2′-фторнуклеозиды
Номер патента: 8609
Опубликовано: 29.06.2007
Авторы: Ши Дзунксинг, Ли Кийеонг, Хонг Дзоон Х., Чу Чунг К., Шинази Рэймонд Ф., Лиотта Деннис К., Макати Джеффри Дж., Чой Йонгсеок






















Формула / Реферат
1. Применение 2'-фтор-b-D-нуклеозида для изготовления лекарственного средства, предназначенного для лечения инфекционного гепатита В у человека, в котором 2'-фтор-b-D-нуклеозид имеет формулу

в которой Base обозначает пуриновое основание;
R1 обозначает ОН, Н, OR3, N3, CN, галоген, CF3, C1-C4алкил, амино, С1-С4алкиламино, ди-С1-С4алкиламино;
R2 обозначает Н, монофосфат, дифосфат, трифосфат, ацил или другую фармацевтически приемлемую отщепляемую группу, которая при введении in vivo способна дать соединение, в котором R2 обозначает Н или фосфат; эфир сульфокислоты, включая C1-С10алкилсульфонил или C1-С10арилалкилсульфонил, в том числе метансульфонил; бензил, в котором фенильная группа необязательно замещена одним или несколькими заместителями, выбираемыми из группы, включающими гидроксил, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфокислоту, сульфат, фосфоновую кислоту, фосфат или фосфонат; и
R3 обозначает ацил, C1-С10алкил, фосфат или другую фармацевтически приемлемую отщепляемую группу, которая при введении in vivo способна отщепляться с образованием исходного соединения или его фармацевтически приемлемой соли, необязательно в сочетании с фармацевтически приемлемым носителем.
2. Применение 2'-фторнуклеозида для изготовления лекарственного средства, предназначенного для лечения инфекционного гепатита С у человека, в котором 2'-фторнуклеозид имеет формулу

в которой Base обозначает пуриновое или пиримидиновое основание;
R1 обозначает ОН, Н, OR3, N3, CN, галоген, CF3, С1-С4алкил, амино, С1-С4алкиламино, ди-С1-С4алкиламино или алкокси;
R2 обозначает Н, монофосфат, дифосфат, трифосфат, ацил или другую фармацевтически приемлемую отщепляемую группу, которая при введении in vivo способна дать соединение, в котором R2 обозначает Н или фосфат; эфир сульфокислоты, включая C1-С10алкилсульфонил или C1-С10арилалкилсульфонил, в том числе метансульфонил; бензил, в котором фенильная группа необязательно замещена одним или несколькими заместителями, включающими гидроксил, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфокислоту, сульфат, фосфоновую кислоту, фосфат или фосфонат; и
R3 обозначает ацил, C1-С10алкил, фосфат или другую фармацевтически приемлемую отщепляемую группу, которая при введении in vivo способна отщепляться с образованием исходного соединения или его фармацевтически приемлемой соли, необязательно в сочетании с фармацевтически приемлемым носителем.
3. Применение по п.1 или 2, где R2 обозначает Н, монофосфат, дифосфат, трифосфат или ацил.
4. Применение по п.1 или 2, где R1 обозначает Н, ОН, OR3, С1-С4алкил или галоген.
5. Применение по пп.1-4, где R3 обозначает ацил.
6. Применение по пп.1-4, где R2 обозначает Н, монофосфат, дифосфат, трифосфат или ацил; R1 обозначает Н, ОН, С1-С4алкил или галоген и R3 обозначает ацил.
7. Применение по п.2, где основание обозначает пуриновое основание.
8. Применение по п.2, где основание обозначает пиримидиновое основание.
9. Применение по п.1, где основание является пуриновым основанием, выбираемым из группы, состоящей из гуанина, аденина, гипоксантина, 2,6-диаминопурина и 6-хлорпурина.
10. Применение по п.1 или 2, где R1 и R2 обозначают водород.
11. Применение по п.1 или 2, где R1 обозначает ОН или OR3.
12. Применение по п.1 или 2, где R1 обозначает ОН.
13. Применение по п.1 или 2, где R1 обозначает Н.
14. Применение по п.1 или 2, где R1 обозначает галоген.
15. Применение по п.1 или 2, где R1 обозначает C1-C4алкил или CF3.
16. Применение по п.1 или 2, где R2 обозначает ацил.
17. Применение по п.1 или 2, где основание обозначает пуриновое основание, выбираемое из аденина, N6-алкилпуринов, N6-ацилпуринов, где ацил является С(О) (алкилом, арилом, алкиларилом или арилалкилом), N6-бензилпурина, N6-галогенпурина, N6-винилпурина, N6-ацетиленпурина, N6-ацилпурина, N6-гидроксиалкилпурина, N6-тиоалкилпурина, N2-алкилпуринов, N2-алкил-6-тиопуринов, гуанина, гипоксантина, 2,6-диаминопурина, 2-хлор-2-аминопурина, инозина или 6-хлорпурина.
18. Применение по п.2, где основание обозначает пиримидиновое основание, выбираемое из тимина, цитозина, 5-фторцитозина, 5-метилцитозина, 6-азапиримидина, в том числе 6-азацитозина, 2- и/или 4-меркаптопиримидина, урацила, 5-галогенурацила, в том числе 5-фторурацила, С5-алкилпиримидинов, С5-бензилпиримидинов, С5-галогенпиримидинов, С5-винилпиримидина, С5-ацетиленпиримидина, С5-ацилпиримидина, С5-гидроксиалкилпурина, С5-амидопиримидина, С5-цианопиримидина, С5-нитропиримидина, С5-аминопиримидина, 5-азацитидинила, 5-азаурацилила, триазолопиридинила, имидазолопиридинила, пирролопиримидинила или пиразолопиримидинила.
19. Применение по п.1 или 2, где лекарственное средство является подходящим для орального введения.
20. Применение по п.1 или 2, где лекарственное средство является подходящим для парентерального введения.
21. Применение по п.1 или 2, где лекарственное средство является подходящим для внутривенного введения.
22. Применение 2'-фтор-b-D-пуринового нуклеозида для изготовления лекарственного средства, предназначенного для лечения инфекционного гепатита В.
23. Применение 2'-фтор-b-D-пуринового или пиримидинового нуклеозида для изготовления лекарственного средства, предназначенного для лечения инфекционного гепатита С.
24. Применение по п.22 или 23, где лекарственное средство является подходящим для орального введения.
25. Применение по п.22 или 23, где лекарственное средство является подходящим для парентерального введения.
26. Применение по п.22 или 23, где лекарственное средство является подходящим для внутривенного введения.
Текст
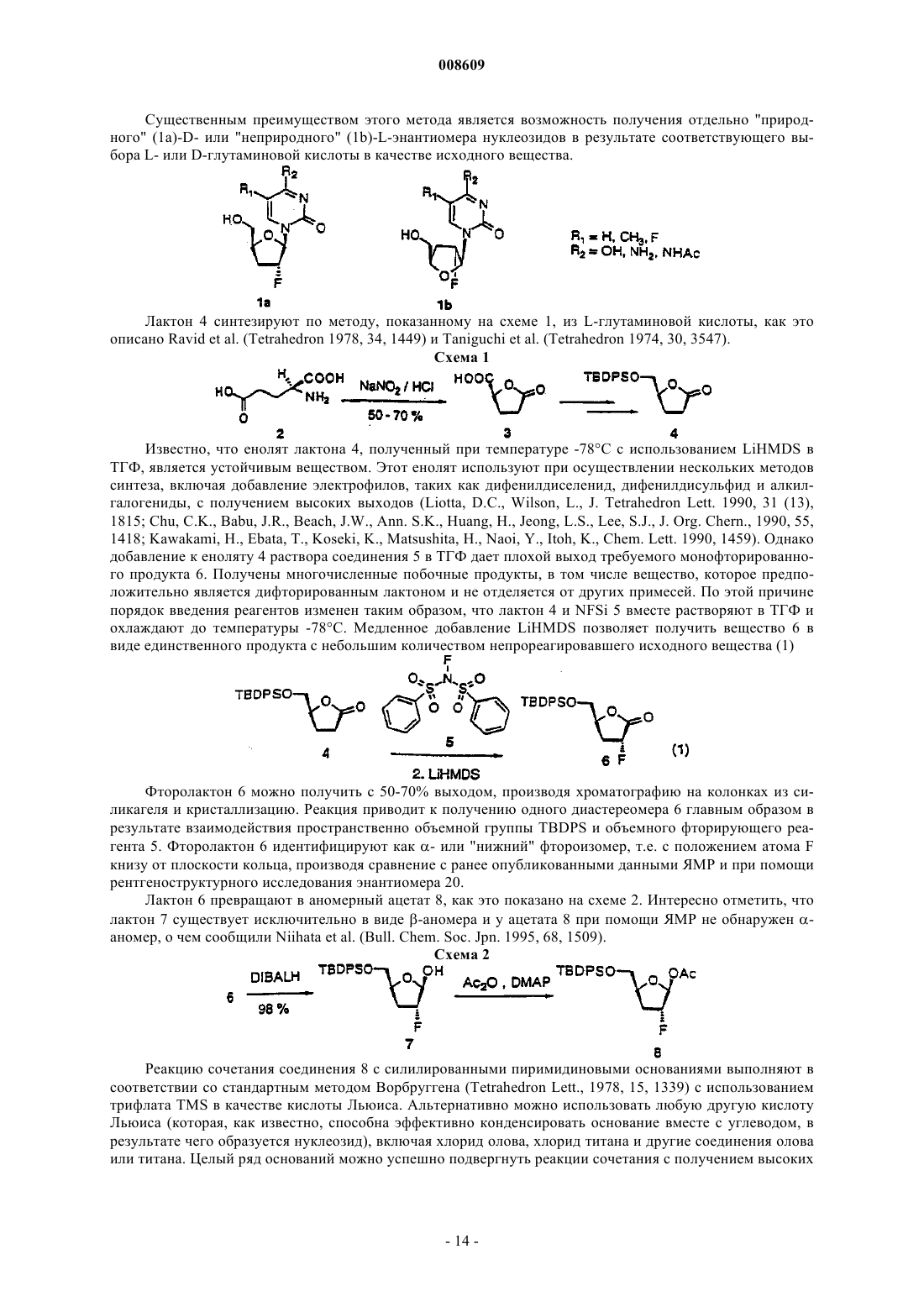
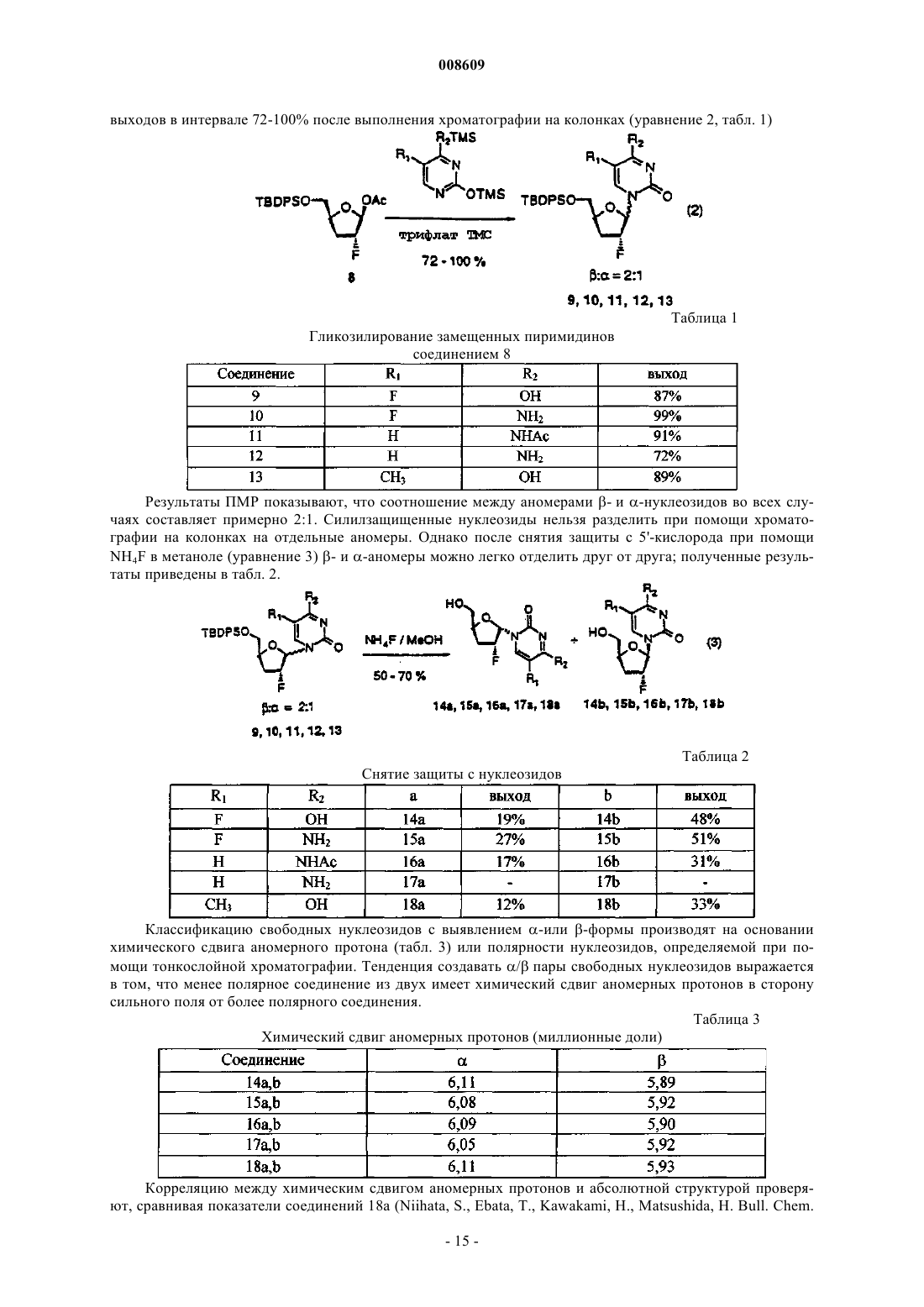
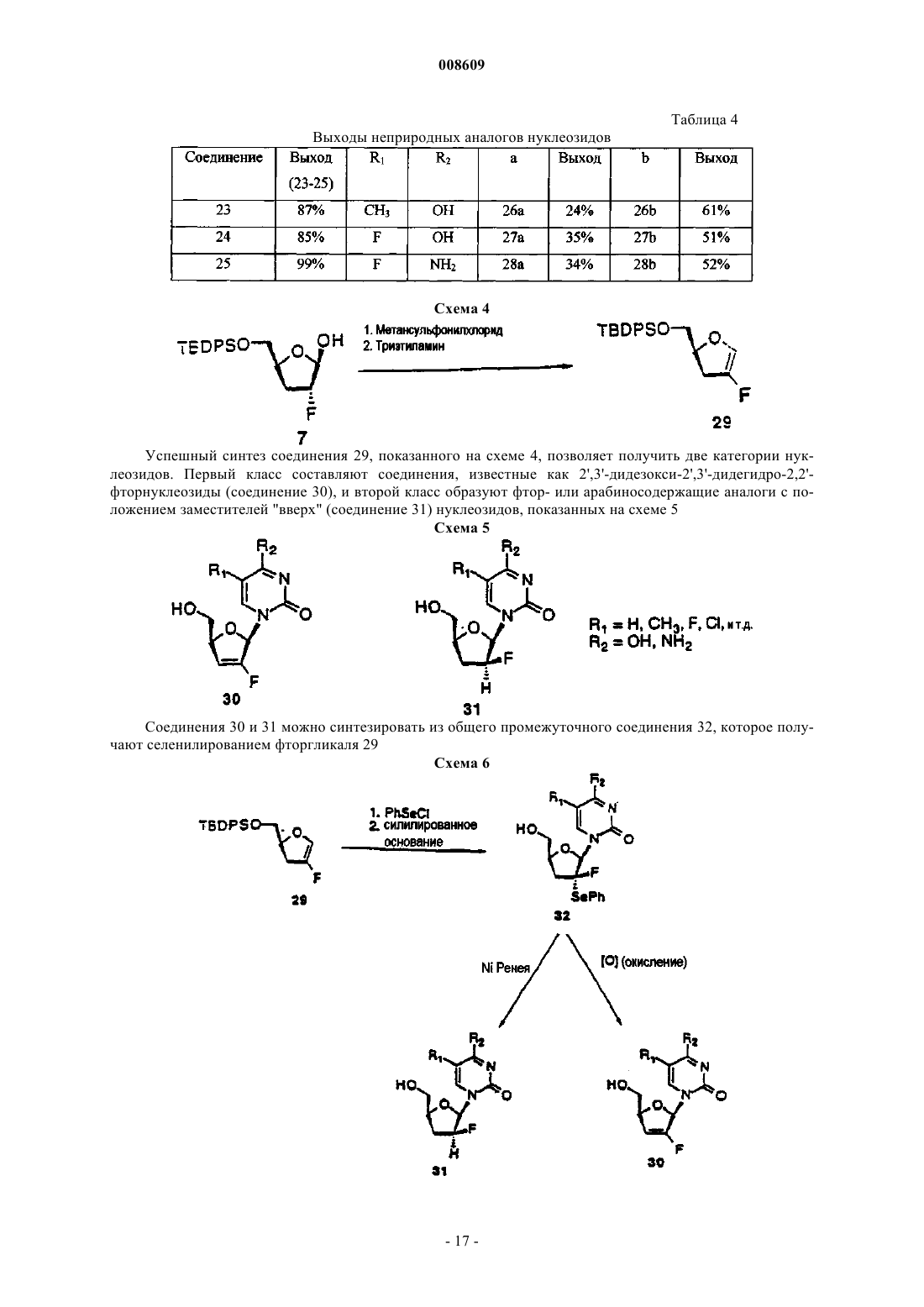
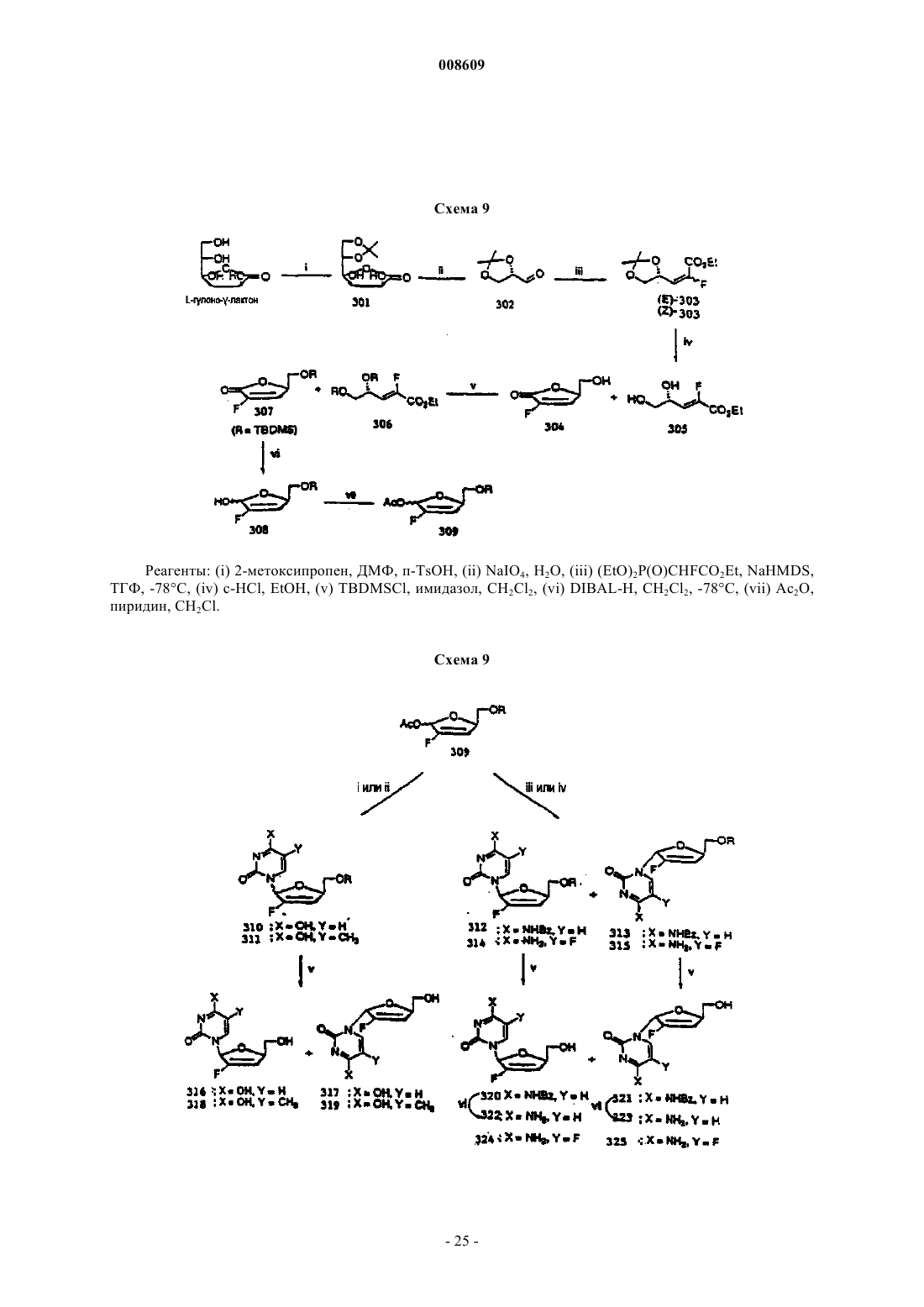
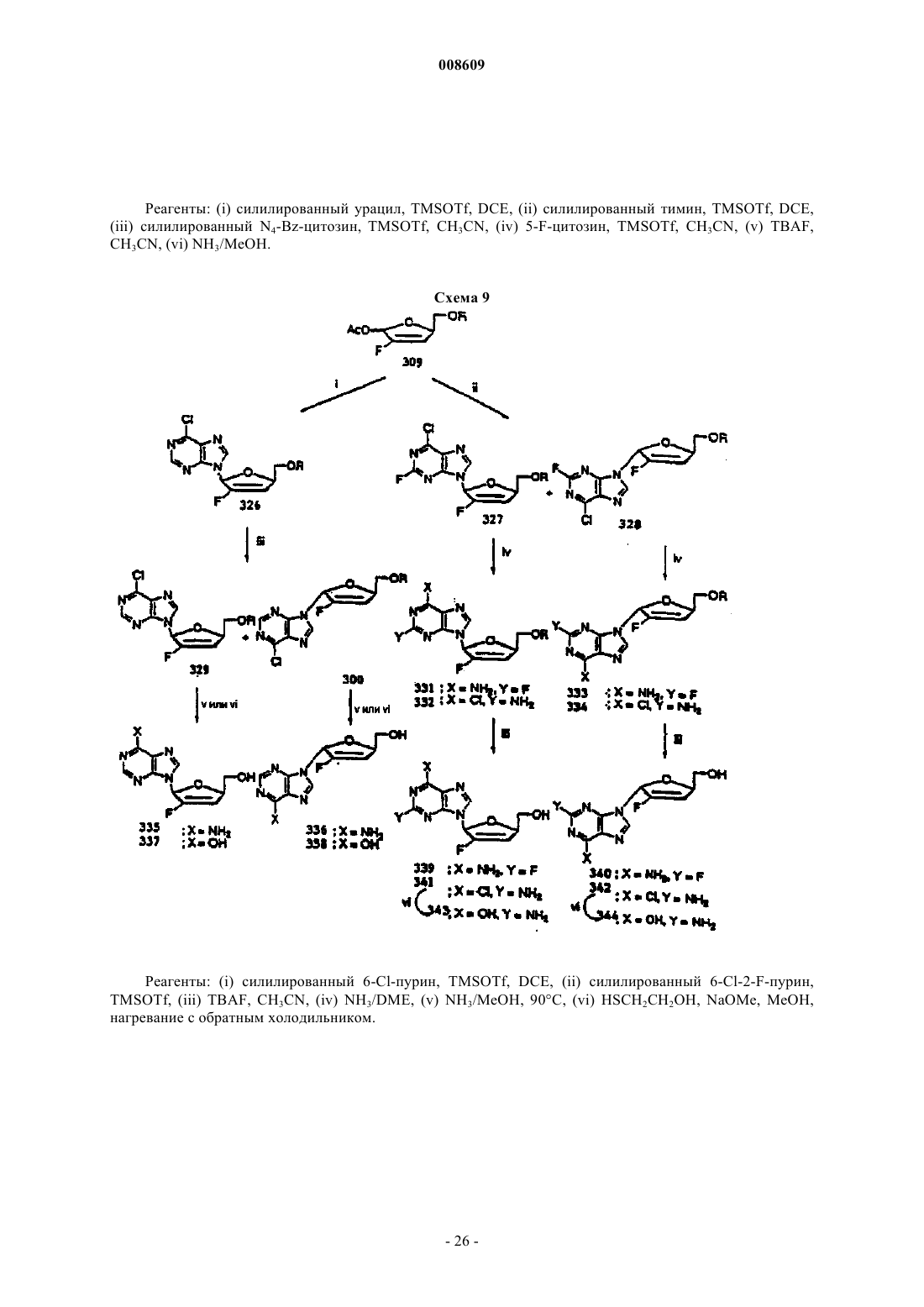
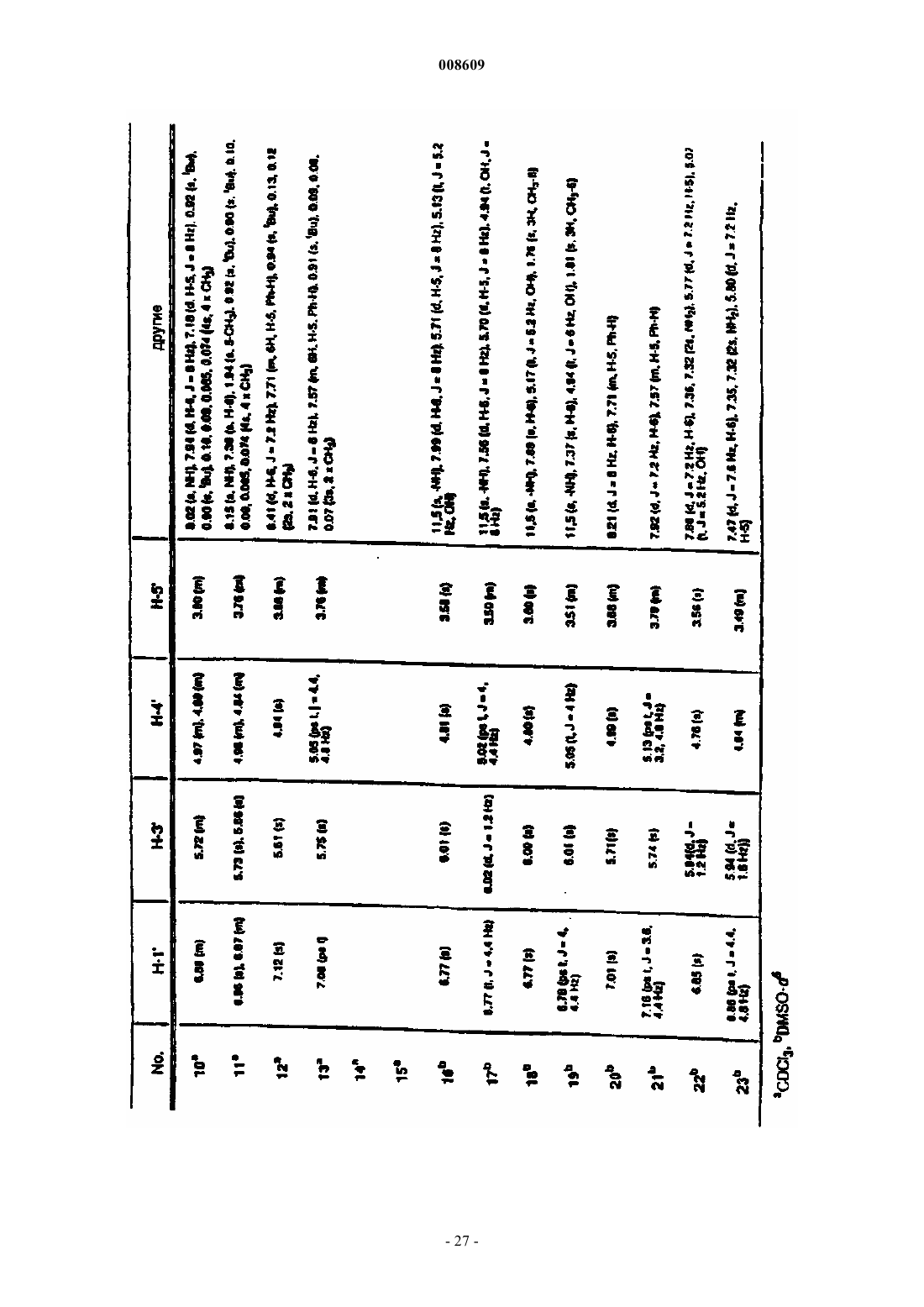
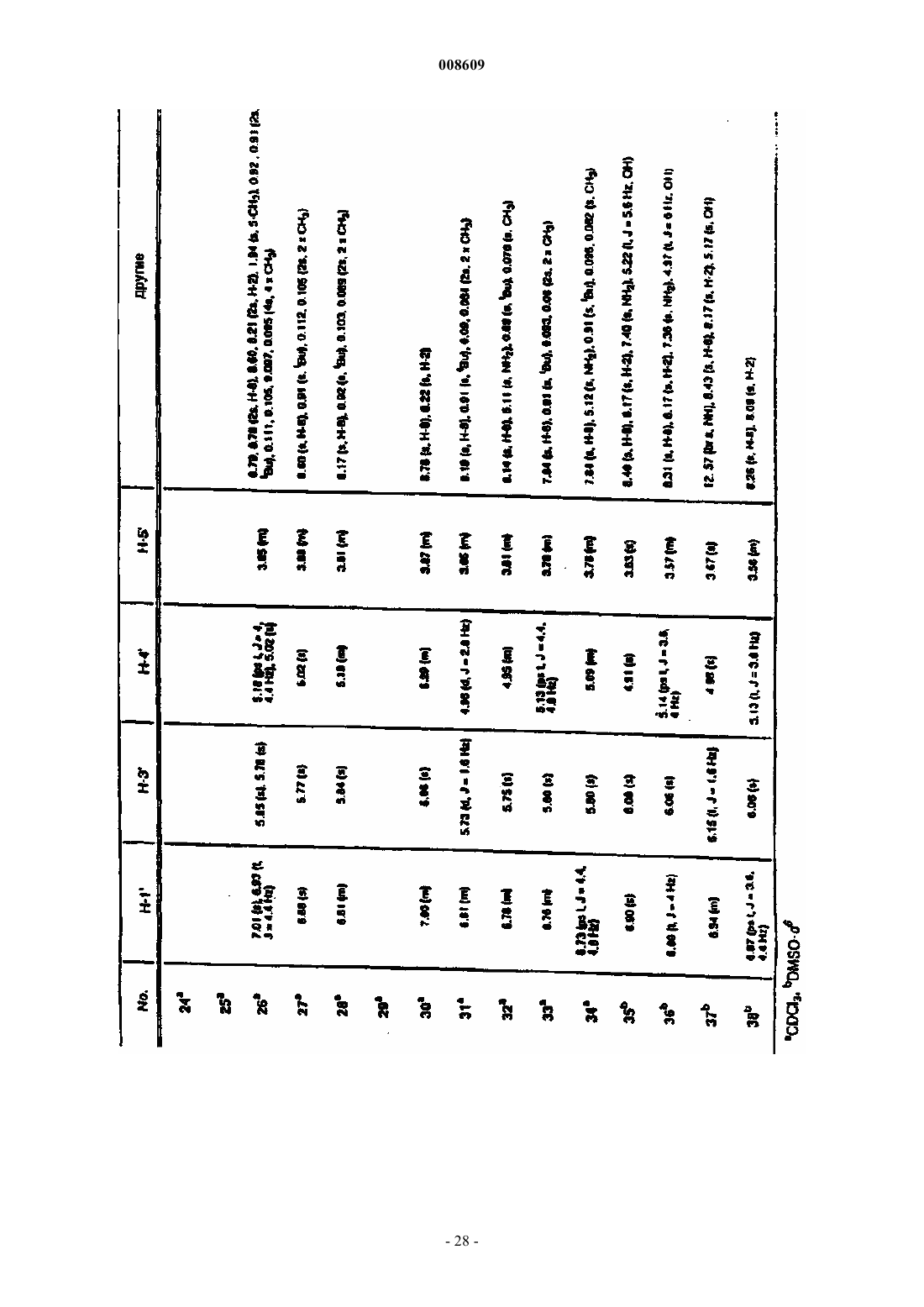
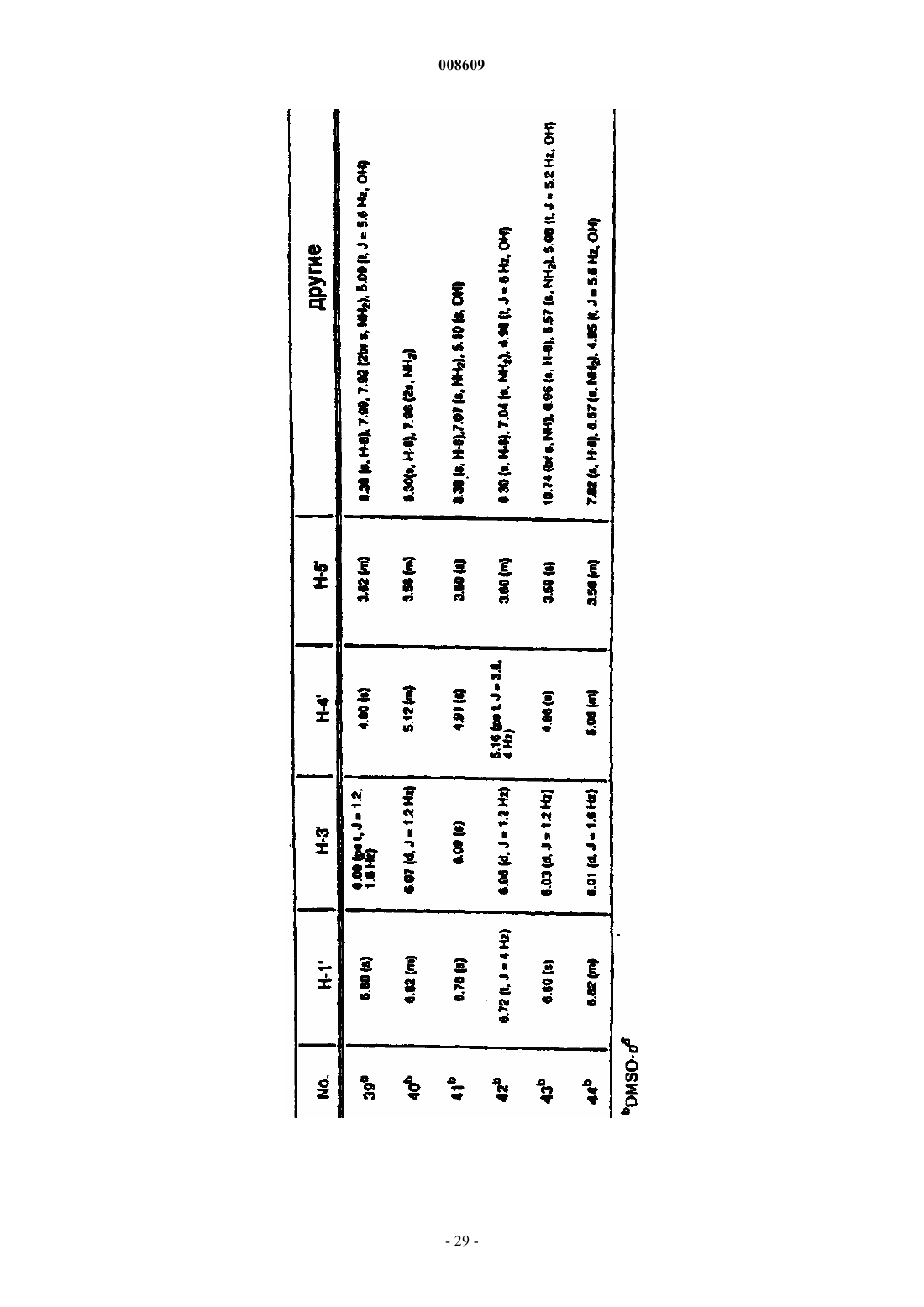
008609 Описываемое изобретение сделано при поддержке правительства на основании грантаAI32351,предоставленного Национальным институтом здравоохранения. Правительство США имеет определенные права на это изобретение. Настоящее изобретение сделано в области фармацевтической химии и относится, в частности, к 2'фторнуклеозидам, способам их получения и применения. Предпосылки создания изобретения Синтетические нуклеозиды, такие как 5-йод-2'-дезоксиуридин и 5-фтор-2'-дезоксиуридин, в течение ряда лет использовались для лечения рака и вирусов герпеса. В 1980-х годах синтетические нуклеозиды привлекли к себе особое внимание в связи с возможностью их применения для лечения вируса иммунодефицита человека (ВИЧ), гепатитов и вируса Эпштейна-Барра. В 1981 г. было установлено, что синдром приобретенного иммунодефицита человека (СПИД) является заболеванием, которое серьезно поражает иммунную систему человека и почти без исключения ведет к смерти. В 1983 г было обнаружено, что этиологической причиной возникновения СПИДа является вирус иммунодефицита человека. В 1985 г. появилась информации о том, что синтетический нуклеозид 3'-азидо-3'-дезокситимидин (AZT) подавляет репликацию вируса иммунодефицита человека. С тех пор был открыт ряд других синтетических нуклеозидов, в том числе 2',3'-дидезоксиинозин (DDI), 2',3'дидезоксицитидин (DDC) и 2',3'-дидезокси-2',3'-дидегидротимидин (D4T), которые эффективно воздействуют на ВИЧ. Синтетические нуклеозиды фосфорилируются в клетке под действием клеточных киназ с образованием 5'-трифосфата и встраиваются в растущую цепь вирусной ДНК, прекращая рост цепи изза отсутствия 3'-гидроксильной группы. Они могут также ингибировать обратную транскриптазу фермента вируса. Успешное применение разных синтетических нуклеозидов для подавления репликации ВИЧ in vivo или in vitro заставило многих исследователей направить усилия на создание и испытание нуклеозидов, в которых атом углерода замещен гетероатомом в 3'-положении нуклеозида. В публикации заявки на европейский патент 0337713 и в патенте США 5041449, выданных BioChem Pharma, Inc., описаны рацемические 2-замещенные-4-замещенные-1,3-диоксоланы, обладающие антивирусным действием. В патенте США 5047407 и в заявке на европейский патент 0382526, выданных также BioChem Pharma,Inc., указано, что ряд нуклеозидов, таких как рацемический 2-замещенный-5-замещенный 1,3-ксатиолан,обладают антивирусной активностью, и особо подчеркнуто, что рацемическая смесь 2-гидроксиметил-5(цитозин-1-ил)-1,3-оксатиолана (именуемого ниже ВСН-189) обладает такой же активностью против ВИЧ, что и AZT, при незначительной токсичности. (-)-Этантиомер рацемата ВСН-189, известный как 3 ТС, который описан в патенте США 5539116, выданном Liotta et al., в настоящее время продается в США как средство для лечения ВИЧ в сочетании с AZT. Кроме того, известно, что цис-2-гидроксиметил-5-(5-фтороцитозин-1-ил)-1,3-оксатиолан ("FTC") обладает сильным действием против ВИЧ. Schinazi, et al. "Selective Inhibition of Human Immunodeficiencyviruses by Racemates and Enantiomers of cis-5-Flouro-l-[2-(Hydroxymethyl)-1,3-Oxathiolane-5-yl] Cytosine",Antimicrobial Agents and Chemotherapy, November 1992, pp. 2423-2431. См. также патент США 5210085; WO 91/11186 и WO 92/14743. Другим вирусом, который создает серьезную опасность для здоровья человека, является вирус гепатита В (далее именуется "HBV"). HBV является второй по значимости причиной возникновения рака у человека после