Способ получения ингибиторов протеазы вируса иммунодефицита человека.
Номер патента: 637
Опубликовано: 29.12.1999
Авторы: Россен Кай, Сейджер Джесс В., Волант Ральф П., Аскин Дэвид, Рейдер Пол Дж.





Формула / Реферат
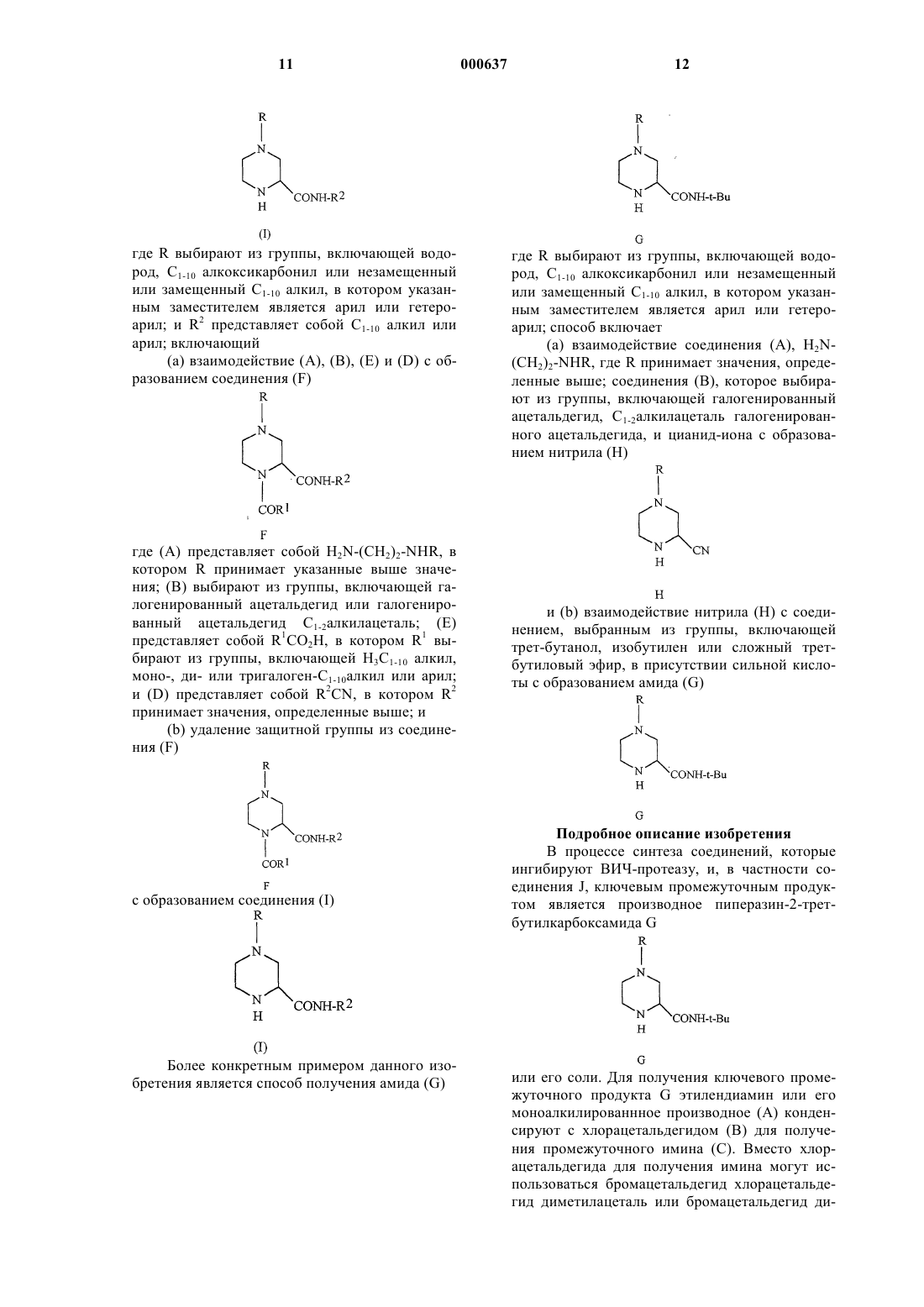
1. Способ получения соединения (I)

где R выбирают из водорода, C1-10 алкоксикарбонила или незамещенного или замещенного C1-10 алкила, где указанным заместителем является арил или гетероарил; и R2 представляет собой C1-10 алкил или арил, отличающийся тем, что включает
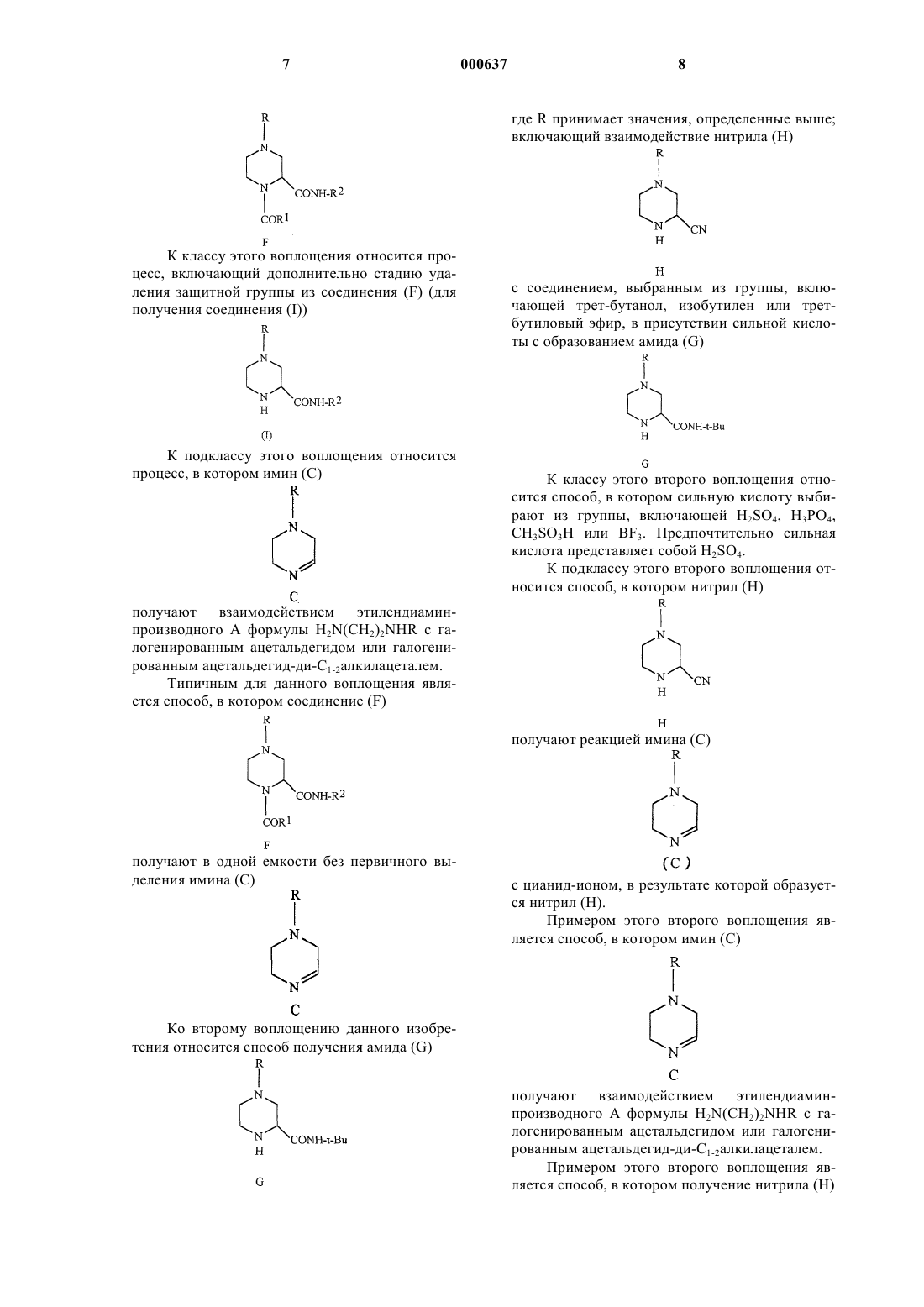
(а) взаимодействие соединений (А), (В), (Е) и (D) для получения соединения (F)

где (А) представляет собой H2N-(CH2)2-NHR, где R принимает значения, определенные выше;
(В) выбирают из галогенированного ацетальдегида или C1-2алкилацеталя галогенированного ацетальдегида;
(Е) представляет собой R1CO2H, где R1 выбирают из водорода, C1-10алкила, моно-, ди- или тригалоген-C1-10алкила или арила, и
(D) представляет собой R2NC, где R2 принимает значения, определенные выше; и
(b) удаление защитной группы из соединения (F)

для получения соединения (I)

2. Способ получения соединения (F)

где R выбирают из водорода, C1-10алкоксикарбонила или незамещенного или замещенного C1-10 алкила, где указанным заместителем является арил или гетероарил;
R1 выбирают из водорода, C1-10алкила, моно-, ди- или тригалоген-C1-10 алкила или арила, и
R2 представляет собой C1-10алкил или арил,
отличающийся тем, что включает взаимодействие имина (С)

с R2NC в присутствии карбоновой кислоты, R1-CO2H, с образованием соединения (F)

3. Способ по п.2, отличающийся тем, что имин (С)

получают взаимодействием этилендиаминового соединения А формулы H2N-(CH2)2-NHR с галогенированным ацетальдегидом или галогенированным ацетальдегид ди-С1-2 алкилацеталем.
4. Способ по п.2, отличающийся тем, что соединение (F)

получают в одном сосуде без первичного выделения имина (С)

5. Способ по п.2, отличающийся тем, что R представляет собой замещенный C1-5алкил, в котором указанным заместителем является гетероарил.
6. Способ по п.5, отличающийся тем, что R представляет собой группу формулы

и R2 представляет собой трет-бутил.
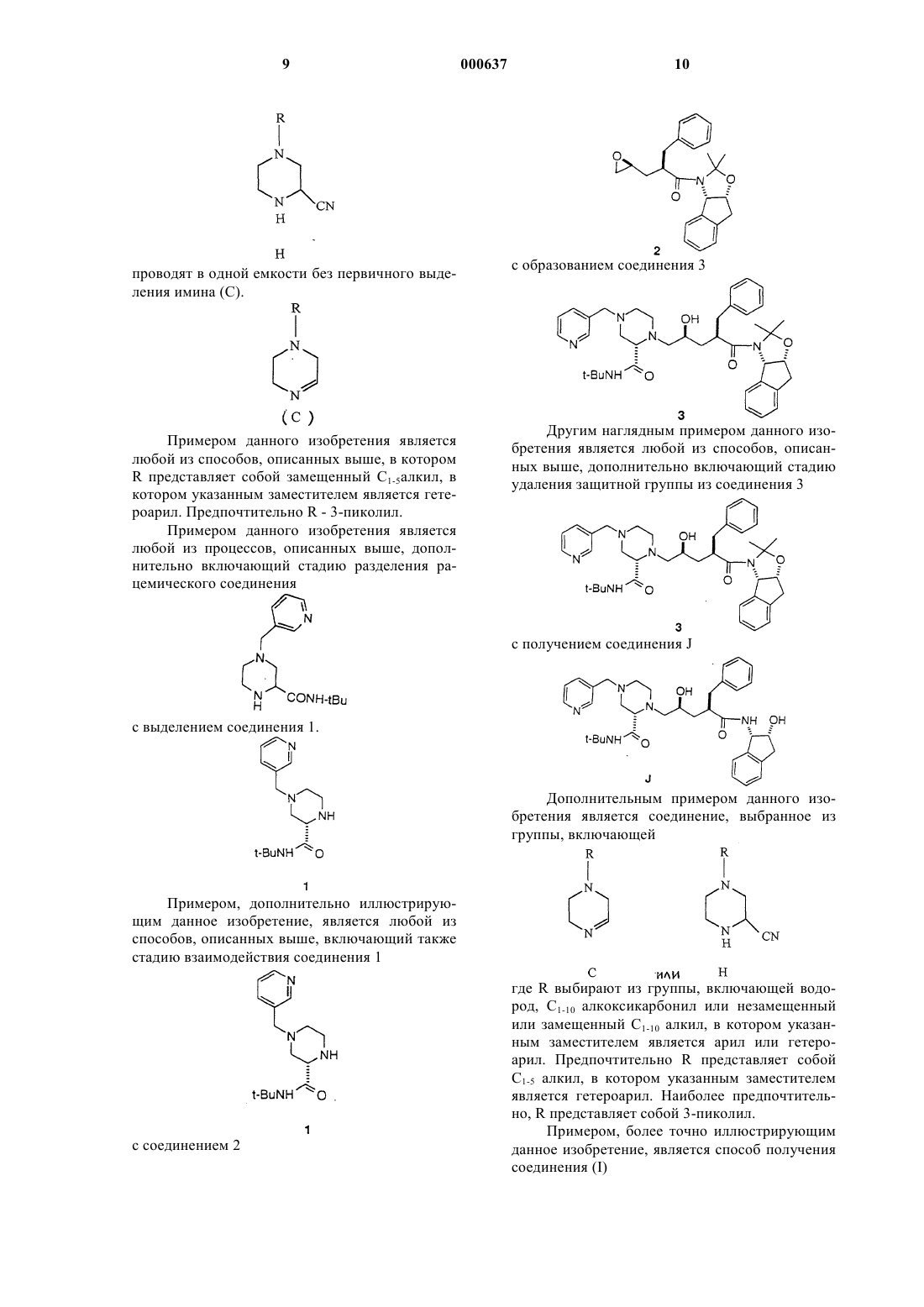
7. Способ получения соединения J

