Аналоги нейропептида y, содержащие по меньшей мере одну замену на синтетическую аминокислоту
Номер патента: 19498
Опубликовано: 30.04.2014
Авторы: Деоливейра Дэниел Б., Дун Чжэн Синь, Чжоу Кевин Л.














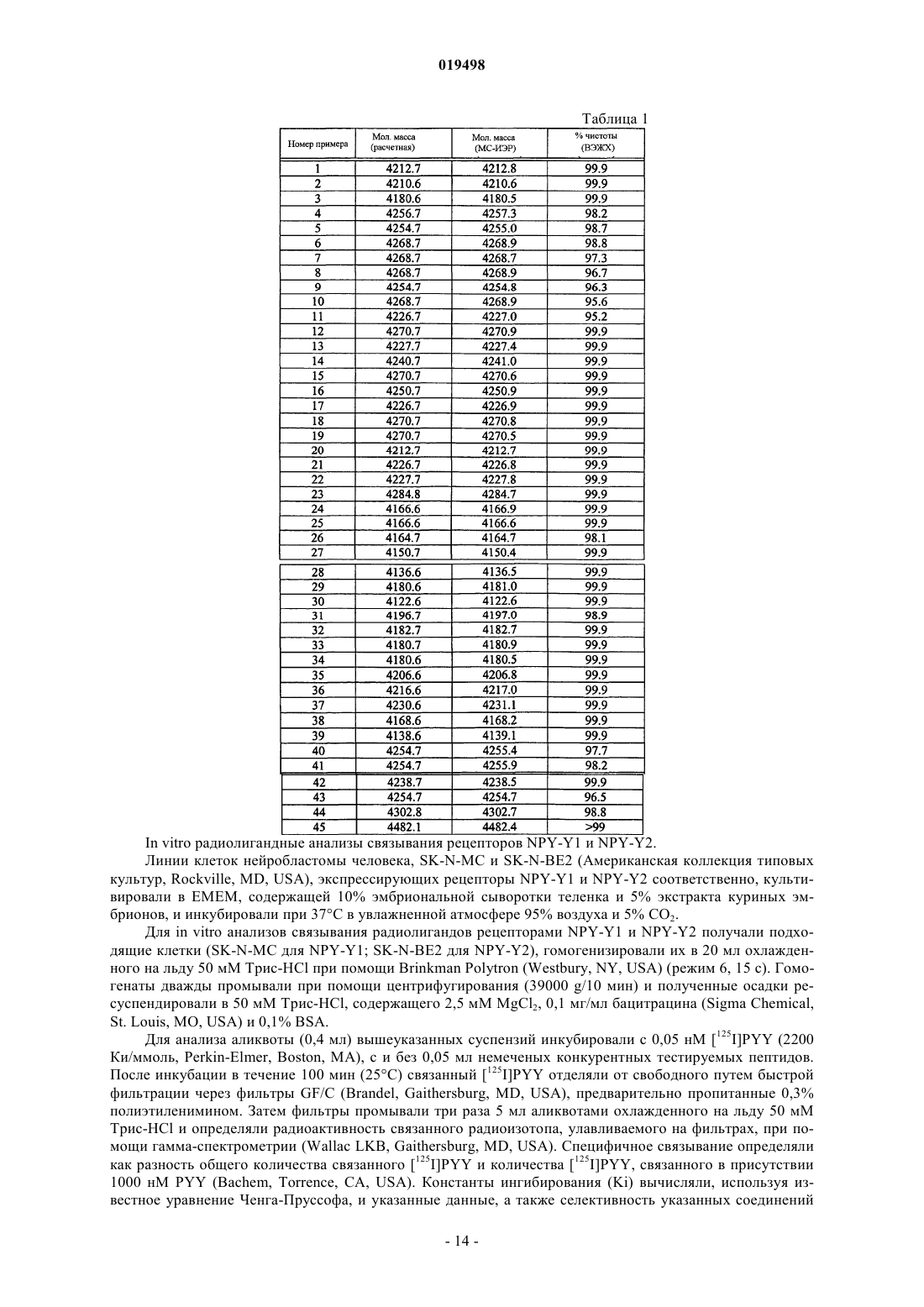














Формула / Реферат
1. Соединение формулы (I) (SEQ ID NO: 2)

где А1 представляет собой Tyr, (X1,X2,X3,X4,X5)Phe или HN-СН((CH2)n-N(R4R5))-С(O);
А2 представляет собой Pro, 3Hyp, цис-3Hyp, 4Hyp или цис-4Hyp;
А3 представляет собой Ser, Abu, Aib, Ala, Thr или HN-CH((CH2)n-N(R4R5))-C(O);
А4 представляет собой Lys, Arg, hArg, Dab, Dap, Orn или HN-CH((CH2)n-N(R4R5))-C(O);
А5 представляет собой Pro, 3Hyp, цис-3Hyp, 4Hyp или цис-4Hyp;
А6 представляет собой Asp, Aib, Asn, Gln, Glu или HN-CH((CH2)n-N(R4R5))-C(O);
А7 представляет собой Asn, Aib, Gln или HN-CH((CH2)n-N(R4R5))-C(O);
А8 представляет собой Pro, 3Hyp, цис-3Hyp, 4Hyp или цис-4Hyp;
А9 представляет собой Gly, Aib или HN-CH((CH2)n-N(R4R5))-С(О);
А10 представляет собой Glu, Aib, Asn, Asp, Gln или HN-CH((CH2)n-N(R4R5))-C(O);
А11 представляет собой Asp, Aib, Asn, Gln, Glu или HN-CH((CH2)n-N(R4R5))-C(O);
А12 представляет собой Ala, Abu, Aib, Nva, Val или HN-CH((CH2)n-N(R4R5))-C(O);
А13 представляет собой Pro, 3Hyp, цис-3Hyp, 4Hyp или цис-4Hyp;
А14 представляет собой Ala, Abu, Aib, Nva, Val или HN-CH((CH2)n-N(R4R5))-C(O);
А15 представляет собой Glu, Aib, Asn, Asp, Gln или HN-CH((CH2)n-N(R4R5))-С(O);
А16 представляет собой Asp, Aib, Asn, Gln, Glu или HN-CH((CH2)n-N(R4R5))-C(O);
А17 представляет собой Met, Acc, Aib, Cha, Ile, Leu, hLeu, Nle, Nva, Tle, Val или HN-CH((CH2)n-N(R4R5))-С(О);
А18 представляет собой Ala, Abu, Aib, Nva, Val или HN-CH((CH2)n-N(R4R5))-C(O);
А19 представляет собой Arg, hArg, Apc, Dab, Dap, Lys, Orn или HN-CH((CH2)n-N(R4R5))-C(O);
А20 представляет собой Tyr, (X1,X2,X3,X4,X5)Phe или HN-СН((CH2)n-N(R4R5))-C(O);
А21 представляет собой Tyr, (X1,X2,X3,X4,X5)Phe или HN-СН((CH2)n-N(R4R5))-C(O);
А22 представляет собой Ser, Abu, Aib, Ala, Thr или HN-CH((CH2)n-N(R4R5))-C(O);
А23 представляет собой Ala, Abu, Aib, Nva, Val или HN-CH((CH2)n-N(R4R5))-C(O);
А24 представляет собой Leu, Acc, Cha, Ile, hLeu, Nle, Nva, Tle, Val или HN-CH((CH2)n-N(R4R5))-С(О);
А25 представляет собой Arg, hArg, Dab, Dap, Lys, Orn или HN-CH((CH2)n-N(R4R5))-C(O);
А26 представляет собой His, 2Pal, 3Pal, 4Pal или HN-CH((CH2)n-N(R4R5))-C(O);
А27 представляет собой Tyr, (X1,X2,X3,X4,X5)Phe или HN-CH((CH2)n-N(R4R5))-C(O);
А28 представляет собой Ile, Acc, Cha, Leu, hLeu, Nle, Nva, Tle, Val или HN-CH((CH2)n-N(R4R5))-С(O);
А29 представляет собой Asn, Aib, Gln или HN-CH((CH2)n-N(R4R5))-C(O);
А30 представляет собой Leu, Acc, Cha, Ile, hLeu, Nle, Nva, Tle, Val или HN-CH((CH2)n-N(R4R5))-С(O);
А31 представляет собой Ile, Acc, Cha, Leu, hLeu, Nle, Nva, Tle, Val или HN-CH((CH2)n-N(R4R5))-С(O);
А32 представляет собой Thr, Aib, Ser или HN-CH((CH2) n-N(R4R5))-C(O);
А33 представляет собой Arg, hArg, Dab, Dap, Lys, Orn или HN-CH((CH2)n-N(R4R5))-C(O);
А34 представляет собой Gln, Asn, Dhp, 3Hyp, цис-3Hyp, 4Hyp, цис-4Hyp, Inp, Ktp, Nip, Oic, hPro, Tic или
HN-CH((CH2)n-N(R4R5))-C(O);
А35 представляет собой Arg, Aic, Apc, hArg, Dab, Dap, Lys, Orn, NH2Phe, NH2CH2Phe или HN-CH((CH2)n-N(R4R5))-С(О);
А36 представляет собой Tyr, Aic, (X1,X2,X3,X4,X5)Phe или HN-CH((CH2)n-N(R4R5))-C(O);
А37 представляет собой HN-CH((CH2)n-N(R4R5))-С(О) или отсутствует;
R1 представляет собой ОН, NH2, (C1-30)алкокси или NH-X6-CH2-X7, где X6 представляет собой (С1-40)алкил или (С2-40)алкенил и где X7 представляет собой Н, ОН, CO2H или C(O)-NH2;
каждый из R2 и R3, независимо в каждом случае, выбран из группы, состоящей из Н, (C1-30)алкила, (C1-30)гетероалкила, (C1-30)ацила, (С2-30)алкенила, (С2-30)алкинила, арил(C1-30)алкила, арил(C1-30)ацила, замещенного (C1-30)алкила, замещенного (C1-30)гетероалкила, замещенного (С2-30)ацила, замещенного (С2-30)алкенила, замещенного (С2-30)алкинила, замещенного арил(C1-30)алкила и замещенного арил(C1-30)ацила;
при условии что, когда R2 представляет собой (С1-30)ацил, арил(C1-30)ацил, замещенный (С2-30)ацил или замещенный арил(C1-30)ацил, R3 представляет собой Н, (C1-30)алкил, (C1-30)гетероалкил, (С2-30)алкенил, (С2-30) алкинил, арил(C1-30)алкил, замещенный (С1-30)алкил, замещенный (C1-30)гетероалкил, замещенный (С2-30)алкенил, замещенный (С2-30)алкинил или замещенный арил(С1-30)алкил;
каждый из R4 и R5, независимо в каждом случае, представляет собой Н, (C1-40)алкил, (С1-40)гетероалкил, (С1-40)ацил, (С2-40)алкенил, (С2-40)алкинил, арил(С1-40)алкил, арил(С1-40)ацил, замещенный (С1-40)алкил, замещенный (C1-40)гетероалкил, замещенный (C1-40)ацил, замещенный (С2-40)алкенил, замещенный (С2-40)алкинил, замещенный арил(С1-40)алкил, замещенный арил(C1-40)ацил, (C1-40)алкилсульфонил или C(NH)-NH2, причем в том случае, когда R4 представляет собой (С1-40)ацил, арил(С1-40)ацил, замещенный (C1-40)ацил, замещенный арил(С1-40)ацил, (С1-40)алкилсульфонил или C(NH)-NH2, R5 представляет собой Н или (С1-40)алкил, (C1-40)гетероалкил, (С2-40)алкенил, (С2-40)алкинил, арил(C1-40)алкил, замещенный (С1-40)алкил, замещенный (С1-40)гетероалкил, замещенный (С2-40)алкенил, замещенный (С2-40)алкинил или замещенный арил(C1-40)алкил;
n, независимо в каждом случае, представляет собой 1, 2, 3, 4 или 5;
каждый из X1, X2, X3, X4 и X5, независимо в каждом случае, представляет собой Н, F, Cl, Br, I, (C1-10)алкил, замещенный (C1-10)алкил, арил, замещенный арил, ОН, СН2NН2, NH2, NO2 или CN;
при условии, что соединение содержит по меньшей мере одну замену на неприродную аминокислоту,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором
А1 представляет собой Tyr;
А2 представляет собой Pro;
А3 представляет собой Ser или Aib;
А4 представляет собой Lys;
А5 представляет собой Pro;
А6 представляет собой Asp или Aib;
А7 представляет собой Asn или Aib;
А8 представляет собой Pro;
А9 представляет собой Gly или Aib;
А10 представляет собой Glu или Aib;
А11 представляет собой Asp или Aib;
А12 представляет собой Ala или Aib;
А13 представляет собой Pro;
А14 представляет собой Ala или Aib;
А15 представляет собой Glu или Aib;
А16 представляет собой Asp или Aib;
А17 представляет собой Met, А6с, Aib или Nle;
А18 представляет собой Ala или Aib;
А19 представляет собой Arg;
А20 представляет собой Tyr;
А21 представляет собой Tyr;
А22 представляет собой Ser или Aib;
А23 представляет собой Ala или Aib;
А24 представляет собой Leu или А6с;
А25 представляет собой Arg;
А26 представляет собой His;
А27 представляет собой Tyr;
А28 представляет собой Ile или А6с;
А29 представляет собой Asn или Aib;
А30 представляет собой Leu или А6с;
А31 представляет собой Ilе, А6с или Leu;
А32 представляет собой Thr или Aib;
А33 представляет собой Arg;
А34 представляет собой Dhp, 4Hyp, Inp, Nip, hPro, Tic или HN-CH((CH2)n-N(R4R5))-C(O);
А35 представляет собой Arg, Apc, Lys, 4NH2Phe или 4NH2CH2Phe;
А36 представляет собой Tyr или Aic;
А37 отсутствует;
R1 представляет собой NH2;
каждый из R2 и R3, независимо в каждом случае, представляет собой Н или (С1-30)ацил;
при условии что, когда R2 представляет собой (C1-30)ацил, R3 представляет собой Н;
каждый из R4 и R5, независимо в каждом случае, представляет собой Н или (С1-40)ацил;
n представляет собой 4 и
каждый из X1, X2, X3, X4 и X5, независимо в каждом случае, представляет собой Н, CH2NH2 или NH2,
или его фармацевтически приемлемая соль.
3. Соединение по п.1 или 2, в котором HN-CH((CH2)n-N(R4R5))-С(O) представляет собой Lys(Ne-C(O)-(СН2)12-СН3), или его фармацевтически приемлемая соль.
4. Соединение по п.1, где указанное соединение представляет собой



или его фармацевтически приемлемая соль.
5. Соединение по любому из пп.1-3, в котором А34 представляет собой 4Hyp, или его фармацевтически приемлемая соль.
6. Соединение по п.5, где указанное соединение представляет собой


или его фармацевтически приемлемая соль.
7. Соединение по п.1 или 2, в котором пептидная связь между А35 и А36 заменена псевдопептидной связью, или его фармацевтически приемлемая соль.
8. Соединение по п.7, в котором А35, А36 представляют собой Lys-y(CH2-NH)Tyr или Lys-y(CH2-N(Ac))Tyr, или его фармацевтически приемлемая соль.
9. Соединение по п.8, где указанное соединение представляет собой

или его фармацевтически приемлемая соль.
10. Фармацевтическая композиция, содержащая эффективное количество соединения по любому из пп.1-9 или его фармацевтически приемлемую соль.
11. Фармацевтическая композиция по п.10, дополнительно содержащая фармацевтически приемлемый носитель.
