Парентеральные композиции пептидов для лечения системной красной волчанки
Номер патента: 9123
Опубликовано: 26.10.2007
Авторы: Клингер Эти, Нафтали Эсмира, Кохен-Веред Шарон, Гилберт Адриан, Вайнштейн Вера























Формула / Реферат
1. Фармацевтическая композиция, содержащая водный носитель, от 0,1 до 20 мг/мл композиции фармацевтически приемлемой соли
a) пептида, содержащего по меньшей мере 12 и самое большее 30 следующих друг за другом аминокислот, имеющих последовательность, соответствующую:
(i) последовательности аминокислот, обнаруживаемой в определяющей комплементарность области (CDR) тяжелой или легкой цепи человеческого моноклонального анти-ДНК 16/6 Id-антитела, или
(ii) последовательности аминокислот, обнаруживаемой в определяющей комплементарность области (CDR) тяжелой или легкой цепи патогенного моноклонального анти-ДНК-антитела, которое индуцирует ответ, подобный заболеванию системной красной волчанкой (СКВ), у мышей, или
b) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательность
(i) TGYYX1X2X3X4X5QSPEKSLEWIG (SEQ ID NO: 11),
где X1 означает Met, Ala или Val; X2 означает Gln, Asp, Glu или Arg; X3 означает Trp или Ala; X4 означает Val или Ser и X5 означает Lys, Glu или Ala;
(ii) EINPSTGGX6X7X8X9X10X11X12KAKAT (SEQ ID NO: 12),
где Х6 и Х7, каждый означает Thr, Val или Ala; Х8 означает Tyr или Phe; X9 означает Asn или Asp; X10 означает Gln или Glu; Х11 означает Lys или Glu и Х12 означает Phe или Tyr;
(iii) YYCARX13X14X15X16PYAX17X18YWGQGS (SEQ ID NO: 13),
где X13 означает Phe, Thr или Gly; X14 означает Leu, Ala или Ser; X15 означает Trp или Ala; X16 означает Glu или Lys; X17 означает Met или Ala и Х18 означает Asp, Lys или Ser;
(iv) GYNX19X20X21X22X23X24SHGX25X26LEWIG (SEQ ID NO: 14),
где X19 означает Met или Ala; X20 означает Asn, Asp или Arg; X21 означает Trp или Ala; X22 означает Val или Ser; X23 означает Lys или Glu; X24 означает Gln или Ala; X25 означает Lys или Glu и Х26 означает Ser или Ala;
(v) YYCARX27X28X29YGX30X31X32GQTL (SEQ ID NO: 15),
где Х27 означает Ser или Phe; X28 означает Gly или Ala; X29 означает Arg, Ala или Glu; X30 означает Asn или Asp; X31 означает Tyr или Phe и Х32 означает Trp, His или Ala;
(vi) X33YYWSWIX34QX35PX36X37GX38EWIG (SEQ ID NO: 16),
где X33 означает Gly или Thr Gly; X34 означает Arg или Lys; X35 означает Pro или Ser; X36 означает Gly или Glu; X37 означает Lys или Asp и Х38 означает Glu, Leu или Ser;
(vii) YYCARX39LLX40X41X42X43X44DVDYX45GX46DV (SEQ ID NO: 17),
где X39 означает Gly или Phe; X40 означает Arg или Ala; X41 означает Gly или Ala; X42 означает Gly или Ala; X43 означает Trp или Ala; X44 означает Asn или Ala; X45 означает Tyr или Trp; Х46 означает Met или Gln;
(viii) FSGYYWS (SEQ ID NO: 8);
(ix) EINHSGSTNYKTSLKS (SEQ ID NO: 9) или
(x) GLLRGGWNDVDYYYGMDV (SEQ ID NO: 10), или
c) пептида, содержащего следующие друг за другом аминокислоты, имеющие любую последовательность из а) и b) или по меньшей мере две последовательности из последовательностей (a)(i), (a)(ii) и (b)(i)-(b)(х), или
d) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательность, содержащую по меньшей мере две идентичные последовательности, включенные в (a)(i), (a)(ii) и (b)(i)-(b)(x); и
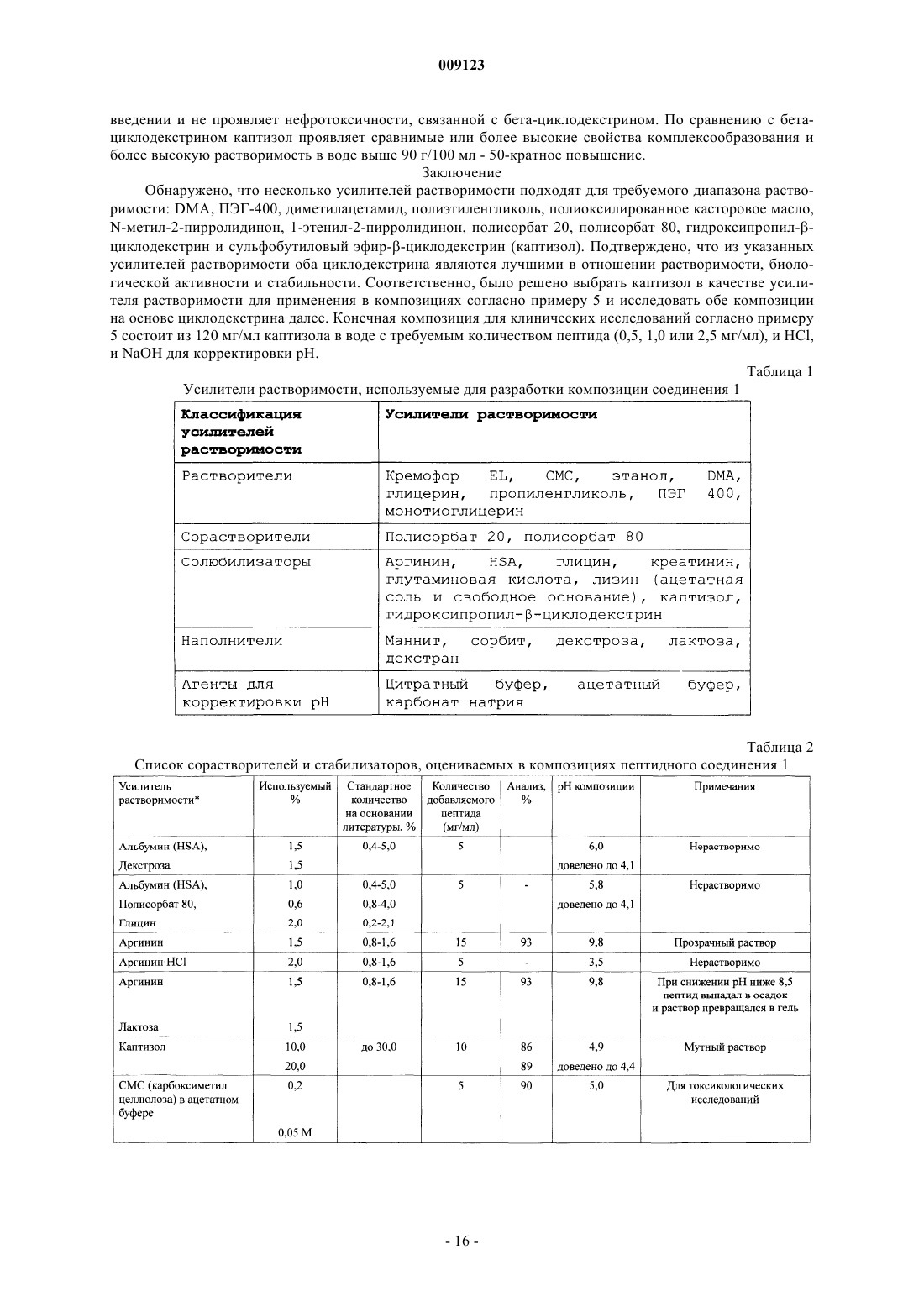
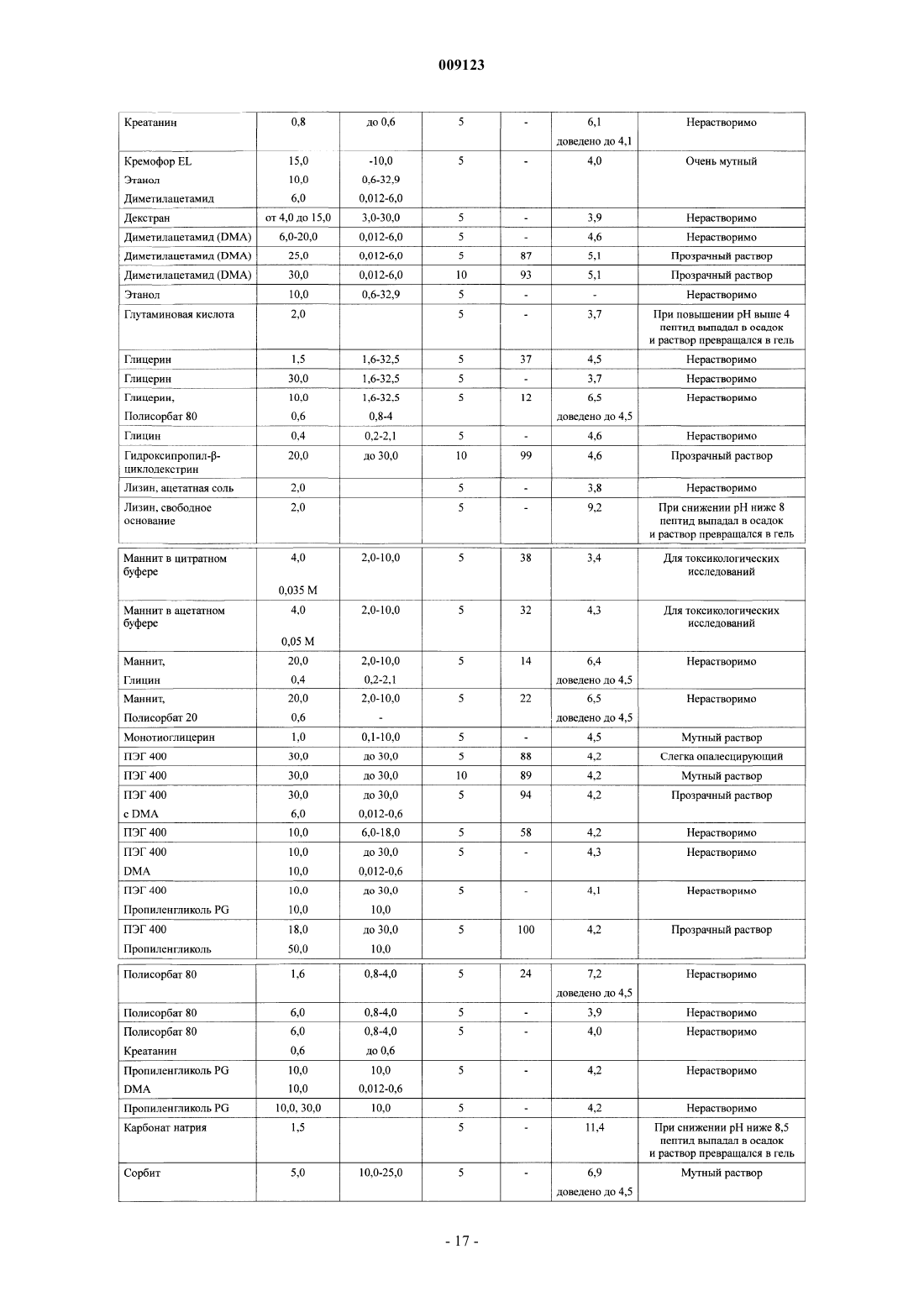
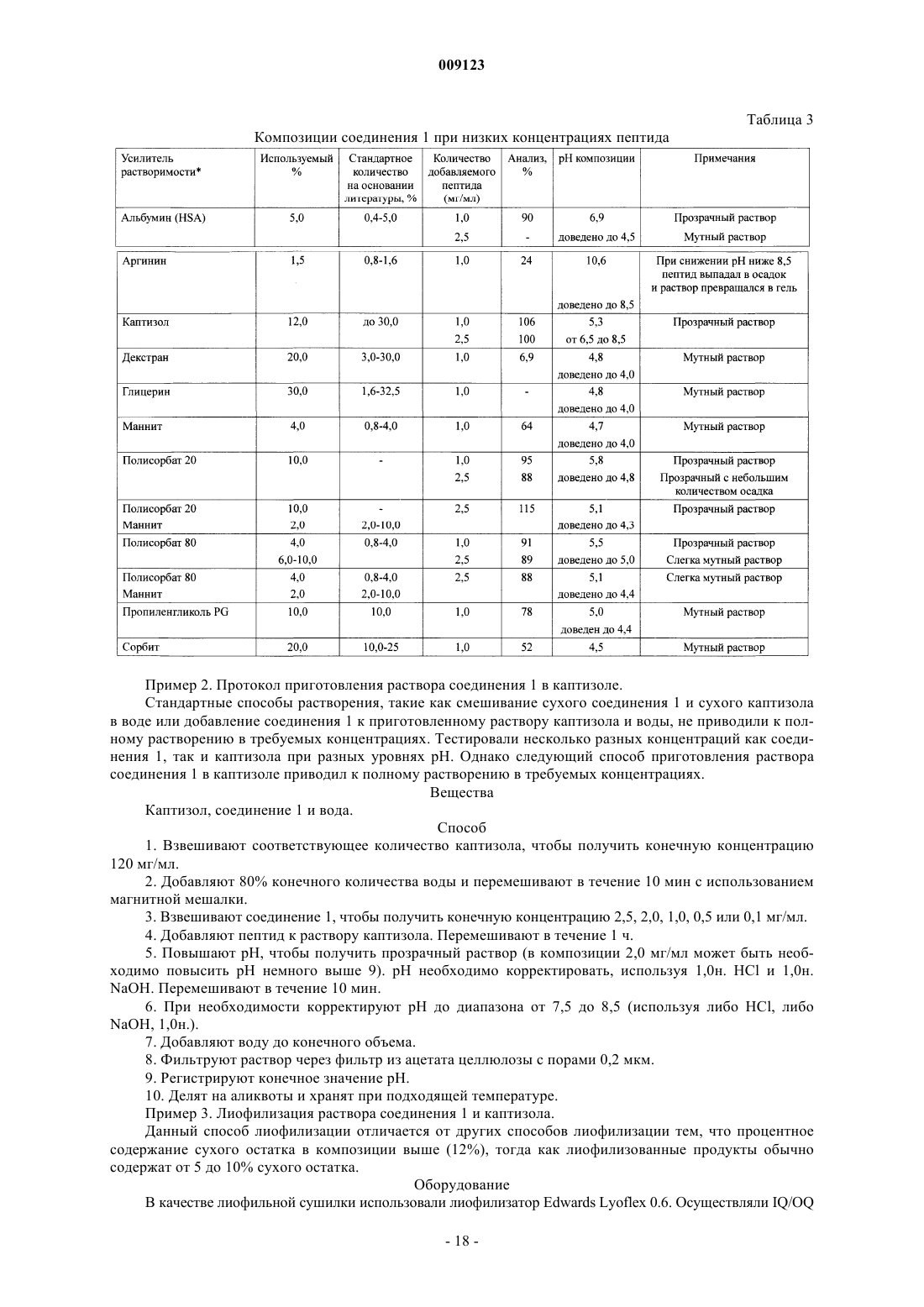
усилитель растворимости, выбранный из группы, состоящей из диметилацетамида, полиэтиленгликоля, полиоксилированного касторового масла, N-мeтил-2-пиppoлидинoнa, 1-этенил-2-пирролидинона, сложных эфиров полиоксиэтиленсорбитана и замещенного b-циклодекстрина,
в которой и пептид, и усилитель растворимости растворены в водном носителе; и
при этом композиция имеет рН от 4 до 9.
2. Фармацевтическая композиция по п.1, в которой пептид имеет последовательность, выбранную из группы, состоящей из
NH2-Thr Gly Tyr Tyr Met Gln Trp Val Lys Gln Ser Pro Glu Lys Ser Leu Glu-Trp Ile Gly-COOH (SEQ ID NO: 1);
NH2-Glu Ile Asn Pro Ser Thr Gly Gly Thr Thr Tyr Asn Gln Lys Phe Lys Ala Lys Ala Thr-COOH (SEQ ID NO: 2);
NH2-Tyr Tyr Cys Ala Arg Phe Leu Trp Glu Pro Tyr Ala Met Asp Tyr Trp Gly Gln Gly Ser-COOH (SEQ ID NO: 3);
NH2-Gly Tyr Asn Met Asn Trp Val Lys Gln Ser His Gly Lys Ser Leu Glu Trp Ile Gly-COOH (SEQ ID NO: 4);
NH2-Tyr Tyr Cys Ala Arg Ser Gly Arg Tyr Gly Asn Tyr Trp Gly Gln Thr Leu-COOH (SEQ ID NO: 5);
NH2-Gly Tyr Tyr Trp Ser Trp Ile Arg Gln Pro Pro Gly Lys Gly Glu Glu Trp Ile Gly-COOH (SEQ ID NO: 6);
NH2-Tyr Tyr Cys Ala Arg Gly Leu Leu Arg Gly Gly Trp Asn Asp Val Asp Tyr Tyr Gly Met Asp Val-COOH (SEQ ID NO: 7);
NH2-Phe Ser Gly Tyr Tyr Тrp Ser-COOH (SEQ ID NO: 8);
NH2-Glu Ile Asn His Ser Gly Ser Thr Asn Tyr Lys Thr Ser Leu Lys Ser-COOH (SEQ ID NO: 9) и
NH2-Gly Leu Leu Arg Gly Gly Trp Asn Asp Val Asp Tyr Tyr Туr Gly Met Asp Val-COOH (SEQ ID NO: 10).
3. Фармацевтическая композиция по п.1, в которой пептид содержит следующие друг за другом аминокислоты, имеющие последовательность
X33YYWSWIX34QX35PX36X37GX38EWIG (SEQ ID NO: 16),
где Х33 означает Gly или Thr Gly; X34 означает Arg или Lys; Х35 означает Pro или Ser; X36 означает Gly или Glu; X37 означает Lys или Asp и Х38 означает Glu, Leu или Ser.
4. Фармацевтическая композиция по любому из пп.1-3, в которой усилителем растворимости является замещенный b-циклодекстрин.
5. Фармацевтическая композиция по п.4, в которой фармацевтически приемлемой солью является ацетатная соль, а замещенным b-циклодекстрином является гепта(сульфобутиловый эфир)-b-циклодекстрин.
