Производные арилсульфонилгидроксамовой кислоты





















Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемая соль, где пунктирная линия представляет необязательную двойную связь;
Х представляет углерод;
Y представляет углерод или серу;
R1, R2, R3, R4, R5, R6 и R9 означают водород;
R7 означает атом водорода, (С1-C6)алкил или -NHCOO(C1-6)алкил;
R8 означает атом водорода или (C1-6)алкил;
Q представляет (С6-С10)арил(С6-С10)арил или (С6-С10)арил(C1-C6)алкокси(С6-С10)арил необязательно замещенный фтором, хлором, (C1-C8)алкилом, (С6-С10)алкокси или перфтор(C1-3)алкилом;
при условии, что когда Y представляет серу, R5 и R6 отсутствуют;
при условии, что когда пунктирная линия представляет двойную связь, R4 и R6 отсутствуют.
2. Соединение по п.1, где Y представляет серу.
3. Соединение по п.1, где Q представляет (С6-С10)арил(C6-С10)арил, (С6-С10)арил(С1-C6)акокси(С6-С10)арил, где каждая терминальная арильная группа необязательно замещена фтором.
4. Соединение по п.1, где R7 представляет водород;
5. Соединение по п.1, где Y представляет углерод;
Q представляет (C6-C10)арил(C1-C6)алкокси(С6-С10)арил.
6. Соединение по п.1, где, по меньшей мере, один из R7-R8 представляет радикал, иной чем водород.
7. Соединение по п.3, где, по меньшей мере, один из R7-R8 представляет радикал, иной чем водород.
8. Соединение по п.1, где, по меньшей мере, один из R7-R8 представляет (C1-C6)алкил.
9. Соединение по п.3, где, по меньшей мере, один из R7-R8 представляет (C1-C6)алкил.
10. Соединение по п.1, где, по меньшей мере, один из R7-R8 представляет метил.
11. Соединение по п.3, где, по меньшей мере, один из R7-R8 представляет метил.
12. Соединение по п.1, где R7 и R8 каждый представляет метил.
13. Соединение по п.3, где R7 и R8 каждый представляет метил.
14. Соединение по п.1, представляющее соединение, выбранное из группы, состоящей из
изопропилового эфира (2R,3S)-(1-[4-(4-(фторбензилокси)бензолсульфонил]-2-гидроксикарбамоилпиперидин-3-ил)карбаминовой кислоты;
гидроксиамида 3-(S)-4-(4'-фторбифенил-4-сульфонил)-2,2-диметилтиоморфолин-3-карбоновой кислоты;
гидроксиамида 3-(S)-4-[4-(4'-фторбензилокси)бензолсульфонил]-2,2-диметилтиоморфолин-3-карбоновой кислоты.
15. Фармацевтическая композиция для (а) лечения состояния, выбранного из группы, состоящей из артрита, злокачественной опухоли, тканевого изъязвления, макулярной дегенерации, рестеноза, периодонтального заболевания, врожденного буллезного эпидермолиза, склерита, в сочетании со стандартными NSAID'S и анальгетиками и в сочетании с цитотоксическими противоопухолевыми агентами, и других заболеваний, характеризующихся металлопротеиназной активностью матрикса, AIDS, сепсисом, септическим шоком и другими заболеваниями, вовлеченными в образование фактора некроза опухолей (TNF) или для (b) ингибирования металлопротеиназ матрикса или образования фактора некроза опухолей (TNF) у млекопитающих, в том числе у человека, включающая соединение по п.1 в количестве, эффективном для такого лечения, и фармацевтически приемлемый носитель.
16. Способ ингибирования металлопротеиназ матрикса или образования фактора некроза опухолей (TNF) у млекопитающих, в том числе у человека, включающий введение указанному млекопитающему эффективного количества соединения по п.1.
17. Способ лечения состояния, выбранного из группы, состоящей из артрита, злокачественной опухоли, тканевого изъязвления, макулярной дегенерации, рестеноза, периодонтального заболевания, врожденного буллезного эпидермолиза, склерита, соединениями формулы I, котрые могут быть использованы в сочетании со стандартными NSAID'S и анальгетиками и в сочетании с цитотоксическими противоопухолевыми агентами, и других заболеваний, вовлеченных в образование фактора некроза опухолей (TNF) у млекопитающих, в том числе у человека, включающий введение указанному млекопитающему соединения по п.1 в количестве, эффективном для лечения такого состояния.
Текст
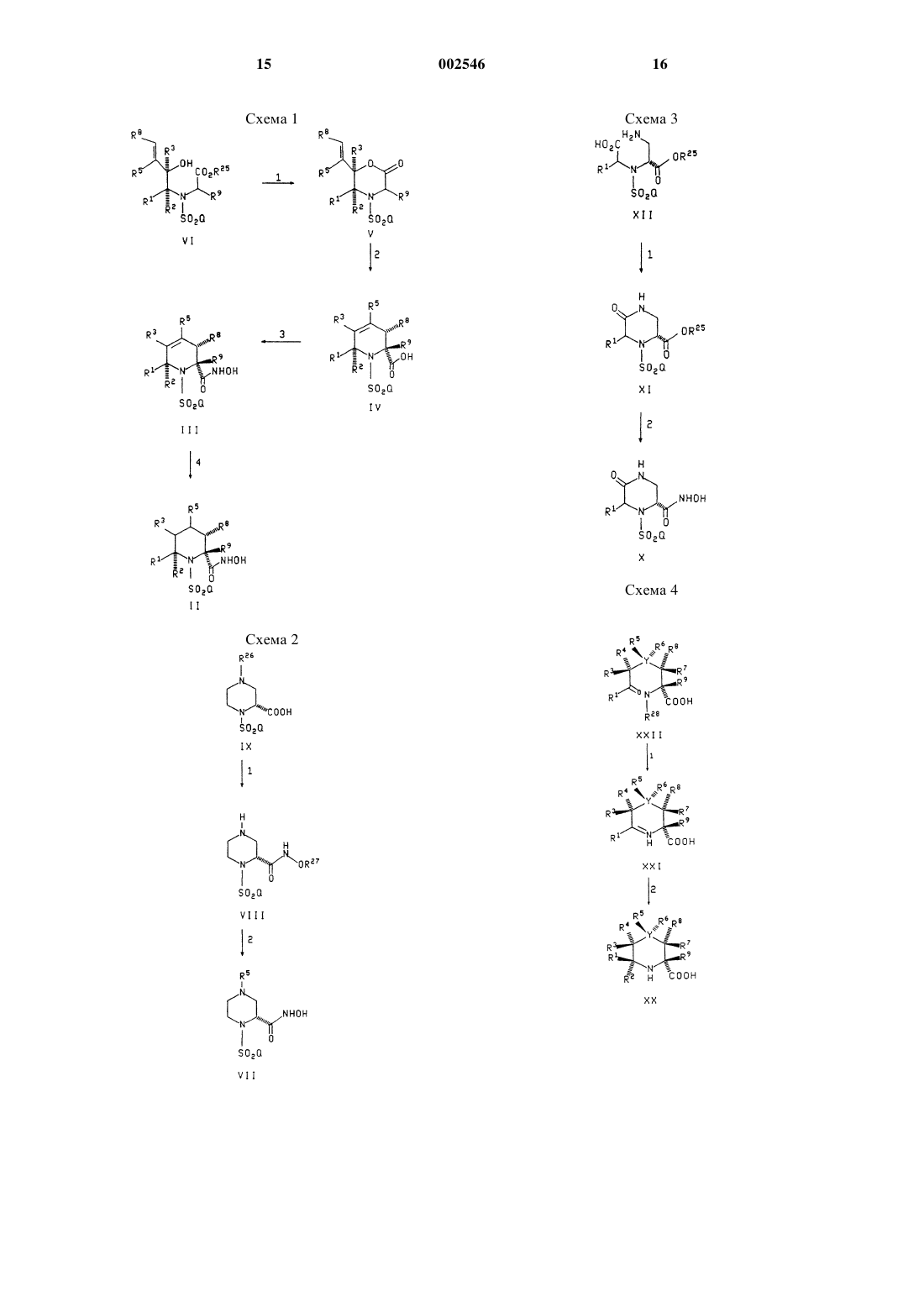
1 Предпосылки изобретения Настоящее изобретение относится к производным арилсульфонилгидроксамовой кислоты, которые являются ингибиторами металлопротеиназ матрикса или ингибиторами образования фактора некроза опухолей (TNF), и в качестве таковых пригодны для лечения состояния, выбранного из группы, состоящей из артрита, злокачественной опухоли, изъязвления ткани, рестеноза, периодонтального заболевания, врожденного буллезного эпидермолиза,склерита и других заболеваний, характеризующихся активностью металлопротеиназ матрикса, AIDS, сепсиса, септического шока и других заболеваний с участием образования TNF. Кроме того, соединения настоящего изобретения могут использоваться в комбинированной терапии со стандартными нестероидными противовоспалительными лекарственными средствами(в дальнейшем NSAID'S) и анальгетиками для лечения артрита, и в сочетании с цитотоксическими лекарственными средствами, такими как адриамицин, дауномицин, цис-платина, этопозид, таксол, таксотер и алкалоидами, такими как винкристин, для лечения злокачественных опухолей. Настоящее изобретение относится также к способу применения таких соединений для лечения вышеуказанных заболеваний у млекопитающих, в частности у людей, и к фармацевтическим композициям, пригодным для этого. Существует ряд ферментов, которые разрушают структурные белки и которые структурно близки металлопротеазам. Металлопротеазы, расщепляющие матрикс, такие как желатиназа, стромелизин и коллагеназа, участвуют в расщеплении матрикса (например, разрушении коллагена) и вовлечены во многие патологические состояния, сопряженные с метаболизмом аномальной соединительной ткани и базальной мембраны матрикса, такие как артрит(например, остеоартрит и ревматоидный артрит), изъязвление ткани (например изъязвление желудка, эпидермиса и роговицы), аномальное заживление ран, периодонтальное заболевание, заболевание костей (например болезнь Педжета и остеопороз), опухолевые метастазы или инвазия, а также HIV-инфекция (J. Leuk.Biol., 52 (2): 244-248, 1992). Известно, что фактор некроза опухолей участвует во многих инфекционных и аутоиммунных заболеваниях (W. Fiers, FEBS Letters,1991, 285, 199). Более того, было показано, чтоTNF является первичным медиатором воспалительного ответа при сепсисе и септическом шоке (С.Е. Spooner с соавт., Clinical Immunology 2 Краткое изложение существа изобретения Настоящее изобретение относится к соединению формулы или к его фармацевтически приемлемой соли, в которой пунктирная линия соответствует необязательной двойной связи; Х представляет углерод, кислород или серу;R1, R2, R3, R4, R5, R6, R7, R8 и R9 выбраны из группы, содержащей водород, (C1-C6)алкил,необязательно замещенный одной или двумя группами, выбранными из (C1-C6)алкилтио, (C1C6)алкокси, трифторметила, галогена, (С 6-С 10) арила, (С 5-С 9)гетероарила, (С 6-С 10)ариламино,(С 6-С 10)арилтио, (С 6-С 10)арилокси, (C5-C9) гетероариламино, (C5-C9)гетероарилтио, (C5-C9) гетероарилокси, (С 6-С 10)арил(С 6-С 10)арила, (С 3-С 6) циклоалкила, гидрокси, пиперазинила, (С 6-С 10) арил(C1-С 6)алкокси,(C5-C9)гетероарил(C1-C6) алкокси, (C1-C6)ациламино, (C1-C6)ацитилтио,(C1-C6)ацилокси, (C1-С 6)алкилсульфинила, (С 6 С 10)арилсульфинила, (C1-C6)алкилсульфонила,(С 6-С 10)арилсульфонила, амино, (C1-C6)алкиламино или C1-C6)алкиламино)2; (С 2-С 6)алкенил, (С 6-С 10)арил(C2-C6)алкенил, (C5-C9)гетероарил(С 2-С 6)алкенил, (C2-C6)алкинил, (С 6-С 10) арил(C2-С 6)алкинил,(C5-C9)гетероарил(С 2-С 6) алкинил, (С 1-C6)алкиламино, (C1-С 6)алкилтио,(C1-С 6)алкокси, перфтор(C1-C6)алкил, (С 6-С 10) арил, (С 5-С 9)гетероарил, (С 6-С 10)ариламино, (С 6 С 10)арилтио, (С 6-С 10)арилокси, (C5-C9)гетероариламино, (С 5-С 9)гетероарилтио, (C5-C9)гетероарилокси, (С 3-С 6)циклоалкил, (C1-С 6)алкилW представляет кислород или NR24, где R24 представляет водород или (C1-C6) алкил;Z представляет собой OR11 или NR24R11,где R24 определен как указано выше, а R11 определен как указано ниже; азетидинильное, пирролидинильное, пиперидинильное, пиперазинильное, морфолинильное, тиоморфолинильное,индолинильное, изоиндолинильное, тетрагидрохинолинильное, тетрагидроизохинолинильное или мостиковое диазабициклоалкильное кольцо,выбранное из группы, состоящей изm равно 1 или 2; р равно 0 или 1; и где каждая гетероциклическая группа может быть необязательно замещена одной или двумя группами, выбранными из гидрокси, (C1 С 6)алкила, (C1-C6)алкокси, (C1-С 10)ацила, (C1 С 10)ацилокси, (С 6-С 10) арила, (С 5-С 9)гетероарила, (С 6-С 10)арил(C1-C6)алкила, (С 5-С 9) гетероарил(C1-C6)алкила, гидрокси (C1-C6)алкила,(C1-C6)алкокси (C1-С 6)алкила, (C1-C6)ацилокси(C1-С 6)алкила, R12R13N, R12R13NSO2, R12R13NCO,R12R13NCO(C1-С 6)алкила, где R12 и R13, каждый,независимо представляют водород, (C1-C6) алкил, (С 6-С 10)арил, (C5-C9)гетероарил, (С 6-С 10) арил (C1-С 6)алкил или (C5-C9)гетероарил(C1-C6) алкил или R12 и R13, взятые вместе с азотом, к которому они присоединены, могут образовывать азетидинильное, пирролидинильное, пиперидинильное, морфолинильное или тиоморфолинильное кольца; R14SO2, R14SO2NH, в котором(C1-C6)алкил или (С 5-С 9)гетероарил (C1-С 6)алкил; R15CONR12, в котором R12 представляет как указано выше, а R15 представляет водород, (C1 С 6)алкил, (C1-C6)алкокси, (С 6-С 10)арил, (C5-C9) гетероарил,(C1-C6)арил(C1-C6)алкил(С 6-С 10) арил(C1-С 6)алкокси или (С 5-С 9)гетероарил(C1C6)алкил; R16OOC, R16OOC (C1-С 6)алкила, в котором R16 представляет (C1-С 6)алкил, (С 6-С 10) арил, (C5-C9)гетероарил, (С 6-С 10)арил(C1-C6) алкил, 5-инданил, CHR17OCOR18, где R17 представляет собой водород или (C1-С 6)алкил, а R18 представляет собой (C1-C6)алкил, (C1-С 6)алкокси или (С 6-С 10)арил; CH2CONR19R20, где R19 иR20, каждый, независимо представляют водород или (C1-C6) алкил или взятые вместе с азотом, к которому они присоединены, могут образовывать азетидинильное, пирролидинильное, пиперидинильное, морфолинильное или тиоморфолинильное кольца; или R21O(C1-C6) алкила, в котором R21 представляет H2N(CHR22)СО, гдеR22 представляет боковую цепь природной Dили L-аминокислоты;COR18 или CH2CONR19R20, где R17, R18, R19 и R20 определены как указано выше; или R1 и R2, или R3 и R4, или R5 и R6, взятые вместе, могут образовывать карбонил; или R1 и R2, или R3 и R4, или R5 и R6, или 7R и R8, взятые вместе, могут