Антитромботические азациклоалкилалканоильные пептиды и псевдопептиды
Номер патента: 1391
Опубликовано: 26.02.2001
Авторы: Чекай Марк, Клейн Скотт И., Гарднер Чарльз Дж., Молино Брюс Ф.




















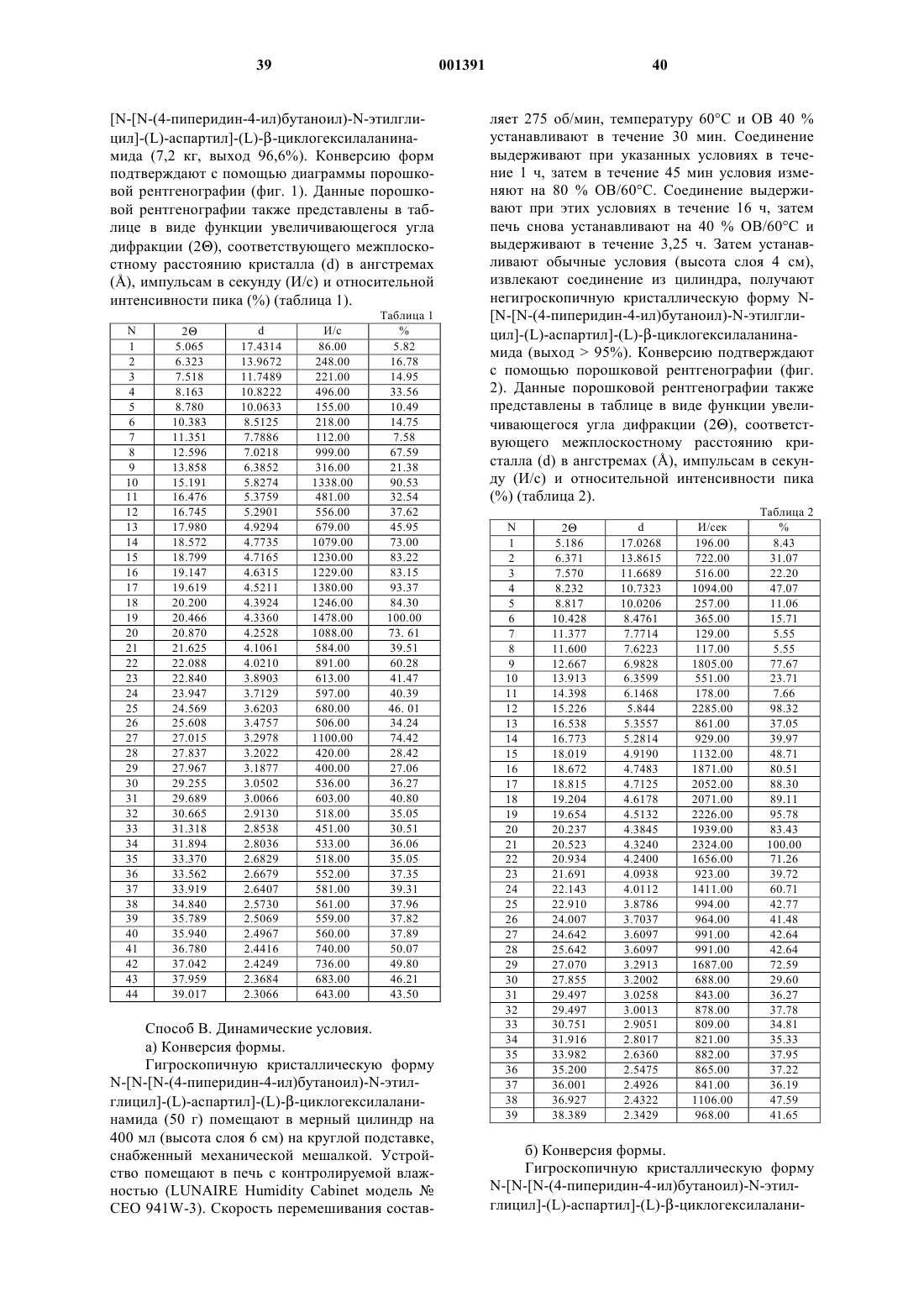
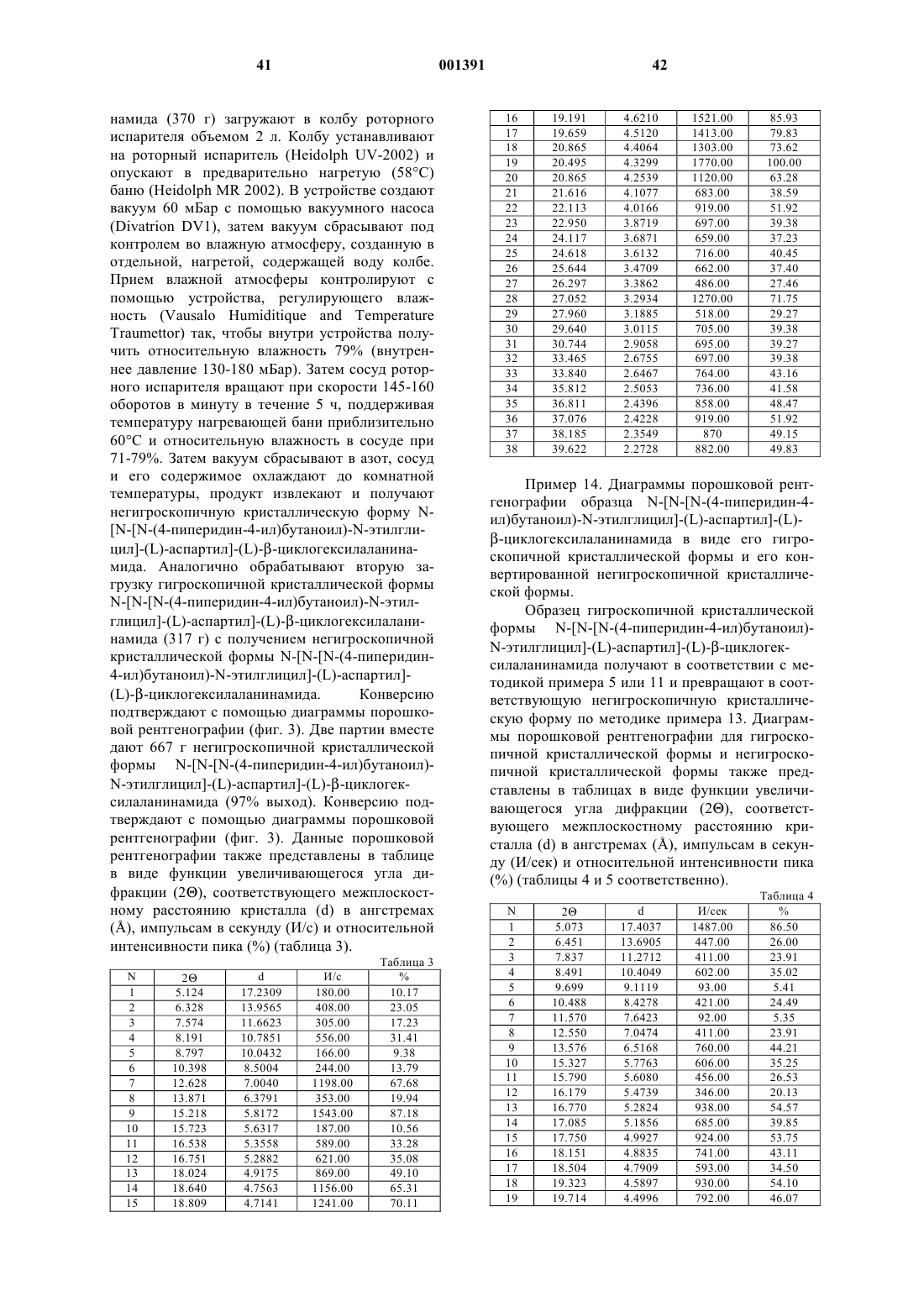
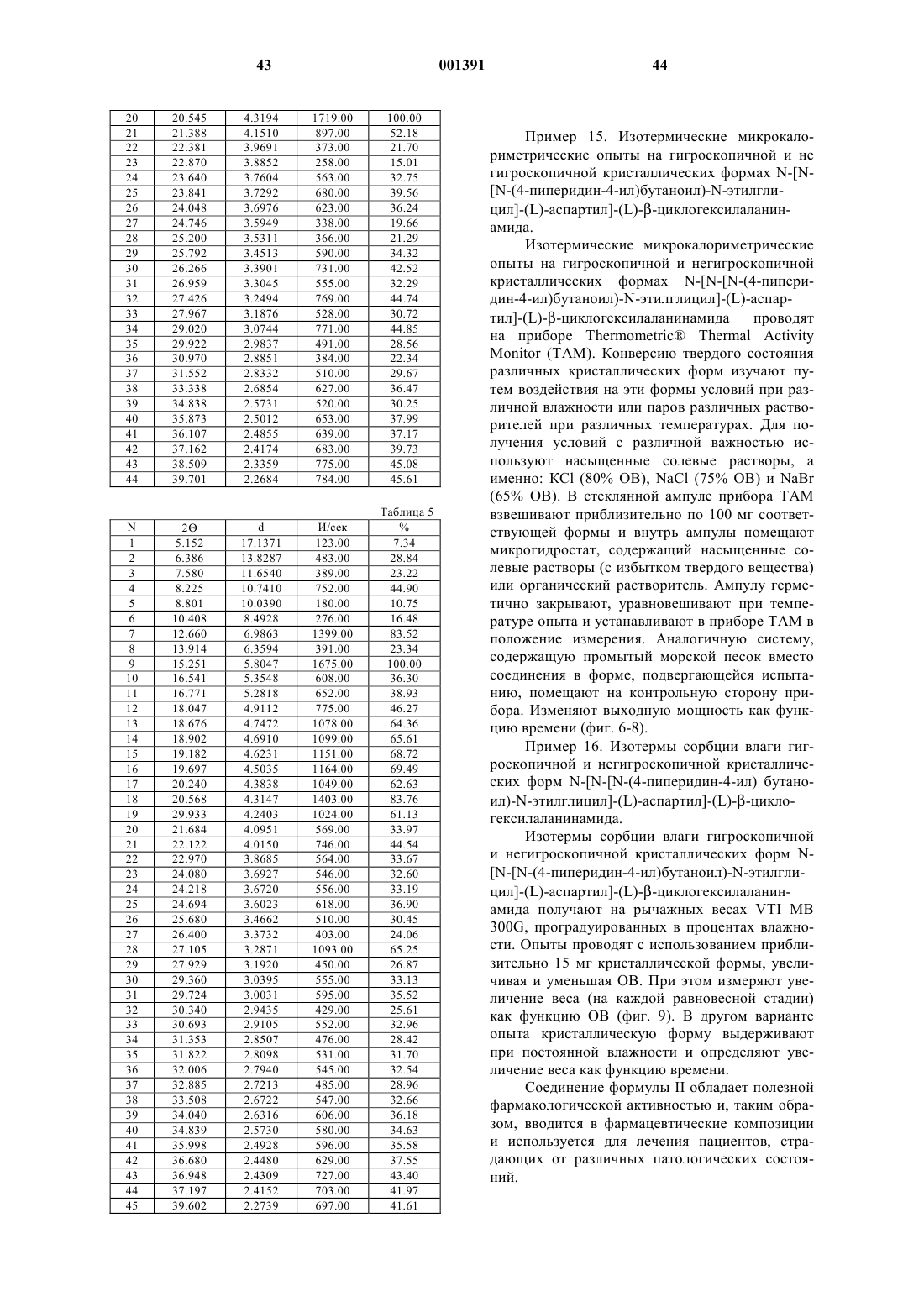




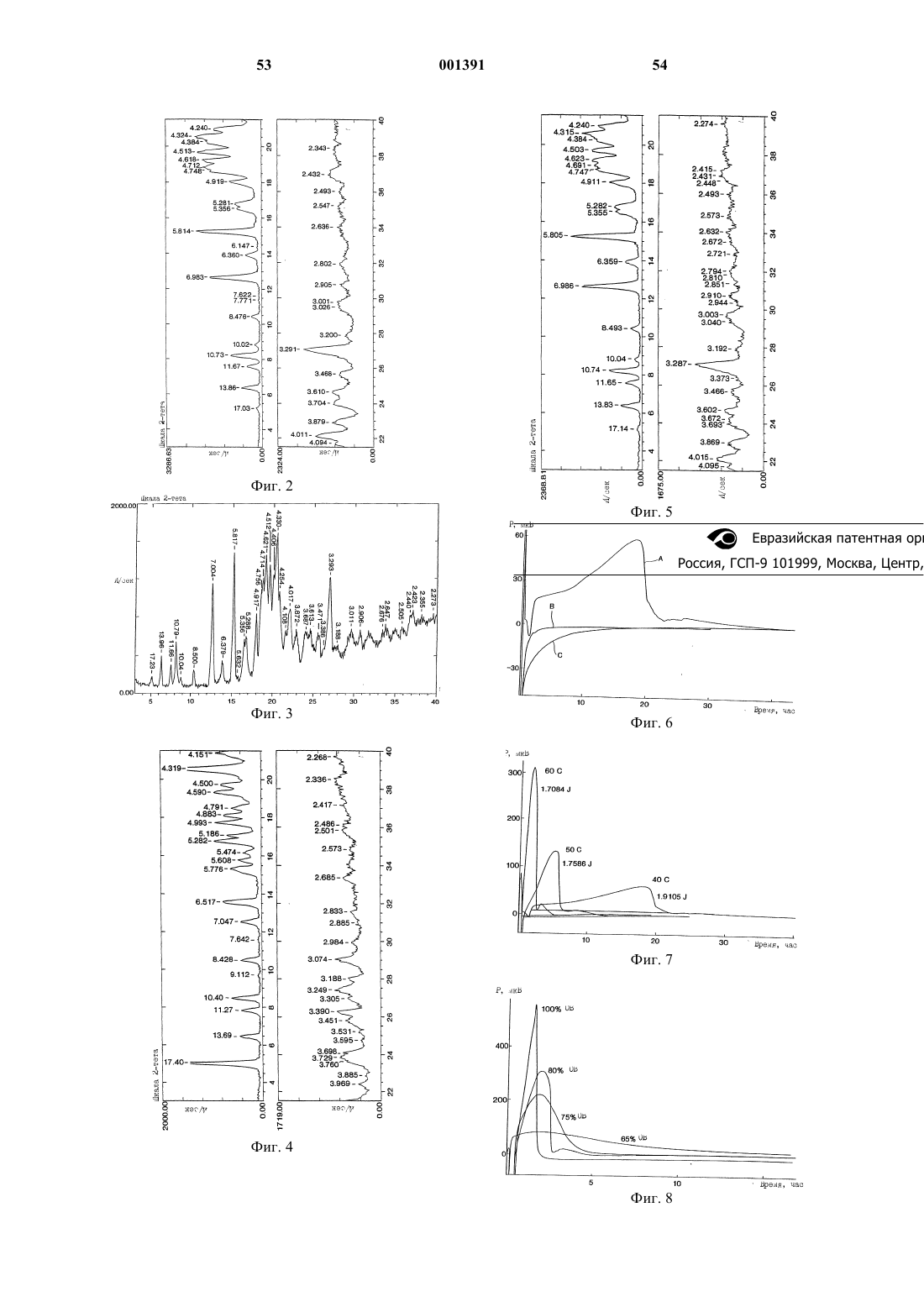

Формула / Реферат
1. Соединение формулы VII

где В представляет собой алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил;
Е1 представляет собой Н;
Е2 представляет собой a -углеродную боковую цепь природной a -аминокислоты, Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, или Е1 и Е2 вместе с атомом азота и атомом углерода, через которые Е1 и Е2 связаны, образуют 4-, 5-, 6- или 7-членное азациклоалкановое кольцо;
G представляет собой OR1 или NR1R2;
R1 и R2 независимо друг от друга представляют собой Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил;
р принимает значения от 1 до 4;
p1 представляет собой неустойчивую при гидрировании защитную группу для кислоты; и
p2' представляет собой p2 или ТФУКЧН-; и
p2 представляет собой неустойчивую в кислоте защитную группу для аминогруппы.
