Способ сульфоксидирования биологически активных соединений
Номер патента: 16297
Опубликовано: 30.03.2012
Авторы: Махал Раджендра Дагесинг, Гхарпур Милинд Морешвар, Майкап Голакчандра Сударшан, Гурджар Мукунд Кешав, Мехта Сатиш Раманлал, Гавари Прашант Садашив, Раджпут Махеш Рамсинг











Формула / Реферат
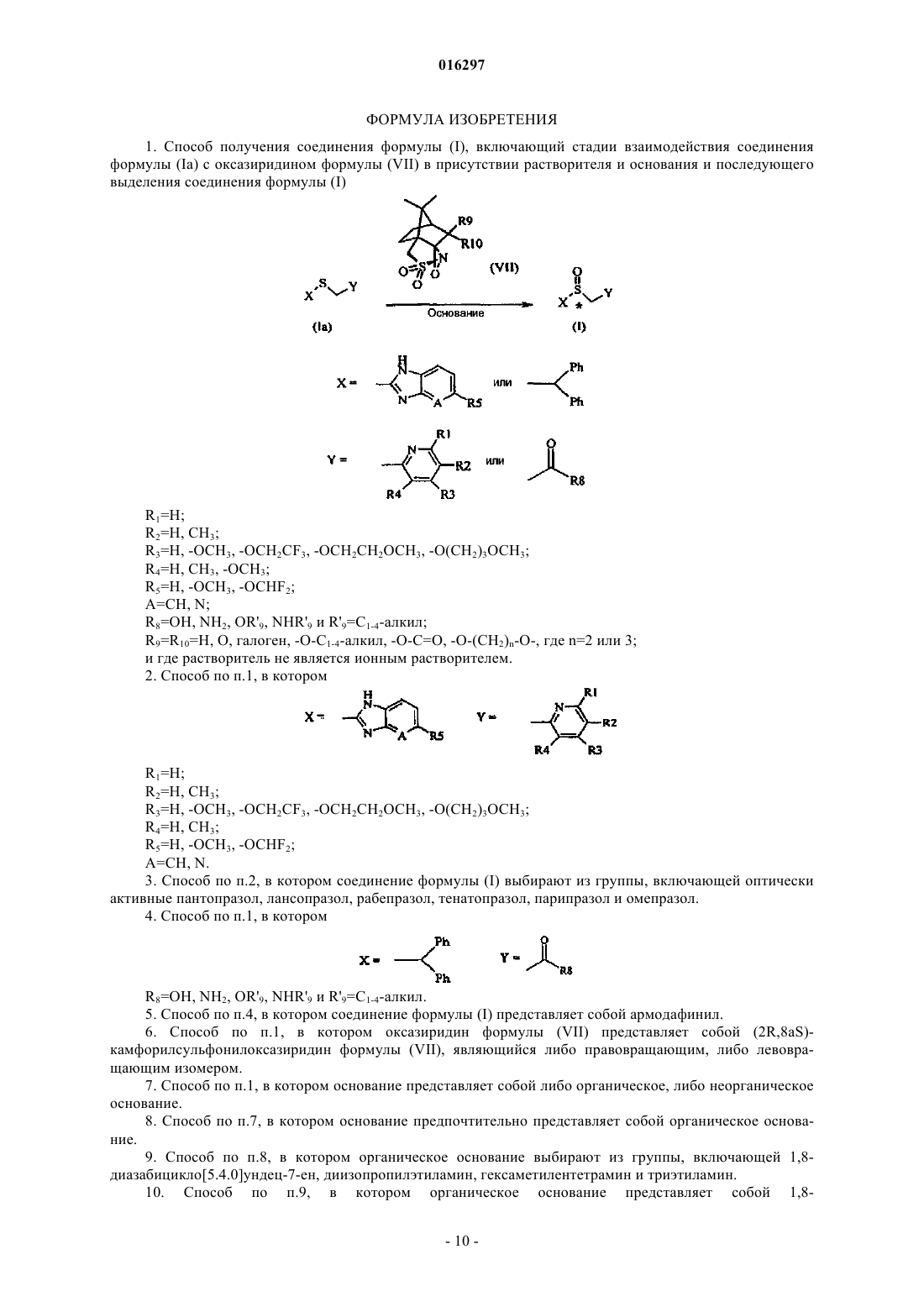
1. Способ получения соединения формулы (I), включающий стадии взаимодействия соединения формулы (Ia) с оксазиридином формулы (VII) в присутствии растворителя и основания и последующего выделения соединения формулы (I)

R1=H;
R2=H, CH3;
R3=H, -OCH3, -OCH2CF3, -OCH2CH2OCH3, -O(CH2)3OCH3;
R4=H, CH3, -OCH3;
R5=H, -OCH3, -OCHF2;
A=CH, N;
R8=OH, NH2, OR'9, NHR'9 и R'9=C1-4-алкил;
R 9= R 10=H, O, галоген, -O-C1-4-алкил, -O-C=O, -O-(CH2)n-O-, где n=2 или 3;
и где растворитель не является ионным растворителем.
2. Способ по п.1, в котором

R1=H;
R2=H, CH3;
R3=H, -OCH3, -OCH2CF3, -OCH2CH2OCH3, -O(CH2)3OCH3;
R4=H, CH3;
R5=H, -OCH3, -OCHF2;
A=CH, N.
3. Способ по п.2, в котором соединение формулы (I) выбирают из группы, включающей оптически активные пантопразол, лансопразол, рабепразол, тенатопразол, парипразол и омепразол.
4. Способ по п.1, в котором

R8=OH, NH2, OR'9, NHR'9 и R'9=C1-4-алкил.
5. Способ по п.4, в котором соединение формулы (I) представляет собой армодафинил.
6. Способ по п.1, в котором оксазиридин формулы (VII) представляет собой (2R,8aS)-камфорилсульфонилоксазиридин формулы (VII), являющийся либо правовращающим, либо левовращающим изомером.
7. Способ по п.1, в котором основание представляет собой либо органическое, либо неорганическое основание.
8. Способ по п.7, в котором основание предпочтительно представляет собой органическое основание.
