоалкила, гидрокси, линейного или разветвленного С 1-С 6 алкокси, и аминогруппы, необязательно замещенной одной или двумя линейными или разветвленнымиC1-C6-алкильными группами. Среди фармацевтически приемлемых кислот можно отметить, но не ограничиваясь только ими, соляную, бромисто-водородную, серную, фосфоновую, уксусную, трифторуксусную, молочную, пировиноградную, малоновую, янтарную, глутаровую, фумаровую, винную, малеиновую, лимонную, аскорбиновую, метансульфоновую, камфорную кислоту и т.д. Среди фармацевтически приемлемых оснований можно отметить, но не ограничиваясь только ими,гидроокись натрия, гидроокись калия, триэтиламин и т.д. Предпочтительными соединениями по изобретению являются соединения формулы (I), которые предпочтительно соответствуют формуле (IA) в которой R1, R2, R3, R4, W1, Z и n имеют значения, указанные для формулы (I). В соответствии со вторым предпочтительным вариантом осуществления изобретения предпочтительными соединениями по изобретению являются соединения формулы (I), которые предпочтительно соответствуют формуле (IB) в которой R1, R2, R3, R4, Z и n имеют значения, указанные выше. В соответствии с третьим предпочтительным вариантом осуществления изобретения предпочтительными соединениями по изобретению являются соединения формулы (I), которые предпочтительно соответствуют формуле (IC) в которой R1, R2, R3, R4, Z и n имеют значения, указанные выше. В одном варианте осуществления изобретения, который представляет интерес, предпочтительно группа Z в соответствии с изобретением представляет собой атом водорода. В другом варианте осуществления изобретения, который представляет интерес, предпочтительно группа R1 в соответствии с изобретением представляет собой атом водорода и группу -C(O)-R5, где R5 более предпочтительно представляет собой атом водорода. Предпочтительно группа R2 предпочтительно в соответствии с изобретением представляет собой атом водорода и группу -CH2CH2O-R8, где R8 более предпочтительно представляет собой атом водорода. Более предпочтительно предпочтительными соединениями по изобретению являются соединения, в которых n представляет собой целое число 2. Наиболее предпочтительно группы R3 и R4 предпочтительно в соответствии с изобретением, которые могут быть одинаковыми или разными, каждая представляет собой независимо друг от друга линейную или разветвленную C1-С 6-алкильную группу. Предпочтительными соединениями в соответствии с изобретением являются 2-[2-(диметиламино)этил]-5-гидроксибензо[а]пирроло[3,4-с]феноксазин-1,3-дион,2-[2-(диэтиламино)этил]-5-гидроксибензо[а]пирроло[3,4-с]феноксазин-1,3-дион,2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)-2,3-дигидробензо[а]пирроло[3,4-с]-феноксазин-8 карбальдегид-1,3-дион,2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)бензо[а]пирроло[3,4-с]феноксазин-1,3-дион,2-[2-(диметиламино)этил]-5-(2-гидроксиэтилметансульфонат)бензо[а]пирроло-[3,4-с]феноксазин 1,3-дион,2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)бензо[е]пиридо[2',3':5,6][1,4]-оксазино[3,2-g]изоиндол-1,3-дион. Энантиомеры, диастереоизомеры, N-оксиды, а также соли присоединения с фармацевтически приемлемой кислотой или основанием предпочтительных соединений составляют неотъемлемую часть изобретения. Настоящее изобретение также относится к способу получения соединений формулы (I), который характеризуется тем, что в качестве исходного вещества применяют соединение формулы (II) в которой W1 и Z имеют значения, указанные для формулы (I),функциональную аминогруппу соединения формулы (II) защищают защитной группой PG, хорошо известной специалисту в данной области, получая соединение формулы (III) в которой PG представляет собой трет-бутоксикарбонильную или феноксикарбонильную группу и W1 и Z имеют значения, указанные выше,соединение формулы (III) подвергают реакции с диизопропиламидом лития, затем дифенилхлорфосфатом, получая соединение формулы (IV) получая соединение формулы (VI) в которой PG, W1 и Z имеют значения, указанные выше,соединение формулы (VI) или обрабатывают в инертной атмосфере диметилацетилендикарбоксилатом, получая соединение формулы (VII) в которой PG, W1 и Z имеют значения, указанные выше,соединение формулы (VIII) подвергают воздействию соляной кислоты, получая соединение формулы (IX) в которой W1 и Z имеют значения, указанные выше,соединение формулы (IX) подвергают воздействию ди-трет-бутилдикарбоната в присутствии 4 диметиламинопиридина, получая соединение формулы (X) в которой - - - представляет собой простую или двойную связь, Boc представляет собой третбутоксикарбонильную группу и W1 и Z имеют значения, указанные выше,соединение формулы (X) подвергают воздействию 2,3-дихлор-5,6-дициано-1,4-бензохинона, получая соединение формулы (XI) в которой Boc, W1 и Z имеют значения, указанные выше,соединение