Терапевтические соединения
Номер патента: 11099
Опубликовано: 30.12.2008
Авторы: Оузман Жаклин, Сейвори Эдвард, Браун Джайлз, Притчард Мартин





Формула / Реферат
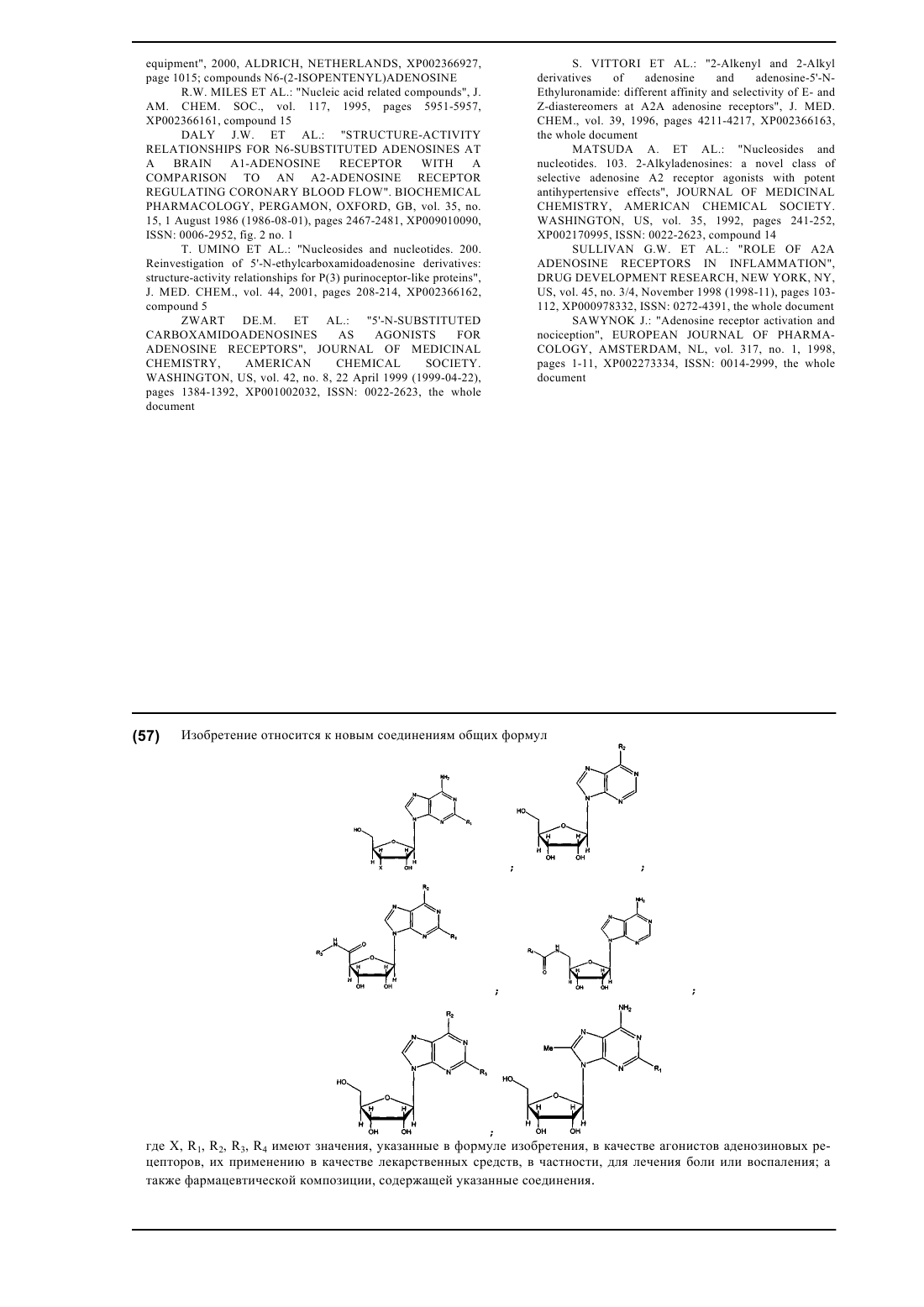
1. Соединение, имеющее формулу

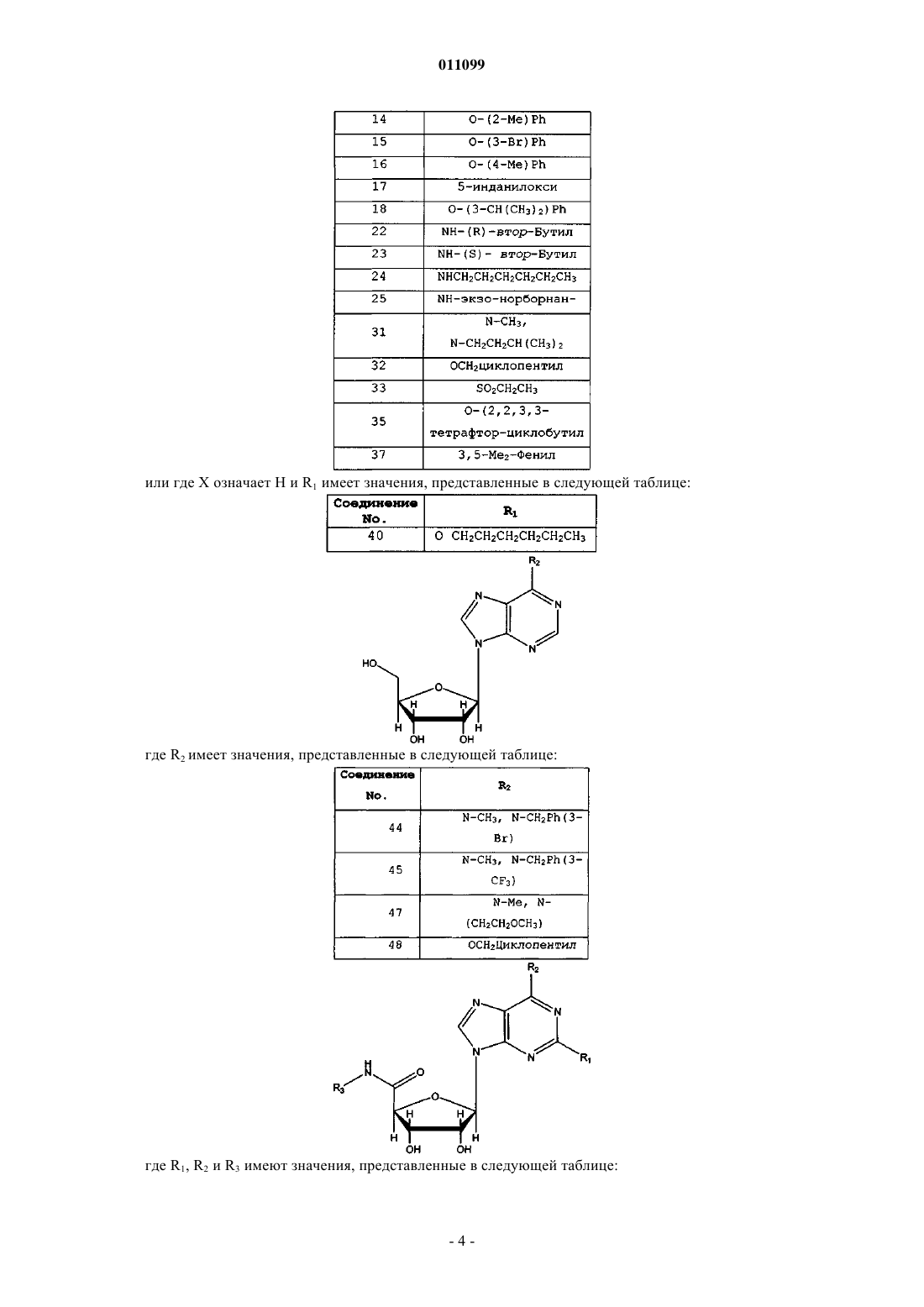
где X означает ОН и R1 имеет значения, представленные в следующей таблице:


или где X означает Н и R1 имеет значения, представленные в следующей таблице:

2. Соединение, имеющее формулу

где R2 имеет значения, представленные в следующей таблице:


3. Соединение, имеющее формулу

где R1, R2 и R3 имеют значения, представленные в следующей таблице:

4. Соединение, имеющее формулу

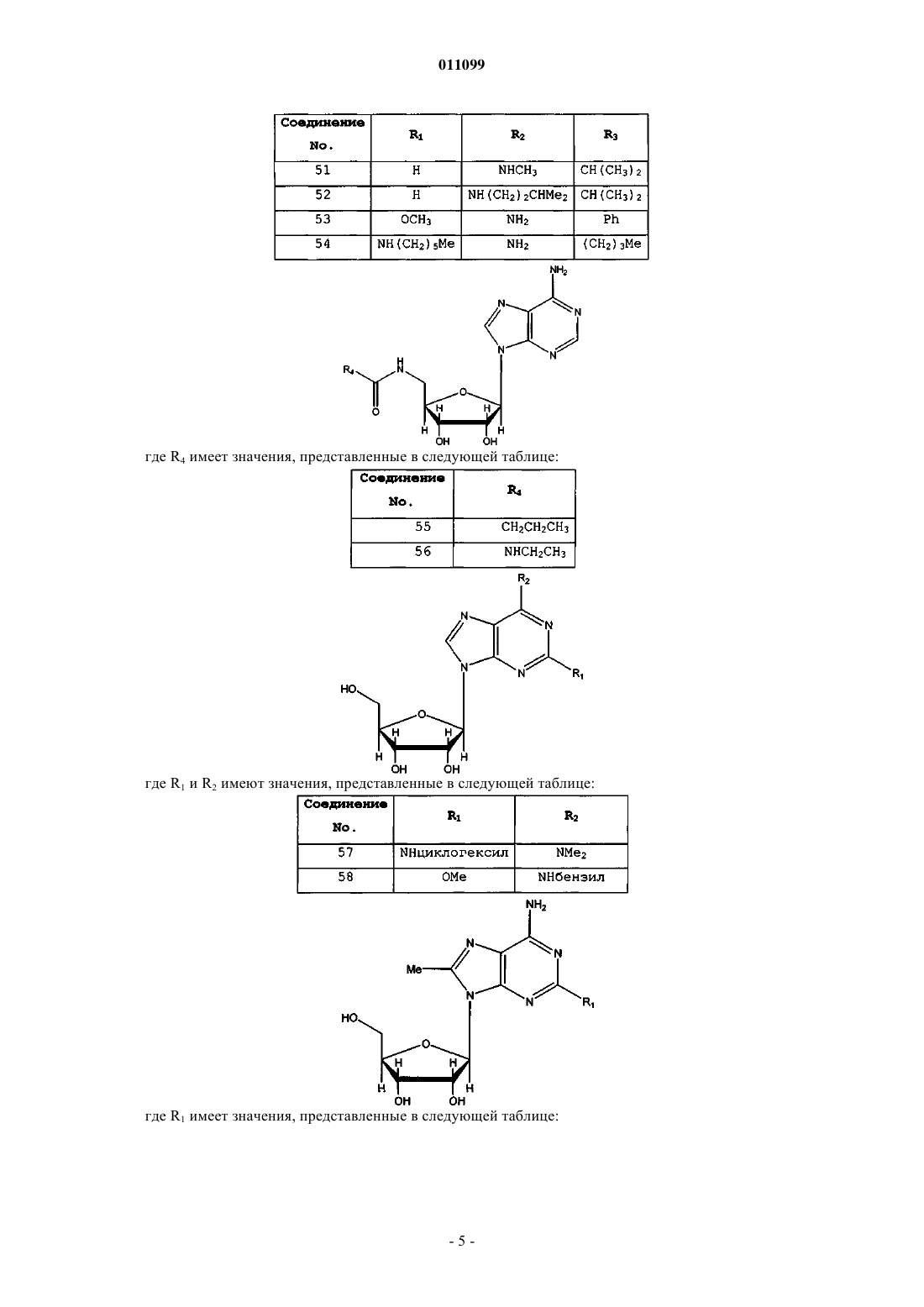
где R4 имеет значения, представленные в следующей таблице:

5. Соединение, имеющее формулу

где R1 и R2 имеют значения, представленные в следующей таблице:

6. Соединение, имеющее формулу

где R1 имеет значения, представленные в следующей таблице:

7. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в качестве лекарственного средства.
8. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в производстве лекарственного средства для профилактики, лечения или облегчения патологического состояния, которое может быть улучшено или предотвращено агонизмом в отношении аденозиновых рецепторов А2А.
9. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в производстве лекарственного средства для профилактики, лечения или облегчения боли.
10. Применение по п.9, где боль представляет собой гипералгезию.
11. Применение по п.10, где гипералгезия представляет собой невропатическую боль.
12. Применение по п.10, где гипералгезия представляет собой боль при воспалении.
13. Применение по любому из пп.9, 10 или 12, где боль вызвана или связана с воспалительным или иммунным заболеванием или является результатом комбинированного воспалительного, аутоиммунного и невропатического повреждения тканей.
14. Применение по п.9 в производстве лекарственного средства для профилактики, лечения или облегчения ишемической боли.
15. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в производстве лекарственного средства для профилактики, лечения или облегчения макро- и микрососудистых осложнений диабета 1 и 2 типа, ретинопатии, невропатии, вегетативной невропатии или повреждения кровеносных сосудов, вызванного ишемией или атеросклерозом.
16. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в производстве лекарственного средства для профилактики, лечения или облегчения воспаления.
17. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в производстве болезнь-модифицирующих (базисных) противоревматических лекарственных средств (DMARD) для замедления развития артропатии.
18. Применение по п.17 в производстве DMARD для замедления развития ревматоидного артрита.
19. Фармацевтическая композиция в единичной дозированной форме, содержащая до 500 мг соединения по любому из пп.1-6, и физиологически приемлемый носитель, эксципиент или разбавитель.
20. Фармацевтическая композиция в единичной дозированной форме, содержащая до 500 мг соединения по любому из пп.1-6 или его фармацевтически приемлемую соль в сочетании с NSAD или DMARD и физиологически приемлемый носитель, эксципиент или разбавитель.
Текст
011099 Настоящее изобретение относится к соединениям, которые являются агонистами аденозиновых рецепторов, и к их применению в качестве терапевтических соединений, в частности в качестве анальгетиков и противовоспалительных соединений или в качестве болезнь-модифицирующих (базисных) противоревматических лекарственных средств (DMARD), помимо этого изобретение относится к фармацевтическим композициям, содержащим указанные соединения. представляет собой повсеместно распространенный локальный горАденозин мон/нейротрансмиттер, который действует на четыре известных рецептора, а именно рецепторы аденозина A1, A2A, А 2 В и A3. Аденозин, в основном, служит для сохранения баланса поступления и потребности энергии в тканях. Например, в сердце высвобождение аденозина замедляет сердцебиение за счет опосредованного рецептором А 1 действия в узлах и предсердиях (Belardinelli, LIsenberg, GAm.J.Physiol. 224, H734-H737), наряду с одновременным расширением коронарной артерии для увеличения поступления энергии (т.е. глюкозы, жира и кислорода) (Knabb et al., Circ.Res. (1983) 53, 33-41). Аналогично, в процессе воспаления аденозин служит для подавления воспалительной активности, тогда как в состояниях избыточной нервной активности (например, эпилепсии) аденозин подавляет нервное возбуждение (Klitgaard et al., Eur J.Pharmacol. (1993) 242, 221-228). Эта система или ее разновидность присутствует во всех тканях. Сам по себе аденозин может применяться для диагностирования и лечения наджелудочковой тахикардии. Известно, что агонисты аденозинового рецептора А 1 действуют как мощные анальгетики(Sawynok, J. Eur J Pharmacol. (1998) 347, 1-11), также известно, что агонисты аденозинового рецептора А 2 А обладают противовоспалительной активностью (см., например, US 5877180 и WO 99/34804). Было показано, что у подопытных животных агонисты рецептора А 2 А оказываются эффективными против широкого круга состояний, включая сепсис, артрит, а также ишемию/реперфузию, вызванную закупоркой почечной, коронарной или церебральной артерии. При данных состояниях общим фактором является уменьшение воспалительной реакции, вызванной ингибирующим действием этого рецептора на большинство, если не на все нервные клетки. Однако повсеместная распространенность аденозиновых рецепторов означает, что введение агонистов аденозиновых рецепторов вызывает неблагоприятные побочные эффекты. Это, в основном, препятствовало развитию способов лечения, основанных на аденозине. Селективные агонисты рецептора А 1 вызывают брадикардию. Агонисты рецептора А 2 А вызывают системную вазодилатацию с последующей гипотензией и тахикардией. Первый селективный агонист рецептора А 2 А (2-[4-(2 карбоксиэтил)фенилэтиламино]-5'-N-этилкарбоксамидоаденозин или CGS21680) был изучен в фазе 2 А клинического испытания в качестве потенциального противогипертензивного средства. Однако введение этого соединения вызывало значительное падение кровяного давления и последующее увеличение минутного объема сердца. Это помешало применению CGS21680 в качестве лекарственного средства. Webb и сотр. (J.Pharmacol Exp Ther (1991) 259, 1203-1212), Casati и сотр. (J Pharmacol Exp Ther (1995) 275(2):914-919) и Bonnizone и сотр. (Hypertension. (1995) 25, 564-9) показывают, что селективные агонисты аденозинового рецептора А 2 А вызывают гипотензию и тахикардию. Степень вызванной тахикардии достаточна для того, чтобы воспрепятствовать их применению в качестве лекарственных средств. Alberti и сотр. (J Cardiovasc Pharmacol. 1997 Sep; 30 (3):320-4) раскрывают, что селективные агонисты аденозинового рецептора А 2 А являются мощными сосудорасширяющими средствами, которые уменьшают давление крови и вызывают заметный прирост частоты сердцебиений, а также активности ренина в плазме. Эти побочные эффекты препятствуют их применению в качестве лекарственных средств.US 5877180 относится к агонистам аденозинового рецептора А 2 А, которые, как установлено, являются эффективными при лечении воспалительных заболеваний. Было выяснено, что предпочтительные агонисты, а именно WRC0090 и SHA 211 (WRC0474), являются более мощными и селективными, чем ранее описанные аналоги аденозина, такие как CGS21680 и CV1808. Введение SHA 211 или WRC0090,как полагают, уменьшает вероятность побочных эффектов, опосредованных связыванием аналогов с другими аденозиновыми рецепторами. Однако в изобретение включены только данные in vitro, относящиеся к активности SHA 211. Не показано, что какое-либо из описанных соединений могло бы быть терапевтически эффективно in vivo без появления серьезных побочных эффектов. Хотя побочные эффекты,опосредованные связыванием мощных и селективных агонистов аденозинового рецептора А 2 А с другими аденозиновыми рецепторами, как ожидается, будут уменьшены за счет применения таких агонистов,повсеместное распространение аденозиновых рецепторов означает, что можно было бы ожидать, что эти соединения активируют аденозиновые рецепторы А 2 А в нормальной ткани и, следовательно, вызовут серьезные побочные эффекты (как, например, гипотензию и рефлекторную тахикардию).US 3936439 раскрывает применение производных 2,6-диаминонебуларина в качестве средств для расширения коронарных сосудов и/или ингибирования агрегации тромбоцитов у млекопитающих. Приведены данные in vivo у собак для подтверждения коронарного сосудорасширяющего действия N2 фенил-2,6-диаминонебуларина,N2-циклогексил-2,6-диаминонебуларина,N2-(п-метоксифенил)-2,62 диаминонебуларина и N -этил-2,6-диаминонебуларина, кроме того данные in vitro подтверждают ингибирующее действие N2-фенил-2,6-диаминонебуларина, N2-циклогексил-2,6-диаминонебуларина, 2,6 диаминонебуларина и N2-этил-2,6-диаминонебуларина в отношении агрегации тромбоцитов. FR 2162128(Takeda Chemical Industries, LTD) раскрывает, что производные аденозина (включая производные 2 алкоксиаденозина, содержащие низшую алкильную группу с не менее, чем двумя атомами углерода) обладают гипотензивной и коронарной сосудорасширяющей активностью. Данные in vivo у собак подтверждают коронарную сосудорасширяющую активность 2-н-пентилоксиаденозина,2-(гидроксиэтокси)аденозина и 2-феноксиаденозина. Однако в US 3936439 или FR 2162128 отсутствует подтверждение того, что какое-либо из описанных соединений могло бы применяться, не вызывая серьезных побочных эффектов. Статья Ribeiro и сотр. (Progress in Neurobiology 68 (2003) 377-392) является обзором аденозиновых рецепторов в нервной системе. В заключительных комментариях этой статьи указано (на стр. 387, правая колонка, строки 4-10 раздела 8), что как уже давно замечено, активация аденозиновых рецепторов на периферии связана с гипотензией, брадикардией и гипотермией Эти побочные эффекты до сих пор значительно ограничивали клиническую применимость агонистов аденозиновых рецепторов. Следовательно, имеется необходимость разработки агонистов аденозинового рецептора, которые могут применяться с минимальными побочными эффектами. Некоторые аспекты настоящего изобретения относятся к лечению боли. Боль имеет два компонента, причем каждый включает активацию сенсорных нейронов. Первый компонент представляет собой первоначальную или немедленную фазу, когда сенсорный нейрон возбуждается, например, в результате действия тепла или давления на кожу. Второй компонент является следствием повышенной чувствительности сенсорных механизмов, передающих нервные импульсы в ткань, которая до того была повреждена. Этот второй компонент называют гипералгезией, и он вовлечен во все формы хронической боли, возникающей при повреждении тканей, но не в первоначальную или немедленную фазу ощущения боли. Таким образом, гипералгезия представляет собой состояние повышенного ощущения боли, вызванное повреждением ткани. Это состояние является естественной реакцией нервной системы, вероятно предназначенной для стимулирования защиты поврежденной ткани травмированного индивидуума, чтобы дать время на восстановление поврежденной ткани. Существует две основных известных причины такого состояния, а именно увеличение активности сенсорных нейронов и изменение в процессинге нейронами болевой информации, которое имеет место в спинном мозге. Гипералгезия может подрывать силы в состояниях хронического воспаления (например, при ревматоидном артрите) и в случаях, когда имеет место повреждение сенсорного нерва (т.е. невропатическая боль). Известно два основных класса анальгетиков: (i) нестероидные противовоспалительные лекарственные средства (NSAID) и родственные ингибиторы СОХ-2 и (ii) опиаты на основе морфина. Анальгетики обоих классов эффективны при регулировании обычной, немедленной или ноцицептивной боли. Однако они менее эффективны при некоторых типах гипералгезической боли, такой как невропатическая боль. Многие практикующие врачи неохотно назначают опиаты в больших дозах, требуемых для воздействия на невропатическую боль, из-за побочных эффектов, вызываемых введением этих соединений (таких как беспокойные состояния, тошнота и рвота), а также из-за возможности того, что пациент может попасть в зависимость от этих препаратов. NSAID обладают значительно менее сильным действием, чем опиаты,поэтому эти соединения требуются даже в больших дозах. Однако это нежелательно, т.к. эти соединения вызывают раздражение желудочно-кишечного тракта. Также существует необходимость в разработке анальгетиков, в частности антигипералгетиков, которые в достаточной степени способны регулировать болевые ощущения при невропатических и других гипералгезических синдромах, и которые не обладают серьезными побочными эффектами или не вызывают зависимости у пациентов. Спонгозин был впервые выделен из тропической морской губки Cryptotethia crypta в 1945 (Bergmann and Feeney, J.Org.Chem. (1951) 16, 981, Ibid (1956) 21, 226), причем он стал первым метоксипурином, найденным в природе. Также он известен как 2-метоксиаденозин или 9 Н-пурин-6-амин, 9Dарабинофуранозил-2-метокси. Первое описание биологической активности спонгозина было дано Bartlett и сотр. (J.Med.Chem. (1981) 24, 947-954). Спонгозин (и другие соединения) были испытаны в отношении их действия в качестве релаксантов скелетных мышц, а также гипотермического, сердечно-сосудистого и противовоспалительного действия у грызунов после перорального введения (противовоспалительную активность оценивали по ингибированию вызванного каррагенаном отека лапы крысы). Спонгозин вызывал 25% ингибирование вызванного каррагенаном отека у крыс при дозировке 20 мг/кг перорально. Однако кроме этого при введении описываемого соединения в указанной дозе наблюдалось снижение среднего кровяного давления (41%) и частоты сердцебиений (25%). Было определено сродство спонгозина к аденозиновым рецепторам крыс А 1 и А 2 А. Полученные (у крыс) значения Kd составляли 340 нМ для рецептора А 1 и 1,4 мкМ для рецептора А 2 А, в то время как величина ЕС 50 для стимулирования рецептора крысы А 2 А, как было показано, составляет 3 мкМ (Daly etal., Pharmacol. (1993) 46, 91-100). У морских свинок эффективность спонгозина испытывали на препаратах изолированного сердца, и полученные значения ЕС 50 составляли 10 мкМ и 0,7 мкМ для аденозиновых рецепторов А 1 и А 2 А соответственно (Ueeda et al. J Med Chem (1991) 34, 1334-1339). Из-за низкой эффективности этого соединения и его плохой селективности к рецепторам его почти совершенно не-2 011099 принимали во внимание, отдавая предпочтение более эффективным и селективным к рецепторам агонистам аденозиновых рецепторов. Неожиданно было обнаружено, что спонгозин является эффективным анальгетиком при дозах уже в сто раз более низких, чем те, которые, могли бы потребоваться, с учетом известной аффинности этого соединения к аденозиновым рецепторам. При этих дозах спонгозин не вызывает значительных побочных эффектов, связанных с более высокими дозами этого соединения или других агонистов аденозиновых рецепторов. Таким образом, терапевтический эффект спонгозина может быть отделен от его побочных эффектов. Активность спонгозина в качестве анальгетика является предметом Международной патентной заявкиPCT/GB03/05379, и активность соединений, родственных спонгозину, в качестве анальгетиков является предметом Международной патентной заявкиPCT/GB04/00935. Применение спонгозина и родственных соединений для лечения воспаления и других расстройств является предметом международной патентной заявкиPCT/GB04/00952. Заявитель обнаружил, что спонгозин и родственные соединения, описанные в PCT/GB04/00935 иPCT/GB04/00952, обладают повышенной аффиностью к аденозиновым рецепторам при значениях pH ниже 7,4. Полагают, что это свойство объясняет неожиданную активность этих соединений при низких дозах. Заявитель смог идентифицировать некоторые другие соединения, которые также обладают повышенной аффинностью к аденозиновым рецепторам при пониженном pH. Ожидается, что эти соединения могут применяться в качестве лекарственных средств без появления серьезных побочных эффектов. В соответствии с настоящим изобретением разработаны новые агонисты аденозинового рецептора следующих формул или их фармацевтически приемлемые соли. Предпочтительными соединениями по настоящему изобретению являются соединения 2, 3, 7-18,22-25, 31-33, 35, 37, 40, 44, 45, 47, 48 или 51-61, как определено в примерах 1-6 ниже по тексту, или их фармацевтически приемлемые соли. Синтез этих соединений описан ниже в примерах 14-30. Кроме того, в настоящем изобретении разработаны способы синтеза соединений 2, 3, 7-18, 22-25,31-33, 35, 37, 40, 44, 45, 47, 48 или 51-61, как указано ниже в формуле изобретения. В некоторых случаях предшественники этих соединений включают одну или несколько защитных групп. Следует принять во внимание, что, если это желательно, для защиты гидроксила вместо указанных могут применяться другие карбоксисодержащие группы. Все соединения по настоящему изобретению, как считают, обладают повышенной аффинностью к аденозиновым рецепторам при значениях pH ниже 7,4. В нормальных тканях млекопитающего значениеpH внеклеточной среды жестко регулируется от 7,35 до 7,45. Некоторые ткани отличаются более низкими значениями pH, в частности полость желудка (pH от 2 до 3) и поверхности некоторых эпителиальных тканей (например, pH поверхности легких составляет приблизительно 6,8). В патологических тканях,например, во время воспаления, ишемии или при поражениях других типов, наблюдается уменьшениеpH. Из-за повышенной аффинности соединений по настоящему изобретению к аденозиновым рецепторам при пониженном pH, полагают, что действие этих соединений