Волокно-предшественник углеродного волокна, способ его получения и способ получения углеродного волокна
Номер патента: 18977
Опубликовано: 30.12.2013
Авторы: Каваками Дайсуке, Эндо Макото, Танака Фумихико





























Формула / Реферат
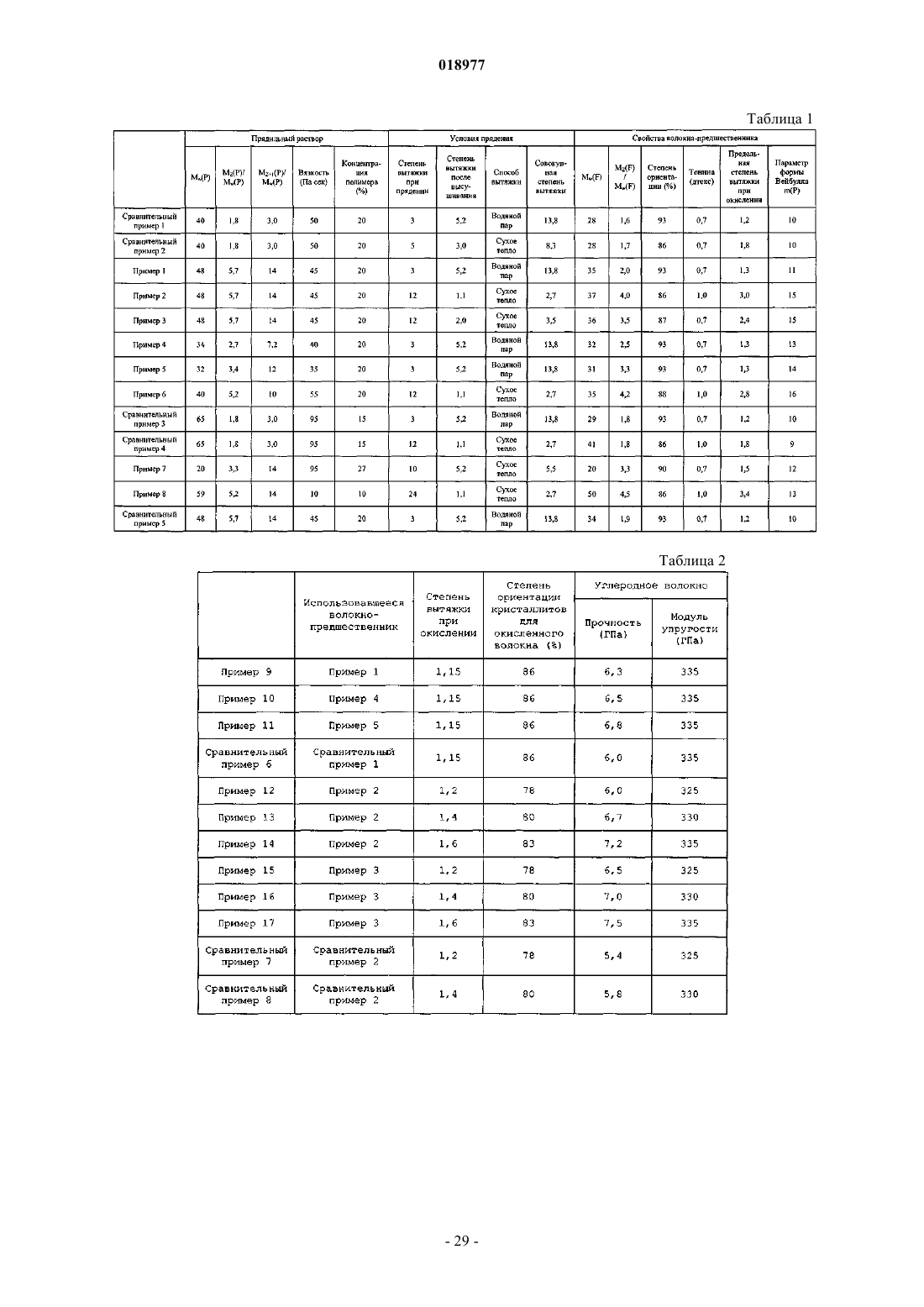
1. Волокно-предшественник углеродного волокна, характеризующееся среднемассовой молекулярной массой Mw(F) в диапазоне от 200000 до 700000 и степенью полидисперсности Mz(F)/Mw(F) (где Mz(F) указывает на Z-среднюю молекулярную массу волокна) в диапазоне от 2 до 5, где данное волокно-предшественник изготовлено из полимера на основе полиакрилонитрила.
2. Волокно-предшественник углеродного волокна по п.1, где параметр формы Вейбулла m(Р) у предела прочности при растяжении для одиночного волокна, полученный в соответствии с методикой JIS R7606 (2000), составляет 11 и более.
3. Волокно-предшественник углеродного волокна по п.1 или 2, характеризующееся степенью ориентации кристаллитов в диапазоне от 85 до 90%.
4. Способ получения волокна-предшественника углеродного волокна, где прядильный раствор подвергают фильтрованию через фильтр, характеризующийся тонкостью фильтрования в диапазоне от 3 до 15 мкм, а после этого прядению для получения набухающего волокна, при этом прядильный раствор получают в результате растворения в растворителе полимера на основе полиакрилонитрила, характеризующегося среднемассовой молекулярной массой Mw(Р) в диапазоне от 200000 до 700000 и степенью полидисперсности Mz(P)/Mw(P) (где Mz(P) указывает на Z-среднюю молекулярную массу полимера в прядильном растворе) в диапазоне от 2,7 до 6, с концентрацией 5 мас.% и более, но менее чем 30 мас.%, и набухающее волокно подвергают первой стадии вытяжки и обработке сухим теплом для получения волокна-предшественника углеродного волокна по п.1.
5. Способ получения волокна-предшественника углеродного волокна по п.4, где после обработки сухим теплом проводят вытяжку при воздействии сухого тепла со степенью вытяжки в диапазоне от 1,1 до 6.
6. Способ получения волокна-предшественника углеродного волокна по п.4, где прядильный раствор подвергают фильтрованию через фильтр, характеризующийся тонкостью фильтрования в диапазоне от 5 до 15 мкм, а после этого прядению.
7. Способ получения волокна-предшественника углеродного волокна по п.6, где фильтрование осуществляют через фильтр, характеризующийся тонкостью фильтрования в диапазоне от 5 до 10 мкм.
8. Способ получения углеродного волокна, где углеродное волокно получают последовательно в результате проведения стадии окисления с окислением волокна-предшественника углеродного волокна по п.1 при одновременном проведении вытяжки со степенью вытяжки в диапазоне от 0,8 до 3 на воздухе при температуре в диапазоне от 200 до 300°С, стадии предварительной карбонизации с предварительной карбонизацией волокна, полученного на стадии окисления, при одновременном проведении вытяжки со степенью вытяжки в диапазоне от 1 до 1,3 в инертной атмосфере при температуре в диапазоне от 300 до 800°С и стадии карбонизации для карбонизации волокна, полученного на стадии предварительной карбонизации, при одновременной вытяжке со степенью вытяжки в диапазоне от 0,96 до 1,05 в инертной атмосфере при температуре в диапазоне от 1000 до 3000°С.
9. Способ получения углеродного волокна по п.8, где стадию окисления проводят таким образом, чтобы волокно, полученное на стадии окисления, характеризовалось степенью ориентации кристаллитов в диапазоне от 78 до 85% при натяжении вытяжки в диапазоне от 0,1 до 0,25 г/дтекс и степени вытяжки в диапазоне от 1,3 до 3.
Текст
ВОЛОКНО-ПРЕДШЕСТВЕННИК УГЛЕРОДНОГО ВОЛОКНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ УГЛЕРОДНОГО ВОЛОКНА Цель настоящего изобретения заключается в предложении волокна-предшественника высокосортного углеродного волокна, которое с меньшей вероятностью вызывает появление пушения без ухудшения производительности при получении. Волокно-предшественник углеродного волокна изготовлено из полимера на основе полиакрилонитрила и характеризуется среднемассовой молекулярной массой Mw(F) в диапазоне от 200000 до 700000 и степенью полидисперсности Mz(F)/Mw(F) (где Mz(F) указывает на Z-среднюю молекулярную массу волокна) в диапазоне от 2 до 5. Предлагается способ получения волокна-предшественника углеродного волокна, соответствующего настоящему изобретению. Согласно этому способу прядильный раствор подвергают фильтрованию через фильтр, характеризующийся тонкостью фильтрования в диапазоне от 3 до 15 мкм, а после этого прядению для получения набухающего волокна,при этом прядильный раствор получают в результате растворения в растворителе полимера на основе полиакрилонитрила, характеризующегося среднемассовой молекулярной массой Mw(P) в диапазоне от 200000 до 700000 и степенью полидисперсности Mz(P)/Mw(P) (где Mz(P) указывает на Z-среднюю молекулярную массу полимера в прядильном растворе) в диапазоне от 2,7 до 6, с концентрацией, равной 5 мас.% и более, но менее чем 30 мас.%, и набухающее волокно подвергают первой стадии вытяжки и обработке сухим теплом для получения вышеуказанного волокна-предшественника углеродного волокна. В способе получения углеродного волокна по изобретению углеродное волокно получают последовательным проведением стадии окисления с окислением волокна-предшественника углеродного волокна при одновременном проведении вытяжки на воздухе при температуре в диапазоне от 200 до 300 С, стадии предварительной карбонизации волокна, полученного на стадии окисления, при одновременном проведении вытяжки в инертной атмосфере при температуре в диапазоне от 300 до 800 С, и стадии карбонизации волокна, полученного на стадии предварительной карбонизации, при одновременной вытяжке в инертной атмосфере при температуре в диапазоне от 1000 до 3000 С. У полученного углеродного волокна размер кристаллитов и параметры, относящиеся к поверхности углеродного волокна, удовлетворяют специфическому соотношению, при этом параметры определяют по методу спектроскопии комбинационного рассеяния. Область техники Изобретение относится к волокну-предшественнику высокосортного углеродного волокна, которое характеризуется превосходной стабильностью при прохождении способа получения углеродного волокна, и способу получения волокна-предшественника углеродного волокна, а также высокотехнологичному и высокосортному углеродному волокну, использующему волокно-предшественник углеродного волокна, и способу получения углеродного волокна. Уровень техники Вследствие демонстрации углеродными волокнами более высоких удельной прочности и удельного модуля упругости в сопоставлении с другими волокнами, углеродные волокна в дополнение к обычным областям применения спортивных товаров и аэрокосмическим областям применения также широко использовались в качестве армирующих волокон для композитных материалов и в общепромышленных вариантах использования, таких как в случае автомобилестроения, гражданского строительства и архитектуры, изготовления контейнера, работающего под давлением, и лопастей ветрового двигателя, и в высшей степени желательными являются как дополнительное улучшение производительности при получении, так и улучшенные эксплуатационные характеристики. В числе углеродных волокон углеродное волокно на основе полиакрилонитрила (который здесь и далее в настоящем документе может быть сокращенно обозначен как ПАН), которое используют наиболее широко, в промышленности получают таким образом, что прядильный раствор, образованный из полимера на основе ПАН, который представляет собой предшественник волокна, подвергают мокрому прядению, сухому прядению или сухомокрому прядению для получения волокна-предшественника углеродного волокна (которое здесь и далее в настоящем документе может быть сокращенно названо волокном-предшественником), после этого волокно-предшественник углеродного волокна превращают в окисленное волокно в результате нагревания в окислительной атмосфере при температуре в диапазоне от 200 до 400 С, а окисленное волокно подвергают карбонизации в результате нагревания в инертной атмосфере притемпературе, равной по меньшей мере 1000 С. Для получения высокотехнологичного углеродного волокна в описывавшемся ранее способе получения зачастую устанавливают повышенные натяжение или степень вытяжки пучка волокон. Однако по мере увеличения степени вытяжки или натяжения более частой становится генерация (образование) пушения или разрыва волокна. В случае возникновения генерации пушения или разрыва волокна сорт и качество ухудшаются, теряемые распушенные или разорванные волокна наматываются на валик или осаждаются в печи и с большей вероятностью повреждают последующий пучок волокон. Таким образом,с точки зрения стабильного получения существует проблема невозможности установления степени вытяжки, достаточно высокой для получения высокотехнологичного волокна, и получение необходимо проводить при компромиссной степени вытяжки, определяемой соотношением взаимоуступок. В частности, предлагались методики, направленные на стабилизацию вытяжки в результате распределения профиля вытяжки в соответствии с прохождением придающей теплостойкость реакции на стадии окисления(патентный документ 1 и патентный документ 2). Однако данные патентные документы предлагают только выбор описывавшейся ранее компромиссной степени вытяжки и не раскрывают каких-либо методик, фундаментально обеспечивающих установление высоких степеней вытяжки на стадии окисления, и в случае выбора на основании данных документов описывавшейся ранее компромиссной степени вытяжки для проведения получения разрыв волокна не может быть уменьшен в достаточных пределах. С другой стороны, улучшение производительности при получении углеродного волокна на основе ПАН рассматривали в разрезе любой из операций, выбираемых из формования, окисления или карбонизации волокон-предшественников углеродного волокна. Прежде всего, обычным методикам, относящимся к улучшению производительности при получении волокон-предшественников, свойственна следующая далее проблема. Говоря более конкретно, при получении волокон-предшественников производительность подвергается ограничениям, обусловленным количеством прядильных отверстий, предельной скоростью приема коагулированных волокон в соответствии со свойствами раствора полимера на основе ПАН и предельной степенью вытяжки (которая может быть названа предельной степенью вытяжки), относящейся к коагулированной структуре (здесь и далее в настоящем документе свойство, указывающее на предельную скорость приема коагулированных волокон, называется прядомостью). Говоря конкретно,при получении волокон-предшественников углеродного волокна, образованных из большого количества одиночных волокон, условия, оказывающие влияние на производительность при получении, необходимо определить в зависимости от того, насколько увеличивают конечную скорость получения волоконпредшественников, определяемую произведением скорости прядения и степени вытяжки. Говоря более конкретно, в случае увеличения скорости прядения в целях улучшения производительности способность к вытяжке уменьшится, и, таким образом, способ получения, вероятно, будет дестабилизирован. С другой стороны, в случае уменьшения скорости прядения способ получения стабилизируется при одновременном уменьшении производительности. Таким образом, существует проблема трудности достижения как улучшенной производительности при получении, так и стабилизации способа получения. Поскольку в отношении описывавшейся ранее проблемы известно то, что на прядомость значи-1 018977 тельное воздействие оказывает способ прядения, для каждого способа прядения будет приведено разъяснение. В способе мокрого прядения прядильный раствор экструдируют из прядильного отверстия в коагуляционной ванне в коагуляционную ванну. Таким образом, непосредственно после экструдирования прядильного раствора из прядильного отверстия протекает коагуляция. Поэтому при увеличении скорости приема увеличивается степень существенной вытяжки при прядении. Увеличение вытяжки при прядении приводит к разрыву волокна на поверхности фильеры, и, таким образом, существует предел увеличения скорости приема. В противоположность этому, в способе сухомокрого прядения прядильный раствор один раз экструдируют в среду воздуха (воздушный зазор), а после этого вводят в коагуляционную ванну, и, таким образом, нить в основном подвергают вытяжке при низком натяжении в воздушном зазоре. Поэтому известно, что существенную вытяжку при прядении в коагуляционной ванне уменьшают, чтобы увеличить прядомость. Например, была предложена методика, по которой концентрацию полимера в прядильном растворе контролируемо выдерживают в целях уменьшения вязкости прядильного раствора, промотирования легкости управления операцией фильтрования и улучшения степени вытяжки при прядении, которая представляет собой соотношение между скоростью приема волокон в коагуляционной ванне и скоростью экструдирования прядильного раствора из фильеры (см. патентный документ 3). В соответствии с этим предложением, хотя признается эффект улучшения в виде степени вытяжки при прядении 10, степень вытяжки при прядении увеличивается только за счет увеличения диаметра отверстия фильеры. Говоря более конкретно, увеличение диаметра отверстия фильеры замедляет линейную скорость экструдирования, увеличивая степень вытяжки при прядении. Однако производительность при получении волокна-предшественника улучшить невозможно, поскольку только в результате увеличения степени вытяжки при прядении прядомость не улучшается. Хотя предложена методика, по которой степень вытяжки при прядении устанавливают на величину в диапазоне от 5 до 50 при использовании раствора для высокоскоростного прядения и формирования специфического воздушного зазора (см. патентный документ 4), данное предложение относится к акриловому волокну для заделывания, у которого количество, по существу, одиночных волокон, формирующих пучок волокон, составляет всего лишь 36 и, таким образом, непригодно для углеродных волокон,полученных в результате окисления и карбонизации пучков волокон, образованных из большого количества одиночных волокон в диапазоне от нескольких тысяч до сотен тысяч. Говоря более конкретно, в каждом из широко известных способов эффект улучшения производительности при получении ограничен. В соответствии с этим существует потребность в методиках улучшения производительности при получении углеродных волокон, которые могут улучшить как прядомость, так и предельную степень вытяжки даже в случае пучков волокон, образованных из большого количества одиночных волокон, а, кроме того, могут подавить генерацию пушения или разрыва волокон,которые ухудшают качество и сорт, а, кроме того, стабильность получения даже в случае использования условий проведения окисления, включающих высокую степень вытяжки. Факт наличия малого пушения у углеродных волокон обуславливает получение преимущества не только в виде стабильности способа в способе получения препрега и способе получения композита, но также и в виде большого числа случаев придания формованному телу, сформованному с использованием углеродных волокон, предела прочности при сжатии для композита, поскольку нарушение выравнивания волокон вследствие пушения и т.п. может быть уменьшено. Получение углеродных волокон, характеризующихся малым пушением, имеет очень большое значение, поскольку предел прочности при сжатии представляет собой важный показатель разработки материала при разработке композитов. Причиной такого пушения считается отчасти наличие дефекта решетки гексагонального углеродного слоя. В принципе дефект решетки гексагонального углеродного слоя можно оценить при использовании спектра комбинационного рассеяния. Хотя в общем имеется множество примеров исследования для оценки углеродных волокон при использовании спектра комбинационного рассеяния (см. патентные документы 5 и 6), можно сказать то, что было проведено множество исследований в отношении структур кристаллитов при одновременном отсутствии какого-либо обсуждения относительно дефектов решетки. В дополнение к этому, в методиках, описывавшихся в данных документах, на основании оценки контролируют только структуру кристаллитов углеродных волокон, а дефект решетки не контролируют. Поэтому, хотя раскрыты методики улучшения средних значений для свойств, какой-либо методики улучшения вариаций свойств описано не было. В дополнение к этому, причина пушения будет рассматриваться при концентрации внимания на пучках углеродных волокон. Так как пушение образуется в результате разрыва слабых волокон, величина вариации прочности соотносится с количеством случаев пушения. На вариацию прочности углеродных волокон во многих случаях указывают параметры Вейбулла(параметр формы Вейбулла и параметр масштаба), и вариация механических свойств углеродных волокон в виде пряди, импрегнированной смолой, несколько улучшается в случае композитного материала,полученного при использовании углеродных волокон, которые обладают идентичными механическими свойствами углеродных волокон в виде пряди, импрегнированной смолой, и отличается по параметру формы Вейбулла. Однако какой-либо пример значительного улучшения среднего значения для физиче-2 018977 ских свойств неизвестен. Например, были предложены углеродные волокна, которые характеризуются распределением предела прочности при растяжении для одиночных волокон, определяемым параметром формы Вейбулла (см. патентные документы 7 и 8). В патентном документе 7 для подавления пушения,которое может быть генерировано в способе графитизации, распределение предела прочности при растяжении для одиночных волокон углеродных волокон, которые до процесса графитизации характеризуются модулем упругости при растяжении для углеродных волокон в виде пряди, импрегнированной смолой, 305 ГПа, контролируемо выдерживают узким (характеризующимся параметром формы Вейбулла в диапазоне от 5 до 6). В соответствии с данной методикой улучшение модуля упругости при растяжении для углеродных волокон в виде пряди, импрегнированной смолой, приводит к появлению морфологии хрупкого разрушения, и, таким образом, с большей вероятностью возникает концентрация напряжений. Таким образом, с большей вероятностью на свойства будут оказывать неблагоприятное воздействие дефекты, что в результате приведет к уменьшению параметра формы Вейбулла. В дополнение к этому, в патентном документе 8 предлагают углеродные волокна, которые являются подходящими для использования в технологии с натягиванием элементарных волокон и демонстрируют превосходные характеристики разрыхления. Как упоминается в патентном документе 8, форму поперечного сечения и морфологию поверхности волокон делают более приемлемыми, прохождение способа переработки улучшают без использования большого количества собирающего агента, и для улучшения прохождения способа важно контролируемо выдерживать параметр формы Вейбулла в диапазоне от 4 до 6. Однако модуль упругости составляет 270 ГПа и менее, и баланс между высоким модулем упругости и узкой вариацией прочности одиночного волокна не достигнут. Патентный документ 1: выложенная японская патентная заявка 62-257422. Патентный документ 2: выложенная японская патентная заявка 58-186614. Патентный документ 3: выложенная японская патентная заявка 64-77618. Патентный документ 4: выложенная японская патентная заявка 11-107034. Патентный документ 5: выложенная японская патентная заявка 3-180514. Патентный документ 6: выложенная японская патентная заявка 9-170170. Патентный документ 7: выложенная японская патентная заявка 4-222229. Патентный документ 8: выложенная японская патентная заявка 2002-266173. Описание изобретения Проблема, решаемая в изобретении. Цель настоящего изобретения заключается в том, чтобы предложить способ получения волокнапредшественника для высокосортного углеродного волокна, который с меньшей вероятностью вызывает появление пушения, без влияния на производительность при получении. В дополнение к этому, цель настоящего изобретения заключается в том, чтобы предложить волокно-предшественник углеродного волокна, из которого может быть получено высокосортное и высококачественное углеродное волокно, без влияния на производительность при получении, с одновременным подавлением появления пушения и разрыва волокна даже в условиях проведения окисления-карбонизации, включающих высокие натяжение или степень вытяжки. Средства решения проблемы. Для достижения описанных выше целей волокно-предшественник углеродного волокна, соответствующее настоящему изобретению, имеет следующую далее конфигурацию. Говоря более конкретно,волокном-предшественником углеродного волокна, соответствующим настоящему изобретению, является волокно-предшественник углеродного волокна, которое характеризуется среднемассовой молекулярной массой Mw(F) в диапазоне от 200000 до 700000 для волокна и степенью полидисперсностиMz(F)/Mw(F) (где Mz(F) указывает на Z-среднюю молекулярную массу волокна) в диапазоне от 2 до 5 и которое изготовлено из полимера на основе полиакрилонитрила. В дополнение к этому, для достижения описывавшихся ранее целей способ получения волокнапредшественника углеродного волокна, соответствующего настоящему изобретению, имеет нижеследующую конфигурацию. Говоря более конкретно, способом получения волокна-предшественника углеродного волокна, согласно настоящему изобретению, является способ получения волокнапредшественника углеродного волокна, по которому прядильный раствор подвергают фильтрованию через фильтр, характеризующийся тонкостью фильтрования в диапазоне от 3 до 15 мкм, а после этого прядению для получения набухающего волокна, при этом прядильный раствор получают в результате растворения в растворителе полимера на основе полиакрилонитрила, характеризующегося среднемассовой молекулярной массой Mw(P) в диапазоне от 200000 до 700000 и степенью полидисперсностиMz(P)/Mw(P) (где Mz(P) указывает на Z-среднюю молекулярную массу полимера в прядильном растворе) в диапазоне от 2,7 до 6, с концентрацией, равной 5 мас.