6. Способ ослабления симптомов системной красной волчанки (СКВ) у человека, включающий в себя введение человеку фармацевтической композиции по любому из пп.1-5 в количестве, эффективном для ослабления симптомов СКВ у человека.
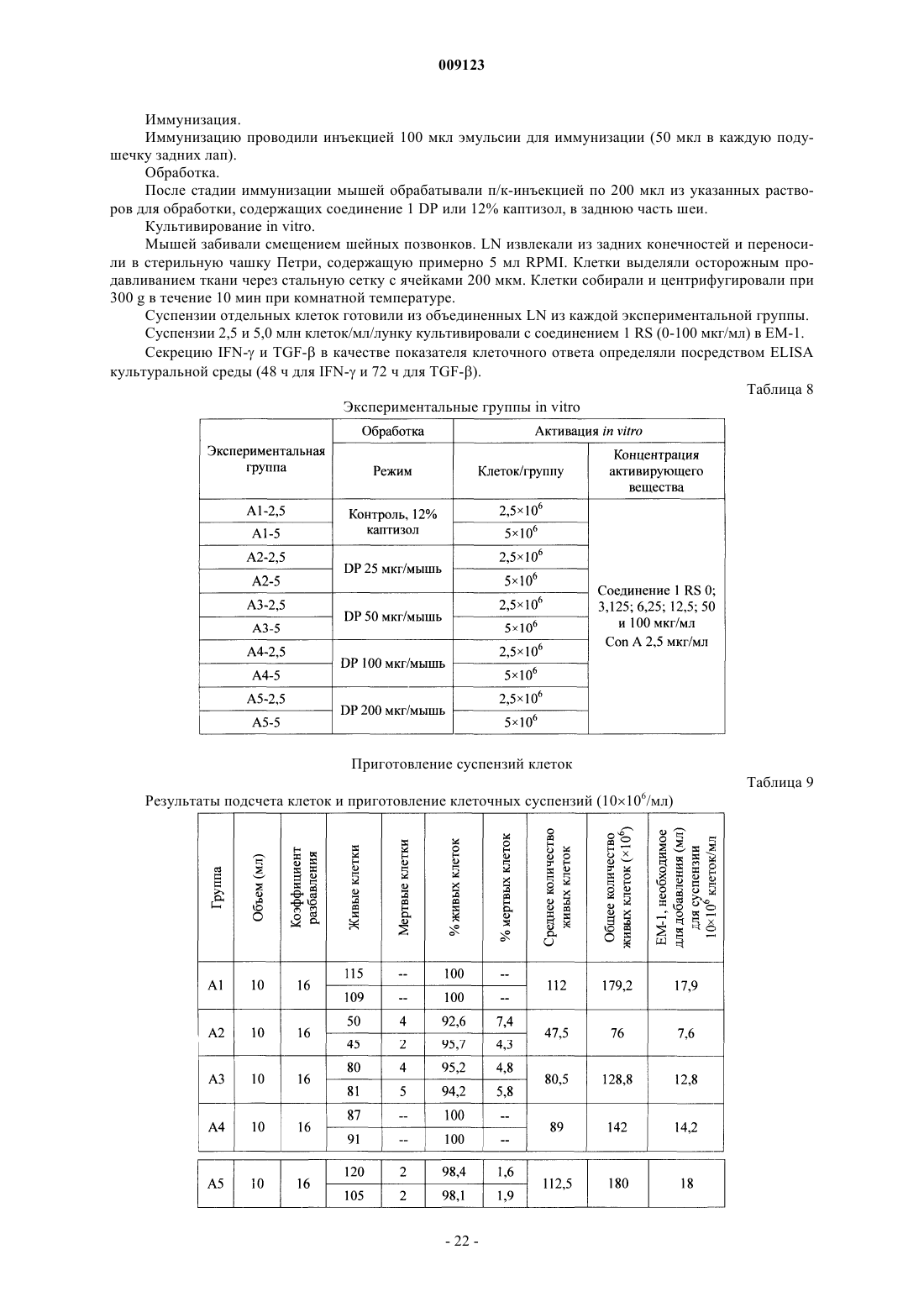
7. Способ получения фармацевтической композиции по любому из пп.1-5, включающий следующие стадии:
a) приготовление раствора диметилацетамида, полиэтиленгликоля, полиоксилированного касторового масла, N-метил-2-пирролидинона, 1-этенил-2-пирролидинона, сложных эфиров полиоксиэтиленсорбитана или замещенного b-циклодекстрина в водном носителе с предварительно определенной концентрацией;
b) добавление предварительно определенного количества фармацевтически приемлемой соли
1) пептида, содержащего по меньшей мере 12 и самое большее 30 следующих друг за другом аминокислот, имеющих последовательность, соответствующую
(i) последовательности аминокислот, обнаруживаемой в определяющей комплементарность области (CDR) тяжелой или легкой цепи человеческого моноклонального анти-ДНК 16/6 Id-антитела, или
(ii) последовательности аминокислот, обнаруживаемой в определяющей комплементарность области (CDR) тяжелой или легкой цепи патогенного моноклонального анти-ДНК-антитела, которое индуцирует ответ, подобный заболеванию системной красной волчанкой (СКВ), у мышей,
2) пептифр, содержащего аминокислоты, имеющие последовательность
(i) TGYYX1X2X3X4X5QSPEKSLEWIG (SEQ ID NO: 11),
где X1 означает Met, Ala или Val; X2 означает Gln, Asp, Glu или Arg; X3 означает Trp или Ala; X4 означает Val или Ser и X5 означает Lys, Glu или Ala;
(ii) EINPSTGGX6X7X8X9X10X11X12KAKAT (SEQ ID NO: 12),
где Х6 и Х7, каждый означает Thr, Val или Ala; Х8 означает Tyr или Phe; X9 означает Asn или Asp; X10 означает Gln или Glu; Х11 означает Lys или Glu и X12 означает Phe или Tyr;
(iii) YYCARX13X14X15X16PYAX17X18YWGQGS (SEQ ID NO: 13),
где X13 означает Phe, Thr или Gly; X14 означает Leu, Ala или Ser; X15 означает Trp или Ala; X16 означает Glu или Lys; X17 означает Met или Ala и X18 означает Asp, Lys или Ser;
(iv) GYNX19X20X21X22X23X24SHGX25X26LEWIG (SEQ ID NO: 14),
где X19 означает Met или Ala; X20 означает Asn, Asp или Arg; X21 означает Trp или Ala; X22 означает Val или Ser; X23 означает Lys или Glu; X24 означает Gln или Ala; X25 означает Lys или Glu и Х26 означает Ser или Ala;
(v) YYCARX27X28X29YGX30X31X32GQTL (SEQ ID NO: 15),
где Х27 означает Ser или Phe; X28 означает Gly или Ala; X29 означает Arg, Ala или Glu; X30 означает Asn или Asp; X31 означает Tyr или Phe и Х32 означает Trp, His или Ala;
(vi) X33YYWSWIX34QX35PX36X37GX38EWIG (SEQ ID NO: 16),
где Х33 означает Gly или Thr Gly; X34 означает Arg или Lys; X35 означает Pro или Ser; X36 означает Gly или Glu; X37 означает Lys или Asp и Х38 означает Glu, Leu или Ser;
(vii) YYCARX39LLX40X41X42X43X44DVDYX45GX46DV (SEQ ID NO: 17),
где Х39 означает Gly или Phe; X40 означает Arg или Ala; X41 означает Gly или Ala; X42 означает Gly или Ala; X43 означает Trp или Ala; X44 означает Asn или Ala; X45 означает Tyr или Trp; X46 означает Met или Gln;
(viii) FSGYYWS (SEQ ID NO: 8);
(ix) EINHSGSTNYKTSLKS (SEQ ID NO: 9) или
(x) GLLRGGWNDVDYYYGMDV (SEQ ID NO: 10), или
3) пептида, содержащего следующие друг за другом аминокислоты, имеющие любую последовательность из а) и b) или по меньшей мере две последовательности из последовательностей (a)(i), (a)(ii) и (b)(i)-(b)(х), или
4) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательность, содержащую по меньшей мере две идентичные последовательности, включенные в (a)(i), (a)(ii) и (b)(i)-(b)(x); и
c) доведение рН раствора со стадии b) до растворения пептида в растворе; и
d) при необходимости доведение рН раствора со стадии с) до рН 4-9, таким образом получая фармацевтическую композицию.
8. Лиофилизованная фармацевтическая композиция, содержащая от 0,1 до 20 мг/мл композиции фармацевтически приемлемой соли
a) пептида, содержащего по меньшей мере 12 и самое большее 30 следующих друг за другом аминокислот, имеющих последовательность, соответствующую
(i) последовательности аминокислот, обнаруживаемой в определяющей комплементарность области (CDR) тяжелой или легкой цепи человеческого моноклонального анти-ДНК 16/6 Id-антитела, или
(ii) последовательности аминокислот, обнаруживаемой в определяющей комплементарность области (CDR) тяжелой или легкой цепи патогенного моноклонального анти-ДНК-антитела, которое индуцирует ответ, подобный заболеванию системной красной волчанкой (СКВ), у мышей, или
b) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательность
(i) TGYYX1X2X3X4X5QSPEKSLEWIG (SEQ ID NO: 11),
где X1 означает Met, Ala или Val; X2 означает Gln, Asp, Glu или Arg; X3 означает Trp или Ala; X4 означает Val или Ser и X5 означает Lys, Glu или Ala;
(ii) EINPSTGGX6X7X8X9X10X11X12KAKAT (SEQ ID NO: 12),
где Х6 и Х7, каждый означает Thr, Val или Ala; X8 означает Tyr или Phe; X9 означает Asn или Asp; X10 означает Gln или Glu; X11 означает Lys или Glu и X12 означает Phe или Tyr;
(iii) YYCARX13X14X15X16PYAX17X18YWGQGS (SEQ ID NO: 13),
где X13 означает Phe, Thr или Gly; X14 означает Leu, Ala или Ser; X15 означает Trp или Ala; X16 означает Glu или Lys; X17 означает Met или Ala и X18 означает Asp, Lys или Ser;
(iv) GYNX19X20X21X22X23X24SHGX25X26LEWIG (SEQ ID NO: 14),
где X19 означает Met или Ala; X20 означает Asn, Asp или Arg; X21 означает Trp или Ala; X22 означает Val или Ser; X23 означает Lys или Glu; X24 означает Gln или Ala; X25 означает Lys или Glu и Х26 означает Ser или Ala;
(v) YYCARX27X28X29YGX30X31X32GQTL (SEQ ID NO: 15),
где Х27 означает Ser или Phe; X28 означает Gly или Ala; X29 означает Tyr или Phe и Х32 означает Trp, His или Ala;
(vi) X33YYWSWIX34QX35PX36X37GX38EWIG (SEQ ID NO: 16), где Х33 означает Gly или Thr Gly; X34 означает Arg или Lys; X35 означает Pro или Ser; X36 означает Gly или Glu; X37 означает Lys или Asp и Х38 означает Glu, Leu или Ser;
(vii) YYCARX39LLX40X41X42X43X44DVDYX45GX46DV (SEQ ID NO: 17),
где Х39 означает Gly или Phe; X40 означает Arg или Ala; X41 означает Gly или Ala; X42 означает Gly или Ala; X43 означает Trp или Ala; X44 означает Asn или Ala; X45 означает Tyr или Trp; X46 означает Met или Gln;
(viii) FSGYYWS (SEQ ID NO: 8);
(ix) EINHSGSTNYKTSLKS (SEQ ID NO: 9); или
(x) GLLRGGWNDVDYYYGMDV (SEQ ID NO: 10), или
c) пептида, содержащего следующие друг за другом аминокислоты, имеющие любую последовательность из а) и b) или по меньшей мере две последовательности из последовательностей (a)(i), (a)(ii) и (b)(i)-(b)(х), или
d) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательность, содержащую по меньшей мере две идентичные последовательности, включенные в (a)(i), (a)(ii) и (b)(i)-(b)(х); и
усилитель растворимости, выбранный из группы, состоящей из диметилацетамида, полиэтиленгликоля, полиоксилированного касторового масла, N-мeтил-2-пиppoлидинoнa, 1-этенил-2-пирролидинона, сложных эфиров полиоксиэтиленсорбитана и замещенного b-циклодекстрина.
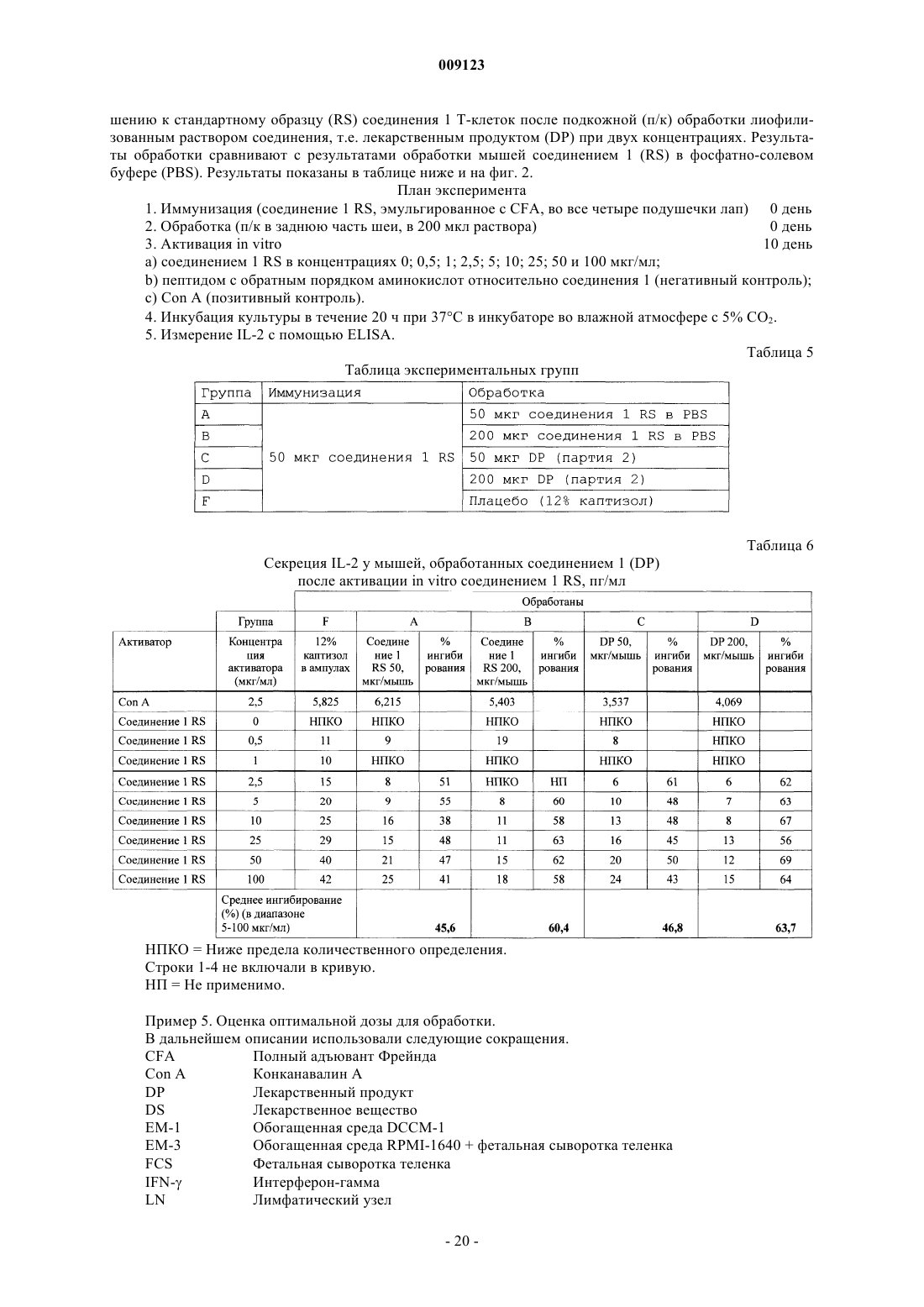
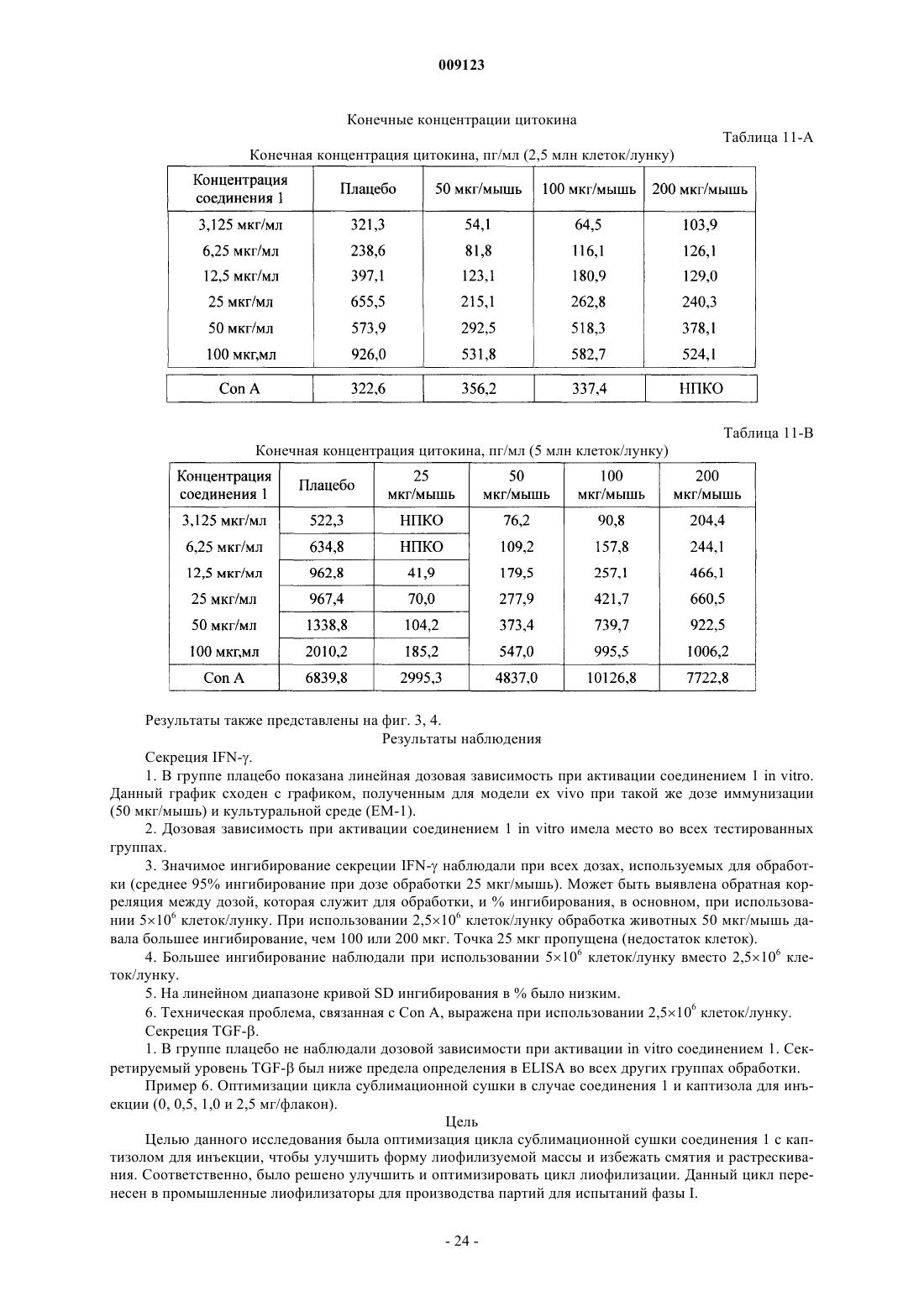
9. Способ лиофилизации фармацевтической композиции по любому из пп.1-5, включающий в себя стадии:
a) снижения температуры фармацевтической композиции до -40шС;
b) поддержания температуры при -40шС в течение предварительно определенного периода времени;
c) повышения температуры раствора до 20шС;
d) поддержания температуры при 20шС в течение предварительно определенного периода времени и
е) снижения давления и поддержание температуры при 20шС в течение предварительно определенного периода времени, таким образом лиофилизируя фармацевтическую композицию.
