Закрепленные на матрице пептидомиметики
Номер патента: 17583
Опубликовано: 30.01.2013
Авторы: Обрехт Даниель, Врейблуд Ян Вим, Меле Керстин, Романьоли Барбара, Хенце Хайко, Демарко Стивен Дж., Лудин Кристиан, Робинсон Джон Энтони, Мухерджи Решми, Гомберт Франк










Формула / Реферат
1. Цикло(-Tyr-His-X-Cys-Ser-Ala-DPro-Dab-Arg-Tyr-Cys-Tyr-Gln-Lys-DPro-Pro), который содержит дисульфидную связь между Cys4 и Cys11, и его фармацевтически приемлемые соли, где X представляет собой Ala или Tyr.
2. Соединение по п.1, отличающееся тем, что имеет антагонистическую активность в отношении CXCR4 и способно либо предотвратить ВИЧ-инфекции у здоровых людей, либо замедлить или прекратить развитие ВИЧ-инфекций у инфицированных пациентов; предотвратить или лечить рак, опосредованный активностью рецептора CXCR4; предотвратить или лечить иммунологическое заболевание, опосредованное активностью рецептора CXCR4; лечить иммуносупрессию или воспаление; или мобилизовать стволовые клетки периферической крови, мезенхимальные стволовые клетки (МСК) или другие стволовые клетки, удержание которых зависит от активности рецептора CXCR4.
3. Применение соединения по п.1 для предотвращения ВИЧ-инфекций у здоровых людей или для замедления или прекращения развития ВИЧ-инфекций у инфицированных пациентов; предотвращения или лечения рака, опосредованного активностью рецептора CXCR4; предотвращения или лечения иммунологического заболевания, опосредованного активностью рецептора CXCR4; лечения иммуносупрессии или воспаления; или мобилизации стволовых клеток периферической крови, мезенхимальных стволовых клеток (МСК) и/или других стволовых клеток, удержание которых зависит от активности рецептора CXCR4.
4. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически инертный носитель.
5. Композиция по п.4 в форме, подходящей для перорального, местного, трансдермального, инъекционного, буккального, назального, легочного или ингаляционного введения.
6. Композиция по п.4 или 5 в форме таблеток, драже, капсул, растворов, жидкостей, гелей, пластырей, кремов, мазей, сиропов, гидросмесей, суспензий, спреев, распылителей или суппозиториев.
7. Применение соединения по п.1 для производства лекарственного средства, обладающего антагонистическим действием в отношении CXCR4.
8. Применение по п.7, где указанное лекарственное средство предназначено для использования в предотвращении ВИЧ-инфекций у здоровых людей или в замедлении или прекращении развития ВИЧ-инфекций у инфицированных пациентов; предотвращении или лечении рака, опосредованного активностью рецептора CXCR4; предотвращении или лечении иммунологического заболевания, опосредованного активностью рецептора CXCR4; лечении иммуносупрессии или воспаления; или мобилизации стволовых клеток периферической крови, мезенхимальных стволовых клеток или других стволовых клеток, удержание которых зависит от активности рецептора CXCR4.
9. Способ производства соединений по п.1, который включает стадии:
(a) связывание активированной специальным образом твердой подложки с соответствующим образом N-защищенной производной Pro;
(b) удаление N-защитной группы из продукта, полученного на стадии (а);
(c) связывание полученного продукта с соответствующим образом N-защищенной производной DPro;
(d) удаление N-защитной группы из продукта, полученного на стадии (с);
(e) связывание полученного таким образом продукта с соответствующим образом N-защищенной производной аминокислоты, которая в полученном конечном продукте находится в положении 14, то есть Lys, причем аминогруппа, находящаяся в ее боковой цепи, также защищена соответствующим образом;
(f) удаление N-защитной группы из продукта, полученного таким образом;
(g) осуществление стадий, по существу, соответствующих стадиям (е) и (f), но с применением соответствующим образом N-защищенных производных аминокислот, которые в желаемом конечном продукте находятся в положениях 13-1, то есть Gln, Tyr, Cys, Tyr, Arg, Dab, DPro, Ala, Ser, Cys, Ala или Tyr, His и Tyr, причем любая функциональная группа, которая может присутствовать в указанных N-защищенных производных аминокислот, также защищена соответствующим образом;
(h) формирование дисульфидного β-стрендового соединения между боковыми цепями цистеиновых остатков в положениях 4 и 11;
(i) отделение полученного таким образом продукта от твердой подложки;
(j) циклизация продукта, отделенного от твердой подложки;
(k) удаление всех защитных групп, связанных с функциональными группами всех членов цепи из аминокислотных остатков; и
(l) при необходимости, превращение полученного таким образом продукта в фармацевтически приемлемые соли или превращение солей, полученных таким образом, в соответствующее свободное соединение или в другую фармацевтически приемлемую соль.
Текст
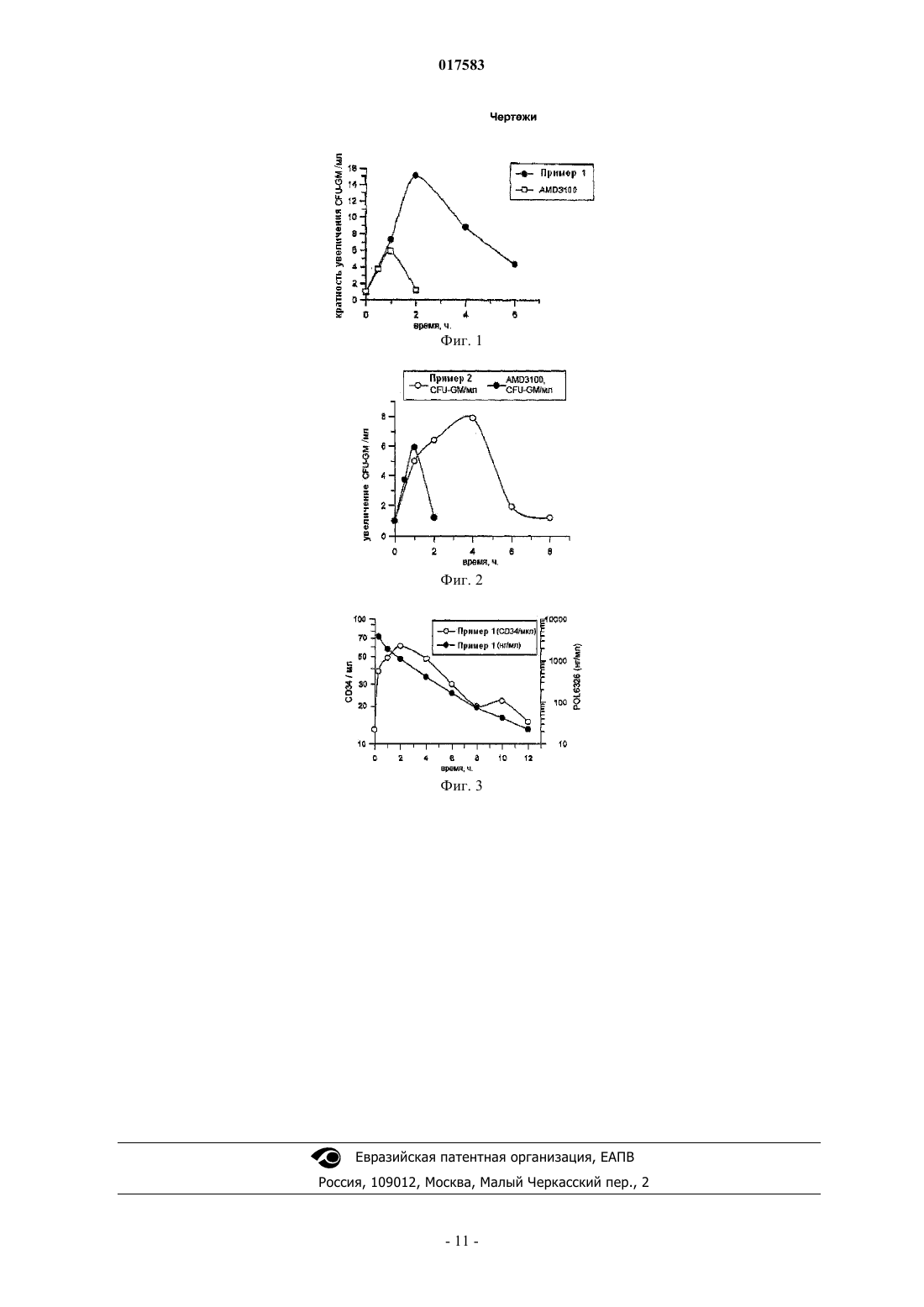
ЗАКРЕПЛЕННЫЕ НА МАТРИЦЕ ПЕПТИДОМИМЕТИКИ Закреплнные на матрице 3-шпилечные пептидомиметики цикло(-Tyr-His-X-Cys-Ser-Ala-DProDab-Arg-Tyr-Cys-Tyr-Gln-Lys-DPro-Pro), дисульфидная связь между Cys4 и Cys11, и их фармацевтически приемлемые соли, где X представляет собой Ala или Tyr, имеют антагонистические свойства в отношении CXCR4 и могут применяться для предотвращения ВИЧ инфекций у здоровых людей или замедления и прекращения развития вируса у инфицированных пациентов; или когда рак опосредован или является результатом активности рецептора CXCR4; или когда иммунологические заболевания опосредованы или являются результатом активности рецептора CXCR4; или для лечения иммуносупрессии; или в частности для мобилизации стволовых клеток периферической крови и/или мезенхимальных стволовых клеток (МСК) и/ или других стволовых клеток, удержание которых зависит от CXCR4 рецептора. Эти 3 шпилечные пептидомиметики могут быть произведены с помощью способа, который основан на стратегии смешанного твердо-жидкофазного синтеза, применяя методы, которые хорошо известны специалистам в пептидной химии.(CH), Гомберт Франк (DE), Лудин Кристиан, Врейблуд Ян Вим (CH) Квашнин В.П., Сапельников Д.А. 017583 Изобретение обеспечивает закреплнные на матрице -шпилечные пептидомиметики, которые имеют антагонистическую активность в отношении CXCR4 и которые, в общем, описаны, но не раскрыты конкретно в WO 2004/096840 А 1.-шпилечные пептидомиметики по изобретению представляют собой цикло(-Tyr-His-X-Cys-SerAla-DPro-Dab-Arg-Tyr-Cys-Tyr-Gln-Lys-DPro-Pro), дисульфидная связь между Cys4 и Cys11, и их фармацевтически приемлемые соли, где X - это Ala или Tyr. В соответствии с изобретением, вышеупомянутые -шпилечные пептидомиметики и их фармацевтически разрешенные соли могут быть приготовлены способом, содержащим следующие этапы:(a) соединение функционализированной специальным образом тврдой подложки с соответствующим образом N-защищнной производной Pro;(b) удаление N-защитной группы из продукта, полученного на стадии (а);(c) соединение полученного таким образом продукта с соответствующим образом N-защищнной производной DPro;(e) соединение полученного таким образом продукта с соответствующим образом N-защищнной производной аминокислоты, которая в желаемом конечном продукте находится в положении 14, то естьLys, причем аминогруппа, находящаяся в ее боковой цепи, также защищена соответствующим образом;(g) осуществление стадий, по существу, соответствующих стадиям (е) и (f), но с применением соответствующим образом N-защищнных производных аминокислот, которые в желаемом конечном продукте находятся в положениях 13-1, то есть Gln, Tyr, Cys, Tyr, Arg, Dab, DPro, Ala, Ser, Cys, Ala или Tyr,His и Tyr, причем любая функциональная группа, которая может присутствовать в указанных Nзащищнных производных аминокислот, также защищена соответствующим образом;(h) формирование дисульфидного -стрендового соединения между боковыми цепями цистеиновых остатков в положениях 4 и 11;(i) отделение полученного таким образом продукта от тврдой подложки;(j) циклизация продукта, отделенного от тврдой подложки;(k) удаление всех защитных групп, связанных с функциональными группами всех членов цепи из аминокислотных остатков; и(l) при желании, превращение полученного таким образом продукта в фармацевтически приемлемые соли или превращение фармацевтически приемлемых или неприемлемых солей, полученных таким образом, в соответствующее свободное соединение или в другую фармацевтически приемлемую соль. Стадии вышеизложенного способа могут быть осуществлены методами, которые хорошо известны всем специалистам в химии пептидов.