% и более, но менее чем 30 мас.%, и набухающее волокно подвергают первой стадии вытяжки и обработке сухим теплом для получения волокнапредшественника углеродного волокна, соответствующего описанному ранее. Кроме того, для достижения описывавшихся ранее целей способ получения углеродного волокна,соответствующего настоящему изобретению, имеет нижеследующую далее конфигурацию. Говоря более конкретно, способом получения углеродного волокна является способ получения углеродного волокна,-3 018977 по которому углеродное волокно получают последовательно в результате проведения стадии окисления для придания волокну-предшественнику углеродного волокна теплостойкости при одновременном проведении вытяжки со степенью вытяжки в диапазоне от 0,8 до 3 на воздухе при температуре в диапазоне от 200 до 300 С, стадии предварительной карбонизации для предварительной карбонизации волокна,полученного на стадии окисления, при одновременном проведении вытяжки со степенью вытяжки в диапазоне от 1 до 1,3 в инертной атмосфере при температуре в диапазоне от 300 до 800 С и стадии карбонизации для карбонизации волокна, полученного на стадии предварительной карбонизации, при одновременной вытяжке со степенью вытяжки в диапазоне от 0,96 до 1,05 в инертной атмосфере при температуре в диапазоне от 1000 до 3000 С. В дополнение к этому, для достижения описывавшихся ранее целей углеродное волокно, соответствующее настоящему изобретению, имеет нижеследующую конфигурацию. Говоря более конкретно, углеродным волокном, соответствующим настоящему изобретению, является углеродное волокно, у которого размер кристаллитов (Lc (нм и параметры (ID/IG, IV/IG, G (см-1, относящиеся к поверхности углеродного волокна, удовлетворяют следующим далее формулам с (1) по (4), при этом параметры определяют по методу спектроскопии комбинационного рассеяния. Выгодный эффект от изобретения. В соответствии с настоящим изобретением может быть получено волокно-предшественник высокосортного углеродного волокна, которое с меньшей вероятностью вызывает появление пушения, без воздействия на производительность. В дополнение к этому, высокосортное и высококачественное углеродное волокно может быть получено без воздействия на производительность при одновременном подавлении появления пушения и разрыва волокна даже в условиях проведения окисления-карбонизации, включающих высокие натяжение или степень вытяжки. Наилучший способ реализации изобретения Изобретатели уже предлагали методику получения волокна-предшественника углеродного волокна,которая обеспечивает достижение превосходной прядомости при использовании полимера на основе ПАН, характеризующегося специфическим молекулярно-массовым распределением (японская патентная заявка 2007-269822). Кроме того, изобретатели рассмотрели методику получения и выявили достижение превосходной стабильности получения на стадии окисления в результате уменьшения изменения молекулярно-массового распределения для волокна-предшественника в сопоставлении с молекулярномассовым распределением для полимера на основе ПАН в прядильном растворе и, тем самым, сделали настоящее изобретение. Необходимо отметить то, что в настоящем изобретении среднемассовую молекулярную массу, Zсреднюю молекулярную массу и Z+1-среднюю молекулярную массу, среднечисленную молекулярную массу сокращенно обозначают символами Mw, Mz, Mz+1 и Mn соответственно, и для различения символовMw, Mz, Mz+1 и Mn добавляют индекс (F) и индекс (Р) в случае отнесения символов ко всем полимерам на основе ПАН, составляющим волокна, и ко всем полимерам на основе ПАН в прядильном растворе соответственно. Волокно-предшественник, соответствующее настоящему изобретению, образовано из полимера на основе ПАН, который имеет среднемассовую молекулярную массу Mw(F) в диапазоне от 200000 до 700000, предпочтительно от 300000 до 500000. В случае образования волокна-предшественника из более низкомолекулярного полимера на основе ПАН, который характеризуется значением Mw(F), меньшим чем 200000, прочность волокна-предшественника уменьшится, что в результате легко приведет к генерации пушения на стадии окисления. В альтернативном варианте, в случае образования волокнапредшественника из более высокомолекулярного полимера на основе ПАН, который характеризуется значением Mw(F), большим чем 700000, среднемассовую молекулярную массу Mw(P) полимеров в прядильном растворе необходимо довести до значения, превышающего 700000. В таком случае повышенное переплетение молекулярных цепей в результате приведет к появлению трудностей при растяжении и,таким образом, уменьшит длину растянутой цепи. Поэтому выгодных эффектов от настоящего изобретения достичь будет невозможно. Хотя значение Mw(F) будет равным или меньшим, чем значение Mw(P),значение Mw(F) можно регулировать при использовании условий на стадии прядения. Это подробно будет описываться дальше. В дополнение к этому, полимер на основе ПАН, составляющий волокно-предшественник, соответствующий настоящему изобретению, предпочтительно характеризуется степенью полидисперсностиMz(F)/Mw(F) (где Mz(F) указывает на Z-среднюю молекулярную массу волокна) в диапазоне от 2 до 5,предпочтительно от 2,5 до 5, более предпочтительно от 3 до 5, а еще более предпочтительно от 3,5 до 5. В настоящем изобретении среднемассовую молекулярную массу Mw(F), Z-среднюю молекулярную массу Mz(F) и среднечисленную молекулярную массу Mn(F) волокна и среднемассовую молекулярную массу Mw(Р), Z-среднюю молекулярную массу Mz(P), Z+1-среднюю молекулярную массу Mz+1(P) и среднечисленную молекулярную массу Mn(P) полимера на основе ПАН при прядении измеряют по методу гель-проникающей хроматографии (здесь и далее в настоящем документе сокращенно называемому методом ГПХ) и представляют в виде значения в пересчете на полистирол. Необходимо отметить, что степень полидисперсности Mz/Mw имеет нижеследующее значение, будь то для волокна или для полимера на основе ПАН. Говоря более конкретно, среднечисленная молекулярная масса Mn представляет собой чувствительный параметр, отражающий вклад низкомолекулярного вещества, содержащегося в полимерном соединении. В противоположность этому, значение Mw отражает вклад высокомолекулярного вещества, значение Mz представляет собой еще более чувствительный параметр, отражающий вклад высокомолекулярного вещества, а значение Mz+1 представляет собой намного более чувствительный параметр, отражающий вклад высокомолекулярного вещества, в сопоставлении со значением Mz. Поэтому молекулярно-массовое распределение Mw/Mn и степени полидисперсности Mz/Mw и Mz+1/Mw могут быть использованы для оценки аспектов ширины молекулярно-массового распределения. Значение Mw/Mn 1 свидетельствует о монодисперсности, а увеличение значения Mw/Mn говорит о более широком молекулярно-массовом распределении в области низкомолекулярного края. С другой стороны, увеличение значения Mz/Mw говорит о более широком молекулярно-массовом распределении в области высокомолекулярного края. В частности, при значительном увеличении значения Mz+1/Mw данный случай говорит о смешивании двух типов полимеров, которые значительно отличаются друг от друга по значению Mw. Как описывалось ранее, аспект молекулярно-массового распределения, определяемый значениемMw/Mn, отличается от того, на что указывает значение Mz/Mw. Таким образом, даже в случае увеличения значения Mw/Mn не всегда верно то, что значение Mz/Mw будет увеличиваться подобным же образом. В настоящем изобретении значение Mw в диапазоне от 200000 до 700000 определяют как обычную молекулярную массу, в то время как значение Mw в диапазоне от 800000 до 15000000 определяют как сверхвысокую молекулярную массу. Несмотря на то что на данном этапе механизм, который обеспечивает достижение эффекта ингибирования генерации пушения на стадии окисления в результате использования волокна-предшественника,соответствующего настоящему изобретению, не определен, механизм оценивают следующим образом. Как широко известно, в принципе, можно было бы получать высокопрочные и высокомодульные волокна на основе ПАН при использовании вытяжки в высокой степени для полимера на основе ПАН, имеющего сверхвысокую молекулярную массу, для получения растянутой цепи молекул полимера на основе ПАН в волокне на основе ПАН и уменьшения количества аморфных фрагментов и концевых групп молекулярных цепей в волокне на основе ПАН тем же самым образом, что и для других органических волокон, типичным примером которых является полиэтиленовое волокно. Однако для эффективного введения в действие данного принципа требуется контролируемое уменьшение переплетения полимера на основе ПАН в растворе полимера на основе ПАН, и для этой цели требуется уменьшить концентрацию полимера на основе ПАН. В случае уменьшения концентрации полимера на основе ПАН производительность при получении уменьшится вследствие усложнения стадии контролируемого выдерживания количества растворителя. В дополнение к этому, в случае необходимости придания огнестойкости волокну на основе ПАН в форме пучка волокон, образованного из одиночных волокон, очень небольшая процентная доля одиночных волокон разрушится вследствие флуктуации прочности среди одиночных волокон, что,таким образом, приведет к генерации пушения. С другой стороны, в сопоставлении с полимером на основе ПАН, имеющим обычную молекулярную массу, в случае полимера на основе ПАН, имеющего сверхвысокую молекулярную массу, требуется более продолжительное время для возращения молекул,трансформированных в результате растяжения и т.п., к первоначальной форме, так называемое время релаксации. Таким образом, небольшое количество сверхвысокомолекулярного полимера на основе ПАН, содержащегося в растворе полимера на основе ПАН, приводит к предпочтительному растяжению сверхвысокомолекулярного полимера на основе ПАН до получения так называемой растянутой цепи. В случае полученного волокна-предшественника с растянутым волокном на основе ПАН, содержащим небольшое количество сверхвысокомолекулярного полимера на основе ПАН, при приложении к волокнупредшественнику растягивающего напряжения растянутая цепь молекул высокопрочного и высокомодульного сверхвысокомолекулярного полимера на основе ПАН в волокне-предшественнике действует так, как если бы это был наполнитель, и когда ориентированный обычный полимер на основе ПАН (матрицы по отношению к наполнителю) приближается к состоянию разрушения, значение вязкости разрушения (сопротивления развитию трещин) увеличивается по следующим причинам от (А) до (С): (А) разрушение прогрессирует, но при этом обходит растянутую цепь сверхвысокомолекулярного полимера на основе ПАН; (В) растянутая цепь сверхвысокомолекулярного полимера на основе ПАН выдерживает напряжение, несущее энергию разрушения; (С) молекулы сверхвысокомолекулярного полимера на основе ПАН выдергиваются. Таким образом, как можно себе представить, одиночные волокна, которые характеризуются низким относительным удлинением при разрушении, из пучка волокон исключаются, что,таким образом, уменьшает генерацию пушения на стадии окисления. Далее будет описываться способ регулирования описывавшегося ранее значения Mz(F)/Mw(F). В настоящем изобретении в качестве пря-5 018977 дильного раствора используют раствор полимера на основе ПАН, растворенного в растворителе, где полимер на основе ПАН имеет среднемассовую молекулярную массу Mw(Р) в диапазоне от 200000 до 700000, предпочтительно от 300000 до 500000. В случае использования раствора низкомолекулярного полимера на основе ПАН, характеризующегося значением Mw(Р), меньшим чем 200000, молекулярная масса в способе получения волокна-предшественника не увеличится, и, таким образом, значение Mw(F) составит менее чем 200000, что в результате приведет к невозможности получения волокнапредшественника для углеродных волокон при превосходной производительности. Говоря более конкретно, в случае использования раствора низкомолекулярного полимера на основе ПАН, характеризующегося значением Mw(P), меньшим чем 200000, прочность полученного волокна-предшественника уменьшится, что в результате приведет к вероятному появлению пушения на стадии окисления. В дополнение к этому, при предпочтительности большего значения Mw(Р) высокомолекулярный полимер на основе ПАН, характеризующийся значением Mw(Р), большим чем 700000, имеет больше переплетений. Таким образом, при вытяжке молекулярная цепь может быть растянута недостаточно. Необходимо отметить, что хотя в целях простого увеличения длины растянутой цепи возможно получить волокнопредшественник углеродного волокна, определенного в п.1 формулы изобретения, путем уменьшения концентрации полимера до получения полуразбавленного раствора, характеризующегося пониженным переплетением, и проведения растяжения, невозможно добиться высокой производительности при получении волокна-предшественника, которая представляет собой еще одну цель настоящего изобретения. В данном случае значение Mw(Р) можно регулировать в результате изменения количеств мономера, инициатора полимеризации, регулятора степени полимеризации и т.п. во время проведения полимеризации для получения полимера на основе ПАН. Степень полидисперсности Mz(P)/Mw(P) для полимера на основе ПАН в прядильном растворе находится в диапазоне от 2,7 до 6, предпочтительно от 3 до 5,8, а более предпочтительно от 3,2 до 5,5. Значение Mz(P)/Mw(P), меньшее чем 2,7, в результате приводит к утрате деформационного упрочнения, как это будет описываться далее, и отсутствию улучшения стабильности экструдирования из фильеры полимера на основе ПАН. С другой стороны, значение Mz(P)/Mw(P), большее чем 6, слишком сильно увеличивает переплетение, что в результате приводит к появлению трудностей при экструдировании из фильеры. Компонент, имеющий более высокую молекулярную массу в растворе полимера на основе ПАН, предпочтительно ориентируется на стадии прядения и несет нагрузку, такую как натяжение вытяжки. В случае превышения нагрузкой энергии связи молекулярной цепи молекулярная цепь разрушится, и пик на высокомолекулярном краю молекулярно-массового распределения, вероятно, уменьшится вследствие предпочтительного прохождения разрушения молекулярной цепи в компоненте, имеющем более высокую молекулярную массу в растворе полимера на основе ПАН. В соответствии с этим, значение Mz/Mw на стадии прядения может уменьшиться, но не увеличится, и должно быть установлено равным или большим значения Mz(F)/Mw(F) для волокна-предшественника. Исходя из этих наблюдений, можно сказать, что использование раствора полимера на основе ПАН, определенного в настоящем изобретении,впервые сделало возможным получение на промышленно успешном уровне масштаба волокнапредшественника, соответствующего настоящему изобретению. В дополнение к этому, полимер на основе ПАН в прядильном растворе предпочтительно характеризуется как значением Mz+1(P) в диапазоне от 3000000 до 10000000, так и степенью полидисперсностиMz+1(P)/Mw(Р) в диапазоне от 6 до 25. Значение Mz+1(P) более предпочтительно находится в диапазоне от 4000000 до 9000000, а еще более предпочтительно от 5000000 до 8500000. В дополнение к этому, значение Mz+1(P)/Mw(P) более предпочтительно находится в диапазоне от 7 до 17, а еще более предпочтительно от 10 до 15. Величина Mz+1(P)/Mw(P) представляет собой показатель, более сильно отражающий присутствие высокомолекулярного вещества в сопоставлении с величиной Mz(P)/Mw(P), и даже в случае разрушения компонента, имеющего высокую молекулярную массу, на стадии прядения высокомолекулярное вещество зачастую может сохраняться в виде компонента, имеющего высокую молекулярную массу, в волокнепредшественнике. До тех пор пока значение Mz+1(P) будет попадать в диапазон от 3000000 до 10000000,значение Mz+1(P)/Mw(P), равное 6 и более, будет приводить к получению достаточного деформационного упрочнения, что в результате создаст достаточный эффект улучшения стабильности экструдирования прядильного раствора, содержащего полимер на основе ПАН (деформационное упрочнение будет разъяснено далее). В дополнение к этому, в случае избыточно большого значения Mz+1(P)/Mw(P) описывающееся далее деформационное упрочнение будет чрезмерно сильным, что в результате может привести к отсутствию эффекта улучшения стабильности экструдирования прядильного раствора, содержащего полимер на основе ПАН. До тех пор пока значение Mz+1(P) будет попадать в диапазон от 3000000 до 10000000, значение Mz+1(P)/Mw(P), равное 25 и менее, может обеспечить достижение достаточной стабильности экструдирования прядильного раствора, содержащего полимер на основе ПАН. В дополнение к этому, в случае попадания значения Mz+1(P)/Mw(P) в диапазон от 6 до 25 значение Mz+1(P), меньшее чем 3000000, может в результате привести к потере прочности полученного волокна-предшественника, а значение Mz+1(P), большее чем 10000000, в результате может привести к появлению трудностей при экструдировании из фильеры прядильного раствора, содержащего полимер на основе ПАН. В дополнение к этому, в молекулярно-массовом распределении предпочтительно использовать полимер на основе ПАН, характеризующийся долей содержания компонентов, которые имеют молекулярную массу, в 5 раз и более превышающую значение Mw(Р), в диапазоне от 1 до 4%. В случае доли содержания молекулярных масс, в 5 раз и более превышающих значение Mw(P), меньшей чем 1%, описывающееся далее деформационное упрочнение может быть слабым, что в результате приведет к недостаточному улучшению стабильности экструдирования из фильеры прядильного раствора, содержащего полимер на основе ПАН. В случае относительной концентрации, большей чем 4%, описывающееся далее деформационное упрочнение может оказаться слишком сильным, что в результате приведет к недостаточному улучшению стабильности экструдирования полимера на основе ПАН. С этой точки зрения доля содержания молекулярных масс, в 5 раз и более превышающих значение Mw(P), предпочтительно находится в диапазоне от 1,2 до 3,8%, а более предпочтительно от 1,5 до 3,6%. Доля содержания компонентов, которые имеют молекулярную массу, в 5 раз и более превышающую значение Mw(P), может быть получена из логарифма молекулярных масс в пересчете на полистирол, который измеряют по методу ГПХ, и кривой молекулярно-массового распределения, построенной по разности показателей преломления, и ее определяют по соотношению между значением интеграла для площади пика, соответствующей молекулярным массам, в 5 раз и более превышающим молекулярную массу в пересчете на полистирол, и значением интеграла для совокупного молекулярно-массового распределения. Разность показателей преломления почти соответствует массе молекул, элюируемых в единицу времени, а значение интеграла для площади пика почти соответствует массовой доле в смеси. Несмотря на то что на данном этапе механизм, который может привести к получению волокнапредшественника углеродного волокна, которое может обеспечить достижение баланса между улучшением производительности при получении и стабилизацией в результате использования описывавшегося ранее полимера на основе ПАН, не определен, механизм оценивают следующим образом. Говоря более конкретно, в способе получения волокна-предшественника углеродного волокна, соответствующего настоящему изобретению, в случае воздействия на раствор полимера на основе ПАН, содержащего сверхвысокомолекулярный полимер на основе ПАН, деформации удлинения для достижения меньшей толщины непосредственно после экструдирования из прядильного отверстия, сверхвысокомолекулярные полимеры на основе ПАН и низкомолекулярные полимеры на основе ПАН переплетаются друг с другом,происходит быстрое увеличение вязкости удлинения, т.