Высокоактивные катализаторы циглера-натта, способ получения катализаторов и их использование
Номер патента: 19316
Опубликовано: 28.02.2014
Авторы: Клендуорт Дуглас Д., Винтер Андреас, Джонсон Кеннет У., Лангхаузер Франц









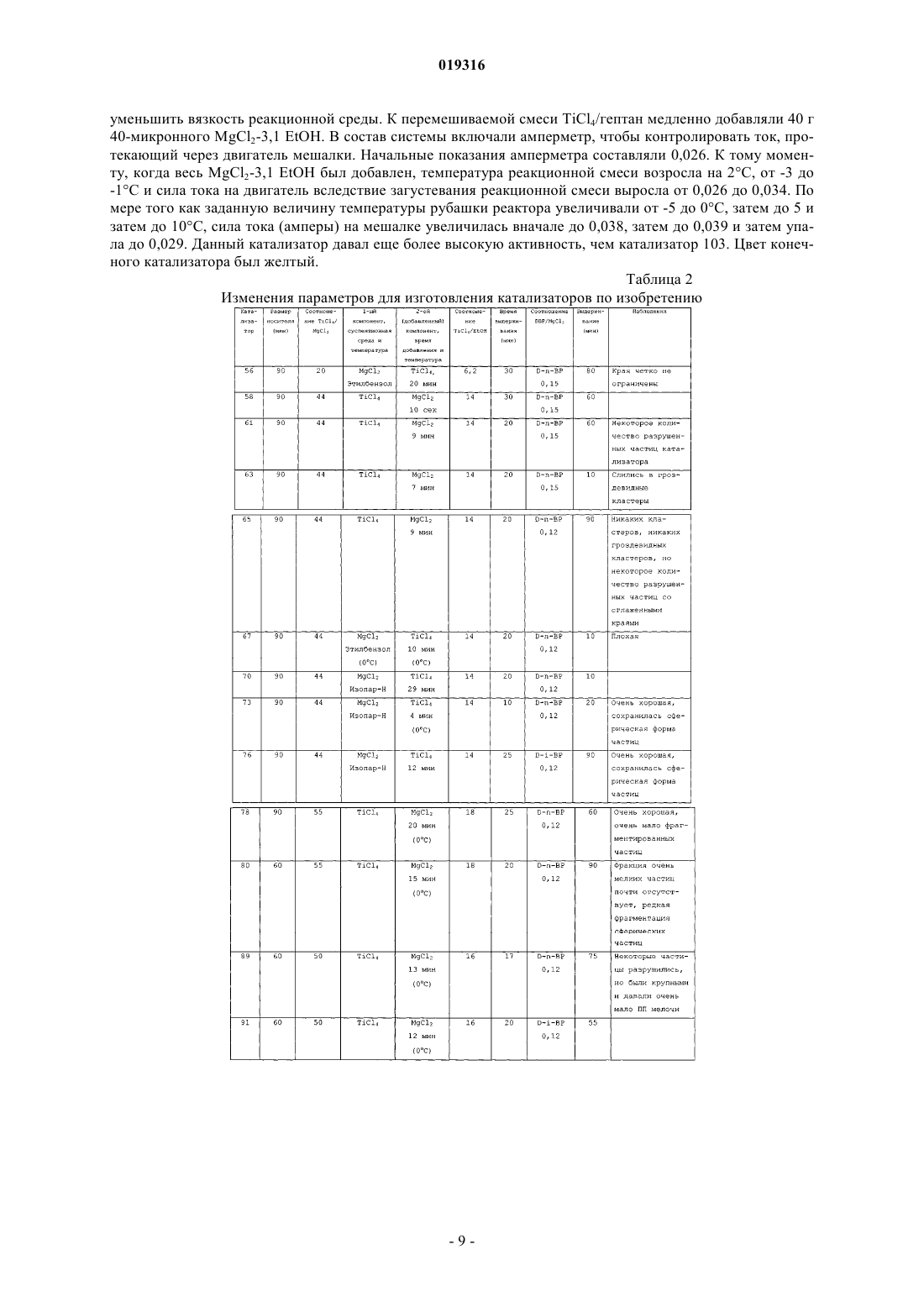
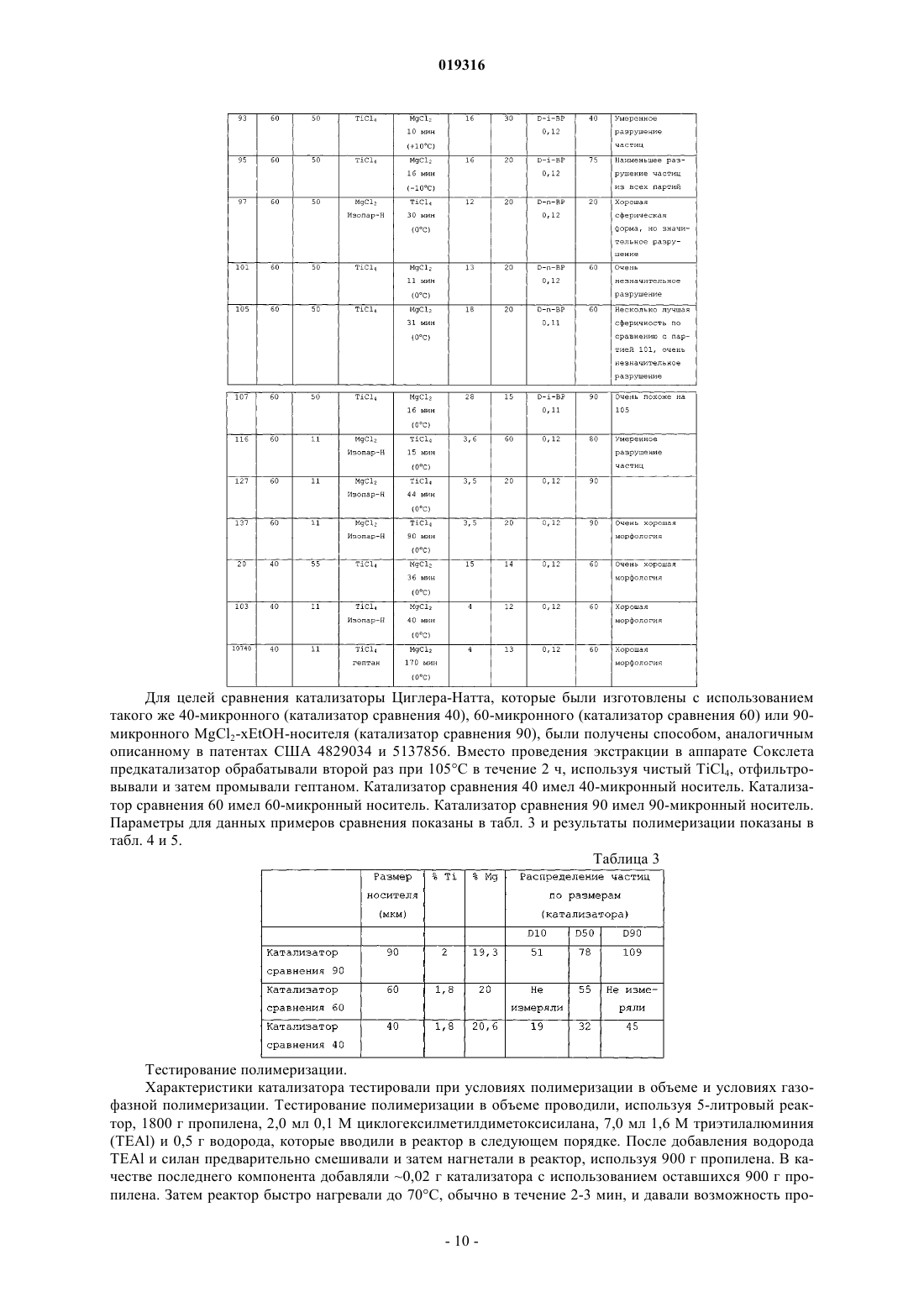

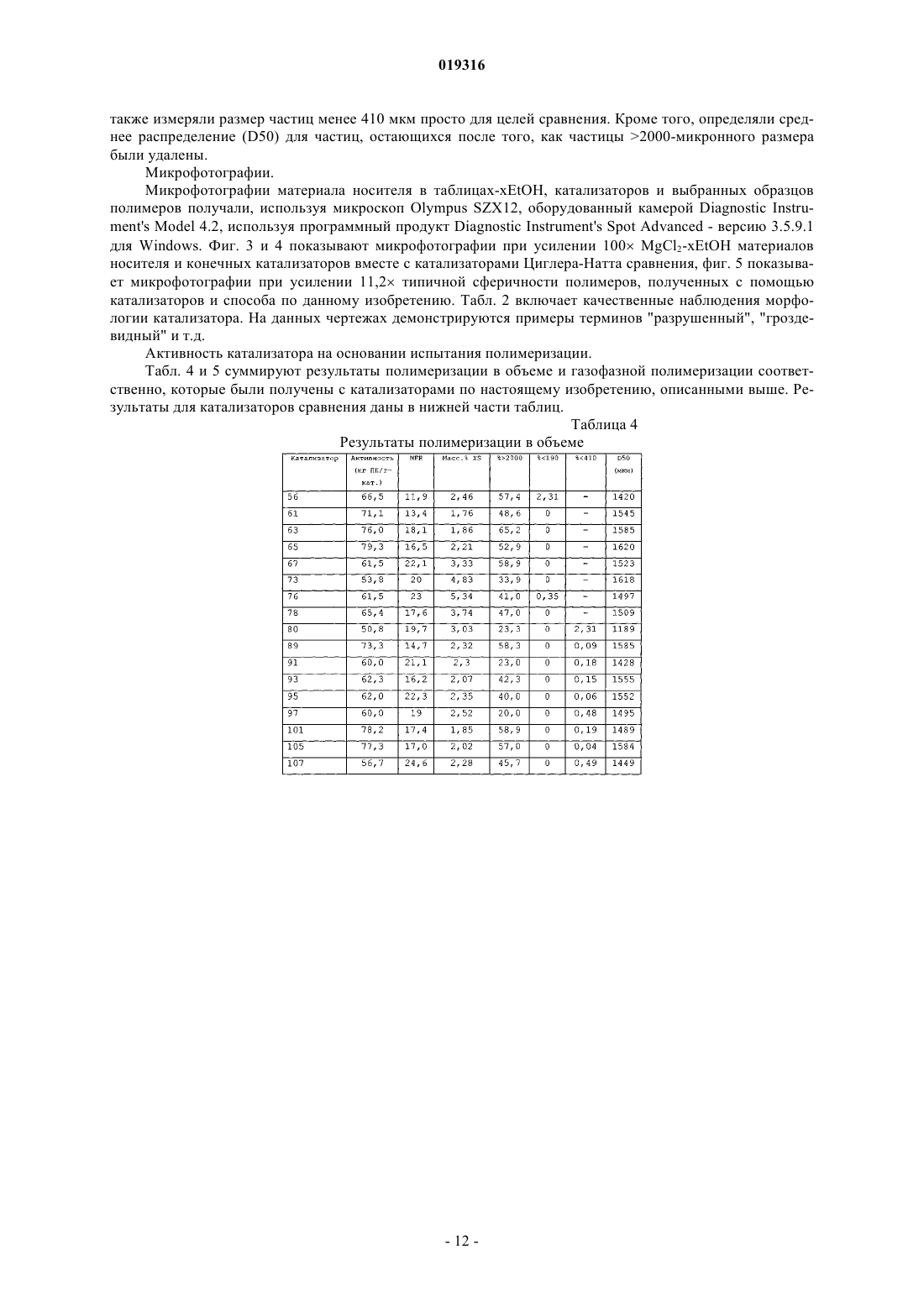
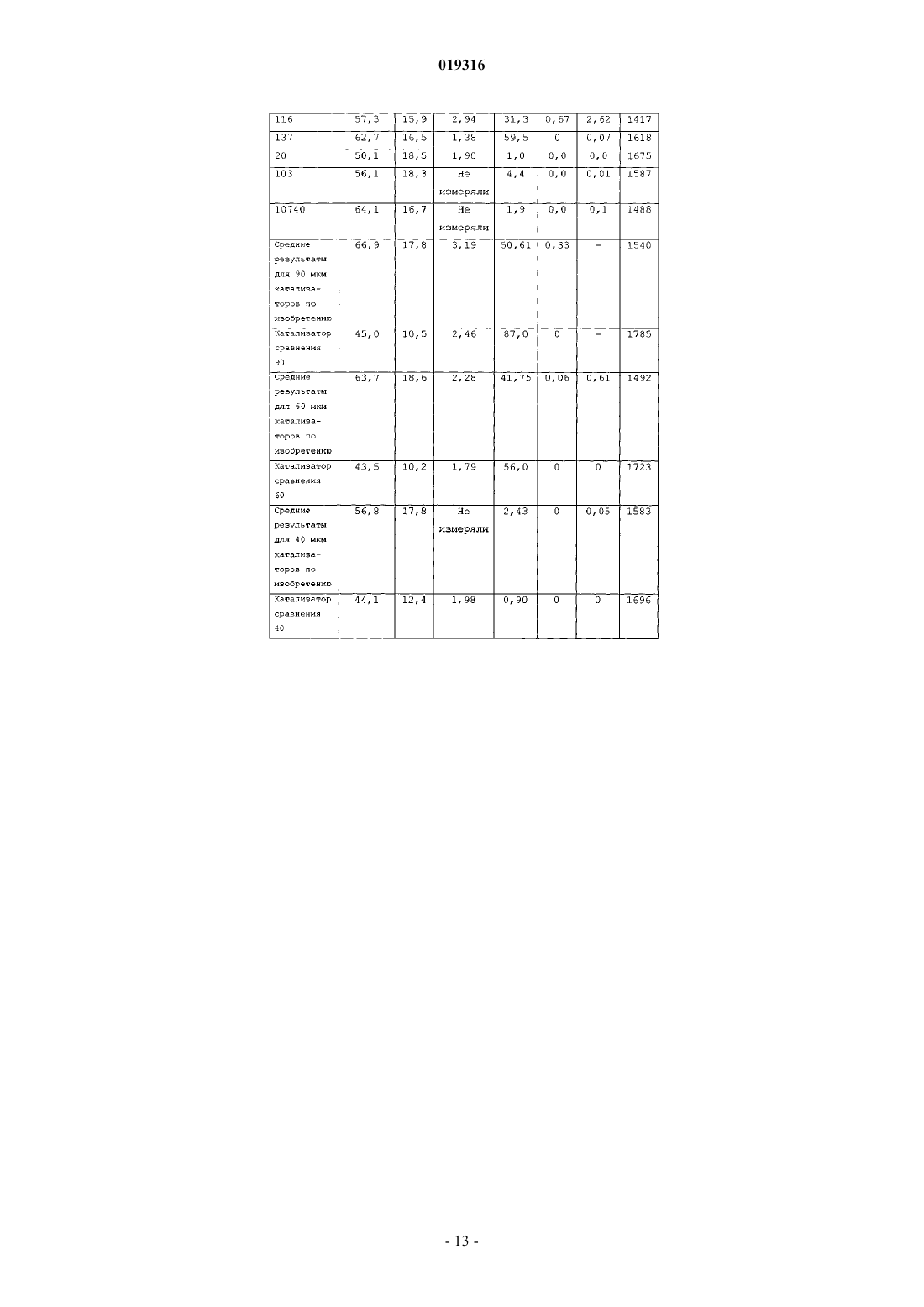







Формула / Реферат
1. Способ получения катализатора Циглера-Натта для полимеризации олефинов, включающий стадии:
a) объединения сферического, охлажденного распылением носителя MgCl2-xROH, где х имеет значение примерно от 1,5 до 6,0 и где ROH представляет собой спирт или смесь спиртов, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 1-10 атомами углерода, с соединением переходного металла в реакторе при температуре примерно от -30 до 40°С;
b) нагревания смеси в реакторе до температуры от примерно 30 до примерно 100°С;
c) одновременно с нагревом в стадии (b) или вслед за достижением температуры стадии (b) добавления внутреннего донора электронов к смеси в реакторе;
d) нагревания полученной в результате смеси до температуры по меньшей мере примерно 80°С и выдерживания полученной в результате смеси при данной температуре примерно в течение времени от 1 до 2 ч, чтобы получить предкатализатор;
e) фильтрования смеси, содержащей предкатализатор, с получением твердого компонента предкатализатора;
f) экстрагирования предкатализатора смесью органического растворителя и соединения переходного металла при температуре по меньшей мере 100°С в течение 1-5 ч с получением катализатора;
g) охлаждения катализатора до комнатной температуры (20°С), промывки катализатора углеводородным растворителем и сушки катализатора под вакуумом и/или при повышенной температуре, равной 30-100°С.
2. Способ по п.1, в котором ROH представляет собой смесь по меньшей мере двух различных спиртов, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 1-10 атомами углерода.
3. Способ по п.1, в котором ROH представляет собой этанол или смесь этанола и высшего спирта, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 3-10 атомами углерода.
4. Способ по п.1, в котором ROH представляет собой этанол или смесь этанола и высшего спирта, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 4-10 атомами углерода.
5. Способ по п.1, в котором ROH представляет собой смесь этанола и бутанола, гексанола, гептанола или октанола.
6. Способ по п.1, в котором ROH представляет собой этанол.
7. Способ по п.1, в котором х имеет значение в интервале примерно от 2,5 до 4,0.
8. Способ по п.1, в котором х имеет значение в интервале примерно от 2,95 до 3,35.
9. Способ по п.1, в котором на стадии (а) соединение переходного металла представляет собой TiCl4 и ROH представляет собой этанол или смесь этанола и высшего спирта, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 3-10 атомами углерода.
10. Способ по п.1, в котором на стадии (а) соединение переходного металла представляет собой TiCl4 и ROH представляет собой этанол.
11. Способ по п.1, в котором стадию экстракции (f) осуществляют, используя метод экстракции в аппарате Сокслета.
12. Способ по п.1, в котором температура реактора для комбинации MgCl2-xROH и соединения переходного металла имеет значение в интервале примерно от -10 до +10°С.
13. Способ по п.1, в котором соединение переходного металла, используемое на стадии (f), представляет собой TiCl4.
14. Способ по п.1, в котором внутренний донор электронов выбирают из группы, состоящей из сложных диэфиров, замещенных циклоалкан-1,2-дикарбоновых кислот, сложных моноэфиров замещенных бензофенон-2-карбоновых кислот, замещенных бензофенон-3-карбоновых кислот, незамещенных и замещенных простых диэфиров (C1-C10-алкил)-1,3-пропана, производных карбоновой кислоты, производных фталевой кислоты и производных сукцинатов.
15. Способ по п.1, в котором внутренний донор электронов выбирают из диизобутилфталата (D-i-BP), ди-н-бутилфталата (D-n-ВР), диизооктилфталата, ди-2-этил,гексилфталата или диизононилфталата.
16. Способ по п.1, в котором смеси со стадии (а) дают возможность взаимодействовать в течение приблизительно от 2 до 300 мин перед обработкой нагреванием со стадии (b).
17. Способ по п.1, в котором полученную в результате смесь из стадии (d) нагревают до температуры от примерно 100 до примерно 135°С.
18. Способ по п.17, в котором полученную в результате смесь из стадии (d) выдерживают при температуре в течение от примерно 1 до примерно 4 ч.
19. Способ по п.1, в котором органический растворитель из стадии (f) представляет собой алифатический или ароматический углеводород.
