Унифицированная дозированная лекарственная форма










Формула / Реферат
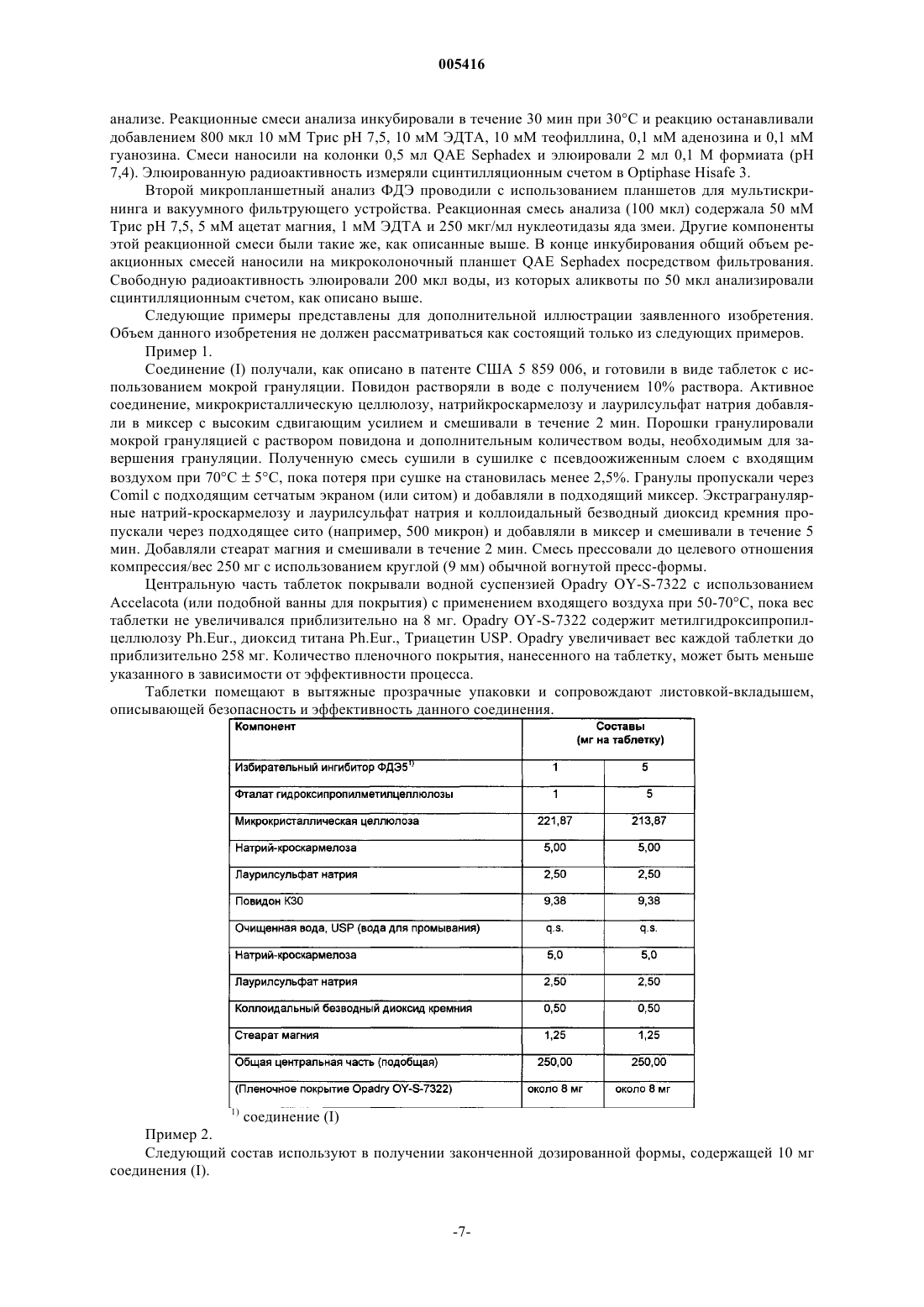
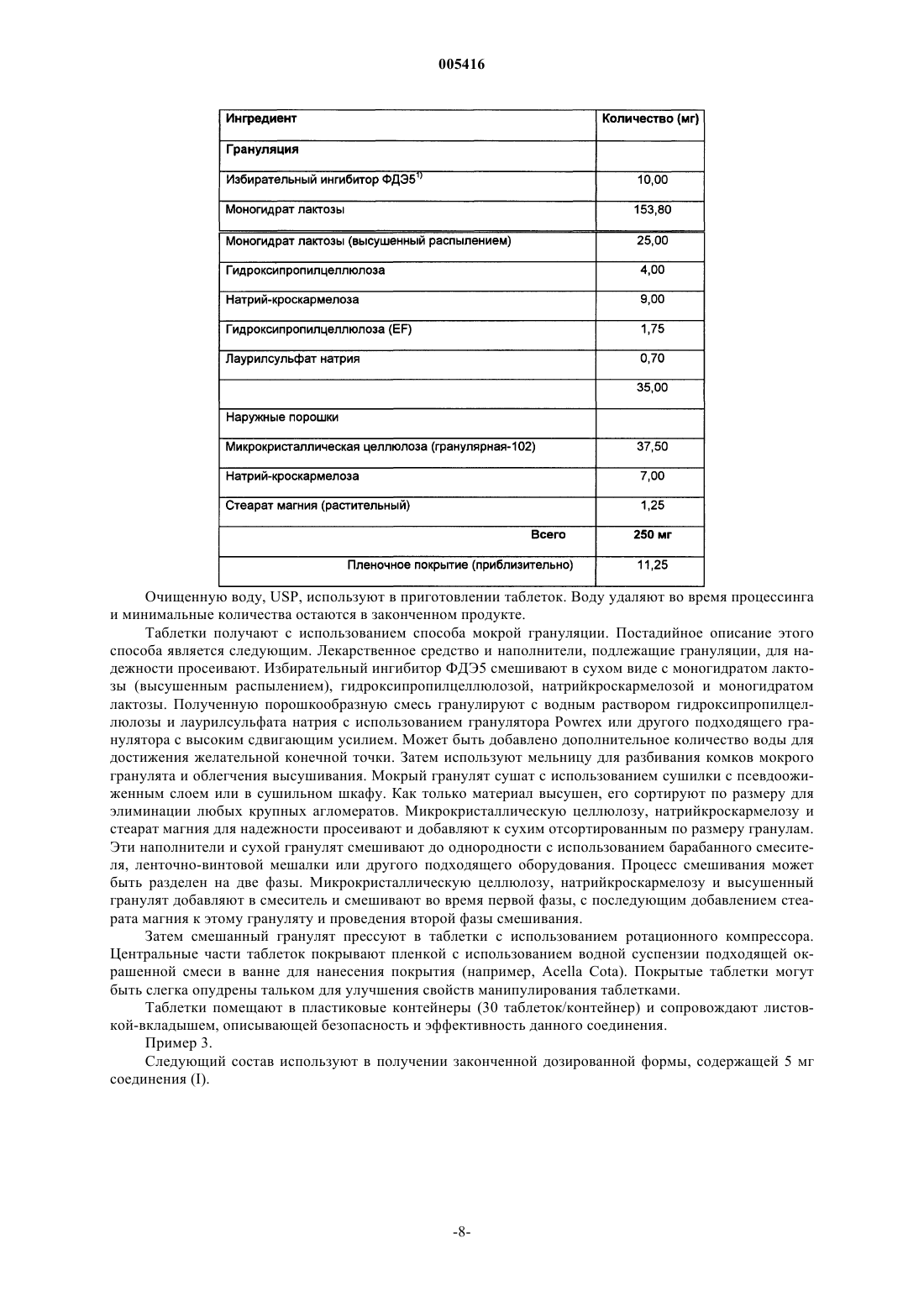
1. Фармацевтическая унифицированная дозированная лекарственная форма, пригодная для перорального введения и содержащая от приблизительно 1 мг до приблизительно 20 мг, до максимальной дозы 20 мг в день соединения, имеющего структурную формулу

2. Фармацевтическая унифицированная дозированная лекарственная форма по п.1, содержащая приблизительно 2-20 мг соединения.
3. Фармацевтическая унифицированная дозированная лекарственная форма по п.1, содержащая приблизительно 5-20 мг соединения.
4. Фармацевтическая унифицированная дозированная лекарственная форма по п.1, содержащая приблизительно 1-5 мг соединения.
5. Фармацевтическая унифицированная дозированная лекарственная форма по п.2, содержащая приблизительно 2,5 мг соединения.
6. Фармацевтическая унифицированная дозированная лекарственная форма по п.3, содержащая приблизительно 5 мг соединения.
7. Фармацевтическая унифицированная дозированная лекарственная форма по п.3, содержащая приблизительно 10 мг соединения.
8. Фармацевтическая унифицированная дозированная лекарственная форма по п.3, содержащая приблизительно 20 мг соединения.
9. Фармацевтическая унифицированная дозированная лекарственная форма по любому из пп.1-8, где унифицированная доза находится в форме, выбранной из группы, состоящей из жидкости, таблетки, капсулы и желатиновой капсулы.
10. Фармацевтическая унифицированная дозированная лекарственная форма по любому из пп.1-8, где унифицированная доза находится в форме таблетки
11. Фармацевтическая унифицированная дозированная лекарственная форма по любому из пп.1-8, пригодная для использования в лечении состояния, в котором желательным является ингибирование ФДЭ5.
12. Фармацевтическая унифицированная дозированная лекарственная форма по п.11, где указанное состояние является сексуальной дисфункцией.
