Способ получения бензоксаборолов
Номер патента: 21532
Опубликовано: 30.07.2015
Авторы: Конде Хосе Х., Заджак Мэттью Аллен, Ковальски Энтони Джон



Формула / Реферат
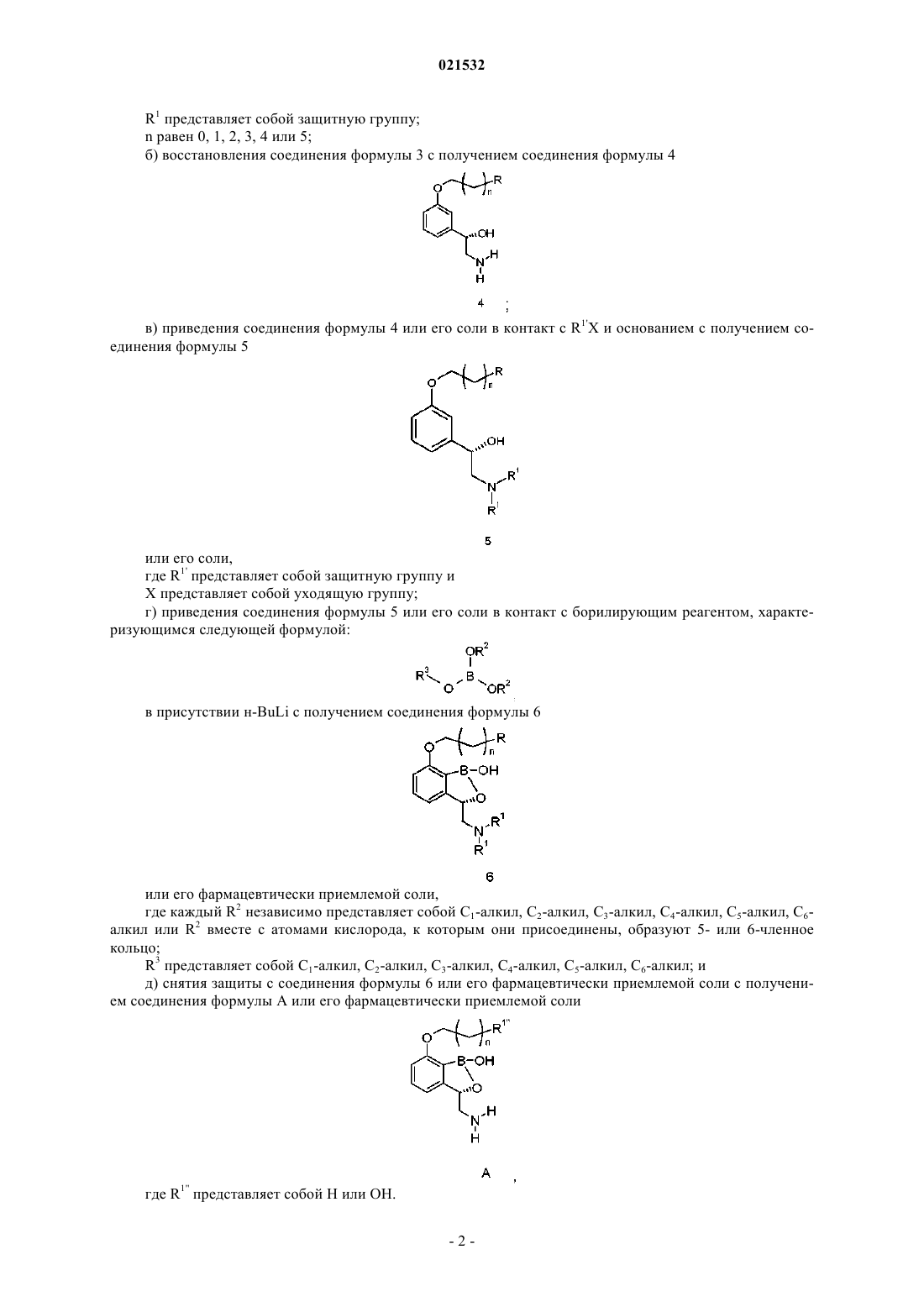
1. Способ получения соединения формулы А, включающий приведение соединения формулы 6

или его фармацевтически приемлемой соли в контакт с реагентом для снятия защиты с получением соединения формулы А

или его фармацевтически приемлемой соли,
где R представляет собой Н или OR1;
каждый R1 и каждый R1' независимо представляют собой защитные группы;
R1'' представляет собой Н или ОН;
n равен 0, 1, 2, 3, 4 или 5,
где соединение формулы 6 получают приведением соединения формулы 5

или его соли в контакт с борилирующим реагентом, характеризующимся следующей формулой:

в присутствии алкиллитиевого реагента,
где каждый R2 независимо представляет собой С1-С6-алкил или R2 вместе с атомами кислорода, к которым они присоединены, образуют 5- или 6-членное кольцо и
R3 представляет собой C1-алкил, С2-алкил, С3-алкил, С4-алкил, C5-алкил, С6-алкил.
2. Способ по п.1, где
R представляет собой OR1;
R1 представляет собой группы -СН(Ra)фенил-(Rb)х, триалкилсилил, тетрагидропиранил, -СН2ОСН3 или -СН2ОСН2СН2ОСН3, где Ra представляет собой Н или метил; Rb представляет собой метокси или C1-алкил, С2-алкил или С3-алкил и х равен 0, 1 или 2;
каждый R1' независимо представляет собой -CH(Rc)фенил-(Rd)y, где Rc представляет собой Н или метил, каждый Rd независимо представляет собой метокси или C1-алкил, С2-алкил или С3-алкил; n равен 0, 1, 2 или 3 и у равен 0, 1 или 2.
3. Способ по п.1, где
R представляет собой OR1;
R1 и каждый R1' представляют собой бензильные группы;
реагент для снятия защиты представляет собой восстановитель;
n равен 1;
соединение формулы 6 восстанавливают в присутствии HCl с получением гидрохлоридной соли соединения формулы А.
4. Способ по п.3, где реагент для снятия защиты представляет собой Pd/C или Pt/C в присутствии Н2 или их смесь, Н2 над гидроксидом палладия или реагент для гидрогенизации в условиях каталитического переноса.
5. Способ по п.1, где борилирующий реагент представляет собой изопропилпинаколборат или три-С1-борат, три-С2-борат или три-С3-борат; n представляет собой число от 0 до 3 и алкиллитиевый реагент представляет собой н-BuLi, н-гексиллитий или втор-BuLi.
6. Способ по п.1 или 5, где борилирующий реагент представляет собой изопропилпинаколборат или триметилборат; n равен 2 и алкиллитиевый реагент представляет собой н-BuLi.
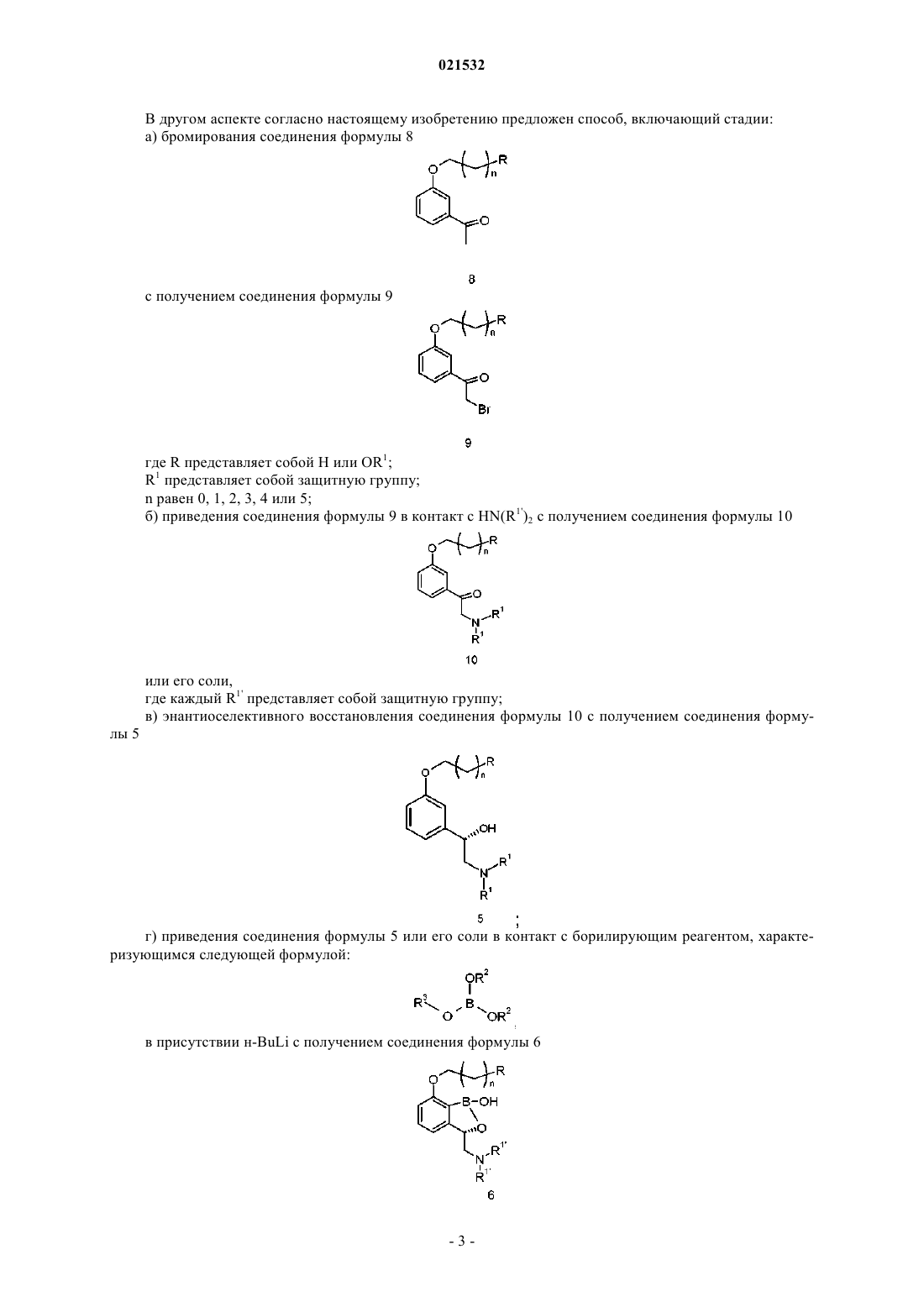
7. Способ по п.1, где соединение формулы 5 или его соль получают
либо энантиоселективным восстановлением соединения формулы 10