е. так называемое деформационное упрочнение в основном вследствие повышенных натяжений молекулярных цепей между переплетенными сверхвысокомолекулярными полимерами на основе ПАН. Вязкость на пределе эластичности для более тонкого фрагмента увеличивается при уменьшении толщины раствора полимера на основе ПАН непосредственно после экструдирования из прядильного отверстия, что стабилизирует текучесть, тем самым, обеспечивая увеличение скорости прядения. В растворе полимера на основе ПАН, предназначенного для использования в настоящем изобретении, относительно более низкомолекулярные полимеры на основе ПАН имеют высокотекучие молекулярные цепи и, таким образом, ориентируются с меньшей вероятностью, тогда как эффект ориентации сверхвысокомолекулярных полимеров на основе ПАН является явно выраженным. Таким образом, использование способа получения волокна-предшественника углеродного волокна, соответствующего настоящему изобретению, приводит к получению выраженного эффекта улучшения прядомости в несколько десятков раз и более в сопоставлении с тем, что имеет место в случае без использования данного способа. В дополнение к этому, меньшее значение Mw(P)/Mn(P) в результате приводит к меньшему уровню содержания низкомолекулярных компонентов, имеющих несколько концевых групп молекулярных цепей на единицу массы, которые, вероятно, вызывают возникновение дефектов решетки в углеродном волокне, полученном в результате окисления-карбонизации полученного волокна-предшественника углеродного волокна. С этой точки зрения меньшее значение Mw(P)/Mn(P) является предпочтительным, а значение Mw(P)/Mn(P) предпочтительно является меньшим, чем значение Mz(P)/Mw(P). Говоря более конкретно, в то время как молекулярно-массовое распределение, которое является равномерно широким в направлении как более высокомолекулярного края, так и более низкомолекулярного края, приводит к небольшому уменьшению стабильности экструдирования раствора полимера на основе ПАН из прядильного отверстия, с точки зрения подавления генерации дефектов решетки в углеродном волокне, полученном в результате окисления-карбонизации полученного волокна-предшественника углеродного волокна,является предпочтительным, чтобы низкомолекулярный край был по возможности наиболее узким (говоря более конкретно, чтобы уровень содержания низкомолекулярных полимеров на основе ПАН был низким), и значение Mz(P)/Mw(P) более предпочтительно является большим по меньшей мере в 1,5 раза и более, а более предпочтительно по меньшей мере в 1,8 раз и более в сопоставлении со значениемMw(P)/Mn(P). В соответствии с представлениями изобретателей, в случае получения полимера на основе полиакрилонитрила в результате проведения радикальной полимеризации, такой как в случае способа водной суспензионной или растворной полимеризации, у молекулярно-массового распределения полимера отсекаются его смещения в направлении низкомолекулярного края, и, таким образом, значениеMw(P)/Mn(P) обычно является большим в сопоставлении со значением Mz(P)/Mw(P). Поэтому при получении раствора полимера на основе ПАН, характеризующегося описывавшимся ранее молекулярно-7 018977 массовым распределением, для использования в способе получения волокна-предшественника углеродного волокна, соответствующего настоящему изобретению, могут быть использованы способ, по которому полимеризацию проводят в указанных условиях, таких как использование типа, соотношения и постадийного добавления инициатора полимеризации, или способ, по которому перемешивают два и более типа полимеров на основе ПАН, которые получают в результате использования обычной радикальной полимеризации и которые характеризуются молекулярно-массовыми распределениями, отличными друг от друга. В числе данных способов простым является последний способ, по которому перемешивают полимеры на основе ПАН, которые характеризуются молекулярно-массовыми распределениями, отличными друг от друга. В данном случае меньшее количество перемешиваемых типов является более простым и двух типов полимеров на основе ПАН зачастую будет достаточно с точки зрения стабильности экструдирования. Что касается значения Mw перемешиваемых полимеров, то в предположении наименования полимера на основе ПАН, характеризующегося более высоким значением Mw, компонентом А при одновременном наименовании полимера на основе ПАН, характеризующегося более низким значением Mw, компонентом В значение Mw компонента А предпочтительно будет находиться в диапазоне от 800000 до 15000000, а более предпочтительно от 1000000 до 5000000, в то время как значение Mw компонента В предпочтительно будет находиться в диапазоне от 150000 до 700000. Чем больше будет разница значений Mw между компонентом А и компонентом В, тем больше будет у перемешиваемых полимеров тенденция к предпочтительному увеличению значений Mz/Mw и Mz+1/Mw. Однако в случае значения Mw компонента А, большего чем 15000000, производительность при получении компонента А может уменьшиться, а в случае значения Mw компонента В, меньшего чем 150000, прочность волокнапредшественника может оказаться недостаточной. Говоря конкретно, соотношение между компонентом А и компонентом В при выражении через среднемассовую молекулярную массу предпочтительно находится в диапазоне от 2 до 45, а более предпочтительно от 20 до 45. В дополнение к этому, массовое соотношение между компонентом А и компонентом В для перемешивания предпочтительно находится в диапазоне от 0,003 до 0,3, более предпочтительно от 0,005 до 0,2, а еще более предпочтительно от 0,01 до 0,1. Массовое соотношение между компонентом А и компонентом В для перемешивания, меньшее чем 0,003, в результате может привести к недостаточному деформационному упрочнению, в то время как массовое соотношение, большее чем 0,3, может слишком сильно увеличить вязкость во время экструдирования полимерного раствора из фильеры, что, тем самым, в результате приведет к появлению трудностей при экструдировании. Соотношение между компонентом А и компонентом В при выражении через среднемассовую молекулярную массу и массовое соотношение между компонентом А и компонентом В для перемешивания измеряют по методу ГПХ. Говоря более конкретно, соотношения измеряют таким образом, что на пике молекулярно-массового распределения, полученного по методу ГПХ, выделяют плечо или секцию пика для вычисления значений Mw каждого из компонентов А и В и соотношения площадей между пиками компонентов А и В. В случае перемешивания полимеров компонента А и компонента В могут быть использованы следующие способы от (D) до (G), а именно: (D) способ, по которому оба полимера смешивают, а после этого разбавляют растворителем; (Е) способ, по которому полимеры, разбавленные в растворителях, смешивают друг с другом; (F) способ, по которому компонент А, который представляет собой более высокомолекулярное вещество, разбавляют в растворителе с последующим смешиванием и растворением компонента В; и (G) способ, по которому компонент А, который представляет собой более высокомолекулярное вещество, растворенное в растворителе, и мономер, который представляет собой материал исходного сырья для компонента В, смешивают для проведения растворной полимеризации мономеров для перемешивания. В качестве способа смешивания, подходящего для использования в данных способах, предпочтительно могут быть использованы следующие способы: способ с перемешиванием в смесительной емкости; способ с определением количества при использовании шестеренчатого насоса и смешиванием при использовании статического смесителя и способ с использованием двухчервячного экструдера. Исходя из той точки зрения, что более высокомолекулярное вещество растворяют гомогенно, предпочтительным является способ, по которому сначала растворяют компонент А, который представляет собой более высокомолекулярное вещество. В частности, в случае использования полимеров для получения предшественника углеродного волокна растворенное состояние компонента А, который имеет более высокую молекулярную массу, является чрезвычайно важным, и в случае присутствия даже небольшого количества нерастворенного вещества нерастворенное вещество будет считаться загрязнителем и отфильтровываться на материале фильтра или может образовывать пустоты в углеродном волокне в случае, когда размер нерастворенного вещества слишком мал для отфильтровывания. Говоря конкретно, по описывавшимся ранее способам (F) и (G) концентрацию полимера компонента А по отношению к растворителю доводят до величины в диапазоне от 0,1 до 5 мас.%, а после этого компонент А смешивают с компонентом В или мономерным материалом исходного сырья для получения компонента В в целях проведения полимеризации. Концентрация полимера компонента А более пред-8 018977 почтительно находится в диапазоне от 0,3 до 3 мас.%, а еще более предпочтительно от 0,5 до 2 мас.%. Концентрацию полимера компонента А по отношению к растворителю в настоящем документе определяют как концентрацию полимера компонента А в растворе в предположении образования раствора только из компонента А и растворителя. Предпочтительно концентрацией полимера компонента А, говоря более конкретно, является концентрация полуразбавленного раствора вещества в виде набора слегка перекрывающихся молекул полимера. В случае смешивания компонента А с компонентом В или мономером, составляющим компонент В, для проведения полимеризации состояние смеси, вероятно, является гомогенным. Таким образом, более предпочтительным вариантом реализации является доведение концентрации полимера компонента А до концентрации разбавленного раствора изолированных цепей. Поскольку, как считается, концентрацию разбавленного раствора определяет межмолекулярный исключенный объем, который определяется молекулярной массой полимера и растворимостью полимера по отношению к растворителю, хотя и невозможно требовать категорически определения, тем не менее, предпочтительно, чтобы концентрация в настоящем изобретении в основном попадала в вышеупомянутый диапазон. В случае вышеупомянутой концентрации полимера, большей чем 5 мас.%, может присутствовать нерастворимое вещество из компонента А. С другой стороны, в случае концентрации полимера,меньшей чем 0,1 мас.%, раствор уже был бы разбавленным раствором, хотя это также зависит и от молекулярной массы, и, таким образом, эффект бы зачастую доходил до предельного значения. В настоящем изобретении, как это описывалось ранее, приемлемым является способ, по которому после контролируемого выдерживания концентрации полимера компонента А по отношению к растворителю в предпочтительном диапазоне от 0,1 до 5 мас.% производят примешивание к смеси и растворение в ней компонента В. С учетом упрощения способа более предпочтительными является способ, по которому высокомолекулярное вещество, разбавленное растворителем, смешивают с мономерным материалом исходного сырья для получения В в целях проведения растворной полимеризации мономера. В качестве способа доведения концентрации полимера компонента А по отношению к растворителю до величины в диапазоне от 0,1 до 5 мас.% может быть использован способ, включающий растворение, или способ, включающий полимеризацию. В случае разбавления важно перемешивать раствор вплоть до возможного гомогенного разбавления раствора, и температура разбавления предпочтительно находится в диапазоне от 50 до 120 С, а период разбавления может быть надлежащим образом установлен в зависимости от температуры разбавления или концентрации до разбавления. В случае температуры разбавления, меньшей чем 50 С, разбавление может протекать в течение продолжительного периода времени, в то время как в случае температуры разбавления, большей чем 120 С, существует вероятность возможного изменения компонента А. В дополнение к этому, с точек зрения уменьшения перекрывания молекул полимера и обеспечения гомогенного смешивания, предпочтительно регулировать концентрацию полимера компонента А по отношению к растворителю в диапазоне от 0,1 до 5 мас.% от получения вышеупомянутого компонента А до начала смешивания с вышеупомянутым компонентом В или вплоть до начала полимеризации мономерного материала исходного сырья для получения компонента В. Говоря конкретно, предпочтительно использовать способ, по которому в случае получения компонента А в результате проведения растворной полимеризации полимеризацию прекращают при концентрации полимера 5 мас.% и менее, по отношению к растворителю, с последующим смешиванием с компонентом В или мономерным материалом исходного сырья для получения компонента В в целях проведения полимеризации мономера. Обычно в случае низкого соотношения между количествами подаваемого мономера и растворителя высокомолекулярное вещество зачастую будет трудно получить в результате проведения растворной полимеризации. Для разрешения данной проблемы долю подаваемого мономера обычно увеличивают, но на этапе концентрации полимера 5 мас.% и менее в системе будет оставаться большое количество непрореагировавшего мономера. Компонент В может быть дополнительно примешан к системе после удаления непрореагировавшего мономера в результате испарения, но с точки зрения упрощения способа непрореагировавший мономер предпочтительно использовать для проведения растворной полимеризации В. Компонент А, предпочтительно использующийся в настоящем изобретении, в желательном варианте является совместимым с ПАН, а предпочтительно с точки зрения совместимости представляет собой полимер на основе ПАН. Композиция компонента А предпочтительно характеризуется концентрацией акрилонитрила (АН) в диапазоне от 93 до 100 мол.%, а более предпочтительно от 98 до 100 мол.%, при расчете на все количество мономеров. Мономер, который может быть сополимеризован с АН, может быть сополимеризован при 7 мол.% и менее. В данном случае при использовании сополимеризуемого компонента, который характеризуется меньшей константой передачи цепи в сопоставлении с АН, предпочитается уменьшать количество сополимеризуемого компонента как можно больше. В качестве мономеров, которые могут быть сополимеризованы с АН, могут быть использованы, например, акриловая кислота, метакриловая кислота, итаконовая кислота и их соли щелочных металлов,аммониевая соль и низшие алкиловые эфиры, акриламид и его производные, аллилсульфоновая кислота,металлилсульфоновая кислота и их соли или алкиловые эфиры и т.п. В настоящем изобретении в качестве способа полимеризации при получении полимера на основе ПАН, который представляет собой компонент А, могут быть выбраны способ растворной полимеризации, способ суспензионной полимеризации, способ эмульсионной полимеризации и т.п. Для целей гомогенной полимеризации АН и сополимеризуемого компонента предпочтительным является использование способа растворной полимеризации. В случае использования для полимеризации способа растворной полимеризации предпочтительным будет использование растворителя, в котором ПАН является растворимым, такого как водный раствор хлорида цинка, диметилсульфоксид, диметилформамид и диметилацетамид, например, такого как водный раствор хлорида цинка, диметилсульфоксид, диметилформамид и диметилацетамид. В случае трудности получения требуемого значения Mw предпочтительным является использование способа полимеризации с применением растворителя, который характеризуется низкой константой передачи цепи, т.е. способа растворной полимеризации с применением водного раствора хлорида цинка или способа суспензионной полимеризации с применением воды. Доля АН, составляющего компонент В, предпочтительно использующийся в настоящем изобретении, предпочтительно находится в диапазоне от 93 до 100 мол.%, а более предпочтительно от 98 до 100 мол.%. Несмотря на возможность сополимеризации мономера, который может быть сополимеризован с АН, в количестве 7 мол.% и менее повышенное количество сополимеризуемого компонента в результате приводит к термическому разложению сополимеризуемого компонента на стадии окисления и к ощутимой деструкции молекулярных цепей, что, таким образом, уменьшает предел прочности при растяжении для углеродных волокон. В качестве мономера, который может быть сополимеризован с АН, может быть использовано соединение, форсирующее придание огнестойкости. Например, в качестве данного соединения могут быть использованы акриловая кислота, метакриловая кислота, итаконовая кислота и их соли щелочных металлов, аммониевая соль и низшие алкиловые эфиры, акриламид и его производные, аллилсульфоновая кислота, металлилсульфоновая кислота и их соли или алкиловые эфиры и т.п. Способ полимеризации для получения компонента В в настоящем изобретении может быть выбран из способа растворной полимеризации, способа суспензионной полимеризации, способа эмульсионной полимеризации и т.п., а для целей гомогенной полимеризации АН и сополимеризуемого компонента предпочтительным является использование способа растворной полимеризации. В случае использования для полимеризации способа растворной полимеризации предпочтительно используют раствор, в котором ПАН является растворимым, такой как водный раствор хлорида цинка, диметилсульфоксид, диметилформамид и диметилацетамид. Прежде всего, предпочтительным является использование для раствора в способе растворной полимеризации диметилсульфоксида, поскольку ПАН характеризуется высокой растворимостью. Все материалы исходного сырья, использующиеся при полимеризации, предпочтительно перепускают через материал фильтра, характеризующийся тонкостью фильтрования 1 мкм и менее, и используют после этого. Полимер на основе ПАН растворяют в органическом растворителе, в котором полимер на основе ПАН является растворимым, таком как диметилсульфоксид, диметилформамид и диметилацетамид, или в неорганическом солевом растворителе, который представляет собой водный раствор неорганической соли, такой как водный раствор хлорида цинка и водный раствор тиоцианата натрия, тем самым, получая прядильный раствор. В случае использования растворной полимеризации в качестве растворителя полимеризации и прядильного растворителя предпочтительно используют один и тот же растворитель, поскольку исключается стадия удаления растворителя и его отделения от полимера на основе ПАН, полученного на стадии полимеризации, и еще раз растворения отделенного полимера на основе ПАН в прядильном растворителе. Концентрация полимера на основе ПАН в прядильном растворе предпочтительно попадает в диапазон от 5 до 30 мас.%, хотя предложение данного диапазона и необязательно будет подходящим по причинам значительного варьирования соотношения между концентрацией полимера и вязкостью в зависимости от растворителя. В случае органического растворителя концентрация более предпочтительно находится в диапазоне от 14 до 25 мас.%, а наиболее предпочтительно от 18 до 23 мас.%. В случае неорганического солевого растворителя концентрация предпочтительно попадает в диапазон от 5 до 18 мас.%. Концентрация полимера менее чем 5 мас.