Производные 1-бутил-2-гидроксиаралкилпиперазина и их применение в качестве лекарственного средства против депрессии
Номер патента: 20716
Опубликовано: 30.01.2015
Авторы: Ли Цзяньци, Цзинь Хуа, Чжен Юнюн, Лв На, Вэн Чжицзе


















Формула / Реферат
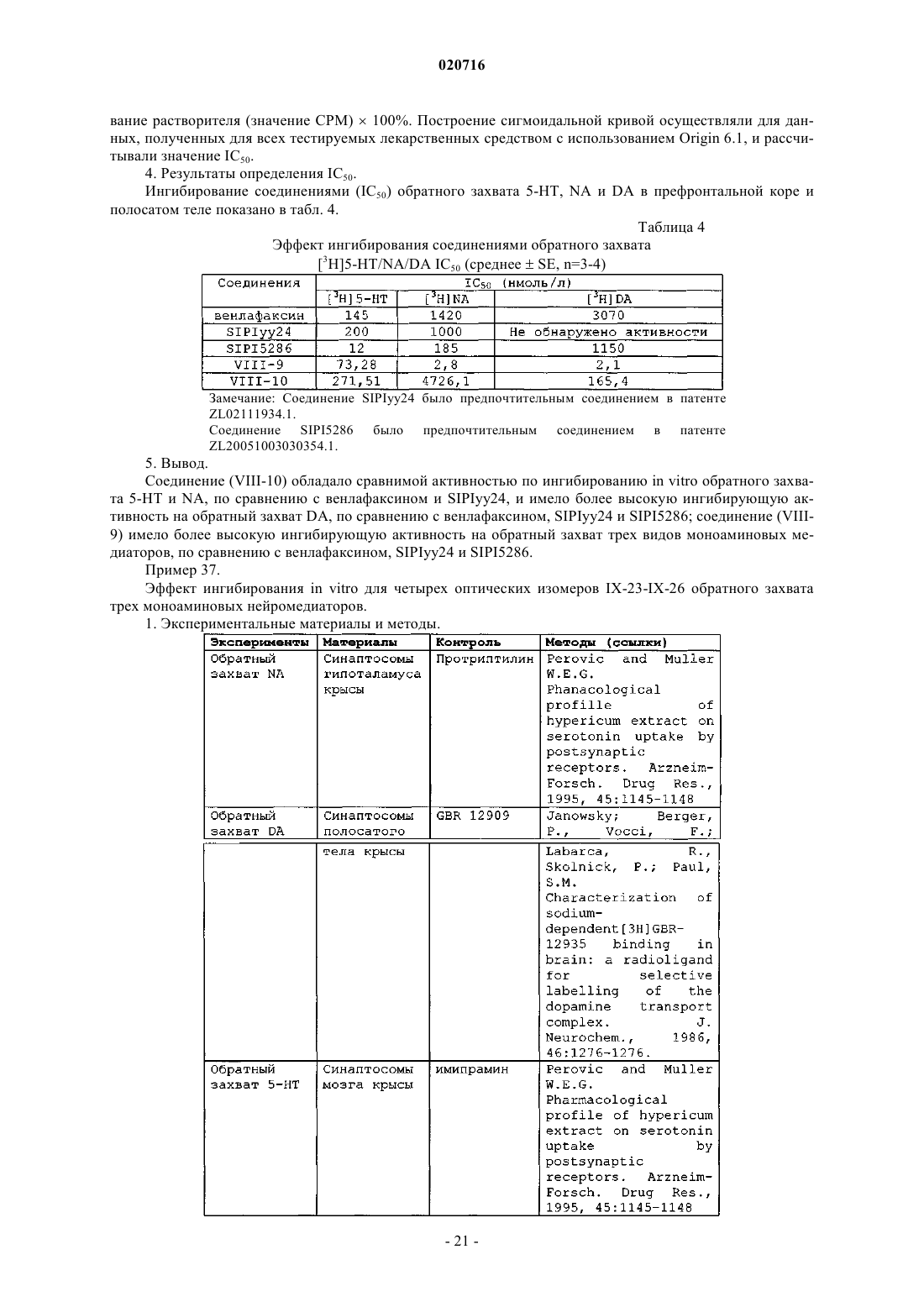
1. Производное 1-бутил-2-гидроксиларалкилпиперазина, отличающееся тем, что представляет собой свободную щелочную форму или соль соединения формулы (1) или свободную щелочную форму или соль оптических изомеров соединения формулы (1)

где соль представляет собой соль, содержащую фармацевтически приемлемые анионы;
Ar1 представляет собой фенил, замещенный фенил или циннаменил, где замещенный фенил содержит от одного до четырех заместителей, представляющих собой галоген, нитро, C1-C4-алкил, C1-C4-алкокси или амино;
m представляет собой целое число от 0 до 5;
R1 представляет собой водород, C1-C5-алкил или фенил.
2. Производное 1-бутил-2-гидроксиларалкилпиперазина по п.1, отличающееся тем, что атомы углерода C1 и C2 в структуре являются хиральными атомами углерода, конфигурация которых является соответственно (1S,2R), (1S,2S), (1R,2S) или (1R,2R), где (1SR,2RS) является эритроизомером и (1SR,2SR) является треоизомером.
3. Производное 1-бутил-2-гидроксиларалкилпиперазина по п.1, отличающееся тем, что соль представляет собой гидрохлорид, гидробромид, сульфат, трифторацетат или метансульфонат.
4. Производное 1-бутил-2-гидроксиларалкилпиперазина по п.1, отличающееся тем, что в соли содержатся 0,5-4 молекулы кристаллизационной воды.
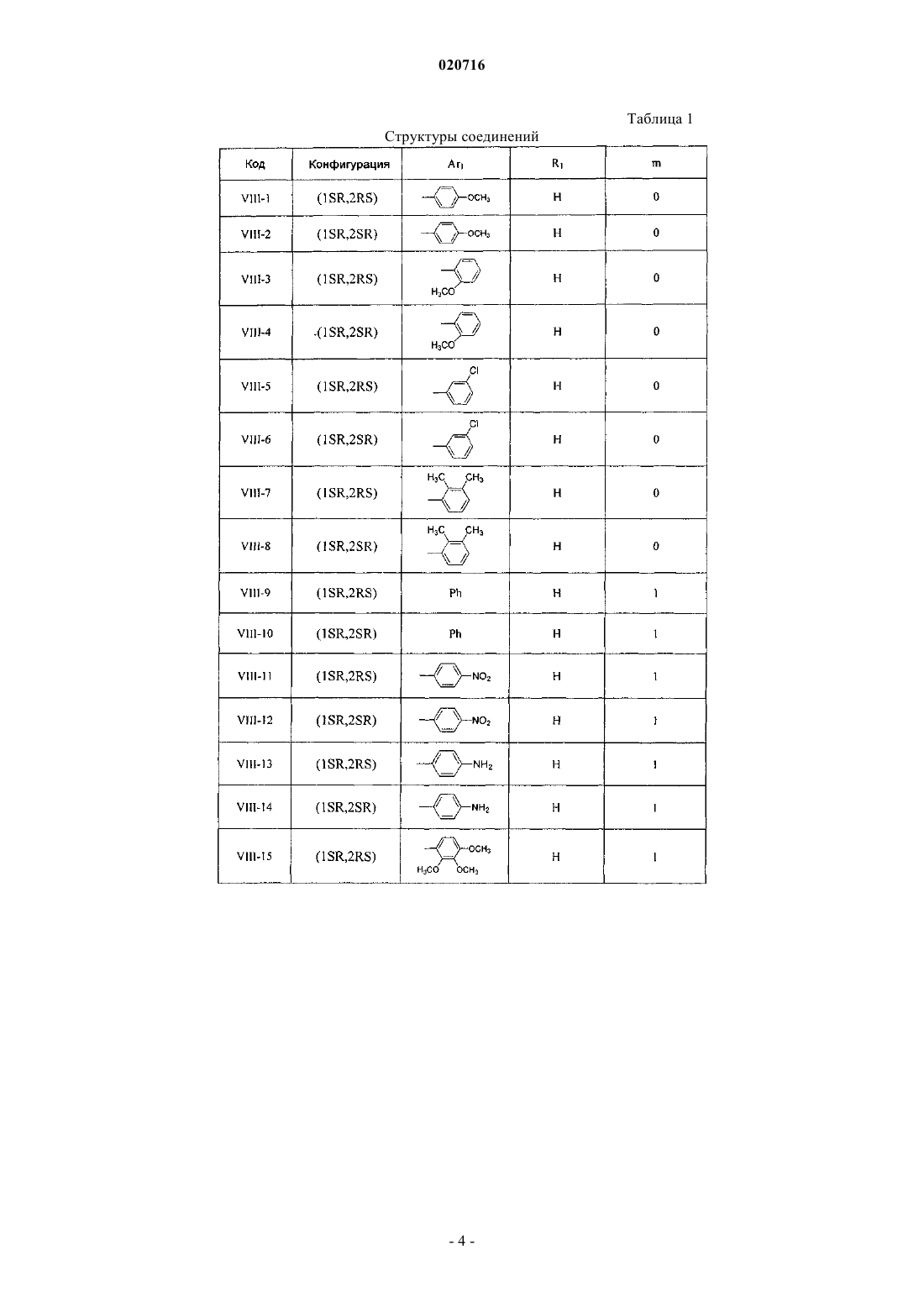
5. Производное 1-бутил-2-гидроксиларалкилпиперазина по любому из пп.1-4, которое выбирают из группы, содержащей:
(1SR,2RS)-N1-п-метоксилфенил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-1;
(1SR,2SR)-N1-п-метоксилфенил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-2;
(1SR,2RS)-N1-о-метоксилфенил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-3;
(1SR,2SR)-N1-о-метоксилфенил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-4;
(1SR,2RS)-N1-м-хлорфенил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-5;
(1SR,2SR)-N1-м-хлорфенил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-6;
(1SR,2RS)-N1-(2,3-диметилфенил)-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-7;
(1SR,2SR)-N1-(2,3-диметилфенил)-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-8;
(1SR,2RS)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-9;
(1SR,2SR)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-10;
(1SR,2RS)-N1-п-нитробензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксилэтил]пиперазин, VIII-11;
(1SR,2SR)-N1-п-нитробензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-12;
(1SR,2RS)-N1-п-аминобензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-13;
(1SR,2SR)-N1-п-аминобензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-14;
(1SR,2RS)-N1-(3',4',5'-триметоксибензил)-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-15;
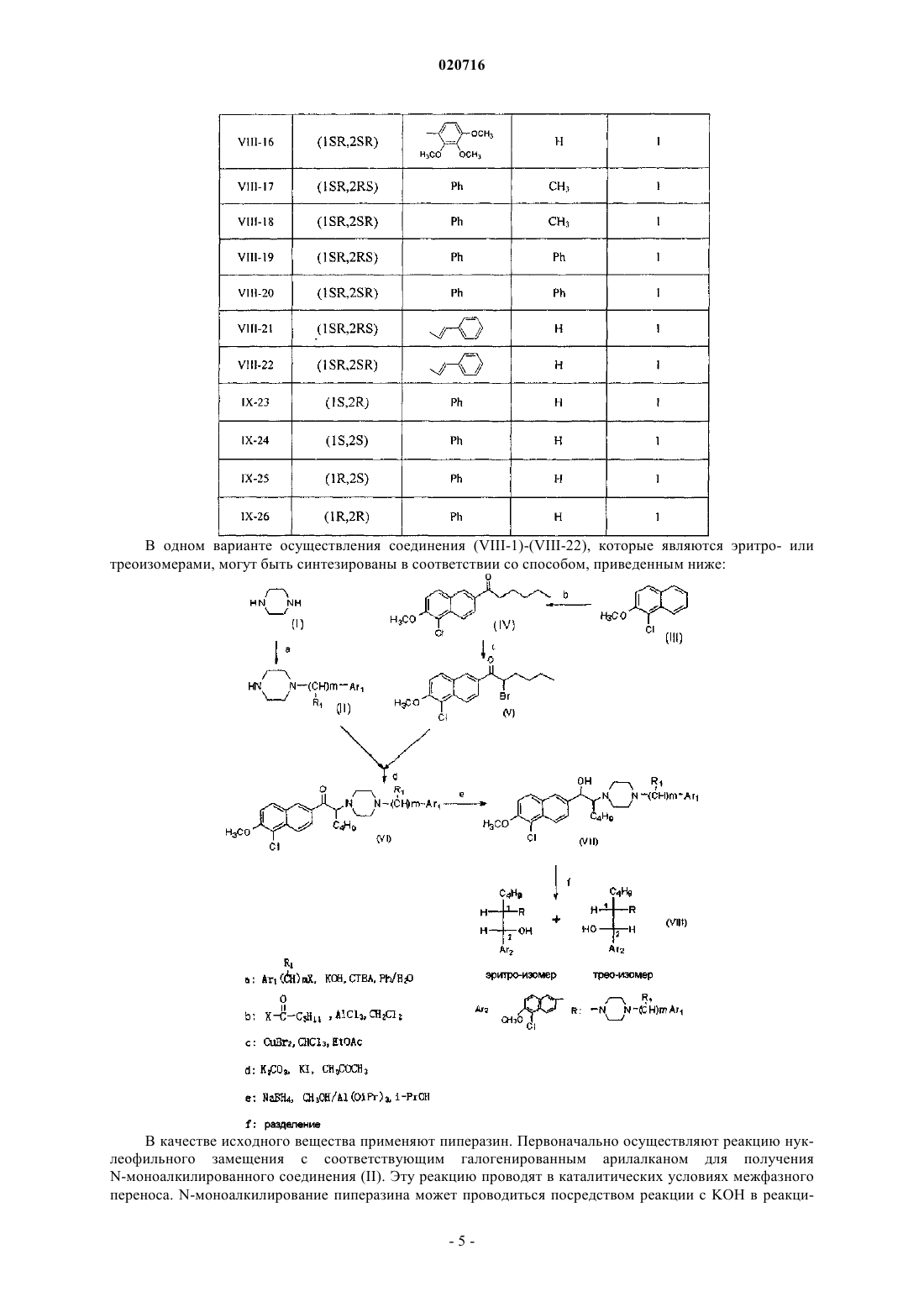
(1SR,2SR)-N1-(3',4',5'-триметоксибензил)-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-16;
(1SR,2RS)-N1-α-фенетил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-17;
(1SR,2SR)-N1-α-фенетил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VII-18;
(1SR,2RS)-N1-бензгидрил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-19;
(1SR,2SR)-N1-бензгидрил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-20;
(1SR,2RS)-N1-циннамил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-21;
(1SR,2SR)-N1-циннамил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, VIII-22;
(1S,2R)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, IX-23;
(1S,2S)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, IX-24;
(1R,2S)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, IX-25 и
(1R,2R)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, IX-26.
6. Фармацевтическая композиция для лечения депрессии, содержащая терапевтически эффективное количество производных 1-бутил-2-гидроксиларалкилпиперазина по любому из пп.1-5, вместе с фармацевтически приемлемыми носителями.
7. Применение производных 1-бутил-2-гидроксиларалкилпиперазина по любому из пп.1-5 в производстве антидепрессанта.
Текст
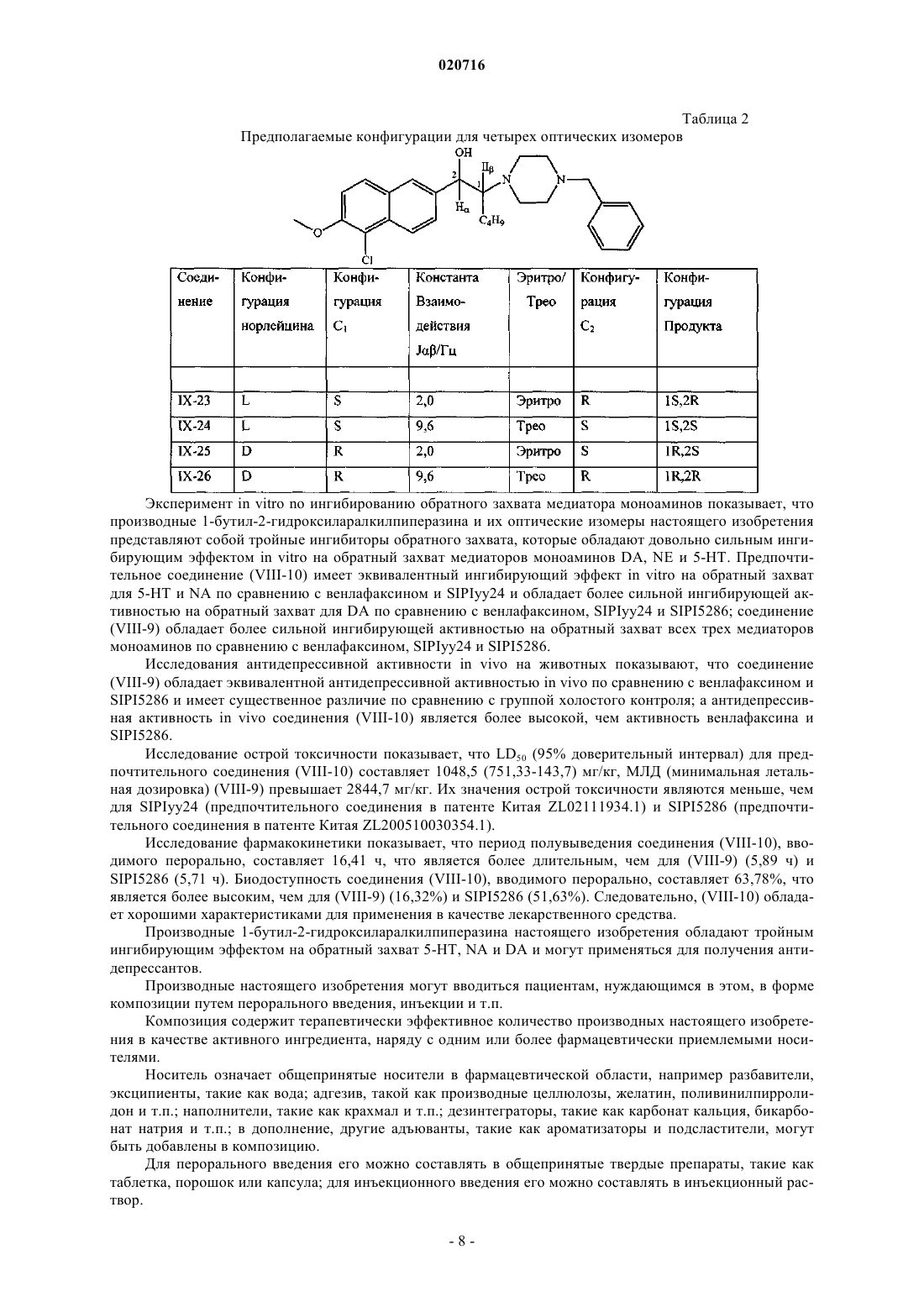
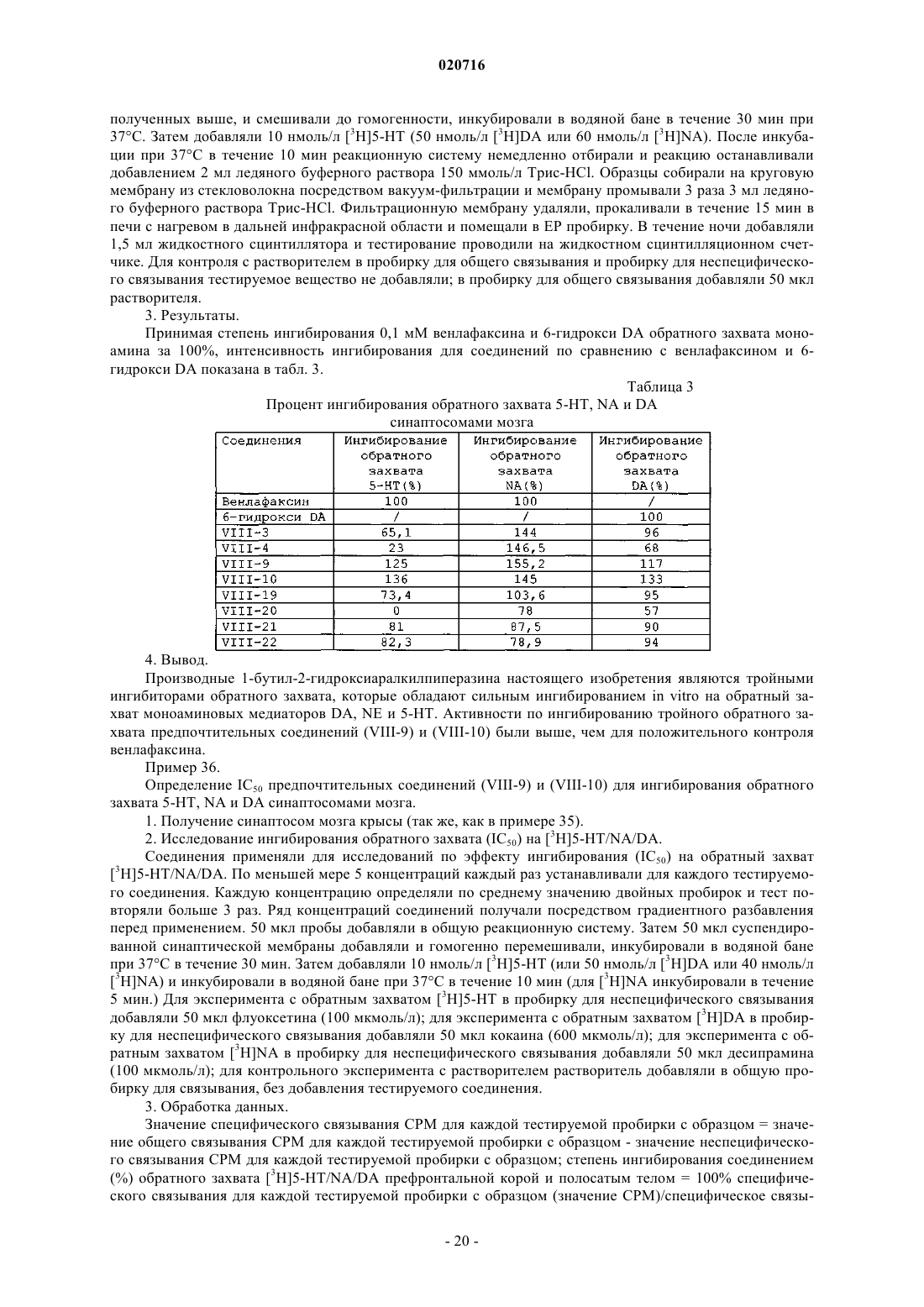
ПРОИЗВОДНЫЕ 1-БУТИЛ-2-ГИДРОКСИАРАЛКИЛПИПЕРАЗИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА ПРОТИВ ДЕПРЕССИИ Изобретение раскрывает производные 1-бутил-2-гидроксиларалкилпиперазина и их применение в качестве антидепрессантов. Производные настоящего изобретения обладают тройным ингибирующим эффектом на обратный захват 5-HT, NA и DA и могут вводиться пациентам,нуждающимся в этом, в форме композиции путем перорального введения, инъекции и т.п. По сравнению с имеющими в настоящее время клиническое применение антидепрессантами двойного назначения (такими, как венлафаксин), указанные производные могут иметь более сильный эффект против депрессии, более широкие показания, более быстрое наступление эффекта и более низкие нейротоксичность и побочные реакции; и указанные производные имеют более высокую активность против депрессии, более низкую токсичность, более высокую биодоступность, более длительный период полувыведения и лучшие характеристики для лекарственного средства по сравнению с производными арилалканолпиперазина и их оптическими изомерами, раскрытыми в предшествующем уровне техники. Производное 1-бутил-2 гидроксиларалкилпиперазина представляет собой свободную щелочную форму или ее соль соединения формулы(71)(73) Заявитель и патентовладелец: СиЭсПиСи ЧЖУНЦИ ФАРМАСЬЮТИКАЛ ТЕКНОЛОДЖИ (ШИЦЗЯЧЖУАН) КО., ЛТД.; ШАНХАЙ ИНСТИТЬЮТ ОФ ФАРМАСЬЮТИКАЛ ИНДАСТРИ (CN) Область техники, к которой относится изобретение Настоящее изобретение относится к производным 1-бутил-2-гидроксиаралкилпиперазина и их применению в качестве антидепрессантов широкого спектра действия. Предшествующий уровень техники Депрессия представляет собой синдром, характеризуемый значительным и продолжительным угнетенным настроением, которое в основном проявляется как аффективное расстройство. Симптомы включают подавленное настроение, неразговорчивость, медленные мышление и двигательную активность и даже попытку суицида. Депрессия, как хроническое психическое заболевание, стало проблемой ужасающего масштаба, которая беспокоит службу здравоохранения в Китае по причинам длительного курса лечения, медленного наступления эффекта и более высокой степени рецидивов, нетрудоспособности и суицидов. В соответствии с "Всемирными докладами о здоровье", опубликованными Всемирной организацией здравоохранения (ВОЗ), депрессия стала четвертым крупнейшим заболеванием в мире, и депрессия может стать второй массовой болезнью после сердечных заболеваний в 2020 г. и, таким образом,становится серьезной проблемой для здоровья человека. До настоящего времени механизм действия антидепрессантов отчетливо не продемонстрирован. Лекарственные средства, обладающие определенным эффектом, действуют на синапсы нервного окончания и проявляют свое лечебное воздействие, регулируя уровень нейромедиаторов в синаптической щели. Биохимическое исследование этиологии показало, что депрессия относится в основном к пяти типам нейромедиаторов, т.е. центральному 5-гидрокситриптамину (5-HT), норадреналину (NA), дофамину(DA), ацетилхолину (Ach) и -аминомасляной кислоте (GABA). Антидепрессанты могут быть разделены на две категории: ранние неселективные антидепрессанты и новые селективные ингибиторы обратного захвата. Неселективные антидепрессанты в основном включают ингибиторы моноаминоксидазы (MAOI) и трициклические антидепрессанты (TCA); селективные ингибиторы обратного захвата в основном включают селективные ингибиторы обратного захвата 5-гидрокситриптамина (5-HT) (SSRI), ингибиторы обратного захвата норадреналина (NA) (NRAI),норадренергические и специфичные ингибиторы обратного захвата 5-HT (NDRI), двойные ингибиторы обратного захвата 5-HT и NA (SNRI), усилители реабсорбции 5-HT и т.п. Ранние ингибиторы моноаминооксидазы и трициклические антидепрессанты обладают серьезными неблагоприятными реакциями; так, для последующих селективных ингибиторов обратного захвата NA и селективных ингибиторов обратного захвата 5-HT, хотя они имеют меньше неблагоприятных реакций,все еще существуют недостатки, такие как медленное наступление эффекта, неопределенная эффективность и т.