13. Фармацевтическая унифицированная дозированная лекарственная форма по п.12, где указанная сексуальная дисфункция является мужской эректильной дисфункцией.
14. Фармацевтическая унифицированная дозированная лекарственная форма по п.12, где указанная сексуальная дисфункция является нарушением возбуждения у женщин.
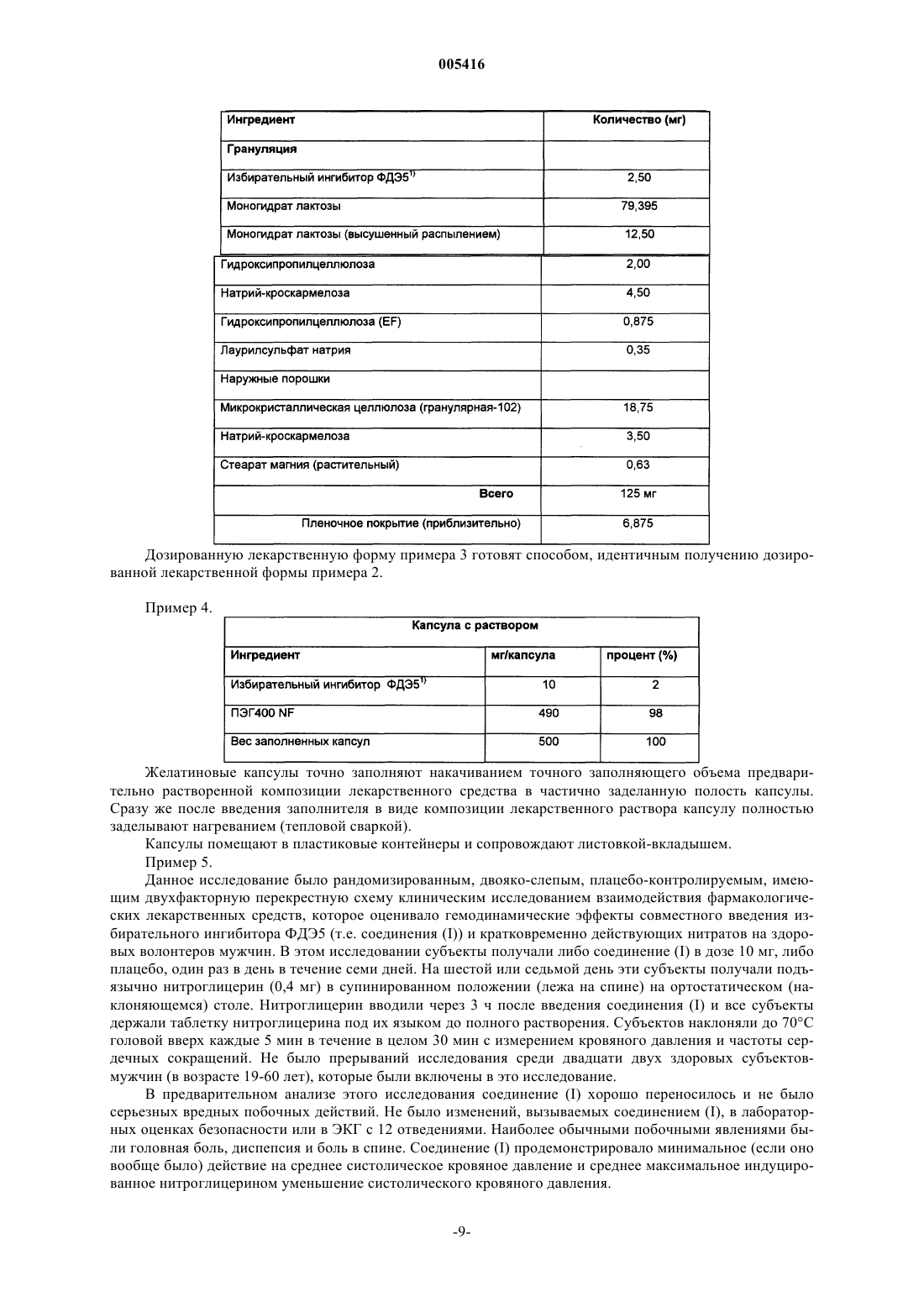
15. Способ лечения сексуальной дисфункции у пациента, нуждающегося в лечении, предусматривающий введение одной или нескольких унифицированных доз, содержащих от приблизительно 1 мг до приблизительно 20 мг, до максимальной дозы 20 мг в день, соединения, имеющего структурную формулу

16. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 2-20 мг указанного соединения.
17. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 5-20 мг указанного соединения.
18. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 1-5 мг указанного соединения.
19. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 2,5 мг указанного соединения.
20. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 5 мг указанного соединения.
21. Способ по п.15, отличающийся тем, что унифицированная доза содержит 10 мг указанного соединения.
22. Способ по п.15, отличающийся тем, что унифицированная доза содержит 20 мг указанного соединения.
23. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 10 мг указанного соединения и вводится один раз в день.
24. Способ по любому из пп.15-23, отличающийся тем, что унифицированная доза находится в форме, выбранной из группы, состоящей из жидкости, таблетки, капсулы и желатиновой капсулы.
25. Способ по любому из пп.15-23, отличающийся тем, что унифицированная доза находится в форме таблетки.
26. Способ по любому из пп.15-25, отличающийся тем, что сексуальная дисфункция является мужской эректильной дисфункцией.
27. Способ по любому из пп.15-25, отличающийся тем, что сексуальная дисфункция является нарушением возбуждения у женщин.
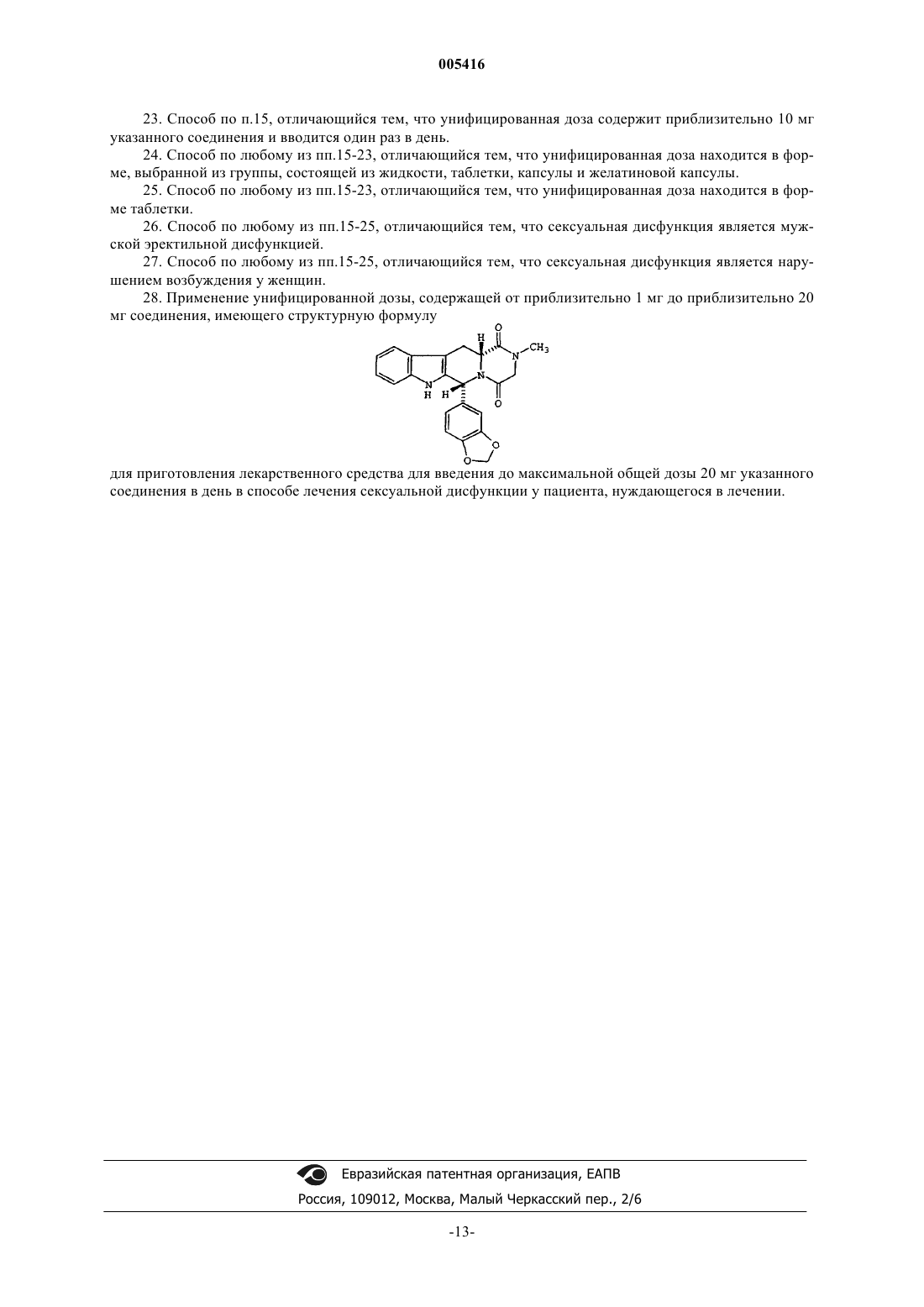
28. Применение унифицированной дозы, содержащей от приблизительно 1 мг до приблизительно 20 мг соединения, имеющего структурную формулу

для приготовления лекарственного средства для введения до максимальной общей дозы 20 мг указанного соединения в день в способе лечения сексуальной дисфункции у пациента, нуждающегося в лечении.
