Производные арилоксиарилсульфониламиногидроксамовой кислоты










Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемые соли, где
R1 представляет собой (С1-С6)алкил;
R2 представляет собой (С1-С6)алкил; или R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют кольцо, выбранное из (С5-С7)циклоалкила, 4-тетрагидропиранила и 4-пиперидинила;
R3 представляет собой водород или (С1-С6)алкил; а
Y представляет собой заместитель по любому из атомов углерода фенильного кольца, способному нести дополнительную связь, независимо выбранный из водорода, фторо, хлоро, трифторметила, (С1-С6)алкокси, трифторметокси, дифторметокси и (С1-С6)алкила.
2. Соединение по п.1, где Y представляет собой водород, фторо или хлоро.
3. Соединение по п.1, где Y представляет собой 4-фторо или 4-хлоро.
4. Соединение по п.1, где R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют циклопентильное кольцо.
5. Соединение по п.3, где R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют циклопентильное кольцо.
6. Соединение по п.1, где R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют 4-тетрагидропиранильное кольцо.
7. Соединение по п.1, где R1 и R2 оба представляют собой метил.
8. Соединение по п.3, где R1 и R2 оба представляют собой метил.
9. Соединение по п.1, где R3 представляет собой водород.
10. Соединение по п.3, где R3 представляет собой водород.
11. Соединение по п.4, где R3 представляет собой водород.
12. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из
этилового эфира 3-[[4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]-пропионовой кислоты,
3-[[4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]-пропионовой кислоты,
этилового эфира 3-[[4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоил-1-метилэтил)амино]-пропионовой кислоты и
3-[[4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоил-1-метилэтил)амино]-пропионовой кислоты.
13. Фармацевтическая композиция для (а) лечения артрита или рака и других заболеваний, характеризующихся активностью матриксной металлопротеиназы-13, или (б) избирательного ингибирования матриксной металлопротеиназы-13 у млекопитающего, включая человека, содержащая количество соединения по п.1 или его фармацевтически приемлемой соли, эффективное при таком лечении или ингибировании, и фармацевтически приемлемый носитель.
14. Способ избирательного ингибирования матриксной металлопротеиназы-13 у млекопитающего, включая человека, при котором указанному млекопитающему вводят эффективное количество соединения по п.1 или его фармацевтически приемлемой соли.
15. Способ лечения артрита или рака и других заболеваний, характеризующихся активностью матриксной металлопротеиназы-13, у млекопитающего, включая человека, при котором указанному млекопитающему вводят количество соединения по п.1 или его фармацевтически приемлемой соли, эффективное при лечении такого состояния.
Текст
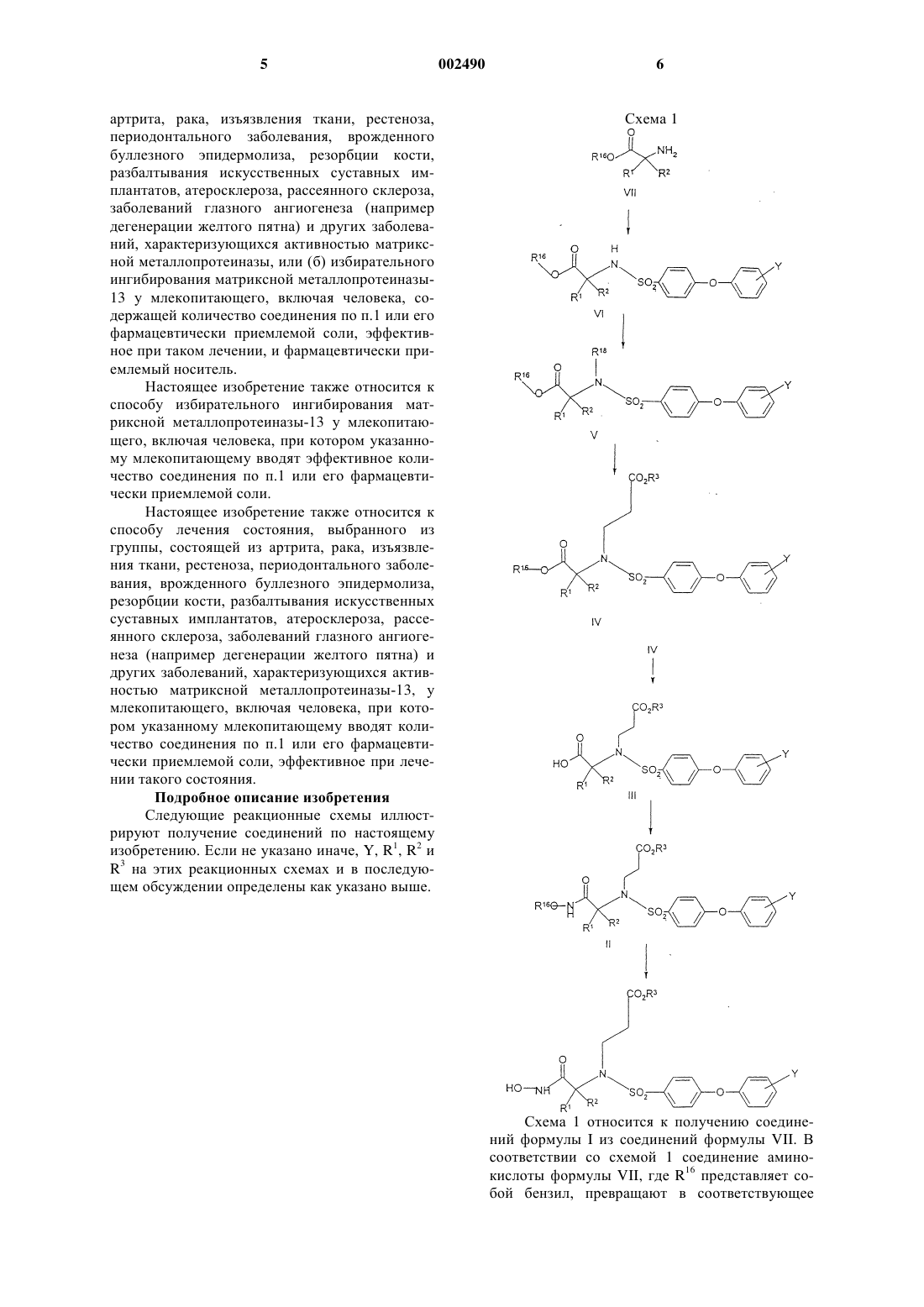
1 Предпосылки изобретения Настоящее изобретение относится к производным арилоксиарилсульфониламиногидроксамовой кислоты. Эти соединения являются избирательно действующими ингибиторами матриксной металлопротеиназы-13 и как таковые полезны при лечении состояния, выбранного из группы, состоящей из артрита, рака, изъязвления ткани, рестеноза, периодонтального заболевания, врожденного буллезного эпидермолиза, резорбции кости, разбалтывания искусственных суставных имплантатов, атеросклероза, рассеянного склероза, заболеваний глазного ангиогенеза (например дегенерации желтого пятна) и других заболеваний, характеризующихся активностью матриксной металлопротеиназы. Данное изобретение относится также к способу использования таких соединений при лечении вышеупомянутых заболеваний у млекопитающих, особенно людей, и к фармацевтическим композициям, пригодным для этого. Существует ряд ферментов, которые осуществляют разрушение структурных белков и которые по структуре родственны металлопротеазам. Матрикс-разрушающие металлопротеиназы, такие как желатиназа, стромелизин и коллагеназа, вовлечены в разрушение тканевого матрикса (например в коллагеновый коллапс) и причастны ко многим патологическим состояниям, в которые вовлечен аномальный метаболизм матрикса соединительной ткани и базальной мембраны, таким как артрит (например остеоартрит и ревматоидный артрит), изъязвление ткани (например роговичное, эпидермальное и желудочное изъязвление), аномальное заживление раны, периодонтальное заболевание, костное заболевание (например болезнь Педжета и остеопороз), метастазирование опухоли или инвазия, а также ВИЧ-инфекция (J. Leuk. Biol.,52(2): 244-248, 1992). Признано, что фактор некроза опухоли вовлечен во многие инфекционные и аутоиммунные заболевания (W. Friers, FEBS letters, 1991,285, 199). Более того, показано, что ФНО (фактор некроза опухоли) является первичным медиатором воспалительной реакции, наблюдаемой при сепсисе и септическом шоке (С.Е.Spooner et al., Clinical Immunology and Immunopathology, 1992,62 S11). Известны (заявка WO 96/27583, опубликованная 12 сентября 1996 г.) некоторые арилсульфониламиногидроксамовые кислоты. Сущность изобретения Настоящее изобретение относится к соединению формулы 2 или его фармацевтически приемлемым солям,гдеR3 представляет собой водород или (С 1 С 6)алкил; аY представляет собой заместитель по любому из атомов углерода фенильного кольца,способному нести дополнительную связь, предпочтительно от 1 до 2 заместителей (более предпочтительно один заместитель, наиболее предпочтительно один заместитель в положении 4) фенильного кольца, независимо выбранный из водорода, фторо, хлоро, трифторметила, (С 1 С 6)алкокси, трифторметокси, дифторметокси и(С 1-С 6)алкила. Термин "алкил", как он используется здесь, если не указано иначе, включает в себя насыщенные одновалентные углеводородные радикалы, имеющие прямые, разветвленные или циклические группировки или их комбинации. Термин "алкокси", как он используется здесь, включает в себя O-алкильные группы, где"алкил" такой, как определено выше. Настоящее изобретение также относится к фармацевтически приемлемым солям присоединения кислоты соединений формулы I. Кислотами, которые используются для получения фармацевтически приемлемых солей присоединения кислоты вышеупомянутых соединенийоснований по этому изобретению, являются те кислоты, которые образуют нетоксичные соли присоединения кислоты, то есть соли, содержащие фармакологически приемлемые анионы,такие как гидрохлорид, гидробромид, гидроиодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат, битартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, паратолуолсульфонат и памоат [то есть 1,1'-метиленбис-(2-гидрокси-3-нафтоат)]. Данное изобретение также относится к солям присоединения основания соединения формулы I. Химическими основаниями, которые могут быть использованы в качестве реагентов для получения фармацевтически приемлемых солей оснований тех соединений формулы I,которые являются кислотными по своей природе, являются те основания, которые образуют нетоксичные соли оснований с такими соединениями. Такие нетоксичные соли оснований включают в себя, но не ограничены ими, соли,полученные путем замещения такими фармакологически приемлемыми катионами, как катионы щелочных металлов (например калия и натрия) и катионы щелочно-земельных металлов 3 водорастворимые аминнные соли присоединения, такие как N-метилглюкамин-(меглюмин), и низшие алканоламмониевые соли, и другие соли оснований фармацевтически приемлемых органических аминов. Соединение формулы I может иметь хиральные центры и, таким образом, существовать в различных энантиомерных формах. Данное изобретение относится ко всем оптическим изомерам и стереоизомерам соединений формулы I и их смесям. Данное изобретение включает в себя также фармацевтические композиции, содержащие и способы лечения или предупреждения, при которых вводят пролекарства соединений формулы I. Соединения формулы I, имеющие свободные амино, амидо, гидрокси или карбоксильные группы, могут быть превращены в пролекарства. Пролекарства включают в себя соединения,где аминокислотный остаток или полипептидная цепь из двух или более чем двух (например двух, трех или четырех) аминокислотных остатков ковалентно связаны посредством пептидных связей со свободными аминогруппами, гидроксигруппами или группами карбоновой кислоты соединений формулы I. Аминокислотные остатки включают в себя 20 встречающихся в природе аминокислот, обычно обозначаемых трехбуквенными символами, и включают в себя также 4-гидроксипролин, гидроксилизин, демозин,изодемозин, 3-метилгистидин, норвалин, бетааланин, гамма-аминомасляную кислоту, цитрулин, гомоцистеин, гомосерин, орнитин и метионинсульфон. Пролекарства включают в себя также соединения, где карбонаты, карбаматы,амиды и сложные алкиловые эфиры, которые ковалентно связаны с вышеуказанными заместителями соединения формулы I через углерод карбонила боковой цепи пролекарства. Пролекарства также включают в себя соединения формулы I, в которых гидроксамовая кислота и карбонильная группировка, взятые вместе, образуют группу формулыI, a U и V независимо представляют собой карбонил, метилен, SO2 или SO3, и b представляет собой целое число от одного до трех, где каждая метиленовая группа возможно замещена гидроксигруппой. Предпочтительные соединения формулы I включают в себя соединения, где Y представляет собой водород, фторо или хлоро, предпочтительно 4-фторо или 4-хлоро. Другие предпочтительные соединения формулы I включают в себя соединения, где R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют циклопентильное или 4-тетрагидропиранильное кольцо. 4 Другие предпочтительные соединения формулы I включают в себя соединения, где R1 и R2 оба представляют собой метил. Другие предпочтительные соединения формулы I включают в себя соединения, где R3 представляет собой водород. Конкретные предпочтительные соединения формулы I включают в себя следующие соединения: этиловый эфир 3-4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]-пропионовой кислоты,3-4-(4-фторфенокси)бензолсульфонил](1-гидроксикарбамоилциклопентил)амино]пропионовую кислоту,этиловый эфир 3-4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоил-1 метилэтил)амино]-пропионовой кислоты и 3-4-(4-фторфенокси)бензолсульфонил](1-гидроксикарбамоил-1-метилэтил)амино]пропионовую кислоту. Другие соединения формулы I включают в себя следующие соединения: 3-4-(4-фторфенокси)бензолсульфонил](4-гидроксикарбамоилтетрагидропиран-4 ил)амино]-пропионовую кислоту,этиловый эфир 3-4-(4-фторфенокси)бензолсульфонил]-(4-гидроксикарбамоилтетрагидропиран-4-ил)амино]-пропионовой кислоты,3-4-(4-хлорфенокси)бензолсульфонил](4-гидроксикарбамоилтетрагидропиран-4 ил)амино]-пропионовую кислоту,этиловый эфир 3-4-(4-хлорфенокси)бензолсульфонил]-(4-гидроксикарбамоилтетрагидропиран-4-ил)амино]-пропионовой кислоты,3-[(4-гидроксикарбамоилтетрагидропиран 4-ил)-(4-феноксибензолсульфонил)амино]пропионовую кислоту,этиловый эфир 3-[(4-гидроксикарбамоилтетрагидропиран-4-ил)-(4-феноксибензолсульфонил)амино]-пропионовой кислоты,этиловый эфир 3-4-(4-фторфенокси)бензолсульфонил]-(4-гидроксикарбамоилпиперидин-4-ил)амино]-пропионовой кислоты,3-4-(4-хлорфенокси)бензолсульфонил](1-гидроксикарбамоил-1-метилэтил)амино]пропионовую кислоту,этиловый эфир 3-4-(4-хлорфенокси)бензолсульфонил]-(1-гидроксикарбамоил-1 метилэтил)амино]-пропионовой кислоты,3-4-(4-фторфенокси)бензолсульфонил](1-гидроксикарбамоилциклогексил)амино]пропионовую кислоту,3-[(1-гидроксикарбамоилциклопентил)-(4 феноксибензолсульфонил)амино]-пропионовую кислоту и 3-4-(4-хлорфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]пропионовую кислоту. Настоящее изобретение относится также к фармацевтической композиции для (а) лечения состояния, выбранного из группы, состоящей из 5 артрита, рака, изъязвления ткани, рестеноза,периодонтального заболевания, врожденного буллезного эпидермолиза, резорбции кости,разбалтывания искусственных суставных имплантатов, атеросклероза, рассеянного склероза,заболеваний глазного ангиогенеза (например дегенерации желтого пятна) и других заболеваний, характеризующихся активностью матриксной металлопротеиназы, или (б) избирательного ингибирования матриксной металлопротеиназы 13 у млекопитающего, включая человека, содержащей количество соединения по п.1 или его фармацевтически приемлемой соли, эффективное при таком лечении, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к способу избирательного ингибирования матриксной металлопротеиназы-13 у млекопитающего, включая человека, при котором указанному млекопитающему вводят эффективное количество соединения по п.1 или его фармацевтически приемлемой соли. Настоящее изобретение также относится к способу лечения состояния, выбранного из группы, состоящей из артрита, рака, изъязвления ткани, рестеноза, периодонтального заболевания, врожденного буллезного эпидермолиза,резорбции кости, разбалтывания искусственных суставных имплантатов, атеросклероза, рассеянного склероза, заболеваний глазного ангиогенеза (например дегенерации желтого пятна) и других заболеваний, характеризующихся активностью матриксной металлопротеиназы-13, у млекопитающего, включая человека, при котором указанному млекопитающему вводят количество соединения по п.1 или его фармацевтически приемлемой соли, эффективное при лечении такого состояния. Подробное описание изобретения Следующие реакционные схемы иллюстрируют получение соединений по настоящему изобретению. Если не указано иначе, Y, R1, R2 иR3 на этих peaкционных схемах и в последующем обсуждении определены как указано выше. Схема 1 относится к получению соединений формулы I из соединений формулы VII. В соответствии со схемой 1 соединение аминокислоты формулы VII, где R16 представляет собой бензил, превращают в соответствующее 7 соединение формулы VI путем взаимодействия с реакционноспособным функциональным производным соединения арилсульфоновой кислоты формулы в присутствии основания, такого как триэтиламин, и полярного растворителя, такого как тетрагидрофуран, 1,2-диметоксиэтан, диоксан, вода или ацетонитрил,предпочтительно 1,2 диметоксиэтан. Реакционную смесь перемешивают при комнатной температуре в течение периода времени между примерно 10 мин и примерно 24 ч, предпочтительно примерно 60 мин. Арилсульфониламиносоединение формулы VI, где R16 представляет собой бензил, превращают в соответствующее соединение формулы V, где R18 представляет собой группу 3 трет-бутилдиметилсиланилоксипропанил, путем взаимодействия с трет-бутил-(3-галогенопропокси)диметилсиланом, предпочтительно иодидным производным, в присутствии основания, такого как карбонат калия, карбонат цезия,гексаметилдисилазид калия или гидрид натрия,предпочтительно гексаметилдисилазид калия. Реакционную смесь перемешивают в полярном растворителе, таком как диметилформамид илиN-метилпирролидин-2-он, при комнатной температуре в течение периода времени между примерно 2 и примерно 48 ч, предпочтительно примерно 18 ч. Соединение формулы V превращают в производное карбоновой кислоты формулы IV путем взаимодействия с эфиратом трифторида бора с образованием промежуточного спирта с последующим окислением и защитой путем этерификации. Конкретно, взаимодействие с эфиратом трифторида бора осуществляют в инертном растворителе, таком как метиленхлорид, хлороформ, предпочтительно метиленхлорид, при комнатной температуре в течение от примерно 15 мин до примерно 4 ч, предпочтительно примерно один час. Окислению спирта способствует использование триоксида хрома в водной серной кислоте (реактив Джонса) при примерно 0 С в течение от примерно одного до примерно 6 ч, предпочтительно примерно 2 ч. Защите карбоновой кислоты способствует обработка свободной кислоты алкилирующим агентом, таким как R3-L, где L представляет собой уходящую группу, такую как иодо, бромо, мезилат или тозилат, предпочтительно иодо, с использованием основания, такого как карбонат калия или карбонат цезия, предпочтительно карбонат калия, в полярном растворителе, таком как диметилформамид, N-метилпирролидин-2 он или тетрагидрофуран, предпочтительно диметилформамид, в течение от примерно 1 до примерно 24 ч, предпочтительно 16 ч, при примерно комнатной температуре. 8 Соединение формулы IV превращают в соединение формулы III путем удаления защитной группы R16 путем гидрогенолиза с использованием палладия на углероде в растворителе, таком как метанол или этанол, в течение периода от примерно 30 мин до примерно 48 ч, предпочтительно 16 ч, при температуре от примерно 20 до примерно 25 С, то есть при комнатной температуре. Соединение карбоновую кислоту формулыIII превращают в производное гидроксамовой кислоты формулы II, где R16 представляет собой бензил, путем активации соединения формулыIII с последующим взаимодействием с бензилгидроксиламином. Соединение формулы III активируют путем обработки (бензотриазол-1 илокси)трис(диметиламино)фосфония гексафторфосфатом в присутствии основания при комнатной температуре в полярном растворителе. Вышеуказанное взаимодействие проводят в течение периода от примерно 15 мин до примерно 4 ч, предпочтительно примерно 1 ч. Активированное соединение, полученное из соединения формулы III, превращают in situ в соединение формулы II путем взаимодействия с гидрохлоридом бензилгидроксиламина. Взаимодействие с гидрохлоридом бензилгидроксиламина проводят в течение от примерно 1 ч до примерно 5 дней, предпочтительно в течение примерно 16 ч, при температуре от примерно 40 до примерно 80 С, предпочтительно примерно 60 С. Подходящие основания включают в себяN-метилморфолин или диизопропилэтиламин,предпочтительно диизопропилэтиламин. Подходящие растворители включают в себя N,Nдиметилформамид или N-метилпирролидин-2 он, предпочтительно N,N-диметилформамид. Соединение формулы II превращают в соединение I путем удаления гидроксиламинной защитной группы. Удаление гидроксиламинной защитной группы осуществляют путем гидрогенолиза бензильной защитной группы с использованием в качестве катализатора палладия на сульфате бария в полярном растворителе при температуре от примерно 20 до примерно 25 С,то есть при комнатной температуре, в течение периода от примерно 1 ч до примерно 5 ч, предпочтительно примерно 3 ч. Соединения формул VII и VIII имеются в продаже или могут быть получены способами,хорошо известными рядовым специалистам в данной области. Фармацевтически приемлемые соли кислотных соединений по изобретению представляют собой соли, образованные с основаниями,а именно катионные соли, такие как соли щелочных и щелочно-земельных металлов, таких как натрий, литий, калий, кальций, магний, а также аммониевые соли, такие как соли аммония, триметиламмония, диэтиламмония и трис(гидроксиметил)-метиламмония. 9 Точно также соли присоединения кислоты,такие как соли присоединения минеральных кислот, органических карбоновых и органических сульфоновых кислот, например соляной кислоты, метансульфоновой кислоты, малеиновой кислоты, также возможны, при условии, что основная группа, такая как пиридил, является составной частью структуры. Соединения формулы I, которые являются основными по своей природе, способны к образованию широкого ряда различных солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения животным, на практике часто желательно вначале выделить соединение формулы I из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто превратить последнюю обратно в соединение-свободное основание путем обработки щелочным реагентом, и после этого превратить данное свободное основание в фармацевтически приемлемую соль присоединения кислоты. Соли присоединения кислоты соединений-оснований по этому изобретению без труда получают путем обработки соединения-основания, по существу, эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе,таком как метанол или этанол. После осторожного выпаривания растворителя получают желаемую твердую соль. Кислотами, которые используют для получения фармацевтически приемлемых солей присоединения кислоты соединений-оснований по настоящему изобретению, являются кислоты,которые образуют нетоксичные соли присоединения кислоты, то есть соли, содержащие фармакологически приемлемые анионы, такие как гидрохлорид, гидробромид, гидроиодид, нитрат,сульфат или бисульфат, фосфат или кислый фосфат, ацетат, лактат, цитрат или кислый цитрат, тартрат или битартрат, сукцинат, малеат,фумарат, глюконат, сахарат, бензоат, метансульфонат и памоат [то есть 1,1'-метилен-бис(2-гидрокси-3-нафтоат)]. Те соединения формулы I, которые также являются кислотными по своей природе, например, где R3 представляет собой водород, способны к образованию солей оснований с различными фармакологически приемлемыми катионами. Примеры таких солей включают в себя соли щелочных металлов или щелочноземельных металлов, и, в частности, соли натрия и калия. Все эти соли получают традиционными способами. Химическими основаниями,которые используют в качестве реагентов для получения фармацевтически приемлемых солей оснований по этому изобретению, являются реагенты, которые образуют нетоксичные соли оснований с описанными здесь кислотными соединениями формулы I. Эти нетоксичные соли 10 оснований включают в себя соли, полученные реакцией замещения такими фармакологически приемлемыми катионами, как натрий, калий,кальций и магний и так далее. Эти соли могут быть легко получены путем обработки соответствующих кислотных соединений водным раствором, содержащим желаемые фармакологически приемлемые катионы, а затем упариванием полученного раствора до сухости, предпочтительно при пониженном давлении. Альтернативно, они могут быть получены также путем смешивания вместе растворов кислотных соединений в низших спиртах и алкоксида желаемого щелочного металла, а затем упариванием полученного раствора до сухости тем же способом, как указано выше. В любом случае предпочтительно используют стехиометрические количества реагентов для того, чтобы гарантировать полноту протекания реакции до конца и максимальные выходы продукта. Способность соединений формулы I или их фармацевтически приемлемых солей (здесь и далее также упоминаемых как селективные в отношении ММП-13 соединения по настоящему изобретению) ингибировать матриксную металлопротеиназу-13 (коллагеназу 3) и, следовательно, проявлять свою эффективность для лечения заболеваний, характеризующихся активностью матриксной металлопротеиназы-13, показана с помощью следующих анализов in vitro. Биологический анализ Ингибирование человеческой коллагеназы(ММР-1) Рекомбинантную коллагеназу человека активируют трипсином, используя следующее соотношение: 10 мг трипсина на 100 мг коллагеназы. Трипсин и коллагеназу инкубируют при комнатной температуре в течение 10 мин, затем добавляют пятикратный избыток (50 мг/10 мг трипсина) ингибитора трипсина из соевых бобов. 10 мМ исходные растворы ингибиторов готовят в диметилсульфоксиде, а затем разводят, используя следующую схему: 10 мМ 120 мкМ 12 мкМ 1,2 мкМ 0,12 мкМ По двадцать пять микролитров раствора каждой концентрации затем добавляют трехкратно в соответствующие лунки 96-луночного микрофлуорометрического планшета. Конечная концентрация ингибитора должна составлять разведение 1:4 после добавления фермента и субстрата. Позитивные контроли (фермент, отсутствие ингибитора) помещают в лунки D1-D6,а слепые контроли (отсутствие фермента, отсутствие ингибиторов) помещают в лунки D7-D12. Коллагеназу разводят до 400 нг/мл и затем 25 мл добавляют в соответствующие лунки микрофлуорометрического планшета. Конечная концентрация коллагеназы в этом анализе составляет 100 нг/мл. Субстрат (DNP-Pro-Cha-Gly-Cys(Me)-His-AlaLys(NMA)-NH2) готовят как 5 мМ исходный раствор в диметилсульфоксиде, а затем разводят до 20 мМ в буфере для анализа. Анализ инициируют путем добавления по 50 мл субстрата на лунку микрофлуорометрического планшета с получением конечной концентрации 10 мМ. Показания флуоресценции (возбуждение 360 нм, эмиссия 460 нм) снимают в момент времени 0, а затем через 20 минутные интервалы. Анализ проводят при комнатной температуре, обычно время анализа составляет 3 ч. Затем строят график флуоресценции против времени как для слепых контролей, так и для образцов, содержащих коллагеназу (данные трехкратных определений усреднены). Точку времени,которая дает хороший сигнал (слепой контроль) и которая находится на линейной части кривой(обычно около 120 мин) выбирают для определения величин ИK50. Нулевое время используют как слепой контроль для каждого соединения при каждой концентрации, и эти величины вычитают из данных для 120 мин. По этим данным строят график концентрации ингибитора против % от контроля(флуоресценция ингибитора, деленная на флуоресценцию одной коллагеназы х 100). ИK50 определяют из концентрации ингибитора, которая дает сигнал, составляющий 50% от контроля. Если определено, что ИК 50 менее 0,03 мМ, то тогда ингибиторы анализируют при концентрациях 0,3 мМ, 0,03 мМ, 0,03 мМ и 0,003 мМ. Ингибирование ММП-13 Рекомбинантную ММП-13 человека активируют 2 мМ АФАР (пара-амино(ацето)ртуть) в течение 1,5 ч при 37 С и разводят до 400 мг/мл в буфере для анализа (50 мМ Трис, рН 7,5, 200 мМ хлорид натрия, 5 мМ хлорид кальция, 20 мМ хлорид цинка,0,02% brij). Добавляют по двадцать пять микролитров разведенного фермента, добавляют на лунку 96-луночного микрофлуоресцентного планшета. Затем в данном анализе фермент разводят в соотношении 1:4 путем добавления ингибитора и суб 12 страта с получением конечной концентрации в анализе 100 мг/мл. 10 мМ исходные растворы ингибиторов готовят в диметилсульфоксиде, а затем разбавляют в буфере для анализа согласно схеме разбавления ингибитора для ингибирования коллагеназы человека (ММП-1). По двадцать пять микролитров каждой концентрации добавляют трехкратно в микрофлуоресцентный планшет. Конечные концентрации в этом анализе составляют 30 мМ, 3 мМ, 0,3 мМ и 0,03 мМ. Субстрат (Dnp-Pro-Cha-Gly-Cys(Me)-His-AlaLys(NMA)-NH2) готовят как для ингибирования коллагеназы человека (ММП-1), и 50 мл добавляют в каждую лунку с получением конечной концентрации для анализа 10 мМ. Показания флуоресценции (возбуждение 360 нм; эмиссия 460 нм) снимают в момент времени 0 и каждые 5 мин в течение часа. Позитивные контроли состоят из фермента и субстрата при отсутствии ингибитора, а слепые контроли состоят только из субстрата. ИК 50 определяют так же, как в случае ингибирования коллагеназы человека (ММП-1). Если определено, что ИК 50 менее 0,03 мМ, то тогда ингибиторы анализируют при конечных концентрациях 0,3 мМ, 0,03 мМ, 0,003 мМ и 0,0003 мМ. Соединения по настоящему изобретению обладают неожиданной избирательной активностью против матриксной металлопротеиназы-13 (коллагеназы 3) по сравнению с матриксной металлопротеиназой-1 (коллагеназой 1). Конкретно, соединения формулы I в 100 раз более избирательно действуют в отношении матриксной металлопротеиназы 13 (коллагеназы 3), чем в отношении матриксной металлопротеиназы-1 (коллагеназы 1) и имеют ИК 50 менее 10 нМ, против матриксной металлопротеиназы-13 (коллагеназы 3). В табл. 1 приведено несколько соединений, которые демонстрируют неожиданную избирательность действия соединений по изобретению. Таблица 1R1 Циклопентил Циклопентил Циклопентил Циклопентил Метил Метил Метил Метил Метил Циклопентил Циклопентил Метил Метил Циклогексил ЦиклогексилR2 Метил Метил Метил Метил Метил Метил Метил Водород ВодородR3 Этил Этил Водород Водород Этил Этил Водород Водород Водород Водород Водород Водород Водород Водород ВодородR 4-Фторфенокси 4-Фторфенокси 4-Фторфенокси 4-Фторфенокси 4-Фторфенокси 4-Фторфенокси 4-Фторфенокси 4-Фторфенокси 4-Фторфенокси Метокси Метокси Метокси Метокси Метокси Метокси 13 Для введения людям в целях ингибирования матриксной металлопротеиназы-13 или для продуцирования фактора некроза опухоли(ФНО), можно использовать различные традиционные пути, включая введение перорально,парентерально или местно. Как правило, активное соединение будут вводить перорально или парентерально в дозировках между примерно 0,1 и 25 мг/кг массы тела субъекта, которого лечат, в день, предпочтительно от примерно 0,3 до примерно 5 мг/кг. Однако обязательно будет иметь место некоторое отклонение в дозировке в зависимости от состояния субъекта, которого лечат. Лицо, ответственное за введение, в любом случае будет определять подходящую дозу для конкретного субъекта. Соединения по настоящему изобретению можно вводить в большом разнообразии различных лекарственных форм. Как правило, терапевтически эффективные соединения по данному изобретению присутствуют в таких лекарственных формах при уровнях концентрации в интервале от примерно 5,0% до примерно 70% по массе. Для перорального введения таблетки, содержащие различные эксципиенты, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, двухзамещенный фосфат кальция и глицин, могут быть использованы вместе с различными разрыхлителями, такими как крахмал (и предпочтительно кукурузный,картофельный или тапиоковый крахмал), альгиновая кислота и некоторые комплексные силикаты, вместе со связывающими веществами для гранулирования, такими как поливинилпирролидон, сахароза, желатин и аравийская камедь. К тому же, смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк,часто очень полезны в целях таблетирования. Твердые композиции подобного типа можно также использовать в качестве наполнителей в желатиновых капсулах; предпочтительные вещества в этой связи также включают в себя лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Когда для перорального введения желательны водные суспензии и/или эликсиры, активный ингредиент может быть объединен с различными подслащивающими агентами или корригентами, красящими веществами или красками, и, при желании, также с эмульгирующими и/или суспендирующими агентами, вместе с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и различные подобные комбинациии их. Для парентерального введения (внутримышечного, внутрибрюшинного, подкожного и внутривенного) обычно готовят стерильный инъекционный раствор активного ингредиента. Можно использовать растворы терапевтического соединения по настоящему изобретению либо в кунжутном или в арахисовом масле, либо в водном пропиленгликоле. Водные растворы 14 должны быть соответственно отрегулированы и забуферены, предпочтительно при рН более 8,если необходимо, а жидкий разбавитель сначала должен быть сделан изотоническим. Эти водные растворы подходят для внутривенных инъекций. Масляные растворы подходят для внутрисуставных, внутримышечных и подкожных инъекций. Получение всех этих растворов в стерильных условиях легко осуществить по стандартным фармацевтическим методикам, хорошо известным специалистам в данной области. Следующие примеры иллюстрируют получение соединений по настоящему изобретению. Точки плавления не откорректированы. Данные ЯМР приведены в миллионых доляхи относятся к сигналу синхронизации дейтерия от растворителя для образца (дейтерийдиметилсульфоксид, если не указано иначе). Коммерческие реагенты используют без дальнейшей очистки. Под ТГФ понимают тетрагидрофуран. Под ДМФ понимают N,N-диметилформамид. Под хроматографией понимают колоночную хроматографию, выполняемую с использованием 3263 мм силикагеля и осуществляемую под давлением азота (флэш-хроматография). Под комнатной температурой или температурой окружающей среды понимают температуру от 20 до 25 С. Все неводные реакции проводили в атмосфере азота для удобства и для максимального увеличения выходов. Концентрирование при пониженном давлении означает, что используют роторный испаритель. Пример 1. Этиловый эфир 3-4-(4 фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]пропионовой кислоты.(A) К раствору соли паратолуолсульфоновой кислоты бензилового эфира 1-аминоциклопентанкарбоновой кислоты (200 г,0,51 моль) и триэтиламина (177 мл, 1,27 моль) в воде (1 л) и 1,2-диметоксиэтане (1 л) добавляют 4-(4-фторфенокси)бензолсульфонилхлорид (161 г, 0,56 моль). Смесь перемешивают при комнатной температуре в течение 16 ч, а затем большую часть растворителя удаляют путем выпаривания в вакууме. Смесь разбавляют этилацетатом и промывают последовательно разбавленным раствором соляной кислоты, водой и солевым раствором. Раствор сушат над сульфатом магния и концентрируют, пока не останется коричневое твердое вещество. Растирают с диэтиловым эфиром с получением бензилового эфира 1-[4-(4-фторфенокси)бензолсульфониламино]-циклопентанкарбоновой кислоты в виде рыжевато-коричневого твердого вещества, 167 г(Б) К раствору бензилового эфира 1-[4-(4 фторфенокси)бензолсульфониламино]-циклопентанкарбоновой кислоты (199 г, 0,42 моль) в безводном N,N-диметилформамиде (2,5 л) при комнатной температуре добавляют гексаметилдисилазид калия (100 г, 0,50 моль) и, через 3 ч, 15 трет-бутил-(3-иодпропокси)диметилсилан (150 г, 0,50 моль). Полученную смесь перемешивают при комнатной температуре в течение 16 ч. Затем добавляют дополнительный трет-бутил-(3 иодпропокси)диметилсилан (20 г, 0,067 моль). Перемешивание при комнатной температуре продолжают в течение дальнейших 3,5 ч. Смесь гасят добавлением насыщенного раствора хлорида аммония.N,N-диметилформамид удаляют выпариванием под вакуумом. Остаток растворяют в диэтиловом эфире и промывают водой и солевым раствором. После высушивания над сульфатом магния диэтиловый эфир выпаривают с получением неочищенного бензилового эфира 1-[3-(третбутил-диметилсиланилокси)-пропил]-[4-(4 фторфенокси)бензолсульфонил]-аминоциклопентанкарбоновой кислоты в виде янтарного масла (279,6 г).(B) К раствору неочищенного бензилового эфира 1-[3-(трет-бутилдиметилсиланилокси)пропил]-[4-(4-фторфенокси)бензолсульфонил]амино-циклопентанкарбоновой кислоты (279 г) в метиленхлориде (1 л) при комнатной температуре добавляют эфират трифторида бора (103 мл, 0,84 моль). Через 1 ч реакцию гасят добавлением последовательно насыщенного раствора хлорида аммония и воды. Органическую фазу отделяют, промывают водой и солевым раствором и сушат над сульфатом магния. Выпаривают растворитель под вакуумом с получением неочищенного бензилового эфира 1-4-(4 фторфенокси)бензолсульфонил]-(3-гидроксипропил)амино]циклопентанкарбоновой кислоты в виде янтарного масла (235 г).(Г) Раствор неочищенного бензилового эфира 1-4-(4-фторфенокси)бензолсульфонил](3-гидроксипропил)амино]циклопентанкарбоновой кислоты (235 г) в ацетоне (2 л) охлаждают на ледяной бане и обрабатывают реактивом Джонса (примерно 200 мл) до тех пор, пока сохраняется оранжевое окрашивание. Смесь перемешивают от 0 С до комнатной температуры в течение 1 ч. После гашения избытка окислителя изопропанолом (10 мл) смесь фильтруют, и фильтрат концентрируют под вакуумом. Остаток растворяют в этилацетате, промывают водой и солевым раствором, сушат над сульфатом магния и концентрируют с получением твердого вещества, которое растирают со смесью диэтилового эфира и гексана с получением бензилового эфира 1-(2-карбоксиэтил)-[4-(4 фторфенокси)бензолсульфонил]-амино-циклопентанкарбоновой кислоты в виде белого твердого вещества (147 г).(Д) К раствору бензилового эфира 1-(2 карбоксиэтил)-[4-(4-фторфенокси)бензолсульфонил]-амино-циклопентанкарбоновой кислоты (147 г) в N,N-диметилформамиде (3 л) при комнатной температуре добавляют карбонат калия (150 г, 1,08 моль) и этилиодид (32,4 мл,0,405 моль). Смесь перемешивают в течение 16 16 ч при комнатной температуре. После фильтрования большую часть растворителя удаляют под вакуумом. Остаток растворяют в воде и подкисляют, используя 6 н. водный раствор хлороводорода. Полученную смесь экстрагируют добавлением диэтилового эфира. Органический экстракт промывают водой и солевым раствором,сушат над сульфатом магния и концентрируют с получением бензилового эфира 1-(2 этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил]-амино-циклопентанкарбоновой кислоты в виде желтого полутвердого вещества(Е) Раствор бензилового эфира 1-(2 этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил]-амино-циклопентанкарбоновой кислоты (74,5 г, 0,13 моль) в этаноле (1,8 л) обрабатывают 10% палладием на активированном угле (7,4 г) и гидрируют в шейкере Раrr при давлении 3 атмосферы в течение 16 ч. После фильтрования через нейлон (размер пор 0,45 мкм) для удаления катализатора растворитель выпаривают с получением 1-(2-этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил]-амино-циклопентанкарбоновой кислоты в виде белой пены. Реакцию повторяют по той же схеме с получением в сумме 125,2 г желаемого продукта.N,N-диметилформамиде (2 л). Смесь перемешивают в течение 1 ч. Затем добавляют дополнительный диизопропилэтиламин (91 мл, 0,52 моля) и O-бензилгидроксиламина гидрохлорид(53,8 г, 0,338 моль), и полученную смесь перемешивают при 60 С в течение 96 ч. После концентрирования под вакуумом остаток растворяют в воде и подкисляют 1 н. водным раствором хлороводорода. Смесь экстрагируют добавлением этилацетата, и экстракт промывают последовательно водой, насыщенным водным раствором бикарбоната натрия и солевым раствором. Раствор сушат над сульфатом магния и концентрируют с получением неочищенного этилового эфира 3-(1-бензилоксикарбамоилциклопентил)-[4-(4-фторфенокси)бензолсульфонил]-амино-пропионовой кислоты в виде желтого масла (164 г).(3) Раствор неочищенного этилового эфира 3-(1-бензилоксикарбамоилциклопентил)-[4-(4 фторфенокси)бензолсульфонил]-аминопропионовой кислоты (164 г) в этаноле (2,4 л) обрабатывают 5% палладием на сульфате бария (50 г) и гидрируют в шейкере Раrr при давлении 3 атмосферы в течение 3 ч. После фильтрования через нейлон (размер пор 0,45 мкм) для удаления катализатора растворитель выпаривают с получением 17 масла. После добавления этилацетата и гексана этиловый эфир 3-4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]-пропионовой кислоты, белое кристаллическое твердое вещество, (73,5 г) собирают путем фильтрования. Фильтрат концентрируют и остаток хроматографируют на силикагеле, элюируя 40% этилацетатом в гексане, для получения большего количества желаемого продукта (32,5 г). Т. пл.: 79-83 С. 1H ЯМР (ДМСО-d6):10.40 (br s, 1H), 8.78 (br s, 1H), 7.80-7.77 (m, 2 Н),7.31-7.03 (m, 6H), 4.02 (q, J = 7,3 Гц, 2 Н), 3.493.45 (m, 2H), 2.70-2.67 (m, 2H), 2.24-2.21 (m,2H), 1.86-1.83 (m, 2H), 1.53-1.50 (m, 4 Н), 1.16 (t,J = 7,3 Гц, 3 Н). МС (масс-спектрометрия) 493(М-1). Анализ: Рассчитано для С 23 Н 27FN2O7SН 2O: С, 53.90; Н, 5.70; N, 5.47. Обнаружено: С, 54.52; Н, 5.63; N, 5.27. Пример 2. 3-4-(4-Фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]пропионовая кислота. Раствор этилового эфира 3-4-(4 фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)-амино]пропионовой кислоты (106 г, 0,214 моль) в этаноле (2,5 л) обрабатывают водным 1 н. раствором гидроксида натрия (856 мл, 0,856 моль) и перемешивают при комнатной температуре в течение 2 ч. Смесь концентрируют для удаления этанола,разбавляют водой, подкисляют 6 н. водным раствором соляной кислоты и экстрагируют добавлением этилацетата. После промывания водой и солевым раствором органический экстракт сушат над сульфатом магния и концентрируют до пены. Кристаллизация из 30% этилацетата в гексане дает 3-4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]пропионовую кислоту в виде белого кристаллического твердого вещества (81.5 г,81%). Т. пл.: 170-172 С. 1 Н ЯМР (ДМСО-d6):12.25 (br s, 1H), 10.40 (br s, 1H), 8.74 (br s, 1H),7.79-7.77 (m, 2H), 7.29-7.03 (m, 6H), 3.45-3.41(m, 2H), 2.61-2.57 (m, 2H), 2.24-2.21 (m, 2H),1.88-1.82 (m, 2H), 1.53-1.50 (m, 4 Н). МС 465 (М 1). Анализ: Рассчитано для C21H23FN2O7S: С,54.07; Н, 4.97; N, 6.00. Обнаружено: С, 54.17; Н, 5.02; N, 6.05. Пример 3. Этиловый эфир 3-4-(4 фторфенокси)бензолсульфонил]-(1-гидроксикарбамоил-1-метилэтил)амино]пропионовой кислоты. Соединение, указанное в заголовке, получают согласно процедуре, аналогичной процедуре, описанной в примере 1, начиная с соли пара-толуолсульфоновой кислоты бензилового эфира 2-амино-2-метилпропионовой кислоты. Т. пл.: 124,8-125 С. 1 Н ЯМР (ДМСО-d6):10.37 (s, 1H), 8.74 (s, 1H), 7.86 (d, 2H, J = 8,9 Гц),7.16-7.30 (m, 4H), 7.04 (d, 2H, J = 8,7 Гц), 3.99 (q, 002490(М-1). Анализ: Рассчитано для C21H25FN2O7S: С,53.84; Н, 5.38; N, 5.98. Обнаружено: С, 54.00; Н, 5.12; N, 5.87. Пример 4. 3-4-(4-Фторфенокси)бензолсульфонил]-(1-гидроксикарбамоил-1-метилэтил)-амино]пропионовая кислота. Соединение, указанное в заголовке, получают из этилового эфира 3-4-(4 фторфенокси)бензолсульфонил]-(1-гидроксикарбамоил-1-метилэтил)амино]пропионовой кислоты согласно процедуре, аналогичной процедуре, описанной в примере 2. Т. пл.: 162-162,5 С. МС: 439 (М-1). 1H ЯМР (ДМСО-d6):12.26 (s, 1H), 10.38 (s, 1H),8.75 (s, 1H), 7.86-7.88 (m, 2H), 7.16-.7.30 (m, 4H),7.03-7.06 (m, 2H), 3.29-3.35 (m, 2H), 2.47-2.59 или его фармацевтически приемлемые соли, гдеR3 представляет собой водород или (С 1 С 6)алкил; аY представляет собой заместитель по любому из атомов углерода фенильного кольца,способному нести дополнительную связь, независимо выбранный из водорода, фторо, хлоро,трифторметила, (С 1-С 6)алкокси, трифторметокси, дифторметокси и (С 1-С 6)алкила. 2. Соединение по п.1, где Y представляет собой водород, фторо или хлоро. 3. Соединение по п.1, где Y представляет собой 4-фторо или 4-хлоро. 4. Соединение по п.1, где R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют циклопентильное кольцо. 5. Соединение по п.3, где R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют циклопентильное кольцо. 6. Соединение по п.1, где R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют 4-тетрагидропиранильное кольцо. 7. Соединение по п.1, где R1 и R2 оба представляют собой метил. 8. Соединение по п.3, где R1 и R2 оба представляют собой метил. 9. Соединение по п.1, где R3 представляет собой водород. 10. Соединение по п.3, где R3 представляет собой водород. 11. Соединение по п.4, где R3 представляет собой водород. 12. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из этилового эфира 3-4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]-пропионовой кислоты,3-4-(4-фторфенокси)бензолсульфонил](1-гидроксикарбамоилциклопентил)амино]пропионовой кислоты,этилового эфира 3-4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоил-1 метилэтил)амино]-пропионовой кислоты и 3-4-(4-фторфенокси)бензолсульфонил](1-гидроксикарбамоил-1-метилэтил)амино]пропионовой кислоты. 13. Фармацевтическая композиция для (а) лечения артрита или рака и других заболеваний, 20 характеризующихся активностью матриксной металлопротеиназы-13, или (б) избирательного ингибирования матриксной металлопротеиназы 13 у млекопитающего, включая человека, содержащая количество соединения по п.1 или его фармацевтически приемлемой соли, эффективное при таком лечении или ингибировании, и фармацевтически приемлемый носитель. 14. Способ избирательного ингибирования матриксной металлопротеиназы-13 у млекопитающего, включая человека, при котором указанному млекопитающему вводят эффективное количество соединения по п.1 или его фармацевтически приемлемой соли. 15. Способ лечения артрита или рака и других заболеваний, характеризующихся активностью матриксной металлопротеиназы-13, у млекопитающего, включая человека, при котором указанному млекопитающему вводят количество соединения по п.1 или его фармацевтически приемлемой соли, эффективное при лечении такого состояния.
МПК / Метки
МПК: C07D 309/14, A61P 35/00, C07C 311/29, A61K 31/16
Метки: кислоты, производные, арилоксиарилсульфониламиногидроксамовой
Код ссылки
<a href="https://eas.patents.su/11-2490-proizvodnye-ariloksiarilsulfonilaminogidroksamovojj-kisloty.html" rel="bookmark" title="База патентов Евразийского Союза">Производные арилоксиарилсульфониламиногидроксамовой кислоты</a>
Замещенные (сульфиновой кислоты, сульфоновой кислоты, сульфониламино или сульфиниламино) n-[(аминоиминометил) фенилалкил] азагетероциклил-амидные производные

Номер патента: 700
Опубликовано: 28.02.2000
Авторы: Паулс Генри В., Евинг Вильям Р., Бекер Майкл Р., Чини Дэниел Л., Чои-Следески Енг Ми, Мэйсон Джонатан Стивен, Спада Альфред П.
МПК: C07D 401/06, A61K 31/44
Метки: сульфониламино, фенилалкил, кислоты, производные, сульфиниламино, замещенные, азагетероциклил-амидные, сульфоновой, сульфиновой, n-[(аминоиминометил
Формула / Реферат:
1. Соединение формулы (1) представляет фенил или моноциклический гетероарил; R представляет водород, необязательно замещенный алкил, необязательно замещенный аралкил, необязательно замещенный гетероаралкил или гидроксиалкил; R1 представляет водород, R3S(O)p или R3R4NS(O)p-; R2 представляет водород или, когда X5 и Х5', взятые вместе, представляют =NR5, R2 представляет водород, необязательно замещенный низший алкил,...
Производные гидроксимовой кислоты и их применение

Номер патента: 278
Опубликовано: 25.02.1999
Авторы: Тошима Норишидже, Ворс Жан-Пьер, Сасаки Норио, Кирио Йоши, Маеда Такако, Гент Даниель Б., Миллигэн Брюс, Перес Жозеф, Саваи Нобумитсу
МПК: C07C 259/04, A01N 37/52
Метки: кислоты, производные, применение, гидроксимовой
Формула / Реферат:
1. Производные гидроксимовой кислоты формулы (I):***где G представляет либо Gi, или G2, или G3, или G4 формулы:***Хь X2, X3 представляют, независимо, водород или атом галогена, гидрокси, меркапто, нитро, тиоцианато, азидогруппу, цианогруппу; алкил, галоидалкил, цианоалкил, алкокси, галоидал-кокси, цианоалкокси, алкилтио, галоидалкил-тио, цианоалкилтио, алкилсульфинил, галоидал-килсульфинил, алкилсульфонил, галоидалкил-сульфонил, причем указанные...
Производные фенилуксусной кислоты

Номер патента: 1380
Опубликовано: 26.02.2001
Авторы: Аммерманн Эберхард, Лоренц Гизела, Харреус Альбрехт, Гроте Томас, Байер Губерт, Штратман Зигфрид, Гётц Норберт, Мюллер Бернд, Заутер Губерт, Мюллер Рут, Харриес Фолкер
МПК: C07D 231/20, A01N 37/36, C07C 251/60...
Метки: фенилуксусной, кислоты, производные
Формула / Реферат:
1. Производные фенилуксусной кислоты формулы I в которой заместители и индекс имеют следующие значения: Х означает NOCH3, СНОСН3 и СНСН3; Y означает NR и О; R, R1 означают водород и С1-C4алкил; R2 означает водород, С1-C6алкил, С1-C4галогеналкил и C3-С6циклоалкил; R3 С2-C6алкинил или С2-C6алкенил, причем эти остатки могут быть частично либо полностью галогенированы или могут нести от одного до трех радикалов из числа следующих: циано, нитро,...
Производные эпоксиянтарной кислоты

Номер патента: 438
Опубликовано: 26.08.1999
Авторы: Хара Каору, Йосино Ясуси, Масаки Мицуо, Такахаси Тосихиро, Номура Ютака
МПК: A61K 31/335, C07D 303/46
Метки: производные, эпоксиянтарной, кислоты
Формула / Реферат:
1. Производные эпоксиянтарной кислоты следующей формулы в которой R1 представляет атом водорода, алкил с 1-30 атомами углерода, арил с 6-40 атомами углерода или аралкил с 7-40 атомами углерода; каждый из R2 и R3 независимо представляет арил с 6-40 атомами углерода, аралкил с 7-20 атомами углерода или алкил с 3-10 атомами углерода; Х представляет -О- или -NR4; R4 представляет атом водорода, алкил с 1-10 атомами углерода или аралкил с 7-20...
Производные 4-алкилтиопиримидин-5-ил-уксусной кислоты

Номер патента: 15
Опубликовано: 30.12.1997
Авторы: Шауб Фриц, Крэйг Джералд Уэйн, Эберле Мартин
МПК: C07D 239/56, A01N 43/54
Метки: кислоты, производные, 4-алкилтиопиримидин-5-ил-уксусной
Формула / Реферат:
1. Производные 2-(4-алкилтиопиримидин-5-ил) уксусной кислоты формулы 1 в которой R1 - С1-4-алкил; R2 - водород, С1-4-алкил или С3-7-циклоалкил; R3 - радикал вида где R4 - водород, С1-4-алкил, С1-4-галоидалкил, арил, арилоксил, С3-5-алкенилоксил, С3-5-алкинилоксил, галоген, циан, арил-С1-4-алкоксил, арилокси-С1-4-алкил, гетероарил, гетероарилоксил, С1-4-алкоксил, С1-4-алкокси-С1-4-алкоксил, циан-С1-4-алкоксил, С3-5-алкенил,...
Предыдущий патент: Носитель записи и устройство для сканирования носителя записи
Следующий патент: Способ уменьшения молекулярной массы сополимеров и терполимеров этилена
Случайный патент: Усовершенствованный протектор для шины для транспортных средств большой грузоподъемности