Соединения 1h-индол-пиридинкарбоксамида и 1н-индол-пиперидинкарбоксамида, способ их получения и фармацевтические композиции, которые их содержат
Номер патента: 14022
Опубликовано: 30.08.2010
Авторы: Вайссман Дина, Брион Жан-Даниель, Разе Рудольф, Ренко Зафиарисоа Долор, Бинтен Фабрис, Пюжоль Жан-Франсуа, Арпе Катрин, Ле Ридан Ален, Разон Патрик, Левуарье Эрик



















Формула / Реферат
1. Соединения формулы (I)

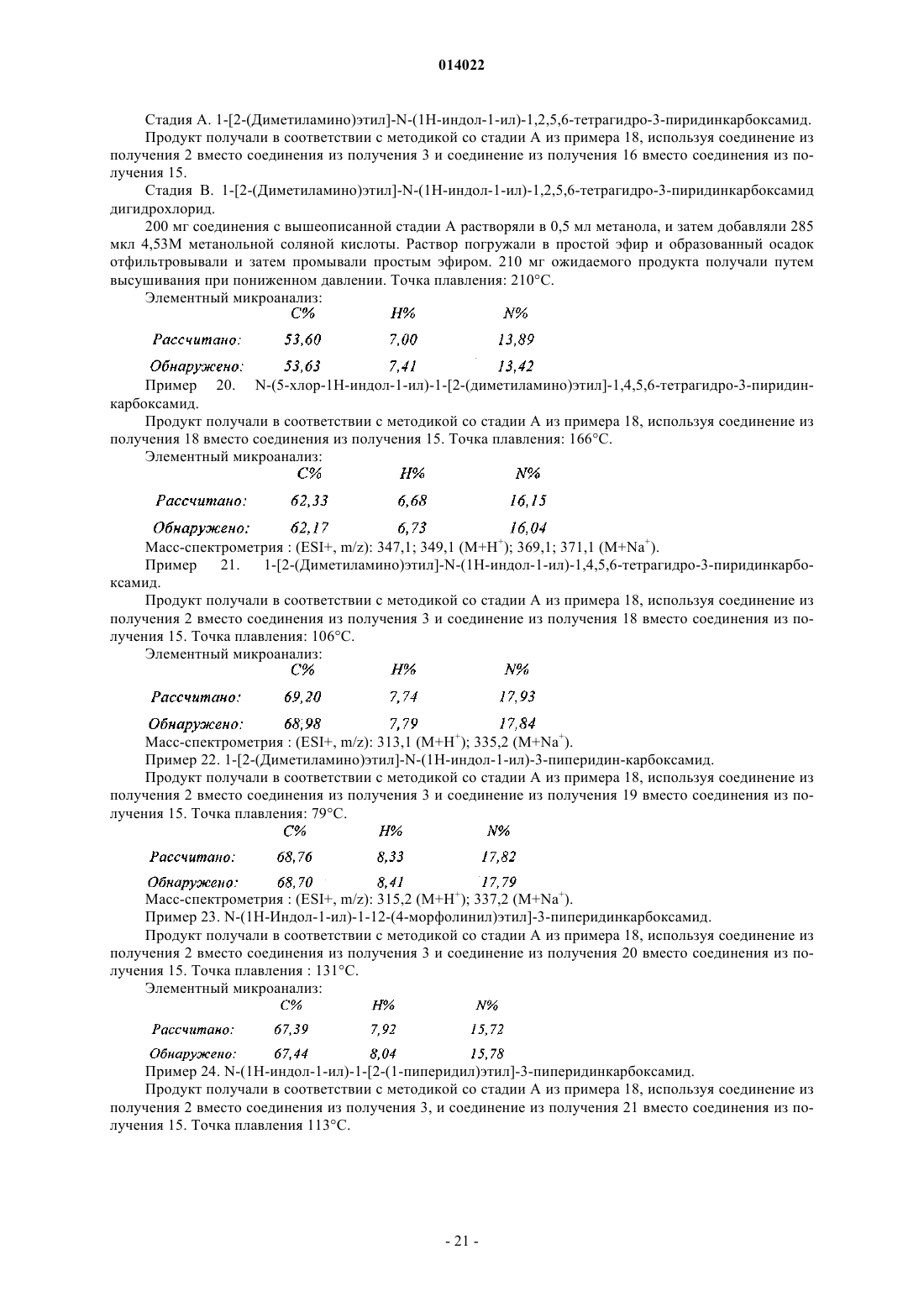
в которой А представляет собой двухвалентный радикал:
![]()
в которой
Z представляет собой атом кислорода или серы,
R6 представляет собой атом водорода,
линейную или разветвленную (С1-С6)алкильную группу, С(О)-AA, где АА представляет собой аминокислотный радикал, линейную или разветвленную (C1-С6)алкоксикарбонильную группу, CHR'-O-C(O)-R", где R' представляет собой атом водорода или линейную или разветвленную (C1-С6)алкильную группу и R" представляет собой линейную или разветвленную (C1-С6)алкильную группу, линейную или разветвленную (C1-С6)алкенильную группу, арильную группу, арил-(C1-С6)алкильную группу, в которой алкильная часть является линейной или разветвленной, линейную или разветвленную (C1-С6)полигалоалкильную группу или линейную или разветвленную (C1-С6)алкильную цепь, замещенную одним или несколькими атомами галогена, одной или несколькими гидроксигруппами, линейными или разветвленными (C1-С6)алкоксигруппами или аминогруппами, необязательно замещенными одной или двумя одинаковыми или различными, линейными или разветвленными (C1-С6)алкильными группами,
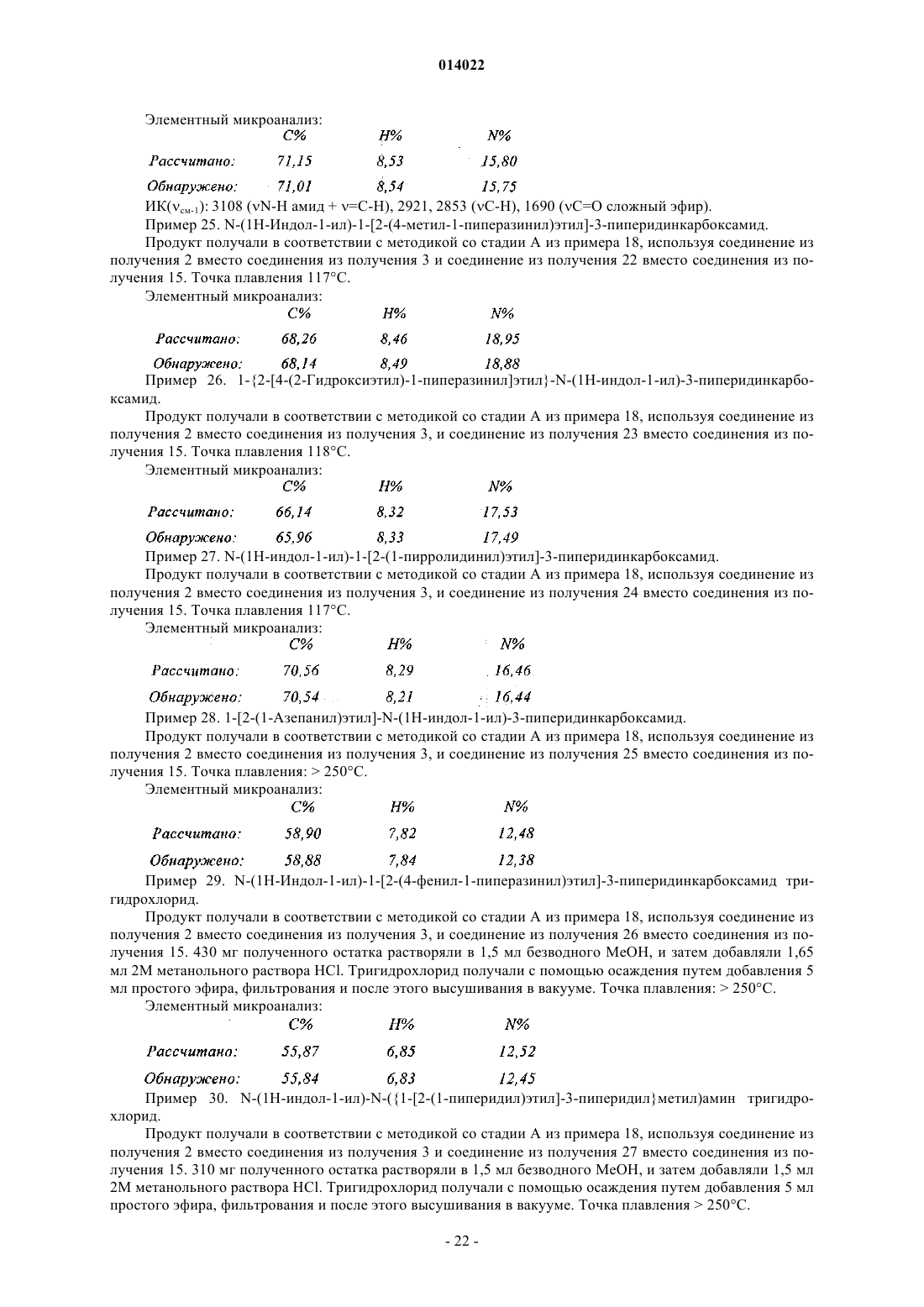
в кольце В ![]() представляет собой простую связь или двойную связь,
представляет собой простую связь или двойную связь,
в кольце С ![]() представляет собой простую связь или двойную связь, кольцо С содержит самое большее только одну двойную связь,
представляет собой простую связь или двойную связь, кольцо С содержит самое большее только одну двойную связь,
R1, R2, R3 и R4, которые могут быть одинаковыми или различными, каждый независимо друг от друга, представляет собой
атом водорода или галогена,
линейную или разветвленную (C1-С6)алкильную группу, линейную или разветвленную (C1-С6)алкоксигруппу, гидроксигруппу, цианогруппу, нитрогруппу, линейную или разветвленную (C1-С6)полигалоалкильную группу, аминогруппу, необязательно замещенную одним или двумя линейными или разветвленными (C1-С6)алкильными и/или линейными или разветвленными (С2-С6)алкенильными группами, алкильные и алкенильные группы могут быть одинаковыми или различными,
или линейную или разветвленную (C1-С6)алкильную цепь, замещенную одним или несколькими атомами галогена, одной или несколькими гидроксигруппами, линейными или разветвленными (C1-С6)алкоксигруппами или аминогруппами, необязательно замещенными одной или двумя одинаковыми или различными, линейными или разветвленными (C1-С6)алкильными группами,
R5 представляет собой атом водорода,
линейную или разветвленную (C1-С6)алкильную группу, аминоалкильную группу, в которой алкильная часть имеет линейную или разветвленную цепь, содержащую от 1 до 6 атомов углерода, или линейную или разветвленную (C1-С6)гидроксиалкильную группу,
X и Y, которые могут быть одинаковыми или различными, каждый независимо друг от друга, представляет собой
атом водорода или линейную или разветвленную (C1-С6)алкильную группу,
Ra, Rb, Rc и Rd, которые могут быть одинаковыми или различными, каждый независимо друг от друга, представляет собой атом водорода или галогена,
линейную или разветвленную (C1-С6)алкильную группу, гидроксигруппу, линейную или разветвленную (C1-С6)алкоксигруппу, цианогруппу, нитрогруппу, линейную или разветвленную (C1-С6)полигалоалкильную группу, аминогруппу (необязательно замещенную одной или двумя, одинаковыми или различными, линейными или разветвленными (C1-С6)алкильными группами), или линейную или разветвленную (C1-С6)алкильную цепь, замещенную одной или несколькими группами, выбранными из галогена, гидрокси, линейного или разветвленного (C1-С6)алкокси, и амино, необязательно замещенного одной или двумя, одинаковыми или различными, линейными или разветвленными (C1-С6)алкильными группами,
и подразумевается, что когда А связан с кольцом С у атома углерода, несущего один из заместителей Ra, Rb, Rc, Rd или Y, и указанный связывающий атом углерода также несет двойную связь, то соответствующий заместитель Ra, Rb, Rc, Rd или Y отсутствует,
Re представляет собой атом водорода,
линейную или разветвленную (C1-С6)алкильную группу; арил-(C1-С6)алкильную группу, в которой алкильная часть является линейной или разветвленной; линейную или разветвленную (С2-С6)алкенильную группу; линейную или разветвленную (С2-С6)алкинильную группу; линейную или разветвленную (C1-С6)алкильную цепь, замещенную одной или несколькими группами, выбранными из гидрокси, амино (необязательно замещенного одной или двумя, одинаковыми или различными, линейными или разветвленными (C1-С6)алкильными группами), линейный или разветвленный (C1-С6)алкокси, и NR7R8, где R7и R8, вместе с атомом азота, к которому они присоединены, образуют необязательно замещенный 4-8-членный гетероцикл, необязательно содержащий одну или несколько двойных связей в пределах гетероцикла и необязательно содержащий в пределах циклической системы второй гетероатом, выбранный из атома кислорода и атома азота; или линейную или разветвленную (C2-С6)алкенильную цепь, замещенную такими же группами, как и алкильная цепь или линейную или разветвленную (С2-C6)алкинильную цепь, замещенную такими же группами, как и алкильная цепь,
их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием,
и подразумевается, что
необязательно замещенный 4-8-членный гетероцикл, необязательно содержащий одну или несколько двойных связей в пределах гетероцикла и необязательно содержащий в пределах циклической системы второй гетероатом, выбранный из атома кислорода и атома азота, может быть замещен одной или несколькими одинаковыми или различными группами, выбранными из линейного или разветвленного (C1-С6)алкила, линейного или разветвленного (C1-С6)гидроксиалкила, линейного или разветвленного (C1-С6)алкокси-(C1-С6)алкила, CO2Rv, СО2-Rw-NRvR'v, CO2-Rw-ORv (где Rv представляет собой атом водорода или линейную или разветвленную (C1-С6)алкильную группу, R'v имеет значения, указанные для Rv и Rw представляет собой линейную или разветвленную (C1-С6)алкиленовую цепь), арила, арилоксикарбонила, линейного или разветвленного арил-(C1-С6)алкоксикарбонила, необязательно замещенного циклоалкила, необязательно замещенного циклоалкилалкила, необязательно замещенного гетероциклоалкила, необязательно замещенного гетероциклоалкилалкила, и аминоалкила, в котором алкильная часть имеет линейную или разветвленную цепь, содержащую от 1 до 6 атомов углерода, и аминочасть необязательно замещена одной или двумя одинаковыми или различными, линейными или разветвленными (C1-С6)алкильными группами,
арил обозначает фенильную или нафтильную группу, каждая необязательно замещена одним или несколькими атомами галогена, нитро, амино, линейными или разветвленными (C1-С6)алкильными или линейными или разветвленными (C1-С6)алкоксигруппами,
циклоалкил обозначает насыщенную 4-8-членную моноциклическую группу,
циклоалкилалкил обозначает циклоалкилалкильную группу, где алкильная группа имеет линейную или разветвленную цепь, содержащую от 1 до 6 атомов углерода, и циклоалкильная группа обозначает насыщенную 4-8-членную моноциклическую группу,
гетероциклоалкил обозначает насыщенную 4-8-членную моноциклическую группу, содержащую 1 или 2 гетероатома, выбранных из азота, кислорода и серы,
гетероциклоалкилалкил обозначает гетероциклоалкилалкильную группу, где алкильная группа имеет линейную или разветвленную цепь, содержащую от 1 до 6 атомов углерода, и гетероциклоалкильная группа обозначает насыщенную 4-8-членную моноциклическую группу, содержащую 1 или 2 гетероатома, выбранных из азота, кислорода и серы,
выражение "необязательно замещенный", когда оно относится к группам циклоалкил, циклоалкилалкил, гетероциклоалкил и гетероциклоалкилалкил, обозначает, что эти группы могут быть замещены одним или несколькими одинаковыми или различными заместителями, выбранными из линейного или разветвленного (C1-С6)алкила, линейного или разветвленного (C1-С6)гидроксиалкила, линейного или разветвленного (C1-С6)алкокси-(C1-С6)алкила, карбокси, линейного или разветвленного (C1-С6)алкоксикарбонила и аминоалкила, в котором алкильная часть имеет линейную или разветвленную цепь, содержащую от 1 до 6 атомов углерода, и аминочасть необязательно замещена одной или двумя одинаковыми или различными, линейными или разветвленными (C1-С6)алкильными группами,
под аминокислотным радикалом подразумеваются радикалы аланил, аргинил, аспарагинил, a-аспартил, цистеинил, a-глутамил, глутаминил, глицил, гистидил, изолейцил, лейцил, лизил, метионил, фенилаланил, пролил, серил, треонил, триптофил, тирозил и валил.
2. Соединения формулы (I) по п.1, отличающиеся тем, что А представляет собой двухвалентный радикал

где R6имеет значения, указанные для формулы (I), и Z представляет собой атом кислорода, их энантиомеры и диастереоизомеры, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
3. Соединения формулы (I) по п.1 или 2, отличающиеся тем, что R6представляет собой атом водорода, их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
4. Соединения формулы (I) по пп.1-3, отличающиеся тем, что R1, R2, R3 и R4 представляют собой атом водорода, атом галогена или линейную или разветвленную (C1-С6)алкоксигруппу, их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
5. Соединения формулы (I) по пп.1-4, отличающиеся тем, что R5 представляет собой атом водорода или линейную или разветвленную (C1-С6)алкильную группу, их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
6. Соединения формулы (I) по пп.1-5, отличающиеся тем, что X и Y представляют собой атом водорода или линейную или разветвленную (C1-С6)алкильную группу, их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
7. Соединения формулы (I) по пп.1-6, отличающиеся тем, что Ra, Rb, Rc и Rd представляют собой атом водорода, их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
8. Соединения формулы (I) по пп.1-7, отличающиеся тем, что Re представляет собой атом водорода или линейную или разветвленную (C1-С6)алкил или линейный или разветвленный (C2-С6)алкенильную группу, их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
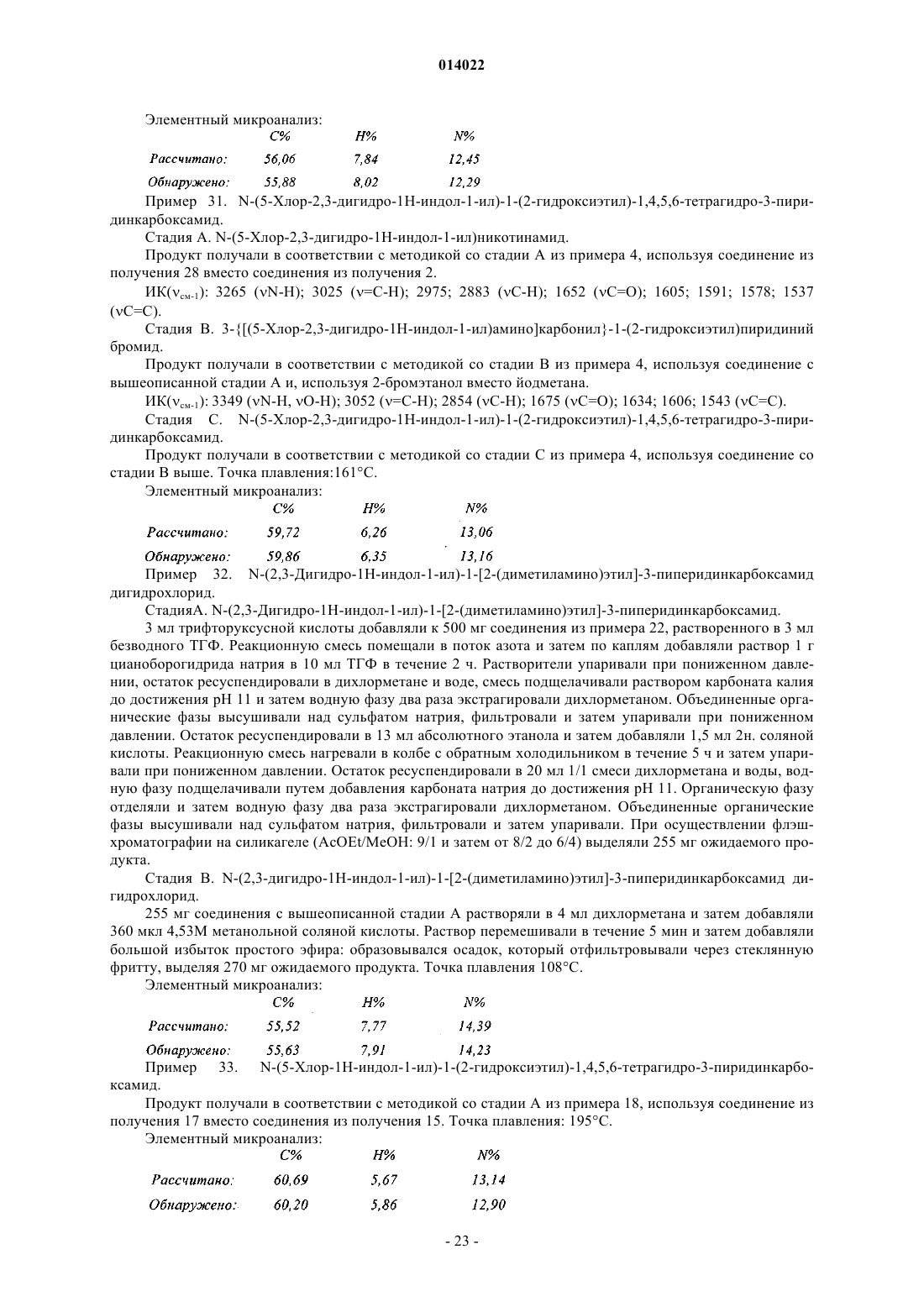
9. Соединения формулы (I) по пп.1-8, отличающиеся тем, что они представляют собой соединения формулы (I/A)