формулы (XI) подвергают воздействию метанолята натрия и затем гидролизуют, получая соединение формулы (XII) в которой R3, R4 и n имеют значения, указанные для формулы (I), получая соединение формулы (I/a), частный случай соединений формулы (I) в которой Boc, R3, R4, W1, Z и n имеют значения, указанные выше,соединение формулы (I/a) необязательно подвергают воздействию в таких же условиях, что и соединение формулы (VIII), получая соединение формулы (I/b), частный случай соединений формулы (I) в которой R3, R4, W1, Z и n имеют значения, указанные выше,или подвергают воздействию в таких же условиях, что и соединение формулы (X), получая соединение формулы (XIV) в которой PG, W1 и Z имеют значения, указанные выше,соединение формулы (XIV) необязательно подвергают воздействию в таких же условиях, что и соединение формулы (XII), получая соединение формулы (I/с), частный случай соединений формулы (I) в которой PG, R3, R4, W1, Z и n имеют значения, указанные выше,соединение формулы (I/с) или необязательно подвергают воздействию муравьиной кислоты, получая соединения формул (I/d) и (I/е), частные случаи соединений формулы (I) в которых R3, R4, W1, Z и n имеют значения, указанные выше,или необязательно подвергают взаимодействию с соединением формулы (XV) в которой G представляет собой уходящую группу и R8a, который не представляет собой атом водорода,имеет такие же значения, как R8 в формуле (I), получая соединение формулы (I/f), частный случай соединений формулы (I) в которой PG, R3, R4,R8a,W1, Z и n имеют значения, указанные выше,-5 009697 с функциональной аминогруппы соединений формулы (I/f) необязательно снимают защиту в соответствии с обычными способами органического синтеза, получая соединение формулы (I/g), частный случай соединений формулы (I) в которой R3, R4, R8a, W1, Z и n имеют значения, указанные выше,соединения формул (I/b), (I/d) и (I/g) составляют соединения формулы (I/h) в которой R2, R3, R4, W1, Z и n имеют значения, указанные выше,соединения формулы (I/h) необязательно подвергают взаимодействию с соединением формулы(XVI) в которой R1a, который не представляет собой атом водорода, имеет такие же значения, как R1 в формуле(I) и G имеет значения, указанные выше,получая соединение формулы (I/i), частный случай соединений формулы (I) в которой PG, W1 и Z имеют значения, указанные выше,соединение формулы (XVII) необязательно подвергают воздействию в таких же условиях, что и соединение формулы (VII), получая соединение формулы (XVIII) в которой PG, W1 И Z имеют значения, указанные выше,соединение формулы (XVIII) необязательно подвергают воздействию в таких же условиях, что и соединение формулы (XII), получая соединение формулы (I/d), как указано выше,-6 009697 соединения формул (I/a)-(I/i), представляющие собой совокупность соединений формулы (I), если это является подходящим, очищают в соответствии с обычными способами очистки, если это является желательным, могут быть разделены на их разные изомеры в соответствии с обычными способами разделения, и, если это является желательным, превращены в их N-оксиды и, если это является подходящим, в их соли присоединения с фармацевтически приемлемой кислотой или основанием. Изобретение также относится к соединениям формулы (X), (XI) и (XIV), которые являются промежуточными продуктами при синтезе для применения для получения соединений формулы (I). Соединения формул (II), (V), (XIII), (XV) и (XVI) являются или коммерчески доступными соединениями, или могут быть получены в соответствии с обычными способами органического синтеза, которые хорошо известны специалисту в данной области техники. Соединения формулы (I) обладают ценными фармакологическими свойствами. Они обладают очень хорошей цитотоксичностью в условиях in vitro не только по отношению к клеточным линиям лейкоза, но также действуют на линии солидных опухолей и, кроме того, обладают действием на клеточный цикл и активны в условиях in vivo на модели лейкоза. Эти свойства делают их пригодными для терапевтического применения в качестве противоопухолевых средств. Среды видов злокачественных новообразований, которые можно лечить при помощи соединений согласно настоящему изобретению, можно отметить, но не ограничиваясь только ими, аденокарциномы,карциномы, саркомы, глиомы и лейкозы. Настоящее изобретение также относится к фармацевтическим композициям, которые содержат продукты формулы (I), их энантиомер, диастереоизомер, N-оксид или одну из их фармацевтически приемлемых солей присоединения с кислотой или основанием, самостоятельно или в сочетании с одним или несколькими инертными, нетоксичными наполнителями или носителями. Среди фармацевтических композиций согласно изобретению особенно можно отметить те, которые являются пригодными для перорального, парентерального, назального