10. Способ по п.9, в котором
стадию а) осуществляют в течение 2 ч;
стадию b) осуществляют в течение 3 ч;
стадию с) осуществляют в течение 13 ч и при давлении 110 мкбар;
стадию d) осуществляют в течение 13 ч и при давлении 110 мкбар и
стадию е) осуществляют в течение 5 ч и давление понижают до 10 мкбар.
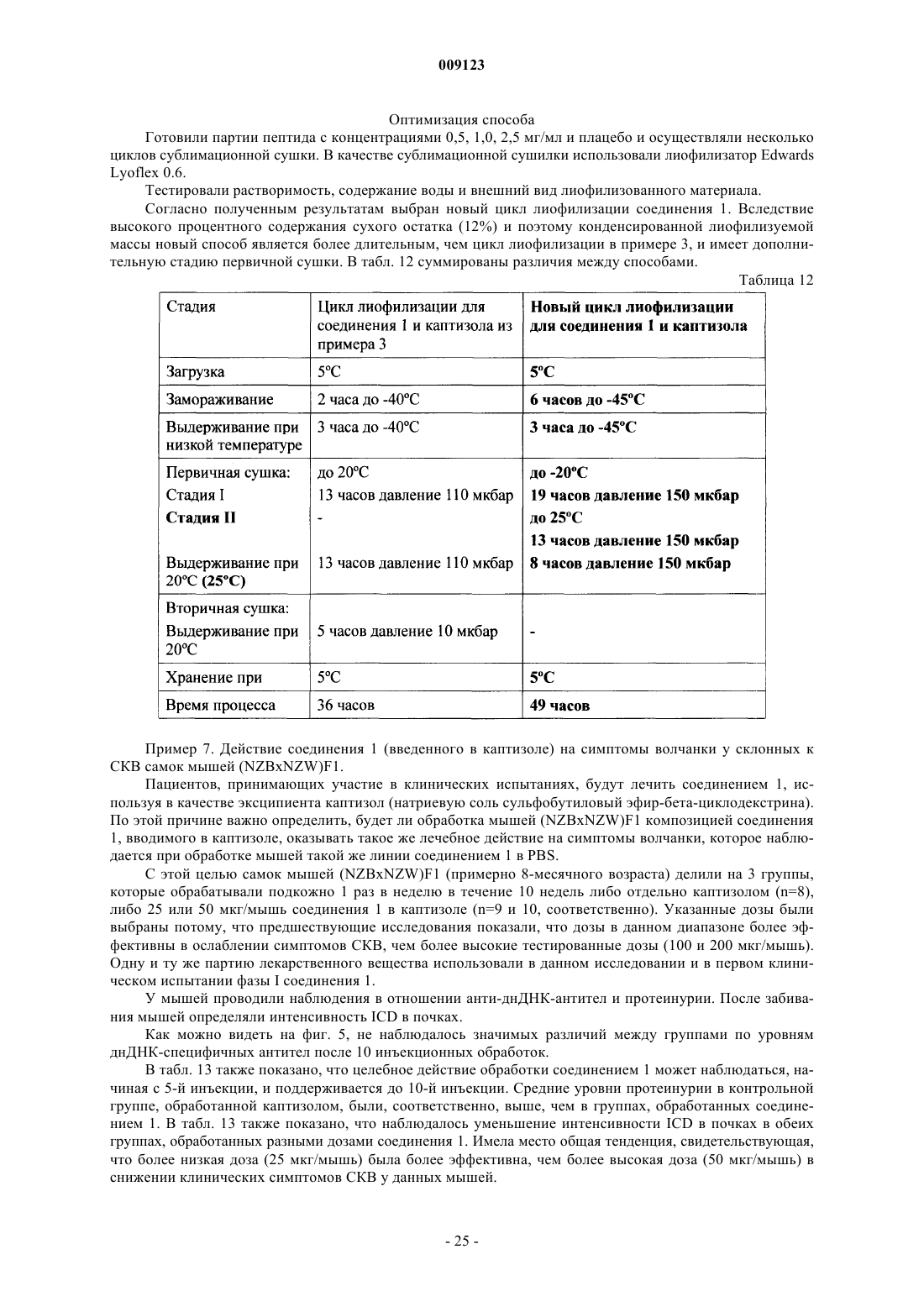
11. Способ лиофилизации фармацевтической композиции по любому из пп.1-5, включающий стадии:
a) снижения температуры фармацевтической композиции до -45шС;
b) поддержания температуры при -45шС в течение предварительно определенного периода времени;
c) повышениятемпературы раствора до -20шС;
d) повышения температуры раствора до 25шС и
e) поддержания температуры при 25шС в течение предварительно определенного периода времени, таким образом лиофилизируя фармацевтическую композицию.
12. Способ по п.11, в котором
стадию а) осуществляют в течение 6 ч;
стадию b) осуществляют в течение 3 ч;
стадию с) осуществляют в течение 19 ч и при давлении 150 мкбар;
стадию d) осуществляют в течение 13 ч и при давлении 150 мкбар и
стадию е) осуществляют в течение 8 ч и при давлении 150 мкбар.
Текст
009123 Данная заявка содержит притязания на приоритет предварительной заявки на выдачу патента США 60/439918, поданной 14 января 2003 г., полное содержание которой, таким образом, включено в виде ссылки. В данной заявке ссылки на различные публикации даны посредством полного цитирования. Описания указанных публикаций в полном объеме включены при этом в данную заявку в виде ссылки, чтобы более полно описать состояние уровня техники, которое известно специалистам на дату изобретения,описанного и заявленного в данном описании. Уровень техники Системная красная волчанка (СКВ) или волчанка является ослабляющим здоровье аутоиммунным заболеванием, характеризующимся наличием множества аутоантител, включая антитела к днДНК, к ядерным антигенам и к рибонуклеопротеинам. СКВ поражает примерно 1 из 2000 человек (в США 1 из 700 женщин). Заболевание, главным образом, поражает молодых женщин при соотношении женщин к мужчинам, составляющем примерно 9:1. Системная красная волчанка может поражать почти любой орган или систему организма. Системная красная волчанка может включать периоды, когда выражено немного симптомов, если они вообще выражены ("ремиссия"), и другие периоды, когда заболевание становится более активным ("вспышка"). Наиболее часто, когда люди упоминают "волчанку", они относят ее к системной форме заболевания. Кортикостероиды являются главной опорой при лечении системных аутоиммунных заболеваний. Угрожающие жизни, приводящие к тяжелой инвалидности проявления СКВ лечат высокими дозами глюкокортикоидов (1-2 мг/кг/сутки). Нежелательные эффекты хронического введения глюкокортикоидов включают множество заметных неблагоприятных эффектов, таких как кушингоидный габитус, ожирение центрального действия, гипертония, инфекция, ломкость капилляров, гирсутизм, усиленный остеопороз,катаракты, сахарный диабет, миопатия и психоз. Кроме токсичности кортикостероидов соблюдение пациентами схемы дозирования также представляет собой серьезную проблему. Для борьбы с активным заболеванием также используют цитотоксические агенты, снижающие частоту вспышек заболевания и уменьшающие потребность в стероидах. Нежелательные побочные эффекты последних включают угнетение костного мозга, повышенную частоту инфекций оппортунистическими организмами, необратимую недостаточность функции яичников, алопецию и повышенный риск злокачественных новообразований. СКВ является воспалительным заболеванием, для которого до настоящего времени не существует определенного способа лечения или средства. Заболевание приводит к острым и хроническим осложнениям. Единственными способами лечения являются паллиативные способы, предназначенные для снятия острых симптомов и предотвращения хронических осложнений, часто с сильными побочными эффектами. Поэтому существует неудовлетворенная потребность в данной области, и врачи, и пациенты были бы рады новым способам лечения, которые потенциально могли бы устранить или уменьшить нежелательные проявления болезни. Несмотря на всестороннее исследование механизмов, лежащих в основе индукции СКВ, информация об этиологии заболевания очень ограничена. До недавнего времени исследования СКВ проводились с использованием лимфоцитов периферической крови (PBL) пациентов на разных клинических стадиях и при использовании разных протоколов лечения. Альтернативно, в качестве модели СКВ исследовали линии мышей, у которых развивается спонтанное СКВ-подобное заболевание. Данный вид анализа приводит к неполным и запутанным интерпретациям роли различных иммунологических и неиммунологических факторов либо в индукции, либо в поддержании заболевания, главным образом, вследствие гетерогенности пациентов, с одной стороны, и невозможности контролировать фазу индукции заболевания в линиях мышей с СКВ, с другой стороны. Несколько лет назад разработана модель СКВ на животных. В указанной модели, основанной на концепции идиотипической сети, формировался широкий спектр связанных с волчанкой аутоантител и клинические проявления (Mendlovic, S. et al. Proc. Natl. Acad. Sci. USA 85: 2260 (1988. Индукцию осуществляли иммунизацией линий мышей, у которых не развивается какое-либо спонтанное аутоиммунное заболевание, с помощью человеческого моноклонального антитела против ДНК (мАт), которое несет общий идиотип, названный 16/6 Id (Shoenfeld, Y. et al., J. Exp. Med. 158: 718 (1983. Человеческое мАт 16/6 является анти-ДНК-антителом исходного изотипа IgM и в культуре переключаемого на IgG1. мАт получали от пациента, и они несут общий идиотип, 16/6 Id (Shoenfeld et al.,1983; Mendlovic et al., 1988). Клетки гибридомы, секретирующие указанное мАт, обычно выращивают в культуре и антитело выделяют из надосадков культур, используя аффинную колонку с белком G, связанным с сефарозой. Человеческое анти-ДНК-мАт 16/6 (IgG1/) описано в Shoenfeld, Y. et al., J. Clin. Invest. 70: 205-208 (1982) и в Waisman, A. et al., Int. Immunol. 7: 689-696 (1995). После иммунизации мыши продуцировали антитела, специфичные по отношению к 16/6 Id, антитела, которые несут 16/6 Id, и антитела, направленные против разных ядерных антигенов (днДНК, онДНК,Sm, рибонуклеопротеидну (РНП), Ro, La и др.). Серологические данные были связаны с лейкопенией,повышенной скоростью оседания эритроцитов, протеинурией, избытком иммунных комплексов в почках и склерозом клубочков (Mendlovic et al., 1988), которые являются типичными проявлениями СКВ. Кроме-1 009123 того, было показано, что экспериментальное заболевание может быть индуцировано мышиным анти-16/6(Waisman, A. et al., Internatl. Immunol. 5: 1293 (1993); Waisman, A. and Mozes, E. Eur. J. Immunol. 23: 1566,(1993. Индукция заболевания регулируется генетически и, таким образом, зависит от линии (Mendlovic,S. et al., Immunology 69: 228 (1990. Указанная уникальная модель индукции экспериментальной СКВ обеспечивает подходящие средства для ясного анализа различных стадий и связанных иммунных параметров, вовлеченных в индукцию и развитие СКВ. СКВ является системным аутоиммунным заболеванием, характеризующимся образованием аутоантител против собственных антигенов, таких как ДНК, Sm, Ro, La, РНП, кардиолипин и гистоны. Этиология СКВ неизвестна, и понимание механизма, посредством которого возникают указанные аутоантитела,дало бы возможность в этом разобраться. Обнаружено, что моноклональные аутоантитела, которые способны вызывать появление антител, которые несут 16/6 Id или взаимодействуют с ним, являются патогенными и, таким образом, способны индуцировать экспериментальную СКВ (Fricke, H. et al., Internatl.Immunol. 2: 225 (1990); Sthoeger, Z.M. et al., J. Clin. Immunol. 13: 127 (1993. Секвенированы вариабельные (V) области девяти аутоантител, которые связываются либо с ДНК, либо с экстрактом ядер (NE)HeLa, выделенные из организма мышей C3H.SW с экспериментальной СКВ (Waisman and Mozes, 1993). Анализировали моноклональные антитела с разной специфичностью, чтобы попытаться определить связи между разными аутоантителами. Обнаружено, что три мАт связываются с ДНК, и было показано, что они имеют признаки последовательности, характерные для патогенных анти-ДНК-антител. Показано, что в одном из указанных мАт, названном 2 С 4 С 2, используется ген V-области тяжелой (Н) цепи (VH), идентичный VH анти-ДНК-мАт, выделенного из других склонных к развитию волчанки мышей, а именно(NZBxNZW)F1. Ген V-области легкой (L) цепи (VL) мАт 2 С 4 С 2 на 98% гомологичен VL другого антиДНК-мАт, также выделенного от мышей (NZBxNZW)F1. Два других анти-ДНК-мАт, названных 5G12-4 и 5G12-6, имеют 93% общих последовательностей VH с последовательностями мАт 2 С 4 С 2. Шесть мАт связывают белки NE HeLa. Девять мАт используют всего пять генов VH и четыре гена VL зародышевой линии, свидетельствуя, что аутоантитела, индуцированные у мышей с экспериментальной СКВ, не происходят из одного клона В-клеток. Три из девяти VH и VL оказались идентичными по последовательности генам зародышевой линии, тогда как по меньшей мере три других имели соматические мутации. Последнее подтверждает, что указанные аутоантитела возникают у мышей как при использовании существующих (непримированных) В-клеток, так и посредством запускаемого антигеном процесса. Кроме того,очевидно, что в случае аутоантител, обнаруженных у мышей с экспериментальной СКВ, используются генетические элементы, сходные с элементами, используемыми в мАт, которые выделяли из линий мышей, у которых волчанка развивается спонтанно. Т-клетки играют важную роль в индукции и развитии экспериментальной СКВ. Так, было показано,что Т-клеточные линии и клоны, специфичные к 16/6 Id, индуцируют экспериментальную СКВ у сингенных реципиентов подобно 16/6-антителу. Поэтому после инокуляции активированных клеток линий у мышей развивались как серологические признаки, так и повреждение почек, которое типично для СКВ(Fricke, H. et al., Immunology 73: 421 (1991. Кроме того, 16/6 Id-специфичная линия Т-клеток, происходящих от мышей C3H.SW, индуцировала СКВ у мышей C57BL/6, которые, как было показано, резистентны к индукции заболевания после инъекций либо 16/6 Id-, либо анти-16/6 Id-мАт (Mendlovic et al.,1990). При попытке идентифицировать патогенную область 16/6 Id были получены (Fab')2-фрагменты 16/6Id-мАт, и обнаружено, что они сохраняют специфичность и патогенную способность целой молекулы 16/6 Id (Ruiz, P.J., et al., Immun. Let. 41: 79 (1994. мАт 5G12, которое было выделено из организма мышей с экспериментальной СКВ и которое, как было показано, связывается с ДНК и несет 16/6 Id, способно индуцировать экспериментальную СКВ у мышей (Waisman, A. et al., Internatl. Immunol. 5: 1293 (1993. Т-клетки, которые специфично реагируют на мАт пролиферацией, вероятно, реагируют на пептиды, представляющие последовательности из их областей, определяющих комплементарность (CDR). Весьма вероятно, что Т-клетки узнают V-области указанных выше антител, так как они не реагируют с другими антителами, которые несут такую же константную область, но имеют другие специфичности. В пределах вариабельной области наиболее вероятно узнаваемыми областями являются CDR, так как они представляют собой области, которые наиболее различаются среди разных антител. CDR-области последовательностей VH девяти патогенных мышиных мАт, указанных выше, которые индуцируют СКВ у мышей, заключены в прямоугольники на фиг. 1 в работе Waisman and Mozes, 1993, на которой представлены полные нуклеотидные и рассчитанные аминокислотные последовательности VH девяти мАт. В экспериментальных моделях СКВ - мыши Balb/c и склонные к СКВ мыши, т.е. мыши(NZBxNZW)F1 - лечение либо пептидами, основанными на mCDR, либо соединением 1 значимо снижало связанные с СКВ показатели, особенно отложения иммунных комплексов (ICD) в почке, протеинурию и лейкопению. Лечение не оказывало влияния на 16/6 Id-специфичный гуморальный ответ (Waisman, A., etCDR1 человека, соединение 1, показанное на фиг. 1, является синтетическим пептидом из 19 аминокислот, основанным на определяющей комплементарность области 1 (CDR1) анти-днДНК-мАт человека, названного 16/6 Id (Waisman, A., et al. "Modulation of murine systemic lupus erythematosus with peptides based on complementarity determining regions of pathogenic anti-DNA monoclonal antibodies". Proc.Natl. Acad. Sci. U.S.A. (1997), 94 (4): 4620-4625). Указанные пептиды, подобно многим пептидам, не очень хорошо растворимы. Поэтому в данной заявке предлагаются композиции, которые улучшают растворимость пептидов. Сущность изобретения Настоящее изобретение относится к фармацевтической композиции, содержащей водный носитель; от 0,1 до 20 мг/мл композиции фармацевтически приемлемой солиa) пептида, содержащего по меньшей мере 12 и самое большее 30 следующих друг за другом аминокислот, имеющих последовательность, соответствующую(CDR) тяжелой или легкой цепи человеческого моноклонального анти-ДНК 16/6 Id-антитела, или(ii) последовательности аминокислот, обнаруживаемой в определяющей комплементарность области (CDR) тяжелой или легкой цепи патогенного моноклонального анти-ДНК-антитела, которое индуцирует ответ, подобный заболеванию системной красной волчанкой (СКВ), у мышей, илиb) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательностьc) пептида, содержащего следующие друг за другом аминокислоты, имеющие любую последовательность из а) и b) или по меньшей мере две последовательности из последовательностей (a)(i), (a)(ii) иd) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательность,содержащую по меньшей мере две идентичные последовательности, включенные в (a)(i), (а)(ii) и (b)(i)(b)(х); и усилитель растворимости, выбранный из группы, состоящей из диметилацетамида, полиэтиленгликоля, полиоксилированного касторового масла, N-мeтил-2-пиppoлидинoнa, 1-этенил-2-пирролидинона,сложных эфиров полиоксиэтиленсорбитана и замещенного -циклодекстрина,в которой и пептид, и усилитель растворимости растворены в водном носителе; и при этом композиция имеет рН от 4 до 9. Настоящее изобретение также относится к способу ослабления симптомов системной красной волчанки (СКВ) у человека, включающему в себя введение человеку любой из фармацевтических компози-3 009123 ций согласно изобретению в количестве, эффективном для ослабления симптомов СКВ у человека. Краткое описание фигур Фиг. 1. CDR1 человека (соединение 1) в виде ацетатной соли - показаны молекулярная и структурная формулы hCDR1, аминокислотная последовательность и физические параметры. Фиг. 2. Секреция IL-2 из клеток, взятых от мышей, обработанных раствором соединения 1 и каптизолом, после того, как затем клетки были активированы раствором соединения 1 в PBS.- 12% каптизол в ампулах. Фиг. 3. Секреция IFN- из клеток, взятых от мышей, обработанных раствором соединения 1, после того, как клетки были затем активированы раствором соединения 1 в ЕМ-1 (2,5106 клеток/лунку).- соединение 1 200 мкг/мышь (лечебная доза). Фиг. 4. Секреция IFN- из клеток, взятых от мышей, обработанных раствором соединения 1, после того, как клетки были затем активированы раствором соединения 1 в ЕМ-1 (5106 клеток/лунку).- соединение 1 25 мкг/мышь. Фиг. 6. Срезы почек от мышей (NZBxNZW)F1, показывающие интенсивность отложений иммунных комплексов. Срезы в верхнем ряду получены от мыши, обработанной каптизолом, срезы среднего ряда от мыши, обработанной 50 мкг/мышь соединения 1, и срезы нижнего ряда от мыши, обработанной 25 мкг/мышь соединения 1. Увеличение - левые: 100, правые: 400. ФИТЦ-иммуногистология. Фиг. 7. Титры антител в сыворотках пациентов с СКВ и контрольных сыворотках здоровых людей при тестировании их сывороток в отношении способности связываться с пептидами Iа, IIa и IIIа, или мАт 5G12, или контрольным пептидом. Фиг. 8. Концентрация человеческих анти-ДНК 16/6 Id-мАт, требуемая для оптимальной стимуляции(0,1-40 мкг/лунку) 16/6 Id-мАт. Концентрацию, дающую самый высокий индекс стимуляции, определяли как оптимальную для запуска пролиферативного ответа. Фиг. 9. Пролиферация PBL от одного пациента с СКВ, стимулированных митогеном фитогемагглютинином (ФГА), в отсутствие или в присутствии hCDR1 или hCDR3. Фиг. 10. Пролиферация PBL от одного пациента с СКВ, стимулированных человеческим мАт 16/6 I,в отсутствие или в присутствии пептидов hCDR1 или hCDR3 человека или пептида mCDR3 мыши. Фиг. 11. Пролиферация PBL от одного пациента с CKB, стимулированных человеческим мАт 16/6 I,в отсутствие или в присутствии пептидов hCDR1 или hCDR3 человека или мышиных пептидов с обратной последовательностью revmCDR1 и revmCDR3. Фиг. 12. Ингибирование секреции IL-2 в PBL пациентов с СКВ, запускаемой мАт 16/6 Id человека, в отсутствие или в присутствии hCDR1 или hCDR3. Фиг. 13. Повышающая регуляция секреции TGF- в PBL одного типичного пациента с СКВ, стимулированной человеческим мАт 16/6 Id, в отсутствие или в присутствии hCDR1 или hCDR3. Фиг. 14. Типичный гель, показывающий активность ММР-2 и ММР-9 в сыворотках пациентов с СКВ и здоровых людей в контроле. Сыворотки (5 мкл) 40 пациентов с СКВ и 25 здоровых людей в качестве контролей анализировали в отношении активности ММР-2 или ММР-9 зимографией в геле. На фи-4 009123 гуре показаны типичные результаты с образцами сыворотки из двух групп. Фиг. 15. График, показывающий количественный анализ активностей ММР-2 и ММР-9 в сыворотках пациентов с СКВ (темные колонки) и здоровых людей в качестве контролей (белые колонки). 