образовывать (С 3 С 6)циклоалкильное, оксациклогексильное, тиоциклогексильное, инданильное или тетралинильное кольца или группу формулы(C1-C6)алкокси(С 6-С 10)арилокси(С 5-С 9)гетероарил, необязательно замещенный фтором, хлором, (C1-C6)алкилом, (C1-C6)алкокси или перфтор(C1-С 3)алкилом; при условии, что Z должен быть замещенным, когда определен как азетидинильное, пирролидинильное, морфолинильное, тиоморфолинильное, индолинильное, изоиндолинильное,тетрагидрохинолинильное, тетрагидроизохинолинильное, пиперазинильное, (C1-С 10)ацилпиперазинильное, (C1-C6)алкилпиперазинильное,(C6-С 10)арилпиперазинильное, (С 5-С 9)гетероарилпиперазинильное или мостиковое диазабициклоалкильное кольцо; при условии, что R7 не представляет водород, только когда R8 радикал, отличный от водорода; при условии, что R6 не представляет водород, только когда R5 радикал, отличный от водорода; при условии, что R3 не представляет водород, только когда R4 радикал, отличный от водорода; при условии, что R2 не представляет водород, только когда R1 радикал, отличный от водорода; при условии, что когда R1, R2 и R9 представляют заместитель, включающий гетероатом,данный гетероатом не может быть непосредственно связан в положениях 2- или 6-; при условии, что когда Х представляет азот, R4 отсутствует; при условии, что когда Х представляет кислород, серу, SO, SO2 или азот и когда одна или несколько групп, состоящие из R1, R2, R5 и R6,представляют заместитель, включающий гетероатом, данный гетероатом не может быть непосредственно связан в положениях 4- или 6-; при условии, что когда Y представляет кислород, серу, SO, SO2 или азот и когда одна или несколько групп, состоящие из R3, R4, R7 и R8,независимо представляют заместитель, включающий гетероатом, данный гетероатом не может быть непосредственно связан в положениях 3- или 5-; при условии, что когда Х представляет кислород, серу, SO или SO2, R3 и R4 отсутствуют; при условии, что когда Y равен 1, a W представляет NR24 или кислород, Z не может представлять гидрокси; при условии, что когда Y представляет кислород, серу, SO или SO2, R5 и R6 отсутствуют; при условии, что когда Y представляет азот, R6 отсутствует; при условии, что когда пунктирная линия представляет двойную связь, R4 и R6 отсутствуют; при условии, что когда R3 и R5 независимо представляют заместитель, включающий гете 002546 6 роатом, когда пунктирная линия представляет двойную связь, данный гетероатом не может быть непосредственно связан с Х и Y; при условии, что когда любой из Х или Y позиция представляет кислород, серу, SO, SO2 или азот, другой из Х или Y представляет углерод; при условии, что когда Х или Y представляет гетероатом, пунктирная линия не является двойной связью; при условии, что, по меньшей мере, один из R1, R2, R3, R4, R5, R6, R7, R8 и R9 должен быть представлен группой формулы II. Термин "алкил", как он используется здесь, если не указано иначе, включает насыщенные моновалентные углеводородные радикалы, имеющие прямую, разветвленную или циклическую составляющую или их сочетания. Термин "алкокси", как он используется здесь, включает O-алкильные группы, где "алкил" определен, как указано выше. Термин "арил", как он используется здесь,если не указано иначе, включает органический радикал, полученный из ароматического углеводорода, путем удаления одного водорода, такой как фенил или нафтил, необязательно замещенный 1-3 заместителями, независимо выбранными из группы, состоящей из фтора, хлора, циано, нитро, трифторметила, (C1-C6) алкокси, (С 6-С 10)арилокси, трифторметокси, дифторметокси и (C1-C6) алкила. Термин "гетероарил", как он используется здесь, если не указано иначе, включает органический радикал, полученный из ароматического гетероциклического соединения, путем удаления одного водорода, такой как пиридил, фурил,пироил, тиенил, изотиазолил, имидазолил, бензимидазолил, тетразолил, пиразинил, пиримидил, хинолил, изохинолил, бензофурил, изобензофурил, бензотиенил, пиразолил, индолил,изоиндолил, пуринил, карбазолил, изоксазолил,тиазолил, оксазолил, бензотиазолил или бензоксазолил, необязательно замещенный 1-2 заместителями, независимо выбранными из группы,состоящей из фтора, хлора, трифторметила, (C1C6)алкокси, (С 6-С 10)арилокси, трифторметокси,дифторметокси и (C1-C6)алкила. Термин "ацил", как он используется здесь,если не указано иначе, включает радикал общей формулы RCO, где R представляет алкил, алкокси (такой как метилоксикарбонил), арил,арилалкил или арилалкилокси, а термин "алкил" или "арил" определен, как указано выше. Термин "ацилокси", как он используется здесь, включает O-ацильные группы, где "ацил" указан выше. Термин "D- или L-аминокислота", как он используется здесь, если не указано иначе,включает глицин, аланин, валин, лейцин, изолейцин, фенилаланин, аспарагин, глутамин,триптофан, пролин, серин, треонин, тирозин,гидроксипролин, цистеин, цистин, метионин, 7 аспарагиновую кислоту, глутаминовую кислоту,лизин, аргинин или гистидин. Нумерация положений в кольце формулыI, как она используется здесь, указана следующим образом: Предпочтительная конформация соединения формулы I включает гидроксамовую кислоту, расположенную по оси во 2-м положении. Соединение формулы I может иметь хиральные центры и по этой причине существовать в разных энантиомерных формах. Настоящее изобретение относится ко всем оптическим изомерам и стереоизомерам соединений формулы I и их смесей. Предпочтительные соединения формулы I включают соединения, в которых Y представляет углерод. Другие предпочтительные соединения формулы I включают соединения, в которых Q представляет (C1-C6)алкокси(С 6-С 10)арил, (С 6 С 10)арил(C1-C6)алкокси(С 6-С 10)арил или (С 6-С 10) арил(C1-C6) алкокси(C1-C6)алкил, где каждая терминальная арильная группа необязательно замещена фтором. Другие предпочтительные соединения формулы I включают соединения, где R2, R3, R6,R7 и R9 представляют водород. Более предпочтительные соединения формулы I включают соединения, где Y представляет углерод; Q представляет (C1-С 6)алкокси(С 6 С 10)арил,(С 6-С 10)арил(C1-C6)алкокси(С 6-С 10) арил, или (С 6-C10)арил(C1-C6)алкокси(C1-C6) алкил. Особые предпочтительные соединения формулы I включают нижеследующие:(2R,4R)-1-(4 метоксибензолсульфонил)-4-(пиперазин-1 карбонил)пиперидин-2-карбоновой кислоты. Другие соединения настоящего изобретения включают: гидроксиамид (3S)-4-[4-(2-хлортиазол-5 илметокси)бензолсульфонил]-2,2-диметилтиоморфолин-3-карбоновой кислоты; гидроксиамид (3S)-2,2-диметил-4-[4-(тиазол-5-илметокси)бензолсульфонил]тиоморфолин-3-карбоновой кислоты; гидроксиамид (3S)-2,2-диметил-4-[4-(пиридин-4-илметокси)бензолсульфонил]тиоморфолин-3-карбоновой кислоты; гидроксиамид (3S)-4-4-[2-(4-фторфенил) этокси]бензолсульфонил-2,2-диметилтмоморфолин-З-карбоновой кислоты; гидроксиамид(2R,4S)-1-(4-бензилоксибензолсульфонил)-4-гидроксипиперидин-2-карбоновой кислоты; гидроксиамид (2R,4S)-1-[4-(4-фторбензилокси)бензолсульфонил]-4-гидроксипиперидин 2-карбоновой кислоты; гидроксиамид 1-(4-бутоксибензолсульфонил)-3-(морфолин-4-карбонил)пиперидин-2-карбоновой кислоты; гидроксиамид 1-[4-(4-фторбензилокси)бензолсульфонил)-3-(морфолин-4-карбонил)пиперидин-2-карбоновой кислоты; гидроксиамид 1-[3-(фторбензилокси)пропан-1-сульфонил]-3-(морфолин-4-карбонил)пиперидин-2-карбоновой кислоты; гидроксиамид 1-(4-бутоксибензолсульфонил)-3-(пирролидон-1-карбонил)пиперидин-2 карбоновой кислоты; гидроксиамид 1-[4-(4-фторбензилокси) бензолсульфонил]-3-(пирролидин-1-карбонил) пиперидин-2-карбоновой кислоты; гидроксиамид 1-[3-(4-фторбензилокси) пропан-1-сульфонил]-3-(пирролидин-1-карбонил)пиперидин-2-карбоновой кислоты; и 1-[4-(4-фторбензилокси)бензолсульфонил]2-гидроксикарбамоилпиперидин-4-карбоновую кислоту. Настоящее изобретение относится также к фармацевтической композиции для (а) лечения состояния, выбранного из группы, состоящей из артрита, злокачественной опухоли, синергичной с цитотоксическими противоопухолевыми агентами, тканевого изъязвления, макулярной дегенерации (дегенерация желтого пятна), рестеноза, периодонтального заболевания, врожденного буллезного эпидермолиза, склерита, в сочетании со стандартными NSAID'S и анальгетиками, и других заболеваний, характеризующихся актив 13 ностью металлопротеиназ матрикса, AIDS, сепсиса, септического шока и других заболеваний с образованием фактора некроза опухолей (TNF),или для (b) ингибирования металлопротеиназ матрикса или образования фактора некроза опухолей (TNF) у млекопитающих, в том числе у человека, включающего любое количество соединения формулы I или его фармацевтически приемлемой соли, эффективное для таких лечений, и фармацевтически приемлемый носитель. Настоящее изобретение относится также к способу ингибирования (а) металлопротеиназ матрикса или (b) образования фактора некроза опухолей (TNF) у млекопитающих, в том числе у человека, включающему введение указанному млекопитающему эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение относится также к способу лечения состояния, выбранного из группы, состоящей из артрита, злокачественной опухоли, тканевого изъязвления, макулярной дегенерации, рестеноза, периодонтального заболевания, врожденного буллезного эпидермолиза, склерита, соединениями формулы I, которые могут быть применимы в сочетании со стандартными NSAID'S и анальгетиками и в сочетании с цитотоксическими противоопухолевыми агентами, и других заболеваний, характеризующихся активностью металлопротеиназ матрикса, AIDS, сепсиса, септического шока и других заболеваний с образованием фактора некроза опухолей (TNF) у млекопитающих, в том числе, у человека, включающему введение указанному млекопитающему соединения формулы I или его фармацевтически приемлемой соли, в количестве, эффективном для лечения такого состояния. Подробное описание настоящего изобретения Схемы нижеследующих реакций иллюстрируют получение соединений настоящего изобретения. Если не указано иначе, R1, R2, R3, R4,R5, R6, R7, R8, R9, n и Аr, приведенные в схемах реакции и в обсуждении, как указано выше. Приготовление 1 относится к получению промежуточных соединений формулы VI. Соединения формулы VI преобразовываются в соединения формулы I в соответствии со способами схемы 1. Исходные материалы формулыXVI могут быть получены в соответствии со