-шпилечные пептидомиметики по настоящему изобретению могут применяться в широком диапазоне применений для предотвращения ВИЧ-инфекции у здоровых людей или замедления и прекращения развития вируса у инфицированных пациентов; или когда рак опосредован или является результатом активности рецептора CXCR4; или когда иммунологические заболевания опосредованы или являются результатом активности рецептора CXCR4; или для лечения иммуносупрессии; или для лечения воспаления, или для мобилизации стволовых клеток периферической крови и/или мезенхимальных стволовых клеток (МСК) и/или других стволовых клеток, удержание которых зависит от CXCR4 рецептора.-шпилечные пептидомиметики по изобретению могут вводиться сами по себе или могут применяться в виде соответствующей композиции вместе с носителями, разбавителями или эксципиентами,хорошо известными в области техники. В частности, -шпилечные пептидомиметики по изобретению могут применяться при лечении для увеличения секреции гемопоэтических стволовых клеток (ГСК) из костного мозга, который будет применяться в аллогенной или аутологической трансплантации. Срочное лечение с помощью инфузированных ГСК широко применяются для восстановления иммунных функций у пациентов, которые подверглись миелоаблативной терапии при лечении таких онкологических заболеваний, таких как множественная миелома и неходжкинская лимфома. Пациенты или доноры подвергаются воздействию веществ, мобилизующих ГСК, таких как соединение по изобретению,и впоследствии клетки собирают из периферической крови посредством афереза. ГСК трансплантируются обратно пациенту (аутологическая трансплантация) после, например, химиотерапии, или от донора реципиенту (аллогенная трансплантация), таким образом, способствуя восстановлению иммунной функции (Frhauf et al., Br.J.Haematol. 122, 360-375 (2003. Другие применения ГСК терапии включает в себя, но не ограничивается этим, терапевтический ангиогенез в случае, например, сердечного приступа (Shepherd RM et al., Blood 2006, 108(12):3662-3667).-шпилечные пептидомиметики по изобретению могут также применяться для лечения или предупреждения ВИЧ-инфекций или рака, такого как рак груди, рак мозга, рак простаты, рак легких, рак почки, нейробластома, неходжкинской лимфома, рак яичников, множественная миелома, хроническая лимфоидная лейкемия, рак поджелудочной железы, меланома, ангиогенез и гемопоэтические ткани; или та-1 017583 ких воспалительных нарушений как астма, аллергический ринит, болезни гиперчувствительности легких,гиперчувствительный пневмонит, эозинофильная пневмония, гиперчувствительность замедленного типа,интерстициальное заболевание легких (ИЗЛ), идиопатический легочный фиброз, ИЗЛ, связанный с ревматоидным артритом, системная красная волчанка, анкилозирующий спондилит, заболевание периферических сосудов, системный склероз, синдром Шегрена, болезнь Гиппеля-Линдау, системные анафилактические реакции или реакции гиперчувствительности, аллергия на лекарства, ревматоидный артрит,псориатический артрит, синдром Бехчета, мукозит, болезнь Крона, множественный склероз, миастения гравис, юношеский диабет, гломерулонефрит, аутоиммунный тиреоидит, отторжение трансплантата,включая отторжение гомотрансплантат или реакцию "трансплантат против хозяина", воспалительные заболевания кишечника, воспалительные дерматозы; либо для лечения иммуносупрессии, включая иммуносупрессию, вызванную отторжением трансплантата.-шпилечные пептидомиметики по изобретению могут вводиться отдельно, в виде смесей более чем одного -шпилечного пептидомиметика, в комбинации с другими агентами, мобилизирующими ГСК, или с анти-ВИЧ агентами, или с противомикробными агентами, или с противовоспалительными препаратами, и/или в комбинации с другими активными фармацевтически активными агентами в зависимости от конкретного случая. Фармацевтические композиции, содержащие -шпилечные пептидомиметики по изобретению, могут быть произведены посредством обычных способов смешивания, растворения, гранулирования, изготовления оболочечных таблеток, растирания в порошок мокрым способом, эмульгации, инкапсуляции,включения или лиофилизации. Фармацевтические композиции могут быть составлены обычным способом, применяя один или более физиологически приемлемых носителей, разбавителей, эксципиентов или вспомогательных веществ, которые способствуют преобразованию -шпилечных пептидомиметиков в препараты, которые могут применяться фармацевтически. Соответствующий состав зависит от выбранного способа введения. Для местного введения -шпилечные пептидомиметики по изобретению могут быть приготовлены в виде растворов, гелей, мазей, кремов, суспензий и т.д., хорошо известных в данной области техники. Системные препараты включают препараты, разработанные для введения посредством инъекции,например подкожного, внутривенного, внутримышечного введения, интратекальная или интраперитонеальная инъекция, а также препараты, разработанные для трансдермального, трансмукозального, перорального или пульмонарного введения. Для инъекций -шпилечные пептидомиметики по изобретению могут быть приготовлены в водных растворах, предпочтительно в физиологически совместимых буферах, таких как раствор Хенкса, раствор Рингера или физиологический раствор. Растворы могут содержать структурирующие агенты, такие как суспензирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, -шпилечные пептидомиметики по изобретению могут быть в форме порошка для соединения с подходящей средой, например апирогенной стерильной водой, перед применением. Для трансмукозального введения в состав могут входить пенетранты, соответствующие барьеру,который необходимо преодолеть, известные в области техники. Для перорального введения, соединения могут быть легко получены путем комбинирования шпилечных пептидомиметиков по изобретению с фармацевтически приемлемыми носителями, хорошо известными в области техники. Такие носители делают возможным получение -шпилечных пептидомиметиков по изобретению в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, гидросмесей, суспензий и т.