п. Следовательно, эффекты для всех видов лекарственных средств, перечисленных выше, при лечении депрессии не являются удовлетворительными. До настоящего времени существующие антидепрессанты еще не могут соответствовать требованиям клинического лечения. Венлафаксин, первый ингибитор двойного обратного захвата 5-HT и NA, появившийся на американском рынке в 1997 г., и дутоксетин, появившийся на рынке в 2004 г., имеют преимущества быстрого наступления действия по сравнению с селективными ингибиторами обратного захвата 5-гидрокситриптамина, такими как флуоксетин, и ингибиторами обратного захвата норадреналина, такими как ребоксетин, и обладают существенными эффектами как на серьезную депрессию, так и на трудноизлечимую депрессию. Начиная с венлафаксина, разработка новых антидепрессантов, которые имеют пути двойного действия на 5-HT и NA, более быстрое наступление действия, меньшее количество побочных эффектов и более сильный эффект становятся основным акцентом исследований и важным направлением разработок. В настоящее время многие исследования указывают, что добавление ингибиторов обратного захвата дофамина (DA) к ингибиторам двойного обратного захвата позволяет получать лучший эффект против депрессии. Ингибиторы тройного селективного обратного захвата 5-HT, NA и DA (также известные как антидепрессанты "широкого спектра действия"), разработанные на основе ингибиторов двойного обратного захвата, в настоящее время все еще находятся на фазе клинических исследований. Например, ингибитор тройного селективного обратного захвата DOV-216303, разработанный DOV Pharmaceutical Inc.,находится на фазе III клинических испытаний; NS-2359, разработанный совместно GlaxoSmithKline иNeuroSearch Inc., находится на фазе II клинических испытаний антидепрессанта. Эти моноаминмедиаторные ингибиторы тройного селективного обратного захвата обладают преимуществами высокой эффективности и быстрого наступления действия и становятся "горячими точками" в разработке антидепрессантов. Заявитель раскрыл производные арилалканолпиперазина и их применение при получении антидепрессантов в патенте Китая ZL02111934.1 Предпочтительное его соединение, N1-бензил-N4-[1-метил-2(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин (IV-19, SIPIyy24, см. формулу А ниже), обладает эффектом двойного ингибирования на обратный захват 5-HT и NA и имеет сильную антидепрессивную биологическую активность на животных. Но дальнейшее исследование выявило, что его антидепрессивный эффект еще не так удовлетворителен, и его побочные реакции являются очевидными. В последующем заявитель раскрыл оптические изомеры соединения SIPIyy24 и его применение в патенте Китая ZL 200510030354.1. Исследование показывает, что (1S,2R) оптический изомер SIPIyy24(код SIPI5286) обладает эффектом ингибирования на обратный захват трех видов медиаторов моноаминов, т.е. 5-HT, NA и DA. Он является новым ингибитором тройного обратного захвата и обладает лучшей антидепрессивной активностью и безопасностью, чем рацемат, и заслуживает право быть новым антидепрессантом. Однако в ходе дополнительных исследований обнаружено, что период полувыведенияSIPI5286 является слишком коротким, и, таким образом, он непригоден для составления в готовую форму лекарственного средства. Описание изобретения Одной из технических проблем для решения в настоящем изобретении является раскрытие производного 1-бутил-2-гидроксиларалкилпиперазина для преодоления недостатков предшествующего уровня техники, т.е. низкой эффективности, значительных побочных эффектов и медленного наступления действия, и, таким образом, решение клинической проблемы и удовлетворение требований клинического применения. Еще одной технической проблемой для решения в настоящем изобретении является раскрытие применения вышеупомянутого производного в получении антидепрессантов. Производное 1-бутил-2 -гидроксиларалкилпиперазина, упомянутое в настоящем изобретении, представляет собой свободную щелочную форму или соль соединения формулы (1) или свободную щелочную форму или соль оптических изомеров соединения формулы (1) где соль представляет собой соль, содержащую фармацевтически приемлемые анионы;Ar1 представляет собой фенил; замещенный фенил или циннаменил, где замещенный фенил содержит от одного до четырех заместителей, представляющих собой галоген, нитро, C1-C4-алкил,C1-C4-алкокси или амино;m представляет собой целое число от 0-5;R1 представляет собой водород, C1-C5-алкил или фенил. Предпочтительно атомы углерода C1 и С 2 в структуре соединений формулы (1) являются хиральными атомами углерода, конфигурация которых является соответственно (1S,2R), (1S,2S), (1R,2S) или(1R,2R), где (1SR,2RS) является эритроизомером и (1SR,2SR) является треоизомером. Когда соединение формулы (1) представляет собой свободную щелочь, оно может образовывать разнообразные соли, содержащие фармацевтически приемлемые анионы, например гидрохлорид, гидробромид, гидройодид, нитрат, сульфат или гидросульфат, фосфат или кислый фосфат, ацетат, лактат, цитрат, тартрат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат или п-толуолсульфонат. В одном из вариантов осуществления изобретения соль представляет собой гидрохлорид, гидробромид, сульфат, трифторацетат или метансульфонат. В еще одном варианте осуществления соль содержит 0,5-4 молекулы кристаллизационной воды. Предпочтительно производные 1-бутил-2-гидроксиларалкилпиперазина выбирают из группы, содержащей:IX-26 (1R,2R)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин. Настоящее изобретение относится также к фармацевтической композиции для лечения депрессии,содержащей терапевтически эффективное количество производных 1-бутил-2 гидроксиларалкилпиперазина вместе с фармацевтически приемлемыми носителями. Настоящее изобретение относится также к применению производных 1-бутил-2 гидроксиларалкилпиперазина в производстве антидепрессанта. Структуры соединений показаны в табл. 1. В одном варианте осуществления соединения (VIII-1)-(VIII-22), которые являются эритро- или треоизомерами, могут быть синтезированы в соответствии со способом, приведенным ниже: В качестве исходного вещества применяют пиперазин. Первоначально осуществляют реакцию нуклеофильного замещения с соответствующим галогенированным арилалканом для полученияN-моноалкилированного соединения (II). Эту реакцию проводят в каталитических условиях межфазного переноса. N-моноалкилирование пиперазина может проводиться посредством реакции с KOH в реакци-5 020716 онной среде бензол/вода, используя бромид цетилтриметиламмония (CTAB) в качестве катализатора межфазного переноса, как изложено в патенте Китая ZL02111934.1. Соединение (III) взаимодействует с соответствующим хлорангидридом кислоты для осуществления реакции Фриделя-Крафтса с получением арилалканона (IV). Эту реакцию проводят при комнатной температуре, используя дихлорметан в качестве растворителя и безводный хлорид алюминия в качестве катализатора. Выход составляет приблизительно 80%. Соединение (IV) бромируют с получением галогенированного арилалканона (V). Эту реакцию осуществляют посредством нагрева в условиях кипячения с обратным холодильником, используя CuBr2 в качестве бромирующего агента и смешанный раствор из хлороформа и этилацетата в качестве растворителя. Выход составляет приблизительно 85%. Соединение (II) может взаимодействовать с соединением (V) для проведения реакцииN4-алкилирования, таким образом обеспечивая соединение арилалканолпиперазина (VI). Реакцию осуществляют при кипячении с обратным холодильником в течение 8-24 ч, применяя K2CO3/ацетон в качестве реакционной системы. Выход составляет 80%. Соединение (VI) взаимодействует с NaBH4 в метаноле при комнатной температуре в течение 0,5-1,0 ч или с изопропоксидом алюминия в изопропаноле при 60-65C в течение 24-48 ч для восстановления карбонильной группы, посредством чего получают соответствующее соединение арилалканолпиперазина (VII). Соединение (VII) разделяют с помощью колоночной хроматографии и получают эритро- и треоизомеры (VIII) соответствующих производных 1-бутил-2-гидроксиларалкилпиперазина. Представляющие интерес соединения (VIII-1)-(VIII-22) могут быть получены с использованием приведенных выше методик. Соединения галогенарилалканов на стадии а, соединение хлорангидрида и соединение (III) на стадии b являются, все, доступными на рынке или могут быть получены общепринятыми способами, изложенными в литературных источниках. Что касается оптических изомеров соединений IX-23-IX-26, то они могут быть синтезированы посредством способа, приведенного ниже. Соединение 3 получают, исходя из хирального норлейцина 1, защищая аминогруппу фталоилом и ацилируя карбоксильную группу оксалилхлоридом; затем соединение 3 подвергают взаимодействию с соединением 4 для осуществления реакции Фриделя-Крафтса с получением, таким образом, соединения 5. Соединение 5 восстанавливают изопропоксидом алюминия и затем гидролизуют с получением соединения 7. Соединение 7 конденсируют с соединением 8 с получением соединения 9, затем представляющий интерес оптически чистый продукт (IX) получают разделением колоночной хроматографией. Детальная схема синтеза показана ниже. В настоящем изобретении применяют прохиральный способ для синтеза четырех оптических изомеров соединения N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазина. Смесь (1S,2R) изомера и (1S,2S) изомера синтезируют, используя L-норлейцин в качестве исходного вещества, затем разделяют колоночной хроматографией с силикагелем или оксидом алюминия в качестве носителя и раствором метиленхлорида и метанола при объемном соотношении, равном 