включающий следующие стадии:
1) взаимодействие нитрила (Н)

где R - группа формулы

c соединением, выбранным из трет-бутанола, изобутилена или трет-бутилового сложного эфира, в присутствии сильной кислоты с образованием соединения формулы

2) разделение рацемического соединения

для выделения соединения 1

3) взаимодействие соединения 1

с соединением 2

для получения соединения 3

4) удаление защитной группы у соединения 3

для получения соединения J

8. Способ получения амида (G)

где R выбирают из водорода, C1-10 алкоксикарбонила или незамещенного или замещенного C1-10 алкила, где указанным заместителем является арил или гетероарил, отличающийся тем, что включает взаимодействие нитрила (Н)

с соединением, выбранным из трет-бутанола, изобутилена или трет-бутилового сложного эфира, в присутствии сильной кислоты с образованием амида (G)

9. Способ по п.8, отличающийся тем, что R представляет собой замещенный C1-5алкил, где указанным заместителем является гетероарил.
10. Способ по п.9, отличающийся тем, что R представляет собой группу формулы

11. Способ по п.10, отличающийся тем, что сильную кислоту выбирают из H2SO4, Н3РO4, СН3SО3Н или ВF3.
12. Способ по п.8, отличающийся тем, что сильная кислота представляет собой H2SO4.
13. Способ по п.8, отличающийся тем, что нитрил (Н)

получают взаимодействием имина (С)

с ионом цианида с образованием нитрила (Н).
14. Способ по п.13, отличающийся тем, что имин (С) получают взаимодействием производного этилендиамина А формулы H2N(СН2)2NHR с галогенированным ацетальдегидом или ди-C1-2 алкилацеталем галогенированного ацетальдегида.
15. Способ по п.13, отличающийся тем, что получение нитрила (Н)

проводят в одном сосуде без первичного выделения имина (С).
16. Соединение формулы (С)

где R выбирают из C1-10алкоксикарбонила или незамещенного или замещенного C1-10алкила, где указанным заместителем является арил или гетероарил.
17. Соединение по п.16, где R представляет собой замещенный C1-5алкил, где указанным заместителем является гетероарил.
18. Соединение по п.17, где R представляет собой группу формулы

19. Соединение формулы (Н)

где R выбирают из водорода, C1-10алкоксикарбонила или незамещенного или замещенного C1-10алкила, где указанным заместителем является арил или гетероарил.
20. Соединение по п. 19, где R представляет собой замещенный C1-5алкил, где указанным заместителем является гетероарил.
21. Соединение по п.20, где R представляет собой

22. Способ получения амида (G)

где R выбирают из водорюфр, C1-10алкоксикарбонила или незамещенного или замещенного C1-10алкила, где указанным заместителем является арил или гетероарил; отличающийся тем, что включает
(а) взаимодействие соединения (A), H2N-(CH2)2-NHR, где R принимает значения, определенные выше, соединения (В), выбираемого из галогенированного ацетальдегида или C1-2алкилацеталя галогенированного ацетальдегида и циианид-иона для получения нитрила (Н)

и (b) взаимодействие нитрила (Н) с соединением, выбираемым из трет-бутанола, изобутилена или сложного трет-бутилового эфира, в присутствии сильной кислоты для получения амида (G)

23. Способ получения соединения 1

включающий следующие стадии:
1) взаимодействие имина (С)

с R2NC в присутствии карбоновой кислоты, R1-СО2Н, с образованием соединения (F)

где R представляет собой группу формулы

и R2 представляет собой трет-бутил;
2) снятие защиты у соединения (F) с образованием рацемического соединения (I)

3) разделение рацемического соединения формулы

для выделения соединения 1

24. Способ получения соединения J

который включает следующие стадии
1) взаимодействие имина (С)

с R2NC в присутствии карбоновой кислоты, R1-СO2Н с образованием соединения (F)

где R представляет собой группу формулы

и R2 представляет собой трет-бутил;
2) снятие защиты у соединения (F) с образованием рацемического соединения (I)

