Способ получения 2-[4-(3- или 2-фторбензилокси)бензиламино]пропанамидов с высокой степенью чистоты
Номер патента: 19898
Опубликовано: 30.07.2014
Авторы: Барбанти Елена, Сальвати Патрисия, Понзини Франческо, Каневотти Ренато, Фаравелли Лаура



























Формула / Реферат
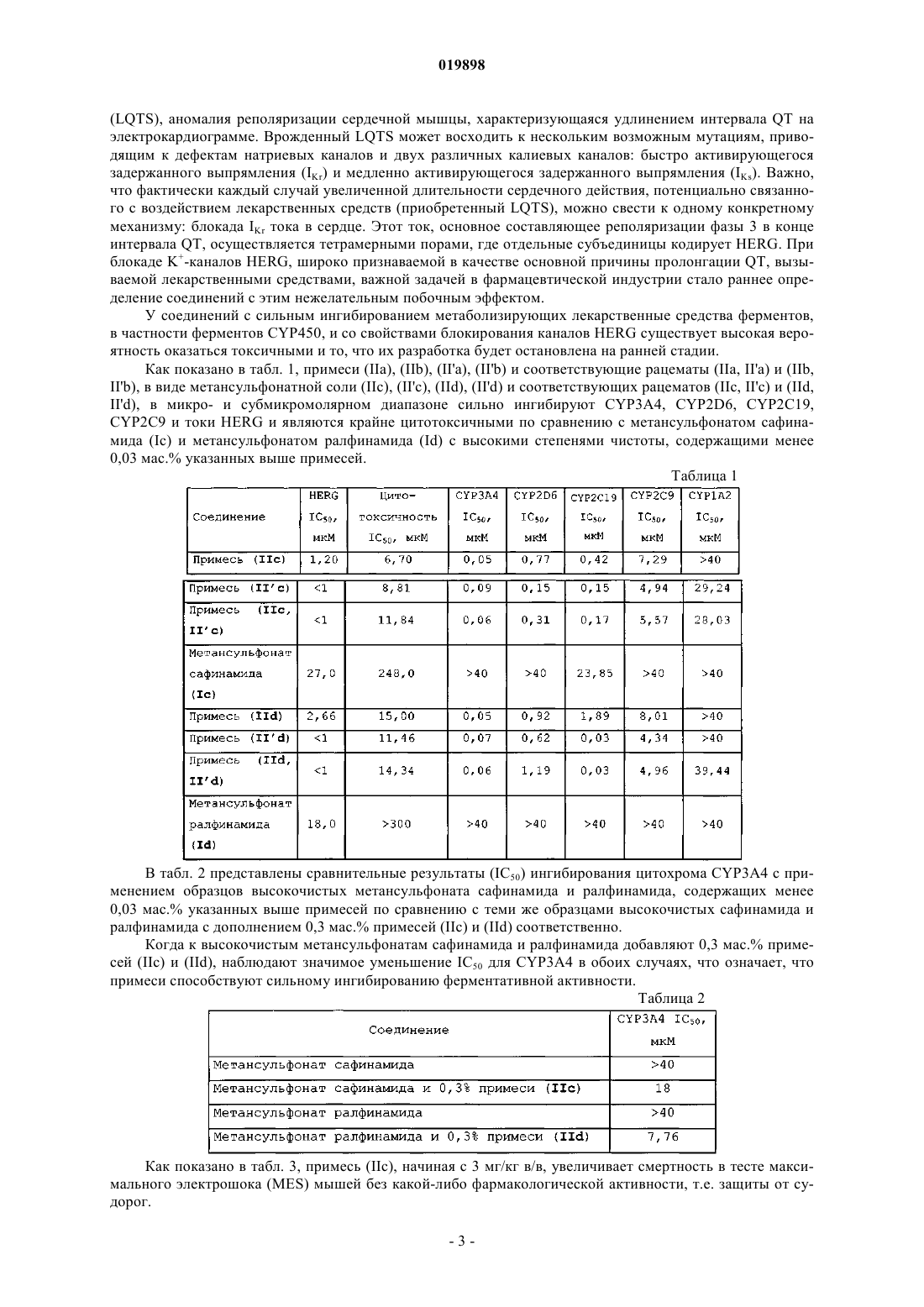
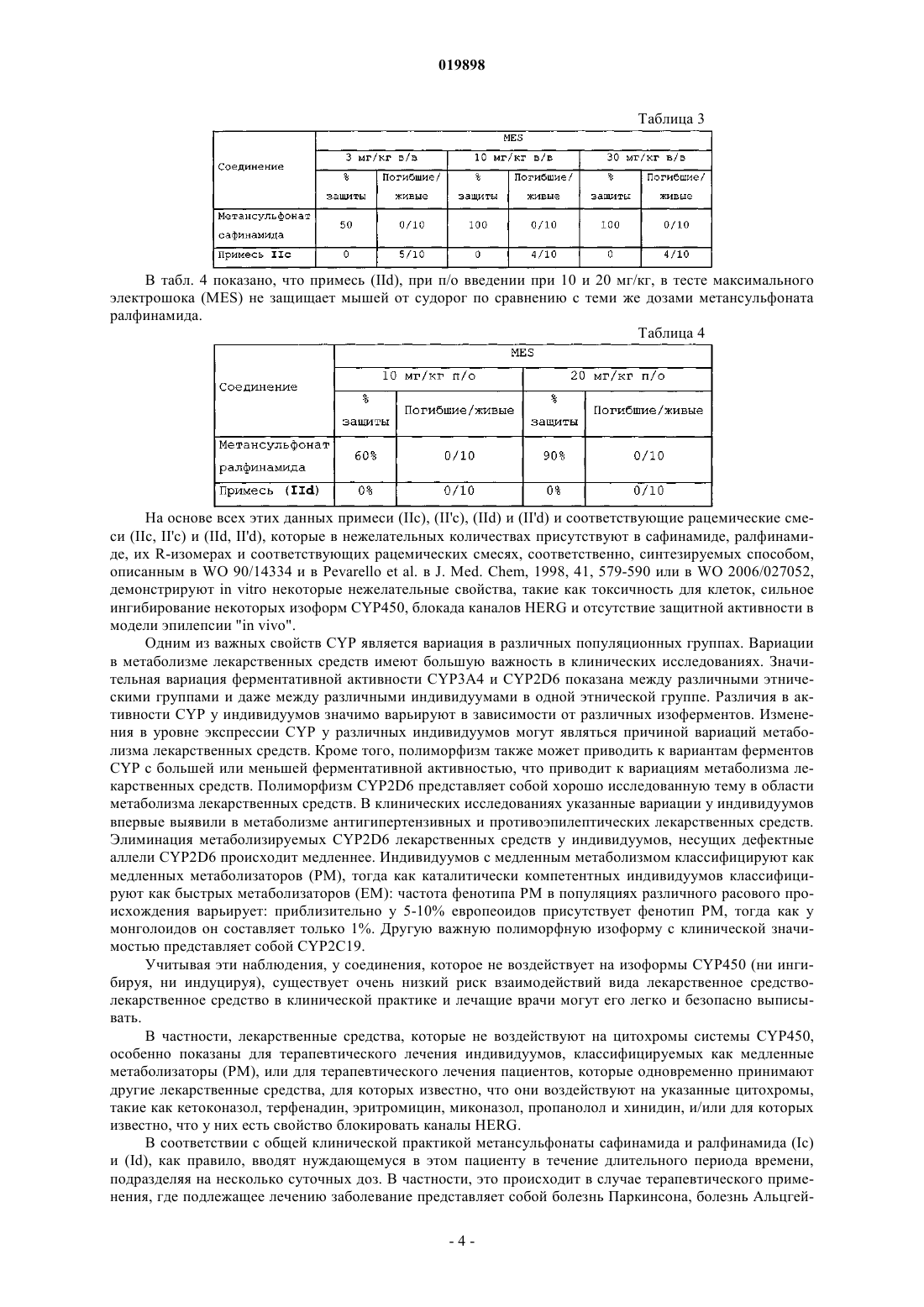
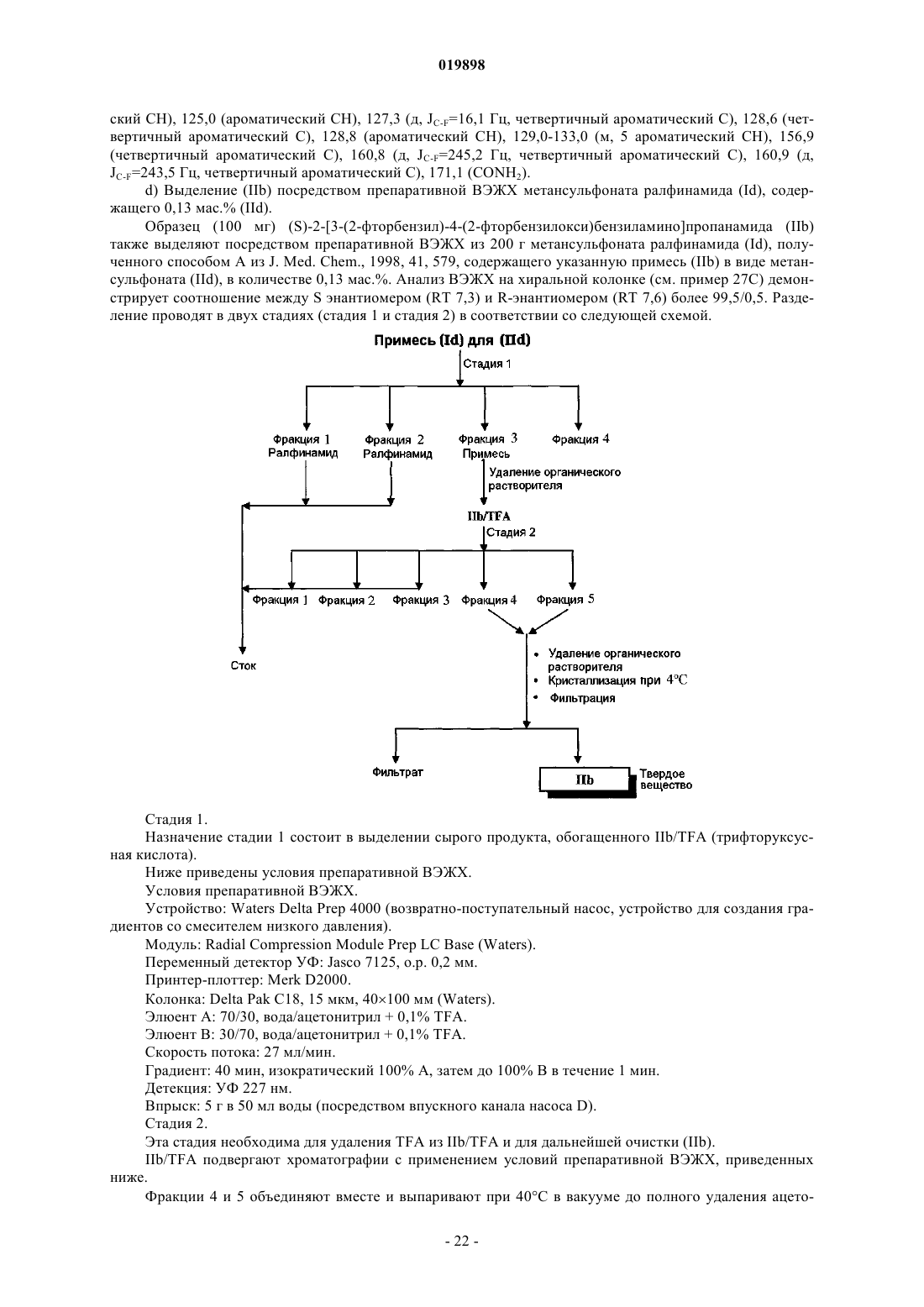
1. Способ получения 2-[4-(3- или 2-фторбензилокси)бензиламино]пропанамидного соединения высокой степени чистоты, выбранного из (S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (сафинамида) формулы (Ia), (S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (ралфинамида) формулы (Ib)

соответствующих R-энантиомеров (I'а) и (I'b), соответствующих рацемических смесей (Ia, I'а) и (Ib, I'b) и их солей с фармацевтически приемлемыми кислотами, где сафинамид (Ia), ралфинамид (Ib), соответствующие R-энантиомеры (I'а) или (I'b) или соответствующие рацемические смеси (Ia, I'а) и (Ib, I'b) или их соли с фармацевтически приемлемой кислотой имеют содержание соответствующих примесей, (S)-2-[3-(3-фторбензил)-4-(3-фторбензилокси)бензиламино]пропанамида (IIa), (S)-2-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамида (IIb)

соответствующих R-энантиомеров (II'а) или (II'b) или соответствующих рацемических смесей (IIa, II'а) или (IIb, II'b) или их солей с фармацевтически приемлемой кислотой, которое составляет менее 0,03 мас.%, характеризующийся тем, что промежуточное основание Шиффа формулы (IIIa), (IIIb)

соответствующие R-энантиомеры (III'а) или (III'b) или соответствующие рацемические смеси (IIIa, III'а) или (IIIb, III'b) (i) получают реакцией иминоалкилирования 4-(3- или 2-фторбензилокси)бензальдегида L-аланинамидом или D-аланинамидом или их рацемической смесью в растворителе, выбранном из (C1-С5)алканолов, в количестве, которое обеспечивает образование суспензии основания Шиффа в насыщенном растворе основания Шиффа в том же растворителе при температуре от 20 до 30°С; и (ii) после завершения реакции иминоалкилирования подвергают реакции восстановления восстановителем, выбранным из боргидрида натрия и боргидрида калия в выбранном количестве органического растворителя, выбранного из (C1-C5)алканолов или их смеси, необязательно с водой в количестве, составляющем менее 1,5 мас.% относительно количества органического растворителя, где отношение органического растворителя к основанию Шиффа обеспечивает получение и присутствие в ходе существенной части реакции восстановления суспензии основания Шиффа в насыщенном растворе основания Шиффа в том же органическом растворителе и составляет от 0,5 до 3,0 л на каждый моль основания Шиффа, где способ дополнительно характеризуется тем, что исходное вещество 4-(3- или 2-фторбензилокси)бензальдегид, применяемое для получения промежуточных оснований Шиффа (IIIa), (IIIb), (III'a), (III'b) или соответствующих рацемических смесей (IIIa, III'а) или (IIIb, III'b), имеет содержание примеси 3-(3- или 2-фторбензил)-4-(3- или 2-фторбензилокси)бензальдегида менее 0,03 мас.%; в результате чего сафинамид (Ia), ралфинамид (Ib), соответствующие R-энантиомеры (I'а) или (I'b) или соответствующие рацемические смеси (Ia, I'а) или (Ib, I'b) получают в форме свободного основания и, необязательно, форму указанного свободного основания преобразуют в их соль с фармацевтически приемлемой кислотой.
2. Способ по п.1, где восстановитель представляет собой боргидрид натрия, а органический растворитель выбран из метанола, этанола и 2-пропанола, необязательно в присутствии воды в количестве, составляющем менее 1,5 мас.% относительно количества органического растворителя.
3. Способ по п.1 или 2, где органический растворитель используют в количестве от 0,7 до 2,5 л на каждый моль основания Шиффа.
4. Способ по п.3, где органический растворитель используют в количестве от 0,8 до 2,0 л на каждый моль основания Шиффа.
5. Способ по любому из пп.1-4, где содержание соответствующих примесей формулы (IIa), (IIb), (II'а), (II'b) или соответствующих рацемических смесей (IIa, II'а) или (IIb, II'b) или их солей с фармацевтически приемлемой кислотой составляет менее 0,01 мас.%.
6. Способ по любому из пп.1-5, где исходное вещество 4-(3- или 2-фторбензилокси)бензальдегид, применяемое для получения промежуточных оснований Шиффа (IIIa), (IIIb), (III'a), (III'b) или соответствующих рацемических смесей (IIIa, III'а) или (IIIb, III'b), имеет содержание примеси 3-(3- или 2-фторбензил)-4-(3- или 2-фторбензилокси)бензальдегида менее 0,01 мас.%.
7. Способ по любому из пп.1-6, где восстановитель представляет собой боргидрид натрия, а органический растворитель представляет собой метанол.
8. Способ по любому из пп.1-7, где рН реакционной смеси перед любым добавлением боргидрида натрия или калия доводят до значений 7-9.
9. Способ по любому из пп.1-8, где восстановитель по отношению к промежуточному основанию Шиффа используется в молекулярном количестве от 0,5 до 1,4.
10. Способ по любому из пп.1-9, где реакцию восстановления проводят при температуре от -10 до 30°С.
11. Способ по п.10, где реакцию проводят при температуре от 5 до 15°С.
12. Способ по любому из пп.1-11, где фармацевтически приемлемая кислота представляет собой метансульфоновую кислоту.
13. Способ по любому из пп.1-12, где промежуточные основания Шиффа формулы (IIIa), (IIIb), (III'a), (III'b) или соответствующие рацемические смеси (IIIa, III'a) или (IIIb, III'b) получают посредством иминоалкилирования 4-(3-фторбензилокси)бензальдегида (IVa) или 4-(2-фторбензилокси)бензальдегида (IVb)

L-аланинамидом, D-аланинамидом или их рацемической смесью в присутствии органического растворителя, выбранного из метанола, этанола и изопропанола, в количестве относительно альдегидов (IVa) или (IVb), которое обеспечивает образование суспензии основания Шиффа в насыщенном растворе основания Шиффа в том же растворителе.
14. Способ по п.13, где L-аланинамид, D-аланинамид или их рацемическую смесь применяют в виде их соли присоединения кислоты в присутствии основания в количестве, достаточном для получения из их солей свободного L-аланинамида, D-аланинамида или их рацемической смеси.
15. Способ по любому из пп.1-14, где реакцию восстановления промежуточного основания Шиффа восстановителем проводят в той же реакционной смеси, образующейся в результате реакции иминоалкилирования в условиях, вызывающих осаждение указанного промежуточного основания Шиффа с получением суспензии указанного промежуточного соединения в том же реакционном растворителе.
16. Способ по любому из пп.1-14, где промежуточное основание Шиффа, образующееся в результате реакции иминоалкилирования, выделяют перед проведением реакции восстановления восстановителем.
17. Способ по любому из пп.1-16, где восстановитель боргидрид натрия или калия в ходе реакции добавляют к смеси основания Шиффа и реакционного растворителя частями, в твердой форме или в форме раствора в метаноле, стабилизированного добавлением гидроксида натрия или калия.
18. Способ по любому из пп.1-17, где исходное вещество 4-(3-фторбензилокси)бензальдегид (IVa) или 4-(2-фторбензилокси)бензальдегид (IVb) для получения промежуточного основания Шиффа (IIIa) (IIIb), (III'a), (III'b) или соответствующей рацемической смеси (IIIa, III'a) или (IIIb, III'b) получают алкилированием 4-гидроксибензальдегида соответственно алкилирующим средством 3-фторбензилом или 2-фторбензилом (Va) или (Vb)

где Y представляет собой уходящую группу, выбранную из Cl, Br, I, OSO2CH3 и OSO2-C6H4-pCH3, в присутствии основания, и необязательно подвергают кристаллизации до использования в последующей стадии реакции с получением исходного вещества 4-(3- или 2-фторбензилокси)бензальдегида (IVa) или (IVb), где содержание примеси 3-(3- или 2-фторбензил)-4-(3- или 2-фторбензилокси)бензальдегида составляет менее 0,03 мас.%.
19. Способ по п.18, где содержание примеси 3-(3- или 2-фторбензил)-4-(3- или 2-фторбензилокси)бензальдегида составляет менее 0,01 мас.%.
20. Способ по п.18 или 19, где Y представляет собой Cl.
21. Способ по любому из пп.18-20, где кристаллизацию проводят добавлением н-гексана к раствору 4-(3-фторбензилокси)бензальдегида (IVa) или 4-(2-фторбензилокси)бензальдегида (IVb) в толуоле.
22. Способ по п.18 или 19, где кристаллизацию проводят, растворяя 4-(3-фторбензилокси)бензальдегид (IVa) или 4-(2-фторбензилокси)бензальдегид (IVb) в горячем растворителе, выбранном из циклогексана и простого ди(С3-С4)алкилового эфира, при кипячении с последующим охлаждением раствора при комнатной температуре или при 10-15°С.
23. Способ по п.22, где растворитель представляет собой простой диизопропиловый эфир.
24. Способ по п.22 или 23, где раствор охлаждают при 10-15°С.
25. Способ по любому из пп.18-24, где реакцию алкилирования проводят в условиях фазового переноса.
26. Способ по п.25, где алкилирование в условиях фазового переноса проводят в системе твердое вещество/жидкость, где реагенты и катализатор фазового переноса растворяют в жидкой органической фазе, а твердая фаза представляет собой неорганическое основание, выбранное из NaOH, KOH, Na2CO3 и K2CO3, или соль 4-гидроксибензальдегида с указанным неорганическим основанием.