ректального, подъязычного, глазного введения или введения в дыхательные пути, и в особенности таблетки или драже, подъязычные таблетки, саше, пакеты, желатиновые капсулы, таблетки для медленного растворения под языком, лепешки,суппозитории, кремы, мази, кожные гели, составы для инъекций или питья, аэрозоли, глазные капли или капли в нос. Таким образом, учитывая фармакологические свойства соединений формулы (I), фармацевтические композиции, которые содержат в качестве активного компонента указанные соединения, в особенности пригодны для лечения злокачественных новообразований. Полезная дозировка изменяется в зависимости от возраста и веса пациента, пути введения, природы терапевтического расстройства и любых сопутствующих видов лечения, и находится в диапазоне от 0,1 до 400 мг в сутки на одно или больше введений. Примеры, представленные ниже, приведены только с целью иллюстрации изобретения и никоим образом его не ограничивают. Используемые исходные материалы являются известными продуктами или продуктами, которые получают в соответствии с известными способами. Структура соединений, описанных в примерах, была определена согласно обычным спектрофотометрическим способам (инфракрасная спектрометрия, ядерный магнитный резонанс, массспектрометрия и т.д). Приготовление А. трет-Бутил 3-[(дифеноксифосфорил)окси]-4H-1,4-бензоксазин-4-карбоксилат. Стадия А. трет-Бутил 2,3-дигидро-4 Н-1,4-бензоксазин-3-он-4-карбоксилат. В инертной атмосфере 73 ммоль 2H-1,4-бензоксазин-3-она растворяли в 100 мл ацетонитрила в присутствии 3,65 ммоль 4-диметиламинопиридина и 80 ммоль ди-трет-бутилдикарбоната. Смесь перемешивали в течение 4 ч. После концентрирования остаток ресуспендировали в этилацетате. Органическую фазу промывали насыщенным раствором хлорида натрия, высушивали над сульфатом магния,фильтровали и концентрировали. После выпаривания растворителя и очищения путем хроматографии на силикагеле (петролейный эфир/этилацетат: 8/2), выделяли ожидаемый продукт. Точка плавления: 72 С. ИК (KBr): C= O = 1713, 1779 см-1; COC = 1148 см-1. Масс-спектр: m/z 250 (М+1). Стадия В. трет-Бутил 3-[(дифеноксифосфорил)окси]-4 Н-1,4-бензоксазин-4-карбоксилат. В инертной атмосфере 12 ммоль TMEDA добавляли к раствору 10 ммоль продукта, полученного на вышеописанной стадии А, в 50 мл безводного ТГФ. После охлаждения раствора до -78 С по каплям добавляли 12 ммоль 2 М LDA (в растворе гептан/ТГФ). После перемешивания в течение 2 ч к реакционной смеси по каплям добавляли 12 ммоль дифенилхлорфосфата, смесь выдерживали при -78 С дополнительно в течение 2 ч. После возвращения температуры до температуры окружающей среды раствор гидролизовали и затем экстрагировали этилацетатом. Органическую фазу высушивали над сульфатом магния,фильтровали и концентрировали. После очищения остатка путем хроматографии на силикагеле (петролейный эфир/этилацетат: 9/1), выделяли ожидаемый продукт. Точка плавления: 64 С.-7 009697 ИК (KBr): C=O = 1732 см-1; P=O 1313 см-1. Масс-спектр: m/z 482 (М+1). Приготовление В. Фенил 3-[(дифеноксифосфорил)окси]-2,3-дигидро-4H-пиридо-[3,2-b][1,4]оксазин-4-карбоксилат. Стадия А. Фенил 2,3-дигидро-4 Н-пиридо[3,2-b][1,4]оксазин-3-он-4-карбоксилат. В инертной атмосфере раствор 10 ммоль 2H-пиридо[3,2-b][1,4]оксазин-3-она в 50 мл тетрагидрофурана охлаждали до -78 С. При этой температуре по каплям добавляли 11 ммоль 1,6 М раствора нбутиллития в гексане. После взаимодействия в течение 30 мин при -78 С, по каплям добавляли 11 ммоль фенилхлорформиата и продолжали перемешивать дополнительно в течение 2 ч. После возвращения температуры до температуры окружающей среды, раствор гидролизовали и затем экстрагировали этилацетатом. Органическую фазу высушивали над сульфатом магния, фильтровали и упаривали. После очищения путем хроматографии на силикагеле (петролейный эфир/этилацетат: 8/2), выделяли ожидаемый продукт. Точка плавления: 97 С. ИК (KBr): C=O = 1717 см-1; 1803 см-1. Масс-спектр: m/z 271 (М+1). Стадия В. Фенил 3-[(дифеноксифосфорил)окси]-4 Н-пиридо[3,2-b][1,4]оксазин-4-карбоксилат. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии В приготовления А, исходя из соединения, полученного на вышеописанной стадии. Точка плавления: 82 С. ИК (KBr): C=O = 1749 см-1; P=O 1294 см-1. Масс-спектр: m/z 503 (М+1). Приготовление С. 8-(Пинаконборонил)-1,4-диоксаспиро[4.5]дец-7-ен. Стадия А. 8-(Трифторметил)сульфонилокси-1,4-диоксаспиро[4.5]дец-7-ен. В инертной атмосфере 2 М раствор 6,4 ммоль LDA в смеси ТГФ/гептан разводили 8 мл ТГФ. Температуру снижали до -78 С и затем медленно добавляли 6,4 ммоль 1,4-диоксаспиро[4.5]декан-8-она, растворенного в 8 мл ТГФ. Реакционную смесь перемешивали в течение 2 ч при этой температуре и добавляли 9,6 ммоль N-фенилтрифторметансульфонимида, растворенного в 8 мл ТГФ. После перемешивания в течение 15 