Применение бензоконденсированных гетероциклических сульфамидных производных для понижения уровней липидов в крови







Формула / Реферат
1. Способ лечения связанного с липидами расстройства, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (I)

где R1 и R2, каждый независимо, выбран из группы, состоящей из атома водорода и C1-4алкила;
R4 выбран из группы, включающей атом водорода и C1-4алкил;
а равно целому числу от 1 до 2;
 представляет собой
представляет собой

где b равно целому числу от 0 до 4;
каждый R5 независимо выбран из группы, состоящей из галогена и C1-4алкила;
или его фармацевтически приемлемой соли.
2. Способ по п.1, включающий введение соединения формулы (I), где
R1 и R2, каждый независимо, выбран из группы, состоящей из атома водорода и C1-4алкила;
R4 выбран из группы, состоящей из атома водорода и C1-4алкила;
а равно целому числу от 1 до 2;
 представляет собой
представляет собой

где b равно целому числу от 0 до 2;
каждый R5 независимо выбран из группы, состоящей из галогена и C1-4алкила;
или его фармацевтически приемлемой соли.
3. Способ по п.2, включающий введение соединения формулы (I), где
R1 и R2, каждый независимо, выбран из группы, состоящей из атома водорода и C1-4алкила;
R4 выбран из группы, состоящей из атома водорода и метила;
а равно целому числу от 1 до 2;
 выбран из группы, состоящей из
выбран из группы, состоящей из
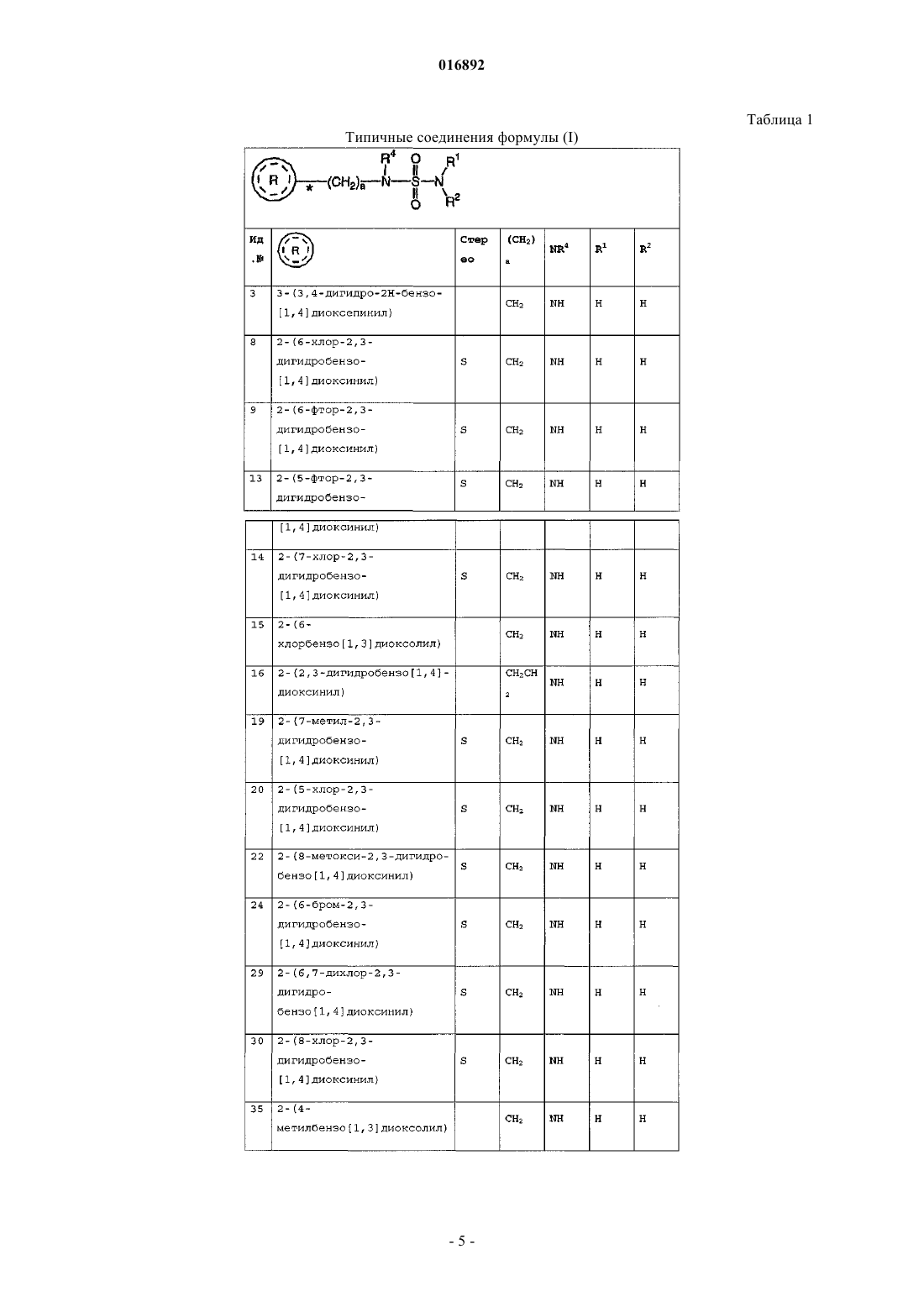
2-(2,3-дигидробензо[1,4]диоксинила),
2-(бензо[1,3]диоксолила),
2-(3,4-дигидро-2Н-бензо[1,4]диоксепинила),
2-(2,3-дигидробензо[1,4]диоксинила),
2-(6-хлор-2,3-дигидробензо[1,4]диоксинила),
2-(6-фтор-2,3-дигидробензо[1,4]диоксинила),
2-(5-фтор-2,3-дигидробензо[1,4]диоксинила),
2-(7-хлор-2,3-дигидробензо[1,4]диоксинила),
2-(6-хлорбензо[1,3]диоксолила),
2-(7-метил-2,3-дигидробензо[1,4]диоксинила),
2-(5-хлор-2,3-дигидробензо[1,4]диоксинила),
2-(6-бром-2,3-дигидробензо[1,4]диоксинила),
2-(6,7-дихлор-2,3-дигидробензо[1,4]диоксинила),
2-(8-хлор-2,3-дигидробензо[1,4]диоксинила);
или его фармацевтически приемлемой соли.
4. Способ по п.3, включающий введение соединения формулы (I), где
R1 и R2, каждый независимо, выбран из группы, состоящей из атома водорода и метила;
R4 выбран из группы, состоящей из атома водорода и метила;
а равно целому числу от 1 до 2;
 выбран из группы, состоящей из
выбран из группы, состоящей из
2-(2,3-дигидробензо[1,4]диоксинила),
2-(6-хлор-2,3-дигидробензо[1,4]диоксинила),
2-(7-хлор-2,3-дигидробензо[1,4]диоксинила),
2-(7-метил-2,3-дигидробензо[1,4]диоксинила),
2-(6-бром-2,3-дигидробензо[1,4]диоксинила) и
2-(6,7-дихлор-2,3-дигидробензо[1,4]диоксинила);
или его фармацевтически приемлемой соли.
5. Способ по п.1, где соединение формулы (I) выбрано из группы, состоящей из (2S)-(-)-N-(6-хлор-2,3-дигидробензо[1,4]диоксин-2-илметил)сульфамида и его фармацевтически приемлемых солей.
6. Способ по п.1, где связанное с липидами расстройство выбрано из группы, включающей повышенные уровни триглицеридов и низкие уровни HLD-холестерина.
7. Способ лечения связанного с липидами расстройства, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения, выбранного из группы, состоящей из
(2S)-(-)-N-(6-хлор-2,3-дигидробензо[1,4]диоксин-2-илметил)сульфамида и его фармацевтически приемлемых солей.
8. Способ по п.7, где связанное с липидами расстройство выбрано из группы, включающей повышенные уровни триглицеридов и низкие уровни HLD-холестерина.
9. Способ лечения связанного с липидами расстройства, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы

или его фармацевтически приемлемой соли;
где связанное с липидами расстройство характеризуется отличными от нормальных уровнями липидов.
Текст
ПРИМЕНЕНИЕ БЕНЗОКОНДЕНСИРОВАННЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СУЛЬФАМИДНЫХ ПРОИЗВОДНЫХ ДЛЯ ПОНИЖЕНИЯ УРОВНЕЙ ЛИПИДОВ В КРОВИ Настоящее изобретение представляет способ лечения расстройств, связанных с липидами,включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества одного или более новых бензоконденсированных гетероциклических сульфамидных производных формулы (I), которые определены в данном описании. 016892 Настоящее изобретение относится к использованию бензоконденсированных гетероциклических сульфамидных производных с целью понижения уровней липидов в крови, улучшения гликемического контроля, метаболического синдрома, гипергликемии и родственных расстройств. Сахарный диабет - это медицинское обозначение наличия повышенного уровня глюкозы в крови. У людей с диабетом либо не продуцируется инсулин, либо продуцируется очень мало инсулина, либо они не реагируют на инсулин, что ведет к увеличению количества глюкозы в крови. Наиболее общей формой диабета является диабет типа II, однажды обозначенный как диабет взрослых или инсулиннезависимый диабет (NIDDM), который может объяснять 90% случаев диабета у взрослых. Однако так как все больше и больше молодых людей имеют излишки веса или ожирение, диабет типа II становится более распространенным среди подростков и детей. Диабет можно также обозначить как гестационный диабет,диабет типа I или аутоиммунный диабет, однажды обозначенный как ювенильный диабет и диабет типаI, также называемый латентным аутоиммунным диабетом взрослых или LADA. Диабет может возникать в результате вредных традиций питания или недостатка физической активности (например, сидячего образа жизни), генетических мутаций, поражения поджелудочной железы, под воздействием лекарства (например, СПИД-терапии), или химических препаратов (например, стероидов), или заболевания (например, кистозного фиброза), синдрома Дауна, синдрома Кушинга). Два редких типа генетических дефектов, ведущих к диабету, называют диабет молодых, появляющийся в период полового созревания(MODY), и атипический сахарный диабет (ADM). Сахарный диабет типа II (инсулиннезависимый сахарный диабет или NIDDM) представляет собой метаболическое расстройство, включающее нарушение регуляции метаболизма глюкозы и резистентность к инсулину, и долгосрочные осложнения, затрагивающие глаза, почки, нервы и кровеносные сосуды. Сахарный диабет типа II обычно развивается у людей во взрослом состоянии (в середине жизни или позднее) и описан как неспособность организма производить достаточно инсулина (аномальная секреция инсулина) или неспособность организма эффективно использовать инсулин (резистентность к действию инсулина в пораженных органах и тканях). Чаще пациенты, страдающие от сахарного диабета типа II,имеют относительный дефицит инсулина. То есть у этих пациентов уровни инсулина в плазме соответствуют значениям от нормальных до высоких в абсолютных значениях, хотя они ниже, чем прогнозируемые для уровня глюкозы, которая присутствует в плазме. Сахарный диабет типа II характеризуется следующими клиническими признаками или симптомами: упорно повышенная концентрация глюкозы в плазме или гипергликемия; полиурия; полидипсия и/или полифагия; хронические микрососудистые осложнения, такие как ретинопатия, нефропатия и нейропатия; и макрососудистые осложнения, такие как гиперлипидемия и гипертензия, которая может вести к слепоте, почечному заболеванию на последней стадии, ампутации конечностей и инфаркту миокарда. Синдром X, также называемый синдромом резистентности к инсулину (IRS), метаболическим синдромом или метаболическим синдромом X, является расстройством, которое представляет факторы риска для развития сахарного диабета типа II и сердечно-сосудистого заболевания, включая непереносимость глюкозы, гиперинсулинемию и резистентность к инсулину, гипертриглицеридемию, гипертензию и ожирение. Диагностирование сахарного диабета типа II включает оценку симптомов и определение глюкозы в моче и крови. Определение уровня глюкозы в крови является необходимым для точного диагноза. Более конкретно, стандартным используемым подходом является определение уровня глюкозы в крови на голодный желудок. Однако считается, что пероральный тест толерантности к глюкозе (OGTT) является более чувствительным, чем уровень глюкозы в крови на голодный желудок. Сахарный диабет типа II связан с ухудшенной пероральной толерантностью к глюкозе (OGT). Таким образом, OGTT может оказать помощь при диагностировании сахарного диабета типа II, хотя обычно не является необходимым для диагноза диабета (Emancipator K, Am. J. Clin. Pathol. 1999 Nov; 112(5):665-74; Type 2 DiabetesMellitus, Decision Resources Inc., March 2000). OGTT позволяет оценить секреторную функцию -клеток поджелудочной железы и чувствительность к инсулину, что помогает при диагностировании сахарного диабета типа II и для оценки тяжести или прогрессирования заболевания (например, Caumo A., BergmanR.N., Cobelli С., J. Clin. Endocrinol Metab 2000, 85(11):4396-402). Чаще OGTT является чрезвычайно полезным для определения степени гипергликемии у пациентов с множественными случаями пограничных уровней глюкозы в крови на голодный желудок, которые не имеют диагноза как диабетики. Кроме того,OGTT пригоден при тестировании пациентов с симптомами сахарного диабета типа II, когда возможный диагноз аномального метаболизма углеводов четко установлен или опровергнут. Таким образом, диагностируют пониженную переносимость глюкозы у индивидуумов, которые имеют уровни глюкозы в крови на голодный желудок ниже уровней, необходимых для диагноза сахарного диабета типа II, но имеют реакцию глюкозы в плазме при проведении OGTT, промежуточную между нормальной и характерной для диабетиков. Пониженная переносимость глюкозы считается преддиабетическим состоянием, и пониженная переносимость глюкозы (как определяют посредством OGTT) является строгим предсказанием развития сахарного диабета типа II (Haffner S.M., Diabet. Med. 1997 Aug; 14Suppl 3:S12-8). Сахарный диабет типа II представляет собой прогрессирующее заболевание, связанное со снижени-1 016892 ем функции поджелудочной железы и/или другими процессами, имеющими отношение к инсулину, усугубленными повышенными уровнями глюкозы в плазме. Таким образом, сахарный диабет типа II обычно имеет длительную преддиабетическую фазу, и различные патофизиологические механизмы могут вести к патологической гипергликемии и пониженной переносимости глюкозы, например аномалиям утилизации и эффективности глюкозы, действия инсулина и/или производства инсулина при преддиабетическом состоянии (Goldberg R.B., Med. Clin. North. Am. 1998 Jul; 82 (4):805-21). Преддиабетическое состояние, связанное с непереносимостью глюкозы, может также быть связано с предрасположенностью к абдоминальному ожирению, резистентности к инсулину, гиперлипидемии и высокому кровяному давлению, то есть синдрому X (Groop L., Forsblom С., Lehtovirta M., Am. J. Hypertens 1997 Sep; 10(9 Pt 2): 172S-180S; Haffner S.M., J. Diabetes Complications 1997 Mar-Apr; ll(2):69-76;Beck-Nielsen H., Henriksen J.E., Alford F., Hother-Nielson O., Diabet Med 1996 Sep; 13(9 Suppl 6):S78-84). Таким образом, дефектный метаболизм углеводов является основным для патогенеза сахарного диабета типа II и пониженной переносимости глюкозы (Dinneen S.F., Diabet Med. 1997 Aug; 14 Suppl 3:S19-24). Действительно, существует непрерывная череда состояний от пониженной переносимости глюкозы и ухудшенных показателей глюкозы на голодный желудок до окончательного сахарного диабета типа II (Ramlo-Halsted B.A., Edelman S.V., Prim Care 1999 Dec; 26 (4): 771-89). Раннее вмешательство у индивидуумов с риском развития сахарного