% неэкономичным образом приводит к использованию большого количества растворителя и может в результате вызывать появление пустот внутри волокна во время коагулирования и, таким образом, ухудшение свойств волокна. С другой стороны, концентрация полимера более чем 30 мас.% в результате приводит к уменьшению вязкости, что дает тенденцию к появлению трудностей при прядении. Концентрацию полимера в прядильном растворе можно контролируемо выдерживать посредством количества использующегося растворителя. Концентрация полимера в настоящем изобретении обозначает массовый процент полимера на основе ПАН, содержащегося в растворе полимера на основе ПАН. Говоря конкретно, после взвешивания раствора полимера на основе ПАН на весах взвешенный раствор полимера на основе ПАН смешивают с растворителем, который не растворяет полимер на основе ПАН, но является совместимым с растворителем, использующимся для раствора полимера на основе ПАН, в целях выведения полимера на основе ПАН из раствора, а после этого полимер на основе ПАН взвешивают. Концентрацию полимера рассчитывают в результате деления массы полимера на основе ПАН после выведения его из раствора на массу полимера на основе ПАН до выведения его из раствора. Вязкость раствора полимера на основе ПАН при температуре 45 С предпочтительно попадает в диапазон от 15 до 200 Пас, более предпочтительно попадает в диапазон от 20 до 100 Пас, а наиболее предпочтительно попадает в диапазон от 25 до 60 Пас. Вязкость раствора менее чем 15 Пас имеет тенденцию к уменьшению прядомости, поскольку сформованная нить, вероятно, приведет к возникновению капиллярного разрушения. Вязкость раствора более чем 200 Пас делает более легким гелеобразование,которое имеет тенденцию к закупориванию материала фильтра. Вязкость прядильного раствора можно регулировать при использовании значения Mw(P), концентрации полимера, типа растворителя и т.п. В настоящем изобретении вязкость раствора полимера на основе ПАН при температуре 45 С может быть измерена при использовании вискозиметра типа В. Говоря конкретно, вязкость измеряютпри использовании вискозиметра типа В после регулирования температуры раствора полимера на основе ПАН,помещенного в химический стакан, в результате погружения химического стакана в теплую водяную баню, температуру которой доводят до температуры 45 С. В качестве вискозиметра типа В для измерения вязкости, например, используют вискозиметр типа B8L, производимый в компании Tokyo Keiki Inc.,таким образом, чтобы вязкость раствора полимера на основе ПАН в диапазоне от 0 до 100 Пас измерять при скорости вращения 6 об/мин для ротора 4, а в случае вязкости раствора в диапазоне от 100 до 1000 Пас ее измеряют при скорости вращения 0,6 об/мин. В настоящем изобретении перед прядением из прядильного раствора прядильный раствор предпочтительно перепускают через материал фильтра для удаления примесей, примешиваемых или генерируемых в материалах исходного сырья для полимера и на каждой стадии. Тонкость фильтрования материала фильтра находится в диапазоне от 3 до 15 мкм, более предпочтительно от 5 до 15 мкм, а еще более предпочтительно от 5 до 10 мкм. В настоящем изобретении тонкость фильтрования материала фильтра определяют как размер (диаметр) частиц для сферических частиц, 95% из которых могут быть собраны во время прохождения через материал фильтра. Поэтому тонкость фильтрования ассоциируют с размером пор, тонкость фильтрования в общем случае увеличивается при уменьшении размера пор. Однако в случае более высокой тонкости фильтрования увеличится скорость сдвига, приложенная к прядильному раствору, что в результате приведет к тенденции к уменьшению значения Mz(P)/Mw(P). Таким образом, в настоящем изобретении является предпочтительным уменьшить тонкость фильтрования. Однако тонкость фильтрования, более высокая, чем 15 мкм, может увеличить количество загрязнителей в полученном прядильном растворе, что, тем самым, в результате приведет к генерации пушения во время растяжения на стадии окисления-карбонизации и растяжения. С другой стороны, тонкость фильтрования менее чем 3 мкм может обеспечить селективное отфильтровывание не только загрязнителей, но также и сверхвысокомолекулярных компонентов, содержащихся в прядильном растворе, тем самым, уменьшая значение Mz(F)/Mw(F). В настоящем изобретении волокно-предшественник углеродного волокна может быть получено в результате прядения из описывавшегося ранее прядильного раствора в соответствии со способом мокрого, сухого или сухомокрого прядения. Прежде всего, предпочтительно использовать способ сухомокрого прядения, поскольку способ сухомокрого прядения в настоящем изобретении приводит к получению свойств полимера на основе ПАН. В каждом случае способа сухомокрого прядения и способа сухого прядения прядение проводят в соответствии с известным способом. Однако в зависимости от установленных условий может быть вызвано расщепление молекулярных цепей, в основном включающих сверхвысокомолекулярные компоненты. Таким образом, будут описаны требующие рассмотрения некоторые моменты получения волокна-предшественника, содержащего сверхвысокомолекулярные компоненты. Диаметр прядильного отверстия, использующийся для прядения, предпочтительно находится в диапазоне от 0,04 до 0,4 мм, а более предпочтительно от 0,1 до 0,15 мм. В случае диаметра прядильного отверстия менее чем 0,04 мм во время экструдирования из фильеры будет прикладываться напряжение сдвига, что, тем самым, не только приведет к утрате межмолекулярного переплетения, но в предельном случае также и вызовет расщепление молекулярных цепей и, таким образом, значение Mz(F)/Mw(F) может быть уменьшено. С другой стороны, в случае диаметра прядильного отверстия более чем 0,4 мм для получения волокна, характеризующегося тониной одиночного волокна 1,5 дтекс и менее, потребуется избыточное растяжение. В случае реализации такого способа может произойти расщепление молекулярных цепей, что уменьшит значение Mz(F)/Mw(F). В способе сухомокрого прядения предпочтительно, чтобы степень вытяжки при прядении для прядильного раствора попадала в диапазон от 2,5 до 15. Степень вытяжки при прядении предпочтительно попадает в диапазон от 5 до 15, а более предпочтительно попадает в диапазон от 10 до 15. Степень вытяжки при прядении в настоящем документе означает величину, полученную в результате деления окружной скорости валика, снабженного источником приведения в движение и первым вводимого в контакт с коагулированными волокнами после покидания коагулированными волокнами фильеры (скорости приема коагулированных волокон), на линейную скорость прядильного растворав прядильном отверстии (линейную скорость экструдирования). Степень вытяжки при прядении выражают следующей формулой: Степень вытяжки при прядении = (скорость приема коагулированного волокна)/(линейная скорость экструдирования) Линейная скорость экструдирования обозначает величину, полученную из объема прядильного раствора, экструдированного в единицу времени через площадь прядильного отверстия. В соответствии с этим линейную степень экструдирования определяют количество экструдированного прядильного раствора и диаметр отверстия фильеры. Прядильный раствор выпускают из прядильного отверстия, после этого сильно деформируют на воздухе, а затем вводят в контакт с коагуляционной ванной, в которой прядильный раствор постепенно коагулирует до получения коагулированных волокон. Вследствие большей легкости растяжения некоагулированного прядильного раствора в сопоставлении с коагулированными волокнами деформирование прядильного раствора в основном происходит на воздухе. Повышенная степень вытяжки при прядении делает более легким получение более тонкого волокна и позволяет устанавливать меньшую степень вытяжки в последующем процессе получения волоконпредшественников. Вытяжка из состояния прядильного раствора является предпочтительной вследствие действия растворителя, уменьшающего переплетение полимеров на основе ПАН и обеспечивающего проведение вытяжки при меньшем натяжении в сопоставлении с вытяжкой в последующем процессе получения волокон-предшественников, что приводит к меньшей вероятности расщепления молекулярных цепей. В случае степени вытяжки при прядении менее чем 2,5 степень вытяжки в последующем процессе получения волокон-предшественников во многих случаях должна быть установлена более высокой. В дополнение к этому, вытяжка при прядении составляет 15 и менее, что достаточно для подавления уменьшения значения Mz(F)/Mw(F). В настоящем изобретении предпочтительно, чтобы коагуляционнная ванна содержала растворитель, использующийся в качестве растворителя в растворе полимера на основе ПАН, такой как диметилсульфоксид, диметилформамид и диметилацетамид, и компонент, промотирующий коагулирование. Компонентом, промотирующим коагулирование, предпочтительно является компонент, который не растворяет описывавшийся ранее полимер на основе ПАН, но является совместимым с растворителем, использовавшимся для раствора полимера на основе ПАН, и, говоря конкретно, предпочтительным является использование воды. В качестве условий для коагуляционной ванны могут быть установлены известные условия, подходящие либо для сухомокрого прядения, либо для мокрого прядения. Раствор полимера на основе ПАН коагулируют в коагуляционной ванне до получения волокон(здесь и далее в настоящем документе называемых набухающим волокном) и волокна сматывают при использовании валика, снабженного источником приведения в движение. Для получения свойств полимера на основе ПАН, подходящих для использования в настоящем изобретении, скорость приема набухающего волокна предпочтительно находится в диапазоне от 20 до 500 м/мин. Скорость приема менее чем 20 м/мин уменьшает производительность при получении, в то время как скорость приема более чем 500 м/мин неизбежно увеличивает напряжение сдвига при прохождении прядильного раствора через материал фильтра или прядильное отверстие, что в результате в некоторых случаях приведет к уменьшению значения Mz(F)/Mw(F). Смотанное набухающее волокно непрерывно подвергают первой стадии вытяжки и обработке сухим теплом, получая тем самым волокно-предшественник углеродного волокна. При необходимости после обработки сухим теплом может быть проведена вторая стадия вытяжки. Первая стадия вытяжки в настоящем изобретении означает вытяжку (стадию вытяжки) на участке от валика приема из коагуляционной ванны до обработки сухим теплом. Первую стадию вытяжки в общем случае проводят на воздухе или в теплой водяной бане. Обычно растворитель, остающийся в коагулированных волокнах, удаляют на стадии промывания водой, а вытяжку после этого проводят в ванне или на воздухе. Необходимо отметить, что коагулированные волокна могут быть подвергнуты непосредственной вытяжке в ванне, а после этого промыванию водой. В дополнение к этому, вторая стадия вытяжки может быть опущена, или в случае проведения второй стадии вытяжки могут быть использованы вытяжка при воздействии сухого тепла или вытяжка в обогревающей среде, или может быть использована комбинация из вытяжки при воздействии сухого тепла и вытяжки в обогревающей среде. Обычно вытяжку в общем случае проводят в обогревающей среде. В настоящем изобретении контролируемое выдерживание натяжения на первой стадии вытяжки или на второй стадии вытяжки делает возможным получение волокна-предшественника углеродного волокна, характеризующееся значением Mz(F)/Mw(F) в описывавшемся ранее диапазоне. На первой стадии вытяжки натяжение находится в диапазоне от 1,5 до 3 мн/дтекс, предпочтительно от 1,8 до 2,8 мн/дтекс, а более предпочтительно от 2 до 2,8 мн/дтекс. Натяжение, увеличенное до более чем 3 мн/дтекс, на первой стадии вытяжки, в результате может привести к невозможности проведения равномерной вытяжки и, таким образом, невозможности сохранения однородности молекулярной ориентации, а зачастую вызывает расщепление молекулярных цепей, что уменьшает значение Mz(F)/Mw(F). В то время как в соответствии с обычными представлениями степень вытяжки увеличивали, в настоящем изобретении важно поддерживать натяжение в ходе всего способа получения волоконпредшественников. Однако натяжение вытяжки, уменьшенное до менее чем 1,5 мн/дтекс на первой ста- 12018977 дии вытяжки, в результате может привести к недостаточной молекулярной ориентации для полученного волокна-предшественника и, таким образом, уменьшить модуль упругости при растяжении для углеродных волокон в виде импрегнированной смолой пряди полученного углеродного волокна. Натяжение на первой стадии вытяжки можно регулировать за счет температуры вытяжки и степени вытяжки, но оно варьируется в зависимости от типа полимера на основе ПАН. В частности, вследствие увеличения натяжения в случае большого значения Mz для полимера на основе ПАН предпочтительно уменьшать степень вытяжки или увеличивать температуру вытяжки. Необходимо отметить, что натяжение на первой стадии вытяжки означает максимальное натяжение среди измеренных значений при измерении натяжения непосредственно перед валиком по отношению к перемещению волокон во время первой стадии вытяжки. В случае проведения первой стадии вытяжки в нескольких ваннах вытяжки в ходе сухомокрого прядения, точка, в которой развивается максимальное натяжение вытяжки, во многих случаях соответствует последней ванне. С другой стороны, в случае мокрого прядения данная точка во многих случаях располагается по соседству с валиком приема, производящим отбор из коагуляционной ванны. Натяжение получают в результате деления нагрузки на волокна на тонину. Нагрузку измеряют при использовании измерителя натяжения, фиксирующего протянутые в промежутке между датчиками волокна. Тонину (дтекс) получают в результате высушивания фиксированной длины подвергаемых измерению волокон способа, а после этого измерения массы постоянной длины волокон. Температура вытяжки на первой стадии вытяжки предпочтительно находится в диапазоне от 60 до 95 С, более предпочтительно от 65 до 85 С, а еще более предпочтительно от 65 до 75 С. С точки зрения уменьшения натяжения температура вытяжки предпочтительно является более высокой. Однако в случае более чем 95 С между одиночными волокнами может возникать адгезия, что ухудшает сортность. С другой стороны, в случае менее чем 60 С может быть ухудшена способность к вытяжке, что уменьшит производительность при получении. В случае проведения первой стадии вытяжки в нескольких ваннах вытяжки температура вытяжки означает максимальную температуру ванн. Степень вытяжки на первой стадии вытяжки означает величину, полученную в результате деления окружной скорости последнего валика на первой стадии вытяжки на окружную скорость валика приема,производящего отбор из коагуляционной ванны. Степень вытяжки на первой стадии вытяжки предпочтительно находится в диапазоне от 1- до 5-кратной, а более предпочтительно от 1- до 3-кратной. Несмотря на предпочтительность меньшей степени вытяжки с точки зрения уменьшения натяжения вытяжки,степень вытяжки менее чем 1 зачастую вызывает релаксацию молекулярной ориентации, что во многих случаях в результате приводит к получению продуктов, характеризующихся ухудшенными как прочностью, так и теплостойкостью. С другой стороны, степень вытяжки более чем 5 в результате приводит к ухудшению стабильности геометрических размеров в способе получения волокон-предшественников и возникновению адгезии между одиночными волокнами, что, тем самым, ухудшает нитеобразующие свойства. Кроме того, в процессе окисления-карбонизации генерируется пушение, что, тем самым, легко приводит к ухудшению свойств. После первой стадии вытяжки в целях предотвращения возникновения адгезии между одиночными волокнами в волокна, подвергаемые первой стадии вытяжки, предпочтительно вводить замасливатель,образованный из силиконового соединения и т.п. В случае использования силиконового замасливателя предпочтительно использовать силиконовый замасливатель, содержащий модифицированный силикон,такой как аминомодифицированный силикон, который характеризуется высокой теплостойкостью. После этого волокна, подвергаемые первой стадии вытяжки, предпочтительно подвергают обработке сухим теплом. Максимальная температура при обработке сухим теплом предпочтительно находится в диапазоне от 160 до 200 С, более предпочтительно от 165 до 198 С, а еще более предпочтительно от 175 до 195 С. Получение предпочтительных результатов при обработке сухим теплом обеспечивает период обработки продолжительностью от 10 до 200 с. В случае понижения максимальной температуры при обработке сухим теплом ниже 160 С, плотность полученного волокна-предшественника углеродного волокна может оказаться недостаточной, что в некоторых случаях в результате приведет к появлению трудностей при достижении выгодных эффектов настоящего изобретения. В альтернативном варианте при превышении максимальной температурой обработки сухим теплом 200 С ощутимым будет сплавление между одиночными волокнами, и в случае получения углеродного волокна может быть уменьшен предел прочности при растяжении для полученного углеродного волокна. При обработке сухим теплом для адаптации к сокращению волокон степень вытяжки может составлять 1 и менее. В дополнение к этому, с точки зрения упрощения способа, вытяжку также предпочтительно проводить в то же самое время,что и обработку сухим теплом (что здесь и далее в настоящем документе называют вытяжкой при воздействии сухого тепла). Необходимо отметить, что описывающуюся далее вторую стадию вытяжки, проводимую в обогревающей среде, и описывающуюся в настоящий момент вытяжку при воздействии сухого тепла в настоящем изобретении считают различными стадиями. Натяжение во время вытяжки при воздействии сухого тепла предпочтительно находится в диапазоне от 1,8 до 10 мн/дтекс. Температура поверхности валика в ходе вытяжки при воздействии сухого тепла предпочтительно находится в диапазоне от 140 до 200 С. Доведение натяжения и температуры до величин в вышеупомянутых диапазонах обеспечивает получение волокна-предшественника, соответствующего настоящему изобретению, без уменьшения значения Mz(F)/Mw(F). Степень вытяжки в ходе вытяжки при воздействии сухого тепла предпочтительно находится в диапазоне от 1,1- до 6-кратной, а более предпочтительно от 2- до 6 кратной. Степень вытяжки менее чем 1,1-кратная в результате может привести к получению у волокнапредшественника недостаточной прочности. С другой стороны, степень вытяжки более чем 6-кратная зачастую приводит к уменьшению значения Mz(F)/Mw(F). Для целей улучшения производительности при получении и улучшения степени ориентации кристаллитов волокно-предшественник углеродного волокна также можно получать и в результате проведения второй стадии вытяжки для волокон в обогревающей среде после проведения для волокон обработки сухим теплом. В качестве обогревающей среды, применяющейся в случае проведения второй стадии вытяжки, предпочтительно используют водяной пар под давлением или перегретый водяной пар ввиду выгодности водяного пара с точки зрения стабильности получения и уменьшения стоимости. В случае использования второй стадии вытяжки натяжение во время второй стадии вытяжки предпочтительно находится в диапазоне от 1,8 до 6 мн/дтекс, более предпочтительно от 3 до 6 мн/дтекс, а еще более предпочтительно от 4 до 5,8 мн/дтекс. Натяжение, увеличенное до более чем 6 мн/дтекс, на второй стадии вытяжки, зачастую приводит к расщеплению молекулярных цепей, что уменьшает значение Mz(F)/Mw(F). Для уменьшения натяжения на второй стадии вытяжки до менее чем 1,8 мн/дтекс существует подход,заключающийся в уменьшении степени вытяжки или увеличении температуры (увеличении давления в случае использования в качестве обогревающей среды водяного пара под давлением). Однако первый вариант ухудшает производительность при получении, в то время как второй вариант, вероятно, приведет к разрыву при вытяжке вследствие сплавления. В случае использования в качестве обогревающей среды водяного пара под давлением натяжение на второй стадии вытяжки можно регулировать за счет степени вытяжки и давления водяного пара под давлением, но оно варьируется в зависимости от типа полимера на основе ПАН, и именно это является предпочтительным для надлежащего регулирования натяжения. Натяжение во время второй стадии