д., для перорального приема пациентом, подлежащим лечению. Для таких пероральных препаратов, как, например, порошки, капсулы и таблетки, подходящие эксципиенты включают наполнители, такие как сахара, например лактоза, сахароза, маннит и сорбит; целлюлозные препараты,такие как кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин,трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрийкарбоксиметилцеллюлоза; гранулирующие агенты; и связывающие агенты. При желании могут быть добавлены дезинтегрирующие агенты, такие как поперечно-связанные поливинилпирролидоны, агар или альгиновая кислота или е соль, такая как альгинат натрия. При желании тврдые лекарственные формы могут быть покрыты сахарной или энтеросолюбильной оболочкой, применяя стандартные методы. Для жидких пероральных препаратов, таких как, например, суспензии, эликсиры и растворы, подходящие носители, эксципиенты и разбавители включают воду, гликоли, масла, спирты и т.д. Помимо этого могут быть добавлены ароматизирующие вещества, консерванты, красители и тому подобное. Для буккального введения композиция может принимать форму таблеток, лепешек и т.д., полученных обычными способами. Для введения путем ингаляции -шпилечные пептидомиметики по изобретению обычно получают в виде аэрозольных спреев в упаковке под давлением или с распылителем, с применением подходящего газа вытеснителя, например, дихлордифторметан, трихлорфторметан, углекислый газ или другой подходящий газ. В случае аэрозоля под давлением для определение единицы дозы может быть создан клапан для доставки измеренного количества. Могут быть созданы капсулы и картриджи, например желатино-2 017583 вые, для применения в ингаляторе или вдувателе, содержащие порошкообразную смесь -шпилечных пептидомиметиков по изобретению и подходящую порошкообразную основу, такую как лактоза или крахмал. Соединения также могут входить в состав ректальных или вагинальных композиций, таких как суппозитории, вместе с подходящими суппозиторными основами, такими как какао масло или другие глицериды. Помимо вышеупомянутых препаратов, -шпилечные пептидомиметики по изобретению могут также быть получены в виде депо-препаратов. Такие долгодействующие препараты могут вводиться путем имплантации (например, подкожно или внутримышечно) или путм внутримышечной инъекции. Для производства таких депо-препаратов -шпилечные пептидомиметики по изобретению могут быть включены в подходящие полимерные или гидрофобные материалы (например, эмульсия в приемлемом масле), или ионообменные смолы, либо могут быть получены в виде умеренно растворимых солей. Более того, могут применяться другие фармацевтические системы доставки, такие как липосомы и эмульсии, хорошо известные в области техники. Также могут применяться определенные органические растворители, такие как диметилсульфоксид. Кроме того, -шпилечные пептидомиметики по изобретению могут доставляться с применением систем с замедленным высвобождением, таких как полупроницаемые матрицы из тврдых полимеров, содержащих терапевтический агент. Различные материалы с замедленным высвобождением были установлены и хорошо известны специалистам в данной области. Капсулы замедленного высвобождения могут, в зависимости от их химической природы, высвобождать соединения от нескольких недель до более чем 100 дней. В зависимости от химической природы и биологической стабильности терапевтического агента, могут быть проведены дополнительные процедуры с целью стабилизации белка. Так как -шпилечные пептидомиметики по изобретению содержат заряженные остатки, они могут быть включены в любые вышеописанные препараты как таковые или в виде фармацевтически приемлемых солей. Фармацевтически приемлемые соли преимущественно более растворимы в воде и других протонных растворителях, чем соответствующая свободная форма. В частности, подходящие фармацевтически приемлемые соли включают в себя соли, например, карбоновой, фосфоновой, сульфоновой и сульфаминовой кислот, например, уксусной кислоты, пропионовой кислоты, каприловой кислоты, каприновой кислоты, додекановой кислоты, гликолевой кислоты, молочной кислоты, фумаровой кислоты,янтарной кислоты, адипиновой кислоты, пимелиновой кислоты, пробковой кислоты, азелаиновой кислоты, яблочной кислоты, винной кислоты, лимонной кислоты, аминокислот, таких как глютаминовой или аспарагиновой кислоты, малеиновой кислоты, гидроксималеиновой кислоты, метилмалеиновой кислоты,циклогексанкарбоновой кислоты, адамантанкарбоновой кислоты, бензойной кислоты, салициловой кислоты, 4-аминосалициловой кислоты, фталевой кислоты, фенилуксусной кислоты, миндальной кислоты,коричной кислоты, метан- или этансульфоновой кислоты, 2-гидроксиэтансульфоновой кислоты, этан-1,2 дисульфоной кислоты, бензолсульфоновой кислоты, 2-нафталенсульфоновой кислоты, 1,5-нафталендисульфоновой кислоты, 2, 3- или 4-метилбензолсульфоновой кислоты, метилсерной кислоты, этилсерной кислоты, додецилсерной кислоты, N-циклогексилсульфаминовой кислоты, N-метил-, N-этил- или Nпропилсульфаминовой кислоты, и других органических протонных кислот, таких как аскорбиновая кислота. Подходящими неорганическими кислотами являются, например, галогеноводородные кислоты,такие как хлороводородная кислота, серная кислота и фосфорная кислоты.-шпилечные пептидомиметики по изобретению либо в свободной форме, либо в форме их фармацевтически приемлемых солей или композиций будут как правило, применяться в количестве, эффективном для достижения необходимого результата. Нужно понимать, что применяемое количество будет зависеть от конкретного применения. Для местного введения для лечения или профилактики ВИЧ-инфекции, терапевтически эффективная доза может быть определена, например, в анализах in vitro, описанных в примерах. Лечение проводиться при явной ВИЧ-инфекции или даже когда ВИЧ-инфекция не очевидна. Специалист в данной области техники будет в состоянии определить терапевтически эффективные количества для местного лечения ВИЧ-инфекции без неоправданного экспериментирования. Для системного введения терапевтически эффективная доза может быть оценена исходя из анализов in vitro. Например, доза может быть сформулирована на основе экспериментов на животных моделях для достижения интервала циркулирующей концентрации -шпилечных пептидомиметиков, который будет включать в себя IC50, определенную в клеточной культуре (т.е. такую концентрацию тестируемого соединения, которая вызывает гибель 50% культуры клеток). Такая информация может применяться для определения более точного определения полезных для человека доз. Начальные дозировки могут быть также определены исходя из данных in vivo, например, животные модели, применяя методы, хорошо известные в области техники. Специалист в данной области техники сможет без труда оптимизировать введение препарата человеку, основываясь на данных экспериментов на животных. Количества доз для применения в качестве анти-ВИЧ агентов могут быть подобраны индивидуаль-3 017583 но для того, чтобы обеспечить достаточный для поддержания терапевтического эффекта уровень шпилечных пептидо-миметиков в плазме. Терапевтически эффективные уровни в сыворотке могут быть достигнуты путем введения многократных доз каждый день. В случаях местного применения или избирательного поглощения, эффективная локальная концентрация -шпилечных пептидомиметиков по изобретению не может быть связана с концентрацией плазмы. Специалист в данной области техники сможет оптимизировать терапевтически эффективные дозы для местного применения, не прибегая к неоправданным экспериментам. Конечно же, количество вводимых -шпилечных пептидомиметиков будет зависеть от самого объекта лечения, от его массы, серьезности заболевания, от способа введения и решений лечащего врача. Анти-ВИЧ терапия может периодически повторяться, если