мин приблизительно при 78 С и последующего возращения температуры до температуры окружающей среды на ночь смесь концентрировали. После очищения на нейтральном геле глинозема(петролейный эфир/этилацетат: 95/5), выделяли ожидаемый продукт. ИК (NaCl пленка): C=C= 1692 см-1; SO2 = 1418 см-1. Стадия В. 8-(Пинаконборонил)-1,4-диоксаспиро[4.5]дец-7-ен. В инертной атмосфере 0,7 ммоль продукта, полученного на вышеописанной стадии А, 1,05 ммоль пинаконборана, 0,028 ммоль хлорида бис(трифенилфосфин)палладия(II), 0,084 ммоль трифениларсина и 2,1 ммоль триэтиламина смешивали в 3 мл толуола и затем нагревали при 80 С в течение 2 ч. После охлаждения остаток ресуспендировали в этилацетате и промывали насыщенным раствором хлорида натрия. Органическую фазу высушивали над сульфатом магния, фильтровали и концентрировали. После очищения остатка путем хроматографии на силикагеле (петролейный эфир/этилацетат: 9/1), выделяли ожидаемый продукт. Точка плавления: 58 С. ИК ( KBr): С=С = 1635 см-1; COC = 1115, 1143 см-1. Масс-спектр: m/z 267 (М+1). Пример 1. 8-(трет-Бутоксикарбонил)-2-[2-(диметиламино)этил]-5-гидрокси-2,3-дигидробензо[а] пирроло[3,4-с]феноксазин-1,3-дион. Стадия А. трет-Бутил 3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)-4 Н-1,4-бензоксазин-4-карбоксилат. В инертной атмосфере 1 М раствор 1 ммоль продукта из приготовления А и 5% хлорид бис(трифенилфосфин)палладия(II) в тетрагидрофуране перемешивали в течение 10 мин при температуре окружающей среды. К реакционной смеси добавляли 1,5 ммоль продукта из приготовления С, несколько капель этанола и 2 ммоль 2 М водного раствора карбоната натрия, затем ее нагревали в колбе с обратным холодильником в течение одного часа. После охлаждения и гидролиза раствор экстрагировали этилацетатом. Органическую фазу высушивали над сульфатом магния, фильтровали и концентрировали. После очищения путем хроматографии на силикагеле (петролейный эфир/этилацетат: 6/4), выделяли ожидаемый продукт. Точка плавления: 92-93 С. ИК (KBr): C=O = 1711 см-1;COC = 1113, 1163 см-1. Масс-спектр: m/z 372 (М+1). Стадия В. Диметил 12-(трет-бутоксикарбонил)-3,3-(1,2-этилендиокси)-1,2,3,4,4 а,6 а-гексагидро-12 Нбензо[а]феноксазин-5,6-дикарбоксилат. В замкнутой системе 8 ммоль продукта, полученного на вышеописанной стадии А, и 40 ммоль диметилацетилендикарбоксилата перемешивали при 80 С в течение 22 ч. В результате очистки путем хро-8 009697 матографии на силикагеле (петролейный эфир/этилацетат: 7/3), выделяли ожидаемый продукт. Точка плавления: 234-235 С. ИК (KBr): C=O = 1728 см-1; COC = 1152 см-1. Масс-спектр: m/z 514 (М+1). Стадия С. Диметил 12-(трет-бутоксикарбонил)-3,3-(1,2-этилендиокси)-1,2,3,4-тетрагидро-12 Нбензо[а]феноксазин-5,6-дикарбоксилат. В инертной атмосфере 0,92 ммоль продукта, полученного на вышеописанной стадии В, и 2,75 ммоль перекристаллизованного N-бромсукцинимида в 23 мл дистиллированного четыреххлористого углерода нагревали в колбе с обратным холодильником в течение 10 мин, используя лампу 60 Вт, в присутствии каталитического количества бензоилпероксида. После охлаждения раствор фильтровали и затем концентрировали. После очищения путем хроматографии на силикагеле (петролейный эфир/этилацетат: 6/4), выделяли ожидаемый продукт. Точка плавления: 50 С. ИК (KBr): C=O = 1701, 1717, 1733 см-1;COC = 1152, 1195 см-1. Масс-спектр: m/z 512 (М+1). Стадия D. Диметил 3-оксо-1,3,4,12-тетрагидро-2 Н-бензо[а]феноксазин-5,6-дикарбоксилат. 3 мл 12 М соляной кислоты по каплям добавляли к 0,6 ммоль продукта, полученного на вышеописанной стадии С, растворенного в 3 мл этанола. Смесь перемешивали в течение 1,5 ч при температуре окружающей среды. После нейтрализации насыщенным раствором гидрокарбоната натрия и экстрагирования этилацетатом, органическую фазу высушивали над сульфатом магния, фильтровали и концентрировали. После очищения путем хроматографии на силикагеле (петролейный эфир/этилацетат: 5/5 до 0/10), выделяли ожидаемый продукт. Точка плавления: 250-251C. ИК (KBr): C=O = 1695, 1720 см-1; NH = 3430 см-1. Масс-спектр: m/z 366 (М+1). Стадия Е. Диметил 12-(трет-бутоксикарбонил)-3-[(трет-бутоксикарбонил)окси]-1,2-дигидро-12 Нбензо[а]феноксазин-5,6-дикарбоксилат. В инертной атмосфере 0,69 ммоль соединения, полученного на вышеописанной стадии D, растворяли в 10 мл тетрагидрофурана. После добавления 1,73 ммоль 4-диметиламинопиридина и 1,73 ммоль ди-трет-бутилдикарбоната, смесь перемешивали в течение 12 ч. После концентрирования остаток ресуспендировали в этилацетате и дважды промывали 1 М раствором соляной кислоты. Органическую фазу высушивали над сульфатом магния, фильтровали и концентрировали, получая ожидаемый продукт. ИК (NaCl пленка): C=O = 1728, 1756 см-1; COC = 1139 см-1. Масс-спектр: m/z 568 (М+1). Стадия F. Диметил 12-(трет-бутоксикарбонил)-3-[(трет-бутоксикарбонил)окси]-12 