в которой ![]() X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
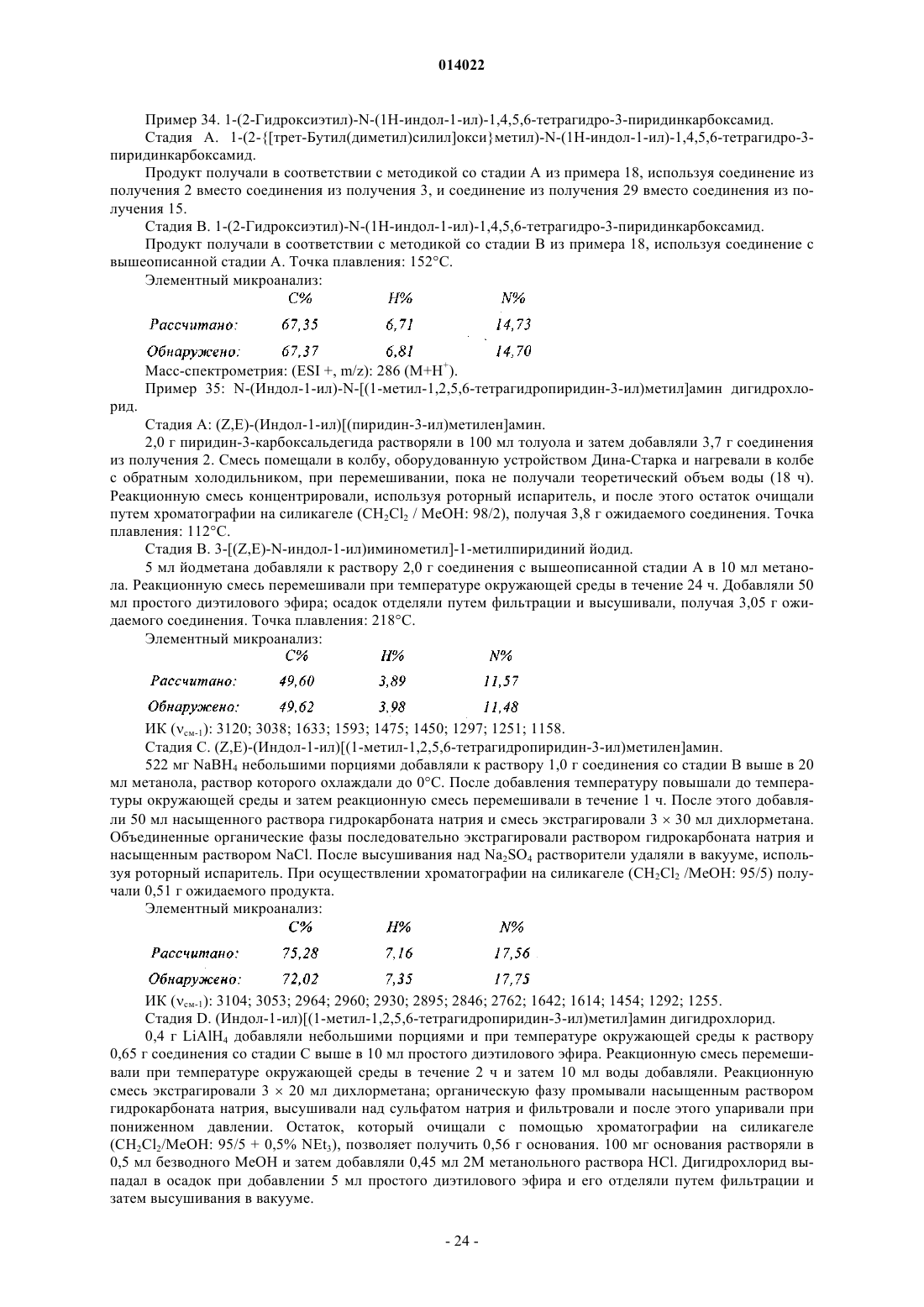
10. Соединения формулы (I) по пп.1-9, отличающиеся тем, что они представляют собой соединения формулы (I/B)

в которой ![]() X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
11. Соединения формулы (I) по пп.1-10, отличающиеся тем, что они представляют собой соединения формулы (I/C)

в которой ![]() X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
12. Соединения формулы (I) по пп.1-11, отличающиеся тем, что они представляют собой соединения формулы (I/D)

в которой ![]() X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re, имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re, имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
13. Соединения формулы (I) по пп.1-12, отличающиеся тем, что они представляют собой соединения формулы (I/E)

в которой ![]() X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
14. Соединения формулы (I) по пп.1-13, отличающиеся тем, что они представляют собой соединения формулы (I/F)

в которой ![]() X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
15. Соединения формулы (I) по пп.1-14, отличающиеся тем, что они представляют собой соединения формулы (I/G)

в которой ![]() X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
16. Соединения формулы (I) по пп.1-15, отличающиеся тем, что они представляют собой соединения формулы (I/H)

в которой ![]() X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
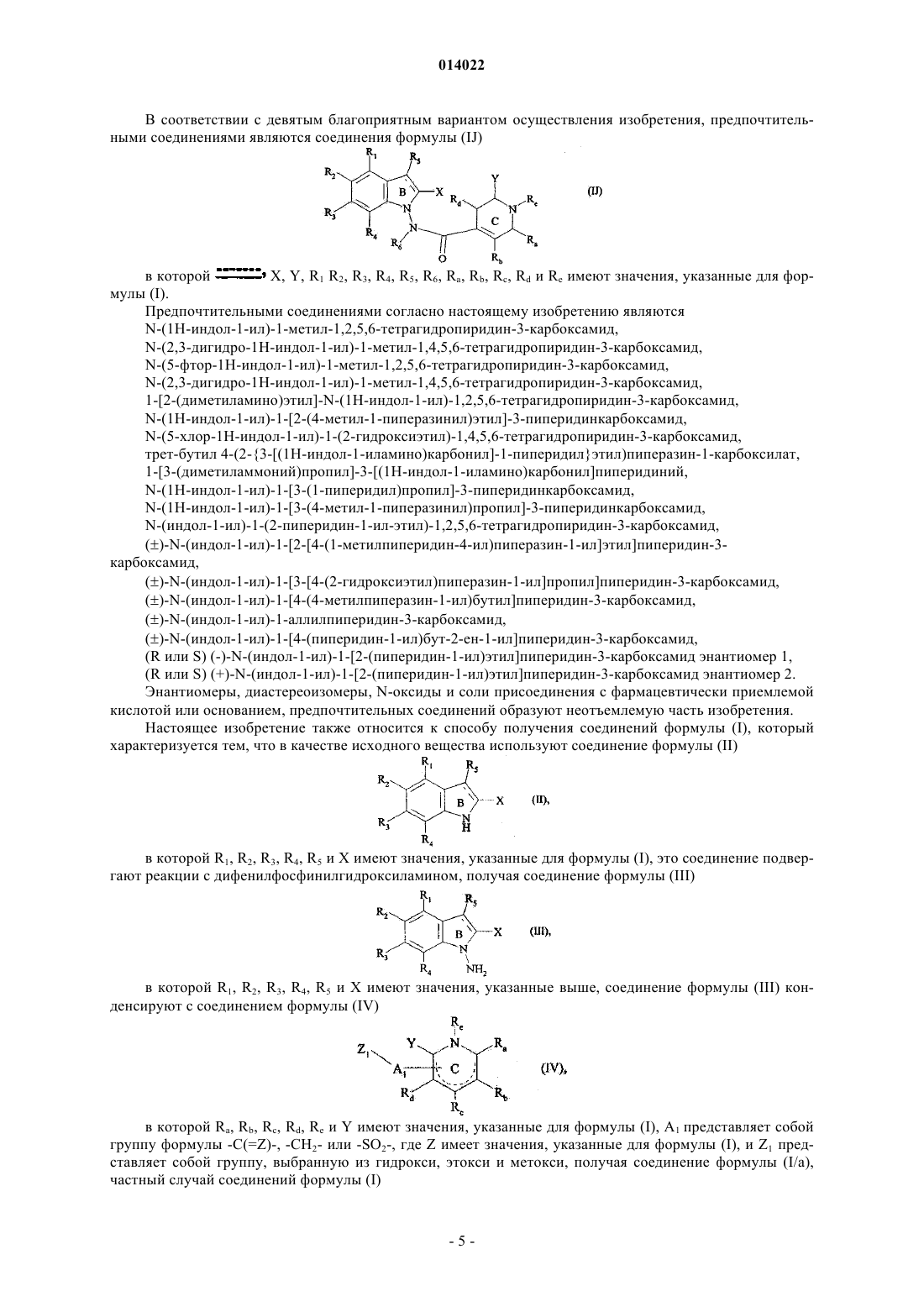
17. Соединения формулы (I) по пп.1-16, отличающиеся тем, что они представляют собой соединения формулы (I/J)

в которой ![]() X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I), их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
18. Соединения формулы (I) по п.1, которые представляют собой
N-(1H-индол-1-ил)-1-метил-1,2,5,6-тетрагидропиридин-3-карбоксамид,
N-(2,3-дигидро-1H-индол-1-ил)-1-метил-1,4,5,6-тетрагидропиридин-3-карбоксамид,
N-(5-фтор-1H-индол-1-ил)-1-метил-1,2,5,6-тетрагидропиридин-3-карбоксамид,
N-(2,3-дигидро-1H-индол-1-ил)-1-метил-1,4,5,6-тетрагидропиридин-3-карбоксамид,
1-[2-(диметиламино)этил]-N-(1H-индол-1-ил)-1,2,5,6-тетрагидропиридин-3-карбоксамид,
N-(1H-индол-1-ил)-1-[2-(4-метил-1-пиперазинил)этил]-3-пиперидинкарбоксамид,
N-(5-хлор-1H-индол-1-ил)-1-(2-гидроксиэтил)-1,4,5,6-тетрагидропиридин-3-карбоксамид,
трет-бутил 4-(2-{3-[(1H-индол-1-иламино)карбонил]-1-пиперидил}этил)пиперазин-1-карбоксилат,
1-[3-(диметиламмоний)пропил]-3-[(1H-индол-1-иламино)карбонил]пиперидиний,
N-(1H-индол-1-ил)-1-[3-(1-пиперидил)пропил]-3-пиперидинкарбоксамид,
N-(1H-индол-1-ил)-1-[3-(4-метил-1-пиперазинил)пропил]-3-пиперидинкарбоксамид,
N-(индол-1-ил)-1-(2-пиперидин-1-ил-этил)-1,2,5,6-тетрагидропиридин-3-карбоксамид,
(±)-N-(индол-1-ил)-1-[2-[4-(1-метилпиперидин-4-ил)пиперазин-1-ил]этил]пиперидин-3-карбоксамид,
(±)-N-(индол-1-ил)-1-[3-[4-(2-гидроксиэтил)пиперазин-1-ил]пропил]пиперидин-3 -карбоксамид,
(±)-N-(индол-1-ил)-1-[4-(4-метилпиперазин-1-ил)бутил]пиперидин-3-карбоксамид,
(±)-N-(индол-1-ил)-1-аллилпиперидин-3-карбоксамид,
(±)-N-(индол-1-ил)-1-[4-(пиперидин-1-ил)бут-2-ен-1-ил]пиперидин-3-карбоксамид,
(R или S) (-)-N-(индол-1-ил)-1-[2-(пиперидин-1-ил)этил]пиперидин-3-карбоксамид энантиомер 1,
(R или S) (+)-N-(индол-1-ил)-1-[2-(пиперидин-1-ил)этил]пиперидин-3-карбоксамид энантиомер 2,
их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием.
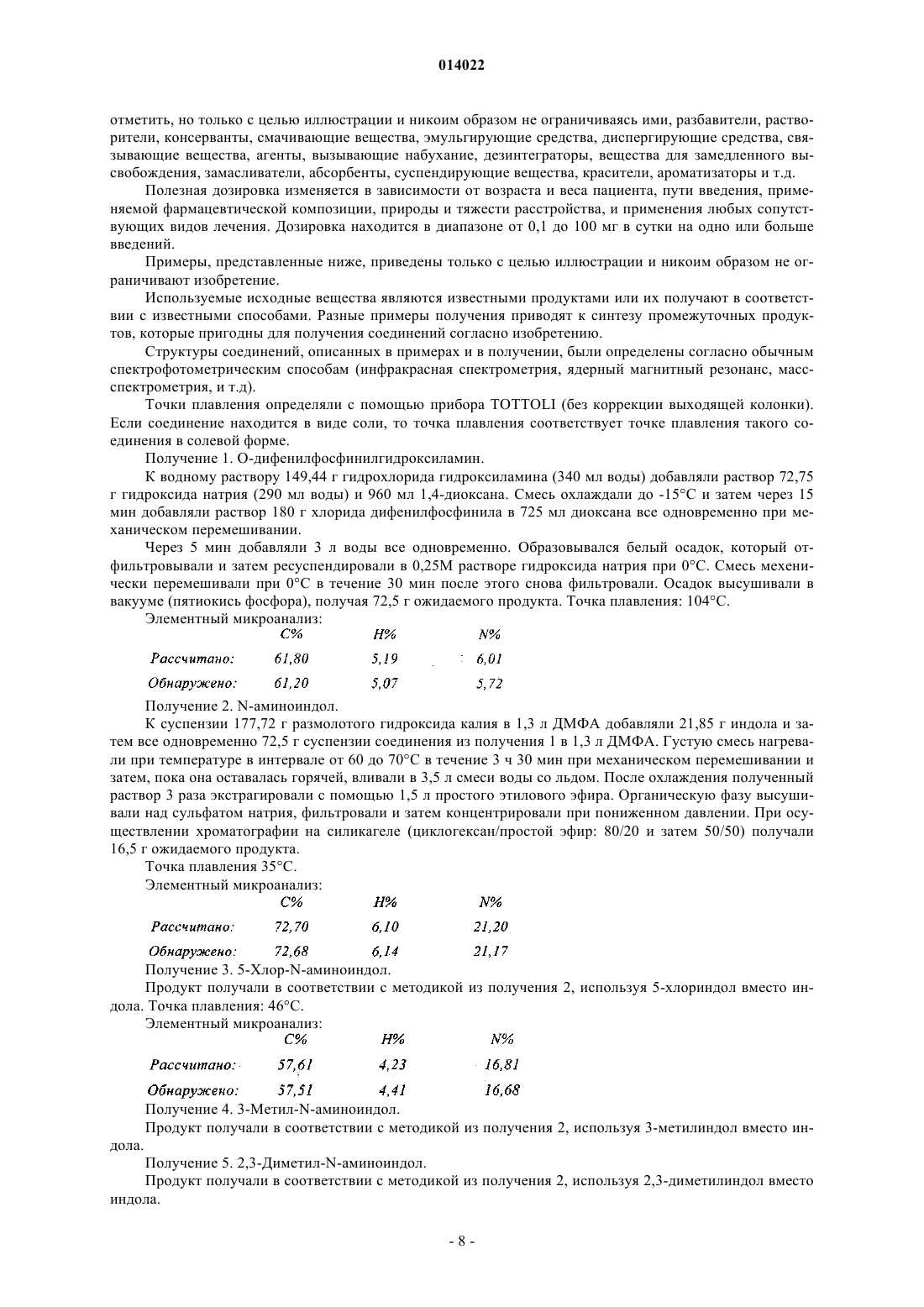
19. Способ получения соединений формулы (I) по п.1, который характеризуется тем, что в качестве исходного вещества используют соединение формулы (II)

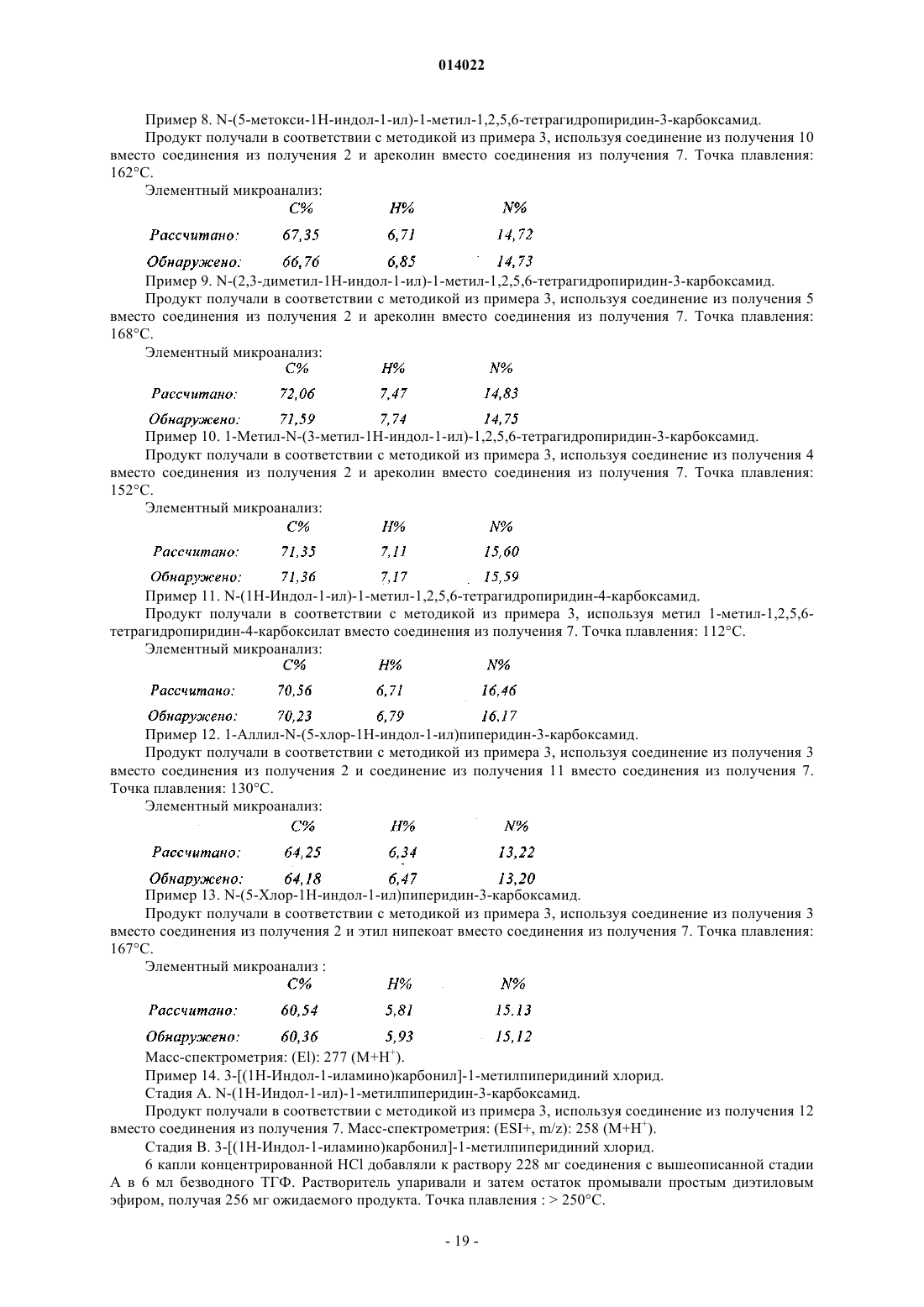
в которой R1, R2, R3, R4, R5 и X имеют значения, указанные для формулы (I), где это соединение в присутствии дифенилфосфинилгидроксиламина превращают в соединение формулы (III)