инфекция обнаруживается или даже если инфекция не обнаруживается. Терапия может проводиться самостоятельно или в комбинации с другими лекарственными средствами, такими как, например, другие анти-ВИЧ агенты или противоопухолевые агенты или другие противомикробные агенты. Как правило, терапевтически эффективная доза -шпилечных пептидомиметиков, описанных здесь,будет обеспечивать терапевтическую пользу, не оказывая существенного токсического эффекта. Токсичность -шпилечных пептидомиметиков по изобретению может быть определена стандартными фармацевтическими методами в клеточных культурах или на экспериментальных животных, например, определением LD50 (доза, смертельная для 50% популяции) или LD100 (доза, смертельная для 100% популяции). Соотношение доз токсического и терапевтического эффектов - терапевтический индекс. Предпочтительными являются соединения с высоким терапевтическим индексом. Данные испытаний на клеточной культуре и исследований на животных могут применяться при подборе нетоксического для людей интервала доз. Дозировка -шпилечных пептидомиметиков по изобретению предпочтительно находится в диапазоне циркулирующих концентраций, который включает эффективную дозу с небольшим токсическим эффектом или без него. Дозировка может измениться в пределах этого диапазона в зависимости от применяемой лекарственной формы и пути введения. Точная рецептура, способ введения и доза могут быть выбраны исходя из состояния пациента непосредственно лечащим врачом (см., например, Fingl et al. 1975, in: The Pharmacological Basis of Therapeutics, Ch.1, p.1). Следующие примеры иллюстрируют изобретение более детально, но не призваны ограничить его объем каким-либо образом. Список применяемых в этих Примерах сокращений: 1. Синтез пептидов. Связывание первого защищенного аминокислотного остатка со смолой 0,5 г 2-хлортритилхлоридной смолы (100-200 меш, сополи(стирол-1% DVB)полимерный матрикс, Cat. No. 01-64-0114, Novabiochem, Merck Biosci-ences Ltd.) (Barlos et al. Tetrahedron Lett. 1989, 30, 3943-3946) (1.4 ммоль/г, 0.7 ммоль) было помещено в сухую колбу. Смолу суспендировали в CH2Cl2 (2,5 мл) и позволили ей набухать при комнатной при постоянном перемешивании в течение 30 мин. Смолу обработали 0,49 ммоль (0,7 экв.) первого соответствующим образом защищенного аминокислотного остатка и 448 мкл (4 экв.) диизопропилэтиламина (DIEA) в CH2Cl2 (2,5 мл), смесь взбалтывали при 25 С в течение 4 ч. 30 мл смолыCH2Cl2 (Ix), DMF (Ix), CH2Cl2 (Ix), MeOH (Ix), CH2Cl2 (Ix), MeOH (Ix), CH2Cl2 (2x), Et2O (2x), а затем сушили в вакууме в течение 6 часов. Загрузка типично составляла 0,6-0,9 ммоль/г. Была приготовлена следующая предварительно нагруженная смола: Fmoc-Pro-2-хлортритиловая смола. Синтез полностью защищенного пептидного фрагмента Синтез проводили в пептидном синтезаторе Syro (MultiSynTech GmbH), применяя 24-96 реакционных сосудов. В каждый сосуд поместили около 60 мг (масса смолы до загрузки) вышеупомянутой смолы. Далее реакционные сосуды запрограммировали и были выполнены нижеследующие стадии: Стадии 3-6 многократно повторяли для добавления каждой аминокислоты. Аналитический способ Время удержания при ВЭЖХ (RT, в минутах) определяли, применяя колонку Jupiter Proteo 90 А,1502.0 мм (cod. 00F-4396-B0 - Phenomenex) со следующими растворителями: А (Н 2 О + 0.1% TFA) и В(CH3CN+ 0.1% TFA) и градиент: 0 мин: 95%А, 5%В; 0.5 мин: 95%А, 5%В; 20 мин: 40%А, 60%В; 21 мин: 0%А, 100%В; 23 мин: 0%А, 100%В; 23.1 мин: 95%А, 5%В; 31 мин: 95%А, 5%В. Формирование дисульфидного -стрендового соединения После завершения синтеза, смолу оставили набухать на 1 ч в 3 мл обезвоженного DMF. Далее в реакцию добавили 10 экв. раствора йода в DMF (6 мл) с последующим перемешиванием в течение 1,5 ч. Смолу отфильтровали, и добавили свежий раствор йода (10 экв.) в DMF (6 мл), с последующим перемешиванием в течение еще 3 ч. Далее смолу отфильтровали и промыли DMF (3 х) и CH2Cl2 (3 х). Расщепление, циклизация основной цепи, снятие защиты и очистка пептида После формирования дисульфидного -стрендового соединения, смолу суспендировали в 1 мл (0,14 ммоль) 1% TFA в CH2Cl2 (об./об.) в течение 3 мин и отфильтровали, фильтрат был нейтрализован при добавлении 1 мл (1,15 ммоль) 20%DIEA в CH2Cl2 (об./об.). Процедуру повторили дважды для того, чтобы расщепление прошло полностью. Смолу промыли трижды в 1 мл CH2Cl2. Слой CH2Cl2 выпарили до сухости. После удаления летучих веществ в пробирку добавили 8 мл обезвоженного DMF. Далее 2 экв.HATU в обезвоженном DMF (1 мл) и 4 экв. DIPEA в обезвоженном DMF (1 мл) были добавлены к пептиду, затем реакционную смесь оставили перемешиваться на 16 ч. Летучие вещества выпарили до сухости. Неочищенные циклизованные пептиды растворили в 7 мл CH2Cl2 и экстрагировали при помощи 10% ацетонитрила в воде (4,5 мл) три раза. Слой CH2Cl2 выпарили до сухости. Для полного снятия защиты с пептида добавили 3 мл смеси для реакции расщепления TFA:TIS:H2O (95:2.5:2.5), и смесь хранили в течение 2,5 ч. Летучие вещества выпарили до сухости и неочищенный пептид растворили в 20% АсОН в воде (7 мл) и экстрагировались при помощи изопропилового эфира (4 мл) трижды. Водный слой собрали и выпарили до сухости, и остаток очистили с помощью препаративного обращеннофазного ВЭЖХ. После лиофилизации получили продукты в виде белых порошков и проанализирован при помощи аналитического способа ВЭЖХ-ESI-MS, описанного выше. Аналитические данные, содержащие чистоту после препаративной ВЭЖХ и ESI-MS, приводятся. Пример 1. Пептид был синтезирован начиная с аминокислоты L-Pro, которая была привита к смоле. Исходная смола Fmoc-Pro-2-хлортритиловая смола была приготовлена, как описано выше. Линейный пептид синтезировали на твердой подложке в соответствии с методикой, описанной выше в следующей последовательности: Смола-Pro-DPro-Lys-Gln-Tyr-Cys-Tyr-Arg-Dab-DPro-Ala-Ser-Cys-Ala-His-Tyr. Дисульфидное -стрендовое соединение вводили как описано выше. Продукт отделили от смолы, циклизовали, сняли и очистили, как указано, с помощью препаративной обращенно-фазной LC-MS. После лиофилизации был получен продукт в виде белого порошка, который проанализировали аналитическим способом ВЭЖХ-ESI-MS, описанным выше ([М+2 Н]2+: 933.1; RT: 10.47; УФ-чистота: 72%). Пример 2. Пептид был синтезирован начиная с аминокислоты L-Pro, которая была привита к смоле. Исходная Fmoc-Pro-2-хлортритиловая смола была получена, как описано выше. Линейный пептид синтезировали на твердой подложке в соответствии с методикой, описанной выше в следующей последовательности: Смола-Pro-DPro-Lys-Gln-Tyr-Cys-Tyr-Arg-Dab-DPro-Ala-Ser-Cys-Ala-His-Tyr. Дисульфидное стрендовое соединение ввели, как описано выше. Продукт отделили от смолы, циклизовали, сняли защиту и очистили, как указано, с помощью препаративной обращенно-фазной LC-MS. После лиофилизации получили продукт в виде белого порошка и проанализировали его с помощью аналитического способа ВЭЖХ-ESI-MS, описанного выше ([М+2 Н]2+: 978,6; RT: 10.95; УФ-чистота: 82%). 2. Биологические способы. 2.1. Получение пептидов. Лиофилизированные пептиды взвесили на весах Microbalance (Mettler МТ 5) и растворили в стерильной воде до конечной концентрации 1 мМ, если иного не установлено. Стоковые растворы хранили при +4 С при защите от света. 