способами, хорошо известными рядовым специалистам в данной области техники. В реакции 1 приготовления 1 соединение формулы XVI преобразовывается в соответствующее гидроксиэфирное соединение формулыVI, сначала взаимодействием XVI с арилсульфонилгалоидом в присутствии триэтиламина и апротонного растворителя, такого как метиленхлорид, тетрагидрофуран или диоксан при температуре между около 20 С и около 30 С, предпочтительно при комнатной температуре. Полученное таким образом соединение далее подвергается взаимодействию с соединением формулы в которой R25 представляет карбобензилокси,(C1-C6)алкил, бензил, аллил или трет-бутил, в присутствии натрийгексаметилдисилазана и смеси растворителей тетрагидрофурандиметилформамид при температуре между около -20 С и около 20 С, предпочтительно около 0 С, с образованием гидроксиэфирного соединения формулы VI. Приготовление 2 относится к альтернативному способу получения соединений формулыVI. Исходные материалы формулы XVIII могут быть получены в соответствии со способами,хорошо известными рядовым специалистам в данной области техники. В реакции 1 приготовления 2 соединение амина формулы XVIII, в 19 которой R25 имеет значения указанные выше,преобразовывается в соответствующее арилсульфониламинное соединение формулы XVII(1) взаимодействием XVIII с арилсульфонилгалоидом в присутствии триэтиламина и апротонного растворителя, такого как метиленхлорид,тетрагидрофуран или диоксан при температуре между около 20 С и около 30 С, предпочтительно при комнатной температуре, (2) взаимодействием полученного таким образом соединения с соединением формулы в присутствии натрийгексаметилдисилазана и смеси растворителей тетрагидрофурандиметилформамид при температуре между около -20 С и около 20 С, предпочтительно около 0 С, и (3) дополнительным взаимодействием данного соединения, полученного таким образом, с озоном в растворе метиленхлоридметанол при температуре между около -90 С и около -70 С, предпочтительно около -78 С. Нестабильное озонидное соединение, полученное таким образом,затем подвергается взаимодействию с трифенилфосфином с образованием арилсульфониламинного соединения формулы XVII. В реакции 2 приготовления 2 данное арилсульфониламинное соединение формулы XVII преобразовывается в соответствующее гидроксиэфирное соединение формулы VI взаимодействием XVII с соединением формулы в которой W представляет литий, магний, медь или хром. Схема 1 относится к получению соединений формулы II, которые представляют соединения формулы I, в которой Х и Y представляют углерод; R4, R6 и R7 представляют водород; а пунктирная линия между Х и Y отсутствует. В реакции 1 схемы 1 соединение формулы VI, где защитная группа R25, представляющая карбобензилокси, (C1-C6)алкил, бензил, аллил или трет-бутил, преобразовывается в соответствующее морфолиноновое соединение формулы V лактонизацией и последующей перегруппировкой Клайзена (Claisen) соединения формулы VI. Данная реакция ускоряется при удалении защитной группы R25 соединения формулы VI и осуществляется в условиях, соответствующих для этой конкретной защитной группы R25. Такие условия включают: (а) обработку водородом и гидрирующим катализатором, таким как 10%ный палладий на угле, где R25 представляет карбобензилокси, (b) омыление, где R25 представляет низший алкил, (с) гидрогенолиз, где R25 представляет бензил, (d) обработку сильной кислотой, такой как трифторуксусная кислота или хлористо-водородная кислота, где R25 представляет трет-бутил, или (е) обработку трибутило 002546 20 ловохлоридом и уксусной кислотой в присутствии катализатора хлорида бис(трифенилфосфин)палладия (II), где R25 представляет аллил. В реакции 2 на схеме 1 морфолиноновое соединение формулы V преобразовывается в соединение карбоновой кислоты формулы IV взаимодействием V с литийгексаметилдисилазаном в апротонном растворителе, таком как тетрагидрофуран, при температуре между около-78 С. Затем в данную реакционную смесь добавляют триметилсилилхлорид, а растворитель тетрагидрофуран удаляют в вакууме и заменяют толуолом. Полученную реакционную смесь нагревают до температуры между около 100 С и около 120 С, предпочтительно около 110 С, и обрабатывают хлористо-водородной кислотой с образованием соединения карбоновой кислоты формулы IV. В реакции 3 на схеме 1 соединение карбоновой кислоты формулы IV преобразовывается в соответствующее соединение гидроксамовой кислоты формулы III обработкой IV 1-(3 диметиламинопропил)-3-этилкарбодиимидом и 1-гидроксибензотриазолом в полярном растворителе, таком как диметилформамид, с последующим добавлением гидроксиламина к данной реакционной смеси через период времени в пределах от около 15 мин до около 1 ч, предпочтительно около 30 мин. Гидроксиламин предпочтительно получают in situ из солевой формы,такой как гидрохлорид гидроксиламина, в присутствии основания, такого как N-метилморфолин. Альтернативно, защищенное производное гидроксиламина или его солевая форма, где гидроксильная группа защищена в виде третбутильного, бензильного или аллильного эфира,может быть использована в присутствии (бензотриазол-1-илокси)трис(диметиламино)фосфонийгексафторфосфата и основания, такого какN-метилморфолин. Удаление гидроксиламинной защитной группы осуществляется гидрогенолизом группы, защищающей бензил или обработкой сильной кислотой, такой как трифторуксусная кислота, по трет-бутильной защитной группе. Аллильная защитная группа может быть удалена обработкой гидридом трибутилолова и уксусной кислотой в присутствии катализатора бис(трифенилфосфин)палладий(II)хлорид. В качестве защищенного гидроксиламинного производного можно также применять N,O-бис(4 метоксибензил)гидроксиламин, где снятие защиты достигается использованием смеси метансульфоновой кислоты и трифторуксусной кислоты. В реакции 4 на схеме 1 соединение гидроксамовой кислоты формулы III превращают при необходимости в соответствующее пиперидиновое соединение формулы II обработкой водородом и катализатором для гидрогенизации,таким как 10%-ный палладий на угле. 21 Схема 2 относится к получению соединений формулы VII, которые представляют соединения формулы I, где Y означает азот; Х представляет углерод; R1, R2, R3, R4, R7 и 8R представляют водород, а R6 отсутствует. Исходные материалы формулы IX могут быть получены в соответствии со способами, хорошо известными рядовым специалистам в данной области техники. В реакции 1 на схеме 2, соединение формулы IX арилсульфонилпиперазин,где R26 представляет карбобензилокси, бензил или карбо-трет-бутилокси, преобразуют в соединение формулы VIII взаимодействием IX с защищенным производным гидроксиламина формулы: R27ONH2-HCl, где R27 представляет трет-бутил, бензил или аллил, в присутствии дициклогексилкарбодиимида, диметиламинопиридина и апротонного растворителя, такого как метиленхлорид. Защитная группа R26 выбрана таким образом, чтобы ее можно было селективно удалить без удаления присутствующей защитной группы R27, в силу чего R26 не может быть такой же, что и R27. Удаление защитной группы R26 из соединения формулы IX осуществляют в условиях, соответствующих этой конкретной защитной группе R26. Такие условия включают: (а) обработку водородом и катализатором для гидрогенизации, таким как 10%-ный палладий на угле, где R26 представляет карбобензилокси, (b) гидрогенолиз, где R26 представляет бензил или (с) обработку сильной кислотой, такой как трифторуксусная кислота или хлористоводородная кислота, где R26 представляет карбо-трет-бутилокси. В реакции 2 на схеме 2 соединение формулы VIII преобразовывается в соответствующее соединение гидроксамовой кислоты формулыVII, где R5 представляет водород или (C1-C6) алкил, взаимодействием, при необходимости,VIII с алкилгалоидом, когда R5 представляет(C1-C6)алкил. Последующее удаление гидроксиламинной защитной группы R27 осуществляют гидрогенолизом по бензильной защитной группе или обработкой сильной кислотой, такой как трифторуксусная кислота, по трет-бутильной защитной группе. Аллильная защитная группа может быть удалена обработкой трибутилоловогидридом и уксусной кислотой в присутствии катализатора бис(трифенилфосфин)палладий(II) хлорида. Схема 3 относится к получению соединений формулы X, которые представляют соединения формулы I, где Y представляет азот; Х представляет углерод; R2, R7, R8 и R9 представляют водород; R3 и R4 взятые вместе представляют карбонил; R5 представляет водород, а R6 отсутствует. В реакции 1 на схеме 3 арилсульфониламинное соединение формулы XII, где R26 определен как указано выше, преобразовывается в соответствующее пиперазиновое соединение формулы XI взаимодействием XII с карбодиимидом и основанием, таким как триэтила 002546 22 мин. Соединение формулы XI дополнительно подвергается взаимодействию с данным соединением гидроксамовой кислоты формулы Х в соответствии с методикой, описанной выше в реакции 3 на схеме 1. Схема 4 относится к получению соединений формулы XIII. Исходные материалы формулы XVIII могут быть получены в соответствии со способами, хорошо известными рядовым специалистам в данной области техники. Соединения формулы XIII представляют соединения формулы I, где Х представляет углерод, а пунктирная линия между Х и Y отсутствует. В реакции 1 на схеме 4 удаляется защитная группаR28, а соединение формулы XXII, в котором Y представляет кислород, серу или углерод, подвергается последующему восстановительному аминированию с получением соответствующего иминного соединения формулы XXI, осуществляемого в надлежащих условиях, при которых используется конкретная защитная группа R28. Такие условия включают условия, примененные выше для удаления защитной группы R26 в реакции 1 на схеме 2. В реакции 2 на схеме 4 иминное соединение формулы XXI преобразуется в соответствующее пиперидиновое соединение формулыXX путем взаимодействия XXI с нуклеофилом формулы R2M, где М представляет литий, галогенид магния или галогенид церия. Данную реакцию осуществляют в эфирных растворителях,таких как диэтиловый эфир или тетрагидрофуран при температуре от около -78 С до около 0 С, предпочтительно около -70C. В реакции 3 на схеме 4 сульфирование пиперидинового соединения формулы XX, с получением соответствующего арилсульфонилпиперидинового соединения формулы XIX, осуществляется путем взаимодействия