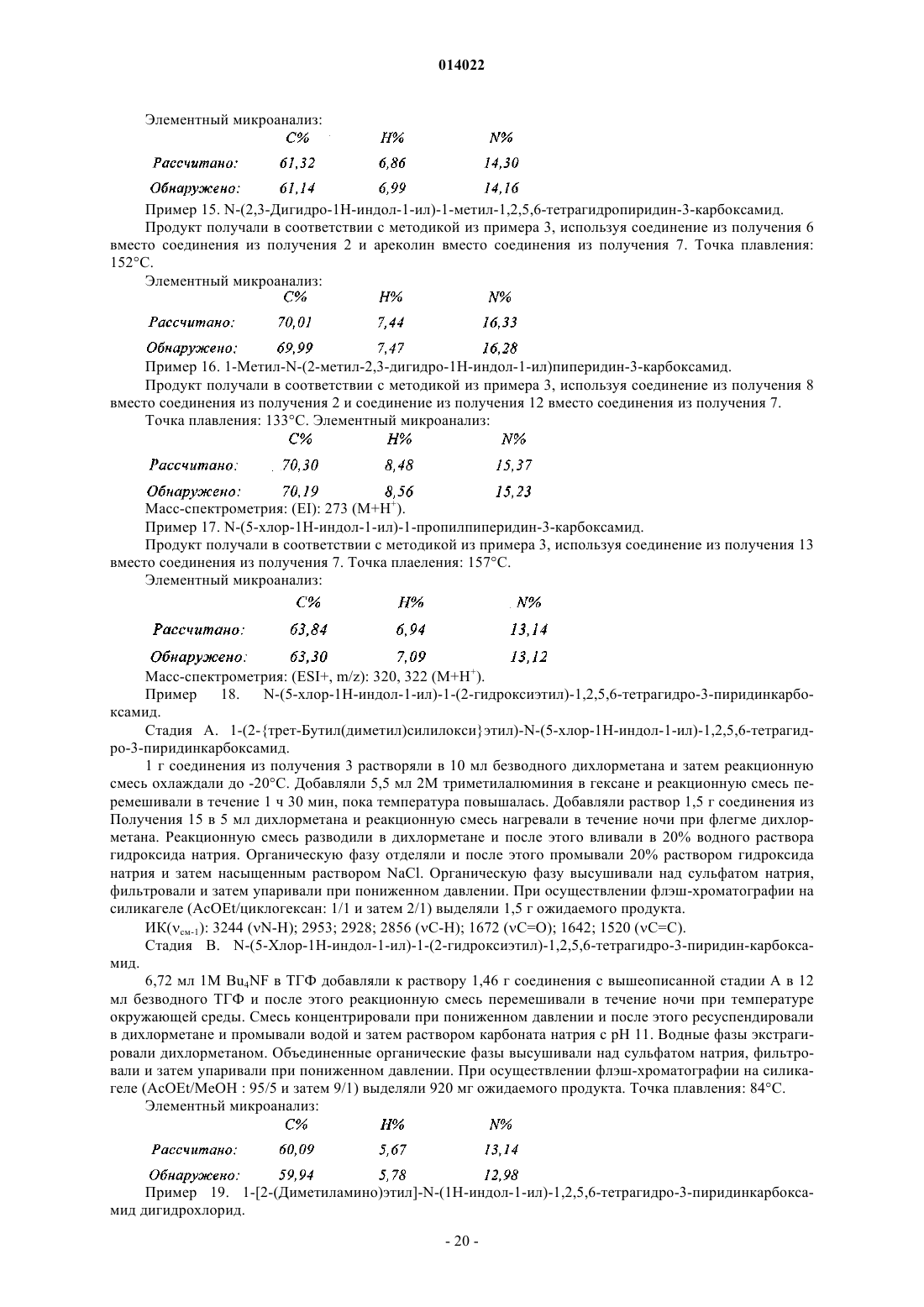
где R1, R2, R3, R4, R5и X имеют значения, указанные выше, где соединение формулы (III) конденсируют с соединением формулы (IV)

в которой Ra, Rb, Rc, Rd, Re и Y имеют значения, указанные для формулы (I), A1 представляет собой группу формулы -C(=Z)-, -СН2- или -SO2-, где Z имеет значения, указанные для формулы (I), и Z1 представляет собой группу, выбранную из гидрокси, этокси и метокси, получая соединение формулы (I/a), частный случай соединений формулы (I)