3) разделение рацемического соединения формулы

для выделения соединения 1

4) взаимодействие соединения 1

с соединением 2

для получения соединения 3

5) снятие защиты у соединения 3 для получения соединения J.
Текст
1 Область техники Данное изобретение относится к новым промежуточным продуктам и способам синтеза соединений, которые ингибируют протеазу, кодированную вирусом иммунодефицита человека(ВИЧ), и в частности соединения, описанного в ЕРО 541168, опубликованной 12 мая 1993 года,как Соединение J, или его фармацевтически приемлемых солей. Соединение J и его аналоги представляют интерес для профилактики и лечения инфекции,вызванной вирусом иммунодефицита человека(ВИЧ-инфекции), и лечения синдрома приобретенного иммунодефицита (СПИД). Эти соединения также полезны для ингибирования ренина и других протеаз. Более точно данное изобретение относится к новым способам синтеза производных пиперазин-трет-буталкарбоксамида,которые являются ключевыми промежуточными продуктами, используемыми для получения ингибитора протеазы, кодированной вирусом иммунодефицита человека (ВИЧ-протеазы), включая соединение J. Предпосылки изобретения Ретровирус, называемый вирусом иммунодефицита человека (ВИЧ), является этиологическим агентом сложного заболевания, которое включает прогрессивную деструкцию иммунной системы (синдром приобретенного иммунодефицита; СПИД) и разрушение центральной и периферической нервной системы. Этот вирус первоначально был известен как LAW (лимфоденопатический вирус), HTVL-III (вирус человеческого Т-клеточного лейкоза) или ARV. Обычной особенностью репликации ретровируса является экстенсивный пост-трансляционный процессинг прекурсорных липопротеинов посредством вирусно кодированной протеазы для генерирования зрелых вирусных белков, необходимых для вирусного микросообщества и для его функционирования. Ингибирование этого процессинга предотвращает нормальное продуцирование инфекционного вируса. Например, в публикации Kohl N.E. et al., Proc. Nat'l Acad.Sci., 85, 4686 (1988) показано, что генетическая инактивация ВИЧ-кодированной протеазы приводит к продуцированию незрелых неинфекционных вирусных частиц. Эти результаты показывают, что ингибирование ВИЧ протеазы представляет собой способ лечения СПИДа и профилактики или лечения ВИЧ-инфекции, который может применяться на практике. Нуклеотидная последовательность ВИЧ показывает присутствие pol-гена в одной откры 000637 2 той рамке считывания [Ratner, L. et al., Nature,313, 277 (1985)]. Аминокислотная последовательная гомология доказывает, что роl последовательно кодирует обратную транскриптазу,эндонуклеазу и ВИЧ протеазу [Toh, H. et al.,EMBO J., 4, 1267 (1985); Power, M.D. et al., Science 231, 1567 (1986); Pearl, L.H. et al., Nature,329, 351 (1987)]. Соединения, представляющие собой конечный продукт, включая соединение J,которое показано в примере 29 ниже и которое может быть получено из новых промежуточных продуктов и способами данного изобретения,являются ингибиторами ВИЧ протеазы и описываются в ЕРО 541168, опубликованной 12 мая 1993 года. Ранее синтез соединения J и родственных соединений осуществлялся посредством 12 стадийного процесса. Эта методика описывается в ЕРО 541168. В используемых ранее способах ингибитор ВИЧ протеазы J получали взаимодействием промежуточного эпоксипроизводного 2 с пиперазинкарбоксамидом 4,содержащим Воc защитную группу, которое приводит к получению связанного промежуточного продукта 5, содержащего Воc защитную группу. Удаление защитной группы из соединения 5 приводит к получению последнего промежуточного продукта - соединения 6, которое подвергают реакции пиколилирования с получением J. Недостатком этого способа получения является то, что для превращения эпоксида 2 в J необходимо проведение трех химических стадий. Таким образом, после удаления защитной группы для осуществления превращения с получением J требуется отдельная стадия пиколилирования. Поскольку более реакционноспособное положение 4 пиперазинкарбоксамида должно 3 быть защищено перед взаимодействием, наиболее эффективный способ защиты для хирального 2-пиперазин-трет-бутилкарбоксамида должен заключаться во введении 3-пиколильного фрагмента в эту точку, т.е. пиперазина 1. Однако не предполагалось, что реакция пиперазина 1 с эпоксидом 2 будет эффективна, так как пиперазин 1 содержит три основные функциональные аминогруппы, способные взаимодействовать с эпоксидом 2. Однако пиперазин 4, содержащий Воc защитную группу, содержит только одну основную функциональную аминогруппу, и таким образом ожидается, что взаимодействие 2 и 4 будет направленным. Ранее был описан обычный способ получения ингибитора ВИЧ-протеазы J из 4 пиколилпиперазинкарбоксамида 1 посредством двухстадийной реакции. Пиперазин 1 конденсируют с эпоксидом 2 с получением связанного продукта 3. Удаление ацетамидной защитной группы из продукта 3 приводит направленно к получению ингибитора ВИЧ-1 протеазы J. Синтез хиральных промежуточных пиперазин-2-карбоксамидов в настоящее время осуществляют поэтапно из ароматической 2 пиперазинкарбоновой кислоты, которая используется в качестве исходного вещества. Сначала получают трет-бутилкарбоксамид из коммерчески доступной пиразин-2-карбоновой кислоты посредством ее активирования с помощью получения хлорида кислоты с последующим взаимодействием с трет-бутиламином, что приводит к образованию пиразин-2-трет-бутилкарбоксамида. Затем пиразиновый цикл восстанавливают гидрированием с получением рацемического пиперазин-трет-бутилкарбоксамида. В том случае, когда необходимо разделение рацемической смеси, оно может быть осуществлено на данном этапе с помощью оптически активных кислот с получением оптически обогащенного материала. В случае получения промежуточного 4 соединения J, разделение проводят на этой стадии (S)-10-камфорсульфоновой кислотой или Lпироглютаминовой кислотой с получением целевого (S)-антипода. Защита для дифференцирования N1 и N4 в пиперазине затем приводит главным образом к целевому 4-защищенному пиперазин-производному. В случае промежуточного соединения J, защиту проводят с помощью ди-трет-бутилдикарбоната и получают N4BOC защищенный продукт и 1,4-бисзащищенный продукт в соотношении 9:1. Если реакцию проводят с 3-пиколилхлоридом для введения 3-пиколил-группы в положение 4 пиперазинового цикла в соединении J, однако,получают очень низкую селективность (4:1 моно/бис алкилирование), что приводит к получению смеси, которая не может быть очищена кристаллизацией. Этот способ получения промежуточных пиперазин-2-трет-бутилкарбоксамидов требует нескольких отдельных стадий и является очень трудоемким и сложным с промышленной точки зрения. Таким образом, сохраняется потребность в более эффективном и упрощенном способе получения ключевых промежуточных пиперазин-2-карбоксамидпроизводных.Comprehensive Organic Synthesis, Vol. 2, page 1083 (B.M. Trost, ed. 1991)] и конденсация по методике Стрекера [D.T. Mowry, Chem. Rev., 42; 42; 236, (1948)] являются хорошо известными способами получения аминокислот и пептидных производных. Характеристическим отличительным признаком реакции Юги является присоединение изоцианида и аниона Х подходящей кислоты НХ к иону иминия (40) с последующей самопроизвольной перегруппировкой Таким образом, продукт получают из четырех разных реагентов, а именно: (4), (5), (43) и НХ, посредством реакции конденсации. Поэтому такую реакцию Юги первоначально называли четырехкомпонентной конденсацией. Несмотря на то, что реакция Юги является очень сильным способом, такая реакция не описана для получения пиперазинкарбоксамидов. Синтез аминокислот по методу Стрекера является доказанно полезным для специалистов в области органического синтеза с момента его открытия в 1850 году. Стрекер обрабатывал ацетальдегидаммоний цианидом водорода и продукт подвергал гидролизу, в результате получал аланин [D.T. Mowry, Chem. Rev., 42, 236 В наиболее общей форме реакция Риттера[L.I. Krimen and D.J. Cota, Organic Reactions,Vol. 17, 213 (1969)] включает нуклеофильное присоединение нитрила к иону карбония в присутствии серной кислоты. 6 пиперазин-2-карбоксамидов. Таким образом,данное изобретение относится к усовершенствованным способам получения ключевых промежуточных продуктов с повышенной простотой и эффективностью, что приводит к экономии капитальных и трудозатрат. Кроме того, в усовершенствованных способах данного изобретения снижается образование побочных продуктов по сравнению с известными ранее способами получения ключевых промежуточных продуктов производных пиперазин-2 карбоксамида. Краткое описание изобретения Данное изобретение относится к способам получения соединений формулы (III) которые являются ключевыми промежуточными продуктами в процессе получения ингибиторов ВИЧ-протеазы, включая соединение J. В соединениях формулы (III) R - выбирают из группы,включающей водород, C1-10 алкоксикарбонил и незамещенный или замещенный C1-10 алкил, в котором указанным заместителем является арил или гетероарил; R2 - C1-10 алкил или арил; и R3 водород или -COR1, где R1 выбирают из группы,включающей водород, C1-10 алкил, моно-, диили тригалогенированный C1-10 алкил или арил. Предпочтительно R2-трет-бутил. В одном воплощении данного изобретения способ получения соединения (F) Последующее разбавление водой приводит к получению амида. Несмотря на то, что синтез Стрекера, как известно, приводит к получению 2-аминокислот после гидролиза соответствующих 2 аминонитрилов, аналогичное взаимодействие аминонитрилов в соответствии с реакцией Риттера для прямого получения 2 аминокарбоксами-производных не является известной реакцией. Более того, не ожидается осуществление такого превращения, поскольку 2-аминонитрилы, как сообщалось, в реакции Риттера превращаются в нежелательные 2 гидроксикарбоксамидные производные [D. Giraud-Clenet and J. Anatol, Compt. Rend., 262, 244(1966)]. В настоящее время установлено, что реакция Юги, а также реакции Стрекера и Риттера,могут применяться в синтезе промежуточных где R, R1 и R2 принимают значения, определенные выше; включает взаимодействие амина (С) 8 где R принимает значения, определенные выше; включающий взаимодействие нитрила (Н) К классу этого воплощения относится процесс, включающий дополнительно стадию удаления защитной группы из соединения (F) (для получения соединения (I К подклассу этого воплощения относится процесс, в котором имин (С) с соединением, выбранным из группы, включающей трет-бутанол, изобутилен или третбутиловый эфир, в присутствии сильной кислоты с образованием амида (G) К классу этого второго воплощения относится способ, в котором сильную кислоту выбирают из группы, включающей Н 2SO4, Н 3 РO4,СН 3SO3 Н или ВF3. Предпочтительно сильная кислота представляет собой H2SO4. К подклассу этого второго воплощения относится способ, в котором нитрил (Н) получают взаимодействием этилендиаминпроизводного А формулы H2N(CH2)2NHR с галогенированным ацетальдегидом или галогенированным ацетальдегид-ди-С 1-2 алкилацеталем. Типичным для данного воплощения является способ, в котором соединение (F) получают реакцией имина (С) получают в одной емкости без первичного выделения имина (С) с цианид-ионом, в результате которой образуется нитрил (Н). Примером этого второго воплощения является способ, в котором имин (С) Ко второму воплощению данного изобретения относится способ получения амида (G) получают взаимодействием этилендиаминпроизводного А формулы H2N(CH2)2NHR с галогенированным ацетальдегидом или галогенированным ацетальдегид-ди-С 1-2 алкилацеталем. Примером этого второго воплощения является способ, в котором получение нитрила (Н) проводят в одной емкости без первичного выделения имина (С). Примером данного изобретения является любой из способов, описанных выше, в которомR представляет собой замещенный C1-5 алкил, в котором указанным заместителем является гетероарил. Предпочтительно R - 3-пиколил. Примером данного изобретения является любой из процессов, описанных выше, дополнительно включающий стадию разделения рацемического соединения Другим наглядным примером данного изобретения является любой из способов, описанных выше, дополнительно включающий стадию удаления защитной группы из соединения 3 Дополнительным примером данного изобретения является соединение, выбранное из группы, включающей Примером, дополнительно иллюстрирующим данное изобретение, является любой из способов, описанных выше, включающий также стадию взаимодействия соединения 1 где R выбирают из группы, включающей водород, C1-10 алкоксикарбонил или незамещенный или замещенный C1-10 алкил, в котором указанным заместителем является арил или гетероарил. Предпочтительно R представляет собойC1-5 алкил, в котором указанным заместителем является гетероарил. Наиболее предпочтительно, R представляет собой 3-пиколил. Примером, более точно иллюстрирующим данное изобретение, является способ получения соединения (I) где R выбирают из группы, включающей водород, C1-10 алкоксикарбонил или незамещенный или замещенный C1-10 алкил, в котором указанным заместителем является арил или гетероарил; и R2 представляет собой C1-10 алкил или арил; включающий где (А) представляет собой H2N-(СН 2)2-NHR, в котором R принимает указанные выше значения; (В) выбирают из группы, включающей галогенированный ацетальдегид или галогенированный ацетальдегид С 1-2 алкилацеталь; (Е) представляет собой R1 СО 2 Н, в котором R1 выбирают из группы, включающей H3C1-10 алкил,моно-, ди- или тригалоген-С 1-10 алкил или арил; и (D) представляет собой R2CN, в котором R2 принимает значения, определенные выше; и(b) удаление защитной группы из соединения (F) Более конкретным примером данного изобретения является способ получения амида (G) где R выбирают из группы, включающей водород, C1-10 алкоксикарбонил или незамещенный или замещенный C1-10 алкил, в котором указанным заместителем является арил или гетероарил; способ включает(а) взаимодействие соединения (A), H2N(CH2)2-NHR, где R принимает значения, определенные выше; соединения (В), которое выбирают из группы, включающей галогенированный ацетальдегид, С 1-2 алкилацеталь галогенированного ацетальдегида, и цианид-иона с образованием нитрила (Н) и (b) взаимодействие нитрила (Н) с соединением, выбранным из группы, включающей трет-бутанол, изобутилен или сложный третбутиловый эфир, в присутствии сильной кислоты с образованием амида (G) Подробное описание изобретения В процессе синтеза соединений, которые ингибируют ВИЧ-протеазу, и, в частности соединения J, ключевым промежуточным продуктом является производное пиперазин-2-третбутилкарбоксамида G или его соли. Для получения ключевого промежуточного продукта G этилендиамин или его моноалкилированнное производное (А) конденсируют с хлорацетальдегидом (В) для получения промежуточного имина (С). Вместо хлорацетальдегида для получения имина могут использоваться бромацетальдегид хлорацетальдегид диметилацеталь или бромацетальдегид ди 13 этилацеталь. Реакция получения имина (С) может быть проведена в присутствии подходящего растворителя при температуре в интервале от 20 до 150 С предпочтительно от 20 до 80 С,наиболее предпочтительно в интервале от 0 до 70 С. Подходящими растворителями являются сложные эфиры, ароматические углеводороды,галогенированные растворители, спирты и вода. Предпочтительно растворитель выбирают из группы включающей воду или спирты; наиболее предпочтительно метанол, этанол, пропанол или их смеси. В варианте данного изобретения,основанном на методике Юги, пиперазиновый цикл и амидная связь образуются в одной реакционной емкости из имина (С) посредством конденсации трет-бутилизоцианида (D) с карбоновой кислотой (Е), в результате чего образуется 1-ацилпиперазин-2-трет-бутилкарбоксамид-производное (F). Если необходимо, карбоксамиды, отличные от трет-бутилпроизводного, могут быть получены с помощью различных C1-10 алкил изоцианидов или арилизоцианидов,используемых вместо третбутилизонитрила (D). Карбоновые кислоты, которые могут применяться в данном процессе,включают, но не ограничиваются только ими,муравьиную кислоту, уксусную кислоту, бензойную кислоту, трифторуксусную кислоту,хлоруксусную, дихлоруксусную или трихлоруксусную кислоту. В соответствии с другим способом, карбоновая кислота может быть получена непосредственно из соли кислоты и НСl или НВr, которые выделяются при алкилировании. Растворители, подходящие для применения в реакции Юги, являются теми же самыми растворителями, которые описаны выше для получения имина (С). Реакция Юги может быть проведена при температурах в интервале от 20 до 150 С, предпочтительно в интервале от 0 до 80 С. Удаление защитной 1-ацильной группы затем приводит к образованию 2-пиперазинтретбутилкарбоксамидного производного (G),как показано на схеме I. Способы удаления 1 ацильной группы хорошо известны специалистам и описаны в литературе (см., например,Greene, T.W.; Wuts, P.G.M., Protective groups in В другом воплощении данного изобретения промежуточный имин (С) взаимодействует с цианид-ионом (например, NaCN, KCN, LiCN, 000637HCN), в соответствии с вариантом данного способа по методике Стрекера, в результате чего получают аминонитрил (Н). Реакцию получения аминонитрила (Н) можно проводить в присутствии подходящего растворителя при температурах в интервале от 20 до 150 С, предпочтительно в интервале от 20 до 80 С, наиболее предпочтительно в интервале от 0 до 70 С. Подходящими растворителями являются сложные эфиры, ароматические углеводороды, галогенированные растворители, спирты и вода. Предпочтительно растворитель выбирают из группы,включающей воду и спирты; наиболее предпочтительно применение метанола, этанола, пропанола или их смесей. Аминонитрил (Н), в свою очередь, легко превращается в пиперазин-2 трет-бутилкарбоксамид-производное (G) посредством взаимодействия с т.