9. Способ по п.8, в котором органическое основание выбирают из группы, включающей 1,8-диазабицикло[5.4.0]ундец-7-ен, диизопропилэтиламин, гексаметилентетрамин и триэтиламин.
10. Способ по п.9, в котором органическое основание представляет собой 1,8-диазабицикло[5.4.0]ундец-7-ен.
11. Способ по п.7, в котором неорганическое основание представляет собой гидроксид щелочного металла.
12. Способ по п.11, в котором неорганическое основание представляет собой гидроксид натрия или гидроксид калия.
13. Способ по п.1, в котором растворитель выбирают из группы, включающей спирт, ароматический углеводород, простой эфир, сложный эфир, амид, нитрил, воду или их смеси.
14. Способ по п.13, в котором растворитель представляет собой спирт или ароматический углеводород.
15. Способ по п.13, в котором растворитель выбран из группы, включающей метанол, этанол, изопропанол, бутанол, простой диизопропиловый эфир, толуол, воду, тетрагидрофуран, ацетонитрил, диметилформамид, диэтилформамид, диметоксиэтан или их комбинации.
16. Способ по п.14, в котором растворитель представляет собой изопропанол или толуол.
Текст
СПОСОБ СУЛЬФОКСИДИРОВАНИЯ БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ Данное изобретение относится к новому способу получения сульфоксидов, предпочтительно стереоселективному получению замещенных или незамещенных хиральных сульфинильных производных 2-(2-пиридилметил)сульфинил-1H-бензимидазола окислением оксазиридином в присутствии подходящего растворителя и основания, при этом растворитель не является ионным растворителем. Гхарпур Милинд Морешвар, Майкап Голакчандра Сударшан, Махал Раджендра Дагесинг, Мехта Сатиш Раманлал, Гурджар Мукунд Кешав,Раджпут Махеш Рамсинг, Гавари Прашант Садашив (IN) Медведев В.Н. (RU)(71)(73) Заявитель и патентовладелец: ЭМКЬЮР ФАРМАСЬЮТИКАЛЗ ЛИМИТЕД (IN) 016297 Область техники, к которой относится изобретение Данное изобретение относится к улучшенному способу получения сульфоксидов, и более конкретно изобретение предусматривает стереоселективное получение замещенных или незамещенных хиральных сульфинильных производных общей формулы (I) окислением оксазиридином в присутствии подходящего растворителя и основания.R1=H; R2R3=H, CH3, -OCH3, -OCH2CF3, -OCH2CH2OCH3, -O(CH2)3OCH3,R4=H, CH3 и R5=H, -OCH3 -OCHF2, R'8=OH, NH2, OR'9, NHR'9 и R'9=C1-4-алкил; A=CH, N. Уровень техники изобретения Сульфоксиды имеют асимметрический центр у атома серы и поэтому существуют в виде двух оптических изомеров, то есть энантиомеров. Желательно получать соединения с улучшенными фармакокинетическими и метаболическими свойствами, которые дадут улучшенный терапевтический показатель,такой как меньшую степень межиндивидуальных колебаний. Соединения общей формулы (I) обычно получают из соответствующих промежуточных сульфидов окислением простой тиоэфирной группы с использованием различных окислителей. Указанные предшествующие способы обычно сопровождаются образованием соответствующих производных сульфонов в качестве загрязняющих примесей. Более того, в конечном продукте в качестве загрязняющих примесей могут также присутствовать не прореагировавшие сульфиды. Весьма сходные физико-химические свойства сопровождающих загрязняющих примесей осложняют их отделение, тем самым делая продукт,представляющий интерес, склонным к загрязнениям. Примесь сульфона образуется во время конверсии сульфоксида, делая завершение окисления опасным. Примесь сульфона вызывается главным образом сверхокислением, которое, в свою очередь, связано с окислителями. Таким образом, окисление следует контролировать. В патенте США 5929244 предложен способ получения эзомепразола путем использования (3'S,2R)(-)-N-(фенилсульфонил)-(3,3-дихлоркамфорил)оксазиридина в присутствии триэтиламина в качестве основания и четыреххлористого углерода в качестве растворителя. Оптическая чистота составляет 94%, но полученный выход только 22%, что делает способ невозможным для коммерческого применения. Он также связан с использованием четыреххлористого углерода в качестве растворителя, что запрещено в промышленном масштабе. В патенте США 5948789 предложен энантиоселективный синтез единственного энантиомера 2-(2 пиридинилметилсульфинил)-1H-бензимидазола, так же как и других структурно родственных сульфоксидов. Процесс окисления ведут в присутствии основания, окислителя, хирального комплекса титана и растворителя. Однако указанный способ страдает основным недостатком: высоким содержанием примеси сульфона, образующегося в процессе. Более того, указанные способы обладают очевидными недостатками и они требуют (применения) изопропилата титана, который не может быть регенерирован и опасен для окружающей среды. Кроме того, реагент, использованный для хиральной индукции, представляет собой диэтилтартрат, который трудно регенерировать из-за эпимеризации и гидролиза во время обработки. Реагент, использованный для окисления, представляет собой гидропероксид кумола, который является взрывчатым и опасным, не говоря уже о его дороговизне. В патенте США 6919459 сделана мимолетная ссылка на использование N-сульфонилоксазиридинов для окисления сульфидов до сульфоксидов. Однако в указанном патенте не доказан и не предложен способ такого окисления N-сульфонилоксазиридинами. В Journal of the Chemical Society, Perkin Transactions 1, 1988, 1753-7 описан способ получения хиральных сульфоксидов с применением (+)-(3-оксокамфорсульфонил)оксазиридина. При этом наблюдали энантиомерный избыток максимум около 60%, что является ограничением для промышленных целей. В Chemical Sciences (1990), 45(12), 1689-94 предложен способ окисления сульфидов до сульфоксидов камфорлактон-сульфонилоксазиридином, 3-эндобромкамфорсульфонилоксазиридином и т.