Промежуточные соединения для использования при получении витамина е








Формула / Реферат
1. Способ получения соединения общей формулы (I)

в которой A представляет собой углеводород от C1 до C20, Y и Z независимо представляют собой углеводород от C1 до C20, который может содержать кислородсодержащую функциональную группу, B представляет OR1 или NHR1, где R1 является либо водородом, либо углеводородом от C1 до C6, при этом указанный способ включает в себя взаимодействие в присутствии катализатора, кислоты Льюиса, соединения общей формулы (II)

в которой Y и Z определены, как указано выше, с соединением общей формулы (III)

или соединением общей формулы (IV)

в которых A определено как указано выше, B' представляет собой O или NR1, где R1 определен, как указано выше, и R представляет собой водород или углеводород от C1 до C6.
2. Способ по п.1, в котором Y представляет собой углеводород от C1 до C6 с кислородной группой, Z представляет собой метил, A представляет собой алифатический углеводород, а R представляет собой CH3.
3. Способ по п.1 или 2, в котором соединения формулы (II) представляют собой 6-метил-6-гептен-2-он; 6-ацетокси-2,5,7,8-тетраметил-2-[(4-метилпент-4-ен)-1-ил]хромен или 6-ацетокси-2,5,7,8-тетраметил-2-[(4-метилпент-4-ен)-1-ил]хроман.
4. Способ по любому из предшествующих пунктов, в котором соединения формулы (III) представляют собой 3-метилбутаналь; имины 3-метилбутаналя; 3,6-диметилоктаналь; имины 3,6-диметилоктаналя; цитраль и имины цитраля.
5. Способ по любому из предшествующих пунктов, в котором соединения формулы (IV) представляют собой ацетали цитраля; ацетали 3-метилбутаналя или ацетали 3,6-диметилоктаналя.
6. Способ по любому из предшествующих пунктов, в котором кислота Льюиса имеет общую формулу M(L)n, в которой M представляет собой алюминий, железо, магний, скандий, иттербий, цинк, титан, кремний и висмут; L представляет собой галогенид, CF3SO3, (CF3SO2)2N, ClO4 или алкил от C1 до C4, а n соответствует электронной валентности M и обычно составляет от 1 до 4, или общую формулу A-H, в которой A представляет собой CF3SO3 или (CF3SO2)2N.
7. Способ по п.6, в котором кислота Льюиса представляет собой треххлористое железо.
8. Способ по любому из предшествующих пунктов, осуществляемый в присутствии основания, выбранного из ароматических аминов, алифатических аминов и карбонатных солей I или II группы Периодической таблицы.
9. Способ по любому из предшествующих пунктов, осуществляемый в присутствии органического растворителя, выбранного из хлорсодержащих растворителей, органических растворителей, эфирных растворителей, нитрильных растворителей и нитрорастворителей.
10. Способ по любому из предшествующих пунктов, осуществляемый при температуре от -80 до +150шC и при атмосферном давлении.
11. Новые соединения, характеризуемые следующими структурами:

12. Способ получения фитона и/или витамина E, который включает в себя гидрогенолиз соединения общей формулы (I), определенного в любом из пп.1-10.
13. Способ по п.12, в котором гидрогенолиз проводят в присутствии катализатора, который представляет собой металл, выбранный из палладия, платины, никеля и цинка, или соли металла - хлорида олова (II) или молибдена (III).
14. Способ по п.13, в котором гидрогенолиз проводят в присутствии палладия на угле.
15. Способ получения витамина E по любому из пп.12-14, который включает в себя гидрогенолиз новых соединений (VII) или (VIII), определенных в п.11.
16. Способ получения фитона по любому из пп.12-14, который включает в себя гидрогенолиз соединений (V) или (VI), определенных в п.11.