табака. Механизм возникновения рака под действием HBV неизвестен. Считается, что он может быть прямым или косвенным фактором образования опухоли вследствие хронического воспалительного процесса, цирроза и регенерации клеток, обусловленной этой инфекционной болезнью. После 2-6-месячного инкубационного периода, в течение которого носитель инфекции не подозревает о существовании опухоли, HBV вызывает острый гепатит и поражение печени, сопровождающееся болями в животе, желтухой и повышенным содержанием в крови определенных ферментов. HBV может вызывать молниеносный гепатит часто с летальным исходом, при котором разрушаются значительные участки печени. Пациенты обычно выздоравливают после острого гепатита. Однако у некоторых пациентов высокие концентрации вирусного антигена сохраняются в крови в течение продолжительного периода времени,являясь причиной возникновения хронического заболевания. Хронические инфекции могут быть причиной хронического персистирующего гепатита. Пациенты, страдающие хроническим персистирующимHBV, особенно часто встречаются в развивающихся странах. К середине 1991 г. только в Азии насчитывалось примерно 225 млн хронических носителей HBV, а во всем мире их число составляло почти 300 млн. Хронический персистирующий гепатит может вызывать утомление, цирроз печени и злокачественную гепатому, основной тип рака печени. В промышленно развитых странах Запада к группам населения с высокой степенью риска заражения HBV относятся носители этого вируса или их кровь. Эпидемиология HBV очень схожа с синдромом приобретенного иммунодефицита и позволяет объяснить тот факт, что заражение HBV широко распространено среди ВИЧ-инфицированных пациентов или больных СПИДом. Однако HBV является более заразным, чем ВИЧ. Как FTC, так и 3 ТС воздействуют на HBV. Furman, et al., "The Anti-Hepatitis В Virus Activities, Cyto-1 008609Cheng, et al. Journal of Biological Chemistry, vol. 267(20), pp. 13938-13942 (1992). Из сыворотки человека получена вакцина для иммунизации пациентов против HBV. Хотя эта вакцина признана эффективной, ее производство является трудоемким процессом из-за ограниченного количества сыворотки, предоставляемого хроническими носителями, при этом процедура очистки является длительной и дорогостоящей. Кроме того, каждую партию вакцины, полученной из другой сыворотки,необходимо испытывать на шимпанзе, чтобы удостовериться в ее безопасности. Вакцины были также получены генноинженерными методами. Ежедневный прием -интерферона, генетически сконструированного методом генной инженерии, также является многообещающим. Вирус гепатита С ("HCV") является основной причиной посттрансфузии и поражения спорадическим гепатитом, не являющимся гепатитом А или В (Alter, H.J. (1990) J. Gastro. Hepatol, 1:78-94, Dienstag,J.L. (1983) Castro 85: 439-462). Несмотря на всестороннее обследование населения, на долю HCV попрежнему приходится по крайней мере 25% случаев острого вирусного гепатита во многих странах(Alter, H.J. (1990) см. выше; Dienstag, J.L. (1983) см. выше; Alter M.J. et al. (1990a) J.A.M.A. 264:22312235; Alter M.J. et al, (1992) N. Engl. J. Med. 327:1899-1905; Alter, M.J. et al. (1990b) N. Engl. J. Med. 321:1494-1500). Поражение HCV протекает без явных симптомов у большого количества хронически инфицированных (и контагеозных) носителей, у которых клинические симптомы не проявляются в течение многих лет. Высокий коэффициент перехода острой формы инфекционного заболевания в хроническую (70-100%) и в болезнь печени (50%), распространение по всему миру и отсутствие вакцины - все это делает HCV серьезной причиной заболеваемости и смертности. Опухоль представляет собой неупорядоченную, дезорганизованную пролиферацию растущих клеток. Опухоль считается злокачественной или раковой, если ее отличают инвазивность и метастазы. Инвазивность - это способность опухоли внедряться в окружающие ткани, проникая через тонкие слои, определяющие границы тканей, и таким образом попадать в систему кровообращения. Метастазы - это способность опухоли мигрировать в другие области тела и вызывать пролиферацию клеток на расстоянии от места первоначального появления опухоли. Рак в настоящее время является второй по значимости причиной смертности в США. Более чем у 8 млн чел. в США был диагностирован рак, и предполагается, что в 1994 г. он будет обнаружен еще у 1 млн 208 тыс. чел. Более 500 тыс. чел. ежегодно умирают в США от этой болезни. На молекулярном уровне природа рака не совсем ясна. Известно, что воздействие на клетку канцерогена, такого как некоторые вирусы, химические вещества или облучение, вызывает изменение ДНК,подавляющее "супрессивный" ген или активирующее "онкоген". Супрессивные гены регулируют рост клетки, но в результате мутации они перестают выполнять эту функцию. Онкогены первоначально являются нормальными генами (именуемыми проонкогенами), которые в результате мутации или изменения условий экспрессии превращаются в трансформирующие гены. Продукты трансформирующих генов вызывают неконтролируемый рост клеток. Более 20 различных нормальных клеточных генов могут стать онкогенами вследствие генетического изменения. Трансформированные клетки отличаются от нормальных клеток во многих отношениях, включая морфологию клетки, взаимодействие между клетками, содержимое мембраны, цитоскелетную структуру, секрецию белка, экспрессию и гибель гена (трансформированные клетки могут расти бесконечно). Практически все типы клеток в организме могут быть трансформированы в клетки доброкачественной или злокачественной опухоли. Наиболее часто опухоли поражают легкие, затем толстую и прямую кишку, молочную железу, предстательную железу, мочевой пузырь, поджелудочную железу и яичники. Другими преобладающими типами рака являются лейкоз, рак центральной нервной системы, в том числе рак мозга, меланома, лимфома, эритролейкоз, рак матки и рак головы и шеи. В настоящее время рак лечат одним или несколькими методами трехкомпонентной терапии: хирургическое вмешательство, облучение и химиотерапия. В результате хирургического вмешательства удаляют основную массу пораженной ткани. Хотя хирургическое вмешательство иногда позволяет эффективно удалять опухоли, расположенные в определенных местах, таких как молочная железа, толстая кишка и кожа, этот метод нельзя использовать для лечения опухолей, расположенных в других местах,таких как позвоночник; он также не подходит для лечения диссеминированных состояний неоплазии,таких как лейкоз. Химиотерапия направлена на прекращение репликации или метаболизма клеток. Этот метод чаще всего используют для лечения лейкоза, а также рака груди, легкого и яичек. В настоящее время для лечения рака используют химиотерапевтические средства пяти основных классов: природные продукты и их производные; антациклины; алкилирующие средства; антипролиферативные средства (именуемые также антиметаболитами) и гормональные средства. Химиотерапевтические средства часто именуются противоопухолевыми средствами. Считается, что алкилирующие средства алкилируют и сшивают гуанин и, вероятно, другие основания в ДНК, прекращая деление клеток. Типичными алкилирующими средствами являются азотсодержащие дихлордиэтилсульфиды, соединения этиленимина, алкилсульфаты, цисплатин и разные нитрозомо-2 008609 чевины. Недостатком этих соединений является то, что они проникают не только в злокачественные клетки, но и в здоровые клетки, которые находятся в процессе естественного деления, например в клетки костного мозга, кожи, слизистой оболочки желудочно-кишечного тракта и эмбриональной ткани. Антиметаболиты обычно являются обратимо или необратимо действующими ингибиторами ферментов либо соединениями, которые другим образом препятствуют репликации, трансляции или транскрипции нуклеиновых кислот. Получено несколько синтетических нуклеозидов, обладающих противораковым действием. Хорошо известным производным нуклеозида, оказывающим сильное противораковое действие, является 5 фторурацил. 5-Фторурацил используется в клинической практике для лечения злокачественных опухолей, включая, например, карциномы, саркомы, рака кожи, рака органов пищеварения и рака молочной железы. Однако 5-фторурацил вызывает серьезные побочные эффекты, такие как тошнота, алопеция,диарея, стоматит, лейкоцитарная тромбоцитопения, анорексия, пигментация и отек. Производные 5-фторурацила, обладающие противораковым действием, описаны в патенте США 4336381 и в публикациях патентов Японии 50-50383, 50-50384, 50-64281, 51-146482 и 53-84981. В патенте США 4000137 указано, что продукт перокисления инозина, аденозина или цитидина метанолом или этанолом обладает активностью против лимфолейкоза. Цитозинарабинозид (именуемый также цитарабин, аrаС и цитозар) является нуклеозидным аналогом дезоксицитидина, который был впервые синтезирован в 1950 г. и введен в клиническую практику в 1963 г. В настоящее время он является важным лекарственным средством, применяемым для лечения острой формы миелоидного лейкоза. Кроме того, это лекарственное средство позволяет эффективно лечить острую форму лимфолейкоза, и в меньшей степени он воздействует на хроническую форму миелоцитарного лейкоза и лимфому, не являющуюся лимфомой Ходжкинса. Основным действием аrаС является ингибирование синтеза ядерной ДНК. Handschumacher, R. and Cheng, Y., "Purine and Pyrimidine Antimetabolites", Cancer Medicine, Chapter XV-1, 3rd Edition, Edited by J. Holland, et al., издатели Lea and Febigol. 5-Азацитидин является аналогом цитидина, который в основном используют для лечения острой формы миелоцитарного лейкоза и синдрома миелодисплазии. 