20. Способ по п.1, в котором органический растворитель из стадии (f) представляет собой ароматический углеводород.
21. Способ по п.1, в котором органический растворитель из стадии (f) представляет собой этилбензол.
22. Способ по п.13, в котором органический растворитель представляет собой этилбензол, объемное отношение TiCl4 и этилбензола составляет примерно 30:70 и время экстракции составляет 1-5 ч при температуре по меньшей мере 100°С.
23. Способ по п.13, в котором органический растворитель представляет собой этилбензол, объемное отношение TiCl4 и этилбензола составляет примерно 20:80 и время экстракции составляет 1-4 ч при 100-135°С.
24. Способ по п.13, в котором органический растворитель представляет собой этилбензол, объемное отношение TiCl4 к этилбензолу составляет примерно 10:90 и время экстракции составляет 1-3 ч при 120-130°С.
25. Способ по п.1, в котором часть ROH удаляют из MgCl2-носителя деалкоголизацией перед взаимодействием с соединением переходного металла.
26. Способ по п.1, в котором на стадии (а) молярное отношение Mg к переходному металлу составляет от примерно 1:100 до примерно 1:5.
27. Способ по п.1, в котором на стадии (а) молярное отношение Mg к переходному металлу составляет от примерно 1:50 до примерно 1:8.
28. Способ по п.1, в котором на стадии (а) молярное отношение Mg к переходному металлу составляет от примерно 1:25 до примерно 1:9.
29. Способ по п.1, в котором внутренний донор электронов представляет собой диизобутилфталат (D-i-BP) или ди-н-бутилфталат (D-n-BP) и молярное отношение D-i-BP или D-n-BP/Mg составляет от примерно 0,12 до примерно 0,15.
30. Способ получения катализатора для полимеризации олефинов, включающий стадии:
a) объединения сферического, охлажденного распылением носителя MgCl2-xEtOH, где х равен от примерно 3,0 до примерно 3,3 и EtOH представляет собой этанол, с TiCl4 в реакторе при температуре примерно от -10 до 10°С;
b) нагревания смеси в реакторе до температуры примерно 40-90°С;
c) добавления диизобутилфталата (D-i-BP) к смеси в реакторе;
d) нагревания полученной в результате смеси примерно до 100-110°С и выдерживания полученной в результате смеси при данной температуре примерно в течение времени от 1 до 2 ч, чтобы получить предкатализатор;
e) фильтрования смеси, содержащей предкатализатор, для получения твердого компонента предкатализатора;
f) экстрагирования предкатализатора с использованием экстракции в аппарате Сокслета смесью этилбензола в качестве органического растворителя и TiCl4 в качестве переходного металла при температуре 120-130°С в течение 1-3 ч для получения катализатора;
g) промывки катализатора растворителем, содержащим гексан или гептан, и сушки катализатора под вакуумом и/или при повышенной температуре, равной 30-100°С.
31. Способ полимеризации олефинов формулы CH2=CHR1, в которой R1 представляет собой водород или углеводородный радикал, содержащий 1-12 атомов углерода, осуществляемый в присутствии катализатора, полученного способом по п.1 или 30.
32. Катализатор, полученный способом по п.1 или 30 и имеющий активность для полимеризации в объеме по полипропилену больше чем 60 кг полипропилена/г кат.×1 ч.
33. Способ получения катализатора Циглера-Натта для полимеризации олефинов, включающий стадии:
a) объединения сферического, охлажденного распылением носителя MgCl2-xROH, сформированного в отсутствие диоксида кремния в качестве затравки кристаллизации, где х имеет значение примерно от 1,5 до 6,0 и где ROH представляет собой спирт или смесь спиртов, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 1-10 атомами углерода, с соединением переходного металла в реакторе при температуре примерно от -30 до +40°С;
b) нагревания смеси в реакторе до температуры от примерно 30 до примерно 100°С;
c) одновременно с нагревом в стадии (b) или вслед за достижением температуры стадии (b) добавления внутреннего донора электронов к смеси в реакторе;
d) нагревания полученной в результате смеси до температуры по меньшей мере примерно 80°С и выдерживания полученной в результате смеси при данной температуре примерно в течение времени от 1 до 2 ч, чтобы получить предкатализатор;
e) фильтрования смеси, содержащей предкатализатор, с получением твердого компонента предкатализатора;
f) экстрагирования предкатализатора смесью органического растворителя и переходного металла при температуре по меньшей мере 100°С в течение 1-5 ч с получением катализатора;
g) охлаждения катализатора до комнатной температуры (20°С), промывки катализатора углеводородным растворителем и сушки катализатора под вакуумом и/или при повышенной температуре, равной 30-100°С.