2.2. Са 2+ анализ: антагонистическая активность пептидов в отношении CXCR4. Увеличение внутриклеточного кальция контролировали, применяя Flexstation 384 (Molecular De-5 017583vices, Sunnyvale, CA) для анализа пептидов на антагонистическую активность в отношении CXCR4 активности в мышиной пре-В клеточной линии 300-19 стабильно трансфецированной CXCR4 человека (см. ссылки 1, 2, 3, приведенные ниже. Клетки массово загружали с Calcium 3 Assay kit (Molecular Devices) в аналитическом буфере (сбалансированный солевой раствор Хенкса, HBSS, 20 мМ HEPES, рН 7.4, 0.1%BSA) в течение 1 ч при комнатной температуре, и затем 200000 меченых клеток были перенесены на 96 луночные аналитические чрные планшеты (Costar No. 3603). 20-кратно концентрированный раствор пептида в аналитическом буфере добавили к клеткам и весь планшет центрифугировали для осаждения клеток на дно лунок. Мобилизацию кальция, индуцированную 10 нМ фактора-1, получаемого из стромальных клеток (SDF-1), измеряли в Flexstation 384 (возбуждение, 485 нМ, эмиссия, 525 нМ) в течение 90 с. Максимальное увеличение флуоресцентного ответа относительно базового уровня применяли для вычисления антагонистической активности. Данные для кривой зависимости эффекта от дозы (концентрация антагониста от процента максимального ответа) были соотнесены с четырьмя параметрами логистического уравнения при помощи SoftmaxPro 4.6 (Molecular Devices), исходя из которого были вычислены значения IC50%. 2.3. FIGS-Assay. Данный анализ выполняли в соответствии со ссылкой 5, приведенной ниже. Стоковые растворы пептидов (10 мкМ) получали путем растворения пептидов в 10 мкМ Tris-HCl при комнатной температуре. Стоковые растворы хранили при +4 С с защитой от света. Рабочие растворы получали непосредственно перед экспериментом путм серии разбавлений в фосфатном солевом буфере (PBS) и добавляли в конечном объеме 10 мкл непосредственно в клеточные культуры. После 48-часовой сокультивации культуры клеток промыли фосфатным солевым буфером и затем в течение 5 мин действовали на них раствором глутаральдегида/формальдегида (0,2%/2%) в PBS. Для фотометрического количественного анализа фиксированные клеточные культуры впоследствии инкубировали с ортонитрофенилгалактопиранозидом(ONPG) в качестве субстрата -галактозидазы, который был ферментативно превращен в хромофор ортонитрофенол (ONP). Данные были получены путем измерения оптической плотности растворов в лунках при длине волны 405 нм, применяя ридер 96 луночных планшетов iEMS. 2.4. Анализ цитотоксичности. Цитотоксическое воздействие пептидов на клетки HELA (Асс 57) и COS-7 (CRL-1651) определялся при помощи анализа уменьшения МТТ (см. ссылки 6 и 7, приведенные ниже). Кратное описание способа: клетки HELA и COS-7 засеяли на 96-луночные планшеты в количествах соответственно 7,0103 и 4,5103 клеток на одну лунку, и оставили на 24 ч при 37 С и 5% содержании СО 2. В этой точке, нулевое время (Tz) определяли с помощью уменьшения МТТ (см. ниже). Из оставшихся лунок удалили супернатант, и в каждую добавили свежую среду и пептиды в серии последовательных разведений: 12,5 мкМ, 25 мкМ и 50 мкМ. Эксперимент с каждой из концентраций провели трижды. Инкубацию клеток продолжали 48 ч при 37 С и 5% СО 2. Лунки промыли PBS один раз и затем в каждую лунку добавили 100 мкл МТТ-реагента (0,5 мг/мл в среде RPMI1640 и, соответственно, DMEM). Далее инкубировали при 37 С в течение 2 ч, затем среда была удалена и в каждую лунку добавили по 100 мкл изопропанола. Поглощение солюбилизированного продукта измеряли при 595 нм (OD595 пептид). Для каждой концентрации было посчитано среднее значение по трем повторениям. Процент роста вычисляли следующим образом: (OD595 пептид-OD595Tz-OD595 Пустая лунка)/(OD595Tz-OD595 Пустая лунка)100% и был нанесен на график для каждой концентрации пептида. Значения LC50 (летальная концентрация, определенная как концентрация, которая убивает 50% клеток) определяли для каждого пептида, применяя функцию линий тренда программы EXCEL (MicrosoftOffice 2000) для концентраций (50, 25, 12.5 и 0 мкМ), соответствующие проценты роста и значение - 50(=TREND(C50:C0,%50:%0,-50. Концентрация GI50 (ингибиро-вание роста) была вычислена для каждого пептида, применяя функцию линий тренда для концентраций (50, 25, 12.5 и 0 мкМ), соответствующие проценты и значение 50 (=TREND(C50:C0,%50:%0,50. 2.5. Клеточная культура. Клетки CCR5 культивировали в среде DMEM при добавлении 4500 мг/мл глюкозы, 10% эмбриональной сыворотки телят (FBS), 50 ед/мл пенициллина и 50 мкг/мл стрептомицина (Пен/Стрепт). КлеткиHut/4-3 поддерживали в среде RPMI, 10% FBS, с добавлением Пен/Стрепт и 10 мМ HEPES. Клетки HELA и CCRF-CEM культивировали в среде RPMI1640 с добавлением 5% FBS, Пен/Стрепт и 2 мМ Lглутамина. Все клеточные линии выращивали при 37 С и 5% СО 2. Клеточные среды, добавки к средам,PBS-буфер, HEPES, Пен/Стрепт, L-глутамин и сыворотки были приобретены у Gibco (Pailsey, UK). Остальные реактивы произведены компанией Merck (Darmstadt, Germany). 2.6. Гемолиз. Пептиды были протестированы на гемолитическую активность против красных клеток крови человека (hRBC). Свежие hRBC трижды промыли соляным фосфатным буфером (PBS) посредством центрифугирования (10 мин, 2000g). Пептиды в концентрации 100 мкМ инкубировали вместе с 20% (объем/объем) hRBC в течение 1 ч при 37 С. Конечная концентрация эритроцитов составила около 0,9109 клеток на 1 мл. Значение 0% относительно 100% лизиса клеток определяли при инкубации hRBC в при-6 017583 сутствии PBS самого по себе и соответственно при добавлении 0,1% раствора Triton-Х 100 в воде соответственно. Образцы центрифугировали и супернатант 20-кратно разбавляли PBS-буфером, и измеряли оптическую плотность (OD) образцов при длине волны 540 нм. При 100% лизисе клеток оптическая плотность (OD540H2O) равнялась приблизительно 1,3-1,8. Процент гемолиза был подсчитан следующим образом: OD540 пептид/OD540H2O)100%. 2.7. Хемотаксический анализ (Анализ миграции клеток). Хемотаксический ответ клеток CCRF-CEM на градиент получаемого из стромальных клеток фактора-1 (SDF-1) был определен при помощи одноразовых аналитических планшетов фирмы Neuroprobe(размер пор 5 мкм) (Gaithersburg, MD) в соответствии с инструкциями производителя и содержащимися здесь ссылками (главным образом ссылка 8, приведенная ниже). Кратко, одну колбу объмом 175 см 3 единожды промыли фосфатным солевым буфером Дульбекко (DPBS) и трипсинизировали в течение 10 мин, либо до тех пока клетки не поднимутся. Трипсин нейтрализовали добавлением свежей среды, содержащей сыворотку, и клетки осадили центрифугированием, промыли единожды DPBS и ресуспендировали при 1-0.5107 кл./мл в RPMI с 0,5% бычьего сывороточного альбумина (BSA). 45 мкл суспензии клеток смешали с 5 мкл 10-кратно концентрированного РЕМ пептида, растворенного в той же аналитической среде. 35 мкл этой смеси нанести на поверхность аналитического фильтра. Далее клетки мигрировали (при 37 С) в нижнюю камеру аналитического планшета, содержащую 1 нМ SDF-1. Спустя 4 ч,фильтр удалили, к мигрировавшим клеткам добавили МТТ до конечной концентрации 0,5 мг/мл и инкубировали ещ 4 ч. После маркирования клеток МТТ вся среда была удалена, и к клеткам было добавлено 100 мкл изопропанола с 10 мМ HCl. Оптическое поглощение при длине волны 595 нм (ABS595) было измерено при помощи планшетного ридера Tecan Genios с применением программного обеспечения Magellan. Число мигрировавших клеток было определено исходя из сравнения значений ABS595 со стандартной кривой, построенной исходя из известного количества клеток в аналитическом планшете, и было соотнесено с концентрацией SDF-1, чтобы получить сигмовидную кривую и определить значенияIC50. Значения IC50 были определены при помощи функции линий тренда в Microsoft Excel путм построения логарифмической кривой по усредннным точкам экспериментальных данных. 