Н-бензо[а]феноксазин-5,6-дикарбоксилат. В инертной атмосфере 0,62 ммоль соединения, полученного на вышеописанной стадии Е, растворяли в 5 мл толуола в присутствии 4,96 ммоль 2,3-дихлор-5,6-дициано-1,4-бензохинона и смесь нагревали при 90 С в течение 24 ч. После охлаждения и концентрирования реакционную смесь ресуспендировали в дихлорметане и промывали 8% раствором гидроокиси натрия. Водную фазу экстрагировали дихлорметаном и органические фазы объединяли, высушивали над сульфатом магния, фильтровали и концентрировали. После очищения путем хроматографии на силикагеле (петролейный эфир/этилацетат: 7/3), выделяли ожидаемый продукт. Точка плавления: 101-102 С. ИК (KBr): C=O = 1731, 1739, 1756, 1766 см-1; COC = 1149 см-1. Масс-спектр: m/z 566 (М+1). Стадия G. Диметил 12-(трет-бутоксикарбонил)-3-гидрокси-12 Н-бензо[а]феноксазин-5,6-дикарбоксилат. В инертной атмосфере 0,28 ммоль соединения, полученного на вышеописанной стадии F, растворяли в 2 мл метанола в присутствии 0,34 ммоль метанолята натрия. Смесь перемешивали при температуре окружающей среды в течение 12 ч. После концентрирования и гидролиза смесь экстрагировали этилацетатом, высушивали над сульфатом магния, фильтровали и упаривали. После очищения путем хроматографии на силикагеле (петролейный эфир/этилацетат: 7/3), выделяли ожидаемый продукт. Точка плавления: 90-91 С (разложение). ИК (KBr): C=O = 1722 см-1; COC = 1152 см-1; vOH = 3442 см-1. Масс-спектр: m/z 466 (М+1). Стадия Н. 8-(трет-Бутоксикарбонил)-2-[2-(диметиламино)этил]-5-гидрокси-2,3-дигидробензо[а] пирроло[3,4-с]феноксазин-1,3-дион. В инертной атмосфере 0,26 ммоль соединения, полученного на вышеописанной стадии G, нагревали при 100 С в 4 мл N,N-диметилэтилендиамина в течение 7 ч. После охлаждения избыток диамина выпаривали. После очищения путем хроматографии на силикагеле (дихлорметан/метанол: 95/5), выделяли-9 009697 ожидаемый продукт. Точка плавления: 190 С (разложение). ИК (KBr):C=O = 1705, 1762 см-1; CO = 1249 см-1; OH = 3446 см-1. Масс-спектр: m/z 490,5 (М+1). Пример 2. Гидрохлорид 2-[2-(диметиламино)этил]-5-гидроксибензо[а]пирроло[3,4-с]феноксазин 1,3-диона. 3 мл 12 М соляной кислоты по каплям добавляли к 0,2 ммоль соединения из примера 1, растворенного в 4 мл этанола. Реакционную смесь перемешивали в течение 1,5 ч при температуре окружающей среды и затем концентрировали. При добавлении диэтилового эфира образовывался осадок, который отфильтровывали, получая ожидаемый продукт. ИК (KBr): C=O = 1686, 1744 см-1; NH,OH = 3431 см-1. Масс-спектр: m/z 390 (М+1). Пример 3. 8-(трет-Бутоксикарбонил)-2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)-2,3-дигидробензо[а]пирроло[3,4-с]феноксазин-1,3-дион. Стадия А. Диметил 12-(трет-бутоксикарбонил)-3-(2-гидроксиэтокси)-12 Н-бензо[а]феноксазин-5,6 дикарбоксилат. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии F примера 1, исходя из соединения со стадии В примера 1. Точка плавления: 87-88 С. ИК (KBr): C=O = 1725 см-1; OH = 3440 см-1. Масс-спектр: m/z 510 (М+1). Стадия В. 8-(трет-Бутоксикарбонил)-2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)-2,3-дигидробензо[а]пирроло[3,4-с]феноксазин-1,3-дион. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии Н примера 1, исходя из соединения с вышеописанной стадии А. Точка плавления: 80 С (разложение). ИК (KBr): C=O = 1707, 1763 см-1; OH = 3447 см-1. Масс-спектр: m/z 534 (М+1). Пример 4. 8-(трет-Бутоксикарбонил)-2-[2-(диметиламино)этил]-5-2-[(метилсульфонил)окси]этокси-2,3-дигидробензо[а]пирроло-[3,4-с]феноксазин-1,3-дион. В инертной атмосфере 0,93 ммоль триэтиламина и затем 0,93 ммоль мезилхлорида добавляли к раствору 0,06 ммоль соединения из примера 3 в 3 мл дихлорметана при 0 С. Продолжали перемешивать при 0 С в течение 8 ч. При температуре окружающей среды раствор гидролизовали и затем экстрагировали дихлорметаном. Органическую фазу высушивали над сульфатом магния, фильтровали и концентрировали. В результате очистки путем хроматографии на силикагеле (дихлорметан/метанол: 95/5), выделяли ожидаемый продукт. Точка плавления: 70-80 С (смола). ИК (KBr): C=O = 1707, 1763 см-1. Масс-спектр: m/z 612 (М+1). Пример 5. 2-[2-(Диметиламино)этил]-5-2-[(метилсульфонил)окси]этокси-1,2,3,8-тетрагидробензо[а]пирроло[3,4-с]феноксазин-1,3-дион. 0,03 ммоль соединения из примера 4 растворяли в 1 мл муравьиной кислоты и перемешивали при температуре окружающей среды в течение 3 ч. После концентрирования остаток ресуспендировали в дихлорметане и промывали 2 М раствором карбоната натрия и затем водой. Органические фазы объединяли, высушивали над сульфатом магния, фильтровали и концентрировали. После очищения путем хроматографии на силикагеле (дихлорметан/метанол: 9/1), выделяли ожидаемый продукт. ИК (KBr): C=O = 1686, 1702 см-1; NH = 3432 см-1. Масс-спектр: m/z 512 (М+1). Пример 6. 