27. Способ по п.25, где алкилирование в условиях фазового переноса проводят в системе жидкость/жидкость, где алкилирующий реагент 3-фторбензильное или 2-фторбензильное производное формулы (Va) или (Vb) растворяют в жидкой органической фазе, а 4-гидроксибензальдегид растворяют в водной фазе в виде соли с указанным неорганическим основанием.
28. Способ по любому из пп.25-27, где катализатор фазового переноса выбран из четвертичных солей аммония, солей фосфония и полиэтиленгликолей низкой молекулярной массы.
29. Способ по п.28, где количество применяемого катализатора фазового переноса составляет от 0,02 до 1 моль на 1 моль 4-гидроксибензальдегида.
30. Способ по п.29, где количество катализатора фазового переноса составляет от 0,1 до 1 моль на 1 моль 4-гидроксибензальдегида.
31. Способ по любому из пп.26-30, где органический растворитель жидкой органической фазы представляет собой простой диалкиловый эфир, выбранный из простого ди-трет-бутилового эфира, простого этил-трет-бутилового эфира, или ароматический углеводород, выбранный из толуола, этилбензола, изопропилбензола и ксилолов.
32. Способ по любому из пп.26-31, где молярное отношение между алкилирующим средством формулы (Va) или (Vb) и 4-гидроксибензальдегидом составляет от 0,6 до 1,5.
33. Способ по любому из пп.26-32, где реакцию проводят при температуре от 60 до 160°С.
34. Способ по любому из пп.26-33, где неорганическое основание выбрано из Na2CO3, K2CO3, NaOH и KOH, температура реакции составляет от 80 до 120°С и молярное отношение между алкилирующим средством формулы (Va) или (Vb) и 4-гидроксибензальдегидом составляет от 0,9 до 1,1.
35. Способ по любому из пп.1-34, где фармацевтически приемлемая кислота представляет собой метансульфоновую кислоту, а содержание соответствующих примесей формулы (IIa), (IIb), (II'а), (II'b) или соответствующих рацемических смесей (IIa, II'а) или (IIb, II'b) в виде соли с метансульфоновой кислотой составляет менее 0,01 мас.%.
36. Выделенное основание Шиффа (R)-2-[4-(3-фторбензилокси)бензилиденамино]пропанамид (III'а) или (R)-2-[4-(2-фторбензилокси)бензилиденамино]пропанамид (III'b).
Текст
Изобретение относится к способу получения терапевтически активных 2-[4-(3- или 2-(фторбензилокси)бензиламино]пропанамидов и их солей с фармацевтически приемлемыми кислотами с высокой степенью чистоты, в частности с содержанием примесей дибензиловых производных менее 0,03 мас.%, предпочтительно менее 0,01 мас.%. Способ осуществляют, подвергая промежуточные основания Шиффа 2-[4-(3- или 2-фторбензилокси)бензилиденамино]пропанамидов реакции восстановления восстановителем,выбранным из боргидрида натрия и боргидрида калия в соответствующем количестве органического растворителя, выбранного из низших (C1-C5)алканолов, обеспечивая образование и присутствие в ходе значительной части реакции восстановления суспензии основания Шиффа в насыщенном растворе основания Шиффа в том же органическом растворителе. Предпосылки изобретения Настоящее изобретение относится к новому способу получения 2-[4-(3 или 2-фторбензилокси)бензиламино]пропанамидного соединения,выбранного из соответствующих R-энантиомеров (I'а) и (I'b), соответствующих рацемических смесей (Ia, I'а) и (Ib, I'b) и их солей с фармацевтически приемлемыми кислотами (Ic), (Id), (I'c), (I'd), а также их рацемических смесей (Ic, I'c) и (Id, I'd) с высоким выходом и с очень высокой энантиомерной и химической чистотой. Данный способ также очень подходит для их получения в больших количествах. Сафинамид (NW-1015, FCE-26743A, PNU-151774E) представляет собой блокатор натриевых каналов, модулятор кальциевых каналов, ингибитор моноаминооксидазы В (МАО-В), ингибитор высвобождения глутамата и модулятор метаболизма дофамина. Сафинамид пригоден для лечения нарушений ЦНС, в частности эпилепсии, болезни Паркинсона,болезни Альцгеймера, депрессии, синдрома беспокойных ног и мигрени (WO 90/14334, WO 2004/089353,WO 2005/102300 и WO 2004/062655). Ралфинамид (NW-1029, FCE-26742A, PNU-0154339E) представляет собой блокатор натриевых каналов, пригодный для лечения характеризующихся болью состояний, включая хроническую боль и невропатическую боль, мигрень, биполярных нарушений, депрессий, сердечно-сосудистых, воспалительных, мочеполовых, метаболических и желудочно-кишечных нарушений (WO 99/35125,WO 03/020273, WO 2004/062655, WO 2005/018627, WO 2005/070405, WO 2005/102300). В частности, сафинамид описан в WO 90/14334. Сафинамид, его R-энантиомер, их рацемическая смесь и их соли с фармацевтически приемлемыми кислотами и их применение для получения фармацевтических композиций, активных в качестве противоэпилептических, противопаркинсонических, нейропротекторных, антидепрессивных, противоспастических и/или снотворных средств, охарактеризованы в формуле изобретения WO 90/14334. Ралфинамид описан в WO 90/14334. Ралфинамид, его R-энантиомер, их рацемическая смесь и их соли с фармацевтически приемлемыми кислотами и их применение для получения фармацевтических композиций, активных в качестве противоэпилептических, противопаркинсонических, нейропротективных, антидепрессорных, противоспастических и/или снотворных средств, охарактеризованы в формуле изобретения WO 90/14334. Кроме того, в формуле изобретения WO 99/035125 представлено применение сафинамида, ралфинамида, соответствующих R-энантиомеров, соответствующих рацемических смесей и их солей с фармацевтически приемлемыми кислотами в качестве анальгетиков. В WO 2006/027052 А 2 описано и содержится в формуле изобретения применение одногоR-энантиомера ралфинамида, т.е. (R)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (I'b) и его солей с фармацевтически приемлемыми кислотами, в качестве селективного модулятора натриевых и кальциевых каналов для селективного лечения патологических повреждений, где патологическую роль играет механизм действия натриевых или кальциевых каналов, включая боль, мигрень, воспалительные процессы, затрагивающие все системы организма, нарушения, затрагивающие кожу и родственную ткань, нарушения дыхательной системы, нарушения иммунной и эндокринной систем, нарушения желудочнокишечной и мочеполовой систем, где терапевтическая активность указанного соединения, по существу,не оказывает какого-либо побочного эффекта в виде ингибирования МАО или демонстрирует значимо сниженный побочный эффект в виде ингибирования МАО. В настоящее время выявлено, что крупномасштабное получение сафинамида и ралфинамида способами, известными из уровня техники, включает две нежелательные примеси, т.е. соответственно(S)-2-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамид (IIb) и их соль, в частности соответствующие метансульфонаты (IIc) и (IId) Такая же ситуация возникает при получении способами, известными из уровня техники,R-энантиомеров (I'а) и (I'b), соответственно, сафинамида и ралфинамида, соответствующих рацемических смесей (Ia, I'а) и (Ib, I'b) и их солей с фармацевтически приемлемыми кислотами, (I'c), (I'd), и соответствующих рацемических смесей (Ic, I'c) и (Id, I'd), в частности метансульфонатов, которые в результате загрязняются соответствующими R-изомерами (II'а), (II'b), (II'с), и (II'd) указанных выше примесей(IIa), (IIb), (IIc) и (IId) или соответствующих рацемических смесей (IIa, II'а), (IIb, II'b), (IIc, II'с) и (IId,II'd). Этот факт имеет особую важность, так как выявлено, что указанные выше примеси демонстрируют очень высокую токсичность в отношении ферментов системы цитохрома Р 450. Многие из кандидатов в лекарственные средства потерпели неудачу в клинических испытаниях вследствие непредусмотренного действия на метаболизм человека или токсичности вследствие наличия нежелательных примесей и, таким образом, удаление таких примесей на ранней преклинической фазе является важным и очень желательным. На преклиническом уровне "способность быть лекарственным средством" новых соединений можно оценивать с применением очень хорошо разработанной группы анализов in vitro, таких как взаимодействие с метаболизирующими лекарственными средствами, ферментами, цитотоксичность, метаболическая стабильность и профилирование, мембранная проницаемость, собственный клиренс и блокада каналов, кодируемых геном человека, родственным гену ether-a-go-go (HERG) и т.д. Система цитохрома Р 450 (CYP450) представляет собой основную систему метаболизма липофильных ксенобиотиков, включая лекарственные средства, канцерогены и загрязнители окружающей среды.CYP450 представляет собой содержащую гем мембраносвязанную мультиферментную систему, представленную во многих тканях, но на наибольшем уровне представленную в печени. Установлено, что в печени человека существует от 15 до 20 различных форм CYP450, метаболизирующих ксенобиотики. До настоящего времени у млекопитающих идентифицировано более четырнадцати семейств генов CYP. В обширных исследованиях выявлено, что несмотря на наличие высокой гомологии, каждое семейство и подсемейство CYP обладает особой ролью в метаболизме ксенобиотиков. Три семейства CYP: CYP1,CYP2 и CYP3, составляют приблизительно 70% CYP микросом печени человека с CYP3, составляющим приблизительно 30%. Эти CYP являются основными ответственными за метаболизм большинства продаваемых лекарственных средств. Семейство CYP1 содержит несколько членов, включая CYP1A1, CYP1A2 и CYP1B1, и они вовлечены в метаболизм ацетаминофена, кломипрамина и имипрамина. Семейство CYP2 содержит несколько подсемейств, включая CYP2A, CYP2B, CYP2C, CYP2D иCYP2E. Подсемейство CYP2C содержит по меньшей мере семь членов. CYP2C9 отвечает за метаболизм ибупрофена, диклофенака, толбутамида и торсемида. CYP2C19 представляет собой основной изофермент, метаболизирующий диазепам и омеопразол. Показано, что CYP2D6 отвечает за метаболизм более 30% лекарственных средств на рынке, включая антидепрессанты и сердечно-сосудистые и антипсихотические лекарственные средства. В семействе CYP3 в печени человека идентифицированы три изоформы. Показано, что CYP3A4 человека является наиболее важной изоформой в метаболизме лекарственных средств. В настоящее время катализируемый CYP3A4 метаболизм представляет собой основной маршрут элиминации почти 50% продаваемых лекарственных средств. Вследствие их важности в метаболизме лекарственных средств, CYP3A4 и CYP2D6 часто вовлечены во взаимодействия лекарственное средство-лекарственное средство и в качестве эффективного ингибитора этих изоформ CYP450 идентифицировано несколько клинически используемых соединений, таких как кетоконазол, терфенадин, эритромицин, миконазол, пропанолол и хинидин соответственно. Это накладывает явное ограничение на использование этих лекарственных средств. Дополнительная проблема, заключающаяся во внезапной смерти в качестве побочного эффекта от действия не противоаритмических лекарственных средств, является главной задачей фармакологической безопасности, стоящей перед фармацевтической индустрией и контролирующими органами в здравоохранении. В последние годы вследствие сообщений о внезапной смерти с рынка отозваны по меньшей мере пять имеющих большой успех лекарственных средств (астемизол, сертиндол, терфенадин, цисаприд, грепафлоксацин). Во всех случаях в качестве предрасполагающего фактора для "трепетаниямерцания", полиморфной желудочковой тахикардии, которая затем может спонтанно ухудшаться до фибрилляции желудочков и вызывать внезапную смерть, участвовал синдром удлинения интервала QT(LQTS), аномалия реполяризации сердечной мышцы, характеризующаяся удлинением интервала QT на электрокардиограмме. Врожденный LQTS может восходить к нескольким возможным мутациям, приводящим к дефектам натриевых каналов и двух различных калиевых каналов: быстро активирующегося задержанного выпрямления (IKr) и медленно активирующегося задержанного выпрямления (IKs). Важно,что фактически каждый случай увеличенной длительности сердечного действия, потенциально связанного с воздействием лекарственных средств (приобретенный LQTS), можно свести к одному конкретному механизму: блокада IKr тока в сердце. Этот ток, основное составляющее реполяризации фазы 3 в конце интервала QT, осуществляется тетрамерными порами, где отдельные субъединицы кодирует HERG. При блокаде K+-каналов HERG, широко признаваемой в качестве основной причины пролонгации QT, вызываемой лекарственными средствами, важной задачей в фармацевтической индустрии стало раннее определение соединений с этим нежелательным побочным эффектом. У соединений с сильным ингибированием метаболизирующих лекарственные средства ферментов,в частности ферментов CYP450, и со свойствами блокирования каналов HERG существует высокая вероятность оказаться токсичными и то, что их разработка будет остановлена на ранней стадии. Как показано в табл. 1, примеси (IIa), (IIb), (II'а), (II'b) и соответствующие рацематы (IIa, II'а) и (IIb,II'b), в виде метансульфонатной соли (IIc), (II'c), (IId), (II'd) и соответствующих рацематов (IIc, II'с) и (IId,II'd), в микро- и субмикромолярном диапазоне сильно ингибируют CYP3A4, CYP2D6, CYP2C19,CYP2C9 и токи HERG и являются крайне цитотоксичными по сравнению с метансульфонатом сафинамида (Ic) и метансульфонатом ралфинамида (Id) с высокими степенями чистоты, содержащими менее 0,03 мас.% указанных выше примесей. Таблица 1 В табл. 2 представлены сравнительные результаты (IC50) ингибирования цитохрома CYP3A4 с применением образцов высокочистых метансульфоната сафинамида и ралфинамида, содержащих менее 0,03 мас.% указанных выше примесей по сравнению с теми же образцами высокочистых сафинамида и ралфинамида с дополнением 0,3 мас.% примесей (IIc) и (IId) соответственно. Когда к высокочистым метансульфонатам сафинамида и ралфинамида добавляют 0,3 мас.% примесей (IIc) и (IId), наблюдают значимое уменьшение IC50 для CYP3A4 в обоих случаях, что означает, что примеси способствуют сильному ингибированию ферментативной активности. Таблица 2 Как показано в табл. 3, примесь (IIc), начиная с 3 мг/кг в/в, увеличивает смертность в тесте максимального электрошока (MES) мышей без какой-либо фармакологической активности, т.е. защиты от судорог. В табл. 4 показано, что примесь (IId), при п/о введении при 10 и 20 мг/кг, в тесте максимального электрошока (MES) не защищает мышей от судорог по сравнению с теми же дозами метансульфоната ралфинамида. Таблица 4 На основе всех этих данных примеси (IIc), (II'с), (IId) и (II'd) и соответствующие рацемические смеси (IIc, II'с) и (IId, II'd), которые в нежелательных количествах присутствуют в сафинамиде, ралфинамиде, их R-изомерах и соответствующих рацемических смесях, соответственно, синтезируемых способом,описанным в WO 90/14334 и в Pevarello et al. в J. Med. Chem, 1998, 41, 579-590 или в WO 2006/027052,демонстрируют in vitro некоторые нежелательные свойства, такие как токсичность для клеток, сильное ингибирование некоторых изоформ CYP450, блокада каналов HERG и отсутствие защитной активности в модели эпилепсии "in vivo". Одним из важных свойств CYP является вариация в различных популяционных группах. Вариации в метаболизме лекарственных средств имеют большую важность в клинических исследованиях. Значительная вариация ферментативной активности CYP3A4 и CYP2D6 показана между различными этническими группами и даже между различными индивидуумами в одной этнической группе. Различия в активности CYP у индивидуумов значимо варьируют в зависимости от различных изоферментов. Изменения в уровне экспрессии CYP у различных индивидуумов могут являться причиной вариаций метаболизма лекарственных средств. Кроме того, полиморфизм также может приводить к вариантам ферментовCYP с большей или меньшей ферментативной активностью, что приводит к вариациям метаболизма лекарственных средств. Полиморфизм CYP2D6 представляет собой хорошо исследованную тему в области метаболизма лекарственных средств. В клинических исследованиях указанные вариации у индивидуумов впервые выявили в метаболизме антигипертензивных и противоэпилептических лекарственных средств. Элиминация метаболизируемых CYP2D6 лекарственных средств у индивидуумов, несущих дефектные аллели CYP2D6 происходит медленнее. Индивидуумов с медленным метаболизмом классифицируют как медленных метаболизаторов (РМ), тогда как каталитически компетентных индивидуумов классифицируют как быстрых метаболизаторов (ЕМ): частота фенотипа РМ в популяциях различного расового происхождения варьирует: приблизительно у 5-10% европеоидов присутствует фенотип РМ, тогда как у монголоидов он составляет только 1%. Другую важную полиморфную изоформу с клинической значимостью представляет собой CYP2C19. Учитывая эти наблюдения, у соединения, которое не воздействует на изоформы CYP450 (ни ингибируя, ни индуцируя), существует очень низкий риск взаимодействий вида лекарственное средстволекарственное средство в клинической практике и лечащие врачи могут его легко и безопасно выписывать. В частности, лекарственные средства, которые не воздействуют на цитохромы системы CYP450,особенно показаны для терапевтического лечения индивидуумов, классифицируемых как медленные метаболизаторы (РМ), или для терапевтического лечения пациентов, которые одновременно принимают другие лекарственные средства, для которых известно, что они воздействуют на указанные цитохромы,такие как кетоконазол, терфенадин, эритромицин, миконазол, пропанолол и хинидин, и/или для которых известно, что у них есть свойство блокировать каналы HERG. В соответствии с общей клинической практикой метансульфонаты сафинамида и ралфинамида (Ic) и (Id), как правило, вводят нуждающемуся в этом пациенту в течение длительного периода времени,подразделяя на несколько суточных доз. В частности, это происходит в случае терапевтического применения, где подлежащее лечению заболевание представляет собой болезнь Паркинсона, болезнь Альцгей-4 019898 мера и синдром беспокойных ног (для применения сафинамида) или хроническая или нейропатическая боль, сердечно-сосудистые или воспалительные нарушения (для применения ралфинамида). Хотя суточное дозирование может варьировать в зависимости от конкретного состояния и потребностей пациентов,суточное дозирование метансульфоната сафинамида, как правило, может варьировать от 10 мг/сутки до 800 мг/сутки, тогда как суточное дозирование метансульфонатов ралфинамида, как правило, может варьировать от 10 мг/сутки до 1 г/сутки. В этих условиях и учитывая представленные выше данные очень желательно сохранить уровень примесей (IIa) и (IIb) или их солей, в частности метансульфонатных солей(IIc) и (IId) в фармацевтических лекарственных формах сафинамида и ралфинамида или их солей, настолько низким, насколько возможно, в любом случае ниже 0,03%, предпочтительно ниже 0,01 мас.% по отношению к количеству соответственно сафинамида и ралфинамида или их солей, в частности метансульфонатных солей. Те же принципы применяют для R-энантиомеров сафинамида и ралфинамида (I'а) и (I'b), соответствующей рацемической смеси (Ia, I'а) и (Ib, I'b) и их солей с фармацевтически приемлемыми кислотами по отношению к соответствующим примесям (II'а), (II'b), соответствующим рацемическим смесям (IIa, II'а) и (IIb, II'b) и их солям с фармацевтически приемлемыми кислотами. Изучение и экспериментальные исследования, проведенные авторами изобретения, показали, что сафинамид, ралфинамид, соответствующие R-энантиомеры, соответствующие рацемические смеси или их соли с фармацевтически приемлемыми кислотами при получении способами, известными из уровня техники, содержат количество соответствующих примесей (IIa), (IIb), их R-энантиомеров (II'а) и (II'b),соответствующих рацемических смесей (IIa, II'а) и (IIb, II'b) или их солей с фармацевтически приемлемыми кислотами (таких как (IIc), (IId), (II'c) и (II'd), или соответствующих рацемических смесей (IIc, II'с) и (IId, II'd, которое составляет более 0,03 мас.