2-Фтораденозин-5'-фосфат (флудара, именуемый также F-araA) является одним из наиболее активных средств, предназначенных для лечения хронической формы лимфоцитарного лейкоза. Это соединение ингибирует синтез ДНК. Воздействие на клетки, оказываемое F-araA, связано с накоплением клеток на границе фаз G1/S и в фазе S; таким образом, это лекарственное средство специфически воздействует на фазу S цикла развития клетки. Введение активного метаболита F-araATP замедляет удлинение цепи ДНК. F-araA является также сильнодействующим ингибитором рибонуклеотид-редуктазы, ключевого фермента, ответственного за образование dATP. 2-Хлордезоксиаденозин является эффективным средством для лечения новообразований В-клеток на начальной стадии, таких как хроническая форма лимфолейкоза, лимфома, не являющаяся лимфомой Ходжкинса, и волосато-клеточный лейкоз. В процессе создания новых биологически активных нуклеозидов было предпринято несколько попыток ввести фторсодержащий заместитель в углеводное кольцо нуклеозида. Фтор был предложен в качестве заместителя, потому что он может быть изополярным и изостерическим имитатором гидроксильной группы, так как длина связи C-F (1,35 ) аналогична длине связи С-O (1,43 ), и фтор является акцептором водородной связи. Фтор способен вызывать значительные электронные изменения в молекуле при минимальных пространственных деформациях. Замещение фтором другой группы в молекуле может вызывать изменения в метаболизме субстрата из-за высокой прочности связи C-F (116 ккал/моль по сравнению с С-Н=100 ккал/моль). В ряде справочных материалов был описан синтез и применение 2'-арабинофторнуклеозидов (нуклеозидов, в которых 2'-фторогруппа имеет "верхнюю" конфигурацию, т.е. вверх от плоскости кольца). В нескольких научных работах были описаны 2-фтор- -D-арабинофуранозилнуклеозиды, обладающие активностью против гепатита В и герпеса. См., например, патент США 4666892, выданный Fox, et al.; патент США 4211773, выданный Lopez, et al.; Su, et al. Nucleosides. 136, "Synthesis and Antiviral Effectsagainst Human Immunodeficiency Virus (HIV-1)," J. Med. Chem. 1990, 33, 2137-2145; Sterzycki, et al. "Synthesis and Anti-HIV Activity of Several 2'-Fluoro-Containing Pyrimidine Nucleosides," J. Med. Chem. 1990,а также заявку на европейский патент 0316017, которая также была подана Sterzycki, et al.; и Montgomery, et al. "9-(2-Deoxy-2-fluoroD-arabinofuranosyl)guanine: A Metabolically Stable Cytotoxic Analogueof 2'-Deoxyguanosine." В патенте США 5246924 описан способ лечения инфекционного гепатита, ко-3 008609 торый включает введение 1-(2'-дезокси-2'-фторD-арабинофуранозил)-3-этилурацила, именуемого также "FEAU". В патенте США 5034518 описаны 2-фтор-9-(2-дезокси-2-фторDарабинофуранозил)адениннуклеозиды, которые обладают противораковым действием, выражающимся в изменении метаболизма адениннуклеозидов, в результате чего это соединение утрачивает способность быть субстратом для аденозина. В заявке на европейский патент 0292023 раскрыты конкретные -D2'-фторарабинонуклеозиды, обладающие активностью против вирусных инфекций. В патенте США 5128458 описаны -D-2',3'-дидезокси-4'-тиорибонуклеозиды в качестве антивирусных средств. В патенте США 5446029 указано, что 2',3'-дидезокси-3'-фторнуклеозиды имеют активность против гепатита. В заявке на европейский патент 0409227 А 2 описаны конкретные 3'-замещенные -Dпиримидин- и пуриннуклеозиды для лечения гепатита В. Кроме того, указано, что L-FMAU (2'-фтор-5-метилL-арабинофуранозилурацил) является сильнодействующим средством против HBV и EBV. См., Chu, et al. "Use of 2'-Fluoro-5-methylLarabinofuranosyluracil as a Novel Antiviral Agent for Hepatitis B Virus and Epstein-Barr Virus" AntimicrobialNovel L-Nucleoside, 2'-Fluoro-5-MethylL-arabinofuranosyl Uracil", Antimicrobial Agents and Chemotherapy, Feb 1996, pp. 380-356; патенты США 5587362; 5567688 и 5565438. В патентах США 5426183 и 5424416 описаны способы получения 2'-дезокси-2',2'дифторнуклеозидов и 2'-дезокси-2'-фторнуклеозидов. См. также "Kinetic Studies of 2',2'difluorodeoxycytidine (Gemcitabine) with Purified Human Deoxycytidine Kinase and Cytidine Deaminase",Biochemical Pharmacology, vol. 45 ( 9), pp. 4857-1861, 1993. В патенте США 5446029, выданном Eriksson, et al., указано, что конкретные 2',3'-дидезокси-3'фторнуклеозиды обладают активностью против гепатита В. В патенте США 5128458 описаны конкретные 2',3'-дидезокси-4'-тиорибонуклеозиды, в которых 3'-заместитель является Н, азидом или фтором. В заявке WO 94/14831 описаны конкретные 3'-фтордигидропиримидин-нуклеозиды, в заявкеWO 92/08727 - -L-2'-дезокси-3'-фтор-5'-замещенные уридиннуклеозиды, предназначенные для лечения простого герпеса 1 и 2. В публикации заявки на европейский патент 0352248 описано большое семейство Lрибофуранозилпуриннуклеозидов, предназначенных для лечения ВИЧ, герпеса и гепатита. Хотя конкретные 2'-фторированные пуриннуклеозиды входят в рамки большого семейства, в этом описании изобретения не рассмотрены способы получения этих соединений, и они специально не описаны и не входят в список предпочтительных нуклеозидов. В этом описании изобретения не описаны способы получения 3'-рибофуранозилфторированных нуклеозидов. Аналогичные соединения представлены в заявкеWO 88/09001, поданной Aktiebolaget Astra. В заявке на европейский патент 0357571 приведена большая группа -D- и -Dпиримидиннуклеозидов, предназначенных для лечения СПИДа, которые включают и нуклеозиды, замещенные в 2'-или 3'-положении фтора группой. Однако этот широкий класс соединений не охватывает 2'фторированные нуклеозиды или способ их получения. В заявке на европейский патент 0463470 приведено описание способа получения (5S)-3 фтортетрагидро-5-[(гидрокси)метил]-2-(3 Н)-фуранона, который является известным промежуточным соединением при получении 2'-фтор-2',3'-дидезоксинуклеозидов,таких как 2'-фтор-2',3'дидезоксицитидин. В U.S.S.N. 07/556713 описаны -D-2'-фторарабинофуранозилнуклеозиды и способ получения указанных веществ, которые являются промежуточными соединениями в процессе синтеза 2',3'-дидезокси 2'-фторарабинозилнуклеозидов. В патенте США 4625020 приведено описание способа получения 1-галоген-2-дезокси-2 фторарабинофуранозильных производных, содержащих защитные сложноэфирные группы из 1,3,5-триO-ацилрибофуранозы. В научной литературе отсутствует описание -L-2'-фторрибофуранозилнуклеозидов, предназначенных для лечения ВИЧ, гепатита (В или С) или заболеваний, обусловленных пролиферацией клеток. По крайней мере в отношении 2'-рибофуранозилнуклеозидов это может быть связано с ранее возникшими трудностями при введении группы фтора в 2'-рибофуранозильную конфигурацию. То же самое верно в отношении L-2'-фтор-2',3'-ненасыщенных пуриннуклеозидов, так как пуриннуклеозиды не устойчивы в кислотных средах, что ведет к расщеплению гликозильной связи. В связи с тем, что распространение синдрома ВИЧ приобретенного иммунодефицита, СПИД ассоциированного комплекса и вирусных гепатитов В и С достигло во всем мире эпидемических масштабов и имеет трагические последствия для инфицированного субъекта, существует большая потребность в создании новых эффективных фармацевтических средств для лечения этих болезней, характеризующихся низкой токсичностью для пациента. Кроме того, существует потребность в новых антипролиферативных средствах. Поэтому целью настоящего изобретения являются способ и композиция для лечения людей, стра-4 008609 дающих вирусными гепатитами В или С. Другой целью настоящего изобретения является применение 2'-фторD-нуклеозида и 2'фторнуклеозида для изготовления лекарственного средства, предназначенного для лечения людей, инфицированных вирусом гепатита В или С соответственно. Изложение сущности изобретения 2'-Фторнуклеозиды являются биологически активными молекулами, позволяющими эффективно лечить гепатит В, гепатит С или ВИЧ. Эти соединения полезны также для лечения аномальной пролиферации клеток, включая опухоли и рак. Спектр активности этого соединения можно легко определить при помощи описанных здесь методов или любого другого подтверждающего анализа. В соответствии с другим вариантом осуществления изобретения активное соединение, его производное или соль, предназначенные для лечения гепатита или ВИЧ, можно вводить в сочетании или поочередно с другим антивирусным средством, таким как средство против ВИЧ или средство против гепатита, включая соединения вышеуказанной формулы. В случае комбинированной терапии эффективные дозы двух или большего количества средств обычно вводят одновременно, а в случае альтернированной терапии эффективные дозы всех средств - последовательно. Дозировка зависит от скорости поглощения,инактивации и выведения лекарственного средства, а также от других факторов, известных специалистам в этой области. Следует отметить, что величина дозы может быть различной в зависимости от серьезности заболевания. Кроме того, вполне понятно, что схемы приема лекарственных средств, разработанные для любого конкретного пациента, должны изменяться с течением времени в соответствии с потребностями субъекта и профессиональным мнением специалиста, наблюдающего за введением этих композиций. Неограничивающие примеры антивирусных средств, которые можно использовать в сочетании с описанными здесь соединениями, включают 2-гидроксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолан(FTC); (-)-энантиомер 2-гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиолана (3 ТС); карбовир, ацикловир,интерферон, фамцикловир, пенцикловир, AZT, DDI, DDC, D4T, абакавир, L-(-)-FMAU, пролекарства на основе фосфата L-DDA и -D-диоксоланнуклеозиды, такие как -D-диоксоланилгуанин (DG), -Dдиоксоланил-2,6-диаминопурин (DAPD) и -D-диоксоланил-6-хлорпурин (АСР), ингибиторы обратной транскриптазы (RT), не являющиеся нуклеозидами, такие как невирапин, МKС-442, DMP-266 (Sustiva), а также ингибиторы протеазы, такие как индинавир, саквинавир, AZT, DMP-450 и др. Эти соединения можно также использовать для лечения вирусных заболеваний, вызываемых вирусом инфекционной анемии лошадей (EIAV), вирусом иммунодефицита кошек и вирусом иммунодефицита обезьян. (Wang, S., Montelaro, R., Schinazi, R.F., Jagerski, В. and Mellors, J.W.: "Activity of nucleoside andD.C., "Equine Infectious Anemia", Vet. Clin. North Am. Equine Pract. United States, 9:321-336, 1993; Philpott,M.S., Ebner, J.P., Hoover, E.A., "Evaluation of 9-(2-phosphonylmethoxyethyl)adenin therapy for feline immunodeficiency virus using a quantitative polymerase chain reaction". Vet. Immunol. Immunopathol. 