2. Соединение по п.1, где
В представляет собой алкил;
Е1 представляет собой Н;
Е2 представляет собой циклоалкилалкил;
G представляет собой NR1R2;
R1 и R2 представляют собой Н; и
р принимает значение 1,
3. Соединение по п.2, где
В представляет собой этил; и
Е2 представляет собой циклогексилметил.
4. Соединение по п.3, где
p1 представляет собой бензил; и
p2' представляет собой ТФУКЧН-.
5. Соединение по п. 3, где
p1 представляет собой бензил; и
p2 представляет собой трет.-бутоксикарбонил.
Текст
1 Настоящая заявка является частичным продолжением заявки США серии 08/476750,поданной 7 января 1995 г. Последняя заявка является продолжающей заявкой, находящейся на рассмотрении заявки США 08/628648,поданной 17 октября 1994 г. Эта заявка является частичным продолжением заявки, находящейся на рассмотрении заявки США 08/138820,поданной 15 октября 1993 г, которая в настоящее время аннулирована. Уровень техники 1. Область техники Изобретение относится к негигроскопичной стабильной кристаллической форме N-[N[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида формулы I Соединение обладает антитромботической активностью, ингибируя в том числе агрегацию тромбоцитов и образование тромбов у млекопитающих и может быть использовано для профилактики и лечения тромбоза, ассоциированного с такими болезненными состояниями, как инфаркт миокарда, удар, периферическое артериальное заболевание и диссеминированное внутрисосудистое свертывание. Кроме того, изобретение относится к способу получения кристаллической формы N-[N[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида, а также к его фармацевтической композиции и его промежуточным соединениям. Изобретение также относится к промежуточным соединениям, которые могут быть использованы при получении тетраазациклоалкилалканоилпептидных или псевдопептидных соединений. Гемостаз, то есть биохимия свертывания крови, представляет собой исключительно сложное явление, в результате которого нормальная цельная кровь и ткани тела самопроизвольно останавливают кровотечение из поврежденных кровеносных сосудов. Для эффективного гемостаза необходима объединенная активность сосудистых, тромбоцитарных и плазматических факторов, а также контролирующий механизм, предупреждающий избыточное свертывание. Дефекты, недостаток или избыток любого из этих компонентов могут привести к кровотечениям или тромбозу. Адгезия тромбоцитов, распространение и агрегация на внеклеточных матрицах являются центральными событиями при образовании тромбов. Эти события опосредуются с помощью семейства адгезивных гликопротеинов, то есть,фибриногена, фибронектина и фактора фон 2 Виллебранда. Фибриноген представляет собой кофактор агрегации тромбоцитов, тогда как фибронектин поддерживает прикрепление тромбоцитов и реакции распределения. Фактор фон Виллебранда имеет большое значение при прикреплении тромбоцитов к субэндотелиальным матрицам и их распределению на этих матрицах. Сайты связывания фибриногена, фибронектина и фактора фон Виллебранда расположены на протеиновом комплексе мембраны тромбоцита, который известен как гликопротеин IIb/IIIa. Адгезивные гликопротеины типа фибриногена не связываются с нормальными находящимися в покое тромбоцитами. Однако когда тромбоцит активирован агонистом, таким как тромбин или аденозиндифосфат, тромбоцит меняет свою форму, вероятно делая сайт связывания GPIIb/IIIa доступным для фибриногена. Соединение настоящего изобретения блокирует рецептор фибриногена и, таким образом, проявляет вышеупомянутую антитромботическую активность. 2. Известные разработки Установлено, что для взаимодействия с клеточным поверхностным рецептором в фибриногене, фибронектине и факторе фон Виллебранда должен присутствовать Arg-Gly-Asp(RGD) (Ruoslahti Е., Pierschbacher, Cell, 1986, 44,517-18). Две другие аминокислотные последовательности, как оказывается, также принимают участие в прикреплении тромбоцитов под действием фибриногена, а именно: последовательность Gly-Pro-Arg и додекапептид, последовательность His-His-Leu-Gly-Gly-Ala-Lys-Gln-AlaGly-Asp-Val. Небольшие синтетические пептиды, содержащие RGD или додекапептид, как было показано, связываются с GPIIb/IIIa рецептором тромбоцита и конкурентно ингибируют связывание фибриногена, фибронектина и фактора фон Виллебранда, а также ингибируют агрегацию активированных тромбоцитов (Plow, etRuggeri, et al., Proc. Natl. Acad. Sci. USA 1986,5709-12; Ginsberg, et al., J. Biol. Chem. 1985, 260,3931-36: и Gartner et al., J. Biol. Chem. 1987, 260,11, 891-94). Индолильные соединения, содержащие гуанидиноалканоил- и гуанидиноалкеноиласпартильные фрагменты, как сообщается, являются ингибиторами агрегации тромбоцитов(Tjoeng, et al., патенты США 5037808 и 4879313). В патенте США 4992463 (Tjoeng, et al.),выданном 12 февраля 1991 г., в общем виде описано, что ряд арил- и аралкилгуанидиноалкилпептидных миметических соединений обладает ингибирующим действием на агрегацию тромбоцитов; конкретно описан ряд моно- и диметоксифенилпептидных миметических соединений и бифенилалкилпептидное миметическое соединение. 3 В патенте США 4857508 (Adams et al.),выданном 15 августа 1989 г., в общем виде описано, что ряд производных гуанидиноалкилпептидов, содержащих концевые аралкильные заместители, проявляет ингибирующую агрегацию тромбоцитов активность, и конкретно описан ряд О-метилтирозин-, бифенил- и нафтилпроизводных, содержащих концевую амидную функциональность.(4), 946-952, 1985) раскрывает ряд синтетических пептидов, в том числе arg-gly-asp-ser и glyarg-gly-asp-ser, которые обладают способностью ингибировать индуцируемую тромбином агрегацию тромбоцитов.Sci. USA 79, 3711-3715, 1982) показали, что тетрапептид глицил-L-пролил-L-аргинил-L-пролин ингибирует связывание фибриногена с тромбоцитами человека. Во Французской заявке 86/17507, поданной 15 декабря 1986 г., указано, что тетра-,пента- и гексапептидные производные, содержащие последовательность -arg-gly-asp-, могут быть использованы в качестве антитроботических агентов. В патенте США 4683291 (Zimmerman etal.), выданном 28 июля 1987 г., показано, что ряд пептидов, состоящих из 6-40 аминокислот,которые содержат последовательность -arg-glyasp-, представляет собой ингибиторы связывания тромбоцитов. В Европейской патентной публикации 0319506, опубликованной 7 июня 1989 г., показано, что ряд тетра-, пета-и гексапептидных производных, содержащих последовательность-arg-gly-asp-, представляет собой ингибиторы агрегации тромбоцитов. Циклические пептидные аналоги, содержащие фрагмент Gly-Asp, как показано в патенте США 5023233, являются антагонистами рецепторов фибриногена. Пептиды и псевдопептиды, содержащие амино-, гуанидино-, имидазалоил- и/или амидиноалканоил- и алкеноил-фрагменты, являются антитромботическими агентами. Они описаны в находящихся на рассмотрении заявках США 07/677066, 07/534385 и 07/460777, поданных 28 марта 1991 г., 7 июня 1990 г. и 4 января 1990 г. соответственно, а также в патенте США 4952562 и международной заявкеPCT/US 90/05448, поданной 25 сентября 1990 г., все права на которые переданы правопреемнику по настоящему изобретению. Пептиды и псевдопептиды, содержащие амино- и гуанидино-алкил- и алкенил- бензоил-,фенилалканоил- и фенилалкеноил-фрагменты,являются антитромботическими агентами, что показано в находящейся на рассмотрении заявке США 07/475043, направленной на рассмотрение 5 февраля 1990 г., и в международной публикацииPCT/US 91/02471, направленной 4 на рассмотрение 11 апреля 1991 г. и опубликованной как Международная публикацияWO 92/13117 29 октября 1992 г., все права на которые переданы правопреемнику по настоящему изобретению. Производные алканоил- и замещенных алканоил-азациклоалкилформиласпарагиновой кислоты описаны как ингибиторы агрегации тромбоцитов в патенте США 5053392, поданном 1 декабря 1989, все права на который переданы правопреемнику по настоящему изобретению и имеющему тех же авторов, что и настоящее изобретение. Производные N-замещенной азациклоалкилкарбонилциклической аминоациласпарагиновой кислоты описаны как антитромботические агенты в патенте США 5064814, поданном 5 апреля 1990 г. теми же изобретателями и все права на который переданы правопреемнику по настоящему изобретению. Производные азациклоалкилформилглициласпарагиновой кислоты представлены как антитромботические агенты в патенте США 5051405, поданном 10 октября 1989, и все права на который переданы правопреемнику по настоящему изобретению. В Европейской патентной заявке 0479481,опубликованной 8 апреля 1992 г., в качестве антагонистов рецепторов фибриногена описаны азациклоалкилалканоилглициласпартиламинокислоты. В Европейской патентной заявке 0478362,опубликованной 1 апреля 1992 г., в качестве антагонистов рецепторов фибриногена описаны азациклоалкилалканоилпептидилаланины. В патентной публикации РСТWO 95/10295 раскрыты азациклоалкилалканоилпептиды и псевдопептиды формулы II и, в частности, N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамид, который ингибирует агрегацию тромбоцитов и образование тромбов у млекопитающих и может быть использован при профилактике и лечении тромбоза. Полученный в соответствии с описанием патентной публикацииWO 95/10295 N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамид является аморфным, гигроскопичным и физически нестабильным, так как он абсорбирует влагу. В патентной публикации РСТWO 95/10295 не описана негигроскопичная стабильная кристаллическая форма N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексил-аланинамида. В патентной публикации РСТWO 95/10295 также показано, что азациклоалкилалканоилпептиды и псевдопептиды обычно получают по методикам стандартного твердофазного 5 пептидного синтеза или пептидного синтеза в жидкой фазе с использованием исходных материалов и/или легко доступных промежуточных соединений, поставляемых химическими компаниями, такими как Aldrich или Sigma (H.(1988. Кроме того, в патентной публикации заявки РСТWO 95/10295 указано, что аморфную и гигроскопичную форму N-[N-[N(4-пиперидин-4-ил)бутаноил)-N-этилглицил](L)-аспартил]-(L)циклогексилаланинамида получают путем последовательного синтеза из С-концевой аминокислоты, как это показано на схеме I. Схема I В публикации РСТ WO 95/10295 не описано образование тетраазациклоалкилалканоилпептидов и псевдопептидов или, в частности, N[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида из основного ди(псевдопептида или пептида), в результате чего оба N- и С-концевых окончания основного ди(псевдопептида или дипептида) сопрягаются с псевдоаминокислотами и/или аминокислотами с образованием тетраазациклоалкил-алканоилпептидов и псевдопептидов. Сущность изобретения Настоящее изобретение относится к негигроскопичной стабильной кристаллической форме Соединение обладает антитромботической активностью, ингибируя в том числе агрегацию 6 тромбоцитов и образование тромбов у млекопитающих и может быть использовано для профилактики и лечения тромбоза, ассоциированного с болезненными состояниями, такими как инфаркт миокарда, удар, периферическое артериальное заболевание и диссеминированное внутрисосудистое свертывание. Кроме того, изобретение относится к фармацевтической композиции негигроскопичной стабильной кристаллической формы N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида и к его промежуточным соединениям. Изобретение также относится к способу получения тетра-азациклоалкилалканоилпептида или псевдопептида формулы II Е 1 представляет собой Н; Е 2 представляет собой -углеродную боковую цепь природной -аминокислоты, Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил,гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, или Е 1 и Е 2 вместе с атомом азота и атомом углерода, через которые Е 1 и Е 2 связаны, образуют 4-, 5-, 6- или 7-членное азациклоалкановое кольцо;R1 и R2 независимо друг от друга представляют собой Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил;m принимает значения от 1 до 5;n принимает значения от 0 до 6; и р принимает значения от 1 до 4, и, в частности, негигроскопичной стабильной формы N[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида. Изобретение также относится к соединению формулы 7 В представляет собой алкил, циклоалкил,циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил; Е 1 представляет собой Н; Е 2 представляет собой -углеродную боковую цепь природной -аминокислоты, Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил,гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, или Е 1 и Е 2 вместе с атомом азота и атомом углерода, через которые Е 1 и Е 2 связаны, образуют 4-, 5-, 6- или 7-членное азациклоалкановое кольцо;R1 и R2 независимо друг от друга представляют собой Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил; р принимает значения от 1 до 4; Р 1 представляет собой неустойчивую при гидрировании кислотную защитную группу; и Р 2' представляет собой Р 2 или ТФУКН-; и представляет собой кислотноР 2 неустойчивую аминозащитную группу. Краткое описание чертежей На фиг. 1 представлена диаграмма порошковой рентгенографии образца негигроскопичной кристаллической формы N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида, полученного в примере 13, способ А. На фиг. 2 представлена диаграмма порошковой рентгенографии образца негигроскопичной кристаллической формы N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида, полученного в примере 13, способ В. На фиг. 3 представлена диаграмма порошковой рентгенографии образца негигроскопичной кристаллической формы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида, полученного в примере 13, способ В(в). На фиг. 4 представлена диаграмма порошковой рентгенографии образца гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида, полученного в примере 14. На фиг. 5 представлена диаграмма порошковой рентгенографии образца негигроскопичной кристаллической формы N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида, полученного в примере 14. На фиг. 6 представлен график выходной мощности при изотермической микрокалори 001391 8 метрии как функции времени для трех различных опытов, которые проведены в соответствии с описанием опыта 15. В опытах оценивается термическая конверсия различных кристаллических форм N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида при экспозиции парами различных растворителей. Линия (А) на фиг. 6 показывает, что сильный экзотермический эффект имеет место в том случае, когда гигроскопичный N-[N-[N-(4-пиперидин-4-ил) бутаноил)-Nэтилглицил]-(L)-аспартил]-(L)циклогексилаланинамид, полученный в соответствии с методиками примеров 5 или 11, выдерживается при 80 %-ной OВ (насыщенный раствор КСl) и 40 С в течение 30 ч; при таком воздействии гигроскопичная форма соединения превращается в негигроскопичную форму соединения. Линия(В) на фиг. 6 указывает на отсутствие экзотермической конверсии, когда гигроскопичный N[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамид, полученный в соответствии с методикой примеров 5 или 11, подвергается воздействию паров метанола при 40 С (растворитель,отличный от воды, в котором соединение растворимо) и, следовательно, на