%. Таким образом, указанные выше продукты непригодны для широкого и безопасного терапевтического применения. В частности, фармацевтические препараты,содержащие сафинамид, ралфинамид, соответствующий R-энантиомер (I'а) или (I'b), соответствующую рацемическую смесь (Ia, I'а) и (Ib, I'b) или их соли с фармацевтически приемлемыми кислотами, где содержание соответствующих примесей (IIa), (IIb), (II'a), (II'b), их рацемических смесей (IIa, II'а) и (IIb, II'b) или их солей с фармацевтически приемлемыми кислотами составляет не ниже 0,03%, предпочтительно 0,01 мас.% относительно указанных выше терапевтически активных веществ, непригодны для использования в качестве лекарственных средств в конкретных группах пациентов, как описано выше. В частности, фармацевтические препараты, содержащие сафинамид, ралфинамид, соответствующиеR-энантиомеры (I'а) или (I'b) или соответствующие рацемические смеси (Ia, I'а) и (Ib, I'b) или их соли с фармацевтически приемлемыми кислотами, где содержание соответствующих примесей (IIa), (IIb), (II'а),(II'b), соответствующих рацемических смесей (IIa, II'а) и (IIb, II'b) или их солей с фармацевтически приемлемыми кислотами не ниже 0,03%, предпочтительно 0,01 мас.% относительно указанных выше активных веществ, непригодны для использования для терапевтического лечения большого множества пациентов, включающих индивидуумов, классифицируемых в качестве медленных метаболизаторов (РМ),или которые одновременно принимают другие лекарственные средства, для которых известно, что они воздействуют на цитохромы системы CYP450. В данных описании и формуле изобретения значения указанных выше пределов, если не указано иначе, следует полагать как обозначающие процентное отношение от массы "активных веществ", т.е. эффективное содержание токсически активных примесей (IIa), (IIb), (II'a), (II'b) или соответствующих рацемических смесей (IIa, II'a) и (IIb, II'b) измеряют относительно эффективного содержания терапевтически активных веществ (Ia), (Ib), (I'a), (I'b) или соответствующих рацемических смесей (IIa, II'а) и (IIb,II'b). Такие выражения, как "высокая чистота", "высокая степень чистоты", "высокая химическая чистота", "высокочистый" и т.п., когда они в данных описании и формуле изобретения относятся к сафинамиду, ралфинамиду, соответствующим R-энантиомерам, соответствующим рацемическим смесям или их солям с фармацевтически приемлемыми кислотами, идентифицируют продукты, содержащие не менее 98,5% (оцениваемых как процент площади способами ВЭЖХ) сафинамида (Ia), ралфинамида (Ib), соответствующих R-энантиомеров (I'a) и (I'b), соответствующих рацемических смесей (Ia, I'a) и (Ib, I'b) или их солей с фармацевтически приемлемыми кислотами, где содержание соответствующей примеси (IIa),(IIb), (II'a), (II'b), соответствующих рацемических смесей (IIa, II'а) и (IIb, II'b) или их солей с фармацевтически приемлемыми кислотами составляет менее 0,03%, предпочтительно менее 0,01 мас.% (относительно "активных веществ"), определяемых способами ВЭЖХ. Другие примеси, с трудом детектируемые, происходят от очень малых количеств бензила,2- и 4-фторбензилхлорида и 3- и 4-фторбензилхлорида, которые содержатся в коммерчески доступном 3-фторбензилхлориде и 2-фторбензилхлориде соответственно, используемых для синтеза 4-(3-фторбензилокси)бензальдегида (IVa) и 4-(2-фторбензилокси)бензальдегида (IVb), промежуточных соединений для получения, соответственно, соединений (Ia), (Ib), (I'a), (I'b) (Ia, I'a) и (Ib, I'b) и их солей с фармацевтически приемлемыми кислотами. Аналогично, указанные выше термины "высокая чистота", "высокая степень чистоты","высокая химическая чистота","высокочистый",когда относятся к промежуточным 4-(3- или 2-фторбензилокси)бензальдегидам (IVa) и (IVb), идентифицируют продукты, содержащие не менее чем 98,5% (оцениваемых как процент площади способами GC) каждого из указанных выше соединений, где содержание соответствующей дибензилированной примеси (VIa) или (VIb) составляет менее 0,03%, предпочтительно менее 0,01 мас.% (оцениваемых способами GC). Описанный в настоящем изобретении способ посредством сильного уменьшения количества примесей обеспечивает продукты с высокой химической чистотой и более безопасным биологическим профилем. В соответствии со способом согласно настоящему изобретению, описанным ниже в разделе"Сущность изобретения", сафинамид, ралфинамид, соответствующие R-энантиомеры (I'а) и (I'b),соответствующие рацемические смеси (Ia, I'a) и (Ib, I'b) и их соли с фармацевтически приемлемыми кислотами, в частности с метансульфоновой кислотой, получают с высокими выходами и высокой чистотой, где содержание соответствующих примесей (S)-2-[3-(3-фторбензил)-4-(3 фторбензилокси)бензиламино]пропанамида(IIa),(S)-2-[3-(2-фторбензил)-4-(2 фторбензилокси)бензиламино]пропанамида (IIb), соответствующих R-энантиомеров (II'а) и (II'b), соответствующих рацемических смесей (IIa, II'а) и (IIb, II'b) и их солей с фармацевтически приемлемыми кислотами, в частности с метансульфоновой кислотой (обобщенно называемых "дибензиловыми производными"), составляет менее 0,03%, предпочтительно 0,01 мас.% относительно "активных веществ". Описанный в настоящем документе способ обеспечивает получение лекарственного средства, содержащего высокочистый (i) сафинамид и его R-энантиомер (I'а), их рацемическую смесь или их соль с фармацевтически приемлемой кислотой, предпочтительно метансульфоновой кислотой или (ii) ралфинамид и его R-энантиомер (I'b), их рацемическую смесь (IIa, II'а) и (IIb, II'b) или их соль с фармацевтически приемлемой кислотой, предпочтительно метансульфоновой кислотой для лечения соответственно(i) эпилепсии, болезни Паркинсона, болезни Альцгеймера, депрессии, синдрома беспокойных ног, боли и мигрени, или (ii) характеризующихся болью состояний, включающих хроническую и нейропатическую боль, мигрень, биполярные нарушения, депрессии, сердечно-сосудистые, воспалительные, мочеполовые,метаболические и желудочно-кишечные нарушения, в условиях, не препятствующих цитохромам системы CYP450, в частности CYP3A4, CYP2D6, CYP2C19, CYP2C9, и не проявляет свойств блокирования каналов HERG. Кроме того, описанный в настоящем документе способ обеспечивает получение лекарственного средства, содержащего высокочистый единственный R-энантиомер ралфинамида или его соль с фармацевтически приемлемой кислотой, предпочтительно метансульфоновой кислотой, для селективного (т.е. где терапевтическая активность активного вещества, которое вводят пациенту, по существу, не обладает каким-либо ингибирующим МАО побочным действием или демонстрирует значительно сниженное ингибирующее МАО побочное действие) лечения патологических аффектаций, где патологическую роль играет механизм(ы) натриевых и/или кальциевых каналов, которые описаны в WO 2006/027052 А 2, такие как боль, мигрень, воспалительные процессы, затрагивающие все системы организма, нарушения, затрагивающие кожу и родственные ткани, нарушения дыхательной системы, нарушения иммунной и эндокринной систем, желудочно-кишечные и мочеполовые нарушения, в условиях, не препятствующих цитохромам системы CYP450, в частности CYP3A4, CYP2D6, CYP2C19, CYP2C9 и не характеризуются свойствами блокирования каналов HERG. Таким образом, способ по данному изобретению обеспечивает получение фармацевтических композиций, содержащих сафинамид, его R-энантиомер (I'а), ралфинамид, его R-энантиомер (I'b), соответствующие рацемические смеси (Ia, I'а) и (Ib, I'b) или их соль с фармацевтически приемлемой кислотой,предпочтительно метансульфоновой кислотой, которые пригодны для лечения указанных выше нарушений у пациентов, которые классифицируют как медленные метаболизаторы (РМ) или для терапевтического лечения пациентов, одновременно принимающих другие лекарственные средства, для которых известно, что они воздействуют на цитохромы системы CYP450, и/или для которых известно, что они обладают свойствами блокирования каналов HERG. Указанные выше фармацевтические составы, кроме сафинамида, ралфинамида, соответствующихR-энантиомеров, соответствующих рацемических смесей или их солей с фармацевтически приемлемой кислотой, предпочтительно метансульфоновой кислотой, с описанной выше высокой степенью чистоты,необязательно могут содержать одно или несколько дополнительных активных средств. Например, фармацевтическая композиция, пригодная для дополнительного лечения болезни Паркинсона или синдрома беспокойных ног, в дополнение к сафинамиду, его R-энантиомеру, их рацемической смеси или их соли с фармацевтически приемлемой кислотой, предпочтительно метансульфоновой кислотой, полученным способом по данному изобретению и с указанной выше высокой степенью чистоты, может содержать одно или несколько дополнительных активных средств против болезни Паркинсона, таких как активные средства, описанные в WO 2004/089353 и WO 2005/102300, предпочтительно агонист дофамина и/или леводопа и/или ингибитор катехол-О-метилтрансферазы (СОМТ). В качестве дополнительного примера фармацевтическая композиция, пригодная для лечения характеризующихся болью состояний, включая хроническую боль и нейропатическую боль и мигрень, в дополнение к ралфинамиду, его R-энантиомеру, их рацемической смеси или их соли с фармацевтически приемлемой кислотой, предпочтительно метансульфоновой кислотой, полученным способом по данному изобретению и с указанной выше высокой степенью чистоты, может необязательно содержать дополнительное активное средство, такое как габапентин и прегабалин или их фармацевтически приемлемую соль, как описано в ЕР 1423168. Аналогично, фармацевтическая композиция, пригодная в качестве лекарственных средств, селективно активных в качестве модуляторов натриевых и/или кальциевых каналов для селективного лечения патологических аффектаций, где патологическую(ие) роль(и) играет(ют) механизм(ы) натриевых и/или кальциевых каналов в соответствии с WO 2006/027052 А 2, таких как боль, мигрень, воспалительные процессы, затрагивающие все системы организма, нарушения, затрагивающие кожу и родственные ткани, нарушения дыхательной системы, нарушения иммунной и эндокринной систем, желудочнокишечные и мочеполовые нарушения, может необязательно содержать дополнительное активное средство. Например, фармацевтическая композиция для лечения характеризующихся болью состояний в дополнение к одному из R-энантиомеров ралфинамида (I'b) или его соли с фармацевтически приемлемой кислотой, предпочтительно метансульфоновой кислотой, полученным способом по данному изобретению и с указанной выше высокой степенью чистоты, может содержать габапентин или родственное габапентину средство. Фармацевтические композиции, содержащие полученные способом по настоящему изобретению сафинамид, ралфинамид, соответствующие R-энантиомеры, соответствующие рацемические смеси или их соли с фармацевтически приемлемыми кислотами высокой степени чистоты, можно получать стандартными способами, известными в данной области, например смешивая активные соединения с веществами фармацевтически, терапевтически инертных органических и/или неорганических носителей. Композиции могут находиться в жидкой форме, например в форме раствора, суспензии, эмульсии; или в твердой форме, например таблетки, пастилки, капсулы, пластыри. Подходящие вещества фармацевтически, терапевтически инертных органических и/или неорганических носителей, пригодные для получения таких композиций, включают, например, воду, желатин,гуммиарабик, лактозу, крахмал, целлюлозу, стеарат магния, тальк, растительные масла, полиалкиленгликоли, циклодекстрины и т.п. Фармацевтические композиции по изобретению можно стерилизовать и они, кроме активного ингредиента(ов), могут содержать дополнительные компоненты, хорошо известные специалистам в данной области, такие как, например, консерванты, стабилизаторы, увлажнители или эмульгаторы, например парафиновое масло, моноолеат маннида, соли для регулировки осмотического давления, буферы и т.п. Известный уровень техники В WO 90/14334, в статье Pevarello et al. in J. Med. Chem., 1998, 41, 579-590 описан двухстадийный способ получения бензилоксибензиламиноалканамидов:b) восстановительное алкилирование -аминоамидов 4-бензилоксибензальдегидами с применением цианоборгидрида натрия или боргидрида натрия в качестве восстановителя, как здесь схематически показано нижеR3, кроме других заместителей, представляет собой СН 3;R4 и R5, кроме других заместителей, представляют собой водород. В частности, поскольку рассматривается получение сафинамида и ралфинамида, восстановительное алкилирование представляет собой восстановительное алкилирование В J. Med. Chem. (Pevarello et al.), 1998, 41, 579-590 сообщалось о выходах в размере 45 и 60% для получения сафинамида и метансульфоната ралфинамида соответственно, начиная от соответствующих Способ, описанный в WO 90/14334 и в процитированной выше статье, является таким же и позволяет однореакторную систему, где иминоалкилирование и восстановление проводят в одном реакторе. К смеси гидрохлорида L-аланинамида, цианоборгидрида натрия, метанола и порошковых молекулярных сит сразу добавляют подходящий альдегид. В соответствии с Pevarello et al., в Org. Prep. Proc. Int. 1996, 28, 179-183 (где описан синтез некоторых производных -бензиламиноамида посредством восстановительного алкилирования) применение-аминоамида в качестве гидрохлорида важно для образования иминиевого иона вместо соответствующего имина, так как иминиевый ион более легко реагирует с цианоборгидридом натрия, чем с карбонильной группой альдегида. В соответствии с утверждением указанных выше авторов, по-видимому, однореакторный способ позволяет избежать проблем с рацемизацией оснований Шиффа, а молекулярные сита ускоряют реакцию(хотя выходы ухудшаются). Цианоборгидрид заявлен как предпочтительное используемое средство, и, по-видимому, этот его выбор сделан вследствие его селективности (см. обзор "Sodium Cyanoborohydride - A Highly SelectiveReducing Agent for Organic Functional Groups". C.F. Lane, Synthesis, 1975, 132-146), что делает его способным различить протонированное основание Шиффа и исходный альдегид. Синтез, описанный в статье Pevarello et al., позволяет выделение продуктов посредством колоночной хроматографии с последующим преобразованием в соответствующие соли посредством обработки кислотами. Информации о энантиомерной и/или химической чистоте сафинамида и ралфинамида и/или их солей не предоставлено. Способ, описанный в современном уровне техники, страдает от многих недостатков, которые ограничивают его применение в большом масштабе; ниже перечислены некоторые примеры указанных недостатков: образование цианидов и цианопроизводных; использование порошковых молекулярных сит, которые физически изменчивы и являются дорогостоящими; выходы, как правило, составляют менее 70%; продукты реакции с низкой чистотой и трудны для очистки; применение больших количеств растворителя (приблизительно от 5 до 7 л на 1 моль), используемого в реакции восстановительного алкилирования, приводящее в низкой конечной концентрации продукта в конечной реакционной смеси (приблизительно 4-6% мас./об.); выделение продукта реакции посредством колоночной хроматографии, которую рассматривают как сложный и дорогостоящий способ выделения в крупномасштабном получении активных средств с применением химического синтеза. Способ получения (R)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (I'b), описанный вWO 2006/027052 А 2, основан на восстановлении боргидридом натрия продукта, являющегося результатом реакции гидрохлорида (R)-аланинамида с 4-(2-фторбензилокси)бензальдегидом (данных относительно степени чистоты данного реагента не приведено) и триэтиламином в сухом метаноле в присутствии молекулярных сит в течение 4 ч. Данных относительно чистоты получаемого конечного соединения не приведено. Также в этом случае недостатками способа, когда его применяют для широкомасштабного получения, являются использование порошковых или гранулированных молекулярных сит, использование больших количеств растворителя и, несмотря на операции очистки, присутствие в конечном продукте (I'b) нежелательной примеси (II'b) в количестве более 0,03 мас.%, что делает активное вещество (I'b),полученное указанным способом, непригодным для терапевтического применения без риска или с низким риском побочных эффектов, вследствие воздействия на цитохромы системы CYP450. Низкая степень чистоты и низкие выходы (30-32 мол.%) конечного продукта способа, описанного вWO 2006/027052, показаны посредством нескольких воспроизводств в различном масштабе описанного в публикации способа, репрезентативный пример которого описан в примере 23.3. Одной из принципиальных характеристик, отличающих способ, описанный в WO 2006/027052, от способа по данному изобретению, и которая, как выявлено, отвечает за необычайно низкие выходы способа на указанном известном уровне техники, состоит в том, что количество органического растворителя(метанола), который используют в указанном способе, относительно молярного количество основания Шиффа составляет приблизительно 5 л на 1 моль основания Шиффа. Выявлено, что эти условия вызывают увеличение нежелательных примесей в конечном продукте, получаемых из молекул, вовлеченных в равновесие между основанием Шиффа и его предшественниками, такими как тот же исходный альдегид,его ацетали и аминоацетали. Иллюстративные примеры, следующие за данным описанием, подтверждают, что продукты, получаемые способами, описанными на современном уровне техники, содержат такое количество примесей(IIa), (IIb), (IIc), (IId), (II'а), (II'b), (II'c), (II'd), (IIa, II'а), (IIb, II'b), (IIc, II'с) или (IId, II'd), которое выше 0,03 мас.% относительно соответствующих терапевтически активных веществ (Ia), (Ib), (Ic), (Id), (I'a),(I'b), (I'c), (I'd), (Ia, I'a), (Ib, I'b), (Ic, I'c) или (Id, I'd). Кроме того, показано, что указанные примеси, при-8 019898 сутствующие в конечном продукте сафинамида, ралфинамида, соответствующих R-энантиомеров, соответствующих рацемических смесях или их солях с фармацевтически приемлемыми кислотами, с применением общеизвестных способов очистки на основе кристаллизации из растворов или хроматографии,которые в любом случае приводят к уменьшению выходов, устранить трудно. Сущность изобретения Целью данного изобретения является способ получения соединения 2-[4-(3- или 2-фторбензилокси)бензиламино]пропанамида высокой степени чистоты, выбранного из соответствующих R-энантиомеров (I'a) и (I'b), соответствующих рацемических смесей (Ia, I'a) и (Ib, I'b) и их солей с фармацевтически приемлемыми кислотами, где сафинамид, ралфинамид, соответствующийR-энантиомер (I'a) или (I'b) или соответствующие рацемические смеси (Ia, I'a) и (Ib, I'b) или их соль с фармацевтически приемлемой кислотой имеют содержание соответствующей примеси,(S)-2-[3-(3-фторбензил)-4-(3-фторбензилокси)бензиламино]пропанамида (IIa), (S)-2-[3-(2-фторбензил)-4(2-фторбензилокси)бензиламино]пропанамида (IIb): соответствующих R-энантиомеров (II'а) или (II'b) или соответствующих рацемических смесей (IIa, II'а) или (IIb, II'b) или их солей с фармацевтически приемлемой кислотой, которое составляет менее 0,03%,предпочтительно менее 0,01 мас.