может быть нацелено на области с низким pH, такие как патологические ткани. Следовательно, дозы этих соединений, требуемые для достижения терапевтических результатов, являются существенно более низкими, чем можно было бы ожидать, исходя из их аффинности к аденозиновым рецепторам при нормальных физиологических внеклеточных значениях pH. Поскольку требуются лишь незначительные дозы соединений, удается избежать серьезных побочных эффектов, связанных с введением агонистов аденозиновых рецепторов, или свести эти эффекты к минимуму. Это имеет неожиданное следствие (вопреки принятым в технике идеям, например, в US 5877180), состоящее в том, что некоторые агонисты аденозиновых рецепторов, которые обладают низкой аффинностью и/или являются неселективными агонистами при физиологических значениях pH (как, например, спонгозин), могут быть терапевтически эффективными без проявления серьезных побочных эффектов. Ранее уже было обнаружено, что некоторые соединения, охватываемые вышеприведенными формулами, являются агонистами аденозиновых рецепторов. Однако не принималось во внимание, что действие данных соединений могло бы быть направлено на патологические ткани или, следовательно, их терапевтические эффекты могли бы быть отделены от их побочных эффектов. С точки зрения данных настоящего изобретения считается, что соединения вышеприведенных формул можно вводить в существенно более низких дозах, чем те, которых можно было бы ожидать на основании их аффинности к аденозиновым рецепторам при pH 7,4, и при этих дозах они вызывают терапевтические эффекты без проявления серьезных побочных эффектов. Таким образом, в соответствии с настоящим изобретением разработано соединение по настоящему изобретению для применения в качестве лекарственного средства. Считается, что заявленные соединения вышеприведенных формул обладают анальгетической и/или противовоспалительной активностью и их можно вводить с уменьшенной вероятностью и тяжестью побочных эффектов по сравнению с другими агонистами аденозиновых рецепторов. В соответствии с настоящим изобретением разработано применение заявленных соединений в производстве лекарственных средств для профилактики, лечения или облегчения боли, в частности гипералгезии. Предпочтительные соединения вышеприведенных формул подробно описаны в примерах. Соединения настоящего изобретения, как полагают, являются эффективными при подавлении ощущения боли у млекопитающих, страдающих от боли, в частности невропатической или воспалительной боли, даже при введении в дозах, которые, как ожидается, дают концентрацию в плазме существенно ниже той, о которой известно, что она активирует аденозиновые рецепторы. Следовательно, считается,что соединения вышеприведенных формул могут лечить боль (в частности, невропатическую и воспалительную боль), не вызывая значительных побочных эффектов, связанных с введением других агонистов аденозиновых рецепторов. Как упоминалось выше, гипералгезия в большинстве случаев является следствием повреждения тканей, либо непосредственно повреждения сенсорного нерва, либо ткани, иннервированной данным-6 011099 сенсорным нервом. Следовательно, существует много состояний, при которых ощущение боли включает компонент гипералгезии. В соответствии с настоящим изобретением разработано применение соединений вышеприведенных формул в качестве анальгетиков (в частности, антигипералгетиков) для профилактики, лечения или облегчения боли (в частности, гипералгезии), вызванной невропатией, включая диабетическую невропатию, полиневропатией, раковых болей, болей при фибромиалгии, миофасциальном болевом синдроме,остеоартрите, панкреатической боли, боли в тазовой области/промежности, болей при постгерпетической невралгии, ревматоидном артрите, ишиасе/поясничной радикулопатии, спинальном стенозе, расстройствах височно-нижнечелюстного сустава, болях при ВИЧ, невралгии тройничного нерва, хронической невропатической боли, боли в нижнем отделе спины, боли при неудачной хирургии спины, боли спины,послеоперационной боли, боли после физической травмы (включая огнестрельные ранения, дорожнотранспортные происшествия, ожоги), сердечные боли, боли грудной клетки, тазовые боли/PID (воспалительные тазовые боли), суставные боли (тендинит, бурсит, острый артрит), шейные боли, кишечные боли, фантомные боли конечностей, акушерские боли (роды/кесарево сечение), боли при почечных коликах, острая боль при герпетическом опоясывающем лишае, острая проникающая боль при панкреатите(раке), боль при дисменорее/эндометриозе. В рамках настоящего изобретения также предложено применение соединений вышеприведенных формул в качестве анальгетиков (в частности, антигипералгетиков) для профилактики, лечения или облегчения боли (в частности, гипералгезии), вызванной воспалительным заболеванием или комбинированным воспалительным, аутоиммунным и невропатическим повреждением тканей, включая ревматоидный артрит, остеоартрит, ревматоидный спондилит, подагрический артрит и другие артритные состояния, рак, ВИЧ, хронические воспалительные заболевания легких, силикоз, патологическое разрастание мягких тканей в легких, заболевания, связанные с резорбцией костей, реперфузионные повреждения(включая повреждения, причиненные органам, вследствие реперфузии, следующей за случаями ишемии,например, инфарктом миокарда, ударом), аутоиммунные поражения (включая рассеянный склероз, синдром Гийена-Барре, тяжелую миастению), отторжение трансплантата организмом хозяина, отторжения аллотрансплантата, лихорадку и миалгию, вызванные инфекцией, СПИД-ассоциированный комплекс(ARC), образование келоида, образование рубцовой ткани, болезнь Крона, язвенный колит и изжогу,синдром раздраженного кишечника, остеопороз, церебральную малярию и бактериальный менингит,кишечную боль, раковую боль, боль спины, фибромиалгию, послеоперационную боль. Кроме этого, было принято во внимание, что спонгозин может быть эффективным при профилактике, лечении или облегчении ишемических болей. Вероятно, что соединения, родственные спонгозину,также могут быть эффективны против ишемической боли. Также было принято во внимание, что соединения настоящего изобретения могут быть эффективны в профилактике, лечении или облегчении ишемической боли. Термин ишемическая боль использован в настоящей заявке для обозначения боли, связанной со снижением поступления крови в ту или иную часть тела. Пониженное поступление крови ограничивает поступление кислорода (гипоксия) и энергии в эту часть тела. Причиной ишемии является плохая перфузия крови в тканях, и поэтому ишемическая боль возникает при заболеваниях коронарных артерий, заболеваниях периферических артерий и состояниях, которые характеризуются недостаточным кровотоком,обычно в результате атеросклероза. Другие сосудистые расстройства также могут приводить к ишемической боли. Эти расстройства включают: гипертрофию левого желудочка, заболевания коронарных артерий, первичную артериальную гипертензию, острый гипертензивный криз, кардиомиопатию, сердечную недостаточность, переносимость физической нагрузки, хроническую сердечную недостаточность, аритмию, сердечную дисритмию, синкопе, артериосклероз, умеренную хроническую сердечную недостаточность, стенокардию, стенокардию (в варианте) Принцметала, стабильную стенокардию и стенокардию,вызванную физической нагрузкой, реокклюзию при сердечном шунтировании, перемежающуюся хромоту (облитерирующий артериосклероз), артериит, диастолическую дисфункцию и систолическую дисфункцию, атеросклероз, повреждение после ишемии/реперфузии, диабет (как типа I, так и типа II), различные виды тромбоэмболии. Кровотечения также могут приводить к ишемической боли. Кроме этого,плохая перфузия может приводить к невропатической и воспалительной боли, которая возникает из-за вызванного гипоксией повреждения нервных клеток (например, при остановке сердца или операции шунтирования, диабете или неонатальном дистрессе). Соединения настоящего изобретения, как считают, являются эффективными в профилактике, лечении или облегчении ишемической боли, даже при введении в дозах, которые, как ожидается, дают концентрации веществ в плазме значительно ниже той, которая по известным данным активирует аденозиновые рецепторы. Полагают, что в указанных дозах данные соединения не вызывают заметных побочных эффектов, связанных с введением более высоких доз спонгозина или других агонистов аденозиновых рецепторов. Далее, в рамках настоящего изобретения разработано применение соединений по настоящему изобретению (т.е. соединений вышеприведенных формул) с целью производства лекарственных средств для профилактики, лечения или облегчения воспаления.-7 011099 В частности, как считают, соединения по настоящему изобретению могут применяться для профилактики, лечения или облегчения воспаления, вызванного или связанного с: раком (таким как различные разновидности лейкемии, лимфомы, карциномы, раком толстой кишки, раком молочной железы, раком легких, раком поджелудочной железы, гепатоцеллюлярной карциномой, раком почек, меланомой, метастазами в печени, легких, молочной железе и простате и т.д.); аутоиммунными заболеваниями (такими как отторжение трансплантированных органов, красная волчанка, реакция трансплантат против хозяина, отторжение аллотрансплантатов, рассеянный склероз, ревматоидный артрит, сахарный диабет I типа, включающий разрушение панкреатических островков, ведущее к диабету и воспалительным последствиям диабета); аутоиммунными повреждениями (включая рассеянный склероз, синдром Гийена-Барре,тяжелую миастению); ожирением; сердечно-сосудистыми состояниями, связанными с плохой перфузией тканей и воспалением (как, например, атеромами, атеросклерозом, ударом, повреждением при ишемической реперфузии, перемежающейся хромотой, повреждением спинного мозга, застойной сердечной недостаточностью, васкулитом, геморрагическим шоком, спазмом сосудов, следующим за субарахноидальным кровотечением, спазмом сосудов, следующим за нарушением мозгового кровообращения, плевритом, перикардитом, сердечно-сосудистыми осложнениями диабета); ишемически-реперфузионным повреждением, ишемией и связанным воспалением, рестенозом, следующим за ангиопластикой и воспалительными аневризмами; эпилепсией, дегенерацией нервов (включая болезнь Альцгеймера), усталостью или спазмом мышц (в частности, спазмом у спортсменов), артритом (таким как ревматоидный артрит, остеоартрит, ревматоидный спондилит, подагрический артрит), фиброзом (например, легких, кожи и печени), рассеянным склерозом, сепсисом, септическим шоком, энцефалитом, инфекционным артритом,реакцией Джариша-Герксгеймера, опоясывающим лишаем, токсическим шоком, церебральной малярией,болезнью Лима, эндотоксическим шоком, грамотрицательным шоком, геморрагическим шоком, гепатитом (возникающим как от повреждения ткани, так и от вирусной инфекции), глубоким тромбозом вен,подагрой; состояниями, связанными с затруднениями дыхания (например, хронической обструктивной болезнью легких, заложенными дыхательными путями, бронхоконстрикцией, легочной вазоконстрикцией, затрудненным дыханием, хроническим воспалительным заболеванием легких, силикозом, разрастанием мягких тканей в легких, кистозным фиброзом, легочной гипертонией, легочной вазоконстрикцией,эмфиземой, бронхиальной аллергией и/или воспалением, астмой, аллергией верхних дыхательных путей,ринитом, весенним конъюктивитом и синдромом нарушения дыхания у взрослых); состояниями, связанными с воспалениями кожи (включая псориаз, экзему, язвы, контактный дерматит); состояниями, связанными с воспалением кишечника (включая болезнь Крона, язвенный колит и изжогу, синдром раздраженного кишечника, воспалительное заболевание кишечника); ВИЧ (в частности, ВИЧ-инфекция), церебральной малярией, бактериальным менингитом, усиленной TNF репликацией ВИЧ, ингибированием активности AZT и DDI под действием TNF, остеопорозом и другими заболеваниями, связанными с резорбцией костей, остеоартритом, ревматоидным артритом, бесплодием при эндометриозе, лихорадкой и миалгией вследствие инфекции, кахексией, вызванной раком, кахексией, вызванной инфекцией или злокачественной опухолью, кахексией, вызванной синдромом приобретенного иммунодефицита (СПИД),СПИД-ассоциированным комплексом (ARC), образованием келоида, образованием рубцовой ткани, неблагоприятными результатами лечения амфотерицином В, неблагоприятными результатами лечения интерлейкином-2, неблагоприятными результатами лечения ОКТ 3 или неблагоприятными результатами лечения GM-CSF, а также другими состояниями, опосредованными избыточной активностью противовоспалительных клеток (включая нейтрофилы, эозинофилы, макрофаги и Т-клетки). Незначительное хроническое воспаление, как известно, связано с ожирением (при наличии или в отсутствии резистентности к инсулину и диабета типа II) (Browning et al. (2004) Metabolism 53, 899-903,увеличенное количество маркеров воспаления в крови женщины с ожирением; Mangge et al. (2004) ExpClin Endocrinol Diabetes 112, 378-382, подростковое ожирение коррелирует с С-реактивным белком маркером воспаления в сыворотке; Maachi et al. Int J Obes Relat Metab Disord. 2004 28, 993-997, системное незначительное воспаление у тучных людей). Возможная причина этого заключается в том, что жировые клетки секретируют TNF-альфа и интерлейкины 1 и 6, которые являются провоспалительными. Соединения по настоящему изобретению, которые относятся к селективным агонистам аденозиновых рецепторов А 2 А и/или A3, являются особенно предпочтительными, благодаря тому, что такие соединения, как полагают, будут обладать сильной противовоспалительной активностью. Под селективными агонистами аденозиновых рецепторов А 2 А и/или A3 подразумеваются агонисты, которые активируют аденозиновые А 2 А и/или A3 рецепторы при более низких концентрациях (предпочтительно от одной тысячной до одной пятой), чем необходимы для активации аденозиновых рецепторов А 1. Кроме того,рецепторы А 1 обладают провоспалительной активностью, поэтому такие эффекты, как ожидается, будут сведены к минимуму для соединений, которые обладают селективностью в отношении рецепторов А 2 А и/или A3. Следует принять во внимание, что любое патологическое состояние, которое может быть предотвращено или улучшено за счет действия агонистов аденозиновых рецепторов А 2 А и/или A3, может быть предотвращено, излечено или облегчено соединениями вышеприведенных формул. В рамках настоящего изобретения разработано применение соединений вышеприведенных формул-8 011099 в производстве лекарственного средства для профилактики, лечения или улучшения патологического состояния, которое может быть улучшено или предотвращено агонизмом в отношении аденозиновых рецепторов А 2 А. Специалист в данной области техники может легко проверить, действует или нет патологическое состояние, которое предотвращают, лечат или улучшают соединениями настоящего изобретения, через аденозиновые рецепторы А 2 А и/или A3. Например, это можно сделать сравнением воздействия соединения в животной модели с патологическим состоянием в присутствии и отсутствии селективного антагониста аденозиновых рецепторов А 2 А и/или A3. Если воздействие соединения в присутствии антагониста снижено или отсутствует по сравнению с воздействием соединения в отсутствии антагониста, делается вывод, что соединение оказывает свое воздействие через аденозиновый рецептор А 2 А и/или A3. Антагонисты аденозиновых рецепторов А 2 А и/или A3 известны специалисту в данной области техники (см.,например, Ongini et al., Farmaco. 