то, что метанол не обладает подвижностью в кристаллах этой формы, чтобы произошла конверсия в негигроскопичную форму. Линия (С) на фиг. 6 показывает, что экзотермическая конверсия отсутствует, когда негигроскопичный N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамид, полученный в соответствии с примером 13, выдерживается при 40 С и 80 %-ной OВ; следовательно, при таких условиях негигроскопичная форма соединения не подвергается конверсии форм, то есть, является стабильной формой. На фиг. 7 представлен график выходной мощности при изотермической микрокалориметрии как функции времени для трех различных опытов, которые проведены в соответствии с описанием опыта 15. В этих опытах оценивается термическая конверсия гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида в его негигроскопичную форму при выдерживании при 80%ной OВ при 40, 50 и 60 С. График показывает,что конверсия протекает в течение приблизительно 24 ч при 40 С, 6,5 ч при 50 С и в течение 3 ч при 60 С. На фиг. 8 представлен график выходной мощности при изотермической микрокалориметрии как функция времени для четырех различных опытов, которые проводятся в соответствии с описанием примера 15. В опытах оценивается термическая конверсия гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин 9 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида в его негигроскопичную форму при выдерживании при 60 С при 65%, 75%, 80% и 100%-ной OВ. Для фиг. 8 характерно то, что более высокая относительная влажность приводит к более быстрой конверсии. Другой характерный признак этого графика состоит в том, что конверсия в негигроскопичную форму соединения имеет место при 100%ной OВ и при 60 С без разжижения, которое наблюдается для гигроскопичной формы при комнатной температуре. На основании этих результатов можно предполагать, что при 60 С скорость конверсии в негигроскопичную форму намного выше, чем скорость разжижения гигроскопичной формы. На фиг. 9 представлено сравнение графиков увеличения веса в зависимости от OВ (в %) для гигроскопичнойи негигроскопичнойформ N-[N-[N-(4-пиперидин-4-ил)бутаноил)-Nэтилглицил]-(L)-аспартил]-(L)циклогексилаланинамида при 25 С, которые получены в соответствии с описанием опыта 16. Фиг. 9 показывает, что при увеличении OВ гигроскопичная форма принимает больше воды, чем негигроскопичная форма, причем это свойство более ярко выражено при относительной влажности свыше 60%. Кроме того, фиг. 9 показывает, что для гигроскопичной формы соединения не характерна десорбция до своего первоначального веса (%), тогда как негигроскопичная форма соединения способна десорбироваться до своего первоначального веса (%). Подробное описание изобретения В описании настоящего изобретения, если не оговорено особо, используются следующие термины, значение которых расшифровывается ниже. В данном описании используются следующие сокращения: ВОС трет.бутилоксикарбонил, CBZ - бензилоксикарбонил, Gly - глицин, Asp - аспарагиновая кислота, Obzl - бензилокси, ТФУК - трифторуксусная кислота, Cha-циклогексилаланин, EtOAc - этилацетет, ДМФА- тетрафторборат (2-1 Н-бензотриазол-1-ил)1,1,3,3-тетраметилурония, ДВ - деионизированная вода, ПНФ - п-нитрофенол, ПФФ - пентафторфенол, ДЦМ -дициклогексидмочевина,NMM - N-метилморфолин, МТБЭ - трет.бутилметиловый эфир, OВ - относительная влажность, ТГФ - тетрагидрофуран, PipBu - 4 пиперидинбутановая кислота и PipBuen - (4 пиперидин)бутилиденилкарбоновая кислота формулы Определение "пациент" относится как к человеку, так и к другим млекопитающим."Фармацевтически приемлемая соль" означает соленую форму исходного соединения формулы I, которая относительно безвредна для пациента при использовании в терапевтических дозах, так что положительные фармацевтические свойства исходного соединения формулы I не ухудшаются из-за побочных эффектов, приписываемых противоиону этой солевой формы. Фармацевтически приемлемая соль также включает цвиттерион и внутреннюю соль соединения формулы I."Алкил" означает насыщенную алифатическую углеводородную группу, которая может быть линейной или разветвленной и содержит приблизительно от 1 до 20 атомов углерода в цепи. Понятие "разветвленный" означает, что низшая алкильная группа, такая как метил, этил и пропил, присоединена к линейной алкильной цепи. Предпочтительными линейными или разветвленными алкильными группами являются"низшие алкильные" группы, которые представляют собой алкильные группы, содержащие от 1 до 10 атомов углерода. Наиболее предпочтительными алкильными группами являются группы, содержащие от 1 до 6 атомов углерода."Циклоалкил" означает насыщенную карбоциклическую группу, содержащую одно или несколько колец и приблизительно от 3 до 10 атомов углерода. Предпочтительными циклоалкильными группами являются циклопропил,циклобутил, циклопентил, циклогексил, циклогептил и декагидронафтил. Определение "циклоалкилалкил" относится к алкильной группе, замещенной циклоалкильной группой. Предпочтительными циклоалкилалкильными группами являются циклопентилметил, циклогексилметил, циклогексилэтил, декагидронафт-1-илметил и декагидронафт-2-илметил."Алкилциклоалкил" означает циклоалкильную группу, замещенную алкильной группой. Примерами алкилциклоалкильных групп являются 1-, 2-, 3- или 4-метил- или этилциклогексил."Aлкилциклоалкилалкил" означает алкильную группу, замещенную алкилциклоалкильной группой. Примерами алкилциклоалкильных групп являются 1-, 2-, 3- или 4 метил- или этилциклогексилметил или 1, 2-, 3 или 4-метил- или этилциклогексилэтил."Азациклоалкан" означает насыщенное алифатическое кольцо, содержащее атом азота. Предпочтительными азациклоалканами являются пиролидин и пиперидин."-Углеродная боковая цепь природной аминокислоты" означает фрагмент, который замещает -углеродный атом природной аминокислоты. Примерами -углеродных боковых цепей природных -аминокислот являются изопропил, метил и карбоксиметил соответственно для валина, аланина и аспарагиновой кислоты. Термин "аминозащитная группа" означает легко удаляемую группу, которая, как известно в данной области, защищает аминогруппу от нежелательной реакции при осуществлении методик синтеза и селективно снимается. Использование защитной группы для защиты аминогруппы от нежелательных реакций при проведении синтезов известно в данной области. Существует много таких защитных групп (см.,например, Т.Н. Greene, P.G.M. Wuts, ProtectiveWiley and Sons, New York (1991), приведена в качестве ссылки). Предпочтительными аминозащитными группами являются ацильные группы, в том числе формильная, ацетильная, хлорацетильная, трихлорацетильная, о-нитрофенилацетильная, о-нитрофеноксиацетильная, трифторацетильная, ацетоацетильная, 4-хлорбутирильная, изобутирильная, о-нитроциннамоильная, пиколиноильная, ацилизотиоцианатная,аминокапроильная, бензоильная и др. подобные группы, а также ацилоксигруппы, в том числе метоксикарбонильная, 9-флуоренилметоксикарбонильная, 2,2,2-трифторэтоксикарбонильная,2-триметилсилилэтоксикарбонильная, винилоксикарбонильная, аллилоксикарбонильная, трет.бутилоксикарбонильная (ВОС), 1,1-диметилпропинилоксикарбонильная, бензилоксикарбонильная(CBZ),п-нитробензилоксикарбонильная, 2,4-дихлорбензилоксикарбонильная и др. аналогичные группы. Понятие "неустойчивая в кислоте аминозащитная группа" означает группу, защищающую аминогруппу, определенную выше, которая легко удаляется при обработке кислотой,оставаясь относительно стабильной при действии других реагентов. Предпочтительной подвижной в кислоте аминозащитной группой является трет.-бутоксикарбонильная группа(ВОС). Термин "неустойчивая при гидрировании аминозащитная группа" означает группу, защищающую аминогруппу, определенную выше,которая легко удаляется при гидрировании, оставаясь относительно стабильной под действием других реагентов. Понятие "защитная кислотная группа" означает легко снимаемую группу, известную в данной области как группа, которая защищает карбоксильную группу (-СО 2 Н) от нежелательной реакции при проведении синтетических реакций и которая селективно снимается. Использование защитных групп для карбоксиль 001391 12 ных групп хорошо известно в данной областиedition, John Wiley and Sons, New York (1991),приведена в качестве справочного материала). Примерами групп, защищающих карбоксильные группы, являются простые эфирные группы,такие как метоксиметил, метилтиометил, тетрагидропиранил, бензилоксиметил, замещенный и незамещенный фенацил, 2,2,2-трихлорэтил,трет.-бутил, циннамил, замещенный и незамещенный бензил, триметилсилил и другие подобные группы, а также амидные и гидразидные группы, включая N,N-диметил, 7-нитроиндолил,гидразид, N-фенилгидразид и др. Понятие "неустойчивая при гидрировании кислотная защитная группа" означает защитную группу для карбоксильной группы, определенную выше, которая легко снимается путем гидрирования, оставаясь относительно стабильной к действию других реагентов. Предпочтительной неустойчивой при гидрировании группой,защищающей карбоксильную группу, является бензильная группа. Понятие "арил" означает фенильную или нафтильную группу."Замещенный арил" означает фенильную или нафтильную группу, замещенную одним или несколькими заместителями для арильной группы, которые могут быть одинаковыми или различными, где "заместители для арильной группы" представляют собой алкил, алкенил,алкинил, арил, аралкил, гидроксигруппу, алкокси-, арилокси-, аралкоксигруппы, гидроксиалкил, ацил, формил, карбоксильную группу, алкеноил, ароил, галоген, нитрогруппу, тригалогенметильную группу, цианогруппу, алкоксикарбонил, арилоксикарбонил, аралкоксикарбонил, ациламино-, ароиламиногруппу, карбамоильную группу, алкилкарбамоильную группу,диалкилкарбамоильную группу, арилкарбамоильную, аралкилкарбамоильную группу, алкилсульфонильную, алкилсульфинильную, арилсульфонильную, арилсульфинильную, аралкилсульфонильную, аралкилсульфинильную группу или -NRaRb, где заместители Ra и Rb независимо друг от друга представляют собой атом водорода, алкил, арил или аралкил."Аралкил" представляет собой алкильную группу, замещенную арильным радикалом. Предпочтительными аралкильными группами являются бензил, нафт-1-илметил, нафт-2 илметил и фенетил."Замещенный аралкил" означает аралкильную группу, замещенную по арильной группе одним или несколькими заместителями для арильной группы."Гетероциклил" означает 4-15-членную моноциклическую или полициклическую систему, в которой один или несколько атомов в кольце представляют собой элемент, отличный от атома углерода, например, атом азота, кисло 13 рода или серы. Предпочтительными гетероциклическими группами являются пиридил, пиримидил и пирролидил. Понятие "замещенный гетероциклил" определяет гетероциклическую группу, замещенную одним или несколькими заместителями для арильной группы."Гетероциклилалкил" и "замещенный гетероциклилалкил" означают алкильную группу,замещенную гетероциклильной или замещенной гетероциклильной группой соответственно. Понятие "гигроскопичность" означает сорбцию, подразумевая при этом приобретенное или уже присутствующее количество воды, которого достаточно для воздействия на физические или химические свойства вещества (Eds. J.Swarbrick, J.C.Boylan, Encyclopedia of Pharmaceutical Technology, Vol. 10, p. 33). Предпочтительные варианты осуществления изобретения Предпочтительное соединение, полученное в соответствии с настоящим изобретением,представлено формулой II, где заместитель Е 2 представляет собой Н, алкил, гидроксиметил, 1 гидроксиэтил, меркаптометил, 2-метилтиоэтил,карбоксиметил, 2-карбоксиэтил, 4-аминобутил,3-гуанидинопропил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, или заместители Е 1 и Е 2 вместе с атомами азота и углерода, через которые Е 1 и Е 2 соединены друг с другом, образуют 4-,5-, 6- или 7-членное азациклоалкановое кольцо,при условии, что гетероциклилалкил не является индол-3-илметилом. Более предпочтительное соединение, полученное в соответствии с настоящим изобретением, описывается формулой II, где заместитель Е 2 представляет собой Н, алкил, гидроксиметил,1-гидроксиэтил, меркаптометил, 2-метилтиоэтил, карбоксиметил, 2-карбоксиэтил, 4-аминобутил, 3-гуанидинопропил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, или заместители Е 1 и Е 2 вместе с атомами азота и углерода, через которые Е 1 и Е 2 соединены друг с другом, образуют 4-,5-, 6- или 7-членное азациклоалкановое кольцо. Еще более предпочтительное соединение настоящего изобретения описывается формулой 14 образуют 4-, 5-, 6- или 7-членное азациклоалкановое кольцо. Также предпочтительным является соединение, которое получено в соответствии с настоящим изобретением и описывается формулой II, где В представляет собой алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил или алкилциклоалкилалкил. Конкретный пример соединения, полученного в соответствии с настоящим изобретением,описывается формулой IIаR1 и R2 независимо друг от друга представляют собой Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил;m принимает значения от 1 до 5;n принимает значения от 2 до 6; и р принимает значения 1 или 2. Более предпочтительный вариант осуществления настоящего изобретения описывается формулой IIа, где В представляет собой алкил, циклоалкил,циклоалкилалкил, алкилциклоалкил или алкилциклоалкилалкил;n принимает значения 3 или 4. Другой предпочтительный вариант осуществления настоящего изобретения описывается формулой IIа, где В представляет собой алкил;R1 и R2 независимо друг от друга представляют собой Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил или алкилциклоалкилалкил;p принимает значение 1. Еще одним предпочтительным соединением, полученным в соответствии с настоящим изобретением, является соединение формулыIIа, которое представляет собой N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамид. 15 Другим вариантом осуществления настоящего изобретения является получение стабильной негигроскопичной кристаллической формыN-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L-)циклогексилаланинамида. В соответствии с изобретением эта форма соединения дает возможность разрабатывать стабильную рецептуру названного соединения. Стабильная негигроскопичная формаN-[N-[N-(4-пиперидин-4-ил)бутаноил)-Nэтилглицил]-(L)-аспартил]-(L)циклогексилаланинамида также имеет высокую температуру плавления и не обладает способностью абсорбировать воду. Стабильная форма также обладает уникальной и неожиданной стабильностью во влажных условиях и при температурах, которые значительно превышают влажность и температуру, обычно встречающиеся при транспортировке и производстве лекарственной формы или при долговременных транспортировке и хранении. Эти свойства также облегчают изготовление лекарственной формы. Конверсия в стабильную форму не сопровождается потерей материала или его чистоты и не оказывает отрицательного влияния на его практические свойства. Предпочтительным вариантом осуществления изобретения является соединение формулы VII, где В представляет собой алкил; Е 1 представляет собой Н; Е 2 представляет собой циклоалкилалкил; G представляет собой NR1R2: и R1 и R2 представляют собой Н; р принимает значение 1. Другой предпочтительный вариант осуществления изобретения представляет собой соединение формулы VII, где В представляет собой этил; Е 1 представляет собой Н; Е 2 представляет собой циклогексилметил; G представляет собой NR1R2; R1 и R2 представляют собой Н; и р принимает значение 1. Еще один предпочтительный вариант осуществления настоящего изобретения соответствует соединению формулы VII, где В представляет собой этил; Е 1 представляет собой Н; Е 2 представляет собой циклогексилметил; G представляет собой NR1R2; R1 и R2 представляют собой Н; р принимает значение 1, Р 1 представляет собой бензил; и Р 2' представляет собой ТФУКН-. Еще один предпочтительный вариант осуществления настоящего изобретения соответствует соединению формулы VII, где В представляет собой этил; Е 1 представляет собой Н; Е 2 представляет собой циклогексилметил; G представляет собой NR1R2; R1 и R2 представляют собой Н; р принимает значение 1, Р 1 представляет собой бензил; и Р 2 представляет собой трет.