%, характеризующийся тем, что промежуточные основания Шиффа формулы (IIIa), (IIIb): соответствующие R-энантиомеры (III'а) или (III'b) или соответствующие рацемические смеси (IIIa, III'а) или (IIIb, III'b), получаемые реакцией иминоалкилирования 4-(3- или 2-фторбензилокси)бензальдегидаL-аланинамидом или D-аланинамидом или их рацемической смесью в растворителе, выбранном из(C1-C5)алканолов, в количестве, которое обеспечивает образование суспензии основания Шиффа в насыщенном растворе основания Шиффа в том же растворителе при температуре от 20 до 30 С, после завершения реакции иминоалкилирования подвергают реакции восстановления с восстановителем, выбранным из боргидрида натрия и боргидрида калия, в выбранном количестве органического растворителя, выбранного из низших (C1-C5)алканолов или их смеси, необязательно с небольшим количеством воды, где отношение органического растворителя к основанию Шиффа обеспечивает образование и присутствие в ходе существенной части реакции восстановления суспензии основания Шиффа в насыщенном растворе основания Шиффа в том же органическом растворителе и составляет от 0,5 до 3,0 л, предпочтительно от 0,7 до 2,5 л, наиболее предпочтительно от 0,8 до 2,0 л на каждый моль основания/Шиффа, причем способ дополнительно характеризуется тем, что исходное вещество 4-(3- или 2-фторбензилокси)бензальдегид, применяемое для получения промежуточных оснований Шиффа (IIIa), (IIIb), (III'а), (III'b) или соответствующих рацемических смесей (IIIa, III'а) или (IIIb, III'b) имеет содержание примеси 3-(3- или 2-фторбензил)-4-(3- или 2-фторбензилокси)бензальдегида менее 0,03%, предпочтительно менее 0,01 мас.%, в результате чего сафинамид, ралфинамид, соответствующиеR-энантиомеры (I'а) или (I'b) или соответствующие рацемические смеси (Ia, I'а) или (Ib, I'b) получают в форме свободного основания и, необязательно, преобразуют указанную форму свободного основания в ее соль с фармацевтически приемлемой кислотой. Формулы (IIIa) и (IIIb), как представлено в данном описании и формуле изобретения, идентифицируют промежуточное основание Шиффа в Е- и в Z-конфигурации. Настоящее изобретение относится также к выделенным промежуточным основаниям Шиффа формулы (III'а) и (III'b). По предпочтительному варианту осуществления данного изобретения способ по настоящему изобретению включает три следующие стадии: а) получение высокочистого исходного вещества, представляющего собой исходное вещество 4-(3- или 2-фторбензилокси)бензальдегид посредством О-бензилирования 4-гидроксибензальдегида производными следующей общей формулы 3- или 2-F-C6H4-CH2-Y (Va) или (Vb), где Y представляет собой уходящую группу (Cl, Br, I, OSO2CH3 и т.д.); где данное O-бензилирование проводят в условиях, которые высокоселективны для О-алкилирования и оно приводит к получению 4-(3 фторбензилокси)бензальдегида или 4-(2-фторбензилокси)бензальдегида высокой чистоты;b) завершение образования промежуточного основания Шиффа конденсацией исходного вещества 4-(3- или 2-фторбензилокси)бензальдегида с L-аланинамидом, D-аланинамидом или их рацемической смесью в форме основания или соли без какого-либо использования молекулярных сит;c) обработка основания Шиффа восстановительной системой, выбранной из боргидрида натрия и боргидрида калия в присутствии органического растворителя, выбранного из (С 1-С 5)алканолов в соответствующем отношении с основанием Шиффа, обеспечивая одновременное присутствие основания Шиффа в твердой форме и форме насыщенного раствора основания Шиффа в указанном растворителе(т.е. суспензии основания Шиффа в насыщенном растворе основания Шиффа в указанном органическом растворителе) в ходе существенной части реакции восстановления для получения после выделения продукта реакции и кристаллизации, соответственно, сафинамида, ралфинамида, соответствующихR-энантиомеров или соответствующей рацемической смеси с очень высоким выходом и с определенной выше химической чистотой, и, необязательно, получение их солей с фармацевтически приемлемыми кислотами известными способами образования солей. Фармацевтически приемлемые кислоты выбирают, например, из азотной, соляной, бромистоводородной, серной, хлорной, фосфорной, метансульфоновой, п-толуолсульфоновой, уксусной, трифторуксусной, пропионовой, гликолевой, молочной, щавелевой, малоновой, яблочной, малеиновой, винной,лимонной, бензойной, коричной, миндальной и салициловой кислот. Синтез исходных веществ 4-(3- или 2-фторбензилокси)бензальдегидов В соответствии с известными способами исходные вещества (фторбензилокси)бензальдегидов, необходимые для получения промежуточных оснований Шиффа (IIIa), (IIIb), (III'а), (III'b) и соответствующих рацемических смесей (IIIa, III'а) и (IIIb, III'b), которые применяют для синтеза, соответственно, сафинамида, ралфинамида, соответствующих R-энантиомеров и соответствующих рацемических смесей по данному изобретению, получают бензилированием 4-гидроксибензальдегида в основной среде. Бензилирование феноловых солей, представляющих собой амбидентные нуклеофилы, приводит к образованию двух разных продуктов, т.е. желаемых О-алкилированных производных и нежелаемых С-алкилированных производных. По существу, выявлено, что фторбензилирование 4-гидроксибензальдегида 3-фторбензилхлоридом,проводимое в соответствии с известным уровнем техники, приводит к образованию 4-(3-фторбензилокси)бензальдегида (IVa) в качестве основного продукта совместно с 3-(3-фторбензил)4-(3-фторбензилокси)бензальдегидом (VIa), который образуется вследствие алкилирования гидроксигруппы в положении 4 и атома углерода в положении 3 4-гидроксибензальдегида. То же самое происходит при фторбензилировании 4-гидроксибензальдегида 2-фторбензилхлоридом по следующей схеме: Восстановление основания Шиффа,образуемого иминоалкилированием 4-(3- или 2-фторбензилокси)бензальдегида L- или D-аланинамидом или их рацемической смесью с исходным веществом альдегидом, содержащим диалкилированную примесь, приводит к образованию конечного продукта формулы (Ia), (Ib), (I'a), (I'b) или соответствующей рацемической смеси (Ia, I'a) или (Ib,I'b), которые также загрязнены соответствующим диалкилированным соединением, дибензиловым производным, (IIa), (IIb), (II'a), (II'b) или соответствующей рацемической смесью (IIa, II'а) или (IIb, II'b), в виде свободного основания или соединения в виде соли, предпочтительно с метансульфоновой кислотой,(IIc), (IId), (II'c), (II'd) или соответствующей рацемической смеси (IIc, II'с) или (IId, II'd), как показано в следующей схеме, демонстрирующей образование дибензилатных примесей (IIc) и (IId), относящихся соответственно к сафинамиду и ралфинамиду: Аналогичным способом получают соответствующий R-энантиомер (II'c), (II'd) и соответствующие рацемические смеси. Вместо предпочтительной метансульфоновой кислоты можно использовать фармацевтически приемлемые кислоты, например азотную, соляную, бромисто-водородную, серную, хлорную,фосфорную, метансульфоновую, п-толуолсульфоновую, уксусную, трифторуксусную, пропионовую,гликолевую, молочную, щавелевую, малоновую, яблочную, малеиновую, винную, лимонную, бензойную, коричную, миндальную и салициловую кислоты. Моноалкилированное производное (сафинамид, ралфинамид, соответствующие R-энантиомеры и соответствующие рацемические смеси) и соответствующие диалкилированные примеси имеют сходные химико-физические свойства, и это делает трудной очистку сафинамида и ралфинамида традиционными способами. Кроме того, известные способы бензилирования, и среди них фторбензилирование, страдают от следующих дополнительных недостатков: 1) использование в качестве растворителя низшего спирта; в основных условиях растворитель, например метанол, может сам действовать в качестве нуклеофильного реагента и приводить к образованию с 3- или 2-фторбензилхлоридом некоторого количества простого метилфторбензилового эфира; 2) экстракция конечного продукта водой-несмешивающимся органическим растворителем возможна только после удаления спиртового реакционного растворителя из реакционной смеси. Выявлено, что, используя указанные выше способы по известному уровню техники, для получения конечного продукта формулы (Ia), (Ib), (I'a), (I'b) или соответствующей рацемической смеси (Ia, I'b) или(Ib, I'b), где содержание примеси (IIa), (IIb), (II'a), (II'b) или соответствующей рацемической смеси (IIa,II'а) или (IIb, II'b) составляет менее 0,03 мас.%, необходимо существенно очистить промежуточный 4-(3-фторбензилокси)бензальдегид (IVa) или 4-(2-фторбензилокси)бензальдегид (IVb) для уменьшения содержания соответствующих примесей формулы (VIa) и (VIb). Указанную очистку предпочтительно проводят, подвергая продукты реакции кристаллизации, более предпочтительно добавляя к раствору неочищенного соединения (IVa) или (IVb) в инертном органическом растворителе смешивающийся инертный органический осадитель. Органический инертный растворитель предпочтительно выбирают из ароматических углеводородов, а более предпочтительно он представляет собой толуол. Смешивающийся инертный органический осадитель предпочтительно выбирают из низших алифатических углеводородов, более предпочтительно он представляет собой н-гексан. Дополнительный способ кристаллизации может состоять из растворения указанных выше соединений(IVa) или (IVb) в горячем растворителе, например циклогексане или простом ди(С 3-С 4)алкиловом эфире,таком как простой диизопропиловый эфир при температуре кипения с обратным холодильником, а затем охлаждения раствора до комнатной температуры, предпочтительно при 10-15 С, наиболее предпочтительно индуцируя кристаллизацию добавлением чистых кристаллов чистого соединения (IVa) или (IVb). По одному из аспектов настоящего изобретения исходное вещество 4-(3- или 2-фторбензилокси)бензальдегид, необходимое для получения промежуточных оснований Шиффа, получают реакцией между алкилирующим средством формулы (Va) или (Vb) (см. схему ниже, где в положении 2 или 3 находится атом F, a Y представляет собой уходящую группу,такую как, например, Cl, Br, I, OSO2CH3, OSO2C6H4-pCH3 и т.д.) и 4-гидроксибензальдегидом, которую проводят в условиях межфазного катализа. В указанных условиях соответствующие 4-(3- или 2-фторбензилокси)бензальдегиды получают с высокими выходами и с очень низкими уровнями Данное новое фторбензилирование 4-гидроксибензальдегида в условиях межфазного катализа можно осуществлять в системе твердое вещество/жидкость, где в жидкой органической фазе растворены реа- 11019898 генты и катализатор фазового переноса, а твердая фаза состоит из неорганического основания или соли 4-гидроксибензальдегида (возможно образующейся in situ из 4-гидроксибензальдегида и самого неорганического основания), и в системе жидкость/жидкость органическая/водная, где неорганическое основание растворяют в водной фазе. Предпочтительная система представляет собой систему твердое вещество/жидкость, где неорганическое основание предпочтительно выбирают из Na2CO3, K2CO3, KOH и NaOH. Органические растворители, используемые в реакции, и в случае системы жидкость/жидкость, и в случае системы твердое вещество/жидкость, могут представлять собой простые диалкиловые эфиры,такие как, например, простой ди-трет-бутиловый эфир, простой этил-трет-бутиловый эфир, или ароматические углеводороды, такие как, например, толуол, этилбензол, изопропилбензол и ксилолы. Все эти растворители можно легко восстановить посредством дистилляции. Применяемые катализаторы фазового переноса могут представлять собой четвертичный аммоний или соли фосфония, такие как, например, бромид тетрабутиламмония, бромид тетрадецилтриметиламмония, бромид гексадецилтрибутилфосфония, хлорид трикаприлилметиламмония (аликват), хлорид метилтри(С 8-С 10)алкиламмония (адоген), где предпочтительным является бромид тетрадецилтриметиламмония. Также в качестве катализаторов фазового переноса можно использовать полиэтиленгликоли низкой молекулярной массы, такие как, например, PEG-200 (CAS 25322-68-3) или PEG-400 (CAS 25322-68-3). Используемое количество катализатора фазового переноса составляет 0,02-1 моль на 1 моль 4-гидроксибензальдегида, предпочтительно 0,1-1 моль на 1 моль 4-гидроксибензальдегида, так как в этих условиях количество C,O-бис-фторбензилированных примесей в результате может составить менее 0,03%, предпочтительно равным 0,01 мас.% или менее. Отношение между алкилирующими средствами формулы (V) и 4-гидроксибензальдегидом составляет от 0,6 до 1,5, предпочтительно от 0,9 до 1,1. Температура реакции составляет от 60 до 160 С, предпочтительно от 80 до 120 С. Время реакции составляет, как правило, от 4 до 8 ч. Выходы реакции являются очень большими, как правило, более 90%. Эффективность реакции, т.е. концентрация продуктов реакции в реакционной смеси, в описанных реакционных условиях является очень высокой, как правило, она больше или равна 25% (мас./об.). Синтез сафинамида и ралфинамида их R-энантиомеров и соответствующих рацемических смесей посредством восстановления оснований шиффа, формируемых реакцией 4-(3- или 2-бензилокси)бензальдегида с L-аланинамидом или D-аланинамидом или их рацемическими смесями и их солями Способ, цель настоящего изобретения, включает две стадии:a) законченное образование промежуточного основания Шиффа;b) восстановление основания Шиффа восстановителем, выбранным из боргидрида натрия и боргидрида калия. Эти две стадии можно проводить последовательно в одном и том же реакторе (однореакторная реакция) с выделением или без выделения основания Шиффа, в обоих случаях с высокими выходами. Образование промежуточных оснований Шиффа включает конденсацию 4-(3- или 2-фторбензилокси)бензальдегида с L-аланинамидом, D-аланинамидом или их рацемической смесью или их солью с кислотой ("аланинамидное соединение"), предпочтительно неорганической кислотой, такой как соляная, бромисто-водородная, серная, метансульфоновая кислоты и т.д. Рацемическую смесь 4-(3- или 2-бензилокси)бензиламинопропанамида получают, когда вместо L- или Dэнантиомера аланинамида применяют их рацемическую смесь. В случае выделения основания Шиффа экспериментальные условия, используемые для его образования, позволяют получать выделенное основание Шиффа в форме осадка с высокими выходами и в очень чистой форме. Получение основания Шиффа надлежащим образом проводят в органическом протонном растворителе, который должен быть инертным относительно реагентов и продуктов, а также быть инертным относительно условий восстановления иминной двойной связи. Если желательно проводить последующую стадию восстановления в той же реакционной среде, подходящими растворителями являются, например,низшие (С 1-С 5)алканолы, предпочтительно метанол, этанол и изопропанол. Образование промежуточного основания Шиффа должно быть полным и это представляет собой важный фактор для получения высоких выходов в последующей стадии восстановления. В соответствии со способом осуществления по данному изобретению перед проведением восстановления иминной двойной связи выделяют промежуточное основание Шиффа (IIIa) или (IIIb) соответствующий R-энантиомер (III'а) или (III'b) или соответствующую рацемическую смесь, являющиеся результатом реакции конденсации между 4-(3- или 2-фторбензилокси)бензальдегидом иL-аланинамидом, D-аланинамидом или их рацемической смесью. Альтернативно, способствовать завершению реакции иминоалкилирования можно, действуя в таких условиях, чтобы вызвать осаждение промежуточных иминосоединений (IIIa), (IIIb), соответствующих R-энантиомеров (III'а) или (III'b) или соответствующей рацемической смеси (IIIa, III'а) или (IIIb,III'b), в реакционном растворителе, а затем подвергнуть суспензию, содержащую указанное промежуточное иминопроизводное, стадии восстановления. Отношение между L-аланинамидом, D-аланинамидом или их рацемической смесью (основанием или солью) и 4-(3- или 2-фторбензилокси)бензальдегидом может составлять 1:1, но также предпочтительно можно использовать 10% избыток аланинамидного соединения. Аланинамидное соединение можно добавлять или в виде свободного основания, или в виде его соли присоединения кислоты. Предпочтительно его добавляют в реакционную смесь в виде соли, наиболее предпочтительно в виде гидрохлорида, вместе со стехиометрическим количеством основания, предпочтительно третичного амина, такого как, например, триэтиламин или диизопропилэтиламин. Температура реакции при получении основания Шиффа составляет от 0 до 60 С, предпочтительно от 20 до 30 С. Время реакции, как правило, составляет от 1 до 15 ч, предпочтительно от 2 до 6 ч. В определенных условиях, когда D- или L-аланинамид используют в виде свободного основания и время реакции иминоалкилирования превышает 8 ч, получаемое в результате основание Шиффа может претерпевать рацемизацию по хиральному центру. Это в особенности верно, когда основание Шиффа не кристаллизуется в ходе реакции иминоалкилирования. Восстановление основания Шиффа восстановителем, выбранным из боргидрида натрия и боргидрида калия, начинают только тогда, когда завершается образование основания Шиффа: если оно начинается до этого, важность, иногда превалируя, приобретают вторичные реакции с потерей в выходе и чистоте. Одна из этих вторичных реакций, наиболее важная, вызывает образование бензиловых спиртов посредством восстановления карбонильной группы выбранного (фторбензилокси)бензальдегида. Завершение образования основания Шиффа можно контролировать известными в данной области аналитическими способами, например посредством анализа количественной GC исходного раствора. Восстановление основания Шиффа представляет собой наиболее важную стадию способа по данному изобретению и его проведение требует определенных условий. Восстановитель боргидрид натрий или калия применяют в молекулярном количестве, которое относительно основания Шиффа находится в диапазоне от 0,5 до 1,4. Применение боргидрида натрия является предпочтительным ввиду его коммерческой доступности и стоимости. Как правило, реакцию проводят в растворителе, который может представлять собой тот же растворитель, в котором после завершения реакции конденсации с аланинамидом основание Шиффа находится в форме суспензии. В таком случае в качестве реакционного растворителя, как правило, применяют низший (С 1-С 5)алканол, такой как метанол, этанол, 1-пропанол и 2-пропанол, предпочтительно метанол. Альтернативно, когда основание Шиффа выделяют из реакционной среды (например, посредством фильтрации или центрифугирования) продукт в виде выделенного основания Шиффа добавляют к выбранному количеству органического растворителя, предпочтительно протонного органического растворителя, такого как низший (С 1-С 5)алканол, предпочтительно метанол, или смеси указанного протонного органического растворителя, необязательно в присутствии небольшого количества воды (предпочтительно, менее 1,5 мас.