36 образцов сыворотки пациентов с СКВ и 15 образцов сыворотки здоровых людей в качестве контролей тестировали в отношении активности ММР-2 или ММР-9, используя специальные наборы для анализа активности. Результаты выражены в виде среднейs.e.m. P=0,0302. Фиг. 16 А-В. Графики, показывающие уровни активности ММР-9 и индексы активности заболевания(SLEDAI) у пациентов с СКВ. 35 образцов сыворотки от 8 пациентов с СКВ мужского пола (фиг. 16 А) и 27 женского пола (фиг. 16 В) тестировали в отношении активности ММР-9 с помощью специального набора для анализа активности. Представлено распределение активности ММР-9 в соответствии с SLEDAI пациентов. Пунктирной линией указана активность ММР-9 у здоровых людей в контроле. Фиг. 17 А-В. Графики, показывающие характер активности ММР-2 (белые кружки) и ММР-9 (черные кружки) в сыворотках двух пациентов с СКВ, собранных в течение 4-6 лет заболевания. Сыворотки тестировали в отношении активностей ММР-2 или ММР-9 с помощью специальных наборов для анализа активности. Анализы осуществляли в двух повторах. Подробное описание Настоящее изобретение относится к фармацевтической композиции, содержащей водный носитель; от 0,1 до 20 мг/мл композиции фармацевтически приемлемой соли а) пептида, содержащего по меньшей мере 12 и самое большее 30 следующих друг за другом аминокислот, имеющих последовательность, соответствующую(CDR) тяжелой или легкой цепи человеческого моноклонального анти-ДНК 16/6 Id-антитела, или(ii) последовательности аминокислот, обнаруживаемой в определяющей комплементарность области (CDR) тяжелой или легкой цепи патогенного моноклонального анти-ДНК-антитела, которое индуцирует ответ, подобный заболеванию системной красной волчанкой (СКВ), у мышей, илиb) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательностьc) пептида, содержащего следующие друг за другом аминокислоты, имеющие любую последовательность из а) и b) или по меньшей мере две последовательности из последовательностей (a)(i), (а)(ii) иd) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательность,содержащую по меньшей мере две идентичные последовательности, включенные в (a)(i), (а)(ii) и (b)(i)(b)(х); и усилитель растворимости, выбранный из группы, состоящей из диметилацетамида, полиэтиленгликоля, полиоксилированного касторового масла, N-мeтил-2-пиppoлидинoнa, 1-этенил-2-пирролидинона,-5 009123 сложных эфиров полиоксиэтиленсорбитана и замещенного -циклодекстрина,в которой и пептид, и усилитель растворимости растворены в водном носителе; и при этом композиция имеет рН от 4 до 9. В одном варианте по меньшей мере 0,5 мг/мл композиции составляет фармацевтически приемлемая соль пептида. В другом варианте пептид имеет последовательность, выбранную из группы, состоящей изNH2-Gly Leu Leu Arg Gly Gly Trp Asn Asp Val Asp Tyr Tyr Туr Gly Met Asp Val-COOH (SEQ ID NO: 10). В одном варианте пептид содержит следующие друг за другом аминокислоты, имеющие последовательностьX33YYWSWIX34QX35PX36X37GX38EWIG (SEQ ID NO: 16),где Х 33 означает Gly или Thr Gly; X34 означает Arg или Lys; Х 35 означает Pro или Ser; X36 означает Gly или Glu; X37 означает Lys или Asp и Х 38 означает Glu, Leu или Ser. В другом варианте любой из описанных выше фармацевтических композиций усилителем растворимости является замещенный -циклодекстрин. В одном варианте замещенным -циклодекстрином является -циклодекстрин, замещенный гидроксипропилом, сульфобутиловым эфиром или сульфопропиловым эфиром. В другом варианте замещенным -циклодекстрином является -циклодекстрин, замещенный сульфобутиловым эфиром. В следующем варианте любой из описанных выше композиций концентрация пептида в растворе составляет по меньшей мере 1,0 мг/мл. В следующем варианте концентрация пептида в растворе составляет по меньшей мере 2,5 мг/мл. В одном варианте концентрация соли пептида составляет от 0,5 до 10 мг/мл. В другом варианте концентрация соли пептида составляет от 0,5 до 2,5 мг/мл. В другом варианте концентрация соли пептида составляет от 2,5 до 5 мг/мл. В другом варианте концентрация соли пептида составляет от 5 до 7 мг/мл. В другом варианте концентрация соли пептида составляет от 7 до 8,5 мг/мл. В другом варианте концентрация соли пептида составляет от 8,5 до 10 мг/мл. В другом варианте концентрация соли пептида составляет от 9 до 10 мг/мл. В другом варианте концентрация соли пептида составляет от 10 до 15 мг/мл. В другом варианте концентрация соли пептида составляет от 15 до 20 мг/мл. В другом варианте концентрация соли пептида составляет 1,0 мг/мл. В другом варианте концентрация соли пептида составляет 2,5 мг/мл. В другом варианте концентрация соли пептида составляет 5 мг/мл. В другом варианте концентрация соли пептида составляет 10 мг/мл. В другом варианте концентрация соли пептида составляет 15 мг/мл. В другом варианте концентрация соли составляет от 0,1 до 0,5 мг/мл. В другом варианте концентрация соли составляет от 0,1 до 0,2 мг/мл. В другом варианте концентрация соли составляет от 0,2 до 0,3 мг/мл. В другом варианте концентрация соли составляет от 0,3 до 0,4 мг/мл. В другом варианте концентрация соли составляет от 0,4 до 0,5 мг/мл. В следующем варианте композиция имеет рН от 6,5 до 8,5. В следующем варианте композиция имеет рН от 7,5 до 8,5. В следующем варианте композиция имеет рН от 4 до 5. В следующем варианте композиция имеет рН от 5 до 6. В следующем варианте композиция имеет рН от 6 до 7. В следующем варианте композиция имеет рН от 7 до 8. В следующем варианте композиция имеет рН от 8 до 9.-6 009123 В другом варианте любой из описанных выше фармацевтических композиций фармацевтически приемлемой солью является ацетатная соль. В следующем варианте фармацевтически приемлемой солью является ацетатная соль, а замещенным -циклодекстрином является гепта(сульфобутиловый эфир)циклодекстрин. В другом варианте композиция, кроме того, содержит фармацевтически приемлемый буфер в количестве и типа, подходящего для получения рН фармацевтической композиции в диапазоне 4-9. Буфером может быть ацетатный буфер, нитратный буфер или карбонат натрия. Настоящее изобретение также относится к способу ослабления симптомов системной красной волчанки (СКВ) у человека, включающему введение человеку любой из указанных выше фармацевтических композиций в количестве, эффективном для ослабления симптомов СКВ у человека. Настоящее изобретение также относится к указанным выше фармацевтическим композициям для применения при лечении СКВ у человека. Настоящее изобретение также относится к способу производства любой из указанных выше фармацевтических композиций, включающему следующие стадии:a) приготовление раствора диметилацетамида, полиэтиленгликоля, полиоксилированного касторового масла, N-метил-2-пирролидинона, 1-этенил-2-пирролидинона, сложных эфиров полиоксиэтиленсорбитана или замещенного -циклодекстрина в водном носителе с предварительно определенной концентрацией;b) добавление предварительно определенного количества фармацевтически приемлемой соли 1) пептида, содержащего по меньшей мере 12 и самое большее 30 следующих друг за другом аминокислот, имеющих последовательность, соответствующую(CDR) тяжелой или легкой цепи человеческого моноклонального анти-ДНК 16/6 Id-антитела, или(ii) последовательности аминокислот, обнаруживаемой в определяющей комплементарность области (CDR) тяжелой или легкой цепи патогенного моноклонального анти-ДНК-антитела, которое индуцирует ответ, подобный заболеванию системной красной волчанкой (СКВ), у мышей,2) пептида, содержащего аминокислоты, имеющие последовательность(x) GLLRGGWNDVDYYYGMDV (SEQ ID NO: 10), или 3) пептида, содержащего следующие друг за другом аминокислоты, имеющие любую последовательность из а) и b) или по меньшей мере две последовательности из последовательностей (a)(i), (а)(ii) и(b)(i)-(b)(х), или 4) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательность,содержащую по меньшей мере две идентичные последовательности, включенные в (a)(i), (а)(ii) и (b)(i)(b)(х); иc) доведение рН раствора со стадии b) вплоть до растворения пептида в растворе; иd) при необходимости доведение рН раствора со стадии с) до рН 4-9, таким образом получая фар-7 009123 мацевтическую композицию. В одном варианте предварительно определенным количеством пептида является такое количество,которое приводит к конечной концентрации пептида в фармацевтической композиции, составляющей по меньшей мере 0,1 мг/мл. В другом варианте предварительно определенным количеством пептида является такое количество,которое приводит к конечной концентрации пептида в фармацевтической композиции, составляющей по меньшей мере 0,5 мг/мл. В следующем варианте предварительно определенным количеством пептида является такое количество, которое приводит к конечной концентрации пептида в фармацевтической композиции, составляющей 2,5, 2,0, 1,0, 0,5 или 0,1 мг/мл. В другом варианте предварительно определенным количеством пептида является такое количество,которое приводит к конечной концентрации пептида в фармацевтической композиции, составляющей 5,10 или 15 мг/мл. В одном варианте способа полученная в результате конечная концентрация замещенного -циклодекстрина в фармацевтической композиции составляет от 70 до 170 мг/мл. В одном варианте способа предварительно определенной концентрацией замещенного -циклодекстрина является такая концентрация, которая приводит к конечной концентрации замещенного -циклодекстрина в фармацевтической композиции от 80 до 170 мг/мл. В одном варианте способа предварительно определенной концентрацией замещенного -циклодекстрина является такая концентрация, которая приводит к конечной концентрации замещенного -циклодекстрина в фармацевтической композиции от 90 до 170 мг/мл. В одном варианте способа предварительно определенной концентрацией замещенного -циклодекстрина является такая концентрация, которая приводит к конечной концентрации замещенного -циклодекстрина в фармацевтической композиции от 100 до 170 мг/мл. В одном варианте способа предварительно определенной концентрацией замещенного -циклодекстрина является такая концентрация, которая приводит к конечной концентрации замещенного -циклодекстрина в фармацевтической композиции от 110 до 170 мг/мл. В одном варианте способа предварительно определенной концентрацией замещенного -циклодекстрина является такая концентрация, которая приводит к конечной концентрации замещенного -циклодекстрина в фармацевтической композиции от 120 до 170 мг/мл. В одном варианте способа предварительно определенной концентрацией замещенного -циклодекстрина является такая концентрация, которая приводит к конечной концентрации замещенного -циклодекстрина в фармацевтической композиции от 130 до 170 мг/мл. В одном варианте способа предварительно определенной концентрацией замещенного -циклодекстрина является такая концентрация, которая приводит к конечной концентрации замещенного -циклодекстрина в фармацевтической композиции от 140 до 170 мг/мл. В одном варианте способа предварительно определенной концентрацией замещенного -циклодекстрина является такая концентрация, которая приводит к конечной концентрации замещенного -циклодекстрина в фармацевтической композиции от 150 до 170 мг/мл. В одном варианте способа предварительно определенной концентрацией замещенного -циклодекстрина является такая концентрация, которая приводит к конечной концентрации замещенного -циклодекстрина в фармацевтической композиции от 160 до 170 мг/мл. В одном варианте способа предварительно определенной концентрацией замещенного -циклодекстрина является такая концентрация, которая приводит к конечной концентрации замещенного -циклодекстрина в фармацевтической композиции, составляющей 120 мг/мл. В следующем варианте стадия b), кроме того, включает перемешивание раствора в течение 1 ч. В другом варианте на стадии с) рН корректируют, используя 1,0 н. НСl или NaOH. В следующем варианте способ, кроме того, включает фильтрование раствора со стадии d) через фильтр из ацетата целлюлозы. В следующем варианте предварительно определенным количеством пептида является такое количество, которое приводит к конечной концентрации пептида в фармацевтической композиции, составляющей 2,5, 2,0, 1,0, 0,5 или 0,1 мг/мл; стадия b), кроме того, включает перемешивание раствора в течение 1 ч; и на стадии с) рН корректируют, используя 1,0 н. НСl или NaOH, и способ, кроме того, включает фильтрование раствора со стадии d) через фильтр из ацетата целлюлозы. Настоящее изобретение также относится к композиции, полученной любым из указанных выше способов. Настоящее изобретение также относится к лиофилизованной фармацевтической композиции, содержащей от 0,1 до 20 мг/мл композиции фармацевтически приемлемой солиa) пептида, содержащего по меньшей мере 12 и самое большее 30 следующих друг за другом ами-8 009123 нокислот, имеющих последовательность, соответствующую(CDR) тяжелой или легкой цепи человеческого моноклонального анти-ДНК 16/6 Id-антитела, или(ii) последовательности аминокислот, обнаруживаемой в определяющей комплементарность области (CDR) тяжелой или легкой цепи патогенного моноклонального анти-ДНК-антитела, которое индуцирует ответ, подобный заболеванию системной красной волчанкой (СКВ), у мышей, илиb) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательностьc) пептида, содержащего следующие друг за другом аминокислоты, имеющие любую последовательность из а) и b) или по меньшей мере две последовательности из последовательностей (a)(i), (a)(ii) иd) пептида, содержащего следующие друг за другом аминокислоты, имеющие последовательность,содержащую по меньшей мере две идентичные последовательности, включенные в (a)(i), (а)(ii) и (b)(i)(b)(x); и усилитель растворимости, выбранный из группы, состоящей из диметилацетамида, полиэтиленгликоля, полиоксилированного касторового масла, N-мeтил-2-пиppoлидинoнa, 1-этенил-2-пирролидинона,сложных эфиров полиоксиэтиленсорбитана и замещенного -циклодекстрина. В одном варианте лиофилизованной фармацевтической композиции по меньшей мере 0,5 мг/мл композиции составляет фармацевтически приемлемая соль пептида. Настоящее изобретение также относится к способу лиофилизации любой из указанных выше фармацевтических композиций, включающему следующие стадии:a) снижение температуры фармацевтической композиции до -40 С;b) поддержание температуры при -40 С в течение предварительно определенного периода времени;c) повышение температуры раствора до 20 С;d) поддержание температуры при 20 С в течение предварительно определенного периода времени иe) снижение давления и поддержание температуры при 20 С в течение предварительно определенного периода времени, таким образом лиофилизируя фармацевтическую композицию. В одном варианте стадию а) осуществляют в течение 2 ч. В другом варианте стадию b) осуществляют в течение 3 ч. В другом варианте стадию с) осуществляют в течение 13 ч. В другом варианте стадию с) осуществляют при давлении 110 мкбар. В другом варианте стадию d) осуществляют в течение 13 ч. В другом варианте стадию d) осуществляют при давлении 110 мкбар. В другом варианте на стадии е) давление понижают до 10 мкбар. В другом варианте стадию е) осуществляют в течение 5 ч. В другом варианте стадию а) осуществляют в течение 2 ч;-9 009123 стадию b) осуществляют в течение 3 ч; стадию с) осуществляют в течение 13 ч и при давлении 110 мкбар; стадию d) осуществляют в течение 13 ч и при давлении 110 мкбар и стадию е) осуществляют в течение 5 ч и давление понижают до 10 мкбар. Настоящее изобретение также относится к лиофилизованной фармацевтической композиции, полученной любым из указанных выше способов. Настоящее изобретение также относится к способу лиофилизации любой из указанных выше фармацевтических композиций, включающему в себя следующие стадии:a) снижение температуры фармацевтической композиции до -45 С;b) поддержание температуры при -45 С в течение предварительно определенного периода времени;c) повышение температуры раствора до -20 С;d) повышение температуры раствора до 25 С иe) поддержание температуры при 25 С в течение предварительно определенного периода времени,таким образом лиофилизируя фармацевтическую композицию. В одном варианте стадию а) осуществляют в течение 6 ч. В другом варианте стадию b) осуществляют в течение 3 ч. В другом варианте стадию с) осуществляют в течение 19 ч. В другом варианте стадию с) осуществляют при давлении 150 мкбар. В другом варианте стадию d) осуществляют в течение 13 ч. В другом варианте стадию d) осуществляют при давлении 150 мкбар. В другом варианте стадию е) осуществляют в течение 8 ч. В другом варианте стадию е) осуществляют при давлении 150 мкбар. В другом варианте стадию а) осуществляют в течение 6 ч; стадию b) осуществляют в течение 3 ч; стадию с) осуществляют в течение 19 ч и при давлении 150 мкбар; стадию d) осуществляют в течение 13 ч и при давлении 150 мкбар и стадию е) осуществляют в течение 8 ч и при давлении 150 мкбар. Настоящее изобретение также относится к лиофилизованной фармацевтической композиции, полученной любым из указанных выше способов. Настоящее изобретение также относится к указанной выше лиофилизованной фармацевтической композиции, в которой содержание воды в композиции составляет менее 5%. В одном варианте содержание воды в композиции составляет менее 4%. В другом варианте содержание воды в композиции составляет менее 3,5%. Настоящее изобретение также относится к упакованной фармацевтической композиции, состоящей из упаковочного материала и предварительно определенного количества любой из указанных выше лиофилизованных фармацевтических