бутанолом в соответствии с реакцией Риттера, которая протекает с применением кислотного катализатора,как показано на схеме II. Более точно, реакцию Риттера проводят через взаимодействие аминонитрила (Н) с изобутиленом, трет-бутанолом или трет-бутиловым эфиром в сочетании с сильной кислотой, такой как H2SO4, Н 3 РО 4,СН 3SО 3 Н или ВF3. Предпочтительно используют серную кислоту. Получение (G) по реакции Риттера из аминонитрила (Н) может быть проведено при температурах в интервале от 20 до 150 С, предпочтительно в интервале от 0 до 50 С, без или в присутствии растворителя, такого как алкан или ароматический растворитель,галогенированный растворитель или сложный эфир. Предпочтительными растворителями являются сами реагенты (т.е. чистые реагенты) или галогенированные растворители, такие как метиленхлорид или хлороформ. Схема II В варианте проведения этой реакции в одной емкости, который приводит непосредственно к образованию (F) по реакции Юги или (Н) по реакции Стрекера, необязательно промежуточным продуктом заявлять имин (С). Возможно также, что сначала имеет место реакция Юги или Стрекера с последующимNалкилированием и закрытием цикла с образованием пиперазина (F) или (Н). Когда образуется пиперазин-2 карбоксамид-производное (G), требуется разделение энантиомеров (S) и (R) для того, чтобы целевой (S)-антипод II мог подвергаться далее 15 обработке для образования соединений - ингибиторов ВИЧ-протеазы, описанных в ЕРО 541168 и, в частности, соединения J. Разделение энантиомеров может выполняться в соответствии с методами, хорошо известными специалистам, например, хиральной ВЭЖХ. Разделение(S) и (R) энантиомеров может проводиться также с помощью получения солей (S)камфорсульфоновой кислоты или(L)пироглютаминовой кислоты и пиперазин-2 карбоксамид-производного из рацемического соединения. Связываание может быть проведено с помощью нагрева соединения II (например, пиперазина 1) и эпоксида 2, чистого или в различных растворителях. Реакция связывания может быть проведена при температурах в интервале от 25 до 150 С, предпочтительно в интервале от 50 до 120 С, причем наиболее предпочтительно в интервале от 65 до 85 С. Растворители, необходимые для этой стадии, включают сложные эфиры, такие как этилацетат, изопропилацетат, нбутилацетат; ацетонитрил; спирты, такие как метанол, этанол, н-пропанол, н-бутанол, третбутанол, трет-амиловый спирт и изопропанол; углеводороды, такие как циклогексан и толуол; простые эфиры, такие как ТГФ и диметиловый эфир; и формамиды, такие как ДМФА. Предпочтительными растворителями являются спирты, причем наиболее предпочтительны метанол и изопропанол. Удаление защитной группы в промежуточном продукте 3 для получения J проводят стандартными способами, например, обработкой сильными кислотами, такими как газообразная НСl в спиртовых растворителях или водная НСl, причем наиболее предпочтительно применение газообразной НСl. Способы и промежуточные продукты данного изобретения полезны для получения соединений, которые используют для ингибирования ВИЧ-протеазы, профилактики или лечения инфекции, вызванной вирусом иммунодефицита 16 человека (ВИЧ), и лечения последующих патологических заболеваний, таких как СПИД. Эти соединения, являющиеся конечными продуктами, и их способность ингибировать ВИЧпротеазу описываются в ЕРО 541168, опубликованной 12 мая 1993 года. Лечение СПИДа или профилактика или лечение ВИЧ-инфекции определяется как лечение, включающее, но без ограничения, лечение широкого спектра состояний, вызванных ВИЧ-инфекцией: СПИД, ARC(комплекс, родственный СПИДу), как симптомный, так и бессимптомный, и действительное или потенциальное воздействие ВИЧ. Например, соединения, которые представляют собой конечный продукт и которые могут быть получены способами и из промежуточных продуктов данного изобретения, являются полезными для лечения ВИЧ-инфекции, после предполагаемого воздействия ВИЧ в результате, например, переливания крови, трансплантации органа, обмена общей воды в организме, покусов, случайного укола или воздействия на кровь пациента в процессе хирургического вмешательства. Ингибиторы ВИЧ-протеазы, которые являются конечными продуктами, полезны также для приготовления и проведения скриннинговых испытаний антивирусных соединений. Например, соединения, представляющие собой конечный продукт, полезны для выделения ферментативных мутантов, которые представляют собой прекрасные инструменты скрининга более мощных противовирусных соединений. Кроме того, такие соединения полезны для установления или определения связывающего сайта других антивирусов к ВИЧ-протеазе, например, посредством конкурентного ингибирования. Таким образом, соединения, представляющие собой конечный продукт и полученные способами и из промежуточных продуктов данного изобретения, являются коммерческими продуктами, предназначенными для продажи для этих целей. Соединение J, представляющее собой конечный продукт и являющееся ингибитором протеазы, имеет строение или его фармацевтически приемлемые соли или их гидраты. Соединение J имеет следующие названия:N-(1(S)-2,3-дигидро-2(R)-гидрокси-1 Нинденил)-4(3)-гидрокси-2(R)-фенилметил-5-[4(3-пиридилметил)-2(S)-трет-бутилкарбамоил)пиперазинил]пентанамид. Соединения, представляющие собой ингибиторы ВИЧ-протеазы, которые могут быть получены способами данного изобретения, описываются в ЕРО 541164. Соединения-ингибиторы ВИЧ-протеазы, могут вводиться пациентам, которые нуждаются в таком лечении, в виде фармацевтических композиций, включающих фармацевтический носитель и терапевтически эффективные количества соединения или его фармацевтически приемлемой соли. В ЕРО 541164 описываются подходящие фармацевтические рецептуры, способы введения, солевые формы и дозировки этих соединений. Соединения данного изобретения могут иметь асимметрические центры и существовать в виде рацематов, рацемических смесей и в виде отдельных диастереомеров или энантиомеров,причем все изомерные формы включаются в данное изобретение. В объем данного изобретения включаются полиморфные модификации и гидраты соединений данного изобретения. Когда любой изменяемый параметр (например, арил) встречается более одного раза в любой составной части, его определение в каждом случае не зависит от его определения в каждом другом случае. Сочетания заместителей и/или изменяемых составных частей также допустимы только в том случае, если такие сочетания приводят к стабильным соединениям. Термин галоген, используемый в описании, включает фтор, хлор, бром и йод, в частности хлор и бром. Если не указано другого, термин алкил используется для обозначения как разветвленных, так и линейных насыщенных алифатических углеводородных групп, содержащих указанное число атомов углерода (Me - метил, Et этил, Pr - пропил, Вu - бутил, t-Bu - трет-бутил). Термин арил в данном описании используется для обозначения фенила (Ph) или нафтила. Термин гетероарил в данном описании используется для обозначения 6-членного ароматического гетероциклического цикла или стабильного 8-10 членного ненасыщенного бициклического гетероцикла, где моно или бициклический гетероцикл состоит из атомов углерода и от одного до трех гетероатомов, выбранных из группы,включающей N, О или S. Например, термин гетероарил должен включать, но без ограничения, следующие фрагменты: В данном описании, в частности на схемах и в примерах, используются следующие аббревиатуры: Вос или ВОС = трет-бутоксикарбонилGC = ГХ = газовая хроматография ВЭЖХ = высокоэффективная жидкостная хроматография НОАс = уксусная кислотаLDS = гексаметил дисилазид лития МеОН = метанолNMR = ЯМР = ядерный магнитный резонанс 1 Н NMR = ПМР = протонный магнитный резонансTHF = ТГФ = тетрагидрофуран ТСХ = тонкослойная хроматография Типичные методики эксперимента с использованием нового способа подробно описаны ниже. Эти методики приведены только в качестве примеров и не ограничивают новый способ данного изобретения. Если не указано другого, все спектры ПМР получены на аппаратуре 250 МГц и все спектры 13 С ЯМР получены на аппаратуре 62 МГц. Пример 1. 1-Фенилкарбокси-4-(3-пиколил)-2-третбутилкарбоксамид-пиперазин. К раствору 3-пиколилэтилендиамина и бензойной кислоты в МеОН добавляют NаНСО 3 и раствор хлорацетальдегида. Смесь нагревают до 40 С в течение 15 мин и добавляют третбутилизоцианид. Реакционную массу перемешивают при температуре 40 С в течение 38 ч,после чего в реакционную смесь добавляют 15 мл 5 N NaOH. Реакционную смесь экстрагируютCH2Cl2 (150 мл) и органическую фазу промывают 10 мл воды. Органическую фазу упаривают досуха и полученное масло очищают методом хроматографии на SiO2 (EtOAc 100% доEtOAc/MeOH 85/15). После концентрирования фракций, содержащих продукт, получают 1 фенилкарбокси-4-(3-пиколил)-2-трет-бутилкарбоксамидпиперазин в виде твердого вещества желтовато-коричневого цвета; т.пл. 134 С; ПМР К раствору 3-пиколилэтилендиамина в 1 пропаноле добавляют формиат натрия, раствор хлорацетальдегида и трет-бутилизоцианид. Реакционную смесь перемешивают в течение 20 ч при температуре 25 С и затем гасят добавлением рассола (75 мл) и 45% КОН (5 мл). Органическую фазу удаляют и промывают снова рассолом (75 мл). После добавления 100 мл изопропилацетата раствор сушат над Na2SO4 и обрабатывают активированным углем Darсo G 60. Отфильтрованный раствор упаривают до получения масла. После хроматографирования наSiO2 (EtOAc/MeOH с 90/10 до ЕtОАс/МеОН 50/50) получают 1-карбокси-4-(3-пиколил)-2 трет-бутилкарбоксамидпиперазин в виде твердого белого вещества, т.пл. 127 С, 13 С ЯМР(CDCl3) 167,5, 162,0, 150,4, 149,2, 136,6, 132,2,123,4, 59,9, 58,0, 52,9, 52,1, 51,5, 43,7, 28,7 (соединение представляет собой смесь двух оптических изомеров в соотношении 2:3, приводятся сдвиги только главного изомера). Пример 3. 4-(3-Пиколил)-2-трет-бутилкарбоксамидпиперазин. Раствор 1-карбокси-4-(3-пиколил)-2-третбутилкарбоксамидпиперазина в ТГФ/Н 2 О/НСl нагревают до 60 С и выдерживают при этой температуре в течение 8 ч. Затем раствор упаривают досуха, в результате получают 4-(3 пиколил)-2-трет-бутилкарбоксамидпиперазин в виде гидрохлорида. 13 С ЯМР (D2O) 163,5, 150,3,143,9, 143,4, 130,6, 128,9, 57,3, 55,6, 53,1, 51,6,48,7, 40,9, 28,3. Пример 4. 4-(3-Пиколил)-2-карбонитрилпиперазин. 3 -Пиколилэтилендиамин Хлорацетальдегид, 50 маc.% в воде Цианид натрия Вода К раствору 3-пиколилэтилендиамина и цианида натрия в воде добавляют водный раствор хлорацетальдегида. Реакционную смесь перемешивают при 25 С в течение 20 ч и для получения насыщенного раствора добавляют твердый NaCl. Реакционную смесь, насыщенную NaCl, затем экстрагируют 400 мл СНСl3,органическую фазу сушат (MgSO4), фильтруют и упаривают. После хроматографирования полученного масла на SiO2 (EtOAc со 100% доEtOAc/MeOH 80/20), упаривания фракций, содержащих продукт, получают 4-(3-пиколил)-2 карбонитрилпиперазин в виде твердого вещества коричневато-желтого цвета, т.пл. 97 С; 13 С ЯМР (СDСl3) 150,1, 149,0, 136,5, 132,9, 123,6,118,9, 59,6, 54,6, 52,7, 46,3, 42,8. Пример 5. 4-(3-Пиколил)-2-трет-бутилкарбоксамидпиперазин. 4-(3-Пиколил)-2-карбонитрилпиперазин 0,283 г (1,4 ммоля) Ледяная уксусная кислота 3,5 мл Серная кислота, 96% 1,6 мл трет-Бутанол 2,5 мл Вода 0,6 мл К раствору 4-(3-пиколил)-2 карбонитрилпиперазина в уксусной кислоте добавляют 0,6 мл Н 2SO4. Образующийся осадок снова переводят в раствор добавлением 0,6 мл Н 20 и 1,5 мл трет-бутанола. ТСХ показывает,что спустя 24 ч происходит только частичное превращение; поэтому добавляют оставшиеся Н 2SO4 (1,0 мл) и трет-бутанол (1,0 мл). Реакция происходит полностью после еще 24 ч. Медленно добавляют водный раствор КОН (до рН 12) и смесь экстрагируют 315 мл CH2Cl2. Соединенные органические фракции сушат надMgSO4, фильтруют и упаривают досуха, в результате получают масло коричневого цвета. После хроматографирования на(EtOAc/MeOH 50/50) получают 4-(3-пиколил)-2 трет-бутилкарбоксамидпиперазин в виде твердого вещества с низкой температурой плавления. 13C ЯМР (СD3 ОD) 172,2, 150,9, 149,0, 139,2,135,3, 125,2, 60,9, 59,7, 56,9, 53,8, 51,9, 44,8,28,9. Пример 6. 4-Бензил-2-карбонитрилпиперазин. Смесь 1-карбокси-4-(3-пиколил)-2-третбутилкарбоксамидпиперазина и (1S)-(+)-10 камфорсульфоновой кислоты нагревают до температуры кипения и кипятят с обратным холодильником в EtOH в течение 12 ч. Реакционную смесь охлаждают до 0 С и медленно добавляют ЕtОАс. Образуется белый осадок, его перемешивают в течение 12 ч, отфильтровывают и сушат, в результате получают соль 1-карбокси-4(3-пиколил)-2-трет-бутилкарбоксамидпиперазина и камфорсульфоновой кислоты. Соль камфорсульфоновой кислоты суспендируют в 20 мл ТГФ и обрабатывают 10 мл 40% раствора NaOH. Фазы разделяют и органическую фазу промывают насыщенным водным раствором К 2 СО 3. Органическую фазу сушат(2S)-1-карбокси-4-(3 пиколил)-2-трет-бутилкарбоксамидпиперазин. Оптическую чистоту определяют на колонкеChiracell AD (0,8 мл/мин, 60% EtOH, 40% гексан, обнаружение при 250 нм). Пример 8. Превращение оксида индена в цис-1 амино-2-инданол. МатериалыN-бензилэтилендиамин Хлорацетальдегид, 50 маc.% в Н 2 О Цианид натрия Гидрокарбонат натрия Вода К раствору N-бензилэтилендиамина, цианида натрия и гидрокарбоната натрия в воде добавляют водный раствор хлорацетальдегида. Реакционную смесь перемешивают при температуре 50 С в течение 2 ч. Реакционную смесь экстрагируют 40 мл СН 2 Сl2, органическую фазу сушат (MgSO4), фильтруют и упаривают. После хроматографирования полученного масла на Удаление защитной группы и разделение 1-карбокси-4-(3-пиколил)-2-трет-бутилкарбоксамидпиперазина. 1-Карбокси-4-(3-пиколил)-2-третбутилкарбоксамидпиперазин Инден оксид Ацетонитрил Вода Конц. H2SO4 5N КОН Граммы или мл 1 мл 10 мл 2,15 мл 0, 92 мл 3,0 мл 15 мл вл.смола 50 мл К 1 мл инден оксида (8,33 ммолей), растворенному в 10 мл ацетонитрила, добавляют 0,15 мл воды (8,33 ммолей). Смесь охлаждают до 0-5 С на ледяной бане. К смеси по каплям добавляют концентрированную серную кислоту,поддерживая температуру смеси ниже 10 С. После того, как вся кислота добавлена, смеси дают нагреться до 20-25 С. Прозрачный раствор выдерживают в течение 30 мин. В эту смесь добавляют 2 мл воды и раствор нагревают в течение 30 мин. Когда весь метилоксазолин полностью превращается в цисаминоинданол, реакционную смесь охлаждают до комнатной температуры. Добавляют 5 N КОН (3 мл, 15 ммолей). Это количество составляет 90% от теории из расчета на серную кислоту. Раствор сохраняет кислую среду в соответствии с пробой лакмусом. Если рH возрастает до 2, происходит реацилирование и выход аминоинданола снижается. Белое твердое вещество (K2SO4) удаляют фильтрованием. 15 мл смолы марки Dowex (смочена ацетонитрилом) добавляют с перемешиванием. Перемешивание смолы продолжают в течение 15 23 мин и отбирают пробу для жидкостной хроматографии (разб.