или его соли,
либо приведением R1'X в основных условиях в контакт с соединением формулы 4

или его солью,
где X представляет собой уходящую группу.
8. Способ по п.7, где соединение формулы 5 или его соль получают с использованием Н2 и комплекса трифенилфосфина (S)-2-[(SP)-2-(дифенилфосфино)ферроценил]-4-изопропил-2-оксазолина и дихлорида рутения(II) или Н2 и (R)-BINAP-Ru-(R,R)-(+)-DPEN Cl ([(R)-2,2'-бис-(дифенилфосфино)-1,1'-бинафтил]-Ru-(R,R)-(+)-[1,2-дифенилэтилендиамин] Cl); n равен 2.
9. Способ по п.7, где соединение формулы 5 или его соль получают приведением соединения формулы 4 или его соли в контакт с бензилбромидом в присутствии карбоната или гидроксида.
10. Способ по п.9, где соединение формулы 4 или его соль получают приведением соединения формулы 2

в контакт с нитрометаном в присутствии хирального реагента с получением соединения формулы 3

с последующим восстановлением нитрогруппы до аминогруппы.
11. Способ по п.10, где хиральный реагент представляет собой дигидрохлорид 1,7,7-триметил-N-(пиридин-2-илметил)бицикло[2.2.1]гептан-2-амина; (4S)-4-этил-2-{1-этил-1-[(4S)-4-(1-метилэтил)-4,5-дигидро-1,3-оксазол-2-ил]пропил}-4,5-дигидро-1,3-оксазол; (S)-4-(трет-бутил)-2-(2-((S)-4-метил-4,5-дигидрооксазол-2-ил)пропан-2-ил)-4,5-дигидрооксазол или N1,N2-бис-[(1R,2R,4R)-1,7,7-триметилбицикло[2.2.1]гепт-2-ил]-1,2-этандиамин в присутствии Cu(OAc)2; n равен 2.
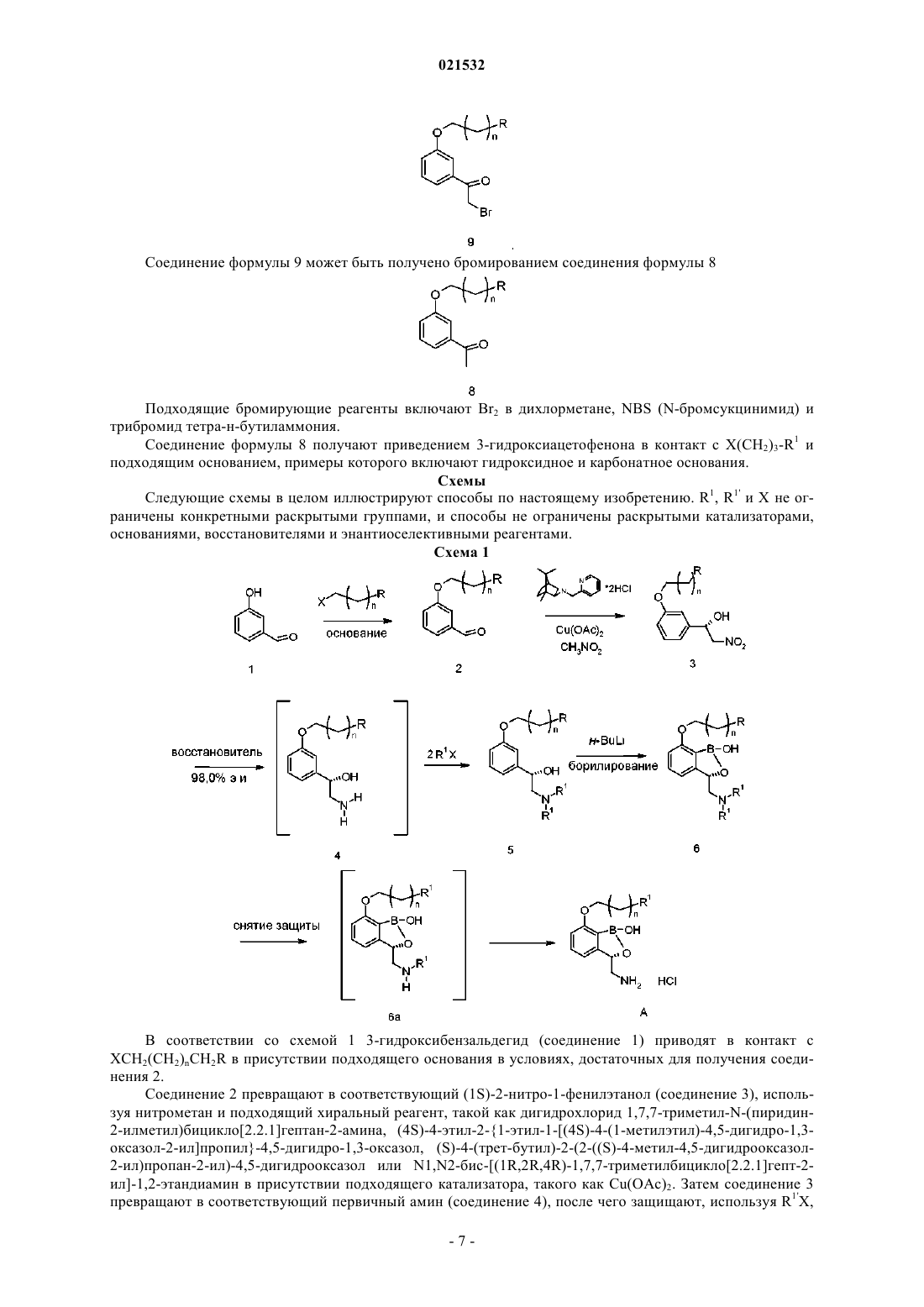
12. Способ по п.7, где соединение формулы 10 или его соль получают приведением HN(R1')2 в контакт с основанием и соединением формулы 9

13. Способ по п.12, где соединение формулы 9 получают бромированием соединения формулы 8

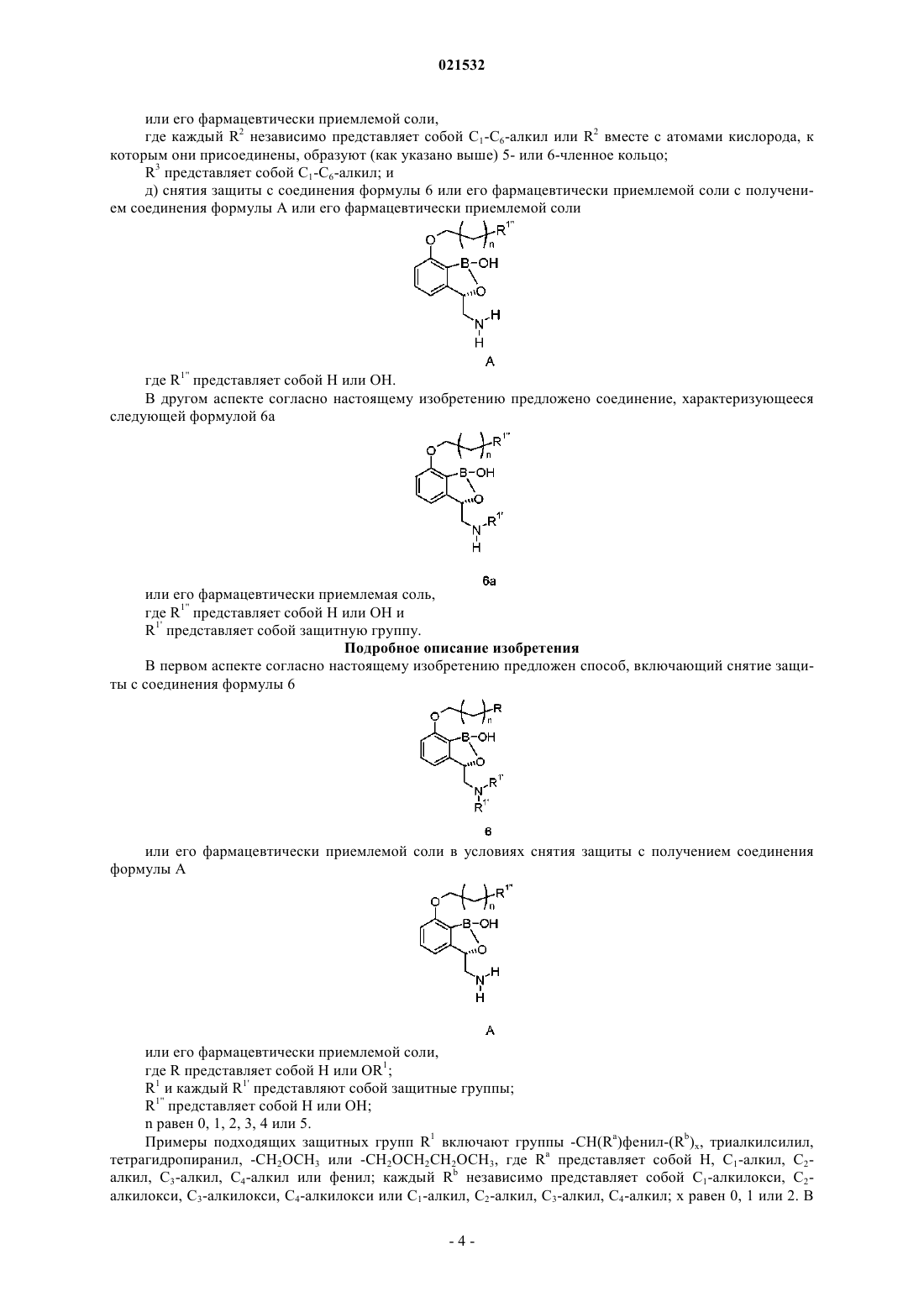
14. Способ получения соединения формулы А, включающий стадии:
а) приведения соединения формулы 2