диабета типа II, направляемое на понижение патологической гипергликемии или ухудшенной переносимости глюкозы, может предупреждать или замедлять прогрессирование к сахарному диабету типа II и связанным осложнениям и/или синдрому X. Следовательно, путем эффективного лечения ухудшенной пероральной толерантности к глюкозе и/или повышенных уровней глюкозы в крови можно предупреждать или ингибировать прогрессирование расстройства до сахарного диабета типа II или синдрома X. Дислипидемия представляет собой группу заболеваний, характеризующихся аномальными изменениями или уровнями концентраций липопротеинов и связанных липидов, таких как триглицериды и холестерин, в крови. Липиды переносятся с током крови в форме липопротеинов, включающих, по существу, ядро из неполярных молекул, таких как, например, триглицериды и холестериновый сложный эфир,окруженное оболочкой из амфипатических липидов, главным образом, фосфолипидов. Приобретенная гиперлипидемия/гиперлипопротеинемия развивается как последствие дисбаланса питания, действия лекарственных средств или соединений или заболевания, такого как недостаточность щитовидной железы или диабет. Семейная гиперлипидемия/гиперлипопротеинемия характеризуется аутосомным наследованием и связана с повышением содержания липопротеинов и липидов в крови. Семейная гиперлипидемия/гиперлипопротеинемия подразделяется на пять категорий (типы I-V) в зависимости от состава и типа частиц липопротеина в крови. Например, при гиперлипопротеинемии типа I и типа IV уровень триглицеридов повышен преимущественно в хиломикронах и VLDL-частицах, соответственно. Вообще, существует обратная зависимость между уровнями HDL-холестерина и триглицеридов,которые вносят вклад в дислипидемию. Если оставить дислипидемию (например, низкий уровень HDLхолестерина и высокие уровни триглицеридов или LDL-холестерина) нелеченной, то можно обострить другие состояния, такие как панкреатит, аномальную переносимость глюкозы, диабет, болезнь коронарной артерии, ишемические болезни сердца, атеросклероз, гепатоспленомегалию и болезнь ожирения печени. Остается потребность обеспечения эффективного лечения связанных с глюкозой расстройств, таких как повышенные уровни глюкозы, сахарный диабет типа II, синдром X и подобные. Остается также потребность обеспечения эффективного лечения связанных с липидами расстройств, таких как повышенные уровни глюкозы, дислипидемия и подобные. Краткое содержание изобретения Настоящее изобретение относится к способу лечения связанных с липидами расстройств, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (I) где R1 и R2, каждый независимо, выбран из группы, состоящей из атома водорода и низшего алкила;R4 выбран из группы, состоящей из атома водорода и низшего алкила; А равно целому числу от 1 до 2; выбран из группы, состоящей из где b равно целому числу от 0 до 4; каждый R5 независимо выбран из группы, состоящей из галогена, низшего алкила;-2 016892 или его фармацевтически приемлемой соли. Настоящее изобретение также относится к способу лечения связанных с липидами расстройств,включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли. В одном примере изобретение относится к способу лечения связанных с липидами расстройств,включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества любого из описанных выше соединений. Подробное описание изобретения Настоящее изобретение относится к способу лечения связанных с липидами расстройств, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, где, a, R1, R2 и R4 такие, как определено в данном описании. Специалист в данной области отдает себе отчет, что лечение связанных с липидами расстройств может также извлекать пользу из лечения сопутствующих болезненных состояний чрезмерного веса и ожирения. Таким образом, в одном варианте способы по настоящему изобретению включают совместную терапию с использованием агента против ожирения и соединения формулы (I) или формулы (II),которые описаны в настоящем описании. Используемый в данном описании термин связанное с липидами расстройство определяют как любое расстройство, которое характеризуется аномальными уровнями липидов. Связанные с липидами расстройства включают повышенные уровни триглицеридов, низкие уровни HDL-холестерина и дислипидемию, предпочтительно повышенные уровни триглицеридов или низкие уровни HDL-холестерина. Лечение связанного с липидами расстройства может включать понижение уровня триглицеридов, повышение HDL-холестерина и/или улучшение соотношения триглицерид/HDL. Используемый в данном описании термин (пока не указано по-другому) антилипидный агент обозначает любой фармацевтический агент, способный понижать уровни триглицеридов, понижать уровни липидов, повышать HDL-уровни или улучшать соотношение триглицерид/HDL-холестерин. Подходящие примеры включают, но не ограничены этим, антилипемические агенты, желчнокислотные смолы, ингибиторы абсорбции холестерина, производные фибриновой кислоты, ингибиторы HMG-CoA редуктазы(то есть статины). Предпочтительным антилипидным агентом является статин, выбранный из группы,включающей аторвастатин (Lipitor), церивастатин (Baycol), флувастатин (Lescol), ловастатин (Mevacor),правастатин (Pravachol), розувастатин (Crestor), симвастатин (Zocor). Используемый в данном описании термин (пока не указано по-другому) агент против ожирения обозначает любой фармацевтический агент, который лечит ожирение, способствует потере веса и/или подавляет аппетит. Подходящие примеры промоторов потери веса включают, но не ограничены этим,римонабант, орлистат, сибутрамин, мазиндол, бензфетамин, фенметразин, фентермин, диэтилпропион,мазиндол, фенилпропаноламин, эфедрин, квипазин, флуоксетин, сертралин, фенфлурамин, дексфенфлурамин, апоморфин, экзендин, дегидроэпиандростерон, этиозоландион, тестостерон, оксандролон, топирамат и подобные. Предпочтительным агентом, промотирующим потерю веса, является римонабант, топирамат, орлистат или сибутрамин. Используемый в данном описании термин субъект относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, который является объектом лечения, наблюдения или эксперимента. Используемый в данном описании термин терапевтически эффективное количество означает, что количество активного соединения или фармацевтического агента вызывает биологический или медицинский ответ в системе тканей у животного или человека, подвергаемого вмешательству исследователя,ветеринара, врача или другого клинициста, который включает облегчение симптомов заболевания или расстройства, подлежащего лечению. В варианте настоящего изобретения R1 выбран из группы, состоящей из атома водорода и метила. В другом варианте настоящего изобретения R2 выбран из группы, состоящей из атома водорода и метила. Еще в одном варианте настоящего изобретения каждый R1 и R2 означает атом водорода, или каждый R1 иR2 означает метил. В варианте настоящего изобретения -(СН 2)а- выбран из группы, состоящей из -СН 2- и -СН 2 СН 2-. В другом варианте настоящего изобретения -(СН 2)а- представляет собой -СН 2-. В варианте настоящего изобретения R4 выбран из группы, состоящей из атома водорода и метила,предпочтительно R4 означает атом водорода. В варианте настоящего изобретения а равно 1. В варианте настоящего изобретения b равно целому числу от 0 до 2. В другом варианте настоящего изобретения с равно целому числу от 0 до 2. В другом варианте настоящего изобретения b равно целому числу от 0 до 1. В другом варианте настоящего изобретения с равно целому числу от 0 до 1. Еще в одном варианте настоящего изобретения сумма b и с равна целому числу от 0 до 2, предпочтительно целому числу от 0 до 1. Еще в одном варианте настоящего изобретения b равно целому числу от 0 до 2 и с равно 0. выбран из группы, состоящей из 2-(2,3-дигидробензо[1,4] В варианте настоящего изобретения диоксинила), 2-(бензо[1,3]диоксолила), 3-(3,4-дигидробензо[1,4]диоксепинила), 2-(6-хлор-2,3-дигидробензо[1,4]диоксинила), 2-(6-фтор-2,3-дигидробензо[1,4]диоксинила), 2-(5-фтор-2,3-дигидробензо[1,4]диоксинила), 2-(7-хлор-2,3-дигидробензо[1,4]диоксинила), 2-(6-хлорбензо[1,3]диоксолила), 2-(7-метил-2,3 дигидробензо[1,4]диоксинила), 2-(5-хлор-2,3-дигидробензо[1,4]диоксинила), 2-(6-бром-2,3-дигидробензо[1,4]диоксинила), 2-(6,7-дихлор-2,3-дигидробензо[1,4]диоксинила), 2-(8-хлор-2,3-дигидробензо[1,4]диоксинила). В другом варианте настоящего изобретения выбран из группы, состоящей из 2-(6-хлор-2,3 дигидробензо[1,4]диоксинила), 2-(7-хлор-2,3-дигидробензо[1,4]диоксинила), 2-(7-метил-2,3-дигидробензо[1,4]диоксинила), 2-(6-бром-2,3-дигидробензо[1,4]диоксинила) и 2-(6,7-дихлор-2,3-дигидробензо[1,4]диоксинила). выбран из группы, состоящей из 2-(7-метил-2,3 В другом варианте настоящего изобретения дигидробензо[1,4]диоксинила) и 2-(6-бром-2,3-дигидробензо[1,4]диоксинила). В варианте настоящего изобретения R5 выбран из группы, состоящей из галогена и низшего алкила. В другом варианте настоящего изобретения R5 выбран из хлора, фтора, брома и метила. В варианте настоящего изобретения стереоцентр на соединении формулы (I) находится в Sконфигурации. В другом варианте настоящего изобретения стереоцентр на соединении формулы (I) находится в R-конфигурации. В варианте настоящего изобретения соединение формулы (I) присутствует в виде энантиомерно обогащенной смеси, где % энантиомерного обогащения (% ее) составляет величину примерно более 75%,предпочтительно примерно более 90%, более предпочтительно примерно более 95%, наиболее предпочтительно примерно более 98%. Дополнительные варианты настоящего изобретения включают такие варианты, где заместители,выбранные для одной или нескольких переменных, определенных в данном описании (то есть R1, R2, R3,R4, X-Y и А), выбраны независимо, представляя собой любые индивидуальные заместители или любое подмножество заместителей, выбранных из полного перечня, который определен в данном описании. Типичные соединения по настоящему изобретению перечислены ниже в табл. 1. Дополнительные соединения по настоящему изобретению перечислены в табл. 3. В приведенных ниже табл. 1 и 2 столбец,озаглавленный стерео, определяет стереоконфигурацию на атоме углерода гетероцикла, присоединенном по отмеченной звездочкой связи. Если обозначение отсутствует, это значит, что соединение получают в виде смеси стереоконфигураций. Где приведено обозначение R или S, там стереоконфигурация указана на основании энантиомерно обогащенного исходного материала.