-бутоксикарбонил. Следует понимать, что настоящее изобретение охватывает все комбинации предпочтительных соединений, предпочтительных вари 001391 16 антов осуществления изобретения и конкретных вариантов его осуществления, описанных здесь. Соединение настоящего изобретения используется в виде свободного основания или свободной кислоты, его цвиттерионовой соли или фармацевтически приемлемой соли. Все эти формы подпадают под объем настоящего изобретения. Когда соединение настоящего изобретения замещено основным остатком, образуется кислотно-аддитивная соль, которая просто является более удобной формой для применения. На практике использование солевой формы соответствует использованию свободного основания. Кислота, которая может быть использована для получения кислотно-аддитивной соли,предпочтительно представляет собой кислоту,которая при смешении со свободным основанием дает фармацевтически приемлемую соль. То есть, соль, анион которой не токсичен для пациента в фармацевтических дозах соли и положительное ингибирующее действие на агрегацию тромбоцитов и образование тромбов, присущее свободному основанию, не ухудшается из-за побочных эффектов, приписываемых аниону. Хотя фармацевтически приемлемые соли указанных основных соединений являются предпочтительными, все кислотно-аддитивные соли могут быть использованы в качестве источника свободного основания, даже если конкретная соль, сама по себе, необходима только как промежуточное соединение. Например, когда соль получают только в целях очистки и идентификации или когда ее используют в качестве промежуточного соединения при получении фармацевтически приемлемой соли с использованием методики ионного обмена. Фармацевтически приемлемые соли настоящего изобретения представляют собой соли, которые получены из следующих кислот: минеральные кислоты, такие как соляная, серная, фосфорная и сульфаминовая кислоты; и органические кислоты, такие как уксусная, лимонная, молочная, винная,малоновая, метансульфоновая, этанcульфоновая, бензолсульфоновая, п-толуолсульфоновая,циклогексилсульфоновая, хинная кислоты и др. К соответствующим кислотно-аддитивным солям относятся следующие соли: гидрогалогениды, например, гидрохлориды и гидробромиды,сульфат, фосфат, нитрат, сульфамат, ацетат,цитрат, лактат, тартрат, малонат, оксалат, салицилат, пропионат, сукцинат, фумарат, малеат,метилен-бисгидроксинафтоаты,гентизаты,мезилаты, изотионаты и ди-р-толуоилтартраты,метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат, циклогексилсульфамат и хинат, соответственно. В соответствии с еще одним объектом настоящего изобретения кислотно-аддитивные соли заявляемых соединений получают реакцией свободного основания с соответствующей кислотой при использовании известных методик 17 или при соответствующей модификации этих методик. Например, кислотно-аддитивные соли соединений настоящего изобретения получают при растворении свободного основания в воде,водно-спиртовом растворителе или в других подходящих растворителях, содержащих соответствующую кислоту, с последующим выделением соли путем упаривания растворителя; или при взаимодействии свободного основания и кислоты в органическом растворителе. В этом случае соль отделяется сразу или может быть выделена после концентрирования раствора. Кислотно-аддитивные соли соединений настоящего изобретения могут быть регенерированы из солей по известным или адаптированным методикам. Например, исходные соединения изобретения могут быть регенерированы из их кислотно-аддитивных солей путем обработки щелочью, например водным раствором бикарбоната натрия или водным раствором аммиака. При замещении соединения настоящего изобретения кислотным остатком могут быть получены основно-аддитивные соли, которые просто более удобны при применении. На практике использование солевой формы соответствует использованию свободной кислоты. Основания, которые могут быть использованы для получения основно-аддитивной соли, предпочтительно представляют собой основание, которое при смешении со свободной кислотой дает фармацевтически приемлемую соль, то есть,соль, катион которой не токсичен для организма в фармацевтических дозах соли и положительное ингибирующее действие на агрегацию тромбоцитов и образование тромбов, присущее свободной кислоте, не ухудшается из-за побочных эффектов, приписываемых катиону. Фармацевтически приемлемые соли, включающие,например, соли щелочных и щелочноземельных металлов, которые подпадают под объем изобретения, представляют собой соли,полученные из следующих оснований: гидрид натрия, гидроксид натрия, гидроксид калия,гидроксид кальция, гидроксид алюминия, гидроксид лития, гидроксид магния, гидроксид цинка, аммиак, этилендиамин, N-метилглюкамин, лизин, аргинин, орнитин, холин, N,N'дибензилэтилендиамин, диамин, хлорпрокаин,диэтаноламин, прокаин, N-бензилфенетиламин,диэтиламин, пиперазин, трис(гидроксиметил)аминометан, гидроксид тетраметиламмония и др. Соли металлов соединений настоящего изобретения могут быть получены путем контактирования гидрида, гидроксида, карбоната или другого реакционноспособного соединения выбранного металла в водном или органическом растворителе со свободной кислотной формой соединения. Используемый водный растворитель может представлять собой воду или он может представлять собой смесь воды с органиче 001391 18 ским растворителем, предпочтительно со спиртом, таким как метанол или этанол, кетоном,таким как ацетон, простым алифатическим эфиром, таким как тетрагидрофуран, или сложным эфиром, таким как этилацетат. Такие реакции обычно проводят при комнатной температуре,однако при необходимости их можно провести и при нагревании. Аминные соли соединений настоящего изобретения могут быть получены путем контактирования амина в водном или органическом растворителе с соединением в виде свободной кислоты. Приемлемыми водными растворителями являются вода или ее смеси со спиртами,такими как метанол или этанол, простыми эфирами, такими как тетрагидрофуран, нитрилами,такими как ацетонитрил, или кетонами, такими как ацетон. Аналогичным образом могут быть получены аминокислотные соли. Основно-аддитивные соли соединений настоящего изобретения могут быть регенерированы из солей с использованием известных или адаптированных способов. Например, исходные соединения настоящего изобретения можно регенерировать из их основно-аддитивных солей при обработке последних кислотой, например,соляной кислотой. Помимо использования в качестве активных соединений, соли соединений настоящего изобретения могут служить для очистки соединений, например, благодаря разнице растворимостей солей и исходных соединений, побочных продуктов и/или исходных материалов. В этом случае используют методики, которые хорошо известны квалифицированным в данной области специалистам. Соединения настоящего изобретения могут иметь асимметричные центры. Такие асимметричные центры независимо друг от друга могут иметь R- или S-конфигурацию. Квалифицированному в данной области специалисту также очевидно, что некоторые соединения формулы I могут иметь геометрическую изомерию. Геометрическими изомерами являются цис- и транс-формы заявляемых соединений,содержащих алкенильный остаток. Настоящее изобретение охватывает отдельные геометрические изомеры и стереоизомеры, а также их смеси. Такие изомеры могут быть выделены из их смесей с помощью известных или адаптированных методик, например, с помощью хроматографии или перекристаллизацией. Или они могут быть получены отдельно из соответствующих изомеров их промежуточных соединений,например, с использованием известных или адаптированных способов, описанных здесь. В заявках США 08/138820 и 08/476750,которые включены в данное описание в качестве ссылок, описаны способы получения аморфного соединения формулы II, и, в частности,аморфного соединения формулы I. 19 Новый способ получения соединения формулы II и, в частности кристаллического соединения формулы I, в соответствии с настоящим изобретением представлен на схеме II, где В, Е 1,Е 2, G, R, m, n и р принимают значения, определенные выше, а Р 1 представляет собой неустойчивую при гидрировании кислотную защитную группу амино, такую как бензил, Р 2 представляет собой неустойчивую в кислоте группу, защищающую аминогруппу, такую как трет.бутоксикарбонил (ВОС), а Р 3 представляет собой неустойчивую при гидрировании группу,защищающую аминогруппу, такую как бензилоксикарбонильная группа (CBZ). Схема II 20 Способ, представленный на схеме II, иллюстрируется на примере получения соединения формулы II, однако следует понимать, что соединение формулы I также может быть получено при использовании соответствующих исходных материалов. При получении соединения формулы I в соответствии со схемой II, В представляет собой этил, Е представляет собой Н, F представляет собой циклогексилметил, G представляет собой NH2, R представляет собой Н, m принимает значение 3, n принимает значение 3,р принимает значение 1, Р 1 представляет собой бензил, Р 2 представляет собой ВОС и Р 3 представляет собой CBZ. Альтернативный способ получения соединения формулы I в соответствии с настоящим изобретением аналогичен способу, представленному на схеме II, за исключением того, что используется соединение формулы III, где значение Р 3 определено выше: вместо соединения формулы IV, гдеR представляет собой Н, m принимает значение 3, n принимает значение 3, р принимает значение 1, а Р 3 представляет собой CBZ, с получением промежуточного соединения формулы V При получении соединений формулы II или их промежуточных соединений может быть также желательно или необходимо предупредить протекание перекрестной реакции между химически активными заместителями, которые присутствуют в природных аминокислотах или псевдоаминокислотах. Такие заместители могут быть защищены с помощью стандартных защитных групп. Защитные группы впоследствии могут быть удалены (или оставлены, если это необходимо) с использованием известных способов и с получением необходимых продуктов или промежуточных соединений (см., например,Green "Protective Groups in Organic Synthesis",Wiley, New York (1981). Селективная защита или селективное снятие защиты могут быть также необходимы или желательны, чтобы преобразовать или удалить существующие заместители, или чтобы провести последующую реакцию с получением конечного продукта. В представляет собой этил, Е 1 представляет собой Н, Е 2 представляет собой циклогексилметил, G представляет собой NH2, р принимает значение 1, Р 1 представляет собой бензил, а Р 3 представляет собой CBZ. На схеме II представлен пятистадийный способ получения соединения настоящего изобретения, начиная от формирования центрального дипептидного промежуточного соединения формулы VI в соответствии с настоящим изобретением В, р, Р 2 и Р 1 принимают значения, определенные выше. В случае получения соединения формулы I, центральное дипептидное промежуточное соединение настоящего изобретения представляет собойBOC-N(Et)-Gly-(L)Asp(OBzl)-ОН. Центральное дипептидное промежуточное соединение получают без защиты свободного остатка карбоновой кислоты. На стадии 2 схемы II, сопряжение с образованием центрального дипептида, может быть осуществлено или в дихлорметане или в смесях 21 этилацетата (с или без ДМФА в качестве сорастворителя) и органических оснований, таких какNMM. Эта реакция может быть проведена при температуре от комнатной температуры и до температуры 40 С. Активация аминокислоты или псевдоаминокислоты следующей формулы для проведения реакции сопряжения может быть осуществлена с использованием не выделяемых активных эфиров с п-нитрофенолом, пентафторфенолом и N-гидроксисукцинимидом под действием дициклогексилкарбодиимида. Время реакции сопряжения лежит в интервале приблизительно от 1 до 20 ч в зависимости от сопрягаемых аминокислот или псевдоаминокислот, активирующего агента,растворителя и температуры. Центральный дипептидный продукт стадии 1 не должен выделяться. Реакционную смесь стадии 1 обычно промывают водой или разбавленной водной кислотой (например, водной НСl) и затем используют непосредственно без сушки на стадии 2. В случае, когда используются активные эфиры на основе фенола, центральный дипептидный продукт экстрагируют из реакционной смеси в щелочную воду, затем реэкстрагируют из подкисленного водного раствора снова в органический растворитель. Полученный раствор используют непосредственно на стадии 2. Дипептидное промежуточное соединение формулы VI используют для получения трипептидного промежуточного соединения формулы представляет собой диссоциацию ТФУК с образованием F3CCO2- и H+, где Н+ протонирует концевую аминогруппу соединения формулыVII, то есть образуется соль ТФУК формулы В случае получения соединения формулы I трипептидное промежуточное соединение настоящего изобретения представляет собой P2'N(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2. На стадии 2 сопряжение аминокислоты или псевдоаминокислоты с образованием центрального дипептида может быть осуществлено в дихлорметане или в смеси этилацетата и ДМФА или ТГФ при комнатной температуре или при температуре, ниже комнатной. Активация центрального дипептида следующей формулы может быть осуществлена с помощью не выделяемых активных эфиров пентафторфенола илиN-гидроксисукцинимида под действием дициклогексилкарбодиимида. Активация также может быть проведена с использованием изопропилхлорформиата. Время реакции может меняться в зависимости аминокислоты или псевдоаминокислоты, которые должны быть сопряжены,активирующего агента, растворителя и температуры и составляет приблизительно от 1 до 20 ч. Когда трипептидное промежуточное соединение не выделяется, реакционную смесь промывают водным органическим основанием, таким как водный N-метилморфолин, и водной кислотой,такой как водная НС 1, и вводят в реакцию на стадии 3 после водных промывок без сушки"как оно есть". На стадии 3 схемы II снятие защитной группы, такой как ВОС, с трипептидного продукта стадии 2 может быть осуществлено при использовании раствора трифторуксусной кислоты в дихлорметане или при использовании смеси НВr в