2001 Jan-Feb; 56 (1-2):87-90; Muller, Curr Top Med Chem. 2003; 3(4):44562). С другой стороны, можно использовать аденозиновый рецептор А 2 А-нокаутированную мышь (OhtaA and Sitkovsky M, Nature 2001; 414:916-20). Например, воздействие соединения на мышь, у которой имеются симптомы патологического состояния, сравнивают с воздействием на аденозин А 2 Анокаутированную мышь, у которой имеются соответствующие симптомы. Если соединение воздействует только на мышь, которая обладает аденозиновыми А 2 А рецепторами, делают вывод, что соединение оказывает свое воздействие через аденозиновые рецепторы А 2 А. Предполагается, что соединения по настоящему изобретению являются значительно более эффективными при низких дозах, чем другие агонисты аденозиновых рецепторов. Так ожидается, что соединения по настоящему изобретению могут эффективно оказывать помощь в дозах, при которых имеется пониженная вероятность и тяжесть побочных эффектов, или при которых побочные эффекты не наблюдаются. Такие соединения предоставляют большие преимущества над значительным большинством других агонистов аденозиновых рецепторов, которые обладают противовоспалительным действием лишь в тех же концентрациях, при которых наблюдаются серьезные побочные эффекты. С другой стороны или в дополнение к сказанному, соединения по настоящему изобретению имеют пониженную вероятность и тяжесть побочных эффектов по сравнению с другими агонистами аденозиновых рецепторов. Кроме этого считают, что соединения по настоящему изобретению могут быть эффективными в качестве базисных противоревматических лекарственных средств (DMARD), в частности для применения в профилактике, лечении или облегчении ревматоидного артрита и, возможно, других заболеваний суставов, таких как остеоартрит. Лекарственные препараты, применяемые для лечения ревматоидного артрита (RA) могут быть разделены на две группы: препараты, которые помогают облегчить симптомы RA; и препараты, которые помогают изменить течение болезни. Препараты, содействующие облегчению симптомов RA, включают нестероидные противовоспалительные лекарственные средства (NSAID), которые облегчают боль и уменьшают воспаление в пораженных суставах, анальгетики (такие как ацетаминофен и наркотические болеутоляющие средства), которые облегчают боль, но не замедляют развитие поражения сустава или не уменьшают воспаление, а также кортикостероиды, которые являются противовоспалительными лекарственными средствами.DMARD помогают облегчить симптомы RA (такие как опухание и болезненность суставов), но кроме этого замедляют развитие повреждения суставов, вызванное RA. Таким образом, хотя не существует средства для лечения RA, DMARD помогают замедлить развитие RA. В прошлом DMARD обычно применялись для лечения RA, после неудовлетворительного результата лечения NSAID. Однако в настоящее время в курсе лечения RA DMARD начинают использовать раньше, так как исследования показали, что раннее применение DMARD приносит существенную пользу. DMARD и NSAID часто применяют в сочетании друг с другом. Результаты клинических исследований показали, что известные DMARD замедляют развитие RA. После 6 месяцев лечения скорость разрушения кости и хряща в суставах пациентов уже начала замедляться. Через 1 год у пациентов наблюдалась очень слабое развитие повреждения сустава, и через 2 года рентгеновское исследование показало, что лишь незначительное число пациентов, принявших участие в исследовании, имели новые повреждения суставов в течение второго года лечения. Примеры известных DMARD включают сульфасалазин, пеницилламин, хлорохин, гидроксихлорохин, золото (с помощью внутримышечной инъекции или перорально в виде ауранофина), метотрексат,циклоспорин, азатиоприн, циклофосфамид, лефлуномид. Позднее были разработаны биологическиеDMARD, которые ингибируют фактор некроза опухолей альфа (TNF-альфа). Одним из примеров является Humira, которая показана для уменьшения признаков и симптомов и для замедления развития структурных повреждений у взрослых с RA от умеренного до весьма активного, которые обладают недостаточной реакцией на один или несколько DMARD. Humira представляет собой антитело против TNFальфа.-9 011099 Многие из известных DMARD вызывают серьезные побочные эффекты. Следовательно, желательно разработать новые DMARD, которые могут применяться с минимальными побочными эффектами. Приведенный ниже по тексту пример 13 демонстрирует способность спонгозина уменьшать вызванное форболовым эфиром высвобождение TNF-альфа в человеческих макрофагах U937. На этом основании предполагается, что спонгозин и родственные соединения вышеприведенных формул также обладают активностью DMARD. В настоящем изобретении разработано применение соединений вышеприведенных формул в производстве лекарственного средства для замедления развития артропатии. Кроме этого, в рамках настоящего изобретения разработан способ замедления развития артропатии,который включает введение соединений вышеприведенных формул субъекту, нуждающемуся в этом. Предпочтительно замедляется развитие RA и, в частности, повреждение сустава, вызванное RA. Соединение по настоящему изобретению может вводиться субъекту на любой стадии курса леченияRA. Соединение по настоящему изобретению могут вводиться в сочетании с одним или несколькимиNSAID или другими DMARD. Предполагается, что соединения по настоящему изобретению являются эффективными в качествеDMARD даже при введении в дозах, которые, как ожидается, дают концентрацию в плазме существенно ниже той, которая, как известно, активирует аденозиновые рецепторы. Считают, что при этих дозах соединения не вызывают заметных побочных эффектов, связанных с применением более высоких доз спонгозина или других агонистов аденозиновых рецепторов. Отдельное преимущество применения соединений по настоящему изобретению в качествеDMARD, как полагают, заключается в том, что они действуют при пероральном приеме, в отличие от антител против TNF-альфа, которые следует вводить с помощью инъекции. Кроме этого был принят во внимание тот факт, что соединения вышеприведенных формул могут быть эффективны при профилактике, лечении или облегчении макро- и микрососудистых осложнений диабета 1 и 2 типов (включая ретинопатию, невропатию, вегетативную невропатию) или повреждения кровеносных сосудов, вызванного ишемией (либо диабетической, либо иной) или атеросклерозом (либо диабетическим либо иным). В соответствии с настоящим изобретением разработано применение соединений вышеприведенных формул в производстве лекарственного средства для профилактики, лечения или облегчения макро- или микрососудистых осложнений диабета 1 или 2 типа, ретинопатии, невропатии, вегетативной невропатии или повреждения кровеносных сосудов, вызванного ишемией или атеросклерозом. Кроме того, в настоящем изобретении разработан способ профилактики, лечения или облегчения макро- или микрососудистых осложнений диабета 1 или 2 типа, ретинопатии, невропатии, вегетативной невропатии или повреждения кровеносных сосудов, вызванного ишемией или атеросклерозом, у субъекта, нуждающегося в такой профилактике, лечении или облегчении, причем способ включает введение субъекту соединений вышеприведенных формул. Полагают, что соединения вышеприведенных формул являются эффективными в профилактике, лечении или облегчении макро- и микрососудистых осложнений диабета 1 и 2 типа, включая ретинопатию,невропатию, вегетативную невропатию, а также повреждения кровеносных сосудов, вызванного ишемией или атеросклерозом (либо диабетическим, либо иным), даже при введении в дозе, которая, как ожидается, дает концентрации в плазме значительно меньше тех, о которых известно, что они активируют аденозиновые рецепторы. Вероятно, что при этих дозах соединения не вызывают значительных побочных эффектов, связанных с введением более высоких доз спонгозина или других агонистов аденозиновых рецепторов. Также считают, что соединения вышеприведенных формул являются эффективными в содействии лечению ран. В рамках настоящего изобретения разработано применение вышеприведенных соединений формул в производстве лекарственного средства для содействия лечению ран. Количество вводимых субъекту соединений настоящего изобретения предпочтительно представляет собой такое количество, которое вызывает увеличение максимальной концентрации в плазме, меньшее, чем значение ЕС 50 данного соединения для аденозиновых рецепторов (предпочтительно при pH 7,4). Следует принять во внимание, что значение ЕС 50 соединения, по-видимому, является различным для различных аденозиновых рецепторов (т.е. аденозиновых рецепторов A1, A2A, А 2 В, A3). Количество соединения, которое следует применять, должно быть рассчитано, исходя из самого низкого значения ЕС 50 данного соединения для различных рецепторов. Таким образом, предпочтительно количество соединения по настоящему изобретению, которое вводят субъекту, должно быть таким количеством, которое дает прирост максимальной концентрации в плазме, меньший, чем самое низкое из значений ЕС 50 соединения для аденозиновых рецепторов. Предпочтительно, максимальная концентрация соединения в плазме составляет от одной десятитысячной до одной второй (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или- 10011099 от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной третьей, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) от наиболее низкого значения EC50. Предпочтительно, количество вводимого соединения по настоящему изобретению дает прирост концентрации в плазме, который сохраняется в течение свыше одного часа от одной десятитысячной до одной второй (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) от самого низкого значения ЕС 50 соединения для аденозиновых рецепторов. Предпочтительно, вводимое количество дает прирост концентрации в плазме, который сохраняется свыше 1 ч, от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной сотой до одной второй, или от одной сотой до одной пятой,или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой от значения ЕС 50 соединения для аденозиновых рецепторов при pH 7,4. Во избежание сомнений, величина ЕС 50 соединения в рамках настоящей заявки определена как концентрация соединения, которая вызывает отклик рецептора равный средней величине между исходным откликом рецептора и максимальным откликом рецептора (что определяют, например, используя кривую доза-отклик). Величину ЕС 50 следует определять в стандартных условиях (уравновешенные солевые растворы,забуференные при pH 7,4). При использовании для определения ЕС 50 изолированных мембран, клеток и тканей, их следует буферировать солевым раствором при pH 7,4 (например, культуральной клеточной среды), как, например у Daly и сотр., (Pharmacol. (1993) 46, 91-100), или, предпочтительно, как у Tilburg и сотр., (J.Med.Chem. (2002) 45, 91-100). Значение ЕС 50, также можно было бы определять in vivo измерением откликов, опосредованных аденозиновыми рецепторами у нормальных здоровых животных или даже в ткани, в которую подаются питательные вещества при нормальных условиях (т.е. кровь, насыщенная кислородом, или изотоническая среда, насыщенная кислородом, также буферированные при pH 7,4) в нормальном здоровом животном. С другой стороны, количество вводимого соединения по настоящему изобретению может являться таким количеством, которое приводит к максимальной концентрации в плазме, меньшей, чем самое низкое или самое высокое значение Kd соединения для аденозиновых рецепторов (т.е. меньше, чем самое низкое или самое высокое значение Kd соединения для аденозиновых рецепторов A1, A2A, А 2 В и A3). Предпочтительно, максимальная концентрация соединения в плазме составляет от одной десятитысячной до одной второй (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной третьей, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) от самого низкого или самого высокого значения Kd. Предпочтительно, количество вводимого соединения является таким количеством, которое приводит к концентрации в плазме, не менее 1 ч сохраняющейся в пределах от одной тысячной до одной второй, или от одной тысячной до одной пятой, более предпочтительно от одной тысячной до одной двадцатой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой от значения Kd соединения для аденозиновых рецепторов. Предпочтительно, количество вводимого соединения является таким количеством, которое приводит к концентрации в плазме, сохраняющейся в течение более 1 ч, в пределах от одной десятитысячной до одной второй (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой, или от одной пятидесятой до одной трети, или от одной десятой до одной второй, или от одной десятой до одной пятой) от наиболее высокого или низкого значения Kd соединения для аденозиновых рецепторов. Значение Kd соединения для каждого рецептора должно быть определено в стандартных условиях с применением плазменных мембран в качестве источника аденозиновых рецепторов, полученных либо из тканей, либо из клеток, эндогенно экспрессирующих эти рецепторы, или из клеток, трансфицированных- 11011099 векторами ДНК, которые кодируют гены аденозиновых рецепторов. С другой стороны могут применяться препараты целых клеток, в которых используются клетки, экспрессирующие аденозиновые рецепторы. Меченные лиганды (например, радиоактивно меченные), селективные для различных рецепторов,следует использовать в буферированных (pH 7,4) солевых растворах (см., например, Tilburg et al.,J.Med.Chem. (2002), 45, 420-429) для определения аффинности к связыванию и, следовательно, Kd соединения для каждого рецептора. С другой стороны, количество вводимого соединения по настоящему изобретению, может являться таким количеством, которое составляет от одной десятитысячной до одной второй (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной трети, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) от минимального количества (или дозы) соединения, которая приводит к появлению побочных эффектов в виде брадикардии, гипотензии или тахикардии у животных тех же видов, как и субъект, которому будут вводить соединение. Предпочтительно, введенное количество дает повышение концентрации в плазме, которое более 1 ч сохраняется в пределах от одной десятитысячной до одной второй (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) от минимального количества соединения, которое вызывает развитие побочных эффектов. Предпочтительно, введенное количество дает повышение концентрации в плазме, которое сохраняется более 1 ч в пределах от одной тысячной до одной второй, или от одной тысячной до одной двадцатой, или от одной сотой или одной пятидесятой и одной второй, или одной сотой или одной пятидесятой и одной пятой от минимальной дозы, которая дает прирост побочных эффектов. С другой стороны количество вводимого соединения по настоящему изобретению может являться таким количеством, которое дает прирост концентрации в плазме, составляющий от одной десятитысячной до одной второй (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной трети, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) от минимальной концентрации соединения в плазме, которая вызывает такие побочные эффекты, как брадикардия, гипотензия или тахикардия, у животных тех же видов, что и субъект, которому будут вводить соединение. Предпочтительно, вводимое количество дает прирост концентрации в плазме, который сохраняется в течение более 1 ч, составляющий от одной десятитысячной до одной второй (или от одной десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) от минимальной концентрации соединения в плазме, которая вызывает побочные эффекты. Предпочтительно, введенное соединение дает прирост концентрации в плазме, который сохраняется более часа, в пределах от одной тысячной до одной второй, или от одной тысячной до одной двадцатой, или от одной сотой или одной пятидесятой до одной второй, или от одной сотой или одной пятидесятой до одной пятой от минимальной концентрации в плазме, которая вызывает побочные эффекты. Подходящая дозировка соединения по настоящему изобретению будет меняться с возрастом, полом, массой и состоянием подвергаемого лечению субъекта, эффективностью соединения (как, например, его значением ЕС 50 для аденозинового рецептора), временем его полужизни, его абсорбцией телом и путем введения и т.д. Однако, подходящая дозировка легко может быть определена специалистом в данной области техники. Удобным путем определения подходящей дозировки является оценка сердечно-сосудистых изменений (например, с помощью ЭКГ и наблюдения за кровяным давлением) при или вблизи от значения ЕС 50 соединения для аденозинового рецептора (предпочтительно, рецептора, к которому оно имеет наибольшую аффинность) для определения максимальной переносимой дозы. После этого, ожидается, что терапевтически эффективная доза составляет от одной десятитысячной до одной второй (или от одной- 12011099 десятитысячной до одной пятой, или от одной десятитысячной до одной двадцатой, или от одной десятитысячной до одной сотой, или от одной десятитысячной до одной тысячной, или от одной тысячной до одной второй, или от одной тысячной до одной пятой, или от одной тысячной до одной двадцатой, или от одной пятидесятой до одной десятой, или от одной сотой до одной второй, или от одной сотой до одной пятой, или от одной пятидесятой до одной второй, или от одной пятидесятой до одной трети, или от одной пятидесятой до одной пятой, или от одной десятой до одной второй, или от одной десятой до одной пятой) от максимальной переносимой дозы. Пример 31 ниже показывает, что для спонгозина доза у людей должна быть менее 28 мг. Эта доза дает увеличение концентраций в плазме от 0,5 до 0,9 мкМ (близкое к Kd для аденозиновых рецепторов А 2 А при pH 7,4 см. ниже). На основе этого результата предпочтительный диапазон дозировок для спонгозина составляет от 0,03 до 0,3 мг/кг. Минимальная концентрация спонгозина в плазме, дающая максимальное обезболивание у вспомогательной крысиной модели артрита, составляла 0,06 мкМ, т.