% относительно количества органического растворителя). Если реакцию конденсации 4-(3- или 2-фторбензилокси)бензальдегида с аланинамидным соединением проводят, добавляя данное последнее вещество в реакционную смесь в виде соли с кислотой, тогда для доведения рН до значения от 7 до 9 добавляют соответствующее количество основания, такого как гидроксид натрия или калия, третичные (С 1-С 4)алкиламины, пирролидин, 4-метилморфолин и т.п. Если в конце реакции иминоалкилирования значение рН реакционной смеси снижается ниже этого интервала, в реакционную смесь, содержащую основание Шиффа, дополнительно добавляют соответствующее количество указанного выше основания, для повторного доведения рН до указанного значения перед любым добавлением восстановителя боргидрида натрия или калия. Восстановитель боргидрид натрия или калия в ходе реакции, как правило, добавляют к смеси основания Шиффа и реакционного растворителя несколькими отдельными частями (как правило, от 15 до 20 частей) в твердой форме, такой как порошок или мелкоизмельченные кристаллы, в контролируемых условиях. Альтернативно, боргидрид натрия или калия добавляют в реакционную смесь частями или по каплям в форме раствора в метаноле, стабилизированного добавлением гидроксида натрия (как правило,приблизительно 15 мас.% гидроксида натрия относительно боргидрида натрия) или гидроксида калия. В соответствии с предпочтительным способом проведения восстановления основания Шиффа стабилизированный раствор боргидрида натрия или калия в метаноле к реакционной смеси, содержащей основание Шиффа и выбранное количество реакционного растворителя, предпочтительно метанола, добавляют частями в количестве от 15 до 25 или по каплям в течение от 1 до 2 ч. Для осуществления стадии восстановления в условиях, где отношение выбранного растворителя к основанию Шиффа обеспечивает одновременное присутствие насыщенного раствора основания Шиффа в указанном растворителе и основания Шиффа в твердой форме, где количество основания Шиффа вне фазы растворителя максимизировано, следует соответствующим образом выбрать количество используемого растворителя. Таким образом, общее количество органического растворителя, используемого на стадии восстановления, может находиться в диапазоне от 0,5 до 3,0 л, предпочтительно от 0,7 до 2,5 л, наиболее предпочтительно от 0,8 до 2,0 л на каждый моль основания Шиффа. В этих условиях значительная часть основания Шиффа, присутствующего в реакционной среде, проходящей стадию восстановления, в ходе существенной части реакции находится в форме твердого вещества. В этих условиях выход конечного продукта является очень высоким и это имеет положительное экономическое влияние на промышленное производство. рН реакционной смеси, в которой проводят стадию восстановления, когда конденсацию между альдегидом и производным аланинамида проводят с его солью, доводят до значения от 7 до 9, предпочтительно от 8 до 8,5 (определяемого непосредственно в реакционной смеси посредством рН-метра), добавляя, если необходимо, соответствующее количество основания, такого как гидроксид натрия или калия,третичные (С 1-С 4)алкиламины, пирролидин, 4-метилморфолин и т.п. Температуру реакции в ходе стадии восстановления поддерживают от -10 до 30 С, предпочтительно от 5 до 15 С. Время восстановления может варьировать от 0,5 до 5 ч, в зависимости от используемого растворителя, температуры, концентрации и т.д., где все факторы хорошо известны специалистам в данной области. Лучшие результаты получают при времени реакции приблизительно 3 ч с применением в качестве восстановителя боргидрида натрия, в качестве растворителя метанола в соотношении от 0,8 до 2,0 л на каждый моль количества основания Шиффа при температуре от 5 до 10 С. В конце реакции реакционный растворитель отгоняют при пониженном давлении, остаток растворяют в водонерастворимом органическом растворителе, а неорганические соли удаляют посредством отмывки водой. Конечный неочищенный продукт, т.е. сафинамид, ралфинамид, соответствующий R-энантиомер или соответствующую рацемическую смесь, восстанавливают, удаляя посредством отгонки органический растворитель, где растворен продукт реакции. Затем неочищенный сафинамид, ралфинамид, соответствующие R-энантиомеры или соответствующую рацемическую смесь очищают посредством кристаллизации. Кристаллизацию можно проводить, добавляя к раствору соответствующего неочищенного соединения формулы (Ia), (Ib), (I'a), (I'b), (Ia,I'a) или (Ib, I'b) в инертном органическом растворителе смешивающийся инертный органический осадитель. Органический инертный растворитель предпочтительно выбирают из ароматических углеводородов, таких как бензол, толуол, диметилбензол и этилбензол и ацетаты низших алкилов, а более предпочтительно он представляет собой этилацетат. Смешивающийся инертный органический осадитель предпочтительно выбирают из низших алифатических углеводородов, таких как гексан и гептан и циклогексан, более предпочтительно он представляет собой н-гексан. Альтернативно кристаллизацию проводят, растворяя конечный неочищенный продукт в горячем органическом растворителе, предпочтительно толуоле или циклогексане, а затем охлаждая раствор при комнатной температуре и восстанавливая чистый продукт посредством фильтрации. Затем основания преобразуют в желаемые соли известными способами, в частности их преобразуют в метансульфонатную соль, физические/химические свойства которой (стабильность, гранулометрические свойства, текучесть и т.д.) подходят для последующего составления в фармацевтический препарат для применения в качестве лекарственного средства. Пример 1. Получение очищенного 4-(2-фторбензилокси)бензальдегида (IVb) посредством межфазного катализа. Смесь 2-фторбензилхлорида (6 кг, 41,50 моль), 4-гидроксибензальдегида (4,7 кг, 38,33 моль), карбоната калия (5,2 кг, 37,33 моль) и бромида тетрадецилтриметиламмония (0,49 кг, 1,46 моль) в толуоле(11,4 кг) при перемешивании и в атмосфере азота медленно доводят до температуры кипения с обратным холодильником и кипятят с обратным холодильником в течение 6 ч. Затем раствор концентрируют при атмосферном давлении, добавляют 3,6 кг толуола и отгоняют и эту процедуру повторяют еще раз. Затем гетерогенную смесь охлаждают до комнатной температуры и твердое вещество удаляют посредством фильтрации. Затем остаточный растворитель удаляют при пониженном давлении и к масляному остатку добавляют 1,4 кг толуола. Смесь нагревают приблизительно до 30-35 С и осаждают несколькими граммами чистого 4-(2-фторбензилокси)бензальдегида. Гетерогенную смесь перемешивают в течение 30 мин при 30-35 С, а затем к смеси, поддерживаемой при перемешивании при 30-35 С, добавляют н-гексан (11 кг) в течение 30 мин. После охлаждения до 0-5 С и перемешивания в течение дополнительного 1 ч при данной температуре твердое вещество собирают посредством фильтрации и высушивают при пониженном давлении с получением 8 кг (89% выход) 4-(2-фторбензилокси)бензальдегида; т.плавл. 56,7 С (DSC, 5 С/мин) с чистотой по GC 98,2 (% площади, см. пример 24 А) и содержанием 3-(2-фторбензил)-4-(2 фторбензилокси)бензальдегида (VIb), определенным посредством GC (см. пример 24 В), 0,01 мас.%. Выходы, приведенные в этом и следующих примерах, если не указано иначе, приведены в виде молярных выходов. 1.1. Дополнительная очистка 4-(2-фторбензилокси)бензальдегида (IVb) посредством кристаллизации. 1 кг продукта, полученного в соответствии со способом, описанным в примере 1, растворяют в 2 кг простого диизопропилового эфира при температуре кипения с обратным холодильником при перемешивании. Раствор охлаждают до 50-55 С в течение 10-15 мин и осаждают с 5 г высокочистого 4-(2-фторбензилокси)бензальдегида (чистота по GC 99,9% площади; см. пример 24 А, и содержание (VIb) менее 0,005%). Суспензию охлаждают до 10-15 С в течение 45-60 мин и перемешивают в течение дополнительного 1 ч. В заключение осадок собирают посредством фильтрации, отмывают холодным простым диизопропиловым эфиром (0,2 кг) и высушивают при пониженном давлении с получением 0,93 кг 4-(2-фторбензилокси)бензальдегида с чистотой по GC 99,8 (% площади, см. пример 24 А) и содержанием 3-(2-фторбензил)-4-(2-фторбензилокси)бензальдегида (VIb), определенными посредством GC в соответствии с примером 24 В, 0,005 мас.%. 1.2. Получение 4-(2-фторбензилокси)бензальдегида (IVb) посредством межфазного катализа (РТС),с применением различных катализаторов. 4-(2-Фторбензилокси)бензальдегид получают алкилированием 4-гидроксибензальдегида (0,39 г) 2-фторбензилхлоридом, следуя той же процедуре из примера 1, но используя три различных катализатора фазового переноса. Результаты приведены в табл. 5. Таблица 5(GC: мас.%), см. пример 24 В.Для снижения содержания примеси (VIb) ниже 0,03 мас.% продукт можно дополнительно очищать способом из примера 1.1 (см. пример 24 В). 1.3. Получение 4-(2-фторбензилокси)бензальдегида (IVb) посредством межфазного катализа (РТС) в ксилоле. 4-(2-Фторбензилокси)бензальдегид получают с 87,2% выходом с содержанием 3-(2-фторбензил)-4(2-фторбензилокси)бензальдегида, определенным посредством GC (см. пример 24 В), 0,02 мас.% реакцией 4-гидроксибензальдегида (47 г) с 2-фторбензилхлоридом тем же способом, как и в примере 1, но с заменой толуола ксилолом в качестве растворителя. 1.4. Получение 4-(2-фторбензилокси)бензальдегида (IVb) посредством межфазного катализа с применением в качестве основания гидроксида калия. 4-(2-Фторбензилокси)бензальдегид получают с 88% выходом с содержанием 3-(2-фторбензил)-4-(2 фторбензилокси)бензальдегида, определенным посредством GC (см. пример 24 В), 0,49 мас.% реакцией 4-гидроксибензальдегида (121 г) с 2-фторбензилхлоридом, тем же способом, как и в примере 1, но с применением гидроксида калия (108,6 г) вместо карбоната калия. Этот продукт дополнительно очищают посредством кристаллизации в соответствии с примером 1.1 для снижения содержания примеси (VIb) ниже 0,03 мас.% (см. пример 24 В). 1.5. Получение 4-(2-фторбензилокси)бензальдегида (IVb) посредством межфазного катализа с применением 2-фторбензилбромида. 4-(2-Фторбензилокси)бензальдегид получают с 89,2% выходом с содержанием 3-(2-фторбензил)-4(2-фторбензилокси)бензальдегида (VIb), определенным посредством GC (см. пример 24 В), 0,06 мас.% реакцией 4-гидроксибензальдегида (161 г) с 2-фторбензилбромидом вместо 2-фторбензилхлорида тем же способом, как и в примере 1. Этот продукт дополнительно очищают посредством кристаллизации в соответствии с примером 1.1 для снижения содержание примеси (VIb) ниже 0,03 мас.% (см. пример 24 В). 1.6. Получение 4-(2-фторбензилокси)бензальдегида (IVb) в изопропаноле. В реактор помещают изопропанол (206 кг), карбонат калия (29,4 кг, 0,21 кмоль), йодид калия(11,4 кг, 0,068 кмоль) и 4-гидроксибензальдегид (26 кг, 0,21 кмоль). Смесь перемешивают при 20-25 С в течение 15 мин. Затем добавляют 2-фторбензилхлорид (33 кг, 0,23 кмоль). Смесь кипятят с обратным холодильником при перемешивании в течение 3 ч. Растворитель удаляют в вакууме до остаточного объема 70 л. Добавляют циклогексан (70 кг) и воду (95 кг), смесь нагревают до 50 С и перемешивают при данной температуре в течение 30 мин. Перемешивание останавливают и фазам позволяют разделиться. Органическую фазу отмывают водой (48 кг) при 50 С. Отделенную органическую фазу концентрируют в вакууме до остаточного объема 60 л. Гетерогенную смесь охлаждают до 20 С приблизительно в течение 2 ч и перемешивают при данной температуре в течение 30 мин. Смесь центрифугируют и твердое вещество отмывают циклогексаном. Влажное твердое вещество сушат в вакууме с получением указанного в заголовке продукта 40,2 кг (0,18 кмоль); выход: 82% с чистотой по GC 99,87 (% площади, см. пример 24 А) и содержанием 3-(2-фторбензил)-4-(2-фторбензилокси)бензальдегида (VIb), определенным посредством GC в соответствии с примером 24 В, 0,063 мас.%. Этот продукт дополнительно очищают посредством кристаллизации в соответствии с примером 1.1 для снижения содержание примеси (VIb) ниже 0,03 мас.% (см. пример 24 В). 1.7. Получение 4-(2-фторбензилокси)бензальдегида (IVb) в этаноле. В реактор помещают 4-гидроксибензальдегид (30,3 г, 248 ммоль), этанол (400 мл),2-фторбензилхлорид (28,92 г, 198 ммоль), карбонат калия (103,8 г, 751 ммоль), йодид натрия (1,34 г,0,05 ммоль). Смесь кипятят с обратным холодильником при перемешивании и в атмосфере азота и поддерживают в этих условиях в течение 5 ч. Смесь охлаждают до комнатной температуры и экстрагируют этилацетатом и отмывают 2 М водным раствором гидроксида натрия (3300 мл). Растворитель выпаривают в вакууме с получением указанного в заголовке соединения в виде желтого масла (40,75 г) с чистотой по GC 91,77 (% площади, см. пример 24 А) и содержанием 3-(2-фторбензил)-4-(2 фторбензилокси)бензальдегида (VIb), определенным в соответствии с примером 24 В, 0,346 мас.%. Этот продукт дополнительно очищают посредством кристаллизации в соответствии с примером 1.1 для снижения содержание примеси (VIb) ниже 0,03 мас.% (см. пример 24 В). Пример 2. Получение (S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (Ib) высокой степени чистоты(однореакторная реакция). а) В реактор при перемешивании помещают метанол (25 л) и гидрохлорид L-аланинамида (2 кг) и смесь перемешивают при 23 С в течение 15 мин (значение рН 3,8); затем к ранее полученному раствору добавляют триэтиламин (1,65 кг) и 4-(2-фторбензилокси)бензальдегид (3,32 кг), полученные в соответствии с примером 1.1, доводя значение рН до 8,3. Смесь перемешивают при 25 С в течение 3 ч (гетерогенной смеси рН 8) и охлаждают при перемешивании до 8 С. К смеси при перемешивании в течение 3 ч добавляют боргидрид натрия (0,53 кг), разделенный на 12 небольших частей, после чего смесь поддерживают в течение дополнительных 30 мин. Реакционную смесь концентрируют в вакууме при 40 С до получения остатка (5,2 л). К реакционной смеси при перемешивании в атмосфере азота добавляют толуол(13,9 кг) и воду (23,0 л). Смесь нагревают до 60 С и поддерживают при данной температуре при перемешивании в течение 30 мин. После разделения фаз органическую фазу отмывают водой (6,4 л) при 60 С и воду выливают. Органическую фазу охлаждают до 18 С в течение 2 ч и поддерживают в этих условиях в течение 1 ч. Гетерогенную смесь фильтруют и твердое вещество отмывают толуолом (31,0 л) и высушивают приблизительно при 40 С в вакууме с выходом 3,96 кг(S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (ралфинамида, Ib) с чистотой по ВЭЖХ 99,4 (% площади), определенной способом из примера 25 А и содержанием C,O-диалкилированного(S)-2-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамида, определенным ВЭЖХ способом из примера 25 В, менее 0,005 мас.%.b) Реакцию проводят в тех же условиях, как описано выше, за исключением того, что боргидрид натрия предварительно растворяют в смеси метанола (приблизительно 5,8 г метанола на каждый грамм боргидрида натрия) и 30% гидроксида натрия (приблизительно 0,5 г 30% гидроксида натрия на каждый грамм боргидрида натрия), а затем по каплям добавляют в течение приблизительно 30 мин в смесь с основанием Шиффа, поддерживая температуру 8 С. Степень чистоты полученного продукта по ВЭЖХ,определенная в соответствии с примером 25 А, составляет 99,5%, а содержание C,O-диалкилированной примеси, определенное ВЭЖХ в соответствии с примером 25 В, - менее 0,005 мас.%.c) К смеси метанола (275 л) и гидрохлорида L-аланинамида (24,4 кг, 0,20 кмоль) при комнатной температуре при перемешивании добавляют безводный триэтиламин (19,8 кг, 0,20 кмоль). К указанной выше смеси в течение приблизительно 20 мин добавляют 4-(2-фторбензилокси)бензальдегид (40 кг,0,17 кмоль), полученный в примере 1.6, и реакционную смесь перемешивают в течение 3 ч при 25 С, после чего температуру снижают до 8 С (смесь А). Во втором реакторе при 0-5 С смешивают метанол(50 л) и гидроксид натрия 30% в воде (3,2 кг). К указанной выше смеси, частями, добавляют порошок боргидрида натрия (6,6 кг, 0,17 кмоль) при 0-5 С. Смесь перемешивают в течение 2 ч при 0-5 С в атмосфере азота (смесь В). Смесь В при перемешивании и в атмосфере азота приблизительно в течение 4 ч добавляют к указанной выше реакционной смеси А, поддерживая температуру 8 С. Реакционную смесь концентрируют в вакууме до остаточного объема 70 л. К остатку при перемешивании и в атмосфере азота добавляют толуол (170 кг) и воду (280 кг) и смесь нагревают до 60-65 С. Органическую фазу отделяют и добавляют воду (70 кг) и двухфазную смесь перемешивают при 60-65 С. Органическую фазу отделяют и постепенно охлаждают до 20 С. Смесь центрифугируют и твердое вещество отмывают толуолом с получением после сушки при пониженном давлении указанного в заголовке продукта (48,4 кг,0,16 кмоль), с выходом: 92%. Чистота продукта по ВЭЖХ составляет 99,87 (% площади, см. пример 25 А),а содержание(S)-2-[3-(2-фторбензил)-4-(2 фторбензилокси)бензиламино]пропанамида составляет менее 0,005 мас.% (см. пример 25 В); т.плавл. 109,5 С (капилляр). Энантиомерный состав ралфинамида, определенный на хиральной колонке ВЭЖХ,представляет собой 100% S-энантиомера (% площади, см. пример 26 А). Пример 3. Получение метансульфоната (S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (Id) высокой степени чистоты. а) Ралфинамид (2,8 кг, 9,26 моль), полученный, как описано в примере 2 а), растворяют в 2-пропаноле (19,5 кг) и поддерживают при 65-70 С и при перемешивании в атмосфере инертного газа. После обработки древесным углем (150 г) и фильтрации раствор осаждают чистым метансульфонатом ралфинамида и в течение 30 мин при перемешивании при 50-55 С добавляют метансульфоновую кислоту (900 г, 9,36 моль). Затем суспензию охлаждают до 15-20 С в течение 2 ч и перемешивание продолжают в течение дополнительного 1 ч. В заключение твердое вещество собирают посредством фильтрации и высушивают при пониженном давлении с получением 3,46 кг (94% выход) метансульфоната ралфинамида. Чистота полученного продукта по ВЭЖХ составляет 99,6 (% площади, см. пример 25 А) и содержание(S)-2-[3-(2-фторбензил)-4-(2 фторбензилокси)бензиламино]пропанамида составляет менее 0,005 мас.% (см. пример 25 В); т.плавл. 240,6 С посредством DSC (5 С/мин). Энантиомерная чистота метансульфоната ралфинамида, определенная на хиральной колонке ВЭЖХ, составляет более 99,9 (% площади, см. пример 26 А).b) Ралфинамид (2,8 кг, 9,26 моль), полученный, как описано в примере 2b), преобразуют в его метансульфонатную соль описанным выше способом. Выход составляет 95,8%. Чистота полученного продукта по ВЭЖХ составляет 99,6 (% площади, см. пример 25 А), а содержание C,O-диалкилированного метансульфоната (S)-2-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамида составляет менее 0,005 мас.% (см. пример 25 В); т.плавл. 240,6 С посредством DSC (5 С/мин). Энантиомерная чистота метансульфоната ралфинамида, определенная на хиральной колонке ВЭЖХ, составляет более 99,8c) Смесь 2-пропанола (385 кг) и (S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (ралфинамид, 48,1 кг, 0,16 кмоль), полученную в примере 2 с), нагревают при перемешивании до 60 С и поддерживают в этих условиях до получения прозрачного раствора. К раствору медленно при 60 С добавляют безводную метансульфоновую кислоту. Гетерогенную смесь охлаждают до 20 С и перемешивают при данной температуре в течение 2 ч. Смесь центрифугируют и твердое вещество отмывают 2-пропанолом с получением, после сушки в вакууме, 61 кг (0,15 кмоль) указанного в заголовке продукта; выход 96%; с чистотой по ВЭЖХ 99,83 (% площади, см. пример 25 А) и менее 0,005 мас.