Текст
005416 Перекрестная ссылка на родственные заявки Данная заявка заявляет преимущество предварительной заявки на патент с порядковым номером 60/132036, поданной 30 апреля 1999 г. Область изобретения Данное изобретение относится к высокоизбирательному ингибитору фермента фосфодиэстеразы(ФДЭ) и к его применению в фармацевтической унифицированной (стандартной) дозированной лекарственной форме. В частности, данное изобретение относится к сильному ингибитору циклический гуанозин-3',5'-монофосфат-специфической фосфодиэстеразы типа 5 (ФДЭ 5), который при включении в фармацевтический продукт применим для лечения сексуальной дисфункции. Описанная здесь унифицированная дозированная лекарственная форма характеризуется избирательным ингибированием ФДЭ 5 и,следовательно, обеспечивает пользу в терапевтических областях, где желательным является ингибирование ФДЭ 5, с минимизацией или элиминацией вредных побочных эффектов, возникающих из ингибирования других фосфодиэстеразных ферментов. Предпосылки изобретения Биохимические, физиологические и клинические эффекты ингибиторов циклический гуанозин-3',5'монофосфат-специфической фосфодиэстеразы (цГМФ-специфической ФДЭ) предполагают их применимость в многочисленных патологических состояниях, в которых является желательным модулирование функции гладких мышц, почечной, гемостатической, воспалительной и/или эндокринной функции. цГМФ-специфическая фосфодиэстераза типа 5 (ФДЭ 5) является основным гидролизующим цГМФ ферментом в гладкой мышце сосудов, и сообщалась ее экспрессия в пещеристом теле полового члена (Taheret al., J. Urol., 149, p. 285A (1993. Таким образом, ФДЭ 5 является привлекательной мишенью в лечении сексуальной дисфункции (Murray, DNP 6(3), pp. 150-56 (1993. Фармацевтический продукт, который обеспечивает ингибитор ФДЭ 5, является в настоящее время доступным и продается под товарным названием ВИАГРА. Активным ингредиентом ВИАГРЫ является силденафил. Этот продукт продается в виде промышленного изделия, включающего в себя таблетки силденафила 25, 50 и 100 мг и листовку-вкладыш в упаковке, содержащую информацию о лекарственном средстве. Листовка-вкладыш указывает, что силденафил является более сильным ингибитором ФДЭ 5,чем другие известные фосфодиэстеразы (более, чем в 80 раз, в отношении ингибирования ФДЭ 1, более,чем в 1000 раз, в отношении ингибирования ФДЭ 2, ФДЭ 3 и ФДЭ 4). Сообщалось, что IC50 для силденафила против ФДЭ 5 равна 3 нМ (Drugs of the Future, 22(2), pp. 138-143 (1997 и 3,9 нМ (Boolel et al., Int. J.of Impotence, 8, pp. 47-52 (1996. Описано, что силденафил имеет в 4000 раз большую селективность в отношении ФДЭ 5 в сравнении с ФДЭ 3 и только в 10 раз большую селективность в отношении ФДЭ 5 в сравнении с ФДЭ 6. Теоретически предполагается, что его относительное отсутствие селективности в отношении ФДЭ 6 является основой для отклонений от нормы, связанных с цветовым зрением (различением цветов). Хотя силденафил имел значительный коммерческий успех, он потерпел крах вследствие его значительных вредных побочных действий, в том числе внезапных приливов крови к лицу (коэффициент встречаемости 10%). Вредные побочные действия ограничивают применение силденафила в случае пациентов, страдающих от отклонений от нормы зрения, гипертензии и, что наиболее важно, применение индивидуумами, которые используют органические нитраты (Welds et al., Amer. J. of Cardiology, 83(5A),pp. 21 (C)-28(C) (1999. Применение силденафила пациентами, принимающими органические нитраты, вызывает клинически значимое падение кровяного давления, которое могло бы создавать опасность для пациента. Поэтому этикетка на упаковке для силденафила дает строгие противопоказания против его применения в комбинации с органическими нитратами (например, нитроглицерином, изосорбидмононитратом, изосорбиднитратом, эритритилтетранитратом) и другими донорами оксида азота в любой форме, либо регулярно,либо периодически, так как силденафил потенциирует гипотензивные эффекты нитратов. См. C.R. Contiet al., Amer. J. of Cardiology, 83 (5A), pp. 29C-34C (1999). Таким образом, даже при доступности силденафила остается потребность в идентификации улучшенных фармацевтических продуктов, которые применимы в лечении сексуальной дисфункции. Патент США 5 859 006 Daugan описывает некоторые тетрациклические производные, которые являются сильными ингибиторами цГМФ-специфической ФДЭ, или ФДЭ 5. Сообщается, что IC50 соединений, описанных в патенте США 5 859 006, находится в диапазоне от 1 нМ до 10 мкМ. Пероральная доза для таких соединений равна 0,58 мг в день для среднего взрослого пациента (70 кг). Так, сообщаются унифицированные (стандартные) дозированные лекарственные формы (таблетки или капсулы) в виде 0,2-400 мг активного соединения. Не описаны значимые вредные побочные действия, присущие соединениям, описанным в патенте США 5 859 006. Заявители обнаружили, что одно такое тетрациклическое производное, (6R, 12 аR)-2,3,6,7,12,12 агексагидро-2-метил-6-(3,4-метилендиоксифенил)-пиразино[2',1':6,1]пиридо[3,4-b]индол-1,4-дион,альтернативно называемое (6R-транс)-6-(1,3-бензодиоксол-5-ил)-2,3,6,7,12,12 а-гексагидро-2-метилпиразино-[1',2':1,6]пиридо[3,4-b]индол-1,4-дионом, и называемое здесь Соединением (I), может быть введено в унифицированной дозе (дозе для одного приема), которая обеспечивает эффективное лечение без по-1 005416 бочных действий, связанных с продаваемым в настоящее время ингибитором ФДЭ 5, силденафилом. До данного изобретения такие побочные действия считали присущими ингибированию ФДЭ 5. Важным является то, что клинические исследования заявителей обнаружили также, что может быть обеспечен эффективный продукт, имеющий пониженную тенденцию к вызыванию внезапных приливов крови к лицу у чувствительных индивидуумов. Наиболее неожиданным было то, что этот продукт может также вводиться с клинически незначимыми побочными эффектами, связанными с комбинированными действиями ингибитора ФДЭ 5 и органического нитрата. Таким образом, противопоказание, которое когда-то считалось необходимым для продукта, содержащего ингибитор ФДЭ 5, является необязательным при введении cоединения (I) в виде унифицированной (стандартной) дозы от приблизительно 1 до приблизительно 20 мг, как описано здесь. Таким образом, данное изобретение обеспечивает эффективную терапию для сексуальной дисфункции у индивидуумов, которые прежде были не поддающимися лечению или страдали от неприемлемых побочных действий, в том числе индивидуумов, имеющих сердечнососудистое заболевание, например, индивидуумов, нуждающихся в терапии нитратами, перенесших инфаркт миокарда за более, чем три месяца, перед началом терапии сексуальной дисфункции, и страдающих от застойной сердечной недостаточности класса 1, или индивидуумов, страдающих от нарушений,связанных со зрением. Данное изобретение обеспечивает соединение (I) в унифицированной (стандартной) дозированной лекарственной форме. То есть, данное изобретение обеспечивает фармацевтическую унифицированную дозированную лекарственную форму, пригодную для перорального введения, содержащую приблизительно 1-20 мг соединения (I). Сущность изобретения Данное изобретение обеспечивает фармацевтическую дозированную лекарственную форму для фармацевтического применения для человека, содержащую от приблизительно 1 до приблизительно 20 мг (6R, 12aR)-2,3,6,7,12,12a-гексагидро-2-метил-6-(3,4-метилендиоксифенил)пиразино[2',1':6,1]пиридо[3,4-b]индол-1,4-диона в унифицированной дозированной лекарственной форме, пригодной для перорального введения. Далее, данное изобретение обеспечивает способ лечения состояний, при которых желательным является ингибирование ФДЭ 5, предусматривающий введение пациенту, нуждающемуся в этом, пероральной дозированной лекарственной формы, содержащей приблизительно 1-20 мг избирательного ингибитора ФДЭ 5, по необходимости, до общей дозы 20 мг в день. Далее, данное изобретение обеспечивает применение пероральной дозированной лекарственной формы, содержащей избирательный ингибитор ФДЭ 5, в дозе приблизительно 1-20 мг, для лечения сексуальной дисфункции. Конкретные состояния, которые могут лечиться данным изобретением, включают в себя, но не ограничиваются ими, мужскую эректильную дисфункцию и женскую сексуальную дисфункцию, в частности, нарушение возбуждения у женщин, также известное как нарушение сексуального возбуждения у женщин. В частности, данное изобретение относится к фармацевтической унифицированной дозированной лекарственной композиции, содержащей приблизительно 1-20 мг соединения, имеющего структурную формулу: причем указанная унифицированная дозированная лекарственная форма пригодна для перорального введения, и к способу лечения сексуальной дисфункции с использованием этой фармацевтической содержащей унифицированную (стандартную) дозу композиции. Подробное описание Для задач данного изобретения, раскрытого и описанного здесь, следующие термины и аббревиатуры определены следующим образом. Термин контейнер обозначает любую емкость и крышку для нее, пригодную для хранения,транспортировки, расфасовки фармацевтического продукта и/или манипулирования