2-[2-(Диметиламино)этил]-5-(2-гидроксиэтокси)бензо[а]пирроло[3,4-с]феноксазин-1,3 дион. Ожидаемый продукт получали в соответствии с методикой из примера 5, исходя из соединения из примера 3. Точка плавления: 216 С (смола). ИК (KBr): vC=O = 1690, 1741 см-1 ; vNH = 3427 см-1. Масс-спектр: m/z 434 (М+1). Пример 7. 8-(Формил)-2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)бензо[а]пирроло[3,4-с]феноксазин-1,3-дион. Ожидаемый продукт получали при очистке путем хроматографии на силикагеле согласно примеру 6. Точка плавления: 202 С. ИК (KBr): C=O = 1693, 1732 см-1; NH = 3428 см-1. Масс-спектр: m/z 462 (М+1).[1,4]оксазино[3,2-g]изоиндол-1,3-дион. Стадия А. Фенил 3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)-4 Н-пиридо[3,2-b'][1,4]оксазин-4-карбоксилат. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии А примера 1, исходя из соединения из приготовления В. ИК (KBr): C=O = 1741 см-1; COC = 1111, 1197 см-1. Масс-спектр: m/z 393 (М+1). Стадия В. 8-(Феноксикарбонил)-5,5-(1,2-этилендиокси)-2-метил-2,3,3 а,3b,4,5,6,7,13 а,13b-декагидробензо[е]пиридо[2',3':5,6][1,4]оксазино[3,2-g]изоиндол-1,3-дион. В замкнутой системе 1 ммоль продукта, полученного на вышеописанной стадии А, и 3 ммоль Nметилимида малеиновой кислоты перемешивали при 95 С в течение 2 ч в присутствии нескольких капель толуола. После очищения путем хроматографии на силикагеле (петролейный эфир/этилацетат: 6/4) выделяли продукт. Точка плавления: 150 С (смола). ИК (KBr): C=O = 1701, 1786 см-1. Масс-спектр: m/z 504 (М+1). Стадия С. 8-(Феноксикарбонил)-5-(2-гидроксиэтокси)-2-метил-2,3-дигидробензо-[е]пиридо[2',3':5,6][1,4]оксазино[3,2-g]изоиндол-l,3-дион. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии С примера 1, исходя из соединения с вышеописанной стадии В. Точка плавления: 250 С (смола). ИК (KBr): C=O = 1707, 1752 см-1; COC = 1191 см-1; OH = 3463 см-1. Масс-спектр: m/z 498 (М+1). Пример 9. 2-[2-(Диметиламино)этил]-5-(2-гидроксиэтокси)бензо[е]пиридо-[2',3':5,6][1,4]оксазино[3,2-g]изоиндол-1,3-дион. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии Н примера 1, исходя из соединения из примера 8. Масс-спектр: m/z 435 (М+1). Пример 10. 8-(трет-Бутоксикарбонил)-2-[2-(диэтиламино)этил]-5-гидрокси-2,3-дигидробензо[а]пирроло[3,4-с]феноксазин-1,3-дион. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии Н примера 1, исходя из соединения со стадии G примера 1 и N,N-диэтилэтилендиамина. Пример 11. 2-[2-(Диэтиламино)этил]-5-гидроксибензо[а]пирроло[3,4-с]феноксазин-1,3-дион. Ожидаемый продукт получали в соответствии с методикой из примера 2, исходя из соединения из примера 10. Фармакологическое исследование соединений по изобретению Пример 12. Действие в условиях in vitro.L1210 лейкоз мыши. Использовали клетки L1210 лейкоза мыши в условиях in vitro. Клетки культивировали в RPMI 1640 полной питательной среде, содержащей 10%-ную фетальную телячью сыворотку, 2 мМ глутамина, 50 ед./мл пенициллина, 50 мкг/мл стрептомицина и 10 мМ Hepes, pH 4. Клетки распределяли на микропланшетах и подвергали действию цитотоксических соединений в течение четырех удвоенных периодов или 48 ч. После этого определяли количество жизнеспособных клеток при помощи колориметрического исследования, используя тетразолий в микрокультуре (J. Carmichael и др., Cancer Res.; 47, 939-942(1987. Результаты выражали в виде значений IC50, концентрации цитотоксического средства, которая ингибирует пролиферацию обработанных клеток на 50%. Все соединения по изобретению обладают хорошей цитотоксичностью по отношению к этой клеточной линии. Например, соединение из примера 2 обнаруживает IC50 0,25 мкМ по отношению к L1210. Линии клеток человека. Соединения по изобретению также исследовали на линиях клеток человека, имеющих происхождение от солидных опухолей, в соответствии с таким же протоколом исследования, который описанный для лейкоза мыши L1210, но с периодами инкубации 4 дня вместо 2 дней. Например, соединение из примера 2 обнаруживает IC50 0,27 мкМ по отношению к DU145 раку предстательной железы, 0,16 мкМ по отношению к А 549 немелкоклеточному раку легких, 0,06 мкМ по отношению к НТ-29 раку ободочной кишки и 0,26 мкМ по отношению к КВ-3-1 плоскоклеточному раку. Эти различные результаты являются очевидным доказательством сильного противоопухолевого действия соединений по изобретению по отношению к лейкозам и солидным опухолям. Пример 13. Действие на клеточный цикл. Клетки L1210 инкубировали в течение 21 ч при 37 С в присутствии различных концентраций исследуемых соединений. Затем клетки фиксировали при помощи 70 об.