Текст
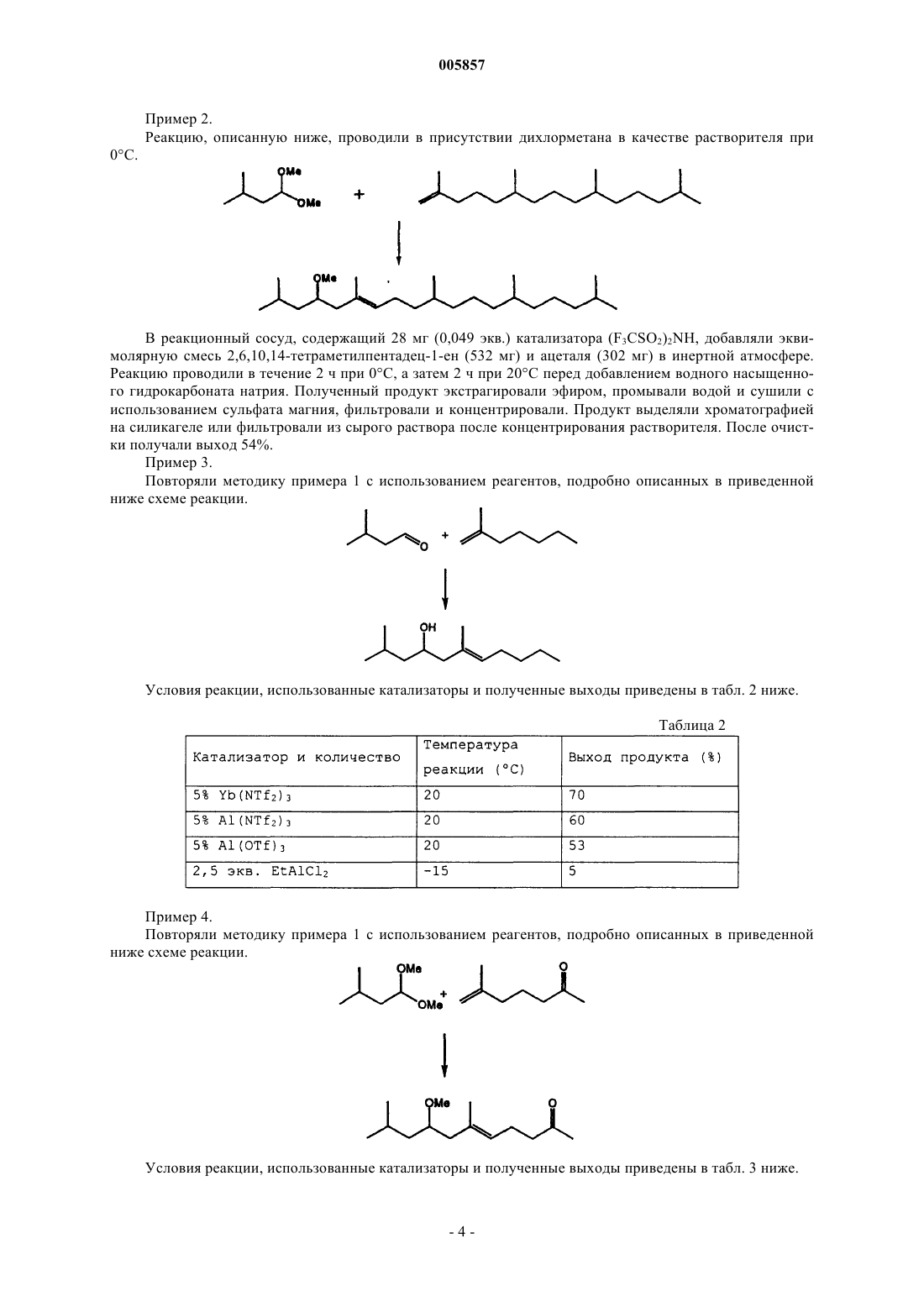
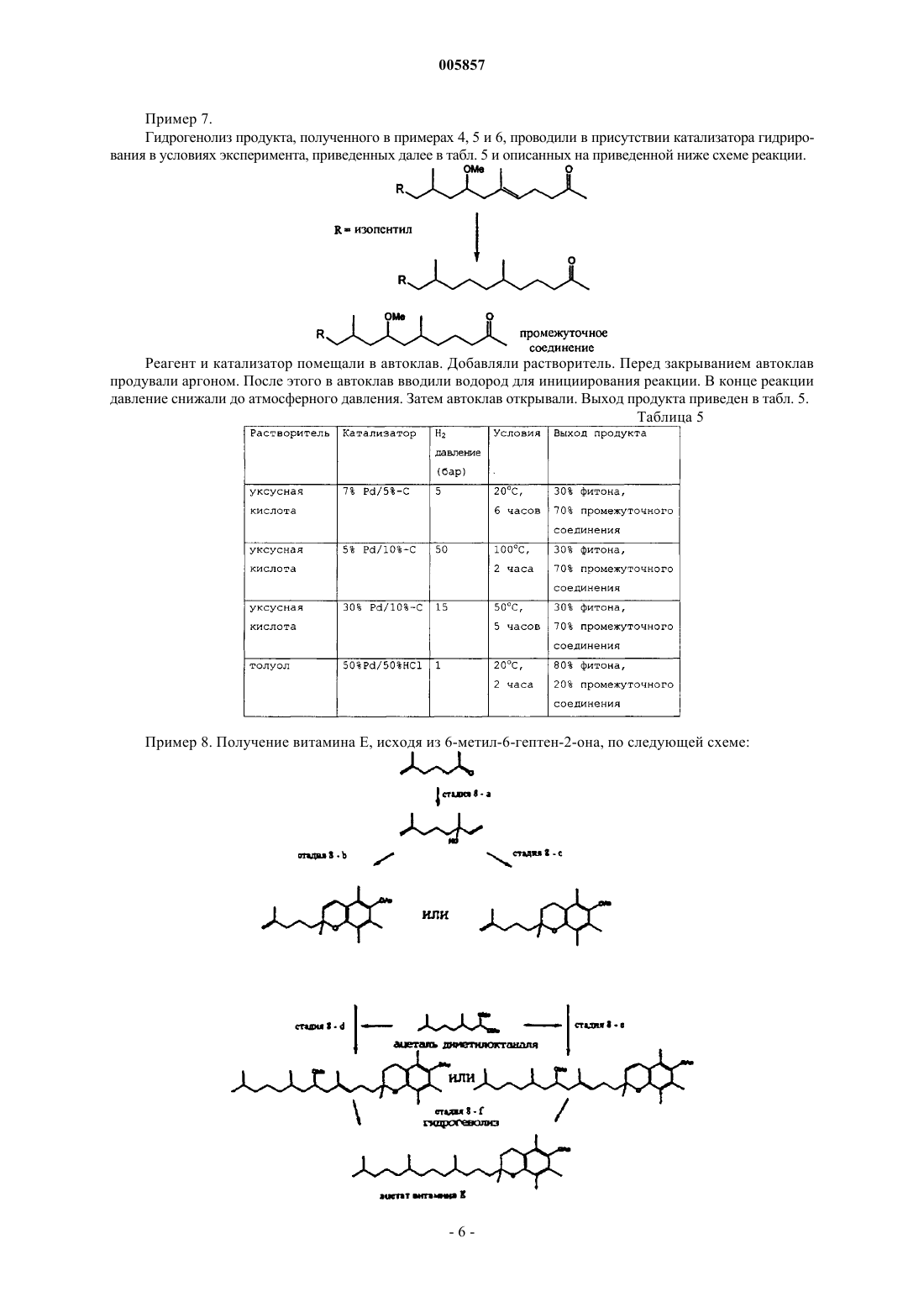
005857 Настоящее изобретение относится к способу получения промежуточных соединений, полезных при получении фитона и/или витамина Е. В течение долгого времени витамин Е получали химически с использованием множества различных способов. Обычно данный витамин получают из промежуточного соединения. В европейском патенте 0544588 описан способ получения витамина Е конденсацией полиненасыщенного производного аллиллового спирта. В патенте США 3867408 описано получение новых кетальных соединений, которые можно использовать при получении фитона, который, в свою очередь, является промежуточным соединением для получения витамина Е. В настоящее время нами найден новый способ получения некоторых бета-олефиновых соединений,которые могут быть использованы для синтеза фитона и которые, в некоторых случаях, могут быть использованы для прямого синтеза витамина Е из этого промежуточного соединения. Соответственно, в настоящем изобретении предлагается способ получения соединения общей формулы (I) в которой А представляет собой углеводород от C1 до C20, Y и Z независимо представляют собой углеводород от C1 до С 20, который может содержать кислородсодержащую функциональную группу, а В представляет собой OR1 или NHR1, где R1 является водородом или углеводородом от C1 до С 6, при этом способ включает в себя взаимодействие в присутствии кислоты Льюиса в качестве катализатора соединения общей формулы (II) где Y и Z определены, как указано выше, с соединением общей формулы (III) или соединением общей формулы (IV) в которой А, В и R определены, как указано выше, a R представляет собой водород или углеводород от C1 до C6. Некоторые соединения общей формулы (I) являются новыми и в качестве таковых также составляют другой аспект данного изобретения. Способ настоящего изобретения включает в себя каталитическую реакцию между соединением общей формулы (II) и соединением общей формулы (III) или общей формулы (IV). Что касается соединения общей формулы (II), то Y и Z представляют собой углеводород от C1 до C20, который может содержать кислородсодержащую функциональную группу. Данная углеводородная группа может быть линейной, циклической, ароматической или алифатической, замещенной или незамещенной. В случае, когда Z представляет собой углеводород,предпочтительным углеводородом является линейный алифатический углеводород, в особенности метил. Соединения общей формулы (II), подходящие для использования в способе настоящего изобретения, включают в себя 6-метил-6-гептен-2-он; 2-метил-1-гептен; 2,6,10,14-тетраметилпентадец-1-ен, 6-ацетокси-2,5,7,8-тетраметил-2[(4-метилпент-4-ен)-1-ил]хромен и 6-ацетокси-2,5,7,8-тетраметил-2-[(4-метилпент-4-ен)-1-ил]хроман. Особенно предпочтительными соединениями общей формулы (II) являются 6-метил-6-гептен-2-он; 6-ацетокси-2,5,7,8-тетраметил-2-[(4-метилпент-4-ен)-1-ил]хромен и 6-ацетокси-2,5,7,8-тетраметил-2-[(4-метилпент-4-ен)-1-ил]хроман. Что касается соединений общей формулы (III), то А представляет собой углеводород от C1 до C20. Данный углеводород может быть линейным или циклическим, замещенным или незамещенным и может быть насыщенным или ненасыщенным. Предпочтительно, А является линейным алифатическим углеводородом, особенно метилом. В представляет собой OR1 или NR1, где R1 является алифатическим линейным или циклическим углеводородом от C1 до С 6 или С 6 ароматическим углеводородом. Соединения общей формулы (III), особенно полезные в способе настоящего