в которой R1, R2, R3, R4, R5, Ra, Rb, Rc, Rd и Re, X и Y имеют значения, указанные выше, и А имеет значения, указанные для формулы (I),
соединения формулы (I/a), образующие совокупность соединений по изобретению, которые очищают при необходимости в соответствии с обычным способом очистки, могут быть разделены, если это является желательным, на их разные изомеры в соответствии с обычным способом разделения и превращены, если это является желательным, в их N-оксиды и, если это является подходящим, в их соли присоединения с фармацевтически приемлемой кислотой или основанием.
20. Фармацевтическая композиция, содержащая в качестве активного компонента по меньшей мере одно соединение по любому из пп.1-18, в комбинации с одним или несколькими инертными, нетоксичными, фармацевтически приемлемыми наполнителями или носителями.
21. Фармацевтическая композиция по п.20, содержащая по меньшей мере один активный компонент - индуктор тирозингидроксилаз по любому из пп.1-18, предназначенная для лечения депрессии, страха, нарушений памяти при старении и/или нейродегенеративных заболеваний, для паллиативного лечения болезни Паркинсона и для адаптации к стрессу.
Текст
СОЕДИНЕНИЯ 1H-ИНДОЛ-ПИРИДИНКАРБОКСАМИДА И 1 Н-ИНДОЛПИПЕРИДИНКАРБОКСАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, КОТОРЫЕ ИХ СОДЕРЖАТ ЛЕ ЛАБОРАТУАР СЕРВЬЕ; САНТР НАСЬОНАЛЬ ДЕ ЛЯ РЕШЕРШ СЬЕНТИФИК; ЮНИВЕРСИТЕ ПАРИ СЮД (FR) в которой А представляет собой двухвалентный радикал в которой Z представляет собой атом кислорода или атом серы; R6 представляет собой атом водорода, алкильную, алкенильную, арилалкильную или полигалоалкильную группу или замещенную, линейную или разветвленную алкильную цепь; представляет собой простую связь или двойную связь; R1, R2, R3 иR4 представляют собой атом водорода или галогена, алкильную, алкокси, гидрокси, циано, нитро, полигалоалкильную или необязательно замещенную аминогруппу, или линейную или разветвленную алкильную цепь, замещенную одной или несколькими группами; R5 представляет собой атом водорода или алкильную,аминоалкильную или гидроксиалкильную группу; X и Y представляют собой атом водорода или алкильную группу; Ra, Rb, Rc и Rd представляют собой атом водорода или галогена, алкил, гидрокси, алкркси, циано,нитро, полигалоалкил, необязательно замещенную аминогруппу или замещенную, линейную или разветвленную алкильную цепь; Re представляет собой атом водорода или алкильную, арилалкильную или алкенильную группу или замещенную, линейную или разветвленную алкильную цепь; их энантиомеры, диастереоизомеры и N-оксиды, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием. Лекарственные средства. 014022 Настоящее изобретение относится к новым соединениям 1H-индолпиридинкарбоксамида и 1Hиндолпиперидинкарбоксамида, к способу их получения и к фармацевтическим композициям, которые их содержат. Из литературы известны различные примеры соединений, обладающих эбурнановой структурой,это в особенности раскрыто в патентной заявке US 3454583, которая относится к винкамину (метил(3,14,16)-(14,15-дигидро-14-гидроксиэбурнаменин-14-карбоксилат) и его соединениям в связи с их сосудорасширяющими свойствами. В патентных заявках FR 2433528 и FR 2381048 раскрыты новые соединения 20,21-диноребурнаменина и в патентной заявке ЕР 0287468 раскрыты новые соединения 17 аза-20,21-диноребурнаменина. В патентной заявке ЕР 0658557 описаны соединения эбурнана, модифицированные в 14-ом и 15-ом положениях эбурнанового скелета. В патентной заявке ЕР 0563916 описаны соединения 1H-индолциклогексанкарбоксамида. Соединения согласно настоящему изобретению являются новыми, кроме того, они обладают очень ценными фармакологическими свойствами. В особенности, было обнаружено, что они являются эффективными селективными или неселективными индукторами тирозингидроксилазы. Более конкретно, настоящее изобретение относится к соединениям формулы (I) в которой А представляет собой двухвалентный радикал : где Z представляет собой атом кислорода или серы,R6 представляет собой атом водорода,линейную или разветвленную (С 1-С 6)алкильную группу, С(О)-АА, где АА представляет собой аминокислотный радикал, линейную или разветвленную (C1-С 6)алкоксикарбонильную группу, CHR'-O-C(O)R", где R' представляет собой атом водорода или линейную или разветвленную (С 1-С 6)алкильную группу и R" представляет собой линейную или разветвленную (С 1-С 6)алкильную группу, линейную или разветвленную (С 2-С 6)алкенильную группу, арильную группу, арил-(С 1-С 6)алкильную группу, в которой алкильная часть является линейной или разветвленной, линейную или разветвленную (C1-С 6)полигалоалкильную группу,или линейную или разветвленную (C1-С 6)алкильную цепь, замещенную одним или несколькими атомами галогена, одной или несколькими гидрокси группами, линейными или разветвленными (C1-С 6) алкокси группами, или аминогруппами, необязательно замещенными одной или двумя одинаковыми или различными, линейными или разветвленными (C1-С 6)алкильными группами,представляет собой простую связь или двойную связь,в кольце В представляет собой простую связь или двойную связь, кольцо С содержит самое в кольце С большее только одну двойную связь,R1, R2, R3 и R4, которые могут быть одинаковыми или различными, каждый независимо друг от друга, представляет собой атом водорода или галогена,линейную или разветвленную (C1-С 6)алкильную группу, линейную или разветвленную (C1-С 6)алкоксигруппу, гидроксигруппу, цианогруппу, нитрогруппу, линейную или разветвленную (C1-С 6)полигалоалкильную группу, аминогруппу (необязательно замещенную одним или двумя линейными или разветвленными (C1-С 6)алкильными и/или линейными или разветвленными (C2-С 6)алкенильными группами, алкильные и алкенильньные группы могут быть одинаковыми или различными), или линейную или разветвленную (C1-С 6)алкильную цепь, замещенную одним или несколькими атомами галогена, одной или несколькими гидроксигруппами, линейными или разветвленными (C1-С 6)алкоксигруппами или аминогруппами, необязательно замещенными одной или двумя одинаковыми или различными, линейными или разветвленными(C1-С 6) алкильными группами,R5 представляет собой атом водорода,линейную или разветвленную (C1-С 6)алкильную группу, аминоалкильную группу, в которой алкильная часть имеет линейную или разветвленную цепь, содержащую от 1 до 6 атомов углерода, или линейную или разветвленную (C1-С 6)гидроксиалкильную группу,X и Y, которые могут быть одинаковыми или различными, каждый независимо друг от друга, представляет собой атом водорода или линейную или разветвленную (C1-С 6)алкильную группу,-1 014022Ra, Rb, Rc и Rd, которые могут быть одинаковыми или различными, каждый независимо друг от друга, представляет собой атом водорода или галогена,линейную или разветвленную (C1-С 6)алкильную группу, гидроксигруппу, линейную или разветвленную (C1-С 6)алкоксигруппу, цианогруппу, нитрогруппу, линейную или разветвленную (C1-С 6)полигалоалкильную группу, аминогруппу (необязательно замещенную одной или двумя, одинаковыми или различными, линейными или разветвленными (C1-С 6)алкильными группами), или линейную или разветвленную (C1-С 6)алкильную цепь, замещенную одной или несколькими группами, выбранными из галогена, гидрокси, линейного или разветвленного (C1-С 6)алкокси, и амино, необязательно замещенного одной или двумя, одинаковыми или различными, линейными или разветвленными (C1-С 6)алкильными группами,и подразумевается, что когда А связан с кольцом С у атома углерода, несущего один из заместителей Ra, Rb, Rc, Rd или Y и указанный связывающий атом углерода также несет двойную связь, то соответствующий заместитель Ra, Rb, Rc, Rd или Y отсутствует,Re представляет собой атом водорода,линейную или разветвленную (C1-С 6)алкильную группу; арил-(C1-С 6)алкильную группу, в которой алкильная часть является линейной или разветвленной; линейную или разветвленную (С 2-С 6)алкенильную группу; линейную или разветвленную (С 2-С 6)алкинильную группу; линейную или разветвленную(C1-С 6)алкильную цепь, замещенную одной или несколькими группами, выбранными из гидрокси, амино(необязательно замещенного одной или двумя, одинаковыми или различными, линейными или разветвленными (C1-С 6)алкильными группами), линейного или разветвленного (C1-С 6)алкокси, и NR7R8, где R7 иR8,вместе с атомом азота, к которому они присоединены, образуют необязательно замещенный 4-8 членный гетероцикл, необязательно содержащий одну или несколько двойных связей в пределах гетероцикла и необязательно содержащий в пределах циклической системы второй гетероатом, выбранный из атома кислорода и атома азота; или линейную или разветвленную (С 2-С 6)алкенильную цепь, замещенную такими же группами, как и алкильная цепь, или линейную или разветвленную (С 2-С 6)алкинильную цепь,замещенную такими же группами, как и алкильная цепь,к их энантиомерам, диастереоизомерам и N-оксидам, а также их солям присоединения с фармацевтически приемлемой кислотой или основанием,и подразумевается, что в качестве необязательно замещенного 4-8-членного гетероцикла, необязательно содержащего одну или несколько двойных связей в пределах гетероцикла и необязательно содержащего в пределах циклической системы второй гетероатом, выбранный из атома кислорода и атома азота, могут быть упомянуты, но никоим образом не ограничиваясь, пирролидин, пиперидин, азепан, пиперазин и морфолин, причем эти гетероциклы необязательно замещены (включая положение на втором атоме азота пиперазина) одной или несколькими одинаковыми или различными группами, выбранными из линейного или разветвленного (C1-С 6)алкила, линейного или разветвленного (C1-С 6)гидроксиалкила, линейного или разветвленного (C1-С 6)алкокси-(С 1-С 6)алкила, CO2Rv, CO2-Rw-NRvR'v, CO2-Rw-ORv (в которой Rv представляет собой атом водорода или линейную или разветвленную (C1-С 6)алкильную группу, R'v имеет значения, указанные для Rv, и Rw представляет собой линейную или разветвленную (C1-С 6)алкиленовую цепь),арила, арилоксикарбонила, линейного или разветвленного арил-(C1-С 6)алкоксикарбонила, необязательно замещенного циклоалкила, необязательно замещенного циклоалкилалкила, необязательно замещенного гетероциклоалкила, необязательно замещенного гетероциклоалкилалкила, и аминоалкила, в котором алкильная часть имеет линейную или разветвленную цепь, содержащую от 1 до 6 атомов углерода, и амино часть необязательно замещена одной или двумя одинаковыми или различными, линейными или разветвленными (C1-С 6)алкильными группами,арил обозначает фенильную или нафтильную группу, каждая необязательно замещена одним или несколькими атомами галогена, нитро, амино, линейными или разветвленными (C1-С 6)алкильными или линейными или разветвленными (C1-С 6)алкоксигруппами,циклоалкил обозначает насыщенную 4-8-членную моноциклическую группу,циклоалкилалкил обозначает циклоалкилалкильную группу, где алкильная группа имеет линейную или разветвленную цепь, содержащую от 1 до 6 атомов углерода, и циклоалкильная группа обозначает насыщенную 4-8-ми членную моноциклическую группу,гетероциклоалкил обозначает насыщенную 4-8-членную моноциклическую группу, содержащую 1 или 2 гетероатома, выбранных из азота, кислорода и серы,гетероциклоалкилалкил обозначает гетероциклоалкилалкильную группу, где алкильная группа имеет линейную или разветвленную цепь, содержащую от 1 до 6 атомов углерода, и гетероциклоалкильная группа обозначает насыщенную 4-8-членную моноциклическую группу, содержащую 1 или 2 гетероатома, выбранных из азота, кислорода и серы, выражение "необязательно замещенный", когда оно относится к группам циклоалкил, циклоалкилалкил, гетероциклоалкил и гетероциклоалкилалкил, обозначает,что эти группы могут быть замещены одним или несколькими одинаковыми или различными заместителями, выбранными из линейного или разветвленного (C1-С 6)алкила, линейного или разветвленного (C1-2 014022 С 6)гидроксиалкила, линейного или разветвленного (C1-С 6)алкокси-(С 1-С 6)алкила, карбокси, линейного или разветвленного (C1-С 6)алкоксикарбонила и аминоалкила, в котором алкильная часть имеет линейную или разветвленную цепь, содержащую от 1 до 6 атомов углерода, и амино часть необязательно замещена одной или двумя одинаковыми или различными, линейными или разветвленными (C1-С 6)алкильными группами,под аминокислотным радикалом подразумеваются радикалы аланил, аргинил, аспарагинил, аспартил, цистеинил, -глутамил, глутаминил, глицил, гистидил, изолейцил, лейцил, лизил, метионил,фенилаланил, пролил, серил, треонил, триптофил, тирозил и валил. Среди фармацевтически приемлемых кислот можно отметить, но не ограничиваясь только ими, соляную кислоту, бромисто-водородную кислоту, серную кислоту, фосфоновую кислоту, уксусную кислоту, трифторуксусную кислоту, молочную кислоту, пировиноградную кислоту, малоновую кислоту, янтарную кислоту, глутаровую кислоту, фумаровую кислоту, винную кислоту, малеиновую кислоту, лимонную кислоту, аскорбиновую кислоту, щавелевую кислоту, метансульфоновую кислоту, камфорную кислоту, лизин и т.д. Среди фармацевтически приемлемых оснований можно отметить, но не ограничиваясь только ими,гидроксид натрия, гидроксид калия, триэтиламин, трет-бутиламин и т.д. Предпочтительными соединения по изобретению являются те соединения, в которых А представляет собой двухвалентный радикал: в котором R6 имеет значения, указанные для формулы (I), и Z представляет собой атом кислорода. В соответствии с изобретением, предпочтительным является атом водорода в качестве заместителя В соответствии с изобретением, предпочтительным является атом водорода, атом галогена или линейная или разветвленная (C1-С 6)алкоксигруппа в качестве заместителей R1, R2, R3 и R4. В соответствии с изобретением, предпочтительным является атом водорода или линейная или разветвленная (С 1-С 6)алкильная группа в качестве заместителя R5. В соответствии с изобретением, предпочтительным является атом водорода или линейная или разветвленная (C1-С 6)алкильная группа в качестве заместителей X и Y. В соответствии с изобретением, предпочтительным является атом водорода в качестве заместителейRa, Rb, Rc и Rd. В соответствии с изобретением, предпочтительным является атом водорода или линейная или разветвленная (C1-С 6)алкильная группа или линейная или разветвленная (С 2-С 6)алкенильная группа в качестве заместителя Re. В соответствии с благоприятным вариантом осуществления изобретения, предпочтительными соединениями являются соединения формулы (IA):X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для форв которой мулы (I). В соответствии со вторым благоприятным вариантом осуществления изобретения, предпочтительными соединениями являются соединения формулы (IB)X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I). В соответствии с третьим благоприятным вариантом осуществления изобретения, предпочтительными соединениями являются соединения формулы (IC)X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I). В соответствии с четвертым благоприятным вариантом осуществления изобретения, предпочтительными соединениями являются соединения формулы (ID)X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I). В соответствии с пятым благоприятным вариантом осуществления изобретения, предпочтительными соединениями являются соединения формулы (IE)X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I). В соответствии с шестым благоприятным вариантом осуществления изобретения, предпочтительными соединениями являются соединения формулы (IF) В соответствии с седьмым благоприятным вариантом осуществления изобретения, предпочтительными соединениями являются соединения формулы (IG)X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для в которой формулы (I). В соответствии с восьмым благоприятным вариантом осуществления изобретения, предпочтительными соединениями являются соединения формулы (IH)X, Y, R1, R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для фор-4 014022 В соответствии с девятым благоприятным вариантом осуществления изобретения, предпочтительными соединениями являются соединения формулы (IJ)X, Y, R1 R2, R3, R4, R5, R6, Ra, Rb, Rc, Rd и Re имеют значения, указанные для формулы (I). Предпочтительными соединениями согласно настоящему изобретению являютсяN-(1H-индол-1-ил)-1-метил-1,2,5,6-тетрагидропиридин-3-карбоксамид,N-(2,3-дигидро-1H-индол-1-ил)-1-метил-1,4,5,6-тетрагидропиридин-3-карбоксамид,N-(5-фтор-1H-индол-1-ил)-1-метил-1,2,5,6-тетрагидропиридин-3-карбоксамид,N-(2,3-дигидро-1H-индол-1-ил)-1-метил-1,4,5,6-тетрагидропиридин-3-карбоксамид,1-[2-(диметиламино)этил]-N-(1H-индол-1-ил)-1,2,5,6-тетрагидропиридин-3-карбоксамид,N-(1H-индол-1-ил)-1-[2-(4-метил-1-пиперазинил)этил]-3-пиперидинкарбоксамид,N-(5-хлор-1H-индол-1-ил)-1-(2-гидроксиэтил)-1,4,5,6-тетрагидропиридин-3-карбоксамид,трет-бутил 4-(2-3-[(1H-индол-1-иламино)карбонил]-1-пиперидилэтил)пиперазин-1-карбоксилат,1-[3-(диметиламмоний)пропил]-3-[(1H-индол-1-иламино)карбонил]пиперидиний,N-(1H-индол-1-ил)-1-[3-(1-пиперидил)пропил]-3-пиперидинкарбоксамид,N-(1H-индол-1-ил)-1-[3-(4-метил-1-пиперазинил)пропил]-3-пиперидинкарбоксамид,N-(индол-1-ил)-1-(2-пиперидин-1-ил-этил)-1,2,5,6-тетрагидропиридин-3-карбоксамид,-N-(индол-1-ил)-1-[2-[4-(1-метилпиперидин-4-ил)пиперазин-1-ил]этил]пиперидин-3 карбоксамид,-N-(индол-1-ил)-1-[3-[4-(2-гидроксиэтил)пиперазин-1-ил]пропил]пиперидин-3-карбоксамид,-N-(индол-1-ил)-1-[4-(4-метилпиперазин-1-ил)бутил]пиперидин-3-карбоксамид,-N-(индол-1-ил)-1-аллилпиперидин-3-карбоксамид,-N-(индол-1-ил)-1-[4-(пиперидин-1-ил)бут-2-ен-1-ил]пиперидин-3-карбоксамид,(R или S) (-)-N-(индол-1-ил)-1-[2-(пиперидин-1-ил)этил]пиперидин-3-карбоксамид энантиомер 1,(R или S) (+)-N-(индол-1-ил)-1-[2-(пиперидин-1-ил)этил]пиперидин-3-карбоксамид энантиомер 2. Энантиомеры, диастереоизомеры, N-оксиды и соли присоединения с фармацевтически приемлемой кислотой или основанием, предпочтительных соединений образуют неотъемлемую часть изобретения. Настоящее изобретение также относится к способу получения соединений формулы (I), который характеризуется тем, что в качестве исходного вещества используют соединение формулы (II) в которой R1, R2, R3, R4, R5 и X имеют значения, указанные для формулы (I), это соединение подвергают реакции с дифенилфосфинилгидроксиламином, получая соединение формулы (III) в которой Ra, Rb, Rc, Rd, Re и Y имеют значения, указанные для формулы (I), A1 представляет собой группу формулы -C(=Z)-, -СН 2- или -SO2-, где Z имеет значения, указанные для формулы (I), и Z1 представляет собой группу, выбранную из гидрокси, этокси и метокси, получая соединение формулы (I/a),частный случай соединений формулы (I) в которой R1, R2, R3, R4, R5, Ra, Rb, Rc, Rd, Re, X и Y имеют значения, указанные выше, и А имеет значения, указанные для формулы (I), соединения формулы (I/a), образующие совокупность соединений по изобретению, которые очищают при необходимости в соответствии с обычным способом очистки,могут быть разделены, если это является желательным, на их разные изомеры в соответствии с обычным способом разделения и превращены, если это является желательным, в их N-оксиды и, если это является подходящим, в их соли присоединения с фармацевтически приемлемой кислотой или основанием. Соединения формул (II) и (IV) являются или коммерчески доступными, или могут быть получены в соответствии с обычными реакциями органического синтеза, которые хорошо известны специалисту в данной области техники. Соединение формулы (IV) в специфическом случае, когда представляет собой двойную связь между атомами углерода, несущими заместители Re и Rd, формулы (IV/a) в которой Ra, Rb, Rc, Re, Y, A1 и Z1 имеют значения, указанные выше, в особенности может быть получено, используя в качестве исходного вещества соединение формулы (V) в которой Ra, Rb, Rc, Y, A1 и Z1 имеют значения, указанные выше, которое подвергают реакции с карбонатом калия и хлорметил хлороформиатом, получая соединение формулы (VI) где Ra, Rb, Rc, Y, A1 и Z1 имеют значения, указанные выше, соединение формулы (VI) подвергают реакции алкилирования в присутствии соединения формулы (VII) в которой Hal представляет собой атом галогена и Re имеет значения, указанные выше, получая соединение формулы (IV/a), как определено выше. представляет собой двойную Соединение формулы (IV) в специфическом случае, когда связь между атомами