% этанола, дважды промывали вPBS и инкубировали в течение 30 мин при 20 С в PBS, содержащем 100 мкг/мл РНКазы и 50 мкг/мл йодида пропидия. Результаты выражали, исходя из процента клеток, которые находились в G2+M фазе через 21 ч, по сравнению с контролем (контроль: 20%). Соединения по изобретению представляют значительный интерес при концентрации ниже 2,5 мкМ, вызывая накопление по крайней мере 80% клеток вG2+M фазе через 21 ч. Пример 14. Действие в условиях in vivo. Противоопухолевое действие на Р 388 лейкоз. Линию Р 388 (лейкоз мышей) получали из Национального онкологического института (Фредерик,США). Клетки опухоли (106 клеток) прививали в день 0 в полость брюшины самкам мышей B6D2F1 (IffaCredo, Франция). В каждой исследуемой группе использовали шесть мышей весом от 18 до 20 г. Продукты вводили внутрибрюшинно в 1-й день. Противоопухолевое действие выражали в виде % О/К Полученные результаты свидетельствуют о значительной активности в условиях in vivo на модели лейкоза Р 388 при значении О/К 210% для дозы 50 мг/кг при низкой токсичности соединений, что указывает на очень хороший терапевтический индекс. Пример 15. Фармацевтическая композиция: раствор для инъекций. Соединение из примера 2 10 мг Дистиллированная вода для инъекций 25 мл ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I)W1 представляет собой вместе с атомами углерода, к которому он присоединен, фенильную группу или пиридильную группу,Z представляет собой водород,R1 представляет собой группу, выбранную из водорода и группы -C(O)-R5, в которой R5 представляет собой группу, выбранную из водорода и таких групп, как линейный или разветвленный С 1-С 6 алкокси и арилокси, где под арилом подразумевают такую группу, как фенил,R2 представляет собой атом водорода или группу формулы -CH2CH2O-R8, в которойR8 представляет собой группу, выбранную из водорода и -S(O)tR6, где R6 представляет собой линейный или разветвленный C1-С 6-алкил и t представляет собой число 2,R3 и R4 каждый представляет собой линейный или разветвленный C1-С 6-алкил,n представляет собой целое число от 1 до 6 включительно,а также их соли присоединения с фармацевтически приемлемой кислотой или основанием. 2. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по п.1, отличающиеся тем, что они представляют собой соединения формулы (IA) в которой R1, R2, R3, R4, W1, Z и n имеют значения, указанные для формулы (I), их соли присоединения с фармацевтически приемлемой кислотой или основанием. 3. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по п.1 или 2, отличающиеся тем, что они представляют собой соединения формулы (IB) в которой R1, R2, R3, R4, Z и n имеют значения, указанные выше, их соли присоединения с фармацевтически приемлемой кислотой или основанием. 4. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по п.1 или 2, отличающиеся тем, что они представляют собой соединения формулы (IC) в которой R1, R2, R3, R4, Z и n имеют значения, указанные выше, их соли присоединения с фармацевтически приемлемой кислотой или основанием. 5. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по любому из пп.1-4,отличающиеся тем, что R1 представляет собой атом водорода или группу -C(O)-R5, где R5 более предпочтительно представляет собой атом водорода, их соли присоединения с фармацевтически приемлемой кислотой или основанием. 6. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по любому из пп.1-5,отличающиеся тем, что R2 представляет собой атом водорода или группу -CH2CH2O-R8, где R8 более предпочтительно представляет собой атом водорода, их соли присоединения с фармацевтически приемлемой кислотой или основанием. 7. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) в соответствии с любым из пп.1-6, отличающиеся тем, что n представляет собой целое число 2, их соли присоединения с фармацевтически приемлемой кислотой или основанием. 8. Замещенные соединения бензо[е][1,4]оксазино[3,2-g]изоиндола формулы (I) по п.1, которые представляют собой 2-[2-(диметиламино)этил]-5-гидроксибензо[а]пирроло[3,4-с]феноксазин-1,3-дион,2-[2-(диэтиламино)этил]-5-гидроксибензо[а]пирроло[3,4-с]феноксазин-1,3-дион,2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)-2,3-дигидробензо[а]пирроло[3,4-с]феноксазин-8 карбальдегид-1,3-дион,2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)бензо[а]пирроло[3,4-с]-феноксазин-1,3-дион,2-[2-(диметиламино)этил]-5-(2-гидроксиэтокси)бензо[е]пиридо[2',3':5,6][1,4]-оксазино[3,2g]изоиндол-1,3-дион,их соли присоединения с фармацевтически приемлемой кислотой или основанием. 9. Фармацевтическая композиция, обладающая противоопухолевой активностью, содержащая в качестве активного компонента по крайней мере одно замещенное соединение бензо[е][1,4]оксазино[3,2g]изоиндола формулы (I) по любому из пп.1-8, самостоятельно или в сочетании с одним или несколькими фармацевтически приемлемыми инертными, нетоксичными наполнителями или носителями. 10. Фармацевтическая композиция по п. 9, пригодная в качестве лекарственного средства для лечения злокачественных опухолей.