х 50). Когда пик аминоинданола на ЖХ исчезает, смолу собирают фильтрованием, промывают ацетонитрилом и затем метанолом. Влажную смолу обрабатывают 50 мл 1 N раствора NН 3 в метаноле и суспензию перемешивают при комнатной температуре в течение 30 мин. Смолу снова выделяют фильтрованием и смесь метанол/NН 3 сохраняют. Другую загрузку 1 N NН 3/МеОН (20 мл) добавляют и смолу повторно суспендируют. После удаления смолы растворы аминоинданола в смеси метанол/NН 3 соединяют и упаривают для удаленияNH3. Анализ конечного метанольного раствора показывает, что цис-1-амино-2-инданол готов для реагента, используемого для разделения, в качестве которого берется винная кислота. Пример 9. Получение рацемического инден оксида. Инден (95%, 122 мл) растворяют в метаноле (812 мл) и ацетонитриле (348 мл), затем фильтруют. Фильтрат разбавляют 0,05 М двухосновным фосфатом натрия (116 мл), затем рH доводят до 10,5 с помощью 1 М водного гидроксида натрия. Водную перекись водорода (35%,105 мл) разбавляют водой (53 мл) и добавляют в течение 3 ч, поддерживая температуру 25 С и рН среды на уровне 10,5 с помощью 1 М водного раствора гидроксида натрия (всего 120 мл). Спустя 6 ч добавляют 1 М водный метабисульфит натрия (26 мл), сохраняя рН свыше 8,3 с помощью 1 М водного NaOH (39 мл). После этого добавляют воду (700 мл) и смесь экстрагируют метиленхлоридом (580 мл и 300 мл). Соединенные органические экстракты, содержащие оксид индена (117 г), упаривают до объема 600 мл. Пример 10. Получение (1S,2R)-инденоксида. Субстрат, (1S, 2R)-инденоксид, получают в соответствии со способом, описанным в публикации D.J. O'Donnell, et al., J.Organic Chemistry,43, 4540 (1978), который введен в список ссылок. Пример 11. Получение цис-1-амино-2-инданола. Инденоксид (117 г) разбавляют метиленхлоридом, доводя общий объем до 600 мл, разбавляют ацетонитрилом (600 мл) и охлаждают до температуры 20 С. После этого добавляют метансульфоновую кислоту (114 мл). Смеси дают нагреться до 25 С и выдерживают в течение 2 ч. Затем добавляют воду (600 мл), смесь нагревают до 45 С и выдерживают при этой температуре в течение 5 ч. Органическую фазу отделяют и водную фазу дополнительно кипятят с обратным холодильником в течение 4 ч с упариванием до получения концентрации 200 г/л. рН раствора доводят до значения 12,5 с помощью 50% водного раствора гидроксида натрия,затем охлаждают до 5 С, фильтруют и сушат(1S, 2R)-инденоксид (85%, исключая ошибки) (250 г, 0,185 моля) растворяют в хлорбензоле (300 мл) и гептане (1200 мл) и медленно добавляют к раствору метансульфоновой кислоты (250 мл, 0,375 моля) в ацетонитриле (1250 мл) при температуре ниже чем приблизительно 10 С. Реакционную смесь нагревают до 22 С и выдерживают в течение часа. К смеси добавляют воду и смесь упаривают отгонкой до тех пор,пока температура смеси не достигнет 100 С. Реакционную смесь выдерживают при температуре 100 С в течение 2-3 ч, затем охлаждают до комнатной температуры. В реакционную смесь добавляют хлорбензол (1000 мл), смесь размешивают и отделяют органическую фазу. рН оставшейся водной фазы, которая содержит 1Sамино-2R-инданол (85%, исключая ошибки, 165 г, 60%), доводят до 12, используя 50% водный раствор гидроксида натрия, продукт выделяют фильтрованием и сушат под вакуумом при температуре 40 С, в результате получают 1Sамино-2R-инданол (85%, исключая ошибки). Пример 13. Получение 1S-амино-2R-инданола.(1S, 2R)-инденоксид (85%, исключая ошибки) (250 г, 0,185 моля) растворяют в хлорбензоле (300 мл) и гептане (1200 мл) и медленно добавляют в раствор дымящей серной кислоты(21% SO3, 184 мл) в ацетонитриле (1250 мл) при температуре ниже приблизительно 10 С. Реакционную смесь нагревают до 22 С и выдерживают при этой температуре в течение 1,0 ч. К смеси добавляют воду и упаривают отгонкой до тех пор, пока температура смеси не достигнет 100 С. Реакционную смесь выдерживают при температуре 100 С в течение 2-3 ч, затем охлаждают до комнатной температуры. К смеси добавляют хлорбензол (1000 мл), смесь размешивают и органическую фазу отделяют. Оставшуюся водную фазу, содержащую 1S-амино-2Rинданол (85%, исключая ошибки), разбавляют равным объемом ацетонитрила. рН доводят до 12,5 с помощью 50%-ного водного раствора гидроксида натрия и органическую фазу отделяют. Оставшуюся водную фазу экстрагируют дополнительным количеством ацетонитрила. Соединенные экстракты ацетонитрила упаривают под вакуумом, в результате получают 1Sамино-2R-инданол (85%, исключая ошибки). В соответствии с другим способом, оставшуюся водную фазу, содержащую 1S-амино-2Rинданол (85%, исключая ошибки) разбавляют равным объемом бутанола, рН доводят до 12,5 водным 50% раствором гидроксида натрия и органическую фазу отделяют. Органическую фазу промывают хлорбензолом. Добавляют Lвинную кислоту, воду удаляют отгонкой для получения кристаллической соли винной кислоты и аминоинданола. Пример 14. Применение бензонитрила. Инденоксид (5 г) растворяют в бензонитриле (50 мл) при температуре 25 С и добавляют серную кислоту (98%, 2,25 мл). Смесь разбавляют 5 М водным раствором гидроксида натрия(50 мл) и экстрагируют метиленхлоридом. Органические экстракты упаривают под вакуумом,в результате получают оксазолин. Пример 15. Выделение цис-1-амино-2-инданола. Цис-1-амино-2-инданол (100 г) растворяют в метаноле (1500 мл) и добавляют раствор Lвинной кислоты (110 г) в метаноле (1500 мл). Смесь нагревают до 60 С, охлаждают до 20 С,фильтруют и продукт сушат под вакуумом. В результате получают соль 1S-амино-2Rинданола и L-винной кислоты в виде метанольного сольвата. Пример 16. Получение 1S-амино-2R-инданола. Метанольный сольват соли 1S-амино-2Rинданола и L-винной кислоты (88 г) растворяют в воде (180 мл) и нагревают до 55-60 С. Раствор осветляют фильтрованием и рН доводят до 12,5 с помощью 50% водного раствора гидроксида натрия. Смесь охлаждают до 0-5 С в течение 2 ч и перемешивают при этой температуре в течение 1 ч, фильтруют, промывают холодной водой и сушат под вакуумом при температуре 40 С, в результате получают 1S-амино-2R-инданол 1,2-индандиол Ацетонитрил Вода Серная кислота 5N КОН Граммы или мл 300 мг 2, 5 мл 0, 04 мл 0,22 мл 1,6 мл 10 мл 30 мл К 300 мг индандиола, растворенным в 3 мл ацетонитрила, содержащим 0,04 мл воды, добавляют по каплям при температуре 0-10 С 0,22 мл концентрированной Н 2SO4. После завершения добавления ледяную баню удаляют и реак 26 ционная масса нагревается до комнатной температуры. После выдерживания в течение 30 мин для анализа методом ЖХ отбирают пробу чистого раствора (разв. Х 500). После того, как весь гликоль израсходован, раствор обрабатывают дополнительно водой и нагревают до кипения на паровой бане для гидролиза оксазолина. Когда Iс анализ показывает полный гидролиз, для нейтрализации серной кислоты добавляют 1,6 мл 5 N КОН. Сульфат калия отфильтровывают из раствора. Фильтрат анализируют на содержание цисаминоинданола, которое составляет 196 мг (66% от теории, что также составляет 75% с учетом непрореагировавшего исходного материала). Раствор пропускают через 10 мл Dowex 504(Н+). На выходе из колонки раствор анализируют на содержание продукта. Анализ показывает,что весь аминоинданол адсорбируется. После промывания смолы метанолом продукт элюируют 1 М раствором NH3 в метаноле (сухой). Аммонийный метанол упаривают для удаленияNH3 и анализируют конечный раствор аминоинданола, готовый к разделению (175 мг или 50% от теории без учета непрореагировавшего гликоля). Пример 18. Получение производных инданола. Соединения -транс-2-бром-1-инданола получают в соответствии со способами, описанными в публикациях S.M. Sutter et al.,J.Am.Chem. Soc., 62, 3473 (1940); и D.R. Daltonet al., J.Org.Chem., 43, 4540 (1978). Пример 19. Получение цис-1-амино-2-инданола из транс-2-бром-1-инданола. Транс-2-бром-1-инданол (10 г, 46,9 ммоля в 100 мл ацетонитрила, содержащих 0,8 мл воды) охлаждают до 5 С и добавляют концентрированную серную кислоту (5,2 мл). Смесь перемешивают в течение 1 ч, затем добавляют 5 М раствор гидроксида калия для доведения рН раствора до 11. Реакционную смесь фильтруют,удаляя таким образом сульфат калия. рН водного ацетонитрильного фильтрата с помощью добавления серной кислоты доводят до значения меньше 2, и нагревают его до 80-100 С, удаляя ацетонитрил отгонкой для получения водного раствора цис-1-аминоинданола. Раствор упаривают до объема 20 мл, затем доводят рН до 12,5 с помощью гидроксида калия. Продукт кристаллизуется, его фильтруют и сушат под вакуумом,в результате получают цис-1-амино-2-инданол. Пример 20. Получение цис-1S-амино-2R-инданола из цис-(1S,2R)-индандиола. 27 Цис-(1S,2R)-индандиол (1 г) растворяют в ацетонитриле (10 мл), охлаждают до 0 С и добавляют концентрированную серную кислоту(1,0 мл). Смесь выдерживают в течение 40 мин с нагреванием до 20 С. Добавляют воду (0,8 мл) и смесь нагревают до температуры кипения. Для доведения рН до значения свыше 11 добавляют водный 5 М раствор гидроксида калия (1,6 мл) и образующийся твердый осадок (сульфат калия) удаляют фильтрованием для получения водного раствора цис-1S-амино-2R-инданола. Пример 21. Получение цис-1-амино-2-инданола из транс-1,2-индандиола. Транс-1,2-индандиол (1,5 г) растворяют в ацетонитриле (25 мл), охлажденном до 0 С, добавляют концентрированную серную кислоту(1,1 мл). Смесь медленно нагревают до 20 С и выдерживают в течение 3 ч. К смеси добавляют воду (2 мл) и нагревают до температуры кипения в колбе с обратным холодильником. Добавляют концентрированный водный раствор гидроксида натрия для доведения рН до 12. Образующийся твердый осадок удаляют фильтрованием для получения водного ацетонитрильного раствора цис-1-амино-2-инданола. Пример 22. Получение цис-1-амино-2-инданола из цис-1,2-индандиола. Цис-1,2-индандиол (1,0 г) растворяют в ацетонитриле (20 мл), охлаждают до 40 С и добавляют дымящую серную кислоту (21% SO3,0,8 мл). Смесь выдерживают в течение 1 ч с постепенным нагреванием до 0 С, добавляют воду, нагревают до 80 С и выдерживают при этой температуре в течение 1 ч для получения водного раствора цис-1-амино-2-инданола. Пример 23. Получение амида 14.(99,7 мас.%, 99,9 % площади,99,5 %, исключая ошибки) 900 г 6,02 моля Карбонат натрия моногидрат 760 г 6,13 моля Диэтоксиметан (DEM) 56,3 л 3-Фенилпропионилхлорид (13) 1,05 кг 6,23 моля Метансульфоновая кислота (MSA) 18,6 г 0,19 моля 2-Метоксипропен (95% ГХ) 1,28 л 13,3 моля 10,8 л 5% водный NaHCO3 Вода 26,2 л 28 40 л DEM и водный раствор карбоната натрия(760 г, 6,13 моля, Nа 2 Со 3H2O в 6,4 л воды) в реакторе объемом 100 л с четырьмя выходами,снабженном термопарой-зондом, механической мешалкой, переходным ниппелем для выпуска азота и барботером, нагревают до 46-47 С и перемешивают в течение 15 мин для растворения твердых веществ. pН водной фазы равна 11,5. В течение 2 ч при температуре 47-59 С добавляют чистый 3-фенилпропионилхлорид 13 (1,05 кг,6,23 моля). Температура внутри реактора возрастает с 47 до 59 С в процессе добавления 13; в процессе добавления хлорангидрида гидроксиамид 12 кристаллизуется из раствора. После завершения добавления реакционную смесь перемешивают при температуре 59 С в течение 0,5 ч, а затем нагревают до 72 С для гарантированного растворения твердых веществ. Температуру повышают до 72 С для растворения гидроксиамида таким образом, что для проведения анализа методом ВЭЖХ может быть отобран гомогенный образец, а также для упрощения деления на фазы. Ход реакции контролируют методом ВЭЖХ: 60:40 ацетонитрил/5,0 мМ каждого из КН 2 РO4 и К 2HРO4. Примерное время удерживания составляет: Время удерживания Вещество После завершения добавления хлорангидрида и перемешивания в течение 0,5 ч при температуре 72 С анализ методом ВЭЖХ показывает 0,6% площади составляет 11, 0,2% сложный эфир амида (побочный продукт) и 98,7% гидроксиамид. Гидроксиамид 12 отделяется не эффективно при выделении ацетонида 14. Водную фазу отделяют и органическую фазу дважды промывают 4,5 л воды. Промытую органическую фазу упаривают и сушат посредством атмосферной азеотропной отгонки. Исходный объем 40 л упаривают до 27 л. В перегонный куб загружают 16 л свежего DEM и загрузку концентрируют при температуре от 88 С до 89 С до 40 л. Высушенную суспензию гидроксиамида 12 в DEM обрабатывают 1,28 л 2-метоксипропена,а затем 18,6 г MSA при температуре 30 С. Добавление MSA в отсутствие 2-метоксипропена приводит к образованию сложного аминового эфира. Эта примесь снова превращается в гидроксиамид 12 в процессе обработки основанием в конце образования ацетонида. рН образца объемом 1,0 мл, разбавленного водой (1,0 мл),составляет, как установлено, 2,8-3,0. Образующуюся смесь выдерживают при температуре 3940 С в течение 3 ч. Контроль образования ацетонида осуществляют методом ВЭЖХ, используя описанные выше условия. Время удерживания составляет приблизительно: Вещество гидроксиамид 12 метиленкеталевая примесь ацетонид 14 амид сложного эфира,побочный продукт Смесь выдерживают при температуре 3840 С до тех пор, пока площадь 12 не достигнет значения 0,4 А%. Типичный профиль площадей пиков хроматограмм ВЭЖХ, %: 0,4% гидроксиамид 12, 96,9% - ацетонид 14, 0,2% побочный продукт - сложный эфир амида, 1,1%- метиленкеталевая примесь. Реакционную смесь охлаждают до 24 С и гасят 10,8 л 5% водного раствора бикарбоната натрия. Водную фазу отделяют и органическую фазу дважды промывают 10,8 л воды, рН которой составляет 7,6. Если рН слишком низок, ацетонидная группа может гидролизоваться обратно с образованием гидроксиамида 12. Промытую органическую фазу (34,2 л) упаривают посредством отгонки под атмосферным давлением в интервале температур 78-80 С до конечного объема 3,5 л. Концентрацию ацетонида доводят до 525 г/л для сведения на минимум потерь при выделении. Теплый раствор 14 в DEM последовательно охлаждают до 57 С, вносят 0,5 г затравочных кристаллов 14, дополнительно охлаждают до 0 С и выдерживают в течение 0,5 ч. Кристаллизация из раствора в загрузке начинается при температуре в интервале 53-55 С. Продукт выделяют фильтрованием, влажный осадок на фильтре промывают охлажденным (0 С) DEM (300 мл). Промытый осадок сушат под вакуумом (26 Нg) при 30 С, в результате получают ацетонид 14(90%,99,5% площади при анализе методом ВЭЖХ). Пример 24. Получение ацетонида 14 из (11-винная кислота) соли.(-)-Цис-1-аминоиндан-2-ол-тартрат,метанольный сольват (44,3 маc.% свободного основания 11) 100 г 297 ммолей Карбонат натрия моногидрат 63,76 г 514 ммолей Диэтоксиметан (DEM) 2,83 л 3-Фенилпропионилхлорид (13) 52,7 г 312 молей Метансульфоновая кислота(-)11 и винной кислоты в виде метанольного сольвата (100 г, 44,3 г свободного основания,297 ммолей) в 2,0 л DEM и водного раствора карбоната натрия (63,8 г, 514 ммолейNa2 СО 3 Н 2O в 316 мл воды) в реакторе объемом 5,0 литров, снабженном четырьмя выходами и термопарой-зондом, механической мешалкой,переходным ниппелем для выхода азота и барботером, нагревают до 50 С. Нагрев реакционной смеси до 60 С не приводит к растворению всех твердых веществ. В течение 30 мин при(52,7 г, 312 ммолей), смесь перемешивают при 50 С в течение 15 мин. Ход реакции отслеживают методом ВЭЖХ: 60:40 ацетонитрил/5,0 мМ каждого из КН 2 РO4 и K2HPO4, 1,0 мл/мин. Время удерживания составляет приблизительно: Время удерживания Вещество 4,1 гидроксиамид 12 6,3 цис-аминоинданол 11 12,5 амид сложного эфира,побочный продукт После завершения добавления хлорангидрида и перемешивания в течение 15 мин при 50 С анализ методом ВЭЖХ суспензионной смеси показывает, что 0,1% площади составляет 11. После этого реакционную смесь нагревают до 75 С. Температуру повышают до 75 С для растворения гидроксиамида 12 в DEM и упрощения разделения фаз. Водную фазу отделяют и органическую фазу дважды промывают водой (250 мл). Тартрат натрия удаляют с водной фазой. Первая водная фаза имеет рН 8,98. рН двух промывных вод составляет 9,1 и 8,1, соответственно. Промытую органическую фазу упаривают и сушат с помощью атмосферной перегонки. Приблизительно 1,0 л дистиллята собирают и 750 мл свежеперегнанного DEM загружают снова в емкость для перегонки. Атмосферную перегонку продолжают до тех пор, пока не будет выделено еще 350 мл дистиллята. KF раствора - 93 мл/л. Высушенный раствор в DEM охлаждают до 30 С и обрабатывают 63 мл 2 метоксипропена, а затем 0,95 г MSA. рН пробы объемом 1,0 мл, разбавленной 1,0 мл воды, равна 3,2. Реакционную смесь выдерживают при 35-42 С в течение 2 ч. Образование ацетонида контролируют методом ВЭЖХ, используя условия, описанные в данном примере. Приблизительное время удерживания - то же, что приведено выше. Смесь выдерживают при 38-40 С до тех пор, пока содержание 12 не будет составлять 0,7% А. Обычный профиль площадей в методе ВЭЖХ: 0,4% площади - гидроксиамид, 96,9% площади - ацетонид 14, 0,2% площади - сложный эфир амида (побочный продукт), 1,1% площади - метиленкеталевые примеси. Реакционную смесь охлаждают до 20 С, фильтруют для получения прозрачного раствора и гасят 520 мл 5% водного раствора бикарбоната натрия. Водную фазу отделяют и органическую фазу промывают 500 мл воды, рН промывной воды составляет 7,4. Промытую органическую фазу(2,0 л) упаривают посредством атмосферной дистилляции при температуре в интервале 7880 С до конечного объема 1,0 л. Концентрация ацетонида сохраняется на уровне около 525 г/л для сведения до минимума потерь при выделении. Теплый раствор 14 в DEM последовательно охлаждают до 50-52 С, вносят 100 мг продукта в качестве затравки и далее охлаждают до 5 С и выдерживают в течение 20 мин. Кристаллизация продукта из раствора начинается при 50 С. Продукт выделяют фильтрованием и влажный осадок на фильтре промывают охлажденным(0 С) DEM (240 мл). Промытый осадок сушат под вакуумом (26 Нg) при 30 С, в результате получают ацетонид 14 (87,9%,99,5% площ. при анализе методом ВЭЖХ). Пример 25. Получение ацетонида 14 (растворитель изопропилацетат). 