(11β,17β)-17-гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3-он и лекарственное средство, его содержащее
Номер патента: 21946
Опубликовано: 30.10.2015
Авторы: Карл Ульрих, Мёллер Карстен, Шведе Вольфганг, Боне Вильхельм, Ротгери Андреа








Формула / Реферат
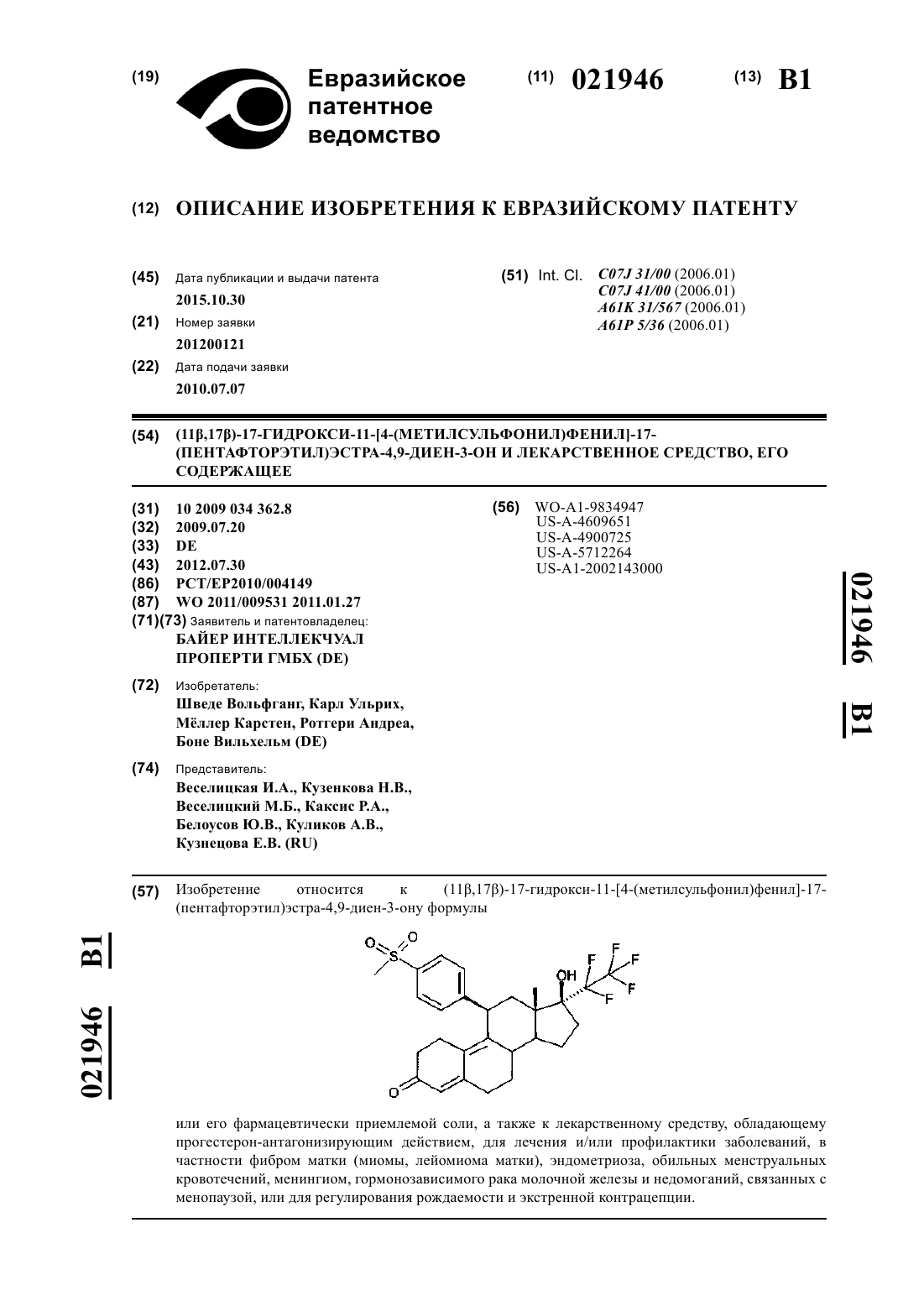
1. (11β,17β)-17-Гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3-он формулы

или его фармацевтически приемлемая соль.
2. Лекарственное средство, обладающее прогестерон-антагонизирующим действием, содержащее соединение или его соль по п.1, в комбинации с инертным, нетоксичным, фармацевтически пригодным наполнителем.