2.8. Стабильность плазмы. 405 мкл раствора плазма/альбумин поместили в полипропиленовую(РР) пробирку, затем смешали с 45 мкл соединения из 100 мкМ раствора В, полученного путм смешивания 135 мкл PBS и 15 мкл 1 мМ раствора пептидов в PBS, рН 7.4. Аликвоты по 150 мкл были помещены в отдельные лунки планшета с фильтром 10 кДа (Millipore MAPPB 1010 Biomax membrane). Для "0-минутных контролей": 270 мкл PBS поместили в полипропиленовую пробирку, добавили 30 мкл стокового раствора В и перемешали на вортексе. 150 мкл контрольного раствора поместили в одну лунку планшета с фильтром и применяли в качестве "отфильтрованного контроля". Ещ 150 мкл контрольного раствора поместили сразу в принимающую лунку (служащей для фильтрата) и применяли в качестве "неотфильтрованного контроля". Целую плашку, включая крышку от испарения, инкубировали 60 мин при 37 С. Образцы плазмы (плазма крысы: Harlan Sera lab UK, плазма человека: Blutspendezentrum Zurich) центрифугировали по меньшей мере в течение 2 ч при 4300 об/мин (3500g) и 15 минут надлежащим образом для получения 100 мкл фильтрата. Для образцов "сывороточного альбумина" (свежеприготовленный альбумин человека: Sigma A-4327, альбумин крысы: Sigma A-6272,оба альбумина при концентрации 40 мг/мл в PBS) достаточно центрифугировать 1 ч. Фильтраты в принимающем полипропиленовом планшете проанализировали при помощи LC/MS следующим образом: Колонка: Jupiter Cl 8 (Phenomenex), подвижная фаза: (А) 0,1% муравьиной кислоты в воде и (В) ацетонитрил, градиент: 5%-100% (Б) за 2 мин, ионизация электроспреем, детекция MRM (тройной квадруполь). Были определены области пиков и значения трх опытов были усреднены. Связывание выражали в проценте от (отфильтрованного и неотфильтрованного в нулевой точке) контроля 1 и 2 по формуле: 100(100 Т 60/T0). Затем вычисляли среднее по этим значениям (см. ссылку 9, приведенную ниже). 3.0. Исследования in vivo. 3.1. Максимальная толерантная доза у мышей. а) В предварительном изучении соединение, описанное в примере 1, диспергированное в воде для инъекций или в 0,9% физиологическом растворе, вводили посредством внутривенной инъекции при следующих уровнях доз: 35, 50, 70, 85, 100, 150, 250 и 500 мг/кг группам, состоящим из одной самки и одного самца мыши (Cr1:CD1(ICR. Помимо этого, две группы, содержащим по 2 самца и 2 самки мыши,получили уровни доз 90 и 100 мг/кг соответственно, и доза 50 мг/кг была повторена в группе, содержащей 1 самца и 1 самку мыши. 2) Исследования максимальной толерантной дозы для соединения примера 2 осуществлялись на мышах CD1 (3 мыши в группе) применяя внутривенное, внутрибрюшинное и подкожное введение. 3.2. Повторные изучения токсичности на мышах. Исследование токсического эффекта и токсикокинетики соединения, описанного в примере 1, проводили путм ежедневного введения внутривенной инъекции мышам в течение, по меньшей мере, 14 дней. Группы из 12 самцов и 12 самок Cr1:CD1(ICR) получали либо контрольную инъекцию (50 мМ на-7 017583 трий дигидроортофосфатный буфер, содержащий 0,9% мас./об. хлорида натрия), либо следующие дозы препарата POL6326: 8, 24 или 40 мг/кг/день, при объеме дозы 5 мл/кг. Исследование каждой дозы проводились на 24 мышах каждого пола на группу. Оценку токсического эффекта проводили исходя из смертности, клинических признаков, массы тела, аппетита, офтальмологического осмотра, клинических и анатомических патологий и токсикокинетических показателей. 3.3. Мобилизация стволовых клеток. а) Мышиная модель. Цель исследования заключалась в оценке способности соединения, описанного в примерах 1 и 2,мобилизовать гемопоэтические клетки-предшественники из костного мозга мыши в периферическую кровь, применяя in vitro исследования гемопоэтической колонии. Для людей точная информация о мобилизации клеток-предшественников обеспечивалась посредством анализа колониеобразующие единицы гранулоцитов-моноцитов (CFU-GM) или путм определения избытка CD34)(+)-клеток при помощиFACS-анализа (см. ссылку 10 ниже). На мышах CD34 не является характерным маркером для стволовых клеток, поэтому на этой модели, как правило, используется CFU-GM (см. ссылку 11, приведенную ниже). Для оценки способности соединений, описанных в примерах 1 и 2, мобилизовать мышиные стволовые клетки (CFU-GM), самки С 3 Н/Hej (Jackson Laboratory) получали подкожную инъекцию (доза 5 мг/кг) соединений, описанных в примерах 1 и 2, и в качестве контроля AMD3100 (Broxmeyer et al., J Exp Med 201, 1307-1318), (в настоящее время проходящий III стадию клинических испытаний) для мобилизации стволовых клеток. В каждой точке времени у 5 животных каждой группы забирали образцы крови и определяли в них количество ядерных клеток с помощью стандартных анализов. Б) Обезьянья модель. Была проведена оценка мобилизации гемопоэтических стволовых клеток периферической крови яванских макак (Масаса fascicularis). Вещество, описанное в примере 1, вводили 4 обезьянам (2 самца и 2 самки) путм медленной болюсной внутривенной инъекции в течение 2 мин, и определяли количествоCD34(+)-клеток при помощи FACS-анализа. Помимо этого осуществляли забор образцов крови для исследования токсикокинетики. 4.0. Результаты. Результаты экспериментов, описанных в п.2.2-2.8, отражены в нижеследующих табл. 1 и 2. Таблица 1 Результаты экспериментов, описанных в п.3.1-3.3., приведены ниже. 4.1. Изучение максимальной толерантной дозы (МТД) на мышах. а) Изучение МТД, соединение примера 1. Было обнаружено, что точный минимальный уровень летальной внутривенной дозы у мышей превышает 90 мг/кг. б) Изучение МТД, соединение примера 2. Самая высокая доза, протестированная для всех трех путей введения, составила болюс 120 мг/кг. При такой дозе все животные выживали и наблюдались только сглаженные, неярко выраженные симптомы. Были выявлены следующие симптомы: небольшая поведенческая депрессия, небольшой цианоз,увеличение глубины дыхания и мышечная релаксация. 4.2. 14-дневное изучение токсичности и токсикокинетики внутривенной инъекции. Уровень NOAEL для соединения, описанного в примере 1, после внутривенного введения мышам составил 40 мг/кг/день. Не было обнаружено никакого значительного влияния лечения на вес тела, изменение веса тела или аппетита, а также значительного влияния лечения не было обнаружено при офтальмологических обследования, которые проводились в течение заключительной недели фазы дозирования. Введение соединения, описанного в примере 1, сопровождалось слегка более высоким количеством-8 017583 белых клеток крови и слегка более высоким абсолютным количеством лимфоцитов у самок, получающих 40 мг/кг/день. У самцов, получавших 40 мг/кг/день, подобные эффекты обнаружены не были, и эти побочные эффекты не рассматривались как неблагоприятные. Введение соединения, описанного в примере 1, не оказало влияния на результаты клинической химии. Увеличение массы органов (почек у самцов,получавших 8 мг/кг/день, и семенных пузырьков у самцов, получавших 24 или 40 мг/кг/день) рассматривали как случайное и не имеющее отношения к лечению. Не было зарегистрировано никаких макроскопических повреждений, связанных с исследуемым соединением. У трх животных (1 самка из контрольной группы, 1 самец, получавший 8 мг/кг/день, и 1 самка, получавшая 24 мг/кг/день) фокальная область вокруг места инъекции (хвост) покраснела и покрылась коркой, что было вызвано уколом от иглы для подкожных инъекций. Не было зарегистрировано никаких микроскопических повреждений, связанных с исследуемым соединением. 