изобретения, включают в себя 3-метилбутаналь; имины 3-метилбутаналя; 3,6-диметилоктаналь; имины 3,6-диметилоктаналя; цитраль и имины цитраля. Под иминами имеются в виду метил-, этил-, пропил-,изопропил-, бутил-, изобутил-, трет-бутил-, фенил-, тозил- и бензилимины, полученные известными способами. Что касается соединений общей формулы (IV), то А представляет собой углеводород от C1 до С 20. Данный углеводород может быть линейным или циклическим, замещенным или незамещенным и может быть насыщенным или ненасыщенным, а R является алифатически линейным или циклическим углеводородом от C1 до C6 или С 6 ароматическим углеводородом. Соединения общей формулы (IV), особенно полезные для способа настоящего изобретения, включают в себя ацетали цитраля; ацетали 3-метилбутаналя; ацетали 3,6-диметилоктаналя. Особенно предпочтительным соединением является 3,6-диметилоктаналь, 3-метилбутаналь и их ацетали. Под ацеталями имеют в виду метил-, этил-, изопропилацетали и гликоль, полученные известными способами. Молярное соотношение соединения общей формулы (II) и соединения общей формулы (III или IV) составляет в подходящем случае от 0,2:1 до 5:1, предпочтительно от0,5:1 до 2:1.-1 005857 Способ настоящего изобретения осуществляют в присутствии кислоты Льюиса. Подходящие кислоты Льюиса включают в себя соединения общей формулы M(L)n, где М представляет алюминий, железо, магний, скандий, иттербий, цинк, титан, кремний и висмут; L представляет собой галогенид, СF3SО 3,(СF3SO2)2N, ClO4 или алкил от C1 до С 4, a n соответствует электронной валентности М и обычно составляет от 1 до 4. Альтернативно, данная кислота Льюиса может представлять собой гидридные соединения общей формулы А-Н, в которой А представляет СF3SО 3 или (СF3SO2)2N. В частности, подходящие для использования в способе настоящего изобретения кислоты Льюиса в соответствии с вышеупомянутыми определениями включают в себя хлорид металла, например хлорид алюминия, железа, висмута, магния, титана, скандия и иттрия, трифторметансульфонаты скандия, иттербия, железа и алюминия; трифторметансульфонамид или его соответствующую соль с металлом - скандием, иттербием, железом и алюминием; и трифторметансульфоновую кислоту. Предпочтительной кислотой Льюиса является треххлористое железо. Количество катализатора, используемого в способе, в удобном случае составляет от 0,001 до 5 мол.экв., предпочтительно от 0,02 до 2,5 мол.экв. Способ настоящего изобретения можно осуществлять в присутствии основания. Подходящие основания можно выбрать из ароматических аминов, например пиридина и 2,6-диметилпиридина; или алифатических аминов, в особенности третичных аминов, например триэтиламина и диизопропилэтиламина; или из неорганического карбоната, в особенности карбоната элемента I группы или II группы периодической таблицы, например карбоната натрия, калия, кальция и магния. Предпочтительным основанием является пиридин. Количество основания, используемого в способе, может составлять от 0 до 1 мол.экв., предпочтительно от 0,1 до 0,5 мол.экв. Реакцию можно осуществлять в присутствии органического растворителя. Подходящие растворители включают в себя хлорсодержащие растворители, такие как дихлорметан, хлороформ или хлорбензол; ароматические растворители, например толуол и ксилол; простые эфиры, такие как тетрагидрофуран, диэтиловый эфир и изопропиловый эфир; нитрильные растворители, такие как ацетонитрил, пропионитрил и бензонитрил, и нитрорастворители, такие как нитрометан и нитроэтан. Количество растворителя, находящегося в реакционной системе, в удобном случае составляет от 0 до 100, предпочтительно от 2 до 10 мас.