-4 016892 Таблица 1 Типичные соединения формулы (I)-5 016892 Таблица 2 Дополнительные соединения по настоящему изобретению Используемый в данном описании, пока не указано по-другому, термин галоген обозначает хлор,бром, фтор и йод. Используемый в данном описании, пока не указано по-другому, термин алкил, сам по себе или как часть группы заместителя, включает линейные и разветвленные цепи. Например, алкильные радикалы включают метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил и подобные. Пока не указано по-другому, используемое с алкилом определение низший обозначает углеродную цепь, состоящую из 1-4 атомов углерода. Используемый в данном описании, пока не указано по-другому, термин алкокси обозначает радикал простого кислородного эфира с описанными выше линейными или разветвленными алкильными группами,например метокси, этокси, н-пропокси, втор-бутокси, трет-бутокси, н-гексилокси и подобными. Используемая в данном описании меткаобозначает наличие стереогенного центра. Если конкретная группа является замещенной (например, алкильной, арильной и др.), то эта группа может иметь один или более заместителей, предпочтительно от одного до пяти заместителей, более предпочтительно от одного до трех заместителей, наиболее предпочтительно от одного до двух заместителей, независимо выбранных из перечня заместителей. Со ссылкой на заместители термин независимо означает, что, если возможно присутствие более одного заместителя, то такие заместители могут быть одинаковыми или разными. По стандартной номенклатуре, используемой на протяжении всей этой заявки, сначала описывается конечная часть обозначенной боковой цепи, затем соседняя фукциональность по направлению к точке присоединения. Таким образом, например, заместитель фенил-алкил-амино-карбонил-алкил относится к группе формулыEDC = этилкарбодиимид,Et3N или ТЭА = триэтиламин,Et2O = диэтиловый эфир,ЭА или EtOAc = этилацетат,EtOH = этанол,IPA = 2-пропанол,Hept = гептан,ГОБТ = 1-гидроксибензотриазол,ВДЖХ = жидкостная хроматография при высоком,ЛАГ = литий алюминий гидрид,М или МеОН = метанол,ЯМР = ядерный магнитный резонанс,Pd-C = катализатор: палладий на угле,ОФ ВДЖХ = жидкостная хроматография при высоком с обращенной фазой,КТ или к.т. = комнатная температура,ТЭА = триэтиламин,ТФУ = трифторуксусная кислота,ТГФ = тетрагидрофуран,ТСХ = тонкослойная хроматография. Когда соединения по этому изобретению имеют по меньшей мере один хиральный центр, то, соответственно, они могут существовать в виде энантиомеров. Когда соединения обладают двумя или более хиральными центрами, то они могут дополнительно существовать в виде диастереомеров. Следует понимать, что все такие изомеры и их смеси включены в область настоящего изобретения. Кроме того, некоторые из кристаллических форм соединений могут существовать в виде полиморфов, и предполагается, что как таковые они включены в настоящее изобретение. Кроме того, некоторые соединения могут образовывать сольваты с водой (то есть гидраты) или обычными органическими растворителями, и предполагается также, что такие сольваты включены в область данного изобретения. Ввиду того, что соли соединений по данному изобретению используются в медицине, они относятся к нетоксическим фармацевтически приемлемым солям. Однако другие соли могут быть пригодны при получении соединений по этому изобретению или их фармацевтически приемлемых солей. Подходящие фармацевтически приемлемые соли соединений включают аддитивные соли кислот, которые могут образовываться, например, при смешивании раствора соединения с раствором фармацевтически приемлемой кислоты, такой как соляная кислота, серная кислота, фумаровая кислота, малеиновая кислота,янтарная кислота, уксусная кислота, бензойная кислота, лимонная кислота, винная кислота, угольная кислота или фосфорная кислота. Кроме того, если соединения по изобретению несут кислотный фрагмент, то их подходящие фармацевтически приемлемые соли могут включать соли щелочных металлов,например соли натрия или калия; соли щелочно-земельных металлов, например соли кальция или магния; и соли, образованные с подходящими органическими лигандами, например четвертичные аммонийные соли. Таким образом, типичные фармацевтически приемлемые соли включают следующие: ацетат,бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, эдетат кальция, камзилат,карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдизилат, эстолат, эзилат, фумарат, глюцептат, глюконат, глутамат, гликоллиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, мандеат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напзилат, нитрат, соль N-метилглюкаминаммония,олеат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, сульфат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид и валерат. Типичные кислоты и основания, которые можно использовать при получении фармацевтически приемлемых солей, включают следующие соединения. Кислоты включают уксусную кислоту, 2,2-дихлоруксусную кислоту, ацилированные аминокислоты, адипиновую кислоту, альгиновую кислоту, аскорбиновую кислоту, L-аспарагиновую кислоту, бензолсульфоновую кислоту, бензойную кислоту, 4-ацетамидобензойную кислоту, (+)-камфорную кислоту,камфорсульфоновую кислоту, (+)-(1S)-камфор-10-сульфоновую кислоту, каприновую кислоту, капроновую кислотую, каприловую кислоту, коричную кислоту, лимонную кислоту, цикламовую кислоту, додецилсерную кислоту, этан-1,2-дисульфоновую кислоту, этансульфоновую кислоту, 2-гидроксиэтансульфоновую кислоту, муравьиную кислоту, фумаровую кислоту, галактаровую кислоту, гентизиновую кислоту, глюкогептоническую кислоту, D-глюконовую кислоту, D-глюкороновую кислоту, Lглутаминовую кислоту, -оксоглутаровую кислоту, гликолевую кислоту, гипуровую кислоту, бромистоводородную кислоту, соляную кислоту, (+)-L-молочную кислоту, -DL-молочную кислоту, лактобионовую кислоту, малеиновую кислоту, (-)-L-яблочную кислоту, малоновую кислоту, -DL-манделовую кислоту, метансульфоновую кислоту, нафталин-2-сульфоновую кислоту, нафталин-1,5-дисульфоновую кислоту, 1-гидрокси-2-нафтойную кислоту, никотиновую кислоту, азотную кислоту, олеиновую кислоту,-7 016892 оротовую кислоту, щавелевую кислоту, пальмитиновую кислоту, памоевую кислоту, фосфорную кислоту, L-пироглутаминовую кислоту, салициловую кислоту, 4-аминосалициловую кислоту, себациновую кислоту, стеариновую кислоту, янтарную кислоту, серную кислоту, дубильную кислоту, (+)-L-винную кислоту, тиоциановую кислоту, паратолуолсульфоновую кислоту и ундециленовую кислоту; и основания включают аммиак, L-аргинин, бенетамин, бензатин, гидроксид кальция, холин, деанол,диэтаноламин, диэтиламин, 2-(диэтиламино)этанол, этаноламин, этилендиамин, N-метилглюкамин, гидрабамин, 1 Н-имидазол, L-лизин, гидроксид магния, 4-(2-гидроксиэтил)морфолин, пиперазин, гидроксид калия, 1-(2-гидроксиэтил)пирролидин, вторичный амин, гидроксид натрия, триэтаноламин, трометамин и гидроксид цинка. Соединения формулы (I) можно получить согласно способу, изображенному на схеме 1. Схема 1 Таким образом, проводят взаимодействие замещенного подходящим образом соединения формулы(X), известного соединения или соединения, полученного известными способами, с сульфамидом, известным соединением, где сульфамид предпочтительно присутствует в количестве примерно от 2 до 5 экв., в органическом растворителе, таком как ТГФ, диоксан и подобные, предпочтительно при повышенной температуре примерно от 50 до 100 С, более предпочтительно примерно при температуре кипения с обратным холодильником, получая соответствующее соединение формулы (Ia). Альтернативно, проводят взаимодействие замещенного подходящим образом соединения формулы(X), известного соединения или соединения, полученного известными способами, с замещенным подходящим образом соединением формулы (XI), известным соединением или соединением, полученным известными способами, в присутствии основания, такого как ТЭА, ДИПЭА, пиридин и подобные, в органическом растворителе, таком как ДМФА, ДМСО и подобном, получая соответствующее соединение формулы (I). Соединения формулы (X), где и где а равно 2, можно получить согласно способу, изображенному на схеме 4. Схема 4 Таким образом, проводят взаимодействие замещенного подходящим образом соединения формулы(XVI), где J1 представляет собой подходящую уходящую группу, такую как Br, Cl, I, тозил, мезил, трифлил и подобные, известного соединения или соединения, полученного известными способами (например,активацией соответствующего соединения, где J1 представляет собой ОН), с цианидом, например цианидом калия, цианидом натрия и подобным, в органическом растворителе, таком как ДМСО, ДМФА, ТГФ и подобные, получая соответствующее соединение формулы (XVII). Соединение формулы (XVII) восстанавливают согласно известным способам, например посредством взаимодействия с подходящим восстановителем, таким как ЛАГ, боран и подобные, получая соответствующее соединение формулы (Хс). Соединения формулы (X), где-8 016892 и где а равно 1, можно получить согласно способу, изображенному на схеме 5. Схема 5 Таким образом, замещенное подходящим образом соединение формулы (XVIII), известное соединение или соединение, полученное известными способами, активируют согласно известному способу,получая соответствующее соединение формулы (XIX), где J2 представляет собой подходящую уходящую группу, например тозилат, Cl, Br, I, мезилат, трифлат и подобные. Проводят взаимодействие соединения формулы (XIX) с фталимидной солью, такой как фталимид калия, фталимид натрия и подобные, в органическом растворителе, например ДМФА, ДМСО, ацетонитриле и подобном, предпочтительно при повышенной температуре в диапазоне от 50 примерно до 200 С,более предпочтительно около температуры кипения с обратным холодильником, получая соответствующее соединение формулы (XX). Проводят взаимодействие соединения формулы (XX) с N2H4, известным соединением, в органическом растворителе, таком как этанол, метанол и подобные, предпочтительно при повышенной температуре в диапазоне примерно от 50 до 100 С, более предпочтительно около температуры кипения с обратным холодильником, получая соответствующее соединение формулы (Xd). Специалист в данной области понимает, что если реакционную стадию по настоящему изобретению можно проводить в различных растворителях или системах растворителей, то указанную реакционную стадию также можно проводить в смеси подходящих растворителей или систем растворителей. Когда способы получения соединений по изобретению дают смесь стереоизомеров, эти изомеры можно разделить, используя обычные методики, например препаративную хроматографию. Соединения можно получить в рацемическом виде или в виде индивидуальных энантиомеров посредством энантиоспецифического синтеза или разделения. Например, можно разделить соединения на составные энантиомеры стандартными методиками, такими как образование диастереомерных пар при образовании соли с оптически активной кислотой, такой как (-)-дипаратолуоил-D-винная кислота и/или (+)-дипаратолуоилL-винная кислота, с последующей фракционированной кристаллизацией и регенерацией свободного основания. Соединения также можно разделить посредством образования диастереомерных сложных эфиров или амидов с последующим хроматографическим разделением и удалением хирального вспомогательного агента. По-другому, соединения можно разделить, используя хиральную ВДЖХ-колонку. В течение какого-либо из процессов получения соединений по настоящему изобретению может быть необходимо и/или желательно защитить чувствительные или реакционноспособные группы на каких-либо из рассматриваемых молекул. Этого можно достичь при помощи обычных защитных групп,таких как группы, описанные в работах Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, PlenumPress, 1973; и T.W. GreeneP.G.M. Wuts, Protective Groups in Organic Synthesis, John WileySons, 1991. Защитные группы можно удалить на последующей обычной стадии, используя способы, известные в данной области. Соединения по настоящему изобретению преимущественно можно вводить в виде отдельных дневных доз, или можно вводить общую дневную дозу, поделив ее на две, три или четыре дозы в день. Кроме того, соединения по настоящему изобретению можно использовать как форму для внутриносового введения посредством подходящих внутриносовых носителей местного применения или как трансдермальные кожные