углерода, несущими заместители Rd и Y, формулы (IV/b) в которой Ra, Rb, Rc, Re, Y, A1 и Z1 имеют значения, указанные выше, в особенности может быть получено, используя в качестве исходного вещества соединение формулы (VIII) в которой Ra, Rb, Rc, Y, A1 и Z1 имеют значения, указанные выше, которое подвергают реакции алкилирования в присутствии соединения формулы (VII), как определено выше, получая соединение формулы (IX)-6 014022 в которой Ra, Rb, Rc, Re, Y, A1 и Z1 имеют значения, указанные выше, и Hal- представляет собой галогенидный анион, соединение формулы (IX) подвергают частичному восстановлению путем реакции с триэтиламином и PtO2, получая соединение формулы (IV/b), как определено выше. представляет собой простую Соединение формулы (IV) в специфическом случае, когда связь, формулы (IV/c) в которой Ra, Rb, Rc, Rd, Re, Y, A1 и Y1 имеют значения, указанные выше, в особенности может быть получено, используя в качестве исходного вещества соединение формулы (X): где Ra, Rb, Re, Rd, Y, A1 и Z1 имеют значения, указанные выше, которое подвергают действию w соединения формулы (VII), как определено выше,получая соединение формулы (IV/c), как определено выше. Соединения формул (II), (IV), (V), (VII), (VIII) и (X) являются или коммерчески доступными или могут быть получены в соответствии с обычными реакциями органического синтеза, которые хорошо известны специалисту в данной области техники. Соединения формулы (I) обладают ценными фармакологическими свойствами, в особенности они являются эффективными индукторами тирозингидроксилазы (TH). Известно, что тирозингидроксилаза является ферментом, лимитирующим скорость реакции, который контролирует в особенности синтез нейромедиаторов в центральных катехоламинергических и допаминергических нейронах. Скорость синтеза этих нейромедиаторов главным образом связана с проявлением нарушением тонических функций головного мозга, включая различные патологии поведения у людей, такие как страх, психозы, депрессия,стресс и др. Благодаря их способности индуцировать тирозингидроксилазу, следовательно, соединения будут применяться в терапии для лечения депрессии, страха, нарушений памяти, связанных со старением и/или нейродегенеративными заболеваниями, и для паллиативного лечения болезни Паркинсона и для адаптации к стрессу. Настоящее изобретение также относится к фармацевтическим композициям, которые содержат в качестве активного компонента по меньшей мере одно соединение формулы (I), его энантиомер, диастереоизомер или N-оксид, или его соль присоединения с фармацевтически приемлемой кислотой или основанием, отдельно или в комбинации с одним или несколькими фармацевтически приемлемыми, инертными, нетоксичными наполнителями или носителями. Фармацевтические композиции, полученные таким образом, обычно находятся в дозированной форме; например, они могут находиться в виде таблеток, драже, капсул, суппозиториев, растворов для инъекций или для питья и могут вводить пероральным, ректальным, внутримышечным или парентеральным путем. Среди фармацевтических композиций согласно изобретению особенно можно отметить те, которые являются пригодными для перорального, парентерального (внутривенного, внутримышечного или подкожного), чрескожного, внутривлагалищного, ректального, назального, подъязычного, буккального,внутриглазного введения или введения в дыхательные пути. Фармацевтические композиции в соответствии с изобретением для парентерального введения включают, главным образом, стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для восстановленных растворов для инъекций или дисперсий. Фармацевтические композиции согласно изобретению для перорального введения в виде твердых форм включают, в особенности, таблетки или драже, подъязычные таблетки, саше, желатиновые капсулы и гранулы, а для перорального, назального, трансбуккального или внутриглазного введения в виде жидких форм включают, в особенности, эмульсии, растворы, суспензии, капли, сиропы и аэрозоли. Фармацевтические композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, а композиции для чрескожного введения включают, в особенности, порошки, аэрозоли, кремы, мази, гели и пластыри. Фармацевтические композиции, указанные выше, приведены только с целью иллюстрации изобретения и никоим образом его не ограничивают. Среди инертных, нетоксичных, фармацевтически приемлемых наполнителей или носителей можно-7 014022 отметить, но только с целью иллюстрации и никоим образом не ограничиваясь ими, разбавители, растворители, консерванты, смачивающие вещества, эмульгирующие средства, диспергирующие средства, связывающие вещества, агенты, вызывающие набухание, дезинтеграторы, вещества для замедленного высвобождения, замасливатели, абсорбенты, суспендирующие вещества, красители, ароматизаторы и т.д. Полезная дозировка изменяется в зависимости от возраста и веса пациента, пути введения, применяемой фармацевтической композиции, природы и тяжести расстройства, и применения любых сопутствующих видов лечения. Дозировка находится в диапазоне от 0,1 до 100 мг в сутки на одно или больше введений. Примеры, представленные ниже, приведены только с целью иллюстрации и никоим образом не ограничивают изобретение. Используемые исходные вещества являются известными продуктами или их получают в соответствии с известными способами. Разные примеры получения приводят к синтезу промежуточных продуктов, которые пригодны для получения соединений согласно изобретению. Структуры соединений, описанных в примерах и в получении, были определены согласно обычным спектрофотометрическим способам (инфракрасная спектрометрия, ядерный магнитный резонанс, массспектрометрия, и т.д). Точки плавления определяли с помощью прибора TOTTOLI (без коррекции выходящей колонки). Если соединение находится в виде соли, то точка плавления соответствует точке плавления такого соединения в солевой форме. Получение 1. O-дифенилфосфинилгидроксиламин. К водному раствору 149,44 г гидрохлорида гидроксиламина (340 мл воды) добавляли раствор 72,75 г гидроксида натрия (290 мл воды) и 960 мл 1,4-диоксана. Смесь охлаждали до -15 С и затем через 15 мин добавляли раствор 180 г хлорида дифенилфосфинила в 725 мл диоксана все одновременно при механическом перемешивании. Через 5 мин добавляли 3 л воды все одновременно. Образовывался белый осадок, который отфильтровывали и затем ресуспендировали в 0,25 М растворе гидроксида натрия при 0 С. Смесь мехенически перемешивали при 0 С в течение 30 мин после этого снова фильтровали. Осадок высушивали в вакууме (пятиокись фосфора), получая 72,5 г ожидаемого продукта. Точка плавления: 104 С. Элементный микроанализ: Получение 2. N-аминоиндол. К суспензии 177,72 г размолотого гидроксида калия в 1,3 л ДМФА добавляли 21,85 г индола и затем все одновременно 72,5 г суспензии соединения из получения 1 в 1,3 л ДМФА. Густую смесь нагревали при температуре в интервале от 60 до 70 С в течение 3 ч 30 мин при механическом перемешивании и затем, пока она оставалась горячей, вливали в 3,5 л смеси воды со льдом. После охлаждения полученный раствор 3 раза экстрагировали с помощью 1,5 л простого этилового эфира. Органическую фазу высушивали над сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении. При осуществлении хроматографии на силикагеле (циклогексан/простой эфир: 80/20 и затем 50/50) получали 16,5 г ожидаемого продукта. Точка плавления 35 С. Элементный микроанализ: Получение 3. 5-Хлор-N-аминоиндол. Продукт получали в соответствии с методикой из получения 2, используя 5-хлориндол вместо индола. Точка плавления: 46C. Элементный микроанализ: Получение 4. 3-Метил-N-аминоиндол. Продукт получали в соответствии с методикой из получения 2, используя 3-метилиндол вместо индола. Получение 5. 2,3-Диметил-N-аминоиндол. Продукт получали в соответствии с методикой из получения 2, используя 2,3-диметилиндол вместо индола.-8 014022 Получение 6. N-Аминоиндолин. Стадия А. 1-Нитрозоиндолин. К раствору, охлажденному до 0 С, 4 г индолина в 100 мл 50% разведенной уксусной кислоты по каплям добавляли раствор 2,32 г NaNO2 в 50 мл воды; затем раствор перемешивали в течение 30 мин при 0 С. После этого суспензию подщелачивали путем добавления 190 мл 20 % водного раствора гидроксида натрия и затем три раза экстрагировали с помощью этилацетата. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали. Получали 4,92 г ожидаемого продукта. Точка плавления: 68 С. Элементный микроанализ: Стадия В. N-Аминоиндолин. 4 г соединения с вышеописанной стадии А растворяли в 50 мл смеси простой диэтиловый эфир/дихлорметан 85/15 и затем по каплям вливали в раствор 1,4 г LiAlH4 1M/Et2O (36 мл) при 0 С. После добавления в течение 30 мин реакционную смесь перемешивали в течение 3 ч при температуре окружающей среды и после этого медленно добавляли 1,5 мл воды, 4,5 мл 15% раствора гидроксида натрия и затем 1,5 мл воды. Соли алюминия отфильтровывали через целит (элюируя с дихлорметаном) и после этого растворители упаривали при пониженном давлении. При осуществлении хроматографии на силикагеле (циклогексан/простой диэтиловый эфир: 7/3) выделяли 2,9 г ожидаемого продукта в виде оранжевого масла. ИК(см-1): 3335 (N-H); 3046; 3025 (=C-H); 2954; 2844 (C-H); 1605 (C=C); 1478 (C-H). Получение 7. Метил-1-метил-1,4,5,6-тетрагидропиридин-3-карбоксилат. Стадия А. 3-(Метоксикарбонил)-1-метилпиридиний йодид. 10 г метил никотината растворяли в 5,2 мл метилйодида и после этого реакционную смесь нагревали при 60 С в течение 24 ч, защищая от света. Остаток очищали путем флэш-хроматографии на силикагеле (CH2Cl2/CH3OH: 9/1 и затем 85/15), выделяя 13,46 г ожидаемого продукта. Стадия В. Метил 1-метил-1,4,5,б-тетрагидропиридин-3-карбоксилат. 4 г соединения с вышеописанной стадии А, разведенного в 100 мл абсолютного метанола, гидрировали при атмосферном давлении в течение 20 ч при 20 С в присутствии 4 мл триэтиламина и 800 мг 10%Pd/C. После отфильтровывания катализатора через целит и промывания (два раза метанолом), фильтрат упаривали в вакууме и получали желтый остаток. Остаток осторожно промывали три раза дистиллированным простым диэтиловым эфиром и затем после упаривания растворителя в вакууме выделяли 2,15 г желтого масла. Получение 8. 2-Метил-1-индолинамин. Продукт получали в соответствии с методикой из получения 6, используя 2-метилиндолин вместо индолина. Получение 9. 5-Фтор-N-аминоиндол. Продукт получали в соответствии с методикой из получения 2, используя 5-фториндол вместо индола. Получение 10. 5-Метокси-N-аминоиндол. Продукт получали в соответствии с методикой из получения 2, используя 5-метоксииндол вместо индола. Получение 11. Этил 1-аллилпиперидин-3-карбоксилат. 2 г этил пиперидин-3-карбоксилата добавляли к 1,6 г аллилбромида и затем добавляли 13 мл бензола. Добавляли 1,08 г карбоната натрия и реакционную смесь перемешивали в течение ночи при температуре флегмы бензола. Реакционную смесь фильтровали и затем упаривали при пониженном давлении. При осуществлении очистки путем перегонки, используя стеклянный термостат, выделяли 1,53 г ожидаемого продукта. Ткипения(810-2 бар): 80-85 С. ИК (см-1): 3078 (=C-H); 2980; 2941; 2866 (C-H); 2791(N-CH2); 1733 (C=O); 1644 (C=C); 1468; 1453 (C-H); 1368 (C-N); 1223; 1182 (C-O). Получение 12. Этил 1-метилпиперидин-3-карбоксилат. 3 г этил пиперидин-3-карбоксилата, 4 мл водного формальдегида, 300 мг 10% Pd/C и 4 мл ледяной уксусной кислоты помещали в атмосферу водорода (1 атм) при 20 С в течение 17 ч. После отфильтровывания катализатора через целит и промывания с помощью 50 мл этанола раствор упаривали при пониженном давлении и получали маслянистый остаток. Остаток разводили в смеси толуол/вода (1/1) и значение рН доводили до 9 путем добавления 20% K2CO3. После разделения двух фаз водную фазу два раза экстрагировали толуолом. Органические фазы промывали водой, высушивали над Na2SO4 и получали слегка окрашенное масло. При осуществлении перегонки при пониженном давлении получали 2,4 г ожи-9 014022 даемого продукта. Точка кипения: 105-110 С (Р = 20 мм рт. ст.) Получение 13. Этил 1-пропилпиперидин-3-карбоксилат. 2 г этил пиперидин-3-карбоксилата добавляли к 1,6 г 1-бромпропана и затем добавляли 13 мл бензола. Добавляли 1,08 г карбоната натрия и реакционную смесь перемешивали в течение ночи при температуре флегмы бензола. Реакционную смесь фильтровали и после этого упаривали при пониженном давлении. При осуществлении очистки путем перегонки, используя стеклянный термостат, выделяли 1,53 г ожидаемого продукта. Получение 14. Метил 1-(2-гидроксиэтил)-1,2,5,6-тетрагидропиридин-3-карбоксилат. Стадия А. Метил 1,2,5,6-тетрагидропиридин-3-карбоксилат. 7 г гидробромида ареколина растворяли в 20 мл воды и затем раствор подщелачивали путем добавления 5,13 г карбоната калия, после этого насыщали с помощью NaCl. Водную фазу три раза экстрагировали простым эфиром. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали до получения 4,7 г бесцветного масла. Масло разводили в 33 мл безводного толуола и затем добавляли 3,92 мл 1-хлорэтил хлороформиата. Образовывался осадок и смесь нагревали в течение ночи при флегме толуола. Осадок отфильтровывали и затем органическую фазу промывали 0,1 М раствором соляной кислоты; водную фазу один раз экстрагировали простым эфиром. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. Остаток ресуспендировали в 25 мл метанола и после этого нагревали в колбе с обратным холодильником в течение 2 ч. Метанол упаривали при пониженном давлении, и получали 3,8 г гидрохлорида метил 1,2,5,6-тетрагидропиридин-3-карбоксилата. Ожидаемый продукт получали путем растворения гидрохлорида метил 1,2,5,6-тетрагидропиридин-3-карбоксилата в воде и затем подщелачивания путем добавления карбоната калия до достижения значения рН, равного 10. Насыщали с помощью хлорида натрия и затем водную фазу три раза экстрагировали простым эфиром; объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали до получения 3 г ожидаемого продукта. Стадия В. Метил 1-(2-гидроксиэтил)-1,2,5,6-тетрагидропиридин-3-карбоксилат. К раствору 1,77 г соединения с вышеописанной стадии А в 17 мл безводного 1,4-диоксана добавляли 5,22 г карбоната калия, 24 мг йодида натрия и затем 900 мкл бромэтанола. Реакционную смесь нагревали в колбе с обратным холодильником в течение ночи при перемешивании и после этого концентрировали при пониженном давлении. Остаток ресуспендировали в воде и водную фазу два раза экстрагировали этилацетатом. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали, выделяя 1,75 г ожидаемого продукта. ИК(см-1): 3398 (O-H); 2950; 2815 (C-H); 1710 (C=O); 1656 (C=C). Получение 15. Метил 1-(2-[трет-бутил(диметил)силил]оксиэтил)-1,2,5,6-тетрагидропиридин-3 карбоксилат. К раствору 1,5 г соединения из получения 14 в 15 мл пиридина добавляли 2,08 г третбутилдиметилсилил хлорида. Раствор перемешивали в течение ночи при температуре окружающей среды. Полученное твердое вещество растворяли в дихлорметане и затем промывали раствором карбоната натрия с рН 11; водную фазу экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (циклогексан/AcOEt: 3/1 и затем 2/1) выделяли 1,9 г ожидаемого продукта. ИК (см-1): 2951; 2929; 2856 (C-H); 1716 (C=O); 1658 (C=C). Получение 16. Метил 1-(2-диметиламиноэтил)-1,2,5,6-тетрагидропиридин-3-карбоксилат. К раствору 1,94 г соединения со стадии А из получения 14 в 27 мл безводного 1,4-диоксана добавляли 2,03 г йодида натрия, 4,3 г карбоната натрия и 1,45 г 2-диметиламиноэтил хлорида. Реакционную смесь перемешивали в течение ночи при флегме диоксана и после этого реакционную смесь фильтровали и промывали дихлорметаном. Фильтрат упаривали при пониженном давлении. Остаток ресуспендировали в дихлорметане, промывали раствором карбоната натрия с рН 10, высушивали над Na2SO4, фильтровали и затем упаривали при пониженном давлении. При осуществлении очистки, используя стеклянный термостат, выделяли 846 мг ожидаемого продукта. ИК (см-1): 2948; 2816 (C-H); 2765 (N-CH3); 1712(C=O); 1657 (C=C). Получение 17. Метил 1-(2-гидроксиэтил)-1,4,5,6-тетрагидропиридин-3-карбоксилат. Стадия А. 1-(2-Гидроксиэтил)-3-(метоксикарбонил)пиридиний бромид. 10 г метил никотината растворяли в 5,2 мл 2-бромэтанола и затем реакционную смесь нагревали при 60 С в течение 24 ч, защищая от света. Остаток очищали путем флэш-хроматографии на силикагеле Стадия В. Метил 1-(2-гидроксиэтил)-1,4,5,6-тетрагидропиридин-3-карбоксилат. К раствору 4 г соединения с вышеописанной стадии А в 100 мл ректифицированного МеОН добавляли 2,73 мл триэтиламина и 400 мг 10% Pd/C. Смесь два раза дегазировали в вакууме и помещали в атмосферу водорода. Реакционную смесь перемешивали в течение 18 ч и после этого фильтровали через целит (элюируя с помощью CH2Cl2). Раствор упаривали при пониженном давлении. Остаток ресуспендировали в 100 мл простого диэтилового эфира и воды; водную фазу два раза экстрагировали простым эфиром, и получали 2,41 г ожидаемого продукта. Точка плавления: 38C. Элементный микроанализ: Получение 18. Метил 1-[(2-диметиламино)этил]-1,4,5,6-тетрагидропиридин-3-карбоксилат. К раствору 2 г соединения из получения 17 в 20 мл безводного дихлорметана добавляли 2 мл триэтиламина и затем 1,1 мл мезилхлорида при 0 С. Реакционную смесь перемешивали в течение 30 мин при температуре окружающей среды и затем добавляли 20 мл воды; органическую фазу разводили в дихлорметане. Водную фазу два раза экстрагировали дихлорметаном. Объединенные органические фазы высушивали над Na2SO4, фильтровали и затем упаривали при пониженном давлении. Остаток ресуспендировали в 20 мл ацетонитрила, и затем добавляли 12,5 мл 2 М диметиламина в МеОН. Реакционную смесь перемешивали в течение 15 ч при температуре окружающей среды и затем в течение 2 ч при 50 С. Реакционную смесь концентрировали при пониженном давлении, ресуспендировали в дихлорметане и промывали раствором K2CO3 с рН 11 и затем насыщенным раствором NaCl. Органическую фазу высушивали над Na2SO4, фильтровали и затем упаривали при пониженном давлении, выделяя 1,42 г ожидаемого продукта. ИК(см-1): 2961; 2853 (C-H); 2771 (N-CH3); 1678 (C=O); 1617 (C=C). Получение 19. Этил 1-[2-(диметиламино)этил]пиперидин-3-карбоксилат. 3,51 г хлорида 2-диметиламиноэтила растворяли в 50 мл безводного 1,4-диоксана и после этого добавляли 9,58 г карбоната натрия и 4,52 г йодида натрия, затем 5,16 г этил пиперидин-3-карбоксилата. Реакционную смесь нагревали в колбе с обратным холодильником в течение 20 ч и затем диоксан упаривали при пониженном давлении. Остаток ресуспендировали в этилацетате и промывали водой и затем насыщенным раствором хлорида натрия. Органическую фазу высушивали над сульфатом натрия и затем упаривали при пониженном давлении. При осуществлении очистки путем перегонки, используя стеклянный термостат, выделяя 3,41 г ожидаемого продукта. Температура кипения: 90 С (5 10-1 бар). ИК(см-1): 2941; 2857; 2815 (C-H); 2766 (N-CH3); 1730 (C=O); 1464 (C-H). Получение 20. Этил 1-[2-(4-морфолинил)этил]пиперидин-3-карбоксилат. 2,0 г соединения из получения 36 и 2,3 мл морфолина растворяли в 15 мл 70% этанола. Реакционную смесь перемешивали при температуре окружающей среды в течение 96 ч. После этого раствор концентрировали и затем ресуспендировали в 30 мл дихлорметана. После этого органическую фазу несколько раз промывали водой, высушивали над Na2SO4, фильтровали и затем концентрировали. При осуществлении хроматографии на силикагеле (CH2Cl2/МеОН: 9/1) выделяли 2,3 г ожидаемого продукта. ИК(см-1): 2943, 2854, 2810 (C-H), 1729 (C=O сложный эфир), 1449 (C-H), 1372 (C-N), 1117 (CO-C). Получение 21. Этил 1-[2-(1-пиперидил)этил]пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя пиперидин вместо морфолина. ИК(см-1): 2939, 2859 (C-H), 1726 (C=O сложный эфир), 1453 (C-H), 1372 (C-N), 1183, 1154 (CO). Получение 22. Этил 1-[2-(4-метил-1-пиперазинил)этил]пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя N-метилпиперазин вместо морфолина. ИК(см-1): 2939, 2799 (C-H), 1731 (C=O сложный эфир), 1452 (C-H), 1373 (C-N). Получение 23. Этил 1-2-[4-(2-гидроксиэтил)-1-пиперазинил]этилпиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя (2-гидроксиэтил)пиперазин вместо морфолина. ИК (см-1): 3235 (O-H), 2940, 2810 (C-H), 1730 (C=O сложный эфир), 1451 (C-H), 1371 (C-N),- 11014022 1222 (C-O). Получение 24. Этил 1-[2-(1-пирролидинил)этил]пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя пирролидин вместо морфолина. ИК (см-1): 2942, 2782 (C-H), 1732 (C=O сложный эфир), 1452 (C-H), 1371 (C-N), 1226 (C-O). Получение 25. Этил 1-[2-(1-азепанил)этил]пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя гексаметиленимин вместо морфолина. ИК (см-1): 2925, 2853, 2810 (C-H), 1733 (C=O сложный эфир), 1450 (С-Н),1370 (C-N), 1225 (CO). Получение 26. Этил 1-[2-(4-фенил-1-пиперазинил)этил]пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя 4-фенил-пиперазин вместо морфолина. ИК(см-1): 2942, 2817 (C-H), 1731 (C=O сложный эфир), 1600 (C=C), 1502, 1451 (C-H). Получение 27. 