200:1, в качестве элюента,для получения(1S,2S)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'нафтил)гидроксиэтил]пиперазина (IX-24) и (1S,2R)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'нафтил)гидроксиэтил]пиперазина (IX-23). Если в качестве исходного вещества применяют D-норлейцин,в соответствии с тем же способом получают (1S,2S)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'нафтил)гидроксиэтил]пиперазин (IX-25) и (1S,2R)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'нафтил)гидроксиэтил]пиперазин (IX-26). Для способа синтеза, приведенного выше, вещества являются доступными, продукты не нуждаются в разделении и получены с высокой оптической чистотой и высоким общим выходом. Чистоту четырех оптических изомеров определяют посредством высокоэффективной жидкостной хроматографии с хиральной колонкой, и их энантиомерный избыток (ее) составляет для всех более 99%. [Условие обнаружения: хиральная колонка OJ-H (от Daicel industries. Co. Ltd., JP) 4,6250 мм; подвижная фаза: н-гексан:этанол:диэтиламин = 40:60:0,1 (об./об./об.): длина волны ультрафиолетового света: УФ 220 нм; температура колонки: 35C]. Конфигурации четырех оптических изомеров могут предполагаться на основании конфигурации исходного вещества и константы взаимодействия продукта. В ходе реакции связи, соединенные с хиральным центром, не разрываются и относительный размер соединенных групп не изменяется, следовательно, конфигурация С 1 связанного с N в конечном продукте является такой же, как у исходного вещества норлейцина. Кроме того, конфигурация С 2 может предполагаться по константе взаимодействия между Н и Н, соединенными с хиральным центром. Конкретно, более низкая константа взаимодействия(1R,2S); более высокая константа взаимодействия J представляет треоформу и соответствующую конфигурацию, которая является (1S,2S) или (1R,2R). Подробности приведены в табл. 2. Таблица 2 Предполагаемые конфигурации для четырех оптических изомеров Эксперимент in vitro no ингибированию обратного захвата медиатора моноаминов показывает, что производные 1-бутил-2-гидроксиларалкилпиперазина и их оптические изомеры настоящего изобретения представляют собой тройные ингибиторы обратного захвата, которые обладают довольно сильным ингибирующим эффектом in vitro на обратный захват медиаторов моноаминов DA, NE и 5-HT. Предпочтительное соединение (VIII-10) имеет эквивалентный ингибирующий эффект in vitro на обратный захват для 5-HT и NA по сравнению с венлафаксином и SIPIyy24 и обладает более сильной ингибирующей активностью на обратный захват для DA по сравнению с венлафаксином, SIPIyy24 и SIPI5286; соединение(VIII-9) обладает более сильной ингибирующей активностью на обратный захват всех трех медиаторов моноаминов по сравнению с венлафаксином, SIPIyy24 и SIPI5286. Исследования антидепрессивной активности in vivo на животных показывают, что соединение(VIII-9) обладает эквивалентной антидепрессивной активностью in vivo по сравнению с венлафаксином иSIPI5286 и имеет существенное различие по сравнению с группой холостого контроля; а антидепрессивная активность in vivo соединения (VIII-10) является более высокой, чем активность венлафаксина иSIPI5286. Исследование острой токсичности показывает, что LD50 (95% доверительный интервал) для предпочтительного соединения (VIII-10) составляет 1048,5 (751,33-143,7) мг/кг, МЛД (минимальная летальная дозировка) (VIII-9) превышает 2844,7 мг/кг. Их значения острой токсичности являются меньше, чем для SIPIyy24 (предпочтительного соединения в патенте Китая ZL02111934.1) и SIPI5286 (предпочтительного соединения в патенте Китая ZL200510030354.1). Исследование фармакокинетики показывает, что период полувыведения соединения (VIII-10), вводимого перорально, составляет 16,41 ч, что является более длительным, чем для (VIII-9) (5,89 ч) иSIPI5286 (5,71 ч). Биодоступность соединения (VIII-10), вводимого перорально, составляет 63,78%, что является более высоким, чем для (VIII-9) (16,32%) и SIPI5286 (51,63%). Следовательно, (VIII-10) обладает хорошими характеристиками для применения в качестве лекарственного средства. Производные 1-бутил-2-гидроксиларалкилпиперазина настоящего изобретения обладают тройным ингибирующим эффектом на обратный захват 5-HT, NA и DA и могут применяться для получения антидепрессантов. Производные настоящего изобретения могут вводиться пациентам, нуждающимся в этом, в форме композиции путем перорального введения, инъекции и т.п. Композиция содержит терапевтически эффективное количество производных настоящего изобретения в качестве активного ингредиента, наряду с одним или более фармацевтически приемлемыми носителями. Носитель означает общепринятые носители в фармацевтической области, например разбавители,эксципиенты, такие как вода; адгезив, такой как производные целлюлозы, желатин, поливинилпирролидон и т.п.; наполнители, такие как крахмал и т.п.; дезинтеграторы, такие как карбонат кальция, бикарбонат натрия и т.п.; в дополнение, другие адъюванты, такие как ароматизаторы и подсластители, могут быть добавлены в композицию. Для перорального введения его можно составлять в общепринятые твердые препараты, такие как таблетка, порошок или капсула; для инъекционного введения его можно составлять в инъекционный раствор. Различные препараты композиции в соответствии с настоящим изобретением могут быть получены с использованием общепринятых способов в фармацевтической области, где содержание активного ингредиента составляет от 0,1 до 99,5% (по массе). Количество, вводимое в настоящем изобретении, может изменяться в соответствии с путем введения, возрастом и массой тела пациента, типом и тяжестью заболевания, подлежащего лечению, и т.п., и суточная доза составляет 5-30 мг/кг массы тела (пероральная) или 1-10 мг/кг массы тела (инъекция). Производные настоящего изобретения показали антагонизм против депрессии при испытаниях на животных. Для преодоления недостатка SIPI5286 структурную модификацию осуществляли, используяSIPI5286 в качестве ведущего соединения. Автор настоящего изобретения обнаружил, что, когда группой заместителя у С 1 является жирный углеводород, с увеличением углеродной цепи от 1 до 4, эритро- и треоизомеры соединений показывают лучшее ингибирование обратного захвата 5-HT, NA и DA, сравнимое с SIPIyy24 и положительным контролем венлафаксином. Когда заместителем является бутил, т.е. для производных 1-бутил-2-гидроксиларалкилпиперазина настоящего изобретения, активность по ингибированию обратного захвата 5-HT, NA и DA его эритро- и треоизомеров достигает максимума, более высокого, чем для SIPIyy24, SIPI5286 и положительного контроля венлафаксина. Однако, когда заместителем С 1 является пентил, ингибирующая активность резко снижается. Когда определение структуры было проведено у других соединений в ZL02111934.1, автор настоящего изобретения обнаружил, что, когда заместителем С 1 является бутил, активность по ингибированию обратного захвата 5-HT, NA и DA достигает максимального значения из всех. Следовательно, автор настоящего изобретения пришел к выводу,что производные 1-бутил-2-гидроксиларалкилпиперазина обладают наивысшей активностью среди производных арилалканолпиперазина. Последующие исследования in vivo на животных также показывают, что по сравнению с производными арилалканолпиперазина, раскрытыми в патенте Китая ZL02111934.1, и оптическим изомером, раскрытым в патенте Китая ZL200510030354.1, производные 1-бутил-2-гидроксиларалкилпиперазина настоящего изобретения имеют преимущества по более высокой активности против депрессии, более низкой токсичности, более высокой биодоступности, более длительному периоду полувыведения и лучшим характеристикам для лекарственного средства. В заключение, производные 1-бутил-2-гидроксиларалкилпиперазина в настоящем изобретении, по сравнению с имеющими в настоящее время клиническое применение антидепрессантами двойного назначения (например, венлафаксином), могут иметь более высокую активность, более широкие показания,более низкую токсичность и меньше нейротоксических побочных реакций. Производные имеют преимущества по более высокой активности против депрессии, более низкой токсичности, более высокой биодоступности, более длительному периоду полувыведения и лучшим характеристикам для лекарственного средства по сравнению с производными арилалканолпиперазина, раскрытыми в патенте КитаяZL02111934.1, и оптическими изомерами, раскрытыми в патенте Китая ZL200510030354.1. Конкретные методы осуществления изобретения. Общий способ 1. Синтез гидрохлорида N-аралкилпиперазина (II). Гексагидрат пиперазина (350 ммоль, от Шанхайской станции химических реактивов), твердый KOH(100 ммоль) и CTAB (бромид гексадецилтриметиламмония, 1 ммоль) добавляли к 18 мл воды и нагревали для растворения. 