композиций. В другом варианте пептид имеет формулу NH2-Gly Tyr Tyr Trp Ser Trp lle Arg Gln Pro Pro Gly LysGly Glu Glu Trp Ile Gly-COOH (SEQ ID NO: 6). Синтетические пептиды согласно данному изобретению основаны на CDR моноклональных патогенных аутоантител, выделенных от мышей с экспериментальной СКВ. Такие моноклональные антитела получают из надосадков гибридом, полученных слиянием, например, клеток селезенки мышей C3H.SW,иммунизированных анти-16/6 Id-мАт, с клетками плазмоцитомы Х 63.653 (Waisman and Mozes, 1993). Примерами таких пептидов являются пептиды формул Ia-Va, приведенных в данном описании, основанные, соответственно, на областях CDR1, CDR2 и CDR3 тяжелой цепи мАт 5G12 и областях CDR1 иCDR3 тяжелой цепи мАт 2 С 4 С 2 (Waisman and Mozes, 1993), и их аналоги. Подразумевается, что пептиды согласно настоящему изобретению включают аналоги пептидов IaVa, включая аналоги, содержащие замену, делецию и добавление, которые приведены в данном описании. Аналоги с заменами имеют замены аминокислот в разных положениях, при этом указанные замены производят на основе объема, гидрофобного-гидрофильного характера и заряда аминокислот. Аминокислоты можно разделить в соответствии с объемом, гидрофобным-гидрофильным характером и зарядом. Что касается объема, то специалистам в данной области понятно, что аминокислотами с наибольшим занимаемым объемом являются Trp, Tyr, Phe, Arg, Lys, Ile, Leu, Met и His, тогда как аминокислотами с наименьшими объемами являются Gly, Ala, Ser, Asp, Thr и Pro, остальные занимают промежуточное положение между ними. Что касается гидрофобного-гидрофильного характера, то хорошо известно, что аминокислоты Gly,Ala, Phe, Val, Leu, Ile, Pro, Met и Trp являются гидрофобными в то время, как все остальные аминокислоты являются гидрофильными. Среди гидрофильных аминокислот Ser, Thr, Gln и Tyr не имеют заряда,тогда как Arg, Lys, His и Asn имеют положительный заряд, a Asp и Glu имеют отрицательные заряды. При отборе пептидов для тестирования в отношении их возможности ингибировать пролиферативный ответ Т-лимфоцитов мышей, которые являются высокоотвечающими на СКВ-индуцирующие ауто- 10009123 антитела, важно, чтобы замены были выбраны из таких замен, которые в совокупности существенно не изменяют объем, гидрофобный-гидрофильный характер и заряд соответствующей части не подвергнутого заменам исходного пептида. Таким образом, гидрофобный остаток может быть заменен гидрофильным остатком или наоборот при условии, что общий эффект существенно не изменит объем, гиброфобный-гидрофильный характер и заряд соответствующего не подвергнутого заменам исходного пептида. Следует понимать, что другие модификации пептидов также входят в объем настоящего изобретения. Таким образом, подразумевается, что пептид согласно настоящему изобретению включает его "химическое производное", которое сохраняет по меньшей мере часть функции пептида, которая дает возможность использовать его для предотвращения или ингибирования пролиферативных ответов Т-клеток и аутоиммунного заболевания."Химическое производное" пептида согласно настоящему изобретению содержит дополнительные химические остатки, обычно не являющиеся частью пептида. В объем данного изобретения включены ковалентные модификации пептида. Такие модификации могут быть введены в молекулу в результате взаимодействия являющихся мишенью аминокислотных остатков пептида с органическим дериватизирующим агентом, который способен взаимодействовать с выбранными боковыми цепями или концевыми остатками. Большое количества таких химических производных и способов их получения хорошо известны в данной области. Также в объем изобретения включены соли пептидов согласно изобретению. В используемом в данном описании смысле термин "соли" относится как к солям карбоксильных групп, так и к кислотноаддитивным солям аминогрупп пептидной молекулы. Соли карбоксильной группы могут быть образованы способами, известными в данной области, и включают неорганические соли, например соли натрия,кальция, аммония, железа и цинка и т.п., и соли с органическими основаниями, такие как соли, образованные, например, с аминами, такими как триэтаноламин, аргинин или лизин, пиперидин, прокаин и т.п. К кислотно-аддитивным солям относятся, например, соли с неорганическими кислотами, такими, например, как хлористо-водородная кислота или серная кислота, и соли с органическими кислотами, такими,например, как уксусная кислота или щавелевая кислота. Такие химические производные и соли предпочтительно используют для модификации фармацевтических свойств пептида, когда речь идет о стабильности, растворимости и т.д. Синтетические пептиды и их аналоги согласно изобретению могут быть выбраны из группы, состоящей из пептидов, имеющих последовательности I-V, приведенные в данном описании, где:Val или Ser и Х 5 означает Lys, Glu или Ala. В одном варианте пептид с последовательностью I имеет формулу (Ia) (SEQ ID NO: 1)(ii) пептид с последовательностью II имеет формулу (SEQ ID NO: 12) Е I N Р S Т G G Х 6 Х 7 X8 X9 Х 10 X11 X12 K А K А Т [II] где каждый из Х 6 и Х 7 является Thr, Val или Ala; X8 является Tyr или Phe; X9 является Asn или Asp; X10 является Gln или Glu; Х 11 является Lys или Glu и X12 является Phe или Tyr. В одном варианте пептид с последовательностью II имеет формулу (IIа) (SEQ ID NO: 2)Glu или Lys; X17 означает Met или Ala и X18 означает Asp, Lys или Ser. В одном варианте пептид с последовательностью III имеет формулу (IIIa) (SEQ ID NO: 3)G Y N X19 Х 20 Х 21 Х 22 Х 23 Х 24 S H G X25 X26 L E W I G [IV] где X19 означает Met или Ala; X20 означает Asn, Asp или Arg; X21 означает Trp или Ala; X22 означает Val или Ser; Х 23 означает Lys или Glu; Х 24 означает Gln или Ala; X25 означает Lys или Glu и Х 26 означает Ser или Ala. В одном варианте пептид с последовательностью IV имеет формулу (IVa) (SEQ ID NO: 4)Y Y С A R Х 27 Х 28 X29 Y G X30 X31 X32 G Q T L [V] где Х 27 означает Ser или Phe; X28 означает Gly или Ala; X29 означает Arg, Ala или Glu; X30 означает Asn или Asp; X31 означает Tyr или Phe и Х 32 означает Trp, His или Ala. В одном варианте пептид с последовательностью V имеет формулу (Va) (SEQ ID NO: 5)and Mozes, 1993). После получения пептида согласно настоящему изобретению его способность ингибировать пролиферативный ответ Т-лимфоцитов мышей, которые являются высокоотвечающими на СКВ-индуцирующие аутоантитела, легко может быть определена специалистами в данной области без того, чтобы прибегнуть к большему, чем это необходимо, экспериментированию, используя такие тесты, которые приведены в данном описании. Одним из тестов, который может быть легко проведен, является тест в отношении способности пептидов с заменами ингибировать in vitro пролиферативные ответы некоторых Т-клеточных линий и клонов, специфичных по отношению к СКВ-индуцирующим аутоантителам. Т-клеточные линии и клоны могут, например, представлять собой Т-клеточные линии и клоны, специфичные по отношению к 16/6 Id-мАт (Fricke et al., 1991), созданные из клеток лимфатических узлов иммунизированных мышей ранее описанным способом (Axelrod, О. and Mozes, E. Immunobiology 172: 99 (1986. На клетки воздействуют стимулирующим антителом, презентированным на облученных сингенных клетках селезенки, в присутствии обогащенной среды, каждые 2 недели. Линии Т-клеток клонируют стандартным способом лимитирующего разведения. Пролиферативные ответы указанных Т-клеточных линий и клонов тестируют, например, способом, описанным в разделе "Материалы и способы" в данном описании. Другим тестом, который может быть проведен для того, чтобы выбрать пептиды, обладающие требуемой активностью, является тест в отношении способности пептидов с заменами ингибировать способность Т-клеточных линий и клонов обеспечивать помощь специфичным по отношению к пептидам Вклеткам в присутствии исходного пептида. Пептиды с заменами также можно тестировать в отношении их способности связываться непосредственно, после биотинилирования с продуктами ММС класса II на антигенпрезентирующих клетках соответствующих линий. С этой целью осуществляют N-концевое биотинилирование соответствующих пептидов при 0 С с избытком биотин-N-гидроксисукцинимида в водном растворе (Mozes, E. et al., EMBO J. 8: 4049 (1989. Прилипающие клетки селезенки мышей или прилипающие клетки лимфоцитов периферической крови (PBL) человека (1106/образец) инкубируют с биотинилированными пептидами в PBS, содержащем 0,1% бычий сывороточный альбумин (PBS/БСА) при 37 С в течение 20 ч с последующей инкубацией с фикоэритринстрептавидином в течение 30 мин при 4 С. После каждой инкубации клетки дважды промывают указанным выше раствором. Затем клетки анализируют проточной цитометрией, используя FACScan. В каждом анализе исследуют, минимум, 5000 клеток (что касается указанных выше способов, см., например, Mozes et al., 1989; Zisman et al., 1991). Следующим тестом, который может быть проведен, является тест в отношении способности пептидов ингибировать секрецию цитокина линией Т-клеток или Т-лимфоцитами мышей, которые являются высокоотвечающими на СКВ-индуцирующие аутоантитела. Цитокины регистрируют следующим образом: активность IL-1 оценивают либо в ELISA, используя пару антител: для захвата и регистрирующее антитело (как описано ниже для IL-4, IL-6, IL-10), или используя анализ LBRM-33(1A5) (Conlon, P.J. J.(ФГА) либо надосадками, либо рекомбинантным IL-1 в разных концентрациях для секреции IL-2. После инкубации в течение ночи надосадки клеток 1 А 5 переносят к IL-2-зависимой линии цитотоксических Тлимфоцитов (CTLL). Стимуляцию линии CTLL посредством IL-2 измеряют через 24 ч по включению 3[Н]-тимидина. IL-2 непосредственно регистрируют, используя IL-2-зависимую линию CTLL или посредством ELISA. Уровни IL-4, IL-6, IL-10, INF- и TNF- в надосадках определяют в ELISA, используя антитела к различным цитокинам (Phamingen/San Diego/Ca./USA) согласно инструкциям производителя. В случае пептидов, которые являются позитивными при тестировании в одном или нескольких указанных тестах in vitro, будут основания предполагать наличие активности in vivo. Однако тесты in vivo также можно проводить, не прибегая к большему, чем это необходимо, экспериментированию. Так, например, можно инъецировать взрослым мышам выбранный для исследования пептид либо в день -3, либо в 0 день. Затем мышей иммунизируют индуцирующим заболевание аутоантителом или пептидом. Спустя 10 дней тестируют клетки лимфатических узлов мышей в отношении их способности пролиферировать в ответ на иммуноген, чтобы выявить ингибирующую способность выбранного для исследования пептида. Другой такой тест на животных in vivo заключается в измерении терапевтической активности непосредственно в мышиной модели in vivo в отношении возникновения СКВ, как описано выше. Пептиды можно инъецировать мышам, у которых экспериментальная СКВ индуцируется различными путями, в разных дозах и в разных временных режимах. Чтобы определить фармакокинетические параметры пептидов, включая объем распределения, захват антигенпрезентирующими клетками и клиренс, можно использовать биотинилированные производные пептидов. Концентрацию растворимой фракции пептидов в различных жидкостях организма можно определить посредством ELISA, используя покрытые авидином планшеты и специфичные антитела против пептидов. Связанные с клетками пептиды можно анализировать посредством FACS, используя конъюгированный с флуорохромом авидин или стрептавидин. Кроме того, обработанных мышей можно периодически тестировать для того, чтобы определить влияние пептидов на ответы в виде аутоантител и на проявления заболевания, вызванного у мышей СКВ-индуцирую- 12009123 щим антителом. Другой способ in vivo заключается в толеризации новорожденных мышей выбранным для исследования пептидом с последующей иммунизацией мышей патогенным аутоантителом, таким как 16/6 Id+,или таким же пептидом и наблюдении за проявлениями заболевания, такими как серологические показатели, связанные с лейкопенией, повышенной скоростью оседания эритроцитов, протеинурией, обилием иммунных комплексов в почках и склерозом клубочков. Таким образом, можно видеть, что, кроме предпочтительных вариантов, которые, как было показано, применимы в примерах, приведенных в данном описании, специалисты в данной области смогут определить дополнительные пептиды, которые также будут применимы, следуя руководствам, приведенным в данном описании, без чрезмерного экспериментирования. Также можно осуществить относительно простой тест in vitro, чтобы проанализировать предполагаемую терапевтическую эффективность любого данного пептида с заменами у любого данного пациента с СКВ. Чтобы оценить достижение конечной цели создания пептидов, которые будут связываться с высокой аффинностью с соответствующими молекулами МНС класса II, но не будут приводить к дальнейшей активации Т-клеток и поэтому будут оказывать терапевтическое действие на пациентов с СКВ,можно анализировать пептиды после их биотинилирования в отношении их способности связываться непосредственно с продуктами HLA класса II на антигенпрезентирующих клетках в лимфоцитах периферической крови пациентов с СКВ. В таких анализах можно использовать здоровых контрольных доноров и контрольные пептиды, чтобы подтвердить их специфичность. В одном варианте терапевтическим агентом согласно изобретению является пептид, выбранный из группы пептидов формул I-V, приведенных в данном описании, включая пептиды Ia-Va и их аналоги с заменами и/или делециями. В другом варианте терапевтическим агентом согласно настоящему изобретению является форма одного полиэпитопного пептида. Так, в предпочтительном варианте двойные пептиды, состоящие из двух разных пептидов, выбранных из группы пептидов формул I-V, приведенных в данном описании,ковалентно связывают друг с другом, например коротким участком из остатков аланина или предполагаемым сайтом для протеолиза катепсином. См., например, патент США 5126249 и европейский патент 495049 в отношении таких сайтов. Это будет индуцировать сайт-специфичный протеолиз предпочтительной формы на два требуемых аналога. Альтернативно, из определенного количества одинаковых или разных пептидов согласно изобретению можно образовать пептидный полимер, например, полимеризацией пептидов с использованием подходящего полимеризующего агента, такого как 0,1% глутаральдегид(Audibert et al. (1981), Nature 289: 593). Полимер предпочтительно будет содержать от 5 до 20 остатков пептидов. Такие полимеры пептидов также можно образовать перекрестным сшиванием пептидов или прикреплением множества пептидов на макромолекулярных носителях. Подходящими макромолекулярными носителями являются, например, белки, такие как столбнячный токсоид, и линейные и разветвленные сополимеры аминокислот, такие как линейный сополимер L-аланина, L-глутаминовой кислоты и Lлизина и разветвленный сополимер L-тирозина, L-глутаминовой кислоты, L-аланина и L-лизина (T,G)-AL-, или многоцепочечный пoли-DL-aлaнин (М. Sela et al. 1955, J. Am. Chem. Soc. 77: 6175). Конъюгаты получают, например, сначала связывая пептид с водорастворимым карбодиимидом, таким как гидрохлорид 1-этил-3-(3'-диметиламинопропил)карбодиимида, и затем осуществляя конъюгацию с макромолекулярным носителем, как описано Muller, G.M. et al. (1982) Proc. Natl. Acad. Sci. USA 79: 569. Содержание связанного пептида в каждом конъюгате определяют анализом аминокислот по сравнению с составом отдельного носителя. Согласно одному варианту настоящего изобретения один или несколько активных пептидов могут быть связаны с подходящим макромолекулярным носителем или могут быть полимеризованы в присутствии глутаральдегида. Пептиды, их полимеры или их конъюгаты с подходящими макромолекулярными носителями будут вводиться пациентам в форме, которая обеспечивает их биодоступность, делая их подходящими для лечения. В том случае, если обнаружено, что более одного пептида обладают значительной ингибирующей активностью, то указанные пептиды будут вводиться пациентам в виде препарата, содержащего смесь данных пептидов. Изобретение, кроме того, относится к фармацевтическим композициям, содержащим по меньшей мере один синтетический пептид согласно изобретению, его конъюгат с подходящим макромолекулярным носителем или его полимер необязательно с фармацевтически приемлемым носителем. В объем изобретения включен любой подходящий путь введения, включая пероральный, внутривенный, подкожный, внутрисуставной, внутримышечный, ингаляционный, интраназальный, интратекальный, внутрибрюшинный, интрадермальный, трансдермальный или другие известные пути, включая энтеральный путь. Пределы доз для введения композиций согласно настоящему изобретению должны быть достаточно большими, чтобы получить эффект, при котором, например, иммунный ответ на СКВ-индуцирующие аутоантитела, который измеряется по пролиферации Т-клеток in vitro, был существенно предотвращен или ингибирован, и в дальнейшем заболевание значимо подвергалось лечению.- 13009123 Дозы не должны быть настолько большими, чтобы вызвать неблагоприятные побочные эффекты,такие как нежелательные перекрестные реакции, генерализованная иммуносупрессия, анафилактические реакции и т.