Способ получения дабигатрана







Формула / Реферат
1. Способ получения дабигатрана формулы VIII

путем взаимодействия хлорида 4-этиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом, восстановления нитрогруппы с последующим взаимодействием с [(4-цианофенил)амино]уксусной кислотой, путем гидролиза и взаимодействия с карбонатом аммония и превращения в дабигатран с помощью реакции с гексил хлорформиатом, отличающийся тем, что продукт реакции хлорида 4-этиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом превращают в гидрохлорид с помощью раствора хлороводорода с получением соединения формулы III-HCl

нитрогруппу которого восстанавливают путем взаимодействия с дитионитом натрия с получением соединения формулы IV

которое подвергают взаимодействию с [(4-цианофенил)амино]уксусной и щавелевой кислотами, продукт данной реакции формулы VI-оксалат

затем гидролизуют и подвергают взаимодействию с карбонатом аммония с получением промежуточного продукта формулы VII-HCl

который затем превращают в дабигатран путем взаимодействия с гексил хлорформиатом.
2. Способ получения дабигатрана по п.1, отличающийся тем, что взаимодействие хлорида 4-метиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом осуществляют в присутствии основания при температуре 40-120°С и затем продукт превращают в гидрохлорид с помощью раствора хлороводорода с получением соединения формулы III-HCl.
3. Способ получения дабигатрана по п.1, отличающийся тем, что взаимодействие соединения формулы III-HCl с дитионитом натрия с получением соединения формулы IV осуществляют при температуре 20-100°С.
4. Способ получения дабигатрана по п.1, отличающийся тем, что взаимодействие соединения формулы IV с 4-цианофенилглицином и щавелевой кислотой осуществляют при температуре 40-120°С с получением соединения формулы VI-оксалат.
5. Гидрохлорид этил-3-[[4-(метиламино)-3-нитробензоил](пиридин-2-ил)амино]пропионата формулы III-HCl

представляющий собой промежуточный продукт для получения дабигатрана.
6. Способ получения гидрохлорида этил-3-[[4-(метиламино)-3-нитробензоил](пиридин-2-ил)амино]пропионата формулы III-HCl, отличающийся тем, что хлорид 4-метиламино-3-нитробензойной кислоты подвергают взаимодействию с этил-3-(пиридин-2-иламино)пропионатом в присутствии основания и продукт реакции превращают в гидрохлорид с помощью раствора хлороводорода.
7. Способ по п.6, отличающийся тем, что продукт реакции хлорида 4-этиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом в форме основания превращают в гидрохлорид с помощью раствора хлороводорода в органическом растворителе, выбранном из C3-C6 простых эфиров, кетонов, сложных эфиров и С1-С5 спиртов.
8. Способ по п.6, отличающийся тем, что продукт реакции хлорида 4-этиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом в форме основания превращают в гидрохлорид с помощью раствора хлороводорода в растворителе, выбранном из диэтилового эфира, этанола, этилацетата и ацетона.
9. Способ по п.6, отличающийся тем, что продукт реакции хлорида 4-этиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом в форме основания превращают в гидрохлорид с помощью раствора хлороводорода в диэтиловом эфире.
10. Способ по любому из пп.6-9, отличающийся тем, что используют органическое или неорганическое основание.
11. Способ по любому из пп.6-9, отличающийся тем, что органическое или неорганическое основание выбрано из аминов, алкоголятов, гидроксидов щелочных металлов, фосфатов и карбонатов.
12. Способ по любому из пп.6-9, отличающийся тем, что в качестве основания используют триэтиламин.
13. Способ по любому из пп.6-9, отличающийся тем, что получаемое соединение формулы III-HCl очищают кристаллизацией.
14. Способ по любому из пп.6-9, отличающийся тем, что получаемое соединение формулы III-HCl очищают кристаллизацией из смеси этанол-ацетонитрил.
15. Способ получения соединения формулы IV

представляющего собой промежуточный продукт для получения дабигатрана, отличающийся тем, что соединение формулы III-HCl подвергают взаимодействию с дитионитом натрия.
16. Способ по п.15, отличающийся тем, что взаимодействие соединения формулы III-HCl с дитионитом натрия проводят в смеси растворителей этанол-вода.