пластыри, хорошо известные специалисту в данной области. Конечно, при трансдермальной системе доставки введение дозы будет скорее непрерывным, чем периодическим, на протяжении всей схемы приема. Например, для перорального приема в виде таблетки или капсулы активный лекарственный компонент можно объединить с нетоксическим фармацевтически приемлемым инертным носителем для перорального приема, таким как этанол, глицерин, вода и подобные. Более того, если желательно или необходимо, можно также включить в смесь подходящие связующие, лубриканты, разрыхлители и красители. Подходящие связующие включают (без ограничения) крахмал, желатин, природные сахара, такие как глюкоза или -лактоза, кукурузные подсластители, природные и синтетические смолы, такие как гуммиарабик, трагакант или олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия,-9 016892 хлорид натрия и подобные. Разрыхлители включают (без ограничения) крахмал, метилцеллюлозу, агар,бентонит, ксантановую смолу и подобные. Жидкие формы в суспендирующих или диспергирующих агентах с подходящими вкусовыми агентами, таких как синтетические и природные смолы, например трагакант, гуммиарабик, метилцеллюлоза и подобные. Для парентерального введения требуются стерильные суспензии и растворы. Когда требуется внутривенное введение, применяют изотонические препараты, которые обычно содержат подходящие консерванты. Соединения по данному изобретению можно вводить в виде любой из описанных выше композиций и согласно схемам приема доз, установленным в данной области, когда бы ни потребовалось лечение депрессии. Дневную дозировку продуктов можно варьировать в широком диапазоне от 0,01 до 200 мг/кг для взрослого человека в день или в любом указанном диапазоне. Для перорального приема предпочтительно получают композиции в виде таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0,100, 150, 200, 250, 500 и 1000 мг активного ингредиента для симптоматического регулирования введения дозы пациенту, принимающему лечение. Эффективное количество лекарства обычно доставляется при уровне доз примерно от 0,01 до 200 мг/кг массы тела в день. Предпочтительный диапазон составляет примерно от 0,1 до 100,0 мг/кг массы тела в день, более предпочтительно примерно от 0,5 до 50 мг/кг,более предпочтительно примерно от 1,0 до 25,0 мг/кг массы тела в день. Соединения можно вводить по схеме от 1 до 4 раз в день. Специалист в данной области может легко определить оптимальные дозы, подлежащие введению, и варьировать их в зависимости от конкретного используемого соединения, способа введения, силы препарата и прогрессирования болезненного состояния. Кроме того, необходимость регулирования дозировки обуславливают факторы, касающиеся подлежащего лечению конкретного пациента, включая возраст,массу, питание пациента и время введения. Специалисту в данной области понятно, что и in vivo, и in vitro испытания, использующие подходящие известные и общепринятые клеточные и/или животные модели, предсказывают способность исследуемого соединения лечить или предупреждать данное расстройство. Кроме того, специалисту в данной области понятно, что клинические испытания на людях, включающие первые исследования на людях диапазона доз и эффективности, можно выполнить на здоровых пациентах и/или пациентах, страдающих от данного расстройства, согласно способам, хорошо известным в клинической и медицинской практике. Следующие примеры приведены в помощь пониманию изобретения и никоим образом не предназначены и не должны толковаться как ограничение изобретения, изложенного в формуле изобретения,которая приведена далее. Пример 1. 3,4-Дигидро-2 Н-бензо[b][1,4]диоксепин-3-ил)метил)сульфамид (соединение 3) Катехол (5,09 г, 46,2 ммоль) и карбонат калия объединяли в ацетонитриле и нагревали при кипячении с обратным холодильником в течение 1 ч. Добавляли 2-хлорметил-3-хлор-1-пропен (5,78 г, 46,2 ммоль) и продолжали взаимодействие при кипячении с обратным холодильником в течение 24 ч. Раствор охлаждали до комнатной температуры и фильтровали. Фильтрат выпаривали и остаток разбавляли водой и экстрагировали диэтиловым эфиром (3). Объединенный органический раствор сушили над MgSO4 и концентрировали. Хроматография (2% этиловый эфир в гексане) давала 3-метилен-3,4-дигидро-2 Нбензо[b][1,4]диоксепин в виде бесцветного масла. МС (ESI): 163,2 (М+Н+). 1 Н ЯМР (300 МГц, CDCl3) : 6,94 (м, 4 Н), 5,07 (с, 2 Н), 4,76 (с, 4 Н). 3-Метилен-3,4-дигидро-2 Н-бензо[b][1,4]диоксепин (5,00 г, 30,8 ммоль) растворяли в сухом ТГФ(100 мл). Добавляли при 0 С боран-ТГФ (1,0 М в ТГФ, 10,3 мл). Реакционную смесь перемешивали при к.т. в течение 5 ч. Добавляли аминосульфоновую кислоту (6,91 г, 61,6 ммоль). Реакционную смесь нагревали при кипячении с обратным холодильником в течение ночи. Охлаждали реакционную смесь до комнатной температуры и добавляли водный гидроксид натрия (3,0 М, 100 мл). Раствор экстрагировали этилацетатом (3100 мл). Объединенный органический раствор сушили над MgSO4. Раствор концентрировали в вакууме и очищали методом хроматографии (от 2 до 8% метанола в дихлорметане) с получением 3,4-дигидро-2 Н-бензо[b][1,4]диоксепин-3-ил)метил)амина в виде бесцветного масла. МС (ESI): 180,1 (М+Н+). 1 Н ЯМР (300 МГц, CDCl3) : 6,92 (м, 4 Н), 4,21 (м, 2 Н), 4,07 (м, 2 Н), 3,33 (широкий, 2 Н), 3,16 (д, J=4 Гц, 1 Н), 2,72 (д, J=4 Гц, 1 Н), 2,30 (м, 1 Н). 3,4-Дигидро-2 Н-бензо[b][1,4]диоксепин-3-ил)метил)амин (2,90 г, 16,2 25 ммоль) и сульфамид(3,11 г, 32,4 ммоль) объединяли в сухом диоксане (60 мл) и нагревали при кипячении с обратным холо- 10016892 дильником в течение ночи. Добавляли хлороформ и осадок удаляли фильтрованием. Фильтрат концентрировали в вакууме и очищали методом хроматографии (от 2 до 8% ацетона в дихлорметане) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества. МС 258,8 (М+Н+). 1 Н ЯМР (300 МГц, ДМСО) : 6,92 (м, 4 Н), 6,71 (широкий, 1 Н), 6,59 (широкий, 2 Н), 4,19 (м, 2 Н),4,04 (м, 2 Н), 3,00 (м, 2 Н), 2,39 (м, 1 Н). Пример 2. N-(2,3-Дигидробензо[1,4]диоксин-2-илметил)сульфамид (соединение 1) Рацемический 2,3-дигидро-1,4-бенздиоксин-2-илметиламин (4,4 г, 26 ммоль) и сульфамид (5,1 г, 53 ммоль) объединяли в 1,4-диоксане (100 мл) и кипятили с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и небольшое количество твердого вещества отфильтровывали и отбрасывали. Фильтрат выпаривали в вакууме и остаток очищали, используя колонку для флэш-хроматографии (ДХМ:метанол=10:1) с получением белого твердого вещества. Твердое вещество перекристаллизовывали из ДХМ с получением указанного в заголовке соединения в виде белого твердого вещества. Т.пл.: 97,5-98,5 С. Элементный анализ: Расчет: С, 44,25; Н, 4,95; N, 11,47; S, 13,13; Найдено: С, 44,28; Н, 4,66; N, 11,21; S, 13,15. 1 Н ЯМР (ДМСО-d6) : 6,85 (м, 4 Н), 6,68 (широкий с, 3 Н, NH), 4,28 (м, 2 Н), 3,97 (дд, J=6,9, 11,4 Гц,1 Н), 3,20 (м, 1 Н), 3,10 (м, 1 Н). Пример 3. (Бензо[1,3]диоксол-2-илметил)сульфамид (соединение 2) Катехол (10,26 г, 93,2 ммоль), метилат натрия (25 мас.% в метаноле, 40,3 г, 186 ммоль) и метилдихлорацетат (13,3 г, 93,2 ммоль) объединяли в сухом метаноле (100 мл). Раствор нагревали при кипячении с обратным холодильником в течение ночи. Реакционную смесь охлаждали до комнатной температуры, подкисляли, добавляя концентрированную соляную кислоту, и затем уменьшали объем примерно до 50 мл в вакууме. Добавляли воду и смесь экстрагировали диэтиловым эфиром (3100 мл). Объединенный органический раствор сушили MgSO4,концентрировали до коричневого твердого вещества и хроматографировали (2% этилацетат в гексане) с получением метилового эфира бензо[1,3]диоксол-2-карбоновой кислоты в виде бесцветного масла. МС (ESI): 195,10 (М+Н+). 1 Н ЯМР (300 МГц, CDCl3) : 6,89 (широкий, 4 Н), 6,29 (с, 1 Н), 4,34 (кв, J=7 Гц, 2 Н), 1,33 (т, J=7 Гц,3 Н). К метиловому эфиру бензо[1,3]диоксол-2-карбоновой кислоты (7,21 г, 40,0 ммоль) добавляли гидроксид аммония (29% в воде, 10 мл) и достаточно ацетонитрила, чтобы смесь была гомогенной (5 мл). Раствор перемешивали в течение 1 ч при комнатной температуре и затем добавляли дистиллированную воду. Амид бензо[1,3]диоксол-2-карбоновой кислоты, выпавший в осадок в виде белого твердого вещества, собирали фильтрованием и использовали без дополнительной очистки. МС (ESI): 160,00 (М+Н+). 1 Н ЯМР (300 МГц, ДМСО) : 7,99 (с, широкий, 1 Н), 7,72 (с, широкий, 1 Н), 6,94 (м, 2 Н) 6,86 (м, 2 Н),6,30 (с, 1 Н). Амид бензо[1,3]диоксол-2-карбоновой кислоты (5,44 г, 32,9 ммоль) растворяли в тетрагидрофуране(ТГФ, 100 мл). Медленно добавляли к раствору при комнатной температуре литийалюминийгидрид(ЛАГ, 1 М в ТГФ, 39,5 мл, 39,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 ч. Добавляли дистиллированную воду для разложения избытка ЛАГ. Добавляли водный гидроксид натрия (3,0 М, 100 мл) и раствор экстрагировали этилацетатом (3100 мл). Объединенный органический раствор промывали водой и сушили над MgSO4. Растворитель выпаривали с получением Сбензо[1,3]диоксол-2-илметиламина в виде бесцветного масла. МС (ESI): 152,1 (М+Н+). 1 Н ЯМР (300 МГц, CDCl3) : 6,87 (м, 4 Н), 6,09 (т, J=4 Гц, 1 Н), 3,13 (д, J=4 Гц, 2H). С-Бензо[1,3]диоксол-2-илметиламин (2,94 г, 19,4 ммоль) и сульфамид (3,74 г, 38,9 ммоль) объединяли в сухом диоксане (50 мл) и раствор нагревали при кипячении с обратным холодильником в течение ночи. Реакционную смесь концентрировали и остаток хроматографировали (от 2 до 10% ацетона в ди- 11016892 хлорметане) с получением указанного в заголовке соединения в виде белого твердого вещества. МС (ESI): 230,0 (М+Н+). 1 Н ЯМР (300 МГц, CDCl3) : 6,87 (м, 4 Н), 6,25 (т, J=4 Гц, 1 Н), 4,79 (широкий, 1 Н), 4,62 (широкий,1 Н), 3,64 (д, J=4 Гц, 2 Н). Пример 4. (2S)-(-)-N-(2,3-дигидробензо[1,4]диоксин-2-илметил)сульфамид (соединение 4) Катехол (13,2 г, 0,12 моль) и карбонат калия (16,6 г, 0,12 моль) перемешивали в ДМФА (250 мл),добавляли (2R)-глицидилтозилат (22,8 г, 0,10 моль) и реакционную смесь перемешивали при 60 С в течение 24 ч. Охлаждали реакционную смесь до комнатной температуры, разбавляли ледяной водой (1 л),и экстрагировали диэтиловым эфиром (4 раза). Объединенный органический раствор промывали 3 раза 10% карбонатом калия, один раз водой, один раз насыщенным раствором соли и выпаривали в вакууме с получением белого твердого вещества, которое очищали методом колоночной флэш-хроматографии(ДХМ:метанол=50:1) с получением 2S)-2,3-дигидробензо[1,4]диоксин-2-ил)метанола в виде твердого вещества. Твердое вещество (13,3 г, 68 ммоль) растворяли в пиридине (85 мл), охлажденном до 0 С, добавляли паратолуолсульфонилхлорид (13,0 г, 68 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 20 ч. Реакционную смесь разбавляли диэтиловым эфиром (1 л) и 1N HCl (1,2 л). Органический слой отделяли и промывали 2 раза 1N HCl (500 мл), 4 раза водой (150 мл), один раз насыщенным раствором соли, сушили (MgSO4) и выпаривали в вакууме с получением белого твердого вещества, которое очищали методом колоночной флэш-хроматографии (Hept:ЭА=2:1), с получением (2S)-2,3 дигидробензо[1,4]диоксин-2-илметилового эфира толуол-4-сульфоновой кислоты в виде белого твердого вещества. Белое твердое вещество объединяли с фталимидом калия (14,4 г, 78 ммоль) в ДМФА (250 мл) и нагревали при кипячении с обратным холодильником в течение 1 ч, охлаждали до комнатной температуры,выливали в энергично перемешиваемую воду (1,5 л) и перемешивали 30 мин. Белое твердое вещество отфильтровывали и твердое вещество промывали несколько раз водой, 2% NaOH и снова водой и сушили на воздухе с получением (2S)-2-(2,3-дигидробензо[1,4]диоксин-2-илметил)изоиндол-1,3-диона в виде белого порошкового твердого вещества. Порошкообразное белое твердое вещество объединяли с гидразином (2,75 г, 86 ммоль) в EtOH (225 мл) и нагревали при кипячении с обратным холодильником в течение 2 ч, охлаждали до комнатной температуры, добавляли 1N HCl до рН 1,0 и перемешивали в течение 15 мин. Белое твердое вещество отфильтровывали и промывали свежим EtOH (твердое вещество отбрасывают), фильтрат выпаривали в вакууме до твердого вещества, которое распределяли между диэтиловым эфиром и разбавленным водным NaOH. Раствор в диэтиловом эфире сушили (Na2SO4) и выпаривали в вакууме с получением светложелтого масла. Масло очищали методом колоночной флэш-хроматографии (ДХМ:МеОН=10:1) с получением масла. Часть масла (4,82 г, 29 ммоль) в 2-пропаноле (250 мл) обрабатывали 1N HCl (30 мл) и нагревали на паровой бане до гомогенности и затем давали охладиться до комнатной температуры. Через 3 ч смесь охлаждали на льду в течение 2 ч. Белое хлопьевидное твердое вещество (соответствующую HClсоль (2S)-C-(2,3-дигидробензо[1,4]диоксин-2-ил)метиламина) отфильтровывали и затем снова перекристаллизовывали из 2-пропанола с получением белого твердого вещества.