XX с арилсульфонилгалоидом в присутствии триэтиламина и апротонного растворителя, такого как метиленхлорид, тетрагидрофуран или диоксан при температуре от около 20 С до около 30 С, предпочтительно при комнатной температуре. В реакции 4 на схеме 4 арилсульфонилпиперидиновое соединение формулы XIX преобразуется в соединение гидроксамовой кислоты формулы XIII в соответствии с методикой, описанной выше в реакции 3 на схеме 1. Схема 5 относится к получению соединений формулы XIV, которые представляют соединения формулы I, где Y представляет азот, Х представляет углерод, пунктирная линия между Х и Y отсутствует, R5 представляет водород, aR6 отсутствует. В реакции 1 на схеме 5 соединение формулы XXVI, где защитные группы R29 иR31, каждая, независимо выбраны из группы,состоящей из карбобензилокси, бензила и карбо-трет-бутокси, а R30 представляет карбобензилокси, (C1-C6)алкил, бензил, аллил или третбутил, преобразуется в соответствующее иминное соединение формулы XXV путем удаления 23 защитной группы R29 и последующего восстановительного аминирования соединения формулы XXVI. Защитная группа R29 выбрана так,чтобы она могла быть селективно удалена в присутствии и без утраты защитной группы R31. Удаление защитной группы R29 из соединения формулы XXVI осуществляется в условиях,подходящих для конкретной защитной группыR29, на использование которой не влияет защитная группа R31. Такие условия включают: (а) обработку водородом и катализатором для гидрогенизации, таким как 10%-ный палладий на угле, где R29 представляет карбобензилокси, аR29 представляет (C1-C6) алкил, а R31 представляет трет-бутил, (с) гидрогенолиз, где R29 представляет бензил, а R31 представляет (C1-C6) алкил или трет-бутил, (d) обработку сильной кислотой, такой как трифторуксусная кислота или хлористо-водородная кислота, где R29 представляет трет-бутил, а R31 представляет (C1-C6)алкил, бензил или аллил, или (е) обработку трибутилоловогидридом и уксусной кислотой в присутствии каталитического бис(трифенилфосфин)палладий(II) хлорида, где R29 представляет аллил, а R31 представляет (C1-C6)алкил, бензил или трет-бутил. Защитная группа R30 может быть выбрана так, чтобы она удалялась на той же реакционной стадии, что и защитная группаR29. В реакции 2 на схеме 5 иминное соединение формулы XXV преобразуется в соответствующее соединение формулы XXIV путем взаимодействия XXV с нуклеофилом формулыR2M, где М представляет литий, магнийгалогенид или кальцийгалогенид. Данная реакция осуществляется в любом растворителе, таком как диэтиловый эфир или тетрагидрофуран, при температуре от около -78 С до около 0 С, предпочтительно около -70 С. В реакции 3 на схеме 5 сульфирование пиперидинового соединения формулы XXIV с получением соответствующего арилсульфонилпиперидинового соединения формулы III осуществляется в соответствии с методикой, описанной выше в реакции 3 на схеме 4. В реакции 4 на схеме 5 арилсульфонилпиперидиновое соединение XXIII преобразовывается в соединение гидроксамовой кислоты формулы XIV путем (1) удаления, при необходимости, защитной группы R30, и защитной группыR31 из XXIII с последующим (2) взаимодействием XXIII в соответствии с методикой, описанной выше в реакции 3 на схеме 1. Удаление защитных групп R30 и R31 из соединения формулыXXIII осуществляется в условиях, подходящих для использования конкретной защитной группы R30 и R31. Такие условия включают условия,примененные выше для удаления защитной группы R25 в реакции 1 на схеме 1. Фармацевтически приемлемые соли кислотных соединений настоящего изобретения 24 представляют соли, полученные с основаниями,а именно катионные соли, такие как соли щелочных и щелочно-земельных металлов, таких как натрий, литий, калий, кальций, магний, а также аммонийные соли, такие как соли аммония, триметиламмония, диэтиламмония и соли трис-(гидроксиметил)метиламмония. Подобно аддитивным солям кислот, таких как минеральные кислоты, органические карбоновые и органические сульфоновые кислоты,например, хлористо-водородная кислота, метансульфоновая кислота, малеиновая кислота, возможно также использование основной группы,такой как пиридил, составляющую часть структуры. Способность соединений формулы I или их фармацевтически приемлемых солей (соединений настоящего изобретения) ингибировать металлопротеиназы матрикса или образование фактора некроза опухолей (TNF) и, следовательно, демонстрировать свою эффективность для лечения заболеваний, связанных с металлопротеиназами матрикса или с образованием фактора некроза опухолей, показано в нижеследующих анализах in vitro. Биологический анализ Ингибирование коллагеназы человека (ММР-1) Рекомбинантную коллагеназу человека активировали с помощью трипсина, применяя нижеследующее соотношение: 10 мкг трипсина на 100 мкг коллагеназы. Трипсин и коллагеназу инкубировали при комнатной температуре в течение 10 мин, а затем добавляли пятикратный избыток (50 мкг/10 мкг трипсина) ингибитора трипсина из соевых бобов. 10 мМ основные [исходные] растворы ингибиторов готовили в диметилсульфоксиде, а затем разбавляли с использованием нижеследующей схемы: 10 мМ 120 мкМ 12 мкМ 1,2 мкМ 0,12 мкМ Двадцать пять микролитров каждой концентрации добавляли, трижды повторяя, в соответствующие лунки 96-луночного микрофлуоресцентного планшета (microfluor). Конечная концентрация ингибитора должна соответствовать разведению 1:4 после добавления фермента и субстрата. Позитивные контроли (фермент,без ингибитора) расположены в лунках D1-D6, а пропуски (без фермента, без ингибитора) расположены в лунках D7-D12. Коллагеназу разбавляли до 400 нг/мл и затем добавляли 25 мкл в соответствующие лунки микрофлуоресцентного планшета. Конечная концентрация коллагеназы в данном испытании составляла 100 нг/мл. Субстрат (DNP-Pro-Cha-Gly-Cys(Me)-HisAla-Lys(NMA)-NH2) готовили в виде 5 мМ основного [исходного] раствора в диметилсульфоксиде и затем разбавляли до 20 мкМ в буфере для анализа. Анализ инициировали путем добавления 50 мкл субстрата на лунку в микро 25 титровальной плашке для получения конечной концентрации 10 мкМ. Показания флуоресценции (360 нм возбуждение, 460 нм эмиссия) были сняты в момент 0 и затем через 20-минутные интервалы. Анализ проводили при комнатной температуре в течение 3-х часов стандартного опыта. Затем время флуоресценции откладывали на графике для холостых образцов и содержащих коллагеназу (данные тройных определений усредняли). Выбирали временную точку, в которой получали хороший сигнал (холостой опыт), и временную точку на линейной части кривой (обычно около 120 мин) для определения значений IC50. Нулевую точку использовали в качестве холостого опыта для каждого соединения в каждой концентрации и эти значения вычитали из данных для 120 мин. Данные откладывали на графике в виде концентрации ингибитора в сравнении с % контроля (ингибированную флуоресценцию делили на флуоресценцию одной коллагеназы х 100). Значения IC50 определяли из концентрации ингибитора, который дает сигнал, равный 50% от контроля. Если значения IС 50 получались 0,03 мкМ,тогда анализируемые ингибиторы составляли концентрации 0,3 мкМ, 0,03 мкМ, 0,03 мкМ и 0,003 мкМ. Ингибирование желатиназы (ММР-2) Ингибирование активности желатиназы определяли с использованием субстрата DnpPro-Cha-Gly-Cys(Me)-His-Ala-Lys(NMA)-NH2(10 мкМ) в тех же условиях, что и ингибирование коллагеназы человека (ММР-1). 72 кД Желатиназу активировали с помощью 1 мМ АРМА (р-аминофенилмеркуриацетата) в течение 15 ч при 4 С и разбавляли для получения конечной концентрации в данном анализе 100 мг/мл. Ингибиторы разбавляли так же, как и для ингибирования коллагеназы человека для получения конечных концентраций в данном анализе 30 мкМ, 3 мкМ, 0,3 мкМ и 0,03 мкМ. Каждую концентрацию давали при троекратном повторении. Показания флуоресценции (360 нм возбуждение, 460 нм эмиссия) снимали в нулевое время и затем с 20-минутными интервалами в течение 4-х часов. Значения IC50 определяли из расчета ингибирования коллагеназы человека (ММР-1). Если получаемые значения IC50 составляли менее, чем 0,03 мкМ, то ингибиторы анализировались в конечных концентрациях 0,3 мкМ, 0,03 мкМ,0,003 мкМ и 0,0003 мкМ. Ингибирование активности стромелизина(ММР-3) Ингибирование активности стромелизина основывается на модифицированном спектрофотометрическом анализе, описанном Weingarten и Feder (Weingarten H. and Feder J., Spectrophotometric Assay for Vertebrate Collagenase,Anal. Biochem. 147, 437-440 (1985. Гидролиз 26 тиопептолидного субстрата [Ас-Pro-Leu-GlySCH[CH2CH(СН 3)2]CO-Leu-Gly-OC2H5] дает меркаптановый фрагмент, который можно проконтролировать в присутствии реагента Эллмана. Рекомбинантный простромелизин человека активировали трипсином с использованием соотношения 1 мкл основного [исходного] раствора трипсина 10 мг/мл на 26 мкг стромелизина. Трипсин и стромелизин инкубировали при 37 С в течение 15 мин с последующим ингибированием 10 мкл ингибитора трипсина из соевых бобов (10 мг/мл) в течение 10 мин при 37 С для подавления активности трипсина. Опыты проводили в общем объеме 250 мкл буфера для анализа (200 мМ натрийхлорида, 50 мМ MES, и 10 мМ кальцийхлорида, рН 6,0) в 96-луночных микролитровых планшетах. Активированный стромелизин разбавляли аналитическим буфером до 25 мкг/мл. Реактив Эллмана(3-карбокси-4-нитрофенилдисульфид) готовили в виде 1 М основного [исходного] раствора в диметилформамиде и разбавляли до 5 мМ аналитическим буфером в 50 мкл на лунку, доводя до 1 мМ конечной концентрации. 10 мМ основные [исходные] растворы ингибиторов готовили в диметилсульфоксиде и разбавляли последовательно в аналитическом буфере так, чтобы добавление 50 мкл в соответствующие лунки давало конечные концентрации 3 мкМ, 0,3 мкМ, 0,003 мкМ и 0,0003 мкМ. Все условия исполняли при троекратном повторении. 