17. Способ получения [4-(цианофенил)амино]уксусной кислоты формулы V

представляющей собой промежуточный продукт для получения дабигатрана, отличающийся тем, что 4-цианоанилин взаимодействует с бромуксусной кислотой.
18. Оксалат 3-([2-[(4-цианофениламино)метил]-1-метил-1H-бензимидазол-5-карбонил]пиридин-2-иламино)этил пропионат формулы VI-оксалат

представляющий собой промежуточный продукт для получения дабигатрана.
19. Способ получения оксалата формулы VI-оксалат, отличающийся тем, что соединение IV подвергают взаимодействию с соединением формулы V и щавелевой кислотой.
20. Способ по п.19, отличающийся тем, что щавелевую кислоту используют в безводной форме либо в форме одного из гидратов.
21. Способ по п.19 или 20, отличающийся тем, что продукт очищают кристаллизацией.
22. Способ по пп.19, 20 или 21, отличающийся тем, что продукт очищают кристаллизацией с использованием растворителя, выбранного из этанола и этилацетата.
Текст
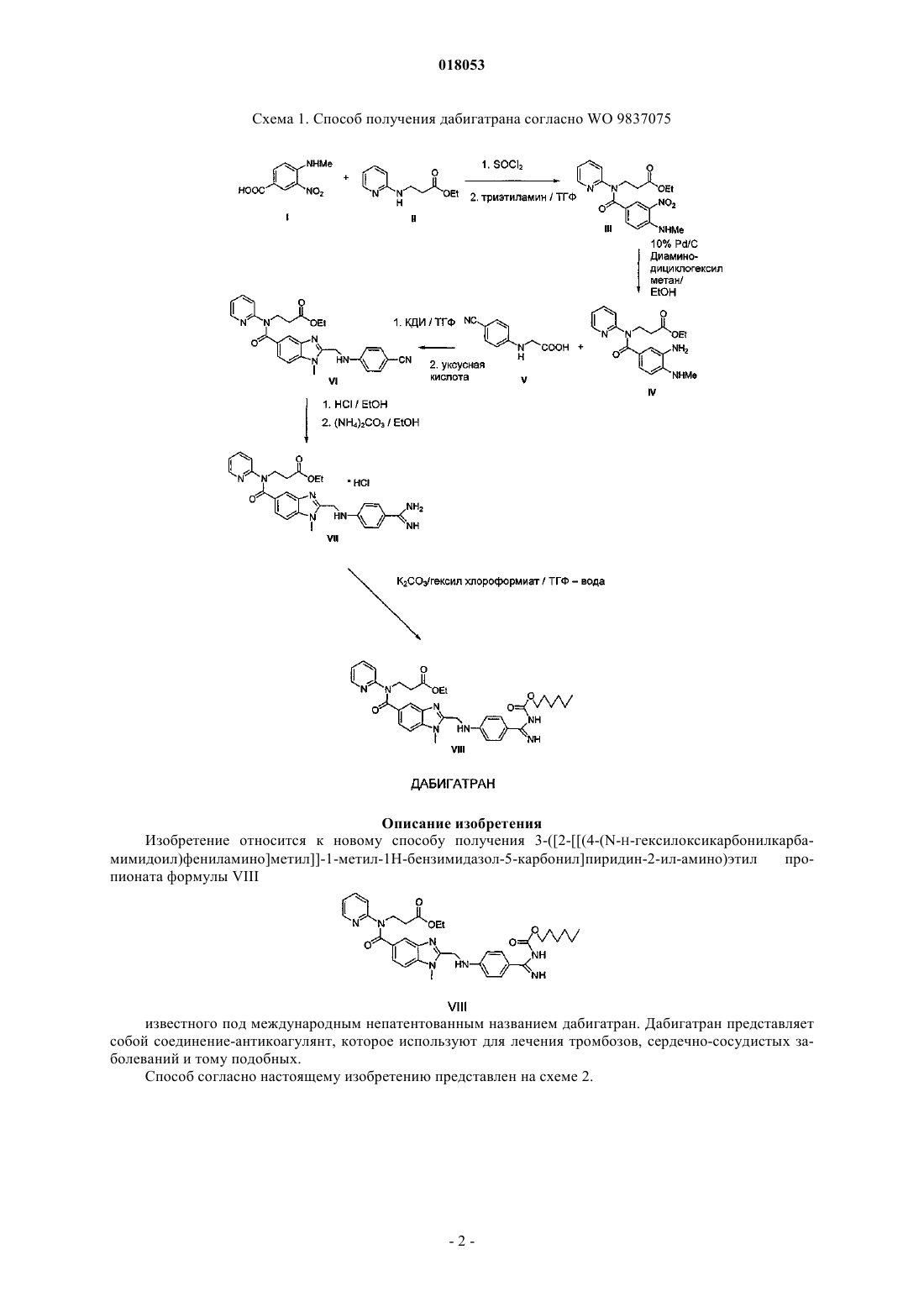
Способ получения дабигатрана формулы VIII, где продукт реакции хлорида 4-этиламино-3 нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом превращают в гидрохлорид с помощью раствора хлороводорода с получением соединения формулы III-HCl, где восстанавливают нитрогруппу посредством реакции с дитионитом натрия, и полученное соединение формулы IV подвергают взаимодействию с [(4-цианофенил)амино]уксусной кислотой и щавелевой кислотой, затем продукт этой реакции VI-оксалат гидролизуют и подвергают взаимодействию с карбонатом аммония с получением промежуточного продукта формулы VII-HCl,который далее превращают в дабигатран реакцией с гексил хлорформиатом. Область техники Изобретение относится к новому способу получения 3-([2-(4-(N-н-гексилоксикарбонилкарбамимидоил)фениламино]метил-1-метил-1H-бензимидазол-5-карбонил]пиридин-2-ил-амино)этил пропионата, известного под международным непатентованным именем дабигатран. Дабигатран представляет собой соединение-антикоагулянт, которое используют для лечения тромбозов, сердечно-сосудистых заболеваний и тому подобных. Предшествующий уровень техники Вещество впервые было описано в документе WO 9837075. Способ его приготовления в соответствии с вышеупомянутым документом показан на схеме 1. На первой стадии получения хлорид соединения I взаимодействует с соединением II в тетрагидрофуране с использованием триэтиламина в качестве основания. Продуктом реакции является соединениеIII. В процессе воспроизведения вышеупомянутого способа соединение III было получено в форме основания. Однако, исходя из нашего опыта и согласно WO 9837075, для дальнейшего применения в производстве высококачественных активных фармацевтических ингредиентов данное соединение требует сложной процедуры очистки, например, с помощью хроматографии. Следующая стадия приготовления представляет собой восстановление нитрогруппы до аминогруппы. В документе WO 9837075 указанную реакцию проводят с помощью каталитического гидрирования с использованием палладия на активном угле при высоком давлении. Однако этот процесс имеет высокие технологические требования и представляет угрозу для безопасности и риск загрязнения окружающей среды. Он требует использования высокого давления и, как следствие, специального оборудования. Используемые катализаторы токсичны и их стоимость вместе со стоимостью оборудования очень негативно отражается на выгодности процесса в целом. Другим недостатком способа является низкое качество продукта формулы IV, полученного этим способом. В соответствии с этим способом продукт получают в форме масла с содержанием примесей от 20 