в контакт с нитрометаном в присутствии хирального реагента с получением соединения формулы 3

где R представляет собой Н или OR1; R1 представляет собой защитную группу и n равен 0, 1, 2, 3, 4 или 5;
б) восстановления соединения формулы 3 с получением соединения формулы 4

или его соли;
в) приведения соединения формулы 4 или его соли в контакт с R1'X и основанием с получением соединения формулы 5

или его соли,
где R1' представляет собой защитную группу и X представляет собой уходящую группу;
г) приведения соединения формулы 5 или его соли в контакт с борилирующим реагентом, характеризующимся следующей формулой:

в присутствии н-BuLi с получением соединения формулы 6

или его фармацевтически приемлемой соли,
где каждый R2 независимо представляет собой C1-алкил, С2-алкил, С3-алкил, С4-алкил, C5-алкил, С6-алкил или R2 вместе с атомами кислорода, к которым они присоединены, образуют 5- или 6-членное кольцо;
R3 представляет собой C1-алкил, С2-алкил, С3-алкил, С4-алкил, C5-алкил, С6-алкил; и
д) приведения соединения формулы 6 или его фармацевтически приемлемой соли в контакт с реагентом для снятия защиты с получением соединения формулы А или его фармацевтически приемлемой соли

где R1'' представляет собой Н или ОН.
15. Способ по п.14, где
R1 и каждый R1' представляют собой бензильные группы;
n равен 2;
хиральный реагент представляет собой дигидрохлорид 1,7,7-триметил-N-(пиридин-2-илметил)бицикло[2.2.1]гептан-2-амина, (4S)-4-этил-2-{1-этил-1-[(4S)-4-(1-метилэтил)-4,5-дигидро-1,3-оксазол-2-ил]пропил}-4,5-дигидро-1,3-оксазол, (S)-4-(трет-бутил)-2-(2-((S)-4-метил-4,5-дигидрооксазол-2-ил)пропан-2-ил)-4,5-дигидрооксазол или N1,N2-бис-[(1R,2R,4R)-1,7,7-триметилбицикло[2.2.1]гепт-2-ил]-1,2-этандиамин в присутствии Cu(OAc)2;
X представляет собой уходящую группу, выбранную из Br, Cl, I, тозила или трифлила;
борилирующий реагент представляет собой изопропилпинаколборат или триметилборат;
агент для снятия защиты представляет собой гидрогенизацию в присутствии катализатора Pd/C, катализатора Pt/C или смеси катализаторов Pd/C и Pt/C.
16. Способ получения соединения формулы А, включающий стадии:
а) бромирования соединения формулы 8

с получением соединения формулы 9

где R представляет собой Н или OR1;
R1 представляет собой защитную группу;
n равен 0, 1, 2, 3, 4 или 5;
б) приведения соединения формулы 9 в контакт с HN(R1')2 с получением соединения формулы 10

или его соли,
где каждый R1' представляет собой защитную группу;
в) энантиоселективного восстановления соединения формулы 10 или его соли с получением соединения формулы 5

или его соли;
г) приведения соединения формулы 5 или его соли в контакт с борилирующим реагентом, характеризующимся следующей формулой:

в присутствии н-BuLi с получением соединения формулы 6

или его фармацевтически приемлемой соли,
где каждый R2 независимо представляет собой C1-алкил, С2-алкил, С3-алкил, С4-алкил, С5-алкил, С6-алкил или R2 вместе с атомами кислорода, к которым они присоединены, образуют 5- или 6-членное кольцо;
R3 представляет собой C1-алкил, С2-алкил, С3-алкил, С4-алкил, C5-алкил, С6-алкил; и
д) приведения соединения формулы 6 или его фармацевтически приемлемой соли в контакт с реагентом для снятия защиты с получением соединения формулы А или его фармацевтически приемлемой соли