фармацевтическим продуктом. Термин IC50 является мерой силы соединения в ингибировании конкретного фермента ФДЭ (например, ФДЭ 1 с, ФДЭ 5 или ФДЭ 6). IС 50 представляет собой концентрацию соединения, которая приводит к 50% ингибированию фермента в эксперименте по типу единственная доза-ответ. Определение величины IC50 для соединения легко проводить по известной in vitro методологии, описанной в Y. Cheng etal., Biochem. Pharmacol., 22, pp. 3099-3108 (1973). Термин листовка-вкладыш упаковки обозначает информацию, сопровождающую этот продукт,которая обеспечивает описание того, как следует вводить продукт, вместе с данными о безопасности и-2 005416 эффективности, необходимыми для врача, фармацевта и пациента, чтобы принять информированное решение в отношении применения данного продукта. Эта листовка-вкладыш обычно рассматривается как этикетка для фармацевтического продукта. Термин пероральная дозированная лекарственная форма используется в общем смысле для ссылки на вводимые перорально фармацевтические продукты. Пероральные дозированные лекарственные формы, признаваемые специалистами в данной области, включают в себя жидкие готовые лекарственные формы, таблетки, капсулы и желатиновые капсулы. Термин нарушения (аномалии) зрения обозначает отклоняющееся от нормы зрение, характеризующееся сине-зеленым зрением, которое, как считают, обусловлено ингибированием ФДЭ 6. Термин покраснение обозначает эпизодическую красноту лица и шеи, приписываемую расширению сосудов, вызываемому приемом внутрь лекарственного средства, обычно сопровождающуюся ощущением тепла на лице и шее и иногда сопровождающуюся потоотделением. Термин свободное лекарственное средство обозначает твердые частицы лекарственного средства,не заключенные в мелко измельченном виде в полимерный соосажденный продукт (копреципитат). Заявляемая теперь дозированная лекарственная форма предпочтительно упакована в виде промышленного изделия для фармацевтического применения человеком, содержащего листовку-вкладыш, контейнер и дозированную лекарственную форму, содержащую приблизительно 1-20 мг соединения (I). Листовка-вкладыш обеспечивает описание того, как вводить фармацевтический продукт, вместе с данными о безопасности и эффективности, необходимыми для врача, фармацевта и пациента, чтобы принять информированное решение в отношении применения этого продукта. Эта листовка-вкладыш обычно рассматривается как этикетка для фармацевтического продукта. Листовка-вкладыш, включенная в это промышленное изделие, указывает, что соединение (I) применимо в лечении состояний, при которых желательным является ингибирование ФДЭ 5. Листовка-вкладыш обеспечивает также инструкции для введения одной или нескольких унифицированных дозированных лекарственных форм приблизительно 1-20 мг, по необходимости, до максимальной общей дозы 20 мг в день. Предпочтительно, эту дозу вводят при приблизительно 5-20 мг/день, более предпочтительно приблизительно 5-15 мг/день. Наиболее предпочтительно дозу 10 мг вводят один раз в день. Предпочтительные подлежащие лечению состояния включают в себя сексуальную дисфункцию (в том числе мужскую эректильную дисфункцию и женскую сексуальную дисфункцию и более предпочтительно нарушение возбуждения у женщин (FAD. Предпочтительным подлежащим лечению состоянием является мужская эректильная дисфункция. Важным является то, что листовка-вкладыш подтверждает применение продукта для лечения сексуальной дисфункции у пациентов, страдающих от заболевания сетчатки, например, диабетической ретинопатии или пигментного ретинита, или у пациентов, которые используют органические нитраты. Таким образом, эта листовка-вкладыш не содержит противопоказаний, связанных с этими состояниями, и, в частности, противопоказаний введения этой дозированной формы с органическим нитратом. Более предпочтительно, листовка-вкладыш не содержит также никаких предостережений или предупреждений, связанных как с заболеваниями сетчатки, в частности, пигментным ретинитом, так и с индивидуумами,склонными к отклонениям от нормы (аномалиям) зрения. Предпочтительно, эта листовка-вкладыш сообщает также коэффициент встречаемости приливов крови к лицу и шее (покраснений) ниже 2%, предпочтительно ниже 1% и наиболее предпочтительно ниже 0,5% пациентов, которым вводят эту дозированную лекарственную форму. Коэффициент встречаемости покраснения демонстрирует явное усовершенствование над прежними фармацевтическими продуктами, содержащими ингибитор ФДЭ 5. Контейнер, используемый в промышленном изделии, является общепринятым в фармацевтической практике. Обычно этот контейнер является вытяжной прозрачной упаковкой, пакетом из фольги, стеклянным или пластиковым флаконом и сопутствующими колпачком или крышкой, или другим подобным изделием, пригодным для применения пациентом или фармацевтом. Предпочтительно, контейнер имеет размер, подходящий для вмещения 1-1000 твердых дозированных форм, предпочтительно 1-500 твердых дозированных форм и, наиболее предпочтительно 5-30 твердых дозированных форм. Пероральные дозированные лекарственные формы, общепринятые для специалистов в данной области, включают в себя например, такие формы, как жидкие композиции, таблетки, капсулы и желатиновые капсулы. Предпочтительно эти дозированные лекарственные формы являются твердыми дозированными лекарственными формами, в частности, таблетками, содержащими приблизительно 1-20 мг соединения (I). Любые фармацевтически приемлемые наполнители для перорального применения пригодны для приготовления таких дозированных лекарственных форм. Подходящие фармацевтические дозированные лекарственные формы включают в себя соосажденные формы, описанные, например, в патенте США 5 985 326 (Butler), включенном здесь в качестве ссылки. В предпочтительных вариантах унифицированная дозированная лекарственная форма данного изобретения является твердым веществом, не содержащим соосажденной формы соединения (I), и предпочтительнее содержит твердое соединение (I) в виде свободного лекарственного средства. Предпочтительно, эти таблетки содержат фармацевтические наполнители, обычно признанные как безопасные, такие как лактоза, микрокристаллическая целлюлоза, крахмал, карбонат кальция, стеарат-3 005416 магния, стеариновая кислота, тальк и коллоидальный диоксид кремния, и их готовят стандартными фармацевтическими способами изготовления, описанными в Remington's Pharmaceutical Sciences, 18th Ed.,Mack Publishing Co., Easton, PA (1990). Такие способы включают в себя, например, мокрую грануляцию с последующим высушиванием, размалыванием и прессованием в таблетки с пленочным покрытием или без него; сухую грануляцию с последующим размалыванием, прессованием в таблетки с пленочным покрытием или без него; сухое смешивание с последующим прессованием в таблетки с пленочным покрытием или без него; отлитые формованием в формах таблетки; мокрую грануляцию с высушиванием и заполнением желатиновых капсул; сухую смесь, помещенную в желатиновые капсулы; или суспензию или раствор, помещенные в желатиновые капсулы. Обычно, твердые дозированные формы имеют идентификационные знаки, которые нанесены непрокрашиванием на ранее окрашенные участки или тиснением на поверхности. Данное изобретение основывается на детальных экспериментах и клинических испытаниях и неожиданных наблюдениях, что побочные действия, которые ранее считали признаком ингибирования ФДЭ 5, могут быть уменьшены до клинически незначимых уровней посредством выбора соединения и унифицированной дозы для одного приема. Это неожиданное наблюдение позволило разработать унифицированную дозированную форму, которая включает в себя соединение (I) в унифицированных дозированных лекарственных формах приблизительно 1-20 мг, которая при пероральном введении минимизирует нежелательные побочные эффекты, ранее считавшиеся неизбежными. Эти побочные эффекты включают в себя покраснение лица, отклонения от нормы зрения и значимое снижение кровяного давления при введении соединения (I) одного или в комбинации с органическим нитратом. Минимальное действие соединения (I), вводимого в виде унифицированных дозированных форм приблизительно 1-20 мг,на ФДЭ 6 позволяет также вводить избирательный ингибитор ФДЭ 5 пациентам, страдающим от заболевания сетчатки, такого как диабетическая ретинопатия или пигментный ретинит. Соединение (I) имеет следующую структурную формулу: Было продемонстрировано в клинических исследованиях на человеке, что соединение структурной формулы (I) проявляет минимальное действие на систолическое кровяное давление при введении с органическими нитратами. В противоположность этому, силденафил демонстрирует в 4 раза более высокое уменьшение систолического кровяного давления над плацебо, что приводит к противопоказаниям в листовке-вкладыше ВИАГРЫ и к предостережениям в отношении определенных пациентов. Нижеследующее иллюстрирует величины IС 50 ФДЭ 5 и ФДЭ 6 для соединения структурной формулы (I), определенные описанными здесь процедурами. СоединениеIC50 ФДЭ 6 (нМ) ФДЭ 6/ФДЭ 5 1 2,5 3400 1300 Соединение структурной формулы (I) дополнительно демонстрирует IC50 против ФДЭ 1 с 10000 и