% C,O-диалкилированного метансульфоната (S)-2-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамида (см. пример 25 В); т.плавл. 237 С (капилляр). Энантиомерный состав метансульфоната ралфинамида, определенный на хиральной колонке ВЭЖХ, представляет собой 100% S-энантиомера (% площади, см. пример 26 А). Пример 4. Получение метансульфоната (R,S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (Id, I'd) высокой степени чистоты с применением основания L-аланинамида (однореакторная реакция).a) (R,S)-2-[4-(2-Фторбензилокси)бензиламино]пропанамид (Ib, I'b). Свободное основание L-аланинамида получают посредством добавления эквимолярного количества метилата натрия к раствору гидрохлорида L-аланинамида (30 г) в этаноле (351 мл). Смесь перемешивают в течение 30 мин в атмосфере азота при комнатной температуре. Твердое вещество фильтруют и растворитель полностью удаляют в вакууме с получением 21,1 г L-аланинамида. В круглодонной колбе 21,1 г 48,8 г 4-(2-фторбензилокси)бензальдегида, полученного в соответствии с примером 1.1, и смесь перемешивают при комнатной температуре в течение 20 ч. Смесь охлаждают до 82 С и к смеси частями добавляют 8 г твердого NaBH4, поддерживая температуру при 82 С. Реакционную смесь перемешивают в течение по меньшей мере 12 ч, затем концентрируют в минимальном объеме. Добавляют толуол (248 мл) и воду (355 мл) и двухфазную смесь перемешивают при 70 С, а затем отделяют органический слой. Органический раствор отмывают водой (70 мл) при 70 С, затем охлаждают при комнатной температуре,получая суспензию, которую фильтруют и отмывают толуолом. Твердое вещество высушивают при 40 С в вакууме с выходом 47,7 г (74,4% выход) указанного в заголовке продукта в виде белого порошка. Чистота полученного продукта по ВЭЖХ составляет 95,85 (% площади, см. пример 25 А), а содержание C,Oдиалкилированного (S)-2-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамида составляет менее 0,005 мас.% (см. пример 25 В). Отношение R/S энантиомеров ралфинамида, определенное на хиральной колонке ВЭЖХ, составляет 52,5/47,5 (% площади, см. пример 26 А). Дополнительный контроль течения реакции иминоалкилирования показывает, что рацемизация происходит на этой стадии.b) Метансульфонат (R,S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (Id, I'd). В круглодонную колбу добавляют 129,5 г 2-пропанола и 16,5 г продукта со стадии а) и нагревают при 703 С при перемешивании до получения полного раствора. Поддерживая температуру при 703 С,по каплям добавляют 5,2 г метансульфоновой кислоты. После перемешивания в течение 30 мин при 703 С смесь медленно охлаждают до 203 С и затем перемешивают в течение 1 ч. Продукт фильтруют,отмывают 2-пропанолом и сушат в вакууме при 40 С с выходом 19,4 г указанного в заголовке продукта в виде белого порошка. Выход: 92%; с чистотой по ВЭЖХ 99,74 (% площади, см. пример 25 А) и менее 0,005 мас.%(R,S)-2-[3-(2-фторбензил)-4-(2 фторбензилокси)бензиламино]пропанамида (см. пример 25 В). Посредством хиральной колонки ВЭЖХ показано, что полученный таким образом (R,S)-ралфинамид представляет собой смесь энантиомеров,S:R = 53,8:47,0 (% площади, см. пример 26 А). Пример 5. Получение метансульфоната (R)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (I'd) высокой степени чистоты (однореакторная реакция).a) (R)-2-[4-(2-Фторбензилокси)бензиламино]пропанамид (I'b). В реактор при перемешивании помещают метанол (28 л) и гидрохлорид D-аланинамида (2,1 кг) и смесь перемешивают при 23 С в течение 15 мин; затем к ранее полученному раствору добавляют триэтиламин (1,65 кг) и 4-(2-фторбензилокси)бензальдегид (3,30 кг), полученный в соответствии с примером 1.1. Смесь перемешивают при 25 С в течение 3 ч и охлаждают при перемешивании до 8 С. Небольшими частями в течение 3 ч при перемешивании добавляют боргидрид натрия (0,50 кг) и смесь перемешивают в течение дополнительных 30 мин. Реакционную смесь концентрируют в вакууме при 40 С до остатка(5,0 л), а затем к реакционной смеси при перемешивании в атмосфере азота добавляют толуол (14 кг) и воду (25,0 л). Смесь нагревают до 60 С и поддерживают при данной температуре при перемешивании в течение 30 мин. После разделения фаз органическую фазу отмывают водой (7,0 л) при 60 С и воду выливают. Органическую фазу охлаждают до 18 С в течение 2 ч и поддерживают в этих условиях в течение 1 ч. Гетерогенную смесь фильтруют и твердое вещество отмывают толуолом (31,2 л) и высушивают приблизительно при 40 С в вакууме с получением 3,90 кг(R)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (I'b) с чистотой по ВЭЖХ, определенной способом из примера 25 А, 99,9 (% площади) и содержанием C,O-диалкилированного (R)-2-[3-(2-фторбензил)4-(2-фторбензилокси)бензиламино]пропанамида, определенным ВЭЖХ способом из примера 25 В, менее 0,005 мас.%.R-энантиомер ралфинамида, полученный в соответствии с приведенным выше примером 5 а), преобразуют в метансульфонатную соль таким же способом, как в примере 3 а. Чистота полученного продукта по ВЭЖХ составляет 99,9 (% площади, см. пример 25 А), а содержание C,O-диалкилированного метансульфоната (R)-2-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамида составляет менее 0,005 мас.% (см. пример 25 В); т.плавл. 241,0 С посредством DSC (5 С/мин). Энантиомерная чистота (I'd), определенная на хиральной колонке ВЭЖХ, составляет более 99,9 (% площади, см. пример 26 В). Пример 6. Получение метансульфоната (S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (Id) высокой степени чистоты с выделением промежуточного основания Шиффа(S)-2-[4-(2 фторбензилокси)бензилиденамино]пропанамида (IIIb). а) (S)-2-[4-(2-Фторбензилокси)бензилиденамино]пропанамид (IIIb). К суспензии 4-(2-фторбензилокси)бензальдегида (60 г, 0,26 моль), полученной как в примере 1.1, и гидрохлорида L-аланинамида (35,7 г, 0,29 моль) в метаноле (280 мл) при комнатной температуре при перемешивании в атмосфере азота добавляют триэтиламин (29,1 г, 0,29 моль). Перемешивание продолжают в течение дополнительного 1 ч. Затем раствор осаждают несколькими мг(S)-2-[4-(2-фторбензилокси)бензилиденамино]пропанамида, температуру снижают до 5-10 С и перемешивание продолжают в течение 2 ч. Твердое вещество собирают посредством фильтрации и отмывают метанолом при 0 С. После сушки при пониженном давлении получают указанное в заголовке соединение с т.плавл. 122 С (капилляр), с 90% выходом. 1 Н-ЯМР (CDCl3, 300 МГц, 298K)(м.д., относительно TMS): 1,46 (3 Н, д, J=7,0 Гц, СН 3), 3,91 (1 Н,кв, J=7,0 Гц, СН-СО), 5,17 (2 Н, с, О-СН 2), 7,02 (2 Н, д, J=8,9 Гц, ароматический Н в орто-положении О-СН 2), 7,09 (1 Н, ддд, JH-F=9,78 Гц, J орто=8,55 Гц, J мета=1,23 Гц, ароматический Н в орто-положении F),7,15 (1 Н, дт, J орто=7,35 Гц, J мета=1,23 Гц, ароматический Н в пара-положении F), 7,27-7,40 (1 Н, м, ароматический Н в пара-положении СН 2), 7,48 (1 Н, дт, J орто=JH-F=7,35 Гц, J мета=1,53 Гц, ароматический Н в орто-положении СН 2), 7,71 (2 Н, д, J=8,9 Гц, ароматический Н в орто-положении CH=N), 8,17 (1 Н, с,C=N). 13 С-ЯМР (CDCl3, 75,4 МГц, 298K)(м.д.): 21,4 (СН 3), 63,8 (ОСН 2), 68,4 (H2NCOCH), 115,0 (д, JCF=22,4 Гц, ароматический СН), 115,5 (д, JC-F=20,7 Гц, ароматический СН), 123,7 (д, JC-F=14,4 Гц, четвертичный ароматический С), 124,5 (ушир.д, ароматический СН), 129,0 (четвертичный ароматический С),129,8 (ушир.д, ароматический СН), 130,1 (ушир.д, 2 ароматический СН), 160,5 (д, JC-F=246,4 Гц, четвертичный ароматический С), 161,1 (ароматический С-О), 161,1 (C=N), 176,9 (CONH2).b) (S)-2-[4-(2-Фторбензилокси)бензиламино]пропанамид (Ib). Смесь (S)-2-[4-(2-фторбензилокси)бензилиденамино]пропанамида (IIIb), полученную, как описано выше, (30 г) и метанола (180 мл) охлаждают при перемешивании до 2-5 С. 18 небольшими частями в течение 90 мин к ранее полученной холодной смеси добавляют боргидрид натрия (3,8 г), поддерживая температуру ниже 5 С. Затем смесь перемешивают в течение дополнительных 10 мин при 5 С. Реакционную смесь концентрируют в вакууме и обрабатывают, как описано в примере 2, с получением 28,75 г(95% выход) (S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (ралфинамид, Ib) с чистотой по ВЭЖХ, определенной способом из примера 25 А, 99,5 (% площади) и содержаниемC,O-диалкилированного (S)-2-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамида, определенным способом ВЭЖХ из примера 25 В, менее 0,005 мас.%. с) Метансульфонат (S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (Id). Проводят реакцию ралфинамида, полученного, как описано в приведенном выше примере 6b), с метансульфоновой кислотой, как описано в примере 3, с получением метансульфонатной соли (Id) с выходом 95%. Чистота полученного продукта по ВЭЖХ составляет 99,9 (% площади, см. пример 25 А), а содержание(S)-2-[3-(2-фторбензил)-4-(2 фторбензилокси)бензиламино]пропанамида составляет менее 0,005 мас.% (см. пример 25 В); т.плавл. 240,6 С посредством DSC (5 С/мин). Энантиомерная чистота метансульфоната ралфинамида, определенная на хиральной колонке ВЭЖХ, составляет более 99,9 (% площади, см. пример 26 А). Пример 7. Получение метансульфоната(I'd) высокой степени чистоты,с выделением промежуточного основания Шиффаa) (R)-2-[4-(2-Фторбензилокси)бензилиденамино]пропанамид (III'b). К суспензии 4-(2-фторбензилокси)бензальдегида (60 г, 0,26 моль), полученной как в примере 1.1, и гидрохлорида D-аланинамида (35,7 г, 0,29 моль) в метаноле (280 мл) при комнатной температуре при перемешивании в атмосфере азота добавляют триэтиламин (29,1 г, 0,29 моль). Перемешивание продолжают в течение дополнительного 1 ч. Затем раствор осаждают несколькими мг(R)-2-[4-(2-фторбензилокси)бензилиденамино]пропанамида, температуру снижают до 5-10 С и перемешивание продолжают в течение 2 ч. Твердое вещество собирают посредством фильтрации и отмывают метанолом при 0 С. После сушки при пониженном давлении получают указанное в заголовке соединение с выходом 91% с т.плавл. 121 С (капилляр). 1 Н-ЯМР (CDCl3, 300 МГц, 298K)(м.д., относительно TMS): 1,46 (3 Н, д, J=7,0 Гц, СН 3), 3,91 (1 Н,кв, J=7,0 Гц, СН-СО), 5,17 (2 Н, с, О-СН 2), 7,02 (2 Н, д, J=8,9 Гц, ароматический Н в орто-положении О-СН 2), 7,09 (1 Н, ддд, JH-F=9,78 Гц, J орто=8,55 Гц, J мета=1,23 Гц, ароматический Н в орто-положении F),7,15 (1 Н, дт, J орто=7,35 Гц, J мета=1,23 Гц, ароматический Н в пара-положении F), 7,27-7,40 (1 Н, м, ароматический Н в пара-положении СН 2), 7,48 (1 Н, дт, J орто=JH-F=7,35 Гц, J мета=1,53 Гц, ароматический Н в орто-положении СН 2), 7,71 (2 Н, д, J=8,9 Гц, ароматический Н в орто-положении CH=N), 8,17 (1 Н, с,C=N). 13 С-ЯМР (CDCl3, 75,4 МГц, 298K)(м.д.): 21,4 (СН 3), 63,8 (ОСН 2), 68,4 (H2NCOCH), 115,0 (д, JCF=22,4 Гц, ароматический СН), 115,5 (д, JC-F=20,7 Гц, ароматический СН), 123,7 (д, JC-F=14,4 Гц, четвертичный ароматический С), 124,5 (ушир.д, ароматический СН), 129,0 (четвертичный ароматический С),129,8 (ушир.д, ароматический СН), 130,1 (ушир.д, 2 ароматический СН), 160,5 (д, JC-F=246,4 Гц, четвертичный ароматический С), 161,1 (ароматический С-О), 161,1 (C=N), 176,9 (CONH2). охлаждают при перемешивании до 2-5 С. 12 небольшими частями в течение 90 мин к ранее полученной холодной смеси добавляют боргидрид натрия (3,8 г), поддерживая температуру ниже 5 С. Затем смесь перемешивают в течение дополнительных 10 мин при 5 С. Реакционную смесь концентрируют в вакууме и обрабатывают, как описано в примере 2, с получением 28,44 г (94% выход(R)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (I'b) с чистотой по ВЭЖХ, определенной способом из примера 25 А, 99,8 (% площади) и содержанием C,O-диалкилированного (R)-2-[3-(2-фторбензил)4-(2-фторбензилокси)бензиламино]пропанамида, определенным способом ВЭЖХ из примера 25 В, менее 0,005 мас.%. с) Получение метансульфоната (R)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (I'd). Проводят реакцию R-энантиомера ралфинамида, полученного в соответствие с приведенным выше примером 7b, с метансульфоновой кислотой, как описано в примере 3 а, с получением метансульфонатной соли (I'd) с выходом 95%. Чистота полученного продукта по ВЭЖХ составляет 99,9 (% площади, см. пример 25 А), а содержание C,O-диалкилированного метансульфоната (R)-2-[3-(2-фторбензил)-4-(2 фторбензилокси)бензиламино]пропанамида составляет менее 0,005 мас.% (см. пример 25 В); т.плавл. 240,6 С посредством DSC (5 С/мин). Энантиомерная чистота метансульфоната ралфинамида, определенная на хиральной колонке ВЭЖХ, составляет более 99,8 (% площади, см. пример 26 В). Пример 7 А. Метансульфонат (R,S) 2-[4-(2-фторбензилокси)бензиламино]пропанамида (Id, I'd). а) Метанол (54 мл) и гидрохлорид (R,S)-аланинамида (10,09 г, 82 ммоль) помещают в 250-мл стеклянный реактор и по каплям при 25 С добавляют безводный триэтиламин (11,36 мл, 96 ммоль). В течение приблизительно 10 мин добавляют 4-(2-фторбензилокси)бензальдегид (15,99 г, 69 ммоль), полученный в примере 1.6, и смесь перемешивают в течение 12 ч при 25 С (смесь А). Во втором реакторе (50 мл) при перемешивании смешивают метанол (20 мл) и гидроксид натрия 30% в воде (1,3 г) и температуру снижают до 0,6 С. К раствору частями при 1 С добавляют порошковый боргидрид натрия (2,61 г,69 ммоль). Смесь перемешивают в течение 2 ч при 1 С в атмосфере азота (смесь В). При перемешивании и в атмосфере азота, в течение приблизительно 30 мин к указанной выше смеси А добавляют смесь В,поддерживая температуру 10 С. Реакционную смесь перемешивают в течение 30 мин при 10 С и концентрируют в вакууме до остаточного объема 20 мл. К остатку при перемешивании и в атмосфере азота добавляют толуол (70 мл) и воду (120 мл) и смесь нагревают до 60-65 С. Органическую фазу отделяют и добавляют воду (20 мл) и смесь перемешивают при 60-65 С. Органическую фазу отделяют и постепенно охлаждают приблизительно до 7 С и поддерживают в этих условиях в течение 3 ч. Смесь фильтруют и твердое вещество отмывают толуолом (35 мл) с получением, после сушки при пониженном давлении,(R,S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (13,59 г); 65,2% выход. Чистота продукта по ВЭЖХ составляет 97,73% (% площади, см. пример 25 А) и содержание С,О-диалкилированного(R,S)-2-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамида составляет 0,020 мас.% (см. пример 25 В). Показано, что полученный таким образом (R,S)-ралфинамид представляет собой смесь энантиомеров S:R = 52,3:47,7 (% площади, см. пример 26 А) посредством хиральной колонки ВЭЖХ.b) Смесь 2-пропанола (102 мл) и (R,S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (10 г,33 ммоль), полученного в примере 7 а), нагревают при перемешивании до 70 С и поддерживают в этих условиях до получения прозрачного раствора. К предыдущему раствору при 60 С медленно добавляют безводную метансульфоновую кислоту(3,18 г, 2,15 мл). Гетерогенную смесь охлаждают до 20 С и перемешивают при данной температуре в течение 2 ч. Смесь центрифугируют и твердое вещество отмывают 2-пропанолом с получением, после сушки в вакууме, 10,77 г указанного в заголовке продукта; 92% выход; с чистотой по ВЭЖХ 99,50 (% площади,см. пример 25 А) и 0,009 мас.% С,O-диалкилированного метансульфоната (R,S)-2-[3-(2-фторбензил)-4-(2 фторбензилокси)бензиламино]пропанамида (см. пример 25 В). Показано, что полученный таким образом(R,S)-ралфинамид представляет собой смесь энантиомеров, S:R = 52,8:47,2 (% площади, см. пример 26 А) посредством хиральной колонки ВЭЖХ.(IId). а) 3-(2-Фторбензил)-4-(2-фторбензилокси)бензальдегид (VIb). В 5-л круглодонную колбу последовательно добавляют 4-гидроксибензальдегид (293 г, 2,407 моль),карбонат калия (315,85 г, 2,287 моль), толуол (900 мл) и 2-фторбензилхлорид (1392 г, 9,628 моль). К реакционной смеси при перемешивании добавляют воду (150 мл). Смесь быстро нагревают до температуры кипения с обратным холодильником и поддерживают в этих условиях в течение трех суток. Анализ GC выявил присутствие 3,2 мас.% (см. пример 24 В) 3-(2-фторбензил)-4-(2-фторбензилокси)бензальдегида(VIb). Смесь охлаждают до 60 С и при перемешивании добавляют воду (1000 мл). Слои разделяют. Ор- 20019898 ганическую фазу отмывают солевым раствором (500 мл); затем растворитель отгоняют при пониженном давлении (10 мм рт.ст.) при 35 С до тех пор, пока растворитель больше не отгоняется. Остаток отгоняют при 3 мм рт.ст., собирая фракцию при 180-220 С. Остаток после холодной отгонки растворяют в CH2Cl2 и растворитель удаляют в вакууме с получением остатка. Чистота по GC составляет 89%, хотя исходный альдегид составляет 0,5%. Хроматография на силикагеле с использованием гексана:этилацетата = 95:5 дает продукт (15,7 г) с чистотой по GC более 99% (% площади; относительно условий GC см. пример 24 В). Т.плавл. продукта составляет 71 С (капилляр). 1 Н-ЯМР (CDCl3, 300 МГц, 298K)(м.д., относительно TMS): 4,06 (2 Н, с, СН 2), 5,23 (2 Н, с, ОСН 2),6,95-7,40 (9 Н, м, ароматический Н), 7,67 (1 Н, ушир.д, J=0,9 Гц, ароматический Н в орто-положении относительно С=O и СН 2), 7,76 (1 Н, дд, J1=2,1 Гц, J2=8,3 Гц, ароматический Н в орто-положении относительно С=O и ароматический СН), 9,84 (1 Н, с, СНО). 13 С-ЯМР (CDCl3, 75,4 МГц, 298K)(м.д.): 29,2 (СН 2), 64,1 (ОСН 2), 111,4 (ароматический СН),115,4 (д, JC-F=22,0 Гц, ароматический СН), 115,5 (д, JC-F=21,1 Гц, ароматический СН), 123,3 (д,JC-F=14,2 Гц, четвертичный ароматический С), 124,1 (д, JC-F=2,6 Гц, ароматический СН), 124,5 (д,JC-F=3,2 Гц, ароматический СН), 126,6 (д, JC-F=15,5 Гц, четвертичный ароматический С), 128,2 (д, JC-F=8,1 Гц, ароматический СН), 129,6 (д, JC-F=6,2 Гц, ароматический СН), 129,6 (четвертичный ароматический С), 130,0 (четвертичный ароматический С), 130,2 (д, JC-F=8,3 Гц, ароматический СН), 131,1 (ароматический СН), 131,3 (д, JC-F=4,1 Гц, ароматический СН), 131,8 (ароматический СН), 160,5 (д, JC-F=246,8 Гц,четвертичный ароматический С), 161,2 (д, JC-F=245,1 Гц, четвертичный ароматический С), 161,3 (четвертичный ароматический С), 191,1 (СНО).b) (S)-2-[3-(2-Фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамид (IIb). К раствору 3-(2-фторбензил)-4-(2-фторбензилокси)бензальдегиду (3,56 г, 0,0105 моль) в 50-мл колбе при комнатной температуре добавляют раствор, ранее полученный посредством осторожного добавления при перемешивании триэтиламина (1,2 г, 0,0119 моль) к 17 мл раствора гидрохлоридаL-аланинамида (1,48 г, 0,0119 моль) в метаноле. Эту реакционную смесь перемешивают в течение 1 ч при комнатной температуре (осаждение происходит соответствующим имином), а затем его переносят в 0,18 л автоклав и к смеси добавляют 0,34 г влажного (50% Н 2 О) 5% Pt/C. Воздух удаляют из автоклава азотом, а затем при 5,0 бар вводят водород. Реакцию проводят при температуре 35 С в течение 3-5 ч. После охлаждения до комнатной температуры и удаления катализатора посредством фильтрации растворитель отгоняют при пониженном давлении до получения остатка приблизительно 6,5 г. К этому остатку добавляют воду (22 мл) и поддерживают при данной температуре при перемешивании в течение по меньшей мере 2 ч. Полученные кристаллы фильтруют и отмывают водой. Получают указанное в заголовке соединение с выходом 83% (0,00872 моль); т.плавл. 