где R1'' представляет собой Н или ОН.
Текст
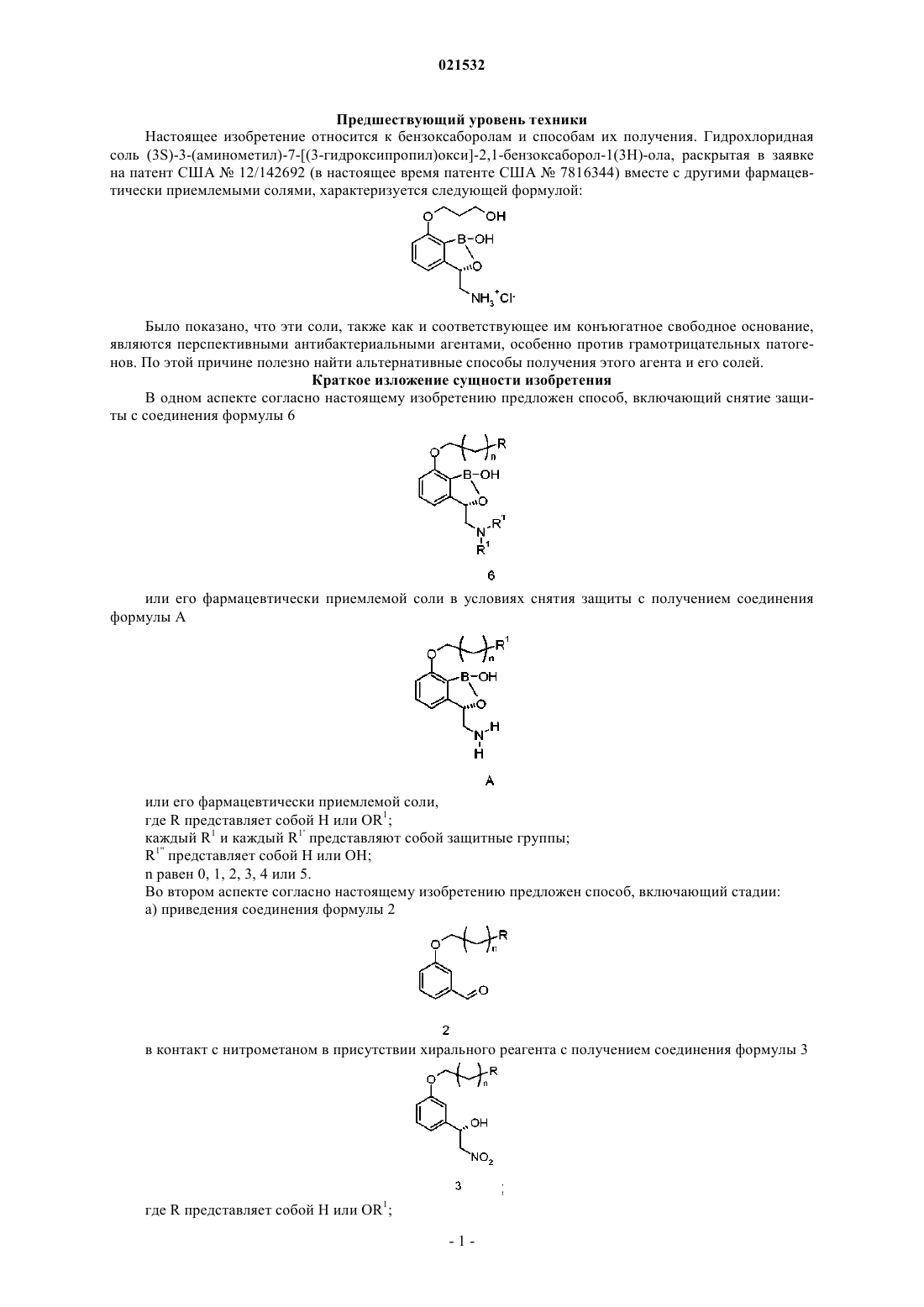
Согласно настоящему изобретению предложен способ, включающий приведение соединения формулы 6 Конде Хосе Х., Ковальски Энтони Джон, Заджак Мэттью Аллен (US) или его фармацевтически приемлемой соли в контакт с реагентом для снятия защиты с получением соединения формулы А или его фармацевтически приемлемой соли; где R представляет собой Н или OR1; R1 и каждый R1' представляют собой защитные группы; R1" представляет собой Н или ОН; n равен 0, 1, 2, 3, 4 или 5. Предшествующий уровень техники Настоящее изобретение относится к бензоксаборолам и способам их получения. Гидрохлоридная соль (3S)-3-(аминометил)-7-[(3-гидроксипропил)окси]-2,1-бензоксаборол-1(3H)-ола, раскрытая в заявке на патент США 12/142692 (в настоящее время патенте США 7816344) вместе с другими фармацевтически приемлемыми солями, характеризуется следующей формулой: Было показано, что эти соли, также как и соответствующее им конъюгатное свободное основание,являются перспективными антибактериальными агентами, особенно против грамотрицательных патогенов. По этой причине полезно найти альтернативные способы получения этого агента и его солей. Краткое изложение сущности изобретения В одном аспекте согласно настоящему изобретению предложен способ, включающий снятие защиты с соединения формулы 6 или его фармацевтически приемлемой соли в условиях снятия защиты с получением соединения формулы А или его фармацевтически приемлемой соли,где R представляет собой Н или OR1; каждый R1 и каждый R1' представляют собой защитные группы;n равен 0, 1, 2, 3, 4 или 5. Во втором аспекте согласно настоящему изобретению предложен способ, включающий стадии: а) приведения соединения формулы 2 в контакт с нитрометаном в присутствии хирального реагента с получением соединения формулы 3R1 представляет собой защитную группу;n равен 0, 1, 2, 3, 4 или 5; б) восстановления соединения формулы 3 с получением соединения формулы 4 в) приведения соединения формулы 4 или его соли в контакт с R1'X и основанием с получением соединения формулы 5 или его соли,где R1' представляет собой защитную группу иX представляет собой уходящую группу; г) приведения соединения формулы 5 или его соли в контакт с борилирующим реагентом, характеризующимся следующей формулой: в присутствии н-BuLi с получением соединения формулы 6 или его фармацевтически приемлемой соли,где каждый R2 независимо представляет собой C1-алкил, С 2-алкил, С 3-алкил, С 4-алкил, C5-алкил, С 6 алкил или R2 вместе с атомами кислорода, к которым они присоединены, образуют 5- или 6-членное кольцо;R3 представляет собой C1-алкил, С 2-алкил, С 3-алкил, С 4-алкил, C5-алкил, С 6-алкил; и д) снятия защиты с соединения формулы 6 или его фармацевтически приемлемой соли с получением соединения формулы А или его фармацевтически приемлемой соли В другом аспекте согласно настоящему изобретению предложен способ, включающий стадии: а) бромирования соединения формулы 8 с получением соединения формулы 9R1 представляет собой защитную группу;n равен 0, 1, 2, 3, 4 или 5; б) приведения соединения формулы 9 в контакт с HN(R1')2 с получением соединения формулы 10 или его соли,где каждый R1' представляет собой защитную группу; в) энантиоселективного восстановления соединения формулы 10 с получением соединения формулы 5 г) приведения соединения формулы 5 или его соли в контакт с борилирующим реагентом, характеризующимся следующей формулой: в присутствии н-BuLi с получением соединения формулы 6 или его фармацевтически приемлемой соли,где каждый R2 независимо представляет собой C1-С 6-алкил или R2 вместе с атомами кислорода, к которым они присоединены, образуют (как указано выше) 5- или 6-членное кольцо;R3 представляет собой C1-С 6-алкил; и д) снятия защиты с соединения формулы 6 или его фармацевтически приемлемой соли с получением соединения формулы А или его фармацевтически приемлемой соли где R1 представляет собой Н или ОН. В другом аспекте согласно настоящему изобретению предложено соединение, характеризующееся следующей формулой 6 а или его фармацевтически приемлемая соль,где R1 представляет собой Н или ОН иR1' представляет собой защитную группу. Подробное описание изобретения В первом аспекте согласно настоящему изобретению предложен способ, включающий снятие защиты с соединения формулы 6 или его фармацевтически приемлемой соли в условиях снятия защиты с получением соединения формулы А или его фармацевтически приемлемой соли,где R представляет собой Н или OR1;R1 и каждый R1' представляют собой защитные группы;n равен 0, 1, 2, 3, 4 или 5. Примеры подходящих защитных групп R1 включают группы -CH(Ra)фенил-(Rb)x, триалкилсилил,тетрагидропиранил, -СН 2 ОСН 3 или -СН 2 ОСН 2 СН 2 ОСН 3, где Ra представляет собой Н, C1-алкил, С 2 алкил, С 3-алкил, С 4-алкил или фенил; каждый Rb независимо представляет собой C1-алкилокси, С 2 алкилокси, С 3-алкилокси, С 4-алкилокси или C1-алкил, С 2-алкил, С 3-алкил, С 4-алкил; х равен 0, 1 или 2. В-4 021532 другом аспекте R1 представляет собой бензил. С 1-С 4-алкил относится к C1-алкилу, С 2-алкилу, С 3-алкилу, С 4-алкилу, например метилу, этилу, нпропилу, изопропилу, н-бутилу, втор-бутилу и трет-бутилу. Аналогично, С 1-С 4-алкокси относится к C1 алкилокси, С 2-алкилокси, С 3-алкилокси, С 4-алкилокси, например метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси и трет-бутокси. C1-С 6-алкил относится к C1-алкилу, С 2-алкилу, С 3-алкилу,С 4-алкилу, C5-алкилу, С 6-алкилу. Примеры подходящих защитных групп R1' включают -СН(Rb)фенил-(Rc)у; примеры таких групп включают группы бензил, 1-фенилэтил, 2-метоксибензил, 3-метоксибензил, 4-метоксибензил, 2,4 диметоксибензил и дифенилметил; в другом аспекте каждый R1' представляет собой бензил. Подходящие способы снятия защиты включают гидрогенизацию, катализируемую металлами, гидрогенизацию с каталитическим переносом или кислотное расщепление. В случаях, когда R1' представляет собой бензил, предпочтительные реагенты для снятия защиты включают Н 2 над Pd/C, H2 над Pt/C, H2 над гидроксидом палладия или формиат аммония и Pd/C. В другом аспекте n равен 0, 1, 2 или 3; предпочтительно n равен 2. При использовании здесь "фармацевтически приемлемая соль" относится к соли присоединения кислоты. Примерами подходящих солей присоединения кислоты являются соли неорганических кислот и органические соли. Примеры подходящих неорганических солей включают соли соляной, бромистоводородной, фосфорной, метафосфорной, азотной и серной кислот; примеры подходящих солей органических кислот включают соли винной, уксусной, трифторуксусной, лимонной, яблочной, молочной, фумаровой, бензойной, муравьиной, пропионовой, гликолевой, глюконовой, малеиновой, янтарной, метансульфоновой, этансульфоновой, стеариновой, бензолсульфоновой, бромбензолсульфоновой и птолуолсульфоновой кислот. Фармацевтически приемлемой солью соединения А предпочтительно является соль HCl. В другом аспекте соединение формулы 6 восстанавливают в присутствии HCl с получением гидрохлоридной соли соединения формулы А. В другом аспекте настоящего изобретения соединение формулы 6 получают приведением соединения формулы 5 или его соли в контакт с борилирующим реагентом в присутствии алкиллития, такого как н-BuLi, нгексиллитий или втор-BuLi. Борилирующий реагент характеризуется следующей формулой: Каждый R2 независимо представляет собой C1-алкил, С 2-алкил, С 3-алкил, С 4-алкил, С 5-алкил, С 6 алкил, и каждый R3 представляет собой C1-алкил, С 2-алкил, С 3-алкил, С 4-алкил, C5-алкил, С 6-алкил. Предпочтительно каждый R2 представляет собой одну и ту же алкильную группу; альтернативно, группыR2 вместе с атомами кислорода, к которым они присоединены, образуют 5- или 6-членное кольцо, которое возможно может быть конденсировано с арильным кольцом. Примеры подходящих борилирующих реагентов включают изопропилпинаколборат, 2-(метокси)-1,3,2-бензодиоксаборол и три-С 1-бораты, триС 2-бораты и три-С 3-бораты, такие как триизопропилборат и триметилборат. В другом аспекте борилирующий реагент представляет собой изопропилпинаколборат или триметилборат. Соединение формулы 5 или его соль могут быть получены приведением R1'X в основных условиях в контакт с соединением формулы 4 или его солью,где X представляет собой подходящую уходящую группу, такую как Br, Cl, I, тозильная или трифлильная группа. Подходящие основания включают карбонаты, такие как карбонаты натрия, калия и цезия, или гидроксиды, такие как гидроксид тетрабутиламмония. Соединение формулы 4 или его соль могут быть получены приведением соединения формулы 2 в контакт с нитрометаном в присутствии хирального реагента с получением соединения формулы 3 с последующим восстановлением нитрогруппы до аминогруппы. Примеры подходящих хиральных реагентов включают дигидрохлорид 1,7,7-триметил-N-(пиридин 2-илметил)бицикло[2.2.1]гептан-2-амина; (4S)-4-этил-2-1-этил-1-[(4S)-4-(1-метилэтил)-4,5-дигидро-1,3 оксазол-2-ил]пропил-4,5-дигидро-1,3-оксазол; (S)-4-(трет-бутил)-2-(2-S)-4-метил-4,5-дигидрооксазол 2-ил)пропан-2-ил)-4,5-дигидрооксазол или N1,N2-бис-[(1R,2R,4R)-1,7,7-триметилбицикло[2.2.1]гепт-2 ил]-1,2-этандиамин в присутствии подходящего катализатора, такого как Со(ОАс)2, Cu(OAc)2, CuCl2,Zn(OTf)2 или Zn(Et)2. Примеры подходящих агентов для восстановления нитрогруппы включают Pd/C, Pt/C или их смесь в присутствии водорода. Соединение формулы 2 легко получают путем взаимодействия R1(CH2)3-X с 3 гидроксибензальдегидом в присутствии подходящего основания, примеры которого включают гидроксидное и карбонатное основания. Альтернативно, соединение формулы 5 или его соль могут быть получены энантиоселективным восстановлением соединения формулы 10 или его соли. Примеры реагентов для энантиоселективного восстановления включают Н 2 и катализатор на основе рутения,родия или иридия,такой как комплекс трифенилфосфина(S)-2-[(Sp)-2(дифенилфосфино)ферроценил]-4-изопропил-2-оксазолина и дихлорида рутения(II) (также известный как катализатор Naud); (R)-BINAP-Ru-(R,R)-(+)-DPEN Cl (также известный как катализатор Noyori), или катализируемое оксазаборолидином восстановление бораном. Соединение формулы 10 или его соль могут быть получены приведением HN(R1')2 в контакт с соединением формулы 9 Соединение формулы 9 может быть получено бромированием соединения формулы 8 Подходящие бромирующие реагенты включают Br2 в дихлорметане, NBS (N-бромсукцинимид) и трибромид тетра-н-бутиламмония. Соединение формулы 8 получают приведением 3-гидроксиацетофенона в контакт с X(CH2)3-R1 и подходящим основанием, примеры которого включают гидроксидное и карбонатное основания. Схемы Следующие схемы в целом иллюстрируют способы по настоящему изобретению. R1, R1' и X не ограничены конкретными раскрытыми группами, и способы не ограничены раскрытыми катализаторами,основаниями, восстановителями и энантиоселективными реагентами. Схема 1 В соответствии со схемой 1 3-гидроксибензальдегид (соединение 1) приводят в контакт сXCH2(CH2)nCH2R в присутствии подходящего основания в условиях, достаточных для получения соединения 2. Соединение 2 превращают в соответствующий (1S)-2-нитро-1-фенилэтанол (соединение 3), используя нитрометан и подходящий хиральный реагент, такой как дигидрохлорид 1,7,7-триметил-N-(пиридин 2-илметил)бицикло[2.2.1]гептан-2-амина, (4S)-4-этил-2-1-этил-1-[(4S)-4-(1-метилэтил)-4,5-дигидро-1,3 оксазол-2-ил]пропил-4,5-дигидро-1,3-оксазол, (S)-4-(трет-бутил)-2-(2-S)-4-метил-4,5-дигидрооксазол 2-ил)пропан-2-ил)-4,5-дигидрооксазол или N1,N2-бис-[(1R,2R,4R)-1,7,7-триметилбицикло[2.2.1]гепт-2 ил]-1,2-этандиамин в присутствии подходящего катализатора, такого как Cu(OAc)2. Затем соединение 3 превращают в соответствующий первичный амин (соединение 4), после чего защищают, используя R1'X,-7 021532 с получением соединения 5; это промежуточное соединение может быть выделено в виде свободного основания или в виде соли, предпочтительно гидрохлоридной соли. Соединение 5 или его соль борилируют подходящим борилирующим реагентом в присутствии сильного основания, такого как н-BuLi, нгексиллитий или втор-BuLi, с получением соединения 6. С этого промежуточного соединения снимают защиту с получением аминометилбензоксаборола, предпочтительно (3S)-3-(аминометил)-7-[(3 гидроксипропил)окси]-2,1-бензоксаборол-1(3H)-ола. Этот продукт предпочтительно выделяют в виде гидрохлоридной соли, и он далее может