п. Эффективные дозы пептидов согласно данному изобретению для применения при лечении СКВ находятся в диапазоне от 1 мкг/кг до 1 мг/кг массы тела. Вводимая доза будет зависеть от возраста, пола,состояния здоровья и массы реципиента, вида сопутствующего лечения, если таковое имеется, частоты лечения и природы требуемого эффекта. Синтетические пептиды согласно изобретению, в частности пептиды с последовательностями I-V,приведенными в данном описании, нацелены на ингибирование или подавление специфичных ответов на антигены у пациентов с СКВ, не влияя на все другие иммунные ответы. Такой подход имеет огромнейшее значение, так как большинство диагностируемых пациентов являются молодыми женщинами, которых необходимо лечить в течение многих лет, а в настоящее время принятое лечение СКВ заключается во введении иммуносупрессирующих агентов, таких как кортикостероиды, и/или цитотоксических лекарственных средств, и те и другие являются неспецифичными и имеют множество неблагоприятных побочных эффектов. Препараты согласно настоящему изобретению могут быть введены парентерально, местно или ректально. Конечно, они могут быть введены в форме, подходящей для каждого пути введения. Например,их можно вводить посредством инъекции, ингаляции, мази, суппозитория и т.д., введение может быть путем инъекции, инфузии или ингаляции; местное - посредством примочки или мази; и ректальное - посредством суппозиториев. Фразы "парентеральное введение" и "введенный парентерально" в используемом в данном описании смысле означают способы введения, отличные от энтерального и местного введения, обычно посредством инъекции, и включают, без ограничения, внутривенную, внутримышечную, внутриартериальную, интратекальную, внутрикапсулярную, внутриглазничную, внутрисердечную, интрадермальную,внутрибрюшинную, через трахею, подкожную, под кутикулу, внутрисуставную, подкапсулярную, субарахноидальную, интраспинальную и внутригрудинную инъекцию и инфузию. Фразы "системное введение" и "введенный системно", "периферическое введение" и "введенный периферически" в используемом в данном описании смысле означают введение соединения, лекарственного средства или другого вещества, отличное от прямого введения в центральную нервную систему так,что оно поступает в систему пациента и таким образом подвергается метаболизму и другим подобным процессам, например подкожное введение. Подробности способов общего приготовления композиций и информацию о дополнительных эксципиентах можно найти в Remington: The Science and Practice of Pharmacy, 20th Edition. Синтетические пептиды могут быть получены, как описано в международной публикации РСТWO 02/067848 или в международной публикации РСТWO 96/30057. Данное изобретение будет более понятным на основании подробного описания экспериментов, которое следует далее. Однако специалист в данной области без труда поймет, что обсуждаемые конкретные способы и результаты являются только иллюстративными для изобретения, которое более полно описано в формуле изобретения, которая следует далее. Подробное описание экспериментов Синтез пептидов I-V и тестирование, показывающее эффективность синтезированных пептидов для лечения СКВ в моделях на животных, описаны в международной публикации РСТWO 96/30057. Дополнительное тестирование на животных описано в международной публикации РСТWO 02/067848. Пример 1. Разработка композиции для соединения 1. Пептиды согласно настоящей заявке описаны в международной патентной публикации WO 02/067848,опубликованной 6 сентября 2002 г., и могут быть получены способами, хорошо известными в данной области (см., например, Peptides: Synthesis, Structure and Applications, ed. by B. Gutte, Academic Press, 1995;Int. J. Pept. Protein Res. (1992) 40: 180-193). Соединение 1 является синтетическим полипептидом, состоящим из 19 аминокислот. Его получают в виде ацетатной соли. Определена растворимость пептида в воде, составляющая менее 0,5 мг/мл. На фиг. 1 показано соединение 1 в виде ацетатной соли. Чтобы разработать композицию с концентрацией пептида, превышающей 2 мг/мл, предпочтительно до 10 мг/мл, проводили эксперименты с несколькими усилителями растворимости. Предварительные эксперименты показали, что концентрации 2 мг/мл нелегко достигнуть. Чтобы разработать композицию для подкожной инъекции, также желательно, чтобы значение рН было в пределах от 4 до 9 и чтобы раствор был изоосмотическим. На основании всестороннего обзора литературы было принято несколько основных подходов, чтобы получить композицию с максимальной растворимостью. Рассматривались следующие факторы: установление рН и буферы; растворители;- 14009123 сорастворители; солюбилизирующие агенты. Способы Соединение 1 растворяли в выбранном растворе усилителя растворимости либо отдельно, либо в комбинации с другими эксципиентами и растворы перемешивали по меньшей мере в течение часа. При необходимости корректировали рН. Растворы исследовали визуально, чтобы оценить растворимость, и отправляли для аналитического определения. В случае нескольких выбранных композиций также тестировали биологическую активность. Результаты В табл. 1 представлен тип усилителей растворимости, используемых для разработки композиции. В табл. 2 и 3 суммированы эксперименты, которые осуществляли с разными усилителями растворимости. В табл. 2 суммирован исходный скрининг, осуществляемый с концентрациями пептида в диапазоне от 5 до 10 мг/мл. Эксперимент, который проводили с более высокой концентрацией пептида, затем повторяли с более низкими дозами (см. табл. 3). Исходные тесты показали, что соединение 1 лучше растворимо в пределах требуемых уровней рН,как кислых, так и основных, но менее стабильно в диапазоне основных значений рН. Соответственно,тестировали несколько буферов и агентов для корректировки рН, включая ацетатный буфер, цитратный буфер и карбонат натрия. Ни один из исходно тестированных буферов не достигал требуемого уровня растворимости пептида. Только выше рН 9,2 и ниже рН 3,0 наблюдались уровни растворимости 2 мг/мл. Однако на начальной стадии были выбраны композиции с ацетатным буфером и цитратным буфером (с маннитом в качестве агента тоничности) для начальных токсикологических исследований. Указанные композиции тестировали в отношении биологической активности, и было доказано, что они активны. Тестировали неводные растворители (см. табл. 1), такие как этанол, глицерин, пропиленгликоль,кремофор и их комбинации, но они не увеличивали растворимость соединения 1. Раствор 30% DMA (диметилацетамида) давал растворимость в требуемых пределах (5-9 мг/мл), но не подходил для фармацевтической композиции из-за своего профиля токсичности. Улучшенную растворимость также наблюдали при использовании 30% (мас./мас.) ПЭГ 400 (5-9 мг/мл). Указанную последнюю композицию выбрали для токсикологических исследований, но показано, что она является неактивной в биологическом анализе и может быть причиной некоторых неблагоприятных эффектов при исследовании токсичности на мышах. Таким образом, было решено больше не заниматься данной композицией. Учитывая предварительные эксперименты, неводные растворители не использовали в данных композициях. Тестировали несколько аминокислот (см. табл. 1), включая L-аргинин, L-глутаминовую кислоту, Lглицин и L-лизин, чтобы повысить растворимость белка. Растворимость пептида в L-аргинине была на требуемом уровне, но конечное значение рН было выше 9. Попытка снизить рН или использовать HClсоль аргинина приводила к выпадению пептида в осадок. Также тестировали сывороточный альбумин человека, и он повышал растворимость пептида при низких концентрациях пептида (1 мг/мл) (см. табл. 3). Однако вследствие его потенциальной иммуногенности и низкой растворимости пептида его не использовали в дальнейших экспериментах. Наполнители (см. табл. 1), включая маннит, сорбит и декстран, тестировали отдельно и в комбинации с другими эксципиентами, но они не повышали растворимость пептида в растворе. Сорастворители (см. табл. 1), включая полисорбат 20 и полисорбат 80, тестировали отдельно и в комбинации с другими эксципиентами. Хотя более низкие концентрации полисорбатов (вплоть до 6%) не увеличивали растворимость пептида, более высокие концентрации (до 10% - см. табл. 2) повышали растворимость пептида до 2 мг/мл. Однако считали, что такие высокие концентрации полисорбатов являются неподходящими для фармацевтических композиций. Также тестировали два типа циклодекстринов, которые оба одобрены для применения в продаваемых парентеральных продуктах: гидроксипропилциклодекстрин и сульфобутиловый эфирциклодекстрин (каптизол). Оба заметно увеличивали растворимость пептида (концентрации на уровне 10 мг/мл для гидроксипропилциклодекстрина и 2,5 для каптизола). Тестировали биологическую активность двух композиций циклодекстрина и обнаружили, что они равны по активности пептиду, взятому отдельно.Captisol (каптизол) является коммерчески доступным полианионным производным -циклодекстрина, в котором соль сульфоната натрия отделена от гидрофобной полости спейсерной группой простого бутилового эфира или простого сульфобутилового эфира (SBE). Каптизол является торговой маркой препарата, гептазамещенного простым сульфобутиловым эфиром -циклодекстрина (SBE7CD) CyDexInc. (www.captisol.com). Структура каптизола позволяет молекулам лекарственного средства войти в гидрофобную полость, таким образом изолируя молекулу лекарственного средства от водного растворителя. Так как наружная поверхность каптизола является гидрофильной, то растворимость готовой молекулы лекарственного средства при этом повышается. Применение циклодекстринов для повышения растворимости молекул лекарственных средств описано в патентах США 5134127 и 5376645, полное содержание которых включено, таким образом, в виде ссылки. Согласно литературным данным каптизол CyDex Inc. является безопасным при парентеральном- 15009123 введении и не проявляет нефротоксичности, связанной с бета-циклодекстрином. По сравнению с бетациклодекстрином каптизол проявляет сравнимые или более высокие свойства комплексообразования и более высокую растворимость в воде выше 90 г/100 мл - 50-кратное повышение. Заключение Обнаружено, что несколько усилителей растворимости подходят для требуемого диапазона растворимости: DMA, ПЭГ-400, диметилацетамид, полиэтиленгликоль, полиоксилированное касторовое масло,N-метил-2-пирролидинон, 1-этенил-2-пирролидинон, полисорбат 20, полисорбат 80, гидроксипропил-циклодекстрин и сульфобутиловый эфирциклодекстрин (каптизол). Подтверждено, что из указанных усилителей растворимости оба циклодекстрина являются лучшими в отношении растворимости, биологической активности и стабильности. Соответственно, было решено выбрать каптизол в качестве усилителя растворимости для применения в композициях согласно примеру 5 и исследовать обе композиции на основе циклодекстрина далее. Конечная композиция для клинических исследований согласно примеру 5 состоит из 120 мг/мл каптизола в воде с требуемым количеством пептида (0,5, 1,0 или 2,5 мг/мл), и НСl,и NaOH для корректировки рН. Таблица 1 Усилители растворимости, используемые для разработки композиции соединения 1 Таблица 2 Список сорастворителей и стабилизаторов, оцениваемых в композициях пептидного соединения 1- 17009123 Таблица 3 Композиции соединения 1 при низких концентрациях пептида Пример 2. Протокол приготовления раствора соединения 1 в каптизоле. Стандартные способы растворения, такие как смешивание сухого соединения 1 и сухого каптизола в воде или добавление соединения 1 к приготовленному раствору каптизола и воды, не приводили к полному растворению в требуемых концентрациях. Тестировали несколько разных концентраций как соединения 1, так и каптизола при разных уровнях рН. Однако следующий способ приготовления раствора соединения 1 в каптизоле приводил к полному растворению в требуемых концентрациях. Вещества Каптизол, соединение 1 и вода. Способ 1. Взвешивают соответствующее количество каптизола, чтобы получить конечную концентрацию 120 мг/мл. 2. Добавляют 80% конечного количества воды и перемешивают в течение 10 мин с использованием магнитной мешалки. 3. Взвешивают соединение 1, чтобы получить конечную концентрацию 2,5, 2,0, 1,0, 0,5 или 0,1 мг/мл. 4. Добавляют пептид к раствору каптизола. Перемешивают в течение 1 ч. 5. Повышают рН, чтобы получить прозрачный раствор (в композиции 2,0 мг/мл может быть необходимо повысить рН немного выше 9). рН необходимо корректировать, используя 1,0 н. НСl и 1,0 н.NaOH. Перемешивают в течение 10 мин. 6. При необходимости корректируют рН до диапазона от 7,5 до 8,5 (используя либо НСl, либоNaOH, 1,0 н.). 7. Добавляют воду до конечного объема. 8. Фильтруют раствор через фильтр из ацетата целлюлозы с порами 0,2 мкм. 9. Регистрируют конечное значение рН. 10. Делят на аликвоты и хранят при подходящей температуре. Пример 3. Лиофилизация раствора соединения 1 и каптизола. Данный способ лиофилизации отличается от других способов лиофилизации тем, что процентное содержание сухого остатка в композиции выше (12%), тогда как лиофилизованные продукты обычно содержат от 5 до 10% сухого остатка. Оборудование В качестве лиофильной сушилки использовали лиофилизатор Edwards Lyoflex 0.6. Осуществляли IQ/OQ- 18009123 оборудования и проверяли в отношении соответствия посредством проверки качества до разработки способа. Готовили растворы соединения 1 и каптизола с концентрациями соединения 1 0,5, 1,0 и 2,5 мг/мл. Загружаемый объем доводили до 1 мл (1,05 г). Основные стадии способа 1. Замораживание. 2. Выдерживание (при низкой температуре). 3. Сушка в вакууме в две стадии: 3.1. первичная сушка - нагревание стеллажа для сушки до верхней поддерживаемой температуры,контроль температуры стеллажа на верхнем поддерживаемом уровне,3.2. вторичная сушка - снижение давления до минимального значения при верхней поддерживаемой температуре стеллажа. Партии 1-3 Замораживание - замораживание проводили от комнатной температуры до -40 С в течение 2 ч. Стеллажи поддерживали при -40 С в течение 3 ч. Сушка - сушку осуществляли при давлении 110 мкбар. Температуру стеллажа увеличивали до 20 С в течение 13 ч и поддерживали при данной температуре еще в течение 13 ч. Общее время процесса составляло 31 ч. Результаты Результаты по содержанию воды: партия 1: 3,8%,партия 2: 4,0% и партия 3: 4,9%. Партии 4 и 5 Так как результаты по содержанию воды при использовании способов, приводящих к получению партий 1, 2 и 3, были выше, чем требуемое значение, было решено добавить стадию вторичной сушки при той же самой температуре и при низком давлении. Сушка - сушку осуществляли при давлении 110 мкбар. Температуру стеллажа увеличивали до 20 С в течение 13 ч и поддерживали при данной температуре еще в течение 13 ч (партия 4) или 8 ч (партия 5). Давление понижали до 10 мкбар дополнительно в течение 5 ч. Общее время процесса составляло 36 ч. Результаты Результаты по содержанию воды: партия 4: плацебо - 3,0%,1 мг/мл - 3,9%,партия 5: плацебо - 4,1%. Заключение Как показано, разработан удовлетворительный способ лиофилизации соединения 1 с каптизолом. Вследствие высокого процентного содержания сухого остатка и поэтому конденсированной лиофилизуемой массы разработанный способ является более длительным, чем имеющиеся современные циклы лиофилизации пептидов, и имеет дополнительную стадию вторичной сушки. В табл. 4 суммирован разработанный способ. Таблица 4 Пример 4. Исследование биологической активности in vivo лиофилизованного раствора соединения(DP, 1 мг/флакон, 12% каптизол). Биологическую активность проверяли по ингибированию секреции IL-2 из специфичных по отно- 19009123 шению к стандартному образцу (RS) соединения 1 Т-клеток после подкожной (п/к) обработки лиофилизованным раствором соединения, т.е. лекарственным продуктом (DP) при двух концентрациях. Результаты обработки сравнивают с результатами обработки мышей соединением 1 (RS) в фосфатно-солевом буфере (PBS). Результаты показаны в таблице ниже и на фиг. 2. План эксперимента 1. Иммунизация (соединение 1 RS, эмульгированное с CFA, во все четыре подушечки лап) 0 день 2. Обработка (п/к в заднюю часть шеи, в 200 мкл раствора) 0 день 3. Активация in vitro 10 деньb) пептидом с обратным порядком аминокислот относительно соединения 1 (негативный контроль);c) Con А (позитивный контроль). 4. Инкубация культуры в течение 20 ч при 37 С в инкубаторе во влажной атмосфере с 5% СО 2. 5. Измерение IL-2 с помощью ELISA. Таблица 5 Таблица экспериментальных групп НПКО = Ниже предела количественного определения. Строки 1-4 не включали в кривую. НП = Не применимо. Пример 5. Оценка оптимальной дозы для обработки. В дальнейшем описании использовали следующие сокращения. Фосфатно-солевой буфер Стандартный образец Подкожное Трипановый синий Трансформирующий фактор роста-бета Вода для инъекции Введение Группу из 20 мышей иммунизировали 50 мкг/мышь соединения 1 RS. Иммунизированных мышей распределяли на пять групп обработки, которые указаны далее: плацебо, 25, 50, 100 и 200 мкг/мышь соединения 1 DP (подкожное введение). Через 10 дней после иммунизации и обработки извлекали LN и готовили суспензию отдельных клеток. Затем измеряли секрецию in vitro IFN- и TGF- культивируемыми клетками в ответ на активацию несколькими концентрациями соединения 1 RS. План эксперимента 1. Иммунизация 0 день 2. Обработка соединением 1 DP 0 день 3. Активация in vitro клеток LN от обработанных мышей 10 день 4. Сбор культуральной среды (для определения IFN-) 12 день 5. Сбор культуральной среды (для определения IGF-) 13 день 6. ELISA в отношении IFN7. ELISA в отношении TGFТаблица 7 Экспериментальные группы Материалы и реагенты Животные. Мыши: 20 самок мышей BALB/c, поставляемых центром разведения животных Harlan, Rehovot. Возраст во время иммунизации (неделя+дни): 10. Средняя масса мышей, включенных в эксперимент: 19,01 г. Вещества. Общие реагенты: 70% этанол получали из 96% этанола разбавлением очищенной Н 2O. Приготовление растворов соединения 1 для иммунизации Эмульсию CFA-соединение 1 RS (500 мкг/мл, 50 мкг/мышь) готовили следующим образом. 