до 40%. Продукт IV, по крайней мере, согласно известному процессу, следует использовать на следующей стадии в загрязненной форме. Открытие альтернативного способа проведения этой реакции определенно существенно повлияет на увеличение эффективности производства дабигатрана. На третьей стадии соединение IV взаимодействует с соединением V с получением соединения VI. Согласно WO 9837075 соединение V получают путем взаимодействия 4-цианофенил анилина с хлоруксусной кислотой в присутствии основания. В процессе воспроизведения способа согласно WO 9837075 авторы настоящего изобретения обнаружили, что реакция протекает с низким выходом (около 50%). Продуктом способа, осуществляемого согласно WO 9837075, является соединение VI в форме основания или ацетата. Оба этих продукта требуют хроматографической очистки, которую сложно реализовывать в промышленных масштабах. На следующей стадии проводят кислотный гидролиз нитрильной группы соединения VI и осуществляют реакцию с карбонатом аммония с получением соединения VII. Последней стадией является взаимодействие промежуточного соединения VII с гексил хлороформиатом с получением дабигатрана. Схема 1. Способ получения дабигатрана согласно WO 9837075 Описание изобретения Изобретение относится к новому способу получения 3-([2-(4-(N-H-гексилоксикарбонилкарбамимидоил)фениламино]метил-1-метил-1H-бензимидазол-5-карбонил]пиридин-2-ил-амино)этил пропионата формулы VIII известного под международным непатентованным названием дабигатран. Дабигатран представляет собой соединение-антикоагулянт, которое используют для лечения тромбозов, сердечно-сосудистых заболеваний и тому подобных. Способ согласно настоящему изобретению представлен на схеме 2. Схема 2. Способ получения дабигатрана согласно настоящему изобретению На первой стадии хлорид соединения I взаимодействует с соединением II в тетрагидрофуране в присутствии основания. Используемое основание выбирают из аминов, алкоголятов, гидроксидов, фосфатов и карбонатов. Триэтиламин представляется наиболее подходящим основанием. Согласно настоящему изобретению, в результате взаимодействия с раствором хлористого водорода в органическом растворителе получают соединение III в форме гидрохлорида. Соединение может очищать простой кристаллизацией, например, из смеси этанол/ацетонитрил, что приводит к высокочистому продукту, причем часто содержание примесей составляет менее 1%, например, содержание целевого продукта составляет 99,3%. Хлористый водород растворяют в органическом растворителе, выбранном, например, из группы простых эфиров, сложных эфиров, кетонов и спиртов. Диэтиловый эфир является наиболее подходящим растворителем. Следующей стадией получения является восстановление нитрогруппы до аминогруппы. Способ согласно настоящему изобретению осуществляют в смеси растворителей этанол и вода. Реагентом является дитионит натрия. Нет необходимости проводить реакцию при повышенном давлении или очень высокой температуре. Данная процедура гораздо менее дорогостоящая и более выгодная с экономической точки зрения. Соединение III вводят в реакцию в форме гидрохлорида, что оказывает положительное влияние на протекание реакции и чистоту продукта. Комбинируя изменение качества и состава исходного соединения с изменением способа восстановления, возможно получать продукт IV с минимальным содержанием примесей, то есть менее 5%, предпочтительно менее 1%. На третьей стадии соединение IV взаимодействует с соединением формулы V с получением соединения формулы VI. В рамках способа согласно настоящему изобретению, соединение VI получают в виде соли с щавелевой