уксусной кислоте и этилацетате. Реакцию можно проводить приблизительно при комнатной температуре в течение приблизительно 1 ч (cпособ с НВr) или приблизительно 2 ч (cпособ с ТФУК). Кислую соль трипептида отфильтровывают в виде кристаллического твердого продукта или непосредственно из реакционной смеси (способ с НВr) или после частичного удаления растворителя путем отгонки и добавления к полученному остатку неполярного растворителя. Еще один способ в соответствии с настоящим изобретением описывается как объединенный способ быстрого и простого получения ТФУКН-N(Еt)Glу-(L)-Asp-(OBzl)-(L)-Cha-NH2 из ВОС-N(Et)Gly-OH. Этот способ представляет собой реакцию, проводимую в одном сосуде и включающую первые две стадии сопряженияcхемы II и обработку ТФУК. ТФУКН-N(Еt)Glу(L)-Asp-(OBzl)-(L)-Cha-NH2 выделяют в виде кристаллов непосредственно из объединенного реакционного раствора. Объединенный способ исключает соответствующие три отдельные реакции, представленные на Схеме II, и решает проблему создания простого, эффективного с точки зрения времени и стоимости синтеза, который может быть использован в промышленности. На cхеме II представлено получение полипептида в обратном порядке, начиная с Nзащищенной аминокислоты с последующим добавлением карбоксильного концевого фрагмента, в отличие от обычного порядка, при котором полипептид получают путем последовательного амидирования аминного конца защищенной С-концевой аминокислоты. Обратный синтетический способ в соответствии с настоя 23 щим изобретением требует введения защитной группы только в первую аминокислоту. Это дает возможность с этого момента использовать последующие аминокислоты, не защищенные ни по аминному, ни по кислотному концам(предполагаемые функциональные группы боковой цепи). Обратный синтетический способ также реализует получение соединения формулы II, в частности соединения формулы I, с помощью поточной технологии производства, в отличие от периодического производства,обычно необходимого при проведении синтеза пептидов в растворе. Новая методика уменьшает стоимость производства, так как нет необходимости покупать аминокислоты, защищенные по аминному концу. При этом не требуется специального оборудования, реагентов или аналитических методик. Еще один способ в соответствии с изобретением представляет собой получение стабильного негигроскопичного кристаллического N[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида, устойчиво получаемого путем нового твердофазного превращения гигроскопичного кристаллического N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида, полученного способами, описываемыми cхемой II и с помощью названных альтернативных реакционных стадий. Гигроскопичная кристаллическая формаN-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида обычно физически нестабильна и при воздействии определенных условий (влажность,температура) превращается в высокостабильную, не гигроскопичную кристаллическую форму N-[N-[N-(4-пиперидин-4-ил)бутаноил)N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида. В соответствии с настоящим изобретением конверсия гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин-4-ил)бутаноил)N-этилглицил]-(L)-аспартил]-(L)циклогексил-аланинамида в высокостабильную негигроскопичную кристаллическую форму N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида может быть осуществлена при статических и динамических условиях. Статическая методика в соответствии с настоящим изобретением описывается как статическая конверсия, так как она включает воздействие на гигроскопичную кристаллическую форму N-[N-[N-(4-пиперидин-4-ил)бутаноил)N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида, помещенную в неподвижный сосуд, такой как пузырек или лоток, определенных условий по температуре и влажности в регулируемой климатической камере. Такую "ста 001391 24 тическую" конверсию проводят при температурах в интервале приблизительно от 20 до 80 С,более предпочтительно приблизительно от 40 до 80 С, и при относительной влажности приблизительно от 40 до 100%, предпочтительно приблизительно от 65 до 80%. Динамическая методика в соответствии с настоящим изобретением описывается как динамическая конверсия, так как включает выдерживание гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин-4-ил)бутаноил)N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида при тех же температурах и относительные влажностях, которые используются в статическом способе, но при перемешивании, в том числе при перемешивании гигроскопичной кристаллической формы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида в колбе роторного испарителя или в цилиндрическом сосуде (в увлажняющей печи) с использованием пропеллерной мешалки. Следующие примеры иллюстрируют изобретение, но не ограничивают его объем. Если не оговорено особо, данные массспектрометического анализа представляют собой данные масс-спектрометрии низкого разрешения при бомбардировке быстрыми атомами,полученные на приборе VG 70SE с расчетной величиной (М+Н)+. Спектры ядерного магнитного резонанса получены на приборе BruckerACF 300 в D2O. Флэш-хроматографию проводят на силикагеле. Высокоэффективную жидкостную хроматографию (ВЭЖХ) проводят на колонках С 18 с обращенной фазой с размерами частиц в интервале 8-15 мкм. Если не оговорено особо, графики порошковой рентгенографии получены путем сканирования порошка образца с помощью дифрактометра Siemens D5000 с Сu-источником излучения (1,8 кВт, 45 кВт и 40 мА). До проведения измерений образцы измельчают, чтобы исключить частицы с размерами, которые влияют на интенсивность пиков. В держатель образца размерами 1,5 х 1 см помещают приблизительно 60 мг образца и сканируют в интервале 3-40 2 тета (2) при величине шага 0,04 и при суммарной экспозиции на каждой стадии 1 с. Пример 1. Получение BOC-N(Et)Gly-(L)Asp-(OBzl)-ОН (стадия 1 схемы II). В трехгорлую круглодонную колбу объемом 1 л загружают 51 г (0,25 моля) BOCN(Et)Gly-OH, 35 г (0,25 моля) ПНФ, 400 млEtOAc и 100 мл ДМФА. Смесь перемешивают до образования раствора и охлаждают до 4-6 С. При температуре приблизительно от 5 до 8 С в течение 10 мин по каплям добавляют раствор 51,5 г (0,25 моля) ДЦК в 125 мл EtOAc. По окончании добавления ДЦК удаляют охлаждающую баню и смесь перемешивают в течение 25 1,5 ч по мере ее нагревания до комнатной температуры (20-22 С). В течение этого времени образуется твердый осадок ДСМ. Окончание образования эфира ПНФ определяют методом аналитической ВЭЖХ (исчезновение BOCN(Et)Gly-OH). Реакционную смесь фильтруют,остающуюся ДЦМ промывают 2 порциямиEtOAc по 50 мл и промывные растворы добавляют к фильтрату. ДЦМ выбрасывают. К отфильтрованному раствору при перемешивании добавляют 67 г (0,3 моля) H2N-(L)Asp(OBzl)-ОН, получают суспензию в 150 мл(138 г, 1,36 моля) NММ. Смесь нагревают до 3840 С и выдерживают при этой температуре в течение 41 ч. Данные аналитической ВЭЖХ указывают на полный расход BOC-N(Et)GlyОПНФ. Реакционную смесь охлаждают до 25 С и непрореагировавший H2N-(L)-Asp(OBzl)-ОН отфильтровывают. Раствор охлаждают и повторно фильтруют, получают еще 1,2 г (выделено 21,7 г; 11,2 г представляют собой добавленные 20% избытка и 10,5 (0,047 моля) представляют собой непрореагировавший материал. Отфильтрованный раствор экстрагируют в воронке Сквибба объемом 2 л одной порцией деионизированной воды в 500 мл, затем 2-мя порциями по 250 мл. Объединенные водные растворы экстрагируют 3-мя порциями по 300 мл смеси трет.-бутилметиловый эфир/EtOAc(1:1) для удаления оставшегося ПНФ (данные ВЭЖХ указывают на следовые количества),затем охлаждают до 5 С и подкисляют от рН 8,9 до рН 1,79 путем добавления по каплям 150 мл концентрированной НСl. Подкисленный водный раствор экстрагируют 2 порциями EtOAc по 200 мл. Данные ВЭЖХ показывают, что в растворе не остается желаемого продукта. ЭкстрактыEtOAc объединяют, сушат над MgSO4, фильтруют и концентрируют на роторном испарителе при 35 С. Полученное бледно-оранжевое масло вакуумируют при 35 С для максимального удаления оставшегося растворителя, получают 85,68 г ВОС-N(Et)Gly-(L)-Asp-(OBzl)-ОН в виде масла (21,3 ммоля, выход 85,5%, без поправки на оставшийся растворитель). Спектр ЯМР 1 Н (250 Мгц):м.д: 7,3 (с),5,1 (с), 3,3 (д.кв), 3,0 (д.кв), 1,4 (с), 1,1 (т). Масс спектр: М=408, М+1 набл=409. ВЭЖХ: 90,79 А% (3,8 А% п-нитрофенола,без поправки на е). Элементный анализ: C20H28N2O7. H, N; Найдено, %: С 57,54, Вычислено, %: С 58,81. Пример 2. Получение BOC-N(Et)Gly-(L)Asp-(OBzl)-(L)-Cha-NH2 (стадия 2 схемы II). Способ А. Изопропилхлорформиатный способ. В EtOAc (6-8 объемов; отношение вес: объем 1:6,5) растворяют один эквивалент BOCN(Et)Gly-(L)-Asp-(OBzl)-ОН и выдерживают при температуре приблизительно от -15 до 0 С. При температуре от -15 до 0 С добавляют NММ 26 при температуре от -15 до 0 С добавляют изопропилхлорформиат (1-1,1 экв.). Реакционную смесь выдерживают при температуре от -15 до 0 С в течение двух-пяти минут. К охлажденному раствору дипептида добавляют раствор H2N(L)-Cha-NH2 (1 экв.) в ТГФ (10 объемов; отношение вес:объем 1:10), температуру поддерживают в интервале от -15 до 0 С. За ходом реакции следят с помощью ВЭЖХ, отбирая образцы через 15 мин, 1 ч и 2 ч, чтобы определить окончание реакции. (Реакция считается законченной,когда количество наблюдаемого дипептида соответствует площади менее 10%, ВЭЖХ). ВОС-трипептидный продукт выпадает в осадок непосредственно из реакционного раствора. Его отфильтровывают из реакционной смеси, промывают EtOAc (2 раза по 1 объему,вес:объем) и сушат в вакууме. Обычно выход составляет 60%, чистота 90% (по площади,ВЭЖХ), содержание аспарагиновой кислоты в виде эпимерного диастереомера менее 1% (по площади). После повторного суспендирования вEtOAc конечный выход BOC-N(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2 составляет приблизительно 60%, степень чистоты повышается до 95% (по площади) при уменьшении содержания диастереомера до менее чем 0,5%. При использовании в изопропилхлорформиатном способе 4,55 г (8,1 ммоля) BOCN(Et)Gly-(L)-Asp-(OBzl)-ОН и общей методики примера, получают BOC-N(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2 в количестве 3,26 г (чистота 97,9%, 0,3% диастереомера), выход 70 % от теоретического. Способ Б. Способ с использованием комплекса Пентафторфенол-ДЦК. В EtOAc (5 объемов; отношение вес:объем 1:5) при комнатной температуре растворяют пентафторфенол (ПФФ, 2,9 экв.) и ДЦК (1 экв.) и охлаждают до температуры -15-0 С. Один эквивалент ВОС-N(Et)Gly-(L)-Asp(OBzl)-ОН растворяют в EtOAc (6 объемов; отношение вес:объем 1:6) и смешивают с одним эквивалентом H2N-(L)-Cha-NH2, которую предварительно растворяют в ДМФА (10 объемов; отношение вес:объем 1:10). Раствор дипептида/Н 2N-(L)Cha-NH2 по каплям добавляют в раствор ПФФ и ДЦК при температуре в интервале -15-0 С. Реакционную массу выдерживают при температуре 15-22 С в течение от пяти до шестнадцати часов. За ходом реакции наблюдают с помощью ВЭЖХ, отбирая контрольные образцы для ВЭЖХ через 1, 2, 3, 4 и 16 ч. (Реакцию считают завершенной, когда согласно ВЭЖХ количество наблюдаемого дипептида составляет менее 2% по площади). Реакционную смесь фильтруют и осадок на фильтре (ДЦМ) промывают EtOAc (2 раза по 0,5 объема: вес:объем). Фильтрат обрабатывают водой (10 объемов, отношение вес:объем 1:10) и водный слой удаляют. Слой EtOAc промывают 27 водой (1 раз 5 объемами, отношение вес:объем 1:5). Слой EtOAc охлаждают, при этом выпадает продукт, который отфильтровывают и промывают EtOAc (2 по 0,4 объема, отношение вес: объем 1:0,4). Мольный выход составляет более 60%, чистота обычно составляет более 90 % (по площади), содержание аспарагиновой кислоты в виде эпимерного диастереомера 1-4% (по площади). После повторного суспедирования в EtOAc конечный выход BOC-N(Et)Gly-(L)-Asp-(OBzl)(L)-Cha-NH2 составляет приблизительно 60%,степень чистоты повышается до более чем 99%(по площади), а содержание диастереомера уменьшается до менее 0,5%. В качестве типичного примера способа на основе комплекса Пентафторфенол-ДЦК с использованием общей методики способа Б и 10 гBOC-N(Et)Gly-(L)-Asp-(OBzl)-ОН и выдерживают при комнатной температуре. К этому раствору добавляют H2N-(L)-Cha-NH2 (1 экв.) и гидроксибензотриазола (ГОБТ, 1 экв.). Полученный раствор охлаждают до температуры приблизительно от 0 до 10 С и добавляют NMM (1-1,1 экв.). В ДМФА (4-5 объемов,отношение вес:объем 1:5) растворяют сшивающий агент ТБТУ (1-1,1 экв.) и добавляют к раствору защищенного дипептида при температуре приблизительно от 0 до 10 С. Этот раствор перемешивают при температуре приблизительно от 10 до 25 С в течение приблизительно 3 ч, до тех пор, пока данные ВЭЖХ не укажут на окончание реакции (менее 2% по площади исходного вещества). Эту реакционную смесь добавляют при перемешивании к смеси 5%-ного водного раствора хлорида натрия (приблизительно 4 объема относительно реакционной смеси) иEtOAc (приблизительно 2 объема относительно реакционной смеси). Фазы разделяют и водную фазу дополнительно экстрагируют порциейEtOAc (приблизительно 1,5 объема относительно реакционной смеси). Органические фазы объединяют и промывают последовательно 0,5 н. водным раствором лимонной кислоты(приблизительно 0,6-0,7 объема относительно органической фазы), 10% водным раствором бикарбоната натрия (две порции, приблизительно 0,6-0,7 объема относительно органической фазы каждая) и 25%-ным водным раствором хлорида натрия (приблизительно 0,3-0,4 объема относительно органической фазы). Полученную органическую фазу концентрируют при пони 001391 28 женном давлении и температуре приблизительно 30-50 С до приблизительно 1/4-1/2 объема от исходного. К полученному теплому раствору добавляют такой же объем гептана. Смесь перемешивают и оставляют охлаждаться до температуры приблизительно от 0 до 20 С до выпадения требуемого трипептида. Твердый продукт отфильтровывают, промывают смесью EtOAc и гептана и сушат. Обычно выход превышает 60%, чистота составляет более 95,7 % (по площади), содержание аспарагиновой кислоты в виде эпимерного диастереомера составляет менее 2% (по площади). В качестве типичного примера способа на основе ГОБТ/ТБТУ при использовании общей методики и 10 г (24,5 ммоля) ВОС-N(Et)Gly-(L)Asp-(OBzl)-OH получено 9,3 г BOC-N(Et)Gly(L)-Asp-(OBzl)-(L)-Cha-NH2 (чистота 96,1%,1,77% диастереомера, Asp), выход 67,7% от теоретического. Масс спектр: Мрасч=560,7: М+1 набл=561. Т. пл. 182,17 (ДСК). Спектр ЯМР 1H (стандарт-ТМС, DMSOd6). , м.д.: 0,89 (м, 1 Н) , 0,94 (м, 1 Н), 1,0 (дт,2 Н), 1,15 (м, 2 Н), 1,06-1,3 (м, 4 Н), 1,36 (д, 9 Н),1,4-1,74 (м, 6 Н), 2,65 (м, 1 Н), 2,85 (м, 1 Н), 3,18BOCN(Et)Gly-(L)-Asp-(OBzl)-(L)-Cha-NH2 (приблизительно 1:12 вес/вес) и к полученному раствору при комнатной температуре добавляют трифторуксусную кислоту (ТФУК). Раствор перемешивают до тех пор, пока данные ВЭЖХ не укажут на окончание реакции (3-5 ч). Раствор концентрируют при температуре 40-45 С приблизительно до 1/2 объема. К полученному теплому раствору при температуре выше 40 С добавляют трет.