32 творителем вместо IРАС не станет метилциклогексан. Объем перед началом атмосферной отгонки составляет 1,65 л. Общий объем метилциклогексана, добавленного для полной заменыIPAC на метилциклогексан, составляет 1,5 л. Температура загрузки в конце отгонки с изменением растворителя равна 101 С, и общий объем в конце составляет 900 мл. Загрузку нагревают до 65-70 С для полного растворения твердых веществ, затем охлаждают до 55 С, вносят затравку продукта и охлаждают до 0 С. Смесь выдерживают при 0 С в течение 15 мин, продукт выделяют фильтрованием и промывают метилциклогексаном (200 мл). Промытый осадок сушат под вакуумом (26 Нg) при температуре 30 С, в результате получают ацетонид 14(87,5%, 99,5% площади согласно анализу методом ВЭЖХ). Пример 26. Смесь (-)-цис-1-аминоиндан-2-ола 11 (80 г,535 ммоля) в 1,2 л IPAC и 560 мл воды обрабатывают 5 (90,8 г, 539 ммолей), поддерживая рН в интервале 8,0-10,5 при 70-72 С 5 N гидроксидом натрия (116 мл, 580 ммолей). Ход реакции отслеживают методом ВЭЖХ: 60:40 ацетонитрил/5,0 мМ каждого из КН 2 РO4 и К 2 НРO4. Время удерживания составляет приблизительно: Время удерживания Вещество По окончании реакции водную фазу отделяют и органическую фазу промывают водой(400 мл) при 72-73 С. рН водной фазы и промывной воды равна 8,1 и 7,9, соответственно. Фазу IPAC, содержащую воду, сушат с помощью атмосферной отгонки. Всего загружают 3,0 л IPAC для снижения KF до 100 мг/л. Конечный объем составляет 1,60 л. Образующуюся суспензию гидроксиамида 12 в IPAC обрабатывают 2-метоксипропеном (119 мл, 1,24 моля),затем MSA (1,1 мл, 3,2 мол.%) при температуре в интервале 35-38 С в течение 4,5 ч. Образование ацетонида отслеживают методом ВЭЖХ,используя условия, описанные выше. Смесь выдерживают при 38-40 С до тех пор, пока в соответствии с данными ВЭЖХ содержание 12 не будет соответствовать 0,4% площади. Реакционную смесь фильтруют для удаления мутного осадка, и фильтрат быстро гасят в охлажденном растворе бикарбоната натрия (950 мл) в течение 15 мин. Водную фазу отделяют и органическую фазу промывают водой (400 мл). Раствор бикарбоната натрия охлаждают до 0-5 С. рН водной фазы и промывной воды, как установлено, равна 7,5 и 7,9, соответственно. Атмосферную отгонку проводят до тех пор, пока рас Ацетонид 14(FMC 9404) Лимонная кислота ТГФ сушенный над ситами Вода 0,3 М Н 2SO4 6% NaHCO3 Кристаллический ацетонид 14 (200 г, 0,622 моля, 99,1 маc.%) растворяют в 1,25 л сухого ТГФ (сушенный над ситами) (KF=11 мг/мл) под атмосферой азота при температуре 25 С при механическом перемешивании. Полученная KF раствора 40 мг/л. Раствор подвергают последовательной обработке вакуум/продувка азотом для тщательного удаления из раствора растворенного кислорода. В раствор добавляют аллилбромид. Полученная KF раствора равна 75 мг/л. Обычно полную конверсию (99,5%) в данной методике получают при значениях KF предварительно полученного LDS раствора 200 мг/л при 10% избытке основания. Затем раствор охлаждают до 20 С. Раствор гексаметилдисилазида лития(LDS, 1,32 М) добавляют к раствору аллилбромид/14 с такой скоростью, чтобы температура реакционной смеси сохранялась на уровне 20 С. Добавление LDS проводят в течение 30 мин. Смесь перемешивают при температуре в интервале от 15 до 20 С и гасят, когда конверсия составляет более 99%. Контроль за ходом реакции осуществляют методом ВЭЖХ. Приблизительное время удерживания: гидроксиацетонид - побочный продукт = 5,3 мин,этилбензол = 5,6 мин, ацетонид 14 = 6,6 мин; аллилацетонид 15 = 11,8 мин; эпи-15 = 13,3 мин. Спустя один час конверсия составляет 99,5%. Реакцию гасят добавлением раствора лимонной кислоты (35,7 г, 0,186 моля) в 186 мл ТГФ. Смесь выдерживают при температуре 15 С в течение 30 мин после добавления лимонной кислоты. Смесь упаривают при пониженном давлении (приблизительно 28 Нg) до получения объема, составляющего приблизительно 30% от исходного при сохранении температуры внутри реактора 11-15 С и собирают 900 мл дистиллята в сухой охлажденный при помощи льда улавливатель. Растворитель затем заменяют, используя всего 2,7 л изопропилацетата (IPAс), продолжая отгонку под вакуумом. Процесс отгонки для замены растворителя прекращают, когда в реакционной массе остается 1 мольного % ТГФ,что определяют методом ПМР (см. описание анализа методом ГХ). Максимальная температура во время отгонки не должна превышать 35 С. Техническую смесь в IPAc промывают 1,05 л дистиллированной воды, 1,18 л 0,3 М серной кислоты и 1,18 л 6% водного раствора бикарбоната натрия. Объем органической фазы после промывки составляет 1,86 л. рН смеси после трех промывок водой составляет 6,5, 1,3 и 8,5, соответственно. Анализ пробы смеси методом ВЭЖХ на данном этапе показывает, что выход 15 составляет 93-94%. Соотношение целевого 15: эпи-15 в соответствии с анализом методом ВЭЖХ (в условиях,описанных выше) составляет 96:4. Анализ методом ГХ показывает, что гексаметилдисилазановый побочный продукт полностью удален на стадии выделения. Пример 27. 34 К раствору аллиламида 15 IPAc, полученному с предыдущей стадии, при 25 С добавляют раствор 36,6 г бикарбоната натрия в 1,03 л дистиллированной воды и полученную двухфазную смесь охлаждают до 5 С. К смеси добавляют твердый N-хлорсукцинимид (141,2 г, 1,06 моля). После добавления NCS экзотермический эффект отсутствует. К этой смеси добавляют водный раствор иодида натрия (158,6 г, 1,06 моля), поддерживая температуру реакционной смеси на уровне 6-11 С. Добавление проводят в течение 30 мин, и смесь приобретает темную окраску. Смеси дают нагреться до 25 С, энергично перемешивают. Ход реакции отслеживают методом ВЭЖХ: система описана выше, приблизительное время удерживания: йодгидрины 16, эпи-16,бис-эпи 16 = 8,1 мин, аллиламид 15 = 11,8 мин. Анализ смеси методом ВЭЖХ спустя 2,25 ч показывает, что конверсия составляет более 99,5%. Приблизительное соотношение диастереомеров 16:эпи-16:бис-эпи 16 в технической смеси на данном этапе составляет примерно 94:2:4 при разделении компонентов с использованием данной системы. Перемешивание прекращают и смесь делят на фракции. К органической фазе добавляют водный раствор сульфита натрия (80 г, 0,635 моля в 400 мл) в течение 1015 мин. Температура смеси возрастает до 2629 С. Смесь перемешивают в течение 40 мин при 25 С. После сульфитной промывки раствор по существу обесцвечивается. Фракции разделяют; KF органической фазы в этой точке составляет 25 г/л. Объем органической фазы - 1,97 л. Количественный анализ смеси методом ВЭЖХ (система описана выше) показывает, что общий выход иодгидрина 16 в данной точке составляет 86% (с учетом совместного элюирования диастереомеров). Пример 28. Раствор йодгидрина 16 упаривают под вакуумом (28 Нg) до получения азеотропно сухой загрузки. Общий объем дистиллята составляет 700 мл, при сохранении температуры реакционной смеси 22-28 С. Отогнанный растворитель заменяют 500 мл IPAc (KF=275 мг/л). Раствор охлажают до 26 С и в течение 10 мин добавляют 25% раствор NaOMe/MeOH(168,1 г). После добавления метоксида натрия температура снижается до 24 С. Смесь стано 35 вится темнее и образуется смолообразное твердое вещество, которое снова растворяется. Смесь выдерживают в течение 1 ч при 25 С. Анализ реакционной смеси проводят методом ВЭЖХ (условия описаны выше), приблизительное время удерживания: эпоксид эпи-2 = 6,5 мин, эпоксид 2, бис-эпи-2 = 7,1 мин, йодогидрин 16 = 8,1 мин. Анализ методом ВЭЖХ показывает, что конверсия йодогидрина в эпоксид составляет 99%. Спустя дополнительные 40 мин добавляют 4,1 г раствора метоксид натрия/метанол. Анализ методом ВЭЖХ через 20 мин показывает 99,5% конверсию. Реакцию гасят добавлением 366 мл воды при 25 С, реакционную массу перемешивают (10 мин) и фракции разделяют. Впоследствии установлено, что более продолжительное выдерживание реакционной смеси и промывка водой с разделением приводит по существу к обратной реакции с образованием йодгидрина в этих условиях в пилотной установке. Эта проблема особенно остро стоит при использовании промывки водой. Для исключения этой проблемы реакцию проводят при температуре 15 С. После того, как достигается конверсия 99% (спустя 1 ч после добавления NaOMe) смесь разбавляют IPAc(40% от объема загрузки) и сначала промывают увеличенным объемом воды (732 мл) при 20 С. Более низкие температуры и более концентрированные смеси могут привести к преждевременному осаждению 2 в процессе промывки. Продолжительность перемешивания и осаждения сохраняют на минимуме (10 и 30 мин, соответственно). В этом случае обратная реакция может быть ограничена до 1%. Технические смеси, содержащие (97:3) эпоксид 2/йодгидрин 16 подвергают очистке для получения эпоксидного продукта, содержащего 0,6% йодгидрина. Эпоксидный продукт, содержащий такое количество йодгидрина, получают без осложнений. Органическую фазу промывают 3% водным раствором сульфата натрия (2750 мл). Объем органической фазы после промывок составляет 1,98 л. рН трех порций промывной воды равен 10,7, 9,4 и 8,6, соответственно. Анализ методом ВЭЖХ показывает, что общий выход эпоксида 2 на данном этапе составляет 86% (с учетом 4% совместно элюированного бис-эпи-2). Раствор эпоксида 2 в IPAc упаривают при пониженном давлении (28 Нg) до объема приблизительно 600 мл, сохраняя температуру реакционной смеси в интервале 15-22 С. Растворитель заменяют на n-РгОН добавлением 750 мл н-пропанола при вакуумной отгонке в реакторе объемом приблизительно 500 мл, поддерживая температуру 30 С. Повышение температуры свыше 35 С в процессе отгонки с заменой растворителя может приводить к образованию побочного продукта простого н-пропилового эфира из эпоксида 2. Анализ реакционной смеси методом ЯМР показывает, что в смеси осталось менее 1% (мол.)IPAc. Густую суспензию охлаждают до температуры 10 С в течение 1 ч и выдерживают 45 мин. Твердый осадок отфильтровывают и промывают 125 мл охлажденного н-РгОН. Продукт сушат в вакуумной печи при 25 С, в результате получают эпоксид 2 (98,9 А%, 97,6 маc.%, 0,8 маc.% эпи-2). Анализ методом ВЭЖХ показывает отсутствие бис-эпи-2 в выделенном твердом продукте. Пример 29.(10 г, 26, 5 ммоля) в изопропиловом спирте (88 мл) нагревают до температуры кипения - до 82 С и кипятят с обратным холодильником в течение 72 ч до полного образования 3. Раствор 3 охлаждают до 0 С и обрабатывают безводным газообразным НСl и выдерживают смесь при температуре 0-5 С в течение 3 ч. Гидролиз гасят медленным добавлением 50%NaOH для доведения рН смеси до 12, поддерживая температуру ниже 25 С. Затем смесь распределяют между изопропилацетатом (200 мл) и водой (50 мл). Смесь перемешивают и фракции разделяют, водную фракцию повторно экстрагируют изопропилацетатом (50 мл). Раствор J в изопропилацетате упаривают до приблизительно 100 г/л, и в насыщенный теплый изопропилацетатный раствор добавляют воду. В смесь вносят затравочные кристаллы и охлаждают для получения соединения J из эпоксида 2. Поскольку в предыдущем описании раскрыты основные положения данного изобретения, а примеры приведены с целью его иллюстрации, следует понимать, что на практике изобретение охватывает все возможные изменения и модификации, как указано в приведенной далее формуле изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения (I) где R выбирают из водорода, C1-10 алкоксикарбонила или незамещенного или замещенногоC1-10 алкила, где указанным заместителем является арил или гетероарил; и R2 представляет собой C1-10 алкил или арил, отличающийся тем,что включает(а) взаимодействие соединений (А), (В),(Е) и (D) для получения соединения (F)(В) выбирают из галогенированного ацетальдегида или C1-2 алкилацеталя галогенированного ацетальдегида;(b) удаление защитной группы из соединения (F)R выбирают из водорода,C1-10 алкоксикарбонила или незамещенного или замещенного C1-10 алкила, где указанным заместителем является арил или гетероарил;R2 представляет собой C1-10 алкил или арил,отличающийся тем, что включает взаимодействие имина (С) получают взаимодействием этилендиаминового соединения А формулы H2N-(CH2)2-NHR с галогенированным ацетальдегидом или галогенированным ацетальдегид ди-С 1-2 алкилацеталем. 4. Способ по п.2, отличающийся тем, что соединение (F) для получения соединения (I) получают в одном сосуде без первичного выделения имина (С) 2. Способ получения соединения (F) 5. Способ по п.2, отличающийся тем, что R представляет собой замещенный C1-5 алкил, в котором указанным заместителем является гетероарил. 6. Способ по п.5, отличающийся тем, что R представляет собой группу формулы и R2 представляет собой трет-бутил. 7. Способ получения соединения J для получения соединения 3 включающий следующие стадии: 1) взаимодействие нитрила (Н) 4) удаление защитной группы у соединения 3 где R - группа формулыc соединением, выбранным из трет-бутанола,изобутилена или трет-бутилового сложного эфира, в присутствии сильной кислоты с образованием соединения формулы для получения соединения J 2) разделение рацемического соединения 8. Способ получения амида (G) для выделения соединения 1 где R выбирают из водорода, C1-10 алкоксикарбонила или незамещенного или замещенногоC1-10 алкила, где указанным заместителем явля 41 ется арил или гетероарил, отличающийся тем,что включает взаимодействие нитрила (Н) с соединением, выбранным из трет-бутанола,изобутилена или трет-бутилового сложного эфира, в присутствии сильной кислоты с образованием амида (G) 9. Способ по п.8, отличающийся тем, что R представляет собой замещенный C1-5 алкил, где указанным заместителем является гетероарил. 10. Способ по п.9, отличающийся тем, чтоR представляет собой группу формулы 11. Способ по п.10, отличающийся тем, что сильную кислоту выбирают из H2SO4, Н 3 РO4,СН 3SО 3 Н или ВF3. 12. Способ по п.8, отличающийся тем, что сильная кислота представляет собой H2SO4. 13. Способ по п.8, отличающийся тем, что нитрил (Н) получают взаимодействием имина (С) с ионом цианида с образованием нитрила (Н). 14. Способ по п.13, отличающийся тем, что имин (С) получают взаимодействием производного этилендиамина А формулы H2N(СН 2)2NHR с галогенированным ацетальдегидом или ди-C1-2 алкилацеталем галогенированного ацетальдегида. 15. Способ по п.13, отличающийся тем, что получение нитрила (Н) проводят в одном сосуде без первичного выделения имина (С). 16. Соединение формулы (С) где R выбирают из C1-10 алкоксикарбонила или незамещенного или замещенного C1-10 алкила,где указанным заместителем является арил или гетероарил. 17. Соединение по п.16, где R представляет собой замещенный C1-5 алкил, где указанным заместителем является гетероарил. 18. Соединение по п.17, где R представляет собой группу формулыR выбирают из водорода,C1-10 алкоксикарбонила или незамещенного или замещенного C1-10 алкила, где указанным заместителем является арил или гетероарил. 20. Соединение по п.19, где R представляет собой замещенный C1-5 алкил, где указанным заместителем является гетероарил. 21. Соединение по п.20, где R представляет собой 22. Способ получения амида (G)R выбирают из водорода,C1-10 алкоксикарбонила или незамещенного или замещенного C1-10 алкила, где указанным замес 43 тителем является арил или гетероарил; отличающийся тем, что включает(а) взаимодействие соединения (A), H2N(CH2)2-NHR, где R принимает значения, определенные выше, соединения (В), выбираемого из галогенированного ацетальдегида илиC1-2 алкилацеталя галогенированного ацетальдегида и циианид-иона для получения нитрила (Н) 44 и R2 представляет собой трет-бутил; 2) снятие защиты у соединения (F) с образованием рацемического соединения (I) 3) разделение рацемического соединения формулы и (b) взаимодействие нитрила (Н) с соединением, выбираемым из трет-бутанола, изобутилена или сложного трет-бутилового эфира, в присутствии сильной кислоты для получения амида (G) для выделения соединения 1 24. Способ получения соединения J 23. Способ получения соединения 1 который включает следующие стадии: 1) взаимодействие имина (С) включающий следующие стадии: 1) взаимодействие имина (С) где R представляет собой группу формулы где R представляет собой группу формулы и R2 представляет собой трет-бутил; 2) снятие защиты у соединения (F) с образованием рацемического соединения (I) 3) разделение рацемического соединения формулы с соединением 2 для выделения соединения 1 для получения соединения 3 5) снятие защиты у соединения 3 для получения соединения J.
МПК / Метки
МПК: C07D 401/06
Метки: вируса, протеазы, способ, человека, ингибиторов, иммунодефицита, получения
Код ссылки
<a href="https://eas.patents.su/24-637-sposob-polucheniya-ingibitorov-proteazy-virusa-immunodeficita-cheloveka.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения ингибиторов протеазы вируса иммунодефицита человека.</a>
Сульфонилалканоиламиногидроксиэтиламиносульфонамид в качестве ингибиторов ретровирусной протеазы