1. 1,874 мг соединения 1 растворяли в 1,87 мл WFI, получая раствор 1 мг/мл. 2. Раствор проверяли с помощью рН-индикаторной полоски и обнаружили, что он имеет рН 5. 3. 1,5 мл раствора эмульгировали с 1,5 мл CFA, получая конечную концентрацию 500 мкг/мл. Приготовление растворов для обработки Обработку проводили п/к-инъекцией 200 мкл раствора. Приготовление 12% раствора каптизола 1,2 г каптизола растворяли в 10 мл WFI, получая 12% раствор каптизола. Последовательность операций при проведении эксперимента Взвешивание мышей. Мышей взвешивали перед иммунизацией. Средняя масса мышей: 19,010,97 г.- 21009123 Иммунизация. Иммунизацию проводили инъекцией 100 мкл эмульсии для иммунизации (50 мкл в каждую подушечку задних лап). Обработка. После стадии иммунизации мышей обрабатывали п/к-инъекцией по 200 мкл из указанных растворов для обработки, содержащих соединение 1 DP или 12% каптизол, в заднюю часть шеи. Культивирование in vitro. Мышей забивали смещением шейных позвонков. LN извлекали из задних конечностей и переносили в стерильную чашку Петри, содержащую примерно 5 мл RPMI. Клетки выделяли осторожным продавливанием ткани через стальную сетку с ячейками 200 мкм. Клетки собирали и центрифугировали при 300 g в течение 10 мин при комнатной температуре. Суспензии отдельных клеток готовили из объединенных LN из каждой экспериментальной группы. Суспензии 2,5 и 5,0 млн клеток/мл/лунку культивировали с соединением 1 RS (0-100 мкг/мл) в ЕМ-1. Секрецию IFN- и TGF- в качестве показателя клеточного ответа определяли посредством ELISA культуральной среды (48 ч для IFN- и 72 ч для TGF-). Таблица 8 Экспериментальные группы in vitro Приготовление суспензий клеток Результаты подсчета клеток и приготовление клеточных суспензий (10106/мл) 009123 Приготовление суспензии клеток (5106/мл) Суспензию 1010 клеток/мл разбавляли 1:2 добавлением 5 мл ЕМ-1 к 5 мл клеточной суспензии. Инкубация культур клеток LN в 48-луночных планшетах Готовили 3 планшета для культуры ткани. В каждый планшет добавляли следующее. Контроль фона (клетки, инкубированные с культуральной средой) 0,5 мл суспензии клеток 0,5 мл культуральной среды (ЕМ-1) Позитивный контроль системы (клетки, стимулированные Con A) 0,5 мл суспензии клеток 0,5 мл Con A 5 мкг/мл в ЕМ-1 (конечная концентрация 2,5 мкг/лунку) Клетки, инкубированные с активирующими растворами соединения 1 (образцы) 0,5 мл суспензии клеток 0,5 мл соединения 1 RS 6,25-200 мкг/мл (конечная концентрация 3,125-100 мкг/мл/лунку) Инкубация культур клеток LN в 96-луночных планшетах После приготовления 48-луночных планшетов готовили 96-луночные планшеты, используя 100 мкл из суспензии клеток и 100 мкл из активирующих растворов. Планшеты для культивирования инкубировали при 37 С в инкубаторе с увлажнением и 5% СO2 в течение 48 или 72 ч. Сбор надосадков Культивируемые планшеты центрифугировали при 300 g в течение 10 мин при комнатной температуре. Надосадки (850 мкл из каждой лунки) переносили либо в зеркальные планшеты, либо в пробирки. Затем надосадок делили на аликвоты для работы (две аликвоты по 200 и одна аликвота объемом 450 мкл),чтобы избежать повторного замораживания/оттаивания образцов. Каждую пробирку помечали соответствующей подробной информацией. 1. Код эксперимента и время после инкубации. 2. Номер группы и образца. 3. Активатор и концентрация. 4. Дата сбора надосадка. Надосадки хранили при -20 С вплоть до использования в ELISA. Результаты Таблица 10 Сводная информация о группах 6- 23009123 Конечные концентрации цитокина Таблица 11-А Конечная концентрация цитокина, пг/мл (2,5 млн клеток/лунку) Результаты также представлены на фиг. 3, 4. Результаты наблюдения Секреция IFN-. 1. В группе плацебо показана линейная дозовая зависимость при активации соединением 1 in vitro. Данный график сходен с графиком, полученным для модели ex vivo при такой же дозе иммунизации(50 мкг/мышь) и культуральной среде (ЕМ-1). 2. Дозовая зависимость при активации соединением 1 in vitro имела место во всех тестированных группах. 3. Значимое ингибирование секреции IFN- наблюдали при всех дозах, используемых для обработки (среднее 95% ингибирование при дозе обработки 25 мкг/мышь). Может быть выявлена обратная корреляция между дозой, которая служит для обработки, и % ингибирования, в основном, при использовании 5106 клеток/лунку. При использовании 2,5106 клеток/лунку обработка животных 50 мкг/мышь давала большее ингибирование, чем 100 или 200 мкг. Точка 25 мкг пропущена (недостаток клеток). 4. Большее ингибирование наблюдали при использовании 5106 клеток/лунку вместо 2,5106 клеток/лунку. 5. На линейном диапазоне кривой SD ингибирования в % было низким. 6. Техническая проблема, связанная с Con А, выражена при использовании 2,5106 клеток/лунку. Секреция TGF-. 1. В группе плацебо не наблюдали дозовой зависимости при активации in vitro соединением 1. Секретируемый уровень TGF- был ниже предела определения в ELISA во всех других группах обработки. Пример 6. Оптимизации цикла сублимационной сушки в случае соединения 1 и каптизола для инъекции (0, 0,5, 1,0 и 2,5 мг/флакон). Цель Целью данного исследования была оптимизация цикла сублимационной сушки соединения 1 с каптизолом для инъекции, чтобы улучшить форму лиофилизуемой массы и избежать смятия и растрескивания. Соответственно, было решено улучшить и оптимизировать цикл лиофилизации. Данный цикл перенесен в промышленные лиофилизаторы для производства партий для испытаний фазы I.- 24009123 Оптимизация способа Готовили партии пептида с концентрациями 0,5, 1,0, 2,5 мг/мл и плацебо и осуществляли несколько циклов сублимационной сушки. В качестве сублимационной сушилки использовали лиофилизатор EdwardsLyoflex 0.6. Тестировали растворимость, содержание воды и внешний вид лиофилизованного материала. Согласно полученным результатам выбран новый цикл лиофилизации соединения 1. Вследствие высокого процентного содержания сухого остатка (12%) и поэтому конденсированной лиофилизуемой массы новый способ является более длительным, чем цикл лиофилизации в примере 3, и имеет дополнительную стадию первичной сушки. В табл. 12 суммированы различия между способами. Таблица 12 Пример 7. Действие соединения 1 (введенного в каптизоле) на симптомы волчанки у склонных к СКВ самок мышей (NZBxNZW)F1. Пациентов, принимающих участие в клинических испытаниях, будут лечить соединением 1, используя в качестве эксципиента каптизол (натриевую соль сульфобутиловый эфир-бета-циклодекстрина). По этой причине важно определить, будет ли обработка мышей (NZBxNZW)F1 композицией соединения 1, вводимого в каптизоле, оказывать такое же лечебное действие на симптомы волчанки, которое наблюдается при обработке мышей такой же линии соединением 1 в PBS. С этой целью самок мышей (NZBxNZW)F1 (примерно 8-месячного возраста) делили на 3 группы,которые обрабатывали подкожно 1 раз в неделю в течение 10 недель либо отдельно каптизолом (n=8),либо 25 или 50 мкг/мышь соединения 1 в каптизоле (n=9 и 10, соответственно). Указанные дозы были выбраны потому, что предшествующие исследования показали, что дозы в данном диапазоне более эффективны в ослаблении симптомов СКВ, чем более высокие тестированные дозы (100 и 200 мкг/мышь). Одну и ту же партию лекарственного вещества использовали в данном исследовании и в первом клиническом испытании фазы I соединения 1. У мышей проводили наблюдения в отношении анти-днДНК-антител и протеинурии. После забивания мышей определяли интенсивность ICD в почках. Как можно видеть на фиг. 5, не наблюдалось значимых различий между группами по уровням днДНК-специфичных антител после 10 инъекционных обработок. В табл. 13 также показано, что целебное действие обработки соединением 1 может наблюдаться, начиная с 5-й инъекции, и поддерживается до 10-й инъекции. Средние уровни протеинурии в контрольной группе, обработанной каптизолом, были, соответственно, выше, чем в группах, обработанных соединением 1. В табл. 13 также показано, что наблюдалось уменьшение интенсивности ICD в почках в обеих группах, обработанных разными дозами соединения 1. Имела место общая тенденция, свидетельствующая,что более низкая доза (25 мкг/мышь) была более эффективна, чем более высокая доза (50 мкг/мышь) в снижении клинических симптомов СКВ у данных мышей.- 25009123 Таблица 13 Клинические симптомы СКВ у мышей (NZBxNZW)F1, обработанных 25 или 50 мкг/мышь соединения 1b Гибель одного животного с высоким уровнем протеинурии привела к более низкому среднему значению в группе. с р 0,05 (по сравнению с контрольными мышами, обработанными каптизолом; Манн-Уитни). На фиг. 6 показаны типичные срезы одной почки из каждой группы обработки. Срезы верхнего ряда получены от мыши, обработанной каптизолом, срезы среднего ряда от мыши, обработанной 50 мкг/мышь соединения 1, и срезы нижнего ряда получены от мыши, обработанной 25 мкг/мышь соединения 1. Можно видеть, что интенсивность отложений иммунных комплексов, наблюдаемая в срезах почек мышей,обработанных соединением 1 (растворенным в каптизоле) при обоих уровнях доз, была намного ниже,чем интенсивность, наблюдаемая в контрольной группе. Пример 8. Клиническое исследование фазы I. Проводимое в нескольких центрах, рандомизированное, двойное слепое, с плацебо-контролем, с использованием однократной дозы, с четырьмя вариантами исследование фазы I, чтобы оценить переносимость и безопасность подкожной инъекции соединения 1 в каптизоле у субъектов с СКВ. Это было первое клиническое исследование с использованием соединения 1 в каптизоле на людях во Франции. Основной целью исследования была оценка переносимости и безопасности соединения 1 в каптизоле, вводимого в виде однократной п/к-инъекции субъектам с СКВ. Второй целью исследования была оценка иммунологических ответов после однократной п/к-дозы соединения 1 в каптизоле у указанных субъектов. В исследовании принимали участие тридцать шесть (36) субъектов. Для включения пациентов с СКВ в исследование должно выполняться по меньшей мере четыре критерия, используемых для диагностики волчанки согласно Американской коллегии ревматологии. Пациенты также должны иметь стабильное, легкое/умеренное заболевание и оценку в баллах, меньше или равную 10, согласно индексу активности заболевания СКВ SLEDAI. Каждый пациент получал однократную п/к-инъекцию перерастворенного соединения 1 для инъекции или соответствующего плацебо (каптизол) в соответствии со следующим распределением по группам: группа А: плацебо (каптизол),группа В: 0,5 мг соединения 1 в каптизоле,группа С: 1 мг соединения 1 в каптизоле,группа D: 2,5 мг соединения 1 в каптизоле. Проводили стандартную группу тестов на безопасность, включая сбор крови и мочи для лабораторных тестов, во время отбора, в день введения дозы, через 24 ч после введения дозы и на 2, 4 и 8 неделе после введения дозы. Перед введением дозы и при последующих намеченных посещениях брали образцы крови для иммунологических тестов, имеющих отношение к СКВ, тестов на антитела против соединения 1 и анализа пролиферации PBL. Выполняли следующие иммунологические тесты: Кумбса (прямой и непрямой),С 3, С 4 и СН 50,общий IgG, IgM и IgA,ANA, анти-днДНК (анализ Фарра), анти-онДНК,анти-ENA (включая анти-La, анти-Ro, анти-РНП, анти-Sm),антитела против кардиолипина,VDRL,антитела FTA,ревматоидный фактор.- 26009123 Безопасность и переносимость соединения 1 в каптизоле в популяции субъектов оценивали на основе следующих критериев: наличие нежелательных явлений (АЕ), включая вспышку СКВ,основные жизненные показатели,ЭКГ в 12 отведениях,изменения при физическом обследовании,стандартные клинические лабораторные тесты,оценка критерия SLEDAI,результаты иммунологических тестов. Подробное описание фазы Iа клинического исследования Основные исполнители исследования и соответствующие места исследования. Шесть (6) исследовательских центров во Франции: проф. Jean Charles Piette (Hopital La PitieCedex), проф. Xavier Mariette (Hopital Bicetre, Kremlin Bicetre). Соединение 1 (в каптизоле), плацебо, вода для инъекций - ампулы, доза и способ введения. Соединение 1 в каптизоле (120 мг/флакон) из флаконов инъецировали подкожно в виде однократной дозы субъекту в следующих дозах: 0,5 мг соединения 1/флакон в каптизоле, 1 мг соединения 1/флакон в каптизоле и 2,5 мг соединения 1/флакон в каптизоле. Плацебо для соединения 1. 120 мг каптизола/флакон (идентичный по внешнему виду с флаконами соединения 1 в каптизоле). Методика. В нескольких центрах проводили рандомизированное, двойное слепое, с плацебо-контролем, с четырьмя вариантами исследование, используя однократную подкожную инъекцию соединения 1 или плацебо. Отбор пациентов с СКВ осуществляли до 21 дня перед исходными процедурами. Каждого выбранного для исследования субъекта случайно распределяли в одну из 4 групп лечения: подкожная инъекция 0,5, 1 или 2,5 мг соединения 1 или соответствующего плацебо. Всех субъектов принимали в клинике в день, предшествующий введению дозы. Каждый субъект получал однократную дозу одного из перечисленных выше средств лечения. Субъектов отпускали из клиники через 24 ч после введения дозы. В дальнейшем проводили наблюдение за субъектами на 2, 4 и 8 неделе после введения дозы. Брали образцы крови (сыворотки и цельной крови) для лабораторных тестов на безопасность при отборе, в день введения дозы (перед дозой), во 2 день (после дозы), на 2, 4 и 8 неделе (последнее посещение). Образцы крови для иммунологических тестов брали при отборе, в день введения дозы (перед дозой) и на 4 и 8 неделе. Пролиферацию лимфоцитов периферической крови (PBL) оценивали в день введения дозы (перед дозой) и на 2, 4 и 8 неделе. Количество субъектов (общее и для каждого режима лечения). Тридцать шесть (36) субъектов случайно распределяли в данном исследовании следующим образом: 9 субъектов в группу лечения 0,5 мг, 9 субъектов в группу лечения 1 мг, 10 субъектов в группу лечения 2,5 мг и 8 субъектов в группу лечения плацебо. Диагностика и основные критерии для включения в исследование. Выбираемые для данного исследования субъекты были пациентами с СКВ, которые удовлетворяли по меньшей мере четырем диагностическим критериям Американской коллегии ревматологии (ACR). Их состояние болезни должно быть стабильным, от слабого до умеренного с оценкой в баллах, равной или меньшей 10, согласно индексу активности заболевания СКВ, обновленному в 2000 г. (SLEDAI 2K). Из участия исключали пациентов с СКВ, которые сообщали об астме нестабильного или тяжелого течения, инсульте, остром инфаркте миокарда, нестабильной ангине, кровоизлиянии в головной мозг и эмболии легких в течение 6 месяцев перед отбором для исследования. Пациенты с СКВ, которые имели какие-либо клинически значимые или нестабильные медицинские или хирургические показания, сахарный диабет, заболевание печени (цирроз, активный гепатит, портальная гипертензия и/или асциты), клинически значимую гипертонию, в истории болезни какую-либо злокачественную опухоль, диализ или хроническое обструктивное заболевание легких (COPD), также исключались из участия в исследовании. Также из участия в исследовании исключали пациентов с СКВ, которые подвергались плазмаферезу или которых лечили в течение 3 месяцев перед отбором одним из лекарственных средств, перечисленных ниже: преднизон 30 мг/сутки или больше (или эквивалентная доза другого кортикостероида), внутривенные кортикостероиды, внутривенный иммуноглобулин G (IgG), пероральные антикоагулянты и любые цитотоксические средства (например, азатиоприн, хлорамбуцил, циклофосфамид, микофенолят мофетила, метотрексат, такролимус). Кроме того, из исследования исключали пациентов с СКВ, приступивших к лечению кортикостероидами (более 10 мг/сутки преднизона или эквивалентная доза другого кортикостероида) и/или противомалярийными средствами в течение последних 3 месяцев перед отбором. Хотя предпринимали попытку сохранить исходно начатое лечение СКВ на протяжении всего курса исследования, однако, исследователи могли изменить лечение участника в любое время в ходе исследо- 27009123 вания, чтобы поддержать и оптимизировать здоровье пациента. Критерии для оценки Безопасность. Оценивали следующие параметры безопасности при отборе, во время госпитализации и при повторных посещениях, включая последнее посещение: основные жизненные показатели (систолическое давление крови, диастолическое давление крови, пульс, насыщение кислородом, температура и масса),ЭКГ в 12 отведениях, изменение при физическом обследовании и стандартные клинические лабораторные тесты на безопасность. Нежелательные явления регистрировали в день введения дозы и при каждом посещении в дальнейшем. Иммунология. Имеющие отношение к СКВ иммунологические тесты проводили при отборе, во время госпитализации и при последующих посещениях, включая последнее посещение. За связанными с лекарственным средством иммунологическими ответами следили, используя анализ пролиферации PBL и анализ антител против соединения 1, в день введения дозы и при последующих посещениях, включая последний визит. Активность заболевания. Оценку активности заболевания с использованием подсчета индекса активности заболевания СКВ,обновленного в 2000 г. (SLEDAI 2K), проводили при отборе, во время госпитализации и при последующих посещениях, включая последний визит. Статистические способы. Использовали компьютерную программу SAS, версия 