Текст
или его фармацевтически приемлемой соли, а также к лекарственному средству, обладающему прогестерон-антагонизирующим действием, для лечения и/или профилактики заболеваний, в частности фибром матки (миомы, лейомиома матки), эндометриоза, обильных менструальных кровотечений, менингиом, гормонозависимого рака молочной железы и недомоганий, связанных с менопаузой, или для регулирования рождаемости и экстренной контрацепции.(71)(73) Заявитель и патентовладелец: БАЙЕР ИНТЕЛЛЕКЧУАЛ ПРОПЕРТИ ГМБХ (DE) Изобретение относится к (11,17)-17-гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3-ону или его фармацевтически приемлемой соли с прогестерон-антагонизирующим действием и лекарственному средству для лечения и/или профилактики заболеваний, в частности фибром матки (миомы, лейомиома матки), эндометриоза, обильных менструальных кровотечений, менингиом, гормонозависимого рака молочной железы и недомоганий, связанных с менопаузой, или для регулирования рождаемости и экстренной контрацепции. Это соединение является важным фармацевтически активным веществом. В частности, его можно использовать для изготовления лекарственных средств для лечения фибром матки или эндометриоза, обильных менструальных кровотечений, менингиом, гормонозависимого рака молочной железы и недомоганий, связанных с менопаузой, или для регулирования рождаемости и экстренной контрацепции. Для лечения фибром матки и эндометриоза соединение согласно изобретению можно также назначать последовательно в комбинации с гестагенами. В такой схеме лечения соединение согласно изобретению можно назначать в течение 1-6 месяцев с последующим перерывом в лечении, или последующим лечением гестагеном в течение 2-6 недель, или последующим лечением пероральным контрацептивом (ПК-комбинации) в течение такого же периода. Эффективность соединения согласно изобретению в качестве антагонистов прогестероновых рецепторов была продемонстрирована in vitro в исследованиях трансактивации и in vivo у крыс (прерывание беременности на раннем сроке). Впервые соединения с антагонистическим действием на прогестероновый рецептор (конкурентные антагонисты прогестероновых рецепторов) стали известны в 1982 (RU 486; ЕР 57115) и с тех пор многие из них были описаны. Антагонисты прогестероновых рецепторов с фторированной 17-боковой цепью были опубликованы в WO 98/34947 и Fuhrmann et al., J. Med. Chem. 43, 5010-5016 (2000). Соединения с фторированной 17-боковой цепью, описанные в WO 98/34947, как правило, обладают очень сильной антагонистической активностью в отношении прогестеронового рецептора. Соединения являются очень сильнодействующими и поэтому предпочтительными в WO 98/34947 являются 11(4-ацетилфенил)-20,20,21,21,21-пентафтор-17-гидрокси-19-нор-17-прегна-4,9-диен-3-он,11-(4 ацетилфенил)-20,20,21,21,21-пентафтор-17-гидрокси-19-нор-17-прегна-4-ен-3-он и 6'-ацетил-9,11 дигидро-17-гидрокси-17-(1,1,2,2,2-пентафторэтил)-4'Н-нафт[3',2',1':10,9,11]естер-4-ен-3-он. Эти соединения в значительной степени трансформируются in vivo в различные метаболиты, некоторые с сильной, а другие со сниженной фармакологической активностью. Метаболизм происходит в основном по 4 заместителю 11-фенильного остатка. Соединения, описанные в WO 2008/058767, являются, по крайней мере, частично метаболитами соединений, описанных в WO 98/34947. Задачей настоящего изобретения является обеспечить доступные сильнодействующие конкурентные антагонисты прогестероновых рецепторов и таким образом создать альтернативы для лечения гинекологических заболеваний. Было обнаружено, что соединение согласно изобретению являются особенно подходящим для решения этой задачи. В частности, соединение с метилсульфонильной группой проявляют очень высокую антагонистическую активность в отношении прогестеронового рецептора, то есть оно ингибирует действие прогестерона на его рецептор. Также было обнаружено, что соединение с метилсульфонильной группой по сравнению, например,с алканоильными группами имеют намного более высокую метаболическую, а также химическую стабильность к температуре, свету и окислительному стрессу. Например, соединение (11,17)-17 гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3-он (пример 4) демонстрирует по сравнению с соответствующим аналогом с алканоильной или гидроксиалканоильной группой(11-(4-ацетилфенил)-20,20,21,21,21-пентафтор-17-гидрокси-19-нор-17-прегна-4,9-диен-3-он или 20,20,21,21,21-пентафтор-17-гидрокси-11-[4-(гидроксиацетил)фенил]-19-нор-17-прегна-4,9-диен-3-он),неожиданно высокую стабильность при тепловой нагрузке под влиянием УФ-света и неожиданно низкую чуствительность к окислению. Также является неожиданным, что указанные соединения с метилсульфонильной группой имеют низкий ингибирующий потенциал в отношении исследуемых CYP-изоферментов CYP1A2, CYP2C8,CYP2C9, CYP2D6 и CYP3A4, и вплоть до максимальной концентрации, растворимой или используемой в анализе (минимум 10 мкМ, максимум 20 мкМ), не достигалось 50% ингибирование в любом исследуемом случае. Эти in vitro полученные данные предполагают для исследуемого вещества особенно низкий риск взаимодействий с совместно вводимыми лекарственными средствами относительно CYP-ингибирования. Кроме того, для (11,17)-17-гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра 4,9-диен-3-она особенно благоприятный профиль безопасности (в кратковременных и постоянных исследованиях) был обнаружен в преклинических исследованиях на грызунах и негрызунах. Настоящее изобретение относится к производным 17-гидрокси-17-пентафторэтил-эстра-4,9(10)диен-11-арила, а именно к (11,17)-17-гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3-ону формулы или его фармацевтически приемлемой соли. Физиологически безвредные соли соединений согласно изобретению являются предпочтительными как соли в рамках настоящего изобретения. Однако соли, которые не являются сами по себе