[]D=-69,6 (c=1,06, EtOH). Белое твердое вещество распределяли между ДХМ и разбавленным NaOH, ДХМ-фракцию сушили[]D=-57,8 (с=1,40, CHCl3). Масло (2,1 г, 12,7 ммоль) и сульфамид (2,44 г, 25,4 ммоль) кипятили с обратным холодильником в диоксане (75 мл) в течение 2 ч и неочищенный продукт очищали методом колоночной флэшхроматографии (ДХМ:МеОН=10:1) с получением белого твердого вещества, которое перекристаллизовывали из ДХМ с получением указанного в заголовке соединения в виде белого кристаллического твердого вещества. Т.пл. 102-103 С; Рацемический 2,3-дигидро-1,4-бенздиоксин-2-илметиламин (8,25 г, 5,0 ммоль) и триэтиламин (1,52 г, 15 ммоль) объединяли в ДМФА (10 мл) и охлаждали на бане со льдом, добавляли диметилсульфамоилхлорид (1,44 г, 10 ммоль). Затем реакционную смесь перемешивали в течение 3 ч при непрерывном охлаждении. Реакционную смесь распределяли между этилацетатом и водой и этилацетатный раствор промывали насыщенным раствором соли, сушили (MgSO4) и выпаривали в вакууме с получением масла. Масло очищали, используя колонку для флэш-хроматографии (этилацетат:гептан=1:1) и получая белое твердое вещество, которое перекристаллизовывали (этилацетат/гексан) с получением указанного в заголовке соединения в виде белого хлопьевидного твердого вещества. Т.пл. 76-78 С; МС 273 (МН+). Элементный анализ: Расчет: С, 48,52; Н, 5,92; N, 10,29; S, 11,78; Найдено: С, 48,63; Н, 5,62; N, 10,20; S, 11,90 1 Н ЯМР (CDCl3) : 6,87 (м, 4 Н), 4,59 (широкий м, 1 Н, NH), 4,35 (м, 1 Н), 4,27 (дд, J=2,3, 11,4 Гц, 1 Н),4,04 (дд, J=7,0, 11,4, 1 Н), 3,36 (м, 2 Н), 2,82 (с, 6 Н). Пример 6. N-(2,3-дигидробензо[1,4]диоксин-2-илметил)-N-метилсульфамид (соединение 7) Рацемический 2,3-дигидро-1,4-бенздиоксин-2-илметиламин (825 мг, 5 ммоль) растворяли в этилформиате (15 мл), кипятили с обратным холодильником в течение 30 мин и выпаривали в вакууме с получением N-(2,3-дигидробензо[1,4]диоксин-2-илметил)формамида в виде масла. Масло в диэтиловом эфире (25 мл) обрабатывали 1 М ЛАГ в ТГФ (9,0 мл, 9,0 ммоль) при 0 С и перемешивали в течение 5 ч при комнатной температуре. Реакционную смесь охлаждали на бане со льдом и гасили водой (0,50 мл) с последующим добавлением 3N NaOH (0,50 мл) и воды (0,50 мл). Затем смесь перемешивали при комнатной температуре в течение 1 ч. Твердое вещество отфильтровывали и выпаривали фильтрат в вакууме, получая остаток, который распределяли между 1N HCl и дизтиловым эфиром. Водную фазу подщелачивали 1N NaOH и экстрагировали диэтиловым эфиром. Органическую фазу сушили (MgSO4) и выпаривали в вакууме с получением (2,3-дигидробензо[1,4]диоксин-2 илметил)метиламина в виде масла. МС 180 (МН+); 1 Н ЯМР (CDCl3) : 6,85 (м, 4 Н), 4,30 (м, 2 Н), 4,02 (дд, J=7,9, 11,6 Гц, 1 Н), 2,85 (м, 2 Н), 2,50 (с, 3 Н). Масло (380 мг, 2,1 ммоль) и сульфамид (820 мг, 8,5 ммоль) объединяли в диоксане (15 мл), кипятили с обратным холодильником в течение 1,5 ч и выпаривали в вакууме, получая сырой остаток. Остаток очищали методом колоночной хроматографии (этилацетат/гептан=1:1) и полученное твердое вещество перекристаллизовывали из смеси этилацетат/гексан с получением указанного в заголовке соединения в виде белого твердого вещества. Т.пл. 97-98 С; МС 257 (М-1). Элементный анализ: Расчет: С, 46,50; Н, 5,46; N, 10,85; S, 12,41; Найдено: С, 46,48; Н, 5,65; N, 10,90; S, 12,07. 1 Н ЯМР (CDCl3) : 6,86 (м, 4 Н), 4,52 (ш.с, 2 Н), 4,46 (м, 1 Н), 4,29 (дд, J=2,3, 11,5 Гц, 1 Н), 4,05 (дд,J=6,5, 11,5 Гц, 1 Н), 3,51 (дд, J=6,7, 14,9 Гц, 1 Н), 3,40 (дд, J=5,9, 14,9 Гц, 1 Н), 2,99 (с, 3 Н). Пример 7. (2S)-(-)-N-(6-хлор-2,3-дигидробензо[1,4]диоксин-2-илметил)сульфамид (соединение 8) Следуя методике, описанной выше в примере 4, проводили взаимодействие 4-хлоркатехола, получая смесь (2S)-С-(7-хлор-2,3-дигидробензо[1,4]диоксин-2-ил)метиламина и (2S)-С-(6-хлор-2,3 дигидробензо[1,4]диоксин-2-ил)метиламина (соотношение изомеров 6-хлор:7-хлор составляло примерно 3:1 по данным ОФ ВДЖХ).- 13016892 Смесь растворяли в 2-пропаноле (100 мл) и добавляли 1N HCl в диэтиловом эфире до достижения рН 1,0. Соль гидрохлорида, которая выпадала в осадок, отфильтровывали (2,65 г) и перекристаллизовывали из смеси метанол/IPA с получением белых кристаллов. Белые кристаллы распределяли между ДХМ и разбавленным NaOH. ДХМ-фазу сушили и выпаривали в вакууме с получением очищенного (2S)-С-(6 хлор-2,3-дигидробензо[1,4]диоксин-2-ил)метиламина в виде масла.[]D=-67,8 (с=1,51, CHCl3). Масло (7,75 ммоль) и сульфамид (1,50 г, 15,5 ммоль) объединяли в диоксане (50 мл) и кипятили с обратным холодильником в течение 2,0 ч, охлаждали до комнатной температуры и выпаривали в вакууме с получением твердого вещества. Продукт очищали методом колоночной флэш-хроматографии, используя смесь ДХМ/метанол=20:1 и получая указанное в заголовке соединение в виде белого твердого вещества. МС 277 (М-1);(дд, J=2,4, 11,5 Гц, 1 Н), 4,05 (дд, J=7,1, 11,5 Гц, 1 Н), 3,45 (м, 2 Н). Элементный анализ: Расчет: С, 38,78; Н, 3,98; N, 10,05; Найдено: С, 38,80; Н, 3,67; N, 9,99. Фильтраты образовавшейся в результате кристаллизации полученной выше соли гидрохлорида(2S)-С-(6-хлор-2,3-дигидробензо[1,4]диоксин-2-ил)метиламина отделяли (содержание изомеров 6-хлор:7 хлор составляло примерно 1:1) и выпаривали в вакууме с получением твердого вещества, которое распределяли между ДХМ (200 мл) и разбавленным NaOH (0,5 М, 50 мл). ДХМ-раствор промывали один раз насыщенным раствором соли, сушили (Na2SO4) и выпаривали в вакууме с получением масла, которое очищали методом ВДЖХ с обращенной фазой (10-50% ACN с 0,16% ТФУ в воде с 0,20% ТФУ) с получением в остатке (2S)-С-(7-хлор-2,3-дигидробензо[1,4]диоксин-2-ил)метиламина. Остаток объединяли с сульфамидом (0,90 г, 9,4 ммоль) в диоксане (25 мл) и кипятили с обратным холодильником в течение 2,5 ч, охлаждали до комнатной температуры и выпаривали в вакууме с получением масла. Масло очищали методом колоночной флэш-хроматографии, используя смесь ДХМ/метанол=10:1 и получая (2S)-(-)-N-(7-хлор-2,3-дигидробензо[1,4]диоксин-2-илметил)сульфамид в виде белого твердого вещества. МС 277 (M-1); 1 Н ЯМР (CDCl3/CD3OD) : 6,88 (д, J=0,7 Гц, 1 Н), 6,81 (м, 2 Н), 4,37 (м, 1 Н), 4,30 (дд, J=2,3, 11,6 Гц,1 Н), 4,04 (дд, J=7,0, 11,6 Гц, 1 Н), 3,38 (м, 2 Н). Пример 8. Хроман-2-илметилсульфамид (соединение 10) Хроман-2-карбоновую кислоту (4,5 г, 25 ммоль) и ГОБТ (3,86 г, ммоль) объединяли в ДХМ (40 мл) и ДМФА (10 мл). Добавляли диметиламинопропилэтилкарбодиимид (EDC, 4,84 г, 25 ммоль) при комнатной температуре и реакционную смесь перемешивали в течение 30 мин. Добавляли гидроксид аммония (2,26 мл, 33,4 ммоль) и перемешивали реакционную смесь в течение 16 ч. Реакционную смесь разбавляли ДХМ (50 мл) и водой (50 мл) и доводили рН смеси примерно до рН 3,0 при помощи 1N HCl. ДХМ-фазу отделяли и водную фазу дважды экстрагировали ДХМ. Объединенную ДХМ-фазу сушили(Na2SO4) и выпаривали в вакууме, получая масло, которое очищали методом колоночной флэшхроматографии (этилацетат) с получением масла. Масло (5,35 г, 30 ммоль) в ТГФ (90 мл) перемешивали, добавляли в это время 1 М ЛАГ в ТГФ (36 мл, 36 ммоль) и затем реакционную смесь перемешивали при комнатной температуре в течение 20 ч. Реакционную смесь гасили водой, перемешивали в течение 2 ч, раствор декантировали, сушили (Na2SO4) и выпаривали в вакууме с получением С-хроман-2-илметиламина в виде маслянистого амина. Маслянистый амин (1,63 г, 10 ммоль) и сульфамид (1,92 г, 20 ммоль) объединяли в диоксане (50 мл) и доводили до кипения с обратным холодильником в течение 2 ч. Раствор охлаждали и выпаривали в вакууме, получая масло, которое очищали методом колоночной хроматографии (ДХМ:метанол 10:1) с получением белого твердого вещества. Твердое вещество перекристаллизовывали из смеси этилацетат/гексан с получением хроман-2-илметилсульфамида в виде белого твердого вещества. Т.пл. 100-101 С; МС 241 (М-1). Элементный анализ: Расчет: С, 49,57; Н, 5,82; N, 11,56; S, 13,23; Найдено: С, 49,57; Н, 5,80; N, 11,75; S, 13,33. Пример 9. 2-(2,3-Дигидробензо[1,4]диоксин-2-ил)этилсульфамид (соединение 16) Добавляли цианид калия (2,05 г, 31,5 ммоль) к 2-бромметил-(2,3-дигидробензо[1,4]диоксину) (6,87 г, 30 ммоль) в ДМСО (90 мл) и перемешивали при температуре окружающей среды в течение 20 ч. Затем реакционную смесь разбавляли водой (250 мл) и дважды экстрагировали диэтиловым эфиром. Раствор в диэтиловом эфире промывали водой, затем дважды промывали насыщенным раствором соли, сушили(Na2SO4) и выпаривали в вакууме с получением 2-цианометил-(2,3 дигидробензо[1,4]диоксина) в виде белого твердого вещества. 1 Н ЯМР (CDCl3) : 6,89 (м, 4 Н), 4,50 (м, 1 Н), 4,31 (дд, J=2,3, 11,5 Гц, 1 Н), 4,08 (дд, J=6,2, 11,6 Гц,1 Н), 2,78 (д, J=6,1, Гц, 2 Н). 2-Цианометил-(2,3 дигидробензо[1,4]диоксин) растворяли в ТГФ (50 мл), добавляли 1 М ВН 3 в ТГФ(80 мл, 80 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 5 ч, затем перемешивали при температуре окружающей среды в течение 16 ч. При охлаждении на бане со льдом добавляли 2N HCl до достижения рН 1,0. Затем реакционную смесь перемешивали в течение 1 ч при комнатной температуре и выпаривали в вакууме с получением масла. Масло распределяли между 3N NaOH и диэтиловым эфиром и раствор в диэтиловом эфире промывали насыщенным раствором соли, сушилиMS (M+H)+ 180. Неочищенный 2-(2,3-дигидробензо[1,4]диоксин-2-ил)этиламин в диоксане (100 мл) объединяли с сульфамидом (3,0 г, 31 ммоль) и нагревали при кипячении с обратным холодильником в течение 2 ч. Раствор охлаждали и выпаривали в вакууме, получая оранжевое твердое вещество, которое очищали методом колоночной хроматографии (ДХМ:МеОН=10:1) с получением белого твердого вещества. Твердое вещество перекристаллизовывали из ДХМ с получением указанного в заголовке соединения в виде твердого вещества. МС (М-1) 257; Т.