е. значительно меньше, чем величина ЕС 50 спонгозина для аденозинового рецептора А 2 А, которая составляет приблизительно 1 мкМ. Предпочтительные уровни дозировки у человека дают максимальные концентрации в плазме от 0,005 до 0,5 мкМ,которые значительно ниже, чем те которые, как ожидалось, дают анальгетический или противовоспалительный эффект за счет действия на этот рецептор. С другой стороны, подходящие терапевтические концентрации соединений по настоящему изобретению, как ожидается, составляют приблизительно 10-20 значений Ki для аденозинового рецептора (рецептора, к которому соединение имеет наибольшую аффинность) при pH 5,5. Таким образом, для спонгозина требуется от 15 до 30 нМ, тогда как при использовании Ki при pH 7,4 можно ожидать требуемой концентрации от 20 до 30 мкМ. Ожидается, что количество вводимого соединения по настоящему изобретению должно составлять 0,001-15 мг/кг. Количество может быть менее 6 мг/кг. Количество может быть по крайней мере 0,001,0,01, 0,1 или 0,2 мг/кг. Количество может быть менее 0,1 или 0,01 мг/кг. Предпочтительные диапазоны составляют 0,001-10; 0,001-5; 0,001-2; 0,001-1; 0,001-0,1; 0,001-0,01; 0,01-15; 0,01-10; 0,01-5; 0,01-2; 0,011; 0,1-10; 0,1-5; 0,1-2; 0,1-1; 0,1-0,5; 0,1-0,4; 0,2-15; 0,2-10; 0,2-5; 0,2-2; 0,2-1,2; 0,2-1; 0,6-1,2 мг/кг. Предпочтительные дозы для человека (например, массой 70 кг) составляют менее 420 мг, предпочтительно менее 28 мг, более предпочтительно менее 21 мг и предпочтительно не менее 0,07, 0,1, 0,7 или 0,8 мг, более предпочтительно не менее 3,5 или 7 мг. Более предпочтительно 7-70 мг, 14-70 мг или 3,5-21 мг. Считают, что указанные выше дозировки значительно ниже (примерно до 1000 раз ниже), чем, как можно было бы ожидать, основываясь на значениях ЕС 50 соединений для аденозинового рецептора А 2 А,потребуются для анальгетического или противовоспалительного эффекта. Указанные выше предпочтительные дозировки предназначены для получения концентраций в плазме, которые составляют примерно от одной сотой до одной второй от значения ЕС 50 соединения для аденозинового рецептора, в отношении которого соединение имеет наивысшую аффинность. Соединение по настоящему изобретению может вводиться отдельно от других терапевтических средств или вместе с ними, например с анальгетиками, или противовоспалительными средствами (такими как опиаты, стероиды, NSAID, каннабиноиды, модуляторы тахикинина или модуляторы брадикинина) или антигипералгетиками (такими как габапентин, прегабалин, каннабиноиды, модуляторы натриевых или кальциевых каналов, противоэпилептические средства и антидепрессанты), или DMARD. В основном, соединение по настоящему изобретению может вводиться известными способами, в любой подходящей композиции, любым удобным путем. Соединения по настоящему изобретению предпочтительно вводят перорально, парентерально, сублингвально, трансдермально, интраректально или через слизистые оболочки. Другие подходящие пути включают внутривенный, внутримышечный, подкожный, ингаляцией и местный. Количество введенного лекарственного средства, как правило, будет выше при пероральном введении, чем, например, при внутривенном. Следует принять во внимание, что соединение по настоящему изобретению может вводиться совместно с физиологически приемлемым носителем, эксципиентом или разбавителем. Для поддержания терапевтически эффективной концентрации в плазме в течение продолжительного периода времени, соединения по настоящему изобретению могут быть включены в композиции с медленным высвобождением. Подходящие композиции, например, для перорального введения, включают твердые единичные дозированные формы и такие же формы, содержащие жидкость, например, для инъекции, такие как таблетки, капсулы, пузырьки и ампулы, в которых действующее начало известными способами включено в композицию с физиологически приемлемым эксципиентом, разбавителем или носителем. Подходящие разбавители и носители известны, и они включают, например, лактозу и тальк, вместе с подходящими связующими средствами и т.д. Единичная доза соединения по настоящему изобретению как правило, включает до 500 мг (например от 1 до 500 мг или (предпочтительно) от 5 до 500 мг) действующего начала. Предпочтительно, действующее начало присутствует в форме фармацевтической композиции, включающей действующее на- 13011099 чало и физиологически приемлемый носитель, эксципиент или разбавитель. Предпочтительные диапазоны дозировок (т.е. предпочтительные количества действующего начала в единичной дозе) составляют 0,001-10; 0,001-5; 0,001-2; 0,001-1; 0,001-0,1; 0,001-0,01; 0,01-15; 0,01-10; 0,01-5; 0,01-2; 0,01-1; 0,1-10; 0,1-5; 0,1-2; 0,1-1; 0,1-0,5; 0,1-0,4; 0,2-15; 0,2-10; 0,2-5; 0,2-2; 0,2-1,2; 0,2-1; 0,5-1; 0,6-1,2, как правило,около 0,2 или 0,6 мг действующего начала на кг субъекта (человека). Предпочтительные количества действующего начала составляют менее 420 мг, предпочтительно менее 28 мг, более предпочтительно менее 21 мг и предпочтительно по крайней мере 0,07, 0,1, 0,7 или 0,8 мг, более предпочтительно по крайней мере 3,5 или 7 мг. Более предпочтительно от 7 до 70 мг, или от 14 до 70 мг, от 3,5 до 21 мг, 0,07-0,7 мг или 0,7-7 мг. Считают, что на этих уровнях можно достичь эффективного лечения, в основном, без сопутствующего падения (например, не более чем 10%) кровяного давления и/или увеличения компенсирующей частоты сердцебиений. Единичная доза соединения по настоящему изобретению может дополнительно включать одно или несколько других лекарственных средств, например, анальгетиков, противовоспалительных средств, антигипералгетиков или DMARD. Предпочтительно соединение по настоящему изобретению вводят с частотой 2 или 3 раза в день. Соединения по настоящему изобретению также могут служить основой для распознавания более эффективных лекарственных средств или лекарственных средств с еще меньшими побочными эффектами. Примерами фармацевтически приемлемых солей являются аддитивные органические соли, образованные с кислотами, дающими физиологически приемлемый анион, например, тозилат, метансульфонат,малат, ацетат, цитрат, малонат, тартрат, сукцинат, бензоат, аскорбат, -кетоглутарат и -глицерофосфат. Кроме этого, можно получать подходящие неорганические соли, включая гидрохлориды, сульфаты, нитраты, бикарбонаты и карбонаты. Фармацевтически приемлемые соли могут быть получены по стандартным методикам, очень хорошо известным в технике, например взаимодействием достаточно основного соединения, такого как амин,с подходящей кислотой, дающей физиологически приемлемый анион. Также могут быть получены соли карбоновых кислот со щелочным (например, натрием, калием или литием) или щелочно-земельным металлом (например, кальцием). Варианты осуществления настоящего изобретения описаны в следующих примерах со ссылками на сопровождающие чертежи, где на фиг. 1 показано воздействие спонгозина (0,6 мг/кг, перорально) на А: кровяное давление у нормальных крыс; В: частоту сердцебиений; на фиг. 2 показано изменение концентрации в плазме в течение определенного времени после введения спонгозина; на фиг. 3 показано антигипералгетическое действие спонгозина (0,6 мг/кг, п.о.) на вызванную каррагенаном гипералгезию. А: развитие во времени (р 0,05, р 0,01 по сравнению с носителем (Sidak's),p0,05 по сравнению с BL (baseline) в течение 5 ч для спонгозина и IND(Dunnett's; В: зависимость антигипералгетического эффекта от дозы; на фиг. 4 показано антигипералгетическое действие спонгозина (0,6 мг/кг, п.о.) в модели невропатической боли, вызываемой повреждением за счет постоянного сжатия (р 0,05, р 0,01 по отношению к носителю (ANOVA Sidak's; на фиг. 5 показано действие спонгозина (0,6 мг/кг, п.о.) при наличии или в отсутствии налоксона в модели невропатической боли, вызываемой повреждением за счет постоянного сжатия; на фиг. 6 показан аддитивный эффект спонгозина и габапентина в модели невропатической боли,вызываемой повреждением за счет постоянного сжатия; на фиг. 7 показано воздействие спонгозина на вызванное LPS высвобождение TNF-альфа в клетках клеточной линии U937 человеческого макрофага; и на фиг. 8 показано, что спонгозин (62,4 и 624 мкг/кг, интраперитонеально) ингибирует вызванное каррагенаном (CGN) воспаление с эффективностью, сравнимой с индометацином (3 мг/кг, п.о.) при концентрациях, которые не оказывают влияния на кровяное давление. Структуры предпочтительных соединений по настоящему изобретению даны в примерах далее по тексту. Значения Ki даны для каждого соединения при pH 5,5 и pH 7,4. Для их расчета стриарные мембраны инкубировали в течение 90 мин при 22 С в присутствии 2 нМ [3 Н]-CGS21680, 1 Ед/мл аденозиндеаминазы и увеличивающихся концентраций исследуемых соединений с последующей фильтрацией и подсчетом сцинтилляции в жидкости. Пример 7. Фиг. 1: Спонгозин (0,624 мг/кг, п.о.) не оказывал значительного влияния на давление крови или частоту сердцебиений. Имплантируемое радиотелеметрическое устройство помещали в брюшные полости 6 крыс на группу. Катетер давления устройства помещали в брюшную аорту и два электрода вводили под кожу в положение lead II (левая сторона брюшной полости/правое плечо). Отдельных крыс помещали в индивидуальные клетки на радиодетектор (DSI) для сбора данных. А: давления крови; В: частоты сердцебиений. Пример 8. Величина ЕС 50 спонгозина для аденозиновых рецепторов (измеренная при pH 7,4) составляет 900 нг/мл (3 мкМ). Фиг. 2 показывает изменение концентрации в плазме по прошествии определенного времени после введения крысе спонгозина в количестве 0,6 мг/кг. Можно видеть, что концентрация в плазме более 3 ч остается выше 2% от величины ЕС 50. Антигипералгетические эффекты наблюдали (без изменения кровяного давления) в том случае, когда максимальная концентрация в плазме составляла от 1 до 30% от величины ЕС 50, определенной in vitro. Если максимальная концентрация в плазме достигает значения ЕС 50, наблюдаются сильные изменения кровяного давления, которые длятся не один час. Пример 9. Фиг. 3: А. Спонгозин (0,624 мг/кг, п.о.) ингибирует вызванную каррагенаном (CGN) термическую гипералгезию (CITH) с эффективностью, сравнимой с индометацином, (3 мг/кг, по). В: зависимость концентрация-реакция для спонгозина через 3 ч после введения. Каррагенан (2%, 10 мкл) вводят в правую заднюю лапу. Помещают источники тепла вблизи от обработанной и необработанной задних лап, причем на чертеже приведена разница во времени задержки отдергивания лап. Спонгозин вводят одновременно с каррагенаном. Пример 10. Фиг. 4: Спонгозин (0,624 мг/кг, п.о) подавляет термическую гипералгезию, вызванную повреждением от постоянного сдавления седалищного нерва крысы. Под анестезией вскрывают седалищный нерв на правой задней лапе и вокруг нервного пучка завязывают четыре неплотных лигатуры. Примерно через две недели на оперированных лапах крыс развивается термическая гипералгезия, которую можно оценить с помощью разницы времени задержки отдергивания лапы для правой и левой лап. Введение спонгозина уменьшает гипералгезию, что демонстрируется сокращением разницы между временами задержки отдергивания лап. Спонгозин был так же или более эффективен по сравнению с карбамазепином(CBZ, 100 мг/кг, подкожно). Пример 11. Фиг. 5: Спонгозин (1,2 мг/кг, п.о.) ингибирует статическую аллодинию, вызванную повреждением от постоянного сдавления седалищного нерва крысы, как в присутствии, так и в отсутствии налоксена (1 мг/кг, подкожно). Под анестезией вскрывают седалищный нерв на правой задней лапе и вокруг нервного пучка завязывают четыре неплотных лигатуры. Примерно через две недели у крыс развивается статическая аллодиния на оперированной лапе, которую можно оценивать по разнице пороговых величин отдергивания лапы для правой и левой лап. Введение спонгозина уменьшает гипералгезию, что демонстрируется увеличением порога отдергивания лапы (PWT) в присутствии и отсутствии налоксена. Veh: носитель.- 18011099 Пример 12. Фиг. 6: Спонгозин и габапентин ингибируют статическую аллодинию, вызванную повреждением от постоянного сдавления седалищного нерва крысы. Спонгозин и габапентин вводили (перорально) в различных соотношениях, как показано на чертеже. Суммарная введенная доза показана на горизонтальной оси и порог отдергивания лапы (PWT) на вертикальной оси. Предсказанный антигипералгезивный эффект (полученный из кривых доза-ответ для каждого средства в отдельности), в предположении, что результаты воздействия двух соединений являются аддитивными, показан символом результаты обозначены символом Очевидно, что наблюдаемые результаты незначительно отличаются от результатов, предсказанных в предположении аддитивности. Пример 13. Клетки клеточной линии человеческих макрофагов U937 выращивали в суспензии до 500 000 клеток/мл, размещенной в 48-луночном планшете, обрабатывали 20 нг/мл РМА и инкубировали в течение 8 ч. Клетки прилипали ко дну лунок, их смывали и оставляли восстанавливаться в течение 36 ч перед использованием. Планшеты преинкубировали со спонгозином в различных концентрациях и через 10 мин добавляли 100 нг/мл LPS для стимулирования выработки TNF. Через 3 ч клеточный супернатант анализировали на содержание TNF-альфа, применяя наборы для ELISA с флуоресцентной меткой. График,показывающий результаты (ингибирование выделения TNF-альфа по отношению к концентрации спонгозина), представлен на фиг. 7. Результаты демонстрируют, что спонгозин ингибирует вызванное LPS выделение TNF, и что это ингибирование чувствительно к ингибиторам аденозиновых рецепторов. Пример 14. Получение соединений 2 и 32 Схема 1 К раствору аденозина (1 экв) в пиридине добавляли бензоилхлорид (7 экв) и полученный раствор кипятили с обратным холодильником при 80 С в течение 4 ч. Растворители удаляли в вакууме, остаток растворяли в EtOAc и промывали водным раствором NaHCO3, насыщенным раствором соли и водой,затем органическую фазу высушивали над MgSO4. Кристаллизация из смеси дихлорметан (DCM)/EtOH привела к получению пентабензоиладенозина в виде белого твердого вещества. К раствору тетраметиламмонийнитрата (TMAN) (1,5 экв) в дихлорметане добавляли трифторуксусный ангидрид (TFAA) (1,5 экв) и полученный раствор перемешивали при комнатной температуре в течение 1 ч. Смесь охлаждали до 0 С и добавляли раствор пентабензоиладенозина (1 экв) в дихлорметане. Полученный раствор оставляли нагреваться до комнатной температуры на 4 ч. Затем раствор промывали водным NaHCO3, насыщенным раствором соли и водой (3 х) и органическую фазу высушивали надMgSO4. Кристаллизация из смеси дихлорметан/EtOH позволила получить пентабензоил-2-нитроаденозин в виде бледно-желтого твердого вещества. К раствору спирта ROH (R=CH2CHF2 (2) или СН 2 циклопентил (32 (1,5 экв) в ТГФ добавляли NaH(1,5 экв) и полученную суспензию перемешивали в течение 1 ч. Полученный раствор добавляли к раствору пентабензоил-2-нитроаденозина (1 экв) в ТГФ и продолжали перемешивание в течение 16 ч. Затем растворители удаляли в вакууме и остаток растворяли в МеОН. Затем добавляли NaOMe (4 экв) и полученную суспензию перемешивали 4 ч, после чего гасили водным раствором лимонной кислоты. Растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая 2-алкоксипроизводное. К суспензии инозина (1 экв) и DMAP (0,1 экв) в MeCN добавляли Et3N (3,8 экв) и уксусный ангидрид (3,5 экв) и полученную смесь перемешивали в течение 1 ч. Затем добавляли МеОН и продолжали перемешивать еще 15 мин. Затем растворители удаляли в вакууме и продукт растирали с изопропанолом. Полученное твердое вещество отделяли фильтрованием и промывали изопропанолом, получая триацетоксиинозин. К раствору триацетоксиинозина (1 экв) в CHCl3 добавляли ДМФА (3 экв) и тионилхлорид (3 экв), и полученный раствор кипятили с обратным холодильником в течение 16 ч. Затем растворители удаляли в вакууме, остаток растворяли в дихлорметане и промывали водным NaHCO3 и насыщенным раствором соли, после чего органическую фазу высушивали над MgSO4, получая триацетокси-6-хлораденозин. К раствору тетраметиламмонийнитрата (TMAN) (1,5 экв) в дихлорметане добавляли трифторуксусный ангидрид (TFAA) (1,5 экв) и полученный раствор перемешивали при комнатной температуре 1 ч. Смесь охлаждали до 0 С и добавляли раствор триацетокси-6-хлораденозина (1 экв) в дихлорметане. Полученный раствор оставляли нагреваться до комнатной температуры в течение 2,5 ч. Затем раствор промывали водн. NaHCO3, насыщенным раствором соли и водой (3 х), и органическую фазу высушивали надMgSO4. Кристаллизация из смеси дихлорметан/EtOH дала триацетокси-6-хлор-2-нитроаденозин в виде бледно-желтого твердого вещества, которое промывали водой и EtOH. К раствору спирта ROH (R=СН 2 циклопропил (3) или 2,2,3,3-тетрафторциклобутан (35 (1,5 экв) в ТГФ добавляли NaH (1,5 экв) и полученную суспензию перемешивали 15 мин. Затем полученный раствор добавляли к раствору триацетокси-6-хлор-2-нитроаденозина (1 экв) в ТГФ и перемешивание продолжали