подхлодящими в фармацевтических целях, но могут, например, быть использованы для выделения или очистки соединений согласно изобретению, также включены. Физиологически безвредные соли соединений согласно изобретению включают, когда они имеют основную функцию, соли с неорганическими или органическими кислотами, в частности минеральные кислоты, карбоновые кислоты и сульфоновые кислоты, например соли хлористо-водородной кислоты,бромисто-водородной кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, этансульфоновой кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, нафталиндисульфоновой кислоты, уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты, молочной кислоты, винной кислоты, яблочной кислоты, лимонной кислоты, фумаровой кислоты, малеиновой кислоты и бензойной кислоты. Физиологически безвредные соли соединений согласно изобретению включают, когда они имеют кислотную функцию, соли щелочных металлов, соли щелочно-земельных металлов или аммониевые соли, такие как те, которые можно получить путем реакции с соответствующими неорганическими или органическими основаниями. Можно упомянуть в качестве примера и предпочтительно соли щелочных металлов (например, натриевые и калиевые соли), соли щелочно-земельных металлов (например, кальциевые и магниевые соли) и аммониевые соли, происходящие от аммиака или органических аминов с 116 атомами углерода, такие как, например и предпочтительно, этиламин, диэтиламин, триэтиламин,этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, бицикло-гексиламин, диметиламино-этанол, прокаин, дибензиламин, N-метилморфолин, аргинин, лизин, этилендиамин, Nметилпиперидин, N-метилглукамин, D-метилглукамин, этидглукамин, 1,6-гексадиамин, глюкозамин, Nметилглицин, 2-амино-1,3-пропандиол, трис-гидроксиметил-аминометан и 1-амино-2,3,4-бутантриол. В рамках настоящего изобретения, если не указанно иначе, заместители имеют следующие значения. Алкил представляет собой линейные или разветвленные алкильные группы с 1-6 атомами углерода,например метил, этил, пропил, изопропил, н-бутил, сек-бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил, гексил, гептил, октил, нонил и децил. Арил представляет собой монотрициклический ароматический, замещенный или незамещенный карбоциклический остаток, например фенил, нафтил, который может быть замещен один или несколько раз галогеном (F, Cl, Br, I), ОН, О-алкилом, СО 2 Н, СО 2-алкилом, NH2, NH(С 1-С 10-алкилом), N(С 1-С 10 алкилом)2, в частности N(СН 3)2, NO2, N3, CN, C1-С 10-алкилом, C1-С 10-перфторалкилом, C1-С 10-ацилом,C1-С 10-ацилокси группами. Гетероарил представляет собой ароматический, моно- или бициклический остаток, как правило, с 510, предпочтительно 5-6 атомами в кольце, и до 5, предпочтительно до 4 гетероатомов из серий S, О и N,например и предпочтительно, для бензофуранила, бензотиофенила, хинолинила, фурила, имидазолила,индазолила, индолила, изохинолинила, оксазолила, пиридазинила, пиридила, пиримидила, пирролила,тиазолила, тиенила, пиразолила, изоксазолила, пиразинила, хинолила или тетразолила. Аралкил представляет собой аралкильные группы, которые могут содержать до 14 атомов углерода,предпочтительно 6-10 атомов углерода в кольце, и 1-8, предпочтительно 1-4 атома углерода в алкильной цепи. Аралкильными остатками, которые могут быть рассмотрены, являются, например, бензил, фенилэтил, нафтилметил, нафтилэтил, фурилметил, тиенилэтил, пиридилпропил. Кольца могут быть замещены один или несколько раз галогеном, ОН, О-алкилом, СО 2 Н, СО 2-алкилом, NH2, NH(С 1-С 10-алкилом),N(С 1-С 10-алкилом)2, NO2, N3, CN, С 1-С 20-алкилом, C1-С 10-перфторалкилом, С 1-С 20-ацилом, С 1-С 20 ацилокси группами. Когда остатки в соединениях согласно изобретению замещены, если не указано иначе, остатки могут быть замещены один или несколько раз. В рамках настоящего изобретения для всех остатков, которые имеют место более одного раза, их значение является независимым друг от друга. Замещение одним,двумя или тремя идентичными или разными заместителями является предпочтительным. Замещение одним заместителем является особенно предпочтительным. Было обнаружено, что соединение согласно изобретению и/или производные демонстрируют хорошее прогестерон-антагонизирующее действие. В нескольких клинических исследованиях было обнаружено, что лечение антагонистами прогестероновых рецепторов (мифепристон, асоприснил, Проэллекс) может привести к значительному уменьшению фибром матки и значительному сокращению симптомов, связанных с указанными фибромами матки. Кроме того, клинические исследования показали, что во время лечения указанным антагонистом прогестероновых рецепторов симптомы, вызванные эндометриозом (особенно боли), могут также значительно сократиться. В вышеприведенной формуле А и В имеют следующее значение: =O; -ОН/-Н или -OH/-C2F5. Принцип получения соединения показан в вышеприведенной схеме. Соединения получают, начиная с (5'R,8'S,10'R,13'S,14'S,17'S)-5,5,13'-триметил-1',2',7',8',12',13',14',15',16',17'декагидро-6'H-спиро[1,3-диоксан-2,3'-[5,10]эпоксициклопента[а]фенантрен]-17'-ола (для изготовления см. Tetrahedron Lett. 26, 2069-2072 (1985) аналогично способу, описанному в WO 98/34947 и в WO 2008/058767. После окисления гидроксильной группы в положении 17 стероидного скелета введение 17-пентафторэтильной боковой цепи в соответствующие 17-кето соединения проводят согласно способам, описанным в WO 98/34947 и в WO 2008/058767. Введение 11-фенильных заместителей проводят путем сопряженного присоединения реагентов - арильных реактивов Гриньяра или ариллития при катализе медью. Получают соединения общей формулы II, где R8 может представляет собой остаток Y или фенильное кольцо, замещенное один или два раза остатком Y, где Y выбран из группы, которая включает SR2, S(O)R3, S(O)2R3,S(O)(NH)R3, S(O)(NR4)R3, S(O)2NR9R10, R2 представляет собой водород или C1-С 6-алкил