300 мМ диметилсульфоксидного основного [исходного] раствора пептидного субстрата разбавляли в 15 мМ буфера для анализа, а сам анализ инициировали добавлением 50 мкл в каждую лунку до получения конечной концентрации субстрата 3 мМ. Холостые опыты содержали пептидный субстрат и реактив Эллмана без фермента. Образование продукта контролировали при 405 нм на спектрофотометре Molecular Devices UVmax. Значения IC50 определяли тем же способом, что и для коллагеназы. Ингибирование ММР-13 Рекомбинантную ММР-13 человека активировали с помощью 2 мМ АРМА (паминофенилмеркуриацетата) в течение 1,5 часов, при 37 С и разбавляли до 400 мг/мл в буфере для анализа (50 мМ Трис, рН 7,5, 200 мМ натрийхлорида, 5 мМ кальцийхлорида, 20 мкМ хлорида цинка, 0,02% Бридж). В лунки 96 луночного микрофлуоресцентного планшета добавляли по двадцать пять микролитров разбавленного фермента. Затем в анализе фермент разбавляли в соотношении 1:4 путем добавления ингибитора и субстрата с получением конечной концентрации в анализе 100 мг/мл. 10 мМ-ные основные [исходные] растворы ингибиторов готовили в диметилсульфоксиде, а 27 затем разбавляли в буфере для анализа согласно схеме разбавления ингибитора для ингибирования коллагеназы человека (ММР-1): Двадцать пять микролитров каждой концентрации добавляли при троекратном повторении в микролитровую плашку. Конечные концентрации в данном анализе составляют 30 мкМ, 3 мкМ., 0,3 мкМ и 0,03 мкМ. Субстрат (Dnp-Pro-Cha-Gly-Cys(Me)-HisAla-Lys(NMA)-NH2) получали как и при ингибировании коллагеназы человека (ММР-1) и 50 мкл его вносили в каждую лунку с получением конечной концентрации в анализе 10 мкМ. Показания флуоресценции (360 нм возбуждение; 450 - эмиссия) снимали в нулевое время и каждые 5 мин в течение 1 ч. Позитивные контроли не содержали фермента и субстрата, а холостые опыты содержали только субстрат. Значения IC50 определяли согласно ингибированию коллагеназы человека (ММР-1). Если получаемые значения IС 50 составляют менее,чем 0,03 мкМ, то ингибиторы анализировали при конечных концентрациях 0,3 мкМ, 0,03 мкМ, 0,003 мкМ и 0,0003 мкМ. Ингибирование образования TNF Способность соединений или их фармацевтически приемлемых солей ингибировать образование TNF и, следовательно, демонстрировать свою эффективность в течение заболеваний с образованием TNF, показаны в нижеследующем испытании in vitro: Моноядерные клетки человека выделяли из некоагулировавшей крови человека с применением одностадийной методики Фиколл-гипак.(2) Выделенные моноядерные клетки промывали три раза солевым раствором Хэнкса (HBSS),уравновешенного двухвалентными катионами, и ресуспендировали до плотности 2 х 106/мл вHBSS, содержащем 1% BSA. Подсчет вели с применением анализатора Abbott Cell Dyn 3500,который свидетельствует, что моноциты в этих препаратах ранжированы в диапазоне от 17 до 24% от общей численности клеток. 180 мкл клеточной суспензий аликвотировали в плоскодонные 96-луночные планшеты(Costar). Добавление соединений и LPS (100 нг/мл конечная концентрация) дает конечный объем 200 мкл. Все условия выполняли при троекратном повторении. После четырех часов инкубации в увлажняемом СO2-инкубаторе планшеты извлекали и центрифугировали (10 минут приблизительно при 250 х g), а супернатанты удаляли и анализировали на TNF с использованием набора RD ELISA. Для введения млекопитающим, в том числе людям, для ингибирования металлопротеиназ матрикса или ингибирования образования фактора некроза опухолей (TNF), можно использовать ряд традиционных путей поступления в организм, в том числе пероральный, парентеральный и топический, обычно соединение на 002546 28 стоящего изобретения должно вводиться перорально или парентерально в дозах от около 0,1 до около 25 мг/кг веса тела пациента, подвергающегося ежедневному лечению, предпочтительно от около 0,3 до 5 мг/кг. Однако некоторые изменения в дозировке имеют место в зависимости от состояния пациента, подвергающегося лечению. Медицинский работник, ответственный за введение лекарственного средства,должен в любом случае определить соответствующую дозу для каждого пациента. Соединения настоящего изобретения могут быть введены в широком ряду отличающихся лекарственных форм. Обычно терапевтически эффективные соединения настоящего изобретения представлены в таких лекарственных формах в концентрациях, колеблющихся в диапазоне от около 5,0% до около 70% по весу. Для перорального введения могут использоваться таблетки, содержащие разные наполнители, такие как микрокристаллическая целлюлоза, натрийцитрат, кальцийкарбонат, дикальцийфосфат и глицин вместе с разными дисинтеграторами, такими как крахмал (и предпочтительно кукурузный, картофельный и крупяной крахмал), альгиновая кислота и некоторые комплексные силикаты, вместе с гранулирующими связывающими веществами, подобными поливинил-пирролидону, сахарозе, желатину и гуммиарабику. Кроме того, при таблетировании очень часто используются смазывающие агенты, такие как магнийстеарат, натрийлаурилсульфат и тальк. Твердые композиции аналогичного вида могут также использоваться в качестве наполнителей в желатиновых капсулах; предпочтительные материалы, в данной связи, включают также лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Если для перорального введения желательны водные суспензии и/или эликсиры,активный ингредиент может быть использован с рядом подслащающих или вкусовых агентов,окрашивающим веществом или красителями и,если потребуется, с эмульгирующим и/или суспендирующим агентами, а также, вместе с такими разбавителями как вода, этанол, пропиленгликоль, глицерин и рядом подобных им сочетаний. В случае животных они преимущественно содержатся в пище животных или в питьевой воде в концентрации 5-5000 млн-1,предпочтительно 25-500 млн-1. Для парентерального введения (внутримышечного, внутриперитонеального, подкожного и внутривенного) обычно готовят стерильный инъецируемый раствор активного ингредиента. Могут употребляться растворы терапевтического соединения настоящего соединения в кунжутном или в арахисовом масле или в водном пропиленгликоле. При необходимости водные растворы должны быть соответственно доведены и забуферены, предпочтительно до рН выше 8,0, а жидкому разбавителю вначале нужно 29 придать изотоничность. Эти водные растворы пригодны для целей внутривенных инъекций. Маслянистые растворы пригодны для целей внутрисуставных, внутримышечных и подкожных инъекций. Получение всех этих растворов легко осуществляется в стерильных условиях обычными фармацевтическими методиками,хорошо известными рядовым специалистам в данной области техники. В случае животных,соединения могут быть введены внутримышечно или подкожно в дозах от около 0,1 до 50 мг/кг/день, преимущественно 0,2-10 мг/кг/день в виде однократной дозы или до 3-х отдельных доз. Настоящее изобретение иллюстрируется нижеследующими примерами, но в деталях не ограничивается ими. Пример 1. Гидрохлорид гидроксиамида(1996), в 30 мл тетрагидрофурана приливали бис(триметилсилил)амид лития (40 мл, 1 М в тетрагидрофуране, 39,8 ммоль). Полученную смесь перемешивали в течение 1 ч при -45 С и затем вновь охлаждали до -78 С. Затем добавляли аллилбромид (5,2 мл, 63,7 ммоль). Через 2 ч данную реакцию гасили добавлением 1 М водного хлористого водорода при -78 С. Полученную смесь экстрагировали диэтиловым эфиром. Объединенные эфирные экстракты промывали насыщенным раствором соли (рассолом) и полученную смесь сушили над сульфатом натрия. После фильтрации и концентрирования полученного фильтрата сырой продукт очищали хроматографией на силикагеле (элюирование смесью этилацетат/гексан 1:5) с получением 1 трет-бутилового эфира 5-метилового эфира(b) Газообразный озон барботировали через перемешиваемый, охлажденный (-78 С) раствор 1-трет-бутилового эфира 5-метилового эфира (2R,4R)-4-аллил-2-бензилоксикарбониламинопентандиовой кислоты (5,0 г, 12,8 ммоль) в 100 мл смеси метанол/ метиленхлорид 10:1, и 0,73 мл уксусной кислоты до сохранения голубой окраски. Затем через этот раствор барботировали газообразный азот до утраты голубой окраски. Данную смесь нагревали до температуры окружающей среды и добавляли диметилсульфид (2,8 мл, 3,83 ммоль). Эту смесь перемешивали в течение 48 ч, разбавляли метиленхлоридом и промывали 10%-ным водным карбонатом натрия, насыщенным раствором соли и полученную смесь сушили над сульфатом на 002546 30 трия. Фильтрация и концентрирование данного фильтрата дает 1-бензиловый эфир 2-третбутиловый эфир 4-метиловый эфир (2R,4S)-6 метоксипиперидин-1,2,4-трикарбоновой кислоты в виде прозрачного масла, которое использовали на последующей стадии без очистки.(c) Смесь 1-бензилового эфира 2-третбутилового эфира 4-метилового эфира (2R,4S)6-метоксипиперидин-1,2,4-трикарбоновой кислоты (4,85 г, 11,9 ммоль) и 10%-ного палладия на угле (500 мг) в 100 мл этанола встряхивали при 45 psi в атмосфере газообразного водорода в течение 1,5 ч. Данную смесь фильтровали через нейлон и полученный фильтрат концентрировали до получения 2-трет-бутилового эфира 4-метилового эфира (2R,4R)-пиперидин-2,4 дикарбоновой кислоты в виде светло-желтого масла, которое использовали на последующей стадии без дополнительной очистки.(2R,4R)-пиперидин-2,4 дикарбоновой кислоты (2,7 г, 11,1 ммоль) и триэтиламина (4,6 мл, 33,3 ммоль) в 30 мл метиленхлорида добавляли 4-метоксибензолсульфонилхлорид (2,3 г, 11,1 ммоль). Полученную смесь нагревали до температуры окружающей среды и перемешивали в течение 4-х часов. Эту реакцию гасили добавлением водного аммонийхлорида и полученную смесь экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли и эту органическую смесь сушили над сульфатом натрия. После фильтрации и концентрирования полученного фильтрата образовавшийся сырой продукт очищали хроматографией на силикагеле (элюирование смесью этилацетат/гексан 3:8) с получением 2-трет-бутилового эфира 4 метилового эфира (2R,4R)-1-(4-метоксибензолсульфонил)пиперидин-2,4-дикарбоновой кислоты.(0 С) раствору 2-трет-бутилового эфира 4 метилового эфира (2R,4R)-1-(4-метокси бензолсульфонил)пиперидин-2,4-дикарбоновой кислоты (4,4 г, 10,6 ммоль) в 30 мл метиленхлорида добавляли по каплям 10 мл трифторуксусной кислоты. Полученную смесь перемешивали в течение 1 ч при 0 С и в течение 8 ч при температуре окружающей среды. Осуществляли концентрирование 4-метилового эфира (2R,4R)-1(4-метоксибензолсульфонил)пиперидин-2,4 дикарбоновой кислоты, который использовали на последующей стадии без очистки.