140 мл раствора аралкилхлорида (100 ммоль, доступен на рынке) в бензоле добавляли по каплям при температуре 70C. После прокапывания реакционную смесь кипятили с обратным холодильником в течение 1,5 ч и дали возможность отстояться и расслоиться. Затем органическую фазу промывали 50 мл воды и 50 мл насыщенного раствора NaCl, соответственно, сушили MgSO4 и отфильтровывали. Растворитель упаривали досуха при пониженном давлении и концентрат затем растворяли в 50 мл абсолютного спирта и доводили до рН 3, добавляя по каплям раствор HCl/C2H5OH. Затем твердое вещество осаждали и фильтровали и сушили. Гидрохлорид N-аралкилпиперазина получали перекристаллизацией из этанола. Выход составлял 80-86%. Общий способ 2. Синтез 2-гексанон-5-хлор-6-метоксилнафталина (IV). Соединение (III) (28,4 ммоль) растворяли в дихлорметане (30 мл). Добавляли AlCl3 (30,8 ммоль),реакционную смесь перемешивали в течение 1 ч при комнатной температуре. При постепенном растворении AlCl3 цвет раствора становился темнее до светло-коричневого. К смеси медленно по каплям добавляли гексаноилхлорид (23,7 ммоль), контролируя температуру ниже 10C. После прокапывания реакционную смесь нагревали естественным путем до комнатной температуры и перемешивали в течение 1 ч. Цвет реакционного раствора становился темнее до коричневого. Реакционный раствор выливали в смесь хлористо-водородная кислота (20 мл)/раздробленный лед (50 г) при перемешивании, и цвет органической фазы становился светлее от светло-желтого до желтого. Органическую фазу отделяли, промывали водой (320 мл) до тех пор, пока водная фаза не станет нейтральной, и сушили безводным Na2SO4 в течение ночи. Осушитель отфильтровывали, остаток промывали небольшим количеством дихлорметана. Затем растворитель фильтрата упаривали и получали светло-желтое маслянистое вещество. Продукт (IV) отделяли в виде светло-желтого масляного продукта колоночной хроматографией (этилацетат:петролейный эфир=1:400-1:60), давали отстояться и отверждали. Выход составлял приблизительно 80%. Общий способ 3. Синтез 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (V). Соединение (IV) (21 ммоль) растворяли в смеси этилацетата (50 мл) и хлороформа (50 мл), затем добавляли CuBr2 (40,2 ммоль), реакцию осуществляли при кипячении с обратным холодильником в течение 3 ч. Полученный CuBr отфильтровали. Фильтрат промывали водой (320 мл) и сушили безводнымNa2SO4 в течение ночи. Осушитель отфильтровывали, причем остаток промывали небольшим количеством этилацетата. Растворитель фильтрата упаривали. Светло-желтое кристаллическое твердое вещество получали перекристаллизацией из этанола. Выход составлял приблизительно 85%. Общий способ 4. Синтез гидрохлорида N1-аралкил-N4-[1-(5'-хлор-6'-метоксил-2'-нафтоил)пентил]пиперазина (VI). Гидрохлорид(10 ммоль),2-(-бромгексанон)-5-хлор-6 метоксилнафталина (V) (12 ммоль), йодид калия (1 ммоль) и безводный K2CO3 (35 ммоль) поместили в ацетон (50 мл). Реакцию осуществляли перемешиванием при кипячении с обратным холодильником в течение от 8 до 12 ч. После фильтрации растворитель упаривали досуха при пониженном давлении. Добавляли 50 мл воды, реакционную смесь экстрагировали EtOAc (3100 мл). Слои сложного эфира отделяли и последовательно промывали 20 мл воды и 30 мл насыщенного раствора NaCl, затем сушилиMgSO4. После фильтрации растворитель упаривали. Концентрат растворяли добавлением 30 мл этанола и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и перекристаллизовывали из смеси этанол/вода или метанола с получением соединения (VI) с выходом 6085%. Общий способ 5. Синтез гидрохлорида N1-аралкил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазина (VII). Изопропоксид алюминия (35 ммоль) растворяли в 80 мл изопропанола и добавляли безводный AlCl3(3,5 ммоль). После нагрева до 45-50C смесь перемешивали в течение 30 мин до прозрачности, добавляли раствор N1-аралкил-N4-аралкилацилалкилпиперазина (10 ммоль) в изопропаноле. Температура возросла до 60-65C, реакцию осуществляли до исчезновения пятна вещества (обнаружение по ТСХ, 6-48 ч). Затем добавляли 15% раствор NaOH (по массе) для доведения рН до приблизительно 7. Экстракцию проводили дихлорметаном или этилацетатом и экстракт промывали насыщенным раствором NaCl (20 мл),сушили MgSO4. После фильтрации растворитель фильтрата упаривали при пониженном давлении. Остаток растворяли в 20 мл этанола и доводили до рН 2 с помощью HCl/C2H5OH. Твердое вещество осаждали и отфильтровывали с выходом 85,95%. Пример 1.(1SR,2RS)-N1-п-Метоксилфенил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'нафтил)гидроксиэтил]пиперазин (эритроформа) (VIII-1). 4,2 г гидрохлорида N1-п-метоксилфенил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1-п-метоксилфенилпиперазин (10 ммоль) и 2-(бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 79%, т.пл.=231,5-233,6C (разл.). Затем осуществляли восстановление карбонила сN1-п-метоксилфенил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазином в соответствии с общим способом 5 и 3,78 г гидрохлорида N1-п-метоксилфенил-N4-[1-бутил-2-гидрокси-2(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 90%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией с получением эритроформы, затем растворяли в этаноле и доводили до рН 2 HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-1) получали перекристаллизацией из смеси этанол/вода или метанола. Элементный анализ соединения показал, что в соединении содержалось 0,5 молекул кристаллической воды. т.пл.=221,6-223,2C (разл.).(1SR,2SR)-N1-п-Метоксилфенил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'нафтил)гидроксиэтил]пиперазин (треоформа) (VIII-2). 4,2 г гидрохлорида N1-п-метоксилфенил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1-п-метоксилфенилпиперазин (10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 79%, т.пл.=231,5-233,6C (разл.). Затем осуществляли восстановление карбонила- 10020716 в N1-п-метоксилфенил-N-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,78 г гидрохлорида N1-п-метоксилфенил-N4-[1-бутил-2-гидрокси-2-(5 хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 90%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Треоформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-2) получали перекристаллизацией из смеси этанол/вода или метанола. Элементный анализ соединения показал, что в соединении содержалось 2 молекулы кристаллической воды. т.пл.=220,4-222,8C (разл.).(1SR,2RS)-N1-о-Метоксилфенил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'нафтил)гидроксиэтил]пиперазин (эритроформа) (VII1-3). 4,03 г гидрохлорида N1-о-метоксилфенил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1-о-метоксилфенилпиперазин (10 ммоль) и 2-(бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 78%, т.пл.=232,4-234,1C (разл.). Затем восстановление карбонила осуществляли в N1-o-метоксилфенил-N4-[1-бутил-2-карбонил-2-(5 хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,58 г гидрохлорида N1-о-метоксилфенил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 89%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Эритроформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/С 2 Н 5 ОН (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-3) получали перекристаллизацией из смеси этанол/вода или метанола. Элементный анализ соединения показал, что в соединении содержалось 2 молекулы кристаллической воды. т.пл.=227,3-229,1C (разл.).(1SR,2SR)-N1-о-Метоксилфенил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'нафтил)гидроксиэтил]пиперазин (треоформа) (VIII-4). 4,03 г гидрохлорида N1-о-метоксилфенил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1-о-метоксилфенилпиперазин (10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 78%, т.пл.=232,4-234,1C (разл.). Затем восстановление карбонила осуществляли в N1-o-метоксилфенил-N4-[1-бутил-2-карбонил-2-(5 хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,58 г гидрохлорида N1-о-метоксилфенил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 89%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Треоформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-4) получали перекристаллизацией из смеси этанол/вода или метанола. Элементный анализ соединения показал, что в соединении содержалось 2 молекулы кристаллической воды. т.пл.=223,5-225,0C (разл.).(10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 79%, т.пл.=231,5-233,6C (разл.). Затем осуществляли восстановление карбонила в N1-м-хлорфенил-N4-[1-бутил-2-карбонил-2-(5 хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,78 г гидрохлорида N1-м-хлорфенил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 90%. Полученное соединение преобразовывали в его свободную щелочную форму и отде- 11020716 ляли колоночной хроматографией. Эритроформу получали и затем растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение(VIII-5) получали перекристаллизацией из смеси этанол/вода или метанола. т.пл.=224,2-226,8C (разл.).(10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 79%, т.пл.=231,5-233,6C (разл.). Затем осуществляли восстановление карбонила в N1-м-хлорфенил-N4-[1-бутил-2-карбонил-2-(5 хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,78 г гидрохлорида N1-м-хлорфенил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 90%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Треоформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-6) получали перекристаллизацией из смеси этанол/вода или метанола. т.