Номер патента: 533
Опубликовано: 28.10.1999
Авторы: Нагараян Сринивазан, Декрессенцо Гэри А., Васкез Майкл Л., Макдонэлд Джозеф Дж., Девадас Балекудру, Фрескос Джон Н., Гетмен Даниел П., Сикорски Джеймс А.
МПК: C07C 311/00, C07D 317/62, A61K 31/34...
Метки: протеазы, ингибиторов, качестве, сульфонилалканоиламиногидроксиэтиламиносульфонамид, ретровирусной
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль, пролекарство или сложный эфир, где символы n и t, каждый независимо друг от друга, равны 0,1 или 2; R1 обозначает водород, алкил с 1-5 атомами углерода, алкенил с 2-5 атомами углерода, алкинил с 2-5 атомами углерода, гидроксиалкил с 1-3 атомами углерода, алкоксиалкил, состоящий из алкила с 1-3 атомами и алкокси с 1-3 атомами углерода, цианоалкил, содержащий алкил с 1-3 атомами...
Способ получения ингибитора протеазы вич из(2-s)-4-пиколил-2-пиперазин-трет-бутилкарбоксамида

Номер патента: 178
Опубликовано: 24.12.1998
Авторы: Воланте Ральф П., Ридер Пол, Енг Кан К., Аскин Дэвид
МПК: C07D 401/06, A61K 31/495
Метки: способ, из(2-s)-4-пиколил-2-пиперазин-трет-бутилкарбоксамида, получения, вич, протеазы, ингибитора
Формула / Реферат:
1. Способ получения ингибитора протеазы ВИЧ структурной формулы включающий следующие стадии: (а) нагревание, по крайней мере, в течение одного часа одного эквивалента (S)-4-(3-пиколил)-2-трет-бутилкарбоксамидпиперазина (1) с приблизительно одним эквивалентом соединения в интервале температур между около 25 и около 150°С, при этом указанная смесь необязательно содержит приемлемый растворитель; (б) снятие защиты у полученного...
Способ “вигесол” коррекции энергетического состояния организма человека