(f) К перемешиваемому раствору из 4 метилового эфира (2R,4R)-1-(4-метоксибензолсульфонил)пиперидин-2,4-дикарбоновой кислоты (4,4 г, 12,3 ммоль), O-бензилгидроксиламингидрохлорида (2,15 г, 13,5 ммоль) и триэтиламина (5,15 мл, 36,9 ммоль) добавляли бензотриазол-1-илокси-трис(диметиламино)фосфонийгексафторфосфат (6,0 г, 12,3 ммоль) при темпе 31 ратуре окружающей среды. Полученную смесь перемешивали в течение 24 ч. Данную смесь разбавляли этилацетатом и промывали 1 М водным раствором хлористого водорода, водным раствором бикарбоната натрия и насыщенным раствором соли. Эту органическую смесь сушили над сульфатом магния, фильтровали и полученный фильтрат концентрировали. Сырой остаток очищали хроматографией на силикагеле(элюирование раствором 5%-ного метанола в метиленхлориде) с получением метилового эфира (2R,4R)-2-бензилокси карбамоил-1-(4 метоксибензолсульфонил)пиперидин-4-карбоновой кислоты в виде бесцветного твердого вещества.(0 С) раствору метилового эфира (2R,4R)-2 бензилоксикарбамоил-1-(4-метокси бензолсульфонил)пиперидин-4-карбоновой кислоты (4,0 г,8,6 ммоль) в 10 мл метанол/вода 9:1 добавляли моногидрат гидроксида лития (1,8 г, 43 ммоль). Полученную смесь перемешивали в течение 2-х часов перед добавлением смолы Амберлит IR120 (96 г). Через 15 мин данную смесь фильтровали и полученный фильтрат концентрировали с получением (2R,4R)-2-бензилоксикарбамоил-1(4-метоксибензолсульфонил)пиперидин-4-карбоновой кислоты, которую использовали на последующей стадии без очистки.(0,47 мл, 3,33 ммоль) добавляли бензотриазол-1 илокси-трис(диметиламино)фосфоний гексафторфосфат (535 мг, 1,21 ммоль) при температуре окружающей среды. Образовавшуюся смесь перемешивали в течение 24 ч. Эту смесь разбавляли этилацетатом и промывали 1 М водным раствором хлористого водорода, водным раствором бикарбоната натрия и насыщенным раствором соли. Данную органическую смесь сушили над сульфатом магния, фильтровали, а полученный фильтрат концентрировали. Полученный сырой остаток очищали хроматографией на силикагеле (элюирование раствором 2% метанола в метиленхлориде) с получением третбутилового эфира (2R, 4R)-[2-бензилоксикарбамоил-1-(4-метоксибензолсульфонил)пиперидин-4-карбонил]пиперазин-1-карбоновой кислоты в виде бесцветного твердого вещества.(2R,4R)-[2-бензилоксикарбамоил-1-(4-метоксибензолсульфонил)пиперидин-4-карбонил]пиперазин-1-карбоновой кислоты (500 мг, 0,81 ммоль) и 5% палладия на сульфате бария (250 мг) в 10 мл метанола встряхивали в атмосфере газообразного водорода 40 psi в течение 1,5 ч. Фильтровали через нейлон и концентрировали данный фильтрат, получая трет-бутиловый эфир 32 золсульфонил)пиперидин-4-карбонил]пиперазин-1-карбоновой кислоты в виде бесцветного твердого вещества, которое использовали на последующей стадии без очистки.(j) Через охлажденный раствор (0 С) третбутилового эфира (2R,4R)-[2-гидроксикарбамоил-1-(4-метоксибензолсульфонил)пиперидин 4-карбонил]пиперазин-1-карбоновой кислоты(420 мг, 0,08 ммоль) барботировали газообразный хлористый водород в течение 10 мин. После дополнительных 20 мин эту смесь концентрировали с получением гидрохлорида гидроксиамида(2R,4R)-1-(4-метоксибензолсульфонил)-4-(пиперазин-1-карбонил)пиперидин-2 карбоновой кислоты в виде бесцветного твердого вещества. Масс-спектр (химическая ионизация при атмосферном давлении; базовый режим) m/z (М+Н) 427, 366; 1(а). К перемешиваемому раствору из 2 трет-бутилового эфира 4-метилового эфира(920 мг, 3,78 ммоль) и триэтиламина (1,58 мл,11,3 ммоль) в 10 мл метиленхлорида добавляли раствор 3-(4-фторфенокси)пропан-1-сульфонилхлорида (1,05 г, 4,16 ммоль) в 2 мл метиленхлорида в атмосфере азота. Данную смесь перемешивали в течение 16 ч при температуре окружающей среды (22 С), затем разбавляли 20 мл 1 н. хлористо-водородной кислоты и 20 мл метиленхлорида. Органический слой удаляли и промывали насыщенным раствором соли и сушили над сульфатом натрия. После фильтрации и концентрирования получали 2,8 г желтого масла, которое очищали флэш-хроматографией(0 С) раствору 2-трет-бутилового эфира 4 метилового эфира (2R,4R)-1-[3-(4-фторфенокси)пропан-1-сульфонил]пиперидин-2,4-дикарбоновой кислоты (1,15 г, 2,5 ммоль) в 10 мл метиленхлорида добавляли 10 мл трифторуксусной кислоты. Данную смесь оставляли нагреваться до температуры окружающей среды(22 С) в течение 16 ч. Эту нагретую смесь концентрировали в вакууме с получением 970 мг сырого 4-метилового эфира (2R,4R)-1-[3-(4 33 фторфенокси)пропан-1-сульфонил]пиперидин 2,4-дикарбоновой кислоты в виде оранжевого твердого вещества.(c) К перемешиваемому раствору 4 метилового эфира (2R,4R)-1-[3-(4-фторфенокси) пропан-1-сульфонил]пиперидин-2,4-дикарбоновой кислоты (970 мг, 2,4 ммоль) в 5 мл метиленхлорида добавляли триэтиламин (1,0 мл, 7,2 ммоль) и гидрохлорид O-бензилгидроксиламина(410 мг, 2,64 ммоль) при температуре окружающей среды (22 С). К образовавшемуся раствору добавляли гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)фосфония(1,17 г, 2,64 ммоль) и данную смесь перемешивали в течение 16 ч в атмосфере азота. Эту смесь разбавляли 25 мл 1 н. хлористоводородной кислоты и 25 мл этилацетата. Органический слой удаляли, а водный слой экстрагировали этилацетатом (2 х). Объединенные органические слои промывали насыщенным водным раствором карбоната натрия (1 х) и насыщенным раствором соли (1 х). Полученный органический слой сушили (сульфат натрия),фильтровали и полученный фильтрат концентрировали в вакууме. Очистка вязкого желтого остатка флэш-хроматографией (элюирование этилацетат/гексаны 1:1) дает 810 мг метилового эфира (2R,4R)-2-бензилоксикарбамоил-1-[3-(4 фторфенокси)пропан-1-сульфонил]пиперидин 4-карбоновой кислоты в виде прозрачного масла.(d) Смесь метилового эфира (2R,4R)-2 бензилоксикарбамоил-1-[3-(4-фторфенокси) пропан-1-сульфонил]пиперидин-4-карбоновой кислоты (800 мг, 1,57 ммоль) и 200 мг 5%-ного палладия на сульфате бария в 15 мл метанола встряхивали в аппарате Парра в атмосфере газообразного водорода 40 psi в течение 2-х ч. Катализатор удаляли пропусканием смеси через 45 мкм-овый нейлоновый фильтр и полученный фильтрат концентрировали с получением 650 мг метилового эфира (2R,4R)-1-[3-(4-фторфенокси) пропан-1-сульфонил]-2-гидроксикарбамоилпиперидин-4-карбоновой кислоты в виде белой пены; MS (химическая ионизация при атмосферном давлении) кислый режим, 417 (М-1); 1H ЯМР (400 МГц, СDСl3)6,94-6,97 (м,2 Н), 6,80-6,83 (м, 2 Н), 4,56 (с, 1 Н), 4,03 (т, 2 Н,J=5,3 Гц), 3,83 (д, 1 Н, J=12,9 Гц), 3,68 (с, 3 Н),3,15-3,28 (м, 3 Н), 2,76 (т, 1 Н, J=11,5 Гц), 2,54 (д,1 Н, J=13,5 Гц), 2,26 (д, 2 Н, J=5,9 Гц), 2,02 (м,1 Н, J=13,0 Гц), 1,73-1,78 (м, 1 Н), 1,56-1,62 (м,1 Н). Пример 3. (2R,4R)-1-[3-(4-Фторфенокси) пропан-1-сульфонил]-2-гидроксикарбамоилпиперидин-4-карбоновая кислота. К перемешиваемому охлажденному (0 С) раствору метилового эфира (2R,4R)-1-[3-(4 фторфенокси)пропан-1-сульфонил]-2-гидрокси карбамоилпиперидин-4-карбоновой кислоты(10:1) добавляли моногидрат гидроксида лития(120 мг, 2,88 ммоль). Через 3 ч при 0 С добавляли предварительно промытую (метанол) смолу Амберлит (4,1 г). Эту смесь фильтровали, а фильтрат концентрировали с получением 370 г(2R,4R)-1-[3-(4-фторфенокси)пропан-1-сульфонил]-2-гидроксикарбамоилпиперидин-4-карбоновой кислоты в виде белой пены; MS (химическая ионизация при атмосферном давлении) кислотный режим, 403 (М-1). Пример 4. Метиловый эфир (2R,4R)-1-[4(4-фторбензилокси)бензолсульфонил]-2-гидроксикарбамоилпиперидин-4-карбоновой кислоты. 4-(4-Фторбензилокси)бензолсульфонилхлорид. MS; 465 (М-1). Названное соединение примера 4 получали способом, аналогичным способу, описанному в примере 2 с использованием определенных реагентов. Пример 5. (2R,4R)-1-[4-(4-Фторбензилокси)бензолсульфонил]-2-гидрокси-карбамоилпиперидин-4-карбоновая кислота. Названное соединение примера 5 получали способом, аналогичным способу, описанному в примере 3, начиная с метилового эфира 1-[4-(4 фторбензилокси)бензолсульфонил]-2-гидроксикарбамоилпиперидин-4-карбоновой кислоты. Пример 6. Изопропиловый эфир 2R,3S-1[4-(4-фторбензилокси)бензолсульфонил]-2-гидроксикарбамоилпиперидин-3-илкарбаминовой кислоты.[2,1-с][1,4]оксазин-9-карбоновой кислоты (8,28 г, 2,86 ммоль) в 100 мл тетрагидрофурана добавляли 2,39 мл концентрированной хлористоводородной кислоты. Через 5 мин данную смесь концентрировали досуха. Полученное твердое вещество суспендировали в этилацетате и смесь перемешивали в течение часа. Твердые частицы собирали фильтрацией, ополаскивали этилацетатом и сушили с получением 9,04 г белого твердого вещества. Два грамма данного твердого вещества растворяли в 26 мл 6 н. хлористо-водородной кислоты и нагревали при температуре кипения с обратным холодильником в течение 6 ч. Полученную смесь охлаждали до 0 С и нейтрализовали 3 н. гидрооксидом натрия и концентрировали в вакууме. Полученные твердые частицы суспендировали в хлороформе и фильтровали через 45 мкм-овый нейлоновый фильтр. Данный фильтрат концентрировали до получения желтого масла, которое очищали флэш-хроматографией (элюирование гексан/этилацетат 2:1 с 1%-ной уксусной кислотой) с получением 802 мг [4S-4,9,9 а]-1-оксо-4-фенилоктагидропиридо[2,1-с][1,4]оксазин-9-карбоновой кислоты в виде белого твердого вещества.[1,4]оксазин-9-карбоновой кислоты (568 мг, 2,06 ммоль) в 15 мл бензола добавляли триэтиламин(0,44 мл, 2,06 ммоль) при 22 С в атмосфере азота. Эту смесь перемешивали при 22 С в течение 45 мин и при температуре кипения с обратным холодильником в течение 50 мин перед добавлением 2-пропанола (3,2 мл, 41,2 ммоль). После дополнительных 20 ч кипения с обратным холодильником данную смесь охлаждали до 22 С и концентрировали в вакууме. Остаток помещали в этилацетат и полученный раствор промывали 5%-ной лимонной кислотой, водой, насыщенным водным раствором бикарбоната натрия, и насыщенным раствором соли. Органический слой сушили (сульфат натрия), фильтровали и полученный фильтрат концентрировали в вакууме. Полученный желтый остаток очищали флэш-хроматографией (элюирование гексаны/этилацетат 3:1) с получением 402 мг изопропилового эфира [4S-4,9,9 а]-1-оксо-4-фенилоктагидропиридо[2,1-с][1,4]оксазин-9-ил)карбаминовой кислоты в виде белого твердого вещества.(c) Смесь изопропилового эфира [4S4,9,9 а]-1-оксо-4-фенилоктагидропиридо(900 мг, 2,71 ммоль) и 20%-ной гидроокиси палладия на угле (920 мг) в 77 мл этанол/вода (10:1) встряхивали в аппарате Парра в атмосфере газообразного водорода при давлении 45 psi в течение 72-х часов. Катализатор удаляли пропусканием данной смеси через 0,45 мкм-овый нейлоновый фильтр и полученный фильтрат концентрировали с получением 610 мг 2R,3S-3 изопропоксикарбониламинопиперидин-2-карбоновой кислоты в виде белого твердого вещества.