пл.=220,4-222,8C (разл.).(1SR,2RS)-N1-(2,3-Диметилфенил)-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'нафтил)гидроксиэтил]пиперазин (эритроформа) (VIII-7). 3,80 г гидрохлорида N1-(2,3-диметилфенил)-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин 2-ил)]этилпиперазина синтезировали, используя N1-(2,3-диметилфенил)пиперазин (10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 80%, т.пл.=232,5-235,6C (разл.). Затем восстановление карбонила осуществляли в N1-(2,3-диметилфенил)-N4-[1-бутил-2-карбонил-2(5-хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,34 г гидрохлорида N1-(2,3-диметилфенил)-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 88%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Эритроформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-7) получали перекристаллизацией из смеси этанол/вода или метанола. т.пл.=218,5-220,8C (разл.).(1SR,2SR)-N1-(2,3-Диметилфенил)-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'нафтил)гидроксиэтил]пиперазин (треоформа) (VIII-8). 3,80 г гидрохлорида N1-(2,3-диметилфенил)-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин 2-ил)]этилпиперазина синтезировали, используя N1-(2,3-диметилфенил)пиперазин (10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 80%, т.пл.=232,5-235,6C (разл.). Затем восстановление карбонила осуществляли в N1-(2,3-диметилфенил)-N4-[1-бутил-2-карбонил-2(5-хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,34 г гидрохлорида N1-(2,3-диметилфенил)-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 88%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Треоформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/С 2 Н 5 ОН (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-8) получали перекристаллизацией из смеси этанол/вода или метанола.N1-бензил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1-бензилпиперазин (10 ммоль) и 2-(-бромгексанон)-5 хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 78%, т.пл.=241,7-243,3C (разл.). Затем восстановление карбонила осуществляли в N1-бензил-N4-[1-бутил-2-карбонил-2-(5-хлор-6 метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,73 г гидрохлоридаN'-бензил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 89%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Эритроформу получали и растворяли в этаноле и доводили до рН 2 с помощьюHCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-9) получали перекристаллизацией из смеси этанол/вода или метанола. Элементный анализ соединения показал, что в соединении содержалось 2 молекулы кристаллической воды. т.пл.=225,0-225,8C (разл.).N1-бензил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1-бензилпиперазин (10 ммоль) и 2-(-бромгексанон)-5 хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 78%, т.пл.=241,7-243,3C (разл.). Затем восстановление карбонила осуществляли в N1-бензил-N4-[1-бутил-2-карбонил-2-(5-хлор-6 метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,73 г гидрохлоридаN1-бензил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 89%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Треоформу получали и растворяли в этаноле и доводили до рН 2 с помощьюHCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-10) получали перекристаллизацией из смеси этанол/вода или метанола. Элементный анализ показал, что в соединении содержалось 2 молекулы кристаллической воды. т.пл.=223,1-224,3C (разл.).(10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 77%, т.пл.=233,5-235,7C (разл.). Затем осуществляли восстановление карбонила в N1-п-нитробензил-N4-[1-бутил-2-карбонил-2-(5 хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,47 г гидрохлорида N1-п-нитробензил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 88%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Эритроформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-11) получали перекристаллизацией из смеси этанол/вода или метанола.(10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 77%, т.пл.=233,5-235,7C (разл.). Затем восстановление карбонила осуществляли в N1-п-нитробензил-N4-[1-бутил-2-карбонил-2-(5 хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5. 3,47 г гидрохлоридаN1-п-нитробензил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 88%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Треоформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-12) получали перекристаллизацией из смеси этанол/вода или метанола. т.пл.=226,8-229,0C (разл.).(эритроформа) (VIII-13). 3,76 г гидрохлорид N1-п-аминобензил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1-п-аминобензилпиперазин (10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 78%, т.пл.=233,6-235,9C (разл.). Затем восстановление карбонила осуществляли в N1-п-аминобензил-N4-[1-бутил-2-карбонил-2-(5 хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,27 г гидрохлорида N1-п-аминобензил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 87%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Эритроформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-13) получали перекристаллизацией из смеси этанол/вода или метанола. т.пл.=219,4-221,0C (разл.).(1SR,2SR)-N1-п-Аминобензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'нафтил)гидроксиэтил]пиперазин (треоформа) (VIII-14). 3,76 г гидрохлорида N1-п-аминобензил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1-п-аминобензилпиперазин (10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 78%, т.пл.=233,6-235,9C (разл.). Затем восстановление карбонила осуществляли в N1-п-аминобензил-N4-[1-бутил-2-карбонил-2-(5 хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,27 г гидрохлорида N1-п-аминобензил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 87%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Треоформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-14) получали перекристаллизацией из смеси этанол/вода или метанола. т.пл.=215,2-218,0C (разл.).N1-(3',4',5'-триметоксибензил)-N4-[1-бутил-2-карбонил-2-(5-хлор-6 метоксинафталин-2-ил)]этилпиперазина синтезировали, используя N1-(3',4',5'-триметоксибензил)пиперазин (10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 75%, т.пл.=235,5-238,6C (разл.). Затем осуществляли восстановление карбонила в N1-(3',4',5'-триметоксибензил)-N4-[1-бутил-2 карбонил-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,63 г гидрохлоридаN1-(3',4',5'-триметоксибензил)-N4-[1-бутил-2-гидрокси-2-(5-хлор-6 метоксинафталин-2-ил)]этилпиперазина получали с выходом 87%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Эритроформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-15) получали перекристаллизацией из смеси этанол/вода или метанола. т.пл.=221,3-223,6C (разл.).N1-(3',4',5'-триметоксибензил)-N4-[1-бутил-2-карбонил-2-(5-хлор-6 метоксинафталин-2-ил)]этилпиперазина синтезировали, используя N1-(3',4',5'-триметоксибензил)пиперазин (10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 75%, т.пл.=235,5-238,6C (разл.). Затем восстановление карбонила осуществляли в N1-(3',4',5'-триметоксибензил)-N4-[1-бутил-2 карбонил-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,63 г гидрохлоридаN1-(3',4',5'-триметоксибензил)-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 87%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Треоформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-16 получали перекристаллизацией из смеси этанол/вода или метанола. т.пл.=226,5-228,8C (разл.).(эритроформа) (VIII-17). 4,22 г гидрохлорида N1 фенетил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1 фенетилпиперазин (10 ммоль) и 2-(-бромгексанон)5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 78%, т.пл.=241,7-243,3C (разл.). Затем осуществляли восстановление карбонила в N1 фенетил-N4-[1-бутил-2-карбонил-2-(5-хлор 6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,75 г гидрохлоридаN1 фенетил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 89%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Эритроформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-17) получали перекристаллизацией из смеси этанол/вода или метанола. т.пл.=235,0-237,6C (разл.).(треоформа) (VII-18). 4,22 г гидрохлорида N1 фенетил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1 фенетилпиперазин (10 ммоль) и 2-(-бромгексанон)5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 78%, т.пл.=241,7-243,3C (разл.). Затем осуществляли восстановление карбонила в N1 фенетил-N4-[1-бутил-2-карбонил-2-(5-хлор 6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 3,75 г гидрохлоридаN1 фенетил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 89%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Треоформу получали и растворяли в этаноле и доводили до рН 2 с помощью HCl/С 2 Н 5 ОН (5 н.) Осажденное твердое вещество отфильтровывали и соединение (VIII-18) получали перекристаллизацией из смеси этанол/вода или метанола, т.