или С 7-С 10 аралкил или арил, R3 представляет собой C1-С 6-алкил или арил, R4 представляет собой группу S(O)2R6, R6 представляет собой фенил или 4-метилфенил, a R9, R10 независимо друг от друга выбраны из группы,которая включает водород, C1-С 10-алкил или арил или в качестве альтернативы представляют собой вместе с атомом азота 3- - 8-членное, насыщенное или ненасыщенное гетероциклическое кольцо и дополнительно может быть гидрокси, C1-С 10-алкокси, бензилокси, C1-С 10-алканоилокси, бензоилокси, силилоксил, алкоксиалкилокси группой Cl, Br, I или группой CmFm+1SO3 с m=1-4 и А и/или В означает карбонильную группу или 17-ОН/17-Н группу или 17-OH/17-C2F5 группу. Затем из соединений общей формулы II можно получить (11,17)-17-гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3-он. Для этого функциональные группы необязательно дополнительно модифицируют. Можно упомянуть, в частности, окисление сульфидов до сульфоксидов или сульфонов с помощью способов, известных специалисту в данной области техники, и образование сульфоксиминов из сульфидов путем добавления Хлорамин-Т-Тригидрат и последующего окисления до соответствующего защищенного сульфоксимина, который потом выделяют, например, путем кислотного расщепления. Однако в качестве альтернативы также можно использовать способы, известные специалисту в данной области техники, начиная с соответствующих сульфоксидов. Для соединений, в которых есть бифенильный остаток в положении 11 стероидного скелета, это можно провести либо непосредственно путем сопряженного присоединения реагентов - диарильных реактивов Гриньяра или диариллития при катализе медью или в качестве альтернативы, например, с помощью катализируемых палладием реакций сочетания соответствующих функционализированных 11-фенильных производных, например, с фенилтрифлатом или фенилнонафлатом. Как правило, вначале можно вводить как 11-фенильный остаток, так и 17 пентафторэтильную боковую цепь. Функциональные группы, особенно 3-кетогруппа, тем временем являются необязательно защищенными, например, в виде кеталя. В качестве кетальной защитной группы можно, например, упомянуть этилендиокси или 2,2-диметилпропилен-1,2-диоксигруппу. Гидроксильные группы являются, например, защищенными в виде метоксиметилового, метоксиэтиловых, тетрагидропираниловых, бензиловых или силил эфиров. Затем на подходящей стадии защитные группы отщепляют с помощью способов, известных специалисту в данной области техники. Во время расщепления 3-кетальной на 3-кетогруппу стероидного скелета, необязательно все еще присутствующую 5-гидроксильную группу удаляют, так что образуется заявленное соединение. Если получение исходных соединений не описано в данном изобретении, способы их получения являются известными специалисту в данной области техники или их можно получить аналогично известным соединениям или способам, описанным в данном изобретении. Смеси изомеров можно разде-3 021946 лить на отдельные соединения обычными способами, например путем кристаллизации, хроматографии или образования солей. Получение солей осуществляют обычным способом путем добавления эквивалентного количества или чрезмерного количества основания или кислоты, необязательно в растворе, к раствору заявленного соединения, с необязательным отделением осадка или обработки раствора обычным способом. Общие определения вышеуказанных остатков или указанных в предпочтительных диапазонах применяют для исходных веществ или промежуточных соединений, необходимых в каждом случае для получения. Соединение согласно изобретению демонстрирует непредвидимый, ценный фармакологический,фармакокинетический и фармакодинамический профиль действия. Поэтому оно является подходящим для применения в качестве лекарственных средства для лечения и/или профилактики заболеваний у людей и животных. Фармацевтическую эффективность соединения согласно изобретению можно объяснить его действием в качестве антагонистов прогестероновых рецепторов и таким образом их антагонизирующим действием на прогестероновый рецептор. Другой объект настоящего изобретения включает лекарственное средство, которое содержит вышеуказанное соединение, в комбинации с инертным, нетоксичным, фармацевтически пригодным наполнителем и дополнительно необязательно по крайней мере одно или несколько других активных веществ, в частности, для лечения и/или профилактики вышеуказанных заболеваний. Для лечения заболеваний, связанных с новообразованиями, можно вводить либо одновременно,либо последовательно, например, следующие активные вещества/классы активных веществ: СМЭР,СДЭРs, антиэстрогены, ингибиторы ароматазы, ингибиторы киназы, ингибиторы ангиогенеза и/или цитостатики. Для лечения фибром матки или эндометриоза соединение согласно изобретению можно объединить одновременно или последовательно с гестагенами или комбинациями эстрогенов и гестагенов. Режимы применения антагонистов прогестероновых рецепторов/гестагена описаны в WO 96/15794(Moller et al., Bayer Schering Pharma AG). Режимы - необязательно повторяющиеся - где антагонист прогестероновых рецепторов вводят в течение 2-4 месяцев с последующим введением гестагена в течение 1-4 недель, являются очень подходящими для лечения фибром матки и эндометриоза. Введение антагониста прогестероновых рецепторов в течение 84 дней с последующим введением гестагена в течение 14 дней - необязательно повторяющееся - является особенно подходящим. Одновременное или последовательное введение соединения согласно изобретению, например, с СМЭР, СДЭРs и эстрогенами можно рассматривать для лечения недомоганий, связанных с менопаузой. СМЭР (селективные модуляторы эстрогеновых рецепторов) представляют собой соединения, которые являются тканеселективными и обладают антиэстрогенным или эстрогенным действием, например, в матке они ингибируют действие эстрогена, но в костях они имеют нейтральное или подобное эстрогену