д. Предпочтительный растворитель представлял собой канцерогенный растворитель, подобный четыреххлористому углероду, поскольку он давал наибольший энантиомерный избыток (до 85%). Другое ограничение"стандартной концентрации" требует более высокого разбавления для предпочтительного растворителя,-1 016297 предложенного в единственном примере. Однако четыреххлористый углерод не используют в фармацевтической промышленности вследствие его канцерогенности и высокой токсичности. Указанный посредственный энантиомерный избыток возможен для субстратов, имеющих значительные различия с обеих сторон сульфидной группы. Для субстратов, таких как бензилфенилсульфид, энантиомерный избыток недопустимо беден (36%). Ограничение связано с использованием четыреххлористого углерода. Однако использование оксазиридина не показано для таких субстратов, как фенилбензилсульфид. Требуемое время для возможного взаимодействия может быть даже 160 ч. При меньшем времени взаимодействия энантиомерный избыток весьма беден, что ниже ограничений, применимых в промышленности. В Journal of Organic Chemistry 1992, 57(26), 7274-7285 предложен способ получения [(8,8 дигалогенокамфорил)сульфонил]оксазиридинов и способ окисления сульфидов до сульфоксидов с использованием дейтерированного хлороформа. Использование дейтерированных растворителей в промышленном масштабе недоступно дорогостояще. Таким образом, указанная ссылка предлагает использование в качестве растворителя четыреххлористого углерода, который промышленно недопустим, как указано ранее. Более того, энантиомерный избыток полученного желаемого изомера находится в интервале 1-75%, что очень мало для получения оптически чистого фармацевтического соединения. Попытка получения указанного энантиомерного избытка также требует для обычного растворителя точной и недоступной температуры взаимодействия -78C. Другой способ проведения энантиоселективного синтеза предусматривает использование хиральных оксазиридинов(III) (Journal of American Chemical Society 1992, 114, 1428-1437). При использовании хиральных оксазиридинов энантиомерный избыток является достижимым для прохиральных сульфидов,например, если прохиральный сульфид имеет весьма объемный заместитель, такой как 9-антрил, и другой (заместитель), такой как метил. Взаимодействие также зависит от растворителя и дает больший энантиомерный избыток в CCl4, который вообще не благоприятен для окружающей среды. Хиральные оксираны, использованные для окисления, представляют собой Энантиомерный избыток больше, если X представляет собой галоген или водород. Таким образом,использование хиральных оксиранов для получения энантиомерно чистых PPIs неизвестно на современном уровне техники и в то же время ограничено из-заa) размера групп прохирального сульфида;c) растворителя и т.д. В Tetrahedron Asymmetry 1995, 6(12), 2911-2914 предложен способ окисления сульфидов до сульфоксидов с применением [(3,3-диметоксикамфорил)сульфонил]оксазиридина(IV) в присутствии пероксида водорода. Использование указанного оксазиридина дает сульфоксиды из неароматических сульфидов с хорошей энантиоселективностью.[(3,3-диметоксикамфорил)сульфонил]имин(IV) Недостатком указанного способа является тот факт, что если окисляют арилсульфиды, энантиомер-2 016297 ный избыток желаемого изомера составляет только 61%, поэтому указанный способ промышленно неосуществим для окисления фармацевтически активных ингредиентов, содержащих ароматические заместители. В Indian Journal of Chemistry 2001, 40B, 1132-1133 указано (схема II), что алкилалкилтиометилсульфиды и арил-арилтиометилсульфиды могут быть окислены до соответствующих сульфоксидов 10-камфорсульфонилоксазиридином с выходом 70-80% и энантиоселективностью 90-95%. Чтобы получить такого рода продукт необходимой степени чистоты, используют препаративную тонкослойную хроматографию. Однако в промышленном масштабе такая очистка невозможна. Способ относится к окислению сульфидной группы, с одной стороны которой находится арильная или алкильная группа, а с другой стороны - алкильная группа. Не существует примера окисления, где у сульфидной группы с другой стороны находится объемная группа, так как объемная группа стремится изменить энантиомерную селективность и существует вероятность, что будет получена низкая энантиомерная селективность. В Tetrahedron Asymmetry 2003, 14, 407-410 предложен способ получения (R)-лансопразола с применением гетерогенной каталитической системы WO3, 30%-ной H2O2 и хинных алкалоидов. Промышленное использование указанной системы весьма ограничено вследствие применения дорогостоящих реагентов, таких как WO3 и хинные алкалоиды. Дополнительное ограничение вызвано также опасностямиTernois James и др. (Tetrahedron Asymmetry 18, 2007, 2959-2964) предлагают способ энантиоселективного окисления сульфидной группы до сульфоксидной с использованием оксазиридина формулы (V),для получения некоторых фармацевтических соединений. Однако, как указано там, с указанным способом связано несколько недостатков. В способе используют четыреххлористый углерод или ионные жидкости и время взаимодействия составляет около 48 ч,что уменьшает эффективность процесса. Применение четыреххлористого углерода имеет несколько вредных воздействий на здоровье, поскольку четыреххлористый углерод представляет собой канцерогенный растворитель, озоноразрушающий агент. Продолжительное действие четыреххлористого углерода может влиять на центральную нервную систему, вызывать повреждения печени и почек и в результате привести к раку. Далее использование ионной жидкости, подобной гексафторфосфату 1-бутил-3-метилимидазолия, которая известна своей нелетучестью, создает серьезные проблемы во время сушки фармацевтически активных ингредиентов. Указанные ионные жидкости могут быть использованы в качестве растворителя для их получения вследствие их нелетучести; кроме того, указанные ионные жидкости требуют применения ультразвукового устройства для расщепления растворов ионных жидкостей на базе имидазолия с пероксидом водорода и уксусной кислотой до относительно безвредных соединений. Более того, энантиомерный избыток, полученный при использовании оксазиридинового реагента, составляет до 78%, что является недостаточным. В Organic Letters 2007, 9(12), 2265-2268 описан способ получения (R)-лансопразола из соответствующего промежуточного сульфида, включающий окисление оксазиридином формулы (VI), полученным из производного азахолестерина. Способ требует низкую температуру около -70C и обеспечивает только 60%-ную конверсию промежуточного сульфида в желаемый продукт. Вследствие жестких температурных условий и низкой конверсии продукта, не считая недостатков окислителей, указанный способ не подходит для промышленных целей. Таким образом, со способами на существующем уровне техники связан ряд проблем, подобных низким выходам и энантиомерной чистоте, регенерации хирального реагента, использованного в процессе, множественным экстракциям, повышенным опасностям в местах производства в отношении высоких количеств пероксидов, использованию дорогостоящих катализаторов.