1-[2-(1-Пиперидил)этил]пиперидин-3-карбальдегид. 0,98 г соединения из получения 18 растворяли в 30 мл безводного ТГФ и затем раствор охлаждали до -80 С. После этого медленно добавляли 2,5 мл раствора DIBAL-H (1,5 М в толуоле) с помощью шприца. Затем раствор перемешивали при -80 С и затем, через 1,5 ч, к смеси дополнительно добавляли 2,5 мл раствора DIBAL-H. После осуществления реакции в течение 3 ч при -80 С добавляли 15 мл воды и после этого температуре позволяли возвратиться обратно до температуры окружающей среды. После этого раствор экстрагировали двумя порциями по 50 мл CH2Cl2 и затем промывали насыщенным растворомNaCl. После высушивания над Na2SO4 и концентрирования, ожидаемый продукт получали и использовали без дополнительной очистки на последующих стадиях. Получение 28. 5-Хлор-N-аминоиндолин. Стадия А. 1-Ацетилиндолин. 23 мл уксусного ангидрида по каплям добавляли к 5 г индолина, в это время температуру смеси поддерживали при 0. Смесь нагревали в колбе с обратным холодильником в течение 4 ч при перемешивании и после этого упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (циклогексан/ AcOEt: 6/4 и затем чистый AcOEt) выделяли 6,4 г ожидаемого продукта. Стадия В. 5-Хлориндолин. 3,84 г SO2Cl2 по каплям добавляли при 0 С к раствору 6,4 г соединения с вышеописанной стадии А в 170 мл четыреххлористого углерода. Образовывался белый осадок, который перемешивали при температуре окружающей среды в течение одного часа. Суспензию разводили в дихлорметане и водой. Органическую фазу экстрагировали и затем промывали 20% водным раствором гидроксида натрия. Органическую фазу высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. Остаток ресуспендировали в 110 мл абсолютного этанола и нагревали в колбе с обратным холодильником до завершения растворения. Добавляли 49 мл 37% HCl и затем раствор перемешивали в колбе с обратным холодильником в течение 3 ч 30 мин. Этанол упаривали при пониженном давлении, и затем водную фазу разводили в 50 мл воды, экстрагировали простым эфиром и затем подщелачивали, используя 20% водного раствора гидроксида натрия. Водную фазу три раза экстрагировали простым эфиром. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (циклогексан/AcOEt: 7/3) выделяли 3,53 г ожидаемого продукта. Стадия С. 5-Хлор-1-нитрозоиндолин. К раствору, охлажденному до 0 С, 3,53 г соединения со стадии В выше в 70 мл 50% разведенной уксусной кислоты по каплям добавляли раствор 1,59 г NaNO2 в 35 мл воды; затем раствор перемешивали в течение 30 мин при 0 С. Затем смесь подщелачивали путем добавления 130 мл 20% водного раствора гидроксида натрия и затем три раза экстрагировали с помощью этилацетата. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении, выделяя 4,15 г ожидаемого продукта. Точка плавления: 89 С. Элементный микроанализ: Стадия D. 5-Хлор-N-аминоиндолин. Суспензию 570 мг LiAlH4 в 15 мл безводного простого эфира перемешивали при флегме простого эфира в течение 45 мин и после этого перемешивали в течение ночи при температуре окружающей среды. 2 г соединения со стадии С выше растворяли в 6 мл безводного ТГФ и затем разводили в 14 мл безводного простого эфира. Таким образом полученный раствор по каплям добавляли к раствору LiAlH4 при 0 С. Раствор перемешивали в течение 3 ч при 0 С и затем 0,5 мл воды, 1,5 мл 15% водного раствора гидроксида натрия и 0,5 мл воды осторожно добавляли в атмосфере азота. Соли алюминия отфильтровы- 12014022 вали через целит (элюируя с помощью дихлор-метан). Раствор упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (циклогексан/AcOEt: 85/15 и затем 8/2 - 7/3) выделяли 1,7 г ожидаемого продукта. Точка плавления: 30 С. Элементный микроанализ: Получение 29. Метил 1-(2-[трет-бутил(диметил)силил]оксиэтил)-1,4,5,6-тетрагидро-3-пиридинкарбоксилат. Продукт получали в соответствии с методикой из получения 15, используя соединение из получения 17 вместо соединения из получения 14. Получение 30. Этил 1-(2-гидроксиэтил)пиперидин-3-карбоксилат. 2 г этил пиперидин-3-карбоксилата добавляли к 1,6 г 2-бромэтанола и затем разводили в 13 мл бензола. Добавляли 1,08 г карбоната натрия и реакционную смесь перемешивали в течение ночи при температуре флегмы бензола. Реакционную смесь фильтровали и затем упаривали при пониженном давлении. При осуществлении очистки путем перегонки, используя стеклянный термостат, выделяли 1,53 г ожидаемого продукта. Температура кипения : 100C (3 10-2 бар) ИК(см-1): 3412 (O-H); 2941; 2808 (C-H); 1730 (C=O); 1468 (С-Н). Получение 31. Этил 1-бензилпиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 30, используя бензил бромид вместо 2 бромэтанола. Получение 32. Метил 4-(4-фторфенил)-1-метилпиперидин-3-карбоксилат. 2,5 г ареколина растворяли в 20 мл безводного простого диэтилового эфира и 25 мл безводного дихлорметана. Раствор охлаждали до -30 С и по каплям добавляли раствора 32,2 мл бромид 1 М (4 фторфенил)магния. Смесь перемешивали в течение 3 ч при температуре от -30 до -35 С и после этого охлаждали до -78 С в течение 30 мин. Затем по каплям добавляли 10 мл 1/1 смеси трифторуксусной кислоты и простого эфира; после этого смесь доводили до 0 С и добавляли 15 мл 1 М раствора соляной кислоты, затем 28% раствор гидроксида аммония для доведения рН до 12. Органическую фазу собирали и водную фазу три раза экстрагировали с помощью этилацетата. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (циклогексан/триэтиламин: 3/1) выделяли ожидаемый продукт. Получение 33. трет-Бутил 4-2-[3-(этоксикарбонил)-1-пиперидил]этилпиперазин-1-карбоксилат. Ожидаемый продукт получали в соответствии с методикой из получения 20, используя трет-бутил пиперазин-1-карбоксилат вместо морфолина. ИК(см-1): 2938, 2811 (C-H); 1731 (C=O сложный эфир); 1698 (C=O NBOc); 1455, 1421, 1373 (CN). Получение 34. Этил 1-[3-(диметиламино)пропил]пиперидин-3-карбоксилат. 1,5 г соединения из получения 24 и 4,8 мл диметиламина растворяли в 10 мл 70% этанола. Реакционную смесь перемешивали при температуре окружающей среды в течение 48 ч. Дополнительно добавляли 4,8 мл диметиламина и после этого реакционную смесь нагревали при 50 С в течение 24 ч. Затем раствор концентрировали, используя роторный испаритель, и после этого ресуспендировали в 50 мл дихлорметана. Затем органическую фазу несколько раз промывали водой, высушивали над Na2SO4, фильтровали и затем концентрировали. При осуществлении хроматографии на силикагеле (CH2Cl2/МеОН : 9/1) выделяли 0,81 г ожидаемого продукта. ИК(см-1): 2942, 2813, 2761 (С-Н); 1730 (C=O сложный эфир); 1466 (С=Н); 1373 (C-N); 1235,1179 (C=O), 1104. Получение 35. Этил 1-(3-гидроксипропил)пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 30, используя 3-бромпропанол вместо 2-бромэтанола. Температура кипения: 125 С (310-2 бар). ИК(СМ-1): 3388 (O-H); 2941; 2861; 2811 (C-H) 1729 (C=O); 1470 (С-Н). Получение 36. Этил 1-(2-хлорэтил)пиперидин-3-карбоксилат. 10,0 г этил 3-пиперидин-3-карбоксилата растворяли в 80 мл ацетона и затем добавляли 11 мл 1 бром-2-хлорэтана и 14 г карбоната калия при температуре окружающей среды. После перемешивания в течение 40 ч растворитель упаривали и затем добавляли 50 мл воды и 100 мл простого диэтилового эфира. Органическую фазу отделяли промывали водой, высушивали над Na2SO4 и затем концентрировали. При осуществлении хроматографии на силикагеле (петролейный эфир/этил простой эфир: 6/4) выделяли 9,24 г ожидаемого продукта. ИК(см-1): 2943, 2808 (C-H), 1729 (C=O сложный эфир), 1468, 1450 (С-Н), 1370 (C-N), 1209,- 13014022 1179 (C-O). Получение 37. Этил 1-(3-хлорпропил)пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 36, используя 1-бром-3-хлорпропан вместо 1-бром-2-хлорпропана. ИК(см-1): 2945, 2808 (C-H), 1730 (C=O сложный эфир), 1469, 1446 (C-H), 1371 (C-N), 1179,1153 (C-O). Получение 38. Этил 1-[3-(1-пиперидил)пропил]пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя пиперидин вместо морфолина и соединение из получения 37 вместо соединения из получения 36. ИК (см-1): 2933, 2854, 2802, 2761 (C-H), 1731 (C=O сложный эфир), 1469, 1463 (С-Н), 1373 (CN), 1271, 1178 (C-O). Получение 39. Этил 1-[3-(4-метил-1-пиперазинил)пропил]пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя N-метилпиперазин вместо морфолина и соединение из получения 37 вместо соединения из получения 36. ИК (см-1): 2939, 2794 (C-H), 1730 (C=O сложный эфир), 1448 (С-Н), 1372 (C-N), 1283, 1150 (CO). Получение 40. Этил 1-(2-[трет-бутил(диметил)силил]оксиэтил)пиперидин-3-карбоксилат. 1,276 г хлорида трет-бутилдиметилсилила добавляли к раствору 1 г соединения из получения 30 в 16 мл пиридина. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды,и затем пиридин упаривали при пониженном давлении (совместное упаривание с толуолом). Остаток ресуспендировали в дихлорметане, два раза промывали водой и затем насыщенным раствором NaCl. Органическую фазу высушивали над сульфатом натрия, фильтровали и затем упаривали. При осуществлении флэш-хроматографии на силикагеле (циклогексан/AcOEt: 3/1) выделяли 1,1 г ожидаемого продукта. ИК(см-1): 2930; 2857 (C-H); 1733 (C=O); 1470 (С-Н). Получение 41. Этил 1-(3-[трет-бутил(диметил)силил]оксипропил)пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 40, используя соединение из получения 35 вместо соединения из получения 30. ИК(см-1): 2951; 2930; 2857 (C-H); 1734 (C=O); 1471 (С-Н). Получение 42. Метил 1-(2-хлорэтил)-1,2,5,6-тетрагидропиридин-3-карбоксилат. Стадия А. Гувацин гидрохлорид. 7 г гидробромида ареколина растворяли в 20 мл воды, раствор подщелачивали путем добавления 5,13 г карбоната калия и после этого насыщали с помощью NaCl. Водную фазу три раза экстрагировали простым диэтиловым эфиром. Объединенные органические фазы высушивали над сульфатом натрия,фильтровали и затем упаривали до получения бесцветного масла весом 4,7 г. Масло ресуспендировали в 20 мл толуола; раствор мутный. После добавления сульфат натрия и фильтрования нерастворимое вещество промывали 13 мл толуола. К органическому раствору добавляли 3,92 мл 1-хлорэтил хлороформиата. Образовывался осадок и реакционную смесь нагревали в течение 12 ч при флегме толуола. Осадок отфильтровывали и затем органическую фазу промывали 0,1 М водным раствором соляной кислоты; водную фазу один раз экстрагировали простым диэтиловым эфиром. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. Остаток ресуспендировали в 25 мл метанола и после этого нагревали в колбе с обратным холодильником в течение 2 ч. Метанол упаривали при пониженном давлении, и 3,8 г ожидаемого продукта получали с выходом 73%. Гувациновое основание получали путем растворения гидрохлорида в воде; водную фазу подщелачивали путем добавления карбонат калия до достижения рН 10, и ее насыщали с помощью NaCl. Водную фазу три раза экстрагировали простым диэтиловым эфиром и объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали до получения 3 г бесцветного масла. Элементный микроанализ: Стадия В. Метил 1-(2-хлорэтил)-1,2,5,6-тетрагидропиридин-3-карбоксилат. К суспензии 530 мг соединения с вышеописанной стадии А в 12 мл ацетона добавляли 2,53 мл триэтиламина и 1,1 мл 1-бром-2-хлорэтана. Смесь перемешивали при температуре окружающей среды в течение 18 ч и затем нагревали в колбе с обратным холодильником в течение 8 ч. Ее упаривали при пониженном давлении, ресуспендировали в дихлорметане и промывали водным раствором карбоната калия. Водную фазу экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. Остаток очищали путем флэш-хроматографии на силикагеле (циклогексан/AcOEt: 7/3), получая 430 мг ожидаемого соединения. ИК(см-1): 2951; 2917; 2811 (C-H); 2767 ( N-CH2); 1709 ( C=O); 1657 ( C=C); 1462; 1436 (C-H); 1375 (C-N); 1261 (C-O). Получение 43. Метил 1-[2-(4-метилпиперазин-1-ил)этил]-1,2,5,6-тетрагидропиридин-3-карбоксилат. К раствору 320 мг соединения из получения 42 в 5 мл 70% водного этанола добавляли 540 мкл 1 метилпиперазина. Раствор перемешивали при температуре окружающей среды в течение 72 ч и после этого упаривали при пониженном давлении. Остаток ресуспендировали в дихлорметане и два раза промывали водным раствором карбоната натрия. Органическую фазу высушивали над сульфатом натрия,фильтровали и упаривали при пониженном давлении, получая 320 мг ожидаемого соединения. ИК (см-1). 2938 (C-H); 2793 (N-CH3); 1712 (C=O); 1657 (C=C); 1438 (C-H); 1373 (C-N); 1262(C-O). Получение 44. 5-Метил-N-аминоиндол. Продукт получали в соответствии с методикой из получения 2, используя 5-метилиндол вместо индола. Получение 45. Метил 1-[2-[4-[2-(трет-бутилдиметилсиланокси)этил]пиперазин-1-ил]этил]-1,2,5,6 тетрагидропиридин-3-карбоксилат. Стадия А. Метил 1-[2-[4-(2-гидроксиэтил)пиперазин-1-ил]этил]-1,2,5,6-тетрагидропиридин-3 карбоксилат. Продукт получали в соответствии с методикой из получения 43, используя 4-(2-гидроксиэтил)пиперазин вместо 1-метилпиперазина. ИК (см-1): 3350 (O-H); 2946 (C-H); 2812 (N-CH2); 1710 (C=O); 1656 (C=C);1437 (C-H); 1351(C-N); 1262 (C-O). Стадия В. Метил 1-[2-[4-[2-(трет-бутилдиметилсиланокси)этил]пиперазин-1-ил]этил]-1,2,5,6-тетрагидропиридин-3-карбоксилат. К раствору 340 мг соединения с вышеописанной стадии А в 5 мл пиридина добавляли 260 мг хлорида трибутилдиметилсилила. Раствор перемешивали при температуре окружающей среды в течение 16 ч и затем пиридин упаривали при пониженном давлении. После растворения остатка в дихлорметана органическую фазу экстрагировали водным раствором карбоната калия. Водную фазу экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле(CH2Cl2/CH3OH : 9/1, затем 85/15) получали 360 мг ожидаемого продукта. Получение 46. Метил 1-[2-(пиперидин-1-ил)этил]-1,2,5,6-тетрагидропиридин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 43, используя пиперидин вместо 1 метилпиперазина. ИК(СМ-1): 2955; 2932; 2872 (C-H); 1736 (C=O); 1661 (C=C); 1434 (С-Н); 1368; 1341 (C-N); 1236; 1194 (C-O). Получение 47. 1,2-Бис[3-(этоксикарбонил)-1,2,5,6-тетрагидропиридин-1-ил]-этан. К раствору 900 мг соединения со стадии А из получения 42 в 10 мл метанола добавляли 0,32 мл 2 бромхлорэтана, затем добавляли 1,6 мл триэтиламина. Смесь нагревали в колбе с обратным холодильником в течение 20 ч и упаривали при пониженном давлении. Остаток растворяли в дихлорметане и экстрагировали насыщенным водным раствором карбоната калия. Водную фазу экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (AcOEt, затем ИК(см-1): 2950; 2908 (C-H); 2807 (N-CH2); 1708 (C=O); 1656 (C=C); 1435 (C-H); 1351 (C-N); 1258 (C-O). Получение 48. Этил -1-[2-[4-[(тетрагидрофуран-2-ил)метил]пиперазин-1-ил]этил]пиперидин-3 карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя 4-[(тетрагидрофуран-2 ил)метил]пиперидин вместо морфолина. При осуществлении хроматографии на силикагеле (CH2Cl2/- 15014022 Получение 49. Этил 1-[2-[4-[2-(диметиламино)этил]пиперазин-1-ил]этил]-пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя N-[2-(диметиламино) этил]пиперидин вместо морфолина. ИК(см-1): 2941; 2810 (C-H); 1730 (C=O сложный эфир); 1466 (C-H); 1304; 1154 (C-О). Получение 50. Метил -1-[2-(4-(2-метоксиэтил)пиперазин-1-ил)этил]-пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя (2-метоксиэтил)пиперидин вместо морфолина. ИК (см-1): 2941; 2809 (C-H); 1734 (C=O сложный эфир); 1451 (C-H); 1305 (C-N); 1155 (C-O); 1014. Получение 51. Этил 1-[2-[4-(1-метилпиперидин-4-ил)пиперазин-1-ил]этил]-пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя (1-метилпиперидин-4 ил)пиперазин вместо морфолина. ИК(см-1): 2936; 2807 (C-H); 1731 (C=O сложный эфир); 1449 (C-H); 1375 (C-N); 1152 (C-O). Получение 52. Этил -1-[3-[4-[2-(трет-бутилдиметилсилилокси)этил]пиперазин-1-ил]пропил]пиперидин-3-карбоксилат. Стадия А. Этил -1-[3-[4-(2-гидроксиэтил)пиперазин-1-ил]пропил]пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя (2-гидроксиэтил)пиперазин вместо морфолина. ИК (см-1): 2940; 2810 (C-H); 1731 (C=O сложный эфир); 1573; 1372; 1154 (C-O). Стадия В. Этил -1-[3-[4-[2-(трет-бутилдиметилсилилокси)этил]пиперазин-1-ил]пропил]пиперидин-3-карбоксилат. 1,0 г соединения с вышеописанной стадии A растворяли в 10 мл пиридина, и добавляли 0,7 гTBDMSCl. Реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч; растворитель удаляли, в присутствии толуола, путем перегонки с помощью роторного испарителя. При осуществлении хроматографии на силикагеле (CH2Cl2/МеОН : 95/5) получали 1,15 г ожидаемого продукта. ИК(см-1): 2935; 2858; 2807 (C-H); 2477; 1731 (C=O сложный эфир); 1460 (C-H); 1409; 1372; 1337; 1314; 1278; 1217; 1181. Получение 53. 3-[N-(Индол-1-ил)аминокарбонил]-1-(4-хлорбутил)пиридиний бромид. Стадия А. 1-(4-Хлорбутил)-3-(метоксикарбонил)пиридиний бромид. 3,0 г метил никотината растворяли в 9 мл метанола, и затем добавляли 5 мл 1-бром-4-хлорбутан. Реакционную смесь нагревали в колбе с обратным холодильником в течение 24 ч. После возвращения до температуры окружающей среды добавляли 25 мл простого диэтилового эфира. Образованное масло выделяли путем отведения супернатанта и два раза промывали 20 мл простого диэтилового эфира. После высушивания при пониженном давлении получали 4,2 г ожидаемого продукта. Стадия В. 3-[N-(Индол-1-ил)аминокарбонил]-1-(4-хлорбутил)пиридиний бромид. 0,28 г соединения из получения 2 растворяли в 4 мл безводного дихлорметана. Раствор охлаждали до -20 С и добавляли 2,1 мл 2 М раствора триметилалюминия в гексане в атмосфере аргона. После повышения температуры реакционной смеси до 0 С в течение 1 ч 30 мин добавляли раствор 0,6 г соединения с вышеописанной стадии А в 4 мл безводного дихлорметана. После этого реакционную смесь нагревали в колбе с обратным холодильником в течение 18 ч. После возвращения до температуры окружающей среды добавляли 40 мл дихлорметана и по каплям добавляли 20% (мас./об.) водного раствора гидроксида натрия до тех пор, пока больше не обнаруживалось метана. Органическую фазу отделяли, последовательно промывали 20% (мас./об.) водного раствора гидроксида натрия и насыщенным раствором NaCl,высушивали над Na2SO4 и концентрировали до объема около 10 мл. Добавляли 50 мл простого диэтилового эфира. Полученное твердое вещество отделяли путем фильтрации и затем несколько раз промывали простым диэтиловым эфиром. После высушивания получали 0,53 г ожидаемого продукта. Точка плавления: 164 С. ИК(см-1): 3028; 1625; 1589; 1556; 1443; 1286; 1210; 1174. Получение 54. Этил -1-[2-(4-бутилпиперазин-1-ил)этил]пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 20, используя 1-бутилпиперазин вместо морфолина. При осуществлении хроматографии на силикагеле (CH2Cl2/CH3OH : 9/1 + 0,5% NEt3) выделяли 1,2 г ожидаемого продукта. ИК (см-1): 2935; 2806 (C-H); 1732 (C=O сложный эфир); 1450 (C-H); 1373 (C-N); 1155 (C-O). Получение 55. Этил -1-(проп-2-инил)пиперидин-3-карбоксилат. Продукт получали в соответствии с методикой из получения 11, используя пропаргилбромид вместо аллилбромида. При осуществлении; флэш-хроматографии на силикагеле (циклогексан/AcOEt: 85/15,затем 8/2) получали 500 мг ожидаемого продукта. Элементный микроанализ: ИК(см-1): 3291 (С-Н); 2940; 2858; 2806 (С-Н); 1727 (C=C); 1629 (C=C); 1468; 1450 (С-Н); 1368; 1310 (N-C); 1223; 1181 (C-O). Получение 56. Этил -1-[4-(пиперидин-1-ил)бут-2-ен-1-ил]пиперидин-3-карбоксилат. В хорошо вентилируемый шкаф последовательно добавляли при температуре окружающей среды к раствору 680 мг 1,4-дибромбутена в 5 мл бензола по каплям раствор 500 мг этил нипекотата в 5 мл бензола и 340 мг карбоната натрия. Реакционную смесь перемешивали при температуре окружающей среды в течение 18 ч, и затем добавляли 1,26 мл пиперидина. Суспензию перемешивали при температуре окружающей среды в течение одного часа и реакционную смесь упаривали при пониженном давлении. Остаток растворяли в дихлорметане и органическую фазу экстрагировали водным раствором карбоната натрия. Водную фазу экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и упаривали при пониженном давлении. При осуществлении флэшхроматографии на силикагеле (CH2Cl2CH3OH : 95/5, затем 9/1) получали 260 мг ожидаемого продукта. ИК(см-1): 2934; 2854 ( C-H); 2796; 2757 (N-CH2); 1731 (C=O); 1467; 1442 (C-H); 1368; 1352 (CN); 1218; 1180 (C-O). Пример 1. N-(1H-индол-1-ил)-1-метил-1,2,5,6-тетрагидропиридин-3-карбоксамид. 