12. Способ лечения нарушения или заболевания, опосредованного связыванием рецептора нейропептида Y, относящегося к сердцу, кровеносным сосудам или почечной системе, такого как спазм сосудов, сердечная недостаточность, шок, гипертрофия сердца, повышенное кровяное давление, стенокардия, инфаркт миокарда, внезапная сердечная смерть, аритмия, болезнь периферических сосудов, нарушение циркуляции жидкости, нарушение массообмена, почечная недостаточность, повышенная активность симпатического нерва, церебральный инфаркт, нейродегенерация, эпилепсия, инсульт, спазм сосудов мозга, кровоизлияние в мозг, депрессия, тревога, шизофрения, деменция, боль, ноцицепция, аномальная моторика и секреция желудочно-кишечного тракта, различные формы непроходимости кишечника, недержание мочи, болезнь Крона, нарушения, связанные с аномальным потреблением напитков и пищи, анорексия, метаболические нарушения, половая дисфункция и репродуктивные нарушения, нарушение или заболевание, связанное с воспалением, респираторное заболевание, астма, бронхоспазм или аномальная секреция лютеинизирующего гормона, гормона роста, инсулина или пролактина, артериальная гипертензия, ожирение, гиперфазия или булимия, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп.1-9 или фармацевтической композиции по п.10 или 11.
13. Способ по п.12, в котором указанный рецептор нейропептида Y представляет собой рецептор NPY-Y1.
14. Способ по п.13, в котором указанное состояние или заболевание является опухолью, экспрессирующей рецептор NPY-Y1.
15. Способ по п.14, в котором указанная опухоль представляет собой рак молочной железы, рак яичников или глиобластому.
Текст
АНАЛОГИ НЕЙРОПЕПТИДА Y, СОДЕРЖАЩИЕ ПО МЕНЬШЕЙ МЕРЕ ОДНУ ЗАМЕНУ НА СИНТЕТИЧЕСКУЮ АМИНОКИСЛОТУ Изобретение относится к новым аналогам нейропептида Y, содержащим их фармацевтическим композициям, содержащим их лекарственным формам, а также к способу лечения заболеваний или состояний, опосредованных связыванием рецептора нейропептида Y. Более конкретно,изобретение связано с новыми аналогами нейропептида Y, включающими по меньшей мере одну аминокислотную замену на неприродную аминокислоту, такую как 4Hyp, в положении 34, которые селективно связываются с нейропептидным рецептором подтипа Y1 по сравнению с рецептором нейропептида подтипа Y2. Область техники, к которой относится изобретение Настоящее изобретение относится к новым аналогам нейропептида Y, содержащим их фармацевтическим композициям, содержащим их лекарственным формам, а также к способу лечения заболеваний или состояний, опосредованных связыванием рецептора нейропептида Y. Более конкретно, настоящее изобретение относится к новым аналогам нейропептида Y, содержащим по меньшей мере одну аминокислотную замену на неприродную аминокислоту, такую как 4Hyp, в положении 34, которые селективно связываются с нейропептидным рецептором подтипа Y1 по сравнению с нейропептидным рецептором подтипа Y2. Уровень техники Нейропептид Y ("NPY"), пептидный нейромедиатор длиной 36 аминокислот, является представителем семейства панкреатических пептидов и обладает значительной гомологией последовательности с панкреатическим полипептидом и пептидом YY. Человеческий нейропептид Y ("hNPY") имеет последовательность H-Tyr-Pro-Ser-Lys-Pro-Asp-Asn-Pro-Gly-Glu-Asp-Ala-Pro-Ala-Glu-Asp-Met-Ala-Arg-Tyr-TyrSer-Ala-Leu-Arg-His-Tyr-Ile-Asn-Leu-Ile-Thr-Arg-Gln-Arg-Tyr-NH2 (SEQ ID NO: 1). NPY был обнаружен,выделен и секвенирован из мозга свиньи и был назван "нейропептидом Y" вследствие его выделения из нервной ткани и присутствия тирозина в качестве N- и С-концевой аминокислоты.NPY и другие представители его семейства пептидов все характеризуются третичной структурой,состоящей из N-концевой полипролиновой спирали и амфифильной -спирали, связанной с -изгибом,образующих шпилькообразную петлю, которую иногда называют "панкреатической полипептидной укладкой". Спирали удерживаются вместе гидрофобными взаимодействиями. Амидированный С-конец выступает из петли шпильки. Вслед за своим открытием, NPY идентифицировали как наиболее распространенный пептид в центральной нервной системе, с широкой распространенностью, включающей кору головного мозга, ствол мозга, гиппокамп, гипоталамус, миндалевидное тело и таламус, а также присутствующий в периферической нервной системе в симпатических нейронах и хромаффинных клетках надпочечников. По-видимому, NPY отвечает главным критериям нейромедиатора, поскольку он накапливается в синаптических гранулах, высвобождается при электрической стимуляции нервов и действует в специфических рецепторах. Очевидно, что NPY сам по себе является важным мессенджером, вероятно в мозге,где NPY мощно ингибирует активность аденилатциклазы и вызывает повышение внутриклеточных уровней кальция. Внутримозговая инъекция NPY вызывает изменения кровяного давления, повышение потребления пищи, увеличение накопления жира, повышение уровня сахара и инсулина в крови, снижение двигательной активности, понижение температуры тела и каталепсию. Вероятно, NPY взаимодействует с семейством близкородственных рецепторов. Указанные рецепторы в целом разделяют на несколько подтипов на основании способности различных тканей и рецепторов связывать различные фрагменты нейропептида Y и близкородственного PYY. Рецептор подтипа Y1 ("рецептор NPY-Y1"), по-видимому, является главным сосудистым рецептором NPY. Рецептор подтипа Y2("рецептор NPY-Y2") также может встречаться постсинаптически на гладких мышцах сосудов. Рецептор подтипа Y3 ("рецептор NPY-Y3"), вероятно, является NPY-специфичным, не связывая при этом PYY. Данный рецептор, возможно, присутствует среди прочих областей в тканях надпочечников, костном мозге, сердце и стволе мозга. В качестве обзора касательно нейропептида Y и рецепторов нейропептида Y см., например, С. Wahlestedt и D. Reis, Annual Review of Pharmacology and Toxicology, 33:309-352 (1993). В публикации по Договору о патентной кооперации (РСТ) WO 95/00161 описан ряд антагонистов и агонистов NPY для регуляции биологических активностей, таких как ожирение и сердечно-сосудистая функция. В европейском патенте 0759441 и патенте США 5576337 указано, что физиологические нарушения,связанные с избытком нейропептида Y, включают нарушения или заболевания, относящиеся к сердцу,кровеносным сосудам или почечной системе, такие как спазм сосудов, сердечная недостаточность, шок,гипертрофия сердца, повышенное кровяное давление, стенокардия, инфаркт миокарда, внезапная сердечная смерть, аритмия, болезнь периферических сосудов и почечные нарушения, такие как нарушение циркуляции жидкости, нарушение массообмена или почечная недостаточность; состояния, связанные с повышенной активностью симпатического нерва, например, во время или после операции на коронарных артериях, а также воздействий и операций в желудочно-кишечном тракте; болезни мозга и заболевания,связанные с центральной нервной системой, такие как церебральный инфаркт, нейродегенерация, эпилепсия, инсульт, и состояния, связанные с инсультом, спазмом сосудов и кровоизлиянием в мозге, депрессией, тревогой, шизофренией и деменцией; состояния, связанные с болью или ноцицепцией; заболевания, связанные с патологической моторикой и секрецией желудочно-кишечного тракта, такие как различные формы непроходимости кишечника, недержание мочи и болезнь Крона; нарушения, связанные с патологией потребления напитков и пищи, такие как анорексия и метаболические нарушения; заболевания, связанные с половой дисфункцией и репродуктивными нарушениями; состояния или нарушения,связанные с воспалением; респираторные заболевания, такие как астма и состояния, связанные с астмой и бронхоспазмом; а также заболевания, связанные с аномальной секрецией гормонов, таких как лютеинизирующий гормон, гормон роста, инсулин и пролактин. В публикации РСТ WO 02/43776 (Reubi) описано применение соединений, которые связывают рецептор NPY-Y1 для приготовления фармацевтической композиции для диагностики или лечения опухолей, экспрессирующих рецептор NPY-Y1, в частности рака молочной железы, рака яичников и глиобластомы. Существуют многочисленные патенты и патентные публикации, в которых раскрыты некоторые аналоги NPY и их применение, такие как патент США 5026685, патент США 5328899, патент США 6511984, публикация РСТ WO 02/43776, публикация РСТ WO 2007/039318 и т.