-бутилметиловый эфир (при весовом отношении приблизительно 1:10 к BOCN(Et)Gly-(L)-Asp-(OBzl)-(L)-Cha-NH2). Смесь медленно охлаждают приблизительно до 5 С и перемешивают в течение 1 ч, чтобы обеспечить полную кристаллизацию. Полученные твердые вещества отфильтровывают и промывают охлажденным трет.-бутилметиловым эфиром. Твердые вещества сушат при пониженном давлении и анализируют на содержание ТФУК-N(Et)Gly(L)-Asp-(OBzl)-(L)-Cha-NH2 (ВЭЖХ, вес:вес). Обычно выход почти количественный, чистота более 95% (по площади). Масс спектр. Мрасч=460 (свободное основание), М+1 набл=461. Элементный анализ С 26 Н 37N4 О 7F3. Вычислено, %: H,N,F, С 54,35%. Найдено, %: С 53,82;. 29 Спектр ЯМР 1 Н (стандарт-ТМС, DMSOd6), , м.д.: 0,9 (м, 2 Н), 1,15 (т, 6 Н), 1,5 (м, 1 Н),1,5-1,8 (м, 6 Н), 2,65 (дд, 1 Н), 2,9 (м, 3 Н), 3,7 (с,2 Н), 3,9 (м, 2 Н), 4,2 (м, 1 Н), 4,75 (м, 1 Н), 5,1 (с,2 Н), 7,0 (с, 1 Н), 7,15 (с, 1 Н), 7,2 (с, 5 Н), 8,13 (д,1 Н), 8,7-8,8 (м, 3 Н). Спектр ЯМР 13 С (отчетливые сигналы,стандарт - ТМС, DMSO-d6), , м.д.: 10,76; 25,49: 25,68; 25,96; 31,66; 33,07: 33,36; 36,25; 38,59; 41,88; 47,02; 49,40; 50,47; 65,71; 127,81-128,34; 135,82; 165,10: 169,34; 173,79. Характерные примеры снятия защиты приведены в таблице А. Лабораторный пример Пример 1 Пример 2Gly-(L)-Asp-(OBzl)-(L)-Cha-NH2 (стадия 4 схемы II). Готовят суспензию приблизительно эквимолярных количеств ТФУКN-(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2, CBZ-PipBu и ТБТУ вEtOAc, ДМФА и воде (объемное отношение 100:8:4, приблизительное отношение общий объем:вес относительно ТФУКN-(Et)Gly-(L)Asp-(OBzl)-(L)-Cha-NH2 11:1). Суспензию охлаждают приблизительно до 0-10 С и добавляют приблизительно 3-4 эквивалента НММ. Смеси дают нагреться до комнатной температуры и перемешивают до тех пор, пока данные ВЭЖХ не укажут на окончание реакции (1-3 ч, за это время образуется раствор). Добавляют воду (2-3 объема изначально добавленной воды) и оставляют для разделения фаз. Водную фазу отделяют и органическую фазу промывают еще двумя порциями воды. Объединенные водные фракции подвергают обратной экстракции EtOAc и объединенные органические фазы промывают 25 %ным водным раствором хлорида натрия. Органическую фазу концентрируют при пониженном давлении приблизительно до 1/2 объема и добавляют трет.-бутилметиловый эфир (приблизительно 1/2 объема раствора). Эту смесь оставляют кристаллизоваться (несколько часов),твердые вещества отфильтровывают, промывают охлажденной смесью EtOAc и трет.бутилметилового эфира. Твердые вещества сушат при пониженном давлении. С помощью ВЭЖХ определяют весовое содержание CBZPipBu-N(Et)Gly-(L)-Asp-(OBzl)-(L)-Cha-NH2. Выход обычно превышает 80%, чистота составляет более 95% (по площади). В конкретном примере приведенной выше методики по общей методике стадии 4 и при использовании 7,25 г ТФУК-N(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2, получают 7,9 г CBZ-PipBu 001391N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида (стадия 5 схемы II). Готовят смесь CBZ-PipBu-N(Et)Gly-(L)Asp-(OBzl)-(L)-Cha-NH2, формиата аммония и 10% Pd/C в смеси спирт: вода, 20:1 (отношение объем:вес 10:1 относительно CBZ-PipBuN(Et)Gly-(L)-Asp-(OBzl)-(L)-Cha-NH2). Смесь нагревают до температуры 40-50 С и перемешивают до тех пор, пора данные ВЭЖХ не укажут на окончание реакции (1-2 ч). Смесь охлаждают до комнатной температуры и фильтруют для удаления катализатора. Полученный раствор нагревают до 40-45 С и добавляют ацетон(объем, приблизительно равный объему отфильтрованного раствора), затем оставляют охлаждаться до температуры 35-40 С. К смеси добавляют затравочные кристаллы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида. При охлаждении до комнатной температуры (несколько часов) из смеси кристаллизуется гигроскопичная форма N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида. Твердые вещества отфильтровывают в атмосфере азота, промывают ацетоном, сушат при пониженном давлении и определяют содержание гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин-4 ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида (анализ с помощью ВЭЖХ вес/вес). Выход составляет 85%, чистота 95% (по площади). В конкретном примере описанного выше способа при использовании общей методики стадии 5 из 5 г CBZ-PipBu-N(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2 получают 3,1 г гигроскопичной кристаллической формы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L) 31 аспартил]-(L)циклогексилаланинамида в виде твердого продукта (чистота 99,6%, по площади),стехиометрический выход 89,4%. Другие соединения, полученные в соответствии с приведенными выше примерами 1-5 и при использовании подходящих исходных материалов, представляют собой:N-[N-[N-(4-пиперидин-4-ил)бутаноил)-Nэтилглицил]аспартил]циклогексилметилаланина этиловый эфир. Пример 6. Получение 4-N-СВZпиперидона. Смесь 40 кг N-бензилоксикарбонилокси) сукцинимида и 26 кг (175 молей) 4 пиперидонНСl.Н 2 О в 38,8 кг воды и 88 кг ТГФ перемешивают при температуре 155 С до полного растворения (приблизительно 5 мин). При перемешивании добавляют NMM (22,8 кг)(экзотерма), поддерживая температуру при 20 С или ниже. Загрузку перемешивают при 205 С в течение 2,5 ч (окончание реакции определяют с помощью ВЭЖХ). Смесь разбавляют 115,2 кг МТБЭ и 38,8 кг воды и перемешивают при 205 С в течение 5 мин. Перемешивание прекращают, слоям дают разделиться, водный слой отделяют и выбрасывают. Органический слой промывают водой (129,6 кг х 2) (перемешивание 5 мин, разделение фаз, отделение/выбрасывание водного (нижнего) слоя). Органический слой промывают 5,2 кг NaCl в 46,8 кг воды (перемешивание 5 мин, разделение фаз, удаление и выбрасывание водного (нижнего) слоя). Органический слой обрабатывают 11,5 кг МgSO4 при перемешивании в течение 1 ч, затем смесь фильтруют. Реактор промывают 8 кг МТБЭ (фильтруют, объединяют с основным фильтратом; суммарное содержание воды в фильтрате 0,52%). Объем смеси уменьшают до половины путем отгонки при пониженном давлении при 30 С. Вакуум сбрасывают в азот, остаток охлаждают до 20 С (содержание воды в остатке 0,43%). Остаток разбавляют 57,6 кг МТБЭ, затем объем смеси снова уменьшают наполовину путем отгонки в вакууме при 30 С. Вакуум сбрасывают в азот, смесь охлаждают до 20 С(содержание воды в остатке -0,25%). Эту операцию повторяют еще 5 раз. Полученный остаток разбавляют 28,8 кг МТБЭ, перемешивают в течение 5 мин, затем определяют содержание воды и содержание 4-N-СВZ-пиперидона (вода 0,05%; анализ 4-N-СВZ-пиперидона вес./вес.33 22,66 вес.%, 35,36 кг, 155 молей, стехиометрический выход 88,6%). Пример 7. Получение PipBu. В токе N2 и при перемешивании получают раствор 53,5 кг 3-карбоксипропилтрифенилфосфонийбромида в 230,1 кг 1,2-диметоксиэтана. В течение 35 мин при температуре 2428 С добавляют трет.-бутоксид калия/ТГФ (20% вес., 141,8 кг в растворе). Смесь перемешивают при этой температуре в течение 0,5 ч (окончание реакции определяют с помощью ВЭЖХ). При перемешивании смесь охлаждают до 102 С, затем к смеси в течение 40 мин добавляют 96,45 кг (титр 1,15 мольных экв.) 4-CBZпиперидона в МТБЭ, так чтобы температура оставалась 122 С. Смесь перемешивают при этой температуре в течение 10 мин, затем нагревают до 202 С и перемешивают при этой температуре в течение 2 ч. К смеси при перемешивании добавляют раствор 22,5 кг концентрированной водной НСl в 245,6 кг воды так,чтобы температура смеси оставалась 202 С; конечное значение рН равно 0,5. Смесь экстрагируют при перемешивании 214,0 кг трет.бутилметилового эфира. Перемешивание прекращают, фазам дают разделиться и водный слой (нижний) отделяют и выбрасывают. Органическую фазу промывают 133,75 кг воды (перемешивание 5 мин, разделение фаз, отделение/выбрасывание водного (нижнего слоя), затем 10,7 кг 50 %-ного NaOH в 126,8 кг воды(перемешивание 10 мин, разделение слоев, отделение/выбрасывание органического (верхнего) слоя). Водный слой экстрагируют EtOAc(123,05 кг х 2) (перемешивание 5 мин, разделение слоев, отделение/выбрасывание органического (верхнего слоя). К водному слою при перемешивании добавляют 13,1 кг концентрированной водной НСl до рН 2,5-3,5 (конечное значение 2,82), затем смесь экстрагируют 123,05 кгEtOAc (перемешивание 5 мин, разделение фаз,отделение/выбрасывание водного (нижнего) слоя). Раствор в EtOAc промывают 133,75 кг воды (перемешивание 5 мин, разделение слоев,отделение/выбрасывание водного (нижнего) слоя) и анализируют на содержание вес/весCBZ-PipBuen [33,22 кг, 108 молей], стехиометрический выход 87,9%). Раствор PipBuen в EtOAc вместе с 6,6 кг 5% Pd/C (50% вес. воды) загружают при перемешивании в автоклав из нержавеющей стали,затем смесь нагревают до 552 С. Добавляют формиат калия (38,2 кг), растворенный в 66,4 кг воды так, чтобы температура реакционной смеси оставалась 552 С (приблизительно 30 мин). Смесь перемешивают при 552 С в течение 2 ч. За это время реакция заканчивается 34 фильтрату). Фильтрат загружают в новый реактор, охлаждают до 20-25 С, дают слоям разделиться, органический слой отделяют и выбрасывают. Водный слой подкисляют 52,1 кг концентрированной водной НСl до рН 2-3 (конечное значение 2,82), затем экстрагируют метиленхлоридом (129,5 кг х 4) (перемешивание 5 мин, разделение слоев, отделение/выбрасывание органического (нижнего) слоя). Водную фазу доводят до рН 6,1 путем добавления при перемешивании 17,85 кг 50%-ного водного раствораNaOH. Смесь фильтруют, получают 224 кг раствора, содержащего 17,6 кг (103 моля) 4-(3'карбоксипропил)пиперидина. Пример 8. Получение CBZ-PipBu. Раствор 224 кг 4-(3'-карбоксипропил) пиперидина в водном NaOH смешивают с 55,3 кг ТГФ и смесь при перемешивании охлаждают до 82 С. Поддерживая температуру 10 С, добавляют NММ (20,9 кг). По окончании добавления доводят температуру до 82 С, затем в течение 1 ч при температуре 15 С добавляют 25,7 кг 1-(бензилоксикарбонил)сукцинимида,растворенного в 49,8 кг ТГФ. При температуре 10-15 С реакция заканчивается через 3 ч (данные ВЭЖХ). Добавляют концентрированную водную НСl (29,9 кг) до рН 2,5-3,5 (конечное значение 3,3), затем добавляют 61,4 кг МТБЭ и смесь перемешивают в течение 5 мин. Перемешивание прекращают, слоям дают разделиться и отделяют водный (нижний) слой (отход). Слой МТБЭ промывают три раза порциями воды по 83,1 кг (время перемешивания 10 мин, затем 5 мин и 5 мин); водный слой в каждом случае отделяют и выбрасывают. Без перемешивания добавляют раствор 8,3 кг 50%-ного водногоNaOH в 95,7 кг воды, затем по окончании добавления смесь перемешивают в течение приблизительно 5 мин. Перемешивание прекращают, фазам дают разделиться, органический слой(верхний) отделяют и выбрасывают. Водный слой возвращают в реактор и экстрагируют трет.-бутилметиловым эфиром (38,4 кг х 2) (перемешивание 5 мин, разделение слоев, отделение и выбрасывание органического (верхнего) слоя). Эту операцию повторяют с использованием 18,5 кг трет.-бутилметилового эфира. Водный слой, возвращенный в реактор, подкисляют до рН 2,5-3,5 (конечное значение 3,37) с помощью 9,9 кг концентрированной водной НСl. Смесь экстрагируют с помощью 76,4 кг трет.бутилметилового эфира (перемешивание 5 мин,разделение слоев, отделение и отбрасывание нижнего (водного) слоя). Органический слой промывают (5 мин перемешивания) раствором 1,1 кг(нижнего) слоя), затем 41,5 кг воды (перемешивание 5 мин, разделение слоев, отделение и выбрасывание водного (нижнего) слоя). В реакторе создают пониженное давление, и летучие 35 растворители удаляют при 55 С до тех пор, пока не прекратится отгонка. Добавляют толуол (32,4 кг) и смесь перегоняют при атмосферном давлении до прекращения отгонки, при этом температура загрузки повышается до 90-95 С. Затем смесь охлаждают до 30-35 С, в реактор загружают гептан (56,85 кг) (две фазы), смесь нагревают до 90-95 С (одна фаза), затем снова охлаждают до 38-42 С. Добавляют зародыш кристаллизации CBZ-PipBu. Из смеси продукт кристаллизуется в течение 1 ч. Твердый продукт отфильтровывают и промывают 19,35 кг смеси толуол:гептан (1:1), затем 33,4 кг гептана. Полученный на фильтре осадок сушат в вакууме при 40 С (до 0,13% потери при сушке), получают 22,4 кг (72,96 моля, стехиометрический выход по 4-пиперидону 42%) CBZ-PipBu. Пример 9. Получение CBZ-PipBuen. К суспензии 82 г 3-карбоксипропилтрифенилфосфонийбромида в 407 мл 1,2-диэтоксиэтана при 14 С в течение 25 мин добавляют 220 г 20%-ного (вес.) трет.-бутоксида калия в тетрагидрофуране, при сохранении температуры реакционной смеси 24-28 С. Смесь перемешивают в течение 1 ч, охлаждают до 10 С, затем в течение 30 мин при охлаждении добавляют 52,5 г 4N-CBZ-пиперидона в 246 мл трет.бутилметилового эфира. По окончании добавления смесь перемешивают при 12 С в течение 10 мин, затем нагревают до 20 С и перемешивают еще 30 мин. Реакционную смесь обрабатывают 410 мл 1 н. водной НСl в течение 10 мин,разбавляют 328 мл трет.-бутилметилового эфира и затем фазам дают разделиться. Органическую фазу промывают 205 мл воды, затем 210 мл 1 н. водного раствора NaOH. Слой NaOH,который содержит продукт, собирают, промывают тремя порциями по 189 г этилацетата, подкисляют до рН 3,48 с помощью концентрированной НСl, затем экстрагируют 189 мл этилацетата. Слой этилацетата отделяют, промывают 211 мл воды, затем сушат в течение 30 мин над 10 г МgSО 4, фильтруют и концентрируют в вакууме. Масляный остаток (50,7 г) кристаллизуют из смеси толуол/гептан, получают 29,46 г(м, 4 Н), 5,15 (с, 2 Н), 5,2 (м, 1 Н), 7,33 (д, 5 Н). Спектр ЯМР 13 С (отчетливые сигналы,стандарт - ТМС, CDCl3), , м.д.: 22,43; 28,2; 34,26; 35,66; 44,88; 45,74; 67,20; 122,02; 127,83; 127,95; 128,45; 128,69; 128,90; 136,17; 136,72; 155,34; 178,39. Пример 10. Получение CBZ-PipBuenN(Et)Gly-(L)-Asp-(OBzl)-(L)-Cha-NH2 (вариант стадии 4 cхемы II). В снабженную кожухом колбу объемом 1 л загружают CBZ-PipBuen (70 г, 0,23 моля) и 36 ДМФА (230 мл) и охлаждают при перемешивании до 0 С. Затем в виде одной порции добавляют ТБТУ (74,9 г, 0,23 моля). При температуре 0 С начинают добавление DIPEA (61,9 г, 0,61 моля). Через 45 мин добавляют трифторацетатN(Et)Gly-(L)-Asp-(OBzl)-(L)-Cha-NH2 (138,7 г,0,24 моля) в виде раствора в ДМФА (230 мл). Путем добавления DIPEA (45 мл) устанавливают рН 7-8 и смеси дают нагреться до комнатной температуры. Через 2 ч реакция завершается(2,5 л) и экстрагируют EtOAc (1 л). Водную фазу повторно экстрагируют EtOAc (0,3 л). Органические слои собирают, промывают водным раствором лимонной кислоты (5% вес., 2 по 1 л), промывают водным раствором NаНСО 3 (5% вес. 2 х 1 л) и промывают водой (2 л). СлоиEtOAc переносят в колбу объемом 2 л и, чтобы вызвать кристаллизацию, добавляют при перемешивании гептан (500 мл). Твердые вещества фильтруют на воронке Бюхнера, промывают смесью EtOAc/гептан (2:1 об./об., 1 л) и сушат до постоянной массы, получают CBZ-PipBuenN(Et)Gly-(L)-Asp-(OBzl)-(L)-Cha-NH2 (143,2 г,0,19 моля, выход 83%). Элементный анализ: C41H57N5O7 Вычислено, %: С 66,02%. Найдено, %: С 65,53: Н, N. Масс спектр: Мрасч=745,91, М+1 набл=746 Спектр ЯМР 1H (стандарт - ТМС, СDСl3) м.д.: 0,86 (кв.д, 1 Н), 0,98 (кв.д, 1 Н), 1,16 (т,2 Н), 1,24 (дт, 6 Н), 1,37 (м, 1 Н), 1,64-1,78 (м, 4 Н),1,86 (кв.