действие. Примерами являются кломифен, ралоксифен, тамоксифен, торимифен, базедоксифен, ласофоксифен и ормелоксифен. Селективные дестабилизаторы эстрогеновых рецепторов (СДЭР) представляют собой фармацевтические препараты, которые полностью антагонизируют эстрогеновый рецептор ("чистые антиэстрогены" без эстрогенного активного компонента) и приводят к понижающей регуляции рецептора (например,фулвестрант, ZK-703 и ZK-253 (Hoffmann J. et al., J. Natl. Cancer Inst. 2004, 96:210-218) и соединения,описанные в WO 98/007740, WO 99/33855 и WO 03/045972). Антиэстрогенами являются соединения, которые полностью антагонизируют эстрогеновый рецептор, например фулвестрант. Ингибиторы ароматазы ингибируют фермент ароматазу и поэтому ароматизацию андрогенов до эстрогенов. Они включают, среди прочего, анастрозол, летрозол, эксеместан, ворозол, форместанс и фадрозол. Ингибиторы киназы представляют собой ферменты, которые переносят фосфатный остаток из АТР в другие субстраты и, в частности, в гидроксильные группы, такие как, например, сорафениб (Нексавар) или иматиниб (Гливек). Ингибиторы ангиогенеза, например авастин, уменьшают или блокируют снабжение сосудов и поэтому и поступление крови в новообразование. Цитостатики, например цисплатин,таксол, Таксотер, являются натуральными или синтетическими веществами, которые ингибируют(подавляют) рост клеток или деление клеток. Гестагены представляют собой в контексте настоящего изобретения либо натуральный прогестерон сам по себе, либо синтетические производные, которые подобно прогестерону связываются с прогестероновым рецептором и, при дозировке выше ингибирующей овуляцию дозы, ингибируют овуляцию. В качестве примеров синтетических производных можно упомянуть дроспиренон, гестоден, левоноргестрел, ципротерон ацетат, десогестрел и 3-кетодесогестрел, норэтистерон, норэтистерон ацетат и диеногест. Комбинации гестагенов и эстрогенов представляют собой комбинации активных веществ, которые содержатся в пероральных контрацептивах, которые являются известными per se, например Ясмин, Фемован, Триквилар, Марвелон, ДЖАЗ и т.д. Соединение согласно изобретению может действовать систематично и/или локально. С этой целью его можно применять подходящим способом, например перо-4 021946 ральным, внутриматочным, интравагинальным, парентеральным, пульмональным, назальным, сублингвальным, лингвальным, буккальным, ректальным, дермальным, трансдермальным, конъюнктивальным,или ушным путем или в виде имплантанта или стента. Внутриматочно означает, в частности, применение с помощью ВМС (внутриматочная система) или ВМК (внутриматочный контрацептив). Интравагинальное применение можно осуществлять, среди прочего, с помощью ИВК/ВКС (интравагинальоем кольцо/вагинальная кольцевая система). Формы для внутриматочного или интравагинального применения (ср., например, WO 01/47490, особенно с. 1, строка 10 - строка 5, строка 13 и строка 7, строка 19 - строка 58, строка 6, или для вагинальных колец: WO 06/010097, особенно с. 10, строка 22 - с. 14, строка 28) могут содержать соединение согласно изобретению и несиликоновые и/или силиконовые полимеры, в частности также эластомеры на основе силоксана(ср. WO 01/47490, особенно с. 7, строка 19 - с. 15, строка 15). Для этих путей введения соединение согласно изобретению можно вводить в пригодных лекарственных формах. Лекарственные формы быстрого высвобождения и/или замедленного высвобождения, функционирующие в соответствии с предыдущим уровнем техники, являются пригодными для перорального введения, которые содержат соединение согласно изобретению в кристаллической, и/или аморфной, и/или расстворенной форме, например таблетки (таблетки, не покрытые оболочкой или таблетки, покрытые оболочкой, например, с энтеросолюбильными покрытием или покрытием замедленного растворения или нерастворимым покрытием, которые контролируют высвобождение соединений согласно изобретению),таблетки или пленки/пластинки, которые быстро распадаются в ротовой полости, пленки/ лиофилизаты,капсулы (например, твердые желатиновые или мягкие желатиновые капсулы), таблетки, покрытые оболочкой, гранулы, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы. Парентеральное применение можно осуществлять при избежании стадии абсорбции (например,внутривенно, внутриартериально, интракардиально, интраспинально или интралюмбарно) или с включением абсорбции (например, внутримышечно, подкожно, интрадермально, перкутанно или интраперитонеально). Инъекционные и инфузионные препараты в форме растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков, среди прочего, являются пригодными в качестве лекарственных форм для парентерального введения. Для других путей введения пригодными являются, например, ингаляционные лекарственные формы (включая порошковые ингаляторы, распылители), назальные капли, растворы и спреи; таблетки для сублингвального, сублингвального или буккального введения, пленки/пластинки или капсулы, суппозитории, ушные или глазные препараты, вагинальные капсулы, водные суспензии (лосьоны, взбалтываемые смеси), липофильные суспензии, мази, крема, трансдермальные терапевтические системы (например, пластыри), молочко, пасты, пены, присыпки, имплантанты или стенты. Соединение согласно изобретению можно преобразовать в вышеуказанные лекарственные формы. Это можно осуществить способом, известным per se, путем смешивания с инертными, нетоксичными,фармацевтически пригодными наполнителями. Эти наполнители включают, среди прочего, веществаносители (например, микрокристаллическая целлюлоза, лактоза, маннит), растворители (например, жидкие полиэтиленгликоли), эмульгаторы и диспергирующие веществи или смачивающие агенты (например,додецилсульфат натрия, полиоксисорбитанолеат), связывающие агенты (например, поливинилпирролидон), синтетические и натуральные полимеры (например, альбумин), стабилизаторы (например, антиоксиданты, например аскорбиновая кислота), красители (например, неорганические пигменты, например оксиды железа) и вкусовые добавки и/или ароматизаторы. Тем не менее, необязательно может быть необходимо отклониться от указанных количеств, а именно в зависимости от веса тела, пути введения, индивидуальной реакции на активное вещество, типа препарата и точки времени или интервала, когда осуществляется применение. Таким образом, в некоторых случаях может быть достаточно применять меньше, чем вышеуказанное минимальное количество, тогда как в других случаях указанный высший предел должен быть превышен. В случае введения большего количества можно порекомендовать распределить его по несколько индивидуальных доз на протяжении дня. Процентные соотношения в следующих исследованиях и примерах представляют собой, если не указано иначе, процентные соотношения по массе; части представляют собой массовые части. Пропорции растворителей, соотношения разбавления и показатели концентрации для жидкостей/жидких растворов всегда относятся к объему. Следующие примеры служат для того, чтобы раскрыть изобретение, никоим образом не ограничивая его. 50 г (5'R,8'S,10'R,13'S,14'S)-5,5,13'-триметил-1',2',6',7',8',12',13',14',15',16'-декагидро-17'Н-спиро[1,3 диоксан-2,3'-[5,10]эпоксициклопента[а]фенантрен]-17'-он (для получения см. Tetrahedron Lett. 26, 20692072 (1985) добавляли к 116 г конденсированного пентафторйодэтана в 500 мл абсолютного толуола при-70 С. 290 мл 1,5-молярного раствора комплекса метиллитий-бромид лития в диэтиловом эфире добавляли к смеси при такой же температуре. Затем смесь перемешивали в течение 1 ч при 0 С. Затем реакционную смесь добавляли к насыщенному водному раствору хлорида аммония и экстрагировали этилацетатом. Органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт растворяли в 200 мл ацетона и добавляли 450 мл воды. Осажденный продукт отфильтровывали и сушили в вакууме. Выход 61,6 г. 1 Н-ЯМР (400 МГц, CDCl3):6.04 brd (1H); 3.60 d (1H); 3.35-3.50 m (3H); 2.51 dbr (1H); 1.06 s (3 Н); 0.93 s (3 Н); 0.85 s (3 Н). 1,23 г магния стружки суспендировали в 5 мл ТГФ и добавляли 50 мкл дибромэтана при перемешивании. Раствор 10,31 г 1-бром-4-(метилтиофенил)бензол в 60 мл ТГФ добавляли к суспензии таким образом, чтобы температура реакции не поднималась выше 55 С. Затем смесь перемешивали в течение дополнительного часа. Затем полученный раствор охлаждали до 0 С. Добавляли 151 мг CuCl и смесь перемешивали в течение дополнительных 15 мин при 0 С. Затем добавляли раствор 5 г вещества, описанного в примере 1 а) в 50 мл ТГФ. Затем реакционной смеси позволяли достичь 23 С в течение приблизительно 3 ч с перемешиванием и затем ее перемешивали при этой температуре в течение дополнительных 10 ч. Затем насыщенный водный раствор NH4Cl добавляли к реакционной смеси с внешним охлаждением. Перемешивание продолжали в течение 30 мин и затем смесь экстрагировали несколько раз этилацетатом. Объединенные органические фазы промывали насыщенным раствором хлорида натрия и сушили над сульфатом натрия. Сырой продукт очищали с помощью хроматографии на силикагеле с последующей кристаллизацией из смеси дихлорметана и диизопропилового эфира. В результате получали 5,72 г указанного в заглавии соединения. 1 Н-ЯМР (300 МГц, CDCl3):7.50 d (2 Н); 7.30 d (2 Н); 4.41 s (1H); 4.28 dbr (1H); 3.40-3.60 m (4H); 2.51 s (3H); 1.05 s (3H); 0.87 s (3H); 0.53 s (3H). 5 г соединения со стадии b) растворяли в смеси 140 мл ТГФ и 140 мл метанола. Раствор 20 г Оксон в 94 мл воды добавляли медленно по каплям при 0 С. Затем смесь перемешивали в течение дополнительных 3,5 ч при 0 С. Затем смесь воды и дихлорметана добавляли к реакционной смеси. Фазы разделяли и водную фазу экстрагировали несколько раз дихлорметаном. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле. В результате получали 3,8 г указанного в заглавии соединения. 1 Н-ЯМР (300 МГц, CDCl3):7.86 d (2H); 7.40 d (2H); 5.81 sbr (1H); 4.50 dbr (1H); 3.07 s (3H); 0.51 s(3H). Пример 2. Антагонистическое действие относительно прогестероновых рецепторов в стабильных трансфектантах человеческих клеток нейробластомы (клетки SK-N-МС) с человеческим прогестероновым рецептором А или прогестероновым рецептором В и репортерной конструкцией MTV-LUC. Клетки SK-N-MC (человеческие клетки нейробластомы), которые были стабильно трансфектированы плазмидами, которые экспрессируют человеческий прогестероновый рецептор В (pRChPR-B-neo) или человеческий прогестероновый рецептор A (pRChPR-A-neo) и репортерную конструкцию (pMMTVLUC), инкубировали в течение 24 ч в отсутствии (отрицательный контроль) или в присутствии увеличивающихся количеств соответстветствующего исследуемого соединения (0,01, 0,1, 1, 10, 100 нмоль/л и 1 мкмоль/л) для определения агонистической эффективности. В качестве положительного контроля генрепортерной индукции клетки обрабатывали синтетическим гестагеном промегестоном (0,01, 0,1, 1, 10,100 нмоль/л и 1 мкмоль/л). Для определения антагонистической активности клетки обрабатывали 0,1 нмоль/л промегестона и дополнительно увеличивающимися количествами соответствующего исследуемого соединения (0,01, 0,1, 1, 10, 100 нмоль/л и 1 мкмоль/л). Активность ген-репортера LUC(LUC=люцифераза) определяли в клеточных лизатах и измеряли как ОСЕ (относительная световая единица). Все измеренные величины представлены в виде процентной эффективности и в виде концентраций ЕС 50 или IC50. 