д. Невзирая на вышесказанное, сохраняется потребность в аналогах NPY, обладающих улучшенной активностью и/или селективностью, и/или in vivo или in vitro характеристиками. Сущность изобретения В одном аспекте настоящее изобретение предоставляет пептидные варианты hNPY следующей формулы (I) (SEQ ID NO: 2):n, независимо в каждом случае, представляет собой 1, 2, 3, 4 или 5; каждый из X1, X2, X3, X4 и X5, независимо в каждом случае, представляет собой Н, F, Cl, Br, I,(C1-10)алкил, замещенный (C1-10)алкил, арил, замещенный арил, О, CH2NH2, NH2, NO2 или CN; и при условии, что соединение содержит по меньшей мере одну замену на неприродную аминокислоту. Подгруппой (IA) соединений, охватываемых вышеуказанной формулой (I), являются соединения, в которых А 1 представляет собой Tyr; А 2 представляет собой Pro; А 3 представляет собой Ser или Aib; А 4 представляет собой Lys; А 5 представляет собой Pro; А 6 представляет собой Asp или Aib; А 7 представляет собой Asn или Aib; А 8 представляет собой Pro; А 9 представляет собой Gly или Aib; А 10 представляет собой Glu или Aib; А 11 представляет собой Asp или Aib; А 12 представляет собой Ala или Aib; А 13 представляет собой Pro; А 14 представляет собой Ala или Aib; А 15 представляет собой Glu или Aib; А 16 представляет собой Asp или Aib; А 17 представляет собой Met, A6c, Aib или Nle; А 18 представляет собой Ala или Aib; А 19 представляет собой Arg; А 20 представляет собой Tyr; А 21 представляет собой Tyr; А 22 представляет собой Ser или Aib; А 23 представляет собой Ala или Aib; А 24 представляет собой Leu или А 6 с; А 25 представляет собой Arg; А 26 представляет собой His; А 27 представляет собой Tyr; А 28 представляет собой Ile или А 6 с; А 29 представляет собой Asn или Aib; А 30 представляет собой Leu или А 6 с; А 31 представляет собой Не, А 6 с или Leu; А 32 представляет собой Thr или Aib; А 33 представляет собой Arg; А 34 представляет собой Dhp, 4Hyp, Inp, Nip, hPro, Tic или HN-CHCH2)n-N(R4R5-C(O); А 35 представляет собой Arg, Apc, Lys, 4NH2Phe или 4NH2CH2Phe; А 36 представляет собой Tyr или Aic; А 37 отсутствует; В формуле (I) или подгруппе (IA) пептидная связь между А 35 и А 36 может быть заменена псевдопептидной связью, где А 35-А 36 может быть Lys-(CH2-NH)Tyr или Lys-(CH2-N(AcTyr. В формуле (I) или подгруппе (IA) А 34 предпочтительно представляет собой 4Hyp. В формуле (I) или подгруппе (IA) HN-CHCH2)n-N(R4R5-С(O) предпочтительно представляет собой Lys (N-C(О)-(СН 2)12-CH3). Предпочтительными соединениями формулы (I) или подгруппы (IA) являются следующие соединения: Подробное описание изобретения Используемый в настоящем изобретении термин "аминокислота" относится к любой природной или неприродной аминокислоте, включая, помимо прочего, -аминокислоты, -аминокислоты или аминокислоты, которая также может являться D- или L-аминокислотой, если не указано иное. За исключением N-концевой аминокислоты, все сокращения аминокислот (например, Ala) в настоящем описании имеют структуру -NH-C(R)(R')-СО-, где все R и R', независимо, являются водородом или боковой цепью аминокислоты (например, в случае Ala R=CH3, a R'=H) или R и R' могут быть соединены с формированием кольцевой системы. В случае N-концевой аминокислоты сокращение обозначает структуру (R2R3)-N-C(R)(R')-СО-, где R2 и R3 имеют значение, определенное в формуле (I). Пептид настоящего изобретения также обозначается согласно другому формату, например[Pro34]hNPY(1-36)-NH2 (SEQ ID NO: 48), причем замещенные аминокислоты в природной последовательности указаны в квадратных скобках, например Pro вместо Gln в hNPY. Обозначение "NH2" в(SEQ ID NO: 49) обозначает форму свободной кислоты. Следующий перечень некоторых сокращений, используемых в настоящем изобретении, представлен для простоты поиска, впрочем, любое сокращение, используемое, но не определенное в настоящем изобретении, не используется в противоречии с его общепризнанными значениями. Греческая буква пси используется в настоящем изобретении для обозначения того, что пептидная связь была заменена псевдопептидной связью. При обозначении аминокислотной последовательности терминиспользуется в формате A-(X-X')-В, где А представляет собой аминоацильный радикал,карбонильная группа которого была модифицирована до X, а В представляет собой аминоацильный радикал, аминогруппа которого была модифицирована до X', X и X' показаны в виде последовательностей символов элементов, разделенных связью, например Lys-(CH2-NH)-Tyr."Алкил" относится к углеводородной группе, содержащей один или несколько атомов углерода, в которой некоторое количество атомов углерода, если таковые присутствуют, соединены одинарными связями, примеры которых включают, помимо прочего, метил, этил, пропил и бутил. Алкильная углеводородная группа может быть линейной или содержать одну или несколько боковых цепей или циклических групп, примеры которых включают, помимо прочего, изопропил и трет-бутил."Замещенный алкил" относится к алкилу, в котором один или несколько атомов водорода углеводородной группы замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена (например, фтора, хлора, брома или йода), ОН, CN, SH, NH2, NHCH3, NO2, (C1-2)алкила, замещенного 1-6 атомами галогена, CF3, OCH3, OCF3 и (СН 2)0-4-СООН. В различных вариантах осуществле-8 019498"Гетероалкил" относится к алкилу, в котором один или несколько атомов углерода в углеводородной группе замещены одним или несколькими из следующих атомов или групп: амино, амидо, О, S, N или карбонил. В различных вариантах осуществления присутствуют 1 или 2 гетероатома."Замещенный гетероалкил" относится к гетероалкилу, в котором один или несколько атомов водорода углеводородной группы замещены одним или несколькими заместителями, выбранными из группы,состоящей из галогена (т.е. фтора, хлора, брома и йода), ОН, CN, SH, NH2, NHCH3, NO2, (C1-2)алкила,замещенного 1-6 атомами галогена, CF3, OCH3, OCF3 и (СН 2)0-4-СООН. В различных вариантах осуществления присутствуют 1, 2, 3 или 4 заместителя."Алкенил" относится к углеводородной группе, состоящей из двух или более атомов углерода, в которой присутствует одна или несколько двойных углерод-углеродных связей, примеры которой включают, помимо прочего, винил, аллил, бутенил и пропенил. Алкенильная углеводородная группа может быть линейной или содержать одну или несколько боковых цепей или циклических групп, примеры которых включают, помимо прочего, н-бутенил или т-бутенил и н-пентенил или циклопентенил."Замещенный алкенил" относится к алкенилу, в котором один или несколько атомов водорода замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена (т.е. фтора, хлора, брома и йода), ОН, CN, SH, NH2, NHCH3, NO2, (С 1-2)алкила, замещенного 1-6 атомами галогена, CF3, OCH3, OCF3 и (СН 2)0-4-СООН. В различных вариантах осуществления присутствуют 1, 2, 3 или 4 заместителя."Арил" относится к необязательно замещенной ароматической группе по меньшей мере с одним кольцом, имеющим конъюгированную -электронную систему, содержащую до двух конъюгированных или конденсированных кольцевых систем. Арил включает, помимо прочего, карбоциклическую арильную, гетероциклическую арильную и биарильную группы. Предпочтительно арил является 5- или 6 членным кольцом. Предпочтительные атомы для гетероциклического арила включают, помимо прочего,один или несколько атомов серы, кислорода и азота. Примеры арила включают, помимо прочего, фенил,1-нафтил, 2-нафтил, индол, хинолин, 2-имидазол и 9-антрацен. Заместители арила выбраны из группы,состоящей из (C1-4)алкила, (C1-4)алкокси, галогена (т.е. фтора, хлора, брома и йода), ОН, CN, SH, NH2,NO2, (С 1-2)алкила, замещенного 1-5 атомами галогена, CF3, OCF3 и (СН 2)0-4-СООН. В различных вариантах осуществления присутствуют 1, 2, 3 или 4 заместителя."Алкиларил" означает "алкил", присоединенный к "арилу", как определено выше. Термин "циклоалкил" включает моноциклоалкильную группу или бициклоалкильную группу с указанным количеством атомов углерода, известные специалистам в данной области. Термин "гетероцикл" включает в себя моноциклические и бициклические системы, содержащие один или несколько гетероатомов, таких как атомы кислорода, азота и/или серы. Указанные кольцевые системы могут быть ароматическими, такими как, например, пиридин, индол, хинолин, пиримидин, тиофен (также известный как тиенил), фуран, бензотиофен, тетразол, дигидроиндол, индазол, Nформилиндол, бензимидазол, тиазол и тиадиазол. Указанные кольцевые системы также могут быть неароматическими, такими как, например, пирролидин, пиперидин, морфолин и т.