д, 1 Н), 2,2 (ушир. д, 4 Н), 2,35 (м, 1 Н),2,4 (м, 2 Н), 2,74 (дд, 1 Н), 3,07 (м, 4 Н), 3,52 (д,1 Н), 3,85 (д, 1 Н), 4,12 (кв, 1H), 4,49 (кв.д, 1 Н),4,68 (дт, 1H), 5,07 (д, 1H), 5,14 (с, 1H), 5,16 (д,1H), 5,22 (т, 2 Н), 6,45 (с, 1H), 7,28 (д, 1H), 7,26(с, 5 Н), 7,35 (с, 5 Н), 7,56 (д, 1H). Спектр ЯМР 13 С (стандарт - ТМС, CDCl3) м.д.: 14,15; 22,68; 24,95; 25,61; 26,03; 26,45; 28,20; 31,71; 32,89; 33,80; 33,89; 34,00; 35,63; 38,37; 44,79; 45,13; 45,65; 50,23; 51,34; 60,40; 66,87; 67,06; 76,50; 77,13; 77,77; 122,46; 126,88; 127,80-128,60; 135,15, 155,19; 170,11; 170,20; 171,61; 173,76; 175,35. Пример 11. Получение гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида (вариант стадии 5 схемы II). В снабженную кожухом колбу объемом 5 л загружают(200 proof (100%), 1260 мл), i-PrOH (70 мл) и воду (ДВ, 70 г). Смесь нагревают до 40-50 С и перемешивают до тех пор, пока данные ВЭЖХ не укажут на окончание реакции (5 ч). Смесь охлаждают до комнатной температуры и фильтруют для удаления катализатора. Полученный раствор нагревают до 40-50 С и добавляют аце 37 тон (объем, приблизительно равный объему отфильтрованного раствора), затем оставляют охлаждаться до температуры 35-40 С. К смеси добавляют затравочные кристаллы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида и при охлаждении до комнатной температуры (несколько часов) из смеси кристаллизуется гигроскопичная форма N-[N-[N-(4-пиперидин-4 ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида. Твердые вещества отфильтровывают на воронке Бюхнера в атмосфере азота, осадок на фильтре промывают ацетоном и сушат на воздухе до постоянной массы,получают N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамид (84,3 г, 0,16 моля, выход 84,4%, чистота 95%). Пример 12. Объединенный способ получения трифторацетата N(Et)Gly-(L)-Asp-(OBzl)(L)-Cha-NH2 (вариант стадий 1-3 схемы II). В снабженную термопарой колбу объемом 500 мл загружают BOC-N(Et)Gly, (20,3 г, 0,1 моля), N-гидроксисукцинимид (11,5 г, 0,1 моля) и дихлорметан (200 мл). Смесь перемешивают при умеренной скорости и к полученному раствору добавляют в одну порцию ДЦК (20,6 г,0,1 моля) в виде твердого вещества. Раствор перемешивают в течение 1 ч, наблюдается выделение небольшого количества тепла (температура поднимается от 20 С до 28 С) и образование осадка ДЦМ. Полученную суспензию фильтруют под вакуумом на воронке Бюхнера с фильтровальной бумагой Whatman 1. Осадок на фильтре промывают дихлорметаном (25 мл х 2). Фильтраты возвращают в исходную колбу объемом 500 мл и затем последовательно добавляют (L)-Asp(OBzl) (22,3 г, 0,1 моля), NMM(33,8 мл, 0,3 моля) и ДМФА (80 г, 1,01 моля). После перемешивания в течение 2 ч при комнатной температуре образование BOC-N(Et)Gly(L)-Asp-(OBzl) заканчивается (данные ВЭЖХ). Реакционную смесь выливают в делительную воронку, содержащую ледяную воду (100 мл). Смесь подкисляют НСl (36%, 25 мл) до рН 1. Слои разделяют, слой дихлорметана промывают ледяной водой (100 мл) и фазы разделяют (рН водной фазы 3-4). Слой дихлорметана возвращают в исходную колбу объемом 500 мл, в которую последовательно в одну порцию в виде твердых веществ добавляют H2N-(L)-Cha-NH2(17 г, 0,1 моля), N-гидроксисукцинимид (11,5 г,0,1 моля), ДЦК (20,6 г, 0,1 моля). После перемешивания в течение 2 ч при комнатной температуре образование BOC-N(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2 заканчивается(данные ВЭЖХ). ДЦМ отделяют путем фильтрования под вакуумом на воронке Бюхнера с фильтровальной бумагой Whatman 1. Осадок на фильтре промывают дихлорметаном (25 мл х 2). Фильтрат переносят в делительную воронку и 38 промывают деионизированной водой (200 мл),содержащей N-метилморфолин (15 мл, рН 8-9). Фазы разделяют, и слой дихлорметана повторно промывают деионизированной водой (150 мл х 2). Дихлорметановую фазу промывают 150 мл 1 н. НСl (рН 1). Фазы разделяют, и слой дихлорметана промывают деионизированной водой(200 мл, рН 3). Раствор BOC-N(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2 в дихлорметане возвращают в чистую колбу объемом 500 мл и добавляют ТФУК (100 мл). После перемешивания в течение 2 ч при комнатной температуре образование ТФУКNH(Et)Gly-(L)-Asp-(OBzl)-(L)-Cha-NH2 заканчивается (данные ВЭЖХ). Реакционную смесь перегоняют в вакууме для удаления дихлорметана и большей части трифторуксусной кислоты, затем для того, чтобы вызвать кристаллизацию, добавляют трет.-бутилметиловый эфир (500 мл) и затравочные кристаллы. Смесь фильтруют под вакуумом на воронке Бюхнера с фильтровальной бумагой Whatman 1. Остаток на фильтре промывают трет.-бутилметиловым эфиром (25 мл х 2) и сушат на воздухе, получают ТФУКNH(Et)Gly-(L)-Asp-(OBzl)-(L)-ChaNH2 (46,8 г, выход 81,5%) в виде белого твердого вещества (чистота 97%,0,2% диастереомера, D-Asp). Пример 13. Получение стабильной негигроскопичной кристаллической формы N-[N-[N(4-пиперидин-4-ил)бутаноил)-N-этилглицил](L)-аспартил]-(L)циклогексилаланинамида. Способ А. Статическая конверсия. Гигроскопичную кристаллическую формуN-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида (7,45 кг) измельчают в молотковой мельнице. Полученный твердый продукт (7,35 кг) помещают в сушильный лоток из нержавеющей стали (90 х 28 см) и лоток накрывают перфорированной алюминиевой фольгой. Лоток затем герметизируют в увлажняющей печи(LUNAIRE Humidity Cabinet модельСЕО 941W-3); печь держат герметично закрытой от начала процесса конверсии форм до извлечения образца. Печь устанавливают при OВ 40% и температуре 60 С, образец выдерживают при этих условиях в течение 1 ч. Увлажняющую печь затем устанавливают при OВ 80%/60 С и выдерживают при этих условиях в течение 12 ч. Образец извлекают через 18 ч выдерживания при 80% ОВ/60 С и анализируют с использованием порошковой рентгенографии для оценки конверсии в негигроскопичную кристаллическую форму N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида. Увлажняющую печь открывают и устанавливают при условиях 40%OВ/60 С и выдерживают в течение 2 ч. Печь снова устанавливают при обычных условиях,затем поддон извлекают из печи и получают негигроскопичную кристаллическую форму N 39[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида (7,2 кг, выход 96,6%). Конверсию форм подтверждают с помощью диаграммы порошковой рентгенографии (фиг. 1). Данные порошковой рентгенографии также представлены в таблице в виде функции увеличивающегося угла дифракции (2), соответствующего межплоскостному расстоянию кристалла (d) в ангстремах, импульсам в секунду (И/с) и относительной интенсивности пика (%) (таблица 1).N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида (50 г) помещают в мерный цилиндр на 400 мл (высота слоя 6 см) на круглой подставке,снабженный механической мешалкой. Устройство помещают в печь с контролируемой влажностью (LUNAIRE Humidity Cabinet модельСЕО 941W-3). Скорость перемешивания состав 40 ляет 275 об/мин, температуру 60 С и OВ 40 % устанавливают в течение 30 мин. Соединение выдерживают при указанных условиях в течение 1 ч, затем в течение 45 мин условия изменяют на 80 % ОВ/60 С. Соединение выдерживают при этих условиях в течение 16 ч, затем печь снова устанавливают на 40 % ОВ/60 С и выдерживают в течение 3,25 ч. Затем устанавливают обычные условия (высота слоя 4 см),извлекают соединение из цилиндра, получают негигроскопичную кристаллическую форму N[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида (выход 95%). Конверсию подтверждают с помощью порошковой рентгенографии (фиг. 2). Данные порошковой рентгенографии также представлены в таблице в виде функции увеличивающегося угла дифракции (2), соответствующего межплоскостному расстоянию кристалла (d) в ангстремах , импульсам в секунду (И/с) и относительной интенсивности пика намида (370 г) загружают в колбу роторного испарителя объемом 2 л. Колбу устанавливают на роторный испаритель (Heidolph UV-2002) и опускают в предварительно нагретую (58 С) баню (Heidolph MR 2002). В устройстве создают вакуум 60 мБар с помощью вакуумного насоса(Divatrion DV1), затем вакуум сбрасывают под контролем во влажную атмосферу, созданную в отдельной, нагретой, содержащей воду колбе. Прием влажной атмосферы контролируют с помощью устройства, регулирующего влажность (Vausalo Humiditique and TemperatureTraumettor) так, чтобы внутри устройства получить относительную влажность 79% (внутреннее давление 130-180 мБар). Затем сосуд роторного испарителя вращают при скорости 145-160 оборотов в минуту в течение 5 ч, поддерживая температуру нагревающей бани приблизительно 60 С и относительную влажность в сосуде при 71-79%. Затем вакуум сбрасывают в азот, сосуд и его содержимое охлаждают до комнатной температуры, продукт извлекают и получают негигроскопичную кристаллическую форму N[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида. Аналогично обрабатывают вторую загрузку гигроскопичной кристаллической формыN-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида (317 г) с получением негигроскопичной кристаллической формы N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида. Конверсию подтверждают с помощью диаграммы порошковой рентгенографии (фиг. 3). Две партии вместе дают 667 г негигроскопичной кристаллической формы N-[N-[N-(4-пиперидин-4-ил)бутаноил)N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида (97% выход). Конверсию подтверждают с помощью диаграммы порошковой рентгенографии (фиг. 3). Данные порошковой рентгенографии также представлены в таблице в виде функции увеличивающегося угла дифракции (2), соответствующего межплоскостному расстоянию кристалла (d) в ангстремах, импульсам в секунду (И/с) и относительной интенсивности пика (%) (таблица 3). Пример 14. Диаграммы порошковой рентгенографии образца N-[N-[N-(4-пиперидин-4 ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида в виде его гигроскопичной кристаллической формы и его конвертированной негигроскопичной кристаллической формы. Образец гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин-4-ил)бутаноил)N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида получают в соответствии с методикой примера 5 или 11 и превращают в соответствующую негигроскопичную кристаллическую форму по методике примера 13. Диаграммы порошковой рентгенографии для гигроскопичной кристаллической формы и негигроскопичной кристаллической формы также представлены в таблицах в виде функции увеличивающегося угла дифракции (2), соответствующего межплоскостному расстоянию кристалла (d) в ангстремах , импульсам в секунду (И/сек) и относительной интенсивности пика Пример 15. Изотермические микрокалориметрические опыты на гигроскопичной и не гигроскопичной кристаллических формах N-[N[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида. Изотермические микрокалориметрические опыты на гигроскопичной и негигроскопичной кристаллических формах N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида проводят на приборе Thermometric Thermal ActivityMonitor (ТАМ). Конверсию твердого состояния различных кристаллических форм изучают путем воздействия на эти формы условий при различной влажности или паров различных растворителей при различных температурах. Для получения условий с различной важностью используют насыщенные солевые растворы, а именно: КСl (80% OB), NaCl (75% OВ) и NaBr(65% OВ). В стеклянной ампуле прибора ТАМ взвешивают приблизительно по 100 мг соответствующей формы и внутрь ампулы помещают микрогидростат, содержащий насыщенные солевые растворы (с избытком твердого вещества) или органический растворитель. Ампулу герметично закрывают, уравновешивают при температуре опыта и устанавливают в приборе ТАМ в положение измерения. Аналогичную систему,содержащую промытый морской песок вместо соединения в форме, подвергающейся испытанию, помещают на контрольную сторону прибора. Изменяют выходную мощность как функцию времени (фиг. 6-8). Пример 16. Изотермы сорбции влаги гигроскопичной и негигроскопичной кристаллических форм N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида. Изотермы сорбции влаги гигроскопичной и негигроскопичной кристаллических форм N[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида получают на рычажных весах VTI MB 300G, проградуированных в процентах влажности. Опыты проводят с использованием приблизительно 15 мг кристаллической формы, увеличивая и уменьшая OB. При этом измеряют увеличение веса (на каждой равновесной стадии) как функцию OВ (фиг. 9). В другом варианте опыта кристаллическую форму выдерживают при постоянной влажности и определяют увеличение веса как функцию времени. Соединение формулы II обладает полезной фармакологической активностью и, таким образом, вводится в фармацевтические композиции и используется для лечения пациентов, страдающих от различных патологических состояний. 45 Настоящее изобретение также относится к способу лечения пациентов, страдающих от или подверженных состояниям, которые могут быть улучшены или предупреждены при введении ингибиторов агрегации тромбоцитов, за счет ингибирования связывания фибриногена и свертывания крови. Кроме того, настоящее изобретение относится к способу профилактики или лечения тромбоза, ассоциированного с некоторыми болезненными состояниями, такими как инфаркт миокарда, удар, периферическое артериальное заболевание и диссеминированное внутрисосудистое свертывание, у людей и других млекопитающих. Следует понимать, что под "лечением" в данном случае подразумевается профилактическая терапия, а также лечение выраженных состояний. Настоящее изобретение также включает в свой объем фармацевтические композиции, которые содержат фармацевтически приемлемое количество, по меньшей мере, одного соединения формулы I в сочетании с фармацевтически приемлемым носителем или наполнителем. На практике соединения и лекарственные композиции в соответствии с настоящим изобретением могут быть введены любым приемлемым способом, например, локально, путем ингаляции, парентерально, ректально или перорально, но предпочтительным является пероральный способ. Соединение формулы II может быть представлено в лекарственных формах, обеспечивающих введение с помощью наиболее предпочтительного способа, и настоящее изобретение также относится к фармацевтическим композициям, содержащим, по меньшей мере, одно заявляемое соединение, которые приемлемы для использования в медицине или ветеринарии. Эти композиции могут быть получены с помощью обычных способов с использованием фармацевтически приемлемых адъювантов или наполнителей. К адъювантам относятся, кроме прочего, разбавители, стерильная водная среда и различные нетоксичные органические растворители. Композиции могут быть получены в форме таблеток, пилюль, капсул, гранул, порошков,водных растворов или суспензий, растворов для инъекций, эликсиров, сиропов и др., и могут содержать один или несколько агентов, выбираемых из группы, включающей подслащивающие вещества, корригенты, красители, стабилизаторы и консерванты, чтобы можно было приготовить фармацевтически приемлемые композиции. Выбор растворителя и содержание активного вещества в растворителе обычно осуществляется с учетом растворимости и химических свойств продукта, конкретного способа введения и условий, которые необходимо