экв. Способ можно осуществлять при температуре от -80 до +150 С, предпочтительно от -50 до +25 С и при атмосферном или повышенном давлении. Предпочтительно реакцию проводят при атмосферном давлении. Способ настоящего изобретения можно осуществлять в течение промежутка времени от 30 мин до 24 ч, предпочтительно от 30 мин до 6 ч в указанных выше условиях реакции для облегчения полного взаимодействия между соединениями общей формулы (II) или (III). Некоторые соединения общей формулы (I) являются новыми и в качестве таковых составляют следующий аспект настоящего изобретения. В частности, соединения формулы V, VI, VII и VIII являются новыми соединениями.-2 005857 Соединения общей формулы (I), полученные способом настоящего изобретения, особенно полезны для использования в качестве исходных веществ в синтезе фитона и/или витамина Е. Таким образом, в соответствии со следующим аспектом настоящего изобретения предлагается способ получения фитона и/или витамина Е, который включает в себя гидрирование соединения общей формулы (I). В конкретном способе воплощения данного синтеза витамин Е можно получить, выбирая исходное вещество из соединения (VII) или (VIII), где Y представляет собой углеводород, содержащий группу хромана или хромена, определенную выше, а фитон можно получить, когда исходное вещество выбирают из соединения (V) или (VI), где Y является линейным C5 кетоном. Стадию гидрирования данного способа можно осуществлять в присутствии газообразного водорода и в присутствии металла или соли металла. Подходящие металлы и соли металлов включают в себя никель Ренея (сплав никеля/алюминия), необязательно в присутствии железа, марганца, кобальта, меди,цинка или хрома; цинк в присутствии уксусной кислоты; хлорид олова (II) и соли молибдена (III). Реакцию можно также проводить в присутствии палладия или платины, которые могут быть нанесены на подходящий инертный носитель, такой как уголь. Гидрирование предпочтительно проводят в присутствии палладия на инертном носителе, таком как уголь. Количество используемого металла или соли металла обычно составляет от 0,01 до 3 мол.экв., предпочтительно от 0,05 до 2 мол.экв. Данную реакцию можно проводить в присутствии растворителя, который можно выбрать из органической кислоты, такой как уксусная кислота; простых эфиров и ароматических углеводородов. Предпочтительными растворителями являются уксусная кислота и толуол. Количество растворителя предпочтительно составляет от 0 до 20 мас.экв. Реакцию можно также проводить в присутствии неорганической кислоты, например НСl или сульфокислоты. Количество неорганической кислоты предпочтительно составляет от 0 до 1 экв., предпочтительно от 0,1 до 0,5 экв. Температура реакции может составлять от 20 до 150 С, предпочтительно от 20 до 90 С, и при давлении от 1 до 50 бар, предпочтительно от 1 до 10 бар. Теперь настоящее изобретение будет далее проиллюстрировано со ссылкой на следующие примеры. Примеры с 1 по 6 направлены на получение промежуточных соединений, пример 7 направлен на получение промежуточного соединения для фитона, а пример 8 направлен на получение витамина Е. В следующих примерах Tf представляет группу F3 СSO2. Пример 1. Реакцию, подробно описанную ниже, проводили в присутствии растворителя - дихлорметана, с использованием различных кислот Льюиса в качестве катализаторов и в условиях, которые указаны в табл. 1. В реакционный сосуд, содержащий катализатор, добавляли эквимолярные количества