п. Синтез. Соединения настоящего изобретения могут быть и были получены с применением методик, раскрытых в настоящих примерах, а также методик, известных из уровня техники. Например, полипептидная область аналога NPY может быть химически или биохимически синтезирована и/или модифицирована; см., например, Stewart, J.M., et al., Solid Phase Synthesis, Pierce Chemical Co., 2-е изд. (1984); а также см., например, Sambrook et al., Molecular Cloning, A Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory Press (1989) по поводу примеров методик биохимического синтеза, включающего введение нуклеиновой кислоты в клетку и экспрессию нуклеиновых кислот. Примеры приводятся в целях иллюстрации и не должны рассматриваться как ограничение объема настоящего изобретения каким-либо образом. Пример 1. [Aib10, 4Hyp34]hNPY(1-36)-NH2. Указанный в заголовке пептид синтезировали с использованием Fmoc-химии. С-концевую часть пептида (остатки 18-36) синтезировали на пептидном синтезаторе ABI 433 (Applied Biosystems, FosterCity, CA, USA) в масштабе 1,0 ммоль. В реакционную ячейку вносили 1,37 г 0,73 ммоль/смолы Rink Amide МВНА (Novabiochem, San Diego, CA, USA). Затем смолу обрабатывали 10 мл NMP в течение 15 мин до набухания смолы. При синтезе пептида использовали методику ABI FastMoc 1,0. Каждый цикл состоял из снятия N-концевых Fmoc-групп с использованием 20% пиперидина с последующей тщательной промывкой в NMP. Затем предварительно снаряженные 1,0 ммоль картриджи с каждой аминокислотой растворяли в 0,45 М HOBT/HBTU. По истечении достаточного времени для растворения аминокислоты ее автоматически переносили в активационную ячейку. Еще два 1,0 ммоль картриджа с аминокислотой растворяли и переносили в активационную ячейку с использованием, таким образом, в общей сложности 3 экв. аминокислоты в стадии присоединения. Затем в активационную ячейку вносили 3 мл 2 М раствора DIPEA, в общей сложности до 6 экв. DIPEA. Затем всю полученную смесь наносили на смолу и проводили перемешивание в течение 15 мин. Реакционную ячейку освобождали, промывали NMP и затем проводили вторую стадию присоединения. После второй стадии присоединения смолу снова тщательно промывали. Каждую последующую аминокислоту присоединяли аналогичным способом. После стадии присоединения первого остатка Tyr в каждой из следующих 4 стадий присоединения, а также в каждой стадии присоединения Arg, смолу блокировали 5 мл блокирующего раствора (0,5 М уксусный ангидрид/0,13 М DIPFA/0,01 М НОВТ) для защиты неацилированных групп смолы. В стадиях присоединения использовали следующие аминокислотные картриджи: цикл 1) Fmoc-Tyr(tBu)-ОН; цикл 2) Fmoc-Arg(Pbf)-ОН; цикл 3) Fmoc-4Hyp-OH; цикл 4)Fmoc-Asn(Trt)-ОН; цикл 9) Fmoc-Ile-OH; цикл 10) Fmoc-Tyr(tBu)-OH; цикл 11) Fmoc-His (Trt)-ОН; цикл 12) Fmoc-Arg(Pbf)-ОН; цикл 13) Fmoc-Leu-OH; цикл 14) Fmoc-Ala-OH; цикл 15) Fmoc-Ser(tBu)-OH; цикл 16) Fmoc-Tyr(tBu)-ОН; цикл 17) Fmoc-Tyr(tBu)-ОН; цикл 18) Fmoc-Arg(Pbf)-ОН и цикл 19) Fmoc-Ala-OH. После последней стадии присоединения смолу промывали NMP, после чего проводили стандартное снятие N-концевой Fmoc-группы с последующей промывкой NMP и затем DCM. После сборки С-концевой части пептидного скелета (остатки 18-36) для синтеза N-концевой части пептида использовали только одну десятую часть смолы (0,1 ммоль), а остальную смолу сохранили. Nконцевую часть указанного в заголовке пептида (остатки 1-17) синтезировали с использованием Fmocхимии при помощи СВЧ на пептидном синтезаторе Liberty (СЕМ, Matthews, NC, USA) в масштабе 0,1 ммоль. Смолу из предыдущего синтеза помещали в коническую пробирку объемом 50 мл вместе с 15 мл ДМФА и вводили в положение смолы на синтезаторе. Затем смолу количественно переносили в реакционную ячейку посредством автоматизированного процесса. Использовали стандартную методику синтезаLiberty для синтеза в масштабе 0,1 ммоль. Данная методика включает снятие N-концевой Fmoc-группы путем первоначальной обработки 7 мл 20% пиперидина, содержащего 0,1 М НОВТ, в ДМФА. Начальную стадию снятия защиты проводили в течение 30 с при СВЧ-облучении (45 Вт, максимальная температура 75 С) и барботировании азотом (цикл 3 с/с перерывом 7 с). Затем реакционную ячейку освобождали и проводили вторую обработку пиперидином, идентичную первой обработке, за исключением того,что ее продолжительность составляла 3 мин. Затем со смолы удаляли пиперидин и тщательно промывали несколько раз ДМФА. Затем добавляли защищенную аминокислоту, Fmoc-Met-OH (2,5 мл, 5 экв.), полученную в виде 0,2 М стокового раствора в ДМФА, с последующим добавлением 1,0 мл 0,45 М (4,5 экв.) HBTU в ДМФА. Затем добавляли 0,5 мл 2 М (10 экв.) DIPEA в NMP. Стадию присоединения проводили в течение 5 мин при использовании СВЧизлучения мощностью 20 Вт, при максимальной температуре 75 С и аналогичного режима барботирования азотом. После первой стадии присоединения реакционную ячейку освобождали от жидкой фазы и повторяли стадию присоединения. Затем аналогично циклу 1 инициировали цикл 2. Все аминокислоты вводили аналогичным образом и в ходе синтеза всей последовательности использовали стратегию двойного присоединения. Присоединение остатков 9-10 (Gly-Aib) включало применение методики блокирования непосредственно после стадии присоединения. Блокирование проводили, добавляя 7 мл 0,5 М уксусного ангидрида, содержащего 0,015 М НОВТ в NMP, вместе с 2 мл 2 М раствора DIPEA, при использовании многостадийной методики с обработкой СВЧ: мощность 50 Вт в течение 30 с (максимальная температура 65 С), с последующим выключением СВЧ-излучения на 30 с, затем второй раунд обработки СВЧ-излучением в течение 30 с (50 Вт), а затем снова выключение СВЧ-излучения на 30 с. После этого со смолы удаляли жидкую фазу и полностью промывали смолу ДМФА. Использовали следующие аминокислоты (Advanced Chemtech,Louisville, KY, USA): цикл 20) Fmoc-Met-OH; цикл 21) Fmoc-Asp(OtBu)-OH; цикл 22) Fmoc-Glu(OtBu)ОН; цикл 23) Fmoc-Ala-OH; цикл 24) Fmoc-Pro-OH; цикл 25) Fmoc-Ala-OH; цикл 26) Fmoc-Asp(OtBu)OH; цикл 27) Fmoc-Aib-OH; цикл 28) Fmoc-Gly-OH; цикл 29) Fmoc-Pro-OH; цикл 30) Fmoc-Asn(Trt)-ОН; цикл 31) Fmoc-Asp(OtBu)-ОН; цикл 32) Fmoc-Pro-OH; цикл 33) Fmoc-Lys(Boc)-ОН; цикл 34) FmocSer(tBu)-ОН; цикл 35) Fmoc-Pro-OH; цикл 36) Fmoc-Tyr(tBu)-OH. После того как пептидный скелет полностью синтезировали, стандартную обработку пиперидином использовали для удаления N-концевой Fmoc-группы при использовании стандартной методики снятия защиты, описанной выше. Затем смолу тщательно промывали ДМФА, после чего переносили обратно в коническую пробирку объемом 50 мл, используя ДМФА в качестве растворителя для переноса. С пептида снимали защитные группы и отсоединяли от смолы при помощи обработки 5 мл следующего реагента: 5% TIS, 2% воды, 5% (мас./об.) DTT и 88% ТФУ, с последующим перемешиванием в течение 3,5 ч. Фильтрат собирали в 45 мл холодного безводного диэтилового эфира. Твердую фазу осаждали в течение 10 мин при 3500 об/мин в охлаждаемой центрифуге. Эфир удаляли с осадка и пептид ресуспендировали в новой порции эфира. Обработку эфиром проводили в общей сложности 2 раза. После последней промывки эфиром пептид оставляли на воздухе для удаления остатков эфира. Осадок пептида ресуспендировали в 8 мл ацетонитрила, а затем в 8 мл деионизированной воды до полного растворения. Далее раствор пептида анализировали с помощью масс-спектрометрии. Масс-спектрометрический анализ с использованием ионизации электрораспылением выявил основной продукт, имеющий массу 4212,1, что соответствовало целевому продукту. Анализ с помощью аналитической ВЭЖХ при использовании колонки С 18 2504,6 мм (Phenomenex, Torrance, CA, USA) с использованием градиента ацетонитрила 2-60% (0,1% ТФУ) в течение 30 мин выявил основной продукт с чистотой 45%. Затем неочищенный пептид очищали с помощью препаративной ВЭЖХ на колонке С 18 в обращенной фазе, используя 1060% ацетонитрил (0,1% ТФУ) в течение 50 мин при скорости потока 10 мл/мин. Очищенный продукт анализировали с помощью ВЭЖХ на предмет чистоты (99%) и с помощью масс-спектрометрии (4212,8 Да), показавшей хорошее соответствие экспериментальной массы расчетной массе 4212,7. Затем пептид лиофилизировали, получив в результате 39 мг очищенного продукта, что соответствует 9% выходу. Пример 2. [Aib17, 4Hyp34]hNPY(1-36)-NH2. Указанный в заголовке пептид синтезировали, используя Fmoc-химию. С-концевую часть пептида(остатки 18-36) синтезировали на пептидном синтезаторе ABI 433A (Applied Biosystems, Foster City, CA,USA) в масштабе 1,0 ммоль. В реакционную ячейку вносили 1,37 г 0,73 