Номер патента: 389
Опубликовано: 24.06.1999
Авторы: Соломатова Галина Владимировна, CОЛОМАТОВА Мария Викторовна, Соломатова Анна Викторовна, Соломатов Виктор Геннадьевич
МПК: A61H 39/04
Метки: энергетического, организма, коррекции, вигесол, человека, состояния, способ
Формула / Реферат:
1. Способ "Вигесол" коррекции энергетического состояния организма человека, включающий диагностику энергетического состояния путем фиксирования болевых ощущений в рефлексогенных областях и последующее седативное или тонизирующее воздействие, отличающийся тем, что на биологически активные точки и/или зоны акупунктуры, а также на сегментарные болевые точки и/или болевые зоны в проекции зоны акупунктуры с недостатком энергии воздействуют...
Способ компьютерной оптической топографии формы тела человека и устройство для его осуществления

Номер патента: 111
Опубликовано: 27.08.1998
Авторы: Садовой Михаил Анатольевич, Сарнадский Владимир Николаевич, Фомичев Николай Гаврилович
МПК: A61B 5/103
Метки: способ, человека, компьютерной, топографии, тела, осуществления, устройство, формы, оптической
Формула / Реферат:
1. Способ компьютерной оптической топографии формы тела человека (пациента) путем проецирования на обследуемую поверхность тела под заданным углом к этой поверхности изображения пространственной системы эквидистантных оптически контрастных прямых линий, видеосъемки изображения этой системы линий под углом, отличным от указанного угла проецирования, аналого-цифровое преобразование видеосигнала изображения, введение его в устройство памяти...
Производные 5-0-дезозаминил-6-0-метилэритронолида а, способ их получения и их применение для получения биологически активных продуктов

Номер патента: 575
Опубликовано: 29.12.1999
Авторы: Дельтиль Мишель, Мазюри Алан, Бонне Алан
МПК: C07H 17/08
Метки: способ, продуктов, биологически, 5-0-дезозаминил-6-0-метилэритронолида, активных, получения, применение, производные
Формула / Реферат:
1. Соединения формулы (I): в которой или R1 представляет радикал алкил, содержащий до 8 атомов углерода, замещенный одним или несколькими радикалами алкила, содержащими до 8 атомов углерода, или одним или несколькими радикалами арила, содержащими до 14 атомов углерода, или R1 представляет радикал арил, содержащий до 14 атомов углерода, который может быть замещен одним или несколькими радикалами алкил, алкенил или алкинил, содержащими до 8...
Предыдущий патент: Тройные радиофармацевтические комплексы
Следующий патент: Синергетическая фунгицидная композиция, содержащая соединение-аналог стробилурина
Случайный патент: Установка для предварительного дробления кусков рубероидно-битумных кровельных покрытий