11-(4-Ацетилфенил)-20,20,21,21,21-пентафтор-17-гидрокси-19-нор-17-прегна-4,9-диен-3-он и 20,20,21,21,21-пентафтор-17-гидрокси-11-[4-(гидроксиацетил)фенил]-19-нор-17-прегна-4,9-диен-3-он,очень сильнодействующие и поэтому предпочтительные примеры из WO 98/34947 и WO 2008/058767,были протестированы в качестве сравнительных соединений вместе с исследуемым соединением.a) Агонистическая активность: ни одно из указанных соединений не демонстрирует агонистической активности.b) Антагонистическая активность. Все указанные соединения демонстрируют 100% антагонистическую эффективность. Антагонистическая эффективность соединений представлена в табл. 1. Таблица 1 Пример 3. Исследование на преждевременное прекращение беременности на самках крыс. Действие прогестерона и прогестеронового рецептора является основным предварительным условием для удачной беременности или гестации у млекопитающих. Прогестерон-антагонистическое действие соединения согласно изобретению исследовали на беременных крысах (6 крыс на группу) на 5-7 день после совокупления в обычных жилищных и кормовых условиях. После успешного ручного спаривания беременных животных (наличие спермы в вагинальном мазке на 1 день беременности=1 д.п.с.) рандомизировали и делили на эксперементальную группу и контрольную группу. Затем каждое животное получало подкожно или перорально 0,15, 0,5, 1,5 или 5 мг/кг исследуемого соединения или 1,0 мл/кг носителя(бензилбензоат/касторовое масло: 1+4 [об./об.]) в сутки с 5 по 7 день (5 д.-7 д.п.с.). Аутопсию осуществляли на 9 день (9 д.п.с.). В качестве характеристики антагонистического действия в отношении прогестероновых рецепторов матку исследовали на наличие сайтов нидации. Полное отсутствие или также наличие патологических, геморрагических или других анормальных сайтов нидации на 9 день (9 д.п.с.) оценивали как аборт. Результаты исследований представлены в табл. 3. Таблица 3 Результаты для крыс (прерывание беременности на раннем сроке)(11,17)-17-гидрокси-11-[4-(метилсульфонил)фенил]-17(пентафторэтил)эстра-4,9-диен-3-она в микросомах печени человека (МПЧ). Для оценки метаболической стабильности соединения использовали выделенные микросомы печени человека (МПЧ). Инкубирования осуществляли с помощью 2,4 мл раствора МПЧ (0,5 мг/мл содержание белка), 30 мкл исследуемого соединения (конечная концентрация 1 мкМ) и 0,6 мл смеси-кофактора (=NADPHгенерирующая система 3 IU глюкоза-6-фосфатдегидрогеназы, 14,6 мг глюкоза-6-фосфата, 1,2 мг NADP) при 37 С в 100 мМ фосфатном буфере при рН значении 7,4. Образцы вносили в 6 моментах времени (260 мин), осаждали с равным объемом метанола и восстановление исследуемых веществ, используемых в супернатанте, определяли с помощью анализа ЖХ-МС/МС. Присущий клиренс вещества в препарате микросом печени можно подсчитать на основании времени полужизни, установленного для распада вещества. На основании этого, вместе с различными физиологическими характеристиками по тщательно перемешанной модели, потом можно установить (метаболический) in vivo клиренс в отношении реакций фазы I. In vivo клиренс (метаболический) у человека, установленный соответственно для исследуемого соединения (11,17)-17-гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3 она, был очень низким: 0,1 Л/ч/кг. Пример 5. Проникновение (11,17)-17-гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3-она в Сасо-2 клетки. Для исследований на проникновения Сасо-2 клетки с количеством клеток 300000 клеток/мл подсчитывали на фильтрующих вкладках Transwell Clear (полиэстер; размер пор 0,4 мкМ) в 12-луночных клеточных культуральных планшетах в течение по крайней мере 14 дней в клеточной культуральной среде (1,5 мл) при 37 С, 5% СО 2 и 95% атмосферной влажности. Перед исследованием для проверки"плотности" клеточного монослоя определяли трансэпителиальную стойкость (TEER значение), которая должна быть больше 300 Ом/см 2. Затем клеточную культуральную среду заменяли горячим буфером для переноса (апикальный 0,5 мл, базолатеральный 1,5 мл) и клетки уравновешивали в нем в течение 5 мин. Исследование на проникновение осуществляли в двух повторностях при концентрации вещества 2 мкМ. В начале эксперимента 100 мкл (Ап 0 мин) вносили из апикальной части и сразу к нему добавляли 100 мкл ледяного останавливающего раствора. Затем фильтры инкубировали при 37 С со слабым взбалтыванием в течение 90 мин, затем 100 мкл снова вносили из апикальной части (Ап 90 мин) и 400 мкл из базолатеральной части (Баз 90 мин) и в каждом случае добавляли одинаковый объем останавливающего раствора. После дополнительного разбавления образцы с 4-кратным объемом останавливающего раствора/транспортирующего буфера (1+1) осаждали в течение ночи при -20 С и супернатант анализировали с помощью ЖХМС/МС. Величину Рарр веществ подсчитывали по следующей формуле:Cres/t - изменение в концентрации вещества через время в рецептороной части.(11,17)-17-Гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3-он продемонстрировал очень высокое проникновение 104 нм/с в этом исследовании. Пример 6. Исследование действия на сердечно-сосудистую систему (вкл. ЭКГ) собак породы бигль под анестезией.(11,17)-17-Гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3-он, растворенный в смеси PEG 400 и HPCD (60% PEG 400, 40% НРCD 30%), вводили внутривенно самкам собак породы бигль под анестезией. Масса тела собак была 9 кг. Было испытано 3 собаки на группу и дополнительно 3 собаки в контрольной группе. 0,1, 0,33 и 1 мг/кг вещества вводили в 3 последовательных инфузиях в каждом случае в течение 30 мин. Максимальное количество носителя составляло 0,4 мл на 1 кг в течение 30 мин. Образцы крови животных брали в разные моменты времени. Наивысшим уровнем плазмы (средним для 3 животных) был 1650 нг/мл по завершении третьей инфузии. В исследуемом диапазоне доз по сравнению с контролем не наблюдалось никакого биологически значимого действия на сердечно-сосудистую систему (давление в легочной артерии, системное артериальное кровяное давление, частота сердцебиений, ЭКГ). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. или его фармацевтически приемлемая соль. 2. Лекарственное средство, обладающее прогестерон-антагонизирующим действием, содержащее соединение или его соль по п.1, в комбинации с инертным, нетоксичным, фармацевтически пригодным наполнителем.