ммоль/смолы Rink Amide МВНА(Novabiochem, San Diego, CA, USA). Затем смолу обрабатывали 10 мл NMP в течение 15 мин до набухания смолы. При синтезе пептида использовали методику ABI FastMoc 1,0. Каждый цикл состоял из снятия N-концевых Fmoc-групп с использованием 20% пиперидина с последующей тщательной промывкой в NMP. Затем предварительно снаряженные 1,0 ммоль картриджи с каждой аминокислотой растворяли в 0,45 М HOBT/HBTU. После растворения аминокислоты ее автоматически переносили в активационную ячейку. Еще два 1,0 ммоль картриджа с аминокислотой растворяли и переносили в активационную ячейку с использованием, таким образом, в общей сложности 3 экв. аминокислоты в стадии присоединения. Затем в активационную ячейку вносили 3 мл 2 М раствора DIPEA, в общей сложности до 6 экв. DIPEA. Затем всю полученную смесь наносили на смолу и проводили перемешивание в течение 15 мин. Реакционную ячейку освобождали, промывали NMP и затем проводили вторую стадию присоединения. После второй стадии присоединения смолу снова тщательно промывали. Каждую последующую аминокислоту присоединяли аналогичным способом. После стадии присоединения первого остатка Tyr в каждой из следующих четырех стадий присоединения, а также в каждой стадии присоединения Arg смолу блокировали 5 мл блокирующего раствора (0,5 М уксусный ангидрид/0,13 М DIPEA/0,01 М НОВТ) для защиты неацилированных групп смолы. В стадиях присоединения использовали следующие аминокислотные картриджи: цикл 1) Fmoc-Tyr(tBu)-ОН; цикл 2) Fmoc-Arg(Pbf)-ОН; цикл 3) Етос-4Hyp-ОН; цикл 4) Fmoc-Arg(Pbf)-ОН; цикл 5) Fmoc-Thr(tBu)-OH; цикл 6) Fmoc-Ile-OH; цикл 7) Fmoc-Leu-OH; цикл 8)Fmoc-Asn(Trt)-ОН; цикл 9) Fmoc-Ile-OH; цикл 10) Fmoc-Tyr (tBu)-ОН; цикл 11) Fmoc-His (Trt)-ОН; цикл 12) Fmoc-Arg(Pbf)-OH; цикл 13) Fmoc-Leu-OH; цикл 14) Fmoc-Ala-OH; цикл 15) Fmoc-Ser(tBu)-OH; цикл 16) Fmoc-Tyr (tBu)-ОН; цикл 17) Fmoc-Tyr (tBu)-ОН; цикл 18) Fmoc-Arg(Pbf)-ОН и цикл 19) Fmoc-AlaOH. После последней стадии присоединения смолу промывали NMP, после чего проводили стандартное снятие N-концевой Fmoc-группы с последующей промывкой NMP и затем DCM. После сборки Сконцевой части пептидного скелета (остатки 18-36) для синтеза N-концевой части пептида использовали одну десятую часть смолы (0,1 ммоль), а остальную смолу сохранили. N-концевую часть указанного в заголовке пептида (остатки 1-17) синтезировали с использованием Fmoc-химии при помощи СВЧ на пептидном синтезаторе Liberty (СЕМ, Matthews, NC, USA) в масштабе 0,1 ммоль. Смолу из предыдущего синтеза помещали в коническую пробирку объемом 50 мл вместе с 15 мл ДМФА и вводили в положение смолы на синтезаторе. Затем смолу количественно переносили в реакционную ячейку посредством автоматизированного процесса. Использовали стандартную методику синтеза Liberty для синтеза в масштабе 0,1 ммоль, включающую снятие N-концевой Fmoc-группы путем первоначальной обработки 7 мл 20% пиперидина, содержащего 0,1 М НОВТ, в ДМФА. Начальная стадия снятия защиты продолжалась в течение 30 с при СВЧ-облучении (45 Вт, максимальная температура 75 С) и барботировании азотом (цикл 3 с/с перерывом 7 с). Затем реакционную ячейку освобождали от жидкой фазы и проводили вторую обработку пиперидином, идентичную первой обработке, в течение 3 мин. Затем смолу осушали и тщательно промывали несколько раз ДМФА. Затем добавляли защищенную аминокислоту, Fmoc-Aib-OH (2,5 мл,5 экв.), полученную в виде 0,2 М стокового раствора в ДМФА, с последующим добавлением 1,0 мл 0,45 М (4,5 экв.) HBTU в ДМФА. Затем добавляли 0,5 мл 2 М (10 экв.) DIPEA в NMP. Стадию присоединения проводили в течение 5 мин при использовании СВЧ-излучения мощностью 20 Вт, при максимальной температуре 75 С и аналогичного режима барботирования азотом. После первой стадии присоединения реакционную ячейку освобождали от жидкой фазы и повторяли стадию присоединения. Затем инициировали цикл 2, аналогичный циклу 1. Все аминокислоты вводили аналогичным образом и в ходе синтеза всей последовательности использовали стратегию двойного присоединения. Присоединение остатков 16-17 (Asp-Aib) включало применение методики блокирования непосредственно после стадии присоединения. Блокирование проводили, добавляя 7 мл 0,5 М уксусного ангидрида, содержащего 0,015 М НОВТ в NMP, вместе с 2 мл 2 М раствора DIPEA, при использовании многостадийной методики с обработкой СВЧ: мощность 50 Вт в течение 30 с (максимальная температура 65 С), с последующим выключением СВЧ-излучения на 30 с, затем второй раунд обработки СВЧ-излучением в течение 30 с (50 Вт), а затем снова выключение СВЧ-излучения на 30 с. После этого смолу осушали и ние 30 с (50 Вт), а затем снова выключение СВЧ-излучения на 30 с. После этого смолу осушали и полностью промывали смолу ДМФА. Использовали следующие аминокислоты (Advanced Chemtech, Louisville,KY, USA): цикл 20) Fmoc-Aib-OH; цикл 21) Fmoc-Asp (OtBu)-ОН; цикл 22) Fmoc-Glu(OtBu)-OH; цикл 23) Fmoc-Ala-OH; цикл 24) Fmoc-Pro-OH; цикл 25) Fmoc-Ala-OH; цикл 26) Fmoc-Asp (OtBu)-ОН; цикл 27) Fmoc-Glu(OtBu)-OH; цикл 28) Fmoc-Gly-OH; цикл 29) Fmoc-Pro-OH; цикл 30) Fmoc-Asn(Trt)-OH; цикл 31) Fmoc-Asp(OtBu)-ОН; цикл 32) Fmoc-Pro-OH; цикл 33) Fmoc-Lys(Boc)-ОН; цикл 34) FmocSer(tBu)-ОН; цикл 35) Fmoc-Pro-OH и цикл 36) Fmoc-Tyr(tBu)-ОН. После того как пептидный скелет полностью синтезировали, стандартную обработку пиперидином использовали для удаления N-концевой Fmoc-группы при использовании стандартной методики снятия защиты, описанной выше. Затем смолу тщательно промывали ДМФА, после чего переносили обратно в коническую пробирку объемом 50 мл, используя ДМФА в качестве растворителя для переноса. С пептида снимали защитные группы и отсоединяли от смолы при помощи обработки 5 мл следующего реагента: 5% TIS, 2% воды, 5% (мас./об.) DTT и 88% ТФУ, с последующим перемешиванием в течение 3,5 ч. Фильтрат собирали в 45 мл холодного безводного диэтилового эфира. Твердую фазу осаждали в течение 10 мин при 3500 об/мин в охлаждаемой центрифуге. Эфир удаляли с осадка и пептид ресуспендировали в новой порции эфира. Обработку эфиром проводили в общей сложности 2 раза. После последней промывки эфиром пептид оставляли на воздухе для удаления остатков эфира. Осадок пептида ресуспендировали в 8 мл ацетонитрила, а затем в 8 мл деионизированной воды до полного растворения. Далее раствор пептида анализировали с помощью масс-спектрометрии. Масс-спектрометрический анализ с использованием ионизации электрораспылением выявил основной продукт, имеющий массу 4210,8, что соответствовало целевому продукту. Анализ с помощью аналитической ВЭЖХ при использовании колонки С 18 2504,6 мм (Phenomenex, Torrance, CA, USA) с использованием градиента ацетонитрила 2-60% (0,1% ТФУ) в течение 30 мин выявил основной продукт с чистотой 54%. Затем неочищенный пептид очищали с помощью препаративной ВЭЖХ на колонке С 18 в обращенной фазе, используя 1060% ацетонитрил (0,1% ТФУ) в течение 50 мин при скорости потока 10 мл/мин. Очищенный продукт анализировали с помощью ВЭЖХ на предмет чистоты (99%) и с помощью масс-спектрометрии (4210,6 Да), показавшей хорошее соответствие экспериментальной массы расчетной массе 4210,6. Затем пептид лиофилизировали, получив в результате 53 мг очищенного продукта, что соответствует 13% выходу. Пример 3. [Aib11,17, 4Hyp34]hNPY(1-36)-NH2. Указанный в заголовке пептид синтезировали, используя Fmoc-химию. С-концевую часть пептида(остатки 18-36) синтезировали на пептидном синтезаторе ABI 433 А (Applied Biosystems, Foster City, CA,USA) в масштабе 1,0 ммоль. В реакционную ячейку вносили 1,37 г 0,73 ммоль/смолы Rink Amide МВНА(Novabiochem, San Diego, CA, USA). Затем смолу обрабатывали 10 мл NMP в течение 15 мин до набухания смолы. При синтезе пептида использовали методику ABI FastMoc 1,0. Каждый цикл состоял из снятия N-концевых Fmoc-групп с использованием 20% пиперидина с последующей тщательной промывкой в NMP. Затем предварительно снаряженные 1,0 ммоль картриджи с каждой аминокислотой растворяли в 0,45 М HOBT/HBTU. После растворения аминокислоты ее автоматически переносили в активационную ячейку. Еще два 1,0 ммоль картриджа с аминокислотой растворяли и переносили в активационную ячейку с использованием, таким образом, в общей сложности 3 экв. аминокислоты в стадии присоединения. Затем в активационную ячейку вносили 3 мл 2 М раствора DIPEA, в общей сложности до 6 экв. DIPEA. Затем всю полученную смесь наносили на смолу и проводили перемешивание в течение 15 мин. Реакционную ячейку освобождали, промывали