35:155166,1992). Кроме того, создан новый и совершенно диастереоселективный метод введения фтора в предшественник кольца безуглеводного сахара. Этот метод включает взаимодействие предшественника хирального кольца безуглеводного сахара (4S)-5-(замещенный окси)-пентан-4-олида, который может быть получен из L-глутаминовой кислоты, с электрофильным источником фтора, которым является, но не ограничивается им, N-фтор-(бис)бензолсульфонимид, с получением основного промежуточного соединения фторлактона 6. Фторлактон восстанавливают в лактон и ацетилируют, что дает аномерный ацетат, который затем используют для синтеза ряда новых -L2'-фторнуклеозидов. Можно также синтезировать соответствующий D-энантиомер, используя D-глутаминовую кислоту в качестве исходного вещества. В соответствии с альтернативным вариантом осуществления изобретения получают фторированный гликаль, который дегидрируют и затем превращают в 2',3'-дидезокси-2',3'-дидегидро-2'-фторнуклеозид или -L- или -D-арабинозил-2'-фторнуклеозид, как это более подробно описано ниже. Кроме того, описан более простой способ получения 2',3'-дидезокси-2',3'-дидегидро-2'фторнуклеозидов, который включает прямую конденсацию силилированного 6-хлорпурина ключевым промежуточным соединением, которое получают из L-2,3-D-изопропилиденглицеральдегида. Изобретение относится к применению 2'-фторD-нуклеозида для изготовления лекарственного средства, предназначенного для лечения инфекционного гепатита В у человека, в котором 2'-фторDнуклеозид имеет формулуR2 обозначает Н, монофосфат, дифосфат, трифосфат, ацил или другую фармацевтически приемлемую отщепляемую группу, которая при введении in vivo способна дать соединение, в котором R2 обозначает Н или фосфат; эфир сульфокислоты, включая C1-С 10 алкилсульфонил или C1-С 10 арилалкилсульфонил, в том числе метансульфонил; бензил, в котором фенильная группа необязательно замещена одним или несколькими заместителями, выбираемыми из группы, включающими гидроксил, амино, алкиламино,ариламино, алкокси, арилокси, нитро, циано, сульфокислоту, сульфат, фосфоновую кислоту, фосфат или фосфонат; иR3 обозначает ацил, C1-C4 алкил, фосфат или другую фармацевтически приемлемую отщепляемую группу, которая при введении in vivo способна отщепляться с образованием исходного соединения или его фармацевтически приемлемой соли, необязательно в сочетании с фармацевтически приемлемым носителем. Предпочтительно, если в указанных выше соединенияхR1 обозначает Н, ОН, OR3, C1-C4 низший алкил или галоген;R3 обозначает ацил,R2 обозначает Н, монофосфат, дифосфат, трифосфат или ацил,R1 обозначает Н, ОН, С 1-С 4 низший алкил или галоген; иR3 обозначает ацил; основание является пуриновым основанием, выбираемым из группы, состоящей из гуанина, аденина,гипоксантина, 2,6-диаминопурина и 6-хлорпурина;R1 обозначает ОН или OR3;R1 обозначает С 1-С 4 низший алкил или CF3;R2 обозначает ацил. Предпочтительно, если в указанных выше соединениях основание обозначает пуриновое основание,выбираемое из аденина, N6-алкилпуринов, N6-ацилпуринов [где ацил является С(О)(алкилом, арилом,алкиларилом или арилалкилом)], N6-бензилпурина, N6-галогенпурина, N6-винилпурина, N6 ацетиленпурина, N6-ацилпурина, N6-гидроксиалкилпурина, N6-тиоалкилпурина, N2-алкилпуринов, N2 алкил-6-тиопуринов, гуанина, гипоксантина, 2,6-диаминопурина, 2-хлор-2-аминопурина, инозина или 6 хлорпурина. Предпочтительно, если лекарственное средство является подходящим для орального, парентерального или внутривенного введения. В другом аспекте изобретение относится к применению 2'-фторнуклеозида для изготовления лекарственного средства, предназначенного для лечения инфекционного гепатита С у человека, в котором 2'фторнуклеозид является соединением, описываемым формулой в которой Base обозначает пуриновое или пиримидиновое основание;R2 обозначает Н, монофосфат, дифосфат, трифосфат, ацил или другую фармацевтически приемлемую отщепляемую группу, которая при введении in vivo способна дать соединение, в котором R2 обозначает Н или фосфат; эфир сульфокислоты, включая C1-С 10 алкилсульфонил или C1 С 10 арилалкилсульфонил, в том числе метансульфонил; бензил, в котором фенильная группа необязательно замещена одним или несколькими заместителями, включающими гидроксил, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфокислоту, сульфат, фосфоновую кислоту, фосфат или фосфонат; иR3 обозначает ацил, C1-С 10 алкил, фосфат или другую фармацевтически приемлемую отщепляемую группу, которая при введении in vivo способна отщепляться с образованием исходного соединения или его фармацевтически приемлемой соли, необязательно в сочетании с фармацевтически приемлемым носителем.R1 обозначает Н, ОН, C1-C4 низший алкил или галоген; иR3 обозначает ацил. Предпочтительно, если R2 обозначает ацил. Предпочтительно, если в указанных выше соединениях основание обозначает пуриновое основание или пиримидиновое основание. Предпочтительно, если основание обозначает пуриновое основание, выбираемое из аденина, N6 алкилпуринов, N6-ацилпуринов [где ацил является С(О) (алкилом, арилом, алкиларилом или арилалкилом)], N6-бензилпурина, N6-галогенпурина, N6-винилпурина, N6-ацетиленпурина, N6-ацилпурина, N6 гидроксиалкилпурина, N6-тиоалкилпурина, N2-алкилпуринов, N -алкил-6-тиопуринов, гуанина, гипоксантина, 2,6-диаминопурина, 2-хлор-2-аминопурина, инозина или 6-хлорпурина. Предпочтительно, если в указанных выше соединениях основание обозначает пиримидиновое основание, выбираемое из тимина, цитозина, 5-фторцитозина, 5-метилцитозина, 6-азапиримидина, в том числе 6-азацитозина, 2- и/или 4-меркаптопиримидина, урацила, 5-галогенурацила, в том числе 5 фторурацила,С 5-алкилпиримидинов,С 5-бензилпиримидинов,С 5-галогенпиримидинов,С 55 5 5 винилпиримидина, С -ацетиленпиримидина, С -ацилпиримидина, С -гидроксиалкилпурина, С 5 амидопиримидина, С 5-цианопиримидина, С 5-нитропиримидина, С 5-аминопиримидина, 5-азацитидинила,5-азаурацилила, триазолопиридинила, имидазолопиридинила, пирролопиримидинила или пиразолопиримидинила. Предпочтительно, если лекарственное средство является подходящим для орального, парентерального или для внутривенного введения. Предлагается применение 2'-фторD-нуклеозида для изготовления лекарственного средства,предназначенного для лечения инфекционного гепатита В или С. Термин "алкил" в используемом здесь значении за исключением особо оговоренных случаев означает насыщенный, с прямой или разветвленной цепью, циклический, первичный, вторичный или третичный углеводород, имеющий C1-С 10 атомов и, в частности, включает метил, этил, пропил, изопропил,циклопропил, бутил, изобутил, трет-бутил, пентил, циклопентил, изопентил, неопентил, гексил, изогексил, циклогексил, циклогексилметил, 3-метилпентил, 2,2-диметилбутил и 2,3-диметилбутил. Алькильная группа может быть необязательно замещена одной или несколькими фрагментами, выбираемыми из группы, включающей гидроксил, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфокислоту, сульфат, фосфоновую кислоту, фосфат или фосфонат, которые могут быть не защищены или при необходимости защищены группами, известными специалистам в этой области, например, описанными в справочнике Greene, et al., Protective Groups in Organic Synthesis, John WileySons, Second Edition, 1991, который включен в это описание изобретения в качестве ссылки. Термин "низший алкил" в используемом здесь значении за исключением особо оговоренных случаев означает С 1-С 4 насыщенную, с прямой или разветвленной цепью, или, когда удобно, циклическую(например, циклопропил) алкильную группу. Термин "алкиламино" или "ариламино" означает аминогруппу, имеющую соответственно один или два алкильных или арильных заместителя. Термин "защищенный" в используемом здесь значении за исключением особо оговоренных случаев означает группу, присоединяемую к атому кислорода, азота или фосфора для предотвращения его дальнейшего взаимодействия или в других целях. Специалистам в области органического синтеза известно большое число защитных групп для атомов кислорода и азота. Термин "арил" в используемом здесь значении за исключением особо оговоренных случаев означает фенил, бифенил или нафтил, предпочтительно фенил. Арильная группа может быть необязательно замещена одной или несколькими фрагментами, выбираемыми из группы, включающей гидроксил, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфокислоту, сульфат, фосфоновую кислоту, фосфат или фосфонат, которые могут быть не защищены или при необходимости защищены группами, известными специалистам в этой области, например, описанными в справочнике Greene, et al.,Protective Groups in Organic Synthesis, John Wiley and Sons, Second Edition, 1991. Термин "алкарил" или "алкиларил" означает алкильную группу с арильным заместителем. Термин"аралкил" или "арилалкил" означает арильную группу с алкильным заместителем. Термин "галоген" в используемом здесь значении означает хлор, бром, йод и фтор. Термин "пуриновое или пиримидиновое основание" означает, но не ограничивается ими, аденин,N6-алкилпурины, N6-ацилпурины [где ацил является С(О) (алкилом, арилом, алкиларилом или арилалкилом)], N6-бензилпурин, N6-галогенпурин, N6-винилпурин, N6-ацетиленпурин, N6-ацилпурин,N6-гидроксиалкилпурин, N6-тиоалкилпурин, N2-алкилпурины, N2-алкил-6-тиопурины, тимин, цитозин,5-фторцитозин, 5-метилцитозин, 6-азапиримидин, в том числе 6-азацитозин, 2- и/или-7 008609 4-меркаптопиримидин, урацил, 5-галогенурацил, в том числе 5-фторурацил, С 5-алкилпиримидины,С 5-бензилпиримидины,С 5-галогенпиримидины,С 5-винилпиримидин,С 5-ацетиленпиримидин,5 5 5 С -ацилпиримидин,С -гидроксиалкилпурин,С -амидопиримидин,С 5-цианопиримидин,5 5 2 2 С -нитропиримидин, С -аминопиримидин, N -алкилпурины, N -алкил-6-тиопурины, 5-азацитидинил,5-азаурацилил, триазолопиридинил, имидазолопиридинил, пирролопиримидинил и пиразолопиримидинил. Пуриновые основания включают, но не ограничиваются ими, гуанин, аденин, гипоксантин,2,6-диаминопурин и 6-хлорпурин. Функциональные кислородные и азотные группы в этом основании могут быть защищены при необходимости или при желании. Приемлемые защитные группы хорошо известны специалистам в этой области и включают триметилсилил, диметилгексилсилил, третбутилдиметилсилил и трет-бутилдифенилсилил, тритил, алкильные группы, ацильные группы, такие как ацетил и пропионил, метансульфонил и п-толуолсульфонил. Активное соединение можно вводить в виде любого производного, которое при введении реципиенту может прямо или косвенно образовывать исходное соединение либо само обладает требуемой активностью. Неограничивающими примерами являются фармацевтически приемлемые соли (альтернативно именуемые "физиологически приемлемыми солями") и соединение, алкилированное или ацилированное в 5'-положении либо в пуриновом или пиримидиновом основании (альтернативно именуемые"фармацевтически приемлемыми производными"). Кроме того, на биологическую активность соединения могут влиять модификации, которые в некоторых случаях повышают активность по сравнению с исходным соединением. Это можно легко определить, получив производное и испытав его на антивирусную активность при помощи описанных здесь методов или любого другого метода, известного специалистам в этой области. Термин "ацил" означает эфир карбоновой кислоты, в котором некарбонильный фрагмент сложноэфирной группы выбирают из группы, включающей алкил или низший алкил с прямой или разветвленной цепью либо циклический алкил или низший алкил, алкоксиалкил, в том числе метоксиметил, аралкил, в том числе бензил, арилоксиалкил, такой как феноксиметил, арил, в том числе фенил, необязательно замещенный галогеном, С 1-С 4 алкил или С 1-С 4 алкокси, эфиры сульфокислоты, такие как алкил или аралкилсульфонил, в том числе метансульфонил, эфир моно-, ди- или трифосфорной кислоты, тритил или монометокситритил, замещенный бензил, триалкилсилил (например, диметил-трет-бутилсилил) или дифенилметилсилил. Арильные группы в сложных эфирах предпочтительно содержат фенильную группу. В используемом здесь значении термин "практически не содержащий" или "практически при отсутствии" означает нуклеозидную композицию, которая содержит по крайней мере 95-98% или более, предпочтительно 99-100% указанного энантиомера этого нуклеозида. Нуклеотидные пролекарственные препараты Все описываемые здесь нуклеозиды можно вводить в виде нуклеотидного пролекарства, что позволяет увеличить активность, биологическую доступность, стабильность или каким-либо другим образом изменить свойства нуклеозида. Известен ряд нуклеотидных пролекарственных лигандов. Как правило,алкилирование, ацилирование или другая липофильная модификация моно-, ди- или трифосфата нуклеозида увеличивает стабильность нуклеотида. Примерами замещающих групп, которые могут заменять один или несколько атомов водорода в фосфатном фрагменте, являются алкил, арил, стероиды, углеводы, в том числе сахара, 1,2-диацилглицерин и спирты. Многие из них описаны в статье R. Jones andN. Bischofberger, Antiviral Research, 27 (1995) 1-17. Все эти группы можно использовать в сочетании с рассматриваемыми нуклеозидами для достижения требуемого эффекта. Активный нуклеозид может быть также получен в виде липида 5'-фосфоэфира или липида 5'-эфира,как это описано в нижеследующих материалах, которые включены в это описание изобретения в качестве ссылки: Kucera, L.S., N. Iyer, E. Leake, A. Raben, Modest E.K., D.L.W., and C. Piantadosi, 1990, "NovelLenting, H. van den Bosch, and D.D. Richman, 1990, "Synthesis and antiretroviral activity of phospholipid analogs of azidothymidine and other antiviral nucleosides", J. Biol. Chem., 265:61127. Неограничивающие примеры патентов США, в которых описаны приемлемые липофильные заместители, которые можно ковалентно вводить в нуклеозид, предпочтительно в 5'-ОН положении нуклеозида или липофильных препаратов, имеют патенты США 5149794 (22 сентября 1992 г., Yatvin et al.); 5194654 (16 марта 1993 г., Hostetler et al.); 5223263 (29 июня 1993 г., Hostetler et al.); 5256641 (26 октября 1993 г., Yatvin et al.); 5411947 (2 мая 1995 г., Hostetler et al.); 5463092 (31 октября 1995 г., Hostetleret al.); 5543389 (6 августа 1996 г., Yatvin et al.); 5543390 (6 августа 1996 г., Yatvin et al.); 5543391 (6 авгу-8 008609 ста 1996 г., Yatvin et al.); и 5554728 (10 сентября 1996 г., Basava et al.), которые включены в данное описание изобретения в качестве ссылки. Иностранными заявками на патент, в которых описаны липофильные заместители, присоединяемые к нуклеозидам по настоящему изобретению, или липофильные препараты, являются WO 89/02733, WO 90/00555, WO 91/16920, WO 91/18914, WO 93/00910, WO 94/26273,WO 96/15132, европейский патент 0350287, европейский патент 93917054.4 и WO 91/19721. Неограничивающие примеры нуклеотидных пролекарств приведены в нижеследующих ссылках: Но, D.H.W. (1973) "Distribution of Kinase and deaminase of 1D-arabinofuranosylcytosine in tissues of man-9 008609 Кислые алкилфосфатные производные анти-ВИЧ агента AZT могут быть менее токсичными по сравнению с аналогом исходного нуклеозида. Antiviral Chem. Chemother. 5, 271-277; Meyer, R.B.,Jr., Shuman, D.A. and Robins, R.K. (1973) "Synthesis of purine nucleoside 3',5'-cyclic phosphoramidates".Lett. 28, 199-202; Shuto, S., Itoh, H., Ueda, S., Imamura, S., Kukukawa, K., Tsujino, M., Matsuda, A. and Ueda,T. (1988) Pharm. Bull. 36, 209-217. Примером полезной фосфатной группой пролекарства является- 10008609 Комбинированная и альтернированная терапия Установлено, что после продолжительного лечения антивирусным средством могут появиться устойчивые к лекарственным средствам варианты ВИЧ и HBV. Устойчивость к лекарственному средству обычно возникает в результате мутации гена, кодирующего фермент, используемый в репликации вируса, причем в случае ВИЧ таким ферментом чаще всего бывает обратная транскриптаза, протеаза или полимераза ДНК, и в случае HBV таким ферментом является полимераза ДНК. Недавно было показано, что действие лекарственного средства против ВИЧ можно продлить, усилить или восстановить путем введения данного соединения в сочетании или поочередно со вторым и, возможно, третьим антивирусным соединением, которое вызывает другую мутацию, отличающуюся от мутации, вызываемой основным лекарственным средством. Кроме того, при помощи такой комбинированной или альтернированной терапии можно изменить фармакокинетику, распределение в организме и другие параметры лекарственного средства. Как правило, комбинированная терапия является более предпочтительной по сравнению с альтернированной терапией, так как она оказывает множественное одновременное действие на вирус. Вторым антивирусным средством, предназначенным для лечения ВИЧ в соответствии с одним вариантом осуществления изобретения, является ингибитор обратной транскриптазы ("RTI"), который может быть или синтетическим нуклеозидом ("NRTI"), или соединением, не являющимся нуклеозидом("NNRTI"). В соответствии с другим вариантом осуществления изобретения в случае ВИЧ вторым (или третьим) антивирусным средством может быть ингибитор протеазы. В соответствии с другими вариантами осуществления изобретения вторым (или третьим) соединением может быть аналог пирофосфата или ингибитор слияния. Данные об устойчивости к лекарственным средствам, полученные in vitro и invivo для ряда антивирусных соединений, приведены в статье "Schinazi, et al., Mutations in retroviral genesassociated with drug resistance", International Antiviral News, 1997. Предпочтительными соединениями для комбинированной или альтернированной терапии, предназначенными для лечения HBV, являются 3 ТС, FTC, L-FMAU, интерферон, -D-диоксоланилгуанин(DXG), -D-диоксоланил-2,6-диаминопурин (DAPD) и -D-диоксоланил-6-хлорпурин (АСР), фамцикловир, пенцикловир, BMS-200475, bis pom PMEA (адефовир, дипивоксил), лобукавир, ганцикловир и рибаварин. Предпочтительными примерами антивирусных средств, которые можно использовать в сочетании или поочередно с описываемыми здесь соединениями для лечения ВИЧ, являются цис-2-гидроксиметил 5-(5-фторцитозин-1-ил)-1,3-оксатиолан (FTC); (-)-энантиомер 2-гидроксиметил-5-(цитозин-1-ил)-1,3 оксатиолан (3 ТС); карбовир, ацикловир, фоскарнет, интерферон, AZT, DDI, DDC, D4T, CS-87 (3'-азидо 2',3'-дидезоксиуридин) и -D-диоксоланнуклеозиды, такие как -D-диоксоланилгуанин (DXG), -Dдиоксоланил-2,6-диаминопурин (DAPD) и -D-диоксоланил-6-хлорпурин (АСР), МKС-442 (6-бензил-1 этоксиметил)-5-изопропилурацил. Предпочтительными ингибиторами протеазы являются криксиван (Merck), нелфинавир (Agouron),ритонавир (Abbott), саквинавир (Roche), DMP-266 (Sustiva) и DMP-450 (DuPont Merck). Более полный список соединений, которые можно вводить в сочетании или поочередно со всеми описываемыми нуклеозидами, включает сукцинатBMS 186318: ингибитор протеазы ВИЧ-1 на основе производного аминодиола (Bristol-MyersSquibb);HIV-1: вирус иммунодефицита человека типа 1;L-735524: ингибитор протеазы ВИЧ-1 на основе гидроксиаминопентанамида (Merck);Ro 31-8959: ингибитор протеазы ВИЧ-1 на основе производного гидроксиэтиламина (Roche);SC-52151: ингибитор протеазы на основе изостера гидроксиэтилмочевины (Searle);SC-55389A: ингибитор протеазы на основе изостера гидроксиэтилмочевины (Searle);VB 11328: ингибитор протеазы на основе гидроксиэтилсульфонамида (Vertex);VX-478: ингибитор протеазы на основе гидроксиэтилсульфонамида (Vertex); ХМ 323: ингибитор протеазы на основе циклической мочевины (Dupont Merck). Комбинированная терапия для лечения пролиферативных состояний В соответствии с еще одним вариантом осуществления изобретения соединения используемые в качестве антипролиферативных средств можно вводить в сочетании с другими соединениями, повышающими эффективность лечения, которые включают, но не ограничиваются ими, антифолат,5-фтор-пиримидин (в том числе 5-фторурацил), аналог цитидина, такой как -L-1,3-диоксоланилцитидин или -L-1,3-диоксоланил-5-фторцитидин, антиметаболиты (в том числе пуриновые антиметаболиты, цитарабин, фударабин, флоксуридин 6-меркаптопурин, метотрексат и 6-тиогуанин), гидроксимочевину,ингибиторы митоза (в том числе СРТ-11, этопозид (VR-21, таксол и винкаалкалоиды, такие как винкристин и винбластин, алкилирующие средства (которые включают, но не ограничиваются ими, бусульфан,- 12008609 хлорамбуцил, циклофосфамид, ифофамид, метахлорамин, мелфалан и тиотепа), неклассические алкилирующие средства, соединения, содержащие платину, блеомицин, противоопухолевый антибиотик, антрациклин, такой как доксорубицин и данномицин, антрацендион, ингибиторы топоизомеразы II, гормональные средства (которые включают, но не ограничиваются ими, кортикостероиды (дексаметазон,преднизон и метилпреднизон), андрогены, такие как флуоксиместерон и метилтестостерон, эстрогены,такие как диэтилстильбэстерол, антиэстрогены, такие как тамоксифен, аналоги LHRH, такие как лейпролид, антиандрогены, такие как флутамид, аминоглютэтимид, мегестролацетат и медроксипрогестерон,аспарагиназу, кармустин, ломустин, гексаметилмеламин, дакарбазин, митотан, стрептозоцин, цисплатин,карбоплатин, левамазол и лейковорин. Соединения по настоящему изобретению можно также использовать в сочетании с ферментными лечебными средствами и модуляторами иммунной системы, такими как интерферон, интерлейкин, фактор опухолевого некроза, фактор, стимулирующий колонию макрофагов, и колониестимулирующий фактор. Способ получения активных соединений В соответствии с одним вариантом осуществления изобретения выполняют диастереоселективную реакцию, обеспечивающую введение фтора в сахарную часть новых аналогов нуклеозида. Этот метод синтеза можно использовать для получения производных пурина и пиримидина. Основной стадией синтеза является фторирование предшественника хирального кольца безуглеводного сахара (4S)-5(защищенный-окси)пентан-4-олида, например, (4S)-5-(трет-бутилдифенилсилокси)пентан-4-олида 4, с использованием источника электрофильного фтора, которым является, но не ограничивается им, N-фтор(бис)бензолсульфонимид 5. Этот относительно новый класс N-фторсульфонимидных реагентов был первоначально получен Барнетом в 1984 г.; с тех пор он был усовершенствован и используется в настоящее время в качестве удобного и реакционноспособного источника электрофильного фтора (Barnette,W.E., J. Am. Chem. Soc. 1984, 106, 452; Davis, F.A.; Han, W., Murphy, C.K, J. Org. Chem. 1995, 60, 4730;Snieckus, V., Beaulieu, F. , Mohri, K., Han, W., Murphy, C.K., Davis, F.A., Tetrahedron Lett. 1994, 35(21),3465). Эти реагенты чаще всего используют для введения фтора в нуклеофилы, такие как еноляты и метилированные ароматические соединения (Davis, F.A., Han, W., Murphy, C.K., J. Org. Chem. 1995, 60,4730). В частности, N-фтор-(бис)бензолсульфонимид (NFSi) является устойчивым на воздухе, простым в обращении твердым веществом с достаточным пространственным объемом для стереоизбирательного фторирования енолята силилзащищенного лактона 4. В качестве неограничивающего примера этого способа ниже подробно описан синтез фторолактона 6 и его использование в качестве промежуточного соединения для синтеза ряда новых -2'-фторнуклеозидов. На основании этого описания специалист в этой области сможет при желании изменить этот способ для достижения поставленной цели и получить требуемое соединение. Можно использовать любой источник электрофильного фтора, который фторирует предшественник, такой как (4S)-5-(защищенный-окси)пентан-4-олид, например, (4S)-5-(трет-бутилдифенилсилокси) пентан-4-олид. Альтернативными источниками электрофильного фтора являютсяJ. Org. Chem. 1995, 60, 4730-4737), 1-фторэтен и его синтетические эквиваленты (Matthews, Tet. Lett.Vol. 35,7, pp. 1027-1030 (1994); фторирующие агенты Accufluor фирмы Allied Signal, Inc., Buffalo Research Laboratory, Buffalo, NewYork, NFTh 1-фтор-4-гидрокси-1,4-диазоабицикло[2.2.2]октан-бис(тетрафторборат), NFPy (пиридингептафтордиборат N-фторпиридиния) и NFSi (N-фторбензолсульфонимид); электрофильные фторирующие реагенты фирмы Aldrich Chemical Company, Inc., в том числе солиN-фторсульфонимиды и амиды, в частности (N-фтор-N-метил-п-толуолсульфонамид, N-фтор-Nпропил-п-толуолсульфонамид и N-фторбензолсульфонимид); фторид N-фторхинуклидиния (J. Chem. Soc. Perkin Trans I 1988, 2805-2811); перфтор-2,3,4,5-тетрагидропиридин и перфтор-(1-метилпирролидин), Banks, Cheng, and Haszeldine,Heterocyclic Polyfluoro-Compounds Part II (1964); 1-фтор-2-пиридон, J. Org. Chem., 1983, 48, 761-762; четвертичные стереогенные центры, имеющие атом фтора (J. Chem. Soc. Perkin Trans, pp. 221-227- 13008609 Существенным преимуществом этого метода является возможность получения отдельно "природного" (1a)-D- или "неприродного" (1b)-L-энантиомера нуклеозидов в результате соответствующего выбора L- или D-глутаминовой кислоты в качестве исходного вещества. Лактон 4 синтезируют по методу, показанному на схеме 1, из L-глутаминовой кислоты, как это описано Ravid et al. (Tetrahedron 1978, 34, 1449) и Taniguchi et al. (Tetrahedron 1974, 30, 3547). Схема 1 Известно, что енолят лактона 4, полученный при температуре -78 С с использованием LiHMDS в ТГФ, является устойчивым веществом. Этот енолят используют при осуществлении нескольких методов синтеза, включая добавление электрофилов, таких как дифенилдиселенид, дифенилдисульфид и алкилгалогениды, с получением высоких выходов (Liotta, D.C., Wilson, L., J. Tetrahedron Lett. 1990, 31 (13),1815; Chu, C.K., Babu, J.R., Beach, J.W., Ann. S.K., Huang, H., Jeong, L.S., Lee, S.J., J. Org. Chern., 1990, 55,1418; Kawakami, H., Ebata, Т., Koseki, K., Matsushita, H., Naoi, Y., Itoh, K., Chem. Lett. 1990, 1459). Однако добавление к еноляту 4 раствора соединения 5 в ТГФ дает плохой выход требуемого монофторированного продукта 6. Получены многочисленные побочные продукты, в том числе вещество, которое предположительно является дифторированным лактоном и не отделяется от других примесей. По этой причине порядок введения реагентов изменен таким образом, что лактон 4 и NFSi 5 вместе растворяют в ТГФ и охлаждают до температуры -78 С. Медленное добавление LiHMDS позволяет получить вещество 6 в виде единственного продукта с небольшим количеством непрореагировавшего исходного вещества (1) Фторолактон 6 можно получить с 50-70% выходом, производя хроматографию на колонках из силикагеля и кристаллизацию. Реакция приводит к получению одного диастереомера 6 главным образом в результате взаимодействия пространственно объемной группы TBDPS и объемного фторирующего реагента 5. Фторолактон 6 идентифицируют как - или "нижний" фтороизомер, т.