1,94 г гидрохлорида метил 1-метил-1,2,5,6-тетрагидропиридин-3-карбоксилата растворяли в 5 мл воды после и этого раствор подщелачивали, используя карбонат калия, для достижения рН 10 и после этого насыщали с помощью NaCl. Водную фазу три раза экстрагировали простым диэтиловым эфиром. Объединенные органические фазы высушивали над Na2SO4, фильтровали и затем упаривали. 1,3 г соединения из получения 2 растворяли в 26 мл безводного дихлорметана и затем после охлаждения до -25 С добавляли 9 мл 2 М раствора триметилалюминий в гексане. Через 1 ч 30 мин добавляли раствор 1,27 г ареколина в 6,5 мл безводного дихлорметана при температуре окружающей среды. Реакционную смесь нагревали в колбе с обратным холодильником в течение ночи и после этого разведенного в дихлорметане и вливали в 50 мл 20% водного раствора гидроксида натрия. Органическую фазу выделяли и водную фазу экстрагировали дихлорметаном. Объединенные органические фазы промывали насыщенным раствором NaCl, высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (CH2Cl2/МеОН: 95/5 и затем 9/1) выделяли 470 мг ожидаемого продукта. Точка плавления: 194 С. Элементный микроанализ: Масс-спектрометрия: (ESI+, m/z): 256,1 (М+Н+); 278,1 (M+Na+). Пример 2. N-(1H-индол-1-ил)-1-метил-1,2,3,6-тетрагидропиридин-3-карбоксамид. Ожидаемый продукт получали при осуществлении очистки соединения из примера 1. 180 мг соединения получали после ресуспендирования остатка в простом диэтиловом эфире, растирания в порошок и фильтрования через стеклянную фритту. Точка плавления:117 С. Элементный микроанализ: Пример 3. N-(1H-индол-1-ил)-1-метил-1,4,5,6-тетрагидропиридин-3-карбоксамид. 1 г соединения из получения 2 растворяли в 10 мл безводного дихлорметана и затем реакционную смесь охлаждали до -20 С. Добавляли 5,5 мл 2 М раствора триметилалюминия в гексане и реакционную смесь перемешивали в течение 1 ч 30 мин, пока температура повышалась. Добавляли раствор 1,5 г соединения из получения 7 в 5 мл дихлорметана и реакционную смесь нагревали в колбе с обратным холодильником в течение 12 ч. Реакционную смесь разводили в дихлорметане и затем вливали в 20% водного раствора гидроксида натрия. Органическую фазу отделяли и промывали, первый раз с помощью 20% раствора гидроксида натрия и затем насыщенным раствором NaCl. Органическую фазу высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (дихлорметан) выделяли 0,640 г ожидаемого продукта. Точка плавления: 207C. Масс-спектрометрия: (ESI+, m/z): 256 (M+H+). Пример 4. N-(2,3-Дигидро-1H-индол-1-ил)метил-1,4,5,6-тетрагидропиридин-3-карбоксамид. Стадия А. N-(2,3-Дигидро-1 Н-индол-1-ил)никотинамид. К 600 мг соединения из получения 2, растворенного в 18 мл безводного дихлорметана, добавляли,при 0 С, 108 мг DMAP и 1,9 мл триэтиламина, затем добавляли 955 мг гидрохлорида никотиноилхлорида. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды и после этого разводили в дихлорметане и затем промывали раствором карбоната калия до достижения рН 11. Водную фазу экстрагировали дихлорметаном. Объединенные органические фазы промывали насыщенным раствором NaCl, высушивали над Na2SO4, фильтровали и затем упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (CH2Cl2 и затем CH2Cl2/СН 3 ОН: 96/4 и 95/5) выделяли 600 мг ожидаемого продукта. ИК (см-1): 3224 (N-H); 3048 (=C-H); 2845 (C-H); 1656 (C=O); 1590; 1532 (C=C). Стадия В. 3-[(2,3-Дигидро-1 Н-индол-1-иламино)карбонил]-1-метилпиридиний йодид. 420 мг соединения с вышеописанной стадии А растворяли в 1,56 мл йодметана. Образовывалось коричневое масло, которое перемешивали в течение 24 ч при температуре окружающей среды, защищая от света. Избыток йодметана удаляли при пониженном давлении. Таким образом получали 670 мг ожидаемого продукта. ИК(см-1): 3224 (N-H); 3048 (=C-H); 2845 (C-H); 1656 (C=O); 1590; 1532 (C=C). Стадия С. N-(2,3-Дигидро-1 Н-индол-1-ил)-1-метил-1,4,5,6-тетрагидропиридин-3-карбоксамид. 1,1 г соединения со стадии В выше растворяли в 15 мл метанола, и затем добавляли 450 мкл триэтиламина и 100 мг PtO2. Реакционную смесь дегазировали и затем помещали в атмосферу водорода(процесс повторяли два раза). Реакционную смесь перемешивали в течение одного часа при температуре окружающей среды и после этого продували азотом. Смесь фильтровали через целит (элюируя с помощью метанола) и затем растворители упаривали при пониженном давлении. При осуществлении флэшхроматографии на силикагеле (AcOEt) выделяли 320 мг ожидаемого продукта. Точка плавления: 127C. Элементный микроанализ: Пример 5. 1-Метил-N-(2-метил-2,3-дигидро-1H-индол-1-ил)-1,2,5,6-тетрагидропиридин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 8 вместо соединения из получения 2 и ареколин гидрохлорид вместо соединения из получения 7. Точка плавления: 178 С. Элементный микроанализ: Масс-спектрометрия: (ESI+, m/z): 272 (М+H+). Пример 6. N-(5-Хлор-1H-индол-1-ил)-1-метил-1,2,5,6-тетрагидропиридин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 3 вместо соединения из получения 2, и ареколин вместо соединения из получения 7. Точка плавления: 155 С. Элементный микроанализ: Пример 7. N-(5-Фтор-1H-индол-1-ил)-1-метил-1,2,5,6-тетрагидропиридин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 9 вместо соединения из получения 2 и ареколин вместо соединения из получения 7. Точка плавления: 105 С. Элементный микроанализ:- 18014022 Пример 8. N-(5-метокси-1H-индол-1-ил)-1-метил-1,2,5,6-тетрагидропиридин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 10 вместо соединения из получения 2 и ареколин вместо соединения из получения 7. Точка плавления: 162 С. Элементный микроанализ: Пример 9. N-(2,3-диметил-1H-индол-1-ил)-1-метил-1,2,5,6-тетрагидропиридин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 5 вместо соединения из получения 2 и ареколин вместо соединения из получения 7. Точка плавления: 168 С. Элементный микроанализ: Пример 10. 1-Метил-N-(3-метил-1H-индол-1-ил)-1,2,5,6-тетрагидропиридин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 4 вместо соединения из получения 2 и ареколин вместо соединения из получения 7. Точка плавления: 152 С. Элементный микроанализ: Пример 11. N-(1H-Индол-1-ил)-1-метил-1,2,5,6-тетрагидропиридин-4-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя метил 1-метил-1,2,5,6 тетрагидропиридин-4-карбоксилат вместо соединения из получения 7. Точка плавления: 112C. Элементный микроанализ: Пример 12. 1-Аллил-N-(5-хлор-1H-индол-1-ил)пиперидин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 3 вместо соединения из получения 2 и соединение из получения 11 вместо соединения из получения 7. Точка плавления: 130C. Элементный микроанализ: Пример 13. N-(5-Хлор-1H-индол-1-ил)пиперидин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 3 вместо соединения из получения 2 и этил нипекоат вместо соединения из получения 7. Точка плавления: 167 С. Элементный микроанализ : Масс-спектрометрия: (El): 277 (М+Н+). Пример 14. 3-[(1H-Индол-1-иламино)карбонил]-1-метилпиперидиний хлорид. Стадия А. N-(1 Н-Индол-1-ил)-1-метилпиперидин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 12 вместо соединения из получения 7. Масс-спектрометрия: (ESI+, m/z): 258 (М+Н+). Стадия В. 3-[(1 Н-Индол-1-иламино)карбонил]-1-метилпиперидиний хлорид. 6 капли концентрированной HCl добавляли к раствору 228 мг соединения с вышеописанной стадии А в 6 мл безводного ТГФ. Растворитель упаривали и затем остаток промывали простым диэтиловым эфиром, получая 256 мг ожидаемого продукта. Точка плавления :250 С. Пример 15. N-(2,3-Дигидро-1H-индол-1-ил)-1-метил-1,2,5,6-тетрагидропиридин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 6 вместо соединения из получения 2 и ареколин вместо соединения из получения 7. Точка плавления: 152C. Элементный микроанализ: Пример 16. 1-Метил-N-(2-метил-2,3-дигидро-1H-индол-1-ил)пиперидин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 8 вместо соединения из получения 2 и соединение из получения 12 вместо соединения из получения 7. Точка плавления: 133 С. Элементный микроанализ: Масс-спектрометрия: (EI): 273 (M+H+). Пример 17. N-(5-хлор-1H-индол-1-ил)-1-пропилпиперидин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 13 вместо соединения из получения 7. Точка плаеления: 157 С. Элементный микроанализ:N-(5-хлор-1H-индол-1-ил)-1-(2-гидроксиэтил)-1,2,5,6-тетрагидро-3-пиридинкарбоксамид. Стадия А. 1-(2-трет-Бутил(диметил)силилоксиэтил)-N-(5-хлор-1 Н-индол-1-ил)-1,2,5,6-тетрагидро-3-пиридинкарбоксамид. 1 г соединения из получения 3 растворяли в 10 мл безводного дихлорметана и затем реакционную смесь охлаждали до -20 С. Добавляли 5,5 мл 2 М триметилалюминия в гексане и реакционную смесь перемешивали в течение 1 ч 30 мин, пока температура повышалась. Добавляли раствор 1,5 г соединения из Получения 15 в 5 мл дихлорметана и реакционную смесь нагревали в течение ночи при флегме дихлорметана. Реакционную смесь разводили в дихлорметане и после этого вливали в 20% водного раствора гидроксида натрия. Органическую фазу отделяли и после этого промывали 20% раствором гидроксида натрия и затем насыщенным раствором NaCl. Органическую фазу высушивали над сульфатом натрия,фильтровали и затем упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (AcOEt/циклогексан: 1/1 и затем 2/1) выделяли 1,5 г ожидаемого продукта. ИК(см-1): 3244 (N-H); 2953; 2928; 2856 (C-H); 1672 (C=O); 1642; 1520 (C=C). Стадия В. N-(5-Хлор-1H-индол-1-ил)-1-(2-гидроксиэтил)-1,2,5,6-тетрагидро-3-пиридин-карбоксамид. 6,72 мл 1 М Bu4NF в ТГФ добавляли к раствору 1,46 г соединения с вышеописанной стадии А в 12 мл безводного ТГФ и после этого реакционную смесь перемешивали в течение ночи при температуре окружающей среды. Смесь концентрировали при пониженном давлении и после этого ресуспендировали в дихлорметане и промывали водой и затем раствором карбоната натрия с рН 11. Водные фазы экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (AcOEt/MeOH : 95/5 и затем 9/1) выделяли 920 мг ожидаемого продукта. Точка плавления: 84C. Элементньй микроанализ:- 20014022 Стадия А. 1-[2-(Диметиламино)этил]-N-(1 Н-индол-1-ил)-1,2,5,6-тетрагидро-3-пиридинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3 и соединение из получения 16 вместо соединения из получения 15. Стадия В. 1-[2-(Диметиламино)этил]-N-(1H-индол-1-ил)-1,2,5,6-тетрагидро-3-пиридинкарбоксамид дигидрохлорид. 200 мг соединения с вышеописанной стадии А растворяли в 0,5 мл метанола, и затем добавляли 285 мкл 4,53 М метанольной соляной кислоты. Раствор погружали в простой эфир и образованный осадок отфильтровывали и затем промывали простым эфиром. 210 мг ожидаемого продукта получали путем высушивания при пониженном давлении. Точка плавления: 210 С. Элементный микроанализ: Пример 20. N-(5-хлор-1H-индол-1-ил)-1-[2-(диметиламино)этил]-1,4,5,6-тетрагидро-3-пиридинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 18 вместо соединения из получения 15. Точка плавления: 166 С. Элементный микроанализ: Масс-спектрометрия : (ESI+, m/z): 347,1; 349,1 (M+H+); 369,1; 371,1 (M+Na+). Пример 21. 1-[2-(Диметиламино)этил]-N-(1H-индол-1-ил)-1,4,5,6-тетрагидро-3-пиридинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3 и соединение из получения 18 вместо соединения из получения 15. Точка плавления: 106 С. Элементный микроанализ: Масс-спектрометрия : (ESI+, m/z): 313,1 (M+H+); 335,2 (M+Na+). Пример 22. 1-[2-(Диметиламино)этил]-N-(1H-индол-1-ил)-3-пиперидин-карбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3 и соединение из получения 19 вместо соединения из получения 15. Точка плавления: 79 С. Масс-спектрометрия : (ESI+, m/z): 315,2 (М+Н+); 337,2 (M+Na+). Пример 23. N-(1H-Индол-1-ил)-1-12-(4-морфолинил)этил]-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3 и соединение из получения 20 вместо соединения из получения 15. Точка плавления : 131 С. Элементный микроанализ: Пример 24. N-(1H-индол-1-ил)-1-[2-(1-пиперидил)этил]-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3, и соединение из получения 21 вместо соединения из получения 15. Точка плавления 113 С. ИК(см-1): 3108 (N-Н амид + =C-H), 2921, 2853 (C-H), 1690 (C=O сложный эфир). Пример 25. N-(1H-Индол-1-ил)-1-[2-(4-метил-1-пиперазинил)этил]-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3 и соединение из получения 22 вместо соединения из получения 15. Точка плавления 117 С. Элементный микроанализ: Пример 26. 1-2-[4-(2-Гидроксиэтил)-1-пиперазинил]этил-N-(1H-индол-1-ил)-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3, и соединение из получения 23 вместо соединения из получения 15. Точка плавления 118 С. Элементный микроанализ: Пример 27. N-(1H-индол-1-ил)-1-[2-(1-пирролидинил)этил]-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3, и соединение из получения 24 вместо соединения из получения 15. Точка плавления 117 С. Элементный микроанализ: Пример 28. 1-[2-(1-Азепанил)этил]-N-(1H-индол-1-ил)-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3, и соединение из получения 25 вместо соединения из получения 15. Точка плавления:250 С. Элементный микроанализ: Пример 29. N-(1H-Индол-1-ил)-1-[2-(4-фенил-1-пиперазинил)этил]-3-пиперидинкарбоксамид тригидрохлорид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3, и соединение из получения 26 вместо соединения из получения 15. 430 мг полученного остатка растворяли в 1,5 мл безводного МеОН, и затем добавляли 1,65 мл 2 М метанольного раствора HCl. Тригидрохлорид получали с помощью осаждения путем добавления 5 мл простого эфира, фильтрования и после этого высушивания в вакууме. Точка плавления:250C. Элементный микроанализ: Пример 30. N-(1H-индол-1-ил)-N-(1-[2-(1-пиперидил)этил]-3-пиперидилметил)амин тригидрохлорид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3 и соединение из получения 27 вместо соединения из получения 15. 310 мг полученного остатка растворяли в 1,5 мл безводного МеОН, и затем добавляли 1,5 мл 2 М метанольного раствора HCl. Тригидрохлорид получали с помощью осаждения путем добавления 5 мл простого эфира, фильтрования и после этого высушивания в вакууме. Точка плавления 250 С. Пример 31. N-(5-Хлор-2,3-дигидро-1H-индол-1-ил)-1-(2-гидроксиэтил)-1,4,5,6-тетрагидро-3-пиридинкарбоксамид. Стадия А. N-(5-Хлор-2,3-дигидро-1H-индол-1-ил)никотинамид. Продукт получали в соответствии с методикой со стадии А из примера 4, используя соединение из получения 28 вместо соединения из получения 2. ИК(см-1): 3265 (N-H); 3025 (=C-H); 2975; 2883 (C-H); 1652 (C=O); 1605; 1591; 1578; 1537(C=C). Стадия В. 3-[(5-Хлор-2,3-дигидро-1H-индол-1-ил)амино]карбонил-1-(2-гидроксиэтил)пиридиний бромид. Продукт получали в соответствии с методикой со стадии В из примера 4, используя соединение с вышеописанной стадии А и, используя 2-бромэтанол вместо йодметана. ИК(см-1): 3349 (N-H, O-H); 3052 (=C-H); 2854 (C-H); 1675 (C=O); 1634; 1606; 1543 (C=C). Стадия С. N-(5-Хлор-2,3-дигидро-1H-индол-1-ил)-1-(2-гидроксиэтил)-1,4,5,6-тетрагидро-3-пиридинкарбоксамид. Продукт получали в соответствии с методикой со стадии С из примера 4, используя соединение со стадии В выше. Точка плавления:161 С. Элементный микроанализ: Пример 32. N-(2,3-Дигидро-1H-индол-1-ил)-1-[2-(диметиламино)этил]-3-пиперидинкарбоксамид дигидрохлорид. СтадияА. N-(2,3-Дигидро-1H-индол-1-ил)-1-[2-(диметиламино)этил]-3-пиперидинкарбоксамид. 3 мл трифторуксусной кислоты добавляли к 500 мг соединения из примера 22, растворенного в 3 мл безводного ТГФ. Реакционную смесь помещали в поток азота и затем по каплям добавляли раствор 1 г цианоборогидрида натрия в 10 мл ТГФ в течение 2 ч. Растворители упаривали при пониженном давлении, остаток ресуспендировали в дихлорметане и воде, смесь подщелачивали раствором карбоната калия до достижения рН 11 и затем водную фазу два раза экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. Остаток ресуспендировали в 13 мл абсолютного этанола и затем добавляли 1,5 мл 2 н. соляной кислоты. Реакционную смесь нагревали в колбе с обратным холодильником в течение 5 ч и затем упаривали при пониженном давлении. Остаток ресуспендировали в 20 мл 1/1 смеси дихлорметана и воды, водную фазу подщелачивали путем добавления карбоната натрия до достижения рН 11. Органическую фазу отделяли и затем водную фазу два раза экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и затем упаривали. При осуществлении флэшхроматографии на силикагеле (AcOEt/MeOH: 9/1 и затем от 8/2 до 6/4) выделяли 255 мг ожидаемого продукта. Стадия B. N-(2,3-дигидро-1H-индол-1-ил)-1-[2-(диметиламино)этил]-3-пиперидинкарбоксамид дигидрохлорид. 255 мг соединения с вышеописанной стадии A растворяли в 4 мл дихлорметана и затем добавляли 360 мкл 4,53 М метанольной соляной кислоты. Раствор перемешивали в течение 5 мин и затем добавляли большой избыток простого эфира: образовывался осадок, который отфильтровывали через стеклянную фритту, выделяя 270 мг ожидаемого продукта. Точка плавления 108 С. Элементный микроанализ: Пример 33. N-(5-Хлор-1H-индол-1-ил)-1-(2-гидроксиэтил)-1,4,5,6-тетрагидро-3-пиридинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 17 вместо соединения из получения 15. Точка плавления: 195C. Элементный микроанализ:- 23014022 Пример 34. 1-(2-Гидроксиэтил)-N-(1H-индол-1-ил)-1,4,5,6-тетрагидро-3-пиридинкарбоксамид. Стадия А. 1-(2-[трет-Бутил(диметил)силил]оксиметил)-N-(1H-индол-1-ил)-1,4,5,6-тетрагидро-3 пиридинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3, и соединение из получения 29 вместо соединения из получения 15. Стадия В. 1-(2-Гидроксиэтил)-N-(1H-индол-1-ил)-1,4,5,6-тетрагидро-3-пиридинкарбоксамид. Продукт получали в соответствии с методикой со стадии В из примера 18, используя соединение с вышеописанной стадии А. Точка плавления: 152 С. Элементный микроанализ: Масс-спектрометрия: (ESI +, m/z): 286 (M+H+). Пример 35: N-(Индол-1-ил)-N-[(1-метил-1,2,5,6-тетрагидропиридин-3-ил)метил]амин дигидрохлорид. Стадия А: (Z,Е)-(Индол-1-ил)[(пиридин-3-ил)метилен]амин. 2,0 г пиридин-3-карбоксальдегида растворяли в 100 мл толуола и затем добавляли 3,7 г соединения из получения 2. Смесь помещали в колбу, оборудованную устройством Дина-Старка и нагревали в колбе с обратным холодильником, при перемешивании, пока не получали теоретический объем воды (18 ч). Реакционную смесь концентрировали, используя роторный испаритель, и после этого остаток очищали путем хроматографии на силикагеле (CH2Cl2 / МеОН: 98/2), получая 3,8 г ожидаемого соединения. Точка плавления: 112 С. Стадия В. 3-[(Z,Е)-N-индол-1-ил)иминометил]-1-метилпиридиний йодид. 5 мл йодметана добавляли к раствору 2,0 г соединения с вышеописанной стадии А в 10 мл метанола. Реакционную смесь перемешивали при температуре окружающей среды в течение 24 ч. Добавляли 50 мл простого диэтилового эфира; осадок отделяли путем фильтрации и высушивали, получая 3,05 г ожидаемого соединения. Точка плавления: 218 С. Элементный микроанализ: ИК (см-1): 3120; 3038; 1633; 1593; 1475; 1450; 1297; 1251; 1158. Стадия C. (Z,Е)-(Индол-1-ил)[(1-метил-1,2,5,6-тетрагидропиридин-3-ил)метилен]амин. 522 мг NaBH4 небольшими порциями добавляли к раствору 1,0 г соединения со стадии В выше в 20 мл метанола, раствор которого охлаждали до 0 С. После добавления температуру повышали до температуры окружающей среды и затем реакционную смесь перемешивали в течение 1 ч. После этого добавляли 50 мл насыщенного раствора гидрокарбоната натрия и смесь экстрагировали 330 мл дихлорметана. Объединенные органические фазы последовательно экстрагировали раствором гидрокарбоната натрия и насыщенным раствором NaCl. После высушивания над Na2SO4 растворители удаляли в вакууме, используя роторный испаритель. При осуществлении хроматографии на силикагеле (CH2Cl2 /МеОН: 95/5) получали 0,51 г ожидаемого продукта. Элементный микроанализ: ИК (см-1): 3104; 3053; 2964; 2960; 2930; 2895; 2846; 2762; 1642; 1614; 1454; 1292; 1255. Стадия D. (Индол-1-ил)[(1-метил-1,2,5,6-тетрагидропиридин-3-ил)метил]амин дигидрохлорид. 