-3 016297 В результате, существует небходимость в разработке простого, эффективного в смысле стоимости,безвредного для окружающей среды способа стереоселективного получения хиральных сульфинильных производных, который подходит для осуществления в коммерческих масштабах, безопасен в работе и дает продукт высокой энантиомерной чистоты. Предмет изобретения Предметом данного изобретения является разработка улучшенного простого, эффективного в смысле стоимости и безвредного для окружающей среды способа стереоселективного получения замещенных или незамещенных хиральных сульфинильных производных общей формулы (I) с хорошим выходом и высокой энантиомерной чистоты. Сущность изобретения Данное изобретение относится к улучшенному, простому, эффективному в смысле стоимости и безвредному для окружающей среды способу стереоселективного получения замещенных или незамещенных хиральных сульфинильных производных с незначительной примесью сульфонов и высокой энантиомерной чистоты окислением оксазиридином в присутствии растворителя и основания. Подробное описание изобретения В соответствии с этим данное изобретение предлагает способ получения соединений формулы (I),предусматривающий стадии взаимодействия соединений формулы (Ia) с оксазиридином формулы (VII) в присутствии растворителя и основания и выделения соединения формулы (I), как требуется для фармацевтических веществR9=R10=H, O, галоген, -O-C1-4 алкил, -O-C=O, -O-(CH2)n-O-, где n=2 или 3. Способ, как рассмотрено выше, вызывает асимметрическое окисление прохирального сульфида для получения энантиомерно обогащенной формы соответствующего сульфоксида. Атом серы сульфида не имеет асимметрии, тогда как в результате стереоселективного окисления образовавшийся сульфоксид представляет собой хиральное соединение. Путем указанного стереоселективного окисления один из энантиомеров может быть получен в энантиомерном избытке по сравнению с другим. Для целей данного изобретения, если где R1=H; R2R3=H, CH3, -OCH3, -OCH2CF3, -OCH2CH2OCH3, -O(CH2)3OCH3; R4=H, CH3 и R5=H, -OCH3,-OCHF2, A=CH, N, то соединение формулы (I) может быть выбрано из группы, включающей оптически активные празолы, такие как пантопразол, лансопразол, рабепразол, тенатопразол, парипразол и омепразол, или соединение, предложенное в патенте США 5776765. Для целей данного изобретения, если где R'8=OH, NH2, OR'9, NHR'9 и R'9=C1-4-алкил, то соединение формулы (I) может представлять собой армодафинил, как предложено в патенте США 4927855. Модафинил имеет стереогенный центр у атома серы и, таким образом, существует в виде двух оптических изомеров, то есть энантиомеров. Модафинил, химически известный как 2-[(дифенилметил) сульфинил]ацетамид, то есть 2-(бензгидрилсульфинил)ацетамид (B), и который может быть получен из соответствующего сульфида (A), представляет собой евгероидный (eugeroic) лекарственный препарат,обычно предназначенный для лечения нарколепсии.R-энантиомер модафинила, который является преимущественным энантиомером, известен как армодафинил и имеет химическое название 2-[(R)-(дифенилметил)сульфинил]ацетамид. Таким образом,способ изобретения может быть использован для получения любого рацемического сульфоксида, примеры которых описаны выше. Для получения рацемических сульфоксидов могут быть использованы рацемические оксазиридины. Использованный окислитель представляет собой соединение формулы (VIII) в которой один или более из R6, R7 и R8 представляют собой хиральные части, имеющие хиральные центры. Предпочтительно он образует циклическую систему. Предпочтительно окислитель, использованный в данном изобретении, представляет собой правовращающий или левовращающий изомер (2R,8aS)-10(камфорилсульфонил)оксазиридина, как изображено в формуле (VII). (+)-Энантиомер, то есть (+)(2R,8aS)-10-(камфорсульфонил)оксазиридин используют для получения соответствующего энантиомера сульфоксида. (-)-Изомер указанного соединения может также быть использован для получения энантиомерно обогащенного соединения.v) R9=R10= кетону. Использованный согласно способу хиральный окислитель представляет собой хиральный оксазиридин, который может быть получен без применения комплексов металлов, таких как тетраизопропилат титана. Тетраизопропилат титана приводит к огромной нагрузке на промывку оборудования и поэтому неблагоприятен для окружающей среды. Реакцию данного изобретения проводят в присутствии органического или неорганического основания. Неорганическое основание выбирают из группы, включающей гидроксиды, алкоголяты и бикарбонаты, карбонаты щелочных или щелочно-земельных металлов и предпочтительные неорганические основания представляют собой гидроксид натрия или гидроксид калия. Однако предпочтительным основанием является органическое основание. Использованное органическое основания выбирают из группы, включающей 1,8 диазабицикло[5.4.0]ундец-7-ен, диизопропилэтиламин, гексаметилентетраамин, триэтиламин и подобные.[5.4.0]ундец-7-ен. Растворитель, использованный для данного изобретения, выбирают из группы органических растворителей, включающей спирты, простые эфиры, сложные эфиры, амиды, нитрилы, ароматические углеводороды, воду и т.