еще 2-6 ч. Затем растворители удаляли в вакууме, добавляли EtOH и водный NH3 и полученный раствор нагревали в запаянной пробирке при 80 С в течение 16 ч. Затем смесь охлаждали, растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая 2 алкоксипроизводное. Пример 16. Получение соединений 7-18 Схема 3(17), или 3-изопропилфенил (18 (1,5 экв) в ТГФ добавляли KOtBu (1,5 экв) и полученную суспензию перемешивали 30 мин. Полученный раствор добавляли к раствору пентабензоил-2-нитроаденозина (см. схему 1) (1 экв) в ТГФ и продолжали перемешивание 16 ч. Затем растворители удаляли в вакууме и остаток растворяли в МеОН. Затем добавляли NaOMe (4 экв) и полученную суспензию перемешивали 4 ч,после чего гасили водным раствором лимонной кислоты. Затем растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая 2-арилоксипроизводное. Раствор 2-хлораденозина в чистом амине RR'NH (RR'N = NH-(R)-втор-бутил (22), или NH-(S)-вторбутил (23), или NH-н-гексил (24), или NH-экзо-норборнан (25), или N(Ме)изоамил (31 либо нагревали при 190 С в микроволновой печи в течение 30 мин, либо нагревали при 40-100 С в течение 16 ч. Затем растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе,получая 2-аминоалкильное производное. Пример 18. Получение соединения 33 Схема 5 К раствору 2-хлораденозина (1 экв) в ДМСО добавляли NaSEt (1,3 экв) и полученный раствор нагревали при 80 С в течение 20 ч. Затем растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая 2-этилтиоаденозин. К раствору 2-этилтиоаденозина в MeCN/H2O (1:1) добавляли метахлорпербензойную кислоту(mCPBA) (3 экв) и продолжали перемешивание в течение 16 ч. Затем растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая соединение 33. Пример 19. Получение соединения 37 Схема 6 Суспензию 2-йодаденозина (1 экв), ArB(OH)2 (Ar=3,5-диметилфенил) (1 экв), Pd(PPh3)4 (0,1 экв),Cs2CO3 (2,2 экв) и Et3N (2,2 экв) в смеси PhMe/EtOH (2:1) нагревали при 110 С в течение 16 ч. Затем суспензию фильтровали, фильтрат концентрировали в вакууме и очищали колоночной хроматографией на обращенной фазе, получая 37. Пример 20. Получение соединения 40 Схема 7- 21011099 К раствору 3'-дезоксиаденозина (1 экв) в пиридине добавляли бензоилхлорид (6 экв) и полученный раствор кипятили при 65 С в течение 4 ч. Растворители удаляли в вакууме, остаток растворяли в EtOAc и промывали водой (х 3) и насыщенным раствором соли, после чего органическую фазу высушивали надMgSO4. Очистка колоночной хроматографией на силикагеле дала 3'-дезокситетрабензоиладенозин. К раствору TMAN (1,5 экв) в дихлорметане добавляли TFAA (1,5 экв) и полученный раствор перемешивали 1 ч при комнатной температуре. Смесь охлаждали до 0 С и добавляли раствор 3'дезокситетрабензоиладенозина (1 экв) в дихлорметане. Полученный раствор оставляли нагреваться до комнатной температуры в течение 16 ч. Затем раствор промывали водой (х 3) и насыщенным раствором соли и органическую фазу высушивали над MgSO4, получая 3'-дезокситетрабензоил-2-нитроаденозин. К раствору н-гексанола (2 экв) в ТГФ добавляли NaH (2,1 экв) и полученную суспензию перемешивали в течение 30 мин. Полученный раствор добавляли к раствору 3'-дезокситетрабензоил-2 нитроаденозина (1 экв) в ТГФ и перемешивание продолжали 1 неделю. Затем растворители удаляли в вакууме, и остаток очищали колоночной хроматографией на силикагеле. К раствору полученного вещества в ТГФ добавляли водный NH3 и полученную суспензию перемешивали 12 ч. Растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая 40. Пример 21. Получение соединений 44, 45 и 47 Схема 8 К раствору 6-хлораденозина (1 экв) в МеОН или ДМСО добавляли амин RR'NH (RR'N =(3-5 экв) и полученный раствор перемешивали 16 ч. Затем растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая 6-диалкиламинопроизводное. Пример 22. Получение соединения 48 Схема 9 К раствору циклопентилметилового спирта (1,5 экв) в ТГФ добавляли NaH (1,5 экв) и полученную суспензию перемешивали 1 ч. Полученный раствор добавляли к раствору триацетокси-6-хлораденозина(см. схема 2) (1 экв) в ТГФ и перемешивание продолжали в течение 16 ч. Затем удаляли растворители в вакууме и остаток растворяли в МеОН. Затем добавляли NaOMe (4 экв) и полученную суспензию перемешивали в течение 4 ч, после чего гасили водным раствором лимонной кислоты. Растворители удаляли в вакууме, и остаток очищали колоночной хроматографией на обращенной фазе, получая 6 алкоксипроизводное. К суспензии 6-хлораденозина в ацетоне при 0 С добавляли HClO4 и продолжали перемешивание в течение 2 ч. Затем добавляли водный NH3 и концентрировали раствор в вакууме. Раствор охлаждали до-20 С и образовавшийся белый осадок 2',3'-О-изопропилиден-6-хлораденозина собирали и промывали ацетоном. К суспензии 2',3'-О-изопропилиден-6-хлораденозина (1 экв) в воде добавляли KOH (2,5 экв) иKMnO4 (2,5 экв) и продолжали перемешивание 4 ч. Затем реакционную смесь гасили пероксидом водорода, концентрировали и охлаждали до -20 С. Полученный осадок собирали и промывали водой, получая 2',3'-О-изопропилиден-6-хлораденозин-5'-карбоновую кислоту. К раствору 2',3'-О-изопропилиден-6-хлораденозин-5'-карбоновой кислоты (1 экв) в ДМСО добавляли RNH2 (R=Me (51) или изоамил (52 (2 экв) и полученный раствор перемешивали 16 ч. Затем растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая соответствующую 2',3'-О-изопропилиден-6-алкиламиноаденозин-5'-карбоновую кислоту. Раствор 2',3'-O-изопропилиден-6-алкиламиноаденозин-5'-карбоновой кислоты (1 экв), реагента Мукияма (1,2 экв), изопропиламина (1,5 экв) и Et3N (2,5 экв) в ДМФА перемешивали в течение 6 ч. Затем растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе. Обработка трифторуксусной кислотой ТФУ/вода (2:1) в течение 2 ч с последующим удалением растворителей в вакууме и очисткой колоночной хроматографией на обращенной фазе позволили получить соединения, указанные в заглавии примера. Пример 24. Получение соединения 53 Схема 11 К суспензии 2-метоксиаденозина в ацетоне при 0 С добавляли HClO4 и продолжали перемешивание в течение 2 ч. Затем добавляли водный NH3, удаляли растворители в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая 2',3'-О-изопропилиден-2-метоксиаденозин. К суспензии 2',3'-O-изопропилиден-2-метоксиаденозина (1 экв) в воде добавляли KOH (2,5 экв) иKMnO4 (2,5 экв) и продолжали перемешивание 2 ч. Добавляли дополнительное количество KOH (0,2 экв) и KMnO4 (0,2 экв) и перемешивание продолжали еще 4 ч. Затем реакционную смесь гасили пероксидом водорода, концентрировали и охлаждали до -20 С.- 23011099 Полученный осадок собирали и промывали водой, получая 2',3'-О-изопропилиден-2 метоксиаденозин-5'-карбоновую кислоту. Раствор 2',3'-О-изопропилиден-2-метоксиаденозин-5'-карбоновой кислоты (1 экв), реагента Мукиямы (1,2,экв), анилина (1,5 экв) и Et3N (2,5 экв) в ДМФА перемешивали в течение 6 ч. Затем добавляли смесь ДМСО/вода (1:1) и полученное белое твердое вещество фильтровали. Этот осадок растворяли в смеси ТФУ/вода (2:1) и перемешивание продолжали в течение 5 ч. Затем растворители удаляли в вакууме, и остаток очищали колоночной хроматографией на обращенной фазе, получая соединение 53. Пример 25. Получение соединения 54 Схема 12(HBTU=гексафторфосфат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония) К суспензии 2-хлораденозина в ацетоне при 0 С добавляли HClO4 и продолжали перемешивание в течение 2 ч. Затем добавляли водный NH3 и раствор концентрировали в вакууме. Раствор охлаждали до-20 С, и полученный белый осадок 2',3'-О-изопропилиден-2-хлораденозина собирали и промывали ацетоном. К суспензии 2',3'-O-изопропилиден-2-хлораденозина (1 экв) в воде добавляли KOH (2,5 экв) иKMnO4 (2,5 экв) и продолжали перемешивание 4 ч. Затем реакционную смесь гасили пероксидом водорода, концентрировали и охлаждали до -20 С. Полученный осадок собирали и промывали водой, получая 2',3'-О-изопропилиден-2-хлораденозин-5'-карбоновую кислоту. Раствор 2',3'-O-изопропилиден-2-хлораденозин-5'-карбоновой кислоты (1 экв) в чистом нгексиламине нагревали до 100 С в запаянной пробирке в течение 24 ч. Удаление растворителей в вакууме и очистка колоночной хроматографией на обращенной фазе дала бледно-коричневое твердое вещество (1 экв), которое растворяли в ДМФА при 0 С. К этому раствору добавляли н-бутиламин (4 экв),DIPEA (диизопропилэтиламин) (2,1 экв) и HBTU (1 экв), и затем продолжали перемешивание при 0 С в течение 4 ч. Затем удаляли растворители в вакууме, остаток растворяли в EtOAc, промывали 0,2 н HCl,водным NaHCO3 и насыщенным раствором соли, и после этого высушивали над MgSO4, получая желтое масло. Это масло растворяли в смеси ТФУ/вода (2:1) и продолжали перемешивание в течение 2 ч. Затем удаляли растворители в вакууме и остаток очищали колоночной хроматографией на обращенной фазе,получая 54. К суспензии аденозина в ацетоне при 0 С добавляли HClO4 и продолжали перемешивание в течение 2 ч. Затем добавляли водный NH3 и раствор концентрировали в вакууме. Раствор охлаждали до -20 С, и полученный белый осадок 2',3'-О-изопропилиденаденозина собирали и промывали ацетоном. К раствору 2',3'-О-изопропилиденаденозина (1 экв), трифенилфосфина (1 экв) и фталимида (1,03 экв) и ТГФ в атмосфере аргона добавляли диэтилазодикарбоксилат (1 экв) и смесь перемешивали в течение 10 ч. Полученный осадок собирали и промывали диэтиловым эфиром. К раствору полученного твердого вещества (1 экв) в EtOH добавляли гидразин (15 экв) и раствор кипятили с обратным холодильником в течение 2 ч, после чего охлаждали до комнатной температуры. Полученный осадок фильтровали,растворяли в воде и доводили pH до 4. Осадок отделяли фильтрованием и pH фильтрата доводили до 10,экстрагировали хлороформом и высушивали над MgSO4, получая 2',3'-О-изопропилиден-5'-аминоаденозин. К раствору масляной кислоты (1 экв) в ДМФА при 0 С добавляли DIPEA (1,2 экв) и TBTU (1 экв), и перемешивали в течение 5 мин. Затем добавляли 2', 3'-О-изопропилиден-5'-аминоаденозин (1 экв) в виде раствора в ДМФА, и полученный раствор перемешивали в течение 3 ч. Затем полученный продукт экстрагировали дихлорметаном, промывали водой (х 3) и высушивали над MgSO4. Обработка смесью ТФУ/вода (2:1) в течение 2 ч с последующим удалением растворителей в вакууме и очисткой колоночной хроматографией на обращенной фазе привела к получению 55. Пример 27. Получение соединения 56 Схема 14 К раствору 2',3'-O-изопропилиден-5'-аминоаденозина (см. схему 13) (1 экв) в дихлорметане добавляли этилизоцианат (1,2 экв) и продолжали перемешивание в течение 16 ч. Затем добавляли полиаминовую смолу, фильтровали, фильтрат концентрировали в вакууме и очищали колоночной хроматографией на обращенной фазе. Полученное твердое вещество растворяли в смеси ТФУ/вода (2:1) и продолжали перемешивание в течение 3 ч. Затем растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая 56. К раствору триацетокси-6-хлор-2-нитроаденозина (см. схему 2) (1 экв) в ТГФ добавляли диметиламин (2 экв) и продолжали перемешивание в течение 4 ч. Затем добавляли циклогексиламин (1 экв) и полученный раствор нагревали до 85 С в течение 2 дней. После этого растворители удаляли в вакууме и остаток растворяли в МеОН. Затем добавляли NaOMe (1 экв) и полученную суспензию перемешивали 16 часов. Растворители удаляли в вакууме, и остаток очищали колоночной хроматографией на обращенной фазе, получая 57. Пример 29. Получение соединения 58 Схема 16 К раствору триацетокси-6-хлор-2-нитроаденозина (см. схему 2) (1 экв) в ДМФА добавляли бензиламин (1 экв) и Et3N (1,5 экв) и продолжали перемешивание в течение 10 мин. Затем растворители удаляли в вакууме, остаток очищали колоночной хроматографией на обращенной фазе и растворяли в МеОН. Затем добавляли NaOMe (2 экв) и полученный раствор перемешивали в течение 4 ч. Растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая 58. Пример 30. Получение соединений 59-61 Схема 17 К раствору 2-хлораденозина (1 экв) в 1 М водн. NaOAc (буферированному при pH 4) добавляли бром (1,2 экв) и полученный раствор перемешивали при комнатной температуре в течение 16 ч. Затем реакционную смесь гасили водным раствором NaHSO3, доводили pH до 7 и охлаждали до 4 С. Полученный осадок 2-хлор-8-бромаденозина собирали, промывали водой и высушивали. Раствор 2-хлор-8-бромаденозина (1 экв) в ГМДС и диоксане кипятили с обратным холодильником при 110 С в течение 8 ч. Затем растворители удаляли в вакууме, добавляли толуол и повторно удаляли растворители в вакууме. Остаток растворяли в N-метилпирролидоне (NMP), добавляли Pd(PPh3)4 (0,04 экв) и SnMe4 (2 экв), и полученную суспензию нагревали при 110 С в течение 16 ч. Затем раствор охлаждали, и растворители удаляли в вакууме. Остаток растворяли в EtOAc, промывали водой и органическую- 26011099 фазу высушивали над MgSO4. К раствору полученного масла в МеОН добавляли K2CO3 и полученную суспензию перемешивали 4 ч. Затем растворители удаляли в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая 2-хлор-8-метиладенозин. Раствор 2-хлор-8-метиладенозина в чистом амине RNH2 (R = циклогексил (59) или циклопентил(60), или н-гексил (61 нагревали при 190 С в микроволновой печи в течение 30 мин. Затем удаляли растворители в вакууме и остаток очищали колоночной хроматографией на обращенной фазе, получая 8 метил-2-аминоалкильное производное. Пример 31. Концентрации спонгозина в плазме определяли после однократного перорального введения у 5 или 6 человек добровольцев. Тахикардию определяли, используя 12-электродную ЭКГ. Минимальная концентрация в плазме, приводящая к анальтезирующему эффекту, составляла у крыс 0,025 мкМ, что предполагало минимальную эффективную дозировку у человека приблизительно 0,8 мг, которая приводила к концентрациям в плазме свыше 0,025 мкМ в течение приблизительно 1,5 ч. Пример 32. Спонгозин (62,4 и 624 мкг/кг, интраперитонеально) ингибирует вызванное каррагенаном (CGN) воспаление с эффективностью, сравнимой с индометацином (3 мг/кг, перорально), при концентрациях,которые не влияют на кровяное давление. Каррагенан (2%, 10 мкл) вводили в правую заднюю лапу крысы и определяли объем лапы плетизмометрией. Спонгозин вводили одновременно с каррагенаном. Результаты показаны на фиг. 8. Спонгозин был столь же эффективен, как и индометацин (индометацин, 3 мг/кг, п.о.). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, имеющее формулу 7. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в качестве лекарственного средства. 8. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в производстве лекарственного средства для профилактики, лечения или облегчения патологического состояния, которое может быть улучшено или предотвращено агонизмом в отношении аденозиновых рецепторов А 2 А. 9. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в производстве лекарственного средства для профилактики, лечения или облегчения боли. 10. Применение по п.9, где боль представляет собой гипералгезию. 11. Применение по п.10, где гипералгезия представляет собой невропатическую боль. 12. Применение по п.10, где гипералгезия представляет собой боль при воспалении. 13. Применение по любому из пп.9, 10 или 12, где боль вызвана или связана с воспалительным или иммунным заболеванием или является результатом комбинированного воспалительного, аутоиммунного и невропатического повреждения тканей. 14. Применение по п.9 в производстве лекарственного средства для профилактики, лечения или облегчения ишемической боли. 15. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в производстве лекарственного средства для профилактики, лечения или облегчения макро- и микрососудистых осложнений диабета 1 и 2 типа, ретинопатии, невропатии, вегетативной невропатии или повреждения кровеносных сосудов, вызванного ишемией или атеросклерозом. 16. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в производстве лекарственного средства для профилактики, лечения или облегчения воспаления. 17. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в производстве болезнь-модифицирующих (базисных) противоревматических лекарственных средств(DMARD) для замедления развития артропатии. 18. Применение по п.17 в производстве DMARD для замедления развития ревматоидного артрита. 19. Фармацевтическая композиция в единичной дозированной форме, содержащая до 500 мг соединения по любому из пп.1-6, и физиологически приемлемый носитель, эксципиент или разбавитель. 20. Фармацевтическая композиция в единичной дозированной форме, содержащая до 500 мг соединения по любому из пп.1-6 или его фармацевтически приемлемую соль в сочетании с NSAD или
МПК / Метки
МПК: A61K 31/7076, C07H 19/16, A61P 29/00, A61P 9/00
Метки: соединения, терапевтические
Код ссылки
<a href="https://eas.patents.su/30-11099-terapevticheskie-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Терапевтические соединения</a>
Радиоактивные терапевтические липосомы