двух реагентов в инертной атмосфере. Реакцию оставляли для взаимодействия на 2 ч, затем добавляли насыщенный водный гидрокарбонат натрия, сульфат магния, фильтровали и концентрировали. Продукт выделяли хроматографией на силикагеле или фильтрованием из сырого раствора после концентрирования. Полученный продукт экстрагировали эфиром, промывали водой и сушили с использованием растворителя. Результаты приведены в табл. 1. Таблица 1-3 005857 Пример 2. Реакцию, описанную ниже, проводили в присутствии дихлорметана в качестве растворителя при 0 С. В реакционный сосуд, содержащий 28 мг (0,049 экв.) катализатора (F3 СSO2)2NН, добавляли эквимолярную смесь 2,6,10,14-тетраметилпентадец-1-ен (532 мг) и ацеталя (302 мг) в инертной атмосфере. Реакцию проводили в течение 2 ч при 0 С, а затем 2 ч при 20 С перед добавлением водного насыщенного гидрокарбоната натрия. Полученный продукт экстрагировали эфиром, промывали водой и сушили с использованием сульфата магния, фильтровали и концентрировали. Продукт выделяли хроматографией на силикагеле или фильтровали из сырого раствора после концентрирования растворителя. После очистки получали выход 54%. Пример 3. Повторяли методику примера 1 с использованием реагентов, подробно описанных в приведенной ниже схеме реакции. Условия реакции, использованные катализаторы и полученные выходы приведены в табл. 2 ниже. Таблица 2 Пример 4. Повторяли методику примера 1 с использованием реагентов, подробно описанных в приведенной ниже схеме реакции. Условия реакции, использованные катализаторы и полученные выходы приведены в табл. 3 ниже. Пример 5. Реакцию, описанную на приведенной ниже схеме реакции, проводили в присутствии дихлорметана в качестве растворителя при 0 С. В реакционный сосуд, содержащий 2,5 мол.экв. катализатора этилдихлоралюминия (в виде 1,8 М раствора в дихлорметане), прибавляли эквимолярную смесь двух реагентов в инертной атмосфере. Реакцию проводили в течение 2 ч при 0 С в насыщенном водном гидрокарбонате натрия. Полученный продукт экстрагировали эфиром, промывали водой и сушили с использованием сульфата магния, фильтровали и концентрировали. Продукт выделяли хроматографией на силикагеле или фильтровали из сырого раствора после концентрирования. Полученный продукт экстрагировали эфиром, промывали водой и сушили с использованием растворителя. Получали выход 10%. Пример 6. Повторяли методику примера 1 с использованием реагентов, описанных на приведенной ниже схеме реакции. Условия реакции, использованные катализаторы и полученные выходы приведены в табл. 4 ниже. Таблица 4-5 005857 Пример 7. Гидрогенолиз продукта, полученного в примерах 4, 5 и 6, проводили в присутствии катализатора гидрирования в условиях эксперимента, приведенных далее в табл. 5 и описанных на приведенной ниже схеме реакции. Реагент и катализатор помещали в автоклав. Добавляли растворитель. Перед закрыванием автоклав продували аргоном. После этого в автоклав вводили водород для инициирования реакции. В конце реакции давление снижали до атмосферного давления. Затем автоклав открывали. Выход продукта приведен в табл. 5. Таблица 5-6 005857 Стадия (а). Получение альфа-линаллола. В двугорлую колбу помещали 24,7 мл 1,7 М раствора винилмагнийхлорида в ТГФ (42 ммоль) в атмосфере аргона. Раствор нагревали до 35 С и по каплям добавляли к магниевому соединению 3,812 г (30 ммоль) 6-метил-6-гептен-2-она в течение 75 мин. После этого смесь выливали в смесь 20 г льда, 24 мл воды и 4 мл 37%-ной хлористо-водородной кислоты. Полученную органическую фазу