д. или их комбинаций. Предпочтительные растворители выбирают из группы,включающей метанол, этанол, изопропанол, бутанол, простой диизопропиловый эфир, толуол, воду, тетрагидрофуран, ацетонитрил, диметилформамид, диэтилформамид, диметоксиэтан или их комбинации и т.д. Растворитель не охватывает ионных растворителей. Реакцию образования сульфоксида проводят при комнатной температуре. Изобретение иллюстрировано следующими примерами, которые служат только для иллюстрации изобретения и не ограничивают его. Все варианты осуществления, очевидные для способа на современном уровне техники, считаются попадающими в рамки данного изобретения. Примеры Пример 1. В колбе емкостью 250 мл 10 г сульфида рабепразола суспендировали в 70 мл изопропилового спирта. К суспензии прибавляли 4,4 г 1,8-диазабицикло[5.4.0]ундец-7-ена и охлаждали до 10-15C. Затем прибавляли 6,6 г (+)-(2R,8aS)-10-(камфорилсульфонил)оксазиридина и перемешивали до окончания взаимодействия сульфида (около 20 ч) при 25-30C. Реакционную смесь фильтровали и твердое вещество промывали изопропиловым спиртом, получая 5,1 г (-)-(камфорсульфонил)имина (извлечение 78%). К фильтрату прибавляли 1,1 г гидроксида натрия и реакционную массу концентрировали в вакууме до образования вязкого масла. Затем прибавляли воду до получения прозрачного раствора. К водному слою прибавляли этилацетат (50 мл) и разбавленной уксусной кислотой доводили pH до 10. Органический слой отделяли и промывали 25 мл воды. Органический слой концентрировали до получения вязкого масла. Затем прибавляли толуол (100 мл) до получения прозрачного раствора. Прибавляли раствор 1,2 г гидроксида натрия в 2 мл воды и перемешивали в течение 15 мин. Твердое вещество отфильтровывали и промывали 25 мл толуола, получая белое твердое вещество. Результат. Выход: 8 г. Химическая чистота, включая R- и S-изомеры: 97,67%.S-изомер: 10,65%. Энантиомерная чистота любого индивидуального изомера, например R-изомера, может быть дополнительно увеличена превращением R-рабепразола в R-рабепразол-натрий и/или растворением Rрабепразол-натрия в воде и установлением pH уксусной кислотой. Очистка R-изомера: 8 г R-рабепразол-натрия, полученного выше, растворяли в 20 мл воды и при температуре от 10 до 15C доводили pH до 9,5, используя уксусную кислоту. Твердое вещество отфильтровывали и промывали 10 мл воды. Результат:S-изомер: 4,91%. Химическая чистота, включая R- и S-изомеры: 99,22%. Пример 2 (армодафинил). В колбу емкостью 250 мл помещали 10 г бензгидрилтиоуксусной кислоты в 100 мл толуола. Реакционную массу охлаждали до 0-5C. Затем к ней прибавляли 5,9 г 1,8-диазабицикло[5.4.0]ундец-7-ена и охлаждали до 0-5C. Реакционную массу перемешивали в течение 30 мин. После этого прибавляли 8,8 г(+)-(2R,8aS)-10-(камфорилсульфонил)оксазиридина и перемешивали при температуре от 25 до 30C до полного завершения взаимодействия. Реакционную смесь фильтровали и твердое вещество промывали водой, получая 8 г хирально или энантиомерно обогащенной армодафиновой кислоты. Выход: 75,25%. Оптическая чистота:S-изомер: 17,83%. Пример 3. К изопропиловому спирту (7 мл) в колбе емкостью 100 мл прибавляли рабепразола сульфид (1 г,0,0029 моль). Колбу охлаждали до температуры от 15 до 20C. Затем прибавляли 1,8 диазабицикло[5.4.0]ундец-7-ен (0,44 г, 0,0029 моль) и перемешивали в течение 10 мин. После этого реакционную массу охлаждали до температуры от 10 до 15C и при 10-15C загружали простой диизопропиловый эфир (3 мл) вместе с (+)-(2R,8aS)-10-(камфорилсульфонил)оксазиридином (0,66 г, 0,0029 моль). Реакционную массу перемешивали (при указанной температуре) в течение 10-15 мин и затем дополни-6 016297 тельно перемешивали при 25-30C. Ход реакции контролировали с помощью ВЭЖХ до завершения взаимодействия. Химическая чистота: 96,20%.S-изомер: 8,66%. Пример 4. К воде (7 мл) в колбе прибавляли рабепразола сульфид (1 г, 0,0029 моль) и охлаждали до 15-20C. Затем прибавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (0,44 г, 0,0029 моль) и перемешивали в течение 5 мин. После этого реакционную массу охлаждали до 10-15C и прибавляли (+)-(2R,8aS)-10(камфорилсульфонил)оксазиридин (0,66 г, 0,0029 моль). Реакционную массу в течение 10-15 мин перемешивали (при указанной температуре) в течение 30 мин и затем дополнительно перемешивали при температуре 25-30C до завершения взаимодействия, что было проконтролировано с помощью ВЭЖХ. Химическая чистота: 62,54%.S-изомер: 29,54%. Пример 5. К диметилформамиду (7 мл) в колбе прибавляли рабепразола сульфид (1 г, 0,0029 моль) и охлаждали до температуры 15-20C. Затем прибавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (0,44 г, 0,0029 моль) и перемешивали в течение 5 мин. После этого реакционную массу охлаждали до температуры 10-15C и прибавляли (+)-(2R,8aS)-10-(камфорилсульфонил)оксазиридин (0,66 г, 0,0029 моль) при 10C. Реакционную массу в течение 30 мин перемешивали при 10-15C и затем при 25-30C до завершения взаимодействия, что было проконтролировано с помощью ВЭЖХ. Химическая чистота: 91,92%.S-изомер: 16,96%. Пример 6. К дихлорметану (7 мл) в колбе прибавляли пантопразола сульфид (1 г, 0,0027 моль) и охлаждали до температуры 15-20C. Затем прибавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (0,41 г, 0,0027 моль) и перемешивали в течение 10 мин. После этого реакционную смесь охлаждали до 10-15C и прибавляли (+)(2R,8aS)-10-(камфорилсульфонил)оксазиридин (0,63 г, 0,0027 моль). Реакционную смесь в течение 30 мин перемешивали при температуре от 10 до 15C и далее дополнительно перемешивали при 25-30C. Реакционную смесь контролировали с помощью ВЭЖХ до завершения взаимодействия, а затем гасили водным гидроксидом натрия и экстрагировали MDC. Органический слой отделяли и концентрировали,получая продукт. Химическая чистота: 83,75%.S-изомер: 19,70%. Пример 7 (S-пантопразол: диизопропилэтиламин в качестве основания и метанол в качестве растворителя). Пантопразола сульфид (1 г, 0,0027 моль) прибавляли к метанолу (7 мл) в колбе емкостью 100 мл и охлаждали до 15-20C. Затем прибавляли диизопропилэтиламин (0,35 г, 0,0027 моль) и перемешивали в течение 10 мин. После этого реакционную