Номер патента: 5624
Опубликовано: 28.04.2005
Авторы: Ларсен Рой Х., Хенриксен Йермунд
МПК: A61K 51/12
Метки: радиоактивные, терапевтические, липосомы
Формула / Реферат:
1. Система конъюгации, отличающаяся тем, что она представляет собой липосому с ионофорами и с хелатообразующим соединением, помещенными внутрь липосомы, причем в указанную липосому дополнительно инкапсулируют тяжелый(ые) радионуклид(ы), излучающий(ие) a-частицы, или 212Pb в качестве генератора для a-излучателя 212Bi. 2. Система конъюгации по п.1, отличающаяся тем, что концентрация хелатообразующего соединения внутри липосом является подходящей...
Модифицированные пептиды как терапевтические агенты

Номер патента: 5404
Опубликовано: 24.02.2005
Авторы: Фейдже Улрих, Лиу Чуан-Фа, Бун Томас Чарлз, Читхэм Дженет
МПК: C12N 15/62, C07K 19/00
Метки: агенты, терапевтические, пептиды, модифицированные
Формула / Реферат:
1. Слитой рекомбинантный белок, применимый в качестве терапевтического агента, имеющий формулу (X1)a-F1-(X2)b и его мультимеры, где F1 обозначает Fc-домен; X1 и X2, каждый независимо, выбран из -(L1)c-P1, -(L1)c-P1-(L2)d-P2, -(L1)c-P1-(L2)d-P2-(L3)e-P3 и -(L1)c-P1-(L2)d-P2-(L3)e-P3-(L4)f-P4; P1, P2, P3 и P4 обозначают, независимо каждая, последовательности рандомизированных пептидов; L1, L2, L3 и L4, каждый независимо, обозначают линкеры и a, b,...
Терапевтические применения вариантов хемокинов