пл.=224,0-226,1C.(10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 76%, т.пл.=212,9-215,2C (разл.). Затем восстановление карбонила осуществляли в N1-бензгидрил-N4-[1-бутил-2-карбонил-2-(5-хлор 6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 4,83 г гидрохлоридаN1-бензгидрил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 85%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Эритроформу получали, растворяли в этаноле и доводили до рН 2 с помощью HCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-19) получали перекристаллизацией из смеси этанол/вода или метанола, т.пл.=183,0-184,6C (разл.).(10 ммоль) и 2-(-бромгексанон)-5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 76%, т.пл.=212,9-215,2C (разл.). Затем осуществляли восстановление карбонила в N1-бензгидрил-N4-[1-бутил-2-карбонил-2-(5-хлор 6-метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 4,13 г гидрохлоридаN1-бензгидрил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 85%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Треоформу получали, растворяли в этаноле и доводили до рН 2 с помощьюHCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-20) получали перекристаллизацией из смеси этанол/вода или метанола, т.пл.=179,3-181,1C (разл.).N1-циннамил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1-циннамилпиперазин (10 ммоль) и 2-(-бромгексанон)5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 82%, т.пл.=232,2-234,0C (разл.). Затем осуществляли восстановление карбонила в N1-циннамил-N4-[1-бутил-2-карбонил-2-(5-хлор-6 метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 4,19 г гидрохлоридаN1-циннамил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил)]этилпиперазина получали с выходом 91%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Эритроформу получали, растворяли в этаноле и доводили до рН 2 с помощью HCl/С 2 Н 5 ОН (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-21) получали перекристаллизацией из смеси этанол/вода или метанола, т.пл.=228,8-230,1C (разл.).N1-циннамил-N4-[1-бутил-2-карбонил-2-(5-хлор-6-метоксинафталин-2 ил)]этилпиперазина синтезировали, используя N1-циннамилпиперазин (10 ммоль) и 2-(-бромгексанон)5-хлор-6-метоксилнафталин (10 ммоль), в соответствии с общим способом 2 - общим способом 4, с выходом 82%, т.пл.=232,2-234,0C (разл.). Затем осуществляли восстановление карбонила в N1-циннамил-N4-[1-бутил-2-карбонил-2-(5-хлор-6 метоксинафталин-2-ил)]этилпиперазине в соответствии с общим способом 5 и 4,19 г гидрохлоридаN1-циннамил-N4-[1-бутил-2-гидрокси-2-(5-хлор-6-метоксинафталин-2-ил]этилпиперазина получали с выходом 91%. Полученное соединение преобразовывали в его свободную щелочную форму и отделяли колоночной хроматографией. Треоформу получали, растворяли в этаноле и доводили до рН 2 с помощьюHCl/C2H5OH (5 н.). Осажденное твердое вещество отфильтровывали и соединение (VIII-22) получали перекристаллизацией из смеси этанол/вода или метанола, т.пл.=194,1-195,6C (разл.).MS: m/z 493 (M+). 1 Н ЯМР (ДМСО-d6):0,36-1,49 (м, 9H, CH2CH2CH2CH3), 2,43-3,27 (м, 11 Н, CH+CH2+пиперазин-Н),3,98 (с, 3 Н, OCH3), 4,5 (д, 1 Н, CH, J=8,0 Гц), 5,18 (с, 1H, ОН), 6,24-6,55 (м, 2 Н, CH=CH), 7,21-8,05 (м,10 Н, Ar-H). Пример 23. Синтез (S)-2-(1,3-дикарбонилизоиндол)гексановой кислоты (2). 6,55 г L-норлейцина (0,05 моль), 7,40 г фталевого ангидрида (0,05 моль), 0,8 мл триэтиламина добавляли в 150 мл толуола при перемешивании при 110C. После проведения реакции посредством кипячения с обратным холодильником в течение 24 ч реакция была завершена. Растворитель упаривали после отстаивания для охлаждения и добавляли 50 мл воды. Экстракцию проводили этилацетатом (350 мл) и этилацетатный слой промывали насыщенным раствором NaCl, сушили безводным MgSO4. Растворитель фильтрата упаривали после фильтрации смеси и 12,28 г белого твердого вещества получали с выходом 90,3%.H ЯМР (ДМСО-d6):0,86-1,37 (м, 9 Н, CH2CH2CH2CH3), 4,46 (м, 1 Н, CH), 7,25-7,86 (м, 4 Н, Ar-H). Пример 24. Получение (R)-2-(1,3-дикарбонилизоиндол)гексановой кислоты проводили так же, как получениеMS: m/z 262 (М+). 1 Н ЯМР (ДМСО-d6):0,90-1,25 (м, 9H, CH2CH2CH2CH3), 4,68 (м, 1 Н, CH), 7,42-7,91 (м, 4 Н, Ar-H). Пример 25. Синтез (S)-2-[2-(1,3-дикарбонилизоиндол)гексаноил]-5-хлор-6-метоксилнафталина (5). 10,45 г соединения 2 (0,04 моль) растворяли в 100 мл дихлорметана при 0C и 8,15 мл (0,1 моль) оксалилхлорида добавляли по каплям в бане со льдом. После прокапывания добавляли 8 капель пиридина. Температура медленно повышалась до комнатной температуры и смесь перемешивали в течение 20 ч. Избыточный хлорангидрид и растворитель упаривали на роторном испарителе при 35C, концентрат растворяли в 100 мл дихлорметана. 9,25 г соединения 4 (0,048 моль) и 6,41 г безводного AlCl3 (0,048 моль) добавляли для взаимодействия в течение 30 ч при комнатной температуре. Реакционную смесь медленно выливали в смесь 1 н. HCl (100 мл)/лед/дихлорметан (100 мл), перемешивали и отстаивали для обеспечения разделения слоев. Водный слой экстрагировали дихлорметаном (2100 мл). Слои дихлорметана отделяли, затем промывали насыщенным раствором NaCl, сушили безводным MgSO4. Растворитель фильтрата упаривали после фильтрации. Получали темно-коричневое маслянистое вещество и отделяли колоночной хроматографией (нейтральный оксид алюминия, петролейный эфир:этилацетат=3:1). 4,68 г светло-желтого масляного вещества получали с выходом 26,8%.MS: m/z 438 (М+). 1 Н ЯМР (ДМСО-d6):1,03-1,62 (м, 9 Н, CH2CH2CH2CH3), 3,10 (с, 3 Н, OCH3), 5,14 (м, 1 Н, CH), 7,358,11 (м, 9 Н, Ar-H). Пример 27. Синтез (2S)-2-[1-гидрокси-2-(1,3-дикарбонилизоиндол)гексил]-5-хлор-6-метоксилнафталина (6). 4,36 г соединения 5 (0,01 моль) растворяли в смеси 19,2 мл толуола и 12,6 мл изопропанола, затем 10,2 г (0,05 моль) изопропоксида алюминия добавляли для взаимодействия при 100C в течение 4 ч. После завершения реакции смесь охлаждали, растворитель упаривали и добавляли 1 н. HCl (50 мл). Экстракцию проводили этилацетатом (350 мл). Этилацетатный слой промывали небольшим количеством воды и насыщенного раствора NaCl, сушили безводным MgSO4. Растворитель упаривали после фильтрации и 4,32 г светло-желтого твердого вещества получали с выходом 98,6%.(0,01 моль). Смесь перемешивали в течение 3 ч при комнатной температуре и получали белое твердое вещество. После фильтрации растворитель фильтрата упаривали и экстракцию проводили смесью вода(20 мл)/дихлорметан (320 мл). Отделенный слой дихлорметана промывали насыщенным растворомNaCl, сушили безводным MgSO4. После фильтрации растворитель фильтрата упаривали и 1,13 г белого твердого вещества получали с выходом 73,4%.(м, 1 Н, CHOH), 7,75-8,29 (м, 5 Н, Ar-H). Пример 31. Получение гидрохлорида (1S,2R)-N1-бензил-N4-[1-бутил-2-гидрокси-2-(5'-хлор-6'-метоксил-2')нафтилэтил]пиперазина (IX-23) и гидрохлорида (1S,2S)-N1-бензил-N4-[1-бутил-2-гидрокси-2-(5'-хлор-6'метоксил-2')нафтилэтил]пиперазина (IX-24). К 0,924 г соединения 7 (0,003 моль) добавляли 10 мл ацетонитрила, 2 мл триэтиламина и 1,624 г соединения 8 (0,007 моль, полученного в соответствии с патентом США 4748726), смесь нагревали и кипятили с обратным холодильником в течение 20 ч, пока завершение реакции не обнаруживали по ТСХ. Ацетонитрил упаривали, экстракцию проводили хлороформом (350 мл) и водой. Слои хлороформа отделяли и сушили MgSO4. Хлороформ упаривали и получали желтое маслянистое вещество, которое представляло собой смесь двух изомеров (9).MS: m/z 465 (M+). Смесь (9) разделяли колоночной хроматографией на силикагеле со смесью дихлорметан:метанол=200:1 в качестве элюента. 0,47 г желтого маслянистого (1S,2S) изомера (выход: 34%) первоначально элюировали и растворяли в 20 мл метанола. Смесь хлористо-водородная кислота/этанол использовали для доведения рН до 2. Белое твердое вещество осаждалось и 0,25 г белого продукта получали фильтрацией. 0,12 г желтого маслянистого (1S,2R) изомера (выход: 8,5%) элюировали позже и растворяли в 10 мл метанола. Смесь хлористо-водородная кислота/этанол применяли для доведения pH до 2. Осаждалось светло-желтое твердое вещество и 0,05 г желтого продукта получали фильтрацией и нагревом досуха. Получение: активный ингредиент смешивали с сахарозой и кукурузным крахмалом, затем смесь смачивали добавлением воды, равномерно перемешивали, сушили, дробили и просеивали, затем добавляли стеарат кальция. Полученную смесь перемешали до гомогенности и затем прессовали в таблетки. Масса таблетки составляла 200 мг на таблетку, содержащую 10 мг активного ингредиента. Пример 34. Инъекция: Получение: активный ингредиент растворяли и равномерно смешивали с водой для инъекций, затем фильтровали. Полученную смесь распределяли в ампулы в стерильных условиях. Масса составляла 10 мг на ампулу, содержащую 2 мг активного ингредиента. Пример 35. Ингибирующий эффект соединений на обратный захват 5-HT, NA и DA синаптосомами мозга. Проводили исследования обратного захвата нейромедиаторов моноаминов синаптосомами мозга,что в настоящее время является важным методом, принятым во всем мире при фармакологических исследованиях центральной нервной системы. Этот метод может не только применяться для исследования механизма действия лекарственного средства, но также использоваться для скрининга новых лекарств,действующих по этому механизму. В настоящем исследовании проводили исследования ингибирующего эффекта соединений на обратный захват 5-HT, NA и DA синаптосомами мозга, применяя венлафаксин(эффективный двойной ингибитор обратного захвата 5-HT и NA) и 6-гидрокси-DA в качестве положительных контролей. 