(d) К перемешиваемому раствору 2R,3S-3 изопропоксикарбониламинопиперидин-2-карбоновой кислоты (320 мг, 1,39 ммоль) в 5 мл метиленхлорида добавляли триэтиламин (0,58 мл,4,17 ммоль) с последующим добавлением 4-(4 фторбензилокси) бензолсульфонилхлорида (460 мг, 1,53 ммоль). Через 16 ч при 22 С данную смесь распределяли между 1 н. хлористоводородной кислотой и этилацетатом. Органический слой удаляли и промывали насыщенным раствором соли и сушили над сульфатом натрия. Фильтрация и концентрирование полученного фильтрата дает 480 мг сырой 2R,3S-1-[4(4-фторбензилокси)бензолсульфонил]-3-изопропоксикарбониламинопиперидин-2-карбоновой кислоты в виде светло-желтого твердого вещества. 36 кислоты (380 мг, 0,77 ммоль) в 5 мл метиленхлорида добавляли триэтиламин (0,32 мл, 2,31 ммоль) с последующим добавлением гексафторфосфата бензотриазол-1-илокси-трис (диметиламино)фосфония (510 мг, 1,15 ммоль). Полученный раствор перемешивали в течение 2-х минут при 0 С в атмосфере азота перед добавлением гидрохлорида О-(триметилсилилэтил) гидроксиламина (195 мг, 1,15 ммоль). Данную смесь оставляли медленно нагреваться до 22 С в течение 14 ч. Смесь концентрировали в вакууме и полученный остаток разбавляли водой и экстрагировали смесью этилацетат/диэтиловый эфир (1:1; 3 х). Объединенные органические экстракты промывали насыщенным водным раствором карбоната натрия (2 х), водой (2 х) и насыщенным раствором соли (1 х). Образовавшийся органический слой сушили (сульфат магния), фильтровали и полученный фильтрат концентрировали в вакууме. Сконцентрированный желтый остаток очищали флэш-хроматографией(элюирование гексаны/этилацетат 65:35) с получением 300 мг изопропилового эфира 2R,3S-[1-[4-(4-фторбензилокси)бензолсульфонил]-2-(2-триметилсиланилэтокси карбамоил)пиперидин-3-ил]карбаминовой кислоты в виде белой пены. MS; 610 (М+1).(0 С) раствору изопропилового эфира 2R, 3S-[1[4-(4-фторбензилокси)бензолсульфонил]-2-(2 триметилсиланилэтоксикарбамоил)пиперидин 3-ил] карбаминовой кислоты (265 мг, 0,44 ммоль) в 4-х мл метиленхлорида добавляли 3 мл трифторуксусной кислоты. Полученный бесцветный раствор оставляли нагреваться до 23 С в течение 2-х часов и перемешивали в течение дополнительных 28 ч. Данную смесь концентрировали в вакууме до твердого вещества/ пены, которую суспендировали в смеси этилацетат/гексаны (1:6) и перемешивали в течение 10 ч. Полученные белые твердые частицы собирали фильтрацией, ополаскивали гексанами и дополнительно очищали флэш-хроматографией(элюирование этилацетат/гексаны 7:3 с 1%-ной уксусной кислотой) с получением 130 мг изопропилового эфира 2R,3S-1-[4-(4-фторбензилокси)бензолсульфонил]-2-гидроксикарбамоилпиперидин-3-илкарбаминовой кислоты в виде белого твердого вещества/пены. MS: 510 (М+1). Пример 7. Гидроксиамид 3-(S)-4-(4'фторбифенил-4-сульфонил)-2,2-диметил тиоморфолин-3-карбоновой кислоты.(a) К перемешиваемому раствору известного (публикация РСТ WO 97/20824) 3-(S)диметилтексилсилил-2,2-диметилтетрагидро 2 Н-1,4-тиазин-3-карбоксилата (1,17 г, 3,70 ммоль) в 6 мл метиленхлорида добавляли триэтиламин (1,02 мл, 7,40 ммоль) с последующим добавлением 4'-фторбифенилсульфонилхлорида(1,0 г, 3,70 ммоль). Полученный раствор перемешивали в течение 56 ч при 23 С. Полученную 37 реакционную смесь разбавляли метиленхлоридом и промывали водой. Органический слой концентрировали в вакууме; остаток растворяли в метаноле и данную смесь нагревали при температуре кипения с обратным холодильником в течение 6 ч. Смесь охлаждали до 23 С и концентрировали в вакууме. Полученный остаток очищали флэш-хроматографией (элюирование смесью этилацетат/гексаны 3:7 с 0,1% уксусной кислотой) с получением 670 мг 3-(S)-4(4'-фторбифенил-4-сульфонил)-2,2-диметилтиоморфолин-3-карбоновой кислоты в виде белой пены/твердого вещества. MS: 427 (М+NH4).(0 С) раствору 3-(S)-4-(4'-фторбифенил-4 сульфонил)-2,2-диметилтиоморфолин-3 карбоновой кислоты (605 мг, 1,48 ммоль) в 5 мл метиленхлорида добавляли триэтиламин (0,62 мл, 4,43 ммоль) в атмосфере азота. Добавляли гексафторфосфат бензотриазол-1-илокси-трис(диметиламино) фосфония (980 мг, 2,22 ммоль) и полученный раствор перемешивали в течение 5 мин перед добавлением гидрохлорида О(триметил-силилэтил)гидроксиламина (376 мг,2,22 ммоль). Ледяную баню удаляли и данную смесь перемешивали в течение 20 ч при 23 С. Перемешанную смесь разбавляли водным раствором хлорида аммония и экстрагировали смесью этилацетат/диэтиловый эфир 1:3 (3 х). Объединенные органические экстракты промывали насыщенным водным раствором карбоната натрия (2 х), водой (1 х) и насыщенным раствором соли (1 х). Образовавшийся органический слой сушили (сульфат магния), фильтровали и полученный фильтрат концентрировали в вакууме. Остаточное желтое масло очищали флэшхроматографией (элюирование этилацетат/гексаны 3:7) с получением 650 мг (2-триметилсиланилэтокси)амид 3-(S)-4-(4'-фторбифенил-4 сульфонил)-2,2-диметилтиоморфолин-3-карбоновой кислоты в виде белой пены. MS: 523 (М 1).(с) Раствор (2-триметилсиланилэтокси) амида 3-(S)-4-(4'-фторбифенил-4-сульфонил)2,2-диметилтиоморфолин-3-карбоновой кислоты (650 мг, 1,24 ммоль) в 8 мл трифторуксусной кислоты перемешивали при 22 С в течение 16 ч. Смесь концентрировали в вакууме, а полученный остаток растирали с метиленхлоридом и диэтиловым эфиром. Растворитель удаляли с получением 550 мг желтовато-коричневого твердого вещества. Полученное твердое вещество суспендировали в смеси диэтиловый эфир/гексаны 1:1 и осторожно перемешивали в течение 20 ч. Полученные твердые частицы собирали фильтрацией (ополаскивая смесью диэтиловый эфир/гексаны 1:1) и сушили с получением 470 мг гидроксиамида 3-(S)-4-(4'-фторбифенил-4-сульфонил)-2,2-диметилтиоморфолин-3-карбоновой кислоты в виде белого твердого вещества. MS: 423(М-1).(0 С) раствору известной (публикация патента Бельгии BE 893025) 2,2-диметилтиоморфолин 3-карбоновой кислоты (600 мг, 3,42 ммоль) в 10 мл смеси вода/диоксан 1:1 добавляли 6 н. раствор гидроксида натрия (1,2 мл, 7,1 ммоль). К полученному раствору добавляли 4-(4-фторбензилокси)бензолсульфонилхлорид (1,08 г,3,77 ммоль). Через 30 и 60 мин дополнительно вносили 1 г 4-(4-фторбензилокси)бензолсульфонилхлорида и 1,2 мл 6 н раствора гидроксида натрия. Полученную смесь (рН приблизительно 12,0) разбавляли водой и экстрагировали диэтиловым эфиром (1 х). Эфирный слой промывали 1 н раствором гидроксида натрия; объединенные основные водные слои подкисляли до рН 3,0 с использованием концентрированной хлористоводородной кислоты и полученную кислую смесь экстрагировали этилацетатом (3 х). Объединенные органические экстракты сушили(сульфат натрия), фильтровали и полученный фильтрат концентрировали в вакууме с получением 820 мг 3-(S)-4-[4-(4-фторбензилокси) бензолсульфонил]-2,2-диметилтиоморфолин-3 карбоновой кислоты в виде белого твердого вещества. MS: 438 (М-1).(0 С) раствору 3-(S)-4-[4-(4-фторбензилокси) бензолсульфонил]-2,2-диметилтиоморфолин-3 карбоновой кислоты (820 мг, 1,87 ммоль) в 5 мл метиленхлорида добавляли в атмосфере азота триэтиламин (0,52 мл, 3,74 ммоль). Добавляли гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)фосфония (1,24 г, 2,81 ммоль) и полученный раствор перемешивали в течение 5 мин перед добавлением О-(трет-бутилдиметилсилил)гидроксиламина (550 мг, 3,74 ммоль). Ледяную баню удаляли и полученную смесь перемешивали в течение 16 ч при 23 С. Эту смесь разбавляли водным раствором хлорида аммония и экстрагировали этилацетатом (3 х). Объединенные органические экстракты промывали водой, насыщенным раствором соли и сушили над сульфатом натрия. Фильтрация и концентрирование полученного фильтрата дает вязкое желтое масло, которое очищали флэшхроматографией (элюент этилацетат/гексаны 1:3) с получением 270 мг (трет-бутилдиметилсилокси)амид 3-(S)-4-[4-(4-фторбензилокси) бензолсульфонил]-2,2-диметилтиоморфолин-3 карбоновой кислоты в виде белой пены. MS: 569(0 С) раствору (трет-бутилдиметилсилокси) амид 3-(S)-4-[4-(4-фторбензилокси)бензол сульфонил]-2,2-диметилтиоморфолин-3-карбоновой кислоты (270 мг, 0,47 ммоль) в 10 мл тетрагидрофурана добавляли две капли концентрированной хлористо-водородной кислоты. Через 30 39 мин смесь разбавляли 15 мл тетрагидрофурана и полученную смесь концентрировали в вакууме до объема приблизительно 5 мл. Объем доводили тетрагидрофураном до приблизительно 25 мл и смесь снова концентрировали до приблизительно 5 мл. Этот процесс повторяли дважды до того, как данную смесь концентрировали досуха. Полученные твердые частицы суспендировали в смеси гексанов и диэтилового эфира и суспендированную смесь перемешивали в течение 16 ч. Твердое вещество собирали фильтрацией, ополаскивали диэтиловым эфиром и сушили с получением 180 мг гидроксиамида 3-(S)4-[4-(4-фторбензилокси)бензолсульфонил]-2,2 диметилтиоморфолин-3-карбоновой кислоты в виде белого твердого вещества. MS: 453 (М-1). 1(д, 1 Н, J=12,7 Гц), 2,78-2,86 (м, 1 Н), 2,44-2,50 (м,1 Н), 1,35 (с, 3 Н), 1,12 (с, 3 Н). Приготовление 1. 4-(4-Фторбензилокси) бензолсульфонилхлорид. К перемешиваемому раствору дигидрата натриевой соли 4-гидроксибензолсульфоновой кислоты (5,13 г, 22,1 ммоль) в 23 мл 1 н. раствора гидроксида натрия добавляли раствор 4 фторбензилбромида (3,3 мл, 26,5 ммоль) в 20 мл этанола. Эту смесь нагревали при температуре кипения с обратным холодильником в течение двух дней, затем охлаждали до температуры окружающей среды (22 С), после чего образовывался осадок белого цвета. Хлопьевидные белые твердые частицы собирали фильтрацией,ополаскивали этилацетатом и диэтиловым эфиром и сушили с получением 4,95 г натриевой соли 4-(4-фторбензилокси)бензол сульфоновой кислоты. Перемешиваемый раствор натриевой соли 4-(4-фторбензилокси)бензолсульфоновой кислоты (13,0 г, 4,27 ммоль) в 50 мл тионилхлорида и двух капель диметилформамида осторожно нагревали при температуре кипения с обратным холодильником в течение 8 ч. Смесь концентрировали до получения твердого желтого вещества, суспендировали в этилацетате и фильтровали. Фильтрат концентрировали до получения 11,2 г 4-(4-фторбензилокси)бензолсульфонилхлорида в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, СDСl3)7,95-7,98 (м,2 Н), 7,38-7,41 (м, 2 Н), 7,08-7,12 (м, 4 Н), 5,12 (с,2 Н). Приготовление 2. 