NMP и затем проводили вторую стадию присоединения. После второй стадии присоединения смолу снова тщательно промывали. Каждую последующую аминокислоту присоединяли аналогичным способом. После стадии присоединения первого остатка Tyr в каждой из следующих четырех стадий присоединения, а также в каждой стадии присоединения Arg смолу блокировали 5 мл блокирующего раствора (0,5 М уксусный ангидрид/0,13 М DIPEA/0,01 М НОВТ) для защиты неацилированных групп смолы. В стадиях присоединения использовали следующие аминокислотные картриджи: цикл 1) Fmoc-Tyr(tBu)-ОН; цикл 2) Fmoc-Arg(Pbf)-ОН; цикл 3) Fmoc-4Hyp-OH; цикл 4) Fmoc-Arg(Pbf)-ОН; цикл 5) Emoc-Thr(tBu)-OH; цикл 6) Fmoc-Ile-OH; цикл 7) Emoc-Leu-OH; цикл 8)Fmoc-Asn(Trt)-ОН; цикл 9) Fmoc-Ile-OH; цикл 10) Fmoc-Tyr(tBu)-ОН; цикл 11) Fmoc-His(Trt)-ОН; цикл 12) Fmoc-Arg(Pbf)-OH; цикл 13) Fmoc-Leu-OH; цикл 14) Fmoc-Ala-OH; цикл 15) Fmoc-Ser(tBu)-OH; цикл 16) Fmoc-Tyr(tBu)-ОН; цикл 17) Fmoc-Tyr(tBu)-ОН; цикл 18) Fmoc-Arg(Pbf)-ОН и цикл 19) Fmoc-Ala-OH. После последней стадии присоединения смолу промывали NMP, после чего проводили стандартное снятие N-концевой Fmoc-группы с последующей промывкой NMP и затем DCM. После сборки С-концевой части пептидного скелета (остатки 18-36) для синтеза N-концевой части пептида использовали только одну десятую часть смолы (0,1 ммоль), а остальную смолу сохранили. Nконцевую часть указанного в заголовке пептида (остатки 1-17) синтезировали с использованием Fmocхимии при помощи СВЧ на пептидном синтезаторе Liberty (СЕМ, Matthews, NC, USA) в масштабе 0,1 ммоль. Смолу из предыдущего синтеза помещали в коническую пробирку объемом 50 мл вместе с 15 мл ДМФА и вводили в положение смолы на синтезаторе. Затем смолу количественно переносили в реакционную ячейку посредством автоматизированного процесса. Использовали стандартную методику синтезаLiberty для синтеза в масштабе 0,1 ммоль. Данная методика включает снятие N-концевой Fmoc-группы путем первоначальной обработки 7 мл 20% пиперидина, содержащего 0,1 М НОВТ, в ДМФА. Начальная стадия снятия защиты продолжалась в течение 30 с при СВЧ-облучении (45 Вт, максимальная температура 75 С) и барботировании азотом (цикл 3 с/с перерывом 7 с). Затем реакционную ячейку освобождали от жидкой фазы и проводили вторую обработку пиперидином, идентичную первой обработке, за исключением того, что продолжительность обработки составляла 3 мин. Затем со смолы удаляли пиперидин и тщательно промывали несколько раз ДМФА. Затем добавляли защищенную аминокислоту, Fmoc-Aib-OH(2,5 мл, 5 экв.), полученную в виде 0,2 М стокового раствора в ДМФА, с последующим добавлением 1,0 мл 0,45 М (4,5 экв.) HBTU в ДМФА. Затем добавляли 0,5 мл 2 М (10 экв.) DIPEA в NMP. Стадию присоединения проводили в течение 5 мин при использовании СВЧ-излучения мощностью 20 Вт, при максимальной температуре 75 С и аналогичного режима барботирования азотом. После первой стадии присоединения реакционную ячейку освобождали от жидкой фазы и повторяли стадию присоединения. Затем инициировали цикл 2, аналогичный циклу 1. Все аминокислоты вводили аналогичным образом, при этом в ходе всего процесса использовали стратегию двойного присоединения. Присоединение остатков 10-11 и 16-17 (Glu-Aib и Asp-Aib) включало применение методики блокирования непосредственно после стадии присоединения. Блокирование проводили, добавляя 7 мл 0,5 М уксусного ангидрида,содержащего 0,015 М НОВТ в NMP, вместе с 2 мл 2 М раствора DIPEA, при использовании многостадийной методики с обработкой СВЧ: мощность 50 Вт в течение 30 с (максимальная температура 65 С) с последующим выключением СВЧ-излучения на 30 с, затем второй раунд обработки СВЧ-излучением в течение 30 с (50 Вт), а затем снова выключение СВЧ-излучения на 30 с. После этого со смолы удаляли жидкую фазу и полностью промывали смолу ДМФА. Использовали следующие аминокислоты (AdvancedChemtech, Louisville, KY, USA): цикл 20) Fmoc-Aib-OH; цикл 21) Fmoc-Asp(OtBu)-OH; цикл 22) FmocGlu(OtBu)-ОН; цикл 23) Fmoc-Ala-OH; цикл 24) Emoc-Pro-OH; цикл 25) Fmoc-Ala-OH; цикл 26) FmocAib-OH; цикл 27) Fmoc-Glu(OtBu)-OH; цикл 28) Fmoc-Gly-OH; цикл 29) Fmoc-Pro-OH; цикл 30) FmocAsn(Trt)-ОН; цикл 31) Fmoc-Asp(OtBu)-OH; цикл 32) Fmoc-Pro-OH; цикл 33) Fmoc-Lys(Boc)-OH; цикл 34) Fmoc-Ser (tBu)-ОН; цикл 35) Fmoc-Pro-OH и цикл 36) Fmoc-Tyr(tBu)-OH. После того как пептидный скелет полностью синтезировали, стандартную обработку пиперидином использовали для удаления N-концевой Fmoc-группы при использовании стандартной методики снятия защиты, описанной выше. Затем смолу тщательно промывали ДМФА, после чего переносили обратно в коническую пробирку объемом 50 мл, используя ДМФА в качестве растворителя для переноса. С пептида снимали защитные группы и отсоединяли от смолы при помощи обработки 5 мл следующего реагента: 5% TIS, 2% воды, 5% (мас./об.) DTT и 88% ТФУ с последующим перемешиванием в течение 3,5 ч. Фильтрат собирали в 45 мл холодного безводного этилового эфира. Твердую фазу осаждали в течение 10 мин при 3500 об/мин в охлаждаемой центрифуге. Эфир удаляли с осадка и пептид ресуспендировали в новой порции эфира. Обработку эфиром проводили в общей сложности 2 раза. После последней промывки эфиром пептид оставляли на воздухе для удаления остатков эфира. Осадок пептида ресуспендировали в 8 мл ацетонитрила, а затем в 8 мл деионизированной воды до полного растворения. Далее раствор пептида анализировали с помощью масс-спектрометрии. Масс-спектрометрический анализ с использованием ионизации электрораспылением выявил основной продукт, имеющий массу 4180,7, что соответствовало целевому продукту. Анализ с помощью аналитической ВЭЖХ при использовании колонки С 18 2504,6 мм (Phenomenex, Torrance, CA, USA) с использованием градиента ацетонитрила 2-60% (0,1% ТФУ) в течение 30 мин выявил основной продукт с чистотой 68%. Затем неочищенный пептид очищали с помощью препаративной ВЭЖХ на колонке С 18 в обращенной фазе, используя 1060% ацетонитрил (0,1% ТФУ) в течение 50 мин при скорости потока 10 мл/мин. Очищенный продукт анализировали с помощью ВЭЖХ на предмет чистоты (99%) и с помощью масс-спектрометрии (4180,5 Да), при этом экспериментальная масса соответствовала расчетной массе 4180,6. Затем пептид лиофилизировали, получив в результате 53 мг очищенного продукта, что соответствует 13% выходу. Другие соединения изобретения могут быть получены средним специалистом в данной области с применением методик синтеза, аналогичных раскрытым в предыдущих примерах. Физические данные соединений, представленных в настоящем изобретении, приведены в табл. 1.In vitro радиолигандные анализы связывания рецепторов NPY-Y1 и NPY-Y2. Линии клеток нейробластомы человека, SK-N-MC и SK-N-BE2 (Американская коллекция типовых культур, Rockville, MD, USA), экспрессирующих рецепторы NPY-Y1 и NPY-Y2 соответственно, культивировали в ЕМЕМ, содержащей 10% эмбриональной сыворотки теленка и 5% экстракта куриных эмбрионов, и инкубировали при 37 С в увлажненной атмосфере 95% воздуха и 5% СО 2. Для in vitro анализов связывания радиолигандов рецепторами NPY-Y1 и NPY-Y2 получали подходящие клетки (SK-N-MC для NPY-Y1; SK-N-BE2 для NPY-Y2), гомогенизировали их в 20 мл охлажденного на льду 50 мМ Трис-HCl при помощи Brinkman Polytron (Westbury, NY, USA) (режим 6, 15 с). Гомогенаты дважды промывали при помощи центрифугирования (39000 g/10 мин) и полученные осадки ресуспендировали в 50 мМ Трис-HCl, содержащего 2,5 мМ MgCl2, 0,1 мг/мл бацитрацина (Sigma Chemical,St. Louis, MO, USA) и 0,1% BSA. Для анализа аликвоты (0,4 мл) вышеуказанных суспензий инкубировали с 0,05 нМ [125I]PYY (2200 Ки/ммоль, Perkin-Elmer, Boston, MA), с и без 0,05 мл немеченых конкурентных тестируемых пептидов. После инкубации в течение 100 мин (25 С) связанный [125I]PYY отделяли от свободного путем быстрой фильтрации через фильтры GF/C (Brandel, Gaithersburg, MD, USA), предварительно пропитанные 0,3% полиэтиленимином. Затем фильтры промывали три раза 5 мл аликвотами охлажденного на льду 50 мМ Трис-HCl и определяли радиоактивность связанного радиоизотопа, улавливаемого на фильтрах, при помощи гамма-спектрометрии (Wallac LKB, Gaithersburg, MD, USA). Специфичное связывание определяли как разность общего количества связанного [125I]PYY и количества [125I]PYY, связанного в присутствии 1000 нМ PYY (Bachem, Torrence, CA, USA). Константы ингибирования (Ki) вычисляли, используя известное уравнение Ченга-Пруссофа, и указанные данные, а также селективность указанных соединений относительно