1. Получение синаптосом мозга крысы. Самцов крыс SD умерщвляли смещением шейных позвонков и затем их мозг быстро отбирали декапитацией и помещали в лед. Относящиеся к исследованию ткани мозга (для эксперимента по обратному захвату [3 Н]5-HT и [3H]NA, брали префронтальную кору; для эксперимента по обратному захвату[3H]DA брали полосатое тело) отделяли и взвешивали. Добавляли 10-кратный избыток (об./М) 0,32 моль/л ледяного раствора сахарозы и гомогенизировали электрически с помощью стекла-тефлона. Гомогенат центрифугировали при 4C при 1000g10 мин. Затем супернатант отбирали и центрифугировали при 4C при 17000g20 мин. Осадок суспендировали в 30 объемах KRH буфера (124 мМ NaCl,4,8 мМ KCl, 1,2 мМ CaCl2, 1,2 мМ MgSO4, 1,0 мМ KH2PO4, 22 мМ NaHCO3, 25 мМ HEPES, 10 мМ глюкозы, 10 мкМ паргилина, 0,2 мг/мл аскорбиновой кислоты) и затем сохраняли в бане со льдом для использования. (Для эксперимента с обратным захватом NA необходимую кору суспендировали в 20 объемахKRH буфера). 2. Эксперименты с обратным захватом [3 Н]-HT/NA/DA. Базовый раствор тестируемого вещества оттаивали непосредственно перед применением и разбавляли буфером KBH до 100 мкмоль/л. 50 мкл его добавляли в 500 мкл общей реакционной системы, и конечная концентрация составляла 10 мкмоль/л. Затем добавляли 50 мкл суспендированных синаптосом,- 19020716 полученных выше, и смешивали до гомогенности, инкубировали в водяной бане в течение 30 мин при 37C. Затем добавляли 10 нмоль/л [3 Н]5-HT (50 нмоль/л [3H]DA или 60 нмоль/л [3H]NA). После инкубации при 37C в течение 10 мин реакционную систему немедленно отбирали и реакцию останавливали добавлением 2 мл ледяного буферного раствора 150 ммоль/л Трис-HCl. Образцы собирали на круговую мембрану из стекловолокна посредством вакуум-фильтрации и мембрану промывали 3 раза 3 мл ледяного буферного раствора Трис-HCl. Фильтрационную мембрану удаляли, прокаливали в течение 15 мин в печи с нагревом в дальней инфракрасной области и помещали в ЕР пробирку. В течение ночи добавляли 1,5 мл жидкостного сцинтиллятора и тестирование проводили на жидкостном сцинтилляционном счетчике. Для контроля с растворителем в пробирку для общего связывания и пробирку для неспецифического связывания тестируемое вещество не добавляли; в пробирку для общего связывания добавляли 50 мкл растворителя. 3. Результаты. Принимая степень ингибирования 0,1 мМ венлафаксина и 6-гидрокси DA обратного захвата моноамина за 100%, интенсивность ингибирования для соединений по сравнению с венлафаксином и 6 гидрокси DA показана в табл. 3. Таблица 3 Процент ингибирования обратного захвата 5-HT, NA и DA синаптосомами мозга 4. Вывод. Производные 1-бутил-2-гидроксиаралкилпиперазина настоящего изобретения являются тройными ингибиторами обратного захвата, которые обладают сильным ингибированием in vitro на обратный захват моноаминовых медиаторов DA, NE и 5-HT. Активности по ингибированию тройного обратного захвата предпочтительных соединений (VIII-9) и (VIII-10) были выше, чем для положительного контроля венлафаксина. Пример 36. Определение IC50 предпочтительных соединений (VIII-9) и (VIII-10) для ингибирования обратного захвата 5-HT, NA и DA синаптосомами мозга. 1. Получение синаптосом мозга крысы (так же, как в примере 35). 2. Исследование ингибирования обратного захвата (IC50) на [3H]5-HT/NA/DA. Соединения применяли для исследований по эффекту ингибирования (IC50) на обратный захват[3 Н]5-HT/NA/DA. По меньшей мере 5 концентраций каждый раз устанавливали для каждого тестируемого соединения. Каждую концентрацию определяли по среднему значению двойных пробирок и тест повторяли больше 3 раз. Ряд концентраций соединений получали посредством градиентного разбавления перед применением. 50 мкл пробы добавляли в общую реакционную систему. Затем 50 мкл суспендированной синаптической мембраны добавляли и гомогенно перемешивали, инкубировали в водяной бане при 37C в течение 30 мин. Затем добавляли 10 нмоль/л [3 Н]5-HT (или 50 нмоль/л [3H]DA или 40 нмоль/л[3H]NA) и инкубировали в водяной бане при 37C в течение 10 мин (для [3H]NA инкубировали в течение 5 мин.) Для эксперимента с обратным захватом [3 Н]5-HT в пробирку для неспецифического связывания добавляли 50 мкл флуоксетина (100 мкмоль/л); для эксперимента с обратным захватом [3H]DA в пробирку для неспецифического связывания добавляли 50 мкл кокаина (600 мкмоль/л); для эксперимента с обратным захватом [3H]NA в пробирку для неспецифического связывания добавляли 50 мкл десипрамина(100 мкмоль/л); для контрольного эксперимента с растворителем растворитель добавляли в общую пробирку для связывания, без добавления тестируемого соединения. 3. Обработка данных. Значение специфического связывания СРМ для каждой тестируемой пробирки с образцом = значение общего связывания СРМ для каждой тестируемой пробирки с образцом - значение неспецифического связывания СРМ для каждой тестируемой пробирки с образцом; степень ингибирования соединением(%) обратного захвата [3 Н]5-HT/NA/DA префронтальной корой и полосатым телом = 100% специфического связывания для каждой тестируемой пробирки с образцом (значение СРМ)/специфическое связы- 20020716 вание растворителя (значение СРМ)100%. Построение сигмоидальной кривой осуществляли для данных, полученных для всех тестируемых лекарственных средством с использованием Origin 6.1, и рассчитывали значение IC50. 4. Результаты определения IC50. Ингибирование соединениями (IC50) обратного захвата 5-HT, NA и DA в префронтальной коре и полосатом теле показано в табл. 4. Таблица 4 Эффект ингибирования соединениями обратного захвата Замечание: Соединение SIPIyy24 было предпочтительным соединением в патентеZL02111934.1. Соединение SIPI5286 было предпочтительным соединением в патенте 5. Вывод. Соединение (VIII-10) обладало сравнимой активностью по ингибированию in vitro обратного захвата 5-HT и NA, по сравнению с венлафаксином и SIPIyy24, и имело более высокую ингибирующую активность на обратный захват DA, по сравнению с венлафаксином, SIPIyy24 и SIPI5286; соединение (VIII9) имело более высокую ингибирующую активность на обратный захват трех видов моноаминовых медиаторов, по сравнению с венлафаксином, SIPIyy24 и SIPI5286. Пример 37. Эффект ингибирования in vitro для четырех оптических изомеров IX-23-IX-26 обратного захвата трех моноаминовых нейромедиаторов. 1. Экспериментальные материалы и методы. 3. Экспериментальные результаты (значение IC50 было получено посредством нелинейного регрессионного анализа кривой ответа степень ингибирования/концентрация). 4. Вывод. Четыре оптических изомера IX-23-IX-26 обладали высокой активностью in vitro по ингибированию трех моноаминовых медиаторов DA, NA и 5-HT и принадлежат к тройным ингибиторам обратного захвата. Пример 38. Исследования in vivo антидепрессивной активности у животных предпочтительных соединений(VIII-9) и (VIII-10). Исследования in vivo антидепрессивного эффекта у животных соединений (VIII-9) и (VIII-10) проводили на мышах с использованием Теста принудительного плавания в эксперименте с приобретенной беспомощностью (Zhang Juntian, Modern Pharmacological Experiments Methods (first volume), BeijingMedical University and Peking Union Medical University Joint Publishing House, 1998: 1064-1066), с венлафаксином в качестве положительного контроля. Таблица 5 Эффект однократного перорального введения на тест по плаванию для ICR мышей (n=12,SD) Результаты плавательного теста для ICR мышей при однократном пероральном введении показали,что соединения (VIII-9) и (VIII-10) обладали эквивалентной антидепрессивной активностью in vivo по сравнению с венлафаксином и SIPI5286, и имели значимое различие по сравнению с группой холостого контроля. ED50 для (VIII-9) составлял 34,3 мг/кг, ED50 для (VIII-10) составлял 11,8 мг/кг и ED50 дляSIPI5286 составлял 28,9 мг/кг. (VIII-10) имело наивысшую антидепрессивную активность in vivo. Таблица 6 Эффект однонедельного перорального введения на тест по плаванию для ICR мышей (n=12,SD) Результаты плавательного теста для ICR мышей при непрерывном пероральном введении в течение одной недели также показали, что соединение (VIII-9) обладает эквивалентной антидепрессивной активностью in vivo по сравнению с венлафаксином и SIPI5286 и имеет значимое различие по сравнению с группой холостого контроля. Антидепрессивная активность in vivo соединения (VIII-10) была выше, чем для венлафаксина и SIPI5286. Пример 39. Исследования острой токсичности соединения (VIII-9) и (VIII-10). Проводили исследования острой токсичности на мышах при пероральном введении соединения Результат. После статистической обработки по методу Блисса, LD50 для VIII-10 (95% доверительный интервал) составлял 1048,5 (751,33-1433,7) мг/кг, минимальная летальная дозировка (МЛД) для (VIII-9) составляла более 2844,7 мг/кг. Их острая токсичность была ниже, чем у предпочтительного соединения SIPIyy24 в патенте ZL02111934.1 и предпочтительного соединения SIPI5286 в патенте ZL20051003030354.1. Пример 40. Исследования фармакокинетики соединения (VIII-9) и (VIII-10). 