отделяли, сушили над сульфатом магния и концентрировали. Выход составлял 4,563 г (98%), а чистота - 95%. Стадия (b). Переход к хроману. В 1 мл этилацетата растворяли 151 мг триметилгидрохинона в атмосфере аргона и нагревали раствор до 75 С. Последовательно прибавляли 16,24 мг хлорида цинка, 4 мкл воды и 2 мкл 37%-ной хлористо-водородной кислоты. В течение 30 мин прибавляли к смеси альфа-линаллол (154 мг). Спустя 10 ч при 75 С альфа-линаллол был израсходован. Добавляли 10 мкл 37%-ной хлористо-водородной кислоты. За образованием хромана наблюдали по пластинкам ТСХ. Спустя 2 ч при 75 С смесь охлаждали до 25 С,разбавляли эфиром, промывали нормальным раствором гидроксида натрия и водой, сушили над сульфатом магния, фильтровали и концентрировали. Полученный при этом сырой хроман разбавляли 5 мл триэтиламина, кипятили в течение 5 ч и выдерживали при 25 С 2 ч при перемешивании. Смесь концентрировали в вакууме и хроматографировали остаток на силикагеле. Требуемый ацетилированный хроман выделяли с 60%-ным выходом (198 мг). Стадия (с). Переход к хромену. В 1 мл этилацетата растворяли 151 мг триметилгидрохинона в атмосфере аргона и нагревали раствор до 75 С. Последовательно прибавляли 16,24 мг хлорида цинка, 4 мкл воды и 2 мкл 37%-ной хлористо-водородной кислоты. В течение 30 мин прибавляли к смеси альфа-линаллол (154 мг). Спустя 10 ч при 75 С смесь охлаждали до 25 С, разбавляли эфиром, промывали нормальным раствором гидроксида натрия и водой, затем сушили над сульфатом магния, фильтровали и концентрировали. Полученный при этом сырой бензохиноновый аддукт разбавляли 5 мл триэтиламина, нагревали до кипения с обратным холодильником в течение 5 ч и охлаждали до 25 С. Добавляли 204 мг уксусного ангидрида и смесь выдерживали при 25 С в течение 2 ч при перемешивании. Смесь концентрировали в вакууме и хроматографировали остаток на силикагеле. Требуемый ацетилированный хромен выделяли с выходом 50% (162 мг). Стадия (d). В 5 мл дихлорметана растворяли 1 ммоль соединения, полученного на стадии (b), и 1 ммоль диметилацеталя диметилоктаналя в инертной атмосфере. Добавляли катализатор (5% мол.экв. трифлата скандия,или треххлористого железа, или трифлата иттербия). Смесь выдерживали при перемешивании в течение 15 ч при температуре окружающей среды. Добавляли 3 мл насыщенного водного раствора гидрокарбоната натрия. Отделяли органическую фазу, сушили над сульфатом магния и концентрировали в вакууме. Сырой требуемый продукт очищали колоночной хроматографией на силикагеле (элюент: пентан/диэтиловый эфир: 9/1 по объему). Стадия (е). В 5 мл дихлорметана растворяли 1 ммоль соединения, полученного на стадии (с), и 1 ммоль диметилацеталя диметилоктаналя в инертной атмосфере. Добавляли катализатор (5% мол.экв. трифлата скандия,или треххлористого железа, или трифлата иттербия). Смесь выдерживали при перемешивании в течение 15 ч при температуре окружающей среды. Добавляли 3 мл насыщенного водного раствора гидрокарбоната натрия. Отделяли органическую фазу, сушили над сульфатом магния и концентрировали в вакууме. Сырой требуемый продукт очищали колоночной хроматографией на силикагеле (элюент: пентан/диэтиловый эфир: 9/1 по объему). Стадия (f). 1 Ммоль продукта, полученного на стадиях (е) и (d), вместе с катализатором (5% палладия на угле; 5% мас.экв.) помещали в автоклав. Добавляли диэтиловый эфир (5 мл). Перед закрытием автоклав продували аргоном. После этого в автоклав вводили водород (от 1 до 5 бар) для инициирования реакции. В конце реакции давление снижали до атмосферного давления. Затем автоклав открывали. После обычной обработки смеси получали витамин Е. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения общей формулы (I) в которой А представляет собой углеводород от C1 до C20, Y и Z независимо представляют собой углеводород от C1 до С 20, который может содержать кислородсодержащую функциональную группу, В представляет OR1 или NHR1, где R1 является либо водородом, либо углеводородом от C1 до С 6, при этом указанный способ включает в себя взаимодействие в присутствии катализатора, кислоты Льюиса, соединения общей формулы (II)-7 005857 в которой Y и Z определены, как указано выше, с соединением общей формулы (III) или соединением общей формулы (IV) в которых А определено как указано выше, В' представляет собой О или NR1, где R1 определен, как указано выше, и R представляет собой водород или углеводород от C1 до С 6. 2. Способ по п.1, в котором Y представляет собой углеводород от C1 до С 6 с кислородной группой, Z представляет собой метил, А представляет собой алифатический углеводород, а R представляет собой СН 3. 3. Способ по п.1 или 2, в котором соединения формулы (II) представляют собой 6-метил-6-гептен-2 он; 6-ацетокси-2,5,7,8-тетраметил-2-[(4-метилпент-4-ен)-1-ил]хромен или 6-ацетокси-2,5,7,8-тетраметил 2-[(4-метилпент-4-ен)-1-ил]хроман. 4. Способ по любому из предшествующих пунктов, в котором соединения формулы (III) представляют собой 3-метилбутаналь; имины 3-метилбутаналя; 3,6-диметилоктаналь; имины 3,6-диметилоктаналя; цитраль и имины цитраля. 5. Способ по любому из предшествующих пунктов, в котором соединения формулы (IV) представляют собой ацетали цитраля; ацетали 3-метилбутаналя или ацетали 3,6-диметилоктаналя. 6. Способ по любому из предшествующих пунктов, в котором кислота Льюиса имеет общую формулу M(L)n, в которой М представляет собой алюминий, железо, магний, скандий, иттербий, цинк, титан,кремний и висмут; L представляет собой галогенид, СF3SО 3, (CF3SO2)2N, ClO4 или алкил от C1 до С 4, а n соответствует электронной валентности М и обычно составляет от 1 до 4, или общую формулу А-Н, в которой А представляет собой СF3SО 3 или (CF3SO2)2N. 7. Способ по п.6, в котором кислота Льюиса представляет собой треххлористое железо. 8. Способ по любому из предшествующих пунктов, осуществляемый в присутствии основания, выбранного из ароматических аминов, алифатических аминов и карбонатных солей I или II группы Периодической таблицы. 9. Способ по любому из предшествующих пунктов, осуществляемый в присутствии органического растворителя, выбранного из хлорсодержащих растворителей, органических растворителей, эфирных растворителей, нитрильных растворителей и нитрорастворителей. 10. Способ по любому из предшествующих пунктов, осуществляемый при температуре от -80 до-8 005857 12. Способ получения фитона и/или витамина Е, который включает в себя гидрогенолиз соединения общей формулы (I), определенного в любом из пп. с 1 по 10. 13. Способ по п.12, в котором гидрогенолиз проводят в присутствии катализатора, который представляет собой металл, выбранный из палладия, платины, никеля и цинка, или соли металла - хлорида олова (II) или молибдена (III). 14. Способ по п.13, в котором гидрогенолиз проводят в присутствии палладия на угле. 15. Способ получения витамина Е по любому из пп. с 12 по 14, который включает в себя гидрогенолиз новых соединений (VII) или (VIII), определенных в п.11. 16. Способ получения фитона по любому из пп. с 12 по 14, который включает в себя гидрогенолиз соединений (V) или (VI), определенных в п.11.
