Новый одноцентровый компонент катализатора с металлсодержащей основной цепью хелата
Номер патента: 11672
Опубликовано: 28.04.2009
Авторы: Хан Кристина, Глэдис Джон, Туба Роберт, Разави Аббас









Формула / Реферат
1. Комплекс, включающий центральный атом М2 и хелатный лиганд с металлсодержащей основной цепью общей формулы (I)

где G представляет собой группу, выбранную из группы СО или монодентатного или бидентатного аминового, или фосфинового, или циклопентадиенилового лиганда, для ингибирования активности металла М1;
металл М1 в основной цепи представляет Re;
металл М2 в комплексе представляет собой металл группы 10 Периодической таблицы;
R1 и R2, каждый независимо, выбраны из гидрокарбилов, имеющих от 1 до 20 углеродных атомов;
А представляет собой О или NR#, где R# выбран из гидрокарбилов, имеющих 4-20 углеродных атомов;
В представляет собой аллил или гидрокарбильную группу, имеющую от 1 до 20 углеродных атомов и пиридиновую группу; и
n - это валентность М1 минус 2.
2. Комплекс по п.1, где М2 представляет собой Ni или Pd.
3. Комплекс по любому из пп.1, 2, где R1 и R2, каждый независимо, представляют собой алкильные группы, имеющие от 1 до 6 углеродных атомов, или арильные группы, имеющие 6-10 углеродных атомов.
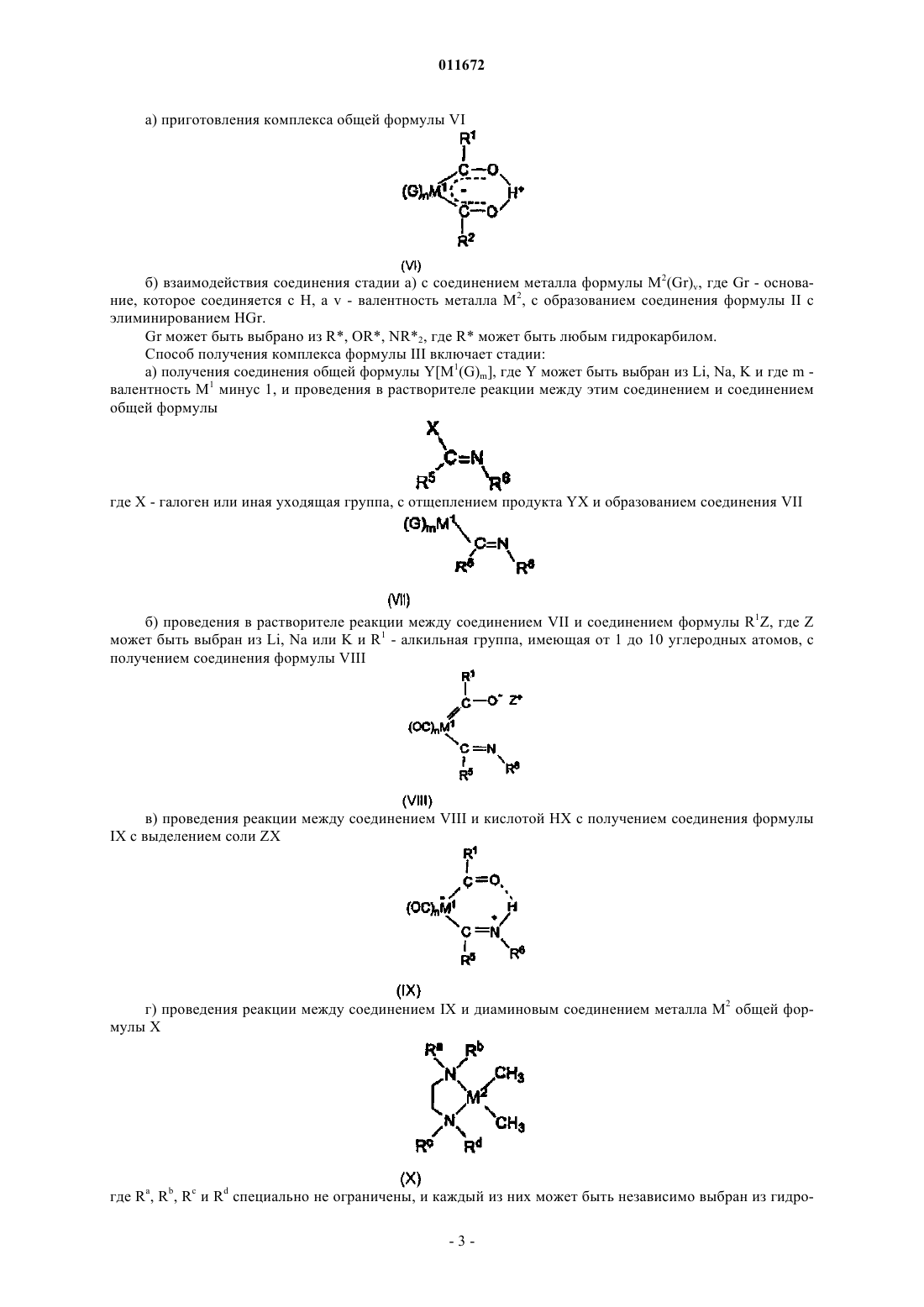
4. Способ приготовления комплекса по любому из пп.1-3, включающий стадии:
а) получения хелатного лиганда общей формулы (V)

б) взаимодействия соединения V с M3Y, где М3 может быть выбран из Li, Na или K, Y - основание и может быть выбрано из R*, OR* или NR*2, где R* может быть любой гидрокарбильной группой, и
в) взаимодействия продукта реакции стадии (б) с LXM2=В, где М2 и В определены выше, X может быть выбран из галогенидов или сульфонатов и L предпочтительно представляет собой фосфин;
г) извлечения комплекса I с элиминированием HY и М3Х.
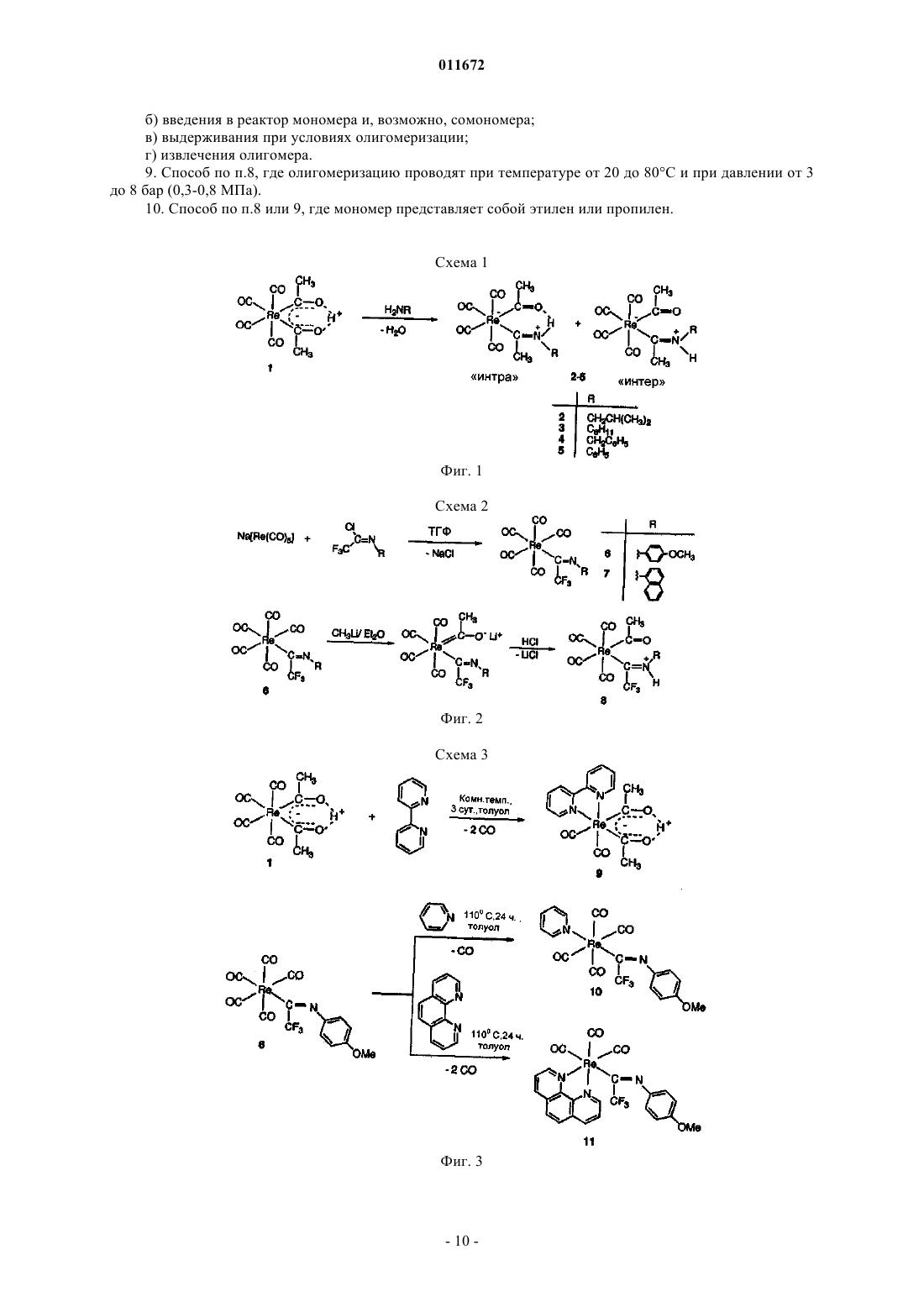
5. Способ приготовления комплекса по любому из пп.1-3, включающий стадии:
а) получения хелатного лиганда общей формулы (V)

б) взаимодействия соединения V с LRxM2=B, где Rx - гидрокарбил, a L - любой нейтральный донорный лиганд, подобный амину или фосфину;
в) извлечения комплекса I с элиминированием HRx и L.
6. Активная каталитическая система, включающая:
а) комплекс по любому из пп.1-3;
б) возможно, активирующий агент;
в) возможно, носитель.
7. Активная каталитическая система по п.6, где активирующий агент представляет собой алюмоксан.
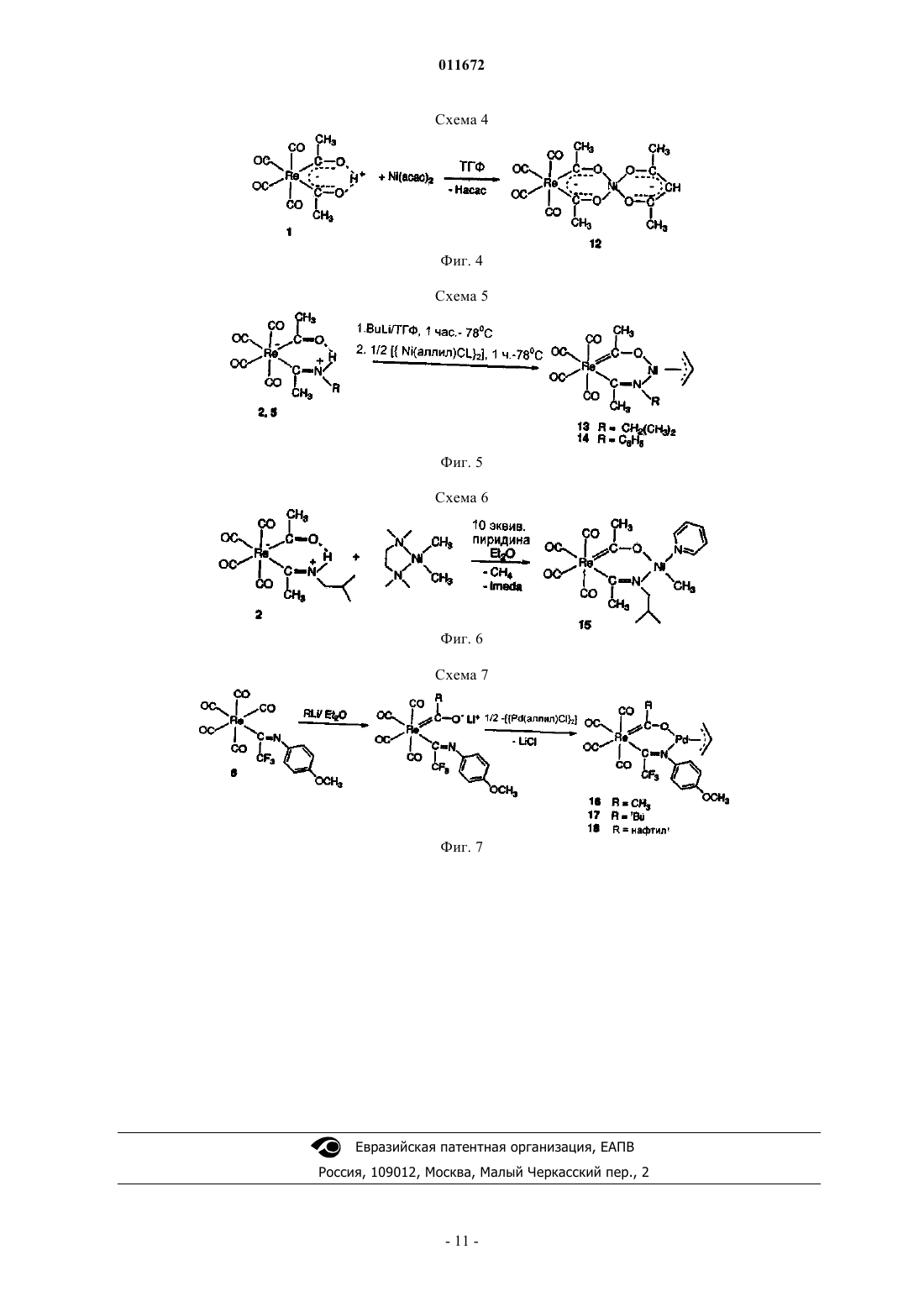
8. Способ олигомеризации альфа-олефинов, включающий стадии:
а) введения в реактор активной каталитической системы по п.6 или 7;
б) введения в реактор мономера и, возможно, сомономера;
в) выдерживания при условиях олигомеризации;
г) извлечения олигомера.
9. Способ по п.8, где олигомеризацию проводят при температуре от 20 до 80шС и при давлении от 3 до 8 бар (0,3-0,8 МПа).
10. Способ по п.8 или 9, где мономер представляет собой этилен или пропилен.
Текст
011672 Настоящее изобретение относится к области каталитических систем на основе новых одноцентровых компонентов катализатора с металлсодержащей основной цепью хелата. Эти комплексы пригодны для олигомеризации олефинов. Комплексы переходных металлов с NO хелатными лигандами интенсивно исследовались в связи с разработкой гомогенных катализаторных систем. Салицилальдиминовые производные рассматриваются как важные лиганды в разнообразных каталитических системах, таких, например, как карбеновые комплексы рутения для метатезиса алкенов, как описано, например, Opstal и Verpoort (Opstal Т., Verpoort F.,in J. Mol. Cat. A: Chem., 2003, 200, 49) или с образованием нейтральных комплексов никеля (II), которые являются высокоактивными катализаторами полимеризации алкенов, как описано, например, Ittel et al.(Ittel S.D., Johnson L.K., Brookhart M., in Chem. Rev. 2000, 100, 1169), и могут также функционировать в однокомпонентном режиме, как описано Younkin et al. (Younkin T.R., Conner E.F., Henderson J.I., Friedrich S.F., Grubbs R.H., Bansleben D.A., in Science, 2000, 287, 460). Различные характеристики хелатных лигандов широко исследовались, например, Hicks et al. (HicksF.A., Jenkins J.C. Brookhart M., in Organometallics 2003, 22, 3533), которые изучали размер NO хелатного кольца. Также были исследованы электронный и стерический эффекты заместителей, влияющие на основные цепи хелатов, например, Shim et al. (Shim С.В., Kim Y.H., Lee B.Y., Dong Y., Yun H., in J. Organomet. Chem. 2003, 675, 72) обнаружили, что очень объемные заместители при N-арильной группе в салицилальдиминатных комплексах играют ключевую роль в образовании полимеров с высокой молекулярной массой и в каталитической активности. Совсем недавно замечательно тонкая электронная регулировка разветвленности и молекулярной массы с помощью отдаленных заместителей при N-арильной группе была разработана и описана Zuideveld et al. (Zuideveld M., Wehrmann P., Rhr C, Mecking S. in Angew. Chem. Int. Ed. 2004, 43, 869). Тем не менее сохраняется необходимость в лучшем понимании роли заместителей и электронного строения этих комплексов для более эффективного осуществления направленного регулирования структуры полимеров, получаемых с этими комплексами. Задачей настоящего изобретения является получение хелатных комплексов с металлсодержащими основными цепями лиганда. Задачей настоящего изобретения также является разработка способа получения этих хелатных комплексов. Задачей настоящего изобретения также является использование этих комплексов для олигомеризации или полимеризации альфа-олефинов. Соответственно, в настоящем изобретении предложены хелатные комплексы с металлсодержащей основной цепью лиганда общей формулы I где G - группа, которая ингибирует активность металла М 1, такая как, например, группа СО или монодентатный или бидентатный амин, или фосфин, или циклопентадиенил, или любой подобный общеупотребительный лиганд; металл М 1 в основной цепи - это металл группы 6-9 Периодической таблицы; металл М 2 в хелатном комплексе - это металл группы 10 Периодической таблицы;R1, R2, каждый независимо, выбраны из гидрокарбилов, имеющих от 1 до 20 углеродных атомов; А - это О или N-R, где R выбран из гидрокарбилов, имеющих 4-20 углеродных атомов; В представляет собой аллил или гидрокарбильную группу, имеющую от 1 до 20 углеродных атомов и пиридин или подобный азотсодержащий гетероцикл, нитрилорганическую или фосфиновую группу; иn - валентность М 1 минус 2. Предпочтительно М 1 выбран из Re или Mn. Предпочтительно М 2 выбран из Ni или Pd. Предпочтительно G - это группа СО. Предпочтительно R1 и R2, каждый независимо, являются алкильными группами, имеющими от 1 до 6 углеродных атомов, или арильными группами, имеющими от 6 до 10 углеродных атомов, более предпочтительно они одинаковы и являются метильными группами. В первом воплощении настоящего изобретения предложены хелатные комплексы общей формулы II где металл М 1, М 2, n, R1 и R2 определены выше; и R3 и R4, каждый независимо, выбраны из гидрокарбилов, имеющих от 1 до 20 углеродных атомов. Предпочтительно R3 и R4, каждый независимо, являются алкильными группами, имеющими от 1 до 6 углеродных атомов, или арильными группами, имеющими от 6 до 10 углеродных атомов, более предпочтительно они одинаковы и являются метильными группами. Во втором воплощении настоящего изобретения предложены хелатные комплексы формулы III где R5 - гидрокарбил, имеющий от 1 до 20 углеродных атомов, R8 - гидрокарбил, имеющий от 1 до 20 углеродных атомов, или полностью или частично галогенированный гидрокарбил, имеющий от 1 до 20 углеродных атомов, и R7 - амин или фосфин. Предпочтительно R5 - арильная группа, более предпочтительно замещенная арильная группа, наиболее предпочтительно замещенная фенильная группа. Предпочтительно R6 - полностью галогенированный алкил, имеющий от 1 до 6 углеродных атомов,более предпочтительно CF3. Предпочтительно R7 - пиридин. В третьем воплощении настоящего изобретения предложены хелатные комплексы формулы IV где все символы описаны выше. В настоящем изобретении также предложен способ получения комплексов формул I, II, III и IV. Существует два общих способа получения комплекса I, исходя из хелатного лиганда общей формулы V В первом способе хелатный лиганд V вводят в реакцию во-первых, с M3Y, где М 3 может быть выбран из Li, Na или K, Y - основание, и оно может быть выбрано из R, OR или NR2, где R может быть любой гидрокарбильной группой, и во-вторых, с LXM2=B, где М 2 и В определены выше, X может быть выбран из галогенидов или сульфонатов и L предпочтительно представляет собой фосфин; с образованием комплекса I с элиминированием HY и М 3 Х. Если В - аллил, то L представляет собой ХМ 2=В, и используется только половина эквивалента. Во втором способе хелатный лиганд V реагирует с LRXM2=B, где RX - гидрокарбил, a L - любой нейтральный донорный лиганд, подобный амину или фосфину, с элиминированием L и HRX. Способ приготовления комплекса формулы II включает стадии:-2 011672 а) приготовления комплекса общей формулы VI б) взаимодействия соединения стадии а) с соединением металла формулы M2(Gr)v, где Gr - основание, которое соединяется с Н, a v - валентность металла М 2, с образованием соединения формулы II с элиминированием HGr.Gr может быть выбрано из R, OR, NR2, где R может быть любым гидрокарбилом. Способ получения комплекса формулы III включает стадии: а) получения соединения общей формулы Y[M1(G)m], где Y может быть выбран из Li, Na, K и где m валентность М 1 минус 1, и проведения в растворителе реакции между этим соединением и соединением общей формулы где X - галоген или иная уходящая группа, с отщеплением продукта YX и образованием соединения VII б) проведения в растворителе реакции между соединением VII и соединением формулы R1Z, где Z может быть выбран из Li, Na или K и R1 - алкильная группа, имеющая от 1 до 10 углеродных атомов, с получением соединения формулы VIII в) проведения реакции между соединением VIII и кислотой НХ с получением соединения формулы г) проведения реакции между соединением IX и диаминовым соединением металла М 2 общей формулы X где Ra, Rb, Rc и Rd специально не ограничены, и каждый из них может быть независимо выбран из гидро-3 011672 карбилов, имеющих от 1 до 20 углеродных атомов, причем указанную реакцию проводят в растворителе и, возможно, со стабилизирующим агентом,д) извлечение соединения формулы III с выделением метана и соответствующего диамина. Обычно растворитель может быть выбран, как правило, из диэтилового эфира (Et2O) или тетрагидрофурана (ТГФ). Предпочтительно все Ra, Rb, Rc и Rd одинаковы и являются метильными группами, в этом случае соответствующий выделяющийся диамин представляет собой тетраметилдиамин. Предпочтительно присутствует стабилизирующий агент, и он выбран из CH3CN или других органических нитрилов, или пиридина, или подобных азотсодержащих гетероциклов. Более предпочтительно это пиридин. Комплекс формулы IV может быть приготовлен различными путями, и способ получения выбирают в зависимости от природы металла М 2. Способ получения комплекса формулы IV, где М 2 - это Ni, включает стадии от а) до в), использованные при получении соединения металла формулы III, с последующим г) взаимодействием в растворителе комплекса формулы IX стадии (в) с соединением R8Z, где R8 - гидрокарбил, имеющий от 1 до 20 углеродных атомов; и с соединением никеля формулы [Ni(аллил)Х 2], где аллил является незамещенным или замещенным, и где X - галоген,при температуре ниже 0 С и в течение периода времени от 30 мин до 2 ч,е) извлечением комплекса никеля общей формулы IV с выделением ZX и алкана R8H. Предпочтительно Z - это литий, X - хлор, a R8 - бутил. Предпочтительно температура составляет менее -10 С, более предпочтительно менее -20 С и наиболее предпочтительно около -78 С. Предпочтительно реакция проводится за период времени около 1 ч. Способ приготовления комплекса формулы IV, где М 2 - Pd, включает стадии от а) до б), используемые при получении соединения металла формулы III, с последующим б) взаимодействием в растворителе комплекса формулы VIII стадии (б) с соединением палладия формулы [Pd(аллил)Х 2], где аллил является незамещенным или замещенным и где X - галоген, при температуре от 0 С до комнатной температуры (около 25 С) и в течение периода времени от 30 мин до 2 ч; в) извлечением комплекса формулы IV с выделением ZX. В последующих воплощениях настоящего изобретения одна или более групп СО, появляющихся в основной цепи хелатных лигандов, может быть заменена аминами, такими как, например, пиридин, дипиридин, фенантролин. Комплексы согласно настоящему изобретению могут быть использованы для олигомеризации альфа-олефинов. Предпочтительно для улучшения активности системы используется активирующий агент. В настоящем изобретении предложена активная каталитическая система олигомеризации, включающая любой из комплексов металла, описанных выше; возможно, активирующий агент; и возможно, носитель. Активирующим агентом может быть алкилалюминий, представленный формулой AIRnX3-n, где R это алкил, имеющий от 1 до 20 углеродных атомов, а X - галоген. Предпочтительные алкилирующие агенты - триизобутилалюминий (ТИБА) или триэтилалюминий (ТЭА). Альтернативно, в качестве активирующего агента может быть использован алюмоксан. Алюмоксаны хорошо известны и предпочтительно включают олигомерные линейные и/или циклические алкилалюмоксаны, представленные формулой для олигомерного, циклического алюмоксана,где n = 1-40, предпочтительно 10-20, m = 3-40, предпочтительно 3-20, a R - С 1-С 8 алкильная группа,предпочтительно метил. Подходящие борсодержащие активирующие агенты могут включать трифенилкарбенийборат, такой как тетракис-пентафторфенил-боратотрифенилкарбений, как описано в ЕР-А-0427696, или имеющие общую формулу [L'-H]+[В Ar1 Ar2 Х 3 Х 4]-, как описано в ЕР-А-0277004 (от с. 6, строка 30 до с. 7, строка 7). Возможно, компонент катализатора может быть нанесен на носитель. Предпочтительные носители включают пористый твердый носитель, такой как тальк, неорганические оксиды и полимерные материа-4 011672 лы носителей, такие как полиолефин. Предпочтительно материал носителей - это неорганический оксид в тонкоизмельченном виде. Подходящие неорганические оксидные материалы хорошо известны в технике. Предпочтительно носитель - это кремнеземный носитель, имеющий площадь поверхности 200-700 м 2/г и объем пор 0,5-3 мл/г. Количество активирующего агента и компонента катализатора, преимущественно применяемых при получении катализатора на твердом носителе, можно варьировать в широком диапазоне, и оно зависит от природы активирующего агента и металла. Как правило, отношение Al/М 2 варьируют от 100 до 2000. В настоящем изобретении также предложен способ олигомеризации альфа-олефинов, который включает стадии: а) введения в реактор активной, возможно нанесенной на носитель, каталитической системы, описанной выше; б) введения в реактор мономера и, возможно, сомономера; в) выдерживания при условиях олигомеризации; г) извлечения олигомера. Условия олигомеризации не особенно ограничены. Температура может составлять от 20 до 80 С,предпочтительно от 30 до 40 С, а давление может составлять от 1 до 10 бар (0,1-1 МПа), предпочтительно от 3 до 8 бар (0,3-0,8 МПа). Олигомеризацию, как правило, проводят в течение по меньшей мере 1 ч. Мономеры и сомономеры, как правило, выбирают из альфа-олефинов, но они могут включать стирол и полярные мономеры, такие как, например, метакрилаты и метилвинилкетоны. Предпочтительно они выбраны из этилена и пропилена. Список чертежей На фиг. 1 представлена схема реакции 1, использованная для приготовления ренакетоиминов,комплексов 2-5; на фиг. 2 - схема реакции 2, использованная для приготовления имидоилпентакарбонилрения, комплексов 6 и 7 и ренаиминового комплекса 8; на фиг. 3 - схема реакции 3, использованная для приготовления бипиридин-ренадикетона (комплекс 9), пиридинимидоилрения (комплекс 10) и фенантролинимидоилрения (комплекс 11); на фиг. 4 - схема реакции 4, использованная для приготовления ренадикетонатоникеля (II), комплекс (12); на фиг. 5 - схема реакции 5, использованная для приготовления ренакетоиминатоаллилникеля(II), комплексы 13 и 14; на фиг. 6 - схема реакции 6, использованная для приготовления ренакетоиминатометилникеля(II), комплекс 15; на фиг. 7 - схема реакции 7, использованная для приготовления ренакетоиминатоаллилпалладия(II), комплексы 16,17 и 18. Примеры Все операции проводили в атмосфере азота. ТГФ, диэтиловый эфир, пентан и толуол были перегнаны с Na/бензофеноном, а CH2Cl2 был перегнан с СаН 2. Первичные амины и пиридин были осушены KOH и перегнаны перед применением. Алкиллитиевые реагенты, бипиридин и фенантролин были получены от Fluka, Aldrich и ACROS, Ni(acac)2 от ABCR, МАО от ATOFINA, [Pd(аллил)Cl2] от Aldrich. Рена-дикетон, трифторацетилимидоилхлориды, [Ni(аллил)Cl2] и [Ni(tmeda)(CH3)2] были приготовлены в соответствии с процедурами, описанными в литературе, соответственно по Lukehart и Zeile (Lukehart С.М., Zeile J.V., in J. Am. Chem. Soc. ,98, 2365, 1976), Tamura et al. (Tamura K., Mizukami H., Maeda K.,Watanabe H., Unayama K., in J. Org. Chem., 58, 32, 1993), Taube et al. (Taube R., Bohme P., Gehrke J.-P., inZ. Anorg. Allg. Chem. 578, 89, 1989) and Kaschube et al. (Kaschube W., Porschke K.R., Wilke G., in J. Organomet Chem., 355, 525, 1988). Спектры ЯМР были записаны на спектрометрах Bruker 300 и 400 MHz, а ИК-спектры были получены на ASI React-IR спектрометре. Газовую хроматографию проводили на приборе ThermoQuest Trace GC 2000. Приготовление ренакетоиминов (комплексы 2-5). Эти синтезы были выполнены в соответствии со схемой 1, представленной на фиг. 1 и описанной у-5 011672 К раствору 0,5 ммоль исходного комплекса 1 в 5 мл CH2Cl2 добавляли 3,0 ммоль RNH2, где R был,соответственно, выбран из СН 2 СН(СН 3)2, С 6 Н 11, СН 2 С 6 Н 5 или С 6 Н 5. Смесь перемешивали при комнатной температуре в течение периода времени, зависящего от природы R. Он составлял, соответственно, 1 ч для R = СН 2 СН(СН 3)2, 6 ч для R = C6H11, 9 ч для R = СН 2 С 6 Н 5 и 48 ч для R = С 6 Н 5. Растворитель удаляли при пониженном давлении. Остаток растворили в пентане и после фильтрации растворитель удалили при пониженном давлении, при этом осталась бледно-желтая смесь масла с твердым веществом. В случае анилина реакцию проводили в Et3N, использованном в качестве растворителя, и добавляли молекулярные сита 4 . Смесь очень медленно перемешивали в течение 2 суток. Растворитель удалили при пониженном давлении. Остаток экстрагировали с примерно 10 мл пентана, и после удаления пентана желтое масло было извлечено в качестве сырого продукта, который очистили хроматографией с силикагелем. Выход находился в диапазоне от 50 до 60%. Был проведен 1 Н ЯМР анализ всех конечных продуктов и получены следующие результаты. Для R = СН 2 СН(СН 3)2 (комплекс 2) 1 Н ЯМР (CDCl3):(интра изомер) 1,07 (d, JHH = 6,7 Hz, 6H,СН 3), 2,05 (m, 1H, CH), 2,50 (s, 3 Н, СН 3 С=О), 2,84 (s, 3 Н, CH3C=N), 3,57 (ps t, JHH = 4 Hz, 2H, CH2), 9,16(s, 3 Н, СН 3 С=О), 2,75 (s, 3 Н, CH3C=N), 4,74 (d, JHH = 5,4 Hz, 2H, CH2), 7,26-7,81 (m, 5 Н, С 6 Н 5), 13,41 (br,1 Н, NH). Для R = С 6 Н 5, (комплекс 5) 1 Н ЯМР (CDCl3):(интер изомер) 2,70 (s, 3 Н, СН 3), 2,77 (s, 3 Н, СН 3),7,15-7,28 (m, 2 Н, Ph), 7,42-7,52 (m, 2 Н, Ph), 14,89 (br, 1 Н, NH). Приготовление имидоилпентакарбонилрения, комплексов 6 и 7, и ренаиминов (комплекс 8). Эти синтезы были выполнены в соответствии с новой схемой 2, представленной на фиг. 2. Раствор 1,65 ммоль Na[Re(CO)5] в 30 мл ТГФ вводили в реакцию с 1,63 ммоль F3CC(Cl)=NR, где R был соответственно выбран из п-СН 3 ОС 6 Н 4 или нафтила. По ходу реакции образовался белый осадок, и раствор стал светло-желтым. Через сутки суспензию отфильтровали и растворитель из фильтрата удалили при пониженном давлении. Желтый остаток растворили в 100 мл пентана, раствор перемешивали в течение 1 ч, осадок отфильтровали и объем фильтрата уменьшили при пониженном давлении. В осадок выпали светло-желтые кристаллы. Выход в обоих случаях был 95%. 1 Н ЯМР и инфракрасный (ИК) анализ были выполнены для всех конечных продуктов, и были получены следующие результаты. Для R = п-СН 3 ОС 6 Н 4 (комплекс 6) 1 Н ЯМР (CDCl3):3,69 (s, 3 Н, ОСН 3), 6,40 (d, JHH = 8 Hz, 2 Н,С 6 Н 4), 6,78 (d, JHH = 8 Hz, 2H, C6H4); 13 С ЯМР (CDCl3):55,6 (s), 114,9 (s), 119,5 (s), 123,2 (q, JCF = 280 Hz,CF3), 151,4 (s), 157,5 (s), 179,6 (s), 180,3 (q, JCF = 34 Hz, ReCCF3), 180,7 (s); ИК (ТГФ, v см-1): 2140, 2056,2011, 1993. Для R = нафтил (комплекс 7) 1 Н ЯМР (C6D6):6,40 (d, JHH = 7,3 Hz, 1H), 7,16 (m, 3 Н), 7,48 (d, JHH = 8,3 Hz, 1H), 7,57 (d, JHH = 8,1 Hz, 1H), 7,73 (d, JHH = 8,1 Hz, 1H); 13C ЯМР (C6D6):112,3 (s), 123,4 (s),125,0 (s), 125,5 (s), 126,0 (s), 126,1 (s), 126,9 (s), 128,1 (s), 134,6 (s), 154,2 (s), 124,2 (q, JCF = 280 Hz, CF3),180,0 (q, JCF = 35,3 Hz, ReCCF3), 179,4 (s), 180,6 (s); ИК (ТГФ, v см-1): 2138, 2026, 2009. 640 мг (1,22 ммоль) комплекса 6, приготовленного на предыдущей стадии, вводили в реакцию с 0,83 мл (1,22 ммоль) CH3Li в 40 мл диэтилового эфира при температуре -78 С. После перемешивания реакционной смеси в течение 2 ч добавили 6 мл (1,22 ммоль) насыщенного раствора HCl в диэтиловом эфире при температуре -78 С и выдержали при перемешивании в течение 15 мин. Затем смесь нагревали до 0 С и перемешивали еще в течение 1 ч при 0 С. Растворитель удалили при пониженном давлении и желтый остаток экстрагировали 120 мл н-гексана при комнатной температуре. После фильтрации раствора и удаления растворителя при пониженном давлении было получено 333 мг желтого масла (комплекс 8) с выходом 50%. Был выполнен ЯМР- и ИК-анализы со следующими результатами. 1 Н ЯМР (CDCl3):2,61 (s, 3 Н, СН 3), 3,74 (s, 3 Н, ОСН 3), 6,85 (d, JHH = 9,0 Hz, 2 Н, С 6 Н 4), 7,09 (d, JHH = 9,0 Hz, 2H, C6H4), 16,11 (s, 1H, NH); 13 С ЯМР (CDCl3):28,69 (s), 54,6 (s), 113,5 (s), 123,2 (s), 128,2 (q, JCF= 280 Hz, CF3), 152,8 (s), 159,0 (s), 186,6 (s), 236,6 (q, ReCCF3); ИК (Et2O, v см-1): 2080,1985,1955,1914. Одна или более CO групп в исходном комплексе 1 или комплексе 6 могут быть заменены азотсодержащими соединениями. Приготовление бипиридин-ренадикетона (комплекс 9). 197 мг (0,51 ммоль) исходного комплекса 1 вводили в реакцию с 85 мг (0,54 ммоль) бипиридина в 14 мл толуола при комнатной температуре в течение 3 суток. В течение этого периода из реакционного раствора постепенно выпадал ярко-желтый порошок. Порошок был собран путем фильтрации, промыт 5-6 011672 мл гексана и осушен под вакуумом. Были получены 70 мг (0,144 ммоль) комплекса 9 с выходом 28%. Были проведены ЯМР- и ИК-анализы и получены следующие результаты. 1 Н ЯМР (CDCl3):1,49 (s, 6H, СН 3), 7,42 (m, 2H, bpy), 7,97 (m, 2H, bpy), 8,05 (m, 2H, bpy), 9,10 (d,JHH = 5 Hz, 2H, bpy), 19,91 (s, 1H, Н); 13 С ЯМР (CDCl3):23,3 (s), 122,6 (s), 126,8 (s), 138,9 (s), 153,3 (s),154,4 (s), 176,8 (s), 179,6 (s); ИК (Et2O, v см-1): 2023, 2014, 1911, 1893. Приготовление пиридин-имидоилрения (комплекс 10). 100 мг (0,189 ммоль) комплекса 6 растворили в 5 мл толуола, к которому добавили 15,7 мг (0,198 ммоль) пиридина. Реакционную смесь дефлегмировали в течение ночи, пока ее цвет не изменился от светло-желтого до вишнево-красного. Толуол удалили при пониженном давлении, а оставшееся красноватое масло промыли 3 мл н-гексана и осушили под вакуумом. Было получено 98 мг (0,17 ммоль) комплекса 10 с выходом 91%. Был проведен ИК-анализ, который дал следующий результат. ИК (CH2Cl2, v см-1): 2086, 1978, 1939, 1893. Приготовление фенантролин-имидоилрения (комплекс 11). 100 мг (0,189 ммоль) комплекса 6 вводили в реакцию с 36 мг (0,2 ммоль) 1,10-фенантролина в 5 мл перегнанного толуола. В течение 1 суток цвет изменился от светло-желтого до темно-оранжевого. После охлаждения образовались оранжево-красные кристаллы, которые были отфильтрованы и промыты 3 мл н-гексана и высушены под вакуумом. Было получено 74 мг (0,11 ммоль) комплекса 11 с выходом 60%. Результаты ЯМР- и ИК-анализов были следующими. 1 Н ЯМР (C6D6):3,40 (s, 3 Н, ОСН 3), 6,15 (d, JHH = 8,8 Hz, 2H, С 6 Н 4), 6,37 (d, JHH = 8,8 Hz, 2H, C6H4),6,41 (m, 2 Н, phen), 6,80 (s, 2H, phen), 7,07 (m, 2H, phen), 8,55 (d, JHH = 5 Hz, 2H, phen); 13C ЯМР (C6D6):55,6 (s), 113,8 (s), 118,9 (s), 124,9 (s), 125,4 (q, JCF = 280 Hz, CF3), 127,2 (s), 130,2 (s), 136,5 (s), 146,4 (s),148,4 (s), 154,0 (s), 155,1 (s), 191,2 (s), 198,2 (s), 201,5 (q, JCF = 31 Hz, ReCCF3); ИК (CH2Cl2, v см-1): 2011,1893. Приготовление комплексов 9, 10 и 11 схематически представлено на фиг. 3. Приготовление комплекса ренадикетонатоникеля (II) (комплекс 12). Раствор 99 мг (0,389 ммоль) Ni(acac)2 в 7 мл ТГФ добавили к раствору 150 мг (0,389 ммоль) комплекса 1 в 5 мл ТГФ. Реакционную смесь перемешивали в течение 3 ч. Растворитель удалили при пониженном давлении, а остаток перекристаллизовали из пентана с получением сине-зеленых твердых кристаллов (комплекс 12). Это приготовление схематически представлено на фиг. 4. Проведенный ЯМР- и ИК-анализ комплекса 12 дал следующие результаты. 1 Н ЯМР (C6D6):1,80 (s, 3H, СН 3), 2,40 (s, 3H, СН 3), 5,34 (s, 1H, СН) и ИК (v см-1): 2081, 2050, 1957,1922, 1590, 1513, 1447, 1386, 1336, 1262, 1131, 1089, 1019, 926, 779. Приготовление ренакетоиминато-аллилникеля (II), комплексы 13 и 14. К раствору 200 мг (0,454 ммоль) комплекса 2 в 7 мл ТГФ добавили 0,283 мл nBuLi (1,6 М в гексане) при температуре -78 С. После перемешивания в течение 1 ч при -78 С добавили раствор 61 мг (0,454 ммоль Ni) [Ni(аллил)Cl2] в 2 мл ТГФ при такой же температуре. Реакционную смесь перемешивали еще 1 ч при -78 С. После удаления растворителя было получено темное красно-коричневое твердое вещество. Таким же образом комплекс 5 последовательно вводили в реакцию с nBuLi и с [Ni(аллил)Cl2],также с получением темного красно-коричневого твердого вещества. Комплексы 13 и 14 схематически представлены на фиг. 5, и они были охарактеризованы 1 Н ЯМР вC6D6. Для R = СН 2 СН(СН 3)2 (комплекс 13) 1 Н ЯМР (C6D6):0,54 (d, 3 Н, СН 3), 0,57 (d, 3 Н, СН 3), 1,38 (s,3 Н, СН 3 СО), 1,96 (s, 3H, CH3CN), 3,98 (d, аллил), 4,63 (m, аллил). Для R = С 6 Н 5 (комплекс 14) 1 Н ЯМР (C6D6):1,27 (s, 3 Н, СН 3 СО), 1,74 (s, 3 Н, CH3CN), 4,03 (d, аллил), 4,73 (m, аллил), 6,49 (m, C6H5), 6,99 (m, C6H5). Приготовление ренакетоиминато-метилникеля (II), комплекс 15. К раствору 420 мг (0,95 ммоль) комплекса 2 и 195 мг (0,95 ммоль) [Ni(tmeda)(CH3)2] в 20 мл диэтилового эфира было добавлено 0,77 мл (9,5 ммоль) пиридина. После перемешивания реакционной смеси в течение 6 ч при комнатной температуре темный красно-коричневый осадок сырого продукта был отфильтрован, а затем промыт пентаном и высушен в вакууме. Этот синтез схематически представлен на фиг. 6. ЯМР анализ дал следующие результаты: 1 Н ЯМР (ТГФ-d8):0,57 (s, 3 Н, NiCH3), 0,84 (m, 6H,СН 2 СН(СН 3)2), 2,17 (s, CH3C=O), 2,57 (s, CH3C=N), 7,54 (t, 2 Н, ру), 7,94 (t, 1H, py), 9,17 (d, 2H, ру). Приготовление ренакетоиминато-аллилпалладия (II), комплексы 16,17 и 18. 107 мг (0,189 ммоль) комплекса 6 вводили в реакцию с 0,128 мл CH3Li в 10 мл ТГФ при температуре 0 С. Смесь выдержали при перемешивании 1 ч, а затем к реакционному раствору добавили 35 мг (0,096 ммоль) [Pd(аллил)Cl2] и смесь перемешивали еще в течение 3 ч. После добавления Pd комплекса цвет реакционного раствора изменился от светло-желтого до темно-зеленого. Растворитель удалили при пониженном давлении, а темный остаток экстрагировали с 70 мл н-пентана. Раствор профильтровали и растворитель удалили из фильтрата при пониженном давлении с образованием зеленого масла (комплекс 16).-7 011672 Используя точно такую же процедуру, комплекс 6 вводили в реакцию с tBuLi и [Pd(аллил)Cl2] также с получением зеленого масла (комплекс 17). Используя точно такую же процедуру, комплекс 6 вводили в реакцию с нафтил-Li и[Pd(аллил)Cl2] также с получением зеленого масла (комплекс 18). Приготовление комплексов 16, 17 и 18 схематически представлено на фиг. 7. Был выполнен ЯМР- и ИК-анализ комплексов 16 и 17 со следующими результатами. 74 мг комплекса 16 (R = СН 3) были получены с выходом 57%. 1 Н ЯМР (C6D6):1,34 (s, 3 Н, СН 3), 3,21 (s, 3 Н, ОСН 3), 2,20 (d, JHH = 13 Hz, 1 Н, С 3 Н 6 (аллил, 2,24 (d,JHH = 13 Hz, 1H, С 3 Н 5 (аллил, 2,81 (d, JHH = 7 Hz, 1H, С 3 Н 5 (аллил, 3,3 (d, JHH = 8 Hz, 1H, С 3 Н 5 (аллил,4,53 (m, 1H, С 3 Н 5 (аллил, 6,54 (d, JHH = 8 Hz, 2 Н, С 6 Н 4), 6,6 (d, JHH = 8 Hz, 2H, C6H4); 13 С ЯМР (C6D6):30,2 (s), 54,9 (s), 60,0 (s), 65,8 (s), 113,9 (s), 115,5 (s), 120,6 (s), 150,7 (s), 157,3 (s), 187,2 (s), 188,7 (s), 217,9; ИК (ТГФ, v см-1): 2103, 2030, 2009, 1989, 1951. 53 мг комплекса 17 (R = C(CH3)3) были получены с выходом 40%. 1 Н ЯМР (C6D6):0,68 (s, 3 Н, С(СН 3, 0,87 (s, 3 Н, С(СН 3, 1,17 (s, 3 Н, С(СН 3; 3,21 (s, 3 Н, ОСН 3),2,21 (d, JHH = 13 Hz, 1H, C3H5 (аллил, 2,24 (d, JHH = 12 Hz, 1H, C3H5 (аллил, 2,82 (d, JHH = 7 Hz, 1H, C3H5(m, 3 Н, нафтил); 13 С ЯМР (C6D6):54,94 (s), 58,9 (s), 68,48 (s), 113,84 (s), 115,23 (s), 120,56 (s), 126,20 (s),126,60 (s), 126,77 (s), 128,84 (s), 129,68 (s), 130,56 (s), 132,63 (s), 134,34 (s), 150,88 (s), 157,33 (s), 184,94 (s),186,07 (s), 187,80 (s), 216,71 (q, JC-C-F = 31 Hz, ReCCF3); ИК (ТГФ, v см-1): 2099, 2005, 1993, 1949. Олигомеризация этилена. В колбу Фишера-Портера, снабженную перемешивающим стержнем, были загружены 20 мл толуола и 1000 экв. МАО (30% в толуоле), и раствор был насыщен этиленом (25 бар этилена). В колбе Шленка 0,020 ммоль соответственно комплексов 1, 2, 3 и 5 растворили в 1 мл ТГФ. При температуре -10 С к раствору был добавлен 1 экв. nBuLi. После перемешивания в течение 30 мин при той же температуре-10 С добавили раствор 0,010 ммоль [Ni(аллил)Cl2] (= 0,020 ммоль Ni) в 1 мл ТГФ. После перемешивания при -10 С в течение времени от 5 до 10 мин растворитель удалили при пониженном давлении с получением темно-коричневого твердого вещества, которое растворили в толуоле и внесли в подготовленную колбу Фишера-Портера при перемешивании и в потоке азота. Температуру отрегулировали на уровне 30 С (3 С) и при давлении этилена 6 бар. В ходе реакции для всех 4 комплексов осаждались темно-коричневые хлопья. Реакционный раствор был проанализирован с помощью 1 Н ЯМР спектроскопии и, следуя методике, описанной Faissner и Huttner (Faissner R., Huttner G., in Eur. J. Inorg. Chem. 2239,2003), были приготовлены ГХ образцы. Обработка MeOH/HCl не дала твердого полимера. Результаты 1 Н ЯМР. Продукты олигомеризации с комплексом 1, 1 Н ЯМР (CDCl3):0,92 (t, JHH = 7 Hz, СН 3), 1,06 (t, JHH = 8 HZ, CH3), 1,68 (m, алифатический Н), 2,12 (m, алифатический H), 4,98 (d, JHH = 10 Hz, виниловый =СН 2),5,05 (d, JHH = 17 Hz, виниловый =СН 2), 5,49 (m, олефиновый =СН), 5,94 (m, виниловый =СН). Продукты олигомеризации с комплексом 3, 1 Н ЯМР (400 MHz, CDCl3):0,92 (t, JHH = 7Hz, CH3),1,00-1,38 (несколько перекрывающихся сигналов), 1,67 (m, алифатический Н), 2,09 (m, алифатический Н), 4,97 (d, JHH = 8,8 Hz, виниловый =СН 2), 5,04 (d, JHH = 17 Hz, виниловый =СН 2), 5,47 (m, олефиновый=СН), 5,92 (m, виниловый =СН). Продукты олигомеризации с комплексом 5, 1 Н ЯМР (400 MHz, CDCl3):0,92-2,18 (несколько перекрывающихся сигналов, алифатический Н), 4,98 (d, JHH = 10 Hz, виниловый =СН 2), 5,05 (d, JHH = 17 Hz,виниловый =СН 2), 5,28 (m, олефиновый =СН), 5,49 (m, олефиновый =СН), 5,94 (m, виниловый =СН). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Комплекс, включающий центральный атом М 2 и хелатный лиганд с металлсодержащей основной цепью общей формулы (I) где G представляет собой группу, выбранную из группы СО или монодентатного или бидентатного аминового, или фосфинового, или циклопентадиенилового лиганда, для ингибирования активности металла М 1; металл М 1 в основной цепи представляет Re; металл М 2 в комплексе представляет собой металл группы 10 Периодической таблицы;R1 и R2, каждый независимо, выбраны из гидрокарбилов, имеющих от 1 до 20 углеродных атомов; А представляет собой О или NR, где R выбран из гидрокарбилов, имеющих 4-20 углеродных атомов; В представляет собой аллил или гидрокарбильную группу, имеющую от 1 до 20 углеродных атомов и пиридиновую группу; иn - это валентность М 1 минус 2. 2. Комплекс по п.1, где М 2 представляет собой Ni или Pd. 3. Комплекс по любому из пп.1, 2, где R1 и R2, каждый независимо, представляют собой алкильные группы, имеющие от 1 до 6 углеродных атомов, или арильные группы, имеющие 6-10 углеродных атомов. 4. Способ приготовления комплекса по любому из пп.1-3, включающий стадии: а) получения хелатного лиганда общей формулы (V) б) взаимодействия соединения V с M3Y, где М 3 может быть выбран из Li, Na или K, Y - основание и может быть выбрано из R, OR или NR2, где R может быть любой гидрокарбильной группой, и в) взаимодействия продукта реакции стадии (б) с LXM2=В, где М 2 и В определены выше, X может быть выбран из галогенидов или сульфонатов и L предпочтительно представляет собой фосфин; г) извлечения комплекса I с элиминированием HY и М 3 Х. 5. Способ приготовления комплекса по любому из пп.1-3, включающий стадии: а) получения хелатного лиганда общей формулы (V) б) взаимодействия соединения V с LRxM2=B, где Rx - гидрокарбил, a L - любой нейтральный донорный лиганд, подобный амину или фосфину; в) извлечения комплекса I с элиминированием HRx и L. 6. Активная каталитическая система, включающая: а) комплекс по любому из пп.1-3; б) возможно, активирующий агент; в) возможно, носитель. 7. Активная каталитическая система по п.6, где активирующий агент представляет собой алюмоксан. 8. Способ олигомеризации альфа-олефинов, включающий стадии: а) введения в реактор активной каталитической системы по п.6 или 7;-9 011672 б) введения в реактор мономера и, возможно, сомономера; в) выдерживания при условиях олигомеризации; г) извлечения олигомера. 9. Способ по п.8, где олигомеризацию проводят при температуре от 20 до 80 С и при давлении от 3 до 8 бар (0,3-0,8 МПа). 10. Способ по п.8 или 9, где мономер представляет собой этилен или пропилен. Схема 1
МПК / Метки
МПК: B01J 31/12, B01J 31/18, B01J 31/22
Метки: новый, хелата, катализатора, компонент, одноцентровый, цепью, металлсодержащей, основной
Код ссылки
<a href="https://eas.patents.su/12-11672-novyjj-odnocentrovyjj-komponent-katalizatora-s-metallsoderzhashhejj-osnovnojj-cepyu-helata.html" rel="bookmark" title="База патентов Евразийского Союза">Новый одноцентровый компонент катализатора с металлсодержащей основной цепью хелата</a>
Усовершенствованный твердый компонент катализатора и способ (со)полимеризации этилена

Номер патента: 8985
Опубликовано: 26.10.2007
Авторы: Мази Франческо, Конти Джузеппе, Адессо Коррадо, Менкони Франческо
МПК: C08F 4/651, C08F 4/654, C08F 210/16...
Метки: компонент, катализатора, твердый, этилена, усовершенствованный, способ, сополимеризации
Формула / Реферат:
1. Твердый компонент катализатора для (со)полимеризации этилена, включающий титан, магний, хлор, органическое кислородсодержащее протонное соединение Dp и нейтральное электронодонорное апротонное соединение D, в следующих диапазонах мольных соотношений: Mg/Ti = 1,0-50; D/Ti = 1,0-15; Cl/Ti = 6,0-100; Dp/D = 0,05-3. 2. Твердый компонент по п.1, дополнительно включающий инертное гранулированное твердое вещество в количестве в диапазоне от 10 до...
Носитель катализатора, очищенный от алюминия, способ получения этого носителя катализатора и способ гидратации с2- или с с3-олефинов водой в присутствии катализатора,представляющего собой этот носитель, пропитанный кислотой

Номер патента: 4067
Опубликовано: 25.12.2003
Авторы: Машмайер Дитрих, Штохниоль Гуидо, Лорэнгель Грегор, Закут Михаэль
МПК: C07C 29/04, B01J 21/16
Метки: собой, катализатора,представляющего, кислотой, катализатора, способ, носитель, этого, с3-олефинов, алюминия, получения, очищенный, пропитанный, присутствии, гидратации, носителя, водой
Формула / Реферат:
1. Носитель катализатора с содержанием алюминия меньше 0,3 вес.%, полученный из слоистых алюминийсодержащих силикатов путем деалюминирования. 2. Носитель катализатора по п.1, отличающийся тем, что носитель катализатора имеет содержание алюминия меньше 0,03 вес.%. 3. Носитель катализатора по п.1 или 2, отличающийся тем, что использованные слоистые силикаты являются сукновальными глинами и/или предпочтительно имеют монтмориллонитовую структуру. 4....
Способ приготовления металлоценового катализатора в виде частиц, содержащего модифицированный алюмоксан, и применение этого катализатора для полимеризации олефинов

Номер патента: 7621
Опубликовано: 29.12.2006
Авторы: Бентсред Пол Кристиан, Линдроос Ярмон, Фредриксен Сив Бодил
МПК: C08F 4/64, C08F 10/00, C07F 17/00...
Метки: полимеризации, приготовления, применение, способ, модифицированный, металлоценового, частиц, содержащего, виде, олефинов, катализатора, алюмоксан, этого
Формула / Реферат:
1. Способ приготовления не нанесенного на носителе катализатора полимеризации олефинов, включающий: а) осуществление взаимодействия алюмоксана и основания Льюиса в возможно галогенированном углеводородном растворителе с образованием суспензии твердых частиц; б) осуществление взаимодействия указанной суспензии с металлоценовым комплексом в возможно галогенированном углеводородном растворителе и в) выделение полученного катализатора полимеризации...
Ингибиторы аминотрансфераз, зависимых от аминокислот с разветвленной цепью, и их применение в лечении диабетической ретинопатии

Номер патента: 6597
Опубликовано: 24.02.2006
Авторы: Ху Лайн-Йен, Велти Девин Франклин, Лиет Эрих, Су Ти-Жи, Райдер Тодд Роберт, Ланоуэ Катрин Фоли, Хутсон Сьюзан М., Брайнс Джастин Стефен, Вустроу Девид Юрген, Рафферти Михаель Франсис
МПК: A61K 31/19, C07C 229/28, A61P 27/02...
Метки: цепью, диабетической, аминотрансфераз, разветвленной, ретинопатии, лечении, применение, зависимых, ингибиторы, аминокислот
Формула / Реферат:
1. Соединение формулы IV где R1-R4 означают водород или C1-C10-алкил; X означает NR5; R5 означает водород или C1-C10-алкил, R6 означает C1-C10-алкил; бензил; C4-C9-арил-C1-C10-алкил, в котором от 1 до 4 кольцевых атомов арила могут быть заменены гетероатомом, выбранным из O, S или N; C1-C10-алкокси; C3-C10-циклоалкил, в котором от 1 до 3 атомов углерода могут быть заменены гетероатомом, выбранным из O, S или NR2, где R2 означает водород или...
Стероиды с фторированной 17α-алкильной цепью, обладающие антигестагенным действием

Номер патента: 3092
Опубликовано: 26.12.2002
Авторы: Клеве Арвед, Клар Ульрих, Шведе Вольфганг, Фурманн Ульрике, Хесс-Штумпп Хольгер, Нееф Гюнтер, Хвалисц Кристоф, Шнейдер Мартин
МПК: A61P 5/36, C07J 41/00, A61K 31/567...
Метки: цепью, действием, антигестагенным, стероиды, 17α-алкильной, фторированной, обладающие
Формула / Реферат:
1. 17a-фторалкильные стероиды общей формулы I в которой R1 представляет собой метильную либо этильную группу, R2 представляет собой остаток формулы СnFmНo, где n равно 2, 3, 4, 5 или 6, m>1 и m+o=2n+1, R3 представляет собой свободную этерифицированную либо эстерифицированную гидроксильную группу, R4 и R5 каждый обозначает атом водорода, а оба вместе образуют дополнительную связь или представляют собой метиленовую группу, St обозначает...
Предыдущий патент: Замещенные производные арила и гетероарила в качестве модуляторов метаболизма и для профилактики и лечения связанных с ним расстройств
Следующий патент: Способ очистки fsh
Случайный патент: Соединение бензимидазола