9.0, чтобы проанализировать и представить данные, собранные во время данного исследования. Не проводили расчетов эффективности и не проводили формальной проверки гипотезы в данном исследовании фазы Ia. Нежелательные явления. Возникновение и частоту нежелательных явлений представляли по классам, соответствующим органам и системам, и с помощью предпочтительной терминологии согласно словарю MedDRA, версия 5.0. Данные сгруппированы по группам обработки. Клинические лабораторные данные. Описательные статистические данные, касающиеся лабораторных показателей, включая количество определений, среднее значение, стандартное отклонение, минимальное и максимальное значения, определяли при отборе, в 1 день (перед дозой), 2 день, на 2, 4 и 8 неделе, и они представлены по группам лечения. Изменения от исходного уровня в каждой временной точке/при посещении также представлены для каждого посещения в соответствии с назначением лечения. Процент аномальных результатов (низких или высоких в соответствующих случаях) представлен на основе параметра в соответствии с группой лечения и посещением/временной точкой. Проводили анализ сдвига от исходного уровня к 24 ч после введения дозы и от исходного уровня к последнему посещению. Основные жизненные показатели. Описательные статистические данные, касающиеся основных жизненных показателей, включая количество определений, среднее значение, стандартное отклонение, медианное, минимальное и максимальное значения, определяли при отборе, в 1 день (перед и после введения дозы и в каждой временной точке), 2 день, на 2, 4 и 8 неделе, и они сгруппированы в соответствии с назначенным лечением. Изменения от исходного уровня в каждой временной точке/при посещении также представлены для посещения в соответствии с назначением лечения. Масса. Описательные статистические данные, касающиеся массы (кг) исходного уровня, в конце исследования и изменение от исходного уровня представлены по группам лечения. ЭКГ. Представлены описательные статистические данные, касающиеся параметров ЭКГ исходного уровня, в конце исследования и изменения от исходного уровня. Представлен анализ сдвига в виде таблиц сдвига от исходного уровня к окончанию исследования между нормальными/аномальными или имеющимися/отсутствующими параметрами ЭКГ. Потенциально клинически значимые (PCS) измерения QTc(по Базетту) идентифицировали согласно предварительно определенным критериям. Представлены таблицы анализа сдвига между PCS и не-PCS абсолютными значениями QTc (по Базетту) и таблица частотыPCS-изменения в QTc (по Базетту) от исходного уровня к любому посещению. Физическое обследование. Результаты физического обследования анализировали по частоте встречаемости субъектов с аномальными или нормальными данными для каждой системы организма на исходном уровне и при последнем визите. Также применяли анализ сдвига от нормальных к аномальным значениям и наоборот. В том случае,когда не происходило изменения относительно исходного уровня, использовали определение "другое". Иммунологические тесты, связанные с соединением 1. В случае иммунологических параметров рассчитывали описательные статистические данные,включая количество определения, среднее значение, стандартное отклонение, медианное, минимальное и- 28009123 максимальное значения, и они представлены по группам лечения и посещениям. Изменение от исходного уровня к каждому последующему визиту также представлено по группам лечения. В соответствующих случаях количество и процент субъектов с негативными/позитивными результатами представлены по группам лечения и посещениям.SLEDAI 2K. Представлены описательные статистические данные, включая среднее значение, стандартное отклонение, медианное, минимальное и максимальное значения SLEDAI 2K. Результаты фазы Iа клинического исследования Распределение субъектов и характеристики СКВ. Тридцать шесть (36) исследуемых субъектов начинали и закончили данное исследование по протоколу. Большинство субъектов (34) во всех группах лечения были женского пола (94,4%) и европеоидной расы (30, 83,3%). Средний возраст во всех группах лечения составлял 35,6 лет (диапазон средних значений от 32 до 39 лет). Большинство субъектов (91,7%) имели диагностические критерии согласно Американской коллегии ревматологии (ACR) от 4 до 6 и среднюю для группы оценку SLEDAI 2K в пределах от 2,1 до 4,1. Результаты по безопасности. Не было заметного различия между исследуемыми группами лечения лекарственным средством и группой, принимавшей плацебо, по частоте встречаемости АЕ. Наиболее распространенными АЕ во всех группах были головная боль, классифицируемая как слабая или умеренная по природе, и реакция в месте инъекции, классифицируемая как слабая по природе. Не наблюдали зависимого от дозы ответа. В ходе исследования не встречались серьезные нежелательные явления (SAE) или АЕ, классифицируемые как тяжелые. Не наблюдали клинически значимого эффекта, который можно приписать исследуемому лекарственному средству, в отношении показателей гематологии, биохимии или анализа мочи. Не наблюдали клинически значимого эффекта, который можно приписать исследуемому лекарственному средству, в отношении параметров основных жизненных показателей (систолического кровяного давления, диастолического кровяного давления, пульса, насыщения кислородом). Не наблюдали клинически значимого эффекта, который можно приписать исследуемому лекарственному средству, в отношении температуры и массы. Не наблюдали клинически значимых различий между группами, которых лечили соединением 1 и плацебо, в отношении измерений ЭКГ по категориям и параметров ЭКГ в цифровой форме. Не было зарегистрировано абсолютного значения PCS-QTc и изменения QTc относительно исходного уровня 60 мс. Сходное количество субъектов в группах, которых лечили соединением 1 и плацебо, имели изменениеQTcB относительно исходного уровня от 30 до 60 мс. Не отмечено клинически значимого действия соединения 1 при физическом обследовании. Иммунологические результаты. Оценка образцов сыворотки всех субъектов показала, что однократное подкожное введение соединения 1 при уровне доз 0,5, 1 и 2,5 мг/пациенту не индуцировало образование антител, специфичных по отношению к соединению 1. У 7 субъектов имел место ответ на соединение 1 выше предела отсечения. Указанные повышенные уровни антител уже имелись перед введением дозы. Не наблюдали повышения уровней антител в последующий период (2 месяца) исследования. Сыворотки указанных субъектов анализировали в отношении изотипа реагирующих антител. Ответ у 2 субъектов был связан с изотипом IgM,и с изотипом IgG - у 2 других. Ни у одного из семи не было специфичных антител IgE. Анализ лимфоцитов периферической крови (PBL) показал, что 50% субъектов (18) были классифицированы как отвечающие (SI2) со сходным распределением во всех группах лечения. Т-клеточный ответ был относительно низким, и невозможно было выявить связи между лечебной дозой соединения 1 или концентрацией, используемой в анализе, и статусом респондера/нереспондера, принимая во внимание, что вводили только однократную п/к-дозу исследуемого лекарственного средства. Также не наблюдалось признаков повышенной частоты встречаемости статуса респондера с течением времени. Анализ столбнячного токсоида (ТТХ), который служит в качестве контроля безопасности, показывает, что ответ на ТТХ сохранялся в течение периода исследования во всех группах лечения, свидетельствуя о том, что соединение 1 в каптизоле не изменяет иммунологический ответ на повторно введенный антиген ТТХ. Полученные иммунологические данные являются результатом введения только однократной дозы исследуемого лекарственного соединения 1. Результаты определения активности заболевания. Не отмечено клинически значимого влияния соединения 1 на индекс SLEDAI (изменение 3, 12 пунктов) во время исследования, за исключением одного субъекта в группе лечения дозой 0,5 мг, у которого регистрировали изменение индекса SLEDAI от 2 до 10 пунктов между исходным уровнем и 4 неделей на основе анализа мочи, показывающего пиурию. Исследователем не подтверждены указанные данные анализа мочи как вспышка волчанки по определению в протоколе, и было решено не изменять лечение. Заключение Данное исследование фазы Iа показало, что однократная подкожная инъецированная доза соедине- 29009123 ния 1, равная 0,5, 1 или 2,5 мг в 120 мг каптизола, была безопасной и хорошо переносимой, и дало возможность для продолжения исследования с многократными дозами в фазе Ib. Пример 9. Клиническое исследование фазы Ib. Проводимое в нескольких центрах, двухнациональное, рандомизированное, двойное слепое, с четырьмя вариантами, с плацебо-контролем, с использованием многократных доз исследование фазы I,чтобы оценить переносимость и безопасность подкожной инъекции соединения 1 в каптизоле у субъектов с СКВ. Данное исследование осуществляется для того, чтобы оценить безопасность и переносимость многократного п/к-введения соединения 1 субъектам с СКВ. Второй целью исследования является оценка иммунологических ответов после многократного п/к-введения соединения 1 в каптизоле у субъектов с СКВ. Соединение 1 вводят в дозах 0,5, 1 или 2,5 мг в каптизоле. Проходящий клиническое испытание продукт вводят через день (исключая выходные дни), всего 12 п/к-инъекций, т.е. 3 дозы в неделю в течение 4 недель. Наблюдение за субъектами проводят при запланированных визитах по графику на 2, 4, 8 и 12 неделе после начала введения доз. Безопасность и переносимость оценивают, используя тесты, сходные с тестами, описанными выше для фазы Ia клинического исследования. Результаты Исследование фазы Ib показывает, что многократные подкожно инъецированные дозы соединения 1, равные 0,5, 1 или 2,5 мг в 120 мг каптизола, являются безопасными и хорошо переносимыми. Пример 10. Выявление антител против пептидов Iа, IIа и IIIа и анти-16/6 Id-антител в сыворотках пациентов с СКВ и здоровых людей в качестве контроля. У людей с СКВ (32 пациента) брали кровь и их сыворотки тестировали в ELISA в отношении их способности связывать пептиды Iа, IIа и IIIа, контрольный пептид р 195-212 (миастеногенный пептид,описанный в публикации РСТWO 94/00148) или мАт 5G12. Выявление антител осуществляли на планшетах, которые покрывали 10 мкг/мл пептидов Iа, IIа иIIIа, р 195-212 или мАт 5G12 в PBS в течение 2 ч, промывали и блокировали 1% овальбумином в PBS дополнительно в течение 2 ч. ELISA продолжали как описано после блокирования в примере 2 международной публикации РСТ 96/30057, используя поликлональное антитело козы против IgG человека,конъюгированное с пероксидазой. Как показано на фиг. 7, у пациентов с СКВ наблюдались значительно более высокие уровни антител, которые связывают либо пептид Ia (незаштрихованные квадраты), IIа (незаштрихованные ромбы),IIIа (незаштрихованные кружки), либо мАт 5G12 (незаштрихованные треугольники), по сравнению со здоровыми людьми в контроле (пептид Iа - здоровые = заштрихованные ромбы; пептид IIа - здоровые = перечеркнутые кружки; пептид IIIа - здоровые = перевернутые незаштрихованные треугольники; 5G12 здоровые = наполовину заштрихованные квадраты). Не наблюдали связывания ни при тестировании сывороток от пациентов, ни при тестировании сывороток в контроле на планшетах, покрытых несоответствующим пептидом р 195-212 (р 195-212 - СКВ = перечеркнутые квадраты; р 195-212 - здоровые = наполовину заштрихованные ромбы). Результаты свидетельствуют о корреляции между целой молекулой антитела и основанными на CDR пептидами по уровню титров антител. Пример 11. Пролиферация PBL от пациентов с СКВ и здоровых людей в качестве контроля в присутствии мАт 16/6 Id человека и пептидов. Лимфоциты периферической крови (PBL) выделяли из крови пациентов с СКВ или здоровых доноров в качестве контроля, используя градиент фикола. Затем PBL инкубировали в присутствии разных концентраций пептидов Iа, IIа или IIIа или мАт 16/6 Id человека в течение 24 ч и в это время брали образцы для измерения IL-2. Анализ продолжали всего в течение 7 дней и 3 Н-тимидин добавляли в последние 16 ч. Пролиферацию определяли, регистрируя количество радиоактивности, включенной в ДНК клеток. Как видно в табл. 14, меньшая часть PBL, взятых от пациентов с СКВ, взаимодействовала с пептидами или с мАт 16/6 Id по сравнению со здоровыми донорами в контроле. Результаты выражены в процентах респондеров (34% на первой строке) и фактическом количестве пациентов (11 из 32: 11/32). Сходные результаты получали в том случае, когда тестировали уровни IL-2, продуцируемого PBL в присутствии пептидов или мАт 16/6 Id, как показано в следующем примере. Таблица 14 Пролиферация PBL от пациентов с СКВ и здоровых доноров в качестве контроля в присутствии мАт 16/6 Id и пептидов Ia-IIIa EMI29.1
МПК / Метки
МПК: A61K 38/10, A61P 29/00, A61K 31/724, A61K 38/16
Метки: парентеральные, лечения, красной, композиции, пептидов, волчанки, системной
Код ссылки
<a href="https://eas.patents.su/30-9123-parenteralnye-kompozicii-peptidov-dlya-lecheniya-sistemnojj-krasnojj-volchanki.html" rel="bookmark" title="База патентов Евразийского Союза">Парентеральные композиции пептидов для лечения системной красной волчанки</a>
Парентеральные композиции пептида для лечения системной красной волчанки

Номер патента: 8438
Опубликовано: 29.06.2007
Авторы: Клингер Эти, Нафтали Эсмира, Гилберт Адриан, Кохен-Веред Шарон, Вайнштейн Вера
МПК: A61K 38/00, A61K 47/40, A61K 51/10...
Метки: красной, парентеральные, композиции, системной, волчанки, лечения, пептида
Формула / Реферат:
1. Фармацевтическая композиция, содержащая водный носитель, от 0,1 до 20 мг/мл композиции фармацевтически приемлемой соли пептида, имеющего структурную формулу NH2-Gly Tyr Tyr Trp Ser Trp Ile Arg Gln Pro Pro Gly Lys Gly Glu Glu Trp Ile Gly-COOH (SEQ ID NO: 1), и замещенный b-циклодекстрин в количестве, эффективном для растворения пептида в водном носителе, при этом композиция имеет рН от 4 до 9. 2. Фармацевтическая композиция по п.1, где...
Композиции и способы для усовершенствованной доставки пептидов, связывающихся с y2-рецепторами, которую осуществляют через слизистые оболочки, и способы лечения и предотвращения ожирения

Номер патента: 8829
Опубликовано: 31.08.2007
Авторы: Клипп Мэри С., Квей Стивен К., Макевилли Конор Дж., Брэндт Гордон
МПК: A61P 43/00, A61K 38/00
Метки: y2-рецепторами, которую, ожирения, доставки, слизистые, лечения, пептидов, связывающихся, композиции, осуществляют, способы, предотвращения, усовершенствованной, оболочки
Формула / Реферат:
1. Применение пептида PYY(3-36) для изготовления медикамента для лечения ожирения или контроля потребления пищи у пациента, причем введение через слизистые оболочки по меньшей мере 50 мкг указанного пептида в указанном медикаменте повышает концентрацию указанного пептида в плазме субъекта по меньшей мере на 5 пмоль/л. 2. Применение по п.1, причем введение медикамента вызывает чувство сытости. 3. Применение по п.1, причем введение медикамента...
Фармацевтические композиции пептидов, имеющих низкую растворимость в физиологической среде

Номер патента: 2227
Опубликовано: 28.02.2002
Автор: Ричардсон Питер
МПК: A61K 38/09
Метки: среде, фармацевтические, растворимость, пептидов, физиологической, имеющих, низкую, композиции
Формула / Реферат:
1. Фармацевтическая композиция, включающая: а) пептидный агонист ЛГРФ, пептидный антагонист ЛГРФ или пептид ГРФ в качестве активного ингредиента; б) никотинамид и с) физиологический водный раствор. 2. Фармацевтическая композиция по п.1, в которой активный ингредиент представляет пептидный антагонист ЛГРФ. 3. Фармацевтическая композиция по любому из предшествующих пунктов, в которой активный ингредиент представляет антид или человеческий ГРФ. 4....
Применение соединения из класса пептидов для лечения аллодинии и других типов хронической или фантомной боли

Номер патента: 7600
Опубликовано: 29.12.2006
Автор: Зельве Норма
МПК: A61P 23/00, A61K 31/165
Метки: класса, лечения, соединения, применение, фантомной, аллодинии, хронической, пептидов, других, типов, боли
Формула / Реферат:
1. Применение соединения формулы (I) где Аr обозначает фенил, который может быть незамещен или замещен по меньшей мере одной галогруппой; Q обозначает (низш.)алкоксигруппу, содержащую 1-3 атома углерода, и Q1 обозначает метил, или его фармацевтически приемлемой соли для приготовления фармацевтической композиции, предназначенной для лечения аллодинии как основного и уникального симптома боли, вне зависимости от природы лежащего в ее основе...
Алатрофлоксациновые парентеральные составы

Номер патента: 2120
Опубликовано: 24.12.2001
Авторы: Валински Стэнли Вальтер, Гвин Роберт Марк, Ламберт Джон Фрэнсис, Гухан Сабраманян Сэм
МПК: C07D 471/04, A61K 38/05, A61P 31/04...
Метки: составы, парентеральные, алатрофлоксациновые
Формула / Реферат:
1. Способ очистки соединения формулы включающий обработку неочищенного продукта реакции, который содержит количество указанного соединения формулы I и менее полярные примеси, гидрофобной смолой. 2. Способ по п.1, в котором указанные менее полярные примеси имеют время удерживания примерно от 2,1 до 30 мин. 3. Способ по п.1, в котором одна из указанных менее полярных примесей имеет время удерживания от 9 до 12 мин. 4. Способ по п.1, в...
Предыдущий патент: Полимерные конъюгаты с пониженными антигенными свойствами, способы их получения и применения
Следующий патент: Антитела против il-20 и связывающие партнеры, а также способы их применения при воспалении
Случайный патент: Усовершенствованный пакер