Экспрессионный вектор и способ продуцирования высоких уровней белков
Номер патента: 11619
Опубликовано: 28.04.2009
Авторы: Сингх Арун К., Гоел Ашиш, Мендиратта Санджив К.


















Формула / Реферат
1. Способ высокой экспрессии представляющих интерес белков с использованием экспрессионного вектора, который включает, по меньшей мере, следующие регуляторные элементы:
a) промотор CMV или его функциональные варианты,
b) интрон,
c) TPL или его функциональные варианты,
d) VA гены или функциональные варианты, и
e) последовательность полиаденилирования гормона роста крупного рогатого скота или функциональные варианты.
2. Способ по п.1, где представляющий интерес белок представляет собой рекомбинантный эритропоэтин.
3. Способ по п.1 для высокой экспрессии гибридного белка TNFR-IgGFc.
4. Способ по п.1 для высокой экспрессии ритуксимаба, трастузумаба, бивакузумаба или других моноклональных антител.
5. Способ по п.1 для высокой экспрессии генов, пригодных для экспрессии в клетках млекопитающих.
6. Экспрессионный вектор млекопитающих, определенный в п.1, который включает по меньшей мере следующие пять регуляторных элементов:
a) промотор CMV или его функциональные варианты,
b) интрон,
c) TPL или его функциональные варианты,
d) VA гены или функциональные варианты и
e) последовательность полиаденилирования гормона роста крупного рогатого скота или функциональные варианты.
7. Вектор по пп.1-6, где интрон представляет собой химерный интрон c SEQ. ID. No.1.
8. Вектор по пп.1-6, где TPL представляет собой регуляторный элемент SEQ. ID. No.4.
9. Вектор по пп.1-6, где VA гены являются имеющими последовательность SEQ. ID. No.5.
10. Новый экспрессионный вектор млекопитающих, определенный в любом из предшествующих пп.1-9, который включает по меньшей мере следующие пять регуляторных элементов:
a) промотор CMV или его функциональные варианты,
b) химерный интрон, имеющий последовательность SEQ. ID. No.1, или его функциональные варианты,
c) TPL, имеющий последовательность SEQ. ID. No.4, или его функциональные варианты,
d) VA гены, имеющие последовательность SEQ. ID. No.5, или их функциональные варианты и
e) последовательность полиаденилирования гормона роста крупного рогатого скота или функциональные варианты.
11. Вектор, определенный в любом из пп.1-10, дополнительно включающий маркер селективности и амплификации, выбранный из дигидрофолатредуктазы, аденозиндезаминазы, орнитиндекарбоксилазы, аспарагинсинтетазы, глутаминсинтетазы или их функциональные варианты.
12. Вектор, определенный в любом из пп.1-11, дополнительно кодирующий ген для эритропоэтина.
13. Вектор, определенный в любом из пп.1-11, дополнительно кодирующий ген для гибридного белка TNFR-IgGFc.
14. Вектор, определенный в любом из пп.1-11, дополнительно кодирующий ген для ритуксимаба, трастузумаба, бивакузумаба или других моноклональных антител.
15. Вектор, определенный в любом из пп.1-11, дополнительно кодирующий соответствующий ген(ы), способный экспрессироваться в клетках млекопитающих.
16. Клетка млекопитающих, трансформированная экспрессионным вектором, определенным в любом из пп.1-15.
17. Вектор, определенный в любом из пп.1-16, где клетки млекопитающих для трансфекции выбраны из Cos, CHO, CHO DHFR-, BHK1 и NS0.
18. Применение вектора, определенного в любом из предшествующих пунктов, для достижения высокой экспрессии представляющих интерес белков.
19. Применение вектора по п.18, где представляющий интерес белок выбран из рекомбинантного эритропоэтина, рекомбинантного гибридного белка TNFR-IgGFc и моноклональных антител, выбранных из ритуксимаба, трастузумаба, бивакузумаба.
20. Способ достижения высокой экспрессии представляющего интерес белка посредством кодирования соответствующего гена в векторе, определенном в любом из предшествующих пп.1-11.

Текст
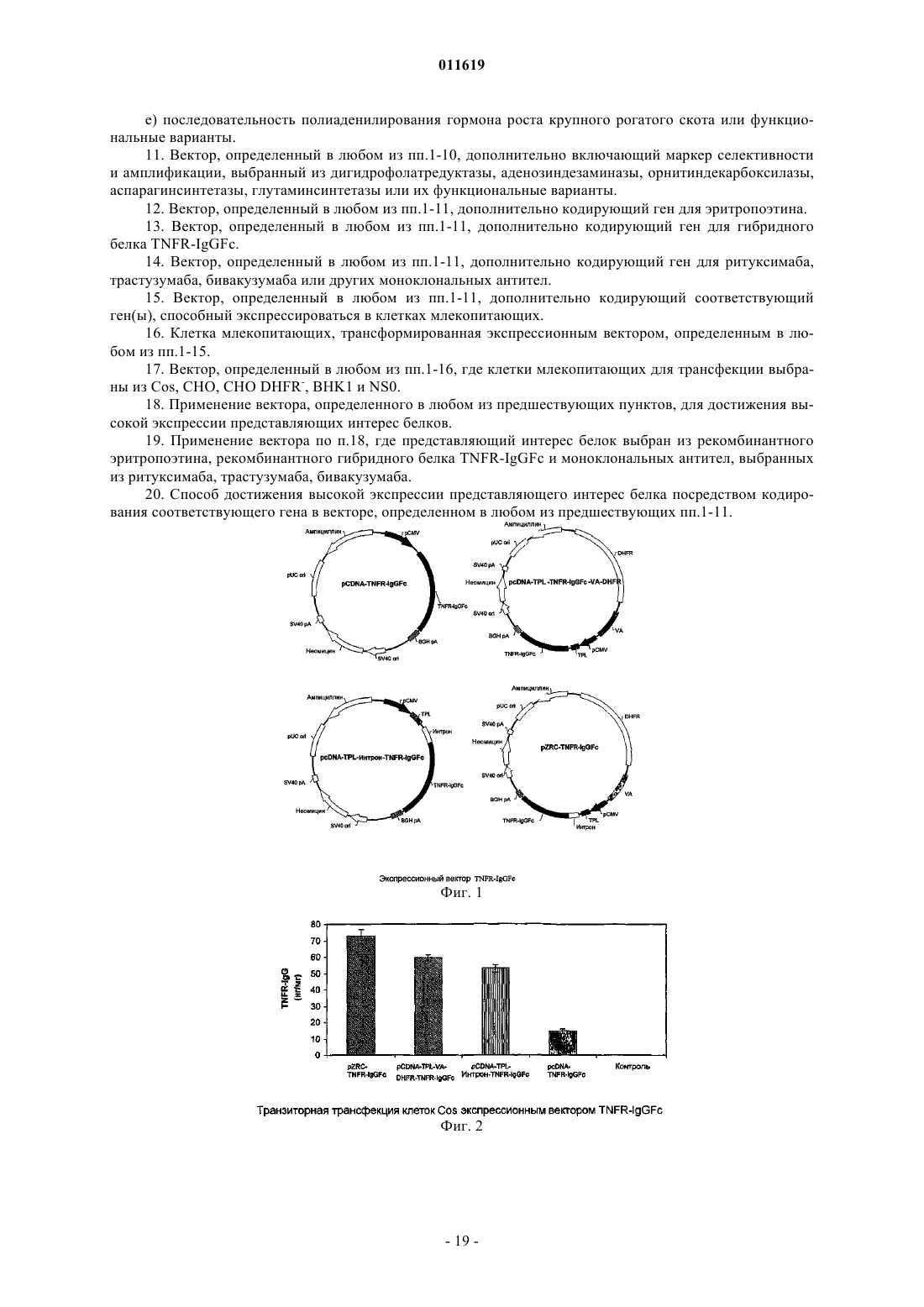
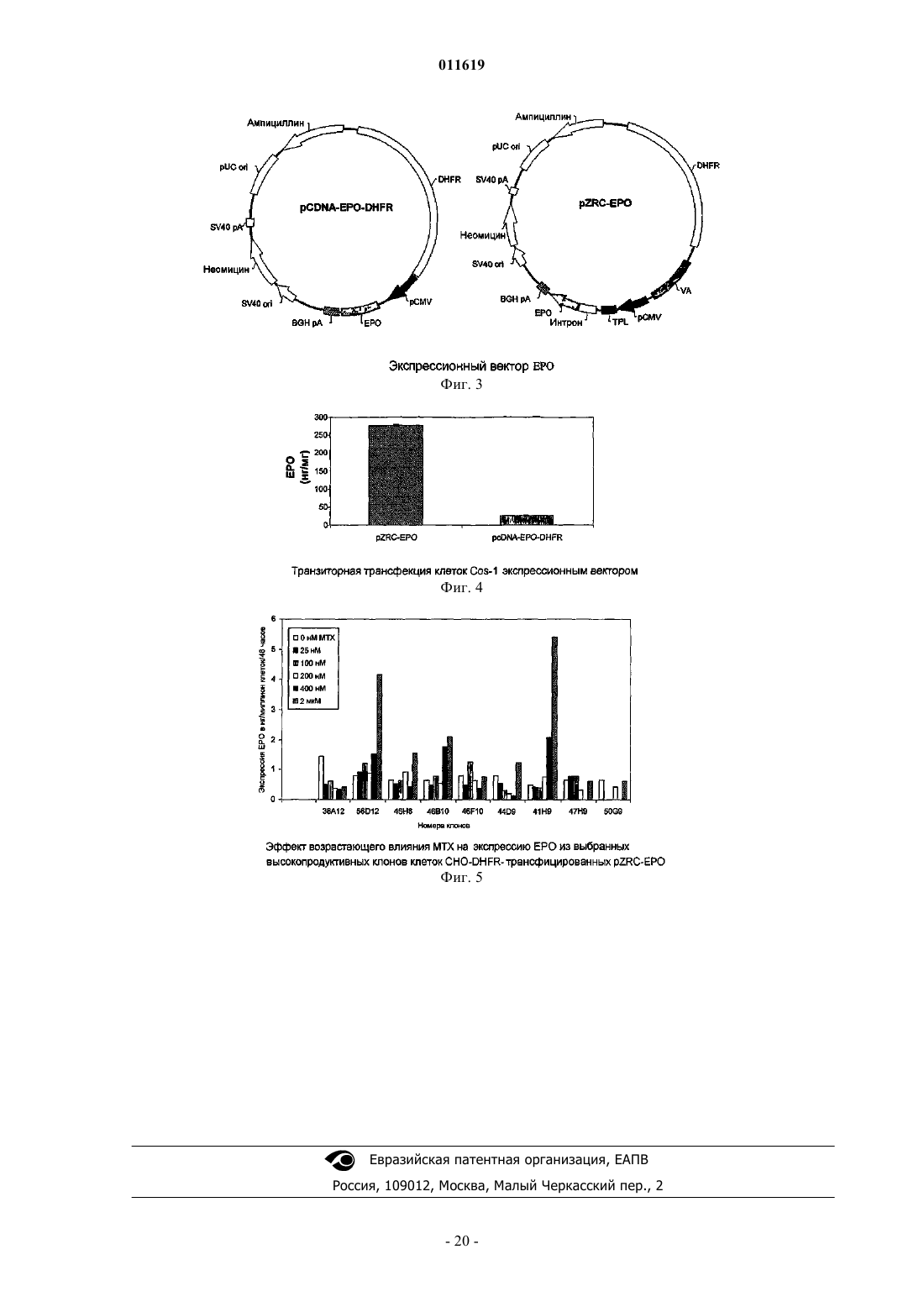
011619 Область техники, к которой относится изобретение Настоящее изобретение относится к новым экспрессионным векторам для продуцирования высоких уровней белка в клетках млекопитающих и способу продуцирования представляющих интерес белков с использованием указанных векторов. Экспрессионные векторы дают экспрессию рекомбинантного белка ЕРО на 80-100% выше, чем некоторые из лучших векторов, описанных в литературе. Предшествующий уровень техники В соответствии с данными IMS, прогнозируется рост биофармацевтической части общего фармацевтического рынка с 6% в 1999 г. до 14% (90 миллиардов долларов) в 2009 г. Такой повышенный интерес к биопрепаратам в основном происходит благодаря их, как правило, высокоспецифичному направленному действию, которое приводит к значительно уменьшенному и строго определенному риску токсичности по сравнению с лекарственными средствами на основе малых молекул. Более того, при использовании рекомбинантных методик для производства указанных биопрепаратов, в отличие от устаревших методов их очистки от тканевых экстрактов или жидкостей организма, могут быть легко получены продукты очень высокой чистоты и с высокой степенью безопасности и со строго определенными физикохимическими характеристиками. Несмотря на обладание всеми такими благоприятными для пациента качествами, большинство рекомбинантных биопрепаратов остается недоступным для большинства людей в мире, потому что они продолжают оставаться непомерно дорогими. Поэтому спасающие жизнь лекарственные средства, подобные эритропоэтину, лекарственные средства, подобные этанерцепту, которые значительно улучшают качество жизни, и многие противораковые лекарственные средства, подобные ритуксимабу, трастузумабу и всем другим моноклональным антителам, и т.д., являются доступными только для очень небольшого процента людей, в то время как подавляющее большинство больных людей повсюду в мире не может использовать их в достаточном количестве. Поэтому существует крайняя необходимость снизить цену на указанные лекарственные средства. Большая составная часть такой высокой цены связана с их производством. Настоящее изобретение обеспечивает решение данной проблемы посредством предоставления экпрессионных векторов, которые могут дать высокую экспрессию белка в клетках-хозяевах из млекопитающих, трансфицированных ими. В последние годы технология рекомбинантных ДНК продвинулась к стадии, где вообще возможно получение желаемого гена, кодирующего желаемый белковый продукт, также называемый биологическим, когда белок впоследствии используется как терапевтический. Когда ген получен, может быть использован широкий круг хозяев для их экспрессии, путем первоначального клонирования гена в любой из множества доступных специфичных по отношению к хозяину экспрессионных векторов, с последующим введением ген-несущего вектора в специфического хозяина путем использования различных способов трансформации и трансфекции. Для того чтобы получить белковую продукцию от таких гентрансформированных клеток-хозяев, различные условия инкубации также являются подходящими. Выбор хозяина и последующие взаимозависимые выборы, например, экспрессионных векторов, способов трансформации и трансфекции и инкубационных методик, зависят от многих факторов, таких как особенности белка, который должен быть продуцирован, конечное использование белка, количество требуемого белка, доступные методы очистки, общая стоимость и доступность технологии, и т.д. Например,относительно интересов данного изобретения, когда продуцируемый рекомбинантный белок предназначен для терапевтического применения, первичная, вторичная и третичная структура белка, степень и качество их гликозилирования, чистота конечного продукта, количество продуцируемого белка, цена, по которой лекарственное средство может быть продано, все способствует вышеприведенному процессу выбора экспрессионного хозяина и выполнения других взаимозависимых выборов. Одно возможное решение, направленное на вышеуказанную проблему высокой стоимости продукции биологических препаратов, состоит в экспрессии их в бактериальных клетках-хозяевах, таких как Е.Liss, New York, 101-127 (1996); Gold, L. Methods Enzymol, 185:11-14 (1990); Hodgson, J., Bio/Technology,11:887-893 (1993); Nicaud et al, J. Biotechnol. 3:255-270 (1986); Olins, P.O., and S.C. Lee., Curr. Opin. Biotechnol. 4:520-525 (1993); Shatzman, A.R., Curr. Opin. Biotechnol, 6:491-493 (1995)]. Обычно используемый бактериальный хозяин, Escherichia coli, является важным организмом-хозяином для продуцирования рекомбинантных белков и широко используется в промышленном масштабе. Это представляет много преимуществ, включая легкое культивирование, низкую стоимость и высокий потенциал продуцированияexpression and Purification 7:335-342 (1996)]. Однако бактериальные хозяева, как правило, не идеальны для продуцирования биологических препаратов, потому что они не имеют необходимого механизма гликозилирования белков [Old R W, and Primrose S.B., Principles of Gene Manipulations, An introduction togenetic Engineering, Blackwell science, United Kingdom. (1994)], и большинство терапевтических белков млекопитающих не являются полностью функциональными без соответствующего гликозилирования. Кроме того, отсутствие секреторного механизма для эффективного высвобождения белка в культуральную среду, ограниченная способность обеспечения образования пространственной дисульфидной связи,неправильный фолдинг, разрушение белка протеазами клеток-хозяев, существенные различия в использовании кодонов, другие модификации, такие как гликации, и т.