NPY-Y1 и NPY-Y2 приведены в табл. 2. Каждое из соединений примеров 1-38 и 40-45 подвергали вышеописанным радиолигандным анализам, и при этом было установлено, что почти все указанные соединения имели Ki ниже 100 нМ, причем некоторые из представленных соединений имели Ki в суб-нМ диапазоне. Также было установлено, что почти все указанные соединения чрезвычайно селективно связываются с NPY-Y1 по сравнению с NPYY2. Таблица 2 Введение. Пептиды настоящего изобретения могут быть предоставлены в форме фармацевтически приемлемых солей. Примеры таких солей включают, помимо прочего, соли, образованные с органическими кислотами (например, уксусной, молочной, малеиновой, лимонной, яблочной, аскорбиновой, янтарной,бензойной, метансульфоновой, толуолсульфоновой или памовой кислотой), неорганическими кислотами(например, соляной кислотой, серной кислотой или фосфорной кислотой), а также полимерными кислотами (например, дигалловая кислота, карбоксилметилцеллюлоза, полимолочная, полигликолевая или сополимеры полимолочной-гликолевой кислот). Типичный способ получения соли пептида настоящего изобретения известен в уровне техники и может быть выполнен с помощью стандартных способов солевого обмена. Соответственно соль ТФУ пептида настоящего изобретения (соль ТФУ получают в результате очистки пептида с использованием препаративной ВЭЖХ при элюции буферными растворами, содержащими ТФУ) может быть превращена в другую соль, такую как ацетат, путем растворения пептида в небольшом количестве 0,25 Н водного раствора уксусной кислоты. Получаемый раствор наносят на полупрепаративную колонку ВЭЖХ (Zorbax, 300 SB, C-8). Колонку элюируют (1) 0,1 Н водным раствором ацетата аммония в течение 0,5 ч, (2) 0,25 Н водным раствором уксусной кислоты в течение 0,5 ч и (3) линейным градиентом (20-100% раствора В в течение 30 мин) при скорости потока 4 мл/мин (раствор А представляет собой 0,25 Н водный раствор уксусной кислоты; раствор В представляет собой 0,25 Н рас- 15019498 твор уксусной кислоты в ацетонитриле/воде, 80:20). Фракции, содержащие пептид, собирают и лиофилизируют до сухого состояния. Дозировка активного компонента в композициях настоящего изобретения может быть различна,впрочем, необходимо, чтобы количество активного компонента было таким, чтобы была получена подходящая дозированная лекарственная форма. Выбранная дозировка зависит от требуемого терапевтического эффекта, пути введения и продолжительности терапии. Как правило, эффективная дозировка относительно активностей настоящего изобретения находится в диапазоне 110-7 - 200 мг/кг/день, предпочтительно 110-4 - 100 мг/кг/день, и может быть введена в виде разовой дозы или разделена на несколько доз. Соединения настоящего изобретения могут быть введены пероральным, перентеральным (например, с помощью внутримышечной, внутрибрюшинной, внутривенной или подкожной инъекции либо импланта), назальным, вагинальным, ректальным, подъязычным или местным путями введения, и могут быть объединены с фармацевтически приемлемыми носителями с получением дозированных лекарственных форм, соответствующих каждому пути введения. Твердые дозированные формы для перорального введения могут включать в себя капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых дозированных формах активное соединение смешано по меньшей мере с одним инертным фармацевтически приемлемым носителем, таким как сахароза, лактоза или крахмал. Такие дозированные формы также могут содержать согласно общепринятым стандартам другие дополнительные вещества, отличные от таких инертных разбавителей, например скользящие вещества, такие как стеарат магния. В случае капсул, таблеток и пилюль дозированные лекарственные формы также могут содержать буферные агенты. Таблетки и пилюли дополнительно могут быть приготовлены с энетеросолюбильными покрытиями. Жидкие дозированные лекарственные формы для перорального введения включают, помимо прочего, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры и т.п., содержащие инертные разбавители, обычно используемые в данной области, такие как вода. Кроме таких инертных разбавителей, композиции также могут включать в себя вспомогательные добавки, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, а также подсластители, ароматизаторы и отдушки. Препараты согласно изобретению для парентерального введения включают, помимо прочего, стерильные водные или неводные растворы, суспензии, эмульсии и т.п. Примерами неводных растворителей или носителей являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и кукурузное масло, желатин, а также инъецируемые органические эфиры, такие как этилолеат. Такие дозированные лекарственные формы также могут содержать вспомогательные добавки, такие как консерванты, смачивающие, эмульгирующие и диспергирующие агенты. Они могут быть стерилизованы, например, путем фильтрации через фильтр, задерживающий бактерии, путем введения стерилизующих агентов, облучения или нагревания указанных композиций. Они могут быть приготовлены в форме стерильных твердых композиций, которые могут быть растворены в стерильной воде или какойлибо другой стерильной инъекционной среде, непосредственно перед применением. Композициями для ректального или вагинального введения предпочтительно являются суппозитории, которые могут содержать, помимо активного вещества, инертные основы, такие как масло какао или воск для суппозиториев. Композиции для назального или подъязычного введения также приготавливают со стандартными инертными основами, хорошо известными в данной области. Кроме того, соединение настоящего изобретения может быть введено в форме композиции с замедленным высвобождением, такой как композиции, описанные в следующих патентах и заявках на патенты. В патенте США 5672659 описаны композиции с замедленным высвобождением, включающие биоактивное средство и полиэфир. В патенте США 5595760 описаны композиции с замедленным высвобождением, включающие биоактивное средство в гелевой форме. В патенте США 5821221 описаны полимерные композиции с замедленным высвобождением, включающие биоактивное средство и хитозан. В патенте США 5916883 описаны композиции с замедленным высвобождением, включающие биоактивное средство и циклодекстрин. В публикации РСТ WO 99/38536 описаны абсорбируемые композиции с замедленным высвобождением биоактивного средства. В публикации РСТ WO 00/04916 описан способ получения микрочастиц, включающих терапевтическое средство, такое как пептид, в процессе "масло в воде". В публикации РСТ WO 00/09166 описаны комплексы, включающие терапевтическое средство,такое как пептид, и фосфорилированный полимер. В публикации РСТ WO 00/25826 описаны комплексы,включающие терапевтическое средство, такое как пептид, и полимер, содержащий неполимеризуемый лактон. Если не определено иное, все технические и научные термины, использованные в настоящем изобретении,имеют такое же значение, в котором они обычно понимаются средним специалистом в данной области техники,к которой относится настоящее изобретение. Кроме того, все публикации, заявки на патенты, патенты и другие источники, указанные в данном изобретении, настоящим полностью включены путем отсылки.n, независимо в каждом случае, представляет собой 1, 2, 3, 4 или 5; каждый из X1, X2, X3, X4 и X5, независимо в каждом случае, представляет собой Н, F, Cl, Br, I,(C1-10)алкил, замещенный (C1-10)алкил, арил, замещенный арил, ОН, CH2NH2, NH2, NO2 или CN; при условии, что соединение содержит по меньшей мере одну замену на неприродную аминокислоту,или его фармацевтически приемлемая соль. 2. Соединение по п.1, в котором А 1 представляет собой Tyr; А 2 представляет собой Pro; А 3 представляет собой Ser или Aib; А 4 представляет собой Lys; А 5 представляет собой Pro; А 6 представляет собой Asp или Aib; А 7 представляет собой Asn или Aib; А 8 представляет собой Pro; А 9 представляет собой Gly или Aib; А 10 представляет собой Glu или Aib; А 11 представляет собой Asp или Aib; А 12 представляет собой Ala или Aib; А 13 представляет собой Pro; А 14 представляет собой Ala или Aib; А 15 представляет собой Glu или Aib; А 16 представляет собой Asp или Aib; А 17 представляет собой Met, А 6 с, Aib или Nle; А 18 представляет собой Ala или Aib; А 19 представляет собой Arg; А 20 представляет собой Tyr; А 21 представляет собой Tyr; А 22 представляет собой Ser или Aib; А 23 представляет собой Ala или Aib; А 24 представляет собой Leu или А 6 с; А 25 представляет собой Arg; А 26 представляет собой His; А 27 