Номер патента: 9414
Опубликовано: 28.12.2007
Авторы: Праудфут Аманда, Джонсон Зо, Шо Джеффри
МПК: A61K 38/19, A61P 29/00, A61P 31/00...
Метки: вариантов, применения, хемокинов, терапевтические
Формула / Реферат:
1. Применение полипептида, содержащего SEQ ID NO:2, для изготовления лекарственного средства для лечения аутоиммунных, воспалительных или инфекционных заболеваний. 2. Применение по п.1, где этот полипептид дополнительно содержит изолейцин в положении 64 SEQ ID NO:2 и таким образом содержит SEQ ID NO:4. 3. Применение по п.1 или 2, дополнительно отличающееся тем, что указанный полипептид не содержит мутацию в положении 9, 10 или 13 в...
Терапевтические средства для лечения боли

Номер патента: 9593
Опубликовано: 28.02.2008
Авторы: Тэфесс Лэйки, Кайл Доналд Дж., Сан Кан
МПК: A61K 31/495, A61P 13/10, A61K 31/44...
Метки: боли, средства, терапевтические, лечения
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль, где Ar1 представляет собой Ar2 представляет собой V представляет собой N или CH; X представляет собой О; Y1 и Y2 представляют собой -CH2- и -CH2-, -О- и -О-, -NH- и -NH-, -S- и -S-, -CH2- и -О-, CH2- и -NH-, -CH2- и -S-, -О- и -CH2-, -NH- и -CH2-, -S- и -CH2-, -О- и -NH-, -NH- и -O-, -S- и -NH- или -NH- и -S- соответственно; R1 представляет собой -H, -галоген, -(С1-С4)алкил,...
Терапевтические комбинации, включающие в себя амлодипин и аторвастатин

Номер патента: 3549
Опубликовано: 26.06.2003
Авторы: Бук Ян, Скотт Роберт Эндрю Доналд
МПК: A61K 31/4422, A61P 9/10, A61K 31/40...
Метки: комбинации, включающие, аторвастатин, терапевтические, амлодипин, себя
Формула / Реферат:
1. Фармацевтическая композиция, содержащая а) некоторое количество амлодипина или его фармацевтически приемлемой соли присоединения кислоты; б) некоторое количество аторвастатина или его фармацевтически приемлемой соли; и в) фармацевтически приемлемый носитель или разбавитель. 2. Фармацевтическая композиция по п.1, содержащая амлодипина бесилат. 3. Фармацевтическая композиция по п.2, содержащая гемикальциевую соль аторвастатина. 4. Применение...
Предыдущий патент: Способы получения производных 4-n, n-диметиламинокротоновой кислоты
Следующий патент: Способ изолирования пространств и изолирующие конструкции, полученные при помощи этого способа
Случайный патент: Способ использования функционального проппанта для определения геометрии подземной трещины