д., вместе делают бактериальные систе-1 011619 мы намного менее привлекательными, нежели системы млекопитающих [Fun, G. et al., J. Biol. Chem, 2 65:3111-3115 (1990); Liang et al., Biochem. J., 229:429-439 (1985); Sarmientos et al., Bio/Technology, 7:495501 (1989); Savvas C. Makrides, Microbiological Reviews, Sept. 1996. 512-538; N. Jenkins and E. M. Curling,Enzyme Microb. Technol, 16:354-364 (1994)]. Поэтому, в большинстве случаев невозможно экспрессировать терапевтические белки в бактериях и большинство биопрепаратов используют эукариотические клетки-хозяева для экспрессии, несмотря на то, что это означает более высокую стоимость производства из-за более низких уровней экспрессии рекомбинантного белка, более жестких условий культивации,более низких скоростей роста и т.д. [(Cornelia Rossmann, Protein Expression and Purification, 7:335-342(1996); Geoff T. Yarranton, Current Opinion in Biotechnology, 1:133-140 (1990)]. Так как системы-хозяева млекопитающих обладают большим преимуществом при продуцировании терапевтических белков,очень важно решить проблему высокой стоимости производства, связанной с ними. Так как стоимость производства может быть снижена посредством увеличения продуктивности, значительные усилия должны быть затрачены на увеличение количества продукта, который может быть продуцирован этими клетками-хозяевами. Факторы, которые обычно влияют на количество продукта, продуцируемого клетками-хозяевами, включают факторы, которые являются внешними по отношению к клетке, такими как условия культивирования, и те, которые являются внутренними для клеток, большинство которых включает факторы, которые регулируют эффективность и качество транскрипции [Foecking and Hofstetter,Gene, 45:101-105 (1986); Kaufman et al, Journal of molecular Biology, 159:601-621 (1985); Wurm et al,PNAS, 1983:5414-5418 (1986); Reiser and Hauser, Drug Research, 37:482-485 (1987); Zettlmeissl et al, Biotechnology, 5:720-725 (1987)] и трансляции [(R. Grabherr and K. Bayer, Food Technol. Biotechnol. 39 (4) 265-269 (2001); Randal J. Kaufman et al. Molecular Biotechnology, 16 (2), 151-160, (2000); Juraj Hlavaty etal., Virology 341, 1-11, (2005); CM. Stenstrom et al Gene. 273(2), 259-65, (2001); M. Ibba and D. Soil, Science, 186, 1893, (1999), и преимущественно обусловлены непосредственно конструкцией экспрессионного вектора. Хотя в литературе сообщается о многих попытках увеличения продуктивности клеток-хозяев посредством улучшения условий культивирования [Palermo D.P. et al., Journal of Biotechnology, 19:35-48Miller, Biotechnology Prog, 18:346-353 (2002); Dezengotita et al., Biotechnology and Bioengineering, 78:741752 (2002); и Sun et al, Biotechnology Prog., 20:576-589 (2004)], улучшение внешних факторов могут увеличить экспрессию только в ограниченной степени и коммерчески неэффективно, до тех пор, пока экспрессионный вектор не будет исходно оптимизирован для достижения идеального базового уровня экспрессии. О большом числе исследований сообщается в уровне техники, которые направлены на внутренние факторы улучшения экспрессии гена. Внутренние факторы, описанные ниже, также известны как регуляторные элементы, которые регулируют генную экспрессию различными путями. Из уровня техники хорошо известно, что для достижения экспрессии представляющего интерес гена из экспрессионного вектора, он должен находиться под влиянием соответствующей 5' и 3' фланкирующей последовательности,которая даст возможность гену стать транскрибированным в мРНК и затем точно транслированным в белок. Описано много важных 5' и 3' фланкирующих последовательностей, таких как ТАТА-боксыDorsch-Hasler, K. et al., PNAS, 82:8325-8329 (1985)], промоторов из числа вирусных промоторов, таких как прямой ранний промотор CMV (цитомегаловирус), ранний или поздний промотор SV40, основной поздний промотор аденовируса [Luigi R., Gene, 168:195-198 (1996); Pizzorno, M.C. et al., J. Virol., 62:11671179 (1988); Okayama and Berg, Mol. Cell Biol, 2:161-171 (1982); Wong et al., Science, 228:810-815 (1985);Foecking and Hofsteffer, Gene, 45:101-105 (1986)] и промоторы млекопитающих, такие как мышиный металлотиониновый промотор, куриный -актиновый промотор [Nicole Israel et al., Gene, 51:197-204 (1987);(1986)], энхансеров, таких как прямой ранний энхансер CMV [Cockett, M.I. et al., Nucleic Acids Research,19:319-325 (1996)], старт- и стоп-кодонов трансляции [Lehninger et al, Principles of Biochemistry - 3rd edition, Worth Publishers, Chapter 27, p1025], и сайтов полиаденилирования, таких как сайты полиаденилирования гормона роста крупного рогатого скота (BGH) и SV40 [Carswell, S. and Alwine, J.C, Mol. Cell. Biol. 9:4248-4258 (1989)]. Интроны являются другим внутренним фактором, который обычно формирует существенную часть эукариотических генов в качестве промежуточных последовательностей между экзонами и тем, что точно удаляется из первичного транскрипта способом, известным как РНК сплайсинг с образованием зрелой мРНК. РНК сплайсинг был показан как отвечающий за стабильность мРНК [Buchman et al, Mol. Cell Biol. 8:4395-404 (1988); Peterson et al, Proc. Natl. Acad. Sci. USA, 83:8883-87 (1986)] и регуляцию генной экспрессии [Brinster et al, Proc. Natl. Acad. Sci. USA, 85:836-40 (1988); Dynan, W.S. andTjian, R., Nature, 316:774-778 (1985)]. Также были разработаны синтетические химерные интроны, как сообщено Huang и др., которые состоят из 5' донорного сайта основного позднего транскрипта аденовируса и 3' сплайсингового сайта мышиного иммуноглобулина [Huang et al, Nucleic Acids Res., 18:937-47(1990)]. Такие химерные интроны поддерживают гетерологичную генную экспрессию лучше, чем обычно используемые интроны. [Huang et al, Nucleic Acids Res., 18:937-47 (1990); Ted Choi et al, Molecular andCellular Biology, 11 (6) : 3070-3074 (1991)]. Как правило, используемым источником для высокоэффективных внутренних факторов, являются вирусы. Вирусы, как общеизвестно, являются наиболее эффективными паразитами в природе, которые используют свои собственные внутренние факторы для манипулирования хозяевами и экспрессии вирусного гена с целью размножения и выживания. Это также широко изучалось для определения их роли в конструкции экспрессионных векторов с целью усовершенствования белковой продукции. Некоторые из наиболее эффективных промоторов, известных в области молекулярной биологии, произведены из вирусов [Luigi R., Gene, 168:195-198 (1996); Pizzorno, M.C. et al., J. Virol, 62:1167-1179 (1988); OkayamaHofsteffer, Gene, 45:01-105 (1986)]. Множество вирусов изучалось в значительной степени на генетическом уровне, и были идентифицированы индивидуальные последовательности, которые могут изменять ядерный и цитоплазматический метаболизм мРНК в клетках-хозяевах. Тройной лидерный элемент из аденовируса (TPL) (GI: 209811) [Akusjarvi G. et al, J Mol Biol., 134 (1) : 143-58 (1979)] является, как известно, одним из таких элементов для улучшения трансляции даже невирусной РНК в инфицированных вирусом клетках, когда непосредственно прикреплен к ней [Berkner K.L. et al, Nucleic Acids Res.,13(3):841-57 (1985)]. Все мРНК, кодируемые частью основной поздней транскрипции аденовируса, имеют общую 5' некодирующую область. Этот элемент может уменьшать период полужизни транскриптов [Huang et al, J.Virol., 2(l):225-35 (1998)]. Этот элемент, как известно, усиливает трансляцию мРНК [Kaufman R. J. et al,Proc Natl AcadSci USA., 82(3):689-93 (1985)]. Другим элементом является вирусные РНК-ассоциированные гены I и II аденовируса(GI: 209811) или их функциональные варианты. VA РНК гены I и II(VA гены), как показано, увеличивают эффективность трансляции гена, содержащего последовательность TPL [Kaufman R. J. et al,Proc Natl Acad Sci USA., 82(3):689-93 (1985)]. VA РНК ген I вовлечен в дефосфорилирование EIF2a и, соответственно,увеличивает скорости белкового синтеза [O'Malley et al, Cell,44:391-400 (1986); Thimmapayya В., et al, Cell, 31:543-551(1982)]. Другим обычно используемым внутренним фактором является количество генных копий, которые являются удобным приемом для увеличения генной экспрессии [Kaufman and Sharp, Journal of molecularSchimke, R.T. (Ed.), Gene Amplification. Cold Spring Harbor Laboratory,Cold Spring Harbor, NY, 1982]. Наиболее общим способом, используемым для увеличения количества генных копий, является выбор клеток для генной амплификации. При таком подходе, например, как описано в ЕР 0045809 или US4634665, клетка-хозяин трансформируется парой генов. Первый ген в паре кодирует желаемый белок, и второй ген кодирует маркер селекции, например, дигидрофолатредуктазу(DHFR) [Alt, F.W. et al., J. Biol. Chem., 253:1337-1370 (1978)]. Указанные два гена представлены на одном экспрессионном векторе или на двух отдельных экспрессионных векторах. После трансфекции клеток указанной парой генов они культивируются в увеличивающихся концентрациях токсического агента,такого как метотрексат в случае метода, использующего ген DHFR в качестве маркера селекции, действие которого нивелируется продуктом гена-маркера селекции. Было обнаружено, что клеточные линии,которые выживают