вытяжки может быть получено при использовании измерителя натяжения, фиксирующего протянутые в промежутке между датчиками волокна непосредственно после покидания ими зоны вытяжки, такой как труба вытяжки, и измеряющего нагрузку, и при делении нагрузки на тонину в точке измерения. Натяжение на второй стадии вытяжки предпочтительно находится в диапазоне от 1,1- до 10-кратного, более предпочтительно от 1,1- до 6 кратного, а еще более предпочтительно от 1,1- до 3-кратного. В случае использования в качестве обогревающей среды для проведения второй стадии вытяжки водяного пара под давлением давление водяного пара у водяного пара под давлением предпочтительно находится в диапазоне от 0,1 до 0,7 МПа, более предпочтительно от 0,1 до 0,5 МПа, а еще более предпочтительно от 0,2 до 0,4 МПа. Необходимо отметить то, что вторую стадию вытяжки предпочтительно не использовать, поскольку по мере увеличения количества стадий вытяжки значение Mz(F)/Mw(F) уменьшится с большей вероятностью. В случае неиспользования второй стадии вытяжки для улучшения производительности при получении предпочтительно проводить описывавшуюся ранее вытяжку при воздействии сухого тепла. По мере увеличения степени вытяжки в ходе всего процесса, включающего первую стадию вытяжки, вытяжку при воздействии сухого тепла и вторую стадию вытяжки, (здесь и далее в настоящем документе называемой совокупной степенью вытяжки) значение Mz(F)/Mw(F) во многих случаях уменьшается. Однако для целей улучшения динамических свойств полученного углеродного волокна совокупную степень вытяжки предпочтительно увеличить, и с точки зрения баланса между обеими характеристиками совокупная степень вытяжки предпочтительно находится в диапазоне от 1- до 15-кратной, более предпочтительно от 2- до 13-кратной, а еще более предпочтительно от 3- до 5-кратной. Тонина одиночного волокна полученного таким образом волокна-предшественника предпочтительно находится в диапазоне от 0,1 до 1,2 дтекс, более предпочтительно от 0,2 до 1,0 дтекс, еще более предпочтительно от 0,3 до 0,8 дтекс. В случае чрезмерно малой тонины одиночного волокна волокнапредшественника стабильность способа в способе получения волокон-предшественников и способе окисления-карбонизации может быть ухудшена вследствие появления разрыва волокна в результате контакта с валиком или направляющим элементом. С другой стороны, в случае чрезмерно большой тонины одиночного волокна может увеличиться различие между внутренними и внешними структурами каждого одиночного волокна после стадии окисления, что, таким образом, приведет к ухудшению технологичности при последующей карбонизации и к уменьшению предела прочности при растяжении и модуля упругости при растяжении. Необходимо отметить то, что тонина одиночного волокна (дтекс) в настоящем изобретении обозначает массу (г), приходящуюся на 10000 м одиночного волокна. В настоящем изобретении степень ориентации кристаллитов для полученного волокнапредшественника предпочтительно находится в диапазоне от 85 до 90%, а более предпочтительно от 85 до 88%. Степень ориентации кристаллитов менее чем 85% может уменьшить у полученного углеродного волокна модуль упругости при растяжении. С другой стороны, степень ориентации кристаллитов более чем 90% может не позволить увеличить степень вытяжки на стадии окисления, тем самым, генерируя пушение. Однако контролируемое выдерживание значения Mz(F)/Mw(F) для волокна-предшественника делает возможным предотвращение генерации пушения на стадии окисления в сопоставлении с волок- 14018977 нами-предшественниками, не соответствующими настоящему изобретению, даже и при сопоставимой степени ориентации кристаллитов. В дополнение к этому, параметр формы Вейбулла m(Р) у предела прочности при растяжении для одиночного волокна волокна-предшественника, соответствующего настоящему изобретению, предпочтительно составляет 11 и более. Параметр формы Вейбулла указывает на вариации предела прочности при растяжении для одиночного волокна и предпочтительно является повышенным, так как это может предотвратить появление пушения на стадии получения углеродного волокна. Параметр формы Вейбулла предпочтительно составляет 13 и более и имеет промышленный предел, доходящий вплоть до 20. Несмотря на существование обычно областей применения, которые требуют наличия небольших вариаций относительного удлинения для одиночного волокна волокон-предшественников, было установлено, что важным является профиль распределения одиночных волокон по прочности, а не величина вариации. Ни одно из волокон-предшественников, полученных в соответствии с обычными подходами, не демонстрирует параметр формы Вейбулла, равный 11 и более. В дополнение к этому, как было установлено, использование волокна-предшественника, характеризующегося высоким параметром формы Вейбулла,демонстрирует тенденцию к увеличению параметра формы Вейбулла для волокон в середине способа окисления-карбонизации, использующего волокно-предшественник, и приводит к получению в качестве конечного продукта углеродного волокна, характеризующегося высоким параметром формы Вейбулла. Поэтому повышенный параметр формы Вейбулла волокна-предшественника приводит к получению углеродного волокна, которое характеризуется превосходной технологичностью в способе окислениякарбонизации и демонстрирует пониженные вариации свойств. Предел прочности при растяжении для одиночного волокна получают в соответствии с документомJIS R7606 (2000) тем же самым образом, как и в случае углеродных волокон. Во-первых, пучок волоконпредшественников длиной в 20 см разделяют на четыре пучка таким образом, чтобы количество одиночных волокон для каждого пучка составляло 255% по отношению к пучку волокон-предшественников, и из каждого из четырех раздельных пучков случайным образом отбирают 100 одиночных волокон в качестве образцов. Отобранные в качестве образцов одиночные волокна фиксируют на перфорированной пластине при использовании клея. Пластину с зафиксированными одиночными волокнами закрепляют в приборе для испытания на растяжение и проводят испытание на растяжение в условиях длины образца 25 мм и скорости растяжения 5 мм/мин. Площадь поперечного сечения волокна рассчитывают как среднюю площадь поперечного сечения, исходя из тонины и плотности, измеренной в соответствии с описывающимся далее методом. Таким образом, полученный предел прочности при растяжении для одиночного волокна используют при получении графика Вейбулла зависимости между двойным логарифмом In прочности и функцией 1/(1-F) от вероятности разрушения F и из наклона кривой на графике рассчитывают параметр формы Вейбулла. Полученное волокно-предшественник углеродного волокна обычно имеет форму непрерывного волокна (элементарного волокна). В дополнение к этому, количество элементарных волокон (одиночных волокон), составляющих один пучок волокон, предпочтительно находится в диапазоне от 1000 до 3000000, более предпочтительно от 12000 до 3000000, еще более предпочтительно от 24000 до 2500000, а наиболее предпочтительно от 24000 до 2000000. Вследствие демонстрации волокном-предшественником углеродного волокна, полученным в соответствии с настоящим изобретением, высокой способности к вытяжке тонина одиночного волокна может быть уменьшена. Поэтому количество одиночных волокон,приходящееся на один пучок волокон, в некоторых случаях увеличивают для получения одного пучка волокон, характеризующегося желательной совокупной тониной. Однако, хотя большее количество одиночных волокон, приходящихся на один пучок волокон, является предпочтительным, в целях улучшения производительности, при его чрезмерно большой величине в некоторых случаях будет невозможно провести однородную окислительную обработку вплоть до внутренних областей пучка. В зависимости от назначения предел прочности при растяжении для одиночного волокна и количество одиночных волокон регулируют надлежащим образом. Далее будет описываться способ получения углеродного волокна по настоящему изобретению. Предлагается способ получения углеродного волокна по настоящему изобретению, который позволяет получить углеродное волокно в результате последовательного проведения стадии окисления, на которой описывавшееся ранее волокно-предшественник углеродного волокна подвергают окислительной обработке при одновременном проведении вытяжки волокна-предшественника углеродного волокна со степенью вытяжки в диапазоне от 0,8 до 3,0 на воздухе при температуре в диапазоне от 200 до 300 С,стадии предварительной карбонизации, на которой волокно, полученное на стадии окисления, подвергают предварительной карбонизации при одновременном проведении вытяжки предпочтительно со степенью вытяжки в диапазоне от 1 до 1,3 в инертной атмосфере при температуре в диапазоне от 300 до 800 С, и стадии карбонизации, на которой волокно, полученное на стадии предварительной карбонизации, подвергают карбонизации при одновременном проведении вытяжки предпочтительно со степенью вытяжки в диапазоне от 0,96 до 1,05 в инертной атмосфере при температуре в диапазоне от 1000 до 3000 С. В способе получения углеродного волокна, соответствующего по настоящему изобретению, придание огнестойкости относится к стадии, на которой для увеличения теплостойкости тепловая обработка в атмосфере, содержащей от 4 до 25 мол.% и более кислорода, при температуре в диапазоне от 200 до 300 С частично превращает или окисляет волокно-предшественник углеродного волокна. Хотя способ получения волокон-предшественников и способ стадии окисления обычно не являются непрерывными,способ получения волокон-предшественников и часть стадии или вся стадия окисления могут быть реализованы и в непрерывном режиме. Степень вытяжки при придании огнестойкости находится в диапазоне от 0,8 до 3, предпочтительно от 1,3 до 3, более предпочтительно от 1,4 до 2. В случае понижения степени вытяжки при придании огнестойкости ниже 0,8 структура с неполной циклизацией у полимера на основе ПАН в окисленном волокне продемонстрирует недостаточную степень ориентации, и у полученного, в конечном счете, углеродного волокна модуль упругости при растяжении уменьшится. В альтернативном варианте в случае превышения степенью вытяжки при придании огнестойкости 3 ухудшится стабильность получения вследствие генерации пушения или разрыва волокна. Использование волокна-предшественника по настоящему изобретению может значительно улучшить степень вытяжки на стадии окисления, что, таким образом,улучшит производительность при получении. В дополнение к этому, натяжение вытяжки на стадии окисления предпочтительно находится в диапазоне от 0,1 до 0,25 г/дтекс. В случае натяжения вытяжки на стадии окисления менее чем 0,1 г/дтекс у полимера на основе ПАН в окисленном волокне трудно будет улучшить степень ориентации для структуры с неполной циклизацией. В случае натяжения вытяжки более чем 0,25 г/дтекс на стадии окисления вероятной будет генерация пушения. Волокно-предшественник по настоящему изобретению обладает структурой, которая способна увеличивать степень вытяжки без увеличения натяжения вытяжки на стадии окисления и, таким образом, является подходящей для улучшения производительности при получении. Степень ориентации кристаллитов для структуры с неполной циклизацией у полимера на основе ПАН в окисленном волокне, соответствующем настоящему изобретению, предпочтительно находится в диапазоне от 78 до 85%, а более предпочтительно от 80 до 85%. Данные степени достигаются в результате установки описывавшихся ранее условий по степени вытяжки и/или натяжению. Говоря более конкретно, степень ориентации кристаллитов может быть увеличена в результате увеличения степени вытяжки и/или натяжения. В случае понижения степени ориентации кристаллитов ниже 78% модуль упругости при растяжении для полученного углеродного волокна может быть уменьшен. С другой стороны, в случае превышения степенью ориентации кристаллитов 85% установка высокой степени вытяжки на стадии окисления может привести к генерации пушения, что, тем самым, уменьшит производительность при получении. Хотя период обработки при окислении может быть подходящим образом выбран в диапазоне от 10 до 100 мин, для целей улучшения стабильности получения на последующей стадии предварительной карбонизации и улучшения динамических свойств углеродного волокна период обработки предпочтительно устанавливать таким образом, чтобы относительная плотность полученного окисленного волокна попадала в диапазон от 1,3 до 1,38. На стадии окисления в качестве средства нагревания волокон используют любые устройства бесконтактного типа, такие как электрический нагреватель, ширильная рама, в которой волокнопредшественник прессуют при использовании воздуха, нагретого водяным паром и т.п., и устройство нагревания инфракрасным излучением, и контактного типа, такие как нагреватель пластинчатого типа и нагреватель барабанного типа. Для улучшения эффективности теплопередачи нагревание предпочтительно проводят, по меньшей мере, частично по способу нагревания контактного типа, а более предпочтительно проводят исключительно по способу нагревания контактного типа. Предварительную карбонизацию и карбонизацию проводят в инертной атмосфере и в качестве использующегося инертного газа применяют, например, азот, аргон и ксенон и т.п. С экономической точки зрения предпочтительно используют азот. В дополнение к этому, будет описываться углеродное волокно, соответствующее настоящему изобретению. Углеродным волокном по настоящему изобретению является углеродное волокно, у которого размер кристаллитов (Lc (нм и параметры (ID/IG, IV/IG, G (см-1, относящиеся к поверхности углеродного волокна, удовлетворяют следующим формулам от (1) до (4) при измерении по методу спектроскопии комбинационного рассеяния: Во-первых, будут описываться различные свойства, подходящие для использования в настоящем изобретении. Углеродное волокно представляет собой поликристалл, образованный из многочисленных кристаллитов графита. В случае увеличения максимальной температуры (здесь и далее в настоящем документе сокращенно называемой температурой карбонизации) карбонизационной обработки для получения углеродного волокна произойдет перегруппировка гексагонального углеродного слоя в углеродном волокне,что, тем самым, будет промотировать увеличение размера кристаллитов и степень ориентации кристаллитов. Таким образом, модуль упругости при растяжении для углеродного волокна увеличится. Говоря более конкретно, существует соотношение, по которому при сохранении других условий постоянными повышенная температура карбонизации увеличивает как размер кристаллитов Lc, так и модуль упругости при растяжении YM. Далее будут описываться параметры, измеренные по методу спектроскопии комбинационного рассеяния. Спектроскопия комбинационного рассеяния представляет собой метод измерения, который является высокочувствительным к дефектам решетки углеродных материалов. Спектр, измеренный по методу спектроскопии комбинационного рассеяния, разделяют на три типа пиков - в области 1360 см-1, в области 1480 см-1 и в области 1600 см-1 - в результате подбора аппроксимирующей кривой при использовании квадратичной функции. Данные три типа пиков называют полосой D (в области 1360 см-1), впадиной между полосой D и полосой G (в области 1480 см-1: данную впадину в настоящем изобретении также называют пиком) и полосой G (в области 1600 см-1), соответственно, которые характеризуются интенсивностями пиков, обозначаемыми символами ID, IV и IG соответственно. Полоса D отображает неупорядоченный углерод, пик в области 1480 см-1 также отображает неупорядоченный углерод, и полоса G отображает саму колебательную моду графита. В случае проведения исследования на основании данной информации зачастую для проведения исследования обычно получают соотношения между интенсивностями пиков. Соотношения ID/IG и IV/IG исключительно хорошо коррелируют с размером кристаллитов(Lc) таким образом, что при увеличении размера кристаллитов значение IG увеличивается, в то время как значения IV и IG уменьшаются. Кроме того, будут подробно описываться значения параметров. ЗначениеID/IG имеет тенденцию к монотонному уменьшению при увеличении температуры карбонизации таким образом, что значение ID/IG имеет порядок величины 2 в случае окисленного волокна, в котором графит почти не наблюдают, и уменьшается до приблизительно 1 при температурах карбонизации в диапазоне от 500 до 900 С, а после этого становится практически нечувствительным к температуре карбонизации. В дополнение к этому, в то время как значение IV/IG демонстрирует сложное поведение по отношению к увеличению температуры карбонизации, значение IV/IG демонстрирует тенденцию к уменьшению от 0,8 до 0,4 при переходе температуры карбонизации от приблизительно 1200 С до приблизительно 1700 С. Говоря более конкретно, формулы от (1) до (3) свидетельствуют о проведении карбонизационной обработки при температуре карбонизации порядка величины в диапазоне от 1200 до 1700 С. В случае увеличения температуры карбонизационной обработки на 100 С значение Lc увеличится на величину порядка 1,5 нм. Далее будет описываться волновое число пика G (см-1) для полосы G. Как представляется, волновое число пика для полосы G исключительно хорошо коррелирует с п-электронными сопряженными структурами, ассоциирующимися с размером кристаллитов в направлении оси А, при этом волновое число пика имеет тенденцию к увеличению по мере увеличения температуры карбонизации в области температуры карбонизации в диапазоне от 1200 до 1700 С. В случае увеличения температуры карбонизационной обработки на 100 С значение G увеличится на величину порядка 3 см-1. Говоря более конкретно, в случае обычных углеродных волокон по мере увеличения температуры карбонизации от 1200 С значение IV/IG уменьшается, в то время как значение G увеличивается, а в случае углеродного волокна, соответствующего по настоящему изобретению, исследование в рамках настоящего изобретения выявило, что при идентичности значения IV/IG как такового сорт углеродного волокна улучшится в случае большего значения G. Как можно себе представить на основании вышеизложенного, величинаIV/IG с идентичным значением и повышенное значение G свидетельствуют о том, что несмотря на сопоставимый размер кристаллитов развились п-электронные сопряженные структуры. С другой стороны, поскольку, как представляется, улучшение сорта углеродного волокна соответствует уменьшению дефектов решетки в углеродном волокне, то согласно оценке углеродное волокно, соответствующее настоящему изобретению, характеризуется повышенным значением G в сравнении со значением IV/IG в сопоставлении с обычными углеродными волокнами, и вследствие наличия данного свойства (для данного размера кристаллитов развивается п-электронная сопряженная структура) сорт углеродного волокна улучшается. Как описывалось ранее, значение IV/IG при увеличении температуры карбонизации демонстрирует тенденцию к уменьшению, в то время как значение G при увеличении температуры карбонизации имеет тенденцию к увеличению. Таким образом, значения IV/IG и G демонстрируют обратную корреляцию. Таким образом, как представляется, в результате умножения значения либо IV/IG, либо G на соответствующий коэффициент и вычисления суммы получают величину в виде показателя, указывающего на соотношение между размером кристаллита углеродного волокна и п-электронными сопряженными структурами. Именно формула (4) в качестве экспериментальной формулы выражает структурный признак углеродного волокна, соответствующего настоящему изобретению. Выражение для обычных волокон в виде формулы (4) представляет собой 1600G+17(IV/IG)1604. Говоря более конкретно, угле- 17018977 родное волокно, соответствующее настоящему изобретению, получают при температуре карбонизации,представляемой формулами от (1) до (3), и оно обладает структурой, которая удовлетворяет соотношению формулы (4). В случае параметра менее чем 1605 сорт полученного углеродного волокна будет только сопоставим с сортами обычных волокон, в то время как данный параметр может превышать 1610,но в промышленности у него имеется верхний предел. Говоря более конкретно, данный параметр составляет 1607 и более. Использование волокна-предшественника, полученного в