пл. 101-103 С (уточненная); 1 Н ЯМР (CDCl3) : 6,86 (м, 4 Н), 4,70 (м, 1 Н), 4,52 (с, 2 Н), 4,30 (м, 2 Н), 3,94 (дд, J=7,4, 11,3 Гц, 1 Н),3,43 (дд, J=6,4, 12,9 Гц, 2 Н), 1,94 (дд, J=6,5, 12,9, 2 Н). Элементный анализ: Найдено: С, 46,48; Н, 5,60; N, 10,81; S, 12,41; Расчет: С, 46,50; Н, 5,46; N, 10,85; S, 12,41. Пример 10. (2S)-(-)-N-(6,7-дихлор-2,3-дигидробензо[1,4]диоксин-2-илметил)сульфамид (соединение 29) 4,5-Дихлоратехол (8,6 г, 48 ммоль) и карбонат калия (6,64 г, 48 ммоль) перемешивали в ДМФА (200 мл). Добавляли (2R)-глицидилтозилат (9,12 г, 40 ммоль) и реакционную смесь перемешивали при 60 С в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры и затем разбавляли ледяной водой (600 мл) и экстрагировали диэтиловым эфиром (4 раза). Объединенный органический раствор промывали 3 раза 10% карбонатом калия, дважды насыщенным раствором соли, сушили (MgSO4) и выпаривали в вакууме с получением вязкого масла (2S)-2-(6,7-дихлор-2,3-дигидробензо[1,4]диоксин)метанола. Масло (2S)-2-(6,7-дихлор-2,3-дигидробензо[1,4]диоксин)метанол (6,4 г, 27 ммоль) растворяли в пиридине (50 мл) и охлаждали до 0 С. Затем добавляли паратолуолсульфонилхлорид (5,2 г, 27 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 20 ч. Реакционную смесь разбавляли диэтиловым эфиром и 1N HCl (750 мл) и органический слой отделяли и промывали 2 раза 1NHCl (250 мл), один раз водой (150 мл), дважды насыщенным раствором соли, сушили (MgSO4) и выпаривали в вакууме с получением светло-желтого твердого вещества (2S)-6,7-дихлор-2,3-дигидробензо(8,0 г, 20,5 ммоль) объединяли с фталимидом калия (6,1 г, 33 ммоль) в ДМФА (75 мл) и нагревали при кипячении с обратным холодильником в течение 1 ч, охлаждали до комнатной температуры, выливали в энергично перемешиваемую воду (0,5 л) и затем перемешивали 30 мин. Белое твердое вещество отфильтровывали и промывали несколько раз водой, 2% NaOH и снова водой и затем сушили на воздухе с- 15016892 получением (2S)-2-(6,7-дихлор-2,3-дигидробензо[1,4]диоксин-2-илметил)изоиндол-1,3-диона (6,0 г, 80%) в виде белого порошкообразного твердого вещества. Белое порошкообразное твердое вещество объединяли с гидразином (1,06 г, 33 ммоль) в EtOH (80 мл) и нагревали при кипячении с обратным холодильником в течение 2 ч, затем охлаждали до комнатной температуры. Добавляли 1N HCl, доводя рН реакционной смеси до рН 1,0, и затем реакционную смесь перемешивали в течение 15 мин. Белое твердое вещество отфильтровывали и промывали свежим EtOH(твердое вещество отбрасывали), фильтрат выпаривали в вакууме, получая твердое вещество, которое распределяли между диэтиловым эфиром и разбавленным водным NaOH. Раствор в диэтиловом эфире сушили (Na2SO4) и выпаривали в вакууме с получением вязкого масла (2S)-2-аминометил-(6,7-дихлор 2,3-дигидробензо[1,4]диоксина). 1 Н ЯМР (CDCl3) : 6,98 (с, 1 Н), 6,96 (с, 1 Н), 4,25 (дд, J=2,0, 11,2 Гц, 1 Н), 4,15 (м, 1 Н), 4,0 (м, 1 Н),2,97 (д, J=5,5 Гц, 2 Н). Часть масла (3,8 г, 16 ммоль) и сульфамид (3,1 г, 32,4 ммоль) кипятили с обратным холодильником в диоксане (100 мл) в течение 2 ч и неочищенный продукт очищали методом колоночной флэшхроматографии (ДХМ:МеОН=20:1) с получением указанного в заголовке соединения в виде белого твердого вещества, которое перекристаллизовывали из смеси этилацетат/гексан с получением указанного в заголовке соединения в виде белого кристаллического твердого вещества. МС [М-Н]- 311,0; Т.пл. 119-121 С; Указанное в заголовке соединение получали согласно методике, описанной выше в примере 4, исходя из 4-метилкатехола и получая белое твердое вещество, которое перекристаллизовывали из смеси этилацетат/гексан с получением указанного в заголовке соединения в виде белого твердого вещества.MS [M-H]- 257; 1 Н ЯМР (CDCl3) : 6,76 (м, 1 Н), 6,66 (м, 2 Н), 4,80 (м, 1 Н), 4,57 (широкий с, 1 Н), 4,40 (м, 1 Н), 4,28 (м,1 Н), 4,03 (дд, J=6,9, 11,4 Гц, 1 Н), 3,45 (м, 2 Н), 2,25 (с, 3 Н). Элементный анализ: Расчет: С, 46,50; Н, 5,46; N, 10,85; S, 12,41; Найдено:С, 46,65; Н, 5,60; N, 10,84; S, 12,61. Пример 13. In vivo исследование на диабетических мышах db/db. Мыши db/db, как известно в данной области, восприимчивы к диабету типа II (Sharma K., McCue P.,Dunn S.R. Am. J. Physiol Renal Physiol. 2003 Jun; 284 (6) :F1138-44). Как известно в данной области, мыши db/db также являются подходящей моделью для дислипидемии. Получали 8-недельных самок мышей db/db (C57BL/6J-Lepdb/db, Jackson Laboratories, Bar Harbor, ME,USA), размещали по одной и кормили в обычном режиме. Отбор крови производили, делая прокол в области хвоста и контролируя глюкозу при помощи глюкометра (OneTouch Basic, Lifescan, Newtown, PA). Мышей в возрасте 10 недель делили случайным образом на подопытные группы на основании уровней глюкозы (первый критерий, среднее значение 250 мг/дл) и массы тела (второй критерий, среднее значение 37 г). Мышам вводили перорально через зонд один раз в день (0,2 мл в 15.00-17.00 ч) контрольный носитель (0,5% метилцеллюлозу, рН 7,4) и носитель, содержащий исследуемое соединение (А 300 мг/кг). На 11 день мыши голодали в течение 4 ч во время светового цикла (пищу убирали в 06.00-10.00 ч), и при помощи глюкометра у них измеряли уровни глюкозы в крови, делая прокол в области хвоста в 10.00 ч. Затем мышей анестезировали пентабарбиталом натрия (1 мл/кг, внутрибрюшинно, Sleepaway,Fort Dodge, Iowa) и отбирали кровь посредством пункции сердца и собирали в гепаринизированные пробирки. Иссекали и взвешивали белый жир (WAT) (ретроперитонеальный жир) и скелетные мышцы (икроножную и камбаловидную мышцы). Образцы плазмы получали центрифугированием при 2000g и 4 С в течение 15 мин и производили определение инсулина, HDL-холестерина и триглицеридов. Приведенные ниже данные представлены в виде среднего значения со стандартной ошибкой, рассчитанного при использовании 9-10 мышей на подопытную группу. Для статистического анализа использовали двусторонние t-тесты Стьюдента. Во всех исследованиях на животных соблюдались указанияInstitutional Animal Care and Use Committee. Соединение 8 оценивали согласно описанной выше методике. Уровни глюкозы в крови самок мышей db/db составляли 25515 мг/дл за 5 дней до экспериментов. В конце эксперимента уровни глюкозы в крови контрольных мышей, обработанных носителем, являлись повышенными - 166% (42022 мг/дл). Уровни глюкозы в крови мышей db/db значительно ниже при обработке соединением 8 по сравнению с мышами, обработанными носителем. Уровни инсулина у животных, обработанных соединением 8, не отличались статистически при сравнении с животными, обработанными носителем. Мыши, обработанные соединением 8, демонстрировали большую массу скелетной мышцы по сравнению с животными, обработанными носителем. Кроме того, отсутствовало существенное снижение массы жира у животных, обработанных соединением 8. Мыши, обработанные соединение 8, также демонстрировали значительное снижение соотношения масс жир:мясо (носитель: 27,91,4 относительно соединения 8: 23,40,9, р 0,01). Кроме того, уровни HDL-холестерина в плазме у мышей db/db, обработанных соединением 8,более высокие по сравнению с мышами, обработанными носителем, тогда как уровни триглицеридов в крови мышей db/db, обработанных соединением 8, ниже, чем у мышей, обработанных носителем. Краткое изложение данных измерений для мышей, обработанных носителем и соединением 8,уровни глюкозы в крови, массы ретроперитонеального жира, скелетных мышц, уровни триглицеридов иHDL-холестерина приведены ниже в табл. 4. Таблица 4 Результаты in vivo исследований на диабетических мышах db/db Таким образом, данные показывают, что соединение 8 эффективно для (а) понижения уровней глюкозы в крови, (b) понижения уровня триглицеридов и (с) повышения уровней HDL-холестерина. Кроме того, животные, обработанные соединением 8, имеют большую массу мышц, чем животные,обработанные носителем, это предполагает, что соединение 8 может сохранять массу мышц, то есть предупреждать диабетическое истощение. Пример 14. Исследование на самках мышей db/db. Соединение 8 суспендировали в 0,5% метоцеле, используя ручной гомогенизатор для уменьшения размера частиц и магнитную мешалку и пластину для поддержания гомогенности суспензии частиц на протяжении всего периода дозирования. В качестве носителя/контроля использовали гидроксипропилметилцеллюлозу (метоцел). Соединение 8 исследовали на обеих диабетических моделях мышей и крыс. Для исследований эффекта понижения уровня глюкозы использовали самок диабетических мышейdb/db с гипергликемией (концентрации глюкозы в крови составляли в среднем 250 мг/дл). Средняя масса тела мышей db/db составляла 37 г. Мыши db/db восприимчивы к диабету типа II. Самок мышей db/db (C57BL/6J-Lepdb/db, Jackson Laboratories, Bar Harbor, ME, USA) 8-недельного возраста размещали группами, по две в каждую клетку, и кормили в обычном режиме (Laboratory rodent diet 5001). Все мыши проходили карантин в течение одной недели до перевода в процедурную комнату для животных. Отбирали каплю крови (около 2 мкл), делая прокол в области хвоста, и определяли концентрацию глюкозы при помощи глюкометра (OneTouch UltraSmart, Lifescan, Milpitas, CA). Мышей в возрасте 10 недель делили случайным образом на три подопытные группы на основании значений уровней глюкозы (первый критерий, среднее значение составляло 250 мг/дл) и массы тела (второй критерий,среднее значение составляло 37 г). Животных делили и размещали по одному по меньшей мере за три дня до обработки лекарством для акклиматизации в новом окружении. Исследование на мышах включало две части: в первой части исследования А с введением разовой дозы 10 мышам, используемым в качестве негативного контроля, вводили носитель (0,5% метоцел); 10 мышам вводили 300 мг/кг соединения- 170168928 JNJ-26489112 в носителе и 10 мышам, используемым в качестве позитивного контроля, вводили 20 мг/кг розиглитазона (сенсибилизатора инсулина, который понижает уровень глюкозы) в носителе. Во второй части исследования А с изучением реакции на дозу 48 мышей делили на 4 подопытные группы по 12 мышей в каждой. Затем животных четырех групп обрабатывали 0,5% метоцелом (носителем), 15 мг/кг соединения 8, 30 мг/кг соединения 8 и 100 мг/кг соединения 8 в носителе соответственно. Мышам вводили перорально через зонд один раз в день (в 15.00-17.00 ч) контрольный носитель(0,5% метоцел, рН 7,4) или носитель, содержащий исследуемое соединение, в течение 10 дней. Дозируемый объем составлял 5 мл/кг массы тела (0,2 мл для мышей массой 40 г). Мышей кормили неограниченно во время всего исследования. Через 18 ч после последнего введения дозы производили вскрытие. При помощи глюкометра измеряли уровни глюкозы в крови в образцах, отобранных посредством прокола в области хвоста в 10.00 ч. Мышей анестезировали пентабарбиталом натрия (1 мл/кг, внутрибрюшинная [i.p.] инъекция, SleepAway, Fort Dodge, Iowa), отбирали кровь посредством пункции сердца,используя шприц на 1 мл, и собирали в гепаринизированные пробирки. Иссекали и взвешивали белую жировую ткань (WAT) (ретроперитонеальные и ингвинальные жировые тела), коричневую жировую ткань (ВАТ), скелетные мышцы (икроножную и камбаловидную мышцы) и содержимое желудка. Образцы плазмы получали центрифугированием цельной крови при 2000-3000g и 4 С в течение 10-20 мин и хранили при -20 С для дальнейшего определения инсулина, HDL, LDL, общего холестерина и триглицеридов. Концентрации инсулина плазмы определяют в двух исследованиях, используя подходящий набор для исследования с использованием связанного с ферментом иммуносорбента (ELISA) (EZRMI-13K,LINCO Research, St. Charles, Missouri) крысиного/мышиного инсулина. Образцы крови