е. с положением атома F книзу от плоскости кольца, производя сравнение с ранее опубликованными данными ЯМР и при помощи рентгеноструктурного исследования энантиомера 20. Лактон 6 превращают в аномерный ацетат 8, как это показано на схеме 2. Интересно отметить, что лактон 7 существует исключительно в виде -аномера и у ацетата 8 при помощи ЯМР не обнаружен аномер, о чем сообщили Niihata et al. (Bull. Chem. Soc. Jpn. 1995, 68, 1509). Схема 2 Реакцию сочетания соединения 8 с силилированными пиримидиновыми основаниями выполняют в соответствии со стандартным методом Ворбруггена (Tetrahedron Lett., 1978, 15, 1339) с использованием трифлата TMS в качестве кислоты Льюиса. Альтернативно можно использовать любую другую кислоту Льюиса (которая, как известно, способна эффективно конденсировать основание вместе с углеводом, в результате чего образуется нуклеозид), включая хлорид олова, хлорид титана и другие соединения олова или титана. Целый ряд оснований можно успешно подвергнуть реакции сочетания с получением высоких- 14008609 выходов в интервале 72-100% после выполнения хроматографии на колонках (уравнение 2, табл. 1) Таблица 1 Гликозилирование замещенных пиримидинов соединением 8 Результаты ПМР показывают, что соотношение между аномерами - и -нуклеозидов во всех случаях составляет примерно 2:1. Силилзащищенные нуклеозиды нельзя разделить при помощи хроматографии на колонках на отдельные аномеры. Однако после снятия защиты с 5'-кислорода при помощиNH4F в метаноле (уравнение 3) - и -аномеры можно легко отделить друг от друга; полученные результаты приведены в табл. 2. Классификацию свободных нуклеозидов с выявлением -или -формы производят на основании химического сдвига аномерного протона (табл. 3) или полярности нуклеозидов, определяемой при помощи тонкослойной хроматографии. Тенденция создавать / пары свободных нуклеозидов выражается в том, что менее полярное соединение из двух имеет химический сдвиг аномерных протонов в сторону сильного поля от более полярного соединения. Таблица 3 Химический сдвиг аномерных протонов (миллионные доли) Корреляцию между химическим сдвигом аномерных протонов и абсолютной структурой проверяют, сравнивая показатели соединений 18 а (Niihata, S., Ebata, Т., Kawakami, H., Matsushida, H. Bull. Chem.Med. Chem. 1989, 32, 1743) с ранее опубликованными спектральными данными и при помощи рентгеноструктурного исследования соединений 14b и 15b. Это открытие противоречит обычной тенденции, характерной для нуклеозидов, в соответствии с которой -аномер обычно является менее полярным из двух аномеров. Предполагается, что у 2'-фторированных нуклеозидов со слабопольным сдвигом сильный диполь связи C-F противоположен диполю аномерной связи C-N в -изомере и уменьшает общий молекулярный диполь. И наоборот, геометрия -аномера позволяет усилить молекулярный диполь в результате добавления диполей связи C-F и C-N. Таким образом, в случае -2'-фторнуклеозидов -аномер является более полярным, чем -изомер.- и -Аномеры соединений 17 а и 17b нельзя разделить хроматографией на колонках, так как из-за наличия свободной аминогруппы нуклеозиды образуют полосы на силикагеле. Поэтому необходимо использовать N4-ацетилцитозин для получения соединения 11 и последующего разделения соединений 16 а и 16b. N4-ацетильную группу количественно удаляют насыщенным раствором аммиака в метаноле, что позволяет разделить соединения 17 а и 17b. Аномеры 15 а и 15b можно легко разделить, используя 5 фторцитозин в качестве основания (соединение 10), при этом на силикагеле отсутствуют полосы. Из десяти нуклеозидов, приведенных в табл. 2, ранее были синтезированы только соединения 17bClercq, E. J. Med. Chem. 1989, 32, 1743). Подобно многочисленным известным аналогам 2'- или фторнуклеозидов с расположением атома фтора вверх от плоскости кольца 14 они синтезированы из природных предшественников (т.е. имеют -D-конфигурацию). По-видимому, в научной литературе ранее не были описаны -L-2'-фторрибофуранозилнуклеозиды. Фтор обычно вводят в эти молекулы путем нуклеофильной атаки на ангидронуклеозид (Mengel, R.,Guschlbauer, W. Angew. Chem., Int. Ed. Engl. 1978, 17, 525) или путем замещения и превращения стереохимически фиксированной гидроксильной группы при помощи трифторида диэтиламиносеры (DAST)(Herdewijn, P., Aerschot, A.V., Kerremans, L., Nucleosides Nucleotides 1989, 8(1), 65). Преимуществом способа по настоящему изобретению является то, что для введения фтора не требуется гидроксильная группа. Таким образом, этот способ не ограничивается использованием природных нуклеозидов или cахаров в качестве исходных веществ и позволяет легко получить неприродные энантиомеры 2'фторнуклеозидов. В соответствии с этой схемой синтезировано несколько неприродных нуклеозидов с использованием D-глутаминовой кислоты 19 в качестве исходного вещества (схема 3). Предшественник кольца сахара 20 фторируют так, как это описано выше, и подвергают реакции сочетания с разными силилированными основаниями (табл. 4). Схема 3- 16008609 Таблица 4 Выходы неприродных аналогов нуклеозидов Успешный синтез соединения 29, показанного на схеме 4, позволяет получить две категории нуклеозидов. Первый класс составляют соединения, известные как 2',3'-дидезокси-2',3'-дидегидро-2,2'фторнуклеозиды (соединение 30), и второй класс образуют фтор- или арабиносодержащие аналоги с положением заместителей "вверх" (соединение 31) нуклеозидов, показанных на схеме 5 Схема 5 Соединения 30 и 31 можно синтезировать из общего промежуточного соединения 32, которое получают селенилированием фторгликаля 29 Схема 6- 17008609 Селенилированное соединение 32 можно превратить во фторсодержащий аналог 31 с положением атома фтора "вверх" в результате восстановления никелем Ренея. Альтернативно окисление селенида 32 при помощи NaIO4 или перекисью водорода и последующее элиминирование промежуточного селеноксида при нагревании позволяют получить соединение 30. Оба эти превращения нефторированных систем хорошо описаны в научной литературе (Wurster, J.A., Ph.D. Thesis, Emory University, 1995; Wilson, L.J.,Ph.D. Thesis, Emory University, 1992). Кроме того, можно также синтезировать энантиомеры нуклеозидов 30 и 31, так как их получают из энантиомера соединения 29. Альтернативный способ получения соединений типа соединения 30 2',3'-дидезокси-2',3'-дидегидро 2'-фторнуклеозидов показан на схеме 7. Этот простой и прямой способ получения данного класса соединений с использованием ряда силилированных оснований оказался весьма успешным. Схема 7 Получение силилкетенацеталя из соединения 6 делает возможным стереоизбирательное введение бромида фенилселения с образованием соединения 36 в виде одного изомера. Восстановление и ацетилирование этого соединения происходит легко и обеспечивает высокий выход соединения 37 в результате выполнения двух стадий. -Ориентация фенилселенильной группой делает возможной стереоизбирательность на последующей стадии гликозилирования, и синтез -изомера нуклеозида 38 происходит с хорошим выходом. Соединение 38 может быть окислено перекисью водорода в дихлорметан с получением продукта элиминирования 39, но опыт работы заявителей показывает,что достаточно адсорбировать соединение 38 силикагелем и оставить выстаиваться на несколько часов,после чего соединение 39 можно элюировать из колонки почти с количественным выходом. Удаление защищенной группы из соединения 39 с получением конечного соединения 30 выполняют в соответствии с приведенным выше описанием, что дает хороший выход (81%) целевого нуклеозида. Схема 8 Тот же порядок химических превращений, который был использован для синтеза соединений 30 и 31, можно использовать для синтеза соединений 34 и 35. Экспериментальная часть Общие способы полученияN-Фтор-(бис)бензолсульфонимид 5 фирмы Allied Signal использовали без дополнительной очистки. Все другие реагенты были предоставлены фирмой Aldrich Chemical Company и использовались без дополнительной очистки. Температуры плавления измеряли в устройстве для определения точки плавления вещества в капилляре Thomas Hoover, и полученные значения не корректировали. Инфракрасные спектры (ИКС) были получены в спектрометре Nicolet Impact 400 FT-IR. Спектры 1 Н ЯМР и 13 С ЯМР определяли в спектрометре NT-360 или Varian 400 МГц. Пластинки для тонкослойной хроматографии (ТСХ) изготовлены из силикагеля 60 F254 (толщиной 0,25 мм) фирмы ЕМ Science. Флеш-хроматографию выполняли на силикагеле 60 (230-400 меш. по стандарту ASTM) фирмы ЕМ Science. Все реакции осуществляли в высушенной пламенем стеклянной посуде в атмосфере сухого аргона. Растворители удаляли испарением в роторном испарителе. Элементные анализы выполнены фирмой Atlantic Microlab, Inc., Atlanta, GA.(2S,4R)-5-(трет-Бутилдифенилсилокси)-2-фторпентан-4-олид (20). В колбу вводят (4R)-5-(трет-бутилдифенилсилокси)пентан-4-олид (20,0 г, 0,564 моль, 1,0 экв.) и Nфтор-(бис)бензолсульфонимид (NFSi) 5 (17,80 г, 0,0564 моль, 1,0 экв.) в 250 мл безводного ТГФ. Раствор охлаждают до -78 С, и 68,0 мл (0,0680 моль, 1,2 экв.) 1,0 М раствора LiHMDS в ТГФ добавляют по каплям в течение 1 ч. Полученную смесь перемешивают при температуре -78 С в течение еще 2 ч, нагревают до комнатной температуры и перемешивают еще 1 ч. После окончания реакции реакционную смесь гасят 10 мл насыщенного раствора NH4Cl. Смесь разбавляют тремя объемами диэтилового эфира и выливают в равный объем насыщенного раствора NаНСО 3. Органический слой дважды промывают насыщенным раствором NaHCO3 и один раз насыщенным раствором NaCl. Органический слой сушат надMgSO4, фильтруют и концентрируют до образования светло-желтого масла. Полученное масло очищают хроматографией на колонках из силикагеля, используя систему растворителей, состоящую из 30% диэтилового эфира/70% гексанов. Полученное белое твердое вещество кристаллизуют из горячих гексанов,что дает 13,04 г (выход 62%) прозрачного кристаллического твердого вещества: Rf (30% диэтилового эфира/70% гексанов)=0,26; т.пл.115-116 С. 1 Н ЯМР (360 МГц, CDCl3) d 7,63-7,60 (м, 4 Н), 7,45-7,35 (м, 6 Н), 5,49 (дт, J=52,9 и 7,9 Гц, 1 Н), 4,69(д, J=186,6 Гц), 77,3 (д, J=5,3 Гц), 65,0, 31,8 (д, J=20,5 Гц), 26,7, 19,1. ИКС (тонкая пленка) 2958, 1796,1252, 1192, 1111, 1016 см-1. Масс-спектрометрия высокого разрешения (МСВР), высчитано для [М+Li] C21H25O3FSiLi: 379,1717. Обнаружено: 379,1713. Элементный анализ, высчитано для CHAFFS: С, 67,71; Н, 6,76. Обнаружено: С, 67,72; Н, 6,78. 5-O-(трет-Бутилдифенилсилил)-2,3-дидезокси-2-фтор-(L)-эритронпентофураноза (21). В колбу вводят лактон 20 (12,12 г, 0,0325 моль, 1,0 экв.) и 240 мл безводного ТГФ. Раствор охлаждают до температуры -78 С, и 65 мл (0,065 моль, 2,0 экв.) 1,0 М раствора DIBALH в гексанах добавляют по каплям в течение 30 мин. Полученную смесь перемешивают при температуре -78 С в течение 3 ч и гасят, медленно добавляя 2,93 мл (0,163 моль, 5,0 экв.) воды. Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают в течение 1 ч, в результате чего в колбе образуется прозрачное желеобразное твердое вещество. Реакционную смесь разбавляют двумя объемами диэтилового эфира и выливают в равный объем насыщенного водного раствора тартрата калия-натрия в колбе Эрленмейера. Полученную смесь перемешивают в течение 20 мин до разрушения эмульсии. Органический слой отделяют, и водный слой трижды экстрагируют 250 мл диэтилового эфира. Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют до образования светло-желтого масла. Полученный продукт очищают хроматографией на колонках из силикагеля, используя систему растворителей, состоящую из гексанов/этилацетата (6:1). Полученное прозрачное масло кристаллизуют из кипящих гексанов, что дает 11,98 г (выход 98%) белого кристаллического твердого вещества: Rf (30% диэтилового эфира/70% гексанов)=0,33; т.пл. 66-67 С. 1 Н ЯМР (360 МГц, CDCl3) d 7,68-7,66 (м, 4 Н), 7,55-7,38 (м, 6 Н), 5,39 (т, J=7,6 Гц, 1 Н), 4,99 (дд,J=52,2 и 4,3 Гц, 1 Н), 4,52 (м, 1 Н), 3,88 (дд, J=10,8 и 2,5 Гц, 1 Н), 3,65 (д, J=7,9 Гц, 1 Н), 3,49 (дд, J=7,9 и 1,8 Гц, 1 Н), 2,44-2,07 (м, 2 Н), 1,07 (с, 9 Н). 13 С ЯМР (100 МГц, CDCl3) d 135,7, 135,5, 132,2, 132,1, 130,2, 130,0, 129,8, 127,9, 127,7,99,8 (д, J=31,1 Гц), 96,6 (д, J=178,3 Гц), 79,4, 64,8, 29,9 (д, J=21,2 Гц), 26,8, 19,2. ИКС (тонкая пленка) 3423, 2932, 1474, 1362, 1113 см-1. МСВР, высчитано для [М+Li] C21H27O3FSiLi: 381,1874. Обнаружено: 381,1877. Элементный анализ, высчитано для C21H27O3FSi: С, 67,35; Н, 7,27. Обнаружено: С, 67,42; Н, 7,31. 1-O-Ацетил-5-O-(трет-бутилдифенилсилил)-2,3-дидезокси-2-фтор-(L)-эритронпентофураноза (22). В колбу вводят лактон 21 (8,50 г, 0,0227 моль, 1,0 экв.) и 170 мл безводного СН 2 Сl2. Затем добавляют DMAP (0,277 г, 0,00277 моль, 0,1 экв.) и уксусный ангидрид (13,5 мл, 0,143 моль, 6,3 экв.) и перемешивают при комнатной температуре в течение ночи. После окончания реакции реакционную смесь выливают в насыщенный раствор NаНСО 3. Органический слой отделяют, и водный слой трижды экстрагируют хлороформом. Объединенные органические слои сушат над MgSO4, фильтруют, и удаляют растворитель с получением светло-желтого масла. Полученное масло очищают хроматографией на колонках из силикагеля, используя систему растворителей, состоящую из гексанов/этилацетата (8:1), что дает 9,85 г(выход 99%) прозрачного бесцветного масла: Rf (30% диэтилового эфира/70% гексанов)=0,44. 1 Н ЯМР (360 МГц, CDCl3) d 7,69-7,67 (м, 4 Н), 7,43-7,38 (м, 6 Н), 6,30 (д, J=10,4 Гц, 1 Н), 5,06 (д,J=54,9 Гц, 1 Н), 4,53 (м, 1 Н), 3,81 (дд, J=10,8 и 4,3 Гц, 1 Н), 3,72 (дд, J=10,8 и 4,3 Гц, 1 Н), 2,38-2,12 (м, 2 Н),1,89 (с, 3 Н), 1,07 (с, 9 Н). 13 С ЯМР (100 МГц, CDCl3) d 169,4, 135,6, 135,5, 133,2, 133,1, 129,8, 129,7, 127,8, 127,7, 99,3 (д,J=34,1 Гц), 95,5 (д, J=178,2 Гц), 81,4, 65,3, 31,6 (д, J=20,5 Гц), 26,8, 21,1, 19,3. ИКС (тонкая пленка) 3074,- 19008609 2860, 1750, 1589, 1229, 1113 см-1. МСВР, высчитано для [М-ОСОСН 3] C21H26O2FSi: 357,1686. Обнаружено: 357,1695. Элементный анализ, высчитано для C23H29O4FSi: С, 66,32; Н, 7,02. Обнаружено: С, 66,30; Н, 7,04. Типичный способ сочетания силилированного основания с соединением 22: (L)-5'-О-(третбутилдифенилсилил)-2',3-дидезокси-2'-фтор-5-фторцитидин (25). В колбу, оснащенную насадкой для молекулярной перегонки, вводят 5-фторцитозин (2,01 г,15,6 ммоль, 5,0 экв.), 35 мл 1,1,1,3,3,3-гексаметилдисилазана (HMDS) и каталитическое количество(1 мг) (NH4)2SO4. Белую суспензию нагревают до температуры кипения в течение 1 ч до силилирования основания и превращения реакционной смеси в прозрачный раствор. Избыток HMDS отгоняют, и оставшийся масляный остаток помещают в вакуум на 1 ч, чтобы удалить последние следы HMDS. Полученное белое твердое вещество растворяют в атмосфере аргона в 5 мл безводного 1,2-дихлорэтана. К этому прозрачному раствору добавляют раствор ацетата 22 (1,30 г, 3,12 ммоль, 1,0 экв.) в 5 мл безводного 1,2 дихлорэтана. К полученной смеси добавляют при комнатной температуре триметилсилилтрифторметансульфонат (3,32 мл, 17,2 ммоль, 5,5 экв.). Ход реакции контролируют ТСХ (10% метанола/90% СН 2 Сl2),по результатам которой реакция заканчивается через 4 ч. Реакционную смесь выливают в насыщенный раствор NаНСО 3. Органический слой отделяют, и водный слой трижды экстрагируют хлороформом. Объединенные органические слои сушат над MgSO4, фильтруют, и удаляют растворитель с получением белой пены. Полученное соединение очищают хроматографией на колонках из силикагеля, используя градиентную систему растворителей от 100% СН 2 Сl2 до 10% метанола в CH2Cl2. Соединение выделяют в виде 1,51 г (выход 99%) белой пены: смесь аномеров Rf (100% EtOAc)=0,36; т.пл. 74-80 С. 1 Н ЯМР (400 МГц, CDCl3) d 8,84 (шс, 1 Н), 8,04 (д, 33,4 Гц, 0,67 Н), 7,67-7,63 (м, 4 Н), 7,51-7,39 (м,6,33 Н), 6,11 (д, J=20 Гц, 0,33 Н), 5,98 (д, J=16,4 Гц, 0,67 Н), 5,88 (шс, 1 Н), 5,41 (д, J=52,4 Гц, 0,33 Н), 5,23(дд, J=50,4 и 4 Гц, 0,67 Н), 4,56 (м, 0,33 Н), 4,45 (м, 0,67 Н), 4,23 (дд, J=12,0 и 1,6 Гц, 0,67 Н), 3,89 (дд,J=11,2 и 3,2 Гц, 0,33 Н), 3,74-3,66 (м, 1 Н), 2,45-1,96 (м, 2 Н), 1,09 (с, 6 Н), 1,06 (с, 3 Н). 13 С ЯМР (100 МГц, CDCl3) d 158,6 (д, J=14,4 Гц), 158,4 (д, J=14,4 Гц), 153,9, 153,8, 136,6 (д,J=240,5 Гц), 136,3 (д, J=239,7 Гц), 135,6, 135,56, 135,5, 135,4, 133,1, 132,9, 132,5, 132,4, 130,1, 130,0, 129,9,127,9, 127,8, 125,8 (д, J=33,4 Гц), 124,6 (д, J=32,6 Гц), 96,5 (д, J=182,0 Гц), 91,7 (д, J=185,1), 90,7 (д,J=35,6 Гц), 87,7 (д, J=15,2 Гц), 81,5, 79,5, 64,9, 63,0, 33,5 (д, J=20,5 Гц), 30,6 (д, J=20,4 Гц), 26,9, 26,8,19,22, 19,18. ИКС (тонкая пленка) 3300, 2960, 1682, 1608, 1513, 1109 см-1. МСВР, высчитано для [М+Li] C25H29N3O3SiF2Li: 492,2106. Обнаружено: 492,2085. Элементный анализ, высчитано для C25H29N3O3SiF21/2 Н 2 О: С, 60,71; Н, 6,11; N, 8,50. Обнаружено: С, 60,67; Н, 6,03; N, 8,44. Типичный способ удаления защитной группы из силилзащищенных нуклеозидов: - и -(L)-2',3'дидезокси-2'-фтор-5-фторцитидин (28 а и 28b). Нуклеозид 25 (1,098 г, 2,26 ммоль, 1,0 экв.) растворяют в 15 мл метанола, в который добавляют фторид аммония (0,838 г, 22,6 ммоль, 10,0 экв.). Полученную смесь интенсивно перемешивают в течение 24 ч и выполняют ТСХ (15% этанола/85% этилацетата), результаты которой показывают, что реакция закончена. Реакционную смесь разбавляют тремя объемами этилацетата и фильтруют через небольшой слой (1 см) силикагеля. Слой промывают 200 мл раствора 15% этанола/85% этилацетата, и удаляют растворитель с получением белой пены. Полученное соединение очищают хроматографией на колонках из силикагеля, используя систему растворителей, состоящую из 15% этанола/85% этилацетата, которая также влияет на разделение - и -аномеров. Выход -аномера в виде белой пены равен 0,190 г(0,768 ммоль, выход 34%) и выход -аномера в виде белой пены равен 0,290 г (1,17 ммоль, выход 52%):-(D)-2',3'-Дидезокси-2'-фторцитидин (17b). Нуклеозид 25 (0,160 г, 0,59 ммоль) растворяют в 10 мл насыщенного раствора аммиака в метаноле. Полученный раствор перемешивают в течение 5 мин, и реакцию прекращают. Раствор аммиака в метаноле удаляют, полученное белое твердое вещество помещают в вакуум и осторожно нагревают на водяной бане с температурой 60 С в течение 2 ч, чтобы удалить сублимацией побочный продукт ацетамид. Белое твердое вещество кристаллизуют из 5% метанола/95% метиленхлорида, что дает количественный выход белого кристаллического твердого вещества. Rf (15% EtOH, 85% EtOAc)=0,18; т.пл.191-195 СRf (100% EtOAc)=0,54; т.пл. 153-156 С. 1 Н ЯМР (400 МГц, CD3OD) d 8,46 (д, J=6,8 Гц, 1 Н), 5,94 (д, J=16,4 Гц, 1 Н), 5,25 (дд, J=51,6 и 4,0 Гц,1 Н), 4,41 (м, 1 Н), 4,05 (дд, J=12,8 и 2,4 Гц, 1 Н), 3,72 (дд, J=12,4 и 2,4 Гц, 1 Н), 2,34-2,09 (м, 2 Н). 13 С ЯМР (100 МГц, CD3OD) d 159,7 (д, J=25,8 Гц), 150,7, 141,8 (д, J=230,6 Гц), 126,3 (д, J=35,7 Гц),98,3 (д, J=184,6 Гц), 91,9 (д, J=36,4 Гц), 83,6, 61,9, 31,9 (д, J=20,5 Гц). ИКС (KBr) 3482, 3037, 1702, 1654,1402, 1103 см-1. МСВР, высчитано для [M+Li] C9H10N2O4F2Li: 255,0769. Обнаружено: 255,0764. Элементный анализ, высчитано для C9H10N2O4F2: С, 43,56; Н, 4,06; N, 11,29. Обнаружено: С, 43,59; Н, 4,06; N, 11,17. Получение L-2'-фтор-2',3'-ненасыщенных нуклеозидов. В настоящее время разработан второй простой метод синтеза ненасыщенных 2'-фторнуклеозидов,который описан ниже. Этот метод синтеза включает взаимодействие защищенного пиримидинового или пуринового основания с основным промежуточным соединением 309 в присутствии кислоты Льюиса,как это показано на схеме 9. В табл. 5, 6 приведены типичные соединения, полученные в соответствии с этим методом синтеза. Растворители; А; EtOAc-гексаны, В; СН 2 Сl2-МеOН, С; СНСl3-МеOН, D; ТГФ-циклогексан, Е; лиофилизован Синтез 2',3'-ненасыщенных D-нуклеозидов ранее осуществляли при помощи реакции элиминирования, используя в качестве исходного вещества легко доступный аналог нуклеозида, который представляет собой модификацию отдельных нуклеозидов с длинной цепью. Некоторые исследователи сообщили о получении D-2'-фтор-2',3'-ненасыщенных пиримидиннуклеозидов путем элиминирования приемлемых аналогов 2'-фторированных нуклеозидов (Martin, J.A., et al., J. Med. Chem. 1990, 33, 2137-2145; Stezycki,- 30
МПК / Метки
МПК: C07H 19/173, A61P 31/18, A61K 31/7064, A61P 35/00, C07D 405/04, A61K 31/7076, A61P 31/20, A61K 31/506, A61K 31/52, C07D 473/00, C07H 19/073
Метки: 2'-фторнуклеозиды
Код ссылки
<a href="https://eas.patents.su/30-8609-2-ftornukleozidy.html" rel="bookmark" title="База патентов Евразийского Союза">2′-фторнуклеозиды</a>
Предыдущий патент: Способ создания моложавой, гладкой кожи
Следующий патент: Суспензионные препараты на основе кристаллического ангидрата тиотропийбромида
Случайный патент: Способ регенерирования катализаторов дегидрирования