соответствии с настоящим изобретением, делает возможным регулирование данного параметра в пределах данного диапазона, тем самым, обеспечивая улучшение сорта углеродного волокна. Далее будет описываться параметр формы Вейбулла m у предела прочности при растяжении для одиночного волокна углеродного волокна. "m" относится к признаку в виде показателя, указывающего на чувствительность к дефектам, и большее значение m означает большую нечувствительность углеродного волокна. Металлические материалы характеризуются параметром формы Вейбулла m, равным приблизительно 20, в случае более высокомодульных материалов концентрация напряжений с большей вероятностью наблюдается на краях дефектов, и обычные пучки углеродных волокон характеризуются параметром формы Вейбулла m, равным приблизительно 5. В числе углеродных волокон углеродные волокна,характеризующиеся модулем упругости низкого уровня приблизительно 41 ГПа, имеют параметр формы Вейбулла m приблизительно 7,9, в то время как углеродные волокна, характеризующиеся модулем упругости высокого уровня приблизительно 940 ГПа, имеют параметр формы Вейбулла m приблизительно 4,2. Таким образом, больший модуль упругости в результате приводит к получению меньшего значенияm. В дополнение к этому, параметр формы Вейбулла m также представляет собой признак, свидетельствующий о размере дефектов и их численной плотности, и он увеличивается тогда, когда размер дефектов и их численная плотность являются более однородными. Например, даже в случае углеродных волокон,содержащих множество дефектов и постоянно разрушающихся при некотором уровне низкой прочности в направлении длины углеродных волокон, и даже вне зависимости от того, какое одиночное волокно рассматривается, значение m увеличивается. На предел прочности при растяжении для углеродного волокна значительное воздействие оказывают значение вязкости разрушения и размеры дефектов. Вследствие включения в высокоэффективные углеродные волокна небольшого количества небольших дефектов размер и форма дефектов с меньшей вероятностью будут однородно распределены между одиночными волокнами. Поэтому значение m имеет тенденцию к относительному увеличению. Необходимо отметить то, что углеродное волокно, соответствующее настоящему изобретению, обычно получают в виде пучка волокон, и, как будет описываться далее, для проведения испытания на растяжение одиночных волокон отбирают образцы одиночных волокон. Углеродное волокно, соответствующее настоящему изобретению, удовлетворяет следующей далее формуле при попадании значения Lc в пределы диапазона от 1,8 до 2,6. Обычно использующиеся углеродные волокна в общем случае характеризуются соотношением 50Lc+150YM50Lc+210 при нахождении значения Lc в диапазоне от 1,8 до 2,6. Для стилизирования ориентации кристаллов при использовании волокон-предшественников обычных волокон в такой степени,чтобы получать углеродные волокна,которые удовлетворяют соотношению 50Lc+210YM50Lc+270, при нахождении значения Lc в диапазоне от 1,8 до 2,6, тепловая обработка в способе окисления-карбонизации должна быть проведена при высоком натяжении. Однако тепловая обработка, проводимая при таком высоком натяжении, вызывает появление пушения, что в результате приводит к потребности частого удаления распушенных волокон, намотанных на валик. В дополнение к этому, размеры дефектов и распределение численной плотности дефектов увеличиваются, в то время как значение m уменьшается. В противоположность этому, волокно-предшественник углеродного волокна,полученное в соответствии с настоящим изобретением, включает длинную последовательность молекулярных цепей и является гомогенным, что, таким образом, делает возможным получение предварительно карбонизованного гомогенного волокна при карбонизационной обработке, проводимой при повышенных натяжениях, и, таким образом, делает возможным получение углеродного волокна, соответствующего настоящему изобретению. Углеродное волокно, соответствующее настоящему изобретению, характеризуется значением m,измеренным по методу, который будет описываться далее, которое составляет 6 и более, предпочтительно 6,1 и более, а более предпочтительно 7 и более. В случае значения m менее чем 6 при использовании в качестве композитного материала увеличивается пушение. В то время как значение m предпочтительно является большим, значению m трудно придать величину 10 и более. Для увеличения значения m важно использовать гомогенное волокно-предшественник, которое незначительно варьируется при переходах между одиночными волокнами. Кроме того, для того, чтобы не уменьшить параметр формы Вейбулла m для волокна, проходящего через каждую стадию процесса окисления-карбонизации при получении углеродного волокна, важно установить такую степень вытяжки, которая характеризуется достаточным запасом по отношению к предельной степени вытяжки в такой степени, чтобы на каждой стадии окислениякарбонизации какого-либо пушения не генерировалось. В случае установления более низкой степени вытяжки, для того чтобы не уменьшить параметр формы Вейбулла m, необходимое значение YM в некоторых случаях не потребуется, и, таким образом, необходимо будет получить более длинные последовательности молекулярных цепей для волокна-предшественника, для того чтобы могла быть установлена более высокая степень вытяжки вплоть до разрушения в процессе окисления-карбонизации. Предел прочности при растяжении для одиночного волокна получают в соответствии с документомJIS R7606 (2000) следующим образом. Во-первых, пучок углеродных волокон длиной в 20 см разделяют на четыре пучка таким образом, чтобы количество одиночных волокон для каждого пучка составляло 25+5% по отношению к пучку волокон-предшественников, и из каждого из четырех раздельных пучков случайным образом отбирают 100 одиночных волокон в качестве образцов. Отобранные в качестве образцов одиночные волокна фиксируют на перфорированной пластине с использованием клея. Пластину с зафиксированными одиночными волокнами закрепляют в приборе для испытания на растяжение, срезают бумагу на боковой поверхности и проводят испытание на растяжение при длине образца 25 мм и скорости растяжения 1 мм/мин. На всех стадиях, таких как отбор образцов, фиксирование на пластине и закрепление в приборе для испытаний, одиночные волокна могут разрушиться до применения прибора для испытания на растяжение. Таким образом, в случае возникновения разрушения во избежание селективного удаления слабых волокон партию подвергают испытанию на растяжение еще раз. Площадь поперечного сечения волокна рассчитывают как среднюю площадь поперечного сечения, исходя из тонины и плотности, измеренной в соответствии с описывающимся далее методом. Полученный таким образом предел прочности при растяжении для одиночного волокна используют для получении графика Вейбулла в виде зависимости двойного логарифма от логарифма прочности от функции 1/(1-F) от вероятности разрушения F и из наклона кривой на графике рассчитывают параметр формы Вейбулла. Второй параметр формы Вейбулла m" в настоящем изобретении определяют как параметр формы Вейбулла, полученный в результате прямолинейной аппроксимации в диапазоне от 0,3 до 1 для вероятности разрушения F. Второй параметр формы Вейбулла m" предпочтительно составляет 5,7 и более. В то время как описывавшееся ранее значение m получают из графика Вейбулла в результате аппроксимации к прямой линии, как было установлено, график Вейбулла для углеродных волокон зачастую является криволинейным. Материал на стороне меньшей интенсивности относительно точки складывания содержит множество дефектов и зачастую характеризуется большим параметром формы Вейбулла, в то время как материал на стороне большей интенсивности относительно точки складывания зачастую характеризуется меньшим параметром формы Вейбулла. Хотя наблюдение за разрушением в виде композитного материала показывает, что разрушение одиночного волокна вызывает концентрацию напряжений в окрестности разрушения, что легко индуцирует разрушение соседних одиночных волокон, разрушение одного одиночного волокна не приводит к разрушению всего композитного материала, и композитный материал зачастую разрушается в случае, когда происходит разрушение одиночных волокон в количестве в диапазоне приблизительно от 10 до 30% от совокупного количества одиночных волокон. Поэтому параметр формы Вейбулла для стороны меньшей интенсивности относительно точки складывания может с меньшей вероятностью оказывать воздействие на прочность композитного материала и, таким образом,часто является важным параметр формы Вейбулла для стороны большей интенсивности относительно точки складывания. В то время как точка складывания варьируется для вероятности разрушения F в диапазоне приблизительно от 0,1 до 0,6, параметр формы Вейбулла, полученный в диапазоне от 0,3 до 1 для вероятности разрушения F, ненамного различается по значению параметра формы Вейбулла, что, таким образом, предотвращает появление какого-либо ложного технического смысла. Значение m" можно регулировать тем же самым образом, что и значение m, и значение m" может быть увеличено в результате увеличения параметра формы Вейбулла для стороны меньшей интенсивности относительно точки складывания, т.е. адаптации параметра формы Вейбулла таким образом, чтобы получить дефекты с однородными и крупными размерами. Значение m" 5,7 и более достигается в результате использования гомогенного волокна-предшественника, характеризующегося по возможности наибольшим уменьшением причин для дефектов и наличием длинных последовательностей молекулярных цепей. Значение m" менее чем 5,7 может в результате привести к увеличению коэффициента вариации (значения KB) предела прочности при растяжении для полученного пластика, армированного углеродным волокном. В настоящем изобретении квадрат коэффициента корреляции для графика Вейбулла, подвергнутого аппроксимации к прямой линии, определяют как R2 в испытании на растяжение одиночного волокна. Значение R2 в настоящем изобретении предпочтительно находится в диапазоне от 0,98 до 1, а более предпочтительно от 0,99 до 1. На графике с 1-F (F: вероятность разрушения) для оси х и S (произведение напряжений под нагрузкой) для оси у максимальное значение S исключительно хорошо коррелирует с пределом прочности при растяжении для однонаправленного пластика, армированного углеродным волокном. В то время как график для S в идеальном случае имеет выпуклые кверху точки изгиба, образующие одну кривую, в случае высокой степени кривизны получают кривую, имеющую множество точек изгиба, таким образом, минимальное значение S невелико при среднем пределе прочности при растяжении для одиночного волокна, что зачастую в результате приводит к невозможности эффективного получения динамических свойств. Как предполагается для значения S, другие волокна несут равную долю напряжения для разрушенного одиночного волокна, и в окрестности разрушенного одиночного во- 19018977 локна возникает концентрация напряжений. Таким образом, значение S неспособно непосредственно указать на свойства композитного материала. Однако значение S эффективно в качестве одного показателя, опосредованно указывающего на свойства композитного материала. Значение R2 указывает на степень кривизны графика Вейбулла, и график Вейбулла демонстрирует повышенную степень кривизны при уменьшении коэффициента корреляции. В случае значения R2 менее чем 0,98 существует тенденция к необходимости улучшения среднего значения динамических свойств углеродного волокна в целях удовлетворения динамических свойств однонаправленного композитного материала. Квадрат R2 коэффициента корреляции может быть доведен до значения, более близкого к 1, в результате уменьшения больших дефектов, отличных от дефектов, распределенных в углеродном волокне. Большие дефекты образуются в результате сплавления во время получения волокна-предшественника, наличия загрязнителей, содержащихся в растворе полимера материала исходного сырья, коррозии во время проведения процесса и т.п., и является предпочтительным уменьшать эти дефекты. Необходимо отметить, что микродефекты и макродефекты, определенные по размерам начальных точек разрушений в поперечном сечении разрушения в испытании на растяжение в результате наблюдения начальных точек в электронный микроскоп, не могут быть классифицированы на группы большей интенсивности и меньшей интенсивности по пределу прочности при растяжении для одиночного волокна и, таким образом, менее вероятно, соотносятся с квадратом R2 коэффициента корреляции. В дополнение к этому, углеродное волокно, соответствующее настоящему изобретению, характеризуется модулем упругости при растяжении для углеродных волокон в виде пряди, импрегнированной смолой, TS в диапазоне от 6 до 9 ГПа. Обычные углеродные волокна характеризуются размером кристаллитов и модулем упругости при растяжении, которые удовлетворяют формуле (5), и в случае значения m 6 и более значение TS является менее чем 6 ГПа. Даже в случае использования углеродного волокна для целей предела прочности при растяжении и сопротивления ударной нагрузке какого-либо экстраординарного эффекта по уменьшению массы конструкционного материала достигнуто не было. Для удовлетворения современных потребностей в данной сфере значение TS предпочтительно составляет 6 ГПа и более, более предпочтительно 6,5 ГПа и более, а еще более предпочтительно 7 ГПа и более. Углеродное волокно, соответствующее настоящему изобретению, характеризуется размером кристаллитов Lc в диапазоне от 1,5 до 2,6 нм. В случае значения Lc для углеродного волокна менее чем 1,5 предел прочности при растяжении будет невелик; в случае значения Lc менее чем 1,8 нм невелика будет степень кристалличности и невелико будет значение YM; а в случае значения Lc более чем 2,6 нм невелик будет предел прочности при сжатии. В каждом случае для конструкционного элемента баланс между модулем упругости при растяжении и пределом прочности при сжатии может оказаться неудовлетворительным. Для улучшения баланса значение Lc предпочтительно находится в диапазоне от 1,8 до 2,6 нм, а более предпочтительно от 2 до 2,4 нм. Значение Lc для углеродного волокна можно контролируемо выдерживать при использовании температуры карбонизации, повышенная температура карбонизации приводит к увеличению значения Lc. Углеродное волокно, соответствующее настоящему изобретению, предпочтительно характеризуется средним диаметром одиночного волокна в диапазоне от 2 до 7 мкм, а более предпочтительно от 5 до 7 мкм. По мере уменьшения среднего диаметра одиночного волокна потенциал среднего предела прочности при растяжении увеличивается. Однако средний диаметр одиночного волокна менее чем 5 мкм приводит к увеличению площади поверхности в сопоставлении с объемом, и, таким образом, после формирования волокна в способе, вероятно, возникнут дефекты, что легко может ухудшить параметр формы Вейбулла. В альтернативном варианте, в случае среднего диаметра одиночного волокна более чем 7 мкм окислительная обработка в одиночных волокнах будет недостаточной и, таким образом, значение YM может быть улучшено с меньшей вероятностью. В дополнение к этому, в углеродном волокне, соответствующем настоящему изобретению, количество одиночных волокон, составляющих пучок волокон, предпочтительно находится в диапазоне от 12000 до 48000, а более предпочтительно от 24000 до 48000. В то время как меньшее количество одиночных волокон создает эффект упрощения однородных технологических обработок высокого порядка,таких как ионная имплантация и плазменная обработка, случай использования крупноразмерного конструкционного материала в результате может приводить к увеличению количеств использующихся волокон и уменьшению эффективности получения. До тех пор пока количество одиночных волокон будет составлять 12000 и более, во многих случаях будет достигаться достаточная эффективность получения. В альтернативном варианте количество одиночных волокон более чем 48000 может привести к проведению негомогенной обработки в способе окисления-карбонизации, что в результате вызовет уменьшение значения m. Кроме того, будет описан способ получения углеродного волокна, соответствующего по настоящему изобретению. Углеродное волокно может быть получено в результате получения окисленного волокна и дополнительных окисления и карбонизации окисленного волокна в соответствии со способом, описывающимся далее. Температура предварительной карбонизации предпочтительно находится в диапазоне от 300 до 800 С. Кроме того, предпочтительно устанавливать скорость увеличения температуры при предвари- 20018977 тельной карбонизации равной 500 С/мин и менее. Степень вытяжки для проведения предварительной карбонизации находится в диапазоне от 1 до 1,3, предпочтительно от 1,1 до 1,3, а более предпочтительно от 1,1 до 1,2. В случае понижения степени вытяжки при проведении предварительной карбонизации ниже 1 степень ориентации для полученных предварительно карбонизованных волокон будет недостаточной, и модуль упругости при растяжении для углеродных волокон в виде пряди, импрегнированной смолой, у углеродных волокон уменьшится. В альтернативном варианте в случае превышения степенью вытяжки для проведения предварительной карбонизации 1,3 генерация пушения или генерация разрыва волокна уменьшат технологичность. Температура карбонизации находится в диапазоне от 1000 до 2000 С, предпочтительно от 1200 до 1800 С, а более предпочтительно от 1300 до 1600 С. Хотя повышенная температура карбонизации увеличивает модуль упругости при растяжении для углеродных волокон в виде пряди, импрегнированной смолой, предел прочности при растяжении достигает максимума при приблизительно 1500 С. Таким образом, температуру карбонизации устанавливают с учетом баланса между обеими характеристиками. Степень вытяжки для проведения карбонизации находится в диапазоне от 0,96 до 1,05, предпочтительно от 0,97 до 1,05, а более предпочтительно от 0,98 до 1,03. В случае понижения степени вытяжки для проведения карбонизации ниже 0,96 степень ориентации и плотность полученного углеродного волокна будут недостаточными, что в результате приведет к уменьшению модуля упругости при растяжении для углеродных волокон в виде пряди, импрегнированной смолой. В альтернативном варианте в случае превышения степенью вытяжки для проведения карбонизации 1,05 генерация пушения или появление разрыва волокна уменьшат технологичность. Полученное углеродное волокно может быть подвергнуто электролитической обработке для модифицирования его поверхности. В качестве электролита, подходящего для использования при электролитической обработке, могут быть использованы раствор кислоты, такой как серная кислота, азотная кислота и хлористо-водородная кислота, и щелочь, такая как гидроксид натрия, гидроксид калия, гидроксид тетраэтиламмония, карбонат аммония и бикарбонат аммония, или их соли в виде водного раствора. В настоящем документе количество электричества, необходимого для электролитической обработки,может быть выбрано надлежащим образом в зависимости от использующейся степени карбонизации углеродного волокна. Электролитическая обработка делает возможным получение надлежащих адгезионных свойств между углеродным волокном и его матрицей в получаемом композитном материале, армированном волокном. Говоря конкретно, решаются следующие далее проблемы: проблема хрупкого разрушения композитного материала, вызванного чрезмерно большой адгезией, проблема уменьшения предела прочности при растяжении в направлении волокна или проблема ухудшенных адгезионных свойств по отношению к смоле, что в результате не приводит к развитию прочностных свойств не в направлении волокна, несмотря на высокий предел прочности при растяжении в направлении волокна. Армированный волокном композитный материал, получаемый в результате проведения электролитической обработки, адаптируют для развития прочностных свойств при хорошем балансе между ними как в направлении волокна, так и не в направлении волокна. После электролитической обработки для придания углеродному волокну единой структуры пучка также может быть проведена и проклеивающая обработка. В качестве проклеивающего агента можно надлежащим образом выбрать проклеивающий агент, который является совместимым со смолой матрицы и т.