4.3. Мобилизация стволовых клеток. а) Мобилизация стволовых клеток у мышей, соединение примера 1. Введение 5 мг/кг соединения примера 1, увеличило число клеток крови CFU-GM, показав максимальный эффект при 120 мин и возврат к базовому уровню по истечение 6 ч после введения (фиг. 1). В том же эксперименте, вещество AMD3100 (в настоящее время проходит III стадию клинических испытаний по мобилизации стволовых клеток) применяли для сравнения в качестве компаратора. В вышеописанном эксперименте была определена зависимость между дозой соединения примера 1 и эффектом на высвобождение CFU-GM (фиг. 1). Прослеживается чткая зависимость эффекта соединения, описанного в примере 1, на высвобождение CFU-GM у мышей с пиковым увеличением уровня при 5 мг/кг. Фиг. 1. Увеличение колониеобразующих единиц на мл крови (CFU-GM) с течением времени как для соединения, описанного в примере 1 (5 мг), так и для ссылочного соединения AMD3100. б) Мобилизация стволовых клеток, соединение примера 2. Введение 5 мг/кг соединения, описанного в примере 2, увеличивало число клеток крови CFU-GM до 6 ч после введения с максимальным эффектом при 240 минутах, при этом введение AMD3100 связано с увеличением частоты и количества клеток-предшественников при 30 и 60 минутах по сравнению с контрольными мышами (фиг. 2). Фиг. 2. Увеличение колониеобразующих единиц на 1 мл крови (CFU-GM) с течением времени как для соединения, описанного в примере 2, так и для ссылочного соединения AMD3100. с) Мобилизация стволовых клеток у обезьян, соединение примера 1: Введение соединения, описанного в примере 1, индуцировало мобилизацию гемопоэтических клеток CD34(+) яванских макак. Как было обнаружено у мышей начало мобилизации было быстрым с пиковым уровнем через два часа. Мобилизация также имела кратковременный характер, и количество стволовых клеток в периферической крови вернулось к базовому уровню с уменьшением в плазме уровней соединения, описанного в примере 1, (фиг. 3). Фиг. 3. Среднее число CD34(+) клеток на 1 мкл крови с течением времени для соединения, описанного в примере 1. Список цитируемой литературы 1. Oberlin E, Amara A, Bachelerie F, Bessia С, Virelizier J-L, Arenzana-Seisdedos F, Schwartz O, HeardProc.Natl.Acad.Sci. 1997. 94:3548-3553. 6. Mossman T. J.lmmunol.Meth. 1983, 65:55-63. 7. Berridge MV, Tan AS. Arch.Biochem.Biophys. 1993, 303:474-482. 8. Frevert CW, Wong VA, Goodman RV, Goodwin R, Martin TR, J.lmmunol.Meth. 1998. 213: 41-52. 9. Singh R., Chang, S.Y., Talor, L.C., Rapid Commun. Mass Spectrom., 1996, 10: 1019-1026. 10. To LB, Haylock DN, Simmons PJ, Juttner CA. Blood 1997, 89(7):2233-2258. 11. Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA et al. J Exp Med 2005. 201(8):1307-1318. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Цикло(-Tyr-His-X-Cys-Ser-Ala-DPro-Dab-Arg-Tyr-Cys-Tyr-Gln-Lys-DPro-Pro), который содержит дисульфидную связь между Cys4 и Cys11, и его фармацевтически приемлемые соли, где X представляет собой Ala или Tyr. 2. Соединение по п.1, отличающееся тем, что имеет антагонистическую активность в отношенииCXCR4 и способно либо предотвратить ВИЧ-инфекции у здоровых людей, либо замедлить или прекратить развитие ВИЧ-инфекций у инфицированных пациентов; предотвратить или лечить рак, опосредо-9 017583 ванный активностью рецептора CXCR4; предотвратить или лечить иммунологическое заболевание, опосредованное активностью рецептора CXCR4; лечить иммуносупрессию или воспаление; или мобилизовать стволовые клетки периферической крови, мезенхимальные стволовые клетки (МСК) или другие стволовые клетки, удержание которых зависит от активности рецептора CXCR4. 3. Применение соединения по п.1 для предотвращения ВИЧ-инфекций у здоровых людей или для замедления или прекращения развития ВИЧ-инфекций у инфицированных пациентов; предотвращения или лечения рака, опосредованного активностью рецептора CXCR4; предотвращения или лечения иммунологического заболевания, опосредованного активностью рецептора CXCR4; лечения иммуносупрессии или воспаления; или мобилизации стволовых клеток периферической крови, мезенхимальных стволовых клеток (МСК) и/или других стволовых клеток, удержание которых зависит от активности рецептораCXCR4. 4. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически инертный носитель. 5. Композиция по п.4 в форме, подходящей для перорального, местного, трансдермального, инъекционного, буккального, назального, лгочного или ингаляционного введения. 6. Композиция по п.4 или 5 в форме таблеток, драже, капсул, растворов, жидкостей, гелей, пластырей, кремов, мазей, сиропов, гидросмесей, суспензий, спреев, распылителей или суппозиториев. 7. Применение соединения по п.1 для производства лекарственного средства, обладающего антагонистическим действием в отношении CXCR4. 8. Применение по п.7, где указанное лекарственное средство предназначено для использования в предотвращении ВИЧ-инфекций у здоровых людей или в замедлении или прекращении развития ВИЧинфекций у инфицированных пациентов; предотвращении или лечении рака, опосредованного активностью рецептора CXCR4; предотвращении или лечении иммунологического заболевания, опосредованного активностью рецептора CXCR4; лечении иммуносупрессии или воспаления; или мобилизации стволовых клеток периферической крови, мезенхимальных стволовых клеток или других стволовых клеток,удержание которых зависит от активности рецептора CXCR4. 9. Способ производства соединений по п.1, который включает стадии:(a) связывание активированной специальным образом тврдой подложки с соответствующим образом N-защищнной производной Pro;(b) удаление N-защитной группы из продукта, полученного на стадии (а);(c) связывание полученного продукта с соответствующим образом N-защищнной производной(d) удаление N-защитной группы из продукта, полученного на стадии (с);(e) связывание полученного таким образом продукта с соответствующим образом N-защищнной производной аминокислоты, которая в полученном конечном продукте находится в положении 14, то есть Lys, причем аминогруппа, находящаяся в ее боковой цепи, также защищена соответствующим образом;(g) осуществление стадий, по существу, соответствующих стадиям (е) и (f), но с применением соответствующим образом N-защищнных производных аминокислот, которые в желаемом конечном продукте находятся в положениях 13-1, то есть Gln, Tyr, Cys, Tyr, Arg, Dab, DPro, Ala, Ser, Cys, Ala или Tyr,His и Tyr, причем любая функциональная группа, которая может присутствовать в указанных Nзащищнных производных аминокислот, также защищена соответствующим образом;(h) формирование дисульфидного -стрендового соединения между боковыми цепями цистеиновых остатков в положениях 4 и 11;(i) отделение полученного таким образом продукта от тврдой подложки;(j) циклизация продукта, отделенного от тврдой подложки;(k) удаление всех защитных групп, связанных с функциональными группами всех членов цепи из аминокислотных остатков; и(l) при необходимости, превращение полученного таким образом продукта в фармацевтически приемлемые соли или превращение солей, полученных таким образом, в соответствующее свободное соединение или в другую фармацевтически приемлемую соль.