3-(4-Фторфенокси)пропан-1-сульфонилхлорид. К перемешиваемому раствору 4-фторфенола (5,0 г, 44,6 ммоль) в 50 мл толуола добавляли гидрид натрия (60%-ная дисперсия в минеральном масле, 1,78 г, 44,6 ммоль) при температуре окружающей среды (22 С). Через 20 мин медленно добавляли раствор 1,3-пропан 002546 40 сульфона (3,9 мл, 44,6 ммоль) в толуоле и эту смесь перемешивали в течение 16 ч. Реакцию гасили добавлением метанола и смесь концентрировали в вакууме до получения беловатого твердого вещества. Это твердое вещество суспендировали в этилацетате, фильтровали, а твердые частицы собирали и сушили с получением 10,9 г беловатого порошка натриевой соли 3-(4-фторфенокси)пропан-1-сульфоновой кислоты. Перемешиваемый раствор натриевой соли 3-(4-фторфенокси)пропан-1-сульфоновой кислоты (2,0 г, 7,8 ммоль) в 10 мл тионилхлорида и одной капли диметилформамида нагревали при температуре кипения с обратным холодильником в течение 16 ч. Затем полученную смесь охлаждали до 0 С, разбавляли 25 мл диэтилового эфира и реакцию гасили путем медленного добавления воды. Образовавшийся органический слой удаляли, а водный слой экстрагировали 25 мл диэтилового эфира. Объединенные органические слои промывали насыщенным раствором соли и сушили над сульфатом натрия. Фильтрование и концентрирование дает 1,75 г 3-(4-фторфенокси)пропан-1-сульфонилхлорида в виде желтого масла; 1 Н ЯМР (400 МГц, CDCl3)6,96-7,00 (м,2 Н), 6,80-6,84 (м, 2 Н), 4,10 (т, 2 Н, J=5,5 Гц), 3,91(т, 2 Н, J=7,5 Гц), 2,47-2,54 (м, 2 Н). Приготовление 3. 4'-Фторбифенилсульфонилхлорид. Хлорсульфоновую кислоту добавляли по каплям к перемешиваемому охлажденному(0 С) 4-фторбифенилу (10,2 г, 59 ммоль). Через 30 мин при 0 С реакционную смесь выливали на лед. Полученный осадок белого цвета собирали фильтрацией и растворяли в хлороформе. Хлороформный раствор промывали водой, насыщенным раствором соли, сушили над сульфатом магния и концентрировали с получением белого твердого вещества. Требуемый 4'-фторбифенилсульфонилхлорид (4,3 г) отделяли от 4'-фторбифенилсульфоновой кислоты кристаллизацией последнего из этилацетата и кристаллизацией оставшегося материала из гексанов. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы или его фармацевтически приемлемая соль, где пунктирная линия представляет необязательную двойную связь; Х представляет углерод;Y представляет углерод или серу;R8 означает атом водорода или (C1-6)алкил;Q представляет (С 6-С 10)арил(С 6-С 10)арил или (С 6-С 10)арил(C1-C6)алкокси(С 6-С 10)арил необязательно замещенный фтором, хлором, (C1C8)алкилом, (С 6-С 10)алкокси или перфтор(C1-3) алкилом; при условии, что когда Y представляет серу, R5 и R6 отсутствуют; при условии, что когда пунктирная линия представляет двойную связь, R4 и R6 отсутствуют. 2. Соединение по п.1, где Y представляет серу. 3. Соединение по п.1, где Q представляет(С 6-С 10)арил(C6-С 10)арил,(С 6-С 10)арил(С 1-C6) акокси(С 6-С 10)арил, где каждая терминальная арильная группа необязательно замещена фтором. 4. Соединение по п.1, где R7 представляет водород; 5. Соединение по п.1, где Y представляет углерод;Q представляет (C6-C10)арил(C1-C6)алкокси(С 6-С 10)арил. 6. Соединение по п.1, где, по меньшей мере, один из R7-R8 представляет радикал, иной чем водород. 7. Соединение по п.3, где, по меньшей мере, один из R7-R8 представляет радикал, иной чем водород. 8. Соединение по п.1, где, по меньшей мере, один из R7-R8 представляет (C1-C6)алкил. 9. Соединение по п.3, где, по меньшей мере, один из R7-R8 представляет (C1-C6)алкил. 10. Соединение по п.1, где, по меньшей мере, один из R7-R8 представляет метил. 11. Соединение по п.3, где, по меньшей мере, один из R7-R8 представляет метил. 12. Соединение по п.1, где R7 и R8 каждый представляет метил. 13. Соединение по п.3, где R7 и R8 каждый представляет метил. 14. Соединение по п.1, представляющее соединение, выбранное из группы, состоящей из изопропилового эфира (2R,3S)-(1-[4-(4(фторбензилокси)бензолсульфонил]-2-гидрокси 42 карбамоилпиперидин-3-ил)карбаминовой кислоты; гидроксиамида 3-(S)-4-(4'-фторбифенил-4 сульфонил)-2,2-диметилтиоморфолин-3-карбоновой кислоты; гидроксиамида 3-(S)-4-[4-(4'-фторбензилокси)бензолсульфонил]-2,2-диметилтиоморфолин-3-карбоновой кислоты. 15. Фармацевтическая композиция для (а) лечения состояния, выбранного из группы, состоящей из артрита, злокачественной опухоли,тканевого изъязвления, макулярной дегенерации, рестеноза, периодонтального заболевания,врожденного буллезного эпидермолиза, склерита, в сочетании со стандартными NSAID'S и анальгетиками и в сочетании с цитотоксическими противоопухолевыми агентами, и других заболеваний, характеризующихся металлопротеиназной активностью матрикса, AIDS, сепсисом, септическим шоком и другими заболеваниями, вовлеченными в образование фактора некроза опухолей (TNF) или для (b) ингибирования металлопротеиназ матрикса или образования фактора некроза опухолей (TNF) у млекопитающих, в том числе у человека, включающая соединение по п.1 в количестве, эффективном для такого лечения, и фармацевтически приемлемый носитель. 16. Способ ингибирования металлопротеиназ матрикса или образования фактора некроза опухолей (TNF) у млекопитающих, в том числе у человека, включающий введение указанному млекопитающему эффективного количества соединения по п.1. 17. Способ лечения состояния, выбранного из группы, состоящей из артрита, злокачественной опухоли, тканевого изъязвления, макулярной дегенерации, рестеноза, периодонтального заболевания, врожденного буллезного эпидермолиза, склерита, соединение формулы I может быть использовано в сочетании со стандартными NSAID'S и анальгетиками и в сочетании с цитотоксическими противоопухолевыми агентами, и других заболеваний, вовлеченных в образование фактора некроза опухолей (TNF) у млекопитающих, в том числе у человека, включающий введение указанному млекопитающему соединения по п.1 в количестве, эффективном для лечения такого состояния.
МПК / Метки
МПК: C07D 211/62, A61K 31/445
Метки: кислоты, арилсульфонилгидроксамовой, производные
Код ссылки
<a href="https://eas.patents.su/22-2546-proizvodnye-arilsulfonilgidroksamovojj-kisloty.html" rel="bookmark" title="База патентов Евразийского Союза">Производные арилсульфонилгидроксамовой кислоты</a>
Замещенные (сульфиновой кислоты, сульфоновой кислоты, сульфониламино или сульфиниламино) n-[(аминоиминометил) фенилалкил] азагетероциклил-амидные производные

Номер патента: 700
Опубликовано: 28.02.2000
Авторы: Чини Дэниел Л., Спада Альфред П., Евинг Вильям Р., Чои-Следески Енг Ми, Мэйсон Джонатан Стивен, Бекер Майкл Р., Паулс Генри В.
МПК: C07D 401/06, A61K 31/44
Метки: n-[(аминоиминометил, сульфоновой, кислоты, сульфиновой, азагетероциклил-амидные, сульфониламино, производные, фенилалкил, сульфиниламино, замещенные
Формула / Реферат:
1. Соединение формулы (1) представляет фенил или моноциклический гетероарил; R представляет водород, необязательно замещенный алкил, необязательно замещенный аралкил, необязательно замещенный гетероаралкил или гидроксиалкил; R1 представляет водород, R3S(O)p или R3R4NS(O)p-; R2 представляет водород или, когда X5 и Х5', взятые вместе, представляют =NR5, R2 представляет водород, необязательно замещенный низший алкил,...
Производные эпоксиянтарной кислоты

Номер патента: 438
Опубликовано: 26.08.1999
Авторы: Такахаси Тосихиро, Номура Ютака, Масаки Мицуо, Хара Каору, Йосино Ясуси
МПК: C07D 303/46, A61K 31/335
Метки: эпоксиянтарной, производные, кислоты
Формула / Реферат:
1. Производные эпоксиянтарной кислоты следующей формулы в которой R1 представляет атом водорода, алкил с 1-30 атомами углерода, арил с 6-40 атомами углерода или аралкил с 7-40 атомами углерода; каждый из R2 и R3 независимо представляет арил с 6-40 атомами углерода, аралкил с 7-20 атомами углерода или алкил с 3-10 атомами углерода; Х представляет -О- или -NR4; R4 представляет атом водорода, алкил с 1-10 атомами углерода или аралкил с 7-20...
Производные арилоксиарилсульфониламиногидроксамовой кислоты

Номер патента: 2490
Опубликовано: 27.06.2002
Автор: Робинсон Ральф Пелтон
МПК: A61P 35/00, A61K 31/16, C07C 311/29...
Метки: кислоты, арилоксиарилсульфониламиногидроксамовой, производные
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемые соли, где R1 представляет собой (С1-С6)алкил; R2 представляет собой (С1-С6)алкил; или R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют кольцо, выбранное из (С5-С7)циклоалкила, 4-тетрагидропиранила и 4-пиперидинила; R3 представляет собой водород или (С1-С6)алкил; а Y представляет собой заместитель по любому из атомов углерода фенильного кольца, способному...
Производные фенилуксусной кислоты

Номер патента: 1380
Опубликовано: 26.02.2001
Авторы: Мюллер Бернд, Аммерманн Эберхард, Мюллер Рут, Штратман Зигфрид, Лоренц Гизела, Заутер Губерт, Харриес Фолкер, Байер Губерт, Харреус Альбрехт, Гётц Норберт, Гроте Томас
МПК: A01N 37/36, C07D 231/20, C07C 251/60...
Метки: кислоты, производные, фенилуксусной
Формула / Реферат:
1. Производные фенилуксусной кислоты формулы I в которой заместители и индекс имеют следующие значения: Х означает NOCH3, СНОСН3 и СНСН3; Y означает NR и О; R, R1 означают водород и С1-C4алкил; R2 означает водород, С1-C6алкил, С1-C4галогеналкил и C3-С6циклоалкил; R3 С2-C6алкинил или С2-C6алкенил, причем эти остатки могут быть частично либо полностью галогенированы или могут нести от одного до трех радикалов из числа следующих: циано, нитро,...
Производные гидроксимовой кислоты и их применение

Номер патента: 278
Опубликовано: 25.02.1999
Авторы: Кирио Йоши, Сасаки Норио, Миллигэн Брюс, Маеда Такако, Тошима Норишидже, Саваи Нобумитсу, Ворс Жан-Пьер, Перес Жозеф, Гент Даниель Б.
МПК: A01N 37/52, C07C 259/04
Метки: гидроксимовой, кислоты, производные, применение
Формула / Реферат:
1. Производные гидроксимовой кислоты формулы (I):***где G представляет либо Gi, или G2, или G3, или G4 формулы:***Хь X2, X3 представляют, независимо, водород или атом галогена, гидрокси, меркапто, нитро, тиоцианато, азидогруппу, цианогруппу; алкил, галоидалкил, цианоалкил, алкокси, галоидал-кокси, цианоалкокси, алкилтио, галоидалкил-тио, цианоалкилтио, алкилсульфинил, галоидал-килсульфинил, алкилсульфонил, галоидалкил-сульфонил, причем указанные...