п., в зависимости от типа использующейся смолы. Углеродное волокно, полученное в соответствии с настоящим изобретением, может быть подвергнуто воздействию широкого ассортимента способов формования, например, автоклавному формованию в случае препрега, трансферному формованию смолы в случае преформы, такой как тканые материалы, и формованию в результате намотки элементарного волокна. Данные формованные изделия предпочтительно используют в качестве авиационных деталей, элементов контейнеров, работающих под давлением, автомобильных деталей или деталей спортивных товаров, таких как удилища или рукоятки клюшек для гольфа. Примеры Далее настоящее изобретение будет дополнительно конкретно описано при обращении к примерам. Затем будут описаны методы измерения различных свойств, использующихся в примерах. Различные типы молекулярной массы: Mz+1, Mz, Mw, Mn. Измеряемый полимер растворяют в диметилформамиде (добавляют бромид лития с концентрацией 0,01 н.), так чтобы концентрация составляла 0,1 мас.%, получая подвергаемый испытанию раствор. В случае проведения измерения для волокна-предшественника волокно-предшественник растворяют в растворителе для получения подвергаемого испытанию раствора. Однако волокно-предшественник с меньшей вероятностью растворяется тогда, когда волокно-предшественник является высокоориентированным и плотным и по мере продления времени растворения и увеличения температуры растворения измерение для волокна-предшественника имеет тенденцию приводить к получению пониженной молекулярной массы. Таким образом, волокно-предшественник подвергали тонкому измельчению и растворяли в тече- 21018977 ние одного дня в растворителе, контролируемо выдерживаемом при 40 С, с одновременным перемешиванием при использовании перемешивающего устройства. Для подвергаемого испытанию полученного раствора кривую молекулярно-массового распределения получали, исходя из кривой ГПХ, измеренной в следующих далее условиях при использовании устройства ГПХ, и вычисляли значения Mz+1, Mz, Mw иMn. Колонка: колонка для ГПХ, относящаяся к типу полярного органического растворителя. Скорость течения: 0,5 мл/мин. Температура: 75 С. Фильтрование образца: мембранный фильтр (фракция 0,45 мкм). Вводимое количество: 200 мкл. Детектор: дифференциальный рефрактометр. При использовании по меньшей мере 6 различных типов характеризующегося одиночным распределением полистирола, различающихся по молекулярной массе, молекулярные массы которых известны,получали калибровочную кривую время элюирования-молекулярная масса и считывали показание по молекулярной массе, определенной для полистирола, которое соответствует времени элюирования на калибровочной кривой, тем самым получая значение Mw. В примерах для получения калибровочной кривой использовали соответственно устройствоTSK-GELM(2), произведенное в компании Tosoh Corp., устройство +TSK-guard Column , произведенное в компании Tosoh Corp., в качестве колонки, диметилформамид и бромид лития, произведенные в компании Wako Pure Chemical Industries, Ltd., устройство 0,45 m-FHLP FILTER, произведенное в компании Millipore Corp., в качестве мембранного фильтра, устройство RID-10AV, произведенное в компании Shimadzu Corp., в качестве дифференциального рефрактометра, и полистиролы, имеющие молекулярные массы 184000, 427000, 791000, 1300000, 1810000 и 4210000 в качестве полистиролов, характеризующихся одиночным распределением. Вязкость прядильного раствора. Для измерения вязкости прядильного раствора в каждом случае при температуре 45 С в качестве вискозиметра типа В использовали вискозиметр типа B8L, произведенный в компании Tokyo Keiki Inc.,таким образом, чтобы вязкость прядильного раствора в диапазоне от 0 до 100 Пас измерять при скорости вращения 6 об/мин для ротора 4, а вязкость в диапазоне от 100 до 1000 Пас измерять при скорости вращения 0,6 об/мин. Степень ориентации кристаллитов для волокна-предшественника и окисленного волокна. Степень ориентации кристаллитов в направлении оси волокна измеряли следующим образом. Пучок волокон разрезали на длины 40 мм, 20 мг пучка волокон точно взвешивали и отбирали в качестве образцов и выравнивали таким образом, чтобы волокно образца было в точности параллельным, а после этого для получения образца пучка волокон, имеющего ширину 1 мм и однородную толщину, использовали шаблон для подравнивания образца. Образец пучка волокон импрегнировали разбавленным раствором коллодия для фиксирования пучка волокон, так чтобы не нарушить его форму, а после этого фиксировали на платформе для измерения широкоугловой дифракции рентгеновского излучения. При использовании в качестве источника рентгеновского излучения излучения Cu-K, сделанного монохроматическим с использованием Ni-фильтра, из полуширины (Н) профиля, простирающегося в меридиональном направлении, включая максимальную интенсивность дифракции, наблюдаемую в окрестности 2=17,получали степень ориентации кристаллитов (%) при использовании следующей формулы: Необходимо отметить, что в качестве вышеупомянутого широкоуглового рентгеновского дифрактометра использовали устройство XRD-6100, произведенное в компании Shimadzu Corp. Тонина одиночного волокна волокна-предшественника. На металлическую рамку с длиной окружности 1 м наматывали 10 витков рулона волокна, образованного из 6000 одиночных волокон, и после этого измеряли массу намотанного волокна, вычисляя массу, приходящуюся на 10000 м, и, тем самым, получая тонину одиночного волокна. Предельная степень вытяжки при окислении. Полученное волокно-предшественник вводили в оборудование с циркуляцией горячего воздуха с температурой атмосферы, выдерживаемой при 240 С, и длиной 7,5 м. Валики для подачи или сматывания волокна-предшественника располагали спереди и позади оборудования, а степень вытяжки измеряли в результате изменения скорости подающего валика при одновременном сохранении скорости сматывающего валика 2,5 м/мин. Скорость валика изменяли на 0,1 в пересчете на степень вытяжки и при каждой скорости рассчитывали количество случаев появления пушения в течение 3 мин, начиная с 9 минут после изменения скорости. Любой вариант, выбираемый из 10 и более случаев появления пушения/м, 10 и более случаев частичного разрыва волокон и разрыва всего пучка волокон, рассматривался в качестве превышения предельной степени вытяжки при окислении, а степень вытяжки, полученную в результате вычитания 0,1 из степени вытяжки при появлении пушения или разрыва волокна, определяли как пре- 22018977 дельную степень вытяжки при окислении. Предел прочности и модуль упругости при растяжении для углеродных волокон в виде пряди, импрегнированной смолой. Предел прочности и модуль упругости при растяжении для пучка углеродных волокон определяют в соответствии с документом JIS R7608 (2007) "Test Method of Resin Impregnated Strand of Carbon Fiber". Импрегнированную смолой прядь углеродного волокна для измерений получали в результате импрегнирования углеродного волокна или графитизированного волокна при использовании 3,4 эпоксициклогексил(метил-3,4-эпоксициклогексилкарбоксилата)(100 мас.ч.)/трифторида борамоноэтиламина (3 мас.ч.)/ацетона (4 мас.ч.) и отверждения при температуре 130 С в течение 30 мин. В дополнение к этому, количество углеродных волокон в виде прядей, импрегнированных смолой, для измерения составляет 6 и среднее значение соответствующих результатов измерений принимают за предел прочности при растяжении. В примерах в качестве 3,4-эпоксициклогексил(метил-3,4 эпоксициклогексилкарбоксилата) использовали "Bakelite" (зарегистрированная торговая марка)ERL4221, произведенный в компании Union Carbide Corp. Параметр формы Вейбулла m, m" и квадрат коэффициента корреляции R2 предела прочности при растяжении для одиночного волокна углеродного волокна. Прочность для одиночного волокна углеродного волокна получали в соответствии с документомJIS R7606 (2000) следующим образом. Во-первых, пучок волокон-предшественников, который имел в длину 20 см, разделяли на четыре пучка таким образом, чтобы количество одиночных волокон для каждого пучка составляло 255% по отношению к пучку волокон-предшественников, и из каждого из четырех раздельных пучков случайным образом отбирали 100 одиночных волокон в качестве образцов. Отобранные в качестве образцов одиночные волокна фиксировали на перфорированной пластине при использовании клея. Пластину с зафиксированными одиночными волокнами закрепляли в приборе для испытания на растяжение и проводили испытание на растяжение в условиях длины образца 25 мм и скорости растяжения 5 мм/мин. Параметр формы Вейбулла получали в соответствии с определением следующей формулы: Символ F указывает на вероятность разрушения, которую получали по методу интегрального распределения для целевого образца. Символы , m и С указывают на предел прочности при растяжении для одиночного волокна (МПа), параметр формы Вейбулла и постоянное число соответственно. Получали график Вейбулла зависимости lnln1/(1-F) от 1n и подвергали его аппроксимации первого порядка для получения из наклона значения m. Корреляционную функцию при значении m, полученном из наклона, обозначают R. В дополнение к этому, зависимость lnln1/(1-F) от ln подвергали аппроксимации первого порядка в диапазоне от 0,3 до 1 для F, получая из наклона значение m". Поперечное сечение одиночного волокна получали в соответствии документом JIS R7607 (2000) в результате деления для измеряемого пучка волокон массы (г/м), приходящейся на единицу длины, на плотность (г/м 3) и, кроме того, на количество одиночных волокон. Параметр формы Вейбулла m(Р) у предела прочности при растяжении для одиночного волокна волокна-предшественника. Параметр формы Вейбулла m(Р) получали тем же самым образом, как и для углеродного волокна,за исключением установления скорости растяжения 5 мм/мин. Размер кристаллитов углеродного волокна. Измеряемые углеродные волокна выравнивали в одном направлении и фиксировали при использовании спиртового раствора коллодия для получения образца для измерений в виде квадратной призмы,имеющего высоту 4 см и длину каждой стороны 1 мм. Для полученного образца для измерений измерение проводили при использовании широкоуглового рентгеновского дифрактометра в следующих условиях. Источник рентгеновского излучения: излучение Cu-K (напряжение на рентгеновской трубке 40 кВ, ток трубки 30 мА). Детектор: гониометр + монохроматор + сцинтилляционный счетчик. Диапазон сканирования: 2 = 10-40. Режим сканирования: пошаговое сканирование, интервал шага 0,02, время счета 2 с. По полученной дифрактограмме получали полуширину пика, наблюдающегося в окрестности 2 = 25-26, и на основании данного значения рассчитывали размер кристаллитов по следующему уравнению Шеррера: Размер кристаллитов (нм) = K/0cosB,где K: 1,0, : 0,15418 нм (длина волны рентгеновского излучения); 0: (E2-12)1/2; Е: наблюдаемая полуширина (измеренное значение), рад; 1: 1,04610-2, рад. В: брэгговский угол дифракции. Необходимо отметить, что в качестве вышеупомянутого широкоуглового рентгеновского дифрак- 23018977 тометра использовали устройство XRD-6100, произведенное в компании Shimadzu Corp. Средние диаметры одиночного волокна волокна-предшественника и углеродного волокна. Для измеряемых пучка волокон-предшественников или пучка углеродных волокон получали массуAf (г/м), приходящуюся на единицу длины, и относительную плотность Bf (г/см 3). При обозначении количества одиночных волокон в измеряемом пучке волокон через Cf по следующему далее уравнению рассчитывали средний диаметр одиночного волокна (мкм) для волокон. Относительную плотность получали по методу Архимеда, по которому при измерении для углеродного волокна использовали одихлорбензол, в то время как при измерении для волокна-предшественника использовали этанол. Спектроскопия комбинационного рассеяния для углеродного волокна. Система измерения и условия проведения измерений представляли собой нижеследующее. Система измерения: микрозонд Ramaonor T-64000 (режим микроскопии), произведенный в компании JobinYvon. Линза объектива: 100. Диаметр луча: 1 мкм. Тип лазера: Ar+ (длина волны возбуждения: 514,5 нм). Мощность лазера: 1 мВт. Конфигурация: тройной монохроматор на 640 мм. Дифракционная решетка: 600 штр./мм (произведена в компании Spectrograph). Дисперсия: единичная, 21 А/мм. Щель: 100 мкм. Детектор CCD (произведен в компании JobinYvon, 1024256). В ходе измерения лазерное излучение собирали на поверхности углеродного волокна, а плоскость поляризации приводили в соответствие с осью волокна. Для каждого образца использовали отличный тип одиночного волокна, проводя измерение при n=6. Для сопоставления и анализа спектров использовали среднее значение для измерений. Спектр комбинационного рассеяния получают в результате проведения коррекции базовой линии посредством прямолинейной аппроксимации в диапазоне от 900 до 2000 см-1. Интенсивность каждой полосы спектра комбинационного рассеяния рассчитывали таким образом, чтобы оценивать точку локального максимума и точку локального минимума, применяя аппроксимацию по методу наименьших квадратов при использовании квадратичной функции для 40 точек экспериментальных данных в области 1360, 1480 и 1600 см-1. Ось волнового числа калибровали таким образом, чтобы эмиссионная линия излучения при 546,1 нм в качестве эмиссионной линии ртутной лампы низкого давления соответствовала 1122,7 см-1. Сравнительный пример 1. В 370 мас.ч. диметилсульфоксида гомогенно растворяли 100 мас.ч. АН, 1 мас.ч. итаконовой кислоты, 4 мас.ч. АИБН в качестве радикального инициатора и 0,1 мас.ч. октилмеркаптана в качестве регулятора степени полимеризации и помещали в реактор, снабженный дефлегматором и перемешивающей лопастью. После вытеснения газовой среды в пространстве реактора азотом вплоть до концентрации кислорода 1000 ч./млн проводили тепловую обработку в следующих далее условиях (называемых условиями проведения полимеризации А) при одновременном перемешивании для проведения полимеризации по способу растворной полимеризации, получая тем самым раствор полимера на основе ПАН.(2) Выдерживание при температуре 60 С в течение 4 ч.(4) Выдерживание при температуре 80 С в течение 6 ч. После приготовления полученного раствора полимера на основе ПАН, имеющего концентрацию полимера 20 мас.%, проводили продувку газообразного аммиака вплоть до достижения значения рН 8,5 для введения в полимер аммониевой группы при одновременной нейтрализации итаконовой кислоты, тем самым получая прядильный раствор. Полимер на основе ПАН в полученном прядильном растворе характеризовался значениями Mw 400000, Mz/Mw 1,8 и Mz+1/Mw 3,0, а вязкость прядильного раствора составляла 50 Пас. Полученный прядильный раствор перепускали через фильтр, характеризующийся тонкостью фильтрования 10 мкм, после этого экструдировали при температуре 40 С сразу в воздух при использовании фильеры, характеризующейся числом отверстий 3000 и диаметром прядильных отверстий 0,12 мм, и перепускали через зазор, равный приблизительно 2 мм, а после этого проводили прядение в условиях степени вытяжки при прядении 4 по способу сухомокрого прядения, который включает введение в коагуляционную ванну, образованную из раствора 20 мас.% диметилсульфоксида и контролируемо выдерживаемую при температуре 3 С, получая тем самым набухающие волокна. Полученные набухающие волокна промывали водой и подвергали первой стадии вытяжки в ванне при натяжении 2,2 мн/дтекс. Температура ванны составляла 65 С, а степень вытяжки была равна 2,7. На волокна, подвергнутые первой стадии вытяжки, наносили силиконовый за- 24018977 масливатель на основе аминомодифицированного силикона, для проведения обработки сухим теплом в течение 30 с использовали валик, нагретый до температуры 165 С, а после этого проводили вторую стадию вытяжки в водяном паре под давлением при натяжении 5,3 мн/дтекс, получая волокнопредшественник углеродного волокна. На второй стадии вытяжки давление водяного пара под давлением устанавливали равным 0,4 МПа, а степень вытяжки устанавливали равной 5,2. Параметр формы Вейбулла m(Р) у полученного волокна-предшественника составлял 10, коэффициент вариации (КВ) прочности одиночного волокна составлял 12%, а коэффициент вариации (КВ) относительного удлинения одиночного волокна составлял 7%. Сравнительный пример 2. Волокно-предшественник углеродного волокна получали тем же самым образом, что и в примере 1,за исключением того, что степень вытяжки при прядении изменяли на 5, на второй стадии вытяжки подачу водяного пара заменяли на подвод сухого тепла и вторую степень вытяжки изменяли на 3,0. Пример 1. 100 мас.ч. АН, 1 мас.ч. итаконовой кислоты и 130 мас.ч. диметилсульфоксида перемешивали и помещали в реактор, снабженный дефлегматором и перемешивающей лопастью. После вытеснения газовой среды в пространстве реактора азотом вплоть до концентрации кислорода 100 ч./млн в качестве радикального инициатора вводили 0,002 мас.ч. 2,2'-азо-бис-изобутилонитрила (АИБН) и проводили тепловую обработку в следующих далее условиях (называемых условиями проведения полимеризации В) при одновременном перемешивании. Выдерживание при температуре 65 С в течение 2 ч. Охлаждение от 65 до 30 С (скорость охлаждения 120 С/ч). После этого отвешивали и вводили в реактор 240 мас.ч. диметилсульфоксида, 0,4 мас.ч. АИБН в качестве инициатора полимеризации и 0,1 мас.ч. октилмеркаптана в качестве регулятора степени полимеризации, и, кроме того, в условиях проведения полимеризации А из сравнительного примера 1 проводили тепловую обработку при одновременном перемешивании для полимеризации оставшегося непрореагировавшего мономера по способу растворной полимеризации, тем самым получая раствор полимера на основе ПАН. После использования и приготовления полученного раствора полимера на основе ПАН, имеющего концентрацию полимера 20 мас.%, проводили продувку газообразного аммиака вплоть до достижения значения рН 8,5 для введения в полимер на основе ПАН аммониевой группы при одновременной нейтрализации итаконовой кислоты, тем самым получая прядильный раствор. Полимер на основе ПАН в полученном прядильном растворе характеризовался значениями Mw 480000, Mz/Mw 5,7 и Mz+1/Mw 14, а вязкость прядильного раствора составляла 45 Пас. Прядение проводили тем же самым образом, что и в сравнительном примере 1, за исключением замены прядильного раствора на прядильный раствор, полученный так, как это описывалось ранее. Полученное волокно-предшественник было превосходным по сорту, и на стадии прядения можно было стабильно производить отбор образцов. Несмотря на уменьшение значения Mz/Mw волокна-предшественника в сопоставлении со значением Mz/Mw прядильного раствора все еще сохранялось значение, более высокое, чем в сравнительном примере 1, что в результате приводило к увеличению предельной степени вытяжки при окислении. Пример 2. Прядение проводили тем же самым образом, что и в примере 1, за исключением изменения степени вытяжки при прядении на 12, на второй стадии вытяжки подачу водяного пара заменяли на подвод сухого тепла и вторую степень вытяжки изменяли на 1,1. Полученное волокно-предшественник было превосходным по сорту, и на стадии прядения можно было стабильно производить отбор образцов. Уменьшение степени вытяжки при прядении сохраняло значение Mz/Mw волокна-предшественника несколько уменьшенным в сопоставлении со значением Mz/Mw прядильного раствора, что в результате приводило к высокой предельной степени вытяжки при окислении. Пример 3. Прядение проводили тем же самым образом, что и в примере 2, за исключением изменения степени вытяжки после высушивания на 2,0. Полученное волокно-предшественник было превосходным по сорту,и на стадии прядения можно было стабильно производить отбор образцов. Несмотря на большее уменьшение значения Mz/Mw волокна-предшественника, чем в примере 2, все еще сохранялось высокое значение, что в результате приводило к высокой предельной степени вытяжки при окислении. Пример 4. Прядильный раствор получали тем же самым образом, что и в примере 1, за исключением изменения количества АИБН при первом введении на 0,001 мас.ч., газовую среду в пространстве реактора вытесняли азотом вплоть до концентрации кислорода 1000 ч./млн, и условия проведения полимеризации А изменяли на условия проведения полимеризации С.