быть очищен путем перекристаллизации. Схема 2 В соответствии со схемой 2 3-гидроксиацетофенон (соединение 7) приводят в контакт сXCH2(CH2)nCH2-R в присутствии подходящего основания в условиях, достаточных для получения соединения 8. Бромирование подходящим бромирующим реагентом приводит к получению бромкетона (соединения 9), который в свою очередь приводят в контакт с HN(R1')2 в условиях, достаточных для получения защищенного амина (соединения 10). Соединение 10 или его соль подвергают энантиоселективному восстановлению до соответствующего (1S)-2-амино-1-фенилэтанола (соединения 5) в условиях энантиоселективного восстановления. Затем соединение 5 или его соль борилируют и снимают с него защиту, как изображено на схеме 1,с получением аминометилбензоксаборола,предпочтительно(3S)-3-(аминометил)-7-[(3 гидроксипропил)окси]-2,1-бензоксаборол-1(3H)-ола, предпочтительно в виде гидрохлоридной соли. Следующие примеры являются иллюстративными для способа по настоящему изобретению и не предназначены ограничивать объем изобретения. Пример 1. Получение гидрохлорида (3S)-3-(аминометил)-7-[(3-гидроксипропил)окси]-2,1 бензоксаборол-1(3 Н)-она. 1 А. Получение 3-(3-[(фенилметил)окси]пропилокси)бензальдегида В реакционный сосуд объемом 3 л загружали карбонат цезия (209,6 г), 3-гидроксибензальдегид(67,6 г), диметилформамид (DMF, 250 мл) и 3-бромпропилфенилметиловый эфир (113,4 г) и перемешивали их при температуре окружающей среды в течение приблизительно 18 ч. Добавляли воду (567 мл), а затем трет-бутилметиловый эфир (907 мл). Нижний водный слой отделяли и органический слой промывали 1 н. гидроксидом натрия (2567 мл) и водой (1567 мл). Органический раствор концентрировали до минимального объема, после чего добавляли этанол (крепость "200 proof" (абсолют), 907 мл) и раствор концентрировали до минимального объема и фильтровали через 1-мкм фильтр. Ацетат меди(II) и бис-HCl соль камфораминометилпиридина (504 мг) загружали в реактор с последующей загрузкой этанола (60 мл) и диизопропилэтиламина (изо-Pr2NEt) (1,16 мл). Содержимое перемешивали в течение 1 ч при комнатной температуре, после чего загружали раствор 3-(3[(фенилметил)окси]пропилокси)бензальдегида в этаноле (15 г в 15 мл). Реакционную смесь охлаждали до температуры от -30 до -40 С; потом к реакционной смеси медленно добавляли нитрометан (33,9 г),поддерживая температуру ниже -30 С, а затем диизопропилэтиламин (359 мг). Температуру реакционной смеси поддерживали при -30 С в течение 24-48 ч. По завершении реакции в реакционную смесь загружали трифторуксусную кислоту (952 мг) и содержимое переносили в отдельный реактор, содержащий раствор 1 н. HCl (75 мл) и трет-бутилметилового эфира (ТВМЕ, 150 мл) при комнатной температуре. После окончания добавления слои оставляли разделяться и водную фазу удаляли. Затем органическую фазу промывали водой (75 мл) и водную фазу удаляли. Потом раствор продукта в ТВМЕ фильтровали через слой силикагеля (15 г), который ополаскивали ТВМЕ. Продукт хранили холодным в виде раствора в ТВМЕ, который перед последующей стадией гидрогенизации следует заменить этанолом. 1 В 1. Получение (1S)-2-амино-1-[3-(3-[(фенилметил)окси]пропилокси)фенил]этанола(1S)-2-Нитро-1-[3-(3-[(фенилметил)окси]пропилокси)фенил]этанол в этанольном растворе (8 г в 160 мл) подвергали гидрогенизации с использованием Pd(4%)/Pt(1%)/С катализатора (1,6 г). После израсходования исходного вещества и гидроксиламинного промежуточного соединения фильтровали продукт реакции в этаноле. 1 В 2. Получение гидрохлорида Порошкообразный карбонат калия (12,8 г) загружали в реактор объемом 250 мл с последующей загрузкой этанольного раствора (1S)-2-амино-1-[3-(3-[(фенилметил)окси]пропилокси)фенил]этанола(12,7 г в 100 мл). В суспензию загружали дополнительное количество этанола (40 мл). В реактор загружали бензилбромид (15,9 г) и суспензию перемешивали при 20-25 С в течение 18-24 ч, после чего твердую фазу отфильтровывали и промывали этанолом (50 мл). Фильтрат разбавляли водой (50 мл), затем нагревали до 50 С. В реактор загружали концентрированную HCl (3,52 мл), что приводило к осаждениюHCl соли продукта. В начале осаждения суспензию выдерживали при 50 С в течение 30 мин, затем суспензию охлаждали до 0 С и выдерживали при 0 С в течение еще 30 мин. Продукт фильтровали и промывали 20%-ным водным раствором этанола (приблизительно 60 мл) при 0 С. В реактор объемом 250 мл загружали гидрохлорид (1S)-2-[бис-(фенилметил)амино]-1-[3-(3[(фенилметил)окси]пропилокси)фенил]этанола (20 г) и толуол (160 мл). Содержимое тщательно перемешивали в течение приблизительно 5 мин, после чего смесь дегазировали путем помещения реактора под вакуум и заполнения его азотом с повторением операций два раза. Суспензию нагревали до 50-55 С,после чего в течение приблизительно 1 ч при интенсивном перемешивании добавляли н-BuLi (16,3 мл,2,6 М). Затем смесь перемешивали в течение приблизительно 1 ч при 50-55 С, после чего наблюдали израсходование исходного вещества. Затем смесь охлаждали до температуры от -20 до -40 С, потом добавляли 1,0 экв. н-BuLi (14,8 мл) с такой скоростью, чтобы поддерживать температуру ниже -20 С. После окончания добавления температуру доводили до -(30-40)С, и настолько быстро, насколько это было возможно (меньше 10 мин), добавляли н-BuLi (43 мл), сохраняя в процессе добавления температуру ниже -20 С. Температуру доводили до -(20-25)С и смесь перемешивали в течение 1 ч. Смесь охлаждали до температуры от -75 до -80 С и непосредственно в CD3OD гасили аликвоту. После полного включения дейтерия, подтвержденного LCMS (жидкостная хроматография - масс-спектрометрия), добавляли THF(тетрагидрофуран) (18 мл), сразу после чего быстро добавляли борат (36 мл). Смесь нагревали до 1525 С в течение от 30 мин до 1 ч, затем добавляли 5%-ный водн. NaHCO3 (200 мл) и смесь интенсивно перемешивали в течение приблизительно 15 мин. Полученную суспензию фильтровали и осадок на фильтре промывали по меньшей мере 30 мл ТВМЕ. Фильтрат оставляли разделяться и затем органический слой промывали три или четыре раза водой (100 мл), оставляя последнюю промывку отстаиваться по меньшей мере на 1 ч. Органический слой концентрировали до приблизительно 40 мл. 1 Д. Получение гидрохлорида (3S)-3-(аминометил)-7-[(3-гидроксипропил)окси]-2,1-бензоксаборол 1(3 Н)-ола К раствору гидрохлорида (3S)-3-(аминометил)-7-[(3-гидроксипропил)окси]-2,1-бензоксаборол 1(3 Н)-она в толуоле (приблизительно 46 мл) добавляли метанол (200 мл), 1 н. HCl в воде (42,5 мл) и катализатор 5% Pd/C (2 г, 11 мас.%). Полученную смесь обрабатывали газообразным водородом при 100 фунт/кв.дюйм (изб.) (примерно 689,4 кПа (изб. и 50 С. После израсходования моно-N-бензиламинного промежуточного соединения реакционный раствор фильтровали через наполнитель для фильтраочистителя с последующим 1-мкм фильтрованием. Затем отфильтрованный раствор неочищенного продукта подвергали дистилляции, добавляли 2-пропанол (200 мл), после чего снова дистиллировали до минимального объема, позволяющего осуществлять перемешивание. Этот концентрированный раствор оставляли кристаллизоваться при комнатной температуре, затем фильтровали и промывали 2-пропанолом. Н ЯМР: (d4-метанол, 400,13 МГц) d (млн-1) 7,48 (t, J=7,81 Гц, 1 Н), 7,01 (d, J=7,58 Гц, 1 Н), 6,93 (d,J=8,21 Гц, 1 Н), 5,37 (dd, J=2,76, 8,77 Гц, 1 Н), 4,18 (t, J=6,11 Гц, 2 Н), 3,78 (t, J=5,47 Гц, 2 Н), 3,59 (dd,J=2,87, 13,29 Гц, 1H), 2,92 (dd, J=8,86, 13,29 Гц, 1H), 2,00 (m, J=6,13 Гц, 2H). Точную массу протонированной молекулы [М + Н]+ измеряли при m/z 238,1247, используя ионизацию электрораспылением с регистрацией положительных ионов. Массу этого иона вычисляли при m/z 238,1251. Используя хиральную HPLC (высокоэффективная жидкостная хроматография), обнаружили,что хиральная чистота составила 99,9%. Пример 2. Получение гидрохлорида (3S)-3-(аминометил)-7-[(3-гидроксипропил)окси]-2,1 бензоксаборол-1(3 Н)-ола. 2 А. Получение 3-(3-[(фенилметил)окси]пропилокси)бензальдегида В реакционный сосуд объемом 3 л загружали карбонат калия (152 г), 3-гидроксибензальдегид (67,6 г), диметилформамид (DMF, 250 мл) и 3-хлорпропилфенилметиловый эфир (96,4 г) и перемешивали их при 90 С в течение приблизительно 18 ч. Добавляли воду (567 мл), а затем трет-бутилметиловый эфир(907 мл). Нижний водный слой отделяли и органический слой промывали 1 н. гидроксидом натрия (2567 мл) и водой (1567 мл). Органический раствор концентрировали до минимального объема, после чего добавляли этанол (крепость "200 proof", 907 мл), и раствор концентрировали до минимального объема и фильтровали через 1-мкм фильтр. 2 Б. Получение (1S)-2-нитро-1-[3-(3-[(фенилметил)окси]пропилокси)фенил]этанола Моногидрат ацетата меди(II) (3,70 г, 0,05 экв.) и бис-камфорэтилендиамин (N1,N2-бис-[(1R,2R,4R)1,7,7-триметилбицикло[2.2.1]гепт-2-ил]-1,2-этандиамин) (7,38 г, 0,06 экв.) загружали в реактор с последующей загрузкой этанола (200 мл, 2 объема). Содержимое нагревали до 50-60 С в течение приблизительно 1 ч или до растворения всей твердой фазы, затем охлаждали до комнатной температуры и перемешивали, после чего загружали раствор 3-(3-[(фенилметил)окси]пропилокси)бензальдегида в этаноле(100 г, 1 экв. в 50 мл, 0,5 об.). Реакционную смесь охлаждали до температуры от -10 до -20 С; потом к реакционной смеси медленно добавляли нитрометан (112,7 г, 5 экв.), поддерживая температуру ниже-10 С, а затем диизопропилэтиламин (1,94 мл, 1,44 г, 0,03 экв.). Температуру реакционной смеси поддерживали при -10 С в течение приблизительно 22-30 ч. По завершении реакции в реакционный сосуд загружали раствор 1 н. HCl (250 мл, 2,5 об.) и трет-бутилметилового эфира (ТВМЕ, 500 мл, 5 об.) при комнатной температуре. После добавления содержимое перемешивали в течение приблизительно 5 мин и доводили температуру до 20 С (приблизительно до комнатной температуры), затем слои оставляли разделяться и водную фазу удаляли. Потом органическую фазу промывали водой (250 мл, 2,5 об.) два раза, причем водную фазу каждый раз удаляли. Затем отгоняли ТМВЕ и заменяли его абсолютным этанолом. 2 В 1. Получение (1S)-2-амино-1-[3-(3-[(фенилметил)окси]пропилокси)фенил]этанола(1S)-2-Нитро-1-[3-(3-[(фенилметил)окси]пропилокси)фенил]этанол в этанольном растворе (8 г в 160 мл) подвергали гидрогенизации с использованием Pd(4%)/Pt(1%)/С катализатора (1,6 г). После израсходования исходного вещества и гидроксиламинного промежуточного соединения фильтровали продукт реакции в этаноле.(1S)-2-[бис-(фенилметил)амино]-1-[3-(3 Порошкообразный карбонат калия (12,8 г) загружали в реактор объемом 250 мл с последующей загрузкой этанольного раствора (1S)-2-амино-1-[3-(3-[(фенилметил)окси]пропилокси)фенил]этанола(12,7 г в 100 мл). В суспензию загружали дополнительное количество этанола (40 мл). В реактор загружали бензилбромид (15,9 г) и суспензию перемешивали при 20-25 С в течение 18-24 ч, после чего твердую фазу отфильтровывали и промывали этанолом (50 мл). Фильтрат разбавляли водой (50 мл), затем нагревали до 50 С. В реактор загружали концентрированную HCl (3,52 мл), что приводило к осаждениюHCl соли продукта. В начале осаждения суспензию выдерживали при 50 С в течение 30 мин, затем суспензию охлаждали до 0 С и выдерживали при 0 С в течение еще 30 мин. Продукт фильтровали и промывали 20%-ным водным раствором этанола (приблизительно 60 мл) при 0 С. 2 Г. Получение (3S)-3-[бис-(фенилметил)амино]метил-7-(3-[(фенилметил)окси]пропилокси)-2,1 бензоксаборол-1(3 Н)-ола В реактор объемом 250 мл загружали гидрохлорид (1S)-2-[бис-(фенилметил)амино]-1-[3-(3[(фенилметил)окси]пропилокси)фенил]этанола (20 г) и толуол (160 мл). Содержимое тщательно перемешивали в течение приблизительно 5 мин, после чего смесь дегазировали путем помещения реактора под вакуум и заполнения его азотом с повторением операций два раза. Суспензию нагревали до 50-55 С,после чего в течение приблизительно 1 ч при интенсивном перемешивании добавляли н-BuLi (16,3 мл,2,6 М). Затем смесь перемешивали в течение приблизительно 1 ч при 50-55 С, после чего наблюдали израсходование исходного вещества. Затем смесь охлаждали до температуры от -20 до -40 С, потом добавляли 1,0 экв. н-BuLi (14,8 мл) с такой скоростью, чтобы поддерживать температуру ниже -20 С. После окончания добавления температуру доводили до -(30-40)С и, настолько быстро, насколько это было возможно (меньше 10 мин), добавляли н-BuLi (43 мл), сохраняя в процессе добавления температуру ниже -20 С. Температуру доводили до -(20-25)С и смесь перемешивали в течение 1 ч. Смесь охлаждали до температуры от -75 до -80 С, и непосредственно в CD3OD гасили аликвоту. После полного включения дейтерия, подтвержденного LCMS, добавляли THF (18 мл), сразу после чего быстро добавляли борат (36 мл). Смесь нагревали до 15-25 С в течение от 30 мин до 1 ч, затем добавляли 5%-ный водн. NaHCO3 (200 мл) и смесь интенсивно перемешивали в течение приблизительно 15 мин. Полученную суспензию фильтровали и осадок на фильтре промывали по меньшей мере 30 мл ТВМЕ. Фильтрат оставляли разделяться и затем органический слой промывали три или четыре раза водой (100 мл), оставляя последнюю промывку отстаиваться по меньшей мере на 1 ч. Органический слой концентрировали до приблизительно 40 мл.