смесь охлаждали до 10-15C и прибавляли (+)-(2R,8aS)-10(камфорилсульфонил)оксазиридин (0,63 г, 0,0027 моль). Реакционную смесь перемешивали при 25-30C до завершения взаимодействия по данным TCX. Затем реакционную смесь гасили разбавленным гидроксидом натрия и экстрагировали дихлорметаном. Органический слой отделяли и концентрировали. Химическая чистота: 98,24%.S-изомер: 25,33%. Пример 8 (пантопразол, используя неорганическое основание в органическом растворителе). Пантопразола сульфид (1 г, 0,0027 моль) прибавляли к метанолу (4 мл) в колбе емкостью 100 мл. Затем колбу охлаждали до 15-20C, прибавляли раствор, гидроксида натрия (0,108 г, 0,0027 моль), перемешивали при 10-15C и загружали (+)-(2R,8aS)-10-камфорилсульфонилоксазиридин (0,63 г, 0,0027 моль). Реакционную смесь перемешивали при 25-30C до завершения взаимодействия по данным TCX и ВЭЖХ. Затем к реакционной смеси прибавляли дихлорметан (5 мл) и отделяли органический слой, который затем концентрировали, получая продукт. Химическая чистота: 98,36%.S-изомер: 30,53%. Пример 9 (другой молярный эквивалент (+)-(2R, 8aS)-10-(камфорилсульфонилоксазиридина). Рабепразола сульфид (40 т, 0,116 моль) прибавляли к изопропиловому спирту (7 мл) в колбе емко-7 016297 стью 100 мл и охлаждали до температуры от 15 до 20C. Затем прибавляли 1,8 диазабицикло[5.4.0]ундец-7-ен (17,9 г, 0,116 моль) и перемешивали при 10-15C. Загружали (+)-(2R,8aS)10-камфорилсульфонилоксазиридин (25,3 г, 0,110 моль). Реакционную массу перемешивали при 25-30C до завершения взаимодействия. Реакционную массу фильтровали и фильтрат концентрировали при пониженном давлении. К остатку прибавляли воду (200 мл) и твердое вещество отделяли фильтрованием. Фильтрат при перемешивании обрабатывали водным раствором гидроксида натрия (12 г в 15 мл воды). Затем при 15-20C уксусной кислотой (25%-ной) устанавливали pH 11. К смеси прибавляли этилацетат и доводили pH до 9,5, используя уксусную кислоту (25%-ную). Органический слой отделяли и частично концентрировали при пониженном давлении, а остаток разбавляли толуолом (120 мл). Полученную смесь при 10-15C перемешивали с раствором гидроксида натрия (4,65 г в 5 мл воды) и твердое вещество отделяли фильтрованием, снова растворяли в воде (200 мл) и промывали дихлорметаном (160 мл). Объединенный органический слой экстрагировали раствором гидроксида натрия (2,5 г в 80 мл воды). Водный слой перемешивали в вакууме и доводили pH до 9,5 уксусной кислотой. Прибавляли этилацетат и раствор перемешивали в течение 30 мин. Полученное твердое вещество отфильтровывали и дважды промывали водой (по 40 мл). Влажное твердое вещество помещали в колбу емкостью 500 мл с 200 мл воды и колбу охлаждали до температуры от 10 до 15C. Затем при 10-15C в перемешиваемую массу загружали раствор гидроксида натрия (4,65 г в 5 мл воды). Уксусной кислотой доводили pH до 9,5. Реакционную массу выдерживали в течение 30 мин при 10-15C, получая в процессе выдерживания твердое вещество. Твердое вещество отфильтровывали и дважды промывали водой (по 200 мл). После сушки и хиральной очистки получали белый продукт, определенный как R-изомер 98,9%ее. Химическая чистота: 99,60%.S-изомер: 1,0%. Пример 10 (взаимодействие любого празола в любом растворителе без неорганического или органического основания в любом растворителе). Рабепразола сульфид (1 г, 0,0029 моль) прибавляли к изопропиловому спирту (7 мл) в колбе емкостью 100 мл и охлаждали до 10-15C. Прибавляли (+)-(2R,8aS)-10-камфорилсульфонилоксазиридин (0,66 г, 0,0029 моль) и реакционную смесь перемешивали при 25-30C до завершения взаимодействия, как показано TCX и ВЭЖХ. К реакционной смеси прибавляли раствор гидроксида натрия и затем дихлорметан(5 мл). Органический слой отделяли и концентрировали, получая продукт. Химическая чистота: 28,47%.S-изомер: 43,86%. Пример 11 (тенатопразол). Тенатопразола сульфид (415 г, 1,2 5 моль) прибавляли к изопропиловому спирту (4980 мл) в колбе. Затем в колбу прибавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (191,3 г, 1,25 моль) и охлаждали до 1015C. Далее в колбу прибавляли 1(R)-(-)-(камфорилсульфонил)оксазиридин (303 г, 1,31 моль) и перемешивали при 25-30C до завершения взаимодействия по данным TCX и ВЭЖХ. Реакционную массу фильтровали и фильтрат концентрировали при пониженном давлении. К остатку прибавляли воду (2075 мл) и твердое вещество отделяли фильтрованием. Фильтрат охлаждали до 15-20C и уксусной кислотой доводили pH до 7,5. Реакционную массу перемешивали при 15-20C и выпавшее твердое вещество отфильтровывали. Влажную лепешку суспендировали в воде (3735 мл). В колбу прибавляли раствор гидроксида натрия (55,2 г в 415 мл воды) и перемешивали в течение 30 мин. Полученное твердое вещество отфильтровывали; фильтрат экстрагировали дихлорметаном (830 мл). Водный слой отделяли и доводилиpH до 7,45 50%-ным раствором уксусной кислоты. Водный слой отделяли и экстрагировали дихлорметаном (830 мл). Органический слой отделяли, частично концентрировали и разбавляли этилацетатом (2905 мл). Смесь частично концентрировали при пониженном давлении и охлаждали до 25-30C. Выпавшее твердое вещество отфильтровывали, промывали этилацетатом (1660 мл) и сушили. Выход: 81,15%. Химическая чистота: 99,38%.S-изомер: 88,81%. Пример 12A (взаимодействие для получения эзомепразола). Омепразола сульфид (50 г; 0,152 моль) прибавляли к изопропиловому спирту (350 мл). К смеси при 10-15C прибавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (23,1 г; 0,152 моль). Затем к смеси прибавляли 1(R)-(-)-(камфорилсульфонил)оксазиридин (34,8 г; 0,151 моль) и оставляли перемешиваться в течение 20 ч до завершения взаимодействия по данным TCX. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. К остатку прибавляли воду (250 мл) и доводили pH примерно до 8,5 уксусной кислотой. Далее прибавляли этилацетат, органический слой отделяли и концентрировали в вакууме. К смеси прибавляли ацетон (150 мл) и фильтровали. Полученную смесь концентрировали и остаток разбавляли толуолом (8 мл) и метанолом (75 мл). К смеси прибавляли метилат натрия (8,2 г) и перемешивали в течение 14 ч. Реакционную смесь фильтровали и фильтрат концентрировали при пони-8 016297 женном давлении. К остатку прибавляли простой диизопропиловый эфир (150 мл) и реакционную смесь отфильтровывали, получая эзомепразол-натрий. Выход: 23,4 г. Выход в %: 46,8%. Хиральная чистота натриевой соли (S)-эзомепразола: 99,64%. Пример 12B. Получение эзопразол-магния. Эзопразол-натрий (5,0 г; 0,1636 моль) растворяли в воде (30 мл) и при комнатной температуре по каплям прибавляли к раствору хлорида магния (0,54 г; 0,0068 моль) в воде (30 мл). Полученную смесь перемешивали в течение 1 ч и фильтровали. Влажную лепешку промывали водой (30 мл) и сушили. Выход: 4,87 г. Выход в %: 96%.(R)-омепразол: 0,36%. Пример 13 (лансопразол; DBU в качестве основания). Лансопразола сульфид (1,0 г; 0,028 моль) при перемешивании прибавляли к изопропиловому спирту (7 мл) при 25-30C. Затем прибавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (0,43 г, 0,0028 моль) и перемешивали при 10-15C. После этого прибавляли (-)-дихлороксазиридин [(-)-(8,8-дихлоркамфорилсульфонил)оксазиридин] (0,8 г; 0,027 моль) и реакционную смесь перемешивали при 25-30C до завершения взаимодействия. Затем реакцию гасили разбавленным раствором гидроксида натрия и экстрагировали дихлорметаном. Органический слой концентрировали, получая продукт. Химическая чистота: 92,43%.(R)-изомер: 9,71%. Пример 14 (дихлороксазиридин без основания). Лансопразола сульфид (1,0 г; 0,0028 моль) при перемешивании прибавляли к изрпропиловому спирту (7 мл) при 25-30C и охлаждали до 10-15 С. Затем прибавляли [(-)-(8,8-дихлоркамфорилсульфонил)оксазиридин (0,8 г; 0,0027 моль) и реакционную смесь перемешивали при 25-30C в течение 15 ч. Реакцию гасили разбавленным раствором гидроксида натрия и экстрагировали дихлорметаном. Органический слой концентрировали, получая продукт. Химическая чистота: 6,85%.(R)-изомер: 31,71%. Пример 15 (S-пантопразола сульфид). Пантопразола сульфид (400 г; 1,089 моль) прибавляли к изопропиловому спирту (3600 мл). К смеси прибавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (164 г) и охлаждали до 10-15C. Затем прибавляли (R)-(-)(камфорилсульфонил)оксазиридин (259,70 г; 1,133 моль), повышали температуру до 25-30C и перемешивали до завершения взаимодействия по данным ВЭЖХ. Реакционную смесь отфильтровывали и фильтрат частично концентрировали при пониженном давлении. После этого прибавляли воду (2000 мл) и остаток фильтровали. Уксусной кислотой доводили pH фильтрата до 9,5 и разбавляли этилацетатом(2000 мл). Далее снова доводили уксусной кислотой pH примерно до 7,5 и отделяли органический слой,который затем частично концентрировали и разбавляли циклогексаном. После этого смесь охлаждали до 10-15C и продукт отделяли фильтрованием. Влажную лепешку прибавляли к этилацетату (2800 мл),нагревали до 70C и частично концентрировали при пониженном давлении. Остаток разбавляли циклогексаном (400 мл) и продукт отделяли фильтрованием при 10-15C. Выход: 243 г. Выход в %: 60%. Химическая чистота: 99,95%. Оптическая чистота: 98,62%. Преимущества данного изобретения приведены ниже.B. Использование окислителя, который может быть легко регенерирован.C. Продукт, полученный по окончании взаимодействия, имеет фармацевтически приемлемую степень чистоты.D. Применение меньшего количества стадий очистки повышает общий выход конечного продукта.E. Экологически безвредный, безопасный и простой способ стереоселективного получения замещенных или незамещенных хиральных сульфинильных производных с меньшими примесями сульфонов и с высокой энантиомерной чистотой.F. Может быть достигнута регенерация хирального реагента.G. Избегание множественных стадий перекристаллизации.-9 016297 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы (I), включающий стадии взаимодействия соединения формулы (Ia) с оксазиридином формулы (VII) в присутствии растворителя и основания и последующего выделения соединения формулы (I)R9=R10=H, O, галоген, -O-C1-4-алкил, -O-C=O, -O-(CH2)n-O-, где n=2 или 3; и где растворитель не является ионным растворителем. 2. Способ по п.1, в которомA=CH, N. 3. Способ по п.2, в котором соединение формулы (I) выбирают из группы, включающей оптически активные пантопразол, лансопразол, рабепразол, тенатопразол, парипразол и омепразол. 4. Способ по п.1, в которомR8=OH, NH2, OR'9, NHR'9 и R'9=C1-4-алкил. 5. Способ по п.4, в котором соединение формулы (I) представляет собой армодафинил. 6. Способ по п.1, в котором оксазиридин формулы (VII) представляет собой (2R,8aS)камфорилсульфонилоксазиридин формулы (VII), являющийся либо правовращающим, либо левовращающим изомером. 7. Способ по п.1, в котором основание представляет собой либо органическое, либо неорганическое основание. 8. Способ по п.7, в котором основание предпочтительно представляет собой органическое основание. 9. Способ по п.8, в котором органическое основание выбирают из группы, включающей 1,8 диазабицикло[5.4.0]ундец-7-ен, диизопропилэтиламин, гексаметилентетрамин и триэтиламин. 10. Способ по п.9, в котором органическое основание представляет собой 1,8- 10016297 диазабицикло[5.4.0]ундец-7-ен. 11. Способ по п.7, в котором неорганическое основание представляет собой гидроксид щелочного металла. 12. Способ по п.11, в котором неорганическое основание представляет собой гидроксид натрия или гидроксид калия. 13. Способ по п.1, в котором растворитель выбирают из группы, включающей спирт, ароматический углеводород, простой эфир, сложный эфир, амид, нитрил, воду или их смеси. 14. Способ по п.13, в котором растворитель представляет собой спирт или ароматический углеводород. 15. Способ по п.13, в котором растворитель выбран из группы, включающей метанол, этанол, изопропанол, бутанол, простой диизопропиловый эфир, толуол, воду, тетрагидрофуран, ацетонитрил, диметилформамид, диэтилформамид, диметоксиэтан или их комбинации. 16. Способ по п.14, в котором растворитель представляет собой изопропанол или толуол.
МПК / Метки
МПК: C07D 401/12, C07C 315/06, C07D 471/04
Метки: сульфоксидирования, активных, биологически, соединений, способ
Код ссылки
<a href="https://eas.patents.su/12-16297-sposob-sulfoksidirovaniya-biologicheski-aktivnyh-soedinenijj.html" rel="bookmark" title="База патентов Евразийского Союза">Способ сульфоксидирования биологически активных соединений</a>
Устройство и способ для ферментативного получения биологически активных соединений

Номер патента: 12052
Опубликовано: 28.08.2009
Авторы: Делле Бернд, Пфаль Михель
Метки: получения, способ, активных, устройство, ферментативного, биологически, соединений
Формула / Реферат:
1. Устройство для ферментативного получения биологически активного соединения, включающее по меньшей мере один изолятор, который содержит ферментер и который окружен рабочей камерой или граничит с ней, причем эта рабочая камера связана с окружающей средой посредством переходного шлюза, отличающееся тем, что в этом изоляторе и в рабочей камере в каждом случае преобладает пониженное давление, причем давление относительно давления окружающей среды...
Микрокапсулы, содержащие суспензии биологически активных соединений, и способ их получения

Номер патента: 219
Опубликовано: 24.12.1998
Авторы: Чен Дзин Линг, Шер Герберт Бенсон
МПК: A01N 25/28
Метки: микрокапсулы, получения, способ, содержащие, биологически, соединений, активных, суспензии
Формула / Реферат:
1. Микрокапсула, содержащая органическую жидкость, включающую чувствительный к ультрафиолетовому свету биологически активный материал и эффективное количество средства защиты от ультрафиолетового света в виде частиц, отличающаяся тем, что средство защиты от ультрафиолетового света выбрано из группы, включающей диоксид титана, оксид цинка и их смеси, суспендированные и тщательно диспергированные в жидкости. 2. Микрокапсула по п.1, отличающаяся...
Способ получения антиоксидантов и биологически активных соединений липидной природы

Номер патента: 3212
Опубликовано: 27.02.2003
Авторы: Деев Сергей Вячеславович, Авчиева Пенкер Бабаевна, Авчиев Марат Исламудинович, Буторова Ирина Анатольевна
МПК: C12P 7/64, C12P 33/00, C12P 23/00...
Метки: природы, антиоксидантов, получения, соединений, липидной, активных, биологически, способ
Формула / Реферат:
Способ последовательного получения антиоксидантов и биологически активных соединений липидной природы, а именно b-каротина, ликопина, фосфолипидов, жирных кислот, эргостерина и убихинонов, заключающийся в том, что осуществляют следующие стадии: а) экстрагируют биомассу гриба Blakeslea trispora с влажностью 7-75% полярным растворителем (например, ацетоном) или смесью полярного и неполярного растворителей (например, ацетона и гексана или ацетона и...
Препарат, включающий по меньшей мере один фунгицидный коназол, способ получения препарата (варианты), его применение для борьбы с фитопатогенными грибами, применение сополимера для получения препарата активных соединений и для стабилизирования водной дисперсии смеси активных соединений

Номер патента: 15876
Опубликовано: 30.12.2011
Авторы: Ёттер Гюнтер, Шроф Вольфганг, Братц Маттиас, Кольтценбург Зебастиан, Крюгер Кристиан, Домбо Петер
МПК: A01N 43/653, A01N 25/14, A01N 25/10...
Метки: сополимера, препарата, активных, борьбы, коназол, включающий, смеси, водной, мере, фунгицидный, препарат, меньшей, способ, фитопатогенными, грибами, один, применение, дисперсии, получения, стабилизирования, соединений, варианты
Формула / Реферат:
1. Препарат, включающий смесь по меньшей мере двух различных средств защиты посевов, гдеа) по меньшей мере одно фунгицидно активное соединение выбрано из группы коназолов (активное соединение 1) иб) по меньшей мере одно дополнительное средство защиты посевов (активное соединение 2), имеющее растворимость в воде при 20°С менее 20 г/л, которое выбрано из группы, состоящей из коназольных фунгицидов, отличных от активного соединения 1,...
Новые производные 5-0-дезозаминил-6-0-метилэритронолида а, способ их получения и их использование при получении биологически активных продуктов

Номер патента: 765
Опубликовано: 24.04.2000
Авторы: Шаппер Бернадетт, Бонне Алан, Лагуарда Жак
МПК: A61K 31/70, C07H 17/00
Метки: получения, использование, 5-0-дезозаминил-6-0-метилэритронолида, получении, продуктов, новые, производные, биологически, активных, способ
Формула / Реферат:
1. Соединения формулы (I) в которых R представляет собой остаток карбоновой кислоты, содержащий до 18 атомов углерода. 2. Соединение формулы (I) по п.1, в котором R представляет собой ацетил. 3. Способ получения соединений формулы (I), определенной в п.1 или 2, отличающийся тем, что соединение формулы (II) в котором R имеет указанное выше значение, подвергают воздействию производного сульфоновой кислоты, затем воздействию основания,...
Предыдущий патент: Система радиочастотных ofdm-mimo передач
Следующий патент: Соединительный профиль и комбинированная шпунтовая стенка
Случайный патент: Игровая система, соответствующие способы и устройства для ее осуществления