Текст
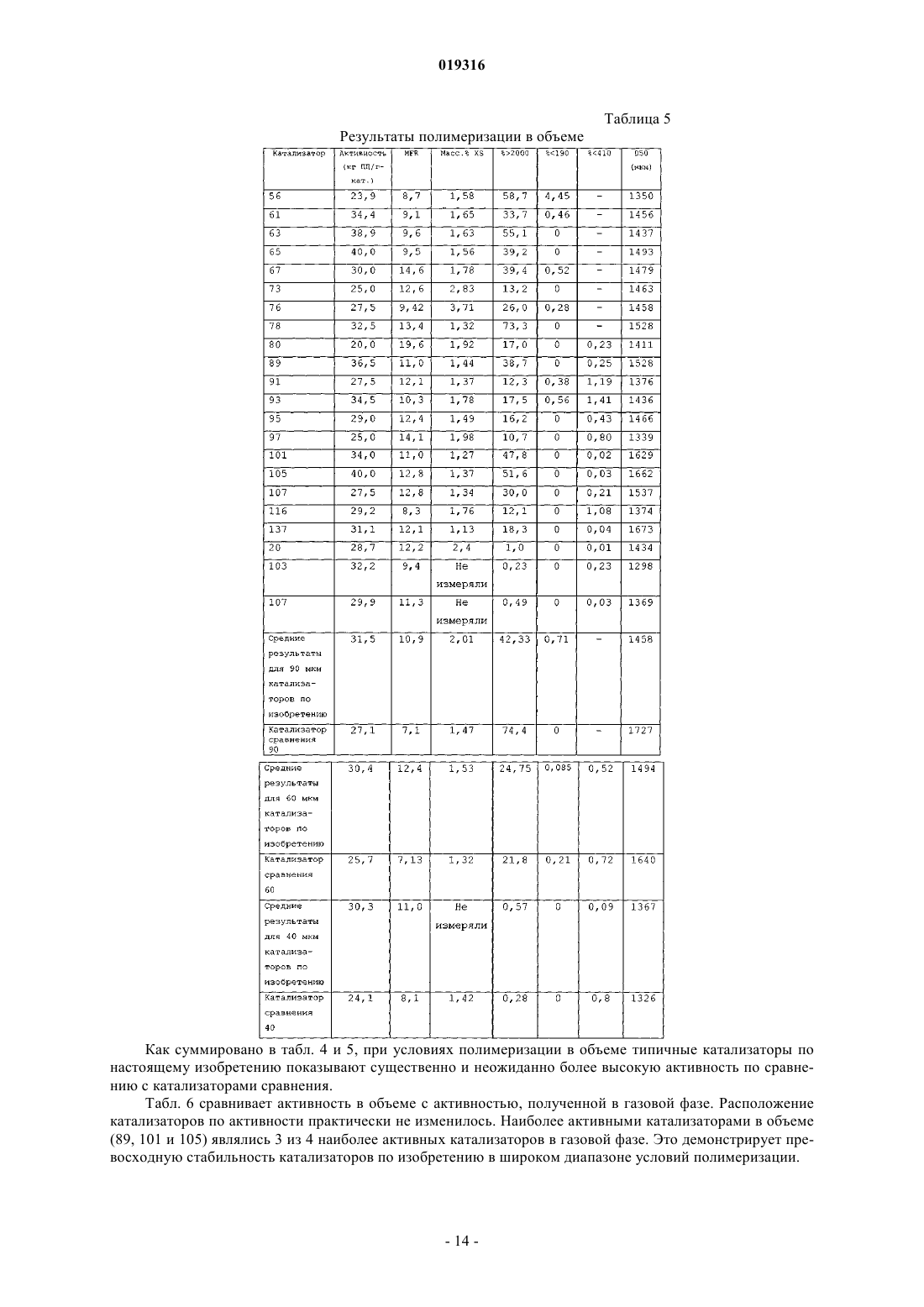
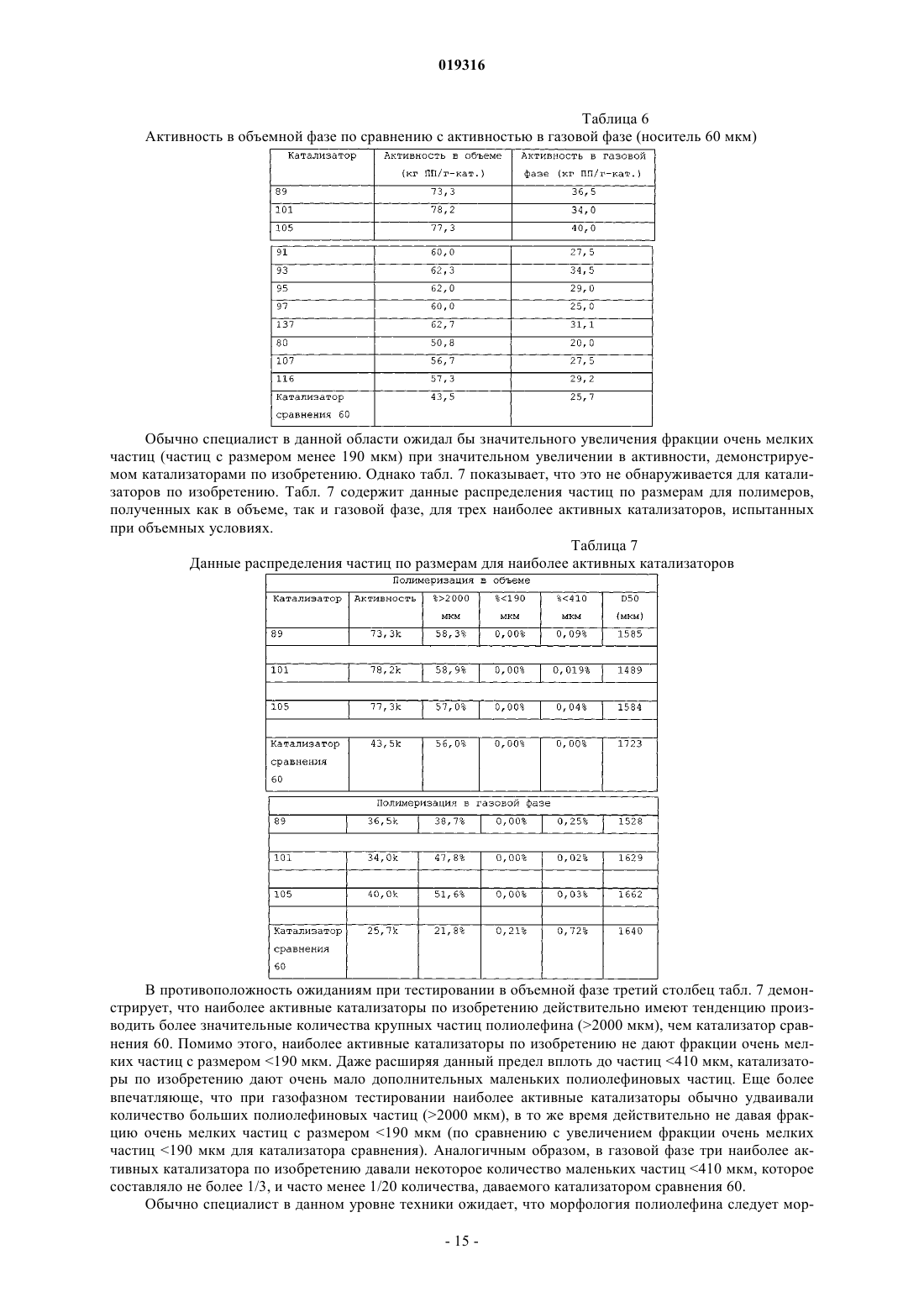
ВЫСОКОАКТИВНЫЕ КАТАЛИЗАТОРЫ ЦИГЛЕРА-НАТТА, СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРОВ И ИХ ИСПОЛЬЗОВАНИЕ В изобретении описываются улучшенные катализаторы Циглера-Натта и способы получения улучшенного катализатора. Катализатор Циглера-Натта получают с использованием сферического(MgCl2-xROH)-носителя, где R представляет собой звено линейного, циклического или разветвленного углеводорода с 1-10 атомами углерода, где ROH представляет собой спирт или смесь по меньшей мере двух различных спиртов и где х находится в диапазоне примерно от 1,5 до 6,0, предпочтительно примерно от 2,5 до 4, более предпочтительно примерно от 2,9 до 3,4 и еще более предпочтительно от 2,95 до 3,35. Катализатор Циглера-Натта включает в себя переходный металл группы 4-8 и внутренний донор. Катализатор имеет улучшенную активность в реакциях полимеризации олефинов, а также хорошую стереорегулярность и чувствительность к водороду.(71)(73) Заявитель и патентовладелец: ЛЮММУС НОВОЛЕН ТЕКНОЛОДЖИ ГМБХ (DE) Притязание на приоритет Настоящая заявка на изобретение заявляет приоритет предварительной заявки США номер 61/060646, поданной 11 июня 2008 года, полное содержание которой настоящим включается ссылкой. Область, к которой относится изобретение Настоящее изобретение относится, в одном аспекте, к улучшенному катализатору Циглера-Натта. В частности, изобретение относится к использованию такого катализатора при полимеризации олефинов в полиолефины и в особенности к улучшенному способу получения катализатора Циглера-Натта. Уровень техники Катализаторы Циглера-Натта обычно состоят из материала носителя катализатора, компонента,представляющего собой переходный металл, и одного или нескольких лигандов, которые соответствуют валентности металла. Компонентом, представляющим собой переходный металл, в типичном случае является переходный металл группы 4-8, причем, как правило, используют титан, цирконий, хром или ванадий. Переходный металл часто предоставляется в виде галогенида металла, например TiCl4. Катализаторы Циглера-Натта используют, чтобы эффективно содействовать полимеризации олефинов с высоким выходом. При полимеризации олефинов катализатор используют в сочетании с сокатализатором на основе алюминийорганического соединения. В случае использования для катализа полимеризации пропилена в катализаторе может быть использован третий компонент. Третий компонент представляет собой донор электронов, используемый для контроля стереорегулярности полимера. Его можно ввести в катализатор в ходе его синтеза (внутренний донор) или его можно добавить в реактор полимеризации в ходе реакции полимеризации (внешний донор). В некоторых реакциях можно использовать как внутренний донор, так и внешний донор. Примерами соединений, которые можно использовать при получении полипропилена, являются сложные ароматические эфиры, простые диэфиры, сукцинаты, алкоксисиланы и стерически затрудненные амины. Одним известным материалом носителя, используемым в некоторых катализаторах Циглера-Натта,является MgCl2. Иногда материал MgCl2 находится в виде комплекса с этанолом (EtOH). EtOH взаимодействует с галогенидом переходных металлов, таким как TiCl4, при приготовлении катализатора. Способы получения комплексов MgCl2-xEtOH, в которых х представляет среднее количество молекул EtOH в материале носителя, описываются в нескольких патентах. Например, патент США 5468698, выданный Коскинену, описывает способы получения материала носителя типа MgCl2-xEtOH. Расплав комплекса MgCl2-xEtOH (х=от 3,3 до 5,5) распыляют в нагретой камере с образованием дисперсного материала MgCl2-xEtOH, в котором х=от 2,0 до 3,2. Коскинен не описывает композицию конкретного катализатора, изготовленного с использованием данного материала носителя. Также описываются катализаторы, использующие MgCl2-xEtOH носители. Например, патент США 4829034, выданный Иисколану, описывает катализатор Циглера-Натта и способ получения данного катализатора с использованием MgCl2-xEtOH-носителя, в котором х равен примерно 3. По Иисколану материал носителя сначала контактирует с внутренним донором, таким как D-i-BP. Затем осуществляют взаимодействие комплекса носителя с D-i-BP и TiCl4 с получением катализатора. Патент США 6020279, выданный Уваи, описывает способ получения катализатора ЦиглераНатта изготовлением MgCl2-xEtOH-носителя, в котором х=от 1,5 до 2,1, и носитель имеет средний диаметр частиц 91 мкм. Носитель объединяют с галогенидом титана, таким как TiCl4, и донором электронов в течение времени от 10 мин до 10 ч при температуре от 120 до 135 С в присутствии алифатического растворителя. В то время как был разработан целый ряд катализаторов Циглера-Натта, вследствие важности полимеризации олефинов по-прежнему существует необходимость в разработке катализаторов, обладающих улучшенной активностью. Улучшение активности катализатора ведет к более высоким выходам продуктов и уменьшает количество катализатора, требующегося для реакции полимеризации олефинов,что снижает стоимость катализатора и количество примесей катализатора в полимере (сниженное содержание золы), в результате давая полимеры с улучшенным профилем эксплуатационных характеристик. Безотносительно к методу, используемому для получения MgCl2-носителя, или даже если такой носитель используется в обычно получаемом катализаторе Циглера-Натта, только уникальный способ объединения трех существенных компонентов катализатора Циглера-Натта, как описывается в данном изобретении, будет давать необычно высокую активность, отклик на водород и стереорегулирующую активность, обнаруживаемые в настоящем изобретении. Сущность изобретения Настоящее изобретение нацелено на улучшенный катализатор Циглера-Натта, который получают,используя улучшенную процедуру в сочетании со сферическим носителем MgCl2-xROH, где R представляет собой звено линейного, циклического или разветвленного углеводорода с 1-10 атомами углерода,где ROH представляет собой спирт или смесь по меньшей мере двух различных спиртов, предпочтительно где ROH представляет собой этанол или смесь этанола и высшего спирта, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 3-10 атомами углерода, предпочтительно 4-10 атомами углерода; и где х находится в диапазоне примерно от 1,5 до 6,0, предпочтительно примерно от 2,5 до 4, более предпочтительно примерно от 2,9 до 3,4 и еще более предпочтительно от 2,95 до 3,35. Катализатор включает в себя переходный металл группы 4-8, такой как Ti, и внутренний донор, например сложные ароматические эфиры, простые диэфиры, сукцинаты или стерически затрудненные амины, предпочтительно диалкилфталаты, аналогичные диизобутилфталату (D-i-BP) или ди-нбутилфталату (D-n-BP). Катализатор по настоящему изобретению имеет улучшенную активность в реакциях полимеризации олефинов, а также хорошую стереорегулярность и чувствительность к водороду. Настоящее изобретение также нацелено на способы получения улучшенного катализатора ЦиглераНатта. Как правило, сферический MgCl2-xROH (x=3,0-3,3) обрабатывают галогенидом переходного металла, таким как TiCl4, при низкой температуре (от -10 до +10 С). Реакционный продукт нагревают примерно до 50 С, и он контактирует с внутренним донором. Полученный в результате предкатализатор нагревают примерно до 105 С и выдерживают при данной температуре в течение некоторого периода времени, предпочтительно примерно от 1 до 2 ч. Реакционную смесь охлаждают до комнатной температуры и твердый катализатор экстрагируют смесью органический растворитель/TiCl4 при повышенной температуре. Катализатор промывают растворителем, таким как гептан, и сушат под вакуумом. Улучшенный катализатор по изобретению можно использовать для получения полипропилена или других полимеризованных олефинов. Катализаторы по изобретению показывают улучшенную активность, в то же время давая полимеры, имеющие высокую стереоспецифичность и морфологию. Краткое описание чертежей Фиг. 1 представляет собой схему оборудования, используемого для получения типовых катализаторов по настоящему изобретению через стадию предкатализатора; фиг. 2 представляет собой схему оборудования, используемого для извлечения активированного катализатора из композиции предкатализатора; фиг. 3 показывает микроснимки (MgCl2-xROH)-носителей; фиг. 4 показывает микроснимки катализаторов по изобретению и фиг. 5 показывает микроснимки полученных в результате полимеров соответственно. Данные микрофотографии показывают типичную сферичность катализаторов и полимеров, полученных способом по данному изобретению, если не указано иным образом в таблице результатов. Подробное описание Настоящее изобретение нацелено на улучшенный катализатор Циглера-Натта, который получают,используя улучшенную процедуру в сочетании со сферическим MgCl2-xROH-носителем, где R представляет собой звено линейного, циклического или разветвленного углеводорода с 1-10 атомами углерода и где ROH представляет собой спирт или смесь по меньшей мере двух различных спиртов, предпочтительно где ROH представляет собой этанол или смесь этанола и высшего спирта, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 3-10 атомами углерода, предпочтительно с 4-10 атомами углерода; и где х находится в диапазоне примерно от 1,5 до 6,0, предпочтительно примерно от 2,5 до 4, более предпочтительно примерно от 2,9 до 3,4 и еще более предпочтительно от 2,95 до 3,35. Данный материал носителя в настоящем описании называют "сферическим MgCl2-носителем". Сферический MgCl2-носитель может иметь любой желаемый размер частиц. В предпочтительных вариантах осуществления сферический MgCl2-носитель имеет средний размер частиц (d50) примерно от 10 до 200 мкм, предпочтительно от 20 до 150 мкм, более предпочтительно от 30 до 120 мкм и еще более предпочтительно от 40 до 90 мкм. Сферический MgCl2-носитель можно получить согласно US 4829034, выданному Иисколану и Коскинену, или US 5905050, выданному Коскинену, струйным охлаждением расплавленного аддукта MgCl2-спирт. Физические характеристики трех предпочтительных твердых носителей суммируются в табл. 1. Таблица 1 Катализатор Циглера-Натта по настоящему изобретению включает переходный металл группы 4-8,предпочтительно переходный металл группы 4-6. В предпочтительных вариантах осуществления катализатор включает Ti, Zr, V или Cr и наиболее предпочтительно Ti. Переходный металл типично обеспечивается в галогенированной форме, такой как хлорид, бромид или йодид. Особенно предпочтительным является хлорид титана. Катализатор Циглера-Натта по настоящему изобретению изготавливают контактом сферическогоMgCl2-носителя с компонентом на основе переходного металла в реакторе при низкой температуре,предпочтительно +10 С или менее, при перемешивании. Сферический MgCl2-носитель и компонент на основе переходного металла можно загружать в реактор в любом порядке, т.