(42,5 мл) и катализатор 5% Pd/C (2 г, 11 мас.%). Полученную смесь обрабатывали газообразным водородом при 100 фунт/кв.дюйм (изб.) (примерно 689,4 кПа (изб. и 50 С. После израсходования моно-Nбензиламинного промежуточного соединения реакционный раствор фильтровали через наполнитель для фильтра-очистителя с последующим 1-мкм фильтрованием. Затем отфильтрованный раствор неочищенного продукта подвергали дистилляции, добавляли 2-пропанол (200 мл), после чего снова дистиллировали до минимального объема, позволяющего осуществлять перемешивание. Этот концентрированный раствор оставляли кристаллизоваться при комнатной температуре, затем фильтровали и промывали 2 пропанолом. 1 Н ЯМР: (d4-метанол, 400,13 МГц) d (млн-1) 7,48 (t, J=7,81 Гц, 1 Н), 7,01 (d, J=7,58 Гц, 1 Н), 6,93 (d,J=8,21 Гц, 1 Н), 5,37 (dd, J=2,76, 8,77 Гц, 1 Н), 4,18 (t, J=6,11 Гц, 2 Н), 3,78 (t, J=5,47 Гц, 2 Н), 3,59 (dd,J=2,87, 13,29 Гц, 1 Н), 2,92 (dd, J=8,86, 13,29 Гц, 1 Н), 2,00 (m, J=6,13 Гц, 2 Н). Точную массу протонированной молекулы [М + Н]+ измеряли при m/z 238,1247, используя ионизацию электрораспылением с регистрацией положительных ионов. Массу этого иона вычисляли при m/z 238,1251. Используя хиральную HPLC, обнаружили, что хиральная чистота составила 99,9%. Пример 3. Альтернативное получение гидрохлорида К раствору 3-гидроксиацетофенона (5,5 г) в диметилформамиде (18 мл) добавляли 3 бромпропилфенилметиловый эфир (9,25 г) при температуре окружающей среды с последующим добавлением карбоната цезия (17 г). Суспензию перемешивали в течение 24 ч и добавляли воду, а затем этилацетат. Органический слой отделяли и водный слой экстрагировали этилацетатом. Органические слои объединяли и промывали два раза 2 н. гидроксидом натрия, четыре раза рассолом и концентрировали в вакууме до масла с получением указанного в заголовке соединения. 3 Б. Получение 2-бром-1-[3-(3-[(фенилметил)окси]пропилокси)фенил]этанона(50 мл) добавляли метанол (50 мл) и раствор трибромида тетрабутиламмония (16 г) в тетрагидрофуране(50 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 45 мин, после чего добавляли трибромид тетрабутиламмония (0,89 г) с последующим дополнительным добавлением 0,6 г. В реакционную смесь добавляли 10%-ный водный тиосульфат натрия и смесь концентрировали в вакууме до минимального объема. Добавляли этилацетат, полученный органический раствор промывали водным тиосульфатом натрия, насыщенным бикарбонатом натрия и рассолом и концентрировали до масла с получением указанного в заголовке соединения. 3 В. Получение 2-[бис-(фенилметил)амино]-1-[3-(3[(фенилметил)окси]пропилокси)фенил]этанона(6,4 г) и дихлорметана (25 мл) добавляли раствор карбоната натрия (7,6 г) в воде (50 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Органический слой отделяли и концентрировали до минимального объема и добавляли трет-бутилметиловый эфир. Органический слой промывали разбавленной уксусной кислотой (4 раза), 0,1 н. гидроксидом натрия (1 раз) и рассолом(1 раз). Органический раствор концентрировали досуха. Масло промывали метанолом и очищали хроматографией с получением указанного в заголовке соединения. 3 Г. Получение Смесь 2-[бис-(фенилметил)амино]-1-[3-(3[(фенилметил)окси]пропилокси)фенил]этанона, катализатора SK-N003-2z (катализатор Naud, 47 мг) и 1 М гидроксида натрия (2,085 мл) в тетрагидрофуране (10 мл) обрабатывали водородом при 150 фунт/кв.дюйм (примерно 1034,1 кПа) в течение 4 ч. Затем реакционную смесь фильтровали через силикагель. Неочищенный фильтрат очищали хроматографией, элюируя трет-бутилметиловым эфиром-гексаном (0-60%) с получением желаемого продукта в виде желтого масла. 3 Д. Получение (3S)-3-[бис-(фенилметил)амино]метил-7-(3-[(фенилметил)окси]пропилокси)2,1-бензоксаборол-1(3 Н)-ола К раствору (1S)-2-[бис-(фенилметил)амино]-1-[3-(3-[(фенилметил)окси]пропилокси)фенил]этанола (240 мг, 0,50 ммоль) в 5 мл толуола при -20 С добавляли 1,75 ммоль н-BuLi в гексане (2,59 М,0,67 мл) в течение приблизительно 5 мин. Затем смесь перемешивали в течение 2 ч, после чего гасили свежеперегнанным триметилборатом (0,28 мл, 259 мг, 2,5 ммоль). Холодную баню удаляли и смесь нагревали до комнатной температуры. Затем реакционную смесь разбавляли 10 мл ТВМЕ, после чего до- 14021532 бавляли 5 мл насыщенного раствора NaHCO3. После интенсивного перемешивания в течение приблизительно 30 мин органический слой промывали 5 мл 0,1 М NaH2PO4, затем 5 мл насыщенного NaHCO3. Потом органический слой сушили над Na2SO4 и концентрировали. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы А, включающий приведение соединения формулы 6 или его фармацевтически приемлемой соли в контакт с реагентом для снятия защиты с получением соединения формулы А или его фармацевтически приемлемой соли,где R представляет собой Н или OR1; каждый R1 и каждый R1' независимо представляют собой защитные группы;n равен 0, 1, 2, 3, 4 или 5,где соединение формулы 6 получают приведением соединения формулы 5 или его соли в контакт с борилирующим реагентом, характеризующимся следующей формулой: в присутствии алкиллитиевого реагента,где каждый R2 независимо представляет собой С 1-С 6-алкил или R2 вместе с атомами кислорода, к которым они присоединены, образуют 5- или 6-членное кольцо иR1 представляет собой группы -СН(Ra)фенил-(Rb)х, триалкилсилил, тетрагидропиранил, -СН 2 ОСН 3 или -СН 2 ОСН 2 СН 2 ОСН 3, где Ra представляет собой Н или метил; Rb представляет собой метокси или C1 алкил, С 2-алкил или С 3-алкил и х равен 0, 1 или 2; каждый R1' независимо представляет собой -CH(Rc)фенил-(Rd)y, где Rc представляет собой Н или метил, каждый Rd независимо представляет собой метокси или C1-алкил, С 2-алкил или С 3-алкил; n равенR1 и каждый R1' представляют собой бензильные группы; реагент для снятия защиты представляет собой восстановитель;n равен 1; соединение формулы 6 восстанавливают в присутствии HCl с получением гидрохлоридной соли соединения формулы А. 4. Способ по п.3, где реагент для снятия защиты представляет собой Pd/C или Pt/C в присутствии Н 2 или их смесь, Н 2 над гидроксидом палладия или реагент для гидрогенизации в условиях каталитического переноса. 5. Способ по п.1, где борилирующий реагент представляет собой изопропилпинаколборат или триС 1-борат, три-С 2-борат или три-С 3-борат; n представляет собой число от 0 до 3 и алкиллитиевый реагент представляет собой н-BuLi, н-гексиллитий или втор-BuLi. 6. Способ по п.1 или 5, где борилирующий реагент представляет собой изопропилпинаколборат или триметилборат; n равен 2 и алкиллитиевый реагент представляет собой н-BuLi. 7. Способ по п.1, где соединение формулы 5 или его соль получают либо энантиоселективным восстановлением соединения формулы 10 или его солью,где X представляет собой уходящую группу. 8. Способ по п.7, где соединение формулы 5 или его соль получают с использованием Н 2 и комплекса трифенилфосфина (S)-2-[(SP)-2-(дифенилфосфино)ферроценил]-4-изопропил-2-оксазолина и дихлорида рутения(II) или Н 2 и (R)-BINAP-Ru-(R,R)-(+)-DPEN Cl ([(R)-2,2'-бис-(дифенилфосфино)-1,1'бинафтил]-Ru-(R,R)-(+)-[1,2-дифенилэтилендиамин] Cl); n равен 2. 9. Способ по п.7, где соединение формулы 5 или его соль получают приведением соединения формулы 4 или его соли в контакт с бензилбромидом в присутствии карбоната или гидроксида. 10. Способ по п.9, где соединение формулы 4 или его соль получают приведением соединения формулы 2 в контакт с нитрометаном в присутствии хирального реагента с получением соединения формулы 3 с последующим восстановлением нитрогруппы до аминогруппы. 11. Способ по п.10, где хиральный реагент представляет собой дигидрохлорид 1,7,7-триметил-N(пиридин-2-илметил)бицикло[2.2.1]гептан-2-амина;N1,N2-бис-[(1R,2R,4R)-1,7,7 триметилбицикло[2.2.1]гепт-2-ил]-1,2-этандиамин в присутствии Cu(OAc)2; n равен 2. 12. Способ по п.7, где соединение формулы 10 или его соль получают приведением HN(R1')2 в контакт с основанием и соединением формулы 9 13. Способ по п.12, где соединение формулы 9 получают бромированием соединения формулы 8 14. Способ получения соединения формулы А, включающий стадии: а) приведения соединения формулы 2 в контакт с нитрометаном в присутствии хирального реагента с получением соединения формулы 3 где R представляет собой Н или OR1; R1 представляет собой защитную группу и n равен 0, 1, 2, 3, 4 или 5; б) восстановления соединения формулы 3 с получением соединения формулы 4 или его соли; в) приведения соединения формулы 4 или его соли в контакт с R1'X и основанием с получением соединения формулы 5 или его соли,где R1' представляет собой защитную группу и X представляет собой уходящую группу; г) приведения соединения формулы 5 или его соли в контакт с борилирующим реагентом, характеризующимся следующей формулой: в присутствии н-BuLi с получением соединения формулы 6 или его фармацевтически приемлемой соли,где каждый R2 независимо представляет собой C1-алкил, С 2-алкил, С 3-алкил, С 4-алкил, C5-алкил, С 6 алкил или R2 вместе с атомами кислорода, к которым они присоединены, образуют 5- или 6-членное кольцо;R3 представляет собой C1-алкил, С 2-алкил, С 3-алкил, С 4-алкил, C5-алкил, С 6-алкил; и д) приведения соединения формулы 6 или его фармацевтически приемлемой соли в контакт с реагентом для снятия защиты с получением соединения формулы А или его фармацевтически приемлемой солиR1 и каждый R1' представляют собой бензильные группы;n равен 2; хиральный реагент представляет собой дигидрохлоридX представляет собой уходящую группу, выбранную из Br, Cl, I, тозила или трифлила; борилирующий реагент представляет собой изопропилпинаколборат или триметилборат; агент для снятия защиты представляет собой гидрогенизацию в присутствии катализатора Pd/C, катализатора Pt/C или смеси катализаторов Pd/C и Pt/C. 16. Способ получения соединения формулы А, включающий стадии: а) бромирования соединения формулы 8 с получением соединения формулы 9R1 представляет собой защитную группу;n равен 0, 1, 2, 3, 4 или 5; б) приведения соединения формулы 9 в контакт с HN(R1')2 с получением соединения формулы 10 или его соли,где каждый R1' представляет собой защитную группу; в) энантиоселективного восстановления соединения формулы 10 или его соли с получением соединения формулы 5 или его соли; г) приведения соединения формулы 5 или его соли в контакт с борилирующим реагентом, характеризующимся следующей формулой: в присутствии н-BuLi с получением соединения формулы 6 или его фармацевтически приемлемой соли,где каждый R2 независимо представляет собой C1-алкил, С 2-алкил, С 3-алкил, С 4-алкил, С 5-алкил, С 6 алкил или R2 вместе с атомами кислорода, к которым они присоединены, образуют 5- или 6-членное кольцо;R3 представляет собой C1-алкил, С 2-алкил, С 3-алкил, С 4-алкил, C5-алкил, С 6-алкил; и д) приведения соединения формулы 6 или его фармацевтически приемлемой соли в контакт с реагентом для снятия защиты с получением соединения формулы А или его фармацевтически приемлемой соли
МПК / Метки
МПК: A61K 31/69, A01N 55/08
Метки: способ, получения, бензоксаборолов
Код ссылки
<a href="https://eas.patents.su/21-21532-sposob-polucheniya-benzoksaborolov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения бензоксаборолов</a>
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Мейллян Пьер, Ансель Жан-Эрик, Бьенейм Юг
МПК: B01J 31/24, A61K 31/355, C07C 39/19...
Метки: замещенных, витамина, получения, использованием, cпособ, фенолов, способ
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Производные 5-0-дезозаминил-6-0-метилэритронолида а, способ их получения и их применение для получения биологически активных продуктов