(1) Выдерживание при температуре 70 С в течение 4 ч.(2) Охлаждение от 70 до 30 С (скорость охлаждения 120 С/ч). Полимер на основе ПАН в полученном прядильном растворе характеризовался значениямиMw 340000, Mz/Mw 2,7 и Mz+1/Mw 7,2, а вязкость прядильного раствора составляла 40 Пас. Прядение проводили тем же самым образом, что и в сравнительном примере 1, за исключением замены прядильного раствора на прядильный раствор, полученный так, как это описывалось ранее. Полученное волокнопредшественник было превосходным по сорту, и на стадии прядения можно было стабильно производить отбор образцов. Несмотря на некоторое уменьшение значения Mz/Mw волокна-предшественника в сопоставлении со значением Mz/Mw прядильного раствора все еще сохранялось более высокое значение в сопоставлении с тем, что имело место в сравнительном примере 1, что в результате приводило к увеличению предельной степени вытяжки при окислении. Параметр формы Вейбулла m(Р) у полученного волокнапредшественника составлял 13, коэффициент вариации (КВ) прочности одиночного волокна составлял 9%, а коэффициент вариации (КВ) относительного удлинения одиночного волокна составлял 7%. Пример 5. Прядильный раствор получали тем же самым образом, что и в примере 4, за исключением изменения количества АИБН при первом введении на 0,002 мас.ч. и равенства времени выдерживания 1,5 часам в условиях проведения полимеризации С. Полимер на основе ПАН в полученном прядильном растворе характеризовался значениями Mw 320000, Mz/Mw 3,4 и Mz+1/Mw 12, а вязкость прядильного раствора составляла 35 Пас. Прядение проводили тем же самым образом, что и в сравнительном примере 1, за исключением замены прядильного раствора на прядильный раствор, полученный так, как это описывалось ранее. Полученное волокно-предшественник было превосходным по сорту, и на стадии прядения можно было стабильно производить отбор образцов. Несмотря на некоторое уменьшение значения Mz/Mw волокна-предшественника в сопоставлении со значением Mz/Mw прядильного раствора все еще сохранялось более высокое значение в сопоставлении с тем, что имело место в сравнительном примере 1, что в результате приводило к увеличению предельной степени вытяжки при окислении. Пример 6. 100 мас.ч. АН, 1 мас.ч. итаконовой кислоты и 360 мас.ч. диметилсульфоксида перемешивали и помещали в реактор, снабженный дефлегматором и перемешивающей лопастью. После вытеснения газовой среды в пространстве реактора азотом вплоть до концентрации кислорода 100 ч./млн в качестве инициатора полимеризации вводили 0,003 мас.ч. АИБН и проводили тепловую обработку в следующих далее условиях при одновременном перемешивании.(1) Выдерживание при температуре 60 С в течение 3,5 ч. После этого отвешивали и вводили в реактор 10 мас.ч. диметилсульфоксида, 0,4 мас.ч. АИБН в качестве инициатора полимеризации и 0,1 мас.ч. октилмеркаптана в качестве регулятора степени полимеризации и, кроме того, в следующих далее условиях проводили тепловую обработку при одновременном перемешивании для полимеризации оставшегося непрореагировавшего мономера по способу растворной полимеризации, тем самым получая раствор полимера на основе ПАН.(2) Выдерживание при температуре 60 С в течение 4 ч.(4) Выдерживание при температуре 80 С в течение 6 ч. После приготовления полученного раствора полимера на основе ПАН, имеющего концентрацию полимера 20 мас.%, проводили продувку газообразного аммиака вплоть до достижения значения рН 8,5 для введения в полимер на основе ПАН аммониевой группы при одновременной нейтрализации итаконовой кислоты, тем самым получая прядильный раствор. Полимер на основе ПАН в полученном прядильном растворе характеризовался значениямиMw 400000, Mz/Mw 5,2 и Mz+1/Mw 10, а вязкость прядильного раствора составляла 55 Пас. Прядение проводили тем же самым образом, что и в примере 1, за исключением замены прядильного раствора на прядильный раствор, полученный так, как это описывалось ранее. Полученное волокно-предшественник было превосходным по сорту, и на стадии прядения можно было стабильно производить отбор образцов. Несмотря на некоторое уменьшение значения Mz/Mw волокна-предшественника в сопоставлении со значением Mz/Mw прядильного раствора все еще сохранялось более высокое значение, что в результате приводило к увеличению предельной степени вытяжки при окислении. Сравнительный пример 3. В 460 мас.ч. диметилсульфоксида гомогенно растворяли 100 мас.ч. АН, 1 мас.ч. итаконовой кислоты и 0,2 мас.ч. АИБН в качестве радикального инициатора и помещали в реактор, снабженный дефлегматором и перемешивающей лопастью. После вытеснения газовой среды в пространстве реактора азотом вплоть до концентрации кислорода 1000 ч./млн проводили тепловую обработку в описывавшихся ранее условиях проведения полимеризации А при одновременном перемешивании для проведения полимеризации по способу растворной полимеризации, тем самым получая раствор полимера на основе ПАН. После приготовления полученного раствора полимера на основе ПАН, имеющего концентрацию полимера 15 мас.%, проводили продувку газообразного аммиака вплоть до достижения значения рН 8,5 для введения в полимер аммониевой группы при одновременной нейтрализации итаконовой кислоты, тем самым получая прядильный раствор. Полимер на основе ПАН в полученном прядильном растворе характеризо- 26018977 вался значениями Mw 650000, Mz/Mw 1,8 и Mz+1/Mw 3,0, а вязкость прядильного раствора составляла 95 Пас. Прядение проводили тем же самым образом, что и в сравнительном примере 1, за исключением замены прядильного раствора на прядильный раствор, полученный так, как это описывалось ранее. Значение Mz/Mw волокна-предшественника оставалось неизменным в сопоставлении со значением Mz/Mw прядильного раствора, что в результате приводило к низкой предельной степени вытяжки при окислении. Сравнительный пример 4. Прядение проводили тем же самым образом, что и в примере 2, за исключением замены прядильного раствора на прядильный раствор, полученный в сравнительном примере 3. Значение Mz/Mw волокнапредшественника было меньшим, и, таким образом, предельная степень вытяжки при окислении была меньшей, чем соответствующие величины в примерах 2 и 6. Экспериментальные условия в описывавшихся ранее примерах и сравнительных примерах и свойства полученных волокон-предшественников суммарно представлены в табл. 1. Пример 8. 100 мас.ч. АН, 1 мас.ч. итаконовой кислоты и 230 мас.ч. диметилсульфоксида перемешивали и помещали в реактор, снабженный дефлегматором и перемешивающей лопастью. После вытеснения газовой среды в пространстве реактора азотом вплоть до концентрации кислорода 1000 ч./млн в качестве инициатора полимеризации вводили 0,002 мас.ч. АИБН, а в качестве регулятора степени полимеризации 0,01 мас.ч. октилмеркаптана и проводили тепловую обработку в условиях проведения полимеризации при одновременном перемешивании.(1) Выдерживание при температуре 65 С в течение 1 ч.(2) Охлаждение от 65 до 30 С (скорость охлаждения 120 С/ч). После этого отвешивали и вводили в реактор 10 мас.ч. диметилсульфоксида, 0,4 мас.ч. АИБН в качестве инициатора полимеризации и 0,3 мас.ч. октилмеркаптана в качестве регулятора степени полимеризации и, кроме того, в условиях проведения полимеризации А из сравнительного примера 1 проводили тепловую обработку при одновременном перемешивании для полимеризации оставшегося непрореагировавшего мономера по способу растворной полимеризации, тем самым получая раствор полимера на основе ПАН. После приготовления полученного раствора полимера на основе ПАН, имеющего концентрацию полимера 27 мас.%, проводили продувку газообразного аммиака вплоть до достижения значения рН 8,5 для введения в полимер аммониевой группы при одновременной нейтрализации итаконовой кислоты,тем самым получая прядильный раствор. Полимер на основе ПАН в полученном прядильном растворе характеризовался значениями Mw 200000, Mz/Mw 3,3 и Mz+1/Mw 14, а вязкость прядильного раствора составляла 95 Пас. Прядение проводили тем же самым образом, что и в сравнительном примере 1, за исключением замены прядильного раствора на прядильный раствор, полученный так, как это описывалось ранее, температуру прядения устанавливали равной 80 С, а условия формования нитей представляли собой то, что продемонстрировано в табл. 1. Полученное волокно-предшественник было превосходным по сорту и характеризовалось высокой предельной степенью вытяжки при окислении. Пример 9. 100 мас.ч. АН, 1 мас.ч. итаконовой кислоты и 130 мас.ч. диметилсульфоксида перемешивали и помещали в реактор, снабженный дефлегматором и перемешивающей лопастью. После вытеснения газовой среды в пространстве реактора азотом вплоть до концентрации кислорода 100 ч./млн в качестве радикального инициатора вводили 0,002 мас.ч. 2,2'-азо-бис-изобутилонитрила (АИБН) и проводили тепловую обработку в следующих далее условиях при одновременном перемешивании.(1) Выдерживание при температуре 65 С в течение 5 ч. Охлаждение от 65 до 30 С (скорость охлаждения 120 С/ч). После этого отвешивали и вводили в реактор 610 мас.ч. диметилсульфоксида, 0,2 мас.ч. АИБН в качестве радикального инициатора и 0,01 мас.ч. октилмеркаптана в качестве регулятора степени полимеризации и, кроме того, в условиях проведения полимеризации А из сравнительного примера 1 проводили тепловую обработку при одновременном перемешивании для полимеризации оставшегося непрореагировавшего мономера по способу растворной полимеризации, тем самым получая раствор полимера на основе ПАН. После использования и приготовления полученного раствора полимера на основе ПАН, имеющего концентрацию полимера 10 мас.%, проводили продувку газообразного аммиака вплоть до достижения значения рН 8,5 для введения в полимер на основе ПАН аммониевой группы при одновременной нейтрализации итаконовой кислоты, тем самым получая прядильный раствор. Полимер на основе ПАН в полученном прядильном растворе характеризовался значениями Mw 590000, Mz/Mw 5,2 и Mz+1/Mw 14, а вязкость прядильного раствора составляла 10 Пас. Прядение проводили тем же самым образом, что и в сравнительном примере 1, за исключением замены прядильного раствора на прядильный раствор, полученный так, как это описывалось ранее, температуру прядения устанавливали равной 20 С, а условия формования нитей представляли собой то, что продемонстрировано в табл. 1. Полученное волокно- 27018977 предшественник было превосходным по сорту и характеризовалось высокой предельной степенью вытяжки при окислении. Сравнительный пример 5. Использовали тот же самый прядильный раствор, что и в примере 1. Прядильный раствор перепускали через фильтр, характеризующийся тонкостью фильтрования 0,5 мкм, после этого экструдировали при температуре 40 С сразу в воздух при использовании фильеры, характеризующейся числом отверстий 6000 и диаметром прядильных отверстий 0,15 мм, и перепускали через зазор, равный приблизительно 2 мм, а после этого проводили прядение по способу сухомокрого прядения, который включает введение в коагуляционную ванну, образованную из раствора 20 мас.% диметилсульфоксида и контролируемо выдерживаемую при температуре 3 С, тем самым получая коагулированные волокна. В дополнение к этому, в условиях степени вытяжки при прядении 4 коагулированные волокна получали и промывали водой,после этого подвергали вытяжке при степени вытяжки в ванне 3 в теплой воде 90 С, кроме того, наносили силиконовый замасливатель на основе аминомодифицированного силикона, для проведения высушивания в течение 30 секунд использовали валик, нагретый до температуры 165 С, и в водяном паре под давлением проводили вытяжку при степени вытяжки 5, получая волокно-предшественник. Несмотря на превосходный сорт полученного волокна-предшественника, предельная степень вытяжки при окислении была сопоставима с соответствующей величиной в сравнительных примерах. Волокна-предшественники, полученные так, как это описывалось ранее, которые продемонстрированы в табл. 2, где количество одиночных волокон, составляющих пучок волокон, выдерживали равным 6000, в течение 90 минут подвергали окислительной обработке при одновременном проведении вытяжки со степенью вытяжки 1,0 на воздухе при распределении температуры в диапазоне от 240 до 260 С, тем самым получая окисленные волокна. После этого полученные окисленные волокна подвергали предварительной карбонизационной обработке при одновременном проведении вытяжки со степенью вытяжки 1,2 в атмосфере азота с распределением температуры в диапазоне от 300 до 700 С, а кроме того, подвергали карбонизационной обработке при степени вытяжки, установленной на 0,97, в атмосфере азота с максимальной температурой 1500 С, тем самым получая непрерывные углеродные волокна. Вследствие наличия у степени вытяжки при окислительной обработке достаточного запаса прохождение способа окисления-карбонизации было надлежащим в любой момент времени. Примеры от 9 до 17, сравнительные примеры от 6 до 8. Волокна-предшественники, полученные так, как это описывалось ранее, которые продемонстрированы в табл. 2, превращали в трощеные нити из 8 волокон-предшественников, так чтобы выдерживать количество одиночных волокон, составляющих пучок волокна, равным 24000, и в течение 90 мин подвергали окислительной обработке при одновременном проведении вытяжки со степенями вытяжки, продемонстрированными в табл. 2, на воздухе с распределением температуры в диапазоне от 240 до 260 С,тем самым получая окисленные волокна. После этого полученные окисленные волокна подвергали предварительной карбонизационной обработке при одновременном проведении вытяжки со степенью вытяжки 1,2 в атмосфере азота с распределением температуры в диапазоне от 300 до 700 С, тем самым получая пучки предварительно карбонизованных волокон. Полученные пучки предварительно карбонизованных волокон подвергали карбонизационной обработке для пучков предварительно карбонизованных волокон при степени вытяжки 0,96 в атмосфере азота с максимальной температурой 1500 С, тем самым получая непрерывные углеродные волокна. Примеры характеризовались почти что отсутствием пушения, наблюдаемого на стадии окисления, стадии предварительной карбонизации или стадии карбонизации, и были подходящими как по стабильности получения, так и по сорту. Сравнительные примеры характеризовались появлением пушения, генерируемого на стадии окисления, стадии предварительной карбонизации и стадии карбонизации, таким образом, едва ли можно сказать то, что сравнительные примеры были подходящими как по стабильности получения, так и по сорту, и между сравнительными примерами и примерами существовали четкие различия. В частности, в сравнительных примерах 6 и 7 имелось мало случаев пушения даже при низкой степени вытяжки несмотря на предельную степень вытяжки при окислении, что в результате приводило к получению неудовлетворительных сортов. Табл. 2 демонстрирует результаты измерения степени ориентации для полученных и окисленных волокон и свойств прядей для пучков углеродных волокон. Примеры от 18 до 20, сравнительные примеры от 9 до 11. Пучок углеродных волокон получали тем же самым образом, что и в примере 17 или сравнительном примере 6, за исключением изменения максимальной температуры при карбонизационной обработке, как это продемонстрировано в табл. 3. Результаты оценки полученного пучка углеродных волокон продемонстрированы в табл. 3.
МПК / Метки
Метки: волокна, углеродного, получения, способ, волокно-предшественник
Код ссылки
<a href="https://eas.patents.su/30-18977-volokno-predshestvennik-uglerodnogo-volokna-sposob-ego-polucheniya-i-sposob-polucheniya-uglerodnogo-volokna.html" rel="bookmark" title="База патентов Евразийского Союза">Волокно-предшественник углеродного волокна, способ его получения и способ получения углеродного волокна</a>
Способы получения полиоксадиазольной нити и полиоксадиазольного волокна, нить и волокно, полученные этими способами

Номер патента: 15707
Опубликовано: 31.10.2011
Автор: undefined
МПК: D01F 6/74, D01D 5/098, D01D 5/12...
Метки: нить, волокна, полиоксадиазольного, способы, нити, полученные, полиоксадиазольной, получения, волокно, этими, способами
Формула / Реферат:
1. Способ получения полиоксадиазольной нити, при котором получают (со)полимерный прядильный раствор, для чего последовательно загружают сначала серную кислоту, содержащую 17-65 мас.% свободного ангидрида, затем 0,02-22,9 мас.% арилендикарбоновой кислоты, представляющей собой терефталевую кислоту с 0,004-0,017 мас.% паратолуиловой кислоты или смесь терефталевой кислоты и 0,5-99,0 мас.% изофталевой кислоты с 0,004-0,017 мас.% смеси паратолуиловой...
Способ изготовления синтетического волокна для использования его в искусственных травяных покрытиях для спортивных площадок и такое синтетическое волокно

Номер патента: 8915
Опубликовано: 31.08.2007
Авторы: Слотвег Герт Бастиан, Ольде Вегхейс Маринус Хендрикус, Ван Дер Гааг Фредерик Ян
МПК: E01C 13/08, D01F 8/06, D01D 5/42...
Метки: изготовления, использования, синтетического, спортивных, покрытиях, искусственных, травяных, способ, волокно, площадок, такое, волокна, синтетическое
Формула / Реферат:
1. Способ изготовления синтетического волокна для использования в искусственном травяном покрытии для спортивных площадок, включающий стадии: i) обеспечения слоя синтетического материала и ii) получения синтетического волокна из слоя синтетического материала, отличающийся тем, что слой синтетического материала образуют по меньшей мере из двух слоев различных синтетических материалов, используя процесс совместной экструзии. 2. Способ по п.1,...
Волокно, имитирующее природное растительное волокно, способ его изготовления и изготовленная из него ткань

Номер патента: 17657
Опубликовано: 28.02.2013
Авторы: Чэнь Хун-Цзэнь, Хуан Тина
МПК: D01F 6/04, D01F 1/10, D03D 15/00...
Метки: природное, изготовления, способ, растительное, ткань, него, волокно, изготовленная, имитирующее
Формула / Реферат:
1. Способ изготовления волокна, имитирующего растительное природное волокно, который включает следующие этапы:(a) подготовку нижеуказанных материалов:(а1) крошку первого полиолефина (10-80 вес.% (в расчете на общий вес материала)), который представляет собой полипропилен с молекулярной массой 3,15´105 г/моль или полиэтилен с молекулярной массой 1,5-2,5´105 г/моль, в качестве субстрата;(а2) микрокапсулы (5-50 вес.%), выполненные из...
Штапельные волокна из политриметилентерефталата и способ их получения

Номер патента: 3017
Опубликовано: 26.12.2002
Авторы: Вандель Дитмар, Мирвальд Ульрих, Келльнер Кристиан, Кордес Инго
МПК: D01F 6/62
Метки: способ, волокна, получения, штапельные, политриметилентерефталата
Формула / Реферат:
1. Штапельные волокна из ПТТ, отличающиеся тем, что они обладают характеристической вязкостью в области от 0,70 до 1,3 дл/г, величиной LASE (10%) от 5 до 12 сН/текс, секущим модулем (Rd-45%) от <1,0 сН/текс на 1%, постоянством извитости >75% и окрашиваются дисперсионными красителями без добавления носителей/средств, способствующих окрашиванию. 2. Штапельные волокна, по п.1, отличающиеся тем, что они обладают характеристической вязкостью в...
Полиэфирные штапельные волокна и способ их получения

Номер патента: 4442
Опубликовано: 29.04.2004
Авторы: Уде Вернер, Вандель Дитмар, Янас Вольфганг, Кордес Инго, Швинд Хельмут
МПК: D01F 6/62
Метки: штапельные, полиэфирные, волокна, способ, получения
Формула / Реферат:
1. Полиэфирные штапельные волокна, отличающиеся тем, что они состоят из a) полиэфира, который содержит, как минимум, 85 мол.% поли(C2-C4-алкилен)терефталата, b) от 0,1 до 2,0 вес.% несмешиваемого, термопластически перерабатываемого, аморфного добавочного полимера, который имеет температуру стеклования от 90 до 170шC и g) от 0 до 5,0 вес.% обычных добавочных веществ, причем сумма из a), b) и g) равна 100%, отношение вязкости расплава...
Предыдущий патент: Производные 1-бензил-3-гидроксиметилиндазола и их применение для лечения заболеваний, основанных на экспрессии мср-1, сх3сr1 и р40
Следующий патент: Гетерофазная полимерная композиция, выполненная из нее труба и способ получения композиции
Случайный патент: Реконструированные сурфактанты, обладающие улучшенными свойствами