при повышенных концентрациях токсического агента, имеют увеличенное количество копий и гена-маркера селекции, и желаемого генного продукта. Выбранная клетка-хозяин, которая имеет увеличенное количество копий релевантного гена, теперь может произвести больше количества желаемого белка, чем исходная клеточная линия. Подобная стратегия для генной амплификации использовалась с другими маркерами селекции, такими как аденозиндезаминаза (ADA), орнитиндекрабоксилаза"Gene Amplification in Mammalian Cells" ed Kellens, R.E., Marcel Dekker Inc., New York 301-311 (1993)]. Такие вышеописанные регуляторные элементы или внутренние факторы по отдельности являются неспособными давать генную экспрессию и должны использоваться в комбинациях. Несмотря на то, что сами элементы были хорошо поняты, эффективность их комбинаций абсолютно не предсказуема для высокой экспрессии. Некоторые комбинации дают очень низкую экспрессию по сравнению с другими. Для примера, Jang и др., используя экспрессионный вектор, состоящий из комбинации промотора SR,лидерной последовательности РНК AMV и DHFR, смогли получить экспрессию эритропоэтина (ЕРО)-3 011619 только 45 МЕ/мл (эквивалентно 0,346 мкг/мл) [Jang H P et al, Biotechnol. Appl. Biochem, 32:167-172,(2000)], в то время как в US5955422 сообщается об уровнях ЕРО от 750 до 1470 Ед/миллион клеток/48 часов (или от 375 до 735 Ед/миллион клеток/24 ч), используя экспрессионный вектор, состоящий из другой комбинации элементов, а именно - промотора SV40 и поли А последовательности и DHFR. Однако другой экспрессионный вектор, описанный в US5888774, и состоящий из комбинации промотора EF1 и элементов ароВ SAR, обеспечивает экспрессию от 1500 до 1700 ME ЕРО/миллион клеток/24 ч. Для других рекомбинантных белков, таких как TNFR-IgGFc (Enbrel), был описан экспрессионный вектор, содержащий комбинацию промотора CMV, TPL, VA I и II и DHFR [США 5605690, Cindy A Jacobs andCraig A Smith]. Несмотря на все достижения, описанные выше, высокая стоимость производства рекомбинантных биопрепаратов, особенно тех, для которых используют экспрессионные системы млекопитающих, все еще остается главной проблемой. Поэтому, несмотря на то, что уровень техники предоставляет большое количество способов улучшения белковой экспрессии посредством изменения экспрессионного вектора,все же желательно создать новый экспрессионный вектор для дополнительного увеличения продуктивности эукариотических клеток-хозяев. Неожиданно, но, несмотря на большой объем знаний, полученный в данной области более чем за два прошедших десятилетия, даже сегодня специалисты в данной области не могут просто собрать и выбрать комбинацию внутренних факторов или регуляторных элементов для построения экспрессионного вектора, который мог бы дать гарантированно высокую экспрессию. Отдельный элемент, когда он добавлен к комбинации, не может обеспечить какого-либо существенного дополнительного или синергического эффекта к экспрессионному потенциалу вектора. Поэтому способ создания нового экспрессионного вектора, который дал бы высокий уровень белковой экспрессии, все еще требует эмпирического тестирования многих возможностей. Авторы предложили новый экспрессионный вектор, который при стабильной трансфекции в клетки CHO-DHFR дает экспрессию от 11,830 МЕ/мл (91 мкг/мл) при 168 часовом культивировании, что эквивалентно от 2366 до 3549 МЕ/106 клеток/24 ч или от 18,2 до 27,3 мкг/106 клеток/24 ч. Неожиданно такой уровень экспрессии для ЕРО является на 80-100% выше, чем для некоторых из лучших векторов, описанных в литературе. Новый экспрессионный вектор будет существенно снижать стоимость продуцирования ЕРО и других рекомбинантных биопрепаратов. Сущность изобретения Настоящее изобретение решает проблему, описанную ранее в уровне техники, предоставлением нового экспрессионного вектора для значительно улучшенного продуцирования рекомбинантных белков в клетках млекопитающих. Оно также предоставляет способ создания таких векторов и способ применения таких векторов для достижения высокого уровня экспрессии белков. Таким образом, один из основных объектов данного изобретения обеспечивает усовершенствованный способ достижения высоких экспрессионных уровней представляющих интерес белков путем применения нового вектора, как описано в данном описании. Следующие последовательности использовали в представленном изобретении. Организм-источник: гибрид аденовирусного генного компонента и мышиного генного компонентаSEQ. ID. NO.3 Тип: ДНК Длина: 583 Название последовательности: кДНК эритропоэтина человека Источник: почечная ДНК человека Настоящее изобретение использует новую комбинацию из пяти элементов, полученных из вирусов и других различных векторных источников для конструирования нового вектора, который дает синергический эффект таких элементов в виде высокой экспрессии желаемых белков в клетках-хозяевах млекопитающих. Более конкретно, данные экспрессионные векторы состоят из новой комбинации следующих пяти элементов: прямой ранний промотор CMV, тройной лидерный элемент аденовируса (TPL)(GI:209811), гибридный (химерный) интрон (SEQ. ID. No.1), вирусные РНК-ассоциированные гены I и II аденовируса (GI:209811) и последовательность полиаденилирования гормона роста крупного рогатого скота. Когда такая новая комбинация из пяти элементов используется в соединении с дополнительными элементами в основном векторе, которые необходимы для функционирования его в качестве вектора,новые векторы, полученные таким образом, демонстрируют синергически более высокую экспрессию,чем другие векторы, которые содержат только некоторые из указанных пяти элементов в соединении с основным вектором. При стабильной трансфекции предложенный новый экспрессионный вектор, также содержащий маркер амплификации DHFR, дал на 80-100% выше экспрессию ЕРО, чем некоторые из лучших векторов, описанных в литературе. Более предпочтительно, новые экспрессионные векторы содержат прямой ранний промотор CMV,тройной лидерный элемент аденовируса (TPL) (SEQ. ID. No.4), гибридный (химерный) интрон (SEQ. ID.No.1), вирусные РНК-ассоциированные гены I и II аденовируса (SEQ. ID. No.5), сайт клонирования, маркер селекции клеток млекопитающего, маркер селекции прокариотической клетки, маркер амплификации/селекции и сайт полиаденилирования гормона роста крупного рогатого скота, как представлено на соответствующих положениях в указанных векторах. Белки, которые могут быть продуцированы с использованием таких векторов, включают, но без ограничения, гормоны, например, FSH, антитела, химерные белки, например, этанерцепт, компоненты крови, например, фактор VII, факторы роста, например, эритропоэтин, цитокины, например, интерфероны,TNF и тому подобное.-6 011619 Описание чертежей На фиг. 1 представлено схематическое изображение различных TNFR-IgGFc экспрессионных векторов, включая новый экспрессионный вектор pZRC-TNFR-IgGFc. На фиг. 2 описана экспрессия белка TNFR-IgGFc из клеток Cos-1, транзиторно трансфицированных различными TNFR-IgGFc экспрессионными векторами, включая новый экспрессионный вектор pZRCTNFR-IgGFc. На фиг. 3 представлено схематическое изображение различных ЕРО экспрессионных векторов,включая новый экспрессионный вектор pZRC-EPO. На фиг. 4 описано сравнение экспрессии ЕРО в клетках Cos-1 транзиторно трансфицированных новым экспрессионным вектором pZRC-EPO и другим кодирующим ЕРО коммерческим экспрессионным вектором pcDNA3.1. На фиг. 5 представлен эффект нарастания воздействия МТХ на экспрессию ЕРО из выбранных высокопродуцирующих клонов CHO-DHFR клеток, стабильно трансфицированных pZRC-EPO. Источник элементов для конструирования нового экспрессионного вектора: Описание изобретения Настоящее изобретение решает проблему высокой стоимости производства, описанную ранее в уровне техники, посредством предоставления новых экспрессионных векторов для высокой продукции рекомбинантных белков в клетках млекопитающих. Такие новые векторы используют широко известные регуляторные элементы, описанные в уровне техники, но содержат уникальную комбинацию таких элементов, которая неожиданно обеспечивает синергически высокую экспрессию. Настоящее изобретение,кроме того, предоставляет способ продуцирования высоких уровней белка, используя данные новые векторы. Новая комбинация элементов, используемых в настоящем изобретении, состоит из:a) прямого раннего промотора CMV;b) тройного лидерного элемента аденовируса (TPL) (SEQ. ID. No.4 и GI:209811);e) сайта полиаденилирования гормона роста крупного рогатого скота. Для того чтобы сконструировать новые векторы, базисные экспрессионные векторы, которые могут быть использованы для присоединения к описанным выше новым комбинациям элементов, включают:b) соответствующий маркер селекции клеток млекопитающих, выбранный из группы, включающей,но, не ограничиваясь ими, устойчивые к лекарственным средствам маркеры, такие как неомицин, гидромицин, пуромицин и тому подобное;c) соответствующий маркер селекции прокариотической клетки, выбранный из группы, включающей, но, не ограничиваясь ими, устойчивые к антибиотикам маркеры, такие как ампициллин, канамицин и тому подобное;d) маркер амплификации/селекции, выбранный из группы, включающей, но, не ограничиваясь ими,-7 011619 дигидрофолатредуктазу, аденозиндезаминазу, орнитиндекарбоксилазу, аспарагининсинтетазу, глутаминсинтетазу