е. сферический MgCl2 носитель можно добавить первым и затем можно добавить компонент на основе переходного металла,или наоборот, но добавление сферического MgCl2-носителя к компоненту на основе переходного металла является предпочтительным. Компонент на основе переходного металла можно разбавить алифатиче-2 019316 ским или ароматическим органическим растворителем, предпочтительно алифатическим углеводородом,наиболее предпочтительно линейным алифатическим углеводородом, аналогичным гептану, или смесью разветвленных углеводородов, аналогичной изопару-Н. Сферический MgCl2-носитель добавляют в реактор в течение периода времени предпочтительно примерно от 4 до 300 мин. Молярное отношение Mg сферического MgCl2-носителя к компоненту на основе переходного металла составляет от 1:100 до 1:5,предпочтительно от 1:50 до 1:8 и наиболее предпочтительно от 1:25 до 1:9. Продукт реакции сферического MgCl2-носителя и компонента на основе переходного металла медленно нагревают до заранее определенной температуры, составляющей примерно от 30 до 100 С. В предпочтительном варианте осуществления реактор нагревают до температуры примерно от 40 до 90 С в течение периода времени, составляющего примерно 2 ч. Донор электронов добавляют в реактор, когда его температура достигнет заранее определенного значения. Затем данный предкатализатор дополнительно нагревают до температуры по меньшей мере 80 С, предпочтительно от 100 до 125 С, более предпочтительно от 100 до 110 С и выдерживают при данной температуре в течение заранее определенного периода времени, предпочтительно от примерно 10 мин до 2 ч. Затем полученную в результате смесь охлаждают до комнатной температуры и фильтруют, чтобы удалить твердый компонент. Твердый компонент экстрагируют при повышенных температурах, используя смесь органического растворителя и переходного металла. Предпочтительно использовать метод экстракции в аппарате Сокслета. Органический растворитель может представлять собой алифатический или ароматический углеводород, предпочтительно ароматический углеводород и наиболее предпочтительно этилбензол, который имеет такую же температуру кипения 136 С, как и TiCl4, что гарантирует постоянное соотношение между TiCl4 и органическим растворителем в газовой фазе и в зоне экстракции. В одном варианте осуществления настоящего изобретения процедура изготовления катализатора Циглера-Натта представляет собой:a) взаимодействие MgCl2-xROH с чистым TiCl4 при температуре от -30 до +40 С, более предпочтительно от -20 до +20 С, еще более предпочтительно от -10 до +10 С, посредством медленного добавления TiCl4 к суспензии MgCl2-xROH/органический растворитель, при обеспечении в то же время непрерывного перемешивания;b) повышение температуры вышеуказанной реакционной смеси до температуры примерно от 30 до 100 С, предпочтительно примерно от 40 до 90 С, после чего следует добавление внутреннего донора электронов и продолжение нагревания смеси по меньшей мере до 80 С в течение примерно от 1 до 2 ч;c) фильтрование реакционной смеси при комнатной температуре для получения твердого предкатализатора;d) экстрагирование предкатализатора с использованием метода экстракции в аппарате Сокслета,применяя TiCl4 и этилбензол (при объемном соотношении примерно 30:70, предпочтительно 20:80, наиболее предпочтительно 10:90), в течение 1-5 ч, предпочтительно 1-4 ч, наиболее предпочтительно 1-3 ч при температуре по меньшей мере 100 С, предпочтительно 100-135 С, наиболее предпочтительно 120130 С; е) охлаждение катализатора до комнатной температуры (20 С), промывку несколько раз углеводородом, например пентаном, гексаном или гептаном, и затем сушку под вакуумом и/или при повышенной температуре, составляющей 30-100 С, предпочтительно 40-90 С, наиболее предпочтительно 50-80 С. Во втором варианте осуществления изобретения способ включает:a) приготовление охлажденной части чистого TiCl4 или TiCl4, разбавленного неароматическим углеводородом;b) взаимодействие чистого или разбавленного TiCl4 при температуре от -30 до +40 С, более предпочтительно от -20 до +20 С, наиболее предпочтительно от -10 до +10 С, посредством медленного добавления предварительно полученных сферических частиц MgCl2-xROH, при обеспечении в то же время непрерывного перемешивания;c) повышение температуры реакционной смеси до температуры примерно от 30 до 100 С, предпочтительно примерно от 40 до 90 С, после чего следует добавление внутреннего донора электронов и продолжение нагревания смеси по меньшей мере до 80 С;d) фильтрование реакционной смеси при комнатной температуре;e) экстрагирование предкатализатора с использованием метода экстракции в аппарате Сокслета с применением TiCl4 и этилбензола (при объемном соотношении примерно 30:70, предпочтительно 20:80,наиболее предпочтительно 10:90), в течение 1-5 ч, предпочтительно 1-4 ч, наиболее предпочтительно 1-3 ч при температуре по меньшей мере 100 С, предпочтительно 100-135 С, наиболее предпочтительно 120130 С;f) охлаждение катализатора до комнатной температуры (20 С), промывку несколько раз углеводородом, например пентаном, гексаном или гептаном, и затем сушку под вакуумом и/или при повышенной температуре, составляющей 30-100 С, предпочтительно 40-90 С, наиболее предпочтительно 50-80 С. Метод экстракции в аппарате Сокслета в целом хорошо известен из уровня техники. В данном случае авторы изобретения взяли предкатализатор и поместили его на пористую стеклообразную фритту. Фритту загружали в главную камеру экстрактора Сокслета. Экстрактор Сокслета помещали на колбу,-3 019316 содержащую растворитель для экстракции, в данном случае TiCl4 и этилбензол. Затем экстрактор Сокслета оборудовали холодильником. Растворитель нагревали до начала орошения. Пары растворителя перемещались вверх в зону дистилляции и заливались в камеру, в которой помещен твердый материал фритты. Холодильник обеспечивает охлаждение паров растворителя и их стекание вниз в стеклянную камеру с рубашкой, в которой помещается твердый материал, температуру которого поддерживают примерно при 100-135 С, наиболее предпочтительно от 120 до 130 С. Камера, содержащая предкатализатор,медленно заполняется теплым растворителем. Любые примеси в предкатализаторе будут тогда растворяться в теплом растворителе и стекать вниз в камеру нагрева, оставляя после себя катализатор. Другие менее предпочтительные методы извлечения примесей из предкатализатора включают, но не ограничиваются этим, стадии промывки смесью органического растворителя и TiCl4 при температуре по меньшей мере 100 С, предпочтительно 100-135 С, наиболее предпочтительно 120-130 С. Органический растворитель может представлять собой алифатический или ароматический углеводород, предпочтительно ароматический углеводород, наиболее предпочтительно этилбензол. В то время как данное описание указывает только на метод экстракции в аппарате Сокслета, авторы изобретения отмечают, что настоящее изобретение действительно с любым методом экстракции, в котором используются органический растворитель и переходный металл в растворе. Сферический (MgCl2-xROH)-носитель является четко определенным, где R представляет собой один или несколько компонентов из нижеследующего (при условии, что общие моли в сумме дают "х"): линейного, циклического или разветвленного углеводорода с 1-10 атомами углерода, и где ROH представляет собой спирт или смесь по меньшей мере двух различных спиртов, предпочтительно где ROH представляет собой этанол или смесь этанола и высшего спирта, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 3-10 атомами углерода, такого как пропанол,бутанол, гексанол, гептанол или октанол, предпочтительно с 4-10 атомами углерода, такого как бутанол,гексанол, гептанол или октанол; и где х находится в диапазоне примерно от 1,5 до 6,0, предпочтительно примерно от 2,5 до 4, более предпочтительно примерно от 2,9 до 3,4 и еще более предпочтительно от 2,95 до 3,35. Если ROH представляет собой смесь этанола и высшего спирта, молярное отношение этанол:высший спирт составляет по меньшей мере 80:20, предпочтительно 90:10, наиболее предпочтительно 95:5. Внутренний донор электронов, на который ссылаются в процедуре, в типичном случае представляет собой основание Льюиса. Подходящие доноры электронов включают сложные диэфиры, простые диэфиры и сукцинаты. Предпочтительные соединения внутренних доноров включают производные карбоновой кислоты и, в частности, производные фталевой кислоты, имеющие общую формулу в которой X и Y, каждый, представляют атом хлора или брома или С 1-С 10-алкоксигруппу или X иY, взятые вместе, представляют атом кислорода, образуя ангидридную функцию. Особенно предпочтительными соединениями внутренних доноров электронов являются сложные эфиры фталевой кислоты формулы (I), в которой X и Y, каждый, представляют собой С 1-С 8-алкоксигруппу, такую как метокси-,этокси-, н-пропокси-, изопропокси-, н-бутокси-, втор-бутокси- или трет-бутоксигруппу. Примеры предпочтительных сложных эфиров фталевой кислоты включают диэтилфталат, ди-н-бутилфталат, диизобутилфталат, ди-н-пентилфталат, ди-н-гексилфталат, (н-пропил)(н-гептил)фталат, (изопропил)(нгептил)фталат, ди-н-гептилфталат, ди-н-октилфталат, динонилфталат, диизононилфталат или ди-2 этилгексилфталат. Дальнейшие примеры предпочтительных соединений внутренних доноров электронов включают сложные диэфиры 3- или 4-членных, необязательно замещенных циклоалкан-1,2-дикарбоновых кислот, а также сложные моноэфиры замещенных бензофенон-2-карбоновых кислот или замещенных бензофенон 3-карбоновых кислот. В качестве гидроксисоединений в реакции этерификации для синтеза данных сложных эфиров используют обычные алканолы, такие как С 1-С 15- или С 5-С 7-циклоалканолы, причем алканолы могут быть необязательно замещены одной или несколькими С 1-С 8-алкильными группами, а также С 1-С 10-фенолы. Дальнейшей группой подходящих внутренних доноров электронов являются незамещенные и замещенные (С 1-С 10-алкил)-1,3-пропан диэфиры и производные группы сукцинатов. Предпочтительно донор электронов представляет собой сложный диэфир, такой как диизобутилфталат (D-i-BP), ди-н-бутилфталат (D-n-BP), (н-пропил)(н-гептил)фталат, (изопропил)(н-гептил)фталат,диизооктилфталат, ди-2-этилгексилфталат и диизононилфталат. Кроме того, при приготовлении твердого каталитического компонента по изобретению можно использовать смеси двух или более соединений внутренних доноров электронов. При использовании для приготовления дисперсного твердого компонента соединение внутреннего донора электронов, как правило, используют в количестве примерно от 0,01 до 2 моль, предпочтительно примерно от 0,04 до 0,6 моль, более предпочтительно примерно от 0,05 до 0,2 моль для каждого 1 моль соединения галогенида магния. Каталитическая система Каталитические системы по изобретению, кроме твердого каталитического компонента, дополнительно включают по меньшей мере одно соединение алюминия в качестве сокатализатора. Кроме соединения(ий) алюминия каталитическая система по изобретению предпочтительно включает по меньшей мере одно соединение внешнего донора электронов. Примеры подходящих соединений алюминия включают триалкилалюминий и его производные, где алкильная группа замещена алкоксигруппой или атомом галогена, например атомом хлора или брома. Алкильные группы могут быть одинаковыми или различными. Алкильные группы могут представлять собой алкильные группы с прямой или разветвленной цепью. Предпочтительными соединениями триалкилалюминия являются соединения, в которых каждая алкильная группа имеет от 1 до 8 атомов углерода, такие как триметилалюминий, триэтилалюминий, триизобутилалюминий, триоктилалюминий или метилдиэтилалюминий. Примеры соединений-внешних доноров электронов, которые можно использовать в каталитической системе по изобретению, включают моно- и полифункциональные карбоновые кислоты, ангидриды карбоновых кислот и сложные эфиры карбоновых кислот, кетоны, простые эфиры, спирты, лактоны, а также органические соединения фосфора и кремния. Кроме того, можно использовать смесь двух или более соединений внешних доноров электронов. Соединение-внешний донор электронов и соединениевнутренний донор электронов, используемые при