МПК / Метки
МПК: A61K 31/535, C07D 498/14, C07D 498/04, C07D 265/38
Метки: содержат, соединения, e][1,4]оксазино[3,2-g]изоиндола, фармацевтические, композиции, бензо, замещенные, которые
Код ссылки
<a href="https://eas.patents.su/14-9697-zameshhennye-soedineniya-benzo-e14oksazino32-gizoindola-i-farmacevticheskie-kompozicii-kotorye-ih-soderzhat.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные соединения бензо [e][1,4]оксазино[3,2-g]изоиндола и фармацевтические композиции, которые их содержат</a>
Замещённые соединения [1,4]бенздиоксино[2,3-e]изоиндола, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 8498
Опубликовано: 29.06.2007
Авторы: Кудер Жерар, Пьерр Ален, Эрб Натали, Рутье Сильвен, Леонс Стефан, Хикман Джон, Лепифр Франк, Ренар Пьер, Сеньар Даньель-Анри
МПК: A61K 31/5365, C07D 487/04, A61P 35/00...
Метки: получения, замещённые, содержат, которые, композиции, способ, фармацевтические, соединения, 1,4]бенздиоксино[2,3-e]изоиндола
Формула / Реферат:
1. Соединения формулы (I) в которой А вместе с атомами углерода, с которыми он связан, представляет собой группу формулы (а) или (b) в которой W1 вместе с атомами углерода, с которыми он связан, представляет собой фенильную группу или пиридильную группу, Z представляет собой группу, выбранную из атомов водорода и галогена и таких групп, как линейный или разветвленный C1-C6-алкил, нитро, циано, гидрокси, линейный или разветвленный...
Соединения пиперазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 9648
Опубликовано: 28.02.2008
Авторы: Гумен Бертран, Дессинже Эме, Пеглион Жан-Луи, Миллан Марк, Маннури-Ла-Кур Клотильда
МПК: A61K 31/495, A61K 31/496, A61K 31/453...
Метки: композиции, фармацевтические, соединения, которые, способ, пиперазина, получения, содержат
Формула / Реферат:
1. Соединение формулы (I) в которой R1, R2, R3 и R4, которые могут быть одинаковыми или разными, каждый представляет собой атом или группу, выбранную из Н, галогена, линейного или разветвленного C1-С6-алкила, линейного или разветвленного C1-С6-алкокси, фенила и циано, X представляет собой связь, атом кислорода или группу, выбранную из -(СН2)m-, -ОСН2- и -NR5-, m представляет собой 1 или 2, R5 представляет собой атом водорода или группу,...
Соединения бензотиазина и бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 7492
Опубликовано: 27.10.2006
Авторы: Десо Патрис, Лестаж Пьер, Корди Алекс
МПК: A61K 31/5415, A61K 31/549, A61P 25/00...
Метки: соединения, получения, фармацевтические, бензотиазина, содержат, способ, которые, композиции, бензотиадиазина
Формула / Реферат:
1. Соединения формулы (I) где R1 представляет собой арильную или гетероарильную группу, R2 представляет собой атом водорода, атом галогена или гидроксильную группу, X представляет собой атом кислорода или атом серы, Y представляет собой атом кислорода, атом серы или группу NR, где R представляет собой атом водорода или линейную или разветвленную C1-С6-алкильную группу, А представляет собой NR4-группу, R3 представляет собой атом водорода,...
Соединения бензотиазина и бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 7227
Опубликовано: 25.08.2006
Авторы: Десо Патрис, Лестаж Пьер, Корди Алекс
МПК: A61P 25/00, A61K 31/549, A61K 31/5415...
Метки: фармацевтические, способ, содержат, бензотиадиазина, бензотиазина, соединения, которые, композиции, получения
Формула / Реферат:
1. Соединения формулы (I) где R1 представляет собой арильную или гетероарильную группу, R2 представляет собой атом водорода, атом галогена или гидроксильную группу, А представляет собой CR4R5 группу или NR4 группу, R3 представляет собой атом водорода, линейную или разветвленную С1-С6-алкильную группу или С3-С7-циклоалкильную группу, R4 представляет собой атом водорода или линейную или разветвленную С1-С6-алкильную группу, или А представляет...
Новые соединения бензоиндолина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 7231
Опубликовано: 25.08.2006
Авторы: Де-Кара Бенжамен, Миллан Марк, Лавьелль Жильбер, Мюлле Оливье, Гобер Ален
МПК: A61P 25/18, A61K 31/496, C07D 209/00...
Метки: фармацевтические, бензоиндолина, соединения, получения, способ, которые, новые, композиции, содержат
Формула / Реферат:
1. Соединения формулы (I) в которой R1 и R2 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино, диалкиламино или группой трифторметила, a R3 и R4 представляют собой атом водорода, или R1 и R4 представляют собой атом водорода, a R2 и R3 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси,...
Предыдущий патент: Замещенные нафтилиндольные производные в качестве ингибиторов ингибитора активатора плазминогена типа 1 (pai-1)
Следующий патент: Новые фторированные соединения бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат
Случайный патент: Вибрационный сепаратор и способ отделения твердых частиц от шлама с использованием указанного сепаратора