0,4 г LiAlH4 добавляли небольшими порциями и при температуре окружающей среды к раствору 0,65 г соединения со стадии С выше в 10 мл простого диэтилового эфира. Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч и затем 10 мл воды добавляли. Реакционную смесь экстрагировали 320 мл дихлорметана; органическую фазу промывали насыщенным раствором гидрокарбоната натрия, высушивали над сульфатом натрия и фильтровали и после этого упаривали при пониженном давлении. Остаток, который очищали с помощью хроматографии на силикагеле(CH2Cl2/MeOH: 95/5 + 0,5% NEt3), позволяет получить 0,56 г основания. 100 мг основания растворяли в 0,5 мл безводного МеОН и затем добавляли 0,45 мл 2 М метанольного раствора HCl. Дигидрохлорид выпадал в осадок при добавлении 5 мл простого диэтилового эфира и его отделяли путем фильтрации и затем высушивания в вакууме. ИК(см-1): 2956; 2437; 2024; 1437; 1376; 1258. Пример 36. 1-Бензил-N-(5-хлор-1H-индол-1-ил)-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 31 вместо соединения из получения 15. Точка плавления : 197C. Элементный микроанализ: Пример 37. 3-[(5-Хлор-1H-индол-1-ил)амино]карбонил-1-метилпиперидиний гидрохлорид. Стадия А. N-(5-Хлор-1H-индол-1-ил)-1-метил-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 12 вместо соединения из получения 15. ИК (см-1): 3117, 3081, 2937, 2464, 1697. Стадия В. 3-[(5-Хлор-1H-индол-1-ил)амино]карбонил-1-метилпиперидиний гидрохлорид. 6 капли концентрированной HCl добавляли к раствору 228 мг соединения с вышеописанной стадии А в 6 мл безводного ТГФ. Растворитель упаривали и затем остаток промывали простым этиловым эфиром, получая 256 мг ожидаемого продукта. Точка плавления :250 С. Элементный микроанализ: Масс-спектрометрия: (ESI +, m/z): 292 (M+H+). Пример 38. (3R,4S)-3-(4-Фторфенил)-N-(1H-индол-1-ил)-1-метилпиперидин-4-карбоксамид. 590 мг соединения из получения 2 растворяли в 6 мл безводного дихлорметана и затем после охлаждения до -20 С добавляли 4,1 мл 2 М триметилалюминия в гексане. Реакционную смесь перемешивали в течение 1 ч 30 мин без контроля температуры. Добавляли 930 мг соединения из получения 32 в 4 мл безводного дихлорметана и затем раствор перемешивали в течение 18 ч при флегме дихлорметана. Раствор разводили в дихлорметане и затем вливали в 20% водного раствора гидроксида натрия. Органическую фазу отделяли и после этого промывали 20% водным раствором гидроксида натрия и насыщенным раствором NaCl. Органическую фазу высушивали над сульфатом натрия, фильтровали и затем упаривали при пониженном давлении. Остаток очищали путем флэш-хроматографии на силикагеле (CH2Cl2/MeOH: 97/3 и затем 95/5 и 9/1). Ожидаемый продукт получали путем осаждения в присутствии простого эфира. Точка плавления: 134C. Масс-спектрометрия: (ESI +, m/z): 352,1 (M+H+). Пример 39. (3R,4S)-3-(4-Фторфенил)-N-(1H-индол-1-ил)-1-метилпиперидин-4-карбоксамид. Ожидаемый продукт получали при осуществлении очистки соединения из примера 38. Точка плавления: 205 С. Элементный микроанализ: Масс-спектрометрия: (ESI +, m/z): 352,1 (M+H+); 374,2 (M+Na+). Пример 40. трет-Бутил 4-(2-3-[(1H-индол-1-иламино)карбонил]-1-пиперидилэтил)пиперазин-1 карбоксилат. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3 и соединение из получения 33 вместо соединения из получения 15. Точка плавления: 48C. Элементный микроанализ: Пример 41. 1-[3-(Диметиламмоний)пропил]-3-[(1H-индол-1-иламино)карбонил]пиперидиний дигидрохлорид. Продукт получали в соответствии с методикой из примера 37, используя соединение из получения 2- 25014022 вместо соединения из получения 3, и соединение из получения 34 вместо соединения из получения 12. Точка плавления :250 С. Элементный микроанализ: Пример 42. N-(1H-Индол-1-ил)-1-[3-(1-пиперидил)пропил]-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3, и соединение из получения 38 вместо соединения из получения 15. Точка плавления: 95 С. Элементный микроанализ: Пример 43. N-(1H-Индол-1-ил)-1-[3-(4-метил-1-пиперазинил)пропил]-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3, и соединение из получения 39 вместо соединения из получения 15. Элементный микроанализ: Пример 44. N-(5-Хлор-1H-индол-1-ил)-1-(2-гидроксиэтил)-3-пиперидинкарбоксамид. Стадия А. 1-(2-[-Бутил(диметил)силил]оксиэтил)-N-(5-хлор-1H-индол-1-ил)-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 40 вместо соединения из получения 15. ИК(см-1): 3225 (N-H); 2930; 2856 (C-H); 1677 (C=O); 1517 (C=C). Стадия B. N-(5-Хлор-1H-индол-1-ил)-1-(2-гидроксиэтил)-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии В из примера 18, используя соединение с вышеописанной стадии А. Точка плавления: 125 С. Элементный микроанализ: Масс-спектрометрия: (ESI+, m/z): 322,1; 324,1 (M+H+); 344,1; 346,1 (M+Na+). Пример 45. 1-(3-Гидроксипропил)-N-(1H-индол-1-ил)-3-пиперидинкарбоксамид. Стадия А. 1-(3-[трет-Бутил(диметил)силил]оксипропил)-N-(1H-индол-1-ил)-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии А из примера 18, используя соединение из получения 2 вместо соединения из получения 3, и соединение из получения 41 вместо соединения из получения 15. Точка плавления: 94 С. ИК (см-1): 2951; 2927; 2855 (C-H); 1706 (C=O); 1614; 1584 (C=C). Стадия В. 1-(3-Гидроксипропил)-N-(1H-индол-1-ил)-3-пиперидинкарбоксамид. Продукт получали в соответствии с методикой со стадии В из примера 18, используя соединение с вышеописанной стадии А. Точка плавления: 51 С. Элементный микроанализ:N-(Индол-1-ил)-1-[2-(4-метилпиперазин-1-ил)этил]-1,2,5,6-тетрагидропиридин-3 карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 43 вместо соединения из получения 7. При осуществлении флэш-хроматографии на силикагеле ИК(см-1): 3054 (=C-H); 2935 (C-H); 2795 (N-CH3); 1681 (C=O); 1646; 1614 (C=C); 1521 (N-H); 1458 (5C-H); 1372 (C-N). Пример 47. N-(5-Хлориндол-1-ил)-1-[2-(4-метилпиперазин-1-ил)этил]-1,2,5,6-тетрагидропиридин-3 карбоксамид. К раствору 782 мг соединения из получения 3 в 12 мл безводного дихлорметана, предварительно охлажденному до -20 С, добавляли 4,30 мл 2 М раствора триметилалюминия в гексане. Раствор перемешивали в атмосфере азота в течение 1 ч 30 мин, позволяя температуре постепенно возвращаться до температуры окружающей среды. Затем добавляли 1,05 г раствора соединения из получения 43 в 8 мл безводного дихлорметана. Смесь нагревали в колбе с обратным холодильником в атмосфере азота в течение 16 ч. К реакционной смеси, охлажденной до 0 С, добавляли 100 мл 1 М водного раствора HCl (медленное добавление в начале), и затем 250 мл воды. При первой экстракции, используя 3100 мл дихлорметана,удаляли избыток N-амино-5-хлориндола. Водную фазу подщелачивали, используя насыщенный раствор карбоната натрия до достижения рН 10, и ее экстрагировали, используя 3100 мл дихлорметана. Органическую фазу высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. При осуществлении хроматографии на силикагелевой колонке (NH4OH/MeOH/CH2Cl2: от 1/1/98 до 2/18/80), с последующей перекристаллизацией из 15 мл смеси uPrOH/AcOEt (1/2) получали 470 мг ожидаемого продукта. Точка плавления: 157 С. Элементный микроанализ: Масс-спектрометрия : (ES1 +, m/z) : 402 (M+1). ИК (см-1): от 2946 до 2814 ( CH); 1681 ( CO); 1650; 1531; 1465. Пример 48. N-(5-Метоксииндол-1-ил)-1-[2-метилпиперазин-1-ил)этил]-1,2,5,6-тетрагидропиридин 3-карбоксамид. Продукт получали в соответствии с методикой из примера 47, используя соединение из получения 10 вместо соединения из получения 3. При осуществлении хроматографии на силикагелевой колонке(NH4OH/MeOH/CH2Cl2: от 1/1/98 до 2/18/80) с последующим растиранием в порошок в 15 мл простого диэтилового эфира в течение 15 мин и затем кристаллизации из 10 мл смеси AcOEt/Et2O (1/2) получали 820 мг ожидаемого продукта. Точка плавления: 121 С. Элементный микроанализ: Масс-спектрометрия : (ESI +, m/z) : 398 (M+1). ИК (см-1): 3216 (NH); 2938 to 2788 ( CH); 1669 ( CO); 1636; 1514; 1477. Пример 49. N-(5-Метоксииндол-1-ил)-1-[2-(4-метилпиперазин-1-ил)этил]-1,2,5,6-тетрагидропиридин-5-карбоксамид. Продукт получали при осуществлении очистки соединения из примера 48. При осуществлении перекристаллизации из 5 мл простого диизопропилового эфира получали 138 мг ожидаемого продукта. Точка плавления: 127C. Элементный микроанализ: Масс-спектрометрия : (ESI +, m/z): 398 (М+1). ИК (см-1): 3102 ( NH); 2932 и 2799 ( CH); 1684 ( CO); 1480; 1445. Пример 50. N-(5-Метилиндол-1-ил)-1-[2-(4-метилпиперазин-1-ил)этил]-1,2,5,6-тетрагидропиридин 3-карбоксамид. Продукт получали в соответствии с методикой из примера 47, используя соединение из получения 44 вместо соединения из получения 3. При осуществлении хроматографии на силикагелевой колонке(NH4OH/MeOH/CH2Cl2 : от 1/1/98 до 2/18/80) с последующим растиранием в порошок в 15 мл простом диэтиловом эфире в течение 15 мин получали 1,16 г ожидаемого продукта. Точка плавления: 140 С. Масс-спектрометрия:(ESI +, m/z) : 382 (М+1). ИК(см-1): от 2945 до 2816 ( CH); 1681 ( CO); 1651; 1533; 1466. Пример 51. N-(5-Метилиндол-1-ил)-1-[2-(4-метилпиперазин-1-ил)этил]-1,2,5,6-тетрагидропиридин 5-карбоксамид. Продукт получали при осуществлении очистки соединения из примера 50. При осуществлении перекристаллизации из 5 мл простого диизопропилового эфира получали 120 мг ожидаемого продукта. Точка плавления: 131 С. Элементный микроанализ: Масс-спектрометрия: (ESI +, m/z) : 382 (М+1). ИК(см-1): 3141 (NH); от 2934 до 2766 ( CH); 1694 ( CO); 1515; 1457. Пример 52. N-(Индол-1-ил)-1-[2-(4-(2-гидроксиэтилпиперазин-1-ил)этил]-1,2,5,6-тетрагидропиридин-3-карбоксамид. Стадия A. N-(Индол-1-ил)-1-[2-[4-[2-(трет-бутилдиметилсиланокси)этил]пиперазин-1-ил]этил]1,2,5,6-тетрагидропиридин-3-карбоксамид. К раствору 200 мг соединения из получения 2 в 5 мл безводного дихлорметана после охлаждения до-20 С добавляли 1,35 мл 2 М раствора триметилалюминия в гексане. Раствор перемешивали в течение 1 ч 30 мин без контроля температуры и затем добавляли 500 мг соединения из получения 45 в 3 мл безводного дихлорметана при температуре окружающей среды. Смесь нагревали в колбе с обратном холодильником в течение 16 ч и после охлаждения разводили в дихлорметане и ее вливали в 20% водного раствора гидроксида натрия. Органическую фазу собирали и водную фазу экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (CH2Cl2/CH3OH : 9/1,затем 8/2) получали 400 мг ожидаемого продукта. ИК (СМ-1): 3244 (N-H); 2949 ; 2929; 2855 (C-H); 2811 (N-CH2); 1674 (C=O); 1644 (C=C),1521; 1505 (N-H); 1460 (5C-H),1360 (C-N); 1256; 1223 (C-O). Стадия В. N-(Индол-1-ил)-1-[2-[4-(2-гидроксиэтил)пиперазин-1-ил]этил]-1,2,5,6-тетрагидропиридин-3-карбоксамид. К раствору 300 мг соединения с вышеописанной стадии А в 4 мл безводного ТГФ добавляли 1,17 мл 1 М раствор фторида тетрабутиламмония в ТГФ. Смесь перемешивали в течение 12 ч при температуре окружающей среды и затем раствор концентрировали при пониженном давлении. После ресуспендирования остатка в дихлорметане органическую фазу экстрагировали с помощью водного раствора гидроксида натрия; водную фазу экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и упаривали при пониженном давлении. При осуществлении флэш-хроматографии на силикагеле (AcOEt/MeOH: 7/3 + 0,1% триэтиламин) получали 180 мг ожидаемого продукта. Точка плавления: 113C. Элементный микроанализ:(5C-H); 1353 (C-N); 1270 ; 1223 (C-O). Пример 53. N-(Индол-1-ил)-1-[(2-пиперидин-1-ил)этил]-1,2,5,6-тетрагидропиридин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 46 вместо соединения из получения 7. При осуществлении флэш-хроматографии на силикагеле (CH2Cl2/CH3OH: 9/1, затем 8/2) выделяли 240 мг ожидаемого продукта. Точка плавления: 65 С. Элементный микроанализ:(C-H); 1370 (C-N). Пример 54. N-(Индол-1-ил)-1-[2-[3-(этоксикарбонил)-1,2,5,6-тетрагидропиридин-1-ил]этил]-1,2,5,6 тетрагидропиридин-3-карбоксамид. Продукт получали в соответствии с методикой из примера 3, используя соединение из получения 47 вместо соединения из получения 7. При осуществлении флэш-хроматографии на силикагеле (AcOEt, затем AcOEt/CHaOH: 95/5) выделяли 180 мг ожидаемого продукта. Точка плавления: 82 С. Элементный микроанализ: ИК(см-1): 3265 (N-H); 2948; 2915; 2802 (C-H); 1708; 1673 (C=O); 1646 ; 1614 ; 1519 (C=C); 1459; 1436 (8C-H); 1371; 1351 (vC-N); 1261; 1223; 1194 (C-O). Пример 55. -N-(Индол-1-ил)-1-[2-[4-[(тетрагидрофуран-2-ил)метил]пиперазин-1-ил]этил]пиперидин-3-карбоксамидил тригидрохлорид. 0,49 г соединения из получения 48 растворяли в 9 мл безводного дихлорметана. Раствор охлаждали до -20 С и затем добавляли 3,5 мл 2 М раствора триметилалюминия в гексане в атмосфере аргона. После доведения температуры реакционной смеси обратно до 0 С в течение 1 ч 30 мин, добавляли раствор 1,1 г соединения из получения 48 в 6 мл безводного дихлорметана. После этого реакционную смесь нагревали в колбе с обратным холодильником в течение 18 ч. После охлаждения добавляли 50 мл дихлорметана; затем 20% (мас./об.) водного раствора гидроксида натрия по каплям добавляли до тех пор, пока больше не выделялось метана. Органическую фазу отделяли последовательно промывали 20% (мас./об.) водным раствором гидроксида натрия и насыщенным раствором NaCl, затем высушивали над Na2SO4 и концентрировали. При осуществлении хроматографии на силикагеле (CH2Cl2/МеОН: 9/1 + 0,5% NEt3) получали 0,46 г ожидаемого амида. 150 мг амида растворяли в 1,0 мл безводного МеОН; 0,6 мл 2 М метанольного раствора HCl добавляли к раствор. Ожидаемый продукт осаждали путем добавления 5 мл простого диэтилового эфира; после этого его отделяли путем фильтрации и высушивали в вакууме. Точка плавления: 250 С. Элементный микроанализ: ИК(см-1): 3385; 2975; 2411; 1694; 1531; 1458; 1325. Пример 56. Этил -N-(индол-1-ил)-1-[2-[4-[2-(диметиламино)этил]пиперазин-1-ил]этил]пиперидин-3-карбоксилат тетрагидрохлорид. Продукт получали в соответствии с методикой из примера 55, используя соединение из получения 49 вместо соединения из получения 48. При осуществлении хроматографии на силикагеле(CH2Cl2/МеОН: от 9/1 до 7/3 + 0,5% NEt3) получали 0,59 г ожидаемого амида. 130 мг амида растворяли в 0,5 мл безводного МеОН; к раствору добавляли 0,7 мл 2 М метанольного раствора HCl. Ожидаемый продукт получали путем осаждения при добавлении 5 мл простого диэтилового эфира; его отделяли путем фильтрации и высушивали в вакууме. Точка плавления: 250 С. Элементный микроанализ: ИК(СМ-1): 2938; 2815; 1689; 1460; 1396; 1315. Пример 57. -N-(Индол-1-ил)-1-[2-[4-(2-метоксиэтил)пиперазин-1-ил)]этил]пиперидин-3-карбоксамид тригидрохлорид. Продукт получали в соответствии с методикой из примера 55, используя соединение из получения 50 вместо соединения из получения 48. При осуществлении хроматографии на силикагеле (CH2Cl2/МеОН: 90/10) получали 0,22 г ожидаемого амида. 200 мг амида растворяли в 1,0 мл безводного МеОН; 0,8 мл 2 М метанольного раствора HCl добавляли к раствор. Ожидаемый продукт получали путем осаждения при добавлении 5 мл простого диэтилового эфира; его отделяли путем фильтрации и высушивали в вакууме. Точка плавления: 250 С. Элементный микроанализ:
МПК / Метки
МПК: C07D 401/12, A61P 25/00, A61K 31/4439, A61K 31/454
Метки: фармацевтические, содержат, композиции, соединения, 1h-индол-пиридинкарбоксамида, которые, способ, получения, 1н-индол-пиперидинкарбоксамида
Код ссылки
<a href="https://eas.patents.su/30-14022-soedineniya-1h-indol-piridinkarboksamida-i-1n-indol-piperidinkarboksamida-sposob-ih-polucheniya-i-farmacevticheskie-kompozicii-kotorye-ih-soderzhat.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения 1h-индол-пиридинкарбоксамида и 1н-индол-пиперидинкарбоксамида, способ их получения и фармацевтические композиции, которые их содержат</a>
Замещённые соединения [1,4]бенздиоксино[2,3-e]изоиндола, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 8498
Опубликовано: 29.06.2007
Авторы: Лепифр Франк, Хикман Джон, Кудер Жерар, Леонс Стефан, Пьерр Ален, Ренар Пьер, Эрб Натали, Сеньар Даньель-Анри, Рутье Сильвен
МПК: A61K 31/5365, C07D 487/04, A61P 35/00...
Метки: 1,4]бенздиоксино[2,3-e]изоиндола, получения, которые, фармацевтические, композиции, соединения, замещённые, способ, содержат
Формула / Реферат:
1. Соединения формулы (I) в которой А вместе с атомами углерода, с которыми он связан, представляет собой группу формулы (а) или (b) в которой W1 вместе с атомами углерода, с которыми он связан, представляет собой фенильную группу или пиридильную группу, Z представляет собой группу, выбранную из атомов водорода и галогена и таких групп, как линейный или разветвленный C1-C6-алкил, нитро, циано, гидрокси, линейный или разветвленный...
Соединения бензотиазина и бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 7492
Опубликовано: 27.10.2006
Авторы: Лестаж Пьер, Корди Алекс, Десо Патрис
МПК: A61K 31/5415, A61K 31/549, A61P 25/00...
Метки: способ, фармацевтические, содержат, получения, соединения, которые, композиции, бензотиазина, бензотиадиазина
Формула / Реферат:
1. Соединения формулы (I) где R1 представляет собой арильную или гетероарильную группу, R2 представляет собой атом водорода, атом галогена или гидроксильную группу, X представляет собой атом кислорода или атом серы, Y представляет собой атом кислорода, атом серы или группу NR, где R представляет собой атом водорода или линейную или разветвленную C1-С6-алкильную группу, А представляет собой NR4-группу, R3 представляет собой атом водорода,...
Новые соединения тиадиазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 9240
Опубликовано: 28.12.2007
Авторы: Бовери Стефан, Ренар Пьер, Кенар Даньель-Анри, Де Тюллио Паскаль, Грэндорж Эмманюэль, Пиротт Бернар, Данобер Лоранс, Лестаж Пьер, Франкотт Пьер
МПК: A61K 31/549, C07D 513/04
Метки: соединения, содержат, получения, способ, фармацевтические, тиадиазина, новые, которые, композиции
Формула / Реферат:
1. Соединения тиадиазина формулы в которой A вместе с двумя атомами углерода, к которым он присоединен, образует тиенильную группу, выбранную из групп A1, A2, A3 где X представляет собой атом серы, Ra и Rb, которые могут быть одинаковыми или разными, каждый независимо друг от друга, представляет собой атом водорода, линейную или разветвленную C1-C6алкильную группу или атом галогена, представляет собой простую или двойную связь, R1...
Соединения бензотиазина и бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 7227
Опубликовано: 25.08.2006
Авторы: Лестаж Пьер, Корди Алекс, Десо Патрис
МПК: A61P 25/00, A61K 31/5415, A61K 31/549...
Метки: которые, получения, содержат, соединения, композиции, бензотиазина, способ, бензотиадиазина, фармацевтические
Формула / Реферат:
1. Соединения формулы (I) где R1 представляет собой арильную или гетероарильную группу, R2 представляет собой атом водорода, атом галогена или гидроксильную группу, А представляет собой CR4R5 группу или NR4 группу, R3 представляет собой атом водорода, линейную или разветвленную С1-С6-алкильную группу или С3-С7-циклоалкильную группу, R4 представляет собой атом водорода или линейную или разветвленную С1-С6-алкильную группу, или А представляет...
Новые соединения бензоиндолина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 7231
Опубликовано: 25.08.2006
Авторы: Лавьелль Жильбер, Гобер Ален, Мюлле Оливье, Де-Кара Бенжамен, Миллан Марк
МПК: A61P 25/18, A61K 31/496, C07D 209/00...
Метки: которые, способ, композиции, соединения, получения, фармацевтические, новые, бензоиндолина, содержат
Формула / Реферат:
1. Соединения формулы (I) в которой R1 и R2 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси, амино, алкиламино, диалкиламино или группой трифторметила, a R3 и R4 представляют собой атом водорода, или R1 и R4 представляют собой атом водорода, a R2 и R3 вместе образуют бензольное кольцо, необязательно замещенное атомом галогена или алкилом, алкокси, циано, нитро, гидрокси,...
Предыдущий патент: Способ получения высокопривитого многофункционального сополимера олефина, полученный сополимер, композиция смазочного масла и способы ее использования
Следующий патент: Производные 5-пиридинил-1-азабицикло [3.2.1] октана, их получение и их применение в терапии
Случайный патент: Промышленный способ получения высокочистого диарилкарбоната, высокочистый дифенилкарбонат и установка для получения высокочистого диарилкарбоната