кислотой. Эту соль можно легко очищать дальнейшей кристаллизацией, что позволяет получать новое промежуточное соединение VI с выходом примерно 80-90%, что приемлемо для промышленного производства. Эту кристаллизацию можно проводить из полярного протонного органического растворителя,предпочтительно из низших С 1-С 5 спиртов, например из этилового спирта. В рамках способа по настоящему изобретению 4-цианофенил анилин подвергают взаимодействию с бромуксусной кислотой. Использование указанного реагента существенно увеличивает выход, примерно до 85-90%, по сравнению с 49% в случае взаимодействия с хлоруксусной кислотой. Возможно, это вызвано тем, что бромид-ион является лучшей уходящей группой в нуклеофильных реакциях по сравнению с хлорид-ионом, что в значительной степени отражается на выходе реакции в случае взаимодействия хлоруксусной или бромуксусной кислоты, соответственно, с относительно слабым нуклеофилом, таким как 4-цианоанилин. Предложенный способ получения позволит реализовать промышленно осуществимое производство дабигатрана. Соединение VI, полученное вышеизложенным способом, может быть в дальнейшем обработано способом согласно WO 9387075. На следующей стадии проводят кислотный гидролиз нитрильной группы соединения VI и взаимодействие с карбонатом аммония с получением соединения формулы VII. Применение исходного соединения VI в форме оксалата в соответствии с настоящим изобретением приводит к соединению VII в форме гидрохлорида. Этот подход оказывает существенное влияние на чистоту продукта, позволяя получить промежуточное соединение VII в высокочистом виде. Последняя стадия представляет собой взаимодействие промежуточного соединения VII с гексил хлорформатом с получением а. Способ согласно настоящему изобретению позволяет получать высококачественный продукт с низким содержанием примесей и относительно высоким выходом. Получение промежуточных соединений III и VI в форме солей значительно упрощает процедуры очистки при производстве дабигатрана; хроматографическая очистка, упомянутая в WO 9387075, не может быть использована при производстве в промышленных масштабах. Указанная процедура значительно увеличит выход процесса. Восстановление нитрогруппы соединения III с помощью дитионита вместо описанного ранее каталитического гидрирования с высокими требованиями к технологии и материалам представляет собой существенное упрощение и положительно повлияет на выгодность процесса в целом. Изобретение показано далее в нескольких примерах. Пример 1. Получение гидрохлорида этил 3-4-(метиламино)-3-нитробензоил](пиридин-2-ил)амино] пропионата (III). Компоненты: Промежуточное соединение I: 100 г-0,51 моль. Промежуточное соединение II: 94 г- 0,48 моль. Хлористый тионил: 850 мл. Сухой триэтиламин: 99 мл. Диметил формамид (сухой): 4 мл. ТГФ: 650 мл. Хлороформ: 500 мл. 850 мл хлористого тионила и 4 мл сухого ДМФ добавляли к 100 г соединения I в инертной атмосфере. Смесь доводили до кипения. В ходе нагревания соединение I растворялось в течение нескольких минут, и образовывался раствор темно-коричневого цвета. Затем раствор кипятили с обратным холодильником в течение еще 40-45 мин. После этого отгоняли избыточный хлористый тионил. К коричневому остатку в инертной атмосфере добавляли 200 мл сухого толуола и удаляли толуол дистилляцией. Данную операцию повторяли ещ раз. Полученный кристаллический коричневый остаток растворяли в сухом ТГФ (600 мл; в инертной атмосфере) при повышенной температуре. После охлаждения раствора кислого хлорида I до температуры 40 С добавляли сухой триэтиламин. К этому раствору по каплям добавляли