и тому подобное. Способ получения желаемого вектора включает выделение различных элементов вектора из других векторных источников или из их биологических источников и включение в другой вектор, который обеспечивает каркас плазмиды. О методиках, используемых для такого конструирования, сообщается в уровне техники, касающейся области конструкции векторов [Sambrook J et al., Molecular Cloning - A laboratory Manual, Cold Spring Harbor Laboratory Press, New York, (1989)] с соответствующими модификациями, в случае необходимости. Для продуцирования желаемого белка сначала получают соответствующей ген, кодирующий белок. Следующие методики являются обычно используемыми, либо как таковые, либо в комбинации, для получения желаемого гена:(1) выделение матричной РНК (мРНК) для желаемого гена и использование его в качестве матрицы для получения комплиментарной ДНК (кДНК) путем обратной транскрипции,(2) выделение природного гена из геномных библиотек ДНК, используя комбинацию подходящих ген-специфических гибридизационных зондов и ферментов рестрикции,(3) выделение гена путем специфической амплификации специфического генного фрагмента полимеразной цепной реакцией (ПЦР), использующей одну или более пар ген-специфических праймеров, и(4) химический синтез гена из составляющих его нуклеотидов. Полученный ген клонируется в сайте клонирования нового вектора способами, хорошо известными в данной области. Образующаяся конструкция трансформируется в подходящую клетку-хозяина млекопитающих. Клетку-хозяина млекопитающих, которая может быть использована для способа, выбирают из группы, включающей, но, не ограничиваясь ими, клеточные линии СНО и СНО DHFR", клеточные линии ВНК 1, NS0, COS и тому подобное. Трансформированные клетки подвергают селекции на основе их способности расти в средах, содержащих соответствующий антибиотик и затем на их способности к экспрессии желаемого белка. Выбранный клон затем выращивают в соответствующих условиях культивирования для роста и продуцирования белка с получением высоких уровней желаемых белков. Количество генных копий и последующая генная экспрессия могут быть увеличены за счет селекции клеток с высоким количеством генных копий при повышенном селективном воздействии специфического цитотоксического лекарственного средства, эффект от которого может быть нивелирован только повышением количества маркера селекции. Так как генная амплификация путем цитотоксического селективного воздействия часто преобразовывает клон в гетерогенную популяцию клеток с вариацией копий представляющего интерес гена, то конечный высокопродуктивный клон выбирают из гетерогенной популяции путем предельного разведения. Пример 1. Конструирование экспрессионного вектора TNFR-IgGFc Экспрессионный вектор млекопитающих pcDNA 3.1 (Invitrogen Life Technologies) был взят как основной каркасный вектор для конструирования всех векторов по изобретению. Данный вектор состоит из прямого раннего промотора CMV и сигнальной последовательности полиаденилирования BGH для контроля транскрипции гена. Два элемента формируют два из пяти элементов новой комбинации. Данный вектор дополнительно состоит из сайта множественного клонирования (MCS), ориджина репликацииpUC для бактерий, устойчивого к ампициллину гена для селекции в Е. Coli и устойчивого к неомицину гена для селекции в клетках млекопитающих. Оставшиеся 3 элемента новой комбинации из пяти элементов, а именно - последовательность тройного лидерного элемента аденовируса, гибридный (химерный) интрон, состоящий из 5' донорного сайта основного позднего транскрипта аденовируса и 3' сплайсингового сайта мышиного иммуноглобулина, и вирусные VA РНК гены I и II аденовируса, вставляли в каркасpcDNA 3.1 в различных комбинациях для того, чтобы найти комбинацию элементов, которые вместе работают синергически, чтобы дать очень высокую экспрессию. кДНК, кодирующие гибридный ген TNFR-IgGFc человека (SEQ. ID. No.2) и ген эритропоэтина человека (SEQ. ID. No.3) использовали в качестве репортеров для изучения влияния комбинации этих элементов на экспрессию белка. На фиг. 1 описан экспрессионный вектор, полученный для TNFR-IgGFc и на фиг. 3 описано то же самое для ЕРО.TNFR-IgGFc представляет собой слитый белок, содержащий 75 кДа TNFR (1-235 а.к.) и Fc фрагмент IgG1 (содержащий СН 2, СН 3 и шарнирную область) [US5605690]. Указанный слитый белок продается как лекарственное средство, называемое Enbrel (Amgen), для лечения ревматоидного артрита. Он действует посредством устранения из циркуляции воспалительного цитокина TNF-. Для того, чтобы клонировать кДНК TNFR I, использовали общую плацентарную ДНК человека (Clontech) в качестве матрицы для основанного на обратнотранскриптазной полимеразной цепной реакции (ОТ-ПЦР) клонирования желаемой последовательности. Двухцепочечную кДНК синтезировали из общей плацентарной РНК с использованием обратной транскриптазы MMLV (MBI Fermentas, США) ген-специфическим праймингом [Maniatis et al., Molecular cloning; A Laboratory Manual (Cold Spring harbor Laboratory Press,Cold Spring Harbor, N.Y.), (1990)]. Полученную кДНК затем подвергали 40 циклам ПЦР амплификации,используя 1000 пикомолей ген-специфических вырожденных олигонуклеотидных праймеров в объеме 100 мкл, содержащем 50 мМ Трис-Cl (рН 8,3), 2,5 мМ MgCl2, 200 мкМ каждого из 4 dNTP и 2,5 единицы-8 011619 полимеразы Pfu. Каждый цикл ПЦР амплификации состоял из инкубации при 94 эээС в течение 30 сек(денатурация), 58 эээС в течение 30 сек (отжиг) и 72 эээС в течение 1 мин (элонгация). Амплифицированный продукт реакции ПЦР разлагали на 1% агарозном геле. Желаемый фрагмент размером приблизительно 705 пар оснований вырезали из геля и очищали, используя набор для выделения из геля Qiagen. Очищенный фрагмент ДНК лигировали в MCS pcDNA 3.1 после рестрикционного расщепления как вектора, так и очищенного продукта ПЦР с EcoRI и PvuII (MBI Fermentas, США). Лигированный продукт трансформировали в Е. coli DH5 и трансформанты отбирали на основании устойчивости к ампициллину. Плазмидную ДНК, выделенную приблизительно из 20 таких колоний, анализировали на наличие кДНК TNFR I рестрикционным расщеплением, используя различные ферменты рестрикции. Одну такую плазмиду секвенировали, используя автоматический ДНК секвенатор (ABI), и она была определена, как имеющая правильную интеграцию и последовательность кДНК TNFR I в векторе pcDNA 3.1. Такая плазмидная ДНК была обозначена как pcDNA-TNFR. Подобным образом выделяли последовательность IgG1Fc, используя плацентарную РНК человека(Clonetech) в качестве матрицы для основанного на ОТ-ПЦР клонирования. ПЦР проводили, используя пару ген-специфических олигонуклеотидных праймеров, соответствующих кодирующей области последовательности IgG1Fc (699 пар оснований). Каждый цикл ПЦР амплификации состоял из инкубации при 94 С в течение 30 с (денатурация), 56 С в течение 30 с (отжиг) и 72 С в течение 1 мин (элонгация). Амплифицированный продукт реакции ПЦР разлагали на 1% агарозном геле. Желаемый фрагмент размером приблизительно 725 пар оснований вырезали из геля и очищали, используя набор для выделения из геляQiagen. Очищенный фрагмент ДНК лигировали в вектор pTZ57R (MBI Fermentas) после линеаризации вектора и расщепления очищенного продукта ПЦР, используя EcoRI и Not I (MBI Fermentas, США). Цитированный продукт трансформировали в Е. coli DH5 и трансформанты отбирали на основании устойчивости к ампициллину. Плазмидную ДНК, выделенную приблизительно из 10 таких колоний, анализировали на наличие IgG1Fc кДНК рестрикционным расщеплением, используя различные ферменты рестрикции. Одну такую плазмиду секвенировали, используя автоматический ДНК секвенатор (ABI), и оно была определена, как имеющая правильную интеграцию и последовательность кДНК IgG1 Fc в вектореpTZ57R. Такая плазмидная ДНК была обозначена как pTZ57R-IgG1Fc.pcDNA-TNFR-IgGFc Слияние генных фрагментов TNFR I и IgG1Fc выполняли следующим способом. Клонированный фрагмент TNFR I выделяли из pcDNA-TNFR полным расщеплением с EcoRI и PvuII. Фрагмент IgG1Fc выделяли из pTZ57R-конструкции путем его расщепления с Pvu II и Not I. Фрагменты выделяли из агарозного геля, используя набор для выделения из геля Qiagen. Оба фрагмента ДНК смешивали с линеализированным вектором pcDNA 3.1, предварительно расщепленным с EcoRI и NotI, в трехкомпонентной лигазной реакции. Лигированный продукт трансформировали в Е. coli DH5 и трансформанты отобрали на основании устойчивости к ампициллину. Плазмидную ДНК, выделенную приблизительно из 10 таких колоний, анализировали на наличие слитого продукта TNFR-IgGFc рестрикционным расщеплением, используя различные ферменты рестрикции. Одну такую плазмиду секвенировали, используя автоматический ДНК секвенатор (ABI), и она была определена, как имеющая правильную интеграцию и последовательность TNFR-IgGFc слитого продукта. Такая векторная конструкция была обозначена как pcDNATNFR-IgGFc (фиг. 1), которая содержала слитый ген, фланкированный промотором CMV на 5'-конце и последовательностью полиаденилирования BGH на 3'-конце.pcDNA-TPL-TNFR-IgGFc-VA-DHFR Для конструирования данного вектора, сначала аденовирусный TPL элемент встраивали ниже промотора CMV и выше гена TNFR-IgG1Fc в pcDNA-TNFR-IgGFc. Это завершалось расщеплениемpCAV/NOT-TNFR (ATCC 68088) с KpnI и Nde I при полном расщеплении с извлечением фрагмента в 700 пар оснований, содержащего TPL, который затем очищали, используя набор для выделения из геляQiagen. Фрагмент из 700 пар оснований лигировали в вектор pcDNA-TNFR-IgGFc, расщепленный KpnI иNde I. Лигированный продукт трансформировали в Е. coli DH5 и трансформанты отбирали на основании устойчивости к ампициллину. Плазмидную ДНК анализировали на наличие и ориентировку TPL в результирующей плазмиде рестрикционным расщеплением, используя различные ферменты рестрикции. Одну такую позитивную плазмидную ДНК, обозначенную как pInt-TNFR-IgG-1, затем использовали для инсерции двух представляющих интерес элементов - одного, в 1000 пар оснований VA РНК гена, выделенного из pCAV/NOT-TNFR двойным расщеплением с EcoRI и Not I, и второго, минигена DHFR, выделенного из плазмиды pDHFR 2.9 (ATCC 37165) [Crouse GF et al, Mol. Cell. Biol., 3:257-266 (1983)] путем полного расщепления с BamHI и SspI. Два фрагмента, фрагмент минигена DHFR и фрагмент VA РНК гена, смешивали с pInt-TNFR-IgG-1, предварительно расщепленным с MunI и BglII, в трехкомпонентной лигазной реакции. Лигированный продукт трансформировали в Е. coli DH5 и трансформанты отбирали на основании устойчивости к ампициллину. Плазмидную ДНК анализировали на наличие и ориентировку VA РНК гена и минигена DHFR в результирующей плазмиде путем рестрикционного расщепления,используя различные ферменты рестрикции. Такая векторная конструкция была обозначена как pcDNATPL-TNFR-IgG1Fc-VA-DHFR (фиг. 1).pcDNA-TPL-Интрон-TNFR-IgGFc Экспрессионный вектор pcDNA-TPL-Интрон-TNFR-IgGFc получали путем вставки химерного интрона ниже аденовирусного TPL элемента и выше гена TNFR-IgGFc в векторе pInt-TNFR-IgG-l. Это завершалось расщеплением pIREShyg3 (Clontech) с BstXI и EcoRI при полном расщеплении с извлечением фрагмента химерного интрона из 350 пар оснований, который дополнительно очищали, используя набор для выделения из геля Qiagen. Фрагмент из 350 пар оснований лигировали в векторную конструкциюpInt-TNFR-IgG-1, предварительно расщепленную с BstXI и EcoRI. Цитированный продукт трансформировали в Е, coli DH5 и трансформанты отбирали на основании устойчивости к ампициллину. Плазмидную ДНК анализировали на наличие и ориентировку TPL, интрона и гена TNFRI-IgG1Fc в результирующей плазмиде путем рестрикционного расщепления, используя различные ферменты рестрикции. Одну такую плазмиду секвенировали, используя автоматический ДНК секвенатор (ABI), и она была определена, как имеющая правильную интеграцию TPL, химерного интрона и гена TNFRI-IgG1Fc в векторе. Такая векторная конструкция была обозначена как pcDNA-TPL-Интрон-TNFR-IgGFc (фиг. 1).pcDNA-TPL-Интрон-TNFR-IgGFc-VA-DHFR (pZRC-TNFR-IgGFc) Описанную выше плазмидную ДНК, pcDNA-TPL-Интрон-TNFR-IgGFc, использовали для вставки двух элементов, из 1000 пар оснований VA РНК гена, выделенного из pCAV/NOT-TNFR путем двойного расщепления с EcoRI и Not I, и минигена DHFR, выделенного из плазмиды pDHFR 2.9 путем полного расщепления с BamHI и SspI. Фрагменты для минигена DHFR и VA РНК гена лигировали в векторную конструкцию pcDNA-TPL-Интрон-TNFR-IgGFc, предварительно расщепленную с MunI и BglII, в трехкомпонентной лигазной реакции. Лигированный продукт трансформировали в Е. coli DH5 и трансформанты отбирали на основании устойчивости к ампициллину. Плазмидную ДНК анализировали на наличие и ориентировку VA РНК гена и минигена DHFR в результирующей плазмиде путем рестрикционного расщепления, используя различные ферменты рестрикции. Дополнительно, интеграцию проверяли и подтверждали ДНК секвенированием. Такая векторная конструкция была обозначена как pcDNA-TPLИнтрон-TNFR-IgGFc-VA-DHFR, также обозначенная как pZRC-TNFR-IgGFc (фиг. 1), имеющая номер доступа МТСС 5277. Пример 2. Сравнение эффективности экспрессии различных TNFR-IgGFc экспрессионных векторов транзисторной трансфекцией Для эксперимента по транзиторной трансфекции использовали клетки Cos 1 (ATCC No. CRL-1650),фибробласто-подобные почечные клетки африканской зеленой мартышки. Клетки постоянно поддерживали в полной ростовой среде (модифицированная по Дульбекко среда Игла с 4 мМ L-глутамина и дополненная 1,5 г/л бикарбоната натрия, 4,5 г/л глюкозы, 10% фетальной телячьей сыворотки (FBS) и антибиотики) при температуре 37 С и в атмосфере 5% диоксида углерода (СО 2). За один день до трансфекции, клетки, растущие в середине логарифмической стадии роста, трипсинизировали и наносили в повторах в 6-луночные планшеты с плотностью в 0,2 миллиона клеток/лунка в 3 мл полной ростовой среды, и инкубировали при 37 С и 5% СО 2. Трансфекцию проводили, используя реагент Qiagen Polyfect и ранее известные протоколы. В день трансфекции трансфекционную смесь готовили, как описано ниже. Общее количество 1,5 мг ДНК, растворенной в не содержащей эндотоксинов воде, фильтровали, используя 0,2 мкм шприцевой фильтр. Для наибольшей конструкции pZRC-TNFR-IgGFc, использовали 1,5 мкг вектора как такового, в то время как для других меньших конструкций эквимолярное количество (в пересчете на ген TNFR-IgGFc) конструкции смешивали с пустым вектором с получением 1,5 мкг общей ДНК. Полученную ДНК сначала растворяли в клеточной ростовой среде, то есть модифицированной по Дульбекко среде Игла с 4 мМ L-глутамина, дополненной 1,5 г/л бикарбоната натрия, 4,5 г/л глюкозы, не содержащей сыворотки или антибиотиков, до общего объема 100 мкл. Надлежащее смешивание проводили путем перемешивания при вращении и последующим кратким вращением раствора в течение нескольких секунд для удаления капель с боковых стенок и крышки пробирки. Затем 10 мкл реагента PolyFect Transfection добавляли к полученному раствору ДНК. Надлежащее смешивание трансфекционной смеси проводили путем всасывания и выдавливания из пипетки 5-7 раз. Затем образцы инкубировали при комнатной температуре (20-25 эээС) в течение 10 мин, для обеспечения образования комплекса. Пока имело место формирование комплекса, полную ростовую среду осторожно аспирировали из 6-луночного планшета. Высеянные клетки промывали 3 мл 1X стерильного забуференного фосфатом солевого раствора(PBS). Это сопровождалось добавлением 1,5 мл свежей полной ростовой среды. После 10 мин формирования комплекса добавляли 0,6 мл полной ростовой среды в реакционную пробирку, содержащую трансфекционные комплексы. Планшеты подвергали осторожному вращению для обеспечения надлежащего смешивания и инкубировали при 25 С, 5% СО 2. Использованную среду удаляли из лунок после 40 и 64 ч трансфекции и оценивали экспрессию TNFR-IgGFc с помощью ELISA, как описано ниже. Необходимое число лунок в 96-луночном титрационном микропланшете (Nunc Maxisorp) покрывали объемом в 100 мкл/лунка козьим антителом против фрагмента IgGFc человека (Calbiochem, каталожный номер 401439) в концентрации 2 мкг/мл в PBS (рН 7,3). Планшеты инкубировали при 4 С в течение ночи во влажных условиях для эффективного покрытия. Спустя 12-18 ч несвязанные покрывающие ан- 10011619 титела удаляли, лунки блокировали блокирующим буфером (1% BSA (бычий сывороточный альбумин),0,05% Твина 20, 5% обезжиренного молока, приготовленного в 1X PBS, рН 7,2) и инкубировали в течение 1 ч при 37 С. После 1 ч блокирования планшеты промывали трижды (250 мкл каждый раз) буферомTNFR-IgGFc, Enbrel (этанерцепт), использовали пробирку многократного использования, содержащую 25 мг/мл белка Enbrel (произведенного Immunex Corporation, США, поставляемого Amgen and WyethPharmaceuticals). Концентрации стандартов, используемых для получения стандартной кривой, составляли 1 нг/мл, 2 нг/мл, 5 нг/мл, 10 нг/мл, 20 нг/мл, 40 нг/мл, и 80 нг/мл. После инкубации в течение 90 мин,что дает возможность связывания антиген-антитело, планшеты промывали трижды (250 мкл каждый раз) буфером PBST и в лунки добавляли 100 мкл детекционного антитела, козьего антитела против IgGFcHRP человека (Pierce, каталожный 31416), разведенного 1:2000 в 1X PBS, содержащем 1% BSA, и инкубировали при 37 С в течение 1 ч. Планшеты промывали трижды для удаления несвязанного конъюгата антитело-HRP. Добавляли 100 мкл субстратного раствора на лунку, содержащего 8 мг порошка OPD [дигидрохлорид о-фенилендиамин(1,2-бензолдиамина)] (Sigma, каталожныйР-1526) и 10 мкл пероксида водорода, приготовленного в цитрат-фосфатном буфере (122,8 мг безводной лимонной кислоты и 188 мгNa2HPO4 в 20 мл дистиллированной воды и рН доведенного до 4,8-5,0) и инкубировали в течение 30 мин в темноте при 37 С. Реакцию останавливали добавлением 50 мкл 1N серной кислоты на лунку. Коэффициент поглощения измеряли на считывающем устройстве для ELISA при 490 нм. Оценку неизвестных образцов производили с помощью программного обеспечения SoftMax Pro (Molecular Devices), используя 4-х параметрическую эмпирическую калибровочную кривую, основанную на уравнении LevebergMarquardt. Результаты На фиг. 2 описаны показательные данные одного такого эксперимента. Самая высокая экспрессия была получена с pZRC-TNFR-IgGFc (72,54,3 нг/мл), которая была на 21,97% выше, чем для pcDNATPL-TNFR-IgGFc-VA-DHFR (59,441,8 нг/мл), на 36,66% выше, чем для pcDNA-TPL-Интрон-TNFRIgGFc (53,052,2 нг/мл) и на 395,55% выше, чем для pcDNA-TNFR-IgGFc (14,631,4 нг/мл). Синергический эффект пяти элементов дополнительно подтверждается тем