1. Цель теста. Исследовать фармакокинетические параметры и биодоступность (VIII-9), (VIII-10) и SIPI5286 при однократном внутривенном и пероральном введении крысам SD. 2. Метод тестирования. 18 самцов крыс были статистически распределены в 6 групп и им вводили три соединения внутривенно и перорально соответственно. Дозировка составляла 10 мг/кг (ВВ) и 50 мг/кг (ПО). Тестируемое вещество растворяли в растворе 5% ДМСО/95% HPCD водном растворе (30%) и вводили животным внутривенно и перорально. Плазму крови собирали в ряд временных отметок после внутривенного и перорального введения и концентрации лекарства в крови определяли посредством ЖХ/МС/МС. Затем на основании концентраций лекарства в крови рассчитывали фармакокинетические параметры. 3. Результаты тестирования. 4. Вывод. Период полувыведения соединения (VIII-10) при пероральном введении составлял 16,41 ч, что является более длительным, чем для соединения (VIII-9) (5,89 ч) и SIPI5286 (5,71 ч). Биодоступность соединения (VIII-10) при пероральном введении составляла 63,78%, что было выше, чем для соединения(VIII-9) (16,32%) и SIPI5286 (51,63%). Соединение (VIII-10) обладает довольно хорошими характеристиками лекарственного средства. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Производное 1-бутил-2-гидроксиларалкилпиперазина, отличающееся тем, что представляет собой свободную щелочную форму или соль соединения формулы (1) или свободную щелочную форму или соль оптических изомеров соединения формулы (1) где соль представляет собой соль, содержащую фармацевтически приемлемые анионы;Ar1 представляет собой фенил, замещенный фенил или циннаменил, где замещенный фенил содержит от одного до четырех заместителей, представляющих собой галоген, нитро, C1-C4-алкил,C1-C4-алкокси или амино;m представляет собой целое число от 0 до 5;R1 представляет собой водород, C1-C5-алкил или фенил. 2. Производное 1-бутил-2-гидроксиларалкилпиперазина по п.1, отличающееся тем, что атомы углерода C1 и C2 в структуре являются хиральными атомами углерода, конфигурация которых является соответственно (1S,2R), (1S,2S), (1R,2S) или (1R,2R), где (1SR,2RS) является эритроизомером и (1SR,2SR) является треоизомером. 3. Производное 1-бутил-2-гидроксиларалкилпиперазина по п.1, отличающееся тем, что соль представляет собой гидрохлорид, гидробромид, сульфат, трифторацетат или метансульфонат. 4. Производное 1-бутил-2-гидроксиларалкилпиперазина по п.1, отличающееся тем, что в соли содержатся 0,5-4 молекулы кристаллизационной воды. 5. Производное 1-бутил-2-гидроксиларалкилпиперазина по любому из пп.1-4, которое выбирают из группы, содержащей:(1R,2R)-N1-бензил-N4-[1-бутил-2-(5'-хлор-6'-метоксил-2'-нафтил)гидроксиэтил]пиперазин, IX-26. 6. Фармацевтическая композиция для лечения депрессии, содержащая терапевтически эффективное количество производных 1-бутил-2-гидроксиларалкилпиперазина по любому из пп.1-5, вместе с фармацевтически приемлемыми носителями. 7. Применение производных 1-бутил-2-гидроксиларалкилпиперазина по любому из пп.1-5 в производстве антидепрессанта.
МПК / Метки
МПК: A61P 25/24, A61K 31/495, C07D 295/084
Метки: лекарственного, 1-бутил-2-гидроксиаралкилпиперазина, качестве, против, производные, депрессии, применение, средства
Код ссылки
<a href="https://eas.patents.su/26-20716-proizvodnye-1-butil-2-gidroksiaralkilpiperazina-i-ih-primenenie-v-kachestve-lekarstvennogo-sredstva-protiv-depressii.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 1-бутил-2-гидроксиаралкилпиперазина и их применение в качестве лекарственного средства против депрессии</a>
Производные имидазолов, способ их получения и их применение в качестве лекарственного средства

Номер патента: 10392
Опубликовано: 29.08.2008
Авторы: Бигг Денни, Пон Доминик, Либератор Анн-Мари
МПК: A61P 35/00, A61K 31/4164, C07D 233/64...
Метки: способ, имидазолов, лекарственного, производные, качестве, средства, получения, применение
Формула / Реферат:
1. Соединение формулы (I) в форме рацемата, энантиомера или в виде любых сочетаний этих форм, в которой X обозначает один или несколько заместителей, одинаковых или различных, выбранных из Н и галогена; Y обозначает -О- или -S-; А обозначает Н или (C1-C6)алкил; Z обозначает один или несколько заместителей, одинаковых или различных, выбранных из (C1-C6)алкила; арила; арил(C1-C6)алкила; гетероарила; Z1-Z'1; NRN-C(O)-Z'2 или Z2-Z'2; Z1 обозначает...
Новое сульфированное соединение сахара и его применение в качестве лекарственного средства

Номер патента: 16931
Опубликовано: 30.08.2012
Авторы: Мори Йоко, Сато Нориюки, Сакагути Кенго, Сахара Хироеки, Ямазаки Такаюки, Масаки Казуйоси, Охта Кейсуке, Мурата Хироси, Миура Масахико, Сугавара Фумио, Такахаси Нобуаки
МПК: A61K 41/00, A61P 35/00, A61K 31/7028...
Метки: лекарственного, соединение, сахара, применение, сульфированное, качестве, средства, новое
Формула / Реферат:
1. Сульфохиновозилацилпропандиольное соединение, представленное общей формулой (I)где R1 представляет собой C1-C22 ацильный остаток жирной кислоты и М представляет собой атом водорода или ион натрия либо кальция, или его фармацевтически приемлемые соли.2. Лекарственное средство, содержащее в качестве активного ингредиента по меньшей мере один активный ингредиент, выбранный из группы, включающей сульфохиновозилацилпропандиольное соединение по...
Производные &beta-d-нуклеозида в качестве лекарственного средства для лечения инфекции вируса гепатита c у хозяина

Номер патента: 7178
Опубликовано: 25.08.2006
Авторы: Соммадосси Жан-Пьер, Лаколла Пауло
МПК: A61K 31/7068, A61K 31/7076, A61P 31/14...
Метки: гепатита, beta-d-нуклеозида, лечения, инфекции, хозяина, производные, качестве, лекарственного, средства, вируса
Формула / Реферат:
1. Применение эффективного против вируса количества производного b-D-нуклеозида структуры или его фармацевтически приемлемой соли или пролекарства, необязательно, в фармацевтически приемлемом носителе или разбавителе при получении лекарственного средства для лечения инфекции вируса гепатита С у хозяина. 2. Применение эффективного против вируса количества производного b-D-нуклеозида структуры или его фармацевтически приемлемой соли или...
Сукцинатная и малонатная соли транс-4-((1r,3s )-6 хлор-3-фенилиндан -1-ил)-1,2,2-триметилпиперазина и их применение в качестве лекарственного средства

Номер патента: 14641
Опубликовано: 30.12.2010
Авторы: Сване Хенрик, Даль Аллан Карстен, Хауэллз Марк, Ринггар Лоне Мюнк, Лопес Де Диего Хейди, Люнгсе Ларс Оле, Бан-Андерсен Бенни, Нильсен Оле
МПК: A61P 25/18, A61K 31/495, C07C 25/22...
Метки: средства, 1-ил)-1,2,2-триметилпиперазина, применение, соли, качестве, транс-4-((1r,3s, хлор-3-фенилиндан, сукцинатная, лекарственного, малонатная
Формула / Реферат:
1. Сукцинатная соль или малонатная соль транс-4-((1R,3S)-6-хлор-3-фенилиндан-1-ил)-1,2,2-триметилпиперазина формулы (I)2. Сукцинатная соль по п.1, которая представляет собой гидросукцинатную соль соединения формулы (I).3. Кристаллическая гидросукцинатная соль соединения I, определенного в п.1.4. Соль по п.3, которая представляет собой кристаллическую альфа-форму.5. Соль по п.3 или 4, кристаллическую форму которой характеризуют порошковой...
Стероиды, замещенные в положении 11, способ их получения, их применение в качестве лекарственного средства и содержащие их фармацевтические композиции

Номер патента: 1868
Опубликовано: 22.10.2001
Авторы: Тетш Жан-Жорж, Ник Франсуа, Ван Де Вельд Патрик, Буали Иамина
МПК: A61P 19/10, C07J 41/00, A61K 31/565...
Метки: содержащие, средства, применение, получения, композиции, способ, замещенные, лекарственного, положении, стероиды, фармацевтические, качестве
Формула / Реферат:
1. Соединения общей формулы в которой n - целое число, равно 2 или 3, либо R1 и R2, идентичные или разные, означают атом водорода или алкил, содержащий от 1 до 4 атомов углерода, либо R1 и R2 образуют вместе с атомом азота, с которым они связаны, моно- или полициклический гетероцикл, имеющий от 5 до 15 звеньев, ароматический или не ароматический, содержащий, при необходимости, от 1 до 3 дополнительных гетероатомов, выбираемых из кислорода,...
Предыдущий патент: Производные 1h-имидазо[4,5-c]хинолинона
Следующий патент: Способ управления реактором гидрирования среднего дистиллята и реактор гидрирования среднего дистиллята
Случайный патент: Оконное стекло с солнцезащитными свойствами