раствор соединения II в сухом ТГФ (50 мл). Время добавления по каплям составляло 15 мин. В процессе прибавления соединения II реакционную смесь охлаждали на водяной бане. Это сопровождалось понижением температуры до 30 С. Затем реакционную смесь перемешивали без нагревания в течение 2 ч. После удаления гидрохлорида триэтиламина упаривали ТГФ. Остаток от упаривания растворяли в 500 мл хлороформа и встряхивали с водой. Отделенный органический слой встряхивали с 2 М HCl и затем с водой. Органический слой сушили над сульфатом натрия и упаривали хлороформ. Выход: неочищенный продукт: 200 г (96%); ВЭЖХ: 88,2%; цвета темного меда. Из этого соединения готовят гидрохлорид. Приготовление гидрохлорида: этилацетат: 850 мл HCl/Et2O. Неочищенный продукт растворяли в 850 мл этилацетата при комнатной температуре. К раствору медленно по каплям добавляли 95 мл раствора HCl в Et2O при интенсивном перемешивании. В процессе перемешивания выделялся желтый осадок. После охлаждения в холодильнике осадок отфильтровывали и сушили (в течение ночи при комнатной температуре, затем в вакууме при 55-75 С, затем в вакууме без нагревания в течение нескольких дней (при этом вес остается практически неизменным). Выход: Неочищенный гидрохлорид: 161,3 г (77,4%); ВЭЖХ: 93,3%. Кристаллизация гидрохлорида: Неочищенный гидрохлорид кристаллизуют из смеси денатурированного несухого этанола с ацетонитрилом в соотношении 9:1. Выход: неочищенный гидрохлорид: 120,2 г (61,6%); ВЭЖХ: 99,3%. Пример 2. Получение этил 3-3-амино-4-(метиламино)бензоил](пиридин-2-ил)амино]пропионата Компоненты: Промежуточное соединение III: 120 г- 0,29 моль; Дитионит натрия: 246 г - 1,2 моль; Этанол: 750 мл; Этилацетат: 600 мл. Соединение III помещали в 1500 мл смеси этанол - вода с соотношении 1:1 и нагревали до 50 С. К полученному таким образом раствору быстро и при интенсивном перемешивании добавляли дитионит натрия. После того, как исходное соединение прореагировало, реакционную смесь концентрировали на вакуумном испарителе. После отделения масла концентрирование прекращали и продукт экстрагировали этилацетатом. После высушивания над сульфатом натрия растворитель упаривали. Продукт получали в виде коричневой, очень вязкой жидкости. Выход: 82 г (81.3%); ВЭЖХ: 95%. Пример 3. 3.2.3. Приготовление [(4-цианофенил)амино]уксусной кислоты (V).G: 211,7 г-1,5 моль; Гидрокарбонат натрия: 35 г, 0,42 моль. Исходные соединения F и G смешивали в 1250 мл воды и полученную суспензию помещали в баню, нагретую до 100-110 С. После трех часов нагревания реакционный сосуд удаляли из бани, охлаждали в холодильнике и отфильтровывали выделившееся вещество. Соединение сушили в вакуумном сушильном шкафу при температуре 100 С. Выход: неочищенный продукт: 122 г (92,8%); ВЭЖХ: 97%. Неочищенный продукт очищали, переводя в натриевую соль с помощью водного раствора гидрокарбоната натрия с последующим закислением. Кислоту получали с использованием разбавленной соляной кислоты (1:1). После фильтрования и промывания водой продукт сушили в вакуумном сушильном шкафу (105 С). Выход: оищенный продукт: 115 г (88%); ВЭЖХ: 99,1%, содержание воды: 0,13%; сульфатная зола: 1,8% Пример 4. Получение оксалата 3-([2-[(4-цианофенил амино)метил]-1-метил-1H-бензимидазол-5 карбонил]пиридин-2-ил-амино)этил пропионата (VI). Компоненты. Промежуточное соединение IV: 82,5 г-0,24 моль; Промежуточное соединение V: 46,4 г - 0,26 моль; 1,1'-карбонилдиимидазол (КДИ): 42,2 г-0,26 моль. Методика. Соединение V и 1,1'-карбонилдиимидазол помещают колбу, продутую инертным газом. К смеси добавляют 2000 мл сухого ТГФ и кипятят в течение 