разводили 1:4 в очищенной древесным углем мышиной сыворотке, которая включена в набор ELISA. Остальную процедуру проводили, следуя инструкции производителей. Общую флуоресценцию детектировали, используя микропланшетный люминометр Orion 1 (Berthold Detection Systems, Pforzheim, Germany). Общий холестерин плазмы, концентрации липопротеина высокой плотности (HDL), липопротеина низкой плотности (LDL) и триглицеридов определяли, используя анализатор химического состава кровиBayer ADVIA 1650 (Bayer Healthcare LLC, Diagnostic Divisions Tarrytown, NY). Согласно протоколу производителей определение холестерина является ферментативным способом, использующим конверсию холестеринэстеразы и холестериноксидазы с последующим определением по конечной точке Триндера; для определения HDL применяли метод удаления (перекиси водорода) под действием каталазы; для определения триглицеридов применяли ферментативный метод определения по конечной точке Триндера. Данные обеих частей исследования (с разовой дозой и откликом на дозу) анализировали, используя стандартный двусторонний t-тест Стьюдента, и выражали в виде средних значений со стандартными ошибками, которые приведены ниже. В части исследования А с введением разовой дозы средние уровни глюкозы в крови db/db мышей составляли 25515 мг/дл до введения дозы. Как показано ниже в табл. 5, в конце эксперимента уровни глюкозы у мышей, обработанных носителем, являлись повышенными - 166% (420122 мг/дл). Уровни глюкозы понижены - 50% у мышей, обработанных соединением 8 при 300 мг/кг, по сравнению с мышами, обработанными носителем. Этот эффект аналогичен эффекту, наблюдаемому при лечении розиглитазоном. Среди мышей, обработанных лекарством, и мышей, обработанных носителем, не наблюдалось изменений концентраций инсулина. Таблица 5 В части исследования А с откликом на дозу, как показано ниже в табл. 6, обработка соединением 8 при дозе 10, 30 или 100 мг/кг не влияла на уровень глюкозы в крови. В противоположность этому гиперинсулинемия у мышей, обработанных 100 мг/кг соединения 8, снижалась на 63,5% по сравнению с мышами, обработанными носителем. В части исследования А с единичной дозой мыши db/db, принимавшие контрольный носитель, демонстрировали дислипидемию с высокими циркулирующими концентрациями триглицеридов и низкими уровнями HDL, что давало в результате высокое соотношение триглицеридов к HDL. Как показано ниже в табл. 7, соединение 8 при дозе 300 мг/кг снижало триглицериды плазмы на 39,4% и повышало HDL на 22,8% по сравнению с мышами, обработанными носителем. Следовательно, соотношение триглицеридов к HDL снижается на 50,2%, что отражает улучшенный липидный профиль. Этот результат аналогичен эффекту, наблюдаемому при обработке розиглитазоном. Для мышей, обработанных соединением 8, розиглитазоном или носителем, не наблюдали изменений уровней LDL. Таблица 7 В части исследования А с откликом на дозу мышей обрабатывали дозой 10, 30 и 100 мг/кг соединения 8. Мыши, обработанные 100 мг/кг соединения 8, демонстрировали значительно пониженнное соотношение триглицеридов к HDL, как показано ниже в табл. 8. Общие концентрации холестерина являлись повышенными и у мышей, обработанных соединением 8 (на 18,7%), и у мышей, обработанных розиглитазоном (на 35,1%), относительно мышей, обработанных носителем, соответственно. Таблица 8 В обеих частях исследования А потребление пищи контролировали каждые три дня в течение периода девяти дней. На первой стадии, как показано в табл. 9, у мышей, которым вводили дозу 300 мг/кг соединения 8, значительно снижалось потребление пищи по сравнению с мышами, обработанными носителем, тогда как обработка розиглитазоном повышала потребление пищи. Несмотря на снижение потребления пищи, не наблюдалось влияния на массу тела мышей, обработанных дозой 300 мг/кг соединения 8. Мыши, обработанные розиглитазоном, имели увеличенную массу тела (прирост массы- 19016892 3,10,4 г, р 0,05) по сравнению с мышами, обработанными носителем (прирост массы: 0,30,2 г). В конце эксперимента масса содержимого желудков была в два раза больше у мышей, обработанных 300 мг/кг соединения 8, чем у мышей, обработанных носителем. Таблица 9 Масса скелетных мышц и коричневой жировой ткани у мышей db/db, обработанных соединением 8 при дозе 300 мг/кг, была больше, чем у мышей, обработанных носителем, как показано ниже в табл. 10. Кроме того, соотношение белой жировой ткани к мышцам было значительно понижено у мышей,обработанных соединением 8, хотя различий в массе ингвинальных и ретроперитонеальных жировых тел между этими группами не обнаружено. Таблица 10 В части исследования А с откликом на дозу, как показано ниже в табл. 11, обработка соединением 8 при дозе 10 мг/кг, 30 мг/кг и 100 мг/кг увеличивала массу коричневой жировой ткани по сравнению с мышами, обработанными носителем. Увеличенную массу содержимого желудка наблюдали у мышей,принимавших дозу 100 мг/кг соединения 8 (обработка лекарством: 0,510,04 г относительно контроля: 0,350,03 г, р 0,05), но не при меньших дозах. Ни в одной из групп не наблюдали различий в потреблении пищи и массе тела. В приведенной ниже табл. 11 соотношение жир/мышцы рассчитывали как отношение: общая масса ингвинального жира и ретроперитонеального жира/мышечная масса. Пример 15. Исследование на диабетических крысах с ожирением. Соединение 8 суспендировали в 0,5% метоцеле, используя ручной гомогенизатор для уменьшения размера частиц и магнитную мешалку и пластину для поддержания гомогенности суспензии частиц на протяжении всего периода дозирования. В качестве носителя/контроля использовали 0,5% гидроксипропилметилцеллюлозу (метоцел). Соединение 8 исследовали на обеих диабетических моделях, мышей и крыс. В этом исследовании отбирали самок диабетических крыс с ожирением линии Zucker (ZDF Gmifa/fa) для исследования эффекта понижения уровня глюкозы и перорального теста толерантности к глюкозе (OGTT). ZDF-крыс (от Charles River Laboratories, Wilmington, MA) 7-недельного возраста размещали индивидуально и кормили кормом С 13004 (полученным от Research Diets, New Brunswick, NJ). Все крысы проходили карантин в течение одной недели перед переводом в процедурную комнату для животных. Отбирали каплю крови (примерно 2 мкл) посредством прокола в области хвоста и определяли концентрацию глюкозы при помощи глюкометра (OneTouch UltraSmart, Lifescan, Milpitas, CA).ZDF-крыс 8-недельного возраста делили случайным образом на четыре подопытные группы на основании уровней глюкозы (первый критерий, среднее значение 150 мг/дл) и массы тела (второй критерий, среднее значение 240 г). 32 крыс делили на 4 подопытные группы по 8 крыс в каждой. Затем четыре группы обрабатывали 0,5% метоцелом (носитель), 10 мг/кг соединения 8, 30 мг/кг соединения 8 и 100 мг/кг соединения 8 в носителе соответственно. Крысам вводили перорально через зонд один раз в день (в 15.00-17.00 ч) контрольный носитель(0,5% метоцел, рН 7,4) или носитель, содержащий соединение 8, в течение 7 дней. Дозируемый объем составлял 5 мл/кг массы тела (1,2 мл для крысы массой 250 г). Крыс кормили неограниченно во время всего исследования. Определяли базальные уровни глюкозы после обработок в день 4 и день 6 у крыс, голодающих в течение 2 ч, при помощи глюкометра (OneTouch UltraSmart, Lifescan, Milpitas, CA) после отбора 2 мкл крови посредством прокола в области хвоста. Пероральный тест толерантности к глюкозе (OGTT) проводили в день 7 на крысах, голодающих в течение 4 ч. Через 2 ч после введения дозы соединения 8 или носителя крысам вводили через зонд 2 г/кг 50% раствора глюкозы непосредственно после определения базальных уровней глюкозы (0 мин). Затем определяли уровни глюкозы в крови через 30, 60, 90 и 120 мин посредством прокола в области хвоста. На день 8 крыс анестезировали посредством внутрибрюшинной [i.p.] инъекции пентобарбитала натрия (1 мл/кг, SleepAway, Fort Dodge, Iowa) и отбирали кровь посредством пункции сердца, применяя шприц на 3 мл и собирая в гепаринизированные пробирки. Образцы плазмы получали центрифугированием цельной крови при 2000-3000g и 4 С в течение 10-20 мин и хранили при -20 С для дальнейшего определения инсулина, HDL, общего холестерина и триглицеридов. Концентрации инсулина в плазме определяли в обоих исследованиях, используя подходящий набор для исследования с использованием связанного с ферментом иммуносорбента (ELISA) (EZRMI-13K,LINCO Research, St. Charles, Missouri) крысиного/мышиного инсулина. Образцы крови разводили 1:4 в очищенной древесным углем мышиной сыворотке, которая включена в набор ELISA. Остальную процедуру проводили, следуя инструкции производителей. Общую флуоресценцию детектировали, используя микропланшетный люминометр Orion 1 (Berthold Detection Systems, Pforzheim, Germany).- 21016892 Общий холестерин плазмы, концентрации липопротеина высокой плотности (HDL), липопротеина низкой плотности (LDL) и триглицеридов определяли, используя Bayer ADVIA 1650 анализатор химического состава крови (Bayer Healthcare LLC, Diagnostic Division, Tarrytown, NY). Согласно протоколу производителей определение холестерина является ферментативным способом, использующим конверсию холестеринэстеразы и холестериноксидазы с последующим определением по конечной точке Триндера; для определения HDL применяли способ с удалением (перекиси водорода) под действием каталазы; для определения триглицеридов применяли ферментативный метод определения по конечной точке Триндера. Данные этого исследования анализировали, используя стандартный двусторонний t-тест Стьюдента, и выражали в виде средних значений со стандартными ошибками, которые приведены ниже. В исследовании В с диабетическими крысами Zucker, как показано в табл. 12, обработка дозой 30 или 100 мг/кг в день соединения 8 давала зависимые от дозы и времени эффекты понижения уровня глюкозы в день 4 и день 6. Таблица 12 Как показано в табл. 13, OGTT, проводимый в день 7, демонстрировал улучшенный профиль глюкозы и инсулина для крыс, обработанных соединением 8, зависимым от дозы образом. После пробы на глюкозу высокие уровни глюкозы сохранялись до 60 мин у крыс, обработанных контрольным носителем,тогда как крысы, обработанные дозой 100 мг/кг соединения 8, имели меньшие уровни глюкозы, которые имели пик через 30 мин. Область под кривой (AUC) для глюкозы в крови также значительно уменьшена у ZDF-крыс, обработанных соединением 8 при дозах 30 и 100 мг/кг, по сравнению с группой,обработанной носителем. Таблица 13 Как показано в табл. 14, обработка дозой 100 мг/кг соединения 8 значительно понижает уровень инсулина в плазме на 32% и уровни триглицеридов в день 8. Соотношение триглицеридов к HDL уменьшается на 59%. Этот результат согласуется с эффектом, наблюдаемым у мышей db/db при обработке соединением 8, кроме того, эффективная доза для крыс ZDF на две трети ниже дозы для мышей db/db. Снижения потребления пищи или потери массы не наблюдали ни в одной из групп при исследовании Пример 16. Исследование на диабетических крысах с ожирением линии Zucker. Соединение 8 суспендировали в 0,5% метоцеле, используя ручной гомогенизатор для уменьшения размера частиц и магнитную мешалку и пластину для поддержания гомогенности суспензии частиц на протяжении всего периода дозирования. В качестве носителя/контроля использовали 0,5% гидроксипропилметилцеллюлозу (метоцел). Соединение 8 исследовали на обеих диабетических моделях, мышей и крыс. В этом исследовании отбирали самок диабетических крыс с ожирением линии Zucker (ZDF Gmifa/fa) для исследования эффекта понижения уровня глюкозы. ZDF-крыс (от Charles River Laboratories,Wilmington, MA) 7-недельного возраста размещали индивидуально и кормили кормом С 13004 (полученным от Research Diets, New Brunswick, NJ). Все крысы проходили карантин в течение одной недели перед переводом в процедурную комнату для животных. Отбирали каплю крови (примерно 2 мкл) посредством прокола в области хвоста и определяли концентрацию глюкозы при помощи глюкометра (OneTouch UltraSmart, Lifescan, Milpitas, CA).ZDF-крыс 8-недельного возраста делили случайным образом на четыре подопытные группы на основании уровней глюкозы (первый критерий, среднее значение 150 мг/дл) и массы тела (второй критерий, среднее значение 240 г). 