161 С (капилляр). 1 Н-ЯМР (CDCl3, 300 МГц, 298K)(м.д., относительно TMS): 1,32 (3 Н, д, J=6,7 Гц, СН 3), 1,97 (1 Н,ушир.с, NH), 3,22 (1 Н, кв, J=6,7 Гц, СН-СО), 3,67 (2 Н, ABq, J=12,8 Гц, диастереотопный Н NCH2), 4,03(S)-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамида. Раствор нагревают при перемешивании при 55-60 С и поддерживают при данной температуре в течение 1 ч. К этому раствору в течение 15 мин добавляют метансульфоновую кислоту (0,00881 моль) и температуру снижают до 20 С в течение 90 мин. После одной ночи твердое вещество собирают посредством фильтрации, высушивают при 50 С при пониженном давлении, а затем кристаллизуют из метанола с получением метансульфонатаd) Выделение (IIb) посредством препаративной ВЭЖХ метансульфоната ралфинамида (Id), содержащего 0,13 мас.% (IId). Образец (100 мг) (S)-2-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамида (IIb) также выделяют посредством препаративной ВЭЖХ из 200 г метансульфоната ралфинамида (Id), полученного способом А из J. Med. Chem., 1998, 41, 579, содержащего указанную примесь (IIb) в виде метансульфоната (IId), в количестве 0,13 мас.%. Анализ ВЭЖХ на хиральной колонке (см. пример 27 С) демонстрирует соотношение между S энантиомером (RT 7,3) и R-энантиомером (RT 7,6) более 99,5/0,5. Разделение проводят в двух стадиях (стадия 1 и стадия 2) в соответствии со следующей схемой. Стадия 1. Назначение стадии 1 состоит в выделении сырого продукта, обогащенного IIb/TFA (трифторуксусная кислота). Ниже приведены условия препаративной ВЭЖХ. Условия препаративной ВЭЖХ. Устройство: Waters Delta Prep 4000 (возвратно-поступательный насос, устройство для создания градиентов со смесителем низкого давления). Модуль: Radial Compression Module Prep LC Base (Waters). Переменный детектор УФ: Jasco 7125, о.р. 0,2 мм. Принтер-плоттер: Merk D2000. Колонка: Delta Pak C18, 15 мкм, 40100 мм (Waters). Элюент А: 70/30, вода/ацетонитрил + 0,1% TFA. Элюент В: 30/70, вода/ацетонитрил + 0,1% TFA. Скорость потока: 27 мл/мин. Градиент: 40 мин, изократический 100% А, затем до 100% В в течение 1 мин. Детекция: УФ 227 нм. Впрыск: 5 г в 50 мл воды (посредством впускного канала насоса D). Стадия 2. Эта стадия необходима для удаления TFA из IIb/TFA и для дальнейшей очистки (IIb).IIb/TFA подвергают хроматографии с применением условий препаративной ВЭЖХ, приведенных ниже. Фракции 4 и 5 объединяют вместе и выпаривают при 40 С в вакууме до полного удаления ацето- 22019898 нитрила. Оставшийся водный раствор поддерживают в холодильнике при 4 С. Нерастворимое вещество выделяют посредством фильтрации и сушат в вакууме при комнатной температуре с получением (IIb)(100 мг; чистота по ВЭЖХ 100%). Условия препаративной ВЭЖХ. Устройство: Waters Delta Prep 4000 (возвратно-поступательный насос, устройство для создания градиентов со смесителем низкого давления). Переменный детектор УФ: Jasco 7125, о.р. 0,2 мм. Принтер-плоттер: Merk D2000. Колонка: Symmetry C18, 7 мкм, 20250 мм (Waters). Элюент А: 70/30, вода/ацетонитрил. Элюент В: 30/70, вода/ацетонитрил. Скорость потока: 15 мл/мин. Градиент: 20 мин, изократический 100% А, затем до 100% В в течение 10 мин. Детекция: УФ 227 нм. Впрыск: 50 мл раствора примеси "IIa/TFA" (посредством впускного канала насоса D). Пример 9. Получение метансульфоната (R)-2-[3-(2-фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамидаa) 3-(2-Фторбензил)-4-(2-фторбензилокси)бензальдегид (VIb). Указанное в заголовке соединение получали в соответствии с примером 8 а).b) (R)-2-[3-(2-Фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамид. Указанное в заголовке соединение получают реакцией 3-(2-фторбензил)-4-(2 фторбензилокси)бензальдегида (VIb), полученного, как в примере 9 а), с гидрохлоридом D-аланинамида способом из примера 8b).(R)-2-[3-(2-Фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамид, полученный на стадии b),преобразуют в указанное в заголовке соединение тем же способом, который описан в примере 8 с). На основе данных 1 Н-ЯМР, 13 С-ЯМР структуру (II'd) приписали полученному таким образом метансульфонату. Спектры 1 Н-ЯМР, 13 С-ЯМР и т.плавл. 190 С (капилляр) полностью совпадают со спектрами 1 Н-ЯМР, 13 С-ЯМР и т.плавл. S-энантиомера (IId) (см. пример 8 с). Анализ ВЭЖХ на хиральной колонке (см. пример 27 С) продемонстрировал отношение между Rэнантиомером (RT 7,6) и S-энантиомером (RT 7,3) более 99,5/0,5. Пример 9 А. Получение метансульфоната(R,S)-2-[3-(2-Фторбензил)-4-(2-фторбензилокси)бензиламино]пропанамид получают из 3-(2-фторбензил)-4-(2-фторбензилокси)бензальдегида (VIb) (5 г), полученного в соответствии с предыдущим примером 8 а), и гидрохлорида (R,S)-аланинамида, как в способе, приведенном в примере 8b). Продукт преобразуют в соль метансульфоновой кислоты способом, описанным в примере 9 с). Соль из (VIb) получают с выходом 70%. Спектроскопические данные полностью совпадают с данными R-энантиомера (II'd) (см. пример 9 с). Анализ ВЭЖХ на хиральной колонке (см. пример 27 С) продемонстрировал отношение между[]25D (с 1% в метаноле): 0. Пример 10. Получение очищенного 4-(3-фторбензилокси)бензальдегида (IVa). а) К смеси 4-гидроксибензальдегида (2,28 кг, 18,68 моль), карбоната калия (2,84 кг, 20,54 моль), йодида калия (0,33 кг, 1,98 моль) в этаноле (21 кг) при перемешивании при комнатной температуре добавляют 3-фторбензилхлорид (2,70 кг, 18,68 моль). Смесь постепенно нагревают до температуры кипения с обратным холодильником, а затем поддерживают при этой температуре в течение 6 ч. Затем реакционной смеси позволяют охлаждаться до 25 С,суспензию фильтруют и твердое вещество отмывают этанолом (1,5 кг); растворы в этаноле объединяют,а затем концентрируют при пониженном давлении до получения остатка приблизительно 6 кг. К этому остатку добавляют толуол (10 кг) и воду (2,5 кг), смесь растворителей энергично перемешивают в течение 30 мин и после отделения водной фазы органический слой выпаривают до сухого состояния при пониженном давлении с получением неочищенного 4-(3-фторбензилокси)бензальдегида. К этому продукту,растворенному в толуоле (3 кг), при перемешивании при 20-25 С добавляют затравку кристаллизации 4-(3-фторбензилокси)бензальдегида, затем в течение 45 мин добавляют н-гексан (6 кг) и смесь охлаждают до 0 С при перемешивании. Через 3 ч твердое вещество фильтруют и отмывают н-гексаном (2 кг). После сушки получают 3,95 кг (92% выход) желаемого продукта с чистотой по газовой хроматографии 99,8 (% площади, см. пример 24 А) и содержанием 3-(3-фторбензил)-4-(3-фторбензилокси)бензальдегида,определенным посредством GC, 0,01 мас.% (% площади, см. пример 24 В); т.плавл. 43,1 С посредством Дальнейшее получение указанного в заголовке соединения проводят следующим образом.(305 г, 1,84 моль) и 4-гидроксибензальдегид (700 г, 5,74 моль). Смесь перемешивают при 20-25 С в течение 15 мин. В реактор помещают 3-фторбензилхлорид (871 г, 6,03 моль) посредством капельной воронки в течение приблизительно 20 мин. Смесь нагревают при температуре кипения с обратным холодильником при перемешивании в течение 3 ч. По истечении этого времени смесь охлаждают приблизительно до 50 С и отбирают образец для контроля в процессе производства. Растворитель удаляют в вакууме до достижения объема приблизительно 1,8 л. Добавляют циклогексан (1,84 кг) и воду (2,5 кг), а затем смесь нагревают до 653 С и перемешивают при данной температуре в течение 30 мин. Перемешивание останавливают и фазам позволяют разделяться в течение 20 мин; водную фазу удаляют. К органической фазе добавляют воду (1,31 кг) и двухфазную смесь перемешивают в течение 30 мин при 653 С. Фазам позволяют разделяться в течение 20 мин. Водную фазу удаляют и органическую фазу концентрируют в вакууме до объема приблизительно 3 л при температуре от 40 до 55 С. Смесь охлаждают приблизительно до 30 С, осаждают и перемешивают при данной температуре в течение 30 мин. Смесь охлаждают до 202 С и перемешивают при данной температуре в течение по меньшей мере 3 ч. Продукт фильтруют в вакууме и твердое вещество отмывают циклогексаном (3150 г). Влажное твердое вещество (1,5 кг) высушивают при 20-25 С при давлении ниже 100 мбар в течение 12 ч с получением 1,17 кг (5,09 моль); 88% выходом с чистотой по газовой хроматографии 99,43 (% площади, см. пример 24 А) и содержанием 3-(3-фторбензил)-4-(3-фторбензилокси)бензальдегида (VIa), определенным посредством GC, 0,063 мас.%(% площади, см. пример 24 В). Этот продукт дополнительно очищают способом, описанным в примере 11.1 с выходом продукта, где содержание примеси (VIa), определенное посредством GC, составляет 0,01 мас.% (см. пример 24 В). Пример 11. Получение 4-(3-фторбензилокси)бензальдегида (IVa) посредством межфазного катализа. Смесь 3-фторбензилхлорида (10 кг, 69,16 моль), 4-гидроксибензальдегида (7,8 кг, 63,88 моль), карбоната калия (9,46 кг, 68,44 моль) и бромида тетрадецилтриметиламмония (1,03 кг, 3,72 моль) в толуоле(30 кг) медленно доводят до температуры кипения с обратным холодильником при перемешивании и в атмосфере азота, а затем кипятят с обратным холодильником в течение 7 ч. Раствор концентрируют при комнатном давлении, а затем добавляют 6 кг толуола и отгоняют. Эту процедуру повторяют еще раз. Затем гетерогенную смесь охлаждают до комнатной температуры и твердое вещество удаляют посредством фильтрации. Оставшийся растворитель удаляют при пониженном давлении, а затем к масляному остатку добавляют 2,6 кг толуола. Смесь перемешивают при 20-25 С и осаждают несколькими граммами чистого 4-(3-фторбензилокси)бензальдегида, а затем к перемешиваемой смеси, поддерживаемой при 2025 С, в течение 45 мин добавляют н-гексан (18 кг). После охлаждения до 3-6 С и перемешивания в течение дополнительного 1 ч при данной температуре твердое вещество собирают посредством фильтрации и высушивают при пониженном давлении с получением 13,5 кг с 86,3% выходом, чистотой по GC 99,8 (% площади, см. пример 24 А) и содержанием 3-(3-фторбензил)-4-(3-фторбензилокси)бензальдегида 0,01 мас.% (см. пример 24 В). 11.1. Дополнительная очистка 4-(3-фторбензилокси)бензальдегида (IVa) посредством кристаллизации. 1 кг 4-(3-фторбензилокси)бензальдегида, полученного в соответствии с примером 10b), растворяют в 2 кг простого диизопропилового эфира при температуре кипения с обратным холодильником при перемешивании. Раствор охлаждают до 50-55 С в течение 10-15 мин и осаждают несколькими граммами чистого 4-(3-фторбензилокси)бензальдегида. Суспензию охлаждают до 10-15 С в течение 45-60 мин и перемешивают в течение дополнительного 1 ч. В заключение осадок собирают посредством фильтрации,отмывают холодным простым диизопропиловым эфиром (0,2 кг) и высушивают при пониженном давлении с получением 0,90 кг 4-(3-фторбензилокси)бензальдегида с чистотой по GC 99,9 (% площади, см. пример 24 А) и содержанием 3-(3-фторбензил)-4-(3-фторбензилокси)бензальдегида, определенным посредством GC, 0,01 мас.% (см. пример 24 В). 11.2. Получение 4-(3-фторбензилокси)бензальдегида (IVa) посредством межфазного катализа с применением 3-фторбензилбромида. 4-(3-Фторбензилокси)бензальдегид получают с 87% выходом с содержанием 3-(3-фторбензил)-4-(3 фторбензилокси)бензальдегида, определенным посредством GC, 0,05 мас.% (см. пример 24 В) реакцией 4-гидроксибензальдегида (26,52 г) с 3-фторбензилбромидом тем же способом, как в примере 11, но с применением вместо 3-фторбензилхлорида 3-фторбензилбромида. Полученный таким образом 4-(3-фторбензилокси)бензальдегид очищают в соответствии с примером 11.1 с выходом указанного в заголовке продукта 95% с содержанием 3-(3-фторбензил)-4-(3-фторбензилокси)бензальдегида, определенным посредством GC, 0,01 мас.% (см. пример 24 В). 11.3. Получение 4-(3-фторбензилокси)бензальдегида (IVa) посредством межфазного катализа с применением метансульфоната 3-фторбензила. 4-(3-Фторбензилокси)бензальдегид получают с 97,5% выходом с содержанием 3-(3-фторбензил)-4(3-фторбензилокси)бензальдегида (VIa), определенным посредством GC, 0,45 мас.% (см. пример 24 В),реакцией 4-гидроксибензальдегида (15,6 г) с метансульфонатом 3-фторбензила вместо 3-фторбензилхлорида тем же способом, как и в примере 11. Этот продукт дополнительно очищают способом из примера 11.1 для снижения содержание примеси (VIa) до 0,01 мас.%. Пример 12. Получение метансульфоната (S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (Ic) высокой степени чистоты (однореакторная реакция). а) Получение (S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (Ia). В 2-л четырехгорлую круглодонную колбу, оборудованную механической мешалкой, термометром,обратным холодильником, и в потоке азота помещают гидрохлорид L-аланинамида (124,6 г, 0,49 моль) и метанол (840 мл) и перемешивают в течение 15 мин при 20 С. Добавляют триэтиламин (49,5 г, 0,49 моль) при такой скорости, что температура остается ниже 30 С. Смесь перемешивают в течение 10 мин, после чего частями в течение приблизительно 30 мин добавляют твердый 4-(3-фторбензилокси)бензальдегид(100 г), полученный в примере 10b). После перемешивания в течение 3 ч при 20 С смесь охлаждают до 5 С и десятью частями с осторожностью в течение 1,5 ч добавляют твердый NaBH4 (16,4 г, 0,44 моль). После окончания добавления смесь перемешивают в течение 30 мин при 5 С. Смесь концентрируют при пониженном давлении до объема 100-150 мл. К остатку добавляют толуол (550 мл) и воду (750 мл) и температуру повышают до 75 С. После перемешивания в течение 30 мин фазы разделяют и органическую фазу отмывают водой (140 мл). После разделения фаз органическую фазу охлаждают до 68 С, осаждают и перемешивают при данной температуре в течение 1 ч. Смесь охлаждают до 20 С приблизительно в течение 2 ч и перемешивают при данной температуре в течение 2 ч. Твердое вещество выделяют посредством фильтрации, отмывают толуолом (240 мл) и сушат в вакууме с выходом 118 г твердого вещества белого цвета; 90% выход. Чистота полученного продукта по ВЭЖХ составляет 99,95 (% площади, см. пример 25 А), а содержание C,O-диалкилированного (S)-2-[3-(3-фторбензил)-4-(3-фторбензилокси)бензиламино]пропанамида составляет 0,008 мас.% (см. пример 25 В). Энантиомерная чистота сафинамида, определенная на хиральной колонке ВЭЖХ, составляет 100%a1) В качестве альтернативного способа восстановление проводят с применением вместо твердогоNaBH4 раствора NaBH4 в метаноле. Получают раствор NaBH4 в метаноле, добавляя NaBH4 (16,4 г) при перемешивании и в атмосфере азота при 0-5 С к смеси метанола (120 мл) и 30% водного раствора NaOH (5,8 мл). В 2-л четырехгорлую круглодонную колбу, оборудованную механической мешалкой, термометром,обратным холодильником, и в потоке азота помещают гидрохлорид L-аланинамида (124,6 г, 0,49 моль) и метанол (720 мл) и перемешивают в течение 15 мин при 20 С. При такой скорости, что температура остается ниже 30 С, добавляют триэтиламин (49,5 г, 0,49 моль). Смесь перемешивают в течение 10 мин,после чего частями в течение приблизительно 30 мин добавляют твердый 4-(3-фторбензилокси)бензальдегид (100 г), полученный в примере 10b). После перемешивания в течение 3 ч при 20 С смесь охлаждают до 5 С и через капельную воронку в течение 1,5 ч осторожно добавляют ранее полученный раствор NaBH4. После окончания добавления смесь перемешивают в течение 30 мин при 5 С. Смесь концентрируют при пониженном давлении до объема 100-150 мл. К остатку добавляют толуол (550 мл) и воду (750 мл) и температуру повышают до 75 С. После перемешивания в течение 30 мин фазы разделяют и органическую фазу отмывают водой (140 мл). После разделения фаз органическую фазу охлаждают до 68 С, осаждают и перемешивают при данной температуре в течение 1 ч. Смесь охлаждают до 20 С приблизительно в течение 2 ч и перемешивают при данной температуре в течение 2 ч. Твердое вещество выделяют посредством фильтрации, отмывают толуолом (240 мл), высушивают при 40 С в вакууме: 116 г твердого вещества белого цвета, 88,5% выход. Чистота продукта по ВЭЖХ составляет 100% (% площади, см. пример 25 А), а содержание ляет 0,009 мас.% (см. пример 25 В). Энантиомерная чистота сафинамида, определенная на хиральной колонке ВЭЖХ, составляет 100%b) Метансульфонат (S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (Ic). Смесь (S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (20 г, 66 ммоль, полученного в примере 12 а) и этилацетата (510 г) нагревают при перемешивании до 65 С и поддерживают в этих условиях до получения прозрачного раствора. К предварительно охлажденному до 55 С раствору в течение 40 мин добавляют метансульфоновую кислоту (7 г, 72,6 ммоль). Смесь постепенно охлаждают до 20 С в течение 3 ч, поддерживают при 20 С в течение 2 ч. Гетерогенную смесь фильтруют, твердое вещество высушивают при пониженном давлении при 40 С с выходом 26,1 г указанного в заголовке соединения в виде порошка белого цвета (99% выход). Чистота полученного продукта по ВЭЖХ составляет 99,94% (% площади, см. пример 25 А), а содержание(S)-2-[3-(3-фторбензил)-4-(3 фторбензилокси)бензиламино]пропанамида составляет 0,005 мас.% (см. пример 25 В). Энантиомерная чистота метансульфоната сафинамида, определенная на хиральной колонке ВЭЖХ,составляет 100% (% площади, см. пример 27 А).b1) (S)-2-[4-(3-Фторбензилокси)бензиламино]пропанамид (20 г, 66 ммоль), полученный в соответствии с примером 12 а 1), преобразуют в метансульфонатную соль (Ic) способом, приведенным в примере 12b), с выходом 26,2 г указанного в заголовке соединения с 99% выходом. Чистота полученного продукта по ВЭЖХ составляет 99,95% (% площади, см. пример 25 А), а содержание(S)-2-[3-(3-фторбензил)-4-(3 фторбензилокси)бензиламино]пропанамида составляет 0,005 мас.% (см. пример 25 В). Энантиомерная чистота метансульфоната сафинамида, определенная на хиральной колонке ВЭЖХ,составляет 100% (% площади, см. пример 27 А). Пример 13. Получение метансульфоната (S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (Ic) высокой степени чистоты. Указанный в заголовке продукт получают с 87% выходом таким же способом, как в примере 12 а 1),за исключением того, что 4-(3-фторбензилокси)бензальдегид получают в соответствии с примером 11 и преобразуют таким образом полученный (S)-2-[4-(3-фторбензилокси)бензиламино]пропанамид в метансульфонат (Ic), с 99,7% (% площади) чистотой, определенной способом из примера 25 А, а содержаниеC,O-диалкилированной примеси (IIa), определенное способом из примера 25 В, составляет 0,005 мас.