40 мин с обратным холодильником, оснащенным хлоркальциевой трубкой. Через 40 мин к реакционной смеси добавляют раствор соединения IV в 330 мл сухого ТГФ и смесь кипятят ещ 5 ч. По истечении времени реакционную смесь слегка охлаждают и удаляют ТГФ испарением в вакууме. К коричневому, похожему на мед остатку добавляют 1000 мл ледяной уксусной кислоты и полученный раствор кипятят 1 ч. Кислоту упаривают, и полученный остаток растворяют в дихлорметане (850 мл) и встряхивают с водой. Отделившийся органический слой сушат над сульфатом натрия и упаривают растворитель. Получают темно-коричневый, похожий на мед продукт. Выход: 119 г (92% в пересчете на ацетат); ВЭЖХ: 82% Приготовление оксалата соединения VI. Неочищенный продукт (119 г) растворяют при кипячении в 650 мл этилацетата. Раствор охлаждают до комнатной температуры и добавляют по каплям при интенсивном перемешивании раствор дигидрата щавелевой кислоты (27,1 г) в 400 мл этилацетата. Сразу выделяется слегка бежевый осадок. Кристаллы отфильтровывают, промывают этанолом и сушат. Выход неочищенного оксалата: 102 г (70%); ВЭЖХ: 90,2%. Кристаллизация неочищенного оксалата соединения VI. 101 г неочищенного продукта растворяют при кипячении в 4100 мл этанола. Раствор охлаждают на водяной бане при одновременном перемешивании. Кристаллическую суспензию охлаждают в холодильнике и затем отфильтровывают кристаллы. Выход кристаллизованного оксалата: 82,5 г (57,2%); ВЭЖХ: 97,8%. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения дабигатрана формулы VIII путем взаимодействия хлорида 4-этиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом, восстановления нитрогруппы с последующим взаимодействием с [(4-цианофенил) амино]уксусной кислотой, путем гидролиза и взаимодействия с карбонатом аммония и превращения в дабигатран с помощью реакции с гексил хлорформиатом, отличающийся тем, что продукт реакции хлорида 4-этиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом превращают в гидрохлорид с помощью раствора хлороводорода с получением соединения формулы III-HCl нитрогруппу которого восстанавливают путем взаимодействия с дитионитом натрия с получением соединения формулы IV которое подвергают взаимодействию с [(4-цианофенил)амино]уксусной и щавелевой кислотами,продукт данной реакции формулы VI-оксалат затем гидролизуют и подвергают взаимодействию с карбонатом аммония с получением промежуточного продукта формулы VII-HCl который затем превращают в дабигатран путем взаимодействия с гексил хлорформиатом. 2. Способ получения дабигатрана по п.1, отличающийся тем, что взаимодействие хлорида 4 метиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом осуществляют в присутствии основания при температуре 40-120 С и затем продукт превращают в гидрохлорид с помощью раствора хлороводорода с получением соединения формулы III-HCl. 3. Способ получения дабигатрана по п.1, отличающийся тем, что взаимодействие соединения формулы III-HCl с дитионитом натрия с получением соединения формулы IV осуществляют при температуре 20-100 С. 4. Способ получения дабигатрана по п.1, отличающийся тем, что взаимодействие соединения формулы IV с 4-цианофенилглицином и щавелевой кислотой осуществляют при температуре 40-120 С с получением соединения формулы VI-оксалат. 5. Гидрохлорид этил-3-4-(метиламино)-3-нитробензоил](пиридин-2-ил)амино]пропионата формулы III-HCl представляющий собой промежуточный продукт для получения дабигатрана. 