13 крыс делили на 4 подопытные группы по 3 крысы в каждой из групп,обрабатываемых соединением 8, и 4 крысы в группе без обработки. Затем три группы обрабатывали дозами 10 мг/кг соединения 8, 30 мг/кг соединения 8 и 100 мг/кг соединения 8 в 0,5 метилцеллюлозе соответственно. Крысам вводили перорально через зонд один раз в день (в период 15.00-17.00 ч) контрольный носитель (0,5% метоцел, рН 7,4) или носитель, содержащий соединение 8, в течение 7 дней. Дозируемый объем составлял 5 мл/кг массы тела (1,2 мл для крысы массой 250 г). Крыс кормили неограниченно во время всего исследования. Затем крысам давали 29-дневный период выведения, когда не вводили дозы. Определяли базальные уровни глюкозы после обработок в день 0 (базисные) и день 36 (после 7 дней приема дозы и 29 дней выведения) у крыс с неограниченным кормлением при помощи глюкометра(OneTouch UltraSmart, Lifescan, Milpitas, CA) после отбора 2 мкл крови посредством прокола в области хвоста. Данные этого исследования анализировали, используя стандартный двусторонний t-тест Стьюдента, и выражали в виде средних значений со стандартными ошибками, которые приведены ниже в табл. 15. Обработка соединением 8 при дозе 30 и 100 мг/кг демонстрировала статистически значительно(р 0,01) пониженные уровни глюкозы в крови даже после 29-дневного периода выведения. Таблица 15 Пример 17. В отдельном варианте композиции для перорального приема 100 мг соединения 8, полученного,как в примере 7, готовили в виде препарата с достаточно мелкодисперсной лактозой, обеспечивая общее количество от 580 до 590 мг для заполнения твердой желатиновой капсулы размера 0. При том, что в предшествующем описании объясняются принципы настоящего изобретения вместе- 23016892 с примерами, приведенными для иллюстрации, понятно, что осуществление изобретения на практике охватывает все обычные вариации, адаптации и/или модификации, которые входят в область следующей далее формулы изобретения, и их эквиваленты. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ лечения связанного с липидами расстройства, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (I) где R1 и R2, каждый независимо, выбран из группы, состоящей из атома водорода и C1-4 алкила;R4 выбран из группы, включающей атом водорода и C1-4 алкил; а равно целому числу от 1 до 2; представляет собой где b равно целому числу от 0 до 4; каждый R5 независимо выбран из группы, состоящей из галогена и C1-4 алкила; или его фармацевтически приемлемой соли. 2. Способ по п.1, включающий введение соединения формулы (I), гдеR1 и R2, каждый независимо, выбран из группы, состоящей из атома водорода и C1-4 алкила;R4 выбран из группы, состоящей из атома водорода и C1-4 алкила; а равно целому числу от 1 до 2; представляет собой где b равно целому числу от 0 до 2; каждый R5 независимо выбран из группы, состоящей из галогена и C1-4 алкила; или его фармацевтически приемлемой соли. 3. Способ по п.2, включающий введение соединения формулы (I), гдеR1 и R2, каждый независимо, выбран из группы, состоящей из атома водорода и C1-4 алкила;R4 выбран из группы, состоящей из атома водорода и метила; а равно целому числу от 1 до 2; выбран из группы, состоящей из 2-(2,3-дигидробензо[1,4]диоксинила),2-(бензо[1,3]диоксолила),2-(3,4-дигидро-2 Н-бензо[1,4]диоксепинила),2-(2,3-дигидробензо[1,4]диоксинила),2-(6-хлор-2,3-дигидробензо[1,4]диоксинила),2-(6-фтор-2,3-дигидробензо[1,4]диоксинила),2-(5-фтор-2,3-дигидробензо[1,4]диоксинила),2-(7-хлор-2,3-дигидробензо[1,4]диоксинила),2-(6-хлорбензо[1,3]диоксолила),2-(7-метил-2,3-дигидробензо[1,4]диоксинила),2-(5-хлор-2,3-дигидробензо[1,4]диоксинила),2-(6-бром-2,3-дигидробензо[1,4]диоксинила),2-(6,7-дихлор-2,3-дигидробензо[1,4]диоксинила),2-(8-хлор-2,3-дигидробензо[1,4]диоксинила); или его фармацевтически приемлемой соли. 4. Способ по п.3, включающий введение соединения формулы (I), гдеR1 и R2, каждый независимо, выбран из группы, состоящей из атома водорода и метила;R4 выбран из группы, состоящей из атома водорода и метила; а равно целому числу от 1 до 2; выбран из группы, состоящей из- 24016892 2-(2,3-дигидробензо[1,4]диоксинила),2-(6-хлор-2,3-дигидробензо[1,4]диоксинила),2-(7-хлор-2,3-дигидробензо[1,4]диоксинила),2-(7-метил-2,3-дигидробензо[1,4]диоксинила),2-(6-бром-2,3-дигидробензо[1,4]диоксинила) и 2-(6,7-дихлор-2,3-дигидробензо[1,4]диоксинила); или его фармацевтически приемлемой соли. 5. Способ по п.1, где соединение формулы (I) выбрано из группы, состоящей из (2S)-(-)-N-(6-хлор 2,3-дигидробензо[1,4]диоксин-2-илметил)сульфамида и его фармацевтически приемлемых солей. 6. Способ по п.1, где связанное с липидами расстройство выбрано из группы, включающей повышенные уровни триглицеридов и низкие уровни HLD-холестерина. 7. Способ лечения связанного с липидами расстройства, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения, выбранного из группы, состоящей из (2S)-(-)-N-(6-хлор-2,3-дигидробензо[1,4]диоксин-2-илметил)сульфамида и его фармацевтически приемлемых солей. 8. Способ по п.7, где связанное с липидами расстройство выбрано из группы, включающей повышенные уровни триглицеридов и низкие уровни HLD-холестерина. 9. Способ лечения связанного с липидами расстройства, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы или его фармацевтически приемлемой соли; где связанное с липидами расстройство характеризуется отличными от нормальных уровнями липидов.
МПК / Метки
МПК: A61K 31/353, A61P 3/10, A61K 31/357, A61K 31/352, A61P 3/06
Метки: липидов, крови, уровней, понижения, бензоконденсированных, производных, гетероциклических, применение, сульфамидных
Код ссылки
<a href="https://eas.patents.su/26-16892-primenenie-benzokondensirovannyh-geterociklicheskih-sulfamidnyh-proizvodnyh-dlya-ponizheniya-urovnejj-lipidov-v-krovi.html" rel="bookmark" title="База патентов Евразийского Союза">Применение бензоконденсированных гетероциклических сульфамидных производных для понижения уровней липидов в крови</a>
Применение бензоконденсированных гетероциклических сульфамидных производных в качестве нейропротективных агентов

Номер патента: 16628
Опубликовано: 30.06.2012
Авторы: Рейтц Аллен Б., Смит-Свинтоски Вирджиния Л.
МПК: A61K 31/357, A61K 31/353, A61P 25/02...
Метки: сульфамидных, гетероциклических, качестве, применение, агентов, производных, бензоконденсированных, нейропротективных
Формула / Реферат:
1. Способ нейропротекции при окислительном стрессе, включающий введение субъекту терапевтически эффективного количества соединения формулы (I)где каждый R1 и R2 независимо выбран из группы, включающей водород и C1-4алкил;R4 выбран из группы, включающей водород и C1-4алкил;а является целым числом 1 или 2;представляет собойгде b является целым числом от 0 до 4;каждый R5 независимо выбран из группы, включающей галоген и C1-4алкил;или его...
Применение бензоконденсированных гетероциклических сульфамидных производных для лечения и профилактики эпилептогенеза

Номер патента: 16610
Опубликовано: 30.06.2012
Автор: Смит-Свинтоски Вирджиния Л.
МПК: A61K 31/353, A61P 25/08, A61K 31/357...
Метки: лечения, гетероциклических, производных, бензоконденсированных, профилактики, сульфамидных, эпилептогенеза, применение
Формула / Реферат:
1. Способ лечения, профилактики, замедления, прекращения или инвертирования эпилептогенеза, включающий введение пациенту терапевтически эффективного количества соединения формулы (I)где R1 и R2, каждый независимо, выбраны из группы, включающей атом водорода и С1-4алкил;R4 выбран из группы, включающей атом водорода и С1-4алкил;а равно целому числу 1 или 2;представляет собой группугде b равно целому числу от 0 до 4;каждый R5 независимо выбран из...
Применение бензоконденсированных гетероциклических сульфамидных производных для лечения маниакального синдрома и биполярного расстройства

Номер патента: 15514
Опубликовано: 31.08.2011
Авторы: Смит-Свинтоски Вирджиния Л., Рейтц Аллен Б.
МПК: A61P 25/18, A61K 31/353, A61K 31/357...
Метки: применение, гетероциклических, расстройства, бензоконденсированных, лечения, производных, сульфамидных, биполярного, синдрома, маниакального
Формула / Реферат:
1. Способ лечения маниакального синдрома, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I)в которой R1 и R2, каждый независимо, выбраны из группы, состоящей из водорода и низшего алкила;R4 выбран из группы, состоящей из водорода и низшего алкила;а представляет собой целое число от 1 до 2;выбран из группы, состоящей изгде b представляет собой целое число от 0 до 4 и где с...
Способы снятия боли с использованием бензоконденсированных гетероциклических сульфамидных производных (варианты)

Номер патента: 16302
Опубликовано: 30.04.2012
Авторы: Смит-Свинтоски Вирджиния Л., Рейтц Аллен Б.
МПК: A61K 31/357, A61K 31/352, A61K 31/353...
Метки: производных, гетероциклических, бензоконденсированных, варианты, снятия, способы, использованием, сульфамидных, боли
Формула / Реферат:
1. Способ снятия боли, включающий введение нуждающемуся субъекту терапевтически эффективного количества соединения формулы (I)где каждый из R1 и R2 независимо выбран из группы, состоящей из водорода и С1-4алкила;R4 выбран из группы, состоящей из водорода и С1-4алкила;a является целым числом 1 или 2;представляет собой;b является целым числом от 0 до 4;каждый R5 независимо выбран из группы, состоящей из галогена и С1-4алкила;или его...
Производные бензоконденсированных гетероциклических оксимов, способ их получения, содержащие их фармацевтическая композиция и комбинация и их применение

Номер патента: 12437
Опубликовано: 30.10.2009
Авторы: Карато Паскаль, Лебеге Никола, Леклерк Вероник, Л'эльгуаль'ш Жан-Марсьяль, Юртеван Орели, Даке Катрин, Бертоло Паскаль, Кторза Ален, Кэньяр Даньель-Анри
МПК: A61K 31/5415, A61K 31/538, A61P 3/06...
Метки: содержащие, фармацевтическая, применение, производные, оксимов, способ, гетероциклических, получения, комбинация, бензоконденсированных, композиция
Формула / Реферат:
1. Соединения формулы (I) в которой X представляет собой атом кислорода или атом серы, А представляет собой (С1-С6)алкиленовую цепь, в которой СН2 группа необязательно может быть заменена атомом кислорода, R1 представляет собой арильную группу, a R2 представляет собой атом водорода либо линейную или разветвленную (С1-С6)алкильную группу, R3 и R4 каждый представляет собой атом водорода, В представляет собой линейную или разветвленную...
Предыдущий патент: Электропроводящее ламинированное остекление
Следующий патент: Объемный катализатор гидрогенизации и его применение
Случайный патент: Полимерная смесь