приготовлении твердого каталитического компонента,могут быть одинаковыми или различными. Предпочтительными соединениями-внешними донорами электронов являются кремнийорганические соединения общей формулы (II) в которой каждый из R1, которые могут быть одинаковыми или различными, представляет С 1-С 20 алкильную группу, 5-7-членную циклическую алкильную группу, необязательно замещенную С 1-С 10 алкильной, С 6-С 18-арильной группой или С 6-С 18-арильной-С 1-С 10-алкильной группой, R2 могут быть одинаковыми или различными и представляют собой С 1-С 20-алкильную группу и n равно целому числу 1, 2 или 3. Предпочтительными соединениями формулы (II) являются диизопропилдиметоксисилан, изобутилизопропилдиметоксисилан, диизобутилдиметоксисилан, дициклопентилдиметоксисилан, циклогексилметилдиметоксисилан, дициклогексилдиметоксисилан, изопропил-трет-бутилдиметоксисилан, изопропил-втор-бутилдиметоксисилан и изобутил-втор-бутилдиметоксисилан. Приготовление каталитической системы. Для приготовления каталитической системы по изобретению можно осуществить контакт соединения алюминия в качестве сокатализатора и соединения-внешнего донора электронов, по отдельности в любом порядке или смешанными вместе, с твердым каталитическим компонентом, обычно при температуре в диапазоне примерно от 0 до 200 С, предпочтительно примерно от 20 до 90 С и давлении примерно от 1 (105 Па) до 100 бар (107 Па), в частности примерно от 1 (105 Па) до 40 бар (4106 Па). Предпочтительно сокатализатор на основе соединения алюминия добавляют в таком количестве,что атомное отношение соединения алюминия к переходному металлу твердого каталитического компонента составляет примерно от 10:1 до 800:1, в частности примерно от 20:1 до 200:1. Каталитические системы по изобретению можно преимущественно использовать при полимеризации алк-1-енов. Подходящие алк-1-ены включают линейные или разветвленные С 2-С 10-алкены, в частности линейные С 2-С 10-алк-1-ены, такие как этилен, пропилен, бут-1-ен, пент-1-ен, гекс-1-ен, гепт-1-ен,окт-1-ен, нон-1-ен, дец-1-ен или 4-метилпент-1-ен. Также можно полимеризовать смеси данных алк-1 енов. Каталитические системы по изобретению, включающие твердые каталитические компоненты и в качестве сокатализатора соединение алюминия или соединение алюминия и соединение-внешний донор электронов, являются превосходными каталитическими системами для использования при производстве пропиленовых полимеров, как гомополимеров пропилена, так и сополимеров пропилена, и одного или нескольких дополнительных алк-1-енов, содержащих вплоть до 10 атомов углерода. Используемый здесь термин "сополимеры" также относится к сополимерам, в которые случайным образом введен дополнительный алк-1-ен, содержащий вплоть до 10 атомов углерода. В данных сополимерах, как правило, содержание сомономера составляет менее чем примерно 15 мас.%. Сополимеры также могут быть в форме так называемых блок- или ударопрочных сополимеров, которые обычно включают, по меньшей мере,матрицу гомополимера пропилена или статистический сополимер пропилена, содержащий менее чем 15 мас.% дополнительного алк-1-ена, имеющего вплоть до 10 атомов углерода, и мягкую фазу сополимера пропилена (эластичную фазу), содержащую от 15 до 80 мас.% дополнительных алк-1-енов, содержащих вплоть до 10 атомов углерода. Кроме того, рассматриваются смеси сомономеров, приводящие, например,к тройным сополимерам пропилена. Полимеризация. Получение полимеров пропилена можно осуществить в любом обычном реакторе, подходящем для полимеризации алк-1-енов, в периодическом или предпочтительно в непрерывном режиме, т.е. в растворе, в виде суспензионной полимеризации, включая полимеризацию в объеме в жидком мономере, или в виде газофазной полимеризации. Примеры подходящих реакторов включают непрерывно действующие реакторы с мешалкой, петлевые реакторы, реакторы с псевдоожиженным слоем или горизонтальные либо вертикальные перемешиваемые реакторы с порошковым слоем. Понятно, что полимеризацию можно осуществить в серии последовательно соединенных реакторов. Время реакции зависит от выбранных реакционных условий. Как правило, время реакции составляет примерно от 0,2 до 20 ч, обычно примерно от 0,5 до 10 ч, наиболее предпочтительно от 0,5 до 2 ч. Как правило, полимеризацию осуществляют при температуре в диапазоне примерно от 20 до 150 С,предпочтительно примерно от 50 до 120 С и более предпочтительно примерно от 60 до 95 С и давлении в диапазоне примерно от 1 (105 Па) до 100 бар (107 Па), предпочтительно примерно от 15 (1,5106 Па) до 50 бар (5106 Па) и более предпочтительно примерно от 20 (2106 Па) до 45 (4,5106 Па) бар. Молекулярную массу полученных в результате полимеров можно контролировать и регулировать в широком интервале, добавляя регулятор степени полимеризации полимера или регулятор молекулярной массы, обычно используемый при полимеризации, такой как водород. Кроме того, можно добавить инертный растворитель, такой как толуол или гексан, или инертный газ, такой как азот или аргон, или меньшие количества порошкообразного полимера, например полипропиленового порошка. Средние (среднемассовые) молекулярные массы полимеров пропилена, полученных с использованием каталитической системы по изобретению, как правило, находятся в диапазоне примерно от 10000 до 2000000 г/моль и скорости течения расплава находятся в диапазоне примерно от 0,01 до 2000 г/10 мин, предпочтительно примерно от 0,1 до 100 г/10 мин. Скорость течения расплава соответствует количеству, которое выдавливается в течение 10 мин из тестирующего инструмента в соответствии с ISO 1133 при температуре 230 С и при нагрузке 2,16 кг. Определенные области использования могут требовать молекулярных масс, отличных от указанных выше, и они, как предполагается, включаются в объем защиты изобретения. Каталитические системы по изобретению дают возможность полимеризовать алк-1-ены, давая полимеры, имеющие хорошую морфологию и высокую объемную плотность по сравнению с каталитическими системами предшествующего уровня техники. Кроме того, каталитические системы по изобретению показывают резкое увеличение производительности. Благодаря своим хорошим механическим свойствам полимеры, получаемые с использованием каталитической системы, включающей в себя твердые каталитические компоненты по изобретению, и, в частности, гомополимеры пропилена или сополимеры пропилена с одним или несколькими дополнительными алк-1-енами, содержащими вплоть до 10 атомов углерода, могут быть использованы предпочтительно для получения пленок, волокон или формованных изделий, в частности для получения пленок. Твердые каталитические компоненты, каталитические системы и полимеры, полученные в нижеприведенных примерах, были охарактеризованы посредством осуществления следующих тестов. Экспериментальный раздел. Синтез катализатора. Несколько образцов катализаторов по настоящему изобретению были получены и испытаны, чтобы продемонстрировать улучшенную активность и определить физические характеристики типичных вариантов осуществления катализатора. Следующее ниже описание типичных вариантов осуществления не имеет намерения каким-либо образом ограничивать объем изобретения. Фиг. 1 показывает оборудование, используемое для получения предкатализатора. Реакционный сосуд (10) включает реакционную камеру (12) и рубашку (14). Рубашка включает впускной канал (16) и выпускной канал (18). Для поддержания выбранной температуры в реакционной камере текучую среду при желательной температуре закачивают в рубашку через впускной канал, она протекает вокруг реакционной камеры и выходит через выпускной канал. Двигатель (20) приводит в действие мешалку (22) внутри реакционной камеры (12). Обратный холодильник (24) обеспечивают источником для продувки азотом (26). Имеется выпускное отверстие (28), чтобы удалять реакционный продукт из реакционной камеры. Имеется дополнительное отверстие (30) с заглушкой (32), чтобы дать возможность добавлять компоненты в реакционную камеру. Далее приводится общая процедура, используемая для получения катализаторов, описанных ниже. Один или несколько параметров варьировали для каждой композиции катализатора, как описано ниже. Описанную процедуру использовали, чтобы создать иллюстративные примеры катализатора по настоящему изобретению, и ее не следует рассматривать в качестве ограничивающей объем изобретения. Катализаторы изготавливали, используя носитель с размером частиц 40 мкм, носитель с размером частиц 60 мкм и носитель с размером частиц 90 мкм на основе MgCl2-xEtOH (см. столбец 2 табл. 2 для информации по конкретному носителю, использованному в конкретной серии). Если не указано иным образом, х в"xEtOH" для 90 мкм составляет 3,2, а для 60 и 40 мкм он составляет 3,1 (см. табл. 1). Сферические носи-6 019316 тели получали от коммерческого поставщика, но также их можно было бы изготовить, как подробно описано в разделе "Уровень техники". Для каждой композиции катализатора сферический MgCl2-носитель (суспендированный в этилбензоле или изопаре-Н, как отмечено в табл. 2) или для альтернативного варианта осуществления TiCl4 в начале загружали в снабженный рубашкой стеклянный реактор (10). Молярное отношение Ti/Mg, используемое в каждой партии катализаторов, дается в столбце 3 табл. 2. Молярное отношение Ti/Mg предпочтительно составляет от 1:100 до 1:5, предпочтительно от 1:50 до 1:8 и наиболее предпочтительно от 1:25 до 1:9. В то время как действительные количества начальных загрузок слегка различались для каждой серии приготовления катализатора, начальная загрузка в целом основывалась на использовании приблизительно 10 г MgCl2-xEtOH (эквивалент 4 г MgCl2). Молярное отношение D-n-BP (ди-н-бутилфталата) илиD-i-ВР (диизобутилфталата) к Mg показано в столбце 8 табл. 2. Молярное отношение DBP/Mg составляет предпочтительно от 0,05 до 3,0, более предпочтительно от 0,1 до 0,2 и еще более предпочтительно примерно от 0,12 до 0,15. Температуру в рубашке понижали до уровня ниже, чем в окружающей среде, что составляло приблизительно +10 С, если не указано иным образом в столбце 5 табл. 2. Затем объединяли MgCl2-xEtOH и TiCl4. Порядок объединения показан в табл. 2, столбцы 4 и 5. Компонент, внесенный в столбец 4, является первым компонентом процедуры, т.е. данный компонент помещают в стеклянный реактор с рубашкой. По причинам безопасности чистый TiCl4 добавляли к твердому MgCl2; вместо этого MgCl2 суспендировали в инертном растворителе, например гептане или изопаре-Н, и затем добавляли. Суспензионная среда (когда она используется) также показана в столбце 4. Компонент в столбце 5 показывает добавленный компонент и время добавления компонента. Если температура появляется в любом из столбцов, она представляет собой температуру данного раствора во время добавления. Температура первого компонента в стеклянном реакторе с рубашкой всегда была 0 С,если не указано иным образом в столбце 4. Температура второго компонента (добавляемого компонента) всегда была комнатной, если не указано иным образом в столбце 5. Добавление осуществляли медленно, чтобы позволить смеси взаимодействовать, обычно в течение периода примерно от 5 с до 300 мин, более предпочтительно примерно от 10 с до 90 мин, еще более предпочтительно примерно от 9 до 45 мин. Время реакции до нагревания указано в столбце 5. Например,в партии катализатора 56 жидкий TiCl4 при 22 С добавляют к комплексу MgCl2-xEtOH, суспендированному в этилбензоле, который находился при температуре 0 С, в течение периода, составляющего 20 мин. В качестве другого примера в партии катализатора 58 MgCl2-xEtOH, суспендированный в этилбензоле и охлажденный до 0 С, добавляли в течение 10 с к жидкому TiCl4, температура которого составляла 0 С. Затем при продувке азотом добавляли второй компонент, т.