6. Способ получения гидрохлорида этил-3-4-(метиламино)-3-нитробензоил](пиридин-2-ил)амино] пропионата формулы III-HCl, отличающийся тем, что хлорид 4-метиламино-3-нитробензойной кислоты подвергают взаимодействию с этил-3-(пиридин-2-иламино)пропионатом в присутствии основания и продукт реакции превращают в гидрохлорид с помощью раствора хлороводорода. 7. Способ по п.6, отличающийся тем, что продукт реакции хлорида 4-этиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом в форме основания превращают в гидрохлорид с помощью раствора хлороводорода в органическом растворителе, выбранном из C3-C6 простых эфиров,кетонов, сложных эфиров и С 1-С 5 спиртов. 8. Способ по п.6, отличающийся тем, что продукт реакции хлорида 4-этиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом в форме основания превращают в гидрохлорид с помощью раствора хлороводорода в растворителе, выбранном из диэтилового эфира, этанола, этилацетата и ацетона. 9. Способ по п.6, отличающийся тем, что продукт реакции хлорида 4-этиламино-3-нитробензойной кислоты с этил-3-(пиридин-2-иламино)пропионатом в форме основания превращают в гидрохлорид с помощью раствора хлороводорода в диэтиловом эфире. 10. Способ по любому из пп.6-9, отличающийся тем, что используют органическое или неорганическое основание. 11. Способ по любому из пп.6-9, отличающийся тем, что органическое или неорганическое основание выбрано из аминов, алкоголятов, гидроксидов щелочных металлов, фосфатов и карбонатов. 12. Способ по любому из пп.6-9, отличающийся тем, что в качестве основания используют триэтиламин. 13. Способ по любому из пп.6-9, отличающийся тем, что получаемое соединение формулы III-HCl очищают кристаллизацией. 14. Способ по любому из пп.6-9, отличающийся тем, что получаемое соединение формулы III-HCl очищают кристаллизацией из смеси этанол-ацетонитрил. 15. Способ получения соединения формулы IV представляющего собой промежуточный продукт для получения дабигатрана, отличающийся тем,что соединение формулы III-HCl подвергают взаимодействию с дитионитом натрия. 16. Способ по п.15, отличающийся тем, что взаимодействие соединения формулы III-HCl с дитионитом натрия проводят в смеси растворителей этанол-вода. 17. Способ получения [4-(цианофенил)амино]уксусной кислоты формулы V представляющей собой промежуточный продукт для получения дабигатрана, отличающийся тем,что 4-цианоанилин взаимодействует с бромуксусной кислотой. 18. Оксалат 3-([2-[(4-цианофениламино)метил]-1-метил-1H-бензимидазол-5-карбонил]пиридин-2 иламино)этил пропионат формулы VI-оксалат представляющий собой промежуточный продукт для получения дабигатрана. 19. Способ получения оксалата формулы VI-оксалат, отличающийся тем, что соединение IV подвергают взаимодействию с соединением формулы V и щавелевой кислотой. 20. Способ по п.19, отличающийся тем, что щавелевую кислоту используют в безводной форме либо в форме одного из гидратов. 21. Способ по п.19 или 20, отличающийся тем, что продукт очищают кристаллизацией. 22. Способ по пп.19, 20 или 21, отличающийся тем, что продукт очищают кристаллизацией с использованием растворителя, выбранного из этанола и этилацетата.
МПК / Метки
МПК: C07D 213/74, C07D 401/12
Метки: способ, получения, дабигатрана
Код ссылки
<a href="https://eas.patents.su/10-18053-sposob-polucheniya-dabigatrana.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения дабигатрана</a>
Способ получения дихлорпропанола, способ получения эпихлоргидрина, способ получения эпоксидных смол и применение оборудования, обладающего коррозионной стойкостью, в способе получения дихлорпропанола

Номер патента: 14241
Опубликовано: 29.10.2010
Авторы: Краффт Филипп, Сметс Валентин, Бальтазар Доминик, Франк Кристиан, Жильбо Патрик
МПК: C07C 29/62, B01J 19/02, C07C 31/36...