соблюдать в фармацевтической практике. Например, при 46 изготовлении таблеток могут быть использованы наполнители, такие как лактоза, цитрат натрия, карбонат кальция, дикальций фосфат, диспергирующие агенты, такие как крахмал, альгиновые кислоты и некоторые комплексные силикагели, смешанные со смазывающими веществами, такими как стеарат магния, лаурилсульфат натрия и тальк. Для получения капсул хорошо использовать лактозу и высокомолекулярные полиэтиленгликоли. При получении водных суспензий можно добавлять эмульгирующие агенты или агенты, которые ускоряют образование суспензии. Могут быть также использованы разбавители, такие как сахароза,этанол, полиэтиленгликоль, пропиленгликоль,глицерин и хлороформ или их смеси, а также другие материалы. Для парентерального введения можно использовать эмульсии, суспензии или растворы соединений настоящего изобретения в растительном масле, например, в кунжутном масле,арахисовом масле, оливковом масле, или в водно-органических растворах, таких как смеси воды и пропиленгликоля, в органических эфирах, подходящих для инъекций, таких как этилолеат, а также стерильные водные растворы фармацевтически приемлемых солей продуктов в соответствии с настоящим изобретением. Растворы солей продуктов настоящего изобретения также могут быть использованы для введения путем внутримышечных или подкожных инъекций. Для внутривенных инъекций могут быть использованы водные растворы, содержащие соли в чистой дистиллированной воде, при следующих условиях: значение рН раствора соответствующим образом подогнано; растворы должны содержать разумный буферный агент; растворы изотоничны благодаря введению достаточного количества глюкозы или хлористого натрия; растворы стерилизованы путем нагревания, облучения и/или микрофильтрации. Также можно использовать местное введение с помощью гелей (на водной или спиртовой основе), кремов или мазей, содержащих соединения настоящего изобретения. Заявляемые соединения могут быть введены в гели или матричные основы для применения в виде пластыря, который должен обеспечивать контролируемое введение соединения через кожный барьер. Твердые композиции для ректального введения включают свечи, приготовленные в соответствии с известными способами и содержащие, по меньшей мере, одно соединение формулы II. Процентное содержание активного ингредиента в композиции в соответствии с настоящим изобретением может меняться так, чтобы обеспечивать соответствующую дозировку лекарства. Очевидно, что несколько стандартных лекарственных форм может быть введено приблизительно в одно и то же время. Необходимая доза может быть определена врачом или квали 47 фицированным медицинским персоналом и зависит от желаемого терапевтического эффекта,способа ведения и продолжительности лечения,а также от состояния пациента. Схема приема лекарственного средства при осуществлении способа настоящего изобретения должна быть такова, чтобы она обеспечивала максимальное терапевтическое действие до тех пор, пока наблюдается улучшение, а с этого момента - эффективный минимальный уровень, который дает облегчение. Обычно пероральная доза может составлять приблизительно от 0,1 мг/кг до 100 мг/кг, предпочтительно от 0,1 до 20 мг/кг, и наиболее предпочтительно приблизительно от 1 до 20 мг/кг, а внутривенная доза - приблизительно от 0,1 мкг/кг до 100 мкг/кг, предпочтительно от 0,1 до 50 мг/кг. В каждом конкретном случае дозировка лекарства определяется с учетом фактором, присущих данному пациенту,таких как возраст, вес, общее состояние здоровья и другие параметры, которые могут влиять на эффективность заявляемого соединения. Кроме того, соединение формулы II может вводиться так часто, как это необходимо для получения желаемого терапевтического действия. У некоторых пациентов может наблюдаться быстрая реакция на более высокие или более низкие дозы и могут быть достаточны более слабые поддерживающие дозы. Для других пациентов может быть необходимо проводить долговременное лечение при норме от 1 до 4 пероральных доз в день, предпочтительно от одного до двух доз в день, в соответствии с физиологическими потребностями каждого конкретного пациента. Обычно активный продукт может быть введен перорально 1-4 раза в день. Конечно, для других пациентов может быть необходимо прописывать не более одной или двух доз в день. Соединения формулы II проявляют значительную фармакологическую активность при проведении описанных в литературе опытов. Результаты этих опытов, как полагают, соответствуют фармакологической активности для человека и других млекопитающих. Ниже приведены результаты фармакологических испытаний in vitro и in vivo, которые характерны для соединения формулы II. В приведенных ниже фармакологических испытаниях оцениваются ингибирующая активность соединения формулы II на опосредуемую фибриногеном агрегацию тромбоцитов, связывание фибриногена с активированными тромбином тромбоцитами, а также ex-vivo ингибирование индуцированной АДФ агрегации тромбоцитов. Результаты этих испытаний коррелируют с ингибирующими свойствами in vivo соединения формулы II. Оценка агрегации тромбоцитов основана на описании, представленном в работе Blood 66(4), 946-952 (1985). Оценка связывания фибриногена соответствует по существу методике, 001391Sci, USA 83, 5708-5712 (1986) и Plow E.F., et al.,Proc. Natl. Acad. Sci, USA 82, 8057-8061 (1985). Изучение ех-vivo ингибирования индуцированной АДФ агрегации тромбоцитов основано на способе Zucker, "Platelet Aggregation MeasuredEnzymology 169, 117-133 (1989). Оценка агрегации тромбоцитов Приготовление фиксированных активированных тромбоцитов Тромбоциты выделяют из концентратов тромбоцитов человека с использованием гельфильтрующей методики, описанной Marguerie(1983). Тромбоциты суспендируют при концентрации 2 х 108 клеток/мл в модифицированном,не содержащем кальция буфере Тирода, содержащем 127 мМ хлористого натрия, 2 мМ хлористого магния, 0,42 мМ Na2HPO4, 11,9 мМ NaHCO3, 2,9 мМ КСl, 5,5 мМ глюкозы, 10 мМ(HSA). Эти промытые тромбоциты активируют путем добавления человеческого a-тромбина при конечной концентрации 2 ед/мл с последующим добавлением ингибитора тромбина I2581 при конечной концентрации 40 мкМ. К активированным тромбоцитам добавляют параформальдегид при конечной концентрации 0,50% и выдерживают при комнатной температуре в течение 30 мин. Фиксированные активированные тромбоциты затем собирают путем центрифугирования в течение 15 мин при 650 хg. Пеллеты тромбоцитов промывают 4 раза указанным буфером Тирода с 0,35% HSA и снова суспендируют при концентрации 2 х 108 клеток/мл в том же буфере. Оценка агрегации тромбоцитов Фиксированные активированные тромбоциты выдерживают в течение одной минуты с выбранной дозой соединения, испытываемого в качестве ингибитора агрегации тромбоцитов, и агрегацию инициируют путем добавления фибриногена человека при конечной концентрации 250 мкг/мл. Для регистрации агрегации тромбоцитов используют устройство для определения профиля агрегации тромбоцитов Model PAP-4. Степень ингибирования агрегации выражают в процентах от скорости агрегации, наблюдаемой в отсутствие ингибитора. Затем для каждого соединения рассчитывают значение IС 50, то есть, количество ингибитора, необходимого для уменьшения скорости агрегации на 50% (см.,например. Plow E.F., et al., Proc. Natl. Acad. Sci,USA 82, 8057-8061 (1985. Оценка связывания фибриногена Тромбоциты промывают от составляющих плазмы с использованием технологии градиента плотности альбумина, описанной Walsh P.N. с 49 соавторами (Br. J. Haematol. 281-296 (1977 и модифицированной Trapani-Lombardo V. с соавторами, (J. Clin. Invest. 76. 1950-1958 (1985. В каждой экспериментальной смеси тромбоциты в модифицированном буфере Тирода (Ruggeri Z. М. et аl., J. Clin. Invest., 72, 1-12 (1983) стимулируют с помощью -тромбина человека при 2225 С в течение 10 мин (3,125 х 1011 тромбоцитов на литр и тромбин в концентрации 0,1 единицы Национального института человека (США)/мл). Затем добавляют гирудин в 25-кратном избытке(единиц/единиц) в течение 5 мин перед добавлением меченного изотопом 125I фибриногена и испытуемого соединения. После этого добавления конечное количество тромбоцитов в смеси составляет 1 х 1011/л. После выдерживания в течение еще 30 мин при 22-25 С связанные и свободные лиганды разделяют путем центрифугирования при 12000 х g в течение 4 мин 50 мкл смеси через 300 мкл 20%ной сахарозы. Пеллеты тромбоцитов отделяют от остатка смеси для определения радиоактивности связанных тромбоцитов. Неспецифическое связывание измеряют в смесях, содержащих избыток немеченного лиганда. При анализе кривых связывания по методу Scatchard неспецифическое связывание получают в виде подогнанного параметра из изотермы связывания с помощью компьютерной программы (MunsonP.J., Method Enzymol., 92, 542-576 (1983. Для определения концентрации каждого соединения, необходимой для 50 %-ного ингибирования связывания фибриногена с активированными тромбином тромбоцитами (IC50), каждое соединение испытывают при 6 или более концентрациях с фибриногеном, меченным 125I в концентрации 0,176 мкмоля/л (60 мкг/мл). IС 50 определяют из графика зависимости остаточного связывания фибриногена и логарифма концентрации соединения.ex-vivo Ингибирование индуцированной АДФ агрегации тромбоцитов Протокол опыта Контрольные образцы крови получают за 5-10 мин до введения испытуемого соединения нечистокровным собакам весом от 10 до 20 кг. Соединения вводят внутрижелудочно через водный зонд или перорально в желатиновых капсулах. Образцы крови (по 5 мл) отбирают через 30-ти минутные интервалы в течение 3 ч и через 6, 42 и 24 ч после введения лекарства. Каждый образец отбирают с помощью венепункции головной вены и собирают непосредственно в пластиковый шприц, содержащий одну часть 3,8 %-ного тринатрий цитрата на девять частей крови.Ex-vivo агрегация тромбоцитов собак Образцы крови центрифугируют при 1000 об/мин в течение 10 мин с получением обогащенной тромбоцитами плазмы (ОТП). После извлечения ОТП образец центрифугируют еще 10 мин при 2000 об/мин с получением плазмы с низким содержанием тромбоцитов (ПНТ). Количество тромбоцитов в ОТП определяют с помощью счетчика Coulter Counter (Соulter Electronics, Hialeah, FL). Если концентрация тромбоцитов в ОТП составляет больше 300000 тромбоцитов/мкл, то ОТП разбавляют с помощью ПНТ до содержания тромбоцитов до 300000 тромбоцитов/мкл. Затем аликвоты ОТП (250 мкл) помещают в кюветы с диффузионным покрытием кремнием (7,25 х 55 мм, Bio/Data Corp.,Horsham, PA). Затем к ОТП добавляют эпинефрин (конечная концентрация 1 мкМ), плазму выдерживают в течение 1 мин при 37 С. К ОТП добавляют стимулятор агрегации тромбоцитов,АДФ, при конечной концентрации 10 мкм. За агрегацией тромбоцитов наблюдают спектрофотометрически с использованием светотрансмиссионного агрегометра (Bio/Date Platelet Aggregation Profiler, Model PAP-4, Bio/Date Corp., Horsham, PA). Для испытуемого соединения дважды записывают скорость изменения (наклон) пропускания света и максимум светопропускания (максимальная агрегация). Данные по агрегации тромбоцитов представляют как процент уменьшения (среднее значение + среднеквадратичная ошибка) наклона или максимальной агрегации в сравнении с данными, полученными в случае контрольной ОТП, которую получают из образцов крови, отобранных до введения испытуемого соединения. Соединения формулы II проявляют высокую активность в описанных выше опытах и рассматриваются как соединения, которые могут быть использованы для лечения и профилактики тромбоза, ассоциированного с некоторыми заболеваниями. Антитромботическая активность в опытах по оценке ex-vivo агрегации тромбоцитов собаки предполагает наличие такой же активности в организме человека (см.,например, Catalfamo, J.L, Dodds W. Jean, "Isolation of Platelets from Laboratory Animals", MethodEnzymol. 169, Part A., 27, (1989. Результаты тестирования соединения формулы II с помощью описанных выше методов представлены ниже в таблице 6. Также в этой таблице представлены результаты сравнительных испытаний для 4-4-(пиперидил)бутаноилглициласпартилтриптофана, то есть, соединения, описанного в европейской патентной публикации 0479481. Ингибирование агрегации фиксированных тромбоцитов (IC50 мкМ) Квалифицированный в данной области специалист легко поймет, что настоящее изобретение хорошо адаптировано для реализации целей настоящего изобретения и получения названных результатов и преимуществ. Соединения, композиции и способы, описанные здесь,представлены в качестве предпочтительных вариантов осуществления изобретения или для пояснения изобретения и не ограничивают его объем. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы VII где В представляет собой алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил; Е 1 представляет собой Н; Е 2 представляет собой -углеродную боковую цепь природной -аминокислоты, Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил,гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил или Е 1 и Е 2 вместе с атомом азота и атомом углерода, через которые Е 1 и Е 2 связаны, обра Фиг. 1ex-vivo ингибирование индуцированной АДФ агрегации тромбоцитов% ex-vivo ингибирования агрегации тромбоцитов После перорального приема 1 ч 3 ч 6 ч 12 ч 24 ч 100 100 100 98 50 53 20 зуют 4-, 5-, 6- или 7-членное азациклоалкановое кольцо;R1 и R2 независимо друг от друга представляют собой Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил; р принимает значения от 1 до 4;p1 представляет собой неустойчивую при гидрировании защитную группу для кислоты; иp2 представляет собой неустойчивую в кислоте защитную группу для аминогруппы. 2. Соединение по п.1, где В представляет собой алкил; Е 1 представляет собой Н; Е 2 представляет собой циклоалкилалкил;
МПК / Метки
Метки: антитромботические, азациклоалкилалканоильные, пептиды, псевдопептиды
Код ссылки
<a href="https://eas.patents.su/29-1391-antitromboticheskie-azacikloalkilalkanoilnye-peptidy-i-psevdopeptidy.html" rel="bookmark" title="База патентов Евразийского Союза">Антитромботические азациклоалкилалканоильные пептиды и псевдопептиды</a>
Пептиды антисекреторного фактора, регулирующие патологические изменения проницаемости

Номер патента: 1201
Опубликовано: 25.12.2000
Авторы: Лённрот Ивар, Лённрот Кристина, Йоханссон Эва, Ланге Стефан, Йеннише Эва
МПК: A61K 38/22, A23K 1/165, A61P 29/00...
Метки: проницаемости, регулирующие, патологические, пептиды, фактора, изменения, антисекреторного
Формула / Реферат:
1. Рекомбинантный антисекреторный фактор, имеющий аминокислотную последовательность SEQ ID No:1, или его гомолог, обладающий, по существу, аналогичной биологической активностью. 2. Фрагмент рекомбинантного антисекреторного фактора по п.1, содержащий, по крайней мере, 7-8 аминокислот и обладающий, по существу, аналогичной биологической активностью. 3. Фрагмент по п.2, состоящий из аминокислот N 35-42 аминокислотной последовательности...
Предыдущий патент: Устройство регулировки фазы
Следующий патент: Синтез бисиндолилмалеимидов
Случайный патент: Автоматическое регулировочное устройство подачи топлива для автомобилей