е. TiCl4 к MgCl2, или наоборот. После предоставления нескольких минут для взаимодействия TiCl4 с MgCl2-носителем, которые указаны как"время добавления" в столбце 5, температуру увеличивали со скоростью примерно 1 С в 1 мин до 50 С. При 50 С добавляли ди-н-бутилфталат (D-n-BP) или диизобутилфталат (D-i-ВР), как указано в столбце 8. Объединенный раствор выдерживали при 50 С в течение времени выдерживания, указанного в столбце 7. Затем температуру рубашки увеличивали приблизительно до 105 С и выдерживали при данной температуре примерно от 1 до 2 ч, причем действительное время указано в столбце 9. После 2 ч при 105 С содержимое реактора переносили в экстрактор Сокслета, фильтровали, пока оно все еще было горячим, и затем промывали гептаном. Как показано на фиг. 2, экстрактор Сокслета использовали для активации катализатора. Экстрагирующее устройство включает первый сосуд (40) с главной камерой (42) и рубашкой (44). Рубашка включает впускной канал (46) и выпускной канал (48). Для поддержания выбранной температуры в главной камере текучую среду при желаемой температуре закачивают в рубашку через впускной канал, она протекает вокруг камеры реактора и выходит через выпускной канал. Двигатель (50) приводит в действие мешалку (52) внутри главной камеры (42). Обратный холодильник (54) обеспечивали источником для продувки азотом (56). Имеется выпускное отверстие(58), чтобы обеспечить возможность удаления текучей среды из главной камеры. Имеется отверстие для ввода (60) с заглушкой (62), чтобы дать возможность добавлять реакционный продукт, представляющий собой предкатализатор, в реакционную камеру. В нижней части главной камеры имеется фильтр (72), чтобы удерживать твердый материал предкатализатора. В нижней части реакционной камеры (42) имеется канал (64), который обеспечивает путь течения в экстракционный сосуд (66). Имеется заглушка (68) для контроля потока из реакционной камеры в экстракционный сосуд. Экстракционный сосуд располагается внутри колбонагревателя (70), который используют для нагревания растворителя (74) в экстракционном сосуде до температуры флегмообразования. Пары растворителя перемещаются через линию дистилляции (76) в главную камеру (42). По мере того как теплый растворитель заполняет главную камеру (42), канал (64) открывается, чтобы дать возможность растворителю, содержащему катализатор, снова слиться в экстракционный сосуд. Каждый из предкатализаторов подвергали экстракции в аппарате Сокслета при 125 С в течение 2 ч, используя 90/10 объемную смесь этилбензола и TiCl4. После экстракции катализатор промывали гептаном и сушили под вакуумом. Любые отклонения от вышеуказанной процедуры отмечаются, как указано ниже. Катализатор 63. D-n-BP добавляли при 85 С, а не при 50 С. Катализатор 101. Часть EtOH сначала удаляли из MgCl2-носителя вакуумной сушкой носителя при 55 С. Образец носителя, т.е. 0,5 г, передавали на ТГА-анализ, который показывает, что на носителе оставалось 2,84 экв. EtOH (=MgCl2-2,84EtOH). Катализатор 105. MgCl2-носитель подвергали деалкоголизации таким же способом, как и катализатор 101, и далее подвергали деалкоголизации, увеличивая температуру масляной бани до 70 С и прикладывая к образцу вакуум. Остаточное содержание спирта до взаимодействия с TiCl4 уменьшали до 2,4 экв.(=MgCl2-2,4EtOH). Катализатор 107. MgCl2-носитель подвергали деалкоголизации таким же способом, как и катализатор 105. Катализатор 116. Вместо термического удаления EtOH из MgCl2 катализатора часть EtOH удаляли взаимодействием с SiCl4. Допуская, что на каждый эквивалент SiCl4 удаляется 1 экв. EtOH, добавляли достаточное количество SiCl4, чтобы уменьшить содержание EtOH от 3,1 до 2,6 экв. (=MgCl2-2,6EtOH). После добавления SiCl4 при 0 С реакционную смесь медленно нагревали до 50 С. При температуре от 25 до 50 С реакционная среда изменилась от хорошо суспендированного твердого вещества, которое выглядело в виде молокообразной жидкости, до взмученного твердого вещества, которое не было суспендированным. Образец отобрали для микрофотографии, которая обнаружила, что сферы MgCl2-носителя слились в "гроздевидные" кластеры. После охлаждения до комнатной температуры жидкость декантировали, твердое вещество промывали гептаном, повторно суспендировали в изопаре-Н и охлаждали до 0 С перед добавлением TiCl4, который также охлаждали до 0 С. После добавления TiCl4 реакционной смеси давали возможность отстояться в течение выходных при комнатной температуре. Катализатор 127. MgCl2-3,1 EtOH (10,26 г) суспендировали в изопаре-Н (200 мл) и охлаждали до-3 С. TiCl4 (50 мл) разбавляли 150 мл изопара-Н и затем охлаждали до 0 С льдом. TiCl4 медленно добавляли к MgCl2 в течение 44 мин. После завершения добавления TiCl4 температуру поддерживали постоянной в течение 10 мин и затем медленно увеличивали до 50 С в течение 100 мин. При 50 С медленно добавляли DBP. Затем раствору дали возможность охладиться до комнатной температуры и продукт суспендировали в течение ночи. На следующий день температуру повышали до 50 С, выдерживали при данной температуре в течение 120 мин, затем температуру повышали до 105 С и выдерживали при данной температуре в течение 90 мин. Затем "предкатализатор" перемещали в активационный сосуд и подвергали экстракции в сосуде Сокслета, как обычно. Катализатор дважды тестировали при условиях полимеризации в объеме, и он показал очень плохую активность. Оказалось, что прерывание процедуры приготовления в течение ночи после добавления DBP является очень вредным для рабочих характеристик катализатора. Катализатор 137. Повторяли процедуру, используемую для приготовления катализатора 127, однако вместо прекращения приготовления в течение вечера после добавления DBP реакционную смесь выдерживали в течение ночи после добавления TiCl4 при комнатной температуре. На следующий день реакционную смесь нагревали до 50 С и добавляли D-n-BP. Остальная часть приготовления катализатора была такой же, как описанная для катализатора 127. Катализаторы 20, 103 и 10740 готовили, используя 40-микронный MgCl2-3,1 EtOH в качестве сферического MgCl2-носителя. Катализатор 20. Такую же процедуру использовали для катализатора 78 (которая следовала стандартной процедуре, описанной выше), за исключением того, что для катализатора 20 использовали 40 микронный MgCl2-3,1 EtOH-носитель, а для катализатора 78 использовали 90-микронный MgCl2-3,2EtOH. MgCl2-3,1 EtOH медленно добавляли к охлажденному TiCl4. Оказалось, что морфология катализатора была очень хорошей, но активность, хотя и хорошая, была слегка ниже, чем у аналогичных катализаторов, изготовленных на 60- и 90-микронных носителях. Цвет катализатора является бледножелтовато-зеленым. Катализатор 103. В данной процедуре целью являлось добавить 40-микронный MgCl2-3,1EtOH/изопар-Н к охлажденному TiCl4 в контролируемой манере и минимизировать количество используемого изопара-Н. 20 г 40-микронного MgCl2-3,1 EtOH объединяли с 23 мл изопара-Н. Суспензия была слишком густой, поэтому добавляли дополнительные 27 мл изопара-Н. После этого оказалось, что суспензия все еще густая, но поддающаяся обработке. В 250-мл колбу добавляли другие 20 г носителя и еще 50 мл изопара-Н. Шприц, оборудованный иглой 12 размера, использовали, чтобы перенести суспензию. В сосуд предкатализатора добавляли 200 мл TiCl4 и затем охлаждали до -5 С. Суспензию MgCl2-3,1EtOH/изопар-Н охлаждали до 0 С, но она была слишком густой, чтобы переносить ее посредством иглы шприца, поэтому использовали 1/8-дюймовую пластмассовую трубку, чтобы по каплям перенести суспензию MgCl2-3,1 EtOH/изопар-Н в сосуд предкатализатора. Данный катализатор показал очень хорошую активность в условиях полимеризации в объеме (56,1 кг ПП/г-кат. час) и газофазной полимеризации(32,2 кг ПП/г-кат. час). Полученный в результате катализатор имел цвет от желтого до бледно-желтого. Катализатор 10740. В данном примере следовали таким же молярным отношениям, как и для катализатора 103. Разница состояла в том, что изопар-Н заменяли гептаном. В реактор предкатализатора загружали 200 мл TiCl4 и 90 мл гептана и затем смесь охлаждали до -3 С. Гептан использовали, чтобы уменьшить вязкость реакционной среды. К перемешиваемой смеси TiCl4/гептан медленно добавляли 40 г 40-микронного MgCl2-3,1 EtOH. В состав системы включали амперметр, чтобы контролировать ток, протекающий через двигатель мешалки. Начальные показания амперметра составляли 0,026. К тому моменту, когда весь MgCl2-3,1 EtOH был добавлен, температура реакционной смеси возросла на 2 С, от -3 до-1 С и сила тока на двигатель вследствие загустевания реакционной смеси выросла от 0,026 до 0,034. По мере того как заданную величину температуры рубашки реактора увеличивали от -5 до 0 С, затем до 5 и затем до 10 С, сила тока (амперы) на мешалке увеличилась вначале до 0,038, затем до 0,039 и затем упала до 0,029. Данный катализатор давал еще более высокую активность, чем катализатор 103. Цвет конечного катализатора был желтый. Таблица 2 Изменения параметров для изготовления катализаторов по изобретению Для целей сравнения катализаторы Циглера-Натта, которые были изготовлены с использованием такого же 40-микронного (катализатор сравнения 40), 60-микронного (катализатор сравнения 60) или 90 микронного MgCl2-xEtOH-носителя (катализатор сравнения 90), были получены способом, аналогичным описанному в патентах США 4829034 и 5137856. Вместо проведения экстракции в аппарате Сокслета предкатализатор обрабатывали второй раз при 105 С в течение 2 ч, используя чистый TiCl4, отфильтровывали и затем промывали гептаном. Катализатор сравнения 40 имел 40-микронный носитель. Катализатор сравнения 60 имел 60-микронный носитель. Катализатор сравнения 90 имел 90-микронный носитель. Параметры для данных примеров сравнения показаны в табл. 3 и результаты полимеризации показаны в табл. 4 и 5. Таблица 3 Тестирование полимеризации. Характеристики катализатора тестировали при условиях полимеризации в объеме и условиях газофазной полимеризации. Тестирование полимеризации в объеме проводили, используя 5-литровый реактор, 1800 г пропилена, 2,0 мл 0,1 М циклогексилметилдиметоксисилана, 7,0 мл 1,6 М триэтилалюминияTEAl и силан предварительно смешивали и затем нагнетали в реактор, используя 900 г пропилена. В качестве последнего компонента добавляли 0,02 г катализатора с использованием оставшихся 900 г пропилена. Затем реактор быстро нагревали до 70 С, обычно в течение 2-3 мин, и давали возможность про- 10019316 текать полимеризации в течение 1 ч. Пропилен остается в жидкой фазе на протяжении всего процесса нагрева. Реактор стендового масштаба, который также можно приспособить для газофазной полимеризации,оборудовали мешалкой. При газофазных условиях порядок добавления был такой же, но загрузки пропилена уменьшали по размеру до 200 г. Аналогично, загрузки TEAl и силана уменьшали на 1/3-1/2, а водород уменьшали на 1/5-1/10 относительно загрузки, используемой при полимеризации в объеме. Катализатор вводили при 40 С и реактор программировали для нагрева до 75 С в течение 10 мин. Газофазные условия поддерживали, контролируя введение пропилена в систему. По мере нагрева системы до конечной температуры пропилен добавляли с такой скоростью, чтобы обеспечить давление в реакционном сосуде таким, что пропилен всегда остается в газовой фазе. Для обеспечения газофазных условий давление в реакторе поддерживали при 400 фунт/кв.