Номер патента: 575
Опубликовано: 29.12.1999
Авторы: Бонне Алан, Мазюри Алан, Дельтиль Мишель
МПК: C07H 17/08
Метки: продуктов, получения, биологически, активных, 5-0-дезозаминил-6-0-метилэритронолида, производные, применение, способ
Формула / Реферат:
1. Соединения формулы (I): в которой или R1 представляет радикал алкил, содержащий до 8 атомов углерода, замещенный одним или несколькими радикалами алкила, содержащими до 8 атомов углерода, или одним или несколькими радикалами арила, содержащими до 14 атомов углерода, или R1 представляет радикал арил, содержащий до 14 атомов углерода, который может быть замещен одним или несколькими радикалами алкил, алкенил или алкинил, содержащими до 8...
Способ получения дихлорпропанола, способ получения эпихлоргидрина, способ получения эпоксидных смол и применение оборудования, обладающего коррозионной стойкостью, в способе получения дихлорпропанола

Номер патента: 14241
Опубликовано: 29.10.2010
Авторы: Франк Кристиан, Краффт Филипп, Сметс Валентин, Бальтазар Доминик, Жильбо Патрик
МПК: C07C 31/36, C07C 29/62, B01J 19/02...
Метки: дихлорпропанола, обладающего, способ, оборудования, смол, способе, эпихлоргидрина, коррозионной, эпоксидных, применение, стойкостью, получения
Формула / Реферат:
1.Способ получения дихлорпропанола, содержащий:(a) стадию, на которой глицерин или сложный эфир глицерина или их смесь вводят во взаимодействие с агентом хлорирования, содержащим хлороводород,(b) по меньшей мере одну другую стадию, осуществляемую на оборудовании, выполненном или имеющем покрытие из материалов, обладающих стойкостью по отношению к агенту хлорирования, в условиях осуществления этой стадии,причем другая стадия является стадией...
Цеолитный катализатор l-типа, способ его получения, способ получения ароматических углеводородов, способ получения бензина

Номер патента: 3559
Опубликовано: 26.06.2003
Авторы: Фукунага Тецуя, Иннес Роберт А., Сугимото Митио
МПК: C07C 5/41, B01J 29/61, C10G 35/095...
Метки: цеолитный, получения, ароматических, бензина, катализатор, способ, углеводородов, l-типа
Формула / Реферат:
1. Цеолитный катализатор L-типа, который получают при нанесении на цеолит L-типа платинового компонента, одного или более галогеновых компонентов и одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, при этом наносимое количество одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, находится в интервале от 0,001 до 3 мас.% из расчета на общую массу катализатора, молярное отношение...
Способ получения защищенного 4-аминометилпирролидин-3-она, промежуточные соединения и способ получения 3-аминометил-4-алкоксииминопирролидина

Номер патента: 2499
Опубликовано: 27.06.2002
Авторы: Чанг Джей Хиок, Моон Кванг Юл, Ким Вон Суп, Ли Тае Хи
МПК: C07D 207/24
Метки: получения, 4-аминометилпирролидин-3-она, соединения, промежуточные, 3-аминометил-4-алкоксииминопирролидина, способ, защищенного
Формула / Реферат:
1. Способ получения соединения формулы (1) в которой Р1 и Р2 обозначают защитные группы; включающий а) восстановление соединения формулы (5) где Р1 такой, как определено для формулы (1); в присутствии катализатора никель Ренея в растворителе в атмосфере водорода в присутствии одной или более добавок с получением соединения формулы (6) где Р1 такой, как определено для формулы (1); b) защиту аминогруппы в присутствии одного или более оснований...
Предыдущий патент: Аппликатор бирок для животных
Следующий патент: Ударная дробилка с горизонтальным валом
Случайный патент: Пиридазиноновые соединения и их применение в качестве ингибиторов daao