представляет собой Tyr; А 28 представляет собой Ile или А 6 с; А 29 представляет собой Asn или Aib; А 30 представляет собой Leu или А 6 с; А 31 представляет собой Ile, А 6 с или Leu; А 32 представляет собой Thr или Aib; А 33 представляет собой Arg; А 34 представляет собой Dhp, 4Hyp, Inp, Nip, hPro, Tic или HN-CHCH2)n-N(R4R5-C(O); А 35 представляет собой Arg, Apc, Lys, 4NH2Phe или 4NH2CH2Phe; А 36 представляет собой Tyr или Aic; А 37 отсутствует;NH2,или его фармацевтически приемлемая соль. 3. Соединение по п.1 или 2, в котором HN-CHCH2)n-N(R4R5-С(O) представляет собойLys(N-C(O)-(СН 2)12-СН 3), или его фармацевтически приемлемая соль. 4. Соединение по п.1, где указанное соединение представляет собой или его фармацевтически приемлемая соль. 5. Соединение по любому из пп.1-3, в котором А 34 представляет собой 4Hyp, или его фармацевтически приемлемая соль. 6. Соединение по п.5, где указанное соединение представляет собой или его фармацевтически приемлемая соль. 7. Соединение по п.1 или 2, в котором пептидная связь между А 35 и А 36 заменена псевдопептидной связью, или его фармацевтически приемлемая соль. 8. Соединение по п.7, в котором А 35, А 36 представляют собой Lys-(CH2-NH)Tyr илиLys-(CH2-N(AcTyr, или его фармацевтически приемлемая соль. 9. Соединение по п.8, где указанное соединение представляет собой или его фармацевтически приемлемая соль. 10. Фармацевтическая композиция, содержащая эффективное количество соединения по любому из пп.1-9 или его фармацевтически приемлемую соль. 11. Фармацевтическая композиция по п.10, дополнительно содержащая фармацевтически приемлемый носитель. 12. Способ лечения нарушения или заболевания, опосредованного связыванием рецептора нейропептида Y, относящегося к сердцу, кровеносным сосудам или почечной системе, такого как спазм сосудов, сердечная недостаточность, шок, гипертрофия сердца, повышенное кровяное давление, стенокардия,- 20019498 инфаркт миокарда, внезапная сердечная смерть, аритмия, болезнь периферических сосудов, нарушение циркуляции жидкости, нарушение массообмена, почечная недостаточность, повышенная активность симпатического нерва, церебральный инфаркт, нейродегенерация, эпилепсия, инсульт, спазм сосудов мозга, кровоизлияние в мозг, депрессия, тревога, шизофрения, деменция, боль, ноцицепция, аномальная моторика и секреция желудочно-кишечного тракта, различные формы непроходимости кишечника, недержание мочи, болезнь Крона, нарушения, связанные с аномальным потреблением напитков и пищи,анорексия, метаболические нарушения, половая дисфункция и репродуктивные нарушения, нарушение или заболевание, связанное с воспалением, респираторное заболевание, астма, бронхоспазм или аномальная секреция лютеинизирующего гормона, гормона роста, инсулина или пролактина, артериальная гипертензия, ожирение, гиперфазия или булимия, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп.1-9 или фармацевтической композиции по п.10 или 11. 13. Способ по п.12, в котором указанный рецептор нейропептида Y представляет собой рецепторNPY-Y1. 14. Способ по п.13, в котором указанное состояние или заболевание является опухолью, экспрессирующей рецептор NPY-Y1. 15. Способ по п.14, в котором указанная опухоль представляет собой рак молочной железы, рак яичников или глиобластому.
МПК / Метки
МПК: A61K 38/00
Метки: содержащие, мере, одну, замену, синтетическую, нейропептида, меньшей, аминокислоту, аналоги
Код ссылки
<a href="https://eas.patents.su/30-19498-analogi-nejjropeptida-y-soderzhashhie-po-menshejj-mere-odnu-zamenu-na-sinteticheskuyu-aminokislotu.html" rel="bookmark" title="База патентов Евразийского Союза">Аналоги нейропептида y, содержащие по меньшей мере одну замену на синтетическую аминокислоту</a>
Полисахариды с антитромботической активноcтью, содержащие по меньшей мере одну ковалентную связь с биотином или производным биотина

Номер патента: 5133
Опубликовано: 30.12.2004
Авторы: Дюшоссуа Филипп, Птиту Морис, Сави Пьер, Эрбер Жан-Марк
МПК: C08B 37/00, A61K 31/715, A61P 7/02...
Метки: производным, биотином, ковалентную, одну, полисахариды, активноcтью, меньшей, связь, биотина, содержащие, антитромботической, мере
Формула / Реферат:
1. Синтетические полисахариды с антитромботической активностью и их фармацевтически приемлемые соли, отличающиеся тем, что они имеют по меньшей мере одну ковалентную связь с биотином или производным биотина. 2. Полисахариды по п.1 формулы где волнистая линия обозначает связь, расположенную либо ниже, либо выше плоскости кольца пиранозы, Po обозначает полисахарид, содержащий n одинаковых или разных моносахаридных единиц, связанных с Pe...
Применение латексной композиции, содержащей по меньшей мере одну уреидогруппу, для адгезии на древесине

Номер патента: 17811
Опубликовано: 29.03.2013
Авторы: Кастен Жан-Кристоф, Бетт Уилльям, Буссо Жан-Ноэль
Метки: применение, мере, латексной, композиции, уреидогруппу, древесине, меньшей, одну, адгезии, содержащей
Формула / Реферат:
1. Применение для поверхностной обработки водостойкой древесины водной дисперсии, которая содержит по меньшей мере один латекс, имеющий по меньшей мере одну уреидогруппу, полученный эмульсионной полимеризацией смеси по меньшей мере одного мономера А, выбранного из стирола или его производных; бутадиена; хлоропрена; сложных эфиров метакриловой кислоты; (мет)акриловых кислот; малеинового ангидрида; сложных виниловых эфиров; виниловых нитрилов и...
Применение сополимера, содержащего по меньшей мере одну привитую алкокси – или гидроксиполиалкиленгликолевую группу, в качестве агента, улучшающего блеск бумаги и получаемые продукты

Номер патента: 12405
Опубликовано: 30.10.2009
Авторы: Кессбергер Михель, Гейн Патрик, Бури Маттиас
МПК: C08F 220/32
Метки: продукты, блеск, меньшей, качестве, привитую, одну, применение, группу, бумаги, алкокси, сополимера, гидроксиполиалкиленгликолевую, получаемые, содержащего, агента, мере, улучшающего
Формула / Реферат:
1. Применение водорастворимого сополимера, несущего низкий ионный заряд, в качестве активатора блеска, не зависящего от угла зрения, т.е. угла в интервале 20-85ш, более конкретно в интервале 45-75ш, отличающееся тем, что указанный сополимер содержит по меньшей мере одну алкокси- или гидроксиполиалкиленгликолевую группу, привитую по меньшей мере к одному мономеру с этиленовой связью, и имеет характеристическую вязкость меньше или равную 100 мл/г,...
Применение сополимера, содержащего по меньшей мере одну привитую алкокси- или гидроксиполиалкиленгликолевую группу, в качестве агента, улучшающего активацию оптического отбеливания, и получаемые продукты

Номер патента: 10797
Опубликовано: 30.12.2008
Авторы: Сюо Жан-Марк, Жакеме Кристиан, Монгуан Жак, Дюпон Франсуа
МПК: C08K 3/00, C09C 3/00, C08F 290/14...
Метки: алкокси, гидроксиполиалкиленгликолевую, меньшей, продукты, мере, агента, группу, сополимера, одну, получаемые, оптического, качестве, отбеливания, активацию, улучшающего, содержащего, применение, привитую
Формула / Реферат:
1. Применение водорастворимого сополимера, содержащего по меньшей мере одну алкокси- или гидроксиполиалкиленгликолевую группу, привитую по меньшей мере к одному мономеру с этиленовой связью в качестве агента, улучшающего активацию оптического отбеливания. 2. Применение по п.1, отличающееся тем, что вышеуказанный сополимер состоит по меньшей мере из одного мономера формулы (I) в которой m и p означают число звеньев алкиленоксида меньше или...
Способ распределения по меньшей мере одного соединения полезных данных по меньшей мере одному мультиплексированному соединению

Номер патента: 12519
Опубликовано: 30.10.2009
Автор: Беллинг Томас
МПК: H04L 29/06
Метки: соединения, одному, мультиплексированному, способ, меньшей, данных, соединению, распределения, одного, полезных, мере
Формула / Реферат:
1. Способ распределения по меньшей мере одного из соединений полезных данных к по меньшей мере одному мультиплексированному соединению, предусмотренному между первым сетевым элементом (MSC-A) и вторым сетевым элементом (MSC-B), отличающийся тем, что посредством первого сетевого элемента (MSC-A) формируется первое сообщение сигнализации и передается на второй сетевой элемент (MSC-B), причем посредством первого сообщения сигнализации второму...
Предыдущий патент: Разделительное устройство для отделения частиц песка и породы
Следующий патент: Производные атропоизомеров 2-пуринил-3-толилхиназолинона и способы применения
Случайный патент: Органоминеральное удобрение и способ его получения