фактом, что наибольшая плазмидаpZRC-TNFR-IgGFc, которая должна показать самую низкую трансфекционную эффективность, все еще давала наивысшую экспрессию. Способность новой комбинации элементов pZRC-TNFR-IgGFc давать высокую экспрессию, дополнительно, подтверждается другими примерами ниже. Пример 3. Конструирование экспрессионных векторов ЕРО Были разработаны два экспрессионных вектора ЕРО. Новый экспрессионный вектор pZRC-EPO подобен новому экспрессионному вектору pZRC-TNFR-IgGFc, предварительно отобранному благодаря его высокой экспрессии посредством экспериментов по транзиторной трансфекции, и кодирует ген ЕРО вместо TNFR-IgGFc. Второй вектор является кодирующим ЕРО коммерческим экспрессионным вектором pcDNA3.1,также несущим ген для DHFR.pcDNA-EPO-DHFR Для того чтобы получить кДНК эритропоэтина человека, общую ДНК, полученную из почки человека (Clontech), использовали в качестве матрицы для ОТ-ПЦР. Двухцепочечную кДНК синтезировали из общей РНК, используя обратную транскриптазу MMLV (MBI Fermentas, США) ген-специфическим праймингом [Maniatis et al., Molecular cloning; A Laboratory Manual (Cold Spring harbor Laboratory Press,Cold Spring Harbor, N.Y.), 1990]. Почечную кДНК затем подвергали 35 циклам ПЦР амплификации, используя 100 пикомолей генспецифических олигонуклеотидных праймеров в объеме 100 мкл, содержащих 50 мМ Трис-Cl (рН 8,3), 2,5 мМ MgCl2, 200 мкМ каждой dNTP и 2,5 единицы полимеразы Pfu. Каждый цикл ПЦР амплификации состоял из инкубации при 94 С в течение 30 с (денатурация), 61 С в течение 1 мин (отжиг) и 72 С в течение 1 мин (элонгация). Амплифицированный продукт ПЦР разлагали на 1% агарозном геле. Желаемый фрагмент приблизительно из 590 пар оснований вырезали из геля и очищали, используя набор для выделения из геля Qiagen. Очищенный фрагмент ДНК лигировали в MCS в рсДНК 3.1 после рестрикционного расщепления как вектора, так и очищенного продукта ПЦР с EcoRI иXbaI (MBI Fermentas, США), образуя, таким образом, липкие концы для направленного клонирования. Лигированный продукт трансформировали в Е. coli DH5 и трансформанты отбирали на основании устойчивости к ампициллину. Плазмидную ДНК, выделенную приблизительно из 20 таких колоний анализировали на наличие кДНК ЕРО рестрикционным расщеплением, используя различные ферменты рестрикции. Одну такую плазмиду секвенировали, используя ДНК секвенатор (ABI), и она была определена,как имеющая правильную интеграцию и последовательность кДНК ЕРО в векторе pcDNA 3.1. Данную плазмиду pcDNA-EPO использовали для вставки минигена DHFR, который выделяли из pDHFR 2.9 путем полного расщепления с Hind III. У минигена DHFR из 2,9 тысяч пар оснований создавали тупые концы, используя полимеразу Кленова (MBI Fermentas, США) и после линеаризации с Mlu I (MBI Fermentas,США) лигировали в pcDNA-EPO с созданием тупых концов, используя полимеразу Кленова (MBI Fer- 11011619mentas, США). Лигированный продукт трансформировали в Е. coli DH5 и трансформанты отбирали на основании устойчивости к ампициллину. Плазмидную ДНК pcDNA-EPO-DHFR (фиг. 3) анализировали на наличие и ориентировку минигена DH.FR в результирующей плазмиде рестрикционным расщеплением, используя различные ферменты рестрикции.pcDNA-TPL-Интрон-EPO-VA-DHFR (pZRC-EPO) Данный экспрессионный вектор получали путем вставки аденовирусного TPL ниже промотораCMV аденовирусного TPL элемента и выше химерного интрона, который был, дополнительно, вставлен выше кДНК ЕРО в конструкции pcDNA-EPO. VA РНК ген и миниген DHFR также были вставлены в данный вектор между геном устойчивости к ампициллину и промотором CMV. Это было достигнуто первичным расщеплением вектора pcDNA-TPL-Интрон-TNFR-IgGFc с EcoRI и XbaI, чтобы удалить фрагмент TNFR-IgGFc. Векторный фрагмент из 6 тыс. пар оснований, полученный таким образом, очищали, используя набор для выделения из геля Qiagen, и лигировали геном ЕРО, имеющим EcoRI и XbaI концы. Лигированный продукт трансформировали в Е. coli DH5 и трансформанты отбирали на основании устойчивости к ампициллину. Плазмидную ДНК анализировали на наличие и ориентировку TPL,химерного интрона и кДНК ЕРО в результирующей плазмиде рестрикционным расщеплением, используя различные ферменты рестрикции. Одну такую плазмиду секвенировали, используя автоматический ДНК секвенатор (ABI), и она была определена, как имеющая правильную интеграцию TPL, химерного интрона и гена ЕРО в векторе. Такую плазмидную ДНК, обозначенную как pInt-EPO-1, использовали для вставки VA РНК гена из 1000 пар оснований, выделенного из pCAV/NOT-TNFR двойным расщеплением с EcoRI и Not I, и минигена DHFR, выделенного из плазмиды pDHFR 2.9 полным расщеплением с BamHI и SspI. Два фрагмента лигировали pInt-EPO-1, предварительно расщепленным с MunI и Bgl II в трехкомпонентной лигазной реакции. Лигированный продукт трансформировали в Е. coli DH5 и трансформанты отбирали на основании устойчивости к ампициллину. Плазмидную ДНК анализировали на наличие и ориентировку VA РНК гена и минигена DHFR в результирующей плазмиде рестрикционным расщеплением, используя различные ферменты рестрикции. Дополнительно, интеграцию проверяли и подтверждали секвенированием ДНК. Такая векторная конструкция была обозначена как pcDNA-TPL-ИнтронЕРО-VA-DHFR, также обозначенная как pZRC-EPO (фиг. 3). Пример 4. Сравнение экспрессионных эффективностей различных экспрессионных векторов ЕРО транзиторной трансфекцией Для эксперимента по транзиторной трансфекции клетки Cos 1 выращивали и обрабатывали, как описано выше. Все трансфекционные смеси изготавливали так же, как в предыдущем примере. Процедуры трансфекции выполняли так же, как описано в примере выше, и использованную среду удаляли от клеток после 40 и 64 ч трансфекции и подвергали ELISA для анализа экспрессии ЕРО, как описано ниже. Сначала 96-луночные титрационные микропланшеты (Nunc Maxisorp) покрывали объемом в 50 мкл/лунка мышиными моноклональными антителами против рекомбинантного ЕРО человека (RD Systems Anti-hEPO Purified Mouse Mab, Clone 9C21D11, каталожный номер МАВ 287) в концентрации 2 мкг/мл в карбонатном буфере, рН 9,6. Планшеты инкубировали при 4 С в течение ночи во влажных условиях для эффективного покрытия. Спустя 12-18 ч несвязанные покрывающие антитела удаляли и лунки блокировали блокирующим буфером, как описано выше. После 1 ч блокирования планшеты промывали трижды (250 мкл каждый раз) буфером PBST. Затем по 50 мкл образцов, соответственно разведенных в 1X PBS, содержащем 1% BSA, добавляли в лунки в повторах. Для стандарта ЕРО использовалиEprex 4000 (рекомбинантный ЕРО человека 4000 МЕ/0,4 мл), произведенный Cilag AG Schaffhausen Швейцария. Концентрации стандартов, используемых для получения стандартной кривой, составляли 4 МЕ/мл, 2 МЕ/мл, 1 МЕ/мл, 0,4 МЕ/мл, 0,2 МЕ/мл и 0,1 МЕ/мл. После инкубации в течение 90 мин, что обеспечивало связывание антиген-антитело, планшеты промывали трижды (250 мкл каждый раз) буфером PBST. Это сопровождалось добавлением 50 мкл объема первичных антител (кроличьих антител против ЕРО человека, очищенных IgG, RD Systems, каталожный номер AB-286-NA) в концентрации 2 мкг/мл в 1X PBS, содержащем 1% BSA, с последующей дополнительной инкубацией в течение 1 ч при 37 С. После 1 ч инкубации планшеты промывали трижды (250 мкл каждый раз) буфером PBST и добавляли 50 мкл детекционного антитела/вторичного антитела (козьего антитела против кроличьего IgGHRP, Bangalore Genei, каталожный номер НРО 020), разведенного 1:8000 в 1X PBS, содержащем 1% BSA,рН 7,2, с последующей дополнительной инкубацией в течение 1 ч при 37 С. Планшеты промывали трижды (250 мкл каждый раз) буфером PBST для удаления несвязанного конъюгата. Это сопровождалось добавлением субстрата. Для этого добавляли 100 мкл субстратного раствора (приготовленного как описано выше) на лунку и инкубировали в течение 30 мин в темноте при 37 С. Реакцию останавливали добавлением 50 мкл 1N серной кислоты на лунку. Коэффициент поглощения измеряли на считывающем устройстве для ELISA при 490 нм. Оценку неизвестных образцов производили программным обеспечением SoftMax Pro (Molecular Devices), используя 4-х параметрическую эмпирическую калибровочную кривую, основанную на уравнении Leveberg-Marquardt. Результаты: На фиг. 4 описаны показательные данные одного такого эксперимента. Самая высокая экспрессия- 12011619 была получена с pZRC-EPO (276,533,09 нг/мл), которая была на 984,85% выше, чем для pcDNA-EPODHFR (25,492,38 нг/мл). Эти данные подтверждают сведения примера 3 о том, что новый экспрессионный вектор, содержащий новую комбинацию пяти элементов, дает очень высокую экспрессию для различных репортерных генов. Превосходная способность новой комбинации элементов в pZRC-EPO была подтверждена в стабильно трансфицированных клетках, как можно видеть в примере 7 ниже. Пример 5. Стабильная трансфекция СНО DHFR- клеток с pZRC-EPO СНО DHFR- клетки (мутанты клеток яичника китайского хомяка за счет гена, кодирующего дигидрофолатредуктазу) постоянно поддерживали в полной среде (МЕМ среда (Sigma), дополненная 10%PBS (Hyclone) и смесью гипоксантина и тимидина). Во время роста клетки для трансфекции в середине логарифмической стадии роста трипсинизировали из флакона Т 25 (25 см 2), один раз промывали в 5 мл полной среды и ресуспендировали в PBS. Пятнадцать микрограммов pZRC-Epo, линеаризированного