%. Пример 14. Получение метансульфоната (R,S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (Ic, I'c) высокой степени чистоты с применением основания L-аланинамида (однореакторная реакция).a) (R,S)-2-[4-(3-Фторбензилокси)бензиламино]пропанамид (Ia, I'a). В 1-л четырехгорлую круглодонную колбу, оборудованную механической мешалкой, термометром,и в потоке азота добавляют гидрохлорид L-аланинамида (59 г, 0,47 моль) и этанол (690 мл) и смесь перемешивают при 203 С в течение 20 мин. В течение приблизительно 15 мин добавляют 30% раствор метилата натрия в метаноле (83,9 г, 0,47 моль). Смесь перемешивают в течение 1 ч при 203 С, твердое вещество (NaCl) отфильтровывают и прозрачный раствор концентрируют при пониженном давлении. Остаток разводят метанолом (640 г, приблизительно 800 мл) и частями в течение периода 30 мин добавляют 4-(3-фторбензилокси)бензальдегид (96,5 г, 0,42 моль), полученный в примере 10b). После перемешивания в течение 20 ч при комнатной температуре прозрачный раствор охлаждают до 52 С и осторожно частями в течение 1,5 ч добавляют твердый NaBH4 (15,8 г, 0,42 моль), поддерживая температуру ниже 10 С. После окончания добавления смесь перемешивают в течение 30 мин при 52 С. Смесь концентрируют при пониженном давлении до объема 100-150 мл. К остатку добавляют толуол (550 мл) и воду (750 мл) и температуру повышают до 752 С. После перемешивания в течение 30 мин фазы разделяют и органическую фазу отмывают водой (140 мл). После разделения фаз органическую фазу охлаждают до 682 С, осаждают и перемешивают при данной температуре в течение 1 ч. Смесь охлаждают до 20 С приблизительно в течение 2 ч и перемешивают при данной температуре в течение 2 ч. Твердое вещество собирают посредством фильтрации в вакууме и отмывают толуолом (240 мл). Влажное твердое вещество высушивают при 40 С в вакууме в течение 12 ч с выходом 70 г (R,S)-2-[4-(3 фторбензилокси)бензиламино]пропанамида с выходом 65% с чистотой по ВЭЖХ 99,88 (% площади, см. пример 25 А) и содержанием (R,S)-2-[3-(3-фторбензил)-4-(3-фторбензилокси)бензиламино]пропанамида,определенным ВЭЖХ, 0,008 мас.% (см. пример 25 В). Анализ продукта на хиральной колонке ВЭЖХ в соответствии с примером 27 А продемонстрировал,что полученное соединение имеет соотношение R:S, составляющее 52:48. Дополнительный контроль течения реакции иминоалкилирования показал, что рацемизация происходит на указанной стадии иминоалкилирования.b) Метансульфонат (R,S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (Ic, I'с). Соединение, полученное в соответствии с примером 14 а), преобразуют в метансульфонатную соль тем же способом, как и в примере 4b), с 85% выходом с чистотой по ВЭЖХ 99,8 (см. пример 25 А). Содержание примеси метансульфоната(R,S)-2-[3-(3-фторбензил)-4-(3 фторбензилокси)бензиламино]пропанамида (IIc, II'с), определенное посредством ВЭЖХ (см. пример 25 В), составляет менее 0,005 мас.%. Пример 15. Получение метансульфоната (R)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (I'с) высокой степени чистоты с применением гидрохлорида D-аланинамида (однореакторная реакция).a) (R)-2-[4-(3-Фторбензилокси)бензиламино]пропанамид (I'а). Соединение получают в соответствии с примером 12 а 1), заменяя гидрохлорид L-аланинамида гидрохлоридом D-аланинамида с получением (R)-2-[4-(3-фторбензилокси)бензиламино]пропанамида с выходом 91% с чистотой по ВЭЖХ 99,8 (% площади, см. пример 25 А), а содержаниеR-энантиомер сафинамида, полученный в соответствии с примером 15 а), преобразуют в метансульфонатную соль (I'с) таким же способом, как в примере 12b с 92% выходом, чистотой по ВЭЖХ 99,9%(R)-2-[3-(3-фторбензил)-4-(3 фторбензилокси)бензиламино]пропанамида (II'с), определенное посредством ВЭЖХ (см. пример 25 В),составляет менее 0,005 мас.%. Указанное в заголовке соединение имеет т.плавл. 216,8 С посредствомDSC (5 С/мин). Энантиомерная чистота, определенная на хиральной колонке ВЭЖХ, составляет более 99,9 (% площади, см. пример 27 В). 1 Н-ЯМР (D2O) (Bruker A V300)(м.д., относительно Н 2 О при 4,7 м.д.): 1,43 (3 Н, д J=7 Гц, СН 3),2,66 (3 Н, с, CH3SO3H), 3,87 (1 Н, кв, J=7 Гц, Н-2), 3,97 (2 Н, ушир.с, CH2NR), 4,89 (2 Н, с, CH2OR), 6,88 и 7,23 (4 Н, АА'ХХ' ароматическая п-дизамещенная система), 6,90-7,22 (4 Н, ароматический Н). 13 С-ЯМР (D2O) (Bruker AV300)м.д.: 15,68 (СН 3), 38,27 (CH3SO3H), 48,99 (CH2NR), 54,81 (СН),69,00 (ОСН 2), 114,15 (д, JC-F=21 Гц, ароматический СН), 114,76 (д, JC-F=20 Гц, ароматический СН), 115,38(ароматический СН), 123,06 (д, JC-F=24 Гц, ароматический СН), 123,24; 130,29 (д, JC-F=6 Гц, ароматический СН), 131,54 (ароматический СН), 138,76 (д, JC-F=7 Гц, ароматический СН), 158,52; 162,89 (д,JC-F=245 Гц, C-F), 171,92 (CONH2). Дальнейшее получение соединения по данному примеру 15 проводили следующим образом.a1) (R)-2-[4-(3-Фторбензилокси)бензиламино]пропанамид (I'a). В круглодонной колбе в 166,8 мл метанола растворяют 12,2 г гидрохлорида D-аланинамида и добавляют последовательно 9,9 г триэтиламина, поддерживая температуру менее 30 С, а затем 20 г 4-(3-фторбензилокси)бензальдегида. Смесь перемешивают при комнатной температуре в течение 3 ч, а затем охлаждают при 82 С и добавляют 3,3 г твердого NaBH4, поддерживая температуру около 8 С. Реакционную смесь перемешивают в течение по меньшей мере 1 ч, концентрируют до минимального объема, а затем добавляют толуол (110 мл) и воду (152 мл). Двухфазную смесь перемешивают при 70 С и отделяют органический слой и отмывают водой (30 мл) при 70 С. Полученный в результате раствор охлаждают до комнатной температуры, фильтруют и отмывают толуолом. Твердое вещество высушивают при 40 С в вакууме с выходом 22,6 г указанного в заголовке продукта в виде порошка белого цветаb1) Метансульфонат (R)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (I'с). В круглодонную колбу добавляют 65 г 2-пропанола и 8,25 г соединения, полученного на стадии а 1) выше, и нагревают при 70 С при перемешивании до получения полного раствора. Поддерживая температуру при 703 С, по каплям добавляют 2,6 г метансульфоновой кислоты. После перемешивания в течение 30 мин при 70 С смесь медленно охлаждают до 20 С, а затем перемешивают в течение 1 ч. Продукт фильтруют, отмывают изопропанолом и сушат в вакууме при 40 С с выходом 10 г указанного в заголовке продукта в виде порошка белого цвета (92% выход); т.плавл. 218,4 С (капилляр); []25D (с 2% в метаноле): +0,6. Чистота полученного продукта по ВЭЖХ составляет 99,88% (% площади, см. пример 25 А), а содержание(R)-2-[3-(3-фторбензил)-4-(3 фторбензилокси)бензиламино]пропанамида составляет 0,006 мас.% (см. пример 25 В); т.плавл. 218,4 С (капилляр). Энантиомерная чистота R-метансульфоната сафинамида, определенная на хиральной колонке ВЭЖХ, составляет 100% (% площади, см. пример 27 В).H-ЯМР (300 МГц, ДМСО-d6): 7,97 (1 Н, ушир.с), 7,70 (1 Н, ушир.с), 7,56-7,47 (3 Н, м), 7,38-7,34 (2 Н,м), 7,27-7,21 (1 Н, дт), 7,17-7,15 (2 Н, д), 5,25 (2 Н, с), 4,10 (2 Н, ушир.с), 3,81-3,79 (1 Н, кв), 2,39 (3 Н, с),1,50-1,48 (3 Н, д). Пример 16. Получение метансульфоната (S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (Ic) высокой степени чистоты,с выделением промежуточного основания Шиффаa) (S)-2-[4-(3-Фторбензилокси)бензилиденамино]пропанамид (IIIa). К суспензии 4-(3-фторбензилокси)бензальдегида (192 г, 0,83 моль), полученной как в примере 10, и гидрохлорида L-аланинамида (114,2 г, 0,93 моль) в метаноле (960 мл) при комнатной температуре при перемешивании в атмосфере азота добавляют триэтиламин (93,12 г, 0,93 моль). Перемешивание продолжают в течение дополнительных 2 ч. Затем раствор осаждают несколькими миллиграммами(S)-2-[4-(3-фторбензилокси)бензилиденамино]пропанамида, температуру снижают до 5-10 С и перемешивание продолжают в течение 3 ч. Твердое вещество собирают посредством фильтрации и отмывают метанолом при 2 С. После сушки при пониженном давлении 190,4 г (76% выход) указанного в заголовке соединения получают с т.плавл. 112,0 С посредством DSC (5 С/мин). 1H-ЯМР (ДМСО-d6) (Bruker AV300)(м.д., относительно TMS при 2,55 м.д.; растворитель ДМСО при 3,35 м.д.): 1,31 (3 Н, д, J=7 Гц, СН 3), 3,86 (1 Н, кв, J=7 Гц, Н-2), 5,18 (2 Н, с, CH2OR), 7,08 и 7,79 (4 Н,АА'ХХ' п-дизамещенная ароматическая система), 7,10-7,50 (4 Н, м, ароматический Н), 8,27 (1 Н, с,CH=NR). 13 С-ЯМР (ДМСО-d6) (Bruken AV300)(м.д.): 20,5 (СН 3), 67,6 (СН), 68,4 (ОСН 2), 114,1 и 114,4 (д, JCF=21 Гц, ароматический СН), 114,5 и 114,8 (д, JC-F=21 Гц; ароматический СН), 114,8 (ароматический СН),123,5 (д, JC-F=2 Гц, ароматический СН), 129,0 и 129,9 (ароматический СН), 130,4 и 130,5 (д, JC-F=7 Гц,ароматический СН), 139,6 и 139,7 (д, JC-F=6 Гц, ароматический четвертичный С), 160,2; 160,5 и 163,8 (д,JC-F=245 Гц C-F), 160,6 (CH=N), 174,8 (СО).b) (S)-2-[4-(3-Фторбензилокси)бензиламино]пропанамид (Ia). Смесь (S)-2-[4-(3-фторбензилокси)бензилиденамино]пропанамида (IIIa) (150 г), полученного, как описано в примере 16 а), и метанола (900 мл) охлаждают при перемешивании до 2-5 С. К ранее полученной холодной смеси небольшими частями в течение 2 ч добавляют боргидрид натрия (19 г), поддерживая температуру ниже 5 С. Затем смесь перемешивают в течение дополнительных 20 мин при 5 С. Реакционную смесь концентрируют в вакууме и обрабатывают, как описано в примере 2, с получением 135 г(89,2% выход) (S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (Ia) с чистотой по ВЭЖХ 98,8 (% площади), определенной способом из примера 25 А, и содержанием С,O-диалкилированного (S)-2-[3-(3 фторбензил)-4-(3-фторбензилокси)бензиламино]пропанамида, определенным способом ВЭЖХ из примера 25 В, 0,005 мас.%.(Ic) таким же способом, как в примере 12b), с 94% выходом с чистотой по ВЭЖХ 99,9% (см. пример 25 А). Содержание примеси метансульфоната(S)-2-[3-(3-фторбензил)-4-(3 фторбензилокси)бензиламино]пропанамида (IIc), определенное посредством ВЭЖХ (см. пример 25 В),составляет менее 0,005 мас.%. Энантиомерная чистота, определенная на хиральной колонке ВЭЖХ, составляет более 99,9 (% площади, см. пример 27 А). Пример 17. Получение метансульфоната (R)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (I'с) высокой степени чистоты,с выделением промежуточного основания Шиффа(80 мл) и перемешивают в течение 15 мин при 20 С. Добавляют триэтиламин (5 г) при такой скорости,что температура остается ниже 30 С. Смесь перемешивают в течение 10 мин, после чего частями в течение приблизительно 30 мин добавляют твердый 4-(3-фторбензилокси)бензальдегид (10 г, пример 10b). После перемешивания в течение 3 ч при 20 С смесь охлаждают до 5 С. После перемешивания в течение 3 ч при данной температуре твердое вещество фильтруют и отмывают небольшим количеством предварительно охлажденного метанола. Влажное твердое вещество сушат в вакууме в течение 12 ч при 25 С с выходом 6,4 г указанного в заголовке соединения в виде твердого вещества белого цвета, с выходом 46,4%; т.плавл. 111,9 С;H-ЯМР (ДМСО-d6) (Bruker AV300)(м.д., относительно TMS при 2,55 м.д.; растворитель ДМСО при 3,35 м.д.): 1,31 (3 Н, д, J=7 Гц, СН 3), 3,86 (1 Н, кв, J=7 Гц, Н-2), 5,18 (2 Н, с, CH2OR), 7,08 и 7,79 (4 Н,АА'ХХ' п-дизамещенная ароматическая система), 7,10-7,50 (4 Н, м, ароматический Н), 8,27 (1 Н, с,CH=NR). 13 С-ЯМР (ДМСО-d6) (Bruken AV300)(м.д.): 20,5 (СН 3), 67,6 (СН), 68,4 (ОСН 2), 114,1 и 114,4 (д, JCF=21 Гц, ароматический СН), 114,5 и 114,8 (д, JC-F=21 Гц; ароматический СН), 114,8 (ароматический СН); 123,5 (д, JC-F=2 Гц, ароматический СН), 129,0 и 129,9 (ароматический СН), 130,4 и 130,5 (д, JC-F=7 Гц,ароматический СН), 139,6 и 139,7 (д, JC-F=6 Гц, ароматический четвертичный С), 160,2; 160,5 и 163,8 (д,JC-F=245 Гц, C-F), 160,6 (CH=N), 174,8 (СО).b) (R)-2-[4-(3-Фторбензилокси)бензиламино]пропанамид (I'а). Соединение получают способом из примера 16b), но с применением (R)-2-[4-(3 фторбензилокси)бензилиденамино]пропанамида (III'а), полученного в примере 17 а), вместо его энантиомера (IIIa).(R)-2-[3-(3-фторбензил)-4-(3 фторбензилокси)бензиламино]пропанамида (II'с), определенное посредством ВЭЖХ (см. пример 25 В),составляет менее 0,005 мас.%. Указанное в заголовке соединение имеет т.плавл. 216,8 С посредствомDSC (5 С/мин). Энантиомерная чистота, определенная на хиральной колонке ВЭЖХ, составляет более 99,9 (% площади, см. пример 27 В). Пример 17 а. Метансульфонат (R,S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (Ic, I'с). а) Метанол (80 мл) и гидрохлорид (R,S)-аланинамида (15,14 г, 123 ммоль) помещают в 1000-мл стеклянный реактор и по каплям при 25 С добавляют безводный триэтиламин (17,04 мл, 144 ммоль). В течение приблизительно 10 мин добавляют 4-(3-фторбензилокси)бензальдегид (23,99 г, 103,5 ммоль),полученный в примере 10b), и смесь перемешивают в течение 10 ч при 25 С (смесь А). Во втором реакторе (100 мл) при перемешивании смешивают метанол (30 мл) и гидроксид натрия 30% в воде (1,3 г) и температуру снижают до 0-6 С. Частями к раствору при 1 С добавляют порошковый боргидрид натрия(3,92 г, 103,5 ммоль). Смесь перемешивают в течение 2 ч при 1-2 С в атмосфере азота (смесь В). Смесь В добавляют при перемешивании и в атмосфере азота в течение приблизительно 30 мин к указанной выше смеси А, поддерживая температуру при 5-10 С. Реакционную смесь перемешивают в течение 30 мин при 5-10 С и концентрируют в вакууме до остаточного объема 20 мл. К остатку при перемешивании и в атмосфере азота добавляют толуол (120 мл) и воду (100 мл) и смесь нагревают до 60-65 С. Органическую фазу отделяют и добавляют воду (30 мл) и смесь перемешивают при 60-65 С. Органическую фазу отделяют и постепенно охлаждают приблизительно до 7 С и поддерживают в этих условиях в течение 3 ч. Смесь фильтруют и твердое вещество отмывают толуолом (310 мл) с получением, после сушки при пониженном давлении, (R,S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (21,40 г).b) 2-Пропанол (65 г) и (R,S)-2-[4-(3-фторбензилокси)бензиламино]пропанамид (8,2 г), полученный в примере 17 а), помещают в реактор. Смесь нагревают при перемешивании до 70 С и поддерживают в этих условиях до получения прозрачного раствора. К предыдущему раствору при 70 С медленно добавляют безводную метансульфоновую кислоту (2,6 г). Гетерогенную смесь охлаждают до 20 С и перемешивают при данной температуре в течение по меньшей мере 2 ч. Смесь центрифугируют и твердое вещество отмывают изопропанолом с получением, после сушки в вакууме, 9,4 г указанного в заголовке продукта с 86,4% выходом с чистотой по ВЭЖХ 99,9 (% площади, см. пример 25 А) и менее 0,005 мас.%(R,S)-2-[3-(3-фторбензил)-4-(3 фторбензилокси)бензиламино]пропанамида (см. пример 25 В). На хиральной колонке ВЭЖХ показано,что у полученного таким образом (R,S)-сафинамида смесь энантиомеров S:R = 50,30:49,70 (% площади,см. пример 27 А). Пример 18. Получение метансульфоната (S)-2-[3-(3-фторбензил)-4-(3-фторбензилокси)бензиламино]пропанамида(IIc). а) 3-(3-Фторбензил)-4-(3-фторбензилокси)бензальдегид (VIa). В 4-л круглодонную колбу, поддерживаемую в атмосфере азота, последовательно добавляют 4-гидроксибензальдегид (400 г, 3,28 моль), карбонат калия (453 г, 3,28 моль), толуол (2 л) и 3-фторбензилхлорид (1400 г, 9,68 моль) и смесь кипятят с обратным холодильником при перемешивании в течение 5 суток. До этой стадии анализ GC выявляет, что реакционная смесь содержит 4-(3-фторбензилокси)бензальдегид и 3-(3-фторбензил)-4-(3-фторбензилокси)бензальдегид в соотношении 91,4:8,6 (площадь/площадь, см. пример 24 А). Реакционную смесь охлаждают до комнатной темпера- 29
МПК / Метки
МПК: C07C 251/24, A61P 25/00, C07C 231/12, A61K 31/165, C07C 237/08
Метки: получения, степенью, высокой, 2-[4-(3, способ, чистоты, 2-фторбензилокси)бензиламино]пропанамидов
Код ссылки
<a href="https://eas.patents.su/30-19898-sposob-polucheniya-2-4-3-ili-2-ftorbenziloksibenzilaminopropanamidov-s-vysokojj-stepenyu-chistoty.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения 2-[4-(3- или 2-фторбензилокси)бензиламино]пропанамидов с высокой степенью чистоты</a>
Способ получения меламина с высоким выходом и с высокой степенью чистоты

Номер патента: 2194
Опубликовано: 28.02.2002
Автор: Ноэ' Серджио
МПК: C07D 251/60
Метки: способ, высоким, выходом, чистоты, высокой, получения, меламина, степенью
Формула / Реферат:
1. Способ получения меламина с высоким выходом и с высокой степенью чистоты из мочевины, включающий следующие стадии: а) подачу мочевины в емкостный реактор непрерывного действия, содержащий расплавленный меламин и работающий при 360-420шС под давлением, превышающим 7 _ 103 кПа, предпочтительно под давлением (8-9) _ 103 кПа, при интенсивном перемешивании выделяющимися газами, б) удаление газообразных продуктов реакции, содержащих в основном...
Способ получения 2-[4-(3- и 2-фторбензилокси)бензиламино]пропанамидов

Номер патента: 17123
Опубликовано: 30.10.2012
Авторы: Веларди Франческо, Каччья Карла, Богонья Луиджи, Барбанти Елена, Сальвати Патричия, Руффилли Тициано
МПК: C07C 231/10, A61K 31/165, C07C 233/13...
Метки: 2-фторбензилокси)бензиламино]пропанамидов, 2-[4-(3, способ, получения
Формула / Реферат:
1. Способ получения (S)-2-[4-(3-фторбензилокси)бензиламино]пропанамида (сафинамид) и (S)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (ралфинамид) высокой степени чистоты формулы (Ia) и (Ib)и их солей фармацевтически приемлемой кислоты, отличающийся тем, что промежуточное соединение основания Шиффа, соответственно формулы (VIa) или (VIb)подвергают каталитической гидрогенизации газообразным водородом в присутствии гетерогенного катализатора в...
Пропиленовый полимер с высокой степенью кристалличности, способ его получения, его применение и содержащая его пленка

Номер патента: 15899
Опубликовано: 30.12.2011
Авторы: Аккерманнс Нина, Туоминен Олли, Оммундсен Эспен, Яскеляйнен Пирьо
МПК: C08F 2/00, C08F 110/06
Метки: полимер, получения, кристалличности, способ, содержащая, степенью, применение, пленка, пропиленовый, высокой
Формула / Реферат:
1. Пропиленовый полимер с высокой кристалличностью, имеющий:а) Тр по меньшей мере 122,5°С иб) Tw по меньшей мере 118°С, гдеТр и Twрассчитывают, исходя из функции термоградиентного элюирования (TREF) пропиленового полимера в интервале от 80 до 140°С, гдеТр представляет собой максимальную температуру TREF функции;Tw представляет собой средневзвешенную температуру TREF функции, определяемую уравнениемTi представляет собой температуру, при которой...
Способ получения алмотриптана высокой чистоты

Номер патента: 16759
Опубликовано: 30.07.2012
Авторы: Петрицкова Гана, Радль Станислав, Ридван Лудек, Затопкова Моника, Груби Петр, Стах Ян, Вослар Михаль, Тисовска Люция
МПК: C07D 401/12
Метки: алмотриптана, получения, способ, высокой, чистоты
Формула / Реферат:
1. Способ получения и очистки 3-[2-(диметиламино)этил]-5-(1-пирролидинилсульфонилметил)-1H-индола формулы (I)отличающийся тем, что:(а) 4-(1-пирролидинилсульфонилметил)фенилгидразин путем взаимодействия с щелочной солью функционального производного 4-хлорбутаналя, выбранной из 4-хлор-1-гидроксибутан-1-сульфоната натрия или калия, превращают в соль, являющуюся нерастворимой или плохо растворимой в воде, которую превращают в...
Способ получения диарилкарбоната высокой чистоты

Номер патента: 10425
Опубликовано: 29.08.2008
Авторы: Хатия Хироси, Фукуока Синсуке, Мацузаки Казухико, Миядзи Хиронори
МПК: B01D 3/00, C07C 68/06, C07C 68/08...
Метки: способ, получения, чистоты, диарилкарбоната, высокой
Формула / Реферат:
1. Способ получения диарилкарбоната высокой чистоты, в котором диарилкарбонат получают, используя в качестве исходного вещества реакционную смесь, содержащую алкиларилкарбонат, полученный реакцией переэтерификации между диалкилкарбонатом и ароматическим моногидроксисоединением, где исходное вещество непрерывно подают в колонну реакционной дистилляции, включающую колонну непрерывной многостадийной дистилляции, в которой присутствует гомогенный...
Предыдущий патент: Композиция для проклеивания минеральной ваты, содержащая моносахарид и/или полисахарид и органическую поликарбоновую кислоту, и полученные изоляционные изделия
Следующий патент: Плазменная свеча зажигания для двигателя внутреннего сгорания
Случайный патент: Устройство для аутентификации защитной маркировки