отношение ФДЭ 1 с/ФДЭ 5 4000. Препараты Препарат ФДЭ 5 человека Рекомбинантное получение ФДЭ 5 человека проводили по существу, как описано в примере 7 патента США 5 702 936, включенного здесь в качестве ссылки, за исключением того, что использовали дрожжевой трансформирующий вектор, происходящий из основной плазмиды ADH2, описанный в V.Price et al., Methods in Enzymology, 1985, pages 308-318 (1990), в который были включены промоторная и терминаторная последовательности ADH2 дрожжей, а не промоторная и терминаторная последовательности ADH1, и хозяин Saccharomyces cerevisiae был протеаза-недостаточным штаммом BJ2-54, депонированным 31 августа 1998 г. Американской Коллекцией Типовых Культур, Manassas, Virginia под номером доступа АТСС 74465. Трансформированные клетки-хозяева выращивали в среде 2 Х SC-leu, pH 6,2, с микроэлементами и витаминами. Спустя 24 ч среду YEP, содержащую глицерин, добавляли до конечной концентрации 2 Х YEP/3% глицерин. Спустя приблизительно 24 ч клетки собирали, промывали и хранили при -70 С. Осадки клеток (29 г) оттаивали на льду с равным объемом лизисного буфера (25 мМ ТрисCl, рН 8, 5 мМ MgCl2, 0,25 мМ дитиотреитол, 1 мМ бензамидин и 10 мкМ ZnSO4). Клетки лизировали в микрофлюидизаторе с N2 при 20000 фунтах на кв. дюйм (138 МПа). Лизат центрифугировали и фильтровали через одноразовые фильтры 0,45 мкм. Фильтрат наносили на колонку 150 мл Q Sepharose Fast Flow(Pharmacia). Колонку промывали 1,5 объемами буфера А (20 мМ Бис-Трис-пропан, рН 6,8, 1 мМ MgCl2,0,25 мМ дитиотреитол, 10 мкМ ZnSO4) и элюировали ступенчатым градиентом 125 мМ NaCl в буфере А с последующим линейным градиентом 125-1000 мМ NaCl в буфере А.-4 005416 Активные фракции из линейного градиента наносили на керамическую гидроксиапатитную колонку 180 мл в буфере В (20 мМ Бис-Трис-пропан (рН 6,8), 1 мМ MgCl2, 0,25 мМ дитиотреитол, 10 мкМZnSO4 и 250 мМ KCl). После нанесения колонку промывали 2 объемами буфера В и элюировали линейным градиентом 0-125 мМ фосфата калия в буфере В. Активные фракции объединяли, осаждали 60% сульфатом аммония и ресуспендировали в буфере С (20 мМ Бис-Трис-пропан, рН 6,8, 125 мМ NaCl, 0,5 мМ дитиотреитол и 10 мкМ ZnSO4). Этот пул наносили на колонку Sephacryl S-300 HR 140 мл и элюировали буфером С. Активные фракции разбавляли 50% глицерином и хранили при -20 С. Полученные препараты имели чистоту около 85% согласно электрофорезу в ДСН-ПААГ. Анализ на ФДЭ-активность Активность ФДЭ 5 может быть измерена в стандартных для данной области анализах. Например,удельная активность любой ФДЭ может быть определена следующим образом. Анализы ФДЭ с использованием способа разделения при помощи активированного угля выполняли по существу, как описано вLoughney et al., (1996), The Journal of Biological Chemistry, 271:796-806. В этом анализе активность ФДЭ 5 превращает [32 Р]цГМФ в [32 Р]5'ГМФ пропорционально количеству присутствующей активности ФДЭ 5. Затем [32 Р]5 ГМФ количественно превращают в свободный [32 Р]фосфат и немеченый аденозин действием 5'-нуклеотидазы яда змеи. Таким образом, количество высвобождающегося [32 Р]фосфата пропорционально активности фермента. Анализ проводят при 30 С в реакционной смеси 100 мкл, содержащей (конечная концентрация) 40 мМ Трис-Cl (рН 8,0), 1 мкМ ZnSO4, 5 мМ MgCl2 и 0,1 мг/мл бычьего сывороточного альбумина. ФДЭ 5 присутствует в количествах, которые дают 30% общий гидролиз субстрата(условия линейного анализа). Реакционную смесь инициируют добавлением субстрата (1 мМ [32 Р]цГМФ) и смесь инкубируют в течение 12 мин. Затем добавляют семьдесят пять (75) мкг яда Crotalus atrox и инкубирование продолжают в течение еще 3 мин (всего 15 мин). Реакцию останавливают добавлением 200 мл активированного угля (суспензии 25 мг/мл в 0,1 М NaH2PO4, рН 4). После центрифугирования (750 хg в течение 3 мин) для осаждения активированного угля берут пробу супернатанта для определения радиоактивности в сцинтилляционном счетчике и рассчитывают удельную активность ФДЭ 5. Препараты имели удельные активности около 3 мкмоль цГМФ, гидролизованного в минуту на миллиграмм белка. Препарат бычьей ФДЭ 6 Бычья ФДЭ 6 была предоставлена доктором N. Virmaux, INSERM U338, Strasbourg. Бычьи сетчатки получали, как описано Virmaux et al., FEBS Letters, 12(6), pp. 325-328 (1971) и см. также A. Sitaramayya etal., Exp. Eye Res., 25, pp. 163-169 (1977). Вкратце, если нет других указаний, все операции проводили на холоде и в тусклом красном свете. Глаза хранили на холоде и в темноте до четырех часов после забивания животных. Получение наружного сегмента бычьей сетчатки (ROS) проводили в основном согласно процедурам, описанным Schichi et al., J. Biol. Chem., 224:529 (1969). В типичном эксперименте 35 бычьих сетчаток растирали в ступке с 35 мл 0,066 М фосфатного буфера, рН 7,0, доведенного до 40% сахарозой, с последующей гомогенизацией в гомогенизаторе Поттера (20 двойных ходов поршня). Суспензию центрифугировали при 25000 х g в течение 20 мин. Осадок гомогенизировали в 7,5 мл 0,006 М фосфатного буфера (40% по сахарозе) и осторожно наслаивали под 7,5 мл фосфатного буфера (не содержащего сахарозы). Центрифугирование проводили в бакетном роторе (со свободно качающимися пробирками) при 45000 х g в течение 20 мин и получали осадок, который является черным в нижней части и имеет также красную полосу на межфазной поверхности 0,066 М фосфат-40% сахароза/0,066 М фосфат (неочищенный ROS). Этот красный материал на межфазной поверхности удаляли, разбавляли фосфатным буфером,откручивали до осадка и перераспределяли в забуференной 40% сахарозе, как описано выше. Эту процедуру повторяли 2 или 3 раза, пока не переставал образовываться осадок. Очищенный ROS промывали в фосфатном буфере и наконец откручивали до осадка при 25000 х g в течение 20 мин. Затем все материалы хранили в замороженном виде до использования. Гипотонические экстракты готовили суспендированием выделенного ROS в 10 мМ Трис-Cl рН 7,5,1 мМ ЭДТА и 1 мМ дитиоэритрите с последующим центрифугированием при 100000 g в течение 30 мин. Сообщалось, что этот препарат имел удельную активность около 35 нмоль цГМФ, гидролизованного в минуту на миллиграмм белка. Препарат ФДЭ 1 с из клеток Spodoptera fugiperda (Sf9) Клеточные осадки (5 г) оттаивали на льду с 20 мл лизисного буфера (50 мМ MOPS рН 7,4, 10 мкМZnSO4, 0,1 мМ CaCl2, 1 мМ ДТТ, 2 мМ бензамидин-HCl, 5 мкг/мл каждого из пепстатина, лейпептина и апротенина). Клетки лизировали пропусканием через French pressure-элемент (SLM-Aminco) при поддержании температур ниже 10 С. Полученный гомогенат клеток центрифугировали при 36000 об/мин при 4 С в течение 45 мин в ультрацентрифуге Бекмана с использованием ротора типа TI45. Супернатант выбрасывали, а полученный осадок ресуспендировали с 40 мл буфера для солюбилизации (лизисный буфер, содержащий 1 М NaCl, 0,1 М MgCl2, 1 мМ CaCl2, 20 мкг/мл кальмодулина и 1% сульфобетаинSB12 (Z3-12) обработкой ультразвуком с использованием устройства настройки VibraCell с микронаконечником в течение 3 х 30 с.) Это выполняли в смеси измельченного льда с солью для охлаждения. После обработки ультразвуком смесь медленно смешивали в течение 30 мин при 4 С для завершения солю-5 005416 билизации мембраносвязанных белков. Эту смесь центрифугировали в ультрацентрифуге Бекмана с использованием ротора типа TI45 при 36000 об/мин в течение 45 мин. Супернатант разбавляли лизисным буфером, содержащим 10 мкг/мл ингибитора I и II калпаина. Осажденный белок центрифугировали в течение 20 мин при 9000 об/мин в роторе Бекмана JA-10. Затем извлеченный супернатант подвергали хроматографии на Mimetic Blue AP-агарозе. Для хроматографии на колонке Mimetic Blue АР-агарозы смолу сначала защищали нанесением 10 колоночных объемов 1% поливинилпирролидина (т.е. MW 40000) для блокирования сайтов неспецифического связывания. Рыхло связанный PVP-40 удаляли промыванием 10 колоночными объемами 2 МNaCl и 10 мМ цитратом натрия рН 3,4. Непосредственно перед добавлением солюбилизированной пробы ФДЭ 1 с 3 колонку уравновешивали 5 колоночными объемами колоночного буфера А (50 мМ MOPS рН 7,4, 10 мкМ ZnSO4, 5 мМ MgCl2, 0,1 мМ CaCl2, 1 мМ ДТТ, 2 мМ бензамидин-HCl). Солюбилизированную пробу наносили на колонку при скорости тока 2 мл/мин с рециркуляцией,так что всю пробу наносили 4-5 раз на протяжении 12 ч. После завершения нанесения колонку промывали 10 колоночными объемами колоночного буфера А, затем 5 колоночными объемами колоночного буфера В (колоночный буфер А, содержащий 20 мМ 5'-АМФ) и затем 5 колоночными объемами колоночного буфера С (50 мМ MOPS рН 7,4, 10 мкМ ZnSO4, 0,1 мМ CaCl2, 1 мМ дитиотреитол и 2 мМ бензамидин-HCl). Фермент элюировали в виде трех последовательных пулов. Первый пул состоял из фермента из промывки 5 колоночными объемами колоночного буфера С, содержащего 1 мМ цАМФ. Второй пул состоял из фермента из промывки 10 колоночными объемами колоночного буфера С, содержащего 1 МNaCl. Конечный пул фермента состоял из промывки 5 колоночными объемами колоночного буфера С,содержащего 1 М NaCl и 20 мМ цАМФ. Активные пулы фермента собирали и циклические нуклеотиды удаляли посредством общепринятой гель-фильтрационной хроматографии или хроматографии на гидроксиапатитных смолах. После удаления циклических нуклеотидов пулы фермента диализовали против диализного буфера, содержащего 25 мМMOPS рН 7,4, 10 мкМ ZnSO4, 500 мМ NaCl, 1 мМ CaCl2, 1 мМ дитиотреитол, 1 мМ бензамидин-HCl, с последующим диализом против диализного буфера, содержащего 50% глицерин. Фермент быстро замораживали при помощи сухого льда и хранили при -70 С. Полученные препараты имели чистоту около 90% согласно электрофорезу в ДСН-ПААГ. Эти препараты имели удельные активности около 0,1-1,0 мкмоль цАМФ, гидролизованного в минуту на миллиграмм белка. Определения IC50 Представляющим интерес параметром в оценке силы конкурентного ингибитора фермента ФДЭ 5 и/или ФДЭ 1 с и ФДЭ 6 является константа ингибирования, т.