дюйм изб. (2,76 кПа изб.), при 75 С, причем газообразный пропилен добавляли через расходомер по мере необходимости. Физические характеристики полипропиленовых полимеров, полученных с использованием различных катализаторов, определяли, используя тесты, описанные ниже. Результаты, полученные из данных тестов, суммируются в табл. 6 и 7. Активность. Результаты по активности, сообщенные на протяжении всего данного исследования, основаны на выходе полимера в граммах, разделенном на массу катализатора, загруженного в реактор в граммах для 1-часовой полимеризации. Авторы изобретения определили, что не было необходимости учитывать потерю активности вследствие остаточного растворителя, захваченного в катализаторах по изобретению,по сравнению с катализатором сравнения, поскольку захваченное количество было фактически равным,как и можно было бы ожидать, поскольку катализаторы были изготовлены с использованием одного и того же носителя. Активности, сообщенные на протяжении всего данного описания, не скорректированы по захваченным растворителям. Таким образом, активность основывается на граммах полимера/грамм катализатора+ растворитель. Чтобы определить активность, основанную только на сухом катализаторе, на нескольких катализаторах проводили термогравиметрический анализ (ТГА). ТГА проводили, помещая 10 мг катализатора в анализатор SDT Q600. Температуру повышали со скоростью 10,0 С/мин до 300 С и массу катализатора непрерывно записывали в виде функции увеличения температуры. Потеря массы от 0 до 190 С соответствует потере остаточного растворителя. Растворимость в ксилоле (мас.% XS). Растворимость в ксилоле измеряли, используя метод проточно-инжекционного анализа полимеров(FIPA) компании Viscotek, который хорошо известен в промышленности. Viscotek опубликовал статью,озаглавленную "FIPA for xylene soluble determination of polypropylene and impact copolymers" (которую можно заказать с веб-сайта Viscotek, http://www.viscotek.com/applications.aspx), демонстрирующую, что метод Viscotek FIPA дает корреляцию 0,994 r2 с методом ASTM D5492-06 (эквивалент ISO 16152) в диапазоне величин растворимой в ксилоле фракции от 0,3 до 20%. Поэтому специалист в данной области смог бы воспроизвести результаты настоящего изобретения, используя метод Viscotek FIPA или методASTM D54 92-06. Массовый процент растворимой в ксилоле фракции в полипропилене является показателем стереорегулирующей способности катализатора - чем выше мас.% XS, тем хуже стереорегулирующая способность катализатора. Измерение скорости течения расплава (MFR). Эффект скорости течения расплава измеряли, используя метод ASTM D 1238-04. На каждые 5 г образца полимера добавляли 0,2 г комплекта стабилизирующих добавок. Комплект добавок состоит из 50 мас.% Irganox 1010 и 50 мас.% Irgafos 168. Поскольку полимер подвергается воздействию воздуха при температуре 230 С в течение нескольких минут в течение испытания, данный комплект добавляют, чтобы ингибировать термическое и окислительное разрушение полимера. Скорость течения расплава дает информацию, касающуюся молекулярной массы и водородного отклика полимера. Чем выше MFR, тем выше скорость отклика на водород для катализатора, производящего полиолефин. Аналогичным образом, чем выше MFR, тем ниже молекулярная масса полимера. Анализ размера частиц. Распределение частиц по размерам определяли в двух стадиях. Сначала, поскольку подающий лоток Malvern Mastersizer оборудован предварительным ситом, которое не будет позволять проходить частицам полимера с размером более 2380 мкм, 10 г каждого образца предварительно просеивали через 2000-микронное сито и записывали мас.% материала с размером более 2000 мкм. Остающийся материал,который прошел через 2000-микронное сито, затем анализировали посредством Malvern Mastersizer. Объемный процент данного материала с размером меньше чем 190 мкм определяли как показатель фракции очень мелких частиц в полимере. Фракции очень мелких частиц с размером менее 190 мкм в катализаторе старались избежать, поскольку мелочь данного размера значительно снижает количество содержания этилена, которое может быть введено в полипропилен-полиэтиленовые сополимеры, полученные с использованием данного катализатора. Однако поскольку почти во всех случаях осуществления изобретения мелкие частицы, которые можно измерить, с размером менее 190 мкм отсутствовали, авторы также измеряли размер частиц менее 410 мкм просто для целей сравнения. Кроме того, определяли среднее распределение (D50) для частиц, остающихся после того, как частицы 2000-микронного размера были удалены. Микрофотографии. Микрофотографии материала носителя в таблицах-xEtOH, катализаторов и выбранных образцов полимеров получали, используя микроскоп Olympus SZX12, оборудованный камерой Diagnostic Instrument's Model 4.2, используя программный продукт Diagnostic Instrument's Spot Advanced - версию 3.5.9.1 для Windows. Фиг. 3 и 4 показывают микрофотографии при усилении 100 MgCl2-xEtOH материалов носителя и конечных катализаторов вместе с катализаторами Циглера-Натта сравнения, фиг. 5 показывает микрофотографии при усилении 11,2 типичной сферичности полимеров, полученных с помощью катализаторов и способа по данному изобретению. Табл. 2 включает качественные наблюдения морфологии катализатора. На данных чертежах демонстрируются примеры терминов "разрушенный", "гроздевидный" и т.д. Активность катализатора на основании испытания полимеризации. Табл. 4 и 5 суммируют результаты полимеризации в объеме и газофазной полимеризации соответственно, которые были получены с катализаторами по настоящему изобретению, описанными выше. Результаты для катализаторов сравнения даны в нижней части таблиц. Таблица 4 Результаты полимеризации в объеме Как суммировано в табл. 4 и 5, при условиях полимеризации в объеме типичные катализаторы по настоящему изобретению показывают существенно и неожиданно более высокую активность по сравнению с катализаторами сравнения. Табл. 6 сравнивает активность в объеме с активностью, полученной в газовой фазе. Расположение катализаторов по активности практически не изменилось. Наиболее активными катализаторами в объеме(89, 101 и 105) являлись 3 из 4 наиболее активных катализаторов в газовой фазе. Это демонстрирует превосходную стабильность катализаторов по изобретению в широком диапазоне условий полимеризации. Таблица 6 Активность в объемной фазе по сравнению с активностью в газовой фазе (носитель 60 мкм) Обычно специалист в данной области ожидал бы значительного увеличения фракции очень мелких частиц (частиц с размером менее 190 мкм) при значительном увеличении в активности, демонстрируемом катализаторами по изобретению. Однако табл. 7 показывает, что это не обнаруживается для катализаторов по изобретению. Табл. 7 содержит данные распределения частиц по размерам для полимеров,полученных как в объеме, так и газовой фазе, для трех наиболее активных катализаторов, испытанных при объемных условиях. Таблица 7 Данные распределения частиц по размерам для наиболее активных катализаторов В противоположность ожиданиям при тестировании в объемной фазе третий столбец табл. 7 демонстрирует, что наиболее активные катализаторы по изобретению действительно имеют тенденцию производить более значительные количества крупных частиц полиолефина (2000 мкм), чем катализатор сравнения 60. Помимо этого, наиболее активные катализаторы по изобретению не дают фракции очень мелких частиц с размером 190 мкм. Даже расширяя данный предел вплоть до частиц 410 мкм, катализаторы по изобретению дают очень мало дополнительных маленьких полиолефиновых частиц. Еще более впечатляюще, что при газофазном тестировании наиболее активные катализаторы обычно удваивали количество больших полиолефиновых частиц (2000 мкм), в то же время действительно не давая фракцию очень мелких частиц с размером 190 мкм (по сравнению с увеличением фракции очень мелких частиц 190 мкм для катализатора сравнения). Аналогичным образом, в газовой фазе три наиболее активных катализатора по изобретению давали некоторое количество маленьких частиц 410 мкм, которое составляло не более 1/3, и часто менее 1/20 количества, даваемого катализатором сравнения 60. Обычно специалист в данном уровне техники ожидает, что морфология полиолефина следует мор- 15019316 фологии катализатора при использовании катализаторов Циглера-Натта. То есть чем более растрескавшиеся частицы катализатора изготавливают, тем выше ожидаемое количество фракции очень мелких частиц и маленьких частиц в конечном полиолефиновом продукте. Неожиданно было обнаружено, что данная взаимосвязь не имеет силы для катализаторов по изобретению. Нет корреляции между наблюдаемыми результатами морфологии из табл. 2 и количеством полученной фракции очень мелких частиц, как демонстрируется в табл. 4 и 5. Специалисты в данном уровне техники поймут, что на основе приведенного здесь описания можно сделать многочисленные изменения и модификации вышеописанных и других вариантов осуществления изобретения без отклонения от его объема патентной защиты, определенной в прилагаемой формуле изобретения. Соответственно данное подробное описание предпочтительных вариантов осуществления необходимо рассматривать в иллюстративном, а не в ограничивающем смысле. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения катализатора Циглера-Натта для полимеризации олефинов, включающий стадии:a) объединения сферического, охлажденного распылением носителя MgCl2-xROH, где х имеет значение примерно от 1,5 до 6,0 и где ROH представляет собой спирт или смесь спиртов, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 1-10 атомами углерода, с соединением переходного металла в реакторе при температуре примерно от -30 до 40 С;b) нагревания смеси в реакторе до температуры от примерно 30 до примерно 100 С;c) одновременно с нагревом в стадии (b) или вслед за достижением температуры стадии (b) добавления внутреннего донора электронов к смеси в реакторе;d) нагревания полученной в результате смеси до температуры по меньшей мере примерно 80 С и выдерживания полученной в результате смеси при данной температуре примерно в течение времени от 1 до 2 ч, чтобы получить предкатализатор;e) фильтрования смеси, содержащей предкатализатор, с получением твердого компонента предкатализатора;f) экстрагирования предкатализатора смесью органического растворителя и соединения переходного металла при температуре по меньшей мере 100 С в течение 1-5 ч с получением катализатора;g) охлаждения катализатора до комнатной температуры (20 С), промывки катализатора углеводородным растворителем и сушки катализатора под вакуумом и/или при повышенной температуре, равной 30-100 С. 2. Способ по п.1, в котором ROH представляет собой смесь по меньшей мере двух различных спиртов, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 110 атомами углерода. 3. Способ по п.1, в котором ROH представляет собой этанол или смесь этанола и высшего спирта,причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 3-10 атомами углерода. 4. Способ по п.1, в котором ROH представляет собой этанол или смесь этанола и высшего спирта,причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 4-10 атомами углерода. 5. Способ по п.1, в котором ROH представляет собой смесь этанола и бутанола, гексанола, гептанола или октанола. 6. Способ по п.1, в котором ROH представляет собой этанол. 7. Способ по п.1, в котором х имеет значение в интервале примерно от 2,5 до 4,0. 