Метки: стойкостью, получения, обладающего, применение, оборудования, способе, дихлорпропанола, смол, эпоксидных, коррозионной, эпихлоргидрина, способ
Формула / Реферат:
1.Способ получения дихлорпропанола, содержащий:(a) стадию, на которой глицерин или сложный эфир глицерина или их смесь вводят во взаимодействие с агентом хлорирования, содержащим хлороводород,(b) по меньшей мере одну другую стадию, осуществляемую на оборудовании, выполненном или имеющем покрытие из материалов, обладающих стойкостью по отношению к агенту хлорирования, в условиях осуществления этой стадии,причем другая стадия является стадией...
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Ларкин Джон Патрик, Колладан Колетт, Крок Вероник, Руссель Патрик
МПК: C07D 487/04
Метки: диазепин-1-карбоновой, кислоты, производные, 10-диоксо-6н-пиридазино, получения, соединений, способ, терапевтически, 1,2, применение, октагидро-6, 1,2-а, активных
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...
Цеолитный катализатор l-типа, способ его получения, способ получения ароматических углеводородов, способ получения бензина

Номер патента: 3559
Опубликовано: 26.06.2003
Авторы: Сугимото Митио, Иннес Роберт А., Фукунага Тецуя
МПК: B01J 29/61, C07C 5/41, C10G 35/095...
Метки: получения, ароматических, углеводородов, бензина, цеолитный, l-типа, способ, катализатор
Формула / Реферат:
1. Цеолитный катализатор L-типа, который получают при нанесении на цеолит L-типа платинового компонента, одного или более галогеновых компонентов и одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, при этом наносимое количество одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, находится в интервале от 0,001 до 3 мас.% из расчета на общую массу катализатора, молярное отношение...
Катализатор для получения сложных эфиров,способ получения сложного эфира и способ получения сложного полиэфира с участием такого катализатора

Номер патента: 11171
Опубликовано: 27.02.2009
Авторы: Макинтош Кэлам Гарри, Хэнратти Алан Джозеф, Партридж Мартин Грэхэм
МПК: C08G 63/85, B01J 31/04
Метки: катализатор, эфира, получения, сложных, катализатора, способ, такого, участием, полиэфира, эфиров,способ, сложного
Формула / Реферат:
1. Катализатор для получения сложного эфира в реакции этерификации, состоящий из продукта взаимодействия: a) соединения титана, циркония или гафния; b) 2-оксикарбоновой кислоты; c) четвертичного аммониевого соединения, выбранного из группы, состоящей из гидроксида тетраэтиламмония и гидроксида тетраметиламмония, и d) соединения цинка. 2. Катализатор по п. 1, в котором соединением титана, циркония или гафния является алкоголят, имеющий формулу...
Способ получения композиции сложного полиэфира, полученная композиция, содержащая ее пленка, раствор для получения композиции и способ его получения

Номер патента: 14016
Опубликовано: 30.08.2010
Авторы: Зайдель Экхард, Отто Бригитта, Хельдманн Карл-Хайнц
МПК: C08K 5/098, C08K 5/00, C08J 5/18...
Метки: сложного, полученная, полиэфира, раствор, способ, содержащая, композиция, композиции, получения, пленка
Формула / Реферат:
1. Способ получения композиции сложного полиэфира, включающий следующие стадии:A) этерификацию дикарбоновой кислоты алкандиолом или переэтерификацию диалкилового эфира дикарбоновой кислоты алкандиолом;B) предварительную поликонденсацию полученного диалкилового эфира дикарбоновой кислоты до форполиконденсата;C) поликонденсацию в расплаве форполиконденсата до сложного полиэфира, причем смесь, которую подвергают поликонденсации, содержит по меньшей...
Предыдущий патент: Диспергаторы асфальтенов на основе фосфоновых кислот
Следующий патент: Способ увеличения адсорбции ингибитора на участке ствола скважины
Случайный патент: Кристаллический несольватированный гидрохлорид 1-(4-(2- пиперидинилэтокси)фенокси)-2-(4-метансульфонилфенил)-6-гидроксинафталина