МПК / Метки
МПК: C07D 311/70, C07C 49/203
Метки: использования, получении, соединения, витамина, промежуточные
Код ссылки
<a href="https://eas.patents.su/10-5857-promezhutochnye-soedineniya-dlya-ispolzovaniya-pri-poluchenii-vitamina-e.html" rel="bookmark" title="База патентов Евразийского Союза">Промежуточные соединения для использования при получении витамина е</a>
Производные витамина d с циклическими субструктурами в боковых цепях, способ и промежуточные продукты для их получения и их применение для получения лекарственных средств

Номер патента: 4703
Опубликовано: 24.06.2004
Авторы: Гизен Клаудиа, Штайнмейер Андреас, Хаберей Мартин, Фэнрих Марианне, Шварц Катика
МПК: C07C 401/00
Метки: витамина, промежуточные, лекарственных, способ, продукты, получения, боковых, средств, субструктурами, цепях, производные, применение, циклическими
Формула / Реферат:
1. Производные витамина D общей формулы I в которой Y1 и Y2, каждый независимо один от другого, обозначает атом водорода или группу -C(O)R5, а Y3 обозначает атом водорода или гидроксигруппу, атом галогена, группу -OC(O)R5 либо группу -OR5, где R5 представляет собой насыщенный C1-C12алкильный радикал и группа Y3 может быть представлена как в 2a-, так и в эпимерной 2b-конфигурации, R1 и R2 каждый обозначает атом водорода или оба вместе...
Нафтильные соединения, промежуточные соединения для их получения, применение нафтильных соединений, способ снижения холестерина

Номер патента: 1600
Опубликовано: 25.06.2001
Авторы: Брайант Генри У., Кроуелл Томас А., Джонс Чарльз Д., Палковиц Алан Д.
МПК: A61K 31/33, C07C 47/546, A61P 19/10...
Метки: соединения, способ, получения, нафтильные, соединений, применение, снижения, холестерина, промежуточные, нафтильных
Формула / Реферат:
1. Соединение формулы I где R1 является -Н, -ОН, -O(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr; где Аr является фенилом или замещенным фенилом, OCO(С1-С6-алкилом), -O(СО)O(С1-С6-алкилом) или -ОSО2(С4-С6-алкилом); R2 является -Н, -F, -Cl, -ОН, -О(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr, где Аr является фенилом или замещенным фенилом, -OCO(С1-С6-алкилом),...
Диароматические соединения пропинила или диенила, промежуточные соединения, фармацевтические и косметические композиции на основе указанных соединений и применение косметических композиций.

Номер патента: 1060
Опубликовано: 30.10.2000
Автор: Бернардон Жан-Мишель
МПК: A61P 17/00, A61K 31/192, C07C 63/66...
Метки: диенила, композиции, соединения, пропинила, косметические, композиций, применение, промежуточные, соединений, указанных, диароматические, основе, фармацевтические, косметических
Формула / Реферат:
1. Диароматические соединения пропинила или диенила общей формулы (I) где R1 является (I) радикалом -СН3, (II) радикалом -СН2-O-R6, (III) радикалом -О-R6, (IV) радикалом -CO-R7, где R6 и R7 имеют значения, приведенные ниже, Аr является радикалом, отвечающим следующей формуле (а): где R5 имеет значения, приведенные ниже, Х является радикалом формулы или или где R8 и R9 имеют значения, приведенные ниже, R2 и R3,...
Четырехциклические конденсированные соединения с гетероатомами, замещенные арилом, промежуточные соединения, способы получения, композиции и методы лечения

Номер патента: 1649
Опубликовано: 25.06.2001
Автор: Гриз Тимоти А.
МПК: A61P 5/30, C07D 471/00, A61K 31/35...
Метки: композиции, замещенные, промежуточные, соединения, получения, конденсированные, четырехциклические, лечения, методы, способы, арилом, гетероатомами
Формула / Реферат:
1. Соединение формулы I или II где Y обозначает -О-, -S-, -СН2-, -СН2СН2-, -СН=СН- или -NR4-; В обозначает -СН2- или -СО-; R1 и R2 обозначают каждый независимо -Н, -ОН, -O(C1-C4алкил), -ОСОС6Н5, -ОСО(С1-С6алкил), -ОSО2(С4-С6алкил), OSO2СF3, Сl или F; n равно 1 или 2; W обозначает -СН2- или >С=O; R3 обозначает 1-пиперидинил, 2-оксо-1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 2-оксо-1-пирролидинил,...
Промежуточные соединения для получения 2-имидазолин-5-онов

Номер патента: 1518
Опубликовано: 23.04.2001
Авторы: Гадрас Ален, Бюфорн Альбер
МПК: C07D 277/36
Метки: соединения, 2-имидазолин-5-онов, получения, промежуточные
Формула / Реферат:
1. Соединение 2-тиотиазолидин-5-он общей формулы (I) в которой R1 означает (C1-С3)-алкильный pадикал или фенильный радикал; R2 означает арильную группу, выбираемую из фенила или пиридила, возможно замещенного 1-3 группами, выбираемыми из атома галогена, нитрогруппы, цианогруппы, (C1-С3)-алкильного радикала, (C1-С3)-алкоксильного радикала; а также его соли, его стереоизомеры, в частности, если радикалы R1 и R2 являются разными, оптические...
Предыдущий патент: Тетрагидротиопиранфталазиноновые производные в качестве ингибиторов pde4
Следующий патент: Инсектицидная композиция, обладающая улучшенной стойкостью при хранении
Случайный патент: Новые соединения бензотиазина и бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат