Оральные композиции левосимендана
Номер патента: 1647
Опубликовано: 25.06.2001
Авторы: Ларма Илькка, Харьюла Маарит, Лехтонен Лассе, Антила Сайла




Формула / Реферат
1. Композиция для орального введения, включающая практически чистую кристаллическую полиморфную форму I левосимендана в качестве активного ингредиента вместе с фармацевтически приемлемым носителем, причем кристаллическая полиморфная форма I левосимендана характеризуется диффрактограммой со следующими положениями пиков:
| Угол 2q (ш) |
| 8,7 |
| 9,5 |
| 12,2 |
| 15,4 |
| 15,9 |
| 17,7 |
| 18,4 |
| 19,2 |
| 20,3 |
| 21,4 |
| 21,8 |
| 23,1 |
| 24,6 |
| 25,7 |
| 27,4 |
2. Препаративная форма в виде таблетки, драже, капсулы, пудры или гранул, материалом которой является композиция по п.1.
3. Композиция по п.1 или 2, в которой количество активного ингредиента в композиции составляет от 0,1 до 20 мас.% композиции.
4. Композиция по п.3, в которой количество активного ингредиента в композиции составляет от 0,5 до 10 мас.% композиции.
5. Композиция по любому из пп.1-4, в которой количество активного ингредиента составляет от 0,1 до 10 мг на дозу.
6. Композиция по любому из пп.1-5, в которой фармацевтически приемлемым носителем является лактоза.
Текст
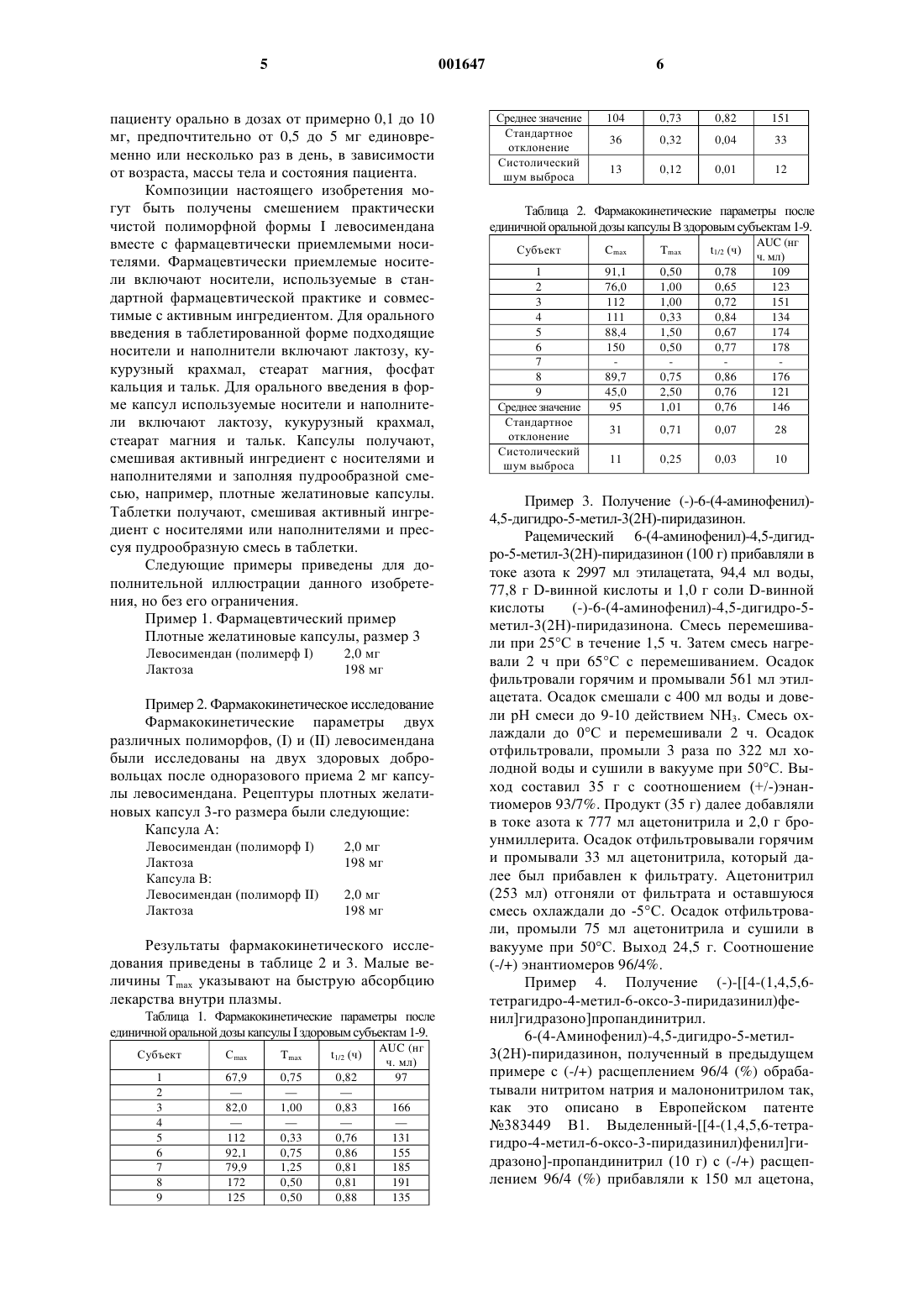
1 Настоящее изобретение относится к фармацевтическим композициям для орального введения, включающим практически чистую полиморфную формуI левосимендана,(-)-энантиомера 4-(1,4,5,6-тетрагидро-4-метил 6-оксо-3-пиридазинил)фенил]гидразоно]пропандинитрила в качестве активного ингредиента. Левосимендан используется в лечении застойной сердечной недостаточности. Рацемическая смесь 4-(1,4,5,6-тетрагидро 4-метил-6-оксо-3-пиридазинил)фенил]гидразоно]пропандинитрила (I) была описана ранее в Европейском патенте на имя заявителя 383449 В 1. Было показано, что соединение (I) является сильнодействующим при лечении застойной сердечной недостаточности и ему присуще значительное кальцийзависимое связывание тропонина. Оптически активные энантиомеры соединения (I) были ранее описаны в Европейском патенте на имя заявителя 565546 В 1. Было показано, что кардиотоническая способность связана в основном с (-)-энантиомером соединения (I), то есть с левосименданом. Было доказано, что оральное введение левосимендана трудно осуществимо в связи с тем,что левосимендан подвержен метаболизму в нижнем желудочно-кишечном тракте. Метаболиты, образующиеся в нижнем желудочнокишечном тракте, могут стать причиной наблюдаемых побочных эффектов при оральном введении левосимендана, таких как цефалгия и учащенное сердцебиение. В связи с этим было бы желательно располагать методами и композициями для орального введения левосимендана, которые могли бы исключить или уменьшить накопление левосимендана в нижнем желудочно-кишечном тракте. В настоящее время найдено, что левосимендан быстро растворяется и абсорбируется в плазме из оральных композиций, включающих практически чистую кристаллическую полиморфную форму I левосимендана в качестве активного ингредиента. Быстрая абсорбция понижает накопление левосимендана в нижнем желудочно-кишечном тракте и тем самым понижает желудочно-кишечный метаболизм левосимендана. Таким образом, в настоящем изобретении предлагается оральная композиция, включающая в качестве активного ингредиента практически чистую кристаллическую полиморфную форму I левосимендана вместе с фармацевтически приемлемым носителем. На фиг. 1 приведены параметры порошковой рентгенограммы в области 3-33 20 полиморфной формы (-)-4-(1,4,5,6-тетрагидро-4 001647 2 метил-6-оксо-3-пиридазинил)фенил]гидразоно]пропандинитрила. В настоящем описании термин "практически чистая кристаллическая полиморфная форма I левосимендана означает (-)-4-(1,4,5,6 тетрагидро-4-метил-6-оксо-3-пиридазинил)фенил]гидразоно]пропандинитрил, в которой, по крайней мере, примерно 90%, предпочтительно,по крайней мере, 95% и более предпочтительно,по крайней мере, 99 мас.%. находится в форме кристаллического полиморфа I. Кристаллическая полиморфная форма I левосимендана может быть получена из соединения (II) разделением рацемата в две отдельные стадии синтеза. Рацемическое соединение (II) может быть получено способами, описанными в литературе(J. Med. Chem., 17, 273-281 (1974. Первоначальная стадия разделения включает взаимодействие рацемической смеси (II) сD- или L-винной кислотой в растворе этилацетата. Оптимально, чтобы раствор этилацетата содержал от 0 до примерно 6 мас.%, предпочтительно от 2 до 4 мас.%, более предпочтительно примерно 3 мас.% воды. Предпочтительно использовать D- или L-винную кислоту и соединение (II) примерно в эквимолярных количествах. Диастереомерные соли(+)-6-(4-аминофенил)-4,5-дигидро-5 метил-3(2 Н)пиридазинона с L-винной кислотой кристаллизуются из этилацетата с достаточно высоким выходом. Кристаллическая диастереомерная соль может быть отфильтрована, а свободное основание выделяется при подщелачивании соли, например, раствором карбоната калия или аммония. После фильтрования маточную жидкость отделяют и обрабатывают повторно для выделения энантиомера, который перед этим не был удален фильтрованием. Обработка может включать, например, охлаждение маточной жидкости и выделение полученной кристаллической диастереомерной соли. Полученный вышеописанным способом продукт содержит, как правило, примерно 90 масс.% требуемого изомера (II). Чистота продукта может быть повышена до примерно 96 масс.% путем перекристаллизации. Предпочтительным растворителем для перекристаллизации является ацетонитрил. Например, продукт,обогащенный (-)энантиомером, перекристаллизовывают путем прибавления продукта к раствору ацетонитрила при кипячении смеси и последующего фильтрования осадка. Если необходимо, фильтрат концентрируют и охлаждают для того, чтобы выкристаллизовать (-)энантиомер (II). 3 Частичное разделение соединения (II) может быть реализовано с использованием других,чем этилацетат, систем растворителей. Такие растворители включают изопропанол, изобутанол, изопропилацетат, бутилацетат, ацетон и ацетонитрил. Использование других, чем D- илиL-винная кислота, кислот, например, бензойной или серной кислот, для разделения также может привести к частичному разделению соединения(II). Однако способ с использованием D-или Lвинной кислоты в этилацетате или в качестве растворителя водного этилацетата дает самую высокую оптическую чистоту для соединения(-)Энантиомер 4-(1,4,5,6-тетрагидро-4 метил-6-оксо-3-пиридазинил)фенил]гидразоно]пропандинитрила (I) получен из 6-(4-аминофенил)-4,5-дигидро-5-метил-3(2 Н)пиридазинона (II),обогащенного (-)энантиомером посредством взаимодействия (II) с нитритом натрия и малонитрилом в кислотных условиях, как это описано в Европейском патенте 383449 В 1. Затем выделяют обогащенное (-)энантиомером соединение (I). Минорный компонент в частично обогащенной энантиомерной смеси соединения (I) может быть отфильтрован от ацетона с остатком главного компонента в его растворе. Это позволяет осуществить регенерацию практически чистого (-)энантиомера соединения (I) из маточного раствора посредством кристаллизации. Таким образом предварительно выделенное соединение (I), обогащенное (-)энантиомером суспендируют в ацетоне как растворителе, предпочтительно содержащем до 2 мас.% воды. Смесь кипятят с обратным холодильником, осадок отфильтровывают. Затем фильтрат,если это необходимо, концентрируют и охлаждают до примерно 0-(-5)С. Выделяют выпавший кристаллический (-)энантиомер. Полученный продукт содержит, как правило, более, чем 90 мас.% нужного (-)энантиомера соединения(I). Кристаллографическая чистота вышеполученной полимерной формы (I) соединения (I) может быть, если это необходимо, повышена посредством нагревания полученного (-)энантиомера соединения (I) при температуре, по крайней мере, примерно 70 С в течение периода времени, необходимого для образования кристаллографически чистой полиморфной формы(I). Походящий для этого температурный интервал находится в области от 70-160 С, предпочтительно 80-130 С. Временной интервал обычно находится в области 1-48 ч, предпочтительно в области 4-24 ч. Эта обработка может быть частью процесса высушивания данного продукта и может проводиться в вакууме. Полиморфная форма I (-)4-(1,4,5,6 тетрагидро-4-метил-6-оксо-3-пиридазинил)фенил]гидразоно]пропандинитрила охарактеризована с помощью метода рентгеновской кристал 001647 4 лографии. Порошковая рентгенограмма полиморфной формы I в области 3-22 20 приведена на фиг. 1, а кристаллографические данные приведены в таблице 1. Параметры дифрактограммы кристаллической решетки были получены на порошковом рентгеновском кристаллографическом оборудовании (XRPD), Siemens D 500 (Siemens AG,Karlsruhe, Germany). Медная рентгенографическая трубка-мишень (длина волны 0,154 нм) трубка находилась в функциональном режиме 40 kVx40mA. Для порошкового рентгенографического анализа образцы свободно крепились в количестве 500 мг порошка к специальному цилиндрическому столику для образца цилиндрической формы диаметром 20 мм и высотой примерно 2 мм. Математический расчет параметров диффрактограмы выполнен с использованием пакета программ математического обеспеченияDiffrac AT V3.1. Основные характеристики диффрактограмы, такие как 2-значения и относительные интенсивности пиков были получены как выходные данные. Таблица 1. Величины углов при рентгенографическом анализе (2) и величины относительных интенсивностей (приведены только величины (%)5%) полиморфной формы I Угол 2 8,7 9,5 12,2 15,4 15,9 17,7 18,4 19,2 20,3 21,4 21,8 23,1 24,6 25,7 27,4 Величины относительных интенсивностей могут существенным образом изменяться из-за различной ориентации кристаллов. Поэтому величины относительных интенсивностей, приведенные в таблице 1, могут рассматриваться как относительные только для, например, немикронной пудры. Настоящее изобретение относится к композиции для орального введения, включающей практически чистую кристаллическую полиморфную форму I левосимендана в качестве активного ингредиента вместе с фармацевтически приемлемым носителем. Композиции настоящего изобретения включают твердые композиции в таких формах, как, например, таблетки, драже, капсулы, пудры и гранулы. Содержание активного вещества в композиции настоящего изобретения составляет, как обычно, от примерно 0,01 до 100 мас.%, предпочтительно от 0,1 до 20 мас.%, наиболее общепринято от 0,5 до 10 мас.%. Обычно левосимендан вводится пациенту орально в дозах от примерно 0,1 до 10 мг, предпочтительно от 0,5 до 5 мг единовременно или несколько раз в день, в зависимости от возраста, массы тела и состояния пациента. Композиции настоящего изобретения могут быть получены смешением практически чистой полиморфной формы I левосимендана вместе с фармацевтически приемлемыми носителями. Фармацевтически приемлемые носители включают носители, используемые в стандартной фармацевтической практике и совместимые с активным ингредиентом. Для орального введения в таблетированной форме подходящие носители и наполнители включают лактозу, кукурузный крахмал, стеарат магния, фосфат кальция и тальк. Для орального введения в форме капсул используемые носители и наполнители включают лактозу, кукурузный крахмал,стеарат магния и тальк. Капсулы получают,смешивая активный ингредиент с носителями и наполнителями и заполняя пудрообразной смесью, например, плотные желатиновые капсулы. Таблетки получают, смешивая активный ингредиент с носителями или наполнителями и прессуя пудрообразную смесь в таблетки. Следующие примеры приведены для дополнительной иллюстрации данного изобретения, но без его ограничения. Пример 1. Фармацевтический пример Плотные желатиновые капсулы, размер 3 Левосимендан (полимерф I) Лактоза Пример 2. Фармакокинетическое исследование Фармакокинетические параметры двух различных полиморфов, (I) и (II) левосимендана были исследованы на двух здоровых добровольцах после одноразового приема 2 мг капсулы левосимендана. Рецептуры плотных желатиновых капсул 3-го размера были следующие: Капсула А: Левосимендан (полиморф I) Лактоза Капсула В: Левосимендан (полиморф II) Лактоза Результаты фармакокинетического исследования приведены в таблице 2 и 3. Малые величины Тmах указывают на быструю абсорбцию лекарства внутри плазмы. Таблица 1. Фармакокинетические параметры после единичной оральной дозы капсулы I здоровым субъектам 1-9. Субъект 6 Среднее значение Стандартное отклонение Систолический шум выброса Таблица 2. Фармакокинетические параметры после единичной оральной дозы капсулы В здоровым субъектам 1-9. Субъект 1 2 3 4 5 6 7 8 9 Среднее значение Стандартное отклонение Систолический шум выброса(-)-6-(4-аминофенил)-4,5-дигидро-5 метил-3(2 Н)-пиридазинона. Смесь перемешивали при 25 С в течение 1,5 ч. Затем смесь нагревали 2 ч при 65 С с перемешиванием. Осадок фильтровали горячим и промывали 561 мл этилацетата. Осадок смешали с 400 мл воды и довели рН смеси до 9-10 действием NН 3. Смесь охлаждали до 0 С и перемешивали 2 ч. Осадок отфильтровали, промыли 3 раза по 322 мл холодной воды и сушили в вакууме при 50 С. Выход составил 35 г с соотношением (+/-)энантиомеров 93/7%. Продукт (35 г) далее добавляли в токе азота к 777 мл ацетонитрила и 2,0 г броунмиллерита. Осадок отфильтровывали горячим и промывали 33 мл ацетонитрила, который далее был прибавлен к фильтрату. Ацетонитрил(253 мл) отгоняли от фильтрата и оставшуюся смесь охлаждали до -5 С. Осадок отфильтровали, промыли 75 мл ацетонитрила и сушили в вакууме при 50 С. Выход 24,5 г. Соотношение(-/+) энантиомеров 96/4%. Пример 4. Получение (-)-4-(1,4,5,6 тетрагидро-4-метил-6-оксо-3-пиридазинил)фенил]гидразоно]пропандинитрил. 6-(4-Аминофенил)-4,5-дигидро-5-метил 3(2 Н)-пиридазинон, полученный в предыдущем примере с (-/+) расщеплением 96/4 (%) обрабатывали нитритом натрия и малононитрилом так,как это описано в Европейском патенте 383449 В 1. Выделенный-4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3-пиридазинил)фенил]гидразоно]-пропандинитрил (10 г) с (-/+) расщеплением 96/4 (%) прибавляли к 150 мл ацетона, 7 0,9 мл воды, 0,2 г активированного угля и 0,4 г броунмиллерита. Смесь кипятили 1 ч с обратным холодильником и фильтровали горячей. Осадок промывали 10 мл горячего ацетонитрила, который далее был прибавлен к фильтрату. Фильтрат кипятили с обратным холодильником 30 мин. Ацетон (61 мл) отгоняли от фильтрата и оставшуюся смесь охлаждали до 0-(-5)С. Смесь отфильтровали и промыли 10 мл холодного ацетона. Кристаллический продукт сушили в вакууме при 100 С в течение 5 ч. Был получен продукт с содержанием более 99% целого (-)энантиомера, и выход составил 6,8 мг. Продукт является практически чистой полиморфной формой I. Энантимерная чистота продуктов определялась ВЭЖХй. Энантиомеры соединения (II) разделялись с использованием хиральной колонки целлюлозного типа (Chiralcel OJ 25 х 0,46 см). Подвижная фаза состояла из этанола. Скорость потока составляла 0,5 мл/мин. Энантиомеры соединения (I) разделялись с использованием колонки с -циклодекстрином (Cyclobond IBeta, 4,6 х 250 мм). Подвижная фаза состояла из раствора 36% метанола в воде, к которому прибавлен буфер, являющийся 1%-ным ацетатом триметиламмония, до рН 6,0. Скорость потока составляла 0,8 мл/мин. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Композиция для орального введения,включающая практически чистую кристаллическую полиморфную форму I левосимендана в качестве активного ингредиента вместе с фармацевтически приемлемым носителем, причем кристаллическая полиморфная форма I левосимендана характеризуется диффрактограммой со следующими положениями пиков: Угол 28,7 9,5 12,2 15,4 15,9 17,7 18,4 19,2 20,3 21,4 21,8 23,1 24,6 25,7 27,4 2. Препаративная форма в виде таблетки,драже, капсулы, пудры или гранул, материалом которой является композиция по п.1. 3. Композиция по п.1 или 2, в которой количество активного ингредиента в композиции составляет от 0,1 до 20 мас.% композиции. 4. Композиция по п.3, в которой количество активного ингредиента в композиции составляет от 0,5 до 10 мас.% композиции. 5. Композиция по любому из пп.1-4, в которой количество активного ингредиента составляет от 0,1 до 10 мг на дозу. 6. Композиция по любому из пп.1-5, в которой фармацевтически приемлемым носителем является лактоза.
МПК / Метки
МПК: A61K 31/50
Метки: левосимендана, оральные, композиции
Код ссылки
<a href="https://eas.patents.su/5-1647-oralnye-kompozicii-levosimendana.html" rel="bookmark" title="База патентов Евразийского Союза">Оральные композиции левосимендана</a>
Бициклические ароматические соединения, фармацевтическая и косметическая композиции на их основе, и применение косметической композиции

Номер патента: 1216
Опубликовано: 25.12.2000
Автор: Бернардон Жан-Мишель
МПК: C07C 69/618, A61K 31/38, C07D 333/24...
Метки: основе, ароматические, косметической, соединения, композиции, косметическая, применение, фармацевтическая, бициклические
Формула / Реферат:
1. Биароматические соединения пропинила общей формулы (I) где R1 является (I) радикалом -СН3; (II) радикалом -CH2-O-R5; (III) радикалом -СО-R6, где R5 и R6 имеют значения, приведенные ниже, Аr является радикалом, выбираемым из радикалов формул (а)-(е): где R5 и R7 имеют значения, приведенные ниже, Х является радикалом формулы где R8 и R9 имеют значения, приведенные ниже, R2 и R3, одинаковые или различные, являются (I) атомом водорода;...
Способ получения композиции поперечно-сшитого биологически совместимого полисахаридного геля, композиции указанного геля и применение композиций

Номер патента: 1500
Опубликовано: 23.04.2001
Автор: Огеруп Бенгт
МПК: A61K 31/715, A61P 5/00, C08B 37/08...
Метки: композиции, биологически, полисахаридного, совместимого, получения, указанного, композиций, способ, применение, геля, поперечно-сшитого
Формула / Реферат:
1. Способ получения композиции поперечно-сшитого биологически совместимого полисахаридного геля, включающий следующие стадии: приготовление водного раствора растворимого в воде поперечно-сшиваемого полисахарида; инициирование реакции поперечного сшивания указанного полисахарида в присутствии полифункционального агента, обеспечивающего сшивание полисахарида; обеспечение стерического затруднения реакции поперечного сшивания до ее завершения перед...
Композиции жидкого алендроната

Номер патента: 1213
Опубликовано: 25.12.2000
Авторы: Остовиц Дразен, Ханке Вильям А., Неруркар Маниш Дж.
МПК: A61P 19/10, A61K 31/66
Метки: алендроната, композиции, жидкого
Формула / Реферат:
1. Водная жидкая фармацевтическая композиция, содержащая: алендроновую кислоту или фармацевтически приемлемую соль в качестве активного ингредиента; достаточное количество буфера так, что А) рН композиции находится между примерно 3,5 и примерно 7,5; и В) 15 мл композиции может увеличить рН 50 мл 0,1 н НСl до рН, по крайней мере, 3; и необязательно, один или более дополнительных реагентов, выбранных из группы, состоящей их консервантов,...
Микробицидные композиции

Номер патента: 503
Опубликовано: 28.10.1999
Авторы: Цеун Рональд, Кнауф-Байтер Гертруде
МПК: A01N 37/02
Метки: микробицидные, композиции
Формула / Реферат:
1. Фитомикробицидная композиция, содержащая компонент I, который является соединением формулы I в которой Х обозначает СН или N; R обозначает CH3 или циклопропил; Y обозначает Н, F, Cl, Br, CF3, CF3O, пропаргилокси; Z обозначает Н, F, Cl, СF3, СF3О; или Y и Z, взятые вместе, представляют метилендиокси, (дифторметилен)диокси, этилендиокси, (трифторэтилен)диокси или бензогруппу и компонент II, который является соединением, выбранным из...
Липосомальная вакцинная композиция, применение липосомальной вакцинной композиции, способ приготовления липосомальной вакцинной композиции и способ лечения млекопитающего.

Номер патента: 839
Опубликовано: 24.04.2000
Авторы: Гарсон Натали Мари-Жозеф Клод, Фрид Мартин
МПК: A61K 39/39
Метки: вакцинной, липосомальная, вакцинная, лечения, способ, липосомальной, применение, приготовления, композиция, композиции, млекопитающего
Формула / Реферат:
1. Липосомальная вакцинная композиция, содержащая антиген или антигенную композицию, иммунологически активную сапониновую фракцию и стерин, характеризующаяся тем, что отношение сапониновая фракция : стерин составляет от 1:1 до 1:100 (по массе). 2. Вакцинная композиция по п.1, отличающаяся тем, что отношение сапониновая фракция : стерин составляет от 1:1 до 1:5 (по массе). 3. Вакцинная композиция по п.1, отличающаяся тем, что отношение...