МПК / Метки
МПК: C07K 7/56, A61K 38/12
Метки: матрице, закрепленные, пептидомиметики
Код ссылки
<a href="https://eas.patents.su/12-17583-zakreplennye-na-matrice-peptidomimetiki.html" rel="bookmark" title="База патентов Евразийского Союза">Закрепленные на матрице пептидомиметики</a>
Закрепленные на матрице пептидомиметики

Номер патента: 15991
Опубликовано: 30.01.2012
Авторы: Робинсон Джон Энтони, Димарко Стив Дж., Гомберт Франк, Врейблуд Вим, Сринивас Нитьякальяни, Обрехт Даниель, Диас Рикардо
МПК: A61K 38/04, A61P 11/00, A61P 19/00...
Метки: матрице, закрепленные, пептидомиметики
Формула / Реферат:
1. Соединения общей формулыгдепредставляет собой DPro-LPro или LPro-DPro и Z является цепочкой из 12 α-аминокислотных остатков, причем положения упомянутых аминокислотных остатков в упомянутой цепи отсчитаны, начиная с N-концевой аминокислоты, при этом данные аминокислотные остатки в положениях Р1-Р12 цепи представляют собойи их энантиомеры и фармацевтически приемлемые соли.2. Соединение по п.1, где матрица представляет собой DPro-LPro и...
Связанные с матрицей пептидомиметики β-шпилечной структуры с активностью антагонистов cxcr4

Номер патента: 10163
Опубликовано: 30.06.2008
Авторы: Лочиуро Серджио, Цумбрунн Юрг, Хенце Хайко, Романьоли Барбара, Обрехт Даниель, Лудин Кристиан, Меле Керстин, Гомберт Франк, Робинсон Джон Энтони, Фрийблуд Ян Вим, Мукерджи Решми, Димарко Стивен Дж.
МПК: C07K 1/04, A61K 38/12, A61P 31/18...
Метки: cxcr4, антагонистов, матрицей, пептидомиметики, связанные, beta;-шпилечной, структуры, активностью
Формула / Реферат:
1. Соединения общей формулы где представляет собой группу, характеризуемую одной из формул A-CO означает DPro, DGln, 4-{2-[2-(метоксиэтокси)этокси]ацетиламино}-DPro или Gly; B-CO означает LPro, LGln, 4-{2-[2-(метоксиэтокси)этокси]ацетиламино}-LPro или Gly; но A-CO и B-CO не могут оба быть Gly; Z означает цепь из n a-аминокислотных остатков, причем n означает целое число 12, 14 или 18, а положения указанных аминокислотных остатков в цепи...
Способ инкапсулирования в целлюлозной матрице

Номер патента: 9256
Опубликовано: 28.12.2007
Авторы: Спир Скотт К., Тернер Меган Б., Роджерс Робин Дон, Сватлоски Ричард Патрик, Холбри Джон Дэвид
МПК: B32B 5/16
Метки: целлюлозной, инкапсулирования, способ, матрице
Формула / Реферат:
1. Способ получения регенерированной целлюлозы с инкапсулированным активным веществом, который включает стадии: (a) получение композиции из целлюлозы, растворенной в расплавленном гидрофильном ионном жидком растворителе, вместе с активным веществом, растворенным или диспергированным, по существу, в этом же растворителе гомогенно, причем вышеуказанный раствор, по существу, свободен от воды, органического растворителя или азотсодержащего основания...
Фармацевтическая композиция, содержащая действующее вещество, диспергированное в матрице

Номер патента: 7151
Опубликовано: 25.08.2006
Авторы: Линдер Рудольф, Дитрих Ранго, Ней Хартмут
МПК: A61K 31/44, A61K 9/20, A61K 9/16...
Метки: фармацевтическая, содержащая, действующее, матрице, вещество, композиция, диспергированное
Формула / Реферат:
1. Композиция, в которой действующее вещество, выбранное из группы ингибиторов ФДЭ 4, практически однородно диспергировано или растворено в матрице эксципиента, содержащей один или несколько эксципиентов, выбранных из группы, включающей жирный спирт, триглицерид, неполный глицерид и эфир жирной кислоты, и при том, что композиция находится в форме, состоящей из микросфер. 2. Композиция по п.1, в которой один или несколько других дополнительных...
Связанные с матрицей бета-шпилечные пептидомиметики с ингибирующей активностью в отношении протеаз

Номер патента: 13814
Опубликовано: 30.08.2010
Авторы: Хенце Хайко, Селлье Одиль, Юнг Франсуаза, Меле Керстин, Демарко Стивен Дж., Гомберт Франк, Обрехт Даниель, Лудин Кристиан
МПК: C07K 7/08, A61K 38/04, C07K 7/06...
Метки: пептидомиметики, связанные, активностью, протеаз, матрицей, ингибирующей, отношении, бета-шпилечные
Формула / Реферат:
1. Соединение общей формулыгдеозначает группу одной из формулгдепредставляет собой Ala; Arg; Asn; Cys; Gln; Gly; His; Ile; Leu; Lys; Met; Phe; Pro; Pro(5RPhe), Ser; Thr; Trp; Tyr; Val; Cit; Orn; tBuA; Sar; t-BuG; 4AmPhe; 3AmPhe; 2AmPhe; Phe(mC(NH2)=NH); Phe(pC(NH2)=NH); Phe(mNHC(NH2)=NH); Phe(pNHC(NH2)=NH); Phg; Cha; C4al; C5al; Nle; 2-Nal; 1-Nal; 4Cl-Phe; 3Cl-Phe; 2Cl-Phe; 3,4Cl2Phe; 4F-Phe; 3F-Phe; 2F-Phe; Tic; Thi; Tza; Mso; AcLys; Dpr; A2Bu;...
Предыдущий патент: Лечение метастатической стадии рака предстательной железы дегареликсом
Следующий патент: Способ получения добавки к мороженому и добавка, полученная этим способом
Случайный патент: Электрод для активных форм кислорода и сенсор, использующий электрод