с ферментом рестрикции SspI, добавляли к 1106 клеток и электропорировали (350 Вольт), используяStratagene Eectroporator 1000. После короткого восстановительного периода в 48 ч в полной среде, клетки подвергали двойной селекции путем содержания их в селективной среде (10% диализованного PBS, дополненного средой МЕМа, без добавления смеси гипоксантина и тимидина, плюс 500 мкг/мл G418(Sigma. Клетки помещали в 5% СО 2, 37 С инкубатор и предоставляли возможность расти в течение двухнедельного периода. Смену среды осуществляли каждые 3 дня. Участки стабильных трансфектантов начинали развиваться, спустя 12 дней после процесса селекции. На 15-й день селекции клетки трипсинизировали. Для выделения единичных клеточных клонов из смешанной популяции стабильных трансфектантов, клетки разводили, используя методику предельного разведения в 96-луночных планшетах. Лунки, содержащие одиночные клетки, обнаруживаемые под микроскопом, метили, и постоянная смена среды приводила к одиночным клеточным клонам. Спустя примерно 12-14 дней, лунки, как было обнаружено, являлись на 70-80% слившимися. Клетки переносили в 24-луночные планшеты и выращивали в течение 48-72 ч перед переносом в 6-луночные планшеты. Одиночные клеточные клоны высевали в 6 луночные планшеты с плотностью 0,1106 клеток на лунку в селективной среде. Использованную среду удаляли из указанных лунок спустя 48 ч и клетки подсчитывали. Удаленную среду анализировали на экспрессию ЕРО с помощью ELISA, как описано выше. Результаты выражали, как общее количество секретированного белка ЕРО/106 клеток/48 ч. Отбирали десять высокоэкспрессирующих клонов и подвергали процессу генной амплификации на основе метотрексата (МТХ), начиная с добавления 25 нМ МТХ к селективной среде. Свежую среду пополняли каждые 3 дня. Клетки выращивали в одной концентрации МТХ в течение приблизительно 20-25 дней до того, как они становились адаптированными. После каждой стадии увеличения концентрации МТХ, клетки анализировали на уровни экспрессии в 6-луночном планшете, как описано выше. Один из таких отобранных 10 клонов не выжил при 25 нМ МТХ. Все остальные девять клонов были подвергнуты постоянному увеличению концентраций МТХ 100 нМ, 400 нМ и, наконец, 2 мкМ МТХ. На фиг. 5 показана экспрессия гетерогенных популяций, полученных из МТХ-амплифицированных клональных популяций в конце различных стадий МТХ амплификации. Селекция с помощью с МТХ преобразует клональную популяцию клеток в гетерогенную популяцию. Следовательно, чтобы выделить высокоэкспрессирующие одиночные клеточные клоны из МТХамплифицированных гетерогенных популяций, сначала на стадии при 400 нМ МТХ отбирают одну гетерогенную популяцию 41 Н 9. Эти клетки были макетом для селекции клонов путем предельного разведения, как описано выше. Спустя приблизительно 14-16 дней, лунки 96-луночного планшета, как было обнаружено, являлись на 70-80% слившимися. Клетки переносили в 24-луночные планшеты и выращивали в течение 48-72 ч перед переносом в 6-луночные планшеты. Одиночные клеточные клоны высевали в 6 луночные планшеты с плотностью 0,1106 клеток на лунку в селективной среде, содержащей 400 нМ МТХ. Использованную среду удаляли из этих лунок спустя 48 ч и клетки подсчитывали. Удаленную среду брали для анализа экспрессии с помощью ELISA, как описано выше. Результаты выражали как общее количество секретированного белка ЕРО/106 клеток/48 ч. В табл. 1 показана экспрессия указанных одиночных клеточных клонов, которые были впервые получены из гетерогенной популяции 41 Н 9. Высокоэкспрессирующие клоны размножали и замораживали в качестве основных клеточных банков для коммерческого производства ЕРО. Пример 6. Продуцирование ЕРО из стабильно экспрессирующих клонов во флаконах Две гетерогенные популяции клеток из числа изображенных на фиг. 5, а именно, 56D12 и 41 Н 9, которые подвергали селекции при 100 нМ МТХ и 400 нМ МТХ, соответственно, испытывали на продукцию белка в масштабе флаконов Т 75. Флаконы Т 75 (75 см 2) инокулировали 2,1 миллионами клеток в среде МЕМ, не содержащей гипоксантин и тимидин, дополненной 10% диализованного PBS (JRH Biosciences) и 600 мкг/мл G418 наряду с подходящей концентрацией МТХ для соответствующей гетерогенной популяции. Флаконы помещали в СО 2 инкубаторы при 37 С, 5% СО 2 на 72 ч. Клеточный монослой,как было обнаружено, был слившимся на приблизительно 80-85% после 72 ч инкубации. В этот момент,старую использованную среду удаляли и клеточный монослой промывали Дулбекко-PBS. К промытым монослоям добавляли 15 мл продуктивной среды [IMDM:Ham F12 (1:1), дополненной 10 мг/л rh-инсулина, 2 мМ глутамина, аминокислотной смесью, 0,2 мМ N-ацетилцистина, 0,25% лактоальбуминна, 2 мг/л FeSO4, 25 мг/л декстрансульфата, 0,05% плюроновой кислоты, 5 мг/л гидрокортизона и 1 мМ Na-бутирата]. Клетки инкубировали в СО 2 инкубаторе при 33 С в течение четырехдневного периода. Спустя четыре дня использованную среду асептически удаляли и анализировали с помощьюELISA. В табл. 2 показана экспрессия двух клонов в продуктивной среде в масштабе флакона Т 75. Таблица 2 Пример 7. Высокий уровень продукции ЕРО в роллерных флаконах Одна из представленных гетерогенных популяций 41 Н 9, описанных выше, которая уже была адаптирована к 2 мкМ МТХ, использовали для исследования роллерного флакона, чтобы проанализировать белковую продукцию в больших масштабах. Клетки 41 Н 9 размножали, используя культуральные флаконы Т 75 и Т 175, для того, чтобы получить достаточное количество клеток для посева в роллерные флаконы. 5 роллерных флаконов инокулировали 30 миллионами клеток каждый в 2 мкМ среду МЕМ, дополненную МТХ, содержащую гипоксантин и тимидин (Sigma), дополненную 10% диализованного PBS(JRH Biosciences) и 600 мкг/мл G418. После перемешивания клеток в среде роллерные флаконы помещали в роллер-флаконный инкубатор при 37 С и скоростью вращения приблизительно 0,35 об./мин для роста. Клетки осматривали спустя 24 ч и были определены как хорошо растущие. Флаконы снова помещали в инкубатор для роста и осматривали под микроскопом каждые 24 ч. После 96 ч инокуляции клеточные монослои были слившимися приблизительно на 80-85%. В этот момент старую использованную среду удаляли и в каждый флакон добавляли 210 мл/флакон продуктивной среды [IMDM:Ham F12 (1:1),дополненной 10 мг/л rh-инсулина, 2 мМ глутамина, аминокислотной смесью, 0,2 мМ N-ацетилцистина,0,25% лактоальбумина, 2 мг/л FeSO4, 25 мг/л декстрансульфата, 0,05% плюроновой кислоты, 5 мг/л гидрокортизона и 1 мМ Na-бутирата]. Клетки инкубировали в роллер-флаконных инкубаторах в течение семидневного периода. По истечении указанного периода использованную среду асептически удаляли из роллерных флаконов, анализировали с помощью ELISA, как предварительно описано и отображено в табл. 3. Этот уровень продукции из гетерогенной, стабильно трансфицированной и МТХамплифицированной популяции, из которой индивидуальные клоны еще не были подвергнуты селекции,показывает экспрессионный уровень, который намного лучше, чем описанный в литературе для индивидуальных стабильных клонов. Например, стабильные клоны, полученные путем трансфекции и генной амплификации вплоть до 20 нМ МТХ, использующие вектор, содержащий промотор SR , AMV РНК 4-х лидерную последовательность, DHFR и резистентность к зеоцину могут дать клон 45 МЕ/мл (эквивалентный 0,346 мкг/мл) [Jang H P et al, Biotechnol. Appl. Biochem, 32:167-172, (2000)]. В патенте США 5955422 показано, что клоны, полученные из клеток СНО, DHFR-, котрансфицированных вектором, содержащим геномную копию гена ЕРО, под контролем промотора SV40 и поли А-последовательности, и вектором, содержащим DHFR, под МТХ генной амплификацией, давали продуцирование от 750 до 1470 Ед/миллион клеток/48 ч (или от 375 до 735 Ед/миллион клеток/24 ч) в бессывороточной продукционной среде в роллерных флаконах. В другом патенте США 5888774 описан клон ЕРО, полученный из клеток СНО-К 1, трансфицированных вектором, содержащим кДНК ЕРО,управляемым промотором EF1 с элементами ароВ SAR и устойчивостью к неомицину, который может продуцировать от 1500 до 1700 ME ЕРО/миллион клеток/24 ч (культура, проанализированная между днем 3 и 4). В отличие от этого, семидневный показательный образец среды гетерогенной популяции 41 Н 9 настоящего изобретения, предварительно адаптированной к 2 мкМ МТХ, как обнаружено, содержал 11830 ЕД/мл в роллерном флаконе, как оценено с помощью ELISA. Основанная на рассчитанных клеточных плотностях от 108 до 1,5108 клеток на роллерный флакон, скорость продукции ЕРО в 7 дней,210 мл культуры составляла от 2366 до 3549 МЕ/106 клеток/24 ч, что значительно выше, чем уровни,описанные выше для других описанных векторов, использующих различные другие комбинации регуляторных элементов. Аналогично, Sung Kwan Yoon и другие [Biotechnology and Bioengineering, 82 (3):289-298, (2003)] сообщают о полностью оптимизированном способе продуцирования ЕРО, использующего клеточную линию, выведенную на клетках СНО DHFR- способом генной амплификации вплоть до 5 мкМ МТХ селективного воздействия, приводящим к выходу приблизительно 50 мкг/мл после культивирования в течение приблизительно 200 ч при 33 С. pZRC-EPO настоящего изобретения стабильно трансфектная, гетерогенная популяция 41 Н 9, которая была адаптирована к сравнительно низкому уровню МТХ (2 мкМ),способна продуцировать 11,830 МЕ/мл (91 мкг/мл) при 168 часовом культивировании, как оценено с помощью ELISA. Уровни экспрессии ЕРО с новым экспрессионным вектором pZRC-EPO на 81-100% выше, чем для описанных лучших векторов в литературе. Способ продуцирования ЕРО, описанный выше для гетерогенной популяции 41 Н 9, также может быть применен к стабильным клонам, описанным в примере 5,табл. 1. Новый вектор настоящего изобретения был депонирован в IMTECH, Chandigarh, India, официально признанным депозитарием по условиям Будапештской конвенции. Номер доступа ожидается. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ высокой экспрессии представляющих интерес белков с использованием экспрессионного вектора, который включает, по меньшей мере, следующие регуляторные элементы:a) промотор CMV или его функциональные варианты,b) интрон,c) TPL или его функциональные варианты,d) VA гены или функциональные варианты, иe) последовательность полиаденилирования гормона роста крупного рогатого скота или функциональные варианты. 2. Способ по п.1, где представляющий интерес белок представляет собой рекомбинантный эритропоэтин. 3. Способ по п.1 для высокой экспрессии гибридного белка TNFR-IgGFc. 4. Способ по п.1 для высокой экспрессии ритуксимаба, трастузумаба, бивакузумаба или других моноклональных антител. 5. Способ по п.1 для высокой экспрессии генов, пригодных для экспрессии в клетках млекопитающих. 6. Экспрессионный вектор млекопитающих, определенный в п.1, который включает по меньшей мере следующие пять регуляторных элементов:a) промотор CMV или его функциональные варианты,b) интрон,c) TPL или его функциональные варианты,d) VA гены или функциональные варианты иe) последовательность полиаденилирования гормона роста крупного рогатого скота или функциональные варианты. 7. Вектор по пп.1-6, где интрон представляет собой химерный интрон c SEQ. ID. No.1. 8. Вектор по пп.1-6, где TPL представляет собой регуляторный элемент SEQ. ID. No.4. 9. Вектор по пп.1-6, где VA гены являются имеющими последовательность SEQ. ID. No.5. 10. Новый экспрессионный вектор млекопитающих, определенный в любом из предшествующих пп.1-9, который включает по меньшей мере следующие пять регуляторных элементов:a) промотор CMV или его функциональные варианты,b) химерный интрон, имеющий последовательность SEQ. ID. No.1, или его функциональные варианты,c) TPL, имеющий последовательность SEQ. ID. No.4, или его функциональные варианты,d) VA гены, имеющие последовательность SEQ. ID. No.5, или их функциональные варианты иe) последовательность полиаденилирования гормона роста крупного рогатого скота или функциональные варианты. 11. Вектор, определенный в любом из пп.1-10, дополнительно включающий маркер селективности и амплификации, выбранный из дигидрофолатредуктазы, аденозиндезаминазы, орнитиндекарбоксилазы,аспарагинсинтетазы, глутаминсинтетазы или их функциональные варианты. 12. Вектор, определенный в любом из пп.1-11, дополнительно кодирующий ген для эритропоэтина. 13. Вектор, определенный в любом из пп.1-11, дополнительно кодирующий ген для гибридного белка TNFR-IgGFc. 14. Вектор, определенный в любом из пп.1-11, дополнительно кодирующий ген для ритуксимаба,трастузумаба, бивакузумаба или других моноклональных антител. 15. Вектор, определенный в любом из пп.1-11, дополнительно кодирующий соответствующий ген(ы), способный экспрессироваться в клетках млекопитающих. 16. Клетка млекопитающих, трансформированная экспрессионным вектором, определенным в любом из пп.1-15. 17. Вектор, определенный в любом из пп.1-16, где клетки млекопитающих для трансфекции выбраны из Cos, CHO, CHO DHFR-, BHK1 и NS0. 18. Применение вектора, определенного в любом из предшествующих пунктов, для достижения высокой экспрессии представляющих интерес белков. 19. Применение вектора по п.18, где представляющий интерес белок выбран из рекомбинантного эритропоэтина, рекомбинантного гибридного белка TNFR-IgGFc и моноклональных антител, выбранных из ритуксимаба, трастузумаба, бивакузумаба. 20. Способ достижения высокой экспрессии представляющего интерес белка посредством кодирования соответствующего гена в векторе, определенном в любом из предшествующих пп.1-11.
МПК / Метки
МПК: C07K 14/715, C07K 14/505, C12N 15/79
Метки: высоких, способ, белков, вектор, продуцирования, экспрессионный, уровней
Код ссылки
<a href="https://eas.patents.su/21-11619-ekspressionnyjj-vektor-i-sposob-producirovaniya-vysokih-urovnejj-belkov.html" rel="bookmark" title="База патентов Евразийского Союза">Экспрессионный вектор и способ продуцирования высоких уровней белков</a>
Белок в1, способный модулировать внутриклеточные каскады воспаления, клеточной гибели или клеточного выживания, кодирующая его молекула днк, вектор, содержащий указанную молекулу днк, штамм клеток, содержащий данный вектор, способы их использования и фармацевтичекая композиция

Номер патента: 4658
Опубликовано: 24.06.2004
Авторы: Малинин Николай, Воллах Дэвид, Болдин Марк
МПК: A61K 38/17, A61P 43/00, C07K 14/47...
Метки: внутриклеточные, фармацевтичекая, днк, вектор, каскады, использования, клеточной, белок, данный, воспаления, клеточного, содержащий, способный, гибели, композиция, молекула, кодирующая, модулировать, способы, выживания, указанную, молекулу, штамм, клеток
Формула / Реферат:
1. Молекула ДНК, кодирующая белок B1 с аминокислотной последовательностью, приведенной на фиг. 3A, способный модулировать внутриклеточные каскады воспаления, клеточной гибели или клеточного выживания, прямо или косвенно ассоциируясь с другими внутриклеточными модуляторами или медиаторами указанных каскадов, а также фрагменты и варианты указанной ДНК, полученные на основе вырожденности генетического кода. 2. Молекула ДНК по п.1, имеющая...
Устройство и способ создания высоких предельных нагрузок на чувствительных висячих весах

Номер патента: 1117
Опубликовано: 30.10.2000
Автор: Грегор Манфред Александер
МПК: G01G 1/22
Метки: чувствительных, весах, предельных, способ, нагрузок, создания, высоких, устройство, висячих
Формула / Реферат:
1. Устройство для создания высоких предельных нагрузок на чувствительных висячих весах для создания и измерения медленных движений и малой энергии с использованием слабой силы взаимодействия масс, отличающееся тем, что оно содержит упругое сдвоенное волокно (3) двойного действия, работающее на растяжение и на изгиб, причем оба волокна поддерживают юстировочную пластину (5), на которую опирается рычаг (4) весов и нагрузка. 2. Устройство по п.1,...
Способ взрывания множества слоев или уровней горной породы

Номер патента: 8615
Опубликовано: 29.06.2007
Авторы: Брент Джеффри, Госвами Тапан
МПК: E21C 41/28, F42D 1/08, F42D 3/04...
Метки: уровней, множества, взрывания, способ, слоев, породы, горной
Формула / Реферат:
1. Способ взрывания множества слоев материала в поле взрыва при открытой добыче извлекаемого минерала, причем поле взрыва включает в себя первый массив материала, содержащий, по меньшей мере, первый слой материала, и второй массив материала, содержащий, по меньшей мере, второй слой материала над первым массивом материала, при этом поле взрыва имеет по меньшей мере одну свободную поверхность на уровне второго массива материала, способ включает...
Химерная изопреноидсинтаза, последовательность днк, экспрессирующий вектор, линия клеток и способ получения химерной изопреноидсинтазы.

Номер патента: 1622
Опубликовано: 25.06.2001
Авторы: Бэк Киоунгвхан, Чэппелл Джозеф
МПК: C07H 21/02, C12N 9/00, C07K 2/00...
Метки: экспрессирующий, получения, изопреноидсинтазы, химерная, линия, последовательность, способ, химерной, клеток, вектор, изопреноидсинтаза, днк
Формула / Реферат:
1. Химерная изопреноидсинтаза, содержащая домен первой изопреноидсинтазы, связанный с доменом второй гетерологичной изопреноидсинтазы, причем указанная химерная изопреноидсинтаза способна катализировать образование изопреноидного продукта реакции, который не образуется в отсутствие домена второй гетерологичной изопреноидсинтазы. 2. Химерная изопреноидсинтаза по п.1, отличающаяся тем, что способна катализировать образование, по крайней мере,...
Выделенная последовательность днк, вектор для экспрессии последовательности днк и способ получения вирусоподобных частиц

Номер патента: 969
Опубликовано: 28.08.2000
Авторы: Хофманн Катрин Дж., Янсен Катрин У., Джойс Джозеф Г., Ниппер Майкл П., Лехман Дэйл И., Джордж Хью Э.
МПК: C07K 16/08, A61K 39/12, A61P 31/20...
Метки: днк, вектор, последовательность, последовательности, выделенная, экспрессии, получения, вирусоподобных, способ, частиц
Формула / Реферат:
1. Выделенная последовательность ДНК, представляющая собой гибридный ген L1 HPV6/11, кодирующий капсидный белок L1 папилломавируса человека типа 11 и не содержащий внутренних сигналов терминации транскрипции, которые распознаются в клетках дрожжей, имеющая следующую структуру: или ее функциональное производное или аналог. 2. Вектор для экспрессии последовательности ДНК по п.1 формулы в клетках-хозяевах, сконструированный на основе...
Предыдущий патент: Применение римонабанта для получения лекарственных средств, пригодных для профилактики и лечения диабета типа 2
Следующий патент: Пищевая добавка
Случайный патент: Одностадийный способ получения 3,4,5-триметокситолуола