МПК / Метки
МПК: A61P 5/36, A61K 31/567, C07J 31/00, C07J 41/00
Метки: лекарственное, средство, 11β,17β)-17-гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3-он, содержащее
Код ссылки
<a href="https://eas.patents.su/10-21946-11beta17beta-17-gidroksi-11-4-metilsulfonilfenil-17-pentaftoretilestra-49-dien-3-on-i-lekarstvennoe-sredstvo-ego-soderzhashhee.html" rel="bookmark" title="База патентов Евразийского Союза">(11β,17β)-17-гидрокси-11-[4-(метилсульфонил)фенил]-17-(пентафторэтил)эстра-4,9-диен-3-он и лекарственное средство, его содержащее</a>
Способ получения 17бета-гидрокси-11бета-{4-(диметиламино)фенил} 17альфа-(проп-1- инил)эстра-4,9-диен-3-она

Номер патента: 14
Опубликовано: 30.12.1997
Авторы: Ряховская Маргарита Игоревна, Морозова Людмила Сергеевна, Гриненко Галина Семеновна, Долгинова Елена Максовна, Климова Людмила Игоревна, Густова Ольга Валериевна, Турчин Константин Федорович, Кочев Дмитрий Михайлович
МПК: A61K 31/565, C07J 1/00
Метки: 17бета-гидрокси-11бета-{4-(диметиламино)фенил, 17альфа-(проп-1, получения, инил)эстра-4,9-диен-3-она, способ
Формула / Реферат:
Способ получения 17b -гидрокси-11b -[4-(диметиламино) фенил]-17a -(проп-1-инил)эстра-4,9-диен-3-она формулы I взаимодействием производного стероида с 4-диметиламинофенилмагнийбромидом в присутствии катализатора в среде тетрагидрофурана, выделением соответствующего арилкеталя с использованием насыщенного водного раствора хлористого аммония, дегидратацией и гидролизом в присутствии кислотного агента в среде растворителя при комнатной...
Лекарственное средство, содержащее производные фенилпиперазина

Номер патента: 11096
Опубликовано: 30.12.2008
Авторы: Мольтсен Айнер Кнуд, Руланд Томас, Пюшль Аск, Андерсен Ким, Смит Гаррик Пол, Банг-Андерсен Бенни
МПК: A61K 31/4409, A61P 25/24, C07D 211/20...
Метки: средство, производные, содержащее, лекарственное, фенилпиперазина
Формула / Реферат:
1. Лекарственное средство для лечения аффективных расстройств, содержащее терапевтически эффективное количество соединения, представленного общей формулой I где Y означает С или СН; X означает S; m означает 1 или 2; р означает 0, 1, 2, 3, 4, 5, 6, 7 или 8; q означает 0, 1, 2, 3 или 4; s означает 1 или 2; пунктирная линия означает возможную связь; каждый R1 независимо выбран из группы, представленной C1-6-алкилом, или два R1, присоединенные к...
Лекарственное средство, содержащее производное сульфопиранозилацилглицерина

Номер патента: 3094
Опубликовано: 26.12.2002
Авторы: Сато Нориюки, Сакагути Кенго, Сугавара Фумио, Ямазаки Такаюки, Масаки Казуйоси, Охта Кейсуке, Сахара Хироеки, Накаяма Котаро, Фудзита Тацуя
МПК: C07H 15/06, A61P 35/00, A61K 31/7032...
Метки: содержащее, производное, сульфопиранозилацилглицерина, лекарственное, средство
Формула / Реферат:
1. Лекарственное средство, полезное в качестве ингибитора ДНК-полимеразы или противоракового агента, содержащее в качестве активного ингредиента, по меньшей мере, одно соединение, выбранное из группы, состоящей из соединений, представленных общей формулой (1) где R101 представляет ацильный остаток ненасыщенной высшей жирной кислоты, a R102 представляет атом водорода или ацильный остаток ненасыщенной высшей жирной кислоты; и их фармацевтически...
Кристаллическая форма [3-(4,5-дигидро-3-изоксазолил)-2-метил-4-(метилсульфонил)фенил]-(5-гидрокси-1-метил-1h-пиразол-4-ил)метанона

Номер патента: 16821
Опубликовано: 30.07.2012
Авторы: Гебхардт Йоахим, Эрк Петер, Крёль Томас, Братц Маттиас, Сакселль Хейди Эмилия
МПК: C07D 413/10, A01N 43/80
Метки: кристаллическая, форма, 3-(4,5-дигидро-3-изоксазолил)-2-метил-4-(метилсульфонил)фенил]-(5-гидрокси-1-метил-1h-пиразол-4-ил)метанона
Формула / Реферат:
1. Кристаллическая форма I [3-(4,5-дигидро-3-изоксазолил)-2-метил-4-(метилсульфонил)фенил]-(5-гидрокси-1-метил-1H-пиразол-4-ил)метанона, которая на рентгеновской порошковой дифрактограмме при 30°С с использованием CuKα облучения демонстрирует по крайней мере 5 из следующих изображений, указанных как 2θ значения: 7,7±0,2°, 10,3±0,2°, 12,7±0,2°, 13,8±0,2°, 16,9±0,2°, 18,8±0,2°, 20,7±0,2°, 22,2±0,2°, 28,0±0,2° и 31,4±0,2°.2....
Фармацевтическая композиция и комбинированное лекарственное средство, содержащее ингибиторы hmg-coa-редуктазы и фосфодиэстеразы 4, и их применение для лечения воспалительных заболеваний легких

Номер патента: 21461
Опубликовано: 30.06.2015
Авторы: Воллин Штефан-Лутц, Вользен Андреа, Маркс Дегенхард, Браун Клеменс
МПК: A61K 31/343, A61K 31/277, A61K 31/22...
Метки: легких, содержащее, средство, фосфодиэстеразы, hmg-coa-редуктазы, фармацевтическая, воспалительных, композиция, заболеваний, комбинированное, лечения, лекарственное, ингибиторы, применение
Формула / Реферат:
1. Фармацевтическая композиция для профилактики или лечения хронических воспалительных заболеваний легких, которая содержит компонент (А) ингибитор фосфодиэстеразы 4 типа (ингибитор PDE4) и компонент (Б) ингибитор редуктазы 3-гидрокси-3-метилглутарил кофермента А (ингибитор HMG-CoA-редуктазы) и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество, где ингибитором PDE4 является...
Предыдущий патент: Способ прогнозирования эффективности лекарственных соединений для пациента
Следующий патент: Переработка углеводородного газа
Случайный патент: Устройство для вытягивания монокристаллов