8. Способ по п.1, в котором х имеет значение в интервале примерно от 2,95 до 3,35. 9. Способ по п.1, в котором на стадии (а) соединение переходного металла представляет собой TiCl4 и ROH представляет собой этанол или смесь этанола и высшего спирта, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 3-10 атомами углерода. 10. Способ по п.1, в котором на стадии (а) соединение переходного металла представляет собойTiCl4 и ROH представляет собой этанол. 11. Способ по п.1, в котором стадию экстракции (f) осуществляют, используя метод экстракции в аппарате Сокслета. 12. Способ по п.1, в котором температура реактора для комбинации MgCl2-xROH и соединения переходного металла имеет значение в интервале примерно от -10 до +10 С. 13. Способ по п.1, в котором соединение переходного металла, используемое на стадии (f), представляет собой TiCl4. 14. Способ по п.1, в котором внутренний донор электронов выбирают из группы, состоящей из сложных диэфиров, замещенных циклоалкан-1,2-дикарбоновых кислот, сложных моноэфиров замещенных бензофенон-2-карбоновых кислот, замещенных бензофенон-3-карбоновых кислот, незамещенных и замещенных простых диэфиров (C1-C10-алкил)-1,3-пропана, производных карбоновой кислоты, произ- 16019316 водных фталевой кислоты и производных сукцинатов. 15. Способ по п.1, в котором внутренний донор электронов выбирают из диизобутилфталата (D-iBP), ди-н-бутилфталата (D-n-ВР), диизооктилфталата, ди-2-этил,гексилфталата или диизононилфталата. 16. Способ по п.1, в котором смеси со стадии (а) дают возможность взаимодействовать в течение приблизительно от 2 до 300 мин перед обработкой нагреванием со стадии (b). 17. Способ по п.1, в котором полученную в результате смесь из стадии (d) нагревают до температуры от примерно 100 до примерно 135 С. 18. Способ по п.17, в котором полученную в результате смесь из стадии (d) выдерживают при температуре в течение от примерно 1 до примерно 4 ч. 19. Способ по п.1, в котором органический растворитель из стадии (f) представляет собой алифатический или ароматический углеводород. 20. Способ по п.1, в котором органический растворитель из стадии (f) представляет собой ароматический углеводород. 21. Способ по п.1, в котором органический растворитель из стадии (f) представляет собой этилбензол. 22. Способ по п.13, в котором органический растворитель представляет собой этилбензол, объемное отношение TiCl4 и этилбензола составляет примерно 30:70 и время экстракции составляет 1-5 ч при температуре по меньшей мере 100 С. 23. Способ по п.13, в котором органический растворитель представляет собой этилбензол, объемное отношение TiCl4 и этилбензола составляет примерно 20:80 и время экстракции составляет 1-4 ч при 100135 С. 24. Способ по п.13, в котором органический растворитель представляет собой этилбензол, объемное отношение TiCl4 к этилбензолу составляет примерно 10:90 и время экстракции составляет 1-3 ч при 120130 С. 25. Способ по п.1, в котором часть ROH удаляют из MgCl2-носителя деалкоголизацией перед взаимодействием с соединением переходного металла. 26. Способ по п.1, в котором на стадии (а) молярное отношение Mg к переходному металлу составляет от примерно 1:100 до примерно 1:5. 27. Способ по п.1, в котором на стадии (а) молярное отношение Mg к переходному металлу составляет от примерно 1:50 до примерно 1:8. 28. Способ по п.1, в котором на стадии (а) молярное отношение Mg к переходному металлу составляет от примерно 1:25 до примерно 1:9. 29. Способ по п.1, в котором внутренний донор электронов представляет собой диизобутилфталат(D-i-BP) или ди-н-бутилфталат (D-n-BP) и молярное отношение D-i-BP или D-n-BP/Mg составляет от примерно 0,12 до примерно 0,15. 30. Способ получения катализатора для полимеризации олефинов, включающий стадии:a) объединения сферического, охлажденного распылением носителя MgCl2-xEtOH, где х равен от примерно 3,0 до примерно 3,3 и EtOH представляет собой этанол, с TiCl4 в реакторе при температуре примерно от -10 до 10 С;b) нагревания смеси в реакторе до температуры примерно 40-90 С;d) нагревания полученной в результате смеси примерно до 100-110 С и выдерживания полученной в результате смеси при данной температуре примерно в течение времени от 1 до 2 ч, чтобы получить предкатализатор;e) фильтрования смеси, содержащей предкатализатор, для получения твердого компонента предкатализатора;f) экстрагирования предкатализатора с использованием экстракции в аппарате Сокслета смесью этилбензола в качестве органического растворителя и TiCl4 в качестве переходного металла при температуре 120-130 С в течение 1-3 ч для получения катализатора;g) промывки катализатора растворителем, содержащим гексан или гептан, и сушки катализатора под вакуумом и/или при повышенной температуре, равной 30-100 С. 31. Способ полимеризации олефинов формулы CH2=CHR1, в которой R1 представляет собой водород или углеводородный радикал, содержащий 1-12 атомов углерода, осуществляемый в присутствии катализатора, полученного способом по п.1 или 30. 32. Катализатор, полученный способом по п.1 или 30 и имеющий активность для полимеризации в объеме по полипропилену больше чем 60 кг полипропилена/г кат.1 ч. 33. Способ получения катализатора Циглера-Натта для полимеризации олефинов, включающий стадии:a) объединения сферического, охлажденного распылением носителя MgCl2-xROH, сформированного в отсутствие диоксида кремния в качестве затравки кристаллизации, где х имеет значение примерно от 1,5 до 6,0 и где ROH представляет собой спирт или смесь спиртов, причем R представляет собой звено линейного, циклического или разветвленного углеводорода с 1-10 атомами углерода, с соединением пе- 17019316 реходного металла в реакторе при температуре примерно от -30 до 40 С;b) нагревания смеси в реакторе до температуры от примерно 30 до примерно 100 С;c) одновременно с нагревом в стадии (b) или вслед за достижением температуры стадии (b) добавления внутреннего донора электронов к смеси в реакторе;d) нагревания полученной в результате смеси до температуры по меньшей мере примерно 80 С и выдерживания полученной в результате смеси при данной температуре примерно в течение времени от 1 до 2 ч, чтобы получить предкатализатор;e) фильтрования смеси, содержащей предкатализатор, с получением твердого компонента предкатализатора;f) экстрагирования предкатализатора смесью органического растворителя и переходного металла при температуре по меньшей мере 100 С в течение 1-5 ч с получением катализатора;g) охлаждения катализатора до комнатной температуры (20 С), промывки катализатора углеводородным растворителем и сушки катализатора под вакуумом и/или при повышенной температуре, равной 30-100 С. Микрофотографии MgCl-xEtOH носителей при увеличении 100 Х Микрофотографии катализаторов по изобретению и катализаторов сравнения при увеличении 100 Х Фиг. 4 Микрофотографии полимеров с использованием катализаторов по изобретению и катализаторов сравнения при увеличении 11,2 Х
МПК / Метки
МПК: B01J 27/138
Метки: способ, использование, получения, катализаторы, циглера-натта, катализаторов, высокоактивные
Код ссылки
<a href="https://eas.patents.su/23-19316-vysokoaktivnye-katalizatory-ciglera-natta-sposob-polucheniya-katalizatorov-i-ih-ispolzovanie.html" rel="bookmark" title="База патентов Евразийского Союза">Высокоактивные катализаторы циглера-натта, способ получения катализаторов и их использование</a>
Способ регулирования морфологии катализаторов циглера-натта

Номер патента: 18919
Опубликовано: 29.11.2013
Авторы: Коффи Тим, Грэй Стивен, Виззини Кайо, Энрикес Генри
МПК: B01J 31/00
Метки: регулирования, катализаторов, способ, морфологии, циглера-натта
Формула / Реферат:
1. Способ регулирования морфологии катализатора Циглера-Натта, имеющего регулируемый размер частиц и распределение частиц по размерам, включающий внесение изменений в процесс осаждения компонента катализатора из раствора для синтеза катализатора, содержащего растворимый предшественник катализатора и агент-осадитель, путем регулирования концентрации агента-осадителя, при этом средний размер частиц компонента катализатора возрастает и...
Способ производства полипропилена с использованием высокопродуктивных катализаторов циглера-натта

Номер патента: 11642
Опубликовано: 28.04.2009
Авторы: Вестберг Торвалд, Йааскелайнен Пирьо
МПК: C08F 10/06, C08F 4/654, C08F 4/651...
Метки: способ, использованием, производства, циглера-натта, катализаторов, полипропилена, высокопродуктивных
Формула / Реферат:
1. Полипропиленовая композиция, содержащая сополимер пропилена с модулем изгиба МИ, выраженным в МПа, в соответствии с отношением: МИ_1700-225Et, где Et - количество этилена в полимере в мас.% и находится в диапазоне 1_Et_3,5. 2. Полипропиленовая композиция по п.1, отличающаяся тем, что количество растворимых компонентов ксилола XS, выраженных в мас.%, соответствует отношению: XS_0,33 Et2+0,33Et+1, где Et - количество этилена в полимере в мас.%...
Катализаторы циглера-натта процессов полимеризации в растворе при высоких температурах

Номер патента: 5891
Опубликовано: 30.06.2005
Автор: Жабер Изам
МПК: C08F 210/02, C08F 4/646, C08F 2/06...
Метки: температурах, катализаторы, высоких, процессов, растворе, полимеризации, циглера-натта
Формула / Реферат:
1. Катализатор, состоящий, по существу, из смеси (i) соединения магния формулы (R2)2Mg, где R2 представляет собой C1-6-алкильный радикал; (ii) алкильного соединения алюминия формулы (R1)3-aAl1(OR5)a, где R1 и R5 независимо выбирают из группы, включающей C1-4-алкильные радикалы, a = 0; (iii) кремнийсодержащего соединения формулы где каждое A независимо выбирают из группы, включающей C1-10-алкильные радикалы; C6-12-арильные радикалы и...
Катализаторы циглера-натта с низким содержанием алюминия и магния для процессов полимеризации олефинов в растворе и способ полимеризации олефинов

Номер патента: 2590
Опубликовано: 27.06.2002
Автор: Джейбер Айсам
МПК: C08F 4/654
Метки: растворе, циглера-натта, низким, катализаторы, олефинов, способ, магния, содержанием, алюминия, полимеризации, процессов
Формула / Реферат:
1. Катализатор, состоящий, по существу, из (i) смеси соединения алкилалюминия с формулой (R1)3Аl и (R2)2Mg, где R1 представляет собой алкильный радикал С1-10 и R2 представляет собой алкильный радикал С1-10, с молярным соотношением Mg и Аl от 4,0:1 до 5,5:1. (ii) галогенида с формулой R3X, где R3 представляет собой алкильный радикал C1-8, a X является галогенидом, выбираемым из группы, состоящей из атомов хлора и брома; (iii) тетрахлорида титана;...
Способ получения катализатора циглера-натта для полимеризации альфа-олефинов в растворе

Номер патента: 2052
Опубликовано: 24.12.2001
Автор: Жабер Изам
МПК: C08F 2/06, B01J 37/00
Метки: способ, циглера-натта, растворе, получения, полимеризации, альфа-олефинов, катализатора
Формула / Реферат:
1. Способ получения катализатора для полимеризации в растворе смеси одного или более линейных С2-12 альфа-олефинов при температуре от 105 до 320шС и давлении от 4 до 20 мПа, содержащего: (i) смесь триалкилалюминиевого соединения формулы (R1)3Al1 и диалкилмагниевого соединения формулы (R2)2Мg, где R1 представляет собой С1-10алкил и R2 представляет собой С1-10алкил при молярном соотношении Мg к Al1 от 4:1 до 8:1; (ii) галогенид формулы R3X, где...
Предыдущий патент: Производные азетидинов, их получение и применение в терапии
Следующий патент: Перемешивающее устройство и способ смешивания газа и раствора
Случайный патент: Имидазопиразиновые ингибиторы syk, фармацевтическая композиция, содержащая имидазопиразиновые ингибиторы syk, и способы их применения