е. Ki. Этот параметр может быть аппроксимирован определением IC50, которая представляет собой концентрацию ингибитора, которая приводит к 50% ингибированию фермента, в эксперименте типа единственная доза-ответ при следующих условиях. Концентрация ингибитора является всегда гораздо более высокой, чем концентрация фермента, так что концентрацию свободного ингибитора (которая неизвестна) аппроксимируют посредством общей концентрации ингибитора (которая известна). Выбирают подходящий диапазон концентраций ингибитора (т.е. концентрации ингибитора по меньшей мере в несколько раз большие и в несколько раз меньшие, чем Ki, присутствуют в этом эксперименте). Обычно концентрации ингибитора находятся в диапазоне от 10 нМ до 10 мкМ. Концентрации фермента и субстрата выбирают таким образом, что менее 20% субстрата потребляется в отсутствие ингибитора (обеспечивая, например, максимальный гидролиз субстрата от 10 до 15%),так что активность фермента является приблизительно постоянной на протяжении анализа. Концентрация субстрата составляет менее 1/10 константы Михаэлиса (Кm). При этих условиях IC50 будет близко приближаться к Ki. Это вытекает из уравнения Cheng-Prusoff, касающегося этих двух параметров: IC50=Ki(1+S/Km), с (1+S/Km) приблизительно 1 при низких величинах S/Km. Величину IC50 оценивают приближенно из точек данных посредством применения этих данных к подходящей модели взаимодействия фермента и ингибитора. Когда известно, что это взаимодействие включает в себя простую конкуренцию ингибитора с субстратом, может быть использована двухпараметрическая модель:Y=A/(1+x/B) где Y обозначает активность фермента, измеренную при концентрации ингибитора х, А обозначает активность фермента в отсутствие ингибитора, а В обозначает IС 50 (см. Y. Cheng et al., Biochem. Pharmacol.,22:3099-3108 (1973. Действия ингибиторов данного изобретения на ферментативную активность препаратов ФДЭ 5 и ФДЭ 6, описанных выше, оценивали в любом из двух анализов, которые отличались один от другого главным образом на основе масштаба и дали по существу одни и те же результаты в отношении величинIC50. Оба анализа включали в себя модификацию процедуры Wells et al., Biochim. Biophys. Acta, 384:430(1975). Первый из анализов выполняли в общем объеме 200 мкл, содержащем 50 мМ Трис рН 7,5, 3 мМ ацетат магния, 1 мМ ЭДТА, 50 мкг/мл нуклеотидазы яда змеи и 50 нМ [3 Н]-цАМФ (Amersham). Соединения данного изобретения растворяли в ДМСО, присутствующем в конечной концентрации 2% в этом-6 005416 анализе. Реакционные смеси анализа инкубировали в течение 30 мин при 30 С и реакцию останавливали добавлением 800 мкл 10 мМ Трис рН 7,5, 10 мМ ЭДТА, 10 мМ теофиллина, 0,1 мМ аденозина и 0,1 мМ гуанозина. Смеси наносили на колонки 0,5 мл QAE Sephadex и элюировали 2 мл 0,1 М формиата (рН 7,4). Элюированную радиоактивность измеряли сцинтилляционным счетом в Optiphase Hisafe 3. Второй микропланшетный анализ ФДЭ проводили с использованием планшетов для мультискрининга и вакуумного фильтрующего устройства. Реакционная смесь анализа (100 мкл) содержала 50 мМ Трис рН 7,5, 5 мМ ацетат магния, 1 мМ ЭДТА и 250 мкг/мл нуклеотидазы яда змеи. Другие компоненты этой реакционной смеси были такие же, как описанные выше. В конце инкубирования общий объем реакционных смесей наносили на микроколоночный планшет QAE Sephadex посредством фильтрования. Свободную радиоактивность элюировали 200 мкл воды, из которых аликвоты по 50 мкл анализировали сцинтилляционным счетом, как описано выше. Следующие примеры представлены для дополнительной иллюстрации заявленного изобретения. Объем данного изобретения не должен рассматриваться как состоящий только из следующих примеров. Пример 1. Соединение (I) получали, как описано в патенте США 5 859 006, и готовили в виде таблеток с использованием мокрой грануляции. Повидон растворяли в воде с получением 10% раствора. Активное соединение, микрокристаллическую целлюлозу, натрийкроскармелозу и лаурилсульфат натрия добавляли в миксер с высоким сдвигающим усилием и смешивали в течение 2 мин. Порошки гранулировали мокрой грануляцией с раствором повидона и дополнительным количеством воды, необходимым для завершения грануляции. Полученную смесь сушили в сушилке с псевдоожиженным слоем с входящим воздухом при 70 С 5 С, пока потеря при сушке на становилась менее 2,5%. Гранулы пропускали черезComil с подходящим сетчатым экраном (или ситом) и добавляли в подходящий миксер. Экстрагранулярные натрий-кроскармелозу и лаурилсульфат натрия и коллоидальный безводный диоксид кремния пропускали через подходящее сито (например, 500 микрон) и добавляли в миксер и смешивали в течение 5 мин. Добавляли стеарат магния и смешивали в течение 2 мин. Смесь прессовали до целевого отношения компрессия/вес 250 мг с использованием круглой (9 мм) обычной вогнутой пресс-формы. Центральную часть таблеток покрывали водной суспензией Opadry OY-S-7322 с использованиемAccelacota (или подобной ванны для покрытия) с применением входящего воздуха при 50-70 С, пока вес таблетки не увеличивался приблизительно на 8 мг. Opadry OY-S-7322 содержит метилгидроксипропилцеллюлозу Ph.Eur., диоксид титана Ph.Eur., Триацетин USP. Opadry увеличивает вес каждой таблетки до приблизительно 258 мг. Количество пленочного покрытия, нанесенного на таблетку, может быть меньше указанного в зависимости от эффективности процесса. Таблетки помещают в вытяжные прозрачные упаковки и сопровождают листовкой-вкладышем,описывающей безопасность и эффективность данного соединения. соединение (I) Пример 2. Следующий состав используют в получении законченной дозированной формы, содержащей 10 мг соединения (I). Очищенную воду, USP, используют в приготовлении таблеток. Воду удаляют во время процессинга и минимальные количества остаются в законченном продукте. Таблетки получают с использованием способа мокрой грануляции. Постадийное описание этого способа является следующим. Лекарственное средство и наполнители, подлежащие грануляции, для надежности просеивают. Избирательный ингибитор ФДЭ 5 смешивают в сухом виде с моногидратом лактозы (высушенным распылением), гидроксипропилцеллюлозой, натрийкроскармелозой и моногидратом лактозы. Полученную порошкообразную смесь гранулируют с водным раствором гидроксипропилцеллюлозы и лаурилсульфата натрия с использованием гранулятора Powrex или другого подходящего гранулятора с высоким сдвигающим усилием. Может быть добавлено дополнительное количество воды для достижения желательной конечной точки. Затем используют мельницу для разбивания комков мокрого гранулята и облегчения высушивания. Мокрый гранулят сушат с использованием сушилки с псевдоожиженным слоем или в сушильном шкафу. Как только материал высушен, его сортируют по размеру для элиминации любых крупных агломератов. Микрокристаллическую целлюлозу, натрийкроскармелозу и стеарат магния для надежности просеивают и добавляют к сухим отсортированным по размеру гранулам. Эти наполнители и сухой гранулят смешивают до однородности с использованием барабанного смесителя, ленточно-винтовой мешалки или другого подходящего оборудования. Процесс смешивания может быть разделен на две фазы. Микрокристаллическую целлюлозу, натрийкроскармелозу и высушенный гранулят добавляют в смеситель и смешивают во время первой фазы, с последующим добавлением стеарата магния к этому грануляту и проведения второй фазы смешивания. Затем смешанный гранулят прессуют в таблетки с использованием ротационного компрессора. Центральные части таблеток покрывают пленкой с использованием водной суспензии подходящей окрашенной смеси в ванне для нанесения покрытия (например, Acella Cota). Покрытые таблетки могут быть слегка опудрены тальком для улучшения свойств манипулирования таблетками. Таблетки помещают в пластиковые контейнеры (30 таблеток/контейнер) и сопровождают листовкой-вкладышем, описывающей безопасность и эффективность данного соединения. Пример 3. Следующий состав используют в получении законченной дозированной формы, содержащей 5 мг соединения (I). Дозированную лекарственную форму примера 3 готовят способом, идентичным получению дозированной лекарственной формы примера 2. Пример 4. Желатиновые капсулы точно заполняют накачиванием точного заполняющего объема предварительно растворенной композиции лекарственного средства в частично заделанную полость капсулы. Сразу же после введения заполнителя в виде композиции лекарственного раствора капсулу полностью заделывают нагреванием (тепловой сваркой). Капсулы помещают в пластиковые контейнеры и сопровождают листовкой-вкладышем. Пример 5. Данное исследование было рандомизированным, двояко-слепым, плацебо-контролируемым, имеющим двухфакторную перекрестную схему клиническим исследованием взаимодействия фармакологических лекарственных средств, которое оценивало гемодинамические эффекты совместного введения избирательного ингибитора ФДЭ 5 (т.е. соединения (I и кратковременно действующих нитратов на здоровых волонтеров мужчин. В этом исследовании субъекты получали либо соединение (I) в дозе 10 мг, либо плацебо, один раз в день в течение семи дней. На шестой или седьмой день эти субъекты получали подъязычно нитроглицерин (0,4 мг) в супинированном положении (лежа на спине) на ортостатическом (наклоняющемся) столе. Нитроглицерин вводили через 3 ч после введения соединения (I) и все субъекты держали таблетку нитроглицерина под их языком до полного растворения. Субъектов наклоняли до 70 С головой вверх каждые 5 мин в течение в целом 30 мин с измерением кровяного давления и частоты сердечных сокращений. Не было прерываний исследования среди двадцати двух здоровых субъектовмужчин (в возрасте 19-60 лет), которые были включены в это исследование. В предварительном анализе этого исследования соединение (I) хорошо переносилось и не было серьезных вредных побочных действий. Не было изменений, вызываемых соединением (I), в лабораторных оценках безопасности или в ЭКГ с 12 отведениями. Наиболее обычными побочными явлениями были головная боль, диспепсия и боль в спине. Соединение (I) продемонстрировало минимальное (если оно вообще было) действие на среднее систолическое кровяное давление и среднее максимальное индуцированное нитроглицерином уменьшение систолического кровяного давления.-9 005416 Пример 6. В двух рандомизированных, двояко-слепых контролируемых плацебо исследованиях соединение (I) вводили пациентам, нуждающимся в этом, в диапазоне доз, как при ежедневном введении, так и при терапии по потребности, для половых сношений и полового акта в домашней обстановке. Дозы от 5 до 20 мг соединения (I) были эффективными и демонстрировали менее 1% приливов крови к лицу и шее и не было сообщений об отклонениях от нормы зрения. Было обнаружено, что доза 10 мг соединения (I) была полностью эффективной и показала минимальные побочные действия. Повышенную эректильную функцию определяли по Международному индексу эректильной функции (IIEF) (Rosen et al., Urology, 49, pp. 822-830 (1997, дневникам сексуальных попыток и вопросу полного удовлетворения. Соединение (I) значимо улучшало процент успешных попыток полового акта, в том числе способности получать и сохранять эрекцию как при режиме введения по потребности, так и при режиме ежедневного введения дозы лекарственного средства. Пример 7. Третье клиническое исследование было рандомизированным, двояко-слепым, контролируемым плацебо исследованием соединения (I), вводимого по потребности пациентам с мужской эректильной дисфункцией. Соединение (I) вводили на протяжении периода восьми недель в лечении мужской эректильной дисфункции (ЭД). Эректильную дисфункцию (ЭД) определяют как стойкую неспособность получать и/или сохранять эрекцию, адекватную для достижения удовлетворительной сексуальной активности. Введение по потребности определяют как дробное (прерывистое) введение соединения (I) перед ожидаемой сексуальной активностью. Популяция исследования состояла из 212 мужчин в возрасте по меньшей мере 18 лет с легкойтяжелой эректильной дисфункцией. Соединение (I) вводили перорально в виде таблеток или соосажденных продуктов, полученных в соответствии с патентом США 5 985 326 (Butler). Соединение (I) вводили в дозах 2 мг, 5 мг, 10 мг и 25 мг по потребности и не более, чем один раз каждые 24 ч. Лечение нитратами, азоловыми противогрибковыми средствами (например, кетоконазолом или итраконазолом), варфарином, эритромицином или антиандрогенами не разрешалось ни в одном случае во время этого исследования. Не разрешались также другие одобренные или экспериментальные лекарственные средства, лечения или приспособления, используемые для лечения ЭД. Сорок одному субъекту вводили плацебо. Двумя первичными переменными эффективности были способность субъекта проникать в его партнера и его способность сохранять эрекцию во время полового акта, измеряемые по Международному индексу эректильной функции (IIEF). Анкета (опросник) IIEF содержит пятнадцать вопросов и является краткой, достоверной мерой эректильной дисфункции (см. R.C. Rosen et al., Urology, 49, pp. 822-830(1997. Вторичными переменными эффективности были балльные оценки согласно IIEF для эректильной функции, оргазменной функции, сексуального желания, удовлетворения половым актом и общего удовлетворения; способности пациента достигать эрекции, способности введения его полового члена во влагалище его партнера, завершения полового акта эякуляцией, удовлетворения степенью твердости его эрекции и общего удовлетворения, причем все эти показатели измерялись при помощи дневника показателей полового акта (SEP); и вопроса общей оценки, задаваемого в конце периода лечения. SEP представляет собой дневник-инструмент пациента, документирующий каждое сексуальное сношение на протяжении хода данного исследования. Аспект безопасности данного исследования включал в себя всех зарегистрированных для участия в исследовании субъектов и его оценивали с использованием оценки всех сообщенных побочных явлений и изменений в клинических лабораторных показателях, жизненно важных признаках, результатах физического обследования и результатах электрокардиограмм. В конечной точке исследования, процент пациентов, которые оценивали их способность проникновения (вопрос 3 IIEF) как почти всегда или всегда, был следующим: 17,5% в группе плацебо, 38,1% в группе 2 мг, 48,8% в группе 5 мг, 51,2% в группе 10 мг и 83,7% в группе 25 мг. Сравнения выявили статистически значимые различия в изменении способности проникновения между плацебо и всеми уровнями доз соединения (I). В конечной точке % пациентов, которые оценивали их способность сохранять эрекцию (вопрос 4IIEF) во время полового акта как почти всегда или всегда, был следующим: 10,0% в группе плацебо,19,5% в группе 2 мг, 32,6% в группе 5 мг, 39,0% в группе 10 мг и 69,0% в группе 25 мг. Сравнение выявило статистически значимые различия в изменении способности проникновения между плацебо и тремя более высокими уровнями доз соединения (I). Это исследование включало в себя также оценку безопасности. Возникающее в ходе лечения побочное действие определяют как состояние, не присутствующее при базовой линии, которое появляется после базовой линии, или состояние, присутствующее при базовой линии, которое увеличивается по тяжести после базовой линии. Наиболее обычными сообщенными возникающими при лечении побочными событиями были головная боль, диспепсия и боль в спине. Встречаемость возникающих в результате лечения побочных явлений, по-видимому, связана с дозой.-10 005416 В целом, данное исследование продемонстрировало, что все четыре дозы соединения (I), а именно,2, 5, 10 и 25 мг, принимаемые по потребности, производили значимое улучшение, относительно плацебо, в сексуальной функции мужчин с эректильной дисфункцией, согласно оценкам с использованиемIIEF, согласно дневникам пациентов, оценивающим частоту успешного полового акта и удовлетворение половым актом, и согласно общей оценке. Объединенные результаты из клинических исследований показали, что введение соединения (I) эффективно лечит мужскую эректильную дисфункцию, как иллюстрируется в следующей таблице.IIEF-область эректильной функции (изменение от базовой линии (фона Доза на один прием Соединения 1)n обозначает число субъектов, SD обозначает стандартное отклонение. Однако на основании объединенных клинических исследований наблюдалось также, что процент возникающих при лечении побочных явлений увеличивался с увеличением дозы для одного приема соединения (I), как показано в следующей таблице. Приведенная выше таблица показывает увеличение побочных явлений при стандартных дозах для одного приема 25 мг-100 мг. Таким образом, даже хотя эффективность в лечении ЭД наблюдалась при дозах 25-100 мг, следует учитывать побочные явления, наблюдаемые от доз 25 мг до доз 100 мг. Согласно данному изобретению, стандартная доза для одного приема приблизительно 1-20 мг,предпочтительно приблизительно 2-20 мг, более предпочтительно приблизительно 5-20 мг и наиболее предпочтительно приблизительно 5-15 мг соединения (I), вводимая максимально до 20 мг на период 24 ч,как эффективно лечит ЭД, так и минимизирует или элиминирует появление вредных побочных эффектов. Важно, что не сообщались отклонения от нормы зрения и по существу элиминировалось покраснение лица и шеи. Неожиданно, кроме лечения ЭД, с дозой соединения (I) приблизительно 1-20 мг, с минимальными вредными побочными эффектами, индивидуумы, подвергающиеся терапии нитратами, могут также лечиться по поводу ЭД с использованием способа и композиции данного изобретения. Принципы, предпочтительные варианты и способы применения данного изобретения были описаны в предыдущем описании. Однако, данное изобретение подразумевает защиту здесь, заключающуюся в том, что оно не должно рассматриваться как ограничиваемое конкретными описанными формами, так как они должны рассматриваться как иллюстративные, а не как ограничительные. Вариации и изменения могут быть произведены специалистами в данной области без отхода от идеи данного изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Фармацевтическая унифицированная дозированная лекарственная форма, пригодная для перорального введения и содержащая от приблизительно 1 мг до приблизительно 20 мг, до максимальной дозы 20 мг в день соединения, имеющего структурную формулу 2. Фармацевтическая унифицированная дозированная лекарственная форма по п.1, содержащая приблизительно 2-20 мг соединения. 3. Фармацевтическая унифицированная дозированная лекарственная форма по п.1, содержащая приблизительно 5-20 мг соединения. 4. Фармацевтическая унифицированная дозированная лекарственная форма по п.1, содержащая приблизительно 1-5 мг соединения. 5. Фармацевтическая унифицированная дозированная лекарственная форма по п.2, содержащая приблизительно 2,5 мг соединения. 6. Фармацевтическая унифицированная дозированная лекарственная форма по п.3, содержащая приблизительно 5 мг соединения. 7. Фармацевтическая унифицированная дозированная лекарственная форма по п.3, содержащая приблизительно 10 мг соединения. 8. Фармацевтическая унифицированная дозированная лекарственная форма по п.3, содержащая приблизительно 20 мг соединения. 9. Фармацевтическая унифицированная дозированная лекарственная форма по любому из пп.1-8,где унифицированная доза находится в форме, выбранной из группы, состоящей из жидкости, таблетки,капсулы и желатиновой капсулы. 10. Фармацевтическая унифицированная дозированная лекарственная форма по любому из пп.1-8,где унифицированная доза находится в форме таблетки. 11. Фармацевтическая унифицированная дозированная лекарственная форма по любому из пп.1-8,пригодная для использования в лечении состояния, в котором желательным является ингибирование ФДЭ 5. 12. Фармацевтическая унифицированная дозированная лекарственная форма по п.11, где указанное состояние является сексуальной дисфункцией. 13. Фармацевтическая унифицированная дозированная лекарственная форма по п.12, где указанная сексуальная дисфункция является мужской эректильной дисфункцией. 14. Фармацевтическая унифицированная дозированная лекарственная форма по п.12, где указанная сексуальная дисфункция является нарушением возбуждения у женщин. 15. Способ лечения сексуальной дисфункции у пациента, нуждающегося в лечении, предусматривающий введение одной или нескольких унифицированных доз, содержащих от приблизительно 1 мг до приблизительно 20 мг, до максимальной дозы 20 мг в день, соединения, имеющего структурную формулу 16. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 2-20 мг указанного соединения. 17. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 5-20 мг указанного соединения. 18. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 1-5 мг указанного соединения. 19. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 2,5 мг указанного соединения. 20. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 5 мг указанного соединения. 21. Способ по п.15, отличающийся тем, что унифицированная доза содержит 10 мг указанного соединения. 22. Способ по п.15, отличающийся тем, что унифицированная доза содержит 20 мг указанного соединения.-12 005416 23. Способ по п.15, отличающийся тем, что унифицированная доза содержит приблизительно 10 мг указанного соединения и вводится один раз в день. 24. Способ по любому из пп.15-23, отличающийся тем, что унифицированная доза находится в форме, выбранной из группы, состоящей из жидкости, таблетки, капсулы и желатиновой капсулы. 25. Способ по любому из пп.15-23, отличающийся тем, что унифицированная доза находится в форме таблетки. 26. Способ по любому из пп.15-25, отличающийся тем, что сексуальная дисфункция является мужской эректильной дисфункцией. 27. Способ по любому из пп.15-25, отличающийся тем, что сексуальная дисфункция является нарушением возбуждения у женщин. 28. Применение унифицированной дозы, содержащей от приблизительно 1 мг до приблизительно 20 мг соединения, имеющего структурную формулу для приготовления лекарственного средства для введения до максимальной общей дозы 20 мг указанного соединения в день в способе лечения сексуальной дисфункции у пациента, нуждающегося в лечении.
МПК / Метки
МПК: A61P 15/10, A61K 31/4985
Метки: дозированная, унифицированная, форма, лекарственная
Код ссылки
<a href="https://eas.patents.su/14-5416-unificirovannaya-dozirovannaya-lekarstvennaya-forma.html" rel="bookmark" title="База патентов Евразийского Союза">Унифицированная дозированная лекарственная форма</a>
Биоадгезивная твердая дозированная лекарственная форма

Номер патента: 818
Опубликовано: 24.04.2000
Автор: Жили Поль Мари Виктор
МПК: A61K 9/00
Метки: форма, твердая, биоадгезивная, лекарственная, дозированная
Формула / Реферат:
1. Биоадгезивная фармацевтическая композиция, содержащая фармацевтически эффективное количество активного ингредиента, от 80 до 98,8 вес.% предварительно превращенного в гель крахмала и от 1 до 10 вес.% гидрофильного образующего матрицу полимера, отличающаяся тем, что композиция дополнительно содержит от 0,2 до 5 вес.% С16-22алкилфумарата щелочного металла в качестве смазывающего вещества. 2. Композиция по п.1, отличающаяся тем, что смазывающее...
Желудочная и/или дуоденальная клейкая лекарственная форма

Номер патента: 3512
Опубликовано: 26.06.2003
Авторы: Нисино Такеси, Инаги Тосио, Ямагути Нориказу, Сираи Хироюки
МПК: A61K 9/30
Метки: клейкая, желудочная, дуоденальная, лекарственная, форма
Формула / Реферат:
1. Лекарственная дозированная форма, прилипающая к желудочной и/или дуоденальной слизистой оболочке, отличающаяся тем, что получена посредством нанесения на композицию, которая содержит лекарственное средство, действующее в желудке или двенадцатиперстной кишке, и водонерастворимый целлюлозный полимер (I), полимера (II), обладающего в кислых условиях адгезионной способностью на поверхности слизистой оболочки пищеварительного тракта и...
Твердая лекарственная форма тербинафина гидрохлорида

Номер патента: 4105
Опубликовано: 25.12.2003
Авторы: Паршуткина Юлиана Евгеньевна, Казакова Галина Львовна, Младенцев Андрей Леонидович, Смирнова Татьяна Васильевна
МПК: A61K 31/137, A61P 31/10
Метки: лекарственная, гидрохлорида, твердая, форма, тербинафина
Формула / Реферат:
Твердая лекарственная форма тербинафина гидрохлорида, содержащая действующее вещество, наполнители, увлажнитель и опудривающий агент, отличающаяся тем, что она содержит в качестве наполнителей микрокристаллическую целлюлозу и аэросил, в качестве увлажнителя водный раствор Коллидона 30, в качестве опудривающих агентов Коллидон CL-M и магния стеарат, а также дополнительно в качестве консерванта содержит натрия бензоат или бензойную кислоту при...
Водно-дисперсная лекарственная форма ивермектина для лечения экто- и эндопаразитозов

Номер патента: 2628
Опубликовано: 29.08.2002
Авторы: Сидоркин Владимир Александрович, Жемеричкин Дмитрий Александрович, Семенов Сергей Вячеславович
МПК: A61P 33/14, A61K 9/10
Метки: ивермектина, водно-дисперсная, лечения, форма, экто, лекарственная, эндопаразитозов
Формула / Реферат:
Водно-дисперсная лекарственная форма ивермектина для лечения экто- и эндопаразитозов, включающая активнодействующее вещество, сорастворитель, поверхностно-активное вещество, консервант, фосфатно-цитратный буфер и дистиллированную воду в качестве растворителя, отличающаяся тем, что она дополнительно содержит витамин Е при следующем содержании компонентов, мас.%: Ивермектин или авермектин 0,1-7,5 Сорастворитель 10-60 Мицеллообразующий...
Лекарственная форма для контролируемого высвобождения ципрофлоксацина для приема один раз в день

Номер патента: 4443
Опубликовано: 29.04.2004
Авторы: Тэлвар Нареш, Стейнифорс Джон, Сен Химадри
МПК: A61K 9/22
Метки: день, высвобождения, форма, один, приема, лекарственная, ципрофлоксацина, контролируемого
Формула / Реферат:
1. Лекарственная форма, принимаемая один раз в день, для контролируемого высвобождения ципрофлоксацина, содержащая фармацевтически эффективное количество ципрофлоксацина, от примерно 0,2 до примерно 0,5% альгината натрия, от примерно 1,0 до примерно 2,0% ксантановой камеди, от примерно 10,0 до примерно 25% бикарбоната натрия и от примерно 5,0 до примерно 20% сшитого поливинилпирролидона, при этом вышеупомянутые проценты являются мас/мас...